Embed Size (px)
DESCRIPTION
Interview Questions for Pharmaceutical Industry Related Jobs
Citation preview

Interview Questions for Pharmaceutical industry related jobs (QA,QC,Production,RA,f&d) for B.pharma /M.pharma (Part 1)
1. "Tell me about yourself."
Don't tell them about your favorite hobbies, health issues, or how much you enjoy playing video games. This is your cue to provide a brief overview (no more than one or two minutes) of the aspects of your experience and background that relate to the position. Tell them about some accomplishments you felt really good about, and how you think they prepared you for the position you're interviewing for.
Example: "I have six years of advertising industry experience, and spent the past three years as the Assistant Production Manager at ABC Corp. overseeing production schedules, staff hiring, and deadlines. During that time, I streamlined the workflow so that we were able to meet the deadline for every monthly print project, and in many cases we went to print well before the actual deadlines. Our efficiency saved the company two weeks worth of staff overtime and expenses. Time management is one of my greatest skills, and I'm sure it that would easily transfer to the Production Manager position you're offering here."
2. "What do you think is your greatest weakness?"
Don't say anything that could eliminate you from consideration for the job. For instance, "I'm slow in adapting to change" is not a wise answer, since change is par for the course in most work environments. Avoid calling attention to any weakness that's one of the critical qualities the hiring manager is looking for. And don't try the old "I'm a workaholic," or "I'm a perfectionist."
The best way to answer this question is honestly--mention a real weakness that won't affect your ability to do the job, or address a skill that you are just learning and want to develop.
Example: "I'm not as strong as I'd like to be on social media, so I'm spending about three hours a week blogging on topics I'm interested in, and reading some perspectives on the business-to-business value of social media. I'm already learning some things I can bring here, and hope to find more ideas on how to use social media as a customer relationship tool."
3. "What did you like least about your last (or current) job?"
Don't vent or focus on the negative with brutally honest answers such as "My boss was a jerk," or "The company culture was too politically correct," or "They just weren't giving me the opportunity to take my career to the next level." Instead, keep the emphasis on the positive, even though there are sure to be things you weren't happy about.
Example: "That's a tough question to answer. I've had lots of opportunity at ABC Company and I work with some outstanding people. I guess if I had to pick one thing, it would be the occasional meeting that goes an hour longer than normal. I like to get stuff done and work with people and that extra hour could have let me to get back to a client more quickly."
4. "Where do you see yourself in 5 years?"
Believe it or not, this question is really disguised as: "Could I count on you to stay with this company long term?"
Since no one knows exactly where they'll be five years from now, the best way to answer this is with a reply that says you hope to be well established as someone who is helping that company succeed. You can also turn the question back to the interviewer, and ask where they see the company in five years. You might not know on a personal level where you'll be, but most companies have goals and plans that look ahead two to five years. Their answer might give you a good idea if it's a company worth sticking around that long for.
5. "Tell me about a time you failed."
Everyone has failed, so don't play dumb or claim you've never messed up. Think of a time when a work-related situation didn't turn out quite as you had hoped. An interviewer is interested in seeing how you took responsibility foryour failure, what you learned from it, and how you would prevent

similar failures from happening again.
Examples:"I once rushed a project to make a shipping deadline but inadvertently skipped a couple of critical steps. Fortunately we discovered the mistake before the customer installed the products, but they weren't pleased. I never made that mistake again."
"I thought my aggressive sales tactics were a great quality until I lost a client for being too pushy. I've since learned to tone things down and really listen to my clients and understand their needs before determining how to help them."
What is dead leg?A dead leg is defined as an area in a piping system where liquid can become stagnant and not be exchanged during flushing.
What is the recommended bio burden limits of purified
water & WFI?Purified water has a recommended bioburden limit of 100 CFU/mL, and water for injection (WFI) has a recommended bio burden limit of 10 CFU/100 mL.
What is significant changes in stability testing?
A 5% change in assay for initial value.
Any degradation products exceeds its acceptance criterion.
Failure to meet acceptance criterion for appearance,physical artributes and functionality test.
Failure to meet acceptance criteria for dissolution for 12 units.
If leak test fail during in process checks what needs to be
done ? Immediately stop packing process and check for
Sealing temperature
Verify for any possible changes like foil width,knurling etc.
Check & quarantine the isolated quantity of packed goods from last passed inprocess.
Collect random samples & do retest.
Blisters from the leak test passed containers shall allow to go further and rest must be deblistered/defoiled accordingly.
Which type of tablets are exempted from Disintegration
testing?

Chewable Tablets
What are the common variables in the manufacturing of
tablets?
Particle size of the drug substance
Bulk density of drug substance/excipients
Powder load in granulator
Amount & concentration of binder
Mixer speed & mixing timings
1. Granulation moisture contentMilling conditionsLubricant blending times
2. Tablet hardness
3. Coating solution spray rate
Whether Bracketing & Validation concept can be applied
in process validation?
Both Matrixing & Bracketing’s can be applied in validation studies.
Matrixing
Different strength of same productDifferent size of same equipment
Bracketting Evaluating extremesLargest and smallest fill volumesFastest and slowest operating speeds
What is the difference between calibration and
Validation?
Calibration is a demonstration that, a particular Instrument or device produces results with in specified limits by comparisons with those produced by a reference or traceable standard over an appropriate range of measurements.

Validation is a documented program that provides high degree of assurance that a specific process, method or system consistently produces a result meeting pre-determined acceptance criteria.
WHAT ARE GOOD MANUFACTURING PRACTICES (GMP)?
A. Good Manufacturing Practices are a set of regulations, codes, and guidelines for the manufacture of: Drug substances and drug products, Medical devices, In vivo and in vitro diagnostic products, FoodsThe term "cGMP" is used by the federal government as current good manufacturing practices. By definition, "cGMP" indicates that the current GMP - which is "state of the art" - can change. "GMP" and "cGMP" are often used interchangeably and essentially they have the same meaning.
Who Enforces Good Manufacturing Practices (GMP)?
Good Manufacturing Practices are enforced in the United States by the FDA (Food and Drug Administration)Good Manufacturing Practices are enforced in the United Kingdom by the Medicines and Healthcare Products Regulatory Agency (MHRA)Good Manufacturing Practices are enforced in Australia by the Therapeutical Goods Administration (TGA)Good Manufacturing Practices are enforced in India by the Ministry of Health, multinational and/or foreign enterprises and those individuals in the following positions:Each of the inspectorates carry out routine GMP inspections to ensure that drug products are produced safely and correctly.
Appearance Defects of Tablets During Compression Activ
ity ? Capping: is the term used, when the upper or lower segment of the tablet separates horizontally, either partially or completely from the main body of a tablet and comes off as a cap, during ejection from the tablet press, or during subsequent handling.
Laminating: is the separation of a tablet into two or more distinct horizontal layers. Sticking/filming: Sticking’ refers to the tablet material adhering to the die wall. Filming is a slow form of sticking and is largely due to excess moisture

in the granulation.
Cracking: Small fine cracks observed on the upper and lower center surface of the tablets, or very rarely on the side wall are referred to as cracks. Chipping: is defined as the breaking of tablet edges, while the tablet leaves the press or during subsequent handling and coating operation. Mottling: is the term used to describe an unequal distribution of colour on a tablet. Double Impression: involves only those punches,which have a monogram or other engraving on them.
What is the standard number of rotations used for
friability test?
100 rotations
What is the fall height of the tablets in the friabilator
during friability testing? 6 inches.Tablets falls from 6 inches height in each turn within the apparatus.
Which capsule is bigger in size - size '0' or size '1'?
'0' size
Water for pharmaceutical use shall be free heavy
metals why ? Heavy metals like lead and arsenic are highly cumulative neurotoxic metals, heavy metals are not eliminated out of our body easily like other drugs and molecules but heavy metals bind with proteins and tend to get accumulated in fatty tissues, nerve tissue is most likely to get damaged by heavy metals, heavy metal causes nervous tissue damage there for water must be free from heavy metals.

Change in the size or shape of the original container
requires any stability study? Change in the size or shape of the original container may not necessitate the initiation of new stability study.
Forced degradation (stress testing) Vs accelerated
stability testing Forced degradation and stress testing are not same. Stress testing is likely to be carried out on a single batch of the drug substance. The testing should include the effect of temperatures (in 10°C increments (e.g., 50°C, 60°C) above that for accelerated testing), humidity (e.g., 75 percent relative humidity or greater) where appropriate, oxidation, and photolysis on the drug substance. The testing should also evaluate the susceptibility of the drug substance to hydrolysis across a wide range of pH values when in solution or suspension. Photo stability testing should be an integral part of stress testing.
According to WHO guidelines what is the storage
condition of climatic zone IVa and zone IVb? Zone IV a: 30°C and 65% RH (hot and humid countries) Zone IV b: 30°C and 75% RH (hot and very humid countries.
What is the main objective of stress testing in stability
studies? Stress testing of the drug substance can help identify the likely degradation products, which can in turn help establish the degradation pathways and the intrinsic stability of the molecule and validate the stability indicating power of the analytical procedures used. The nature of the stress testing will depend on the individual drug substance and the type of drug product involved.
What is the formula for calculating number of air
changes in an area? Number of air changes/hour in an area is

= Total Room Airflow In CFM x 60/Total Volume of room in cubic feet For calculating Total Room Airflow in CFM, first calculate air flow of individual filter. Formula is given below. Air flow (in cfm) = Avg.air velocity in feet/Minute x Effective area of filter
Then find Total air flow. Formula is as follow- Total Air flow = Sum of air flow of individual filter. Air flow Velocity can be measured with the help of Anemometer.
How many Tablets shall be taken for checking friability?
For tablets with unit mass equal or less than 650 mg, take sample of whole tablets corresponding to 6.5g.For tablets with unit mass more than 650mg,take a sample of 10 whole tablets.
What is the formula for calculating weight loss during
friability test? %Weight loss = Initial Weight - Final Weight X 100/Initial Weight
What is the pass or fail criteria for friability test?
Generally the test is run for once.If any cracked,cleaved or broken tablets present in the tablet sample after tumbling,the tablets fails the test.If the results are doubtful,or weight loss is grater than the targeted value,the test should be repeated twice and the mean of the three tests determined.A mean weight loss from the three samples of not more than 1.0% is considered acceptable for most of the products.
What is the standard number of rotations used for
friability test? 100 rotations

What is the fall height of the tablets in the friabilator
during friability testing? 6 inches.Tablets falls from 6 inches eight in each turn within the apparatus.
Why do we check hardness during inprocess checks?
To determine need for the pressure adjustments on the tableting machine. Hardness can affect the disintegration time.If tablet is too hard, it may not disintegrate in the required period of time. And if tablet is too soft it will not withstand handling and subsequent processing such as coating,packing etc.
What are the factors which influence tablet hardness?
1.compression force 2.Binder quantity(More binder more hardness) 3.Moisture content
What is the recommended temperature for checking DT of a
dispersible tablet? 25 ±10C (IP) & 15 – 250C (BP)
What is mesh aperture of DT apparatus ?
1.8 -2.2mm (#10)
What is the pass/fail criteria for disintegration test?
If one or two tablets/capsules fails to disintegrate completely, repeat the test on another 12 additional dosage units. The requirement is meet if not fewer than 16 out of 18 tablets/capsules tested are disintegrated completely.
What is the recommended storage conditions for empty
hard gelatin capsules? 15 - 250C & 35 -55% RH

What is the recommended upward and downward
movement frequency of a basket-rack assembly in a DT apparatus? 28 – 32 cycles/minute
In a tablet manufacturing facility ‘positive’ pressure is
maintained in processing area or service corridors? In tablet manufacturing facilities, pressure gradients are maintained to avoid cross contamination of products through air. Usually processing areas are maintained under positive pressure with respect to service corridors.
What checks shall be carried out, while calibrating DT
apparatus? While calibrating DT apparatus, following checks shall be performed.1) Number of strokes per minute (Limit:29-32 cycles/min)2) Temperature by probe & standard thermometer (Limit: 37 ± 1 OC).3) Distance travelled by basket (Limit:53 -57mm)
What is the difference between disintegration and
dissolution? Disintegration is a disaggregation process, in which an oral dosage form falls apart in to smaller aggregates.(Disintegration time is the ‘break up’ time of a solid dosage form). Where as dissolution is a process by which solid substance enters in the solvent to yield a solution.It is controlled by the affinity between the solid substance and the solvent. That is why it is said that disintegration is a subset of dissolution.
Why do we calibrate a qualified equipment/instrument
on definite intervals?

An equipment or instrument can ‘drift’ out of accuracy between the time of qualification and actual use. So it is recommended to calibrate and recalibrate the measuring devices and instruments on predetermined time intervals, to gain confidence on the accuracy of the data.
Why do we consider three consecutive runs/batches for
process validation? Why not two or four? The number of batches produced in the validation exercise should be sufficient to allow the normal extent of variation and trends to be established and to provide sufficient data for evaluation and reproducibility.
First batch quality is accidental (co-incidental),
Second batch quality is regular (accidental),
Third batch quality is validation(conformation).In 2 batch we cannot assure the reproducibility of data,4 batches can be taken but the time and cost are involved.
Explain about revalidation criteria of AHU system?
AHU system shall be revalidated periodically as mentioned in the regulatory standards. AHU shall be revalidated in following cases also.
When basic design of AHU is changed,
When clean room volume is changed,
When new equipment is installed
When a construction is carried out, that calls for reconstruction of AHU system.
What needs to be checked during AHU validation?
During AHU validation, following tests shall be carried out
Filter efficiency test,
Air velocity & number of air changes,
Air flow pattern (visualization)
Differential pressure, temperature and RH
Static condition area qualification
Dynamic condition qualification
Non-viable count
Microbial monitoring
Area recovery and power failure study

What is the difference between calibration and
Validation? Calibration is a demonstration that, a particular Instrument or device produces results with in specified limits by comparisons with those produced by a reference or traceable standard over an appropriate range of measurements. Validation is a documented program that provides high degree of assurance that a specific process, method or system consistently produces a result meeting pre-determined acceptance criteria. In calibration performance of an instrument or device is comparing against a reference standard. But in validation such reference standard is not using. Calibration ensures that instrument or measuring devices producing accurate results. Whereas validation demonstrates that a process, equipment, method or system produces consistent results (in other words, it ensures that uniforms batches are produced).
10. Q Which capsule is bigger in size - size '0' or size '1'? A. '0' size
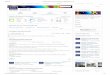
11. Define process flow of API manufacturing?

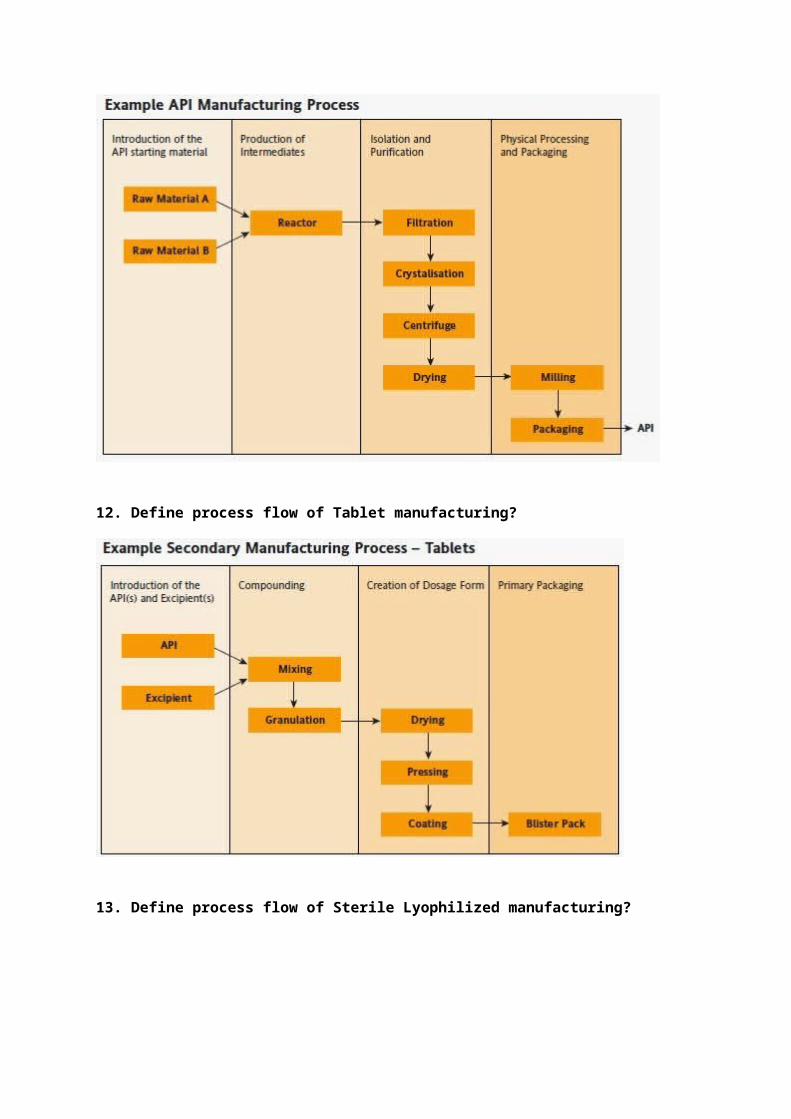
12. Define process flow of Tablet manufacturing?
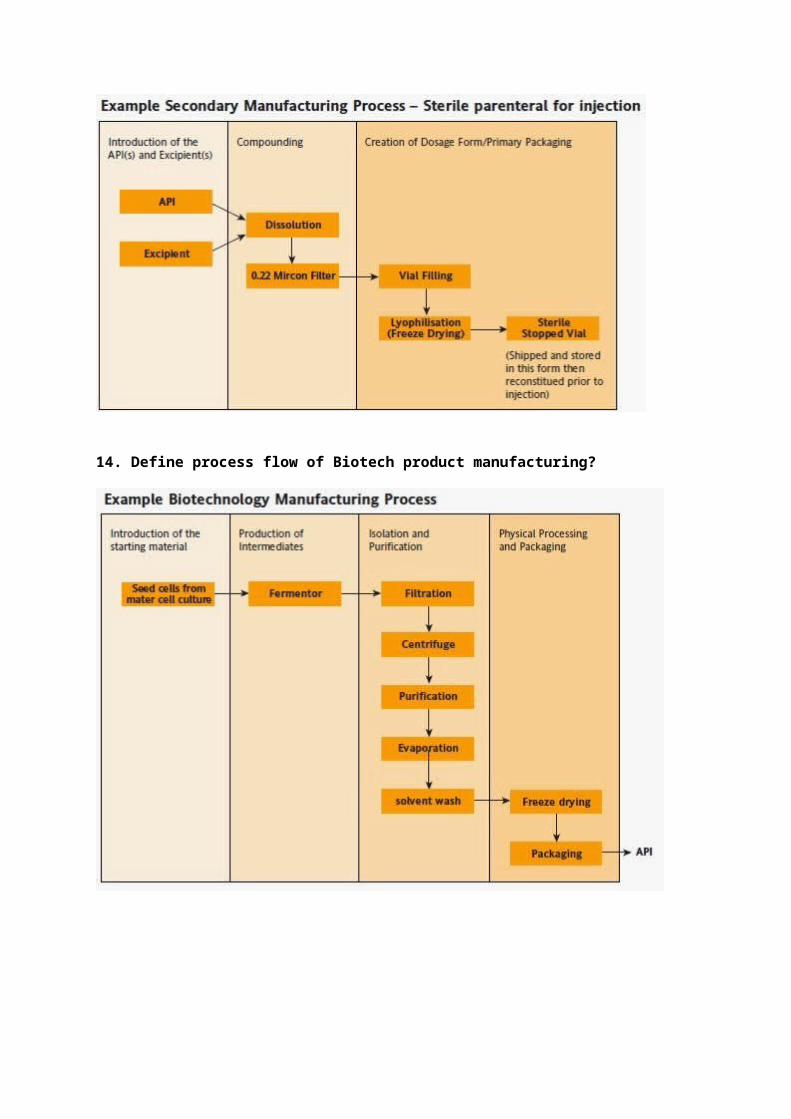
13. Define process flow of Sterile Lyophilized manufacturing?
14. Define process flow of Biotech product manufacturing?

Interview Question and Answer for Production Pharmacist
Q. What are the factors which influence tablet hardness?Ans: 1.compression force2.Binder quantity(More binder more hardness)
3.Moisture contentQ. Which type of tablets are exempted from Disintegration testing?
Ans: Chewable Tablets
Q. What is the recommended temperature for checking DT of a dispersible tablet?
Ans: 25 ±10C (IP) & 15 – 250C (BP)
Q. List out the appearance defects of tables during compression activity ?
1. Capping:- ‘Capping’ is the term used, when the upper or lower segment of the tablet separates horizontally, either partially or completely from the main body of a tablet and comes off as a cap, during ejection from the tablet press, or during subsequent handling.
2. Lamination / Laminating:- ‘Lamination’ is the separation of a tablet into two or more distinct
horizontal layers.
3. Sticking/filming: ‘ Sticking’ refers to the tablet material adhering to the die wall. Filming is a slow form of sticking and is largely due to excess moisture in the granulation.
4. Cracking:- Small fine cracks observed on the upper and lower center surface of the tablets, or very rarely on the side wall are referred to as cracks.
5. Chipping:- ‘ Chipping’ is defined as the breaking of tablet edges, while the tablet leaves the press or during subsequent handling and coating operation.
6. Mottling:‘ Mottling’ is the term used to describe an unequal distribution of colour on a tablet.
7. Double Impression: ‘ Double impression’ involves only those punches,which have a monogram or other engraving on them.
Q. What is the recommended storage conditions for empty hard gelatin capsules?
Ans: 15 - 250C & 35 -55% RH
Q. When performing the ‘uniformity of weight’ of the dosage unit, how many tablet/capsule can deviate the established limit?
Ans: Not more than two of the individual weights can deviates from the average weight by more than the percentage given in the pharmacopeia,and none can deviates more than twice that percentage.Weight Variation limits for Tablets
IP/BP Limit USP
80 mg or less 10% 130mg or less
More than 80mg or Less than 250mg 7.5% 130mg to 324mg
250mg or more 5% More than 324mg
Weight Variation limits for Capsules
Q. If sticking observed during tablet compression what may the probable reason for the same?
Ans: 1.If the granules are not dried properly sticking can occur.
2.Too little or improper lubrication.
3.Too much binder
4.Hygroscopic granularQ. What is the difference between calibration and validation?
Ans: In tablet manufacturing facilities, pressure gradients are maintained to avoid cross
BP Limit
Less than 300mg 10%
300mg or More 7.5%
contamination of products through air. Usually processing areas are maintained under positive pressure with respect to service corridors.
Q. What are the basic differences between Elixir and Suspention?
Ans: In an elixir, the active ingredients are mixed with a liquid, usually a kind of syrup or alcohol, in
which they can dissolve.
In a suspension, the medicine is mixed with a liquid, usually water, in which it cannot dissolve and
therefore remains intact in the form of small particles. The important thing to remember is that you
have to shake a suspension before giving each dose so that the medicine particles are evenly
distributed throughout the liquid.
Q. What do you mean by DQ, IQ, OQ, & PQ ?
Ans: Design Qualification (DQ): documented verification that the proposed design of the facilities,
equipment, or systems is suitable for the intended purpose.
Installation Qualification (IQ): documented verification that the equipment or systems are installed or
modified & comply with the approved design of the manufacturer’s recommendations and/or user
requirements.
Operational Qualification (OQ): documented verification that the equipment or systems are installed or
modified & perform as intended throughout the anticipated operating ranges.
Performance Qualification (PQ): documented verification that the equipment and ancillary systems are
connected & can perform effectively and reproducibly based on the approved process method and
specifications.
Q. Write the functions & use of HVAC system.
Ans: Heating, Ventilating and Air conditioning system is used for temperature and humidity control
within a manufacturing environment. It includes air handling units, air distribution network, air-cooling
and heating system, air filtration, equipment control system, monitoring and alarm decreases
Q. What is Clean Room & Aseptic Area?
Ans: Clean Room
A room in which the concentration of airborne particles is controlled to meet specified airborne
particulate cleanliness class. In addition, the concentration of microorganisms in the environment is

monitored; is cleanliness class defined is also assigned a microbial level for air, surface, and
personnel gear.
Aseptic Area
Any area in an aseptic process system for which airborne particulate and microorganism levels are
controlled to specific levels appropriate to the activities conducted within that environment.
Q. Which type of water are used in sterile preparation.
Ans: Water for Injection(Bulk)-
Characters-A clear, colorless, odorless and tasteless liquid.
o Nitrates-Not more than 0.2 ppm
o Heavy metals-Not more than 0.1 ppm
o Conductivity-Conductivity at 20°C not more than 1.1 µS.cm-1.
o Microbial Contamination-Total viable aerobic count should less than 10 micro-organism per 100 ml.
Absence of E.
o Coli, Salmonella, Staphylococcus aureus & Pseudomonas.
o Bacterial endotoxins-Less than 0.25 IU per ml.
Q. The addition of a 0.01 gm equivalent of sodium hydroxide to 0.25 L of a buffer solution
produced a PH of 0.50. What will be the buffer capacity?
Ans: (0.01/0.25)/0.50= 0.08(Eq/L)/ PH chnage
Q. Accurately weigh an amount of Lithum Citrate USP (Containing 2.5% moisture as stated in
certificate of analysis) to obtain 200 mEq of Lithium. (Note: 1 mEq Lithium is equivalent to
0.00694 gm of Lithium)
Ans:
W= Weight in gram of Lithium Citrate USP
A= 200 eEq of Li+ or 1.39 gm of Li+
B= 282 g/mole
D= 0.975
E= 3 x 6.94 g/mole or 20.8 g/mole
W= 1.39 g (282 g/Mole)/ 0.975 (20.8 g/mole) = 19.3 gm
Q. What are the packaging component of Aerosol?
Ans:
o Propellant

o Container
o Valve and actuator
o Product concentrate
Q. How many grams of dextrose required to prepare 4000 ml of 8% solution ?
Ans. g. solute = 4000 x = 200 g
S g. = 100 ml , X = 4000 ml
X = 4000 x S/100 = 200 g
Q. Peppermint spirit contains 10% (v/v) of peppermint oil. What volume of spirit contain 75 ml of active
ingredient ?
Ans. Volume in ml = 75/0.1 =750 ml
Q. What are the differences between Ointment & Cream?
Ans.Ointment Cream
An ointment is 80 percent oil and 20 percent water.
A cream is 50 percent oil and 50 percent water.
Ointment is an emulsion of water/Oil A cream is an emulsion of Oil/water
Thick Thin
Ointment has very moisturizing effect Cream has moderate in moisturizing
Preferable for Dry skin Preferable for moist skin
Q. What s Vortex?
Ans. In fluid dynamics, a vortex is a region within a fluid where the flow is mostly a spinning motion
about an imaginary axis, straight or curved.
Q. Write down the tablet manufacturing process.
Ans.WET GRANULATION DRY GRANULATION DIRECT
COMPRESSION
1.Millingand mixing of drugs and excipients
1.Millingand mixing of drugs and excipients
1. Milling and mixing of drugs and excipients
2.Preparationof binder solution
2.Compressioninto slugs or roll compaction
2.Compression of tablet

3.Wetmassing by addition of binder solutionor granulating solvent
3.Millingand screening of slugs and compacted powder
4.Screeningof wet mass
4.Mixingwith lubricant and disintegrant
5.Dryingof the wet granules
5.Compressionof tablet
6.Screeningof dry granules
7.Blendingwith lubricant and disintegrant to produce “running powder”
8.Compressionof tablet
Q. How many Methods of Sterilization involved in pharmaceutical plant?
Ans.
1.Physical Method
a.Thermal (Heat) methods
b.Radiation method
c.Filtration method
2.Chemical Method
a. Gaseous method
Q. What is BFS technology?
Ans. BFS means Blow Fill Seal. This technology usually used for sterilized product like ophthalmic
drops, where container preparation, material fill and sealing all are happen at a time.
Q. What are the main components of Effervescent tablets?
Ans. The effervescent tablet mainly consists of three components:
• Active ingredient;

• Acid source;
• Alkaline compound, constituted by a carbonate or bicarbonate
Q. What is Lyophilization?
Ans: Lyophilization, or freeze drying, there is a water is frozen, followed by its removal from the
sample, initially by sublimation (primary drying) and then by desorption (secondary drying). In this
process, the moisture content of the product is reduced to such a low level that does not support
biological growth or chemical reactions which gives the stability to the formulation. This technique
useful in formulation development of drugs which are thermolabile and/or unstable in aqueous
medium.
Q. Define primary and secondary packaging.
Ans: The primary packaging consist of those packaging components which have a direct contact with
the product (i.e. bottle, cap, cap liner, label etc).
The secondary packaging mainly provides the additional physical protection necessary to endure the
safe warehousing and for refill packaging.
Q. Define Strip package and Blister package.
Ans. Strip packages have at least one sealed pocket of material with each pocket containing a single
dose of the product. The package is made of two layers of film or laminate material. The nature and
level of protection which is required by the contained product will affect the composition of these
layers.
Blister packages are composed of a base layer, with cavities called blisters which contain the
pharmaceutical product, and a lid. This lid is sealed to the base layer by heat, pressure or both. They
are more rigid than strip packages and are not used for powders or semi-solids. In tropical areas
blister packages with an additional aluminium membrane is used which provide greater protection
against high humidity.
Q. PEG is …………. surfactant.
A. anionic
B. anionic & cationic depending on pH
C. non anionic
D. cationic

Q. The alcohol content in an elixir is not more from
A. 70%
B. 60%
C. 50%
D. 40%
Q. Water no. of a formulation can be increased by addition of:
A. Alcohol
B. Glycerine
C. Sufactant
D. Emulsion
Q. Alcoholic solution is milky with the compound:
A. Glutathemide
B. Paraldehyde
C. Chlorambucil
D. Chloral hydrate
Q. Rancidity of a fat is due to :
A. Oxidation
B. Hydrolysis
C. Saponification
D. Neutralisation
Quality Assurance Interview Questions1. What is an SOP ?
A Standard Operating Procedure (SOP) is a certain type of document that describes in a step-by-step outline form how to perform a particular task or operation. Everyone in a company must follow the same procedures to assure that tasks are performed consistently and correctly. Most companies have a wide variety of SOPs that describe how to do different tasks. In many companies technicians and operators are trained in how to follow individual SOPs and their training record specifies which SOPs they are trained on and are authorized to use.

2. What is 21 CFR part 11 ?
Title 21 CFR Part 11 of the Code of Federal Regulations deals with the Food and Drug Administration (FDA) guidelines on electronic records and electronic signatures in the United States. Part 11, as it is commonly called, defines the criteria under which electronic records and electronic signatures are considered to be trustworthy, reliable and equivalent to paper records
3. What are user requirements ?
User Requirements Specification describes what users require from the System. User requirementspecifications are written early in the validation process, typically before the system is created. It is written by the System Owner and End Users, with input from Quality Assurance. Requirements outlined in the URS are usually tested in the Performance Qualification. User Requirements Specifications are not intended to be a technical document; readers with only a general knowledge of the system should be able to understand the requirements outlined in the URS.
4. What is a validation plan ?
Validation Plans define the scope and goals of a validation project. Validation plans are written before a validation project and are specific to a single validation project. Validation Plans can include:
Deliverables (Documents) to be generated during the validation process Resources/Departments/Personnel to participate in the validation project Time-Line for completing the validation project
5. What is an IQ document ?
Installation Qualifications are a collection of test cases used to verify the proper installation of a System. The requirement to properly install the system was defined in the Design Specification. Installation Qualifications must be performed before completing Operational Qualification or Performance Qualification.
6. What is an OQ Document ?
Operational Qualifications are a collection of test cases used to verify the proper functioning of a System. The operational qualification tests requirements defined in the Functional Requirements. Operational Qualifications are usually performed before the system is released for use.
7. What is a PQ Document ?

Performance Qualifications are a collection of test cases used to verify that a System performs as expected under simulated real-world conditions. The performance qualification tests requirements that were defined in the User Requirement Specification (or possibly the Functional Requirements). Due to the nature of performance qualifications, these tests are sometime conducted with power users as the system is being released.
8. What is a Validation Summary Report ?
Validation Summary Reports provide an overview of the entire validation project. When regulatory auditors review validation projects, they typically begin by reviewing the summary report. The validation summary report should include:
A description of the validation project All test cases performed, including if those test cases passed without issue All deviations reported, including how those deviations were resolved
9. What is a Change Request ?
Change Control is a general term describing the process of managing how changes are introduced into a controlled System. In validation, this means how changes are made to the validated system. Change control is required to demonstrate to regulatory authorities that validated systems remain under control after system changes. Change Control systems are a favorite target of regulatory auditors because they vividly demonstrate an organization capacity to control its systems.
Q. Why water for pharmaceutical use is always kept in close loop in
continuous circulation ?
A. Water is a best medium for many microorganisms, microorganism can be a
highly pathogenic which causes serious diseases(many diseases are water
born), these pathogens infect after consumption of contaminated
water, microorganisms tend to settle on a surface if water is allowed to stand in a
stagnant position for few hours, these settled microorganism form a film over the
surface of vessel and piping, such film formed by microorganisms is also called
as biofilm, biofilms are very difficult of remove, once a biofilm is formed at a
particular point then that point may form a biofilm again even after cleaning very
easily as seed from this point is may not completely get removed effectively.
Biofilms then can become a source of microbial contaminations;
therefore purified water after collection in a distribution system is always kept in a
closed loop in a continuous circulation.
A continuous circulation is also not enough at some points, therefore it is aided
with high temperature range from 65 °C to 80°C, a minimum temperature of 65
°C is considered a self sanitizing, but better assurance is obtained with a
temperature of 80°C .
Purified water collected should be stored in a stainless still vessel which must
facilitate distribution to the point of use in a closed loop of continuous circulation,
tank should be made of corrosion free material of construction, and must facilitate
sanitization and easy cleaning.
Q. Water for pharmaceutical use shall be free cations,anions and other
impurities why ?
A.Water for pharmaceutical must be free from inorganic as well as organic
impurities, minerals, andheavy metals. Some impurities like calcium, magnesium,
ferrous are responsible for degradation of drug molecule, many cations like
ferrous and calcium magnesium act as catalysts in degradation reaction of drug
molecule, anions like chloride are highly active they participate in nucliophylic
substitution reactions, where in they break a double bond between -C=C- in to a
single bond as CL –CH-CH2- , which a reason why we observe that color dies
tend to fed in presence of chlorine as most of the dies used are diazo compounds
which has plenty of places for nucliophylic substitution reactions, which is also a
reason why stability of drug is drastically affected in presence of cations and
anions from mineral origin present in water.
Q. Water for pharmaceutical use shall be free heavy metals why ?
A. Heavy metals like lead and arsenic are highly cumulative neurotoxic
metals, heavy metals are not eliminated out of our body easily like other drugs
and molecules but heavy metals bind with proteins and tend to get accumulated
in fatty tissues, nerve tissue is most likely to get damaged byheavy metals, heavy
metal causes nervous tissue damage there for water must be free from heavy
metals.
Q. Brazil falls under which climatic zone ?
A. Zone IVB (30 degree celsius and 75% relative humidity)
Q. Change in the size or shape of the original container requires any
stability study?
A. Change in the size or shape of the original container may not necessitate the
initiation of new stability study.
Q. Forced degradation(stress testing) and accelerated stability testing are
same?
A. Forced degradation and stress testing are not same. Stress testing is likely to
be carried out on a single batch of the drug substance. The testing should
include the effect of temperatures (in 10°C increments (e.g., 50°C, 60°C) above
that for accelerated testing), humidity (e.g., 75 percent relative humidity or
greater) where appropriate, oxidation, and photolysis on the drug substance. The
testing should also evaluate the susceptibility of the drug substance to hydrolysis
across a wide range of pH values when in solution or suspension. Photo
stability testing should be an integral part of stress testing.
Q. According to WHO guidelines what is the storage condition of climatic
zone IVa and zone IVb?
A. Zone IV a: 30°C and 65% RH (hot and humid countries)
Zone IV b: 30°C and 75% RH (hot and very humid countries
Q. Countries comes under climatic zone IVb?

A.Brazil,Cuba,China,Brunei,Cambodia,Indonesia,Malaysia,Myanmar,Philippines,
Singapore,Thailand
Q. What is the purpose of stress testing in stability studies?
A. Stress testing of the drug substance can help identify the likely degradation
products, which can in turn help establish the degradation pathways and the
intrinsic stability of the molecule and validate the stability indicating power of the
analytical procedures used. The nature of the stress testing will depend on the
individual drug substance and the type of drug product involved.
Q. What is the formula for calculating number of air changes in an area?
A. Number of air changes/hour in an area is
= Total Room Airflow In CFM x 60
Total Volume of room in cubic feet
For calculating Total Room Airflow in CFM, first calculate air flow of individual
filter. Formula is given below.
Air flow (in cfm) = Avg.air velocity in feet/Minute x Effective area of filter
Then find Total air flow. Formula is
Total Air flow = Sum of air flow of individual filter.
Air flow Velocity can be measured with the help of Anemometer.
Q. What is dead leg?
A. A dead leg is defined as an area in a piping system where liquid can become
stagnant and not be exchanged during flushing.
Q. What is the recommended bio burden limits of purified water & WFI?
A. Purified water has a recommended bioburden limit of 100 CFU/mL, and water

for injection (WFI) has a recommended bio burden limit of 10 CFU/100 mL.
Q. Brief about ICH stabilty guidelines?
A. Q1A- Stability testing of new drug substance & products
Q1B- Photo stability testing of new drug substances & products
Q1C-Stability testing of new dosage forms
Q1D-Bracketing & Matrixing designs for testing of new drug substances and
products
Q1E-Evaluation of stability data
Q1F-Stability data package for registration applications in climatic zone III & IV
(Withdrawed)
Q. What is significant changes in stability testing?
A.
1. A 5% change in assay for initial value.
2. Any degradation products exceeds its acceptance
criterion.
3. Failure to meet acceptance criterion for
appearance,physical artributes and functionality
test.
4. Failure to meet acceptance criteria for dissolution
for 12 units.
Q. If leak test fail during in process checks what needs to be done ?
A.
Immediately stop packing process and check for
1.Sealing temperature
2.Verify for any possible changes like foil width,knurling etc.
3.Check & quarantine the isolated quantity of packed goods from last passed

inprocess.
4.Collect random samples & do retest.
5.Blisters from the leak test passed containers shall allow to go further and rest
must be deblistered/defoiled accordingly.
Q. How many Tablets shall be taken for checking friability?
A. For tablets with unit mass equal or less than 650 mg, take sample of whole
tablets corresponding to 6.5g.For tablets with unit mass more than 650mg,take a
sample of 10 whole tablets.
Q. What is the formula for calculating weight loss during friability test?
A. %Weight loss = Initial Weight - Final Weight X 100
Initial Weight
Q. What is the pass or fail criteria for friability test?
A. Generally the test is run for once.If any cracked,cleaved or broken tablets
present in the tablet sample after tumbling,the tablets fails the test.If the results
are doubtful,or weight loss is grater than the targeted value,the test should be
repeated twice and the mean of the three tests determined.A mean weight loss
from the three samples of not more than 1.0% is considered acceptable for most
of the products.
Q. What is the standard number of rotations used for friability test?
A. 100 rotations
Q. What is the fall height of the tablets in the friabilator during friability
testing?
A. 6 inches.Tablets falls from 6 inches eight in each turn within the apparatus.
Q. Why do we check hardness during inprocess checks?
A. To determine need for the pressure adjustments on the tableting machine.

Hardness can affect the disintegration time.If tablet is too hard, it may not
disintegrate in the required period of time. And if tablet is too soft it will not
withstand handling and subsequent processing such as coating,packing etc.
Q. What are the factors which influence tablet hardness?
A.
1.compression force
2.Binder quantity(More binder more hardness)
3.Moisture content
Q. Which type of tablets are exempted from Disintegration testing?
A. Chewable Tablets
Q. Which capsule is bigger in size - size '0' or size '1'?
A. '0' size
Q. What is the recommended temperature for checking DT of a dispersible
tablet?
A. 25 ±10C (IP) & 15 – 250C (BP)
Q. What is mesh aperture of DT apparatus ?
A. 1.8 -2.2mm (#10)
Q. What is the pass/fail criteria for disintegration test?
A. If one or two tablets/capsules fails to disintegrate completely, repeat the test
on another 12 additional dosage units. The requirement is meet if not fewer than
16 out of 18 tablets/capsules tested are disintegrated completely.
Q. What is the recommended storage conditions for empty hard gelatin
capsules?

A. 15 - 250C & 35 -55% RH
Q. Which method is employed for checking “Uniformity of dosage unit”?
A.
A.)Content uniformity
B.) Weight Variation
Weight variation is applicable for following dosage forms;Hard gelatin
capsules,uncoated or film coated tablets,containing 25mg or more of a drug
substance comprising 25% or more by weight of dosage unit.
Q. What is the recommended upward and downward movement frequency
of a basket-rack assembly in a DT apparatus?
A. 28 – 32 cycles per minute.
Q. When performing the ‘uniformity of weight’ of the dosage unit, how
many tablet/capsule can deviate the established limit?
A. Not more than two of the individual weights can deviates from the average
weight by more than the percentage given in the pharmacopeia,and none can
deviates more than twice that percentage.
Weight Variation limits for Tablets
IP/BP Limit USP80 mg or less 10% 130mg or lessMore than 80mg or Less than 250mg
7.5% 130mg to 324mg
250mg or more 5% More than 324mg
Weight Variation limits for Capsules
Q. What needs to be checked during inprocess QA checks?
A.
IP LimitLess than 300mg 10%300mg or More 7.5%

a.) Environmental Monitoring
b.) Measured values obtained from the process equipment (ex:temperature,RPM
etc.)
c.) Measured values obtained from persons (ex:timmings,entries etc.)
d.) Process attributes (Ex:weight,hardness,friability etc.)
Q. What precautions shall be taken while collecting inprocess samples ?
A. While collecting inprocess samples, avoid contamination of the product being
sampled (Don’t collect samples with bare hands) & avoid contamination of
sample taken.
Q. In a tablet manufacturing facility ‘positive’ pressure is maintained in
processing area or service corridors?
A. In tablet manufacturing facilities, pressure gradients are maintained to avoid
cross contamination of products through air. Usually processing areas are
maintained under positive pressure with respect to service corridors.
Q. If sticking observed during tablet compression what may the probable
reason for the same?
A.
1.If the granules are not dried properly sticking can
occur.
2.Too little or improper lubrication can also leads to
sticking.
3.Sticking can occur because of too much binder or
hygroscopic granular.
Q. What checks shall be carried out, while calibrating DT apparatus?
A. While calibrating DT apparatus, following checks shall be performed.
1.) Number of strokes per minute (Limit:29-32 cycles/min)
2.) Temperature by probe & standard thermometer

(Limit: 37 ± 1 OC).
3). Distance travelled by basket (Limit:53 -57mm)
Q. What is In process checks?
A. In process checks are checks performed during an activity,In order to monitor
and,if necessary,to adjust the process to ensure that product confirms to its
specification.
Q. What is the difference between disintegration and dissolution?
A. Disintegration is a disaggregation process, in which an oral dosage form falls
apart in to smaller aggregates.(Disintegration time is the ‘break up’ time of a solid
dosage form).
Where as dissolution is a process by which solid substance enters in the solvent
to yield a solution.It is controlled by the affinity between the solid substance and
the solvent.
In other word disintegration is a subset of dissolution.
Q. Why do we calibrate a qualified equipment/instrument on definite
intervals?
A. An equipment or instrument can ‘drift’ out of accuracy between the time of
qualification and actual use.So it is recommended to calibrate and recalibrate the
measuring devices and instruments on predetermined time intervals, to gain
confidence on the accuracy of the data.
Q. Why do we consider three consecutive runs/batches for process
validation? Why not two or four?
A. The number of batches produced in the validation exercise should be sufficient
to allow the normal extent of variation and trends to be established and to provide
sufficient data for evaluation and reproducibility.

· First batch quality is accidental (co-incidental),
· Second batch quality is regular (accidental),
· Third batch quality is validation(conformation).
In 2 batch we cannot assure the reproducibility of data,4 batches can be taken
but the time and cost are involved.
Q. Explain about revalidation criteria of AHU system?
A. AHU system shall be revalidated periodically as mentioned in the regulatory
standards. AHU shall be revalidated in following cases also.
· When basic design of AHU is changed,
· When clean room volume is changed,
· When new equipment is installed
· When a construction is carried out, that calls for reconstruction of AHU
system.
Q. What needs to be checked during AHU validation?
A. During AHU validation, following tests shall be carried out
· Filter efficiency test,
· Air velocity & number of air changes,
· Air flow pattern (visualization)
· Differential pressure, temperature and RH
· Static condition area qualification
· Dynamic condition qualification
· Non-viable count
· Microbial monitoring
· Area recovery and power failure study.
Q. Position of oblong tablets to be placed in hardness tester to determine
the hardness? Lengthwise / widthwise?
A. Position of oblong tablets should be length wise because the probability of
breakage is more in this position.

Q. Explain in detail about qualification of pharmaceutical water system?
A. Qualification of pharmaceutical water system involves three phases
· Phase -1
· Phase -2
· Phase -3
Phase -1
A test period of 2-4 weeks should be spent for monitoring the system
intensively. During this period the system should operate continuously without
failure or performance deviation.Water cannot be used for pharmaceutical
manufacturing in this phase.The following should be included in testing approach.
· Under take chemical & microbiological testing in accordance with a defined
plan.
· Sample incoming feed water daily to verify its quality.
· Sample each step of purification process daily.
· Sample each point of use daily.
· Develop appropriate operating ranges.
· Demonstrate production and delivery of product water of required quantity
and quality.
· Use and refine the SOP’s for operation,maintenance,sanitization and trouble
shooting.
· Verify provisional alert and action levels.
· Develop and refine test failure procedure.
Phase -2
A further test period of 2-4 weeks. Sampling scheme will be same as Phase –
1.Water can be used for manufacturing process in this phase.
Approach should also
· Demonstrate consistent operation within established ranges.
· Demonstrate consistent production & delivery of water of required quality
and quantity.

Phase - 3
Phase 3 runs for one year after satisfactory completion of phase-2.Water can be
used for manufacturing process during this process.
Objectives & Features of Phase -3
· Demonstrate extensive reliable performance.
· Ensure that seasonal variations are evaluated.
· The sample locations, sampling frequencies and test should be reduced to
the normal routine pattern based on established procedures proven during Phase
-1 & phase - 2.
Q. What are the recommended environmental monitoring limits for
microbial contamination?
Q. What is the difference between calibration and Validation?
A. Calibration is a demonstration that, a particular
Instrument or device produces results with in specified limits by comparisons with
those produced by a reference or traceable standard over an appropriate range
of measurements.
Where as Validation is a documented program that provides high degree of
assurance that a specific process, method or system consistently produces a
result meeting pre-determined acceptance criteria.
In calibration performance of an instrument or device is comparing against a
reference standard. But in validation such reference standard is not using.

Calibration ensures that instrument or measuring devices producing accurate
results. Whereas validation demonstrates that a process, equipment, method or
system produces consistent results (in other words, it ensures that uniforms
batches are produced).
Q. Briefly explain about ICH climatic zones for stability testing & long term
storage conditions?
A.ICH STABILITY ZONESZone Type of ClimateZone I Temperate zoneZone II Mediterranean/subtropical zoneZone III Hot dry zoneZone IVa Hot humid/tropical zoneZone IVb ASEAN testing conditions hot/higher humidity
Long term Storage conditionClimatic Zone Temperature Humidity Minimum
DurationZone I 21ºC ± 2ºC 45% rH ± 5% rH12 MonthsZone II 25ºC ± 2ºC 60% rH ± 5% rH12 MonthsZone III 30ºC ± 2ºC 35% rH ± 5% rH12 MonthsZone IV 30ºC ± 2ºC 65% rH ± 5% rH12 MonthsZone IVb 30ºC ± 2ºC 75% rH ± 5% rH12 MonthsRefrigerated 5ºC ± 3ºC No Humidity 12 MonthsFrozen -15ºC ± 5ºC No Humidity 12 Months
Q. What is bracketing & matrixing in stability testing?
A.Both Matrixing & Bracketing’s are reduced stability testing designs
Bracketing
The design of a stability schedule, such that only samples of extremes of certain
design factors (ex:strength,package size) are tested at all time points as in full
design.The designs assumes that the stability of any intermediate level is
represented by the stability of extremes tested.
Matrixing

The design of a stability schedule, such that a selected subset of possible
samples for all factor combinations is tested at a specified time point.At a
subsequent time point another subset of samples for all factor combination is
tested.The design assumes that the stability of each subset samples tested
represents the stability of all samples at a given time point.
There for a given time point other than initial & final ones not every batch on
stability needs to be tested.
Q.What are the common variables in the manufacturing of tablets?
A.
· Particle size of the drug substance
· Bulk density of drug substance/excipients
· Powder load in granulator
· Amount & concentration of binder
· Mixer speed & mixing timings
· Granulation moisture content
· Milling conditions
· Lubricant blending times
· Tablet hardness
· Coating solution spray rate
Q. Whether bracketing & validation concept can be applied in process
validation?
A.Both Matrixing & Bracketing’s can be applied in validation studies.
Matrixing
Different strength of same product
Different size of same equipment
Bracketting - Evaluating extremes
Largest and smallest fill volumes
Fastest and slowest operating speeds