Embed Size (px)
Citation preview

JtMed Genet 1997;34:783-786
Interstitial duplication of the short arm ofchromosome 2: report of a new case and review
A Megarbane, N Souraty, M Prieur, D Theophile, P Chedid, J Auge, M Vekemans
AbstractAn 18 month old girl was referred to usbecause of dysmorphic features and psy-chomotor and growth retardation. Onphysical examination, she was found tohave microcephaly, open fontanelies, aprominent forehead, a flat occiput, hyper-telorism, sparse eyebrows, a small nosewith a depressed nasal bridge, a bulgingphiltrum, a thin upper lip, a high archedpalate, low set and posteriorly rotatedears, a small mandible, a short neck with alow hair line, and eye malformations.High resolution chromosome analysisidentified a de novo direct duplication ofthe 2p21.00-.p24.2 region. The phenotypeof de novo partial trisomy 2p is discussed.(JMed Genet 1997;34:783-786)
Keywords: dysmorphology; growth retardation; mentalretardation; partial duplication 2p
Unite de GenetiqueM6dicale, Laboratoirede BiologieMoleculaire etCytog6netique, Facult6de Medecine,UniversiteSaint-Joseph, Beirut,LebanonA MegarbaneN Souraty
Service deCytogenetique, H6pitalNecker-EnfantsMalades, Paris, FranceM PrieurD TheophileJ AugeM Vekemans
Service de P6diatrie,Hopital Rizk, Beirut,LebanonP Chedid
Correspondence to:Dr Megarbane, Unite deGenetique Medicale,Laboratoire deCytogenetique et BiologieMoleculaire, Faculte deMedecine, UniversiteSaint-Joseph, 42 rue deGrenelle, 75007 Paris,France.
Received 26 November 1996Revised version accepted forpublication 2 April 1997
Up to the present, approximately 30 patientswith an interstitial duplication of the short armof chromosome 2 (dup(2p)) have been re-
ported. In most cases, the duplication resultsfrom an unbalanced product of a parentaltranslocation involving the short arm ofchromosome 2 and another chromosome.However, three patients presented a terminalduplication associated with a deletion of theshort arm of an acrocentric chromosome.' 2Only five cases reported carried a de novointerstitial duplication of the short arm ofchromosome 2`-7 and one a pure interstitial tri-somy secondary to a malsegregation of amaternal balanced chromosomalrearrangement.'
Here, we report a new case of de novo inter-stitial duplication of the short arm of chromo-some 2.
Case reportThe proband, a newborn girl, is the fourthchild of healthy, non-consanguineous Leba-nese parents. At birth, the mother was 35 yearsold and the father 36. The father is a schoolteacher and the mother a housewife. The fam-ily history was unremarkable. Two previouspregnancies ended at 2 and 3 months of gesta-tion for unknown reasons.Medical follow up during pregnancy was not
performed. Delivery at term by cephalicpresentation was normal, as was the amnioticfluid. Birth weight was 2100 g, length 46 cm
(-3rd centile), and head circumference (OFC)36.3 cm (-3rd centile). The Apgar score was
B
7e,> _Vt.
p~~ ~~~~~~~~~~~~-W...
: -L Ir
Figure 1(A) Facial appearance at 18 months. (B) Profileview showing the prominentforehead, bulging philtrum,and low set, posteriorly rotated ears. (Photographsreproduced with permission.)
not available. The baby was immediately breastfed and discharged from hospital on the thirdday.At 18 months the child was referred to us
because of dysmorphic features and psycho-motor and growth retardation. Her weight was7800 g, length 70 cm (-3rd centile), and OFC42.2 cm (-3rd centile). She could stand andwalk with support. On physical examination,the baby was microcephalic, the fontanelleswere open, there was a large and prominentforehead, a wide glabella, a flat occiput, sparsehair, hypertelorism, a regular slant of thepalpebral fissures, sparse eyebrows, a smallnose with a depressed nasal bridge, a bulgingphiltrum, a thin upper lip, a high arched palate,and normal teeth. The ears were low set andposteriorly rotated with a prominent anthelix.A small mandible and a short neck with a lowhair line at the back were also noted (fig 1).Dermatoglyphics on the fingertips and palmswere unremarkable. Ophthalmological evalua-tion showed mild microcornea in both eyes,anisometropia, bilateral hypermetropic astig-matism, and left exotropia and amblyopia.There was no evidence of optic atrophy. Noother malformation was present except mildpectus excavatum and widely spaced nipples.
Radiological examination of the skeleton,abdominal ultrasound, echocardiography, anda cerebral CT scan were normal. Completeblood count, blood glucose, urine analysis,amino acid studies of plasma and urine as wellas liver and thyroid function studies were alsounremarkable.From birth to the time of examination, the
clinical course was uneventful except for a seri-ous episode of respiratory distress owing toinfection at the age of 6 months, which wastreated by antibiotics.
783
PIP..L
on Novem
ber 24, 2021 by guest. Protected by copyright.
http://jmg.bm
j.com/
J Med G
enet: first published as 10.1136/jmg.34.9.783 on 1 S
eptember 1997. D
ownloaded from
Migarbane et al
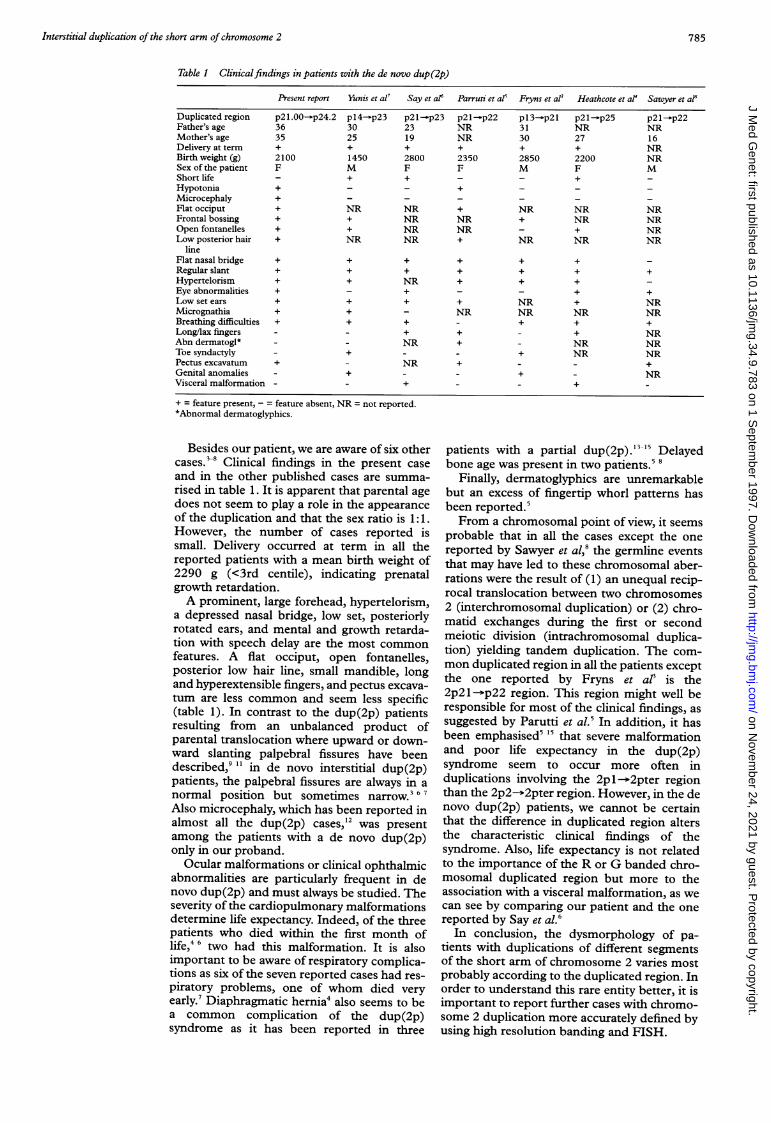
Figure 2 FISH using chromosome 2 painting on a metaphase of the proband. Note thatboth chromosomes 2 are completely coloured.
Figure 3 FISH of two cosmidchromosome and (B) cosmid c(chromosome.
CYTOGENETIC STUDIES
High resolution chromosome analysis was per-formed using G and R banding after lym-phocyte culture. An abnormal short arm ofchromosome 2 was found in all 30 metaphasesexamined. In order to confirm the cytogeneticfindings, in situ hybridisation using a chromo-
Figure 4 (A) R and G banded partial karyotype ofproband's chromosome 2 with (B) ideograms of the normaland duplicated 2p. Duplicated chromosome is on the right.The duplicated region is marked with arrows(46,XM,dup (2) (p21p24)).some 2 paint was performed and confirmed thechromosome 2 origin of the additional material
' (fig 2). In addition, three cosmids, cCI 2-111,cCI 2-10, and cCI 2-610, were used for FISHstudies. Cosmids cCI 2-111 and cCI 2-10,which map in the 2p2l and 2p2l-p22 regionrespectively, were duplicated. However, cosmidcCI 2-610, which maps in the 2p25.1 region,did not show any duplicated signal on theabnormal chromosome (fig 3A, B). Therefore,the karyotype of the patient was interpreted as46,XX,dup(2) (p2 1p24) (fig 4A, B).The chromosome abnormality occurred de
___j novo, as both parents had a normal karyotype.__ The possibility of non-paternity was ruled out_ by blood and HLA typing.
EBV transformed lymphocytes of the patient- . @are available on request.
DiscussionThe pertinent features and the rearrangementof the patient's chromosomes reported here arecompatible with the partial duplication of theshort arm of chromosome 2 syndrome firstdefined by Francke9 in 1978. However, as par-tial monosomy of the derivative chromosomealso influences the phenotype,' "°a detailedstudy of carriers of de novo interstitial duplica-tion of the short arm of chromosome 2 is very
I I2-610showsno duplicatedsignalon the abnormal helpful in delineating the clinical phenotypeassociated with this chromosome aberration.
784 on N
ovember 24, 2021 by guest. P
rotected by copyright.http://jm
g.bmj.com
/J M
ed Genet: first published as 10.1136/jm
g.34.9.783 on 1 Septem
ber 1997. Dow
nloaded from
Interstitial duplication of the short arm ofchromosome 2
Table 1 Clinicalfindings in patients with the de novo dup(2p)
Present report Yunis et al7 Say et al' Parruti et al5 Fryns et al3 Heathcote et aP Sawyer et at'
Duplicated region p2l.00-p24.2 pl4-'p23 p21-p23 p2l-p22 pl3- p21 p2l-p25 p21-p22Father's age 36 30 23 NR 31 NR NRMother's age 35 25 19 NR 30 27 16Delivery at term + + + + + + NRBirth weight (g) 2100 1450 2800 2350 2850 2200 NRSex of the patient F M F F M F MShortlife - + + - - +Hypotonia + - - +Microcephaly +Flat occiput + NR NR + NR NR NRFrontal bossing + + NR NR + NR NROpen fontanelles + + NR NR - + NRLow posterior hair + NR NR + NR NR NR
lineFlat nasal bridge + + + + + +Regular slant + + + + + + +Hypertelorism + + NR + + +Eye abnormalities + - + - - + +Low set ears + + + + NR + NRMicrognathia + + - NR NR NR NRBreathing difficulties + + + - + + +Long/lax fingers - - + + - + NRAbn dermatogl* - - NR + - NR NRToe syndactyly - + - - + NR NRPectus excavatum + - NR + - - +Genital anomalies - + - - + - NRVisceral malformation - - + - - +
+ = feature present, - = feature absent, NR = not reported.*Abnormal dermatoglyphics.
Besides our patient, we are aware of six othercases."8 Clinical findings in the present caseand in the other published cases are summa-rised in table 1. It is apparent that parental agedoes not seem to play a role in the appearanceof the duplication and that the sex ratio is 1:1.However, the number of cases reported issmall. Delivery occurred at term in all thereported patients with a mean birth weight of2290 g (<3rd centile), indicating prenatalgrowth retardation.A prominent, large forehead, hypertelorism,
a depressed nasal bridge, low set, posteriorlyrotated ears, and mental and growth retarda-tion with speech delay are the most commonfeatures. A flat occiput, open fontanelles,posterior low hair line, small mandible, longand hyperextensible fingers, and pectus excava-tum are less common and seem less specific(table 1). In contrast to the dup(2p) patientsresulting from an unbalanced product ofparental translocation where upward or down-ward slanting palpebral fissures have beendescribed,9 11 in de novo interstitial dup(2p)patients, the palpebral fissures are always in anormal position but sometimes narrow.3 67Also microcephaly, which has been reported inalmost all the dup(2p) cases,12 was presentamong the patients with a de novo dup(2p)only in our proband.
Ocular malformations or clinical ophthalmicabnormalities are particularly frequent in denovo dup(2p) and must always be studied. Theseverity of the cardiopulmonary malformationsdetermine life expectancy. Indeed, of the threepatients who died within the first month oflife,4 6 two had this malformation. It is alsoimportant to be aware of respiratory complica-tions as six of the seven reported cases had res-piratory problems, one of whom died veryearly.7 Diaphragmatic hernia4 also seems to bea common complication of the dup(2p)syndrome as it has been reported in three
patients with a partial dup(2p).-3 15 Delayedbone age was present in two patients.' 8
Finally, dermatoglyphics are unremarkablebut an excess of fingertip whorl patterns hasbeen reported.'From a chromosomal point of view, it seems
probable that in all the cases except the onereported by Sawyer et al,8 the germline eventsthat may have led to these chromosomal aber-rations were the result of (1) an unequal recip-rocal translocation between two chromosomes2 (interchromosomal duplication) or (2) chro-matid exchanges during the first or secondmeiotic division (intrachromosomal duplica-tion) yielding tandem duplication. The com-mon duplicated region in all the patients exceptthe one reported by Fryns et al is the2p21--p22 region. This region might well beresponsible for most of the clinical findings, assuggested by Parutti et al.5 In addition, it hasbeen emphasised' " that severe malformationand poor life expectancy in the dup(2p)syndrome seem to occur more often induplications involving the 2pl ->2pter regionthan the 2p2-*2pter region. However, in the denovo dup(2p) patients, we cannot be certainthat the difference in duplicated region altersthe characteristic clinical findings of thesyndrome. Also, life expectancy is not relatedto the importance of the R or G banded chro-mosomal duplicated region but more to theassociation with a visceral malformation, as wecan see by comparing our patient and the onereported by Say et al.6
In conclusion, the dysmorphology of pa-tients with duplications of different segmentsof the short arm of chromosome 2 varies mostprobably according to the duplicated region. Inorder to understand this rare entity better, it isimportant to report further cases with chromo-some 2 duplication more accurately defined byusing high resolution banding and FISH.
785
on Novem
ber 24, 2021 by guest. Protected by copyright.
http://jmg.bm
j.com/
J Med G
enet: first published as 10.1136/jmg.34.9.783 on 1 S
eptember 1997. D
ownloaded from

Megarbane et al
We wish to thank Alexandre Nieder and Maya Samaha for theirtechnical help, and Nancy Groot for kindly providing thecosmids. This work was supported by grants from the LebaneseNational Council for Scientific Research.
1 Fineman R, Buyse M, Morgan M. Variable phenotype asso-ciated with duplication of different regions of 2p.Am JrMedGenet 1983;15:451-6.
2 Stoll C, Messer J, Vors J. Translocation t(2;14) equilibr&echez une mere et trisomie partielle d'une partie du brascourt d'un chromosome no 2 chez deux de ses enfants. AnnGenet (Paris) 1974;17:193-6.
3 Fryns JP, Kleczkowska A, Kenbis H, Decock P, Van DenBerghe H. Partial duplication of the short arm of chromo-some 2 (dup(2)(p l3-p21) associated with mental retarda-tion and an Aarskog-like phenotype. Ann Genet (Paris)1989;3: 174-6.
4 Heathcote GJ, Sholdice J, Walton J, Willis RN, Sergovitch F.
Anterior segment mesenchymal dysgenesis associated withpartial duplication of the short arm of chromosome 2. CanOphthalmol 1991;26:35-43.
5 Parruti G, Di Ilio C, Calabrese G, et al. A new case of par-tial 2p trisomy due to de novo interstitial duplication 2p21-22. Ann Genet (Paris) 1989;32:55-8.
6 Say B, Carpenter NJ, Giacoia G, Jegathesan S. Agenesis ofthe lung associated with a chromosome abnormality. JfMedGenet 1980;17:477-8.
7 Yunis E, Gonzalez J, Zuniga R, Torres De Caballero OM,Mondragon A. Direct duplication 2pl4-2p23. Hum Genet1979;48:241-2.
8 Sawyer JR, Jones E, Hawks F, Quirk JG, Cuniff C. Duplica-tion and deletion of chromosome band 2(p21p22)resulting from a familial interstitial insertion (2;11)(p21;p 15). Am Med Genet 1994;49:422-7.
9 Francke U. Clinical syndromes associated with partialduplications of chromosomes 2 and 3: dup(2p), dup(2q),dup(3p), dup(3q). Birth Defects 1978;XIV(6C):191-217.
10 Larson L, Wasdahl W, Saumur J, et al. Familial reciprocaltranslocation, t(2;10)(p24;q26), resulting in duplication 2pand deletion 10q. Clin Genet 1982;21:187-95.
11 Francke U, Jones KL. The 2p partial trisomy syndrome:duplication of region 2p23-2pter in 2 members of a t(2;7)translocation kindred. Am Dis Child 1976;130: 1244-9.
12 Gorlin R, Cohen M, Levin LS. Syndromes of the head andneck. 3rd ed. Oxford: Oxford University Press, 1990:70.
13 Bender K, Reinwein H, Gorman L, Wolf U. Familiare 2/C-translokation: 46,XY,t(2p-;Cp+) und 46,XX,Cp+. Hu-mangenetik 1969;8:94-104.
14 Mayer U, Schwanitz G, Grosse K, Etzold R. Partial trisomy2p due to a familial translocation 2/6. Cytogenetics andclinical case with special reference to ophthalmologicchanges. Ann Genet (Paris) 1978;21:172-6.
15 Sarda P, Lefort G, Devaux P, Humeau C, Rieu D. Multiplecongenital anomalies due to partial 2p13-2pter duplica-tion resulting from an unbalanced X;2 translocation. AnnGenet (Paris) 1992;35:117-20.
786
on Novem
ber 24, 2021 by guest. Protected by copyright.
http://jmg.bm
j.com/
J Med G
enet: first published as 10.1136/jmg.34.9.783 on 1 S
eptember 1997. D
ownloaded from





























