Embed Size (px)
Citation preview

3
INTRODUCTION
Pharmaceutical chemistry – is the chemistry of drugs and the science which is
developing rapidly. Quality control is integral to all modern processes and the
pharmaceutical industry is no exception. Testing a pharmaceutical product involves
chemical, physical and sometimes microbiological analyses. Each year lots of new
biologically active compounds are synthesized and new drugs are appeared on the
pharmaceutical market, and methods of synthesis and analysis of the traditional
medicines (so called "generics") are improved.
That is why the authors did not want to cover absolutely all the methods of
preparation or analysis of drugs, and made their generalizations to provide concrete
material for example, giving the most typical representatives of certain groups of
drugs that will allow students to study program material creatively.
This textbook has been designed primarily for students of pharmaceutical
universities and departments of pharmacy in higher educational establishments.
During writing this textbook it was assumed that students of III-V years
already have sufficient knowledge of chemistry, to avoid excessive increase in the
volume of the book, the authors refused to repeat certain reactions.
In the section "Identification" and "Limit tests," which refers determination of
certain ions (when the opposite ion is unknown or not named) interact with a specific
method given in substance (substances), we consider that it is appropriate to give the
reactions in ion-molecular form. In other sections reactions are given mainly in
molecular form. Some complex reactions of organic substances are schematic and not
balanced.
The textbook consists of two parts. In the first part analysis of medicinal
substances of inorganic, aliphatic and aromatic structure is given. The second part
involves main methods of analyses for medicinal substances of heterocyclic structure
and natural origin. Main methods for identification and assay are given according to
European Pharmacopoeia and State Pharmacopoeia of Ukraine, but at the same time
the most widely used and specific reactions not included into Pharmacopoeias are
also given.
In the first and second parts of the textbook features of the pharmaceutical
analysis of drugs in groups according to the composition and chemical structure are
discussed: inorganic - by groups of periodic table, organic - in accordance with the
presence of functional groups, the natural biologically active compounds - by the
chemical structure and biological effects (alkaloids, glycosides, vitamins, hormones,
antibiotics, etc.).
Each group of drugs is described in a separate section by the following plan:
general characteristic, physical and chemical properties, ways or schemes of
preparation, reactions and methods of identification, tests for purity, and the most
widely used methods for quantitative determination, special storage conditions,
pharmacological action and usage in medical practice. The most attention is paid to
the modern medicines and methods for their research.
The authors are grateful for critical comments and suggestions, which will be
considered in future work on the textbook of pharmaceutical chemistry.

4
Chapter 1. SUBJECT OF PHARMACEUTICAL CHEMISTRY
IDENTIFICATION AND TESTS FOR PURITY
Plan
1. Subject of pharmaceutical chemistry.
2. Structure of Pharmacopoeia and of a monograph.
3. General reactions for identification of inorganic preparations.
4. Tests for purity:
general questions on impurity determination;
clarity and degree of opalescence of liquids;
degree of coloration of liquids;
determination of pH;
loss on drying and determination of water;
limit tests.
Subject of pharmaceutical chemistry
Pharmaceutical chemistry studies chemical structure, preparation, physical
and chemical properties of medicinal substances, their identification, assay and
determination of impurities, storage of the preparations and their use in medicine,
methods of standardization of medicines, structure-activity relationships for the
preparations.
Pharmaceutical chemistry is one of the most important disciplines for the
pharmacists. It gives very important knowledge about chemical properties of
medicinal preparations. These properties have great influence on the pharmacological
activity because the behaviour of preparations depend on their solubility in different
solvents, possibility to interact with biological systems and other preparations, to
participate in biochemical processes, to form different metabolites etc.
Pharmaceutical chemistry is based on the other chemical sciences such as inorganic,
organic, analytical, physical, colloidal chemistry, and pharmacology.
Pharmaceutical analysis is one of the main parts of pharmaceutical chemistry.
Its objects are medicinal substances and medicinal preparations produced on
pharmaceutical plants or in the chemist’s.
Substance is the standardized biologically active compound or standardized
mixture of such compounds used for the medicines’ preparation.
The main document standardizing the quality of the medicinal substances and
medicinal preparations is Pharmacopoeia. European pharmacopoeia is legislative for
the many countries of Europe. State Pharmacopoeia of Ukraine is legislative in
Ukraine. Pharmacopoeia is a legal act, which contains general requirements for the
preparations, monographs, and methods of medicines standardization.
Pharmacopoeia’s requirements are obligatory for all the enterprises of the country,
which produce, analyze, store, and realise the medicaments.

5
Structure of Pharmacopoeia and of a monograph
Pharmacopoeia has two large parts: general methods and monographs for the
substances (in European Pharmacopoeia there are no monographs for the dosage
forms because their quantity nowadays is great).
All monographs have the same structure.
At the beginning of each monograph, there are international and chemical
names of the substance. Then graphic and molecular formulas of the substance and its
molecular weight are given.
Definition contains the limits of content of a substance. An upper limit
exceeding 100 % may be stated, e.g. 101 %. It means that the result of the assay is
not more than 101 %, calculated in terms of the equivalent content of compound.
Characters give the appearance of the substance and its solubility.
Identification is the important part of the monograph. The tests given in this
part of the monograph do not establish absolute proof of identity, but they provide a
means of verifying that the identity of the material being examined is in accordance
with the label on the container. Depending on the chemical nature of the substance,
the combination of different physico-chemical methods with chemical reactions is
used for the identification.
Certain monographs have subdivisions “First identification” and “Second
identification”. The tests of “Second identification” can be used instead of the test or
tests of the “First identification” provided it could be demonstrated that the substance
or preparation is fully traceable to a batch certified to comply with all the
requirements of the monograph.
Each substance can contain different foreign compounds, called impurities.
That is why another very important part of a monograph is tests: appearance of the
solution (clarity and degree of opalescence of liquids and degree of coloration of
liquids), acidity or alkalinity, tests of permitted limits of certain impurities, etc. The
concentration of certain impurities is given either as a percentage or in parts per
million by weight (ppm). Quantity of such impurities should not exceed the stated
limit. There are also impurities, which could not be discovered at all by the methods
described (limits for them are not stated).
The assay gives the method for the quantitative analysis of a substance in order
to know the limits of its content.
Storage expresses conditions of storage of a substance.
At the end of a monograph, there are labelling and action and use.
General reactions for identification of inorganic preparations
Among other methods of analyses given in European Pharmacopoeia, there are
general reactions of ions (anions and cations) and functional groups, given into the
general article “Identification reactions of ions and functional groups”.
In this part, we will discuss identification reactions for inorganic ions only.

6
ALUMINIUM
No precipitate is formed after adding of thioacetamide reagent to the
aluminium salt solution in the presence of dilute hydrochloric acid (absence of heavy
metals salts). After adding of sodium hydroxide solution a gelatinous white
precipitate is formed which dissolves on further addition of the reagent:
Al3+
+ 3NaOH Al(OH)3+ 3Na+
Al(OH)3 + 3NaOH Na3[Al(OH)6]
The gelatinous white precipitate is reformed after adding of ammonium
chloride solution.
Na3[Al(OH)6] + 3NH4Cl Al(OH)3 +3NH4OH +3NaCl
AMMONIUM SALTS
For the emission of ammonium from its salts, magnesium oxide is added to the
prescribed solution of the substance. Then a current of air is passed through the
mixture and the escaping gas is directed on the surface of a mixture of 0,1M
hydrochloric acid and methyl red solution. The colour of the indicator changing to
yellow shows the presence of ammonium salts.
NH3 + HCl NH4Cl
The solution obtained forms yellow precipitate in reaction with sodium
cobaltinitrite solution:
2NH4Cl + Na3[Co(NO2)6] (NH4)2Na[Co(NO2)6] + 2NaCl
AMMONIUM SALTS AND SALTS OF VOLATILE BASES
After heating of the preparation with sodium hydroxide solution, the reaction
mixture gives off vapour that can be identified by its odour and its alkaline reaction. to
NH4+ + OH
- NH3 + H2O
ARSENIC
a) After heating of the prescribed solution on a water-bath with an equal
volume of hypophosphorous reagent a brown precipitate is formed.
NaH2PO
2 + HCl → H3PO2 + NaCl
As2O3 + 3H3PO2 → 2As + 3H3PO3
As2O5 + 5 H3PO2 → 2As + 5 H3PO3
The State Pharmacopoeia of Ukraine additionally uses the following two
identification reactions of arsenic compounds:
b) Identification of arsenic (III) (arsenites):
The solution of the substance gives yellow precipitate after adding of dilute
hydrochloric acid and sodium sulphide solution. The formed precipitate dissolves on
addition of ammonia.
As2O3 +6HCl 2AsCl3 + 3H2O
2AsCl3 + 3Na2S As2S3 + 6NaCl
As2S3 + 6NH4OH (NH4)3AsO3 + (NH4)3AsS3 + 3H2O
c) Identification of arsenic (V) (arsenates):

7
The prescribed solution reacts with ammonium chloride, ammonia and
magnesium sulphate solution forming a white, crystalline precipitate, which dissolves
on addition of dilute hydrochloric acid. (This reaction is used for distinction arsenates
from arsenites).
Na2HAsO4 + MgSO4 + NH4OH ClNH4 NH4MgAsO4 + Na2SO4+ H2O
BISMUTH
a) Bismuth preparations react with sodium sulphide solution giving brown
precipitate. Reaction is carried out after dissolving of preparation in hydrochloric
acid and heating to boiling and cooling the mixture (filtering if necessary). Then after
adding of water, a white or slightly yellow precipitate is formed (due to the formation
of the oxychloride BiOCl). After adding of reagent, it turns brown.
2Bi3+
+ 3Na2S Bi2S3 + 6Na+
b) For this reaction the solution is boiled with dilute nitric acid. The solution
obtained reacts with thiourea solution forming a yellowish-orange colour or an
orange precipitate. After adding of sodium fluoride solution, the obtained solution is
not decolourised within 30 min.
Bi3 3(NH2)2CS [Bi((NH2)2CS)3]
3
BROMIDES
a) Silver nitrate solution in the presence of dilute nitric acid gives a curdled,
pale yellow precipitate, which dissolves with difficulty in ammonia.
Br- + AgNO3 AgBr + NO3
-
AgBr + 2NH4OH [Ag(NH3)2]Br + 2H2O
b) In this reaction free bromine is formed after interaction with lead dioxide in
the presence of acetic acid. Free bromine brominates fuchsin (this reaction is carried
out on the filter paper strip impregnated by decolorized fuchsin solution). Due to the
forming of bromosubstituted fuchsin a violet colour appears on the filter paper.
c) The State Pharmacopoeia of Ukraine additionally uses the following
reaction. Bromides are oxidized by chloramine solution in the presence of dilute
hydrochloric acid and chloroform with the forming of free bromine. Due to the
extraction of bromine chloroform layer is coloured yellowish-brown.
CH
3
SO2N
Cl
Na
CH3
SO2NH
2
+ 2HCl + Cl2 + NaCl
2Br - + Cl2 Br2 + 2 Cl -

8
CALCIUM
a) Reaction with glyoxal hydroxyanile alcohol solution. This reaction is carried
out in basic conditions (presence of dilute sodium hydroxide solution and sodium
carbonate solution). After shaking of the reaction mixture with chloroform and
adding water the chloroform layer is coloured red.
Ca2+
H+
N
OH
N
OH
CH CH N
O
N
O
CH CH
Ca
+ +2
NaOH
Na2CO3
CHCl3
b) Substance is dissolved in acetic acid. After adding of potassium
ferrocyanide solution the solution remains clear (absence of iron salts). On addition
of ammonium chloride a white, crystalline precipitate is formed:
Ca2+
+ 2NH4+ + K4[Fe(CN)6] (NH4)2Ca[Fe(CN)6] + 4K
+
The next two reactions of calcium ions are additionally used by the State
Pharmacopoeia of Ukraine, reaction c) is used also in EP for the test for calcium.
c) After adding of ammonium oxalate solution to the prescribed solution a
white precipitate of calcium oxalate is formed. It does not dissolve in dilute acetic
acid and ammonia and dissolves in dilute mineral acids.
Ca2+
+ (NH4)2C2O4 CaC2O4 + 2NH4+
d) Calcium salt, treated with hydrochloric acid and put into the colourless
flame, makes it orange-red.
CARBONATES AND BICARBONATES a) After adding of dilute acetic acid to the solution (or the suspension) of the
substance colourless and odourless gas (carbon dioxide) is formed by both carbonates
and hydrocarbonates.
CO32-
+ 2CH3COOH CO2 + H2O + 2CH3COO-
HCO3- + CH3COOH CO2 + H2O + CH3COO
-
This gas collected in barium hydroxide solution gives a white precipitate that
dissolves on addition of an excess of hydrochloric acid.
CO2 + Ba(OH)2 BaCO3 + H2O
BaCO3 + 2HCl BaCl2 + CO2 + H2O
There are two reactions, used by the State Pharmacopoeia of Ukraine, which
help to distinguish hydrocarbonates from carbonates:
b) In reaction with magnesium sulphate solutions of carbonates give a white
precipitate. Bicarbonates give a precipitate only after boiling:
4Na2CO3 + 4MgSO4 + 4H2O 3MgCO3∙Mg(OH)2∙3H2O↓ + 4Na2SO4 + CO2↑
2HCO3- + MgSO4 → Mg(HCO3) + SO4
2-
tº
Mg(HCO3) → MgCO3↓ + CO2↑ + H2O
c) Carbonates solutions become violet-red after adding of phenolphthalein.
Bicarbonates solutions stay colourless.

9
CHLORIDES
a) Chlorides give curdled, white precipitate with silver nitrate solution in the
presence of dilute nitric acid. Precipitate dissolves easily in ammonia.
Cl- + AgNO3 AgCl + NO3
-
AgCl + 2NH4OH [Ag(NH3)2]Cl + 2H2O
b) After chlorides interaction with potassium dichromate and sulphuric acid the
chromyl chloride is formed. This gas colours a filter-paper strip impregnated with
diphenylcarbazide solution into violet-red.
4Cl- + K2Cr2O7 + 3H2SO4 2CrO2Cl2 + K2SO4 + 3H2O + 2SO4
2-
O=CNH-NH-C6H5
NH-NH-C6H5
O=CNH-NH-C6H5
N=N-C6H5
CrO2Cl2
N=N-C6H5
C OH
N-NH-C6H5Ñr
2+ +
N=N-C6H5
C OH
N-NH-C6H5
Cr2 + 2
N=N-C6H5
C O
N-NH-C6H5
Cr
C6H5-N=N
CO
C6H5-HN-N
+ 2H
IODIDES a) Silver nitrate solution in the presence of dilute nitric acid gives a curdled,
pale-yellow precipitate, which does not dissolve in ammonia:
I- + AgNO3 AgI + NO3
-
b) Free iodine is formed after reaction of iodides with potassium dichromate
solution in the presence of dilute sulphuric acid. After shaking of reaction mixture
with chloroform the chloroform layer is coloured violet or violet-red.
6KI + K2Cr2O7 +7H2SO4 3I2 + Cr2(SO4)3 + 4K2SO4 + 7H2O
IRON
In EP there are one reaction for Fe2+
identification and two reactions for Fe3+
.
a) Potassium ferricyanide solution gives with ferrous (Fe2+
) salts a blue
precipitate that does not dissolve after adding of dilute hydrochloric acid:
3Fe2+
+ 2K3[Fe(CN)6]3‾
Fe3[Fe(CN)6]2↓ + 6K+
b) Potassium thiocyanate solution gives with ferric (Fe3+
) salts complex of red
colour:
Fe3+
+ 3KSCN [Fe(SCN)3] + 3K+
After adding of isoamyl alcohol or ether to one portion of reaction mixture
organic layer is coloured pink. After adding of mercuric chloride solution to the
second portion of red mixture the red colour disappears:
2[Fe(SCN)3 ]+ 3HgCl2 3[Hg(SCN)2] + 2FeCl3
c) Potassium ferrocyanide solution gives with ferric salts a blue precipitate that
does not dissolve after adding of dilute hydrochloric acid:
4Fe3+
+ 3K4[Fe(CN)6] Fe4[Fe(CN)6]3↓ + 12K+

10
LEAD
a) Reaction with potassium chromate solution with formation of yellow
precipitate that dissolves on addition of strong sodium hydroxide:
Pb2+
+ K2CrO4 PbCrO4 + 2K+
PbCrO4↓ + 4NaOH Na2[Pb(OH)4] + Na2CrO4
b) Potassium iodide solution gives a yellow precipitate, disappearing on
boiling and re-forming after cooling, as glistening, yellow plates.
Pb2+
+ 2KI PbI2 + 2K+
MAGNESIUM
Dilute ammonia gives with magnesium ions a white precipitate that dissolves
on addition of ammonium chloride solution:
Mg2+
+ 2OH- Mg(OH)2
Mg(OH)2 + 2NH4Cl MgCl2 + 2NH4OH
Then disodium hydrogen phosphate solution gives a white crystalline
precipitate: NH4Cl
Mg2+
+ Na2HPO4 + NH4OH NH4MgPO4 + H2O + 2Na+
MERCURY
a) When the solution of mercuric salts is placed on well-scraped copper foil, a
dark-grey stain is formed. It becomes shiny on rubbing. After drying and heating of
the foil in a test-tube the spot disappears (metallic mercury sublimates).
Hg2+
+ Cu Hg↓ + Cu2+
b) In strongly alkaline conditions mercury salts form dense yellow precipitate:
Hg2+
+ 2NaOH HgO + 2Na+ + H2O
c) The State Pharmacopoeia of Ukraine additionally uses the following
reaction:
potassium iodide solution, being added dropwise gives a red precipitate that
dissolves in excess of reagent:
Hg2+
+ 2I‾ HgI2↓
HgI2 + 2KI K2[HgI4]
NITRATES
a) Powdered substance containing nitrates is added to a mixture of
nitrobenzene and sulphuric acid. After adding water, strong sodium hydroxide
solution and acetone the layer of organic solvent is coloured deep violet.
The State Pharmacopoeia of Ukraine additionally uses two other reactions.
b) Nitrates do not discolour potassium permanganate solution acidified with
dilute sulphuric acid (distinction from nitrites).
c) Nitrites in the reaction with phenazone in the presence of dilute hydrochloric
acid colour the solution green.

11
C6H
5
NNO
C6H
5
NNO
ONCH3
CH3
CH3
CH3
NaNO2
HCl
PHOSPHATES (ORTHOPHOSPHATES)
a) A yellow precipitate is formed in reaction of phosphates with silver nitrate
solution. This precipitate dissolves on addition of ammonia.
PO43-
+ 3AgNO3 Ag3PO4 + 3NO3-
Ag3PO4 + 6NH4OH → [Ag(NH3)2]3PO4 + 6H2O
b) A yellow colour develops after interaction with molybdovanadic reagent:
PO43-
+ HVO3 + 11H2MoO4 + 4NH4+ (NH4)4[PO4(MoO3)11VO3]+ 11H2O + H
+
POTASSIUM
a) After heating potassium salts with sodium carbonate solution no precipitate
is formed (distinction from alkaline-earth metals). Then to the hot solution sodium
sulphide solution is added, and no precipitate is formed (distinction from heavy
metals). After cooling in iced water and adding solution of tartaric acid a white,
crystalline precipitate is formed. COOH
CH OH
CH OH
COOH
COOK
CH OH
CH OH
COOH
+ K + + H +
b) A yellow or orange-yellow precipitate is formed after interaction with
sodium cobaltinitrite solution in the presence of dilute acetic acid:
2K+ + Na3[Co(NO2)6] K2Na[Co(NO2)6] + 2Na
+
c) This reaction is additionally used by the State Pharmacopoeia of Ukraine
Potassium salt put into the colourless flame makes it violet (the colour appears red
through an indigo prism).
SILVER
Silver salt solution forms a curdled, white precipitate after adding of
hydrochloric acid. Precipitate dissolves on addition of dilute ammonia.
Ag+ + HCl AgCl + H
+
AgCl + 2NH4OH [Ag(NH3)2]Cl + 2H2O
SODIUM
a) After heating sodium salts with potassium carbonate solution to boiling no
precipitate is formed (distinction from alkaline-earth metals). Adding of potassium
pyroantimonate solution and heating to boiling with following cooling yields a dense
white precipitate:
Na+ + K[Sb(OH)6] Na[Sb(OH)6] + K
+
b) A voluminous, white, crystalline precipitate is formed in the reaction with
methoxyphenylacetic reagent. After placing in water at 20°C and stirring for 5 min

12
the precipitate does not disappear, but it dissolves completely on adding of dilute
ammonia. After adding of dilute ammonium carbonate solution no precipitate is
formed.
Na+
N(CH3)
4
N(CH3)
4 CH
OCH3
COO
CH
OCH3
COONa
+ +
+
+-
c) Sodium salt, treated with hydrochloric acid and put into the colourless flame
makes it yellow.
SULPHATES
a) Barium chloride solution in the presence of dilute hydrochloric acid gives a
white precipitate:
SO42-
+ BaCl2 BaSO4 + 2Cl-
b) When to the suspension obtained during reaction (a) iodine solution is added
the suspension remains yellow (distinction from sulphites SO3- and dithionites S2O4
2-
), but is decolorized by adding dropwise stannous chloride solution (distinction from
iodates IO3-).
2I2 + 2SnCl2 → SnCl4 + SnI4
On boiling the mixture, no coloured precipitate is formed (distinction from
selenates SeO42-
and tungstates WO42-
).
SULPHITES
The State Pharmacopoeia of Ukraine, unlike European Pharmacopoeia, gives
two general reactions for sulphites identification.
a) To the prescribed solution containing sulphite ions dilute hydrochloric acid
is added. Gradually the solution gives off a gas with strong odour (sulphurous
dioxide).
SO32-
+ 2HCl SO2 + H2O + 2Cl-
b) After interacting with iodine the solution is decolourized:
SO32-
+ I2 + H2O SO42-
+ 2HI
ZINC
a) After adding of strong sodium hydroxide solution a white precipitate is
formed.
Zn2+
+2NaOH Zn(OH)2 + 2Na+
After adding of a further quantity of this solution the precipitate dissolves and
remains clear on addition of ammonium chloride solution.
Zn(OH)2 + 2NaOH Na2ZnO2 + 2H2O
After adding of sodium sulphide solution a flocculent white precipitate is
formed:
Zn2+
+ Na2S ZnS + 2Na+
b) Potassium ferrocyanide solution gives with zinc salts a white precipitate that
does not dissolve after adding of dilute hydrochloric acid:
Zn2+
+ 2K4[Fe(CN)6] K2Zn3[Fe(CN)6]2↓ + 6K+

13
Tests for purity
General questions on impurity determination
The very important part of the pharmacopoeial analysis is impurities
determination and limit tests.
Impurities are certain chemical substances, which can be present in small
amounts together with the main compound. Their presence in the preparation can be
explained either by the process of Preparation or by the storage.
During Preparation of the preparations, impurities can get into them from the
raw materials from which the substances are prepared, from the reagents, or from the
apparatuses. On the other hand, they can be the semi-products or side-products of
synthesis.
Among the impurities that get into the preparations from the apparatuses,
materials and reagents, we can meet inorganic ions – iron, magnesium, zinc, arsenic,
heavy metals; anions of the acids usually used for acidifying of the reaction mixture –
chlorides, sulphates, phosphates and so on. Such impurities are named non-specific
and their determination is the same in many preparations. That is why methods for
their determination are described in general part of the Pharmacopoeia as “Limit
tests”. In monographs, only preparation of the solution to be examined is noted. Other
impurities which can get into the preparation during their Preparation are
characteristic for only one preparation or group of preparations. These impurities are
named specific. It means that their determinations are described in the monographs.
Very often chromatographic methods (gas chromatography or high-pressure liquid
chromatography) are used for their determining. Sometimes simple chemical
reactions are used.
Impurities can get into the preparation during their storage too. For example,
some substances decompose when kept, particularly in presence of air and light. Such
impurities also can be divided into specific and non-specific. Non-specific impurities
can get into the preparation from the air. It may be, for example, ammonium salts.
These impurities also are determined according to general methods. Specific
impurities usually are the products of the different chemical processes going into the
preparations during the storage. In this case, the determination of impurities is carried
out by the method described in the monograph. Example of such impurities is acetic
acid in Aspirin.
A choice of the method for impurity determination depends of the processes of
medicines manufacture, the composition of the raw materials, the properties of the
substance and its behaviour during storage.
Impurities in the substances can be determined by chemical or physico-
chemical methods.
1. Chemical methods involve carrying out different chemical reactions with the
use of standard solutions or without them. Such general quantitative or limit tests are
laid down for a number of non-specific impurities. Limit tests use simple
comparisons of opalescence or colour with fixed standards. Standard solution is the
solution containing exact quantity of certain ion. For example, chloride standard
solution contains an exact quantity of sodium chloride in water solution. Usually

14
concentration of a standard solution is expressed in ppm and standard solutions of
one impurity with different concentrations are used for different preparations.
A choice of the reagent and the reaction for the determination is very
important. The analytical reaction chosen has to be sensitive in the certain test,
specific and reproducible.
Results of the reactions are compared colorimetrically or nephelometrically in
the solutions of the substance and in the standard solution after adding of
corresponding reagents. If result of the reaction in the solution examined (colour,
opalescence, or spot) is less than that in the standard we can say that amount of
impurity is less than in this standard solution.
2. Physico-chemical methods. Different chromatographic methods are often
used in the monographs (thin-layer chromatography, gas chromatography, liquid
chromatography, etc.) for impurities determination.
Clarity and degree of opalescence of liquids
This test is carried out by comparing the liquid to be examined with a reference
suspension or with the solvent.
A liquid is considered clear if its clarity is the same as that of water R or of the
solvent, or if its opalescence is not more pronounced than that of reference
suspension I.
There are reference suspensions I-IV, they should be freshly prepared from the
standard of opalescence and water R. Standard of opalescence is obtained by dilution
of primary opalescent suspension which in its turn is obtained by mixing up two
solutions: hydrazine sulphate solution and hexamethylenetetramine solution (they
give a white precipitate in a form of opalescent suspension).
The solutions should be compared using identical test tubes of colourless,
transparent, neutral glass with a flat base and an internal diameter of 15 mm to
25 mm, the depth of the layer being 40 mm, in diffused daylight 5 min after
preparation of the reference suspension, viewing vertically against a black
background.
Degree of coloration of liquids
The examination of the colour intensities of liquids in the range brown-yellow-
red is carried out by one of the two methods (I or II), which differ by the size of the
tubes, quantity of liquids and the way of viewing. Test is carried out using identical
tubes of colourless, transparent, neutral glass, in diffused daylight, looking against a
white background.
A solution is colourless if it has the appearance of water R or the solvent or is
not more intensely coloured than reference solution B9.
There are reference solutions of the scales B (brown, B1-B9), BY (brownish-
yellow, BY1-BY7), Y (yellow, Y1-Y7), GY (greenish-yellow, GY1-GY7), R (red, R1-
R9), and corresponding five standard solutions. Each reference solution is prepared
using prescribed volumes of a standard solution and hydrochloric acid 10g/l.
Standard solutions B, BY, Y, GY, R are prepared by mixing three primary
solutions: yellow (ferric chloride solution), red (cobalt chloride solution), and blue

15
(copper sulphate solution) in different proportions with hydrochloric acid 10g/l.
Primary solutions themselves are acidified with hydrochloric acid.
Method I: Liquids to be examined are compared in identical tubes of 12 mm
external diameter, taking volumes of 2 ml, viewing horizontally.
Method II: Liquids to be examined are compared in identical tubes with a flat
base and an internal diameter of 15 to 25 mm, the depth of the layer being 40 mm.
The comparison is made by looking vertically downwards through the columns of
liquid in the tubes.
Determination of pH
pH value of a solution gives the important information about purity and
identity of a preparation, because pH shows the presence of acidic or alkaline
impurities, and it is one of the chemical characteristics of a substance. Storage and
usage of solutions depend on their pH. Solutions for injections should have pH value
close to pH value of blood.
European Pharmacopoeia recommends using two methods for pH
determination: electrometric (potentiometric) and colorimetric, using indicators.
1. Potentiometric determination is used to measure the exact value of pH.
The potentiometric determination of pH is made by measuring the potential
difference between 2 appropriate electrodes immersed in the solution to be examined:
one of these electrodes is sensitive to hydrogen ions (usually a glass electrode) and
the other is the reference electrode (for example, a saturated calomel electrode), the
measuring apparatus is a voltmeter.
2. Colorimetric determination is made according to the table describing
relationship between reaction of solution, approximate pH and colour of certain
indicators.
Loss on drying and determination of water
These tests can be prescribed to control the quantity of water in the substance.
Loss on drying is the loss of mass expressed as per cent m/m. To check the
loss on drying the prescribed quantity of the substance should be placed in a
weighing bottle previously dried under the conditions prescribed. Then the substance
should be dried to constant mass or for the prescribed time over:
o diphosphorus pentoxide: “in a desiccator”, at atmospheric pressure or “in
vacuo” (pressure of 1.5 kPa to 2.5 kPa), or “under high vacuum” (at a pressure not
exceeding 0.1 kPa), at room temperature or within the temperature range prescribed
in the monograph;
o in an oven within a specified temperature range.
The term “dried to constant mass” means that two consecutive weighings do
not differ by more than 0.5 mg, the second weighing following an additional period
of drying.

16
Determination of water by distillation
Distillation of water is carried out with the help of toluene, which form
aseotropic mixture with water. After the distillation, when the water and toluene have
completely separated, the volume of water is read and calculation of the content of
water present in the substance as ml per kg is made. This method is widely used for
the analysis of medicinal plants preparations.
Semi-micro determination of water
The volumetric method of titration of water is based upon the quantitative
reaction of water with iodosulphurous reagent, being the mixture of sulphur dioxide
and iodine in an anhydrous medium of ethyleneglycol monomethyl ether in the
presence of a base – anhydrous pyridine.
SO2 + I2 + 2H2O H2SO4 + 2HI
The titration vessel is fitted with two platinum electrodes, a nitrogen inlet tube,
a stopper which accommodates the burette tip, and a vent-tube protected by a
desiccant. The substance to be examined is introduced through a side-arm which can
be closed by a ground stopper. Stirring is effected magnetically or by means of a
stream of dried nitrogen passed through the solution during the titration. The end-
point is determined by amperometry.
Pyridine reacts with hydroiodic acid and sulphur dioxide (reducing its
volatility).
N
H
I2
SO2
OH2 I
N
SOO
ON
+ + +3 + + +2
According to a monograph, either direct (by iodosulphurous reagent) or back
(excess of iodosulphurous reagent is titrated by anhydrous methanol) titration can be
used.
N
H
N
SOO
O
CH3OH CH
3SO
4++
+-
Individual determinations can be carried out successively if each component of
the test mixture is compatible with the other components and no other reactions take
place.
Micro determination of water
It is the coulometric titration of water, reactions which take place are similar to
the semi-micro determination of water. Iodine is produced electrochemically in the
reaction cell by oxidation of iodide. The iodine produced at the anode reacts
immediately with the water and the sulphur dioxide contained in the reaction cell.
The amount of water in the substance is directly proportional to the quantity of

17
electricity up until the titration end-point. When all of the water in the cell has been
consumed, the end-point is reached and thus an excess of iodine appears.
This method is used for the quantitative determination of small amounts of
water, a range of 10 µg up to 10 mg of water is recommended.
Limit tests
TEST FOR CHLORIDES
A solution of a substance is acidified with nitric acid, and this mixture as a
single addition is poured into the silver nitrate solution: HNO3
Cl- + AgNO3 AgCl + NO3
-
A white curdled precipitate or only opalescence (because a very small amount
of chlorides can be present) is formed. The opalescence is compared with the
opalescence produced by addition of a standard solution (containing a definite
quantity of sodium chloride and acidified with nitric acid) to the silver nitrate
solution.The liquids should be stirred and set aside protected from light for 5 minutes
before comparison, since the full opalescence is not developed immediately. After
standing for 5 min, any opalescence in the test solution should be not more intense
than that in the standard. The test-tubes should be compared laterally against a black
background.
Why the medium for this test is dilute nitric acid? If the medium is alkaline,
silver hydroxide (AgOH) may be formed. In this case we would obtain white
precipitate or opalescence even if there are no chlorides in the preparation. If the
medium is neutral the carbon dioxide presented in water would react with the silver
nitrate solution with the formation of white precipitate of Ag2CO
3. Nitric acid is
used for acidifying of the solution because another acids can form precipitates with
silver ions (for example, Ag2SO4).
TEST FOR SULPHATES
The sulphates impurity determination is based on the reaction with the barium
chloride by formation of white precipitate in the presence of acetic acid, which helps
to prevent precipitation of another anions of the second analytical group: CH3COOH
SO42-
+ BaCl2 BaSO4 + 2Cl-
In order to make this reaction more sensitive the reagent is prepared at first
from the solution of barium chloride and a small amount of sulphate standard solution
containing alcohol. Then solution to be examined and acetic acid are added to this
reagent. After 5 minutes the opalescence is compared with the opalescence produced
by addition of sulphate standard solution (containing a definite quantity of potassium
sulphate) and acetic acid to the above-mentioned mixture, containing barium
chloride. Any opalescence in the test solution should be not more intense than that in
the standard. Sensitivity of this test increases due to the presence of a small amount
of sulphate standard solution in each of the tubes, giving ionic concentrations which

18
exceed the solubility product of barium sulphate. The presence of alcohol helps to
prevent supersaturation.
TEST FOR AMMONIUM
Method A. This method is based on the reaction of ammonium with alkaline
potassium tetraiodomercurate solution (Nessler reagent):
NH2
I-Hg
I-Hg
NH3 + 2K2[HgI4] + KOH
+
I + 5KI + H2O-
The prescribed quantity of the substance to be examined is dissolved in water
in a test-tube, made alkaline if necessary by the addition of dilute sodium hydroxide
solution and mixed with alkaline potassium tetraiodomercurate solution. A standard
for comparison is prepared in the same manner, using ammonium standard solution
(containing a definite quantity of ammonium chloride) diluted with water, instead of
a solution of a substance. Both test tubes are stopped. After 5 min, any yellow colour
in the test solution should be not more intense than that in the standard.
Method B. To the prescribed quantity of the substance to be examined
dissolved or suspended in 1 ml of water heavy magnesium oxide is added. Then jar is
closed immediately after placing a wetted piece of silver manganese paper under the
polyethylene cap. It is allowed to stand at 40 °C for 30 min.
Heavy magnesium oxide makes medium alkaline and during the heating
ammonia is evolved. Then ammonia reacts with silver manganese paper (containing
silver nitrate and manganese sulphate):
2AgNO3 + MnSO4 + 4NH3 + 2H2O 2Ag + MnO2+ (NH4)2SO4 +
NH4NO3If the silver manganese paper shows a grey colour, it should be not more
intense than that of a paper prepared at the same time and in the same manner using
the ammonium standard solution instead of a solution (or suspension) of the
substance.
Methods C and D are described additionally by the State Pharmacopoeia of
Ukraine.
Method C. This method is used for the determination of ammonium impurity
in the preparations containing alkaline-earth and/or heavy metals. Sodium hydroxide
and sodium carbonate solutions are added to precipitate alkaline-earth and/or heavy
metals in the form of carbonates. After filtering the precipitates off, the reaction with
alkaline potassium tetraiodomercurate solution is carried out with the filtrate.
Method D. If the preparation contains ferric salts they should be bound in the
complex by adding of sodium-potassium tartrate solution in the presence of sodium
hydroxide solution. A complex is forming:

19
Apparatus for the limit test A
Dimensions in millimeters
CH CH C
O
COO
O
Fe
OO
-
Then the reaction with alkaline potassium tetraiodomercurate solution is
carried out.
TEST FOR ARSENIC
Pharmacopoeia describes two methods for arsenic impurity determination.
Method A is based on the reduction of arsenic compounds to gaseous
arsenious hydride, or arsine, by the use of zinc and dilute acid:
6Zn + As2O3 + 12HCl → 2AsH3+ 3H2O + 6 ZnCl2The solution is
previously treated with stannous chloride solution (reducing agent)
to convert compounds of arsenic (V) into compounds of arsenic
(III).
The apparatus for arsenic determination consists of a 100 ml
conical flask closed with a ground-glass stopper, through which
passes a glass tube. This tube is open at the upper end. The lower
end is drawn out to a diameter of 1 mm. There is a hole in the side
of this tube, 15 mm from the lower end. Such peculiarity in the
construction in made to remove condensed moisture from the
constricted part. The tube is loosely packed with lead acetate cotton
(or a small plug of cotton and a rolled piece of lead acetate paper).
Lead acetate reacts with hydrogen sulphide that could be possibly
emitted together with arsine from the sample, forming on the cotton
a precipitate of lead sulphide.
The upper end of the tube has a perfectly flat, ground surface.
Over the top of this tube a disc or a small square of mercuric
bromide paper is fixed (large enough to cover the orifice of the
tube), in such a way that gas evolved from the bottle passes through
a mercuric bromide paper. Pharmacopoeia suggests fixing this
paper using a second glass tube of the same diameter,
with a similar flat ground surface, placed in contact with
the first, and held in position by two spiral springs.
Arsine being formed reacts with mercury bromide on the paper, forming a stain,
intensity of colouration of which depends on the impurity concentration:
AsH3 + HgBr2 → AsH2(HgBr) + HBr
AsH3 + 2HgBr2 → AsH(HgBr)2 + 2HBr
AsH3 + 3HgBr2 → As(HgBr)3 + 3HBr
AsH3 + As(HgBr)3 → As2Hg3 + 3HBr
The solution to be examined is placed in the flask, hydrochloric acid, stannous
chloride solution and potassium iodide solution are added, and allowed to stand for
15 min. Then activated zinc is introduced, and two parts of the apparatus are

20
assembled immediately. The flask is immersed in a water bath at a temperature such
that a uniform evolution of gas is maintained. The reaction is allowed to go on for 2
hours. Standard is prepared in the same manner, using arsenic standard solution
(prepared by dissolving a definite quantity of arsenic trioxide in sodium hydroxide),
diluted with water. The mercuric bromide paper is then removed, and the stain is
compared with the standard stain.
The stain produced on the mercuric bromide paper in the test should be not
more intense than that in the standard.
Method B is based on the reduction of arsenic compounds by the use of
hypophosphorus reagent, in acidic medium. Hypophosphorus reagent, which is
solution of sodium hypophosphite and hydrochloric acid, reduces compounds of
arsenic (III) and (V) to a brown precipitate of free arsenic.
The substance to be examined is introduced into a test-tube containing
hydrochloric acid and potassium iodide and then mixed with 3 ml of hypophosphorus
reagent. Mixture is heated on a water-bath for 15 minutes and compared with a
standard, prepared in the same way, containing known quantity of arsenic standard
solution. Any colour in the test solution should be not more intense than that in the
standard.
NaH2PO2 + HCl NaCl + H3PO2
As2O3 + 3H3PO2 2As↓ + 3H3PO3
As2O5 +5H3PO2 2As↓ + 5H3PO3
The process of reduction has two stages, and phosphine can be considered a
real reducing agent:
2H3PO2 H3PO4 + PH3
As2O3 + PH3 2As↓ + H3PO3
This method can be also used for determination of selenium and tellurium
impurities, which are in the same way reduced to the free state. It is used often for the
arsenic determination in the preparations containing bismuth, mercury, silver and
antimony as far as sulphides and sulphites.
TEST FOR HEAVY METALS
Heavy metals are the metals with density equal or exceeding 5 g/cm3 (Pb, Cd,
Cu, Fe, Ni, Zn, Hg and others). They are the very undesirable impurities in medicinal
substances.
Method A. Concentrated solutions of heavy metals salts yield, on addition of
thioacetamide reagent in acidic medium, a black (or dark) precipitates of metal
sulphides. With very dilute solutions a brown coloration is produced. The intensity of
it varies according to the quantity of heavy metals present.
CH3
C
S
NH2
CH3COO NH
4+ + ++-2H2O H2S
Pb
2+ + H2S → PbS + 2H
+
Medium must be acidic to prevent the formation of the precipitates of metals
hydroxides and carbonates. The solutions are made acidic with buffer solution pH

21
3,5. Colouration in the test solution should be compared after 2 minutes with
colouration in a standard prepared by adding of buffer solution pH 3,5 and
thioacetamide reagent to lead standard solution (containing a definite quantity of lead
(II) nitrate). A blank should also be prepared, using a mixture of water and the
solution to be examined. Compared to the blank, the standard shows a slight brown
colour.
After 2 min, any brown colour in the test solution should be not more intense
than that in the standard.
Methods B-F describe especial conditions of sample Preparation but the main
conditions are the same.
TEST FOR CALCIUM
Reaction is based on the possibility of calcium-ions to form a white precipitate
with ammonium oxalate solution in the medium of dilute acetic acid.
CH3COOH
Ca2+
+ (NH4)2C2O4 CaC2O4 + 2NH4+
Dilute acetic acid and a solution of a substance is added to the previously
prepared mixture, containing a small amount of alcoholic calcium standard solution
(the principle of this is the same as in the test for sulphates, above) and the main
reagent, ammonium oxalate solution. After 15 minutes the opalescence is compared
with the opalescence produced by addition of aqueous calcium standard solution
(prepared by dissolving of known quantity of calcium carbonate in acetic acid, then
diluted with water) and dilute acetic acid to the mixture, containing ammonium
oxalate. Any opalescence in the test solution should be not more intense than that in
the standard.
We can’t choose the alkaline medium because of calcium hydroxide
precipitation. If the pH=7, calcium carbonate can form. The choice of acetic acid can
be explained by solubility of calcium oxalate in mineral acids.
TEST FOR MAGNESIUM
8-Hydroxyquinoline in the alkaline medium forms a yellowish-green complex
with the impurity at the presence of chloroform:
N
OH
Mg2+
N
O
N
OMg
H+CHCl
3+2 + 2
Magnesium is extracted from aqueous solution by the chloroform solution of
the main reagent, 8-hydroxyquinoline. The chloroform layer is then used for
comparison with the standard prepared in the same manner, using magnesium
standard solution, containing a definite quantity of magnesium sulphate. Any colour
in the solution obtained from the substance should be not more intense than that in
the standard.
22
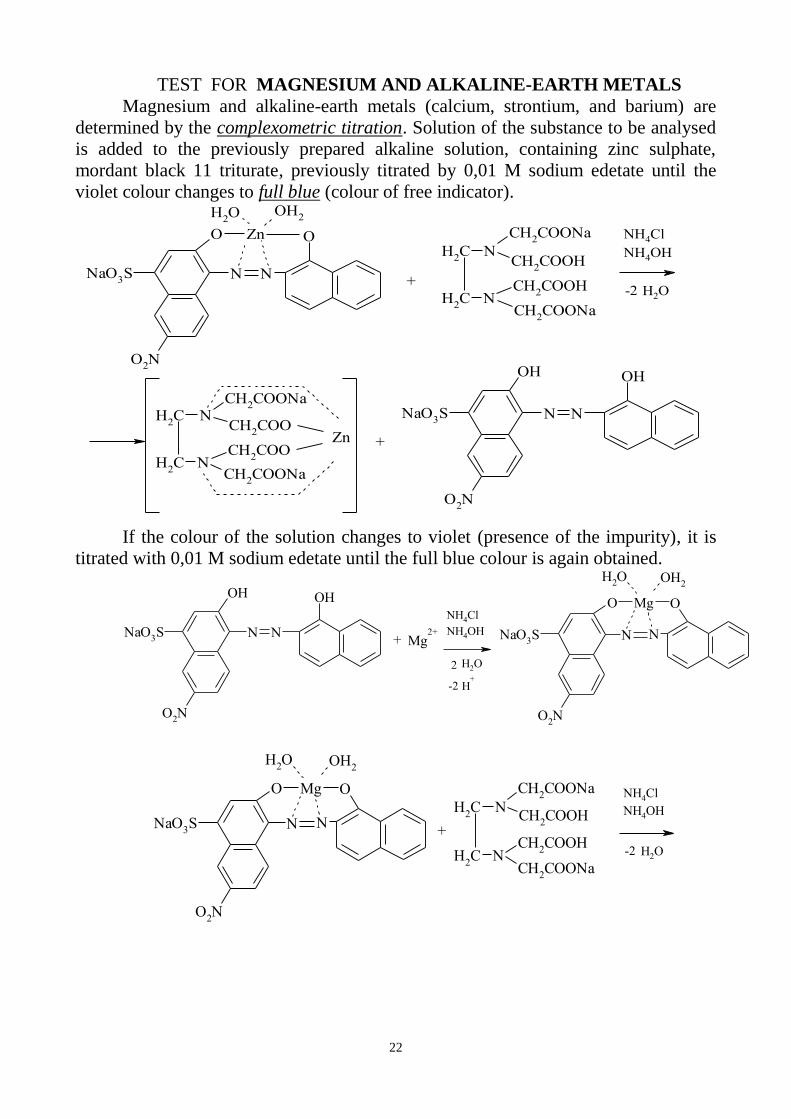
TEST FOR MAGNESIUM AND ALKALINE-EARTH METALS
Magnesium and alkaline-earth metals (calcium, strontium, and barium) are
determined by the complexometric titration. Solution of the substance to be analysed
is added to the previously prepared alkaline solution, containing zinc sulphate,
mordant black 11 triturate, previously titrated by 0,01 M sodium edetate until the
violet colour changes to full blue (colour of free indicator).
N
O
NaO3S
O2N
N
OZn
OH2H
2O
CH2
CH2
N
N
CH2COONa
CH2COO
CH2COO
CH2COONa
Zn
CH2
CH2
N
N
CH2COONa
CH2COOH
CH2COOH
CH2COONa
N
OH
NaO3S
O2N
N
OH
+
+
-2 H2O
NH4Cl
NH4OH
If the colour of the solution changes to violet (presence of the impurity), it is
titrated with 0,01 M sodium edetate until the full blue colour is again obtained.
N
O
NaO3S
O2N
N
Mg O
OH2
N
OH
NaO3S
O2N
N
OH
Mg2+
H+
H2O
+
-2
2 H2O
NH4Cl
NH4OH
CH2
CH2
N
N
CH2COONa
CH2COOH
CH2COOH
CH2COONa
N
O
NaO3S
O2N
N
Mg O
OH2
H2O
+
-2 H2O
NH4Cl
NH4OH
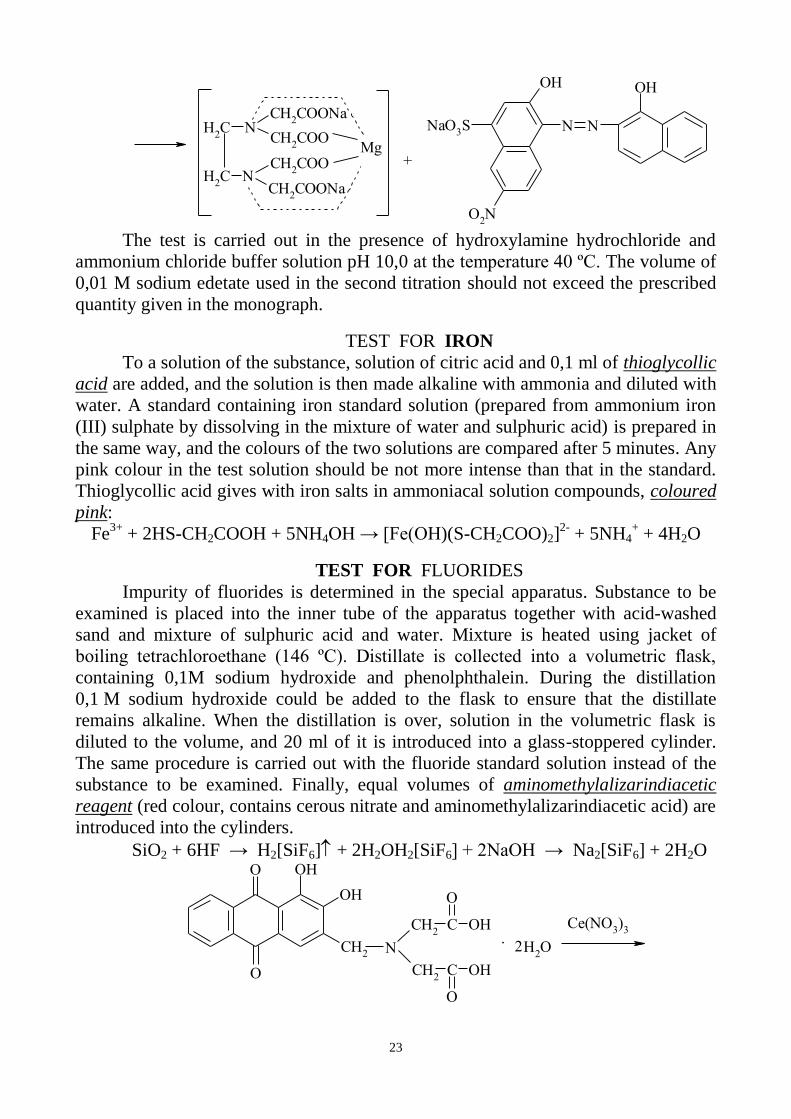
23
CH2
CH2
N
N
CH2COONa
CH2COO
CH2COO
CH2COONa
Mg
N
OH
NaO3S
O2N
N
OH
+
The test is carried out in the presence of hydroxylamine hydrochloride and
ammonium chloride buffer solution pH 10,0 at the temperature 40 ºC. The volume of
0,01 M sodium edetate used in the second titration should not exceed the prescribed
quantity given in the monograph.
TEST FOR IRON
To a solution of the substance, solution of citric acid and 0,1 ml of thioglycollic
acid are added, and the solution is then made alkaline with ammonia and diluted with
water. A standard containing iron standard solution (prepared from ammonium iron
(III) sulphate by dissolving in the mixture of water and sulphuric acid) is prepared in
the same way, and the colours of the two solutions are compared after 5 minutes. Any
pink colour in the test solution should be not more intense than that in the standard.
Thioglycollic acid gives with iron salts in ammoniacal solution compounds, coloured
pink:
Fe3+
+ 2HS-CH2COOH + 5NH4OH → [Fe(OH)(S-CH2COO)2]2-
+ 5NH4+ + 4H2O
TEST FOR FLUORIDES
Impurity of fluorides is determined in the special apparatus. Substance to be
examined is placed into the inner tube of the apparatus together with acid-washed
sand and mixture of sulphuric acid and water. Mixture is heated using jacket of
boiling tetrachloroethane (146 ºC). Distillate is collected into a volumetric flask,
containing 0,1M sodium hydroxide and phenolphthalein. During the distillation
0,1 M sodium hydroxide could be added to the flask to ensure that the distillate
remains alkaline. When the distillation is over, solution in the volumetric flask is
diluted to the volume, and 20 ml of it is introduced into a glass-stoppered cylinder.
The same procedure is carried out with the fluoride standard solution instead of the
substance to be examined. Finally, equal volumes of aminomethylalizarindiacetic
reagent (red colour, contains cerous nitrate and aminomethylalizarindiacetic acid) are
introduced into the cylinders.
SiO2 + 6HF → H2[SiF6] + 2H2OH2[SiF6] + 2NaOH → Na2[SiF6] + 2H2O O
O
OH
OH
CH2 N
CH2
C
O
OH
CH2
C
O
OH
OH2
Ce(NO3)
3.
2

24
O
O
OH
O
CH2
O
CeO
H2O
H2O
N
CH2
CH2
C
O
C
O
Na2[SiF
6]
NaOH
O
O
ONa
ONa
CH2 N
CH2
C
O
CH2
C
O
O
O
Ce
H2O
H2O
F
Any blue colour in the test solution should be less intense than that in the
standard.
TEST FOR PHOSPHATES
Test for phosphates is carried out with sulphomolybdic reagent (solution of
ammonium molybdate and sulphuric acid) at the presence of stannous chloride
solution:
PO43-
+ 12(NH4)2MoO4 + 27H+ → H7[P(Mo2O7)6] + 24NH4
+ + 10 H2O
To compare blue colouration of the test solution and of a standard, prepared in
the same manner using phosphate standard solution (containing a known quantity of
potassium dihydrogen phosphate), volumes of 20 ml each are used, after 10 minutes.
Any yellow colour in the test solution should be not more intense than that in the
standard.
TEST FOR POTASSIUM
Freshly prepared solution of sodium tetraphenylborate gives with potassium a
white precipitate:
K+ + NaВ(C6H5)4 KB(C6H5)4 + Na
+
After 5 minutes, any opalescence in the test solution should be not more
intense than that in the standard, containing potassium standard solution (solution of a
known quantity of potassium sulphate) and the solution of sodium tetraphenylborate.
TEST FOR ALUMINIUM
Similar to the test for magnesium, solution of 8-hydroxyquinoline in
chloroform is used for the extraction of aluminium. This reagent gives fluorescent
complex with aluminium:

25
N
OH
Al3+
N
O
N
OAl
H+CHCl
3
N
O
+3 + 3
The intensity of the fluorescence of the test solution (I1), of the standard (I2)
and also of the blank (I3) is measured using fluorimeter (an excitant beam at 392 nm).
The fluorescence (I1–I3) of the test solution should be not greater than that of the
standard (I2–I3).
TEST FOR ZINC
Impurity of zinc is determined with the potassium ferrocyanide solution in the
presence of hydrochloric acid (according to the State Pharmacopoeia of Ukraine). In
the presence of zinc ions a white precipitate is forming:
HCl
3Zn2+
+ 2K4[Fe(CN)6] K2Zn3[Fe(CN)6]2 + 6K+
Acidic medium is used because zinc is amphotheric and in alkaline medium it
can be reformed into zincate ZnO22-
. If the substance to be examined contains
additionally the iron impurity (blue colour appears after adding of potassium
ferrocyanide solution) iron should be removed using dilute ammonia, in the form of
hydroxide that is filtered off. Then the filtrate is acidified (zincate is transformed into
Zn2+
) and the test is carried out.
After 10 min, any opalescence in the test solution should be not more intense
than that in the standard.

26
Chapter 2. MEDICINAL PREPARATIONS CONTAINING
THE ELEMENTS OF 7TH
AND 6TH
GROUPS OF
D.I. MENDELEYEV PERIODIC SYSTEM
Plan
1. Preparations of the halogens and hydrogen (hydrochloric acid,
concentrated).
2. Preparations of hypochlorous and hydrochloric acids’ salts (chlorinated
lime).
3. Preparations of the halides (sodium chloride, potassium chloride, sodium
bromide, potassium bromide, sodium iodide, potassium iodide).
4. Preparations of iodine (iodine, iodine solution in alcohol 5% and 10%,
iodinole).
5. Preparations of manganese (potassium permanganate).
6. Preparations of hydrogen peroxide (hydrogen peroxide solution 3 and 30
per cent, hydroperite, magnesium peroxide).
7. Preparations of sulphur (sodium thiosulphate, sodium sulphate decahydrate,
sulphur for external use).
Preparations of the halogens and hydrogen
Hydrochloric acid, concentrated, E.P.
(Acidum hydrochloricum concentratum)
HCl
Preparation
Pure hydrochloric acid is usually made by direct synthesis from hydrogen and
chlorine produced by electrolysis of sodium chloride solution:
NaCl Na+ + Cl
-.
Cathode: Anode:
H2O + ē → Ho + OH
- 2Cl
- − 2ē → 2Cl
o
Ho + H
o → H2 2Cl
o → Cl2
2NaCl + 2H2O Cl2 + H2 + 2NaOH
The chlorine is burned in hydrogen in large-diameter silica tubes.
H2 + C12 → 2HC1
The hydrogen chloride so formed is absorbed in water:
Properties
Pharmacopoeial preparation is a clear, colourless, fuming liquid, miscible with
water. It has a relative density of about 1.18.
Concentrated hydrochloric acid contains not less than 35 % and not more than
39% of hydrogen chloride. This acid fumes because hydrogen chloride combines
with ammonia (always present in the air) forming ammonium chloride (and it is
exactly the very minute particles of this compound that form the fume).

27
Identification
1. Diluted with water, the solution of the preparation is strongly acid. This
property is confirmed with correspondent indicator (E.P.).
2. Hydrochloric acid is recognized by the identification reactions for chlorides
(above) (E.P.).
3. It complies with the limits of the assay (E.P.).
4. After heating of HCl with manganese dioxide free chlorine evolves, being is
detected by its odour: to
4HCl + MnO2 Cl
2+ MnCl
2 + 2H
2O
Tests for purity
Free chlorine is detected by liberation of iodine from potassium iodide in the
presence of iodide-free starch solution. The iodine colours starch solution blue.
Cl2 + 2KI I
2 + 2KCl
Blue colour should disappear on the addition of 0.2 ml of 0.01 M sodium
thiosulphate solution.
Assay (quantitative determination)
1. Hydrochloric acid is assayed by titration with sodium hydroxide, using
methyl red as indicator (alkalimetry, s = 1) (E.P.).
HCl + NaOH NaCl + H2O
2. According to the density of hydrochloric acid, because a definite density
corresponds to a definite acid concentration.
Storage In an airtight container made of glass or another inert material, at a
temperature below 30 C.
Usage Concentrated hydrochloric acid is used for Preparation dilute
hydrochloric acid. The latter is administered orally to improve gastric acidity.
Preparations of hypochlorous and hydrochloric acids’ salts
Chlorinated lime (Calcaria chlorata)
CaOCl
Cl
Ca(OH)2 H
2O. . n
Chlorinated Lime is a mixed calcium salt of hypochlorous and hydrochloric
acids (calcium chloro-hypochlorite), associated with varying proportions of calcium
hydroxide and moisture.
Preparation
CaO + H2O Ca(OH)2
Ca(OH)2 + Cl2 CaOCl2 + H2O

28
Properties
Chlorinated lime is a dull white or light greyish powder with chlorine’s odour,
is partly solvable in water. It slowly decomposes on exposure to air due to the action
of atmospheric carbon dioxide and moisture.
Identification
1. Solution of chlorinated lime 1:10, added to the red litmus, colours it blue
(рН>7), the blue colour then disappears owing indicator’s destruction by chlorine:
CaOCl2 + H
2O Ca(OH)
2 + Cl
2
2. Under the effect of hydrochloric acid, chlorinated lime decomposes with the
evolution of free chlorine:
CaOCl2 + 2HCl → CaCl2 + Cl2 + H2O
After adding of potassium iodide solution the yellow colour of iodine appears:
Cl2 + 2KI I
2 + 2KCl
3. For the calcium identification the solution of ammonium oxalate is added
after chlorine evolved. The latter is reached by the boiling of the substance with
acetic acid. Acetic acid, like other acids, liberates chlorine from the chlorinated lime:
2CaOCl2 + 2CH3COOH → (CH3COO)2Ca + CaCl2 + Cl2↑ + H2O
(CH3COO)
2Ca + (NH
4)
2C
2O
4 CaC
2O
4+ 2CH
3COONH
4
Assay
Iodometric titration. For the assay, an aqueous suspension of the substance is
treated with acetic acid in the presence of excess of potassium iodide. The liberated
chlorine displaced an equivalent amount of iodine from potassium iodide. The iodine
produced in this way is titrated with sodium thiosulphate, using starch solution as
indicator (s = 1/2).
2CaOCl2 + 2HCl + 2KI → I2 + CaCl2+ 2KCl + H2O
I2 + 2Na2S2O3 2NaI + Na2S4O6
Chlorinated lime should contain not less than 32 % of available chlorine.
Storage In an airtight containers at cool, protected from light place.
Usage Disinfectant.
Preparations of the halides
Sodium chloride (Natrii chloridum), EP
NaCl
Potassium chloride (Kalii chloridum), EP
KCl
Preparation
Sodium chloride occurs widely in nature as massive underground deposits of
rock salt. This is exactly the main source for the production of the Preparation.

29
“Common salt” has been manufactured also for hundreds of years by evaporation of
seawater in shallow pans.
During the manufacture the brine obtained is purified in several stages. The
reagents for purification are barium chloride solution, sodium carbonate solution and
hydrochloric acid:
Na2SO4 + BaCl2 BaSO4 + 2NaCl
Na2HPO4 + BaCl2 BaHPO4 + 2NaCl
MgCl2 + Na2CO3 MgCO3 + 2NaCl
CaCl2 + Na2CO3 CaCO3 + 2NaCl
BaCl2 + Na2CO3 BaCO3 + 2NaCl
Na2CO3 + 2HCl 2NaCl + CO2 + H2O
The precipitates are filtered off after the adding of BaCl2, then after adding of
Na2CO3. Then the solution is purified by recrystallization.
Potassium chloride is made from carnallite, KCl·MgCl2·6H2O by a process of
fusion whereby the liquefied magnesium chloride hexahydrate can be separated from
the solid potassium chloride; or by a process of crystallization. It also can be prepared
from the silvinite KCl·NaCl.
Properties
Both the preparations are white, crystalline powders or colourless crystals (or
white pearls for NaCl). They are freely soluble in water, practically insoluble in
ethanol.
Identification is carried out using general identification reactions (EP) for
sodium, potassium and chlorides (above).
Tests for purity
Bromides. This impurity is determined with chloramine and sodium
thiosulphate in the presence of phenol red solution. The absorbance of the solution
measured at 590 nm is not greater than that of a standard prepared using solution of
potassium bromide. CH
3
SO2N
Cl
Na
CH3
SO2NH
2
+ 2HCl + Cl2 + NaCl
2Br - + Cl2 Br2 + 2 Cl -
Ferrocyanides (in sodium chloride). Ferrocyanides are detected by formation
of a blue precipitate (Prussian blue) after the adding of ferric ammonium sulphate in
sulphuric acid and ferrous sulphate solution. Blue colour should not develop within
10 min.
3Na4[Fe(CN)
6] + 4FeNH4(SO4)2 Fe4[Fe(CN)
6]3 + 6Na2SO4 + 2(NH4)2SO4

30
Iodides. In this test the crystalline substance is moistened by the acidified
mixture of sodium nitrite solution, being an oxidizing agent, and iodide-free starch
solution. An oxidizing agent liberates iodine from iodides, and blue colour is
producing. The substance should not show any blue colour.
2NaI + 2NaNO2 + 2H2SO4 I2 + 2NO + 2Na2SO4 + 2H2O
Barium. The test for barium is included to guard against the possible presence
of this impurity resulting from the use of barium chloride in process of purification
(see Preparation). The reagent is dilute sulphuric acid. After 2 h, any opalescence in
the solution should be not more intense than that in a mixture of solution S and
distilled water.
BaCl2 + H2SO4 BaSO4↓ + 2HCl
Potassium and sodium are pharmacological antagonists, that’s why impurity of
potassium is determined in sodium preparations and impurity of sodium is determined
in potassium preparations, by atomic emission spectrometry.
Assay
1. Argentometry (EP). A weighed quantity of substance is dissolved in water
and treated with a measured volume of 0,1 M silver nitrate determining the end-point
potentiometrically; s=1. Calculation is made with reference to the dried substance.
NaCl + AgNO3 AgCl + NaNO3
2. Back argentometry by Volhard in the presence of dibuthyl phthalate (EP).
A weighed quantity is dissolved in water, acidified with nitric acid, and treated
with an excess of silver nitrate solution in the presence of dibuthyl phthalate:
KCl + AgNO3 AgCl + KNO3
The excess of silver nitrate is then determined by titration with ammonium
thiocyanate, using ferric ammonium sulphate as indicator (s = 1).
Calculation is made with reference to the dried substance.
AgNO3 + NH4SCN AgSCN + NH4NO3
3NH4SCN + Fe(NH4)(SO4)2 [Fe(SCN)3] + 2(NH4)2SO4
3. Mercurimetric method. Standard solution is mercury nitrate, indicator is
diphenylcarbazone that forms red-violet complex with excessive amount of mercury
nitrate (s=2). Calculation is made with reference to the dried substance.
2NaCl + Hg(NO3)2 HgCl2 + NaNO3
CO
NH
N N
NH C6H
5
C6H
5
Hg(NO3)
2 CO
NH
N N
C6H
5
N
C6H
5
C O
NN
C6H
5
N
C6H
5
NH
Hg
HNO3
2 +
2_
The end-point can be also determined with sodium nitroprusside:
Na2[Fe(CN)5NO] + Hg(NO3)2 Hg[Fe(CN)5NO] +2NaNO3
Storage In airtight containers.

31
Usage The most important function of the sodium chloride is providing a
constant blood pressure. Isotonic (0,9%) sodium chloride solution is widely used as a
solvent for different injection solutions and as sodium chloride intravenous infusion.
Potassium chloride is used as antiarrythmic and the source of potassium ions.
Sodium bromide (Natrii bromidum), EP
NaBr
Potassium bromide (Kalii bromidum), EP
KBr
Preparation
Sodium and potassium bromides are manufactured from a bromide of iron,
FeBr2·2FeBr3, or Fe3Br8, which is produced by the action of bromine on moist iron
borings.
Fe + Br2 FeBr2
3FeBr2 + Br2 Fe3Br8 (FeBr2 . 2FeBr3)
Then this compound is boiled with the sodium or potassium carbonate solution
and filtered, and the filtrate is evaporated to dryness: t
о
Fe3Br8 + 4Na2CO3 + 4H2O 8NaBr + Fe(OH)2+ 2Fe(OH)3+ 4CO2
(K2CO3) (KBr)
The residue is then extracted with water and recrystallized.
Properties
Sodium bromide is a white, granular powder or small, colourless, transparent
or opaque crystals, slightly hygroscopic, freely soluble in water, soluble in alcohol.
Potassium chloride is a white, crystalline powder or colourless crystals, freely soluble
in water and in glycerol, slightly soluble in alcohol.
Identification is carried out using general identification reactions (EP) for
sodium, potassium and the reaction (a) for bromides with silver nitrate solution
(above).
There is one more non-pharmacopoeial reaction for bromides identification.
This is formation of a black precipitate when the copper sulphate (in the presence of
concentrated sulphuric acid) is added to the substance:
Cu2+
+ 2Br- CuBr2
This precipitate is destructed on addition of water.
Tests for purity
Bromates are determined by the interaction of bromate and bromide in the
presence of dilute sulphuric acid; with liberation of free bromine, which reacts with
potassium iodide in the presence of starch solution. Blue or violet colour should not
develop after 5 min.
BrO3- + 5Br
- + 6H
+ 3Br2 + 3H2O
Br2 + 2KI → I2 + 2KBr

32
Chlorides are determined by argentometric titration (Volhard method) after
boiling the solution with hydrogen peroxide concentrated solution in the presence of
dilute nitric acid. Bromides are more easily oxidized than chlorides, that is why
bromides are oxidized to bromine and only chlorides react with silver nitrate.
Iodides are determined by the adding of ferric chloride solution and the
coloration of chloroform layer. It should stay colourless.
2Fe3+
+ 2I- 2Fe
2+ + I2
Assay
Back argentometry by Volhard in the presence of dibuthyl phthalate (EP)
(s=1). Calculation is made with reference to the dried substance.
In calculating the result, a correction must be made to allow for the volume of
silver nitrate used in reacting with any chloride present (as determined by the test for
chloride):
The percentage content of NaBr is calculated from the expression:
a − 2,902 b
a = percentage content of NaBr and NaCl obtained in the assay and calculated
as NaBr; b = percentage content of Cl in the test for chlorides.
The percentage content of KBr is calculated from the expression:
a − 3,357 b
a = percentage content of KBr and KCl obtained in the assay and calculated as
KBr; b = percentage content of Cl in the test for chlorides.
The coefficients equal to the relations of molecular mass of the preparations to
the atomic mass of Cl.
Storage In airtight containers.
Usage The tranquillising preparations.
Sodium iodide (Natrii iodidum), EP
NaI
Potassium iodide (Kalii iodidum), EP
KI
Preparation
Sodium iodide and potassium iodide are prepared by processes exactly
analogous to those described for sodium (and potassium) bromide, but with iodine
instead of bromine.
Properties
Both preparations are white crystalline powders or colourless crystals. They are
very soluble in water, freely soluble in alcohol. Sodium iodide is deliquescent.
Identification is carried out using general identification reactions (E.P.) for
sodium, potassium and iodides (above).

33
Iodides can be also recognized by oxidation with the sodium nitrite solution:
2NaI + 2NaNO2 + 2H2SO4 I2 + 2NO + 2Na2SO4 + 2H2O
Ferric chloride solution can be also used as oxidizing agent:
2I- + 2Fe
3+ I2 + 2Fe
2+
Free iodine colours the chloroform layer violet.
Tests for purity
Iodates. The test for iodates is similar in principle to the test for bromate in
sodium bromide. To the solutions of preparations iodide-free starch solution and
dilute sulphuric acid are added. Blue colour should not develop.
IO3- + 5I
- + 6H
+ 3I2 + 3H2O
Thiosulphates are determined by the colour of the solution after adding of
starch solution and iodine. A blue colour should be produced.
I2 + 2Na2S2O3 2NaI + Na2S4O6
Assay
1. Iodatometry (EP). The solution is acidified with hydrochloric acid and
titrated with 0.05 M potassium iodate (until the colour changes from red to yellow).
This results first in liberation of iodine, but on further addition of iodate the iodine is
converted into iodine monochloride:
KIO3 + 5KI + 6HCl 3I2 + 6KCl + 3H2O
KIO3 + 2I2 + 6HCl 5ICl + KCl + 3H2O
Equation, which summarize the results of the reactions:
2KI + KIO3 + 6HCl 3ICl + 3KCl + 3H2O
To determine the end-point, the titration is continued, with vigorous shaking,
until most of the iodine has been converted into monochloride, as shown by the
decoloration of the added chloroform (s = 2). Calculation is made with reference to
the dried substance.
2. Argentometry (by Fajans). The solution of the substance is acidified with
acetic acid and then treated with 0,1 M silver nitrate using sodium eosinate as
indicator:
NaI + AgNO3 AgI + NaNO3
At the end-point the yellow precipitate of AgI colours pink due to the
adsorption of the indicator molecules on it (s = 1). Calculation is made with reference
to the dried substance.
3. Mercurymetric method (without indicator). The solution is titrated with
mercury nitrate solution (s = 4). At the end-point the red precipitate or mercury (II)
iodide is formed:
4KI + Hg(NO3)2 K2[HgI4] + 2KNO3
K2[HgI4] + Hg(NO3)2 2HgI2 + 2KNO3
Calculation is made with reference to the dried substance.
Storage Protected from light.
Usage Iodides are administered at the cases of a lack of iodine in the organism,

34
including endemic goitre. There are several preparations of eye drops, used for
treating of eye diseases (cataract etc.)
The preparations of iodine
Iodine (Iodum), EP
I2
Preparation
Iodine can be obtained from oil water or seaweeds (contain 0,5 % of iodine).
Professor Magidson O.Y. elaborated the process of iodine manufacturing from
oil water. This process consists of several stages:
1. Purification of oil water from the impurities of oil and naphthenic acids.
2. Oxidation of iodides containing in it into free iodine by sodium nitrite in the
presence of sulphuric acid:
2NaI + 2NaNO2 + 2H2SO4 I2 + NO + 2Na2SO4 + 2H2O
3. Adsorption of iodine by activated charcoal.
4. Desorption of iodine with the help of sodium hydroxide:
3I2 + 6NaOH 5NaI + NaIO3 + 3H2O
5. Subsequent oxidation of the iodides into free iodine by the action of
chlorine:
2NaI + Cl2 I2 + 2NaCl
6. Purification of iodine by the process of sublimation.
Properties
Iodine looks like greyish-violet, brittle plates or small crystals with a metallic
sheen. Preparation is very slightly soluble in water, soluble in alcohol, slightly
soluble in glycerol, very soluble in concentrated solutions of iodides. Solutions of
iodine in chloroform and carbon disulphide are violet (the same colour as iodine
vapour), solutions in water, alcohol, and aqueous iodides are reddish-brown.
Identification
1. A few fragments are heated in a test-tube. Violet vapour is evolved and a
bluish-black crystalline sublimate is formed (EP).
2. Iodine gives with starch solution a blue colour which disappears on warming
but reappears on cooling (EP). This is a very characteristic reaction of iodine.
Tests for purity
Bromides and chlorides. This test depends on the fact that silver iodide is
almost insoluble in dilute ammonia, but silver chloride is freely and silver bromide is
appreciably soluble, being reprecipitated on addition of excess of nitric acid.
Cl- + AgNO3 AgCl + NO3
-
AgCl + 2NH4OH [Ag(NH3)2]Cl + 2H2O
[Ag(NH3)2]Cl + 2HNO3 AgCl + 2NH4NO3

35
The solution obtained in this way is compared to the standard containing
0,01M hydrochloric acid mixed with water, dilute nitric acid and silver nitrate
solution.
Assay
A solution of the substance in aqueous potassium iodide is slightly acidified
with dilute acetic acid and titrated with 0,1M sodium thiosulphate, using starch
solution as indicator (s=1/2):
I2 + 2Na2S2O3 2NaI + Na2S4O6
Taking into consideration reaction of iodine with potassium iodide, the
equations can be represented as:
I2 + KI K[I3]
K[I3] + 2Na2S2O3 KI + 2NaI + Na2S4O6
Storage In airtight glass containers fitted with glass stoppers since the iodine
vapour attacks both cork and rubber.
Usage Antiseptic.
Iodine solution in alcohol 5% (Solutio Iodi spirituosa 5%)
Composition: Iodine – 5,0 g;
Potassium Iodide – 2,0 g;
Alcohol (96 %) – 41,0 g;
Purificated water to 100 g.
This solution contains 47,5 to 52,5 mg/ml of iodine, and 19,2 to 21,0 mg/ml of
potassium iodide.
Properties
A clear, reddish-brown liquid.
Identification
1. After adding of starch solution to the preparation blue-violet colour appear,
showing the presence of iodine.
2. Chloroform is added to the preparation till its decolouration (extraction of
iodine), then potassium is recognized with tartaric acid solution.
3. After the extraction of iodine (see 2) iodide identification is carried out using
the reaction (b) for iodides (above).
4. Iodine can be recognized by formation of iodoform, being the yellow
precipitate with characteristic odour:
C2H5OH + 4I2 + 6NaOH CHI3 + 5NaI + HCOONa + 5H2O
Assay
Quantity of iodine is determined by sodium thiosulphate titration using starch
solution as indicator; s =1/2:
I2 + 2Na2S2O3 2NaI + Na2S4O6

36
Quantity of potassium iodide is determined by Fajans method in the solution
obtained (s=1):
2NaI + KI + 3AgNO3 3AgI + KNO
3 + 2NaNO
3
Quantity of potassium iodide is calculated according to the formula:
analysisfor
OSNaAgNO
V
ТKVКVKI
100)(% 3223
Storage In an airtight amber bottles, protected from light.
Usage Antiseptic.
Iodine solution in alcohol 10% (Solutio Iodi spirituosa 10%)
Composition: Iodine – 100 g;
Alcohol (96 %) to 1 l.
This solution contains 9,5 to 10,5 % of iodine.
Properties
A clear, reddish-brown liquid.
Identification
See the identification (1) of iodine solution in alcohol 5%.
Tests for purity
Hydroiodic acid can form during the storage of the preparation:
I2 + C2H5OH → 2HI + CH3COH
This impurity is determined by the alkalimetric titration.
Assay As described under iodine (above).
Storage In an airtight amber bottles, protected from light. The preparation can
be stored for a month.
Usage Antiseptic.
Iodinole (Iodinolum)
Composition: Iodine – 1 g;
Potassium Iodide – 3 g;
Polyvinyl alcohol – 9 g;
Water to 1 l.
Iodinole is investigated as to the iodine solution in alcohol 5%.

37
Preparations of manganese
Potassium permanganate (Kalii permangаnas), EP
KMnO4
Preparation Manganese dioxide is fused with excess of potassium hydroxide in the
presence of a free supply of air. Then chlorine is passed through the solution:
2MnO4 +4KOH +O2 2K2MnO4 + 2H2O
2K2MnO4 + Cl2 2KMnO4 + 2KCl
Properties
A dark purple or brownish-black, granular powder or dark purple or almost
black crystals, usually having a metallic lustre, soluble in cold water, freely soluble in
boiling water. It decomposes on contact with certain organic substances.
Identification
1. After adding of dilute sodium hydroxide solution in the presence of alcohol a
green colour develops. After heating to boiling a dark brown precipitate is formed
(EP).
2KMnO4 + 3C2H5OH 2KOH + 2MnO2 + 3CH3CHO + 2H2O
2. After filtering the mixture obtained, the filtrate gives reaction of potassium
with sodium cobaltinitrite (EP, above).
3. After adding of hydrogen peroxide solution and dilute sulphuric acid to the
preparation it gets colourless:
2KMnO4 + 5H2O2 + 3H2SO4 2MnSO4 + K2SO4 + 5O2↑ + 8H2O
Assay
Iodometric titration after addition of potassium iodide, starch solution as
indicator (s=2/10 or 0,2):
2KMnO4 + 10KI +8H2SO4 2MnSO4 + 6K2SO4 + 5I2 + 8H2O
I2 + 2Na2S2O3 2NaI + Na2S4O6
Storage In an airtight container.
Usage Antiseptic.
Preparations of hydrogen peroxide
Hydrogen peroxide solution (3 per cent)
(Hydrogenii peroxydum 3 per centum)
Hydrogen peroxide solution (30 per cent)
(Hydrogenii peroxydum 30 per centum), EP
H2O2

38
Preparation
1. The part of hydrogen peroxide now produced is made by electrolytic
process. Sulphuric acid (40-68 %) is electrolyzed to give persulphuric acid; by
vacuum distillation (70-75C) this is hydrolyzed with formation of hydrogen
peroxide, which is removed as vapour.
H2SO
4 + HOH H
3O
+ + HSO
4
-
Cathode: Anode:
2H3O+ + 2e 2H2O 2HSO4
- - 2e 2HSO4
2H3O 2H2O + H2 2HSO4 H2S2O8
HOH
HOH
SO
O
OH
O
O
SO
OOH
H2O
2H
2SO
4+ + 2
2. Industrial manufacture of hydrogen peroxide is also carried out by air
autooxydation of alkylanthrahydroquinones (R = 2-ethyl, 2-tert-butyl, 2-penthyl): OH
OH
R
O
O
RO
2
H2O
2+
During the process the mixture of benzene with secondary alcohols is used,
hydrogen peroxide is extracted with water, distillated and rectificated.
Alkylanthraquinones formed in the process are reduced and used again.
Pharmacopeial preparation 3 % contains 2,5 to 3,5 per cent m/m of hydrogen
peroxide, yielding about 10 times its volume of oxygen on thermal decomposition.
Pharmacopeial preparation 30 % contains 29,0 to 31,0 per cent m/m of hydrogen
peroxide, yielding about 110 times its volume of oxygen on thermal decomposition.
Properties
Water solutions of hydrogen peroxide are colourless, clear liquids. Hydrogen
peroxide decomposes more or less rapidly in alkaline solutions, by the action of light
and heat, or under catalytic influences, particularly of iron, copper, or manganese
ions.
2H2O2 2H2O + O2↑
Stability of hydrogen peroxide is increased by addition of very small quantities
of inorganic or organic preservatives or stabilisers, such as sodium benzoate, boric
acid, urea etc.
Identification
1. After adding of dilute sulphuric acid and potassium permanganate solution
the solution becomes colourless or slightly pink (EP).
2KMnO4 + 5H2O2 + 3H2SO4 2MnSO4 + K2SO4 + 8H2O +5O2↑

39
2. The very characteristic and very delicate test for hydrogen peroxide consists
in adding ether and potassium chromate solution in the presence of dilute sulphuric
acid to the Preparation. A blue colour is being communicated to the ether on shaking.
The blue colour is due to perchromic acids formation (EP):
K2Cr2O7 + H2SO4 H2Cr2O7 + K2SO4
Cr OOH
O
O
Cr
O
O
OH H2O
2CrOH
O
O
O O Cr
O
O
OH H 2O
CrOH
O
O
O O Cr
O
O
OH H2O
2CrOH O O
O
O O
O
Cr OH
O O
O O
H 2O
+ +
+ 5 + 5
On staying the blue colour of ether layer gets green due to reduction Cr6+
Cr3+
.
3. It complies with the requirement for the content of H2O2 (EP).
Tests for purity
Test for acidity; organic stabilizers; and non-volatile residue.
Assay
Titration with potassium permanganate solution in the presence of dilute
sulphuric acid (EP) until a pink colour obtained (s=5/2 or 2,5).
5H2O2 + 2KMnO4 + 3H2SO4 2MnSO4 + K2SO4 + 8H2O +5O2
Storage Protected from light; if the solution does not contain a stabiliser, store
at a temperature below 15 °C. The label should state the name of the stabilizer.
Usage Antiseptic.
Hydroperite (Hydroperitum)
CONH
2
NH2
H2O
2.
Preparation
The preparation is obtained from the equal quantities of urea and hydrogen
peroxide. It is produced in tablets of white colour.
Properties
The citric acid solution 0,08 % is added as preservative. One tablet of
hydroperite is equivalent to 15 ml of hydrogen peroxide solution 3 %.
Identification
1. Urea in hydroperite solution is determined by the biuret test:

40
CONH
2
NH2
NH C O NH3
NH C OCONH
2
NH2
NH2
C
O
NH
C
O
NH2
O C
NH2
NH
C
NH
OHO C
NH2
NH
C
NH2
OKOH
Cu2+
O C
H2N
NH
C
N
OK
Cu
+
+
2
2. The presence of hydrogen peroxide is confirmed by perchromic acids
formation.
Assay
The same method as for the hydrogen peroxide solution. The preparation
contains 35% of hydrogen peroxide.
Storage Protected from light.
Usage Antiseptic.
Magnesium peroxide (Magnesii peroxydum)
(MgO2 + MgO)
Preparation
MgCl2 + 2KOH 2KCl + MgO + H2O
MgO + H2O2 MgO2 + H2O
Properties White odourless powder, practically insoluble in water, soluble in mineral
acids and boiling acetic acid. The preparation contains not less than 25,9 % of
magnesium peroxide.
Identification
1. Identification reaction for Mg2+
after dissolving the preparation in the
hydrochloric acid.
MgCl2 + Na2HPO4 + NH3 MgNH4PO4 + 2NaCl
2. Reaction of perchromic acids formation (above).
Assay
Permanganatometric titration (without indicator, s=5/2 or 2,5):
MgO2 + H2SO4 MgSO4 + H2O2
5H2O2 + 2KMnO4 +3H2SO4 2MnSO4 + K2SO4 + 8H2O + 5O2↑
Storage In an airtight container.
Usage Antacidic and disinfectant for the treatment of gastro-intestinal diseases.

41
Preparations of sulphur
Sodium thiosulphate (Natrii thiosulphas), EP
Na2S2O3 · 5H2O
Preparation
Oxidation of polisulphides:
2CaS2 + 3O2 2CaS2O3
CaS2O3 + Na2SO4 Na2S2O3 + CaSO4
Properties
Transparent, colourless crystals, efflorescent in dry air, very soluble in water,
practically insoluble in alcohol. It dissolves in its water of crystallisation at about 49
°C.
Identification
1. It decolourises iodinated potassium iodide solution (EP).
I2 + 2Na2S2O3 2NaI + Na2S4O6
2. After adding to the preparation of silver nitrate excess the white precipitate
is formed. It rapidly becomes yellowish, and then black (E.P.):
Na2S2O3 + 2AgNO3 Ag2S2O3 + 2NaNO3 (White)
Ag2S2O3 Ag2SO3 + S (Yellow)
Ag2SO3 + S + H2O Ag2S + H2SO4 (Black)
3. After adding to the preparation of water and hydrochloric acid a precipitate
of sulphur is formed and gas is evolved which gives a blue colour to starch iodate
paper (EP):
Na2S
2O
3 + 2HCl 2NaCl + SO
2 + S + H
2O
5SO2 + 2KIO3 → I2 + 4SO3 + K2SO4
4. Identification reaction (a) for sodium with potassium pyroantimonate (EP,
above).
Tests for purity
Sulphates and sulphites. After the adding of iodinated potassium iodide
solution to the substance sulphites are oxidized to sulphates. Then test for sulphates is
performed, using sulphates standard solution.
Sulphides are determined by reaction with sodium nitroprusside. The solution
should not become violet.
Na2S + Na2[Fe(CN)5NO] Na4[Fe(CN)5NOS]
Assay
Iodometric titration using starch solution as indicator, s=2:
I2 + 2Na2S2O3 2NaI + Na2S4O6
Storage In an airtight container.

42
Usage Detoxicant and desencibilisant as injections, sometimes as an external
insecticide.
Sodium sulphate decahydrate (Natrii sulphas decahydricus), EP
Na2SO4 10 H2O
Preparation
Anhydrous sodium sulphate is manufactured by heating sodium chloride with
concentrated sulphuric acid. The second stage requires a dull red heat:
NaCl + H2SO4 → NaHSO4 + HCl
NaCl + NaHSO4 → Na2SO4 + HCl
The decahydrate, which is also known as Glauber’s salt, is produced from
anhydrous sodium sulphate by crystallization from water.
Properties
White, crystalline powder or colourless, transparent crystals; freely soluble in
water, practically insoluble in alcohol. It partly dissolves in its own water of
crystallisation at about 33 °C.
Identification is carried out using general identification reactions (E.P.) for
sodium and sulphates (above). The substance should also comply with the test for
loss on drying (EP).
Assay
1. The substance is dissolved in water and titrated with 0,1M lead nitrate in the
presence of hydrochloric acid and methanol, determining the end-point
potentiometrically (EP). Calculation is made with reference to the dried substance
(s=1).
Na2SO4 + Pb(NO3)2 → PbSO4↓ + 2NaNO3
2. Gravimetric determination of sulphate by precipitation with barium chloride:
Na2SO4 + BaCl2 → BaSO4↓ + 2NaCl
Storage In an airtight container.
Usage Laxative preparation.
Sulphur for external use (Sulfur ad usum externum), EP
S
Preparation
The preparation is made by grinding of purificated sulphur (Sulphur
depuratum).
Properties
A yellow powder, practically insoluble in water, soluble in carbon disulphide,
slightly soluble in vegetable oils. The size of most of the particles is not greater than

43
20 µm and that of almost all the particles is not greater than 40 µm. It melts at about
120 °C.
Identification
1. Heated in the presence of air, it burns with a blue flame, emitting sulphur
dioxide which changes the colour of moistened blue litmus paper to red (EP).
S + O2 → SO2
SO2 + H2O → H+ + HSO3
-
2. The substance is oxidized with bromine water. The solution obtained gives
reaction (a) of sulphates with barium chloride (E.P., above).
3. The substance is dissolved in hot pyridine in the presence of sodium
hydrocarbonate. On boiling the solution, it colors bluish or green.
Assay
1. The oxygen-flask method (EP).
Sulphur is combusted to sulphuric acid (hydrogen peroxide solution is used as
an absorbent). The acid is titrated with 0,1M sodium hydroxide using phenolphthalein
solution as indicator. Blank titration is carried out (s=1/2).
S + O2 → SO2
SO2 + H2O2 → H2SO4
H2SO4 + NaOH → Na2SO4 + H2O
2. Alkalimetric back titration.
The substance is dissolved in standard potassium hydroxide alcohol solution:
12S + 6KOH → 2K2S5 + K2S2O3 + 3H2O
Then alcohol is distilled off, and the water and hydrogen peroxide concentrated
solution are added.
K2S5 + 8KOH + 16 H2O2 → 5K2SO4 + 20 H2O
K2S2O3 + 2KOH + 4H2O2 → 2K2SO4 + 4H2O
The equations can be summarize as:
S + 2KOH + 3H2O2 → K2SO4 + 4H2O
The excess of potassium hydroxide is titrated with hydrochloric acid solution,
using methyl orange as indicator. Blank titration is carried out (s=1/2).
KOH + HCl → KCl + H2O
Storage Protected from light.
Usage For the treatment of skin diseases, such as psoriasis, itch etc. Sometimes
it is used against intestinal worms.

44
Chapter 3. MEDICINAL PREPARATIONS CONTAINING
THE ELEMENTS OF 5TH
, 4TH
AND 3RD
GROUPS OF
D.I. MENDELEYEV PERIODIC SYSTEM
Plan
1. Preparations of nitrogen: (nitrous oxide, sodium nitrite, ammonia solution
concentrated).
2. Preparations of arsenic and bismuth: (arsenious trioxide for homoeopathic
preparations, bismuth subnitrate).
3. Preparations of carbon (activated charcoal, sodium hydrogen carbonate).
4. Preparations of boron and aluminium (boric acid, borax, aluminium oxide
hydrated).
Preparation of nitrogen
Nitrous oxide (Nitrogenii oxydum), EP
N2O
Preparation Nitrous oxide is produced from ammonium nitrate by thermic decomposition.
NH4NO3 t o
H2O + N2O 2
Then nitrous oxide is liquefied under 150 atmospheres pressure.
Properties Nitrous oxide is a colourless gas, heavier than the air, with a specific odour.
One part of nitrous oxide dissolves in about 1,5 volumes of water at 20 C and at a
pressure of 101 kPa.
Identification 1. Infrared absorption spectrophotometry, comparing with the reference
spectrum of nitrous oxide (EP).
2. Place a glowing splinter of wood in the substance to be examined. The
splinter bursts into flame (EP).
3. After introducing the substance into alkaline pyrogallol solution a brown
colour does not develop (EP).
4. After mixing of nitrous oxide with nitrogen oxide the red smoke doesn’t
appear (distinction from oxygen).
Tests for purity
Carbon dioxide, carbon monoxide, nitrogen monoxide, nitrogen dioxide, water.
Tests for purity are carried out by chromatographic methods.
Assay Method of gas chromatography (EP).

45
Storage Store liquefied under pressure in suitable containers (full metal
cylinders of grey colour). The taps and valves should not be greased or oiled.
Usage For inhalation as general anaesthetic in the mixture with oxygen (N2O –
80%, O2 – 20%).
Sodium nitrite (Natrii nitris), EP
NaNO2
Preparation Sodium nitrite is made by heating the sodium nitrate with metallic lead. The
product after cooling is extracted with water, and the solution is filtered from the
insoluble lead oxide and evaporated to crystallisation.
NaNO3 + Pb PbO + NaNO
2
Properties Colourless crystals or mass or yellowish rods, hygroscopic, freely soluble in
water, soluble in alcohol. Preparation is destructed on heating, or in the presence of
sand or glass:
3HNO2 HNO
3 + 2NO + H
2O
Identification
1. Formation of azodye (EP).
To the solution of the substance sulphanilic acid solution is added to form
diazonic salt. After adding of -naphthol solution and dilute sodium hydroxide
solution an intense red colour develops, showing the azodye formation.
NH2
SO3H
NaNO2
N+
N
SO3H
OH
NaOH
N
SO3H
N
NaO
Cl-
2. Identification reaction (b, EP) for sodium (above).
3. Identification reactions for nitrites:
a) the specific reaction for nitrite is formation of green-coloured product after
adding of phenazone and dilute sulphuric acid (EP):
C6H
5
NNO
C6H
5
NNO
ONCH3
CH3
CH3
CH3
NaNO2
HCl
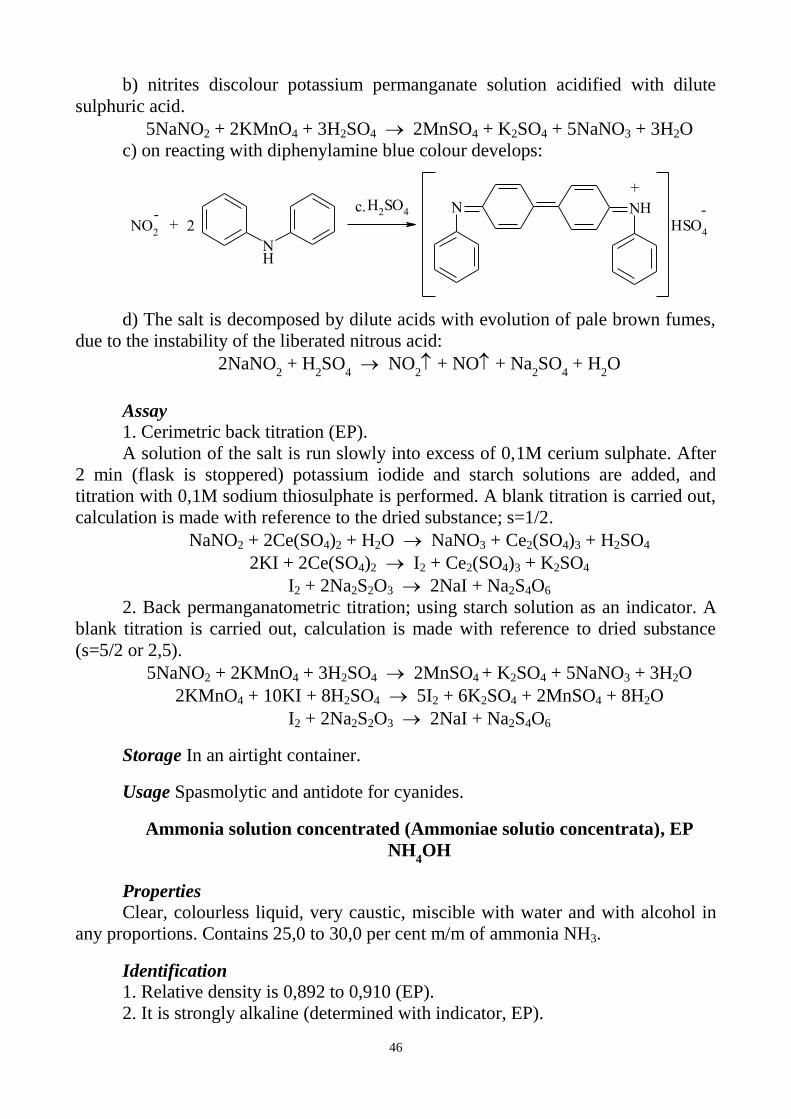
46
b) nitrites discolour potassium permanganate solution acidified with dilute
sulphuric acid.
5NaNO2 + 2KMnO4 + 3H2SO4 2MnSO4 + K2SO4 + 5NaNO3 + 3H2O
c) on reacting with diphenylamine blue colour develops:
NH
NO2
H2SO
4 NN
HSO4
2+- H
+
-c.
d) The salt is decomposed by dilute acids with evolution of pale brown fumes,
due to the instability of the liberated nitrous acid:
2NaNO2 + H
2SO
4 NO
2 + NO + Na
2SO
4 + H
2O
Assay
1. Cerimetric back titration (EP).
A solution of the salt is run slowly into excess of 0,1M cerium sulphate. After
2 min (flask is stoppered) potassium iodide and starch solutions are added, and
titration with 0,1M sodium thiosulphate is performed. A blank titration is carried out,
calculation is made with reference to the dried substance; s=1/2.
NaNO2 + 2Ce(SO4)2 + H2O NaNO3 + Ce2(SO4)3 + H2SO4
2KI + 2Ce(SO4)2 I2 + Ce2(SO4)3 + K2SO4
I2 + 2Na2S2O3 2NaI + Na2S4O6
2. Back permanganatometric titration; using starch solution as an indicator. A
blank titration is carried out, calculation is made with reference to dried substance
(s=5/2 or 2,5).
5NaNO2 + 2KMnO4 + 3H2SO4 2MnSO4 + K2SO4 + 5NaNO3 + 3H2O
2KMnO4 + 10KI + 8H2SO4 5I2 + 6K2SO4 + 2MnSO4 + 8H2O
I2 + 2Na2S2O3 2NaI + Na2S4O6
Storage In an airtight container.
Usage Spasmolytic and antidote for cyanides.
Ammonia solution concentrated (Ammoniae solutio concentrata), EP
NH4OH
Properties Clear, colourless liquid, very caustic, miscible with water and with alcohol in
any proportions. Contains 25,0 to 30,0 per cent m/m of ammonia NH3.
Identification 1. Relative density is 0,892 to 0,910 (EP).
2. It is strongly alkaline (determined with indicator, EP).

47
3. Through the solution air is bubbled. The gaseous mixture obtained is leaded
over the surface of a solution containing hydrochloric acid and methyl red solution.
The colour changes from red to yellow.
NH3 + HCl NH4Cl
After adding of sodium cobaltinitrite solution a yellow precipitate is formed.
2NH4Cl + Na3[Co(NO2)6] (NH4)2Na[Co(NO2)6] + 2NaCl
Assay
Back acidimetric titration or neutralization method. The measured volume of
the preparation is added to the weighed flask containing excess of hydrochloric acid,
which is then reweighed. The excess of the acid is titrated with 0,1M sodium
hydroxide using methyl red as indicator (s=1, EP).
NH4OH + HCl NH4Cl + H2O
HCl + NaOH NaCl + H2O
Storage Store protected from air, at a temperature not exceeding 20 °C.
Usage For faint and collapse, a person is given to inhale the vapour of the
solution.
Preparations of arsenic and bismuth
Arsenious trioxide for homoeopathic preparations
(Arsenii trioxidum ad praeparationes homoeopathicae), EP
As2O3
Preparation The preparation is manufactured by burning of orpiment As2S3, or arsenical
iron pyrites FeAsS in the current of air:
2As2S3 + 9O2 2As2O3 + 6SO2
2FeAsS + 5O2 As2O3 + 2SO2 + Fe2O3
Properties White or almost white powder, practically insoluble to sparingly soluble in
water. It dissolves in solutions of alkali hydroxides and carbonates, with formation of
the corresponding meta-arsenites, e.g.:
As2O3 + 2NaOH 2NaAsO2 + H2O
As2O3 + Na2CO3 2NaAsO2 + CO2
It dissolves also in concentrated hydrochloric acid (exhibiting weak basic
properties):
As2O3 + 6HCl 2AsCl3 + H2O
Identification 1. Sodium sulphide solution, added to a solution of the preparation in dilute
hydrochloric acid, gives a yellow precipitate, which is soluble in dilute ammonia
(EP).

48
As2O3 + 6HCl 2AsCl3 + 3H2O
2AsCl3 + 3Na2S As2S3+ 6NaCl
2. Hypophosphorous reagent, added to a solution of the preparation in dilute
hydrochloric acid and heated on a water-bath, gives a black precipitate (EP):
As2O3 + 6HCl 2AsCl3 + 3H2O
NaH2PO2 + HCl H3PO2 + NaCl
2AsCl3 + 3H3PO2 + 3H2O 2As + 3H3PO3 + 6HCl
3. After adding to the solution of the preparation silver nitrate solution a yellow
precipitate is formed. It dissolves on addition of nitric acid and ammonia.
Na3AsO3 + 3AgNO3 Ag3AsO3 + 3NaNO3
Assay
1. Iodometric titration (EP).
The substance is dissolved in dilute sodium hydroxide solution. Then the
mixture is neutralized with dilute hydrochloric acid, and sodium hydrogen carbonate
is added (for interaction with hydroiodic acid). The solution is titrated with iodine
using starch solution as indicator; s=1/2.
As2O3 + 2I2 + 2H2O As2O5 + 4HI
HI + NaHCO3 NaI + CO2 + H2O
2. Direct bromatometric titration using methyl red is an indicator. At the end-
point indicator gets colourless due to its oxidation; s=3/2 or 1,5.
3As2O3 + 2KBrO3 3As2O5 + 2KBr
5KBr + KBrO3 + 6HCl 3Br2 + 6KCl + 3H2O
Storage In an airtight container.
Usage In homoeopathic preparations, sometimes in stomatology.
Bismuth subnitrate, heavy (Bismuthi subnitras, ponderosum), EP
4[BiNO3(OH)2], BiO(OH)
Bismuth subnitrate is a basic salt of variable composition:
Bi OH
OH
NO3
BiO
OHBi
O
NO3
BiONO
3
BiOOH
, ,,
Preparation Bismuth ochre Bi2O3 is heated with charcoal to give metallic bismuth. Then
metallic bismuth is dissolved in nitric acid.
Bi2O3 + 3C 2Bi + 3CO
Bi + 4HNO3 Bi(NO3)3 + NO + 2H2O

49
Bi
NO3
NO3
NO3
- HNO3
Bi
NO3
NO3
OH - HNO3
BiO
NO3
- HNO3
Bi OH
OH
NO3
- HNO3
BiO
OH
H2O
H2O
Properties This is a white powder, practically insoluble in water and in alcohol. It
dissolves in mineral acids with decomposition.
Wet with water powder makes blue litmus paper red (pH7).
BiONO
3
BiOOH
BiO
OH+ H2O HNO3 + 2
Identification 1. Potassium iodide, added to a solution of the preparation, gives a black
precipitate of bismuth iodide, soluble in excess of precipitant to form an orange
solution containing potassium iodobismuthite (EP).
Bi(NO3)3 + 3KI BiI3 + 3KNO3
BiI3 + KI K[BiI4]
2. Solutions of bismuth salts acidified with nitric acid give a yellowish-orange
colour with thiourea (EP):
Bi3 3(NH2)2CS [Bi((NH2)2CS)3]
3
3. Nitrates are determined according to general identification reactions (EP,
above).
4. The pH of the solution of the preparation should be measured (EP).
5. After heating of the preparation yellow-brown fumes and bright yellow
precipitate are formed:
Bi O BiOOH
NO3
Bi2O
3 NO O2
H 2O+ + +
t 0
2 2 1/22
Assay
Complexonometric titration (EP). The substance is dissolved in a mixture of
perchloric acid and water and titrated with 0,1M sodium edetate, using xylenol
orange triturate as indicator; s=1.
Bi3+
SO3Na
CH3
OH
CH2
CH3
O
CH2
N
CH2COONa
CH2COONa
N
NaOOCH2C
NaOOCH2C 3Na +-
+
(or Na3Ind)
BiInd
HClO4

50
Bi3+
CH2
CH2
N
N
CH2COO
CH2COO
CH2COO
CH2COONa
Bi
CH2
CH2
N
N
CH2COONa
CH2COOH
CH2COOH
CH2COONa
H+
Na+
+ + 2+
CH2
CH2
N
N
CH2COONa
CH2COOH
CH2COOH
CH2COONa
CH2
CH2
N
N
CH2COO
CH2COO
CH2COO
CH2COONa
Bi
H
+
Na+
Ind3-
+ +BiInd
2
_
_
The percentage of Bi is calculated, with reference to the dried substance.
Storage In an airtight container.
Usage Antiseptic, for the treatment of certain gastro-intestinal diseases.
Preparations of carbon
Charcoal, activated (Carbo activatus), EP
C
Preparation
Activated charcoal is obtained from vegetable matter by suitable carbonisation
processes intended to confer a high adsorption power. This preparation is usually
obtained by a dry distillation of foliar trees without air presence. For more adsorb
properties medicinal charcoal is steamed at 180C. During this process the resinous
substances are removed. The best charcoal’s brands are obtained by treating with zinc
chloride, magnesium chloride, sodium hydroxide or phosphoric acid solutions with
the following heating to 300-400C. Then charcoal is carefully purified by washing,
and dried.
In the preparation there are micropores (we can see them in microscope, their
diameter is 10-1
to 10-3
cm) and ultrapores (we can’t see them in microscope, their
diameter is about 9,2.10-7
cm). Ultrapores are essential for the process of adsorption.
The general pores surface of 1 g of activated charcoal is about 103 m
2.
Properties
A black, light powder free from grittiness, practically insoluble in all usual
solvents.
Identification
1. When heated to redness it burns slowly without a flame (EP).

51
2. It complies with the test for adsorption power (EP).
Tests for purity
Adsorption power. Adsorption of phenazone by the weighed quantity of
charcoal, mixed with water is measured. After shaking for 15 min the quantity of
phenazone that has not been absorbed is determined by bromatometric titration, using
methyl red as indicator. Blank titration is carried out.
Not less than 40 g of phenazone is adsorbed per 100 g of activated charcoal,
calculated with reference to the dried substance.
The adsorption power of preparation can also be determined with the
methylene blue.
Since activated charcoal is usually used at high dosage Pharmacopoeia
demands high purity of this Preparation. The preparation can’t contain sulphides
(determined using hydrochloric acid and lead acetate paper).
The quantities of copper, lead and zinc are determined by atomic absorption
spectrometry.
Storage Store in an airtight container.
Usage Preparation is used at different intoxications.
Sodium hydrogen carbonate (Natrii hydrocarbonas), EP
NaHCO3
Preparation Sodium hydrogen carbonate is manufactured by the ammonia-soda process (or
method by Solve). Strong brine containing a high concentration of ammonia is passed
through a “carbonating tower” where it is saturated with carbon dioxide under
pressure. The ammonia and carbon dioxide react to form ammonium bicarbonate, and
this, undergoing decomposition with the sodium chloride, causes the precipitation of
sodium bicarbonate, which is not very soluble in water. The reactions are reversible,
but the greater part of the sodium chloride is eventually converted into sodium
bicarbonate. Then it is separated by filtration.
NH3 + CO2 + H2O NH4HCO3
NH4HCO3 + NaCl NH4Cl + NaHCO3
Properties
Preparation is a white crystalline powder. Preparation is soluble in water,
practically insoluble in alcohol. When heated in the dry state or in solution, it
gradually changes into sodium carbonate.
Identification
It is carried out using general identification reactions (EP) for sodium,
carbonates and bicarbonates (above).
After adding of phenolphthalein to the water solution of the preparation a pale
pink colour is produced. After heating gas is evolved, and the solution becomes red
(EP).

52
NaHCO3 + H2O NaOH + CO2 + H2O
Tests for purity
Appearance of solution, carbonates, chlorides, sulphates, ammonium, arsenic,
calcium, iron, and heavy metals. The test for carbonates requires that a 5% w/v
solution shall have a pH not greater than 8,6.
Assay
Acidimetric titration, using methyl orange as indicator (EP, s=1):
NaHCO3 + HCl NaCl + CO2 + H2O
Storage In an airtight container.
Usage Antacidic.
Preparations of boron and aluminium
Boric acid (Acidum boricum), EP
H3BO3
Preparation
1. Boric acid is produced by decomposing of crude borax or tincal, Na2B4O7
10H2O, of kernite, Na2B4O7 4H2O with sulphuric acid. Mixture of concentrated
sulphuric acid and water is added to a boiling solution of borax in water.
Na2B4O7 10H2O + H2SO4 4H3BO3 + Na2SO4 + 5H2O
The hot liquid is filtered and set aside to crystallize. The boric acid is then
filtered off, washed until free from sulphate, and allowed to dry at ordinary
temperatures.
2. Boric acid can be also manufactured from asharite: 110 °C
B2O3 2MgO + 2H2SO4 → 2MgSO4 + 2H3BO3
Properties This is a white, crystalline powder, colourless, shiny plates greasy to the touch,
or white crystals, soluble in water and in alcohol, freely soluble in boiling water and
in glycerol (85 per cent). When heated, boric (orthoboric) acid loses water in three
stages: 100 C
H3BO3 HBO2 + H2O metaboric acid
160 C
4HBO2 H2B4O7 + H2O pyroboric acid
red heat
H2B4O7 2B2O3 + H2O boron trioxide
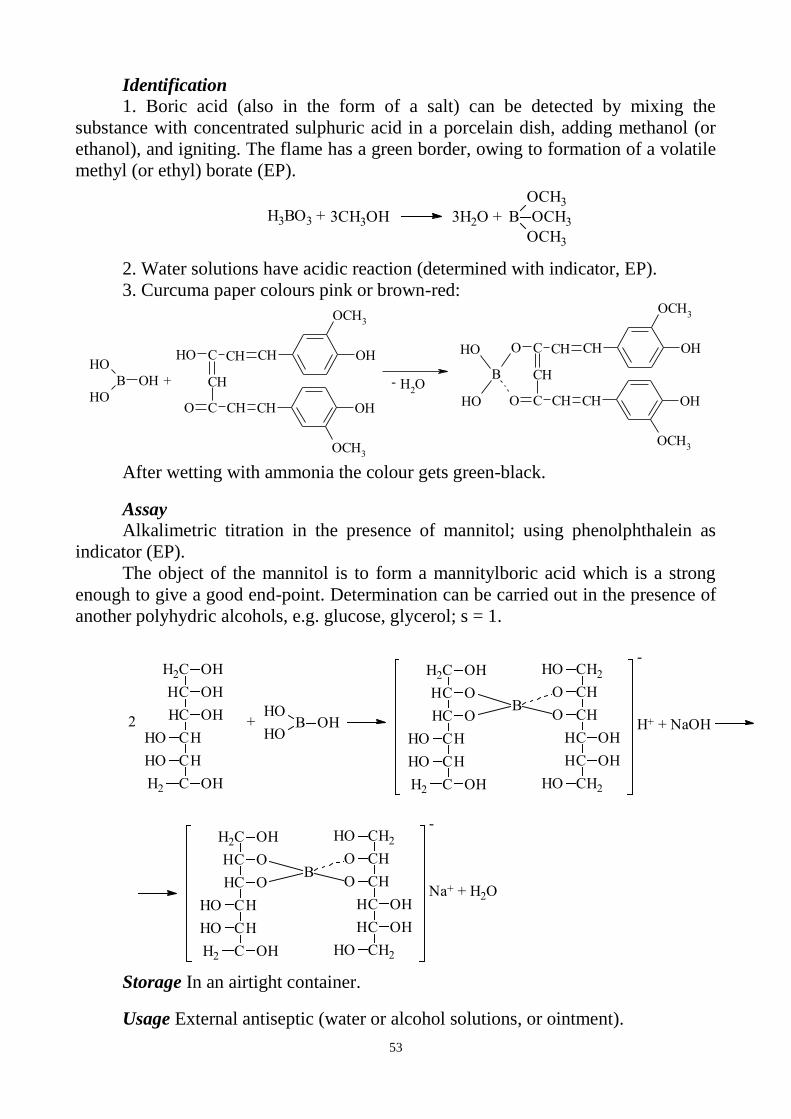
53
Identification 1. Boric acid (also in the form of a salt) can be detected by mixing the
substance with concentrated sulphuric acid in a porcelain dish, adding methanol (or
ethanol), and igniting. The flame has a green border, owing to formation of a volatile
methyl (or ethyl) borate (EP).
H3BO3 + 3CH3OH 3H2O + B
OCH3
OCH3
OCH3
2. Water solutions have acidic reaction (determined with indicator, EP).
3. Curcuma paper colours pink or brown-red:
B OH
OH
OH- OH
2
CH OHCHC
CH
OH
OCH3
CH OHCHCO
OCH3
CH OHCHC
CH
O
OCH3
B
O CH OHCHC
OCH3
OH
OH
+
After wetting with ammonia the colour gets green-black.
Assay
Alkalimetric titration in the presence of mannitol; using phenolphthalein as
indicator (EP).
The object of the mannitol is to form a mannitylboric acid which is a strong
enough to give a good end-point. Determination can be carried out in the presence of
another polyhydric alcohols, e.g. glucose, glycerol; s = 1.
H2C
C
C
C
C
C OHH2
HO
HO
OH
OH
OH
H
H
H
H
2 + B OHHO
HO
H
H
H
H
H
H
B
OH
OH
HO
O
O
CH2
C
C
CH
CH
CH2
O
O
OH
HO
HO
H2 OHC
C
C
C
C
H2C HO-
H+ + NaOH
-
B
OH
OH
HO
O
O
CH2
C
C
CH
CH
CH2
O
O
OH
HO
HO
H2 OHC
C
C
C
C
H2C HO
H
H
H
H
H
H
Na+ + H2O
Storage In an airtight container.
Usage External antiseptic (water or alcohol solutions, or ointment).

54
Borax (Borax), EP
Na2B4O7 1OH2O
Preparation
Preparation is obtained from native calcium borate by a process involving
decomposition with hot sodium carbonate solution. Calcium carbonate is filtered off
and cold solution is set aside for crystallization of sodium tetraborate.
CaB4O7 + Na2CO3 Na2B4O7 + CaCO3
Properties
A white, crystalline powder, colourless crystals or crystalline masses,
efflorescent, soluble in water, very soluble in boiling water, freely soluble in glycerol.
Identification 1. See identification of boric acid (EP, above).
2. After adding phenolphthalein solution into the solution of preparation it is
red (alkaline medium). On the addition of glycerol (85 per cent) the colour disappears
(solution becomes acidic, E.P.).
3. Sodium is determined using general identification reactions (EP) for sodium
(above).
Assay
1. Alkalimetric titration of mannitol neutralised solutions using
phenolphthalein as an indicator; (EP, s=1/2):
Na2B
4O
7
CH2OH
CH OH
COH H
CH OH
C OHH
CH2OH
CH2OH
CH O
CH O
COH H
C HOH
CH2OH
B
CH2OH
C HO
C HO
C OHH
CH OH
CH2OH
CH2OH
CH O
CH O
COH H
C HOH
CH2OH
B
CH2OH
C HO
C HO
C OHH
CH OH
CH2OH
+ 4 2Na+
+ 2H+
Na2B4O7 + 7H2O 4H3BO3 + 2NaOH
H2C
C
C
OH
OH
OH
H
H2
+ H3BO3
CH2
CH
CH2
HO
HO
HO H2
H
H2C
C
C
OH
O
OB
O
O
HO
CH2
CH
CH2
-
H+ NaOH
-H2C
C
C
OH
O
OB
O
O
HO
CH2
CH
CH2
H
H2
Na+

55
CH2OH
CH O
CH O
COH H
C HOH
CH2OH
B
CH2OH
C HO
C HO
C OHH
CH OH
CH2OH
CH2OH
CH O
CH O
COH H
C HOH
CH2OH
B
CH2OH
C HO
C HO
C OHH
CH OH
CH2OH
2H+
+ 2NaOH 2Na+
2. Acidimetric titration, using methyl orange as indicator (s=1/2):
Na2B
4O
7 + 2HCl + 5H
2O 4H
3BO
3 + 2NaCl
Storage In an airtight container.
Usage Antiseptic.
Preparations of aluminium
Aluminium oxide hydrated (Aluminii oxydum hydricum), EP
Al(OH)3
Preparation
Al2(SO
4)
3 + 6NH
4OH 2Al(OH)
3 + (NH
4)
2SO
4 60 ºC
or 2KAl(SO4)
2 + 6NH4OH 2Al(OH)
3 + K
2SO
4 + 3(NH
4)
2SO
4
alum
Properties
A white, amorphous powder, practically insoluble in water. The preparation
dissolves in dilute mineral acids and in solutions of alkali hydroxides.
Identification 1. Aluminium is determined using general identification reactions (EP) for
aluminium (above).
2. After roasting of the preparation with cobalt nitrate solution cobalt aluminate
(cobalt blue) is formed:
2Al(OH)3 Al2O3 + 3H2O
2Co(NO3)2 2CoO +4NO2 + O2
Al2O3 + CoO Co(AlO2)2
Assay
1 Complexometric back titration of aluminium (EP, s=1/2).
An excess of sodium edetate is added to the neutral solution of the preparation
(prepared using HCl and NH4OH), then it is titrated with 0,1 M zinc sulphate, using
solution of dithizone in ethanol as indicator (colour changes from greenish-blue to
reddish-violet); medium of ammonium acetate and dilute acetic acid. Calculation is
made for Al2O3.
56
Al3+
CH2
CH2
N
N
CH2COO
CH2COO
CH2COO
CH2COONa
AlCH
2
CH2
N
N
CH2COONa
CH2COOH
CH2COOH
CH2COONa
H+
Na+
+
-
+
2
CH3COOH
CH3COONH4
CH2
CH2
N
N
CH2COONa
CH2COOH
CH2COOH
CH2COONa
CH2
CH2
N
N
CH2COONa
CH2COO
CH2COO
CH2COONa
Zn+ ZnSO4
CH3COOH
CH3COONH4
- H2SO4
NN
NHNH
CS
NN
Zn
NN
CSZnSO4+
- H2SO4
CH3COOH
CH3COONH4
2. Gravimetric method. The substance is roasted to give aluminium oxide.
Storage In an airtight container, at a temperature below 30ºC.
Usage Antacidic.

57
Chapter 4. MEDICINAL PREPARATIONS CONTAINING THE
ELEMENTS OF 2ND
GROUP OF
D.I. MENDELEYEV PERIODIC SYSTEM
Plan
1. Preparations of magnesium (light and heavy magnesium oxides,
magnesium sulphate heptahydrate, light and heavy magnesium carbonates).
2. Preparations of calcium (calcium chlorides: dihydrate and hexahydrate).
3. Preparation of barium (barium sulphate).
4. Preparations of zinc (zinc oxide, zinc sulphate heptahydrate).
5. Preparations of mercury (mercuric chloride, mercury oxide, mercury
oxicyanide).
Preparations of magnesium
Magnesium is widely distributed, the most important native sources are the
minerals dolomite (MgCO3.CaCO
3); magnesite (MgCO
3), talc (3MgO.4SiO
2.H
2O),
and carnallite (MgCl2KCl6H2O). The magnesium salts are also found in seawater
and waters of many mineral springs.
Magnesium oxide, light (Magnesii oxydum leve)
Magnesium oxide, heavy (Magnesii oxydum ponderosum), EP
MgO
Preparation Magnesium oxide (light) is produced by heating of magnesium light carbonate
at 250о-350
оС until carbon dioxide is no longer evolved:
3MgCO3 . Mg(OH)
2 . 3H
2O 4MgO + 3CO
2 + 4H
2O
Properties Magnesium oxide is fine, white, amorphous powder, practically insoluble in
water. It dissolves in dilute acids with at most slight effervescence.
Identification 1. 15 g of magnesium oxides has an apparent volume before settling of about
150 ml (light oxide) or 30 ml (heavy oxide) (EP).
2. Preparation is identified after dissolving in nitric acid with following
neutralisation with sodium hydroxide (EP):
MgO + 2HNO3 Mg(NO3)2 + H2O
Mg2+
+ 2OH- Mg(OH)2
Mg(OH)2 + 2NH4+ Mg
2+ + 2NH4OH
Mg2+
+ Na2HPO4 + NH4OH MgNH4PO4 + 2Na+ + H2O
3. It complies with the test for loss on ignition (EP).
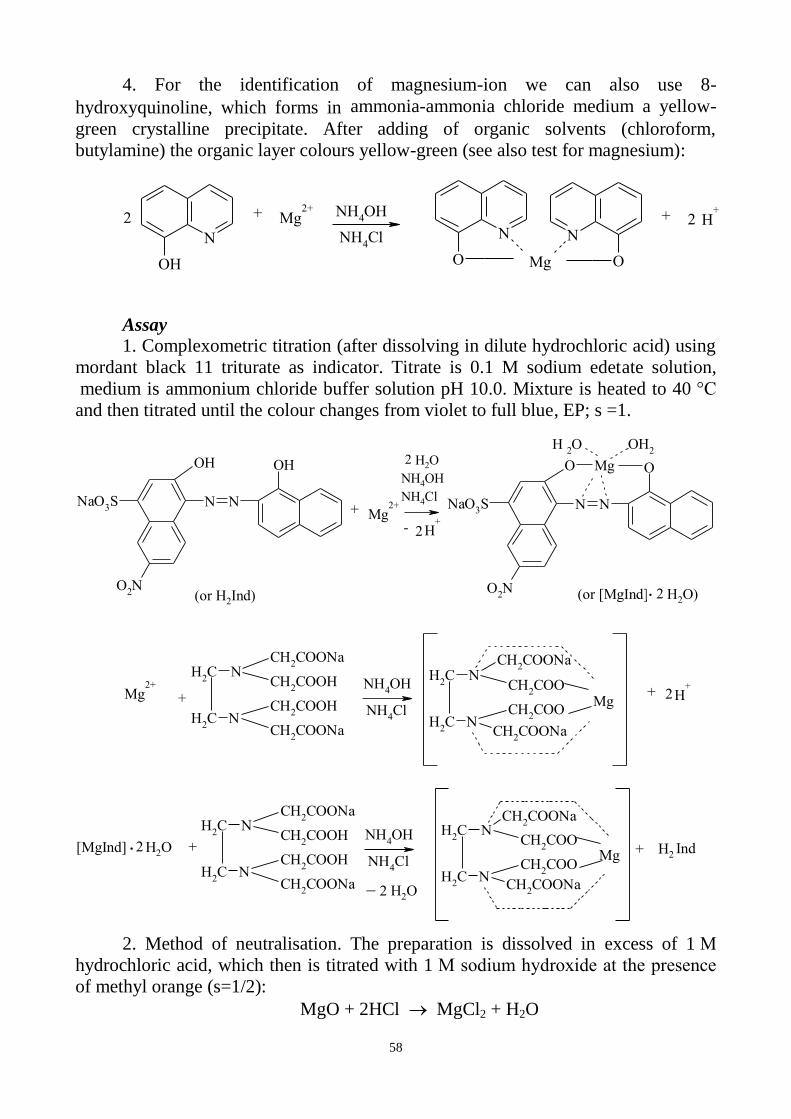
58
4. For the identification of magnesium-ion we can also use 8-
hydroxyquinoline, which forms in ammonia-ammonia chloride medium a yellow-
green crystalline precipitate. After adding of organic solvents (chloroform,
butylamine) the organic layer colours yellow-green (see also test for magnesium):
N
OH
Mg2+
N
O
N
OMg
H+NH
4OH
NH4Cl
+2 + 2
Assay
1. Complexometric titration (after dissolving in dilute hydrochloric acid) using
mordant black 11 triturate as indicator. Titrate is 0.1 M sodium edetate solution,
medium is ammonium chloride buffer solution pH 10.0. Mixture is heated to 40 °C
and then titrated until the colour changes from violet to full blue, EP; s =1.
N
O
NaO3S
O2N
N
OMg
H 2O
N
OH
NaO3S
O2N
N
OH
Mg2+
OH2
H+
(or [MgInd] H2O) 2.
+
2-
(or H2Ind)
H2O
NH4OH
NH4Cl
2
CH2
CH2
N
N
CH2COONa
CH2COO
CH2COO
CH2COONa
Mg
CH2
CH2
N
N
CH2COONa
CH2COOH
CH2COOH
CH2COONa
Mg2+
H+NH
4OH
NH4Cl
++ 2
CH2
CH2
N
N
CH2COONa
CH2COO
CH2COO
CH2COONa
MgInd
CH2
CH2
N
N
CH2COONa
CH2COOH
CH2COOH
CH2COONa
NH4OH
NH4Cl
+ + H2[MgInd] H2O 2.
_ 2 H2O
2. Method of neutralisation. The preparation is dissolved in excess of 1 М
hydrochloric acid, which then is titrated with 1 М sodium hydroxide at the presence
of methyl orange (s=1/2):
MgO + 2HCl MgCl2 + H2O

59
HCl + NaOH NaCl + H2O
Storage Preparation should be stored in an airtight container, because
magnesium oxide can react with atmospheric carbon dioxide and water, with the
formation of magnesium carbonate and hydroxide respectively:
MgO + CO2 MgCO3↓
MgO + H2O Mg(OH)2↓
Usage Antacidic.
Magnesium sulphate heptahydrate (Magnesii sulfas heptahydricus), EP
MgSO4 · 7H2O
Preparation Preparation is manufactured by the action of sulphuric acid on magnesite:
MgCO3 + H2SO4 MgSO4 + CO2 + H2O
The liquid is filtrated and filtrate is evaporated to crystallisation. The excess of
sulphuric acid is taken to avoid hydrolysis of magnesium sulphate and formation of
basic salts, e.g. Mg(OH)OSO3H.
Properties Preparation is a white, crystalline powder or brilliant, colourless crystals, freely
soluble in water, very soluble in boiling water, practically insoluble in alcohol,
effloresce slightly in warm air.
Identification Identification is carried out using general identification reactions (EP) for
magnesium and sulphates (above).
Assay
Complexometric titration (see magnesium oxide, light) The calculation is made
with reference to the dried substance (s = 1, EP).
Storage In an airtight container.
Usage Prescribed as sedative, spasmolytic, analgesic, anticonvulsant, tocolytic,
for the treatment of hypertension and eclampsia (intramuscular, intravenous
injections); as antidote when poisoning of heavy metals, arsenic, tetraethyl lead,
soluble barium salts. Prescribed orally, it has holagogic and laxative action.
Magnesium carbonate, light (Magnesii subcarbonas levis), EP
Magnesium carbonate, heavy (Magnesii subcarbonas ponderosus), EP
Hydrated basic magnesium carbonate
3MgCO3 · Mg(OH)2 · 3H2O

60
Preparation
4MgSO4 + 4Na2CO3 + 4H2O 3MgCO3 Mg(OH)2 3H2O + 4Na2SO4 +
CO2
To prepare magnesium carbonate heavy, magnesium sulphate and sodium
carbonate are dissolved separately in boiling water. The solutions are mixed and
evaporated to dryness, and the residue is digested with boiling water. The insoluble
carbonate is then separated, washed until free from sulphate and dried.
To prepare magnesium carbonate light, cold solutions of magnesium sulphate
and sodium carbonate are mixed and boiled for 15 min, and the precipitate of
magnesium carbonate is then separated, washed until free from sulphate, and dried.
Properties Preparations are white powders, practically insoluble in water. They dissolve in
dilute acids with strong effervescence.
Identification 1. 15 g of magnesium carbonate has an apparent volume before settling of
about 180 ml (light carbonate) or 30 ml (heavy carbonate) (EP).
2. Identification is carried out using general identification reactions (EP) for
magnesium (after dissolving in dilute nitric acid and neutralizing with dilute sodium
hydroxide solution) and carbonates (above).
Assay
Complexometric titration (see Magnesium oxide, light) after dissolving
preparation in a mixture of water and dilute hydrochloric acid (s = 1, EP). The
content is calculated as MgO.
Storage In an airtight container.
Usage Astringent and antacidic.
Preparations of calcium
Calcium occurs in nature as CaCO3, in such minerals as chalk, marble,
limestone, and associated with MgCO3 in dolomite. CaSO4 occurs as gypsum (CaSO4
2H2O); the fluoride, CaF2, occurs as fluorspar. Bones contain a high percentage of
calcium phosphate.
Calcium chloride dihydrate (Calcii chloridum dihydricum), EP
CaCl2 2H2O
Calcium chloride hexahydrate (Calcii chloridum hexahydricum), EP
CaCl2 6H2O

61
Preparation Calcium chloride usually is prepared by the dissolving of calcium carbonate in
slight excess of hot hydrochloric acid, filtering when the reaction is complete, and
evaporating to a syrup which crystallizes when cooled.
CaCO3 + 2HCl CaCl2 + H2O + CO2
Impurities of magnesium and ferrous salts, which can be presented in certain
minerals, in the process of Preparation change into chlorides MgCl2 and FeCl2.
Solution obtained is saturated with gaseous chlorine (FeCl2 is oxidized into FeCl3),
and then an excess of calcium hydroxide is added:
2FeCl3 + 3Ca(OH)2 2Fe(OH)3 + 3CaCl2
MgCl2 + Ca(OH)2 Mg(OH)2 + CaCl2
During this process the concentration of calcium chloride increases. Excess of
calcium hydroxide is modified into calcium chloride with hydrochloric acid:
Ca(OH)2 + 2HCl CaCl2 +2H2O
Properties White, crystalline mass or colourless crystals, very soluble in water, freely
soluble in alcohol. Preparations freeze at about 29C. The crystals deliquesce in moist
air, effloresce in dry air, and melt in their water of crystallization at about 34C.
Identification
1. Identification is carried out using general identification reactions (EP) for
calcium and chlorides (above).
2. The preparations comply with the limits of the assay (EP).
Assay
1. Complexometric titration using calconecarboxylic acid triturate as
indicator
(s = 1, EP):
N N
OHHOOC OH
SO3HCa N N
ONaOOC O
SO3Na
Ca
H 2O OH
2
NaOH
H+
2 H2O
(or [CaInd] 2 H2O).
2+ +
2_
(or H2Ind)
CH2
CH2
N
N
CH2COONa
CH2COO
CH2COO
CH2COONa
Ca
CH2
CH2
N
N
CH2COONa
CH2COOH
CH2COOH
CH2COONa
Ca2+
H+NaOH
++ 2

62
Titrate is 0,1M sodium edetate, medium is strong sodium hydroxide solution.
Solution is titrated until the colour changes from violet to full blue.
CH2
CH2
N
N
CH2COONa
CH2COO
CH2COO
CH2COONa
Ca Ind
CH2
CH2
N
N
CH2COONa
CH2COOH
CH2COOH
CH2COONa
NaOH+ + H2
[CaInd] H2O 2._
2 H2O
2. Argentometry; s=1/2.
3. Mercurimetry; s=1.
Storage In an airtight container.
Usage Calcium chloride solution is prescribed for allergic diseases; it has
haemostatic and anti-inflammatory properties, and reduces the penetrability of blood
vessel wall. Calcium chloride is also a specific antidote for poisoning with
magnesium preparations; it is used for hypofunction of parathyroid glands. Calcium
chloride solution is prescribed as intravenous injections or orally (being injected
intramuscular, it causes necrosis).
Preparation of barium
From the barium salts only barium sulphate is used in medicine, because it is
not soluble either in water or in acids and alkalis (the soluble barium salts are toxic).
In nature barium occurs in form of sulphate – heavy spar or barytes (BaSO4) and
carbonate – witherite (BaCO3).
Barium sulphate (Barii sulfas), EP
BaSO4
Preparation
Soluble barium chloride is prepared from the minerals:
BaSO4 + 4C → BaS + 4CO↑
BaS + 2HCl →BaCl2 + H2S↑
BaCO3 + 2HCl → BaCl2 + H2O + CO2↑
Then the substance is prepared by precipitation from a solution of barium
chloride by addition of a soluble sulphate (or sulphuric acid):
BaCl2 + Na2SO4 BaSO4 + 2NaCl
To obtain fine dispersed barium sulphate solutions during precipitation should
be much diluted, and a protective colloid (e.g. linseed infusion) should be added.
Properties A fine, heavy, white powder, free from gritty particles, practically insoluble in
water and in organic solvents. It is very slightly soluble in acids and in solutions of
alkali hydroxides.

63
Identification 1. To identify barium sulphate it has to be transformed into soluble salts.
Preparation is heated with sodium carbonate solution: to
BaSO4 + Na2CO3 BaCO3 + Na2SO4
The precipitate is filtered off. The filtrate is acidified with dilute hydrochloric
acid and then used for the sulphate identification (above, EP).
2. The residue collected in the preceding test is washed with water, then treated
with dilute hydrochloric acid, and filtered. Then dilute sulphuric acid is added to the
filtrate to determine barium ion. A white precipitate of barium sulphate is insoluble in
dilute sodium hydroxide solution.
BaCO3 + 2HCl BaCl2 + H2O + CO2
BaCl2 + H2SO4 BaSO4 + 2HCl
Tests for purity
Barium sulphate is used internally in high dosage (50-100 g) therefore
pharmacopoeia gives strict requirements to the purity of the preparation:
tests for acidity or alkalinity, acid-soluble substances, oxidisable sulphur compounds,
soluble barium salts, phosphates, arsenic, heavy metals, loss on ignition; and
sedimentation.
Oxidisable sulphur compounds are determined using the reaction with
potassium iodide and potassium iodate solutions in the presence of starch solution.
5SO2 + 2KIO3 → I2 + 4SO3 + K2SO4
5KI + KIO3 3I2 + 3H2O + 6KCl
The blue colour of the solution should be more intense than that of a standard
prepared without potassium iodate.
In the test for soluble barium salts an acetic acid extract (solution S) after the
adding of dilute sulphuric acid is compared to the mixture of solution S and distilled
water.
In the sedimentation test, which controls the particle size, a weighed quantity
of the sample is shaken up with a measured volume of water in a measuring cylinder
of specified dimensions. The suspension is allowed to stand for 15 min, when the
process of settling must not have been below a defined mark.
Assay Is not required by Pharmacopoeia.
Storage In well-glued double paper packages (internal package is made of oil-
paper). It must be kept away from the carbonates to avoid barium carbonate
formation.
Usage For the X-rays diagnostics (roentgenology).

64
Preparations of zinc
Zinc is found in nature as minerals zinc blend ZnS; calamine ZnCO3. Usage of
zinc compounds in medicine is based on their possibility to form compounds with
proteins called albumates, which act as weak astringent and cauterizing agents.
Insoluble albuminates are used for treatment of wounds, forming protective film on
them. Zinc is a synergist of vitamins and promote their action.
Zinc oxide (Zinci oxydum), EP
ZnO
Preparation
1. Zinc oxide may be prepared by the heating of native zinc carbonate:
ZnCO3 ZnO + CO2
2. A solution of zinc sulphate is added to a boiling solution of sodium
carbonate. The precipitated basic carbonate of zinc is collected, washed until free
from sulphate, dried, and ignited; it then loses carbon dioxide and water, leaving the
oxide. to
5ZnSO4 + 5Na2CO3 + 3H2O 2ZnCO3 . 3Zn(OH)2 + 5Na2SO4 + 3CO2 to
2ZnCO3 . 3Zn(OH)
2 5ZnO + 2CO
2 + 3H
2O
Properties Zinc oxide is a soft, white or faintly yellowish-white, amorphous powder, free
from gritty particles, practically insoluble in water and in alcohol. It dissolves in
dilute mineral acids.
Identification 1. It becomes yellow when strongly heated; the yellow colour disappears on
cooling (EP).
2. Identification of zinc ions is carried out using general identification reactions
(EP, above).
3. Reaction of formation Reenman’s green. Zinc oxide is heated with cobalt
nitrate. Green colour is obtained.
ZnO + Co(NO3)2 CoZnO2 + 2NO2 + O
Assay
Complexometric titration (after dissolving preparation in dilute acetic acid)
using xylenol orange triturate as indicator; (s = 1, EP).
65
Zn2+
SO3Na
CH3
OH
CH2
CH3
O
CH2
N
CH2COONa
CH2COONa
Zn Ind
N
NaOOCH2C
NaOOCH2C 2Na +-
+
(CH2)6N4
(or Na2Ind)
Titrate is 0,1M sodium edetate, in the presence of hexamethylenetetramine.
Mixture is titrated until the violet-pink colour changes to yellow.
Zn2+
CH2
CH2
N
N
CH2COONa
CH2COO
CH2COO
CH2COONa
Zn
CH2
CH2
N
N
CH2COONa
CH2COOH
CH2COOH
CH2COONa
H+
+ + 2
(CH2)6N4
CH2
CH2
N
N
CH2COONa
CH2COO
CH2COO
CH2COONa
ZnInd
CH2
CH2
N
N
CH2COONa
CH2COOH
CH2COOH
CH2COONa
Zn Ind
+ +
(CH2)6N4
H2
Storage Preparation should be stored in an airtight container, because it can
absorb atmospheric carbon dioxide and perform into zinc carbonate.
Usage Preparation is used externally in ointments, powders and other forms as
astringent, drying and disinfectant for skin diseases.
Zinc sulphate heptahydrate (Zinci sulfas heptahydricus), EP
ZnSO4 7H2O
Preparation
The substance is prepared by the action of diluted sulphuric acid on metallic
zinc or zinc oxide. Then the liquids are filtered and evaporated to crystallization. to
Zn + H2SO4 ZnSO4 + H2
ZnO + H2SO4 ZnSO4 + H2O
Properties
A white, crystalline powder or colourless, transparent crystals, efflorescent,
very soluble in water, practically insoluble in alcohol.

66
Identification 1.Identification is carried out using general identification reactions (EP) for
zinc and sulphates (above).
2. The preparation complies with the limits of the assay (EP).
Assay Complexometric titration (see zinc oxide). (s=1).
Storage In a non-metallic, airtight container.
Usage Preparation is used externally as antiseptic and astringent agent: eye
drops 0,1; 0,25; 0,5%; 0,25-0,5 % solutions for laryngology; 0,1-0,5 % solutions for
urology and gynaecology.
Preparations of mercury Most of the mercury compounds is prepared from the chief ore of mercury –
cinnabar, HgS.
Mercuric chloride (Hydrargyri dichloridum), EP
HgCl2
Preparation
1. Metallic mercury is dissolved in sulphuric acid and heated in the presence of
nitric acid. Then solution is evaporated to dryness, and the residue is mixed with
sodium chloride and manganese peroxide. The mixture is heated again, and mercury
dichloride sublime:
Hg + 2H2SO4 HgSO4 + 2H2O + SO2
HgSO4 + 2NaCl HgCl2 + Na2SO4
2. Preparation can be prepared by heating the mixture of mercury and chlorine
at 335-340 ºC. In this method mercurous chloride (Hg2Cl2) may be obtained as by-
product. To purify the preparation, it is sublimed and then crystallised from alcohol
or water, in which mercury monochloride does not dissolve.
Hg + Cl2 HgCl2
Properties A white, crystalline powder or colourless or white crystals or heavy crystalline
masses, soluble in water, in ether and in glycerol, freely soluble in alcohol.
Identification Identification is carried out using general identification reactions
(EP) for mercury and chlorides (above).
Tests for purity Mercurous chloride is determined by dissolving the
preparation in ether. The solution obtained should not show any opalescence.
Assay
1. Complexonometric titration by substitute. At first 0.1 M sodium edetate and
buffer solution pH 10.9 are added. Mercury ions form complexes with the titrate.
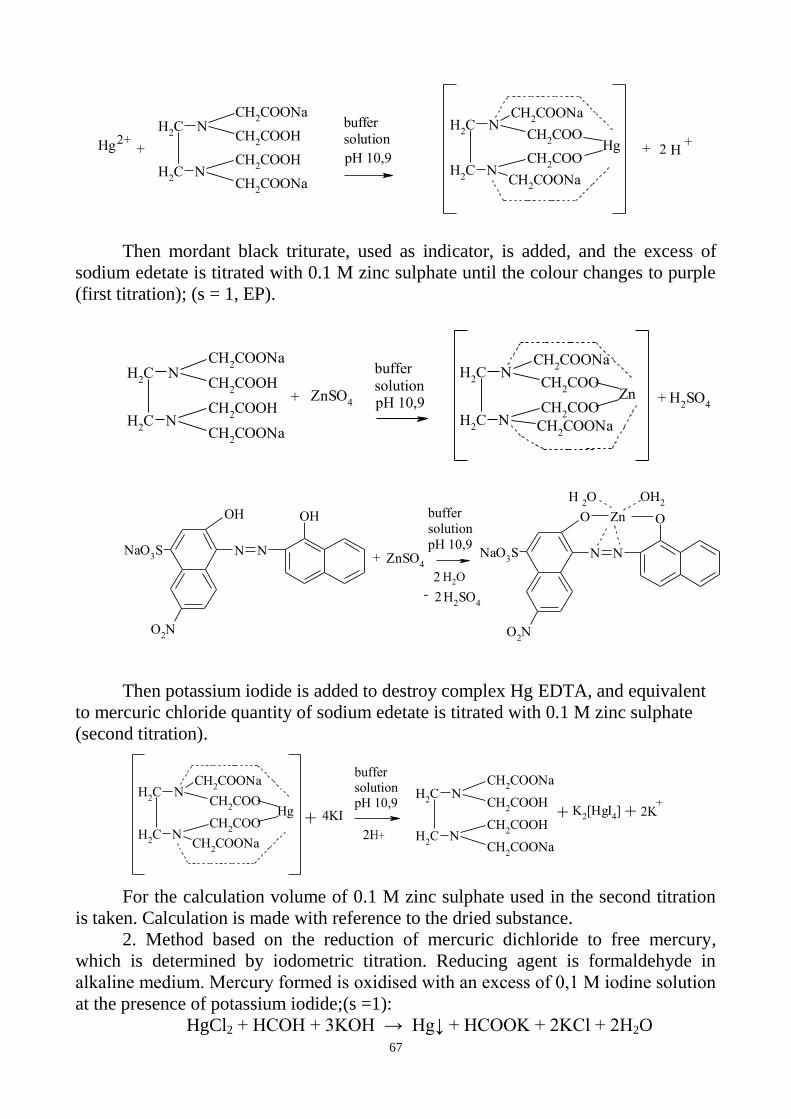
67
Hg
CH2
CH2
N
N
CH2COONa
CH2COOH
CH2COOH
CH2COONa
CH2
CH2
N
N
CH2COONa
CH2COO
CH2COO
CH2COONa
Hg
22+
+ H++
pH 10,9
buffer
solution
Then mordant black triturate, used as indicator, is added, and the excess of
sodium edetate is titrated with 0.1 M zinc sulphate until the colour changes to purple
(first titration); (s = 1, EP).
ZnSO4
CH2
CH2
N
N
CH2COONa
CH2COOH
CH2COOH
CH2COONa
CH2
CH2
N
N
CH2COONa
CH2COO
CH2COO
CH2COONa
Zn
H2SO
4+ +pH 10,9
buffer
solution
N
O
NaO3S
O2N
N
OZn
H 2O
N
OH
NaO3S
O2N
N
OH
OH2
H2SO
4
ZnSO4
+
2-
H2O2
buffer
solution
pH 10,9
Then potassium iodide is added to destroy complex Hg EDTA, and equivalent
to mercuric chloride quantity of sodium edetate is titrated with 0.1 M zinc sulphate
(second titration).
CH2
CH2
N
N
CH2COONa
CH2COOH
CH2COOH
CH2COONa
CH2
CH2
N
N
CH2COONa
CH2COO
CH2COO
CH2COONa
Hg
K2[HgI
4]
buffer
solution
pH 10,9
+ 4KI + + 2K+
2H+
For the calculation volume of 0.1 M zinc sulphate used in the second titration
is taken. Calculation is made with reference to the dried substance.
2. Method based on the reduction of mercuric dichloride to free mercury,
which is determined by iodometric titration. Reducing agent is formaldehyde in
alkaline medium. Mercury formed is oxidised with an excess of 0,1 М iodine solution
at the presence of potassium iodide;(s =1):
HgCl2 + HCOH + 3KOH → Hg↓ + HCOOK + 2KCl + 2H2O

68
Hg + I2 HgI2
HgI2 + 2KI K2[HgI4]
Excess of iodine is titrated with sodium thiosulphate solution, using starch
solution as indicator:
I2 + 2Na2S2O3 2NaI + Na2S4O6
Storage In an airtight container.
Usage Preparation is used as antiseptic. It is very toxic and not used internally.
When mercury dichloride gets on skin, it also causes poisoning. Pills and solutions of
preparation are usually coloured with eosin to distinguish them from other
preparations (by bright pink colour). The preparation forms precipitates with proteins
and alkaloids, so strong tea and milk can be used as first aid for mercury poisoning.
Yellow mercuric oxide (Hydrargyri oxydum flavum)
HgO
Preparation It is prepared by precipitation, using solutions of mercuric chloride and sodium
hydroxide. The yellow precipitate is left to settle, washed by decantation, and then
washed on the filter until free from chloride. It is then allowed to dry at air
temperature.
HgCl2 + 2NaOH HgO + 2NaCl + H2O
The colour of the preparation varies from yellow to orange depending on the
temperatures at which it is precipitated, and is due to the size of the particles. The
process of Preparation can’t be carried out at high temperatures, because this give the
red oxide, which is not used in medicine. To obtain preparation free from by-products
(e.g. mercuric subchloride HgOHCl), solution of mercuric chloride is added to the
alkali solution.
Properties Yellow or orange-yellow, heavy, fine amorphous powder. Preparation darkens
slowly being exposed to light. Practically insoluble in water and alcohol, easy soluble
in dilute hydrochloric, nitric and acetic acids.
Identification After dissolving in hydrochloric acid identification is carried out with
potassium iodide solution, being added dropwise. In this case a red precipitate that
dissolves in excess of reagent is formed.
HgO + 2HCl HgCl2 + H2O
HgCl2 + 2KI HgI2↓ + 2KCl
HgI2↓ + 2KI K2[HgI4]
Tests for purity
Mercurous salts are discovered with hydrochloric acid:
Hg22+
+ 2Cl- Hg2Cl2 (white)

69
Assay
1. Indirect acidimetric titration. Mercuric oxide is weighed and dissolved in
water with excess of potassium iodide. Potassium hydroxide obtained is titrated with
hydrochloric acid solution at the presence of methyl red as indicator (s=1/2):
HgO + 4KI + H2O K2HgI4 + 2KOH
KOH + HCl KCl + H2O
2. Back alkalimetric titration (s=1/2).
HgO + 2HCl → HgCl2 + H2O
HCl + NaOH → NaCl + H2O
Storage In an airtight amber bottles.
Usage Preparation is used as external antiseptic in eye ointments. Very toxic!
Mercuric oxycyanide (Hydrargyri oxycyanidum)
HgO Hg(CN)2
Properties
White or slight yellowish powder, very slightly soluble in water. Water
solutions have alkaline reaction.
Identification Potassium iodide, ferrous sulphate, and ferric chloride solutions are added to
the solution of Preparation. After acidifying with hydrochloric acid red precipitate of
mercuric iodide, HgI2, appears. After adding more potassium iodide red precipitate
disappears, and a blue precipitate is formed (Prussian blue).
HgO Hg(CN)2 + 4KI + H2O 2HgI2↓ + 2KCN + 2KOH
HgI2↓ + 2KI K2[HgI4]
18KCN + 3FeSO4 + 4FeCl3 → Fe4[Fe(CN)6]3↓ + 3K2SO4 + 12 HCl
Assay
Mercury oxide is determined by direct acidimetry, using methyl orange as
indicator (s=1/2). NaCl
HgO + 2HCl HgCl2 + H2O
Preparation contains no less than 45% of HgO.
Then mercuric cyanide is assayed by indirect acidimetry (or acidimetry by
substituent). To the solution obtained potassium iodide is added, and solution is
titrated with 0,1M hydrochloric acid (s=1/2).
Hg(CN)2 + 4KI K2HgI4 + 2KCN
KCN + 2H2O 2HCN + 2KOH
2KOH + 2HCl 2KCl + 2H2O
Preparation contains no less than 53,3 % of Hg(CN)2.
Storage In an airtight amber bottles.
Usage Preparation is used as external antiseptic. Very toxic!

70
Chapter 5. MEDICINAL PREPARATIONS CONTAINING THE
ELEMENTS OF 1ST
AND 8TH
GROUPS OF
D.I. MENDELEYEV PERIODIC SYSTEM
Plan
1. Preparations of copper (copper sulphate pentahydrate).
2. Preparations of silver (silver nitrate and colloidal preparations of silver).
3. Preparations of iron (ferrous sulphate heptahydrate).
Preparation of copper In nature copper is found as copper pyrites, CuFeS2 (from which it is extracted
in metallurgy), cuprite, Cu2O, copper glance Cu2S, and malachite, CuCO3 Cu(OH)2.
Copper forms two series of compounds, the cuprous, derived from cuprous
hydroxide, CuOH, and the cupric, derived from cupric hydroxide, Cu(OH)2.
Copper sulphate pentahydrate (Cupri sulfas pentahydricus), EP
CuSO4 5H2O
Preparation Copper sulphate is prepared by the dissolving of metal copper in concentrated
sulphuric acid in the presence of nitric acid:
3Cu + 3H2SO4 + 2HNO3 3CuSO4 + 4H2O + 2NO
Solution obtained is evaporated (sulphuric acid, nitric acid and nitrogen oxide
are evolved) and CuSO4 . 5H
2O is crystallised from the solution.
Properties A blue, crystalline powder or transparent, blue crystals, freely soluble in water,
soluble in methanol, practically insoluble in alcohol.
Identification 1. Ammonia solution, added to a solution of a cupric salt, gives a blue
precipitate, which dissolves in excess of the precipitant to form a deep blue solution
(EP):
2CuSO4 + 2NH4OH (NH4)2SO4 + Cu2(OH)2SO4
Cu2(OH)2SO4 + 6NH4OH + (NH4)2SO4 2[Cu(NH3)4]SO4 + 8H2O
The colour is due to the presence of the cuprammonium cation, [Cu(NH3)4]+.
2. Water solution of copper sulphate (1:20) in reaction with metallic iron
covers it with thin red coating of metal copper:
CuSO4 + Fe FeSO4 + Cu
3. Solution of the preparation gives with sodium sulphide a black precipitate of
cupric sulphide, which dissolves in dilute nitric acid on boiling.
CuSO4 + Na2S CuS + Na2SO4 t
o
3CuS + 8HNO3 3Cu(NO3)2 + 3S + 2NO + 4H2O

71
4. Solution of copper sulphate, acidified with acetic acid, gives with potassium
ferrocyanide reddish-brown precipitate of copper ferrocyanide, soluble in ammonia.
2CuSO4 + K4[Fe(CN)6] Cu2[Fe(CN)6] + 2K2SO4
5. Reaction with polyalcohols and oxyacids (glycerol, gluconic acid, etc.) is
specific for cupric salts.
6. The solution of the preparation gives reaction (a) of sulphates (EP, above).
7. The preparation complies with the test for loss on drying (EP).
Assay
Iodometric titration after reducing Cu2+
to Cu+1
with potassium iodide in the
presence of sulphuric acid. Iodine is titrated with 0,1M sodium thiosulphate, using
starch solution (added towards the end of the titration) as indicator (s=1).
2CuSO4 + 4KI 2CuI +I2 + 2K2SO4
I2 +2Na2S2O3 2NaI + Na2S4O6
Storage In an airtight container.
Usage Water solution of the preparation is used as external antiseptic an
astringent, in the combined preparations of vitamines and minerals as source of
copper, sometimes it is used internally as emetic.
Preparations of silver Silver occurs in the free state, also as horn silver, AgCl, and as silver glance,
Ag2S. Silver forms only one series of compounds, in which the metal is univalent.
Silver nitrate (Argenti nitras), EP
AgNO3
Preparation Preparation is manufactured by the dissolving of copper-silver alloy in nitric
acid on heating: t
o
Ag Cu + 4HNO3 AgNO3 + Cu(NO3)2 + NO + 2H2O
Properties A white, crystalline powder or transparent, colourless crystals, very soluble in
water, soluble in alcohol.
Identification 1. Identification is carried out using general identification reactions (EP) for
silver and nitrates (above).
2. Silver can be recognized by the reaction of “silver mirror”:
AgNO3 + 2NH4OH [Ag(NH3)2]NO3 + 2H2O
[Ag(NH3)2]NO
3 R CO
H
H 2O Ag R C
O
ONH4
NH3 NH
4NO
32 + + 2 + + + 2

72
3. Diphenylamine gives a blue colouration with nitrates as far as with nitrites
(see sodium nitrite, above).
Tests for purity
Appearance of solution, acidity or alkalinity, foreign salts, aluminium, lead,
copper and bismuth.
Foreign salts are determined after precipitating of silver with dilute
hydrochloric acid and filtering the precipitate off. The filtrate is evaporated to dryness
and the residue is weighed.
The test for aluminium, lead, copper and bismuth consists in adding
concentrated ammonia to the solution of the Preparation. At first, it produces a dark
precipitate of Ag2O, but the precipitate dissolves in excess of ammonia, and the
solution should be then clear and colourless. Lead, bismuth and aluminium cause a
white turbidity due to precipitation of their hydroxides (which are not soluble in
excess of ammonia), and copper gives a blue solution due to the formation of
cuprammonium ion.
Assay
Thiocyanatometric titration after dissolving the preparation in water and
acidifying with dilute nitric acid, using ferric ammonium sulphate solution as
indicator until reddish-yellow colour is obtained (s = 1, EP).
AgNO3 + NH4SCN AgSCN + NH4NO3
3NH4SCN + FeNH4(SO4)2 Fe(SCN)3 + 2(NH4)2SO4
Storage As silver nitrate slowly decomposes in light, turning black, it should
be stored protected from light; in a non-metallic container.
Usage Silver nitrate solution is used as external antiseptic, for the
cauterizations.
Colloidal preparations of silver
Colloidal preparations of silver (collargol and protargol) contain metallic silver
in the colloidal state bound with peptides obtained from egg or milk proteins.
Collargol and protargol are used as external antiseptics, due to the presence of silver.
Collargol (Collargolum, Argentum colloidale)
Preparation
Colloidal preparations of silver are manufactured from egg protein or casein
(milk protein) and silver nitrate. Protein is steamed or treated with acids (or alkalies)
forming lisalbinic and protalbinic acids (or their salts, e.g. sodium salts), which have
reducing properties.
Silver nitrate is converted into silver oxide by the sodium hydroxide solution:
AgNO3 + NaOH → AgOH↓ + NaNO3
2AgOH → Ag2O↓ + H2O

73
Then silver oxide is purified and mixed with sodium lisalbinate and sodium
protalbinate solutions. As a result of reduction metallic silver is obtained and in the
colloidal state it forms coordination bonds with peptides.
Properties
Greenish or bluish-black plaites with a metallic sheen, soluble in water with
formation of a colloidal solution. Contains not less than 70 % of silver.
Identification
1. It is carbonized when heated, giving the characteristic odour of “burned
horn”.
2. The residue obtained in the reaction (1) is dissolved in nitric acid and
filtrated. Hydrochloric acid gives with the filtrate a white precipitate of silver
chloride:
AgNO3 + HCl → AgCl↓ + HNO3
3. Biuret test for identification of peptide part of preparation (see Hydroperite).
4. Reaction of distinction from protargol. Colloidal solution of the preparation
(1:50) on adding dilute hydrochloric acid gives brown precipitate. The precipitate
dissolves after addition of alkali, and the colloidal solution forms again.
Assay.
The preparation is weighed and carbonized by concentrated sulphuric acid and
nitric acids during 15 min. Then silver nitrate obtained is determined by
thiocyanatometric titration (see silver nitrate, s=1).
mlg
T
TAgNO
CSNNH
golcollarCSNNH /01541,0
70
10001079,0
70
1003
4
4
Storage In an airtight amber bottles, protected from light.
Usage Water solutions of the preparation are used as external antiseptics in the
forms of eye drops, ointments, solutions for irrigations of purulent wounds and
bladder.
Protargol (Protargolum)
Properties
Brownish-yellow powder, very soluble in water, practically insoluble in
alcohol and ether, hygroscopic. Contains not less than 7,8 % and not more than 8,3 %
of silver.
Preparation, identification, assay and storage See Collargol.
Usage Antiseptic, astringent, anti-inflammatory in the forms of 1-5 % solutions
(application on mucous membranes of upper parts of respiratory tract and in urology),
and 1-3 % eye drops. Solutions are prepared ex tempore.

74
Preparations of iron
Ores of iron are very widely distributed in nature and include haematite, Fe2O3,
limonite, 2Fe3O3 3H2O, magnetic iron ore, Fe3O4, and spathic iron ore, FeCO3. Iron
forms two series of salts, the ferrous salts (solutions are coloured very pale green),
derived from ferrous hydroxide, Fe(OH)2, and the ferric salts (solutions are coloured
yellow, brown, or red), derived from ferric hydroxide, Fe(OH)3.
Ferrous sulphate heptahydrate (Ferrosi sulfas heptahydricus), EP
FeSO4 ∙ 7H2O
Preparation
Ferrous sulphate is prepared by dissolving of an excess of metallic iron in 25-
30% sulphuric acid on heating at 80 C:
Fe + H2SO4 → FeSO4 + H2↑
Solution obtained is evaporated to dryness and dried at 30 C, because at 64 C
preparation melts in its own water of crystallization.
Properties
Light green, crystalline powder or bluish-green crystals, efflorescent in air,
freely soluble in water, very soluble in boiling water, practically insoluble in alcohol.
The pH of the solution is 3.0 to 4.0, owing to hydrolysis.
Identification
1. Identification is carried out using general identification reactions (EP) for
iron (reaction a) and sulphates (above).
2. The substance complies with the limits of the assay (EP).
3. Dimethylglyoxime gives with ferrous salts red complex.
Tests for purity
Appearance of solution, pH, chlorides, ferric ions, manganese, zinc and heavy
metals.
Ferric ions are determined by titration with 0,1M sodium thiosulphate after
interaction with potassium iodide (indicator is starch solution). EP states the limit of
the titrate.
2Fe3+
+ 2I‾ → 2Fe2+
+ I2
I2 + 2Na2S2O3 → 2NaI + Na2S4O6
Manganese compounds are determined by oxidation to red-coloured
permanganate ion MnO4- and comparing with the standard solution prepared using
0,02M potassium permanganate.
Assay
1. Cerimetric titration after dissolving the preparation in the previously
prepared mixture of sodium hydrogen carbonate, water and sulphuric acid. Titrate is
0,1M ammonium and cerium nitrate, indicator is ferroin. Solution is titrated until the
red colour disappears (s=1, EP).
3FeSO4+3(NH4)2Ce(NO3)6 → Fe2(SO4)3+Fe(NO3)3+3Ce(NO3)3+6NH4NO3

75
N
N
Fe
Ce4+
N
N
Fe
Ce3+
2 +
+
3 +
+
3 3
red green
2. Permanganatometric titration, without indicator. Titration is carried out until
pink coloration of the solution (s=10/2 or 5).
10FeSO4 + 2KMnO4 + 8H2SO4 → 5Fe2(SO4)3 + K2SO4 + 2MnSO4 + 8H2O
Storage In an airtight container.
Usage As a source of iron for iron-lacking anemia.

76
Chapter 6. RADIOPHARMACEUTICAL PREPARATIONS
(RADIOPHARMACEUTICA)
Radiopharmaceutical preparation is any medicinal product which, when ready
for use, contains one or more radionuclides (radioactive isotopes) included for a
medicinal purpose.
A nuclide is a species of atom characterised by the number of protons and
neutrons in its nucleus (and hence by its atomic number Z, and mass number A) and
also by its nuclear energy state. Isotopes of an element are nuclides with the same
atomic number but different mass numbers. Nuclides containing an unstable
arrangement of protons and neutrons will transform spontaneously to either a stable
or another unstable combination of protons and neutrons with a constant statistical
probability. Such nuclides are said to be radioactive and are called radionuclides. The
initial unstable nuclide is referred to as the parent radionuclide and the resulting
nuclide as the daughter nuclide.
The radioactive decay or transformation may involve the emission of charged
particles, electron capture (EC) or isomeric transition (IT). The charged particles
emitted from the nucleus may be alpha particles (helium nucleus of mass number 4)
or beta particles (negatively charged, generally called electrons or positively charged,
generally called positrons). The emission of charged particles from the nucleus may
be accompanied by the emission of gamma rays. Gamma rays are also emitted in the
process of isomeric transition.
The decay of a radionuclide is governed by the laws of probability with a
characteristic decay constant and follows an exponential law. The time in which a
given quantity of a radionuclide decays to half its initial value is termed the half-life
(T1/2).
The penetrating power of each radiation varies considerably according to its
nature and its energy. Alpha particles are completely absorbed in a thickness of a few
micrometers to some tens of micrometers of matter. Beta particles are completely
absorbed in a thickness of several millimetres to several centimetres of matter.
Gamma rays are not completely absorbed but only attenuated and a tenfold reduction
may require, for example, several centimetres of lead. For most absorbents, the
denser the absorbent, the shorter the range of alpha and beta particles and the greater
the attenuation of gamma rays.
Each radionuclide is characterised by an invariable half-life, expressed in units
of time and by the nature and energy of its radiation or radiations. The energy is
expressed in electronvolts (eV), kilo-electronvolts (keV) or mega-electronvolts
(MeV).
Generally the term “radioactivity” is used to describe the phenomenon of
radioactive decay and to express the physical quantity (activity) of this phenomenon.
The radioactivity of a preparation is the number of nuclear disintegrations or
transformations per unit time.
In the International System (SI), radioactivity is expressed in becquerel (Bq)
which is one nuclear transformation per second. Absolute radioactivity measurements

77
require a specialised laboratory but identification and measurement of radiation can
be carried out relatively by comparing with standardised preparations.
Identification of padiopharmaceutical preparation is carried out by comparing
the beta-ray spectrum or gamma-ray spectrum obtained of the preparation with that of
a standartizated radionuclide.
Identification is also carried out by examining the chromatogram obtained in
the test for radiochemical purity. The distribution of radioactivity should contribute
to the identification of the preparation.
Tests for purity in radiopharmaceutical preparations usually contain
radionuclidic purity and radiochemical purity.
Radionuclidic purity: the ratio, expressed as a percentage, of the radioactivity
of the radionuclide concerned to the total radioactivity of the radiopharmaceutical
preparation. The relevant radionuclidic impurities are listed with their limits in the
individual monographs.
The most generally useful method for examination of radionuclidic purity is
that of gamma spectrometry (for example, the gamma-ray spectrum does not
significantly differ from that of a standardised preparation). Due to differences in the
half-lives of the different radionuclides present in a radiopharmaceutical preparation,
the radionuclidic purity changes with time.
Radiochemical purity: the ratio, expressed as a percentage, of the radioactivity
of the radionuclide concerned which is present in the radiopharmaceutical preparation
in the stated chemical form, to the total radioactivity of that radionuclide present in
the radiopharmaceutical preparation. The relevant radiochemical impurities are listed
with their limits in the individual monographs.
The determination of radiochemical purity requires separation of the different
chemical substances containing the radionuclide and estimating the percentage of
radioactivity associated with the declared chemical substance. The monographs for
radiopharmaceutical products may include paper chromatography, thin-layer
chromatography, electrophoresis, gas chromatography and liquid chromatography.
Moreover certain precautions special to radioactivity must also be taken for radiation
protection.
Each monograph for radiopharmaceutical preparation has a part
“Radioactivity”.
Measurement of radioactivity.
The radioactivity of a preparation is stated at a given date and, if necessary,
time.
It is common to carry out the measurement with the aid of a primary standard
source. Primary standards may not be available for short-lived radionuclides e.g.
positron emitters. Ionisation chambers and Geiger-Müller counters may be used to
measure beta and beta/gamma emitters; scintillation or semiconductor counters or
ionisation chambers may be used for measuring gamma emitters; low-energy beta
emitters require a liquid-scintillation counter. For the detection and measurement of
alpha emitters, specialised equipment and techniques are required.
The radioactivity of a solution is expressed per unit volume to give the
radioactive concentration.

78
Storage Store in an airtight container in a place that is sufficiently shielded to
protect personnel from irradiation by primary or secondary emissions.
Radiopharmaceutical preparations are intended for use within a short time and
the end of the period of validity must be clearly stated.
Sodium phosphate 32
P injection
(Natrii phosphatis (32
P) solutio iniectabilis), EP
Preparation
Phosphorus-32 is a radioactive isotope of phosphorus and may be produced by
neutron irradiation of sulphur.
Properties
A clear, colourless solution.
Sodium phosphate (32
P) injection is a sterile solution of disodium and
monosodium (32
P) orthophosphates made isotonic by the addition of sodium chloride.
The injection contains not less than 90.0 per cent and not more than 110.0 per cent of
the declared phosphorus-32 radioactivity at the date and hour stated on the label. Not
less than 95 per cent of the radioactivity corresponds to phosphorus-32 in the form of
orthophosphate ion. The specific radioactivity is not less than 11.1 MBq of
phosphorus-32 per milligram of orthophosphate ion.
Phosphorus-32 has a half-life of 14.3 days and emits beta radiation.
Identification
1. The beta-ray spectrum or the beta-ray absorption curve does not differ
significantly from that of a standardised phosphorus-32 solution obtained under the
same conditions. The maximum energy of the beta radiation is 1.71 MeV (E.P.)
2. On the chromatogram obtained in the test for radiochemical purity, the
distribution of radioactivity contributes to the identification of the preparation (E.P.).
3. Zirconium nitrate in the concentrated nitric acid gives with the preparation a
white friable precipitate.
Usage Anti-tumour for the treatment of blood diseases and for the tumour
diagnostics.
Sodium iodohippurate (131
I) injection
(Natrii iodohippurati (131
I) solutio iniectabilis), EP
Preparation
Iodine-131 is a radioactive isotope of iodine and may be obtained by neutron
irradiation of tellurium or by extraction from uranium fission products.
Properties
A clear, colourless liquid.
Sodium iodohippurate (131
I) injection is a sterile solution of sodium 2-(2-
[131
I]iodobenzamido)acetate. It may contain a suitable buffer and a suitable
antimicrobial preservative such as benzyl alcohol. The injection contains not less than
90.0 per cent and not more than 110.0 per cent of the declared iodine-131

79
radioactivity at the date and hour stated on the label. Not less than 96 per cent of the
iodine-131 is in the form of sodium 2-iodohippurate. The specific radioactivity is
0.74 GBq to 7.4 GBq of iodine-131 per gram of sodium 2-iodohippurate.
Iodine-131 has a half-life of 8.04 days and emits beta and gamma radiation.
Identification
1. The gamma-ray spectrum does not differ significantly from that of a
standardised iodine-131 solution. The most prominent gamma photon of iodine-131
has an energy of 0.365 MeV (E.P.).
2. On the chromatograms obtained in the test for radiochemical purity, the
main peak of radioactivity in the chromatogram obtained with the test solution is
similar in position to the spot corresponding to 2-iodohippuric acid in the
chromatogram obtained with the reference solution (EP).
Usage For the kidney diseases diagnostics.

80
Chapter 7. GENERAL METHODS OF ANALYSIS
OF ORGANIC MEDICINAL SUBSTANCES
Plan
1. Classification of organic medicinal substances.
2. Determination of basic physical constants: (melting point, boiling point,
density, refractive index, specific optical rotation).
3. Bases of chemical analysis of organic substances: (element analysis, analysis
by functional groups).
Organic medicinal preparations are classified on:
1. Aliphatic:
• alkanes and their haloderivatives;
• alcohols;
• aldehydes;
• carbonic acids, oxy- and aminoacids;
• ethers and esters.
2. Cyclic:
• terpenoids;
• derivatives of cyclopropane, derivatives of adamantane.
3. Aromatic;
• phenols;
• aromatic amines and the acyl-derivatives;
• oxy- and aminoacids of aromatic origin;
• derivatives of aromatic sulphoacids.
4. Heterocyclic:
• are classified according to the structure of their heterocycles.
5. Biologically active natural compounds;
• alkaloids;
• glycosides;
• hormones;
• vitamins;
• antibiotics.
The general methods of analysis of organic medicinal substances include
determination of their physical constants.
Determination of melting point
Melting point is very important physical constant for organic medicinal
substances, which is used for their identification (in pure condition or as derivatives:
oximes, amides, hydrazones and etc.).
The melting point is a temperature, at which a hard phase of substance is in
equilibrium with fusion.
There are three methods for determining of melting point (according to State
Pharmacopoeia of Ukraine).
I. Melting point – capillary method.

81
The melting point determined by the capillary method is the temperature at
which the last solid particle of a compact column of a substance in a tube passes into
the liquid phase.
When prescribed in the monograph, the same apparatus and method are used
for the determination of other factors, such as meniscus formation or melting range,
that characterise the melting behaviour of a substance.
Apparatus. The apparatus consists of:
—a suitable glass vessel containing a liquid bath (for example, water, liquid
paraffin or silicone oil) and fitted with a suitable means of heating,
—a suitable means of stirring, ensuring uniformity of temperature within the
bath,
—a suitable thermometer with graduation at not more than 0.5 °C intervals and
provided with an immersion mark. The range of the thermometer is not more than
100 °C,
—alkali-free hard-glass capillary tubes of internal diameter 0.9 mm to 1.1 mm
with a wall 0.10 mm to 0.15 mm thick and sealed at one end.
Method. Unless otherwise prescribed, dry the finely powdered substance in
vacuo and over anhydrous silica gelR for 24 h. Introduce a sufficient quantity into a
capillary tube to give a compact column 4 mm to 6 mm in height. Raise the
temperature of the bath to about 10 C below the presumed melting point and then
adjust the rate of heating to about 1 C per minute. When the temperature is 5 °C
below the presumed melting point, correctly introduce the capillary tube into the
instrument. For the apparatus described above, immerse the capillary tube so that the
closed end is near the centre of the bulb of the thermometer, the immersion mark of
which is at the level of the surface of the liquid. Record the temperature at which the
last particle passes into the liquid phase.
II. Melting point – open capillary method.
For certain substances, the following method is used to determine the melting
point (also referred to as slip point and rising melting point when determined by this
method).
Use glass capillary tubes open at both ends, about 80 mm long, having an
external diameter of 1.4 mm to 1.5 mm and an internal diameter of 1.0 mm to
1.2 mm.
Introduce into each of 5 capillary tubes a sufficient amount of the substance,
previously treated as described, to form in each tube a column about 10 mm high and
allow the tubes to stand for the appropriate time and at the prescribed temperature.
Unless otherwise prescribed, substances with a waxy consistency are carefully
and completely melted on a water-bath before introduction into the capillary tubes.
Allow the tubes to stand at 2-8 °C for 2 h.
Attach one of the tubes to a thermometer graduated in 0.5 °C so that the
substance is close to the bulb of the thermometer. Introduce the thermometer with the
attached tube into a beaker so that the distance between the bottom of the beaker and
the lower part of the bulb of the thermometer is 1 cm. Fill the beaker with water to a
depth of 5 cm. Increase the temperature of the water gradually at a rate of 1 °C/min.

82
The temperature at which the substance begins to rise in the capillary tube is
regarded as the melting point.
Repeat the operation with the other 4 capillary tubes and calculate the result as
the mean of the 5 readings.
III. Melting point – instantaneous method.
The instantaneous melting point is calculated using the expression:
,2
)( 21 tt
in which t1 is the first temperature and t2 the second temperature read under the
conditions stated below.
Apparatus. The apparatus consists of a metal block resistant to the substance to
be examined, of good heat-conducting capacity, such as brass, with a carefully
polished plane upper surface. The block is uniformly heated throughout its mass by
means of a micro-adjustable gas heater or an electric heating device with fine
adjustment. The block has a cylindrical cavity, wide enough to accomodate a
thermometer, which should be maintained with the mercury column in the same
position during the calibration of the apparatus and the determination of the melting
point of the substance to be examined. The cylindrical cavity is parallel to the upper
polished surface of the block and about 3 mm from it. The apparatus is calibrated
using appropriate substances of known melting point.
Method. Heat the block at a suitably rapid rate to a temperature about 10 °C
below the presumed melting temperature, then adjust the heating rate to about 1 °C
per minute. At regular intervals drop a few particles of powdered and, where
appropriate, dried substance, prepared as for the capillary tube method, onto the block
in the vicinity of the thermometer bulb, cleaning the surface after each test. Record
the temperature t1 at which the substance melts instantaneously for the first time in
contact with the metal. Stop the heating. During cooling drop a few particles of the
substance at regular intervals on the block, cleaning the surface after each test.
Record the temperature t2 at which the substance ceases to melt instantaneously when
it comes in contact with the metal
Determination of boiling point
The boiling point is the corrected temperature at which the vapour pressure of a
liquid is equal to 101.3 kPa.
Apparatus. The apparatus is that used for Distillation Range with the exception
that the thermometer is inserted in the neck of the flask so that the lower end of the
mercury reservoir is level with the lower end of the neck of the distillation flask and
that the flask is placed on a plate of isolating material pierced by a hole 35 mm in
diameter.

83
Method. Place in the flask (A) 20 ml of the liquid to be examined and a few
pieces of porous material. Heat the flask so that boiling is rapidly achieved and record
the temperature at which liquid runs from the side-arm into the condenser.
Correct the observed temperature for barometric pressure by means of the
formula:
t1 = t2 + (101.3 - b)
t1 – the correct temperature,
t2 – the observed temperature at barometric pressure b,
k – the correction factor as shown in Tables,
b – the barometric pressure, in kPa, at the time of determination.
Determination of density
The relative density of a substance is the ratio of the mass of a certain volume
of the substance to the mass of an equal volume of water, both weighed at 20 °C.
Value of density confirms identification, and on occasion (an ethyl alcohol,
glycerol etc) - and quantitative maintenance of medicinal substances.
According to EP and SPU density is determined by tree methods:
1. By pyknometer (gravimetric) accuracy - 0,001.
0012,0)(
99703,0)(
01
02
mm
mm
where that is
mo - mass of empty pyknometer;
m1 - is mass of pyknometer with water;
m2-; it is mass of pyknometer with the explored liquid;
0,0012 is air density at 20°С and 1011 kPa;

84
0,99703 is water density at 20°С (with air density).
2. By areometer (accuracy 0,01) for determination of liquids density.
3. Method for determination of waxes and fats.
0012,0)()(
99703,0)(
321
02
mmmm
mm
m - is mass of empty pyknometer;
m1 - is mass of pyknometer with water;
m2 - is mass of pyknometer with fat;
m3 - is mass of pyknometer with water and fat.
Determination of refractive index (refractometry)
The refractive index of a medium with reference to air is equal to the ratio of
the sine of the angle of incidence of a beam of light in air to the sine of the angle of
refraction of the refracted beam in the given medium.
The index of refraction depends on a temperature, wavelength light, and in
solutions - from nature of solvent and concentration. Dependence of refractive index
is expressed by formulas:
n=no + C . F
,)( 0
F
nnC
where
C- is concentration of solution (%);
no -is index of refraction of solvent;
n - is index of refraction of solution;
F - is correction factor (value of increasing of refractive index at increasing of
concentration on 1%). Determination is carried out at the temperature of 20°С and
wave-length D of sodium spectrum (589.3 nm).
Index of water refraction:
nD20
= 1,3330
If the solution consisting of a few ingredients
n= n0 +n1 +n2 +……..+ ni
or
n= n0 + C1.F1 + C2
.F2 +……..+Ci.Fi
Concentration of one of components in mixture can be expressed by formula:
1
220
1
]........)[(
F
FCFCnnC ii
Determination of specific rotation (polarimetry) Optical rotation is the property displayed by chiral substances of rotation the
plane of polarised light.
Optical rotation is considered to be positive (+) for dextrorotatory substances
(i.e. those rotate the plane of polarisation in a clockwise direction) and negative (-)
for laevorotatory substances.

85
The specific optical rotation [αm]t
λ is the rotation expressed in radians (rad) ,
measured at the temperature t and at the wavelength λ given by a 1 m thickness of
liquid or a solution containing 1 kg/m 3 of optically active substance.
For the liquid individual substances the specific rotation is found out by
formula:
lD
20][ , where
α- is angle of rotation in degrees read at 20°C;
ρ -is density at 20°C in grams per cubic centimeter;
l - is length in decimeters of the polarimeter tube.
For solutions:
lCD
100][ 20
, l
CD
20][
100
C- is concentration of solution (%).
For qualitative and quantitative analysis of organic substances the spectral
methods – such as UV-, IR- and NMR, photoelectrocolorimetry, fluorimetry are often
used.
For chemical analysis of this group of substances the element analysis and
analysis by functional groups is used.
Element analysis of organic substances
The element analysis allows defining the structural elements of substances.
Determination is based on preliminary mineralization - destruction of substance with
formation of simple inorganic substances.
Determination of carbon and hydrogen
Burning with copper oxide:
the substance is mixed with copper oxide, put into the tube with barium
hydroxide water and heated on flame; a carbon forms carbon dioxide, which causes
dimness of barium hydroxide water:
CO2 + Ba(OH)2 BaCO3 + H2O
The presence of hydrogen conduces to formation of water found out as drops in
overhead part of the test tube. Colourless crystals of copper sulphate get dark blue.
CuSO4 . H2O + 4H2O CuSO4 . 5H2O
On thermic decomposition of substance with sulphur, sodium sulphite or
thiosulphate hydrogen associates into hydrosulphur, which is determined with leaden
paper:
H2S + (CH3COO)2Pb PbS + 2CH3COOH
Determination of oxygen
Oxygen is usually determined by functional groups. The determination of
oxygen is often related with difficulties and often confirmed only after the
determination of functional groups (carboxy-, hydroxy-, nitro- and etc).
Determination of nitrogen
Nitrogen can be determined by the smell of the burnt horn at incineration.

86
1. Method of Kal - burning with the mixture of thiosulphate and carbonate of
sodium. In result of this reaction forms sodium thiocyanate, which gives reaction
with salts of iron (III). The red painting appears:
3SCN- + Fe3+
Fe(SCN)3
2. Incineration with metallic sodium and reaction of Berlin azure formation:
2NaCN + FeSO4 Fe(CN)2 + Na2SO4
Fe(CN)2 + 4NaCN Na4[Fe(CN)6]
3Na4[Fe(CN)6] + 4FeCl3 Fe4[Fe(CN)6]3 + 12NaCl
This method is very sensitive.
Detection of sulphur
Sulphur can be detected by the Lassaigne test after heating with metallic
sodium. Organically integrated sulphur gets into sodium sulphide, which is detected
by appearance of the red-violet colouring of sodium nitroprusside:
Na2S + Na2[Fe(CN)5NO] Na4[Fe(CN)5NOS]
Detection of halogens
Chlorine, bromine or iodine can often be detected by heating the substance on a
piece of clean, oxidised copper wire, owing to the formation volatile chloride,
bromide, or iodide of copper, the flame becomes green (Beilstein's test ).
CuO + HalR CuHal2 + CO2 + H2O
On heating halogens of copper (II) are reduced:
CuHal2 Cu2Hal2
Fluorine derivatives can’t be determined by the colouring of flame, because
Cu2F2;- is non-volatile substance.
It is possible to detect chloride, bromide, or iodide by ordinary analytical
reactions after mineralization.
If halogen exists as a salt of halogenderivative acid (in ionised condition), these
reactions can be conducted without mineralization.
Analysis by functional groups
Functional groups are atoms or groups of atoms, which stipulate properties of
substances and determine character of reactions of their identification.
The most general are resulted below:
Alcohol hydroxyl
1. Reaction of esterification in the presence of anhydrous sulphuric acid: O
OH
O
ORR-OH + R`C
-H2OR`C
The products of reaction usually have the characteristic fruit smell.
2. Oxidation.
O
H
O
OHR-CH2OH RC
[O]RC
[O]
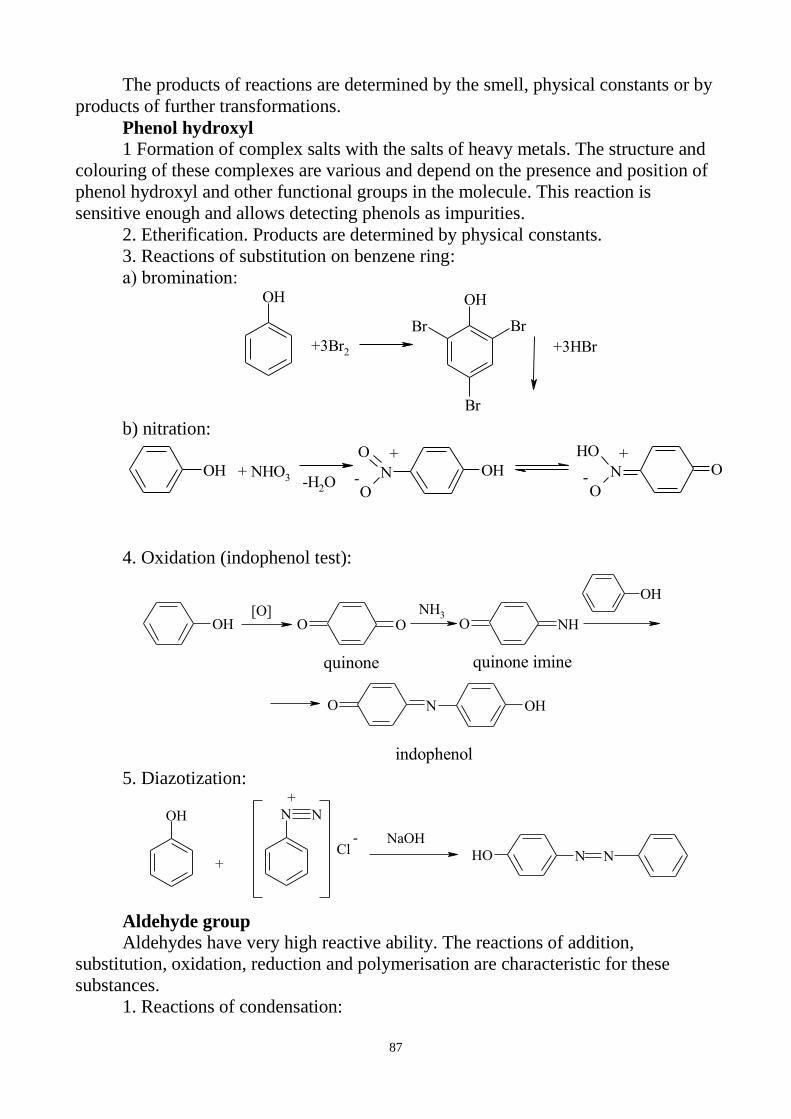
87
The products of reactions are determined by the smell, physical constants or by
products of further transformations.
Phenol hydroxyl
1 Formation of complex salts with the salts of heavy metals. The structure and
colouring of these complexes are various and depend on the presence and position of
phenol hydroxyl and other functional groups in the molecule. This reaction is
sensitive enough and allows detecting phenols as impurities.
2. Etherification. Products are determined by physical constants.
3. Reactions of substitution on benzene ring:
а) bromination: OH OH
Br Br
Br
+3Br2 +3HBr
b) nitration:
OH N OH
O
O
N O
O
OH
+ NHO3
+
-
+
--H2O
4. Oxidation (indophenol test):
OH O O O NH
OH
OHO N
[O] NH3
quinone quinone imine
indophenol 5. Diazotization:
OH
N
N N
OH N+
Cl
+
- NaOH
Aldehyde group
Aldehydes have very high reactive ability. The reactions of addition,
substitution, oxidation, reduction and polymerisation are characteristic for these
substances.
1. Reactions of condensation:

88
O
HN-R`RC +R`NH2 -H2O
R-CH
It is used for determining of primary aromatic nitrogen (lignin test).
O
HN-NH-R`RC +R`NHNH2 -H2O
R-CH
2. Reactions of oxidation are reduction:
a) reaction with ammonium silver nitrate solution (Tollense): O
HRC + H2O2[Ag(NH3)2]NO3 2 Ag + 2NH4NO3 + RCOONH4 + NH3+
The raid of metallic silver appears on the walls of test tube. This reaction is
given by all the aldehydes.
b) cupri-tartaric solution (Fehling reagent):
Cu2+
+ 2OH- Cu(OH)2 COONa
CHOH
CHOH
COOK
COO
CHOH
CHOH
COOK
OOC
HOHC
HOHC
NaOOC
Cu
2 +Cu(OH)2+NaOH+KOH
2KNa[Cu(C4H4O6)2]+RCOH+3NaOH+2KOH 2CuOH +2RCOONa+4KNaC4H4O6+2H2O
2CuOH
t o
Cu2O +H2O
On heating red precipitate forms only in the presence of aliphatic aldehydes.
This reaction often used in the analysis of sugars.
d) with alkaline potassium tetraiodomercurate solution (Nessler reagent): O
H
O
OKRCRC K2[HgI4] + 3KOH +Hg +4KI + 2H2O +
Reaction is very sensitive and often used for determining of aldehydes as
impurities.
Carboxyl group
1. Reactions with the salts of heavy metals.
2. Esterification.
3. At adding of sodium hydrogen carbonate solution (unlike alcohols and
phenols) carbon dioxide is educed.
Ester group
1. Hydrolysis. O
OR`R-COOH + R`OH+H2ORC
The products of reaction are determined by physical constants.
2. Reaction with hydroxylamine. Complexes with iron have red-violet colour.
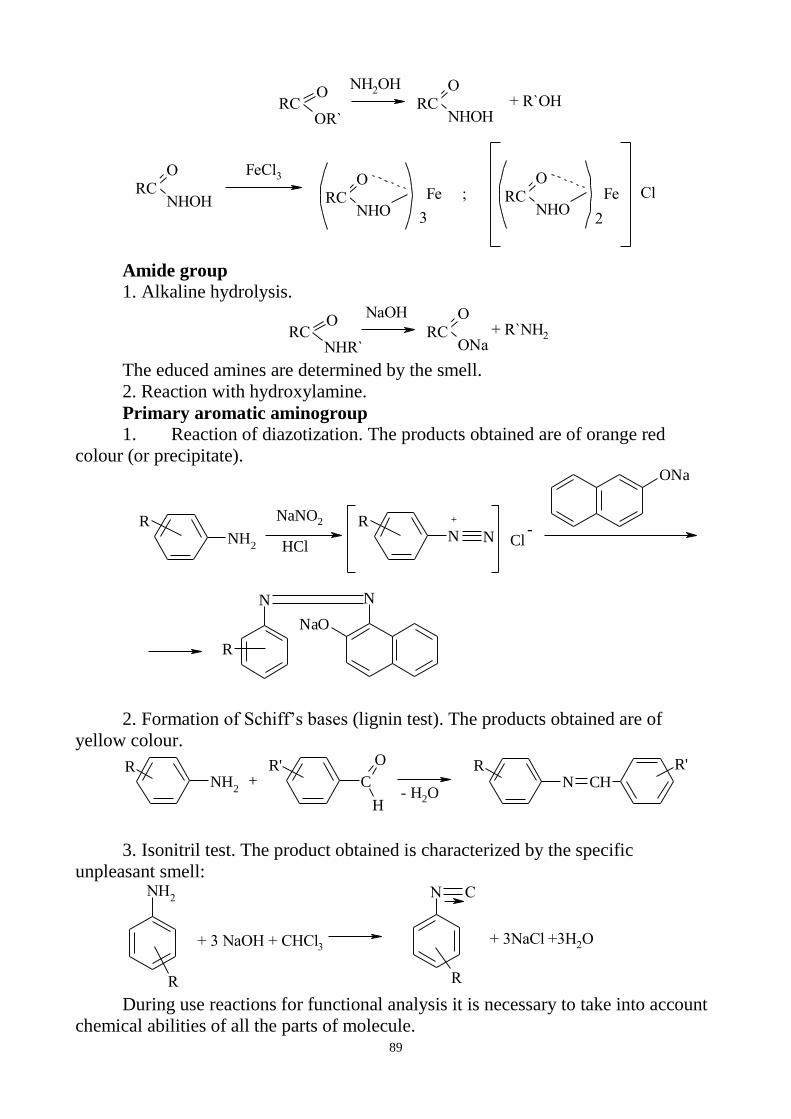
89
O
OR`
O
NHOH+ R`OHRC
NH2OH
RC
O
NHOH
O
NHO
O
NHO
RC
FeCl3
RC RC
3
Fe
2
Fe Cl;
Amide group
1. Alkaline hydrolysis.
O
NHR`
O
ONa+ R`NH2RC
NaOH
RC
The educed amines are determined by the smell.
2. Reaction with hydroxylamine.
Primary aromatic aminogroup
1. Reaction of diazotization. The products obtained are of orange red
colour (or precipitate).
NH2
RN
RN
ONa
N
R
N
NaO
NaNO2
HCl
+
Cl-
2. Formation of Schiff’s bases (lignin test). The products obtained are of
yellow colour.
NH2
R R'C
H
O RN CH
R'+
- H2O
3. Isonitril test. The product obtained is characterized by the specific
unpleasant smell: N CNH
2
RR
+ 3 NaOH + CHCl3 + 3NaCl +3H2O
During use reactions for functional analysis it is necessary to take into account
chemical abilities of all the parts of molecule.

90
Chapter 8. MEDICINAL SUBSTANCES, WHICH ARE THE
DERIVATIVES OF SATURATED HALOCARBONS
AND ALCOHOLS OF ALIPHATIC ORIGIN
Plan
1. Medicinal substances with organic halogen (chloroform, iodoform,
ethyl chloride, halothane (phthorotanum)).
2. Medicinal substances – aliphatic alcohols (ethanol, glycerol).
Halocarbons are formed when one or some atoms of hydrogen in hydrocarbon
molecule substituted by halogens.
The general stage in analysis of these compounds is determination of halogen.
For this purpose is usually used the Beilstein’s test. For confirming of halogen by
analytical reactions it is necessary to transfer it into ionised condition. During
mineralization the simple inorganic substances are formed.
This class of compounds includes such substances as: chloroform, iodoform,
ethylene chloride and halothane.
Chloroform (Chloroformium)
CHCl3
Trichloromethane
Preparation
Electrolysis of sodium chloride in the presence of alcohol or acetone:
2NaOH + Cl2 NaClO + NaCl + H2O
C
H
O
CH3
C2H5OH + NaClO + NaCl + H2O
C
H
O
CH3
C
H
O
Cl3C+ 3NaOCl + 3NaOH
C
H
O
Cl3C + NaOH CHCl3 + HCOONa
Properties Colourless, transparent, heavy and mobile volatile liquid with the
characteristic odour and sweet taste. It is mixed in all proportions with an anhydrous
ethanol, ether and petrol, essential and fatty oils. Non-mixed with glycerol.
During storage can be oxidized.
C
Cl
Cl
O2CHCl3 + O2 2HOCCl32 + 2HCl

91
C
Cl
Cl
O2 + O2 2CO2 + 2Cl2
To avoid oxidation for chloroform is added ethanol (0,6 – 1%), which used as
preservative substance:
C
Cl
Cl
O C
OC2H
5
OC2H
5
O
C
H
O
CH3
+ 2C2H5OH + 2HCl
HCl + C2H5OH C2H5Cl + H2O
Cl2 + C2H5OH + 2HCl
CCl Cl
O
2CHCl3 + O2 2CCl3(OH) 2 + 2HCl
CCl Cl
O
2 + O2 2CO2 + 2Cl2
Identification
Physical constants: boiling point and density.
According to Pharmacopoeia it is necessary to make quantification of
ethanol:
3C2H5OH + 2K2Cr2O7 +16HNO3 4Cr(NO3)3 + 3CH3COOH + 4KNO3 + 11H2O
excess
K2Cr2O7 + 6KI + 14HNO3 8KNO3 + 2Cr(NO3)3 + 3I2 + 7H2O
I2 + 2Na2S2O3 2NaI + Na2S4O6
S=3/2.The indicator is starch.
Purity Free chlorine: Cl2 + 2KI I2 + 2KCl
Aldehydes: O
H
O
OKRCRC K2[HgI4] + 3KOH +Hg +4KI + 2H2O +
Usage External medicine for massages at neuralgia.
At poisonings with arsine, for laboratory works as preservative.
Storage In well-stoppered orange glass bottles, in cool place.

92
Iodoform (Iodoformium)
CHI3
Triiodomethane
Preparation
C2H5OH + 4I2 + 3Na2CO3 CHI3 + 5NaI + HCOONa + 2H2O +
3CO2
Properties Yellow powder with the specific odour. It melts at first, and then
decomposes with educing of violet steams of iodine. Volatile already at room
temperature, distillated with water vapour. Solutions of substance quickly decompose
under action of light and air with educing of iodine.
Identification On heating a violet steams of iodine are educed:
2CHI3 + 2O2 3I2 + CO + CO2 + H2O
Assay Argentometry (Volhard method). The substance is dissolved in
water-alcohol mixture, heated with silver nitrate solution in the presence of nitric
acid. The excess of silver nitrate is titrated with ammonia thiocyanate using
ferric-ammonia sulphate solution as an indicator. S=1/3.
to
CHI3 + 3AgNO3 + H2O 3AgI + 3HNO3 + CO
AgNO3 + NH4SCN AgSCN + NH4NO3
Fe3+
+ 3SCN- Fe(SCN)3
Carry out a blank titration.
Usage Antiseptic. Use externally as powder, ointments, pastes for
treatment of wounds and ulcers.
Storage In well-closed containers, protected from light.
Ethyl chloride (Aethylii chloridum)
C2H5Cl
Preparation
1. Chlorination of ethane: C2H6 + Cl2 C2H5Cl + HCl
2. Hydrochloration of ethylene: CH2CH2 + HCl CH3CH2Cl
Properties Colourless, very volatile liquid. Catches fire easily. It burns with a
green flame. The boiling temperature is 12º C .
Identification
On heating with alkali:
C2H5Cl + KOH C2H5OH + KCl

93
carry out reactions on alcohol:
C2H5OH + 4I2 + 6KOH CHI3 + 5KI + HCOOK + 5H2O yellow
and reactions of chlorides.
Quality is confirmed by physical constants – boiling point and density.
Purity Impurity of ethanol is determined by iodoform test (see above).
Usage For short-term narcosis or local anaesthesia, due to cooling of tissues.
Storage In ampules, or in bottles with special stoppers, in cool and protected
from light action place.
Halothane (Phthorotanum)
CF3-CHClBr
(RS)-2-bromo-2-chloro-1,1,1-trifluoroethane
Preparation F
3C CH
2Cl F
3C CHClBr+ Br2 + HBr
Properties A clear, colourless, mobile, heavy, non-flammable liquid, slightly
soluble in water, miscible with ethanol, with ether and with trichloroethylene.
It contains 0,01% of thymol as a stabiliser.
Identification
1. IR-spectrum.
2. To the substance add 2-methyl-2-propanol in a test-tube. Add copper edetate
solution, concentrated ammonia and a mixture of strong hydrogen peroxide solution
and water (solution a). Prepare a blank at the same time (solution b). Place both tubes
in a water-bath, cool, and then add glacial acetic acid. To the each of solutions add a
mixture of freshly prepared alizarin solution and zirconyl nitrate solution. Solution
(a) is yellow and solution (b) is red.
The obtained fluorides decompose the complex of alizarin with zirconium (in
the examined test the red colouring becomes yellow; in the blank test the red
colouring remains):
O
O
O
OH
SO3Na
O
O
OH
OH
SO3Na
Zr/4
+ 6F-
+ [ZrF6]2 -
To 1 ml of each of solutions add buffer solution pH 5.2, phenol red solution
and chloramine solution. Solution (a) is bluish-violet and solution (b) is yellow.

94
At pH 5.2 the phenol red has a yellow colouring. In the presence of bromides
and under action of chloramine it becomes bromothymol blue, which has a bluish-
violet colouring:
SO3H
O
C
HO
Br
SO3H
O
Br
BrC
HO
Br
+ 4Br2 +4HBr
To 2 ml of each of solutions add sulphuric acid, acetone and potassium
bromate solution. Warm the tubes in a water-bath, cool and add nitric acid and silver
nitrate solution. Solution (a) is opalescent and a white precipitate is formed after a
few minutes; solution (b) remains clear.
Bromides are oxidised to free bromine by interaction with potassium bromate:
5Br-+BrO3
-+6H
+3Br2+3H2O;
and then they are deleted as pentabromoacetone:
CH3COCH3+5Br2CBr3COCHBr2+HBr;
chlorides in the solution are detected with silver nitrate solution:
Cl-+Ag
+AgCl
Purity Contents of thymol is determined by gas-chromatography.
Usage General anaesthetic.
Storage Store in an airtight container, protected from light, at temperature
25C. The choice of material for the container is made taking into account the
particular reactivity of halothane with certain metals. In the end of each 6 months of
storage the substance is re-examined.
Alcohols – are organic compounds with the general formula R-OH. They are
classified as primary, secondary or tertiary according to the kind of carbon containing
the –OH group.
Alcohols are enough inert chemically; they have weak-acid properties, inclined
to oxidation and enter into reactions of substitution (for example, esterification).
The main pharmacological action of low-molecular alcohols is influence on
central nervous system. High-molecular alcohols (more than 16 atoms of carbon)
practically do not influence on organism.
The simplest representatives are ethanol and glycerol.

95
Ethanol (Spiritus aethylicus)
CH3-CH2OH
Preparation
Alcohol fermentation of starch:
During manufacturing of alcohol can be obtained such side products as pyruvic
acid, acetaldehyde, glycerol, and fusel oil. For purifying ethyl alcohol must be
distilled.
Properties Colourless, clear, volatile, flammable liquid, hygroscopic. Miscible
with water and with methylene chloride. It burns with a blue, smokeless flame.
Boiling point 78ºC.
Identification
1. Relative density (0.805- 0.812).
2. IR-spectrum.
3. Mix the substance with solution of potassium permanganate and dilute
sulphuric acid. Cover immediately with a filter paper moistened with a freshly
prepared solution containing of sodium nitroprusside and piperazine hydrate in water.
After a few minutes an intense blue colour appears on the paper and becomes paler
after 10-15 minutes.
4. To the substance add water and dilute sodium hydroxide solution, then
slowly add 0.05 M iodine. A yellow precipitate is formed within 30 min (iodoform
test):
C2H5OH + 4I2 + 6KOH CHI3 + 5KI + HCOOK + 5H2O
Purity Volatile impurities determine by gas-chromatography method.
Assay By Pharmacopoeia no assay is proposed.
Non-Pharmacopoeial method – dichromatometry:
3C2H5OH + 2K2Cr2O7 +16HNO3 4Cr(NO3)3 + 3CH3COOH + 4KNO3 + 11H2O excess
K2Cr2O7 + 6KI + 14HNO3 8KNO3 + 2Cr(NO3)3 + 3I2 + 7H2O
I2 + 2Na2S2O3 2NaI + Na2S4O6
nC6H
12O
6(C
6H
10O
5)n nC
12H
22O
11+ OnH
22H
nC2H
5OH
O
nCO2+
starch
amylase, 60 C
maltose
, maltase
glucose
zymase , 30-33 C
fermentation
2
2
2
o
o
2
5C2H5OH +2KMnO4+3H2SO4
CH3 C
O
H + K2SO4 + 2MnSO4 8H2O+

96
S=3/2.The indicator is starch.
Usage External antiseptic and irritant substance for massages and compresses.
It is manufactured as 95%, 90%, 70%, 40% aqueous solutions.
Storage In well-closed containers, protected from light.
Glycerol (Glycerinum)
CH
2
CH
C
OH
OH
OHH2
Propane-1,2,3-triol
Preparation By saponification of fats:
CH2
CH
C
O
O
O
C
C
C
O
O
O
R
R
R
CH2
CH
C
OH
OH
OHH2
OH-
H2
+ 3RCOO-
Properties Syrupy liquid, unctuous to the touch, colourless or almost
colourless, clear, very hygroscopic. Miscible with water and with alcohol, slightly
soluble in acetone, practically insoluble in fatty oils and in essential oils.
Identification
1. Refractive index (1.470-1.475).
2. IR-spectrum.
3. Reaction with nitric acid and potassium dichromate solution.
A blue colouring ring develops at the interface of the liquids within 10 min, the
blue colour does not diffuse into the lower layer.
4. Heat the substance with potassium hydrogen sulphate in an evaporating dish.
Vapours are evolved which blacken filter paper impregnated with alkaline potassium
tetraiodomercurate solution:
CH2OH
CH2OH
KICHOHKOH
C HO
CHK
2HgI
4
CH2
O
CH2
CH ++ 2+ OHg H
C OK,2KHSO4 3
2
Assay Indirect alkalimetry method.
Mix the substance with water. Add sulphuric acid and sodium periodate. Allow
to stand protected from light for 15 min. Add the solution of ethylene glycol and
allow to stand protected from light for 20 min. Using phenolphthalein as indicator,
titrate with 0.1 M sodium hydroxide. S=1. Carry out a blank titration.

97
+ ++ + O2
CH2
CH2
CH NaIO4
OH
OH
OH
HC
H
O
NaIO3
HCOOH H2 2 2
The educed formic acid is titrated with sodium hydroxide solution:
HCOOH NaOH HCOONa OH2++
2. Acetilation:
CH2
CH
C
OH
OH
OH
CH2
CH
C
O
O
O
COCH3
COCH3
COCH3
H2
+ 3(CH3CO)2O
H2
+ 3CH3COOH
After destruction of acetic anhydride the solution analyzed is neutralized with
an alkali (the indicator is phenolphthalein) and boiled with the sodium hydroxide
solution: CH
2
CH
C
O
O
O
COCH3
COCH3
COCH3
CH2
CH
C
OH
OH
OHH2
+ 3NaOH
H2
+ 3CH3COONa
The excess of the alkali is titrated with hydrochloric acid; S=1/3.
NaOH + HCl NaCl + H2O
3. Back dichromatometry; s=3/7. CH
2
CH
C
OH
OH
OHH2
3 + 7K2Cr2O7 + 28H2SO4 9CO2+ 7K2SO4 + 7Cr2(SO4)3 + 40H2O
The excess of potassium dichromate is titrated with FeSO4·(NH4)2SO4·6H2O: K2Cr2O7 + 6Fe(NH4)2(SO4)2 + 7H2SO4 Cr2(SO4)3 + 3Fe2(SO4)3 + K2SO4 + 6(NH4)2SO4 + 7H2O
Usage It is a base for ointments and solutions. Anhydrous glycerol can cause
burns.
Storage In an airtight container.

98
Chapter 9. MEDICINAL SUBSTANCES DERIVATIVES OF
ALDEHYDES AND CARBOXYLIC ACIDS OF ALIPHATIC ORIGIN
Plan
1. Medicinal substances derivatives of aldehydes (solution of formaldehyde,
methenamine, chloral hydrate).
2. Salts of aliphatic carboxylic acids (potassium acetate, calcium lactate,
calcium gluconate, sodium citrate for injections, sodium hydrogen citrate).
Aldehydes – are compounds, which contain the aldehyde (carbonyl) group.
Aldehydes have strong reactive ability. They can get into reactions of oxidation
(sometimes reduction), addition, and polymerisation.
This chemical class of substances includes formaldehyde, hexamine and
chloral hydrate.
Medicinal substances derivatives of aldehydes
Formaldehyde solution 35% ( 35% Solutio Formaldehydi)
C
H
O
H
Preparation
Oxidation of methyl alcohol:
C
H
O
H2CH3OH + O2
500 - 600O
C
Kt2 + 2H2O
Oxidation of methane (Medvedev method):
C
H
O
H2CH4 + O22CH3OH
O2
+ 2H2O2
Properties A clear, colourless liquid, miscible with water and with alcohol. It
may be cloudy after storage.
Methyl alcohol is added as a stabiliser (to 15%) to avoid paraformaldehyde
formation.
Identification
1. Reaction with chromotropic acid in the presence of sulphuric acid. A violet-
blue or violet-red colour develops within 5 min:
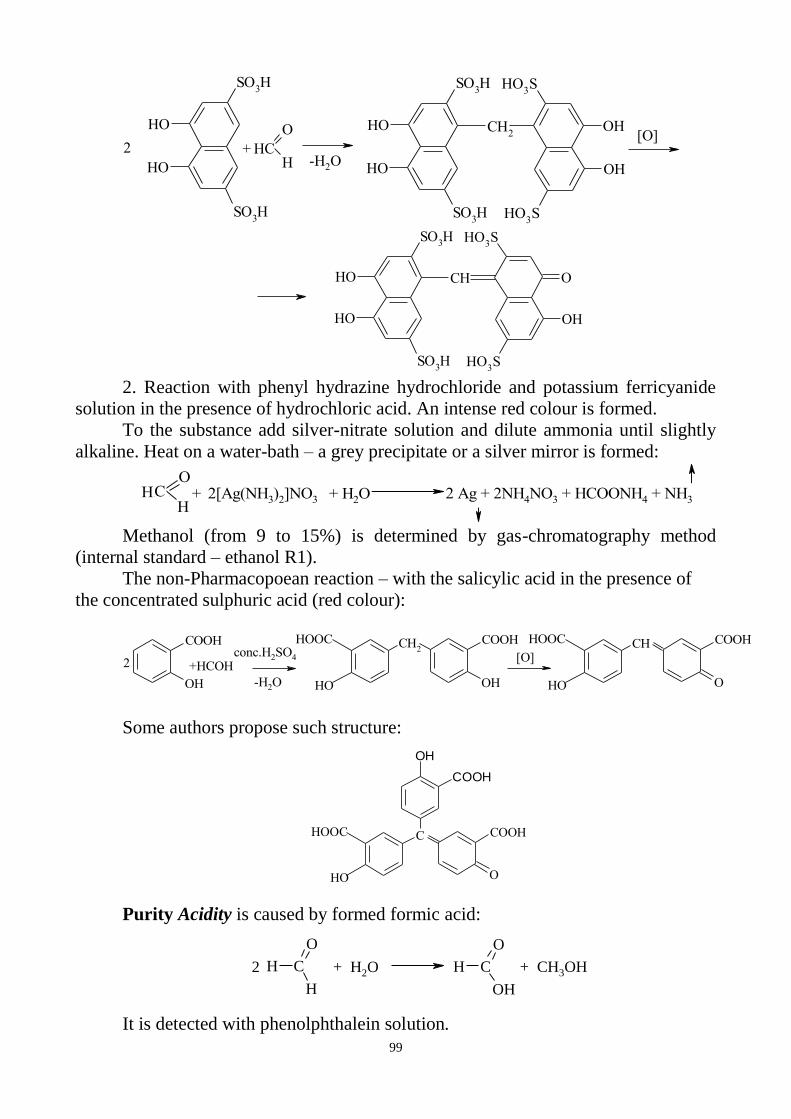
99
OH
SO3H
SO3H
OH
O
H
OH
SO3H
SO3H
OH
CH2
HO3S
OH
HO3S
OH
HC+-H2O
[O]2
HO
3S
O
HO3S
OH
OH
SO3H
SO3H
OH
CH
2. Reaction with phenyl hydrazine hydrochloride and potassium ferricyanide
solution in the presence of hydrochloric acid. An intense red colour is formed.
To the substance add silver-nitrate solution and dilute ammonia until slightly
alkaline. Heat on a water-bath – a grey precipitate or a silver mirror is formed:
O
HRC + H2O2[Ag(NH3)2]NO3 2 Ag + 2NH4NO3 + HCOONH4 + NH3+H
Methanol (from 9 to 15%) is determined by gas-chromatography method
(internal standard – ethanol R1).
The non-Pharmacopoean reaction – with the salicylic acid in the presence of
the concentrated sulphuric acid (red colour):
OH
COOH CH2
OH
COOHHOOC
HO
CHHOOC
HO
COOH
O
2 +HCOHconc.H2SO4
-H2O
[O]
Some authors propose such structure:
HOOC
HO
COOH
O
C
OH
COOH
Purity Acidity is caused by formed formic acid:
C
H
O
H C
OH
O
H+ H2O + CH3OH2
It is detected with phenolphthalein solution.

100
Methanol is determined by gas chromatography.
Assay
1. Iodometry. S=1.
To the solution of substance add sodium hydroxide solution, iodine and dilute
sulphuric acid. Titrate with sodium thiosulphate using starch solution as indicator.
C
H
O
HC
H
O
H + I2 + H2O + 2HI
Sodium hydroxide solution is added because HI in acid medium can reduce
formic acid to formaldehyde.
I2 + 2NaOH NaI + NaOI + H2O
C
H
O
H C
OH
O
HNaOI + NaI +
NaI + NaOI + H2SO4 I2 + Na2SO4 + H2O
I2 +2Na2S2O3 2NaI + Na2S4O6
2. Oxidation with hydrogen peroxide in the alkaline medium (S =1):
C
H
O
H C
ONa
O
H+ H2O2 + NaOH + H2O
NaOH + HCl NaCl + H2O
3. Titration with the sodium sulphite (for quantification of formaldehyde in the
«Formidron»); (S =1):
C
H
O
H C
SO3Na
OH
H H+ NaSO3 + H2O + NaOH
NaOH + HCl NaCl + H2O
4. Refractometry.
Usage Used in the treatment of warts and as antiseptic, disinfectant and
deodorant; in preservation of anatomical specimens. Formalin is a protoplasmatic
poison!
Storage In well-closed containers, protected from light, at a temperature of
15ºC to 25ºC.
Methenamine (Hexamethylentetraminum)
N
N
NN
1,3,5,7-tetra-azotricyclo[3.3.1.13,7]decane

101
Preparation Mix the solutions of formaldehyde and ammonia.
N
N
NN
CH
H
O
CH
H
OH
NH2
CH2
NHNH
NH
NH
CH
H
O
+ NH3- H2O NH3
3
Summary:
N
N
NN
CH
H
O
6 + 4NH3 + 6H2O
Properties A white, crystalline powder or colourless crystals, freely soluble in
water, soluble in alcohol and in methylene chloride.
Identification
1. IR-spectrum.
2. To the solution of substance add sulphuric acid and immediately heat to
boiling. Allow to cool, then to the solution add water and acetylacetone reagent. Heat
for 5 min. An intense yellow colour develops.
3. To the solution of substance add dilute sulphuric acid and immediately
heat to boiling. The solution gives reaction of ammonium salt and salts of volatile
bases.
Dissolve the substance in water and acidify with dilute hydrochloric acid. Add
potassium iodobismuthate solution. An orange precipitate is formed immediately.
4. With silver nitrate solution – white precipitate appears:
N
N
NN N
N
NN
2 + 3AgNO32 3AgNO3
.
Assay
1. Dissolve the substance in methanol. Titrate with 0.1 M perchloric acid,
determining the end-point potentiometrically. S=1.
2. Acid-base titration. The indicator is methyl red solution. S =1/2:
(CH2 )6N4+ 2H2SO4
+ 6H2O 6HC H
O
+ 2(NH4)2SO4
(NH4)2SO4 + 2NaOHto
Na2SO4+ 2NH4OH
+NH4OH NH3 H2O
(CH2 )6N4+HClO4 (CH2)6N4
. HClO4

102
N
N
NN
CH
H
O
+ 2H2SO4 + 6H2O 6 + 2(NH4)2SO4
tO
H2SO4 + 2NaOH Na2SO4 + 2H2O
3. Acidimetry (with the mixed indicator – methyl orange and methyl blue).
S=1:
N
N
NN N
N
NN
+ HCl . HCl
4. Iodine monochloride method (iodchlorimetry). Back titration, the indicator is
starch solution. S=1/2:
N
N
NN N
N
NN
+ 2ICl . 2ICl
ICl + KI I2 + KCl
I2 + 2Na2S2O3 2NaI + Na2S4O6
5. Argentometry by Volhard method. Back titration; the excess of silver nitrate
is titrated with ammonia thiocyanate using ferric-ammonia sulphate as an indicator.
S=2/3:
N
N
NN N
N
NN
2 + 3AgNO32 3AgNO3
.
AgNO3 + NH4SCN AgSCN + NH4NO3
3NH4SCN + Fe(NH4)(SO4)2 Fe(SCN)3 + 2(NH4)2SO4
Usage Hexamine is used as a urinary antiseptic. It is used at poisonings by salts
of heavy metals as antidote.
Storage In well-stoppered bottles, protected from light.
Chloral hydrate (Chloralum hydratum)
Cl3C C
OH
OH
H
2,2,2-trichloroethane-1,1-diol

103
Preparation
C
H
O
CH3
C
OC2H
5
OH
CH3
H C
OC2H
5
OH
Cl3C H
C
H
O
Cl3C C
OH
OH
CH3
H
C2H5OHCl2, FeCl3 C2H5OH Cl2
- HCl
H2SO4
H2O
trichloracetic aldehyde
Properties Transparent, colourless crystals, very soluble in water, freely
soluble in alcohol and in ether.
Under light action slowly decomposes (oxidation):
C
OH
OH
Cl3C H C
H
O
C
Cl
Cl
H Cl3C COOH+ + HCl + H2O 2
Identification
1. Add dilute sodium hydroxide solution – the mixture becomes cloudy
and, when heated, gives off an odour of chloroform:
Cl3C C
OH
OH
H + NaOH CHCl3 + HCOONa + H2O
2. To the solution of substance add sodium sulphide solution – a yellow
colour develops which quickly becomes reddish-brown. On standing for a short time,
a red precipitate may be formed.
The non – Pharmacopoeial reaction – with silver ammonia-nitrate solution:
O
HCCl3C + H2O2[Ag(NH3)2]NO3 2 Ag + 2NH4NO3 + CCl3COONH4 + NH3+
Purity Chloral alcoholate is the intermediate product of synthesis; it is detected
with sodium hydroxide solution on adding of iodine (iodoform test).
CCl3CH(OH)OC2H5 + NaOH CHCl3 + HCOONa + C2H5OH
C2H5OH + 4I2 + 6NaOH CHI3 + 5NaI + HCOONa + 5H2O
Assay
1. Dissolve the substance in water and add sodium hydroxide solution, titrate
with sulphuric acid using phenolphthalein as indicator.
Titrate the neutralised solution with silver nitrate, using potassium chromate
solution as indicator. S =1.
CCl3CH(OH)2 + NaOH CHCl3 + HCOONa + H2O excess
2NaOH + Н2SO4 Na2SO4 +2H2O
CHCl3 + 4NaOH 3NaCl +HCOONa + 2H2O

104
The educed sodium chloride is titrated with silver nitrate solution:
NaCl +AgNO3 AgCl +NaNO3
AgNO3 + K2CrO4 Ag2CrO4 + 2KNO3
Volume of titrant = ])[(342 AgNOSOHNaOH VVV
2. Iodometry (indicator is starch). S=1.
2CCl3CH(OH)2 + 2I2 + 3Na2CO3 2CCl3COONa + 4NaI + 3H2O + 3CO2
3. Acid-base titration, the indicator is phenolphthalein solution. Carry out a
dlank titration. S=1.
CCl3CH(OH)2 + NaOH CHCl3 + HCOONa + H2O excess
NaOH + HCl NaCl + H2O
Usage Hypnotic.
Storage In an airtight container.
Derivatives of carboxylic acids Carboxylic acids have in their molecules carboxyl group -COOH.
General properties of this substances are: ability to react with alkali, to form
precipitates with the salts of heavy metals, to get into reactions of esterification with
alcohols etc.
In free condition these substances are not used in medicine because of their
irritant action. In the most causes use their salts: potassium acetate, calcium lactate,
calcium gluconate, sodium citrate etc.
Potassium acetate (Kalii acetas)
CH3COOK
Preparation Neutralisation of acetic acid with potassium carbonate:
2CH3COOH + K2CO3 2CH3COOK + H2O + CO2
Properties A white, crystalline powder or colourless crystals, deliquescent,
very soluble in water, freely soluble in alcohol.
Identification
1. Reactions of acetate-ion:
a) reaction of ester forming with ethanol ( the specific fruit odour):
CH3COOK + C2H5OH + conc. H2SO4 CH3COOC2H5 + K2SO4 + H2O
b) interaction with the salts of ferric. Brown-red colouring:
9CH3COOK + 3FeCl3 + 2H2O 2CH3COOH [Fe3(OH)2(CH3COO)6]+CH3COO-+9KCl
c) it gives the reaction of acetyl:

105
La3+
+ 3CH3COO- + 2H2O La(OH)2CH3COO + 2CH3COOH
Blue
2. Reactions of К+:
a) reaction with tartaric acid:
K+ + HC4H4O6
- KHC4H4O6
b) reaction with sodium cobaltinitrite:
2K+ + Na
+ + [Co(NO2)6]
3- K2Na[Co(NO2)6]
c) colouring of flame (a violet colour).
Assay
1. Non-aqueous titration.
Dissolve the substance in anhydrous acetic acid. Add naphtholbenzein solution.
Titrate with 0.1 M perchloric acid. Carry out a blank titration. S=1.
CH3COOK + CH3COOH (CH3COOKH)+ . CH3COO-
HClO4 + CH3COOH (CH3COOH2)+ . ClO4
-
(CH3COOKH)+ . CH3COO- + (CH3COOH2)
+ . ClO4
- KClO4 +
3CH3COOH
Summary: CH3COOK + HClO4 CH3COOH + KClO4
2. Acidimetry (in liquid medicines). The indicator is tropaeolin-O. S=1.
CH3COOK + HCl CH3COOH + KCl
Usage Used in solutions for dialysis.
Storage In well-closed containers, protected from moisture.
Calcium lactate (Calcii lactas)
CH3
CH C
O
O
OH
-Ca
2
. 5H2O
Calcium bis(2-hydroxypropanoate)
Preparation Fermentation of sugary materials by pure cultures of lactic acid
bacteria in the presence of calcium carbonate.
C6H
12O
6 CH3CHCOOH
OH
CaCO3
CH3CH(OH)COO
Ca CO
2H
2O
2
+ +2
Properties A white or almost white, crystalline or granular powder, slightly
efflorescent, soluble in water, freely soluble in boiling water, very soluble in alcohol.
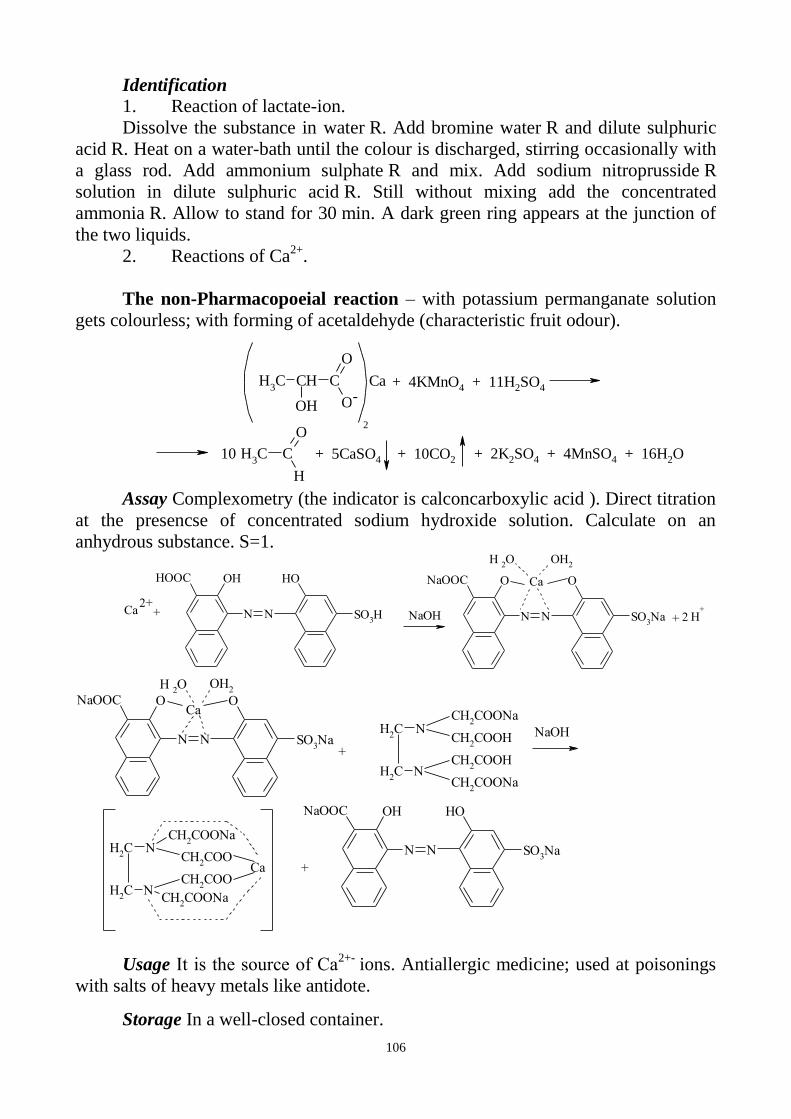
106
Identification
1. Reaction of lactate-ion.
Dissolve the substance in water R. Add bromine water R and dilute sulphuric
acid R. Heat on a water-bath until the colour is discharged, stirring occasionally with
a glass rod. Add ammonium sulphate R and mix. Add sodium nitroprusside R
solution in dilute sulphuric acid R. Still without mixing add the concentrated
ammonia R. Allow to stand for 30 min. A dark green ring appears at the junction of
the two liquids.
2. Reactions of Ca2+
.
The non-Pharmacopoeial reaction – with potassium permanganate solution
gets colourless; with forming of acetaldehyde (characteristic fruit odour).
CH3
CH C
O
O
OH
C
H
O
CH3
-Ca
2
+ 4KMnO4 + 11H2SO4
10 + 5CaSO4 + 10CO2 + 2K2SO4 + 4MnSO4 + 16H2O
Assay Complexometry (the indicator is calconcarboxylic acid ). Direct titration
at the presencse of concentrated sodium hydroxide solution. Calculate on an
anhydrous substance. S=1.
N N
OHHOOC OH
SO3HCa
N N
ONaOOC O
SO3Na
Ca
H 2O OH
2
NaOH H+
2++
+ 2
CH2
CH2
N
N
CH2COONa
CH2COO
CH2COO
CH2COONa
Ca
N N
ONaOOC O
SO3Na
Ca
H 2O OH
2
CH2
CH2
N
N
CH2COONa
CH2COOH
CH2COOH
CH2COONa
NaOH
N N
OHNaOOC OH
SO3Na
+
+
Usage It is the source of Са2+-
ions. Antiallergic medicine; used at poisonings
with salts of heavy metals like antidote.
Storage In a well-closed container.

107
Calcium gluconate (Calcii gluconas)
CH2OH
(CHOH)4
COO-2
Ca OH2
.
Calcium D-gluconate monohydrate
Preparation Electrochemical oxidation of glucose in the presence of calcium
carbonate and bromine:
CH2OH
(CHOH)4
COO-2
Ca
CH2OH
(CHOH)4
COH
Br2, H
2O
-HBr
CH2OH
(CHOH)4
COOH
CaCO3
-CO2
Properties A white, crystalline or granular powder, sparingly soluble in water,
freely soluble in boiling water.
Identification
1. Thin-layer chromatography.
2. Reactions of Ca2+
.
The non –Pharmacopoeial reaction: with FeCl3 solution. A light-green
colouring develops (reaction of gluconate-ion).
Purity Sucrose and reducing sugars are determined with cupri-tartaric solution
(Fehling reagent). No red precipitate is formed.
Assay Complexonometry (see calcium lactate).
Usage Used in treatment of calcium deficiency.
Storage In well-closed containers.
Sodium citrate for injections (Natrii citras pro injectionibus)
CH
2
C
CH2
OH
COONa
COONa
COONa
. 5 1/2H2O
Trisodium 2-hydroxypropane-1,2,3-tricarboxylate
Calculate with reference to the anhydrous substance.

108
Preparation
CH2
C
CH2
OH
COOH
COOH
COOH
CH2
C
CH2
OH
COONa
COONa
COONa
2 + 3Na2CO3 + 3H2O + 3CO22
Properties A white, crystalline powder or white, granular crystals, slightly
deliquescent in moist air, freely soluble in water, practically insoluble in alcohol.
Identification 1. Reaction of citrates.
Dissolve the substance in water R. Add sulphuric acid R and potassium
permanganate solution R. Warm until the colour of the permanganate is discharged.
Add sodium nitroprusside R solution in dilute sulphuric acid R and 4 g of sulphamic
acid R. Make alkaline with concentrated ammonia R, added dropwise until all the
sulphamic acid has dissolved. Addition of an excess of concentrated ammonia R
produces a violet colour, turning to violet-blue:
CH3
CH COO-
OH
CH3
C
O
H
Na2[Fe(CN)
5NO]
CH3
CO
HNa
2[Fe(CN)
5C
O
H]
[O], H+
+ CO2
CH2 2. Reactions of Na
+.
Non-Pharmacopoeial reactions:
a) forming of pentabromacetone:
C
CH3
CH3
O C
CHBr2
CBr3
O
CH2
C
CH2
OH
COONa
COONa
COONa
CH2
C
CH2
OH
COOH
COOH
COOH
H+ KMnO4
- CO2
Br2
Acetone white
b) on heating a white precipitate develops, which and dissolves on cooling:
CH2
C
CH2
OH
COO
COO
COO
CH2
C
CH2
OH
COO
COO
COO
2
-
-
-
-
-
-
2
Ca3+ 3CaCl2
+ 6Cl-t0
Purity
Oxalates. Dissolve the substance in water, add hydrochloric acid and
granulated zinc. Heat on a water-bath. Add phenylhydrazine hydrochloride and to
boiling. Cool rapidly, add hydrochloric acid and potassium ferricyanide solution.
Any pink colour in the solution is not more intense than that in a standard.

109
Assay
1. Non-aqueous titration. Titrate with perchloric acid in the medium of
anhydrous acetic acid, indicator – naphtholbenzein. S=1/3.
2. Ion-exchange chromatography:
CH
2
C
CH2
OH
COOH
COOH
COOH
CH2
C
CH2
OH
COONa
COONa
COONa
[Kat(H+)]
Citric acid is neutralised with an alkali; s=1/3:
CH
2
C
CH2
OH
COONa
COONa
COONa
CH2
C
CH2
OH
COOH
COOH
COOH
+ 3NaOH + 3H2O
3. Back argentometry by Volhard method; s=1/3:
CH
2
C
CH2
OH
COONa
COONa
COONa
CH2
C
CH2
OH
COONa
COONa
COONa
+ 3AgNO3 + 3NaNO3
AgNO3 + NH4SCN + NH4NO3AgSCN
FeNH4(SO4)2 + 3NH4SCN Fe(SCN)3 + 2(NH4)2SO4
Usage It is used for containing of blood.
Storage Store in an air-tight container.
Sodium hydrogen citrate (Natrii hydrocitras pro injectionibus)
CH
2
C
CH2
OH
COONa
COONa
COOH . H2O1,5
Disodium hydrogen 2-hydroxypropane-1,2,3-tricarboxylate sesquihydrate
Identification, assay, usage and storage – see sodium citrate.
+ HClO4
CH3COOH
+ NaClO4
CH2
C
CH2
OH
COONa
COONa
COONa
CH2
C
CH2
OH
COOH
COOH
COOH
3 3

110
Chapter 10. MEDICINAL SUBSTANCES WHICH ARE
DERIVATIVES OF ALIPHATIC AMINO ACIDS
Plan
1. General characteristic of aminoacids.
2. Medicinal substances, which are the derivatives of aliphatic amino acids.
Amino acids are derivatives of carboxylic acids, which contain in their
molecules one or more amino groups. -Amino acids are structural elements of
proteins and commonly occur in nature. More than 20 -amino acids have been
obtained from hydrolysates of proteins. They have general formula:
CH NH2
R
COOH
Amino acids can be divided into 2 groups:
-«Non-essential» amino acids can be synthesised by living organism in enough
quantities for supplement of normal functions;
-«Essential» amino acids – which are not synthesised by living organism or
synthesised in not enough quantities and have to be obtained with the food.
-Amino acids (except the simplest glycine H2N-CH2-COOH) exist in
optically active forms. Aliphatic aminoacids are soluble in water and non-soluble in
organic solvents. All aminoacids are amphoteric compounds due to acidic group (-
COOH) and basic group (-NH2). They can form salts with acids and bases.
R CH C
NH3
OH
O
R CH C
NH2
OH
O
R CH C
ONa
O
NH2
+
-ClHCl NaOH
- H2O
Aminoacids are amphoteric electrolytes; they exist as dipole ions in water
solutions and in solid condition.
Aminoacids are classified to -, -, - etc. in dependence of disposition of
carboxylic and aminogroup and have different chemical properties.
Heating:
-aminoacids form 2,5-diketopiperazines by intermolecular condensation:
R CH
C
NH2
OH
O
RCH
C
NH2O
OH
NH
NHR
R
O
O
tO
- 2H2O
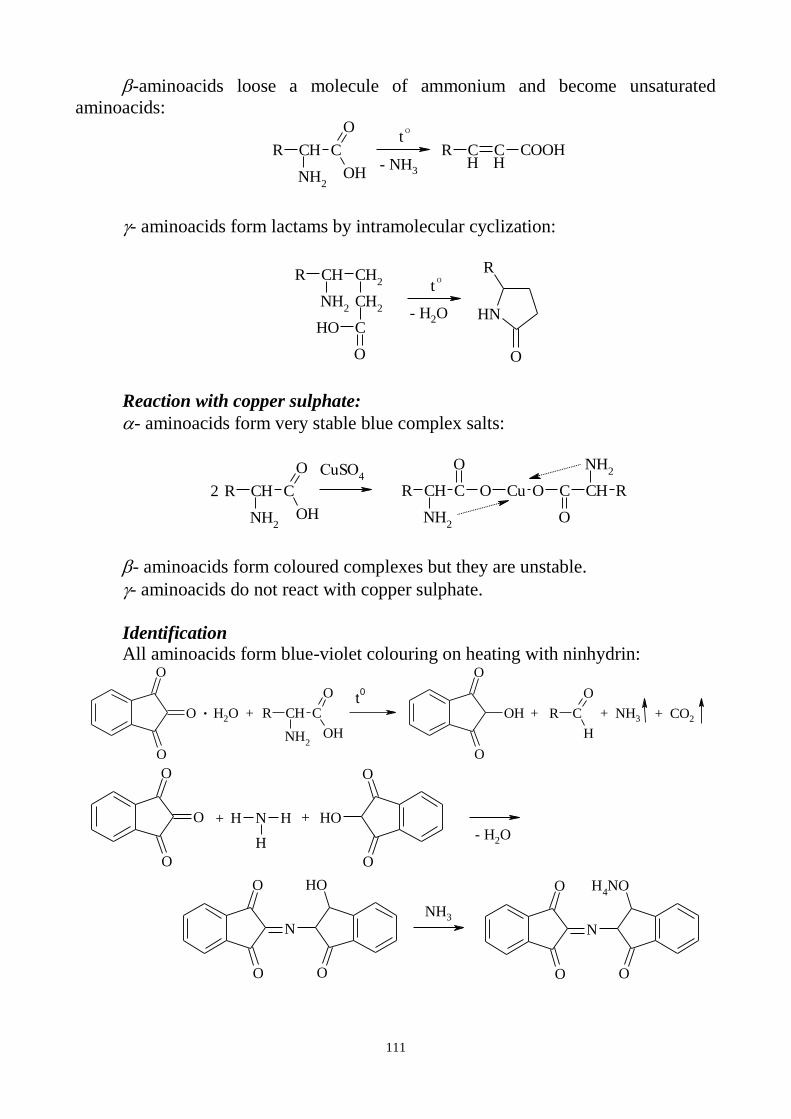
111
-aminoacids loose a molecule of ammonium and become unsaturated
aminoacids:
R CH C
NH2
OH
O
R CH
CH
COOHt
O
- NH3
- aminoacids form lactams by intramolecular cyclization:
R CH CH2
CH2
C
O
OH
NH2
NH
R
O
tO
- H2O
Reaction with copper sulphate:
- aminoacids form very stable blue complex salts:
R CH C
NH2
OH
O
R CH C O Cu O C CH R
NH2
NH2
O
O
CuSO4
2
- aminoacids form coloured complexes but they are unstable.
- aminoacids do not react with copper sulphate.
Identification
All aminoacids form blue-violet colouring on heating with ninhydrin: O
O
O
R CH C
NH2
OH
O
O
OH
O
R C
O
H
. H2O + + + NH3 + CO2
t0
O
O
O
H N H
H
O
O
OH
OH
O
N
O
O
H4NO
O
N
O
O
+ +- H2O
NH3

112
Assay
1. Determination of nitrogen by sulphuric acid digestion (Kjeldahl method)
– detection of nitrogen in organic compounds is required by Pharmacopoeia for many
substances of this group. This method consist of two stages:
a) mineralization of organic substance (boiling in the special apparatus in
the presence of K2SO4, CuSO4 and conc. H2SO4);
R CH C
NH2
OH
O
H2SO
4
CO2 + H
2O + NH
4HSO
4
[O]
Then 30% NaOH solution is added:
NH4HSO4 + 2NaOH NH3 + 2H2O + Na2SO4
The educed ammonium gets into the flask with hydrochloric acid. Titrate the
distillate with 0.01 M sodium hydroxide, using methyl red solution as indicator.
b) acid-base titration:
NH3 + HCl NH4Cl
HCl + NaOH NaCl +H2O.
excess
Repeat the test using about 50 mg of glucose in place of the substance to be
examined (n2 - volume of 0.01 M sodium hydroxide in the blank, ml; n1- volume of
0.01 M sodium hydroxide in the test, ml).
.
)(01401,0,% 12
m
nnN
2. Formol titration (the non-Pharmacopoeial method).
Formalin neutralised by phenolphthalein is added into amino acid solution. N-
methylene derivative with free carboxyl is formed, which then is titrated with an
alkali:
H
COONa
CH2
COOC CHR
N
+
NaOH
2O
O
H
H H+
+ +R RCH COOH
NN CH2
CH2
CH2OH
R CH COOH
NH2
The titration of -aminoacids is difficult, because they exist as intramolecular
salts.
3. Acid-base titration in non-aqueous solvents.
Usage of amino acids in medicinal practice is based on their ability to take part
in the synthesis of proteins, hormones, peptides and ferments.
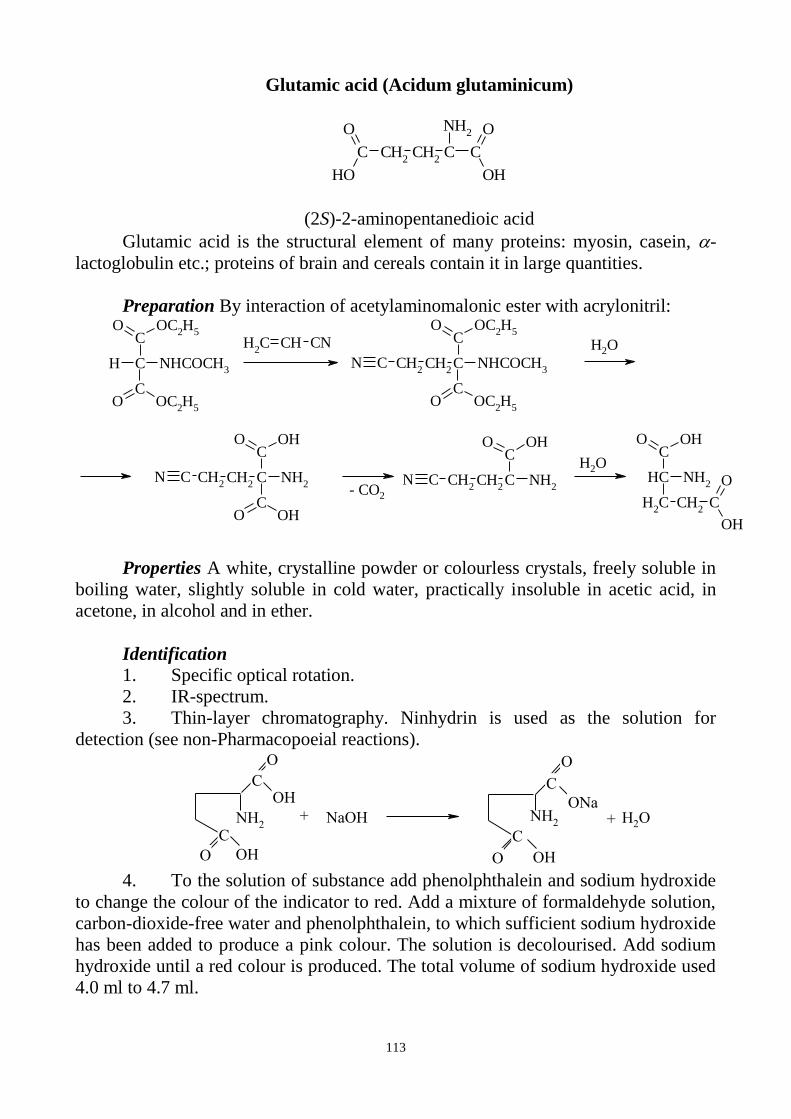
113
Glutamic acid (Acidum glutaminicum)
C CH2
CH2
C C
O
OH OH
ONH2
(2S)-2-aminopentanedioic acid
Glutamic acid is the structural element of many proteins: myosin, casein, -
lactoglobulin etc.; proteins of brain and cereals contain it in large quantities.
Preparation By interaction of acetylaminomalonic ester with acrylonitril:
C
C
C
OC2H
5O
NHCOCH3
O OC2H
5
H
CH2
CH CN
CN
C
C
C
OC2H
5O
NHCOCH3
O OC2H
5
CH2
CH2
CN
C
C
C
OHO
NH2
O OH
CH2
CH2
C
CH
CH2
OHO
NH2
CH2
C
O
OH
CN
C
C
OHO
NH2
CH2
CH2
H2O
- CO2
H2O
Properties A white, crystalline powder or colourless crystals, freely soluble in
boiling water, slightly soluble in cold water, practically insoluble in acetic acid, in
acetone, in alcohol and in ether.
Identification
1. Specific optical rotation.
2. IR-spectrum.
3. Thin-layer chromatography. Ninhydrin is used as the solution for
detection (see non-Pharmacopoeial reactions).
NH2
NH2
OH OH
C
O
O
COH
+ NaOH
C
O
ONa
C
O
+ H2O
4. To the solution of substance add phenolphthalein and sodium hydroxide
to change the colour of the indicator to red. Add a mixture of formaldehyde solution,
carbon-dioxide-free water and phenolphthalein, to which sufficient sodium hydroxide
has been added to produce a pink colour. The solution is decolourised. Add sodium
hydroxide until a red colour is produced. The total volume of sodium hydroxide used
4.0 ml to 4.7 ml.
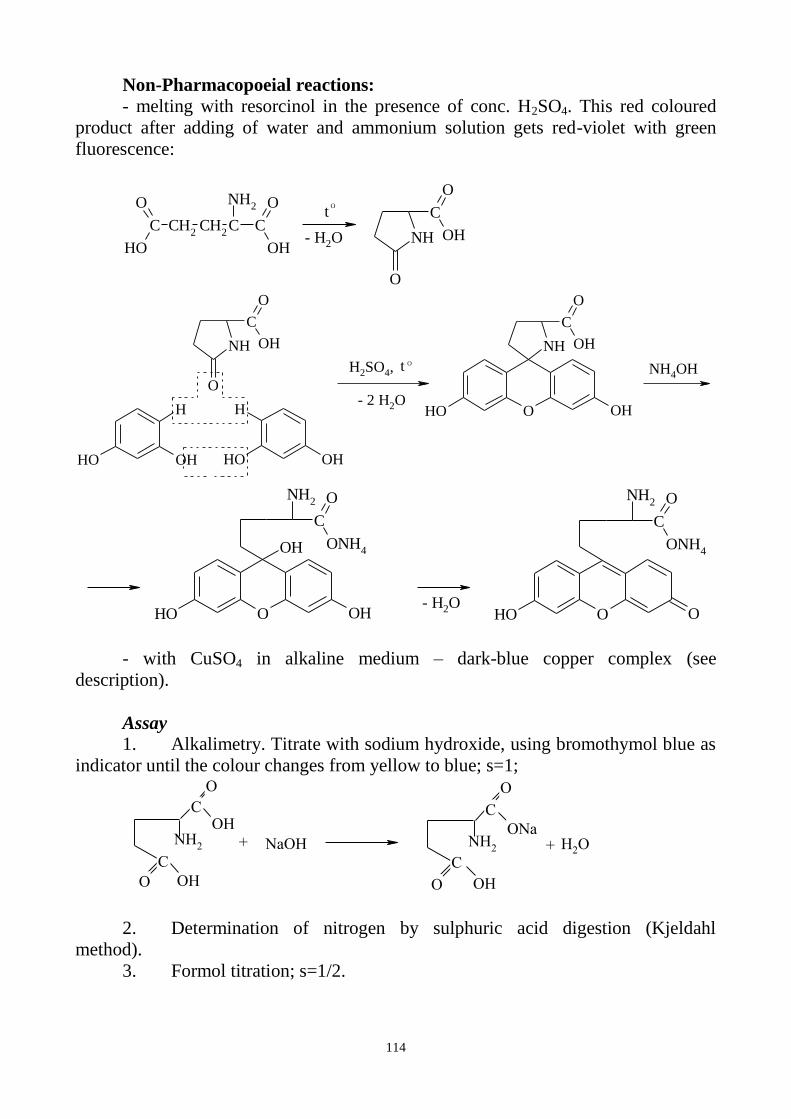
114
Non-Pharmacopoeial reactions:
- melting with resorcinol in the presence of conc. H2SO4. This red coloured
product after adding of water and ammonium solution gets red-violet with green
fluorescence:
C CH2
CH2C C
O
OH OH
ONH2
NH
O
C
OH
O
tO
- H2O
NH
O
C
OH
O
H
OHOH
H
OH OH
NH
C
OH
O
O OHOH
t OH2SO4,
- 2 H2O
NH4OH
ONH4
O
C
NH2
O OOH
C
NH2
ONH4
O
OH
O OHOH- H2O
- with CuSO4 in alkaline medium – dark-blue copper complex (see
description).
Assay
1. Alkalimetry. Titrate with sodium hydroxide, using bromothymol blue as
indicator until the colour changes from yellow to blue; s=1;
NH2 NH
2
OH OH
C
O
O
COH
+ NaOH
C
O
ONa
C
O
+ H2O
2. Determination of nitrogen by sulphuric acid digestion (Kjeldahl
method).
3. Formol titration; s=1/2.

115
NH2
OH OH
C
O
O
CONa
+ NaOH
C
O
OHN
C+
O
HCOH CH2
C
O
ONa
NC
OO
CH2
Na
Usage In medicine glutamic acid is usually used for treatment of CNS-
diseases, epilepsy psychoses, reactive conditions and in paediatrics.
Storage In well-closed containers, protected from light.
Меthionine (Methioninum)
(2S)-2-amino-4-(methylsulphanyl)butanoic acid
Blood proteins, protoplasm, albumin and casein contain methionine.
Preparation Methionine is obtained by condensation of acetylaminomalonic
ester and β-methylthioethanol with further hydrolysis of the intermediate product:
Properties A white or almost white, crystalline powder or colourless crystals,
soluble in water, very slightly soluble in alcohol, practically insoluble in ether.
Identification
1. Optical rotation.
2. IR-spectrum.
3. Thin-layer chromatography; then ninhydrin is used as solution for
detection.
4. The substance and glycine dissolve in dilute sodium hydroxide solution.
Add solution of sodium nitroprusside and heat. Add phosphoric and hydrochloric
acids a dark-red colouring appears:
H2S + Na2[Fe(CN)5NO] Na2H2[Fe(CN)5NOS]
COOC2H5
HC
COOC2H5
NHCOCH3
HOCH2CH2SCH3CH3 S CH2CH2 C NHCOCH3
COOC2H5
COOC2H5
H2O
CH3 S CH2CH2 CH COOH
NH2
+ CO2 + CH3COOH + 2C2H5OH
CH3 S CH2CH2 CH C
O
NH2OH
+ N a 2 S + C H 3 O H + 2 N H 3 + 2 H O C H 2 C H 2 C H C O O N a
O H
H 3 C S N a
t + 5 N a O H
S C H 2 C H 2 C H C O O H
N H 2
2 C H 3

116
Non-Pharmacopoeial reactions:
- after acidifying of the product melted the smell of mercaptan and hydrogen
sulphide appears:
Na2S + H3CSNa + 2H2SO4 H2S + CH3SH + Na2SO4 + NaHSO4
- with СuSO4 in the presence of sodium acetate a lilac-blue precipitate is
formed.
Assay
1. Non-aqueous titration. Dissolve the substance in anhydrous formic acid,
add anhydrous acetic acid. Titrate with perchloric acid. The end-point is stated by
potentiometric method; s=1.
CH3-S-CH2-CH2-CH-COOH + HClO4
HCOOH
NH2NH3
CH3-S-CH2-CH2-CH-COOH ClO4
2. Determination of nitrogen by sulphuric acid digestion (Kjeldahl
method).
3. Iodometry in the medium of phosphate buffer and in the presence of
potassium iodide. The excess of iodine is titrated with Na2S2O3. (The indicator is
starch). S=2.
4. Iodine monochloride titration (iodchlorimetry); the indicator is starch.
S=1.
5. Formol titration. S=1.
Usage For treating and prevention of liver diseases and toxic defeats.
Storage Store in a well-closed container, protected from light.
Aminalon (Aminalonum)
Acidum γ-aminobutyricum
Preparation By alkaline hydrolysis of pyrrolidone-2:
S CH2CH2 CH
NH2
H3C COOH + ICl + H2O
S CH2CH2 CH
NH2
H3C COOH
O
+ HI + HCl
ICl + HI I2 + HCl
I2 + 2Na2S2O3 2NaI + Na2S4O6
H2N CH2 CH2 CH2 COOH
C2H5OH
CH3COOHCOOK
CH2
CH2 CH2 NH2
C2H5OH
KOH
N
H
OCH2
CH2 CH2 NH2
COOH
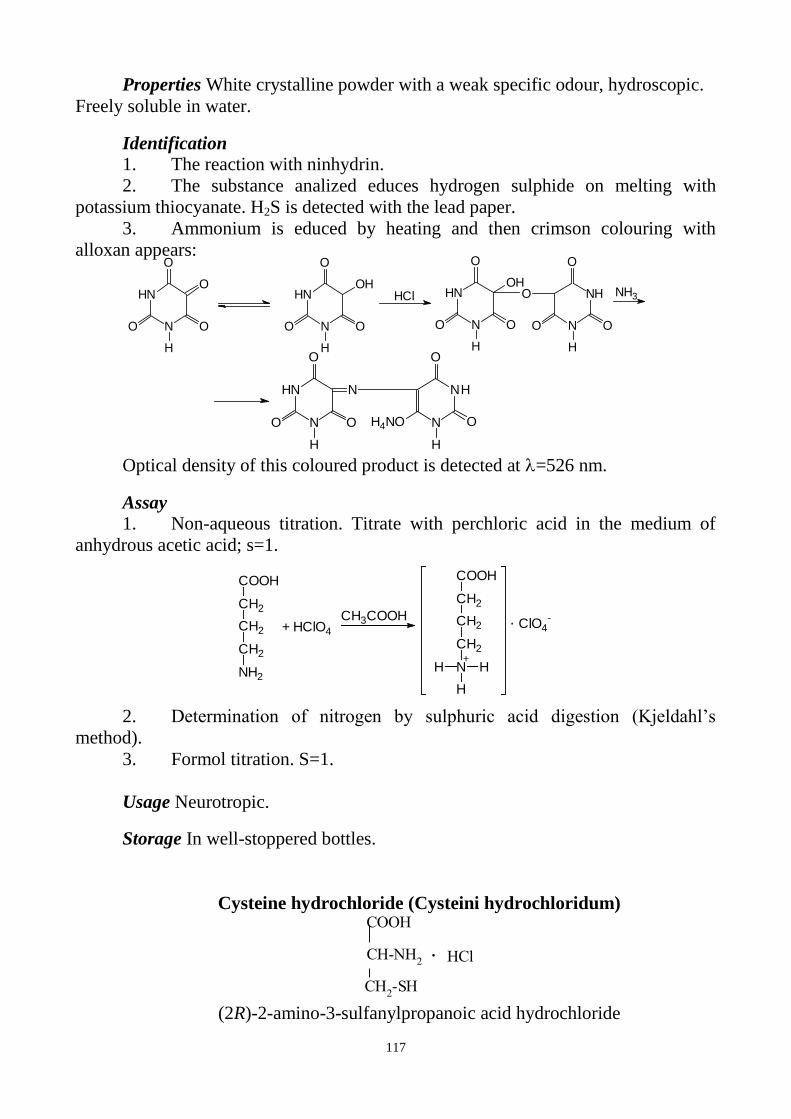
117
Properties White crystalline powder with a weak specific odour, hydroscopic.
Freely soluble in water.
Identification 1. The reaction with ninhydrin.
2. The substance analized educes hydrogen sulphide on melting with
potassium thiocyanate. H2S is detected with the lead paper.
3. Ammonium is educed by heating and then crimson colouring with
alloxan appears:
Optical density of this coloured product is detected at =526 nm.
Assay
1. Non-aqueous titration. Titrate with perchloric acid in the medium of
anhydrous acetic acid; s=1.
2. Determination of nitrogen by sulphuric acid digestion (Kjeldahl’s
method).
3. Formol titration. S=1.
Usage Neurotropic.
Storage In well-stoppered bottles.
Cysteine hydrochloride (Cysteini hydrochloridum)
CH-NH2
CH2-SH
COOH
. HCl
(2R)-2-amino-3-sulfanylpropanoic acid hydrochloride
COOH
CH2
CH2
CH2
NH2
+ HClO4
CH3COOH
COOH
CH2
CH2
CH2
N H
H
H+
. ClO4-
NH3
N
NH
O
OO
H
OHN
N
O
OO
H
OHHClN
N
O
OO
H
OHHHN
N
O
O
OO
H
HN
N
O
OO
H
N
N
O
H
N
H4NO O
H

118
Preparation Homocystine is reduced by hydrogen in the presence of catalysts:
S-CH2-CH-COOH
S-CH2-CH-COOH
NH2
NH2
CH-NH2
CH2-SH
COOH
CH-NH2
CH2-SH
COOH[H]
Zn2
HCl. HCl
Properties A white, crystalline powder or colourless crystals, freely soluble in
water, slightly soluble in alcohol.
Identification
1. Specific optical rotation.
2. IR-spectrum.
3. Thin-layer chromatography (the detector is ninhydrin).
4. Dissolve the substance in water, add dilute sodium hydroxide solution
and sodium nitroprusside solution. A yellow colour develops which becomes red.
5. The reaction of chlorides.
Non-Pharmacopoein reactions:
-mix the substance with conc. H2O2 solution, and with FeCl3 solution. Cool,
add dilute hydrochloric acid and barium chloride solution - a white precipitate forms.
(oxidise to sulphate and precipitate with BaCl2 is formed).
-the reaction with copper sulphate (black precipitate).
-the reduction of phosphorwolframic acid (blue colouring).
-the reaction with FeCl3- blue colour is formed.
Assay
1. Iodometry. Dissolve the substance in dilute hydrochloric acid, cool and
add potassium iodide and 0.05 M iodine solution. Titrate with sodium thiosulphate,
using starch solution as indicator. Carry out a blank titration. S=2.
2. Determination of nitrogen by H2SO4 digestion (Kjeldahl method).
3. Formol titration. S=1.
4. Alkalimetry. S=1.
5. Argentometry. S=1.
Usage For treating cataract; for electrophoresis (5% aqueous solution).
Storage In well-closed containers, protected from light.
+ I22 S CH2 CH COOH
NH2
COOHCHCH2S
NH2
COOH
CH NH2
CH2 SH
+ 2HI

119
Acetylcysteine (Acetylcysteinum)
HC COOH
NH CH3
O
HSCH2
(2R)-2-(acetylamino)-3-sulfanylpropanoic acid
Calculated with reference to the dried substance.
Properties A white, crystalline powder or colourless crystals, freely soluble in
water and in alcohol, practically insoluble in methylene chloride.
Identification
1. The specific optical rotation.
2. Melting point: 104 °C to 110 °C.
3. IR spectrophotometry.
4. Thin layer chromatography.
5. With sodium nitroprusside solution and concentrated ammonia a dark
violet colour develops.
Assay Iodometry. Titrate with 0.05 M iodine solution, using starch solution as
indicator; s=2:
HC COOH
NH CH3
O
HSCH2
HC COOH
NH CH3
O
SCH2
SCH2
CH
NH
O
H3C
2 + I2 HOOC + 2 HI
Storage Store protected from light.
Usage Mucolytic; antidote for paracetamol poisoning.
Sodium calcium edetate 10% solution for injections
(Solutio tetacini calcii 10% pro injectionibus)
Disodium [(ethylenedinitrilo)tetra-acetato]calciate(2-)
Properties A white or almost white powder, hygroscopic, freely soluble in
water, practically insoluble in alcohol and in ether.
CH2
N
CH2COONa
CH2COO
Ca
CH2COO
CH2COONaN
CH2

120
Identification
1. IR-spectrum.
2. Dissolve the substance, add lead nitrate solution and potassium iodide
solution. No yellow precipitate is formed. Make alkaline to red litmus paper by the
addition of dilute ammonia, add ammonium oxalate solution – a white precipitate is
formed.
CH2
CH2
N
N
CH2COO
CH2COO
CH2COONa
CH2COONa
Ca + Pb(NO3)2 Ca(NO3)2+
CH2
CH2
N
N
CH2COO
CH2COO
CH2COONa
CH2COONa
Pb
Ca(NO3)2 + (NH4)2C2O4CaC2O4 + 2NH4NO3
Dissolve the substance. Make alkaline to red litmus paper by the addition of
dilute ammonia, add ammonium oxalate solution – at most, a slight precipitate is
formed.
3. Ignite. The residue gives reactions of Ca2+
.
4. Reactions of Na+
.
The non –Pharmacopoeial reaction: add the substance into the mixture of
FeCl3 and NH4CNS –a colouring gets yellow.
Assay Dissolve the substance, add hexamethylenetetramine and hydrochloric
acid. Titrate with lead nitrate. (Indicator – xylenole orange triturate). S=1.
CH2
CH2
N
N
CH2COO
CH2COO
CH2COONa
CH2COONa
Ca + Pb(NO3)2 Ca(NO3)2+
CH2
CH2
N
N
CH2COO
CH2COO
CH2COONa
CH2COONa
Pb
Usage Chelating substance, used in treatment of lead poisoning.
Storage In a well-closed container.

121
Chapter 11. MEDICINAL SUBSTANCES DERIVATIVES OF
ETHERS AND ESTERS
Plan
1. General characteristic of ethers.
2. Medicinal substances derivatives of aliphatic and arylaliphatic ethers – ether,
diphenhydramine hydrochloride (dimedrolum); Preparation, identification and purity,
methods of quantitative definition, usage and storage.
3. General characteristic of esters of mineral acids.
4. Medicinal substances of mineral acid esters – glycerol trinitrate solution,
pentaaerythrityl tetranitrate, calcium glycerophosphate, phytinum, busulfan;
Preparation, identification and purity, methods of quantitative definition, usage and
storage.
5. Medicinal substances derivatives of esters of arylaliphatic acids.
Ethers are organic compounds with general formula: R – O – R'.
The lower ethers are very volatile liquids with the characteristic odour. On the
air or by influence of oxidisers they easy form various explosive peroxides and
hydrogen peroxides. It is necessary to take into consideration during Preparation,
storage, analysis and usage.
Pharmacopoeial medicines from the group of ethers are: ether and
diphenhydramine.
Medicinal substances derivatives of ethers
Ether (Aether)
Ether, anaesthetic (Aether anaestheticus)
C2H5 – O – C2H5
Preparation On heating a mixture of ethyl alcohol and concentrated sulphuric
acid to 135o C:
C2H5OH + S
HO
HO
O
OS
O
O
HO
H5C2O
+ H2O
S
O
O
HO
H5C2O
+ C2H5OH H5C2 O C2H5 + H2SO4
It is necessary to support the optimal temperature regimen (130-140C) for the
maximum yield because of forming side products, which can be divided according to
their chemical properties into 4 groups:
- acids (CH3COOH, H2SO3 and H2SO4);
- peroxides (hydrogen peroxide, dioxyethyl peroxide, oxyethyl
hydroperoxide, ethylidene peroxide);
- unsaturated compounds (ethylene, vinyl alcohol);

122
- aldehydes (acetaldehyde).
During storage of diethyl ether (especially at inappropriate conditions) under
light and oxygen action analogous side products are formed. Diethyl ether can
contain impurities of water and ethyl alcohol.
During analysis of diethyl ether, storage and working with it, it is necessary to
keep safety precautions (Inflammable! Explosive!).
There are 2 substances in Pharmacopoeia, which differs by their purity: ether
and anaesthetic ether (Aether anaestheticus).
Properties A clear, colourless, very mobile, highly flammable, volatile liquid.
Soluble in 15 parts of water, miscible with alcohol and fatty oils.
Vapours of ether form the explosive mixture in certain proportions with air,
oxygen and nitrogen protoxide.
Identification
1. Relative density (0.714-0.716).
2. Test for distillation range (it distils completely between 34.0C and 35.0C).
Purity
1. Acids (detected with bromothymol blue).
2. Acetone and aldehydes are determined with alkaline potassium
tetraiodomercurate solution (Nessler reagent). The lower layer shows only a slight
opalescence (for anaesthetic ether) and it may show yellow or reddish-brown
opalescence but not grey or black opalescence (for ether):
CH3C
O
H
+ K2[HgI4] + 3KOH Hg + CH3COOK + 4KI + 2H2O
3. Peroxides (add KI and starch solutions – no colour is produced):
H5C2 – O – O – C2H5 + 2KI + H2O I2 + H5C2 – O –C2H5 + 2KOH;
In the substance the side smell and non-volatile residue are determined.
Usage As a solvent for tinctures, extracts and some other external medicines; it
is used in analytical practice. Ether is very limited used as an anaesthetic medicine.
Storage Store in an airtight container, protected from light, at the temperature
of 8C to 15C. The contents of a partly filled container may deteriorate rapidly.
Diphenhydramine hydrochloride (Dimedrolum)
CH O CH2
CH2 N
CH3
CH3
H5C6
H5C6
. HCl
2-(Diphenylmethoxy)-N,N-dimethylethanamine hydrochloride
Preparation Interaction of benzhydrol and β-dimethylamine ethyl chloride
hydrochloride in the presence of alkali:

123
Cl CH2
CH2 N
CH3
CH3
CH OH CH O CH2
CH2 N
CH3
CH3
CH O CH2
CH2 N
CH3
CH3
+ . HCl
H5C6
H5C6
NaOHH5C6
H5C6
HCl
H5C6
H5C6
. HCl
Properties A white or almost white, crystalline powder, very soluble in water,
freely soluble in alcohol, practically insoluble in ether.
Identification
1. Melting point (168˚С -172˚С).
2. UV-spectrum.
3. IR-spectrum.
4. With conc. H2SO4 - an intense yellow colouring develops.
CH O CH2
CH2 N
CH3
CH3
CH O CH2
CH2 N
CH3
CH3HH
H5C6
H5C6
. HCl
conc.H2SO4H5C6
H5C6
HCl
+ +
SO4 +2-
It becomes red by the addition of HNO3. Then water and chloroform are added,
and an intense violet colour develops in the chloroform layer.
5. Reactions of chlorides.
The non-Pharmacopoeial reaction: acidic hydrolysis.
Melting point of the benzhydrol formed after recrystallising is 62-67˚C.
CH O CH2
CH2 N
CH3
CH3
CH OH OH CH2
CH2 N
CH3
CH3
H5C6
H5C6
. HCl
to
, HCl, H2OH5C6
H5C6
+ . HCl
Assay
1. Alkalimety. Dissolve the substance in alcohol, add 5.0 ml of hydrochloric
acid Carry out a potentiometric titration, using 0.1 sodium hydroxide solution. Read
the volume added between the 2 points of inflexion; s=1.
HCl + NaOH NaCl + H2O H
5C
6
CHH
5C
6
O CH2CH
2N
CH3
CH3
H5C
6
CHH
5C
6
O CH2CH
2N
CH3
CH3
. HCl + +
C2H5OH
NaOH NaCl+H2O
2. Non-aqueous titration.
The substance is titrated with perchloric acid in the medium of anhydrous
acetic acid in the presence of (CH3COO)2Hg till blue-green colouring. Indicator-
crystal violet solution; s=1. Carry out a blank titration.

124
CH O CH2
CH2 N
CH3
CH3
CH O CH2
CH2 N
CH3
CH3H
H5C6
H5C6
. HClCH3COOH
H5C6
H5C6
HgCl2 + 2CH3COOH+
ClO4 + -
2 + 2HClO4 + (CH3COO)2Hg
2.
3. Back iodchlorimetry using starch solution as an indicator; s=1.
CH O CH2
CH2 N
CH3
CH3
CH O CH2
CH2 N
CH3
CH3
H5C6
H5C6
. HCl
ICl H5C6
H5C6
HCl. . ICl
IСl + KI I2 + KCl
I2 + 2Na2S2O3 2NaI + Na2S4O6
4. Back argentometry by Volhard method; s S=1.
Usage Histamine H1-receptor antagonist.
Storage In well-closed containers, protected from light.
Medicinal substances of mineral acids esters
Esters of mineral acids can be determined as oxyacids that have an atom of
hydrogen substituted by an organic radical.
1% Glycerol trinitrate solution (Solutio Nitroglycerini 1%)
Propane-1,2,3-triyl trinitrate Preparation It is prepared by slowly addition of glycerol into an ice cooled
(–15o C) mixture of conc. H2SO4, and HNO3:
Properties A clear, colourless or slightly yellow solution. Miscible with
acetone and with ethanol. Practically insoluble in water, freely soluble in ethanol.
Identification
1. IR-spectrum.
CH2
CH
CH2
O
O
O
NO2
NO2
NO2
CH2
CH
CH2
OH
OH
OH
+ 3HNO3H2SO4 + 3H2O
C H 2 O N O 2
C H
C H 2
O
O
N O 2
N O 2

125
2. Thin-layer chromatography.
3. It complies with the limits of assay.
4. Non-Pharmacopoeial reactions:
a) reaction with diphenylamine. A blue colouring appears.
b) reaction of glycerol-residue after saponification on heating with KHSO4.
Assay
1. Test solution. Prepare a solution of the substance in methanol.
Reference solution. Dissolve sodium nitrite in methanol.
Into 3 flasks introduce 10 ml of the test solution, 10 ml of the reference
solution and 10 ml of methanol as a blank. To each flask add dilute sodium hydroxide
solution, mix and allow to stand for 30 min. Add sulphanilic acid solution and dilute
hydrochloric acid. After exactly 4 min read the absorbance of the test solution and the
reference solution at 540 nm using a blank titration as a compensation liquid.
Calculate the amount of glyceryl trinitrate in mg in the test solution from the
follow expression:
1008.60
TR
sT
mA
CmA
AT = absorption of the test solution,
mT = mass of the substance to be examined, in milligrams,
C = percentage content of sodium nitrite used as reference,
Ar = absorption of the reference solution,
mS = mass of sodium nitrite, in milligrams. CH2ONO2
CHONO2
CH2ONO2
+ 5NaOH CH3COONa + HCOONa + 2NaNO2 + NaNO3 + 3H2O
NH2
SO3H
NaNO2
HCl
SO3H
N N
Cl
NHCH2CH2NH2
NHCH2CH2NH2
N=N SO3H
2. Nitrate is determined by colorimetry (it is based on interaction with phenol-
2,4-disulphonic acid). The concentration of glycerol is stated by calibration diagram:
(NH4
+)3
OO
N SO3
SO3
O3NH4OH
OH
SO3H
SO3H
O2N
CH3COOH
KNO3
OH
SO3H
SO3H
. H S O 4 - +
N N
H
H 2 S O 4
N O 3 -
N O 3 -
2 N H N H N H

126
3. Reaction of saponification (in the presence of H2O2 to avoid reducing
processes). S=1/5.
The excess of alkali titrated with HCl solution by phenolphthalein. A blank
titration is carried out.
4. Alkalimetry in the medium of pyridine. Titrate with dibutylammonium
hydroxide. End-point is stated by potentiometry. S=1/3. H2C O-NO2
HC
H2C O-NO2
O-NO2 + 3[(C4H9)4N]OH
H2C OH
HC
H2C OH
OH + 3[(C4H9)4N]NO3ï ³ðèäèí
Usage Antispasmodic. It is used for treatment certain diseases of the heart.
Storage Store dilute solutions protected from light, at a temperature of 2C to
15C.
The substance must be cautiously proposed and kept because of its explosive
properties.
Pentaaerythrityl tetranitrate (Erynitum)
2,2-bis(hydroxymethyl)propane-1,3-diol trinitrate
Preparation Esterification of pentaaerytithritol with HNO3:
Properties A white or slightly yellowish powder, soluble in acetone, slightly
soluble in alcohol, practically insoluble in water.
Identification
1. Melting point138-142 oС.
2. IR-spectrum.
3. Thin-layer chromatography.
4. Non-Pharmacopoeial reactions:
a) reaction for nitrates after hydrolysis (with diphenylamine).
b) after hydrolysis pentaaerytritol interacts with benzoyl chloride and forms
pentaaerythritil benzoate with melting point 99-101oС.
C3H5(ONO2)3 + 5NaOH NaNO3 +2NaNO2 + CH3COONa + HCOONa + 3H2OH2O2
4C3H5(ONO2)3 6N2 + 12CO2 + O2 +10H2O
C
CH2 O NO2
CH2 O NO2
H2C
H2C
O
O
O2N
O2N
C
CH2 O NO2
CH2 O NO2
H2C
H2C
O
O
O2N
O2NH2SO4
+ 4HNO3C
CH2OH
CH2OH
HOH2C
HOH2C+ 4H2O
Pyridine

127
Assay For the substance is the liquid chromatography.
For tablets is gravimetry. To take into account the contents of stearic acid
(titration with an alkali in the dimethylformamide or acetone medium - see glyceryl
trinitrate).
Usage Spasmolytic.
Storage Protected from light and heat. Explosive!
Calcium glycerophosphate (Calcii glycerophosphas)
Preparation Esterification of glycerol with excess of sodium monophosphate
in the presence of H2SO4. The formed diglycerophosphates are hydrolysed with
alkali. Calcium glycerophosphate is precipitated with calcium acetate in the presence
of alcohol.
Properties A white powder, hygroscopic, sparingly soluble in water, practically
insoluble in alcohol.
Identification 1. Mix with potassium hydrogen sulphate, heat and direct the white vapour
towards a piece of filter paper impregnated with a freshly prepared solution of
sodium nitroprusside – develops a blue colour in contact with piperidine.
2. Ignite and carry out reaction of phosphates with molybdovanadic reagent
– a yellow colour develops:
Reactions of Ca2+
.
Non-Pharmacopoeial reactions:
a) reaction of glycerophosphoric acid:
CaPO3 O C3H5(OH)2 nH2O.
CH2
CH
CH2
OH
OH
O P O
O
O
Ca
H 3 P O 4 + 1 2 ( N H 4 ) 2 M o O 4 + 2 1 H N O 3 ( N H 4 ) 3 P O 4 . 1 2 M o O 3 + 2 1 N H 4 N O 3 + 1 2 H 2 O
CH2
CH
CH2
OH
OH
O P
O
O
O Ca
+ (CH3COO)2Pb
CH2
CH
CH2
OH
OH
O P
O
O
O Pb
+ (CH3COO)2Ca
white precipitate
CH2
CH2 OH
O
OH
P
Ca
O
CH
O
O + 2HNO3+ H2O
t o
CH2
CH2 OH
O
OHCH
H
H3PO4Ca(NO3)2
+ +

128
b) reaction of glycerol (on heating with KHSO4 the smell of acrolein appears).
Assay Compexonometry; s=1.
Usage For hypotrophy, rachitis and disfunction of nervous system.
StorageIn well-stoppered bottles.
Phytinum
Phytinum occurs in various plants: pea, lentil, hemp, potato etc. It is the
mixture of calcium and magnesium salts of inositol phosphoric acids, mainly of
inositol hexaphosphoric acid:
It is obtained from the scarps during producing of vegetable oil and starch. The
solution is purified from proteins and neutralised with NH4OH or Na2CO3 – insoluble
phytinum is educed. By adding of HCl this precipitate becomes soluble acidic salt
and then precipitated with ethanol as the soluble in water phytinum.
Identification
1. Reactions of Ca2+
in solution of acetic acid.
2. Reactions of phosphoric acid.
Assay It is based on interaction of phytinum with copper sulphate solution:
copper sulphate is detected by iodometry after reducing copper salt of inositol
phosphoric acids (indicator - starch). S=1.
Carry out a blank titration.
Calculate on P2O5 .
Usage Phytinum is the stimulant of blood-forming processes; it is used at low
concentrations of phosphorus in organism and improves functions of central nervous
system.
Storage In well-stoppered bottles.
(HO)OPO
OMg
O
OPO(OH)2OPO(OH)2
CaOO
OPO(OH)
OPO(OH)
OPO(OH)
I2 + 2Na2S2O3 2NaI + Na2S4O6
2CuSO4 + 4KI Cu2I2 + 2K2SO4 + I2
CH2
CH2 OH
O
OH
P
Ca
O
CH
O
O + KHSO4CaHPO4
-
CH2
CH2 OHO
CH
OSO3K
OSO3K
C
CH
CH2
Ht
o

129
Busulfan (Myelosanum)
Butane-1,4-diyl di(methanesulphonate)
Properties A white or almost white, crystalline powder, very slightly soluble in
water, freely soluble in acetone and acetonitrile, very slightly soluble in alcohol and
in ether. It melts at about 116C.
Identification
1. IR-spectrum.
2. Thin-layer chromatography.
3. Add sodium hydroxide to the substance. Heat until a clear solution is
obtained, add potassium permanganate solution. The colour changes to blue and
finally to green. Filter and add ammoniacal silver nitrate solution – a precipitate is
formed.
Add potassium nitrate and sodium hydroxide, mix and heat to fusion. After
adding dilute hydrochloric acid the solution gives reaction of sulphates (with barium
chloride).
4. The Non-Pharmacopoeial reaction: on heating with alcoholic alkali
alcohol solution the white precipitate forms:
Assay Alkalimetry. To the substance water is added; solution is shaken, boiled
under a reflux condenser for 30 min. Allow to cool. Titrate with sodium hydroxide
until a pink colour is obtained. Indicator – phenolphthalein; s=1/2.
Usage For treatment of leukaemia.
Storage In an airtight container, protected from light.
CH3SO2 O (CH2)4 O SO2 CH3NaOH
2CH3SO3Na + HO (CH2)4 OH
CH3 SO2 O (CH2)4 O SO2 CH3
H2O
tº2CH3SO3H + HO(CH2)4OH
CH3SO3H + NaOH CH3SO3Na + H2O
CH3 SO2 O (CH2)4 O SO2 CH3
CH3 S ONa
O
O
+ CH3OH Na2SO4 K2MnO4NaOH Na2MnO42KMnO4+ + + + H2O
CH3OH + 2KMnO4 3H C
O
H
+ 2MnO4+ 2KOH 2H2O+
H C
O
H
+ 2[Ag(NH3)2]NO3 + H2O HCOONH4+ 2Ag + NH3
+ 2NH4NO3
+
CH3SO2 O (CH2)4 O SO2 CH3NaOH
2CH3SO3Na + HO (CH2)4 OH

130
During working with the substance keep safety precautions (to avoid its action
on skin and mucous membrane).
Esters of arylaliphatic acids
Aprofene (Aprophenum)
-Diethylaminoethyl ester 1,1-diphenylpropionic acid hydrochloride
Preparation Interaction of diphenylpropionic acid and -diethylaminoethyl
chloride.
Properties White crystalline powder. Readily soluble in water, 95% alcohol
and chloroform. Soluble in acetone and benzol.
Identification
1. Reactions of Cl-.
2. Dissolve in conc. H2SO4 - a green-yellow colouring appears, that after
shaking of solution remains on the walls of the test-tube during several minutes.
3. 7 drops of K2Cr2O7 solution in H2SO4 solution add to the substance. The
test-tube cover with the paper, impregnated with sodium nitroprusside solution and 1
drop of piperidine; heat. The blue spot appears (reaction for methyl group, bonded
with tertiary nitrogen atom).
4. Reaction with hydroxylamine and FeCl3 (for ester group).
5. At addition of CuSO4 and NH4CNS solutions the brown precipitate appears.
6. Reaction with Marquis reagent (HCOH in conc. H2SO4) – a yellow colour
develops.
7. Evaporate the substance with conc. HNO3 and add KOH alcoholic solution -
a violet colour develops (reaction of 1,1-diphenylpropionic acid, formed during
hydrolysis):
8. After action of ammonium vanadate solution in conc. H2SO4 a green colour
developed becomes brown.
Assay
1. Non-aqueous titration in the presence of (CH3COO)2Hg (indicator is
crystal violet). Carry out a blank titration. S=1.
C
CH3
C OCH2 CH2 N
C2H5
C2H5
O
. HCl
C
CH3
C OCH2 CH2 N
C2H5
C2H5
O
H+
tºC
CH3
COOH + HO(CH2)2N
C2H5
C2H5

131
2. In solution for injections (Solutio Apropheni 1% pro injectionibus) and
in pills (Tabulettae Apropheni – 0,025) aprofene is determined by alkalimetry
(indicator is phenolphthalein). S=1.
3. Argentometry (Volhard method). S=1.
4. Mercurimetry. S=1.
Usage Cholinolytic, spasmolytic.
StorageIn well-stoppered bottles.
Adiphenine hydrochloride (Spasmolytinum)
-Diethylaminoethyl ester 1,1-diphenylacetic acid hydrochloride
Properties White crystalline powder, odourless. Freely soluble in water and in
alcohol, soluble in chloroform.
Identification
1. Hydrolysis:
Diphenylacetic acid gets into ether layer and then its melting point is detected.
2. Reactions of Cl-.
Assay
1. Non-aqueous titration in the presence of (CH3COO)2Hg. S=1.
2. Alkalimetry in the presence of organic solvent. S=1.
3. Argentometry (Volhard method). S=1.
Usage Cholinolytic, spasmolytic.
Storage In well-stoppered bottles, protected from light and humidity action.
CH C O
O
CH2CH2 N
C2H5
C2H5
. HCl
CH C O
O
CH2CH2 N
C2H5
C2H5tº
H2OCH C
O
OH
+ HOCH2CH2N
C2H5
C2H5

132
Chapter 12. AMIDATED DERIVATIVES OF CARBONIC ACID
AND DI-(β-CHLORETHYL)–AMINE DERIVATIVES
Plan
1. Amidated derivatives of carbonic acid:
а) general characteristic;
b) urethanes. Medicinal substances. Meprobamate: Preparation, identification
and assay, usage, storage.
c) acyclic ureides. Medicinal substances. Bromisoval: Preparation,
identification and assay, usage, storage.
2. Derivatives of di–(β-chlorethyl)–amine.
а) general characteristic;
b) medicinal substances, derivatives of di–(β-chlorethyl)–amine:
novembichinum: Preparation, identification and assay;
cyclopyhosphamide: Preparation, identification and assay Preparation;
sarcolysin: Preparation, identification and assay, usage and storage;
chlorambucil: Preparation, identification, assay, storage and usage.
Carbonic acid can form salts, esters, amides and other derivatives. For
medicine amides are very important because their derivatives are medicinal
substances.
This class of compounds includes urethanes and ureides (acyclic and cyclic).
There are two amides of carbonic acid:
1. Carbamic acid – the product of substitution one hydroxyl group by
aminogroup;
2. Carbamide or urea – the product of substitution two hydroxyl groups by
aminogroups (full amide).
Urethanes are esters of carbamic acid:
Ureides are acyl derivatives of urea or products of interaction with acids. They
can be cyclic and acyclic. Ureides are usually made by interaction of urea with
anhydrides, acyl chlorides, or (most commonly) esters of suitable organic acid:
From dicarboxylic acids there are derived ureido-acids and cyclic ureides:
R O C NH2
O
O C
NH2
NH C
O
COOH_
H2OHOOC
+O C
NH2
NH2
O C
NH2
NH C CH2R
O
_H2O
O C CH2R
OH
O C
NH2
NH2
HOOC
O C
OH
OH
O C
OH
NH2
O C
NH2
NH2

133
Hydrogens of methylene group in barbituric acid can be substituted by various
radicals. This ability is in the base of synthesis of such medicinal substances as
barbiturates with hypnotic effect.
Urethanes
General scheme of Preparation:
Chemical properties:
Meprobamate (Meprotanum)
2-methyl-2-propylpropane-1,3-diyl dicarbamate
Preparation By the scheme:
Properties A white or almost white amorphous or crystalline powder, slightly
soluble in water and in ether, freely soluble in 95% alcohol and in acetone.
Identification.
1. Melting point 104-108C.
2. IR-spectrum.
3. Add acetic anhydride and sulphuric acid. The melting point of diacetyl
derivative is 124 – 128С.
R OH + Cl C NH2
O
R O C NH2
O
+ HCl
H3C C CH2 CH2 CH3
CH2 O C
CH2 O C NH2
NH2
O
O
+ 2NaOHR O C NH2
O
1)
2) R O C NH2
O
2 + H2SO4 + 2H2O 2ROH + 2CO2 + (NH4)2SO4
ROH + NH3 + Na2CO3
tº
tº
H3C C CH2CH2CH3
CH2OH
CH2OH
+ 2Cl C
O
NH2 H3C C CH2CH2CH3
CH2
CH2
O
O
C
C
NH2
NH2
O
O
+ 2HCl
карбамоилхлоридcarbamoyl chloride
2-methyl-2-propylpropane diol-1,3 meprobamate
+O C
NH2
NH2
HOOCCH2
HOOC
_2H2O
O C
NH
NH
C
C
CH2
O
O
barbituric acid

134
4. Dissolve in alcoholic potassium hydroxide and boil under a reflux
condenser, add glacial acetic acid and solution of cobalt nitrate in ethanol. A deep-
blue colour develops.
Non-Pharmacopoeial reactions:
a) on heating with alkali NH3 appears, which is detected by the smell and the
wet red litmus paper gets blue.
b) on heating with dil. H2SO4 educes СО2 (Ca(OH)2 solution gets turbid):
Assay Determination of nitrogen by sulphuric acid digestion (see aminoacids).
Usage Anxiolytic (tranquilliser).
Storage In well-closed bottles.
H3C C CH2 CH2 CH3
CH2 O C
CH2 O C NH2
NH2
O
O
+ 4NaOHtº
H3C C CH2 CH2 CH3
CH2
CH2
OH
OH
+ 2NH3 + 2Na2CO3
+
Ca(OH)2 + CO2 CaCO3 + H2O
+ 2CO2 + (NH4)2SO4H3C C CH2 CH2 CH3
CH2
CH2
OH
OH
2H2OH3C C CH2 CH2 CH3
CH2 O C
CH2 O C NH2
NH2
O
O
+ H2SO4
+ 2 C H 3 C O O H H 3 C C C H 2 C H 2 C H 3
C H 2 O C
C H 2 O C
O
O
N H
N H
C O C H 3
C O C H 3
H 3 C C
O
O C H 3 C
O
+ 2 H 3 C C C H 2 C H 2 C H 3
C H 2 O C
C H 2 O C N H 2
N H 2
O
O
c . H 2 S O 4
C
CH2
CH2
CH2 O C NH2
O
CH2 O C NH2
O
H3C
CH3(CH2)2
(CH2)2H3C+ 4KOH C
CH2
C
O
CH2
O
H3C
(CH2)2H3C
OH
OHCo(NO3)2
CH2
CH2
H3C
(CH2)2H3C
OH
Co
HOCH3
C

135
Acyclic ureides
General scheme of synthesis
The bromoanhydride of α–bromoderivative of aliphatic acid obtain and then it
is interacts with urea.
Bromisoval (Bromisovalum)
N-(-bromoisovalerianyl)-urea
The product of condensation of urea with monobromoisovalerianic acid.
Preparation
Properties White crystalline powder with the bitterish taste and weak odour.
Very slightly soluble in water, soluble in ethanol.
Identification
1. On heating with alkali educes NH3 (the specific odour).
2. On heating with the mixture (H2O + conc. H2SO4) the irritant odour of
isovalerianic acid appears.
3. After adding of HCl in the solution Br- are detected.
The chloroform layer gets brown-yellow (free bromine).
R CH2 COOHPBr5
R CH COBr
Br
C O
H2N
H2NR CH C NH C NH2
Br O O
CH C NH C NH2
Br O O
CH
H3C
H3C
SO2N(Na)Cl + 2HCl C6H5C6H5 SO2NH2 + Cl2 + NaCl
2HBr + Cl2 Br2 + 2HCl
+ (NH4)2SO4 + CO2 + HBrCH
H3C
H3C
CH2 COOHH2O
H2SO4CH C NH C NH2
Br O O
CH
H3C
H3C
, tº
tºCH C NH C NH2
Br O O
CH
H3C
H3C
4NaOHCH
H3C
H3C
CH COONa
OH
+ 2NH3 + Na2CO3 + NaBr
CH C NH C NH2
Br O O
CH
H3C
H3C
CH
H3C
H3C
CH2COClPBr5
CH
H3C
H3C
CH
Br
C
O
Br
C OH2N
H2N
хлорангидрид изовалериановокислоты
бромангидрид бромизовалериановойкислоты
бромизовал
chloranhydride of isovalerianic acid bromanhydride of -bromisovalerianic acid
bromisoval

136
4. On heating with CuSO4 solution in alkaline medium a pink colouring
appears (or by adding of CuSO4 excess - a red-violet colour develops – biuret test).
Assay Back argentometry by Volhard method. Heat with an alkali (for
obtaining Br-). The excess of AgNO3 is titrated with NH4SCN (indicator is
NH4Fe(SO4)2). Carry out a blank titration. S=1.
NaBr + AgNO3 AgBr + NaNO3
AgNO3 + NH4SCN AgSCN + NH4NO3
3NH4SCN + NH4Fe(SO4)2 Fe(SCN)3 +2(NH4)2SO4
Usage Soft tranquilliser and hypnotic.
Storage In well-closed orange glass bottles.
Di-(β-chlorethyl)-amine derivatives
The general structure of these substances:
Where R can be aliphatic, aromatic or heterocyclic radical.
Di-(β-chlorethyl)-amine derivatives have alkylative properties. They can react
with the nucleophylic centres of protein molecules (nucleinic acids, ferments) with
oppressing of DNA and RNA synthesis and as the result – with blockade of cells
mitosis. The nuclei of tumour tissues and lymphoid tissues are very sensitive for the
action of these substances. This ability was in the base for creation of cytostatics.
Di-(β-chlorethyl)-amines can easily react with nucleoproteids of cells of
blood-forming tissues and so they oppress blood-forming process.
For the synthesis of these substances as the initial product are usually used
aminoderivatives (aliphatic, aromatic or heterocyclic) and then by β–chlorethanol or
ethylene oxide the oxyethyl group is added:
tº
CH C NH C NH2
Br O O
R NaOH R CH C ONa
OH O+ C O
H2N
H2N
R N
CH2CH2Cl
CH2CH2Cl
H2
O C NH C ONa
N N
Cu/2
CuSO4; NaOH
tºH2N C NH C NH2
O O
H2N C NH2
O
+HN C O
ClCH2 CH2OHR NH2 R N
CH2CH2OH
CH2CH2OH
H2N C NH2
O
NaOHNH3 + NH C O

137
Substitution of oxygroups by chlorine is carried out with thionyl chloride
[sulphur dichlorooxide (IV)]:
The nitrogen atom gives the basic character for the derivatives of di-(β-
chlorethyl)-amine. The medicinal substances of these group are bases or
hydrochlorides:
For identification of substances various reactions used: of aliphatic, aromatic
part of molecule bonded with di-(β-chlorethyl)-amine. The choice of reaction
depends on the structure of substance. General reaction for determining of di-(β-
chlorethyl)-amine are based on interaction with nicotinic acid and benzidine; with
diethylamide of pyridine carboxylic acid and other reagents. If the substance is the
salt (hydrochloride) – chloride-ion is detected with silver nitrate solution.
Novembichinum, sarcolysin, chlorambucil, cyclophosphamide are used in
oncology.
Novembichinum
Cl
CH2-CH
2-Cl
CH2-CH
2-Cl
H3C-CH-CH2-N. HCl
2-Chloropropyl-di-(β-chlorethyl)-amine hydrochloride
Preparation
Properties White powder, soluble in water and in alcohol, insoluble in ether.
Identification
1. With Dragendorff reagent (KBiI4 in KI + dil. H2SO4) the substance
forms an orange precipitate.
2. Reactions for Cl-.
R N
CH2CH2Cl
CH2CH2Cl
R N
CH2CH2Cl
CH2CH2Cl
. HCl
R N
CH2CH2Cl
CH2CH2ClR N
CH2CH2OH
CH2CH2OH
SOCl2
2-chlorpropyl-N-di(-oxyethyl)-amine
SOCl2
CH3
CHCl
CH2
NCH2CH2OH
CH2CH2OH
H2C CH2
OCH3
CHCl
CH2NH2
HClCH3
CH
CH2
NH
CH3
CHCl
CH2 N
CH2CH2Cl
CH2CH2Cl
HClCH3
CHCl
CH2 N
CH2CH2Cl
CH2CH2Cl
. HCl
propyleneimine 2-chlorpropylamine
2-chlorpropyl-N-di(-chlorethyl)-amine

138
Assay Back argentometry by Volhard method. Indicator is NH4Fe(SO4)2. S=1.
Usage For inhibition of pathological cell growth. To keep safety precautions
during the work with this substance, to avoid its action on skin and mucous
membrane.
Storage In well-stoppered bottles or ampules, in cool place.
Cyclophosphamide (Cyclophosphanum)
(2RS)-N,N-bis(2-chloroethyl)tetrahydro-2H-1,3,2-oxazaphosphorin-2-amine 2-
oxide
Preparation
Properties A white or almost white, crystalline powder, soluble in water,
freely soluble in ethanol, slightly soluble in ether.
Identification
1. Melting point (51C).
2. IR- spectrum.
3. Thin-layer chromatography.
4. After boiling with alkali Cl- are detected:
5. Non-Pharmacopoeial reactions:
a) the substance is heated with conc. H2SO4 then it is cooled and 20% NaOH is
added. On heating NH3 educed, the wet red litmus paper gets blue (reaction for amide
nitrogen);
b) the substance is heated with the mixture of conc. H2SO4 and HNO3 till the
oxides of nitrogen removed and the solution gets colourless. Then into this solution
(NH4)2MoO4 is added, intense yellow colour develops and in time the yellow
precipitate appears:
H3PO4 + 12(NH4)2MoO4+21HNO3(NH4)3PO4.12MoO3+21NH4NO3+12H2O
O
P
NH
O
N
CH2CH2Cl
CH2CH2Cl
. H2O
3 N H 4 S C N + N H 4 F e ( S O 4 ) 2 2 ( N H 4 ) 2 ( S O 4 ) + F e ( S C N ) 3
N H 4 N O 3 + A g S C N
+ A g C l
. H N O 3
C H 3
C H C l
C H 2 N
C H 2 C H 2 C l
C H 2 C H 2 C l
. H C l
C H 3
C H C l
C H 2 N
C H 2 C H 2 C l
C H 2 C H 2 C l
e x c . A g N O 3 + N H 4 S C N
+ e x c. . A g N O 3
пропаноламин3
ClCH2CH2N
ClCH2CH2POCl2 + HO (CH2) NH2
O
P
NH
O
N
CH2CH2Cl
CH2CH2Cl
-бис-( -хлорэтил)-амиддихлорангидрид фосфорной
циклофосфанN-di-(-chlorethyl)-amide
dichloranhydride of phosphoric acid
propanolamine
cyclophosphamide
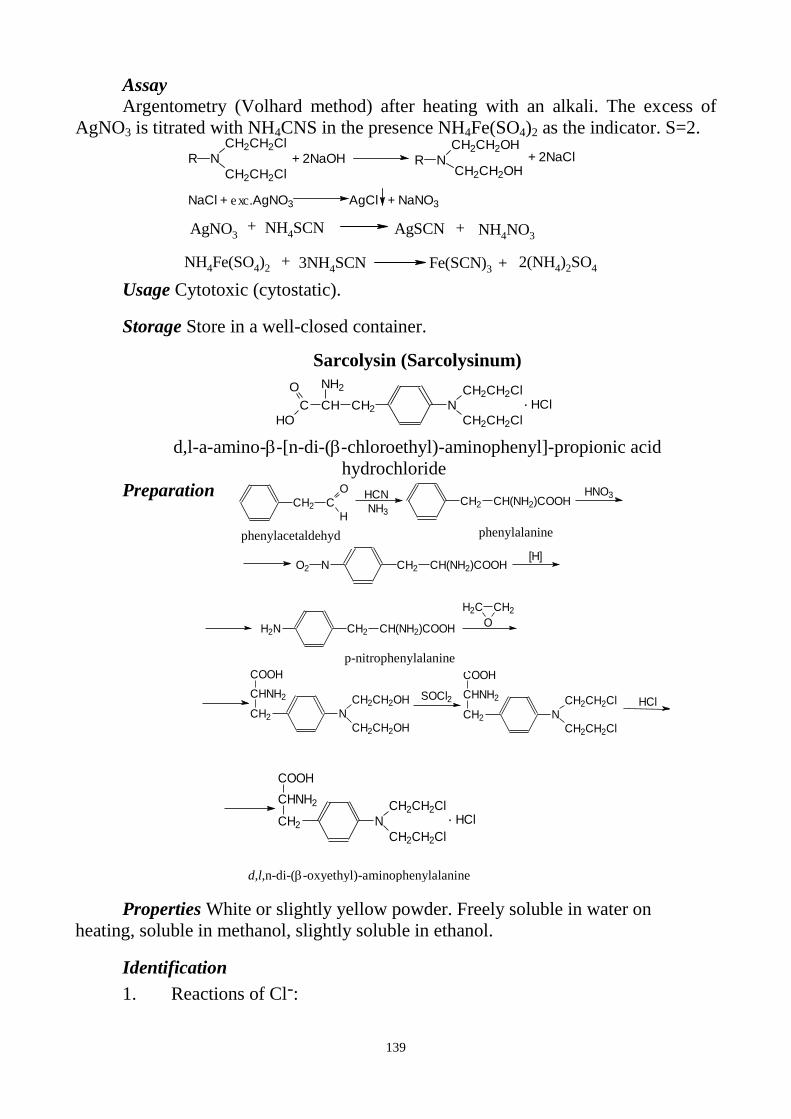
139
Assay
Argentometry (Volhard method) after heating with an alkali. The excess of
AgNO3 is titrated with NH4CNS in the presence NH4Fe(SO4)2 as the indicator. S=2.
Usage Cytotoxic (cytostatic).
Storage Store in a well-closed container.
Sarcolysin (Sarcolysinum)
d,l-a-amino--[n-di-(-chloroethyl)-aminophenyl]-propionic acid
hydrochloride
Preparation
Properties White or slightly yellow powder. Freely soluble in water on
heating, soluble in methanol, slightly soluble in ethanol.
Identification
1. Reactions of Cl-:
C CH CH2 N
CH2CH2Cl
CH2CH2Cl
NH2O
HO
. HCl
CH2
CHNH2
COOH
N
CH2CH2Cl
CH2CH2Cl
. HCl
HClCH2
CHNH2
COOH
N
CH2CH2Cl
CH2CH2Cl
SOCl2
CH2
CHNH2
COOH
N
CH2CH2OH
CH2CH2OH
H2C CH2
OCH2 CH(NH2)COOHH2N
[H]NO2 CH2 CH(NH2)COOH
HNO3CH2 CH(NH2)COOH
NH3
HCNCH2 C
O
H
phenylacetaldehyd
e
phenylalanine
p-nitrophenylalanine
d,l,n-di-(-oxyethyl)-aminophenylalanine
AgNO3+ NH4SCN AgSCN + NH4NO3
NH4Fe(SO4)2+ 3NH4SCN Fe(SCN)3 + 2(NH4)2SO4
R N
C H 2 C H 2 C l
C H 2 C H 2 C l
+ 2 N a O H R N
C H 2 C H 2 O H
C H 2 C H 2 O H
+ 2 N a C l
N a C l + e x c . A g N O 3 A g C l + N a N O 3
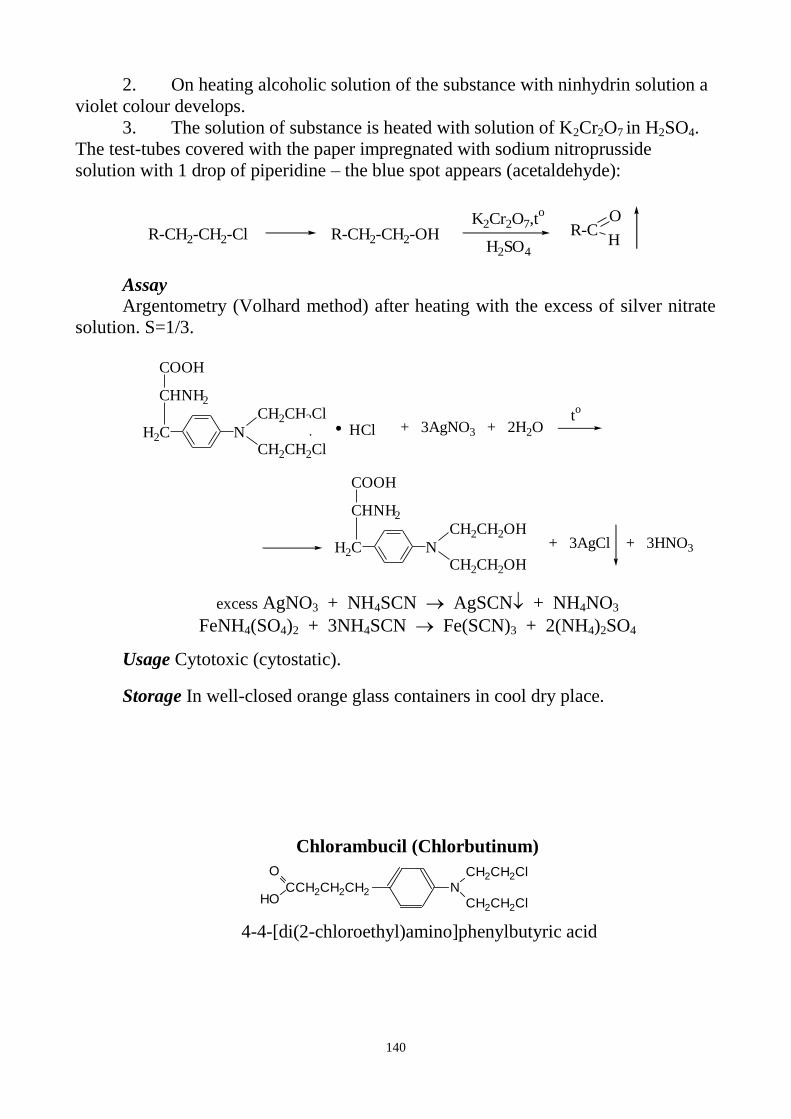
140
2. On heating alcoholic solution of the substance with ninhydrin solution a
violet colour develops.
3. The solution of substance is heated with solution of K2Cr2O7 in H2SO4.
The test-tubes covered with the paper impregnated with sodium nitroprusside
solution with 1 drop of piperidine – the blue spot appears (acetaldehyde):
R-CH2-CH2-Cl R-CH2-CH2-OHK2Cr2O7,t
o
H2SO4
R-CO
H
Assay
Argentometry (Volhard method) after heating with the excess of silver nitrate
solution. S=1/3.
COOH
CHNH2
H2C N
CH2CH2Cl
CH2CH2Cl
+ 3AgNO3 + 2H2Oto
COOH
CHNH2
H2C N
CH2CH2OH
CH2CH2OH
HCl
+ 3AgCl + 3HNO3
excess AgNO3 + NH4SCN AgSCN + NH4NO3
FeNH4(SO4)2 + 3NH4SCN Fe(SCN)3 + 2(NH4)2SO4
Usage Cytotoxic (cytostatic).
Storage In well-closed orange glass containers in cool dry place.
Chlorambucil (Chlorbutinum)
4-4-[di(2-chloroethyl)amino]phenylbutyric acid
N
CH2CH2Cl
CH2CH2Cl
CH2CH2CH2C
O
HO
.

141
Preparation
Properties A white, crystalline powder, practically insoluble in water, freely
soluble in alcohol and in acetone.
Identification
1. Melting point: 64C to 67C.
2. IR-spectrum.
3. Add dilute hydrochloric acid, filter and wash precipitate. To one of
combined filtrate and washings add potassium tetraiodomercurate solution. A pale-
brown precipitate is formed. To another part add potassium permanganate solution –
the colour of the latter is discharges immediately.
4. Dissolve the substance in acetone and dilute with water. Add dilute nitric
acid and silver nitrate solution. No opalescence is produced immediately. Heat the
solution on a water-bath - an opalescence develops:
Non-Pharmacopoeial reaction: for the substance the solution of K2Cr2O7 in
H2SO4 is added. The test-tube is covered with the paper impregnated with sodium
nitroprusside solution with 1 drop of piperidine and heated; the blue spot appears.
(CH2)3 COOHHNO3
(CH2)3 COOHO2N[H]
ROH
N
HOCH2CH2
HOCH2CH2
(CH2)3 COORSOCl2
H2N (CH2)3 COORHO (CH2)2 Cl
N
ClCH2CH2
ClCH2CH2
(CH2)3 COOH
H2ON
ClCH2CH2
ClCH2CH2
(CH2)3 COOR
-phenylbutiric acid p-nitrophenylbutiric acid
monochlorethanol
ester of p-di-(-oxyethyl) of aminophenylbutiric acid
chlorambucil
COOH
COOH
N CH2CH2CH2
CH2Cl CH2
CH2Cl CH2
2AgNO3 2H2O
N CH2CH2CH2
CH2 CH2
CH2 CH2HO
HO
2AgCl 2HNO3++
+ +

142
Assay
1. The substance is dissolved in acetone neutralised by phenolphthalein
and titrated with NaOH (indicator is- phenolphthalein). S=1.
2. The substance is heated with the excess of AgNO3 (with a reflux
condenser). The excess of AgNO3 is titrated with NH4CNS (Volhard method).
S=1/2.Carry out a blank titration.
Usage Cytotoxic (cytostatic).
Storage In well-closed containers, protected from light.
For the analysis of di-(β-chlorethyl)-amine derivatives optical methods are
used. Sarcolysin and chlorambucil can be identified and quantified by UV-
spectrophotometry.
Photometry of these substances is carried out by coloured product of their
reaction with diethylamide β-pyridine carboxylic acid. For sarcolysin extraction
photometry sodium eosinate is used; cyclophosphamide is detected as complex with
Fe(CNS)3.
IR-spectrophotometry is used for identification and quantification of these
substances.
COOH
COONa
+ NaOHN CH2CH2CH2
CH2 CH2
CH2 CH2
N CH2CH2CH2
CH2
CH2 CH2
CH2
+ H2O
Cl
Cl
Cl
Cl

143
Chapter 13. MEDICINAL SUBSTANCES DERIVATIVES OF
CYCLOALKANES AND TERPENOIDS
Plan
1. Medicinal substances derivatives of cycloalkanes.
а) general characteristic.
b) cyclopropane, amantadine hydrochloride, remantadine hydrochloride,
gludantanum. Preparation, identification, assay, usage and storage.
2. General characteristic of terpenoids.
а) medicinal substances derivatives of monocyclic terpenoids (menthol,
validolum, terpinum hydratum).
b) medicinal substances derivatives of bicycle terpenoids (camphor, camphora
monobromata, acid sulphocamphoric).
Among the various cycloalkanes cyclopropane and adamantane derivatives are
used in medicinal practice.
Cyclopropane (Cyclopropanum)
Preparation In industry it is obtained from allyl chloride:
Properties Colourless, combustible gas with the characteristic odour and
irritant taste. Boiling point –34.5C, in forms flammable mixtures with air, oxygen
and nitrogen protoxide.
Amantadine hydrochloride (Midantanum)
Tricyclo[3.3.1.13,7
]decan-1-amine
Preparation
Properties A white or almost white, crystalline powder, freely soluble in water
and in alcohol, practically insoluble in ether. It sublimes being heated.
H2C CH2
CH2
ClCH2 CH CH2
HBrClCH2 CH2 CH2 Br
Zn
NH2 . HCl
NH2 . HClBr NH2
Br2, 100 -150º
-HBr -HBr
NH3 HCl
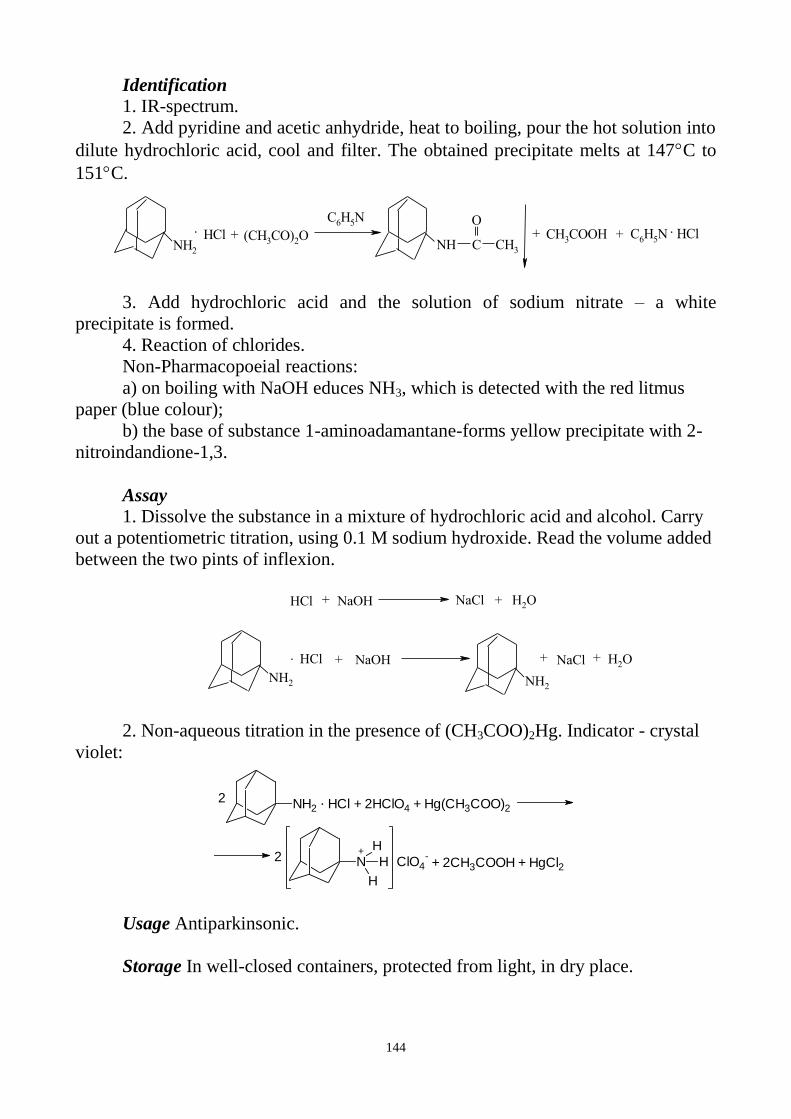
144
Identification
1. IR-spectrum.
2. Add pyridine and acetic anhydride, heat to boiling, pour the hot solution into
dilute hydrochloric acid, cool and filter. The obtained precipitate melts at 147C to
151C.
3. Add hydrochloric acid and the solution of sodium nitrate – a white
precipitate is formed.
4. Reaction of chlorides.
Non-Pharmacopoeial reactions:
a) on boiling with NaOH educes NH3, which is detected with the red litmus
paper (blue colour);
b) the base of substance 1-aminoadamantane-forms yellow precipitate with 2-
nitroindandione-1,3.
Assay
1. Dissolve the substance in a mixture of hydrochloric acid and alcohol. Carry
out a potentiometric titration, using 0.1 M sodium hydroxide. Read the volume added
between the two pints of inflexion.
. HCl
HCl NaOH NaCl H2O+ +
+ NaOH
NH2
+ NaCl + H2O
NH2
2. Non-aqueous titration in the presence of (CH3COO)2Hg. Indicator - crystal
violet:
Usage Antiparkinsonic.
Storage In well-closed containers, protected from light, in dry place.
+ 2CH3COOH + HgCl2ClO4-
+N
H
H
H
+ 2HClO4 + Hg(CH3COO)22 NH2 . HCl
2
NH2
.HCl (CH3CO)2O+
C6H5N
NH C CH3
O+ CH3COOH + C6H5N HCl.

145
Remantadine hydrochloride (Remantadinum)
-methyl-1-adamantylmethylamine hydrochloride
Preparation Reductive amination of adamantyl ketone with formamide:
Properties White crystalline powder, odourless, with the bitter taste. Slightly
soluble in water, soluble in alcohol.
Identification
1. Solution of the substance by action of sodium nitroprusside in the presence
of acetone and sodium carbonate gets violet.
2. Reaction of Cl-.
Assay Non-aqueous titration (see amantadine hydrochloride).
Usage Remantadine has antiviral activity. It is used for treatment and
prevention of influenza.
Storage In well-closed containers, protected from light, in dry place.
Gludantanum
Glucuronide l-aminoadamantane
Properties White or slightly yellowish crystalline powder, odourless. Slightly
soluble in water.
Identification
1. Reaction of glucuronide – with cupri-tartaric solution (Fehling’s reagent).
2. Reaction with sodium nitroprusside – a green colouring develops.
Assay Non-aqueous titration.
Usage Antiparkinsonic.
Storage In well-closed containers, protected from light.
CH CH3
NH2
. HCl
CH CH3
NH2
. HClHCl
CH CH3
NH C
O
H
C
O
NH2
H
C
O
CH3
NHO
COOH
HH
H
H
OH
OH
OH

146
Medicinal substances derivatives of terpenoids Terpenoids are hydrocarbons and their oxy-derivatives (alcohols, aldehydes,
ketones, etc.); essential oils and resins of coniferwood contain such class of
substances.
In the base of terpenoids is molecule of isoprene:
Medicinal substances of monocyclic terpenoids
Levomеnthol (Mentholum) CH
3
OH
CHCH
3CH
3
**
*
(1R,2S,5R)-5-methyl-2-(1-methylethyl)cyclohexanol
The molecule of levomenthol contains 3 asymmetric carbon atoms, so there are
23=8 optically active isomers and 4 racemates.
Levomenthol occurs in the essential oil of Mentha piperita (or other species) in
free condition and as ester of acetic acid.
Preparation
1. Partial freezing - for essential volatile oil with contents of menthol up to
80% after fractional distillation. The fraction, boiling at 208-212C, is cooled till -
20C and crystals of levomenthol develop.
2. Essential oil with contents of levomenthol about 50-60% is heated with boric
acid under low pressure.
OH O
B3+
+ H3BO3+ 3H2O
3
The educed ester of boric acid has a high boiling point - it is allows to separate
it form the other parts of oil. Then by distillation with water steam after
saponification levomenthol is educed.
The synthetic menthol is obtained by two methods:
1. Reduction of thymol under the pressure and in the presence of catalyst:
H2C C CH CH2
CH3
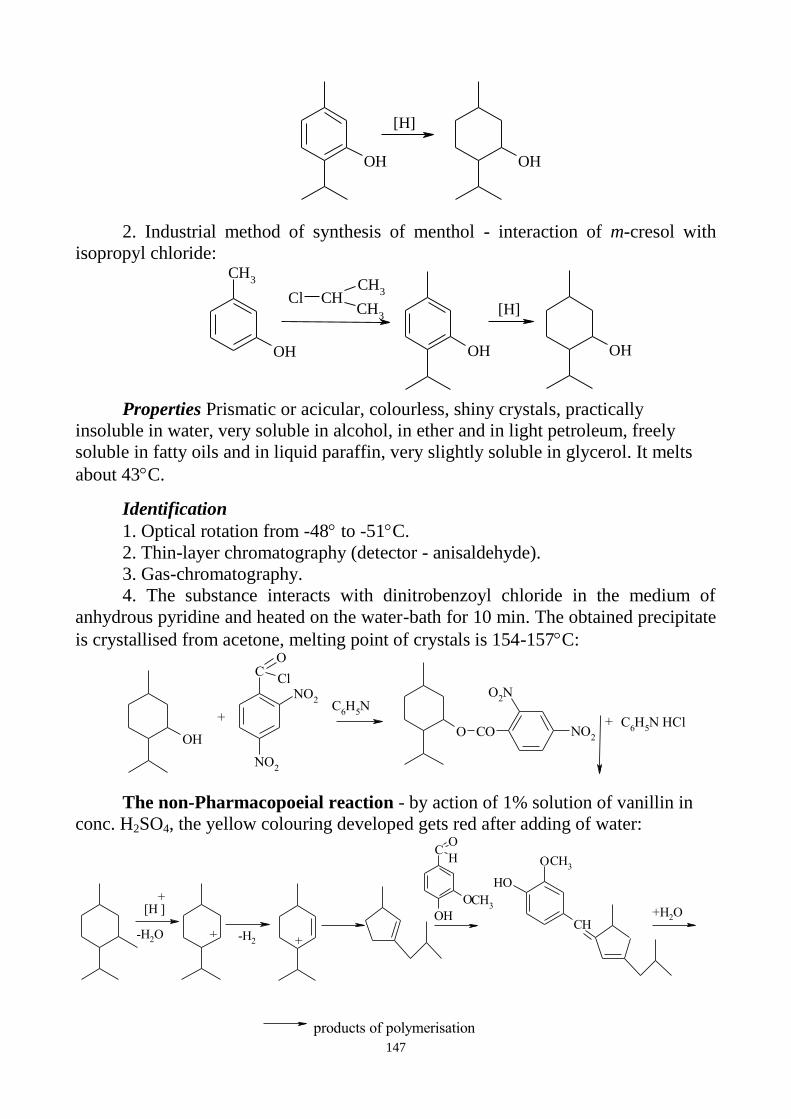
147
OH OH
[H]
2. Industrial method of synthesis of menthol - interaction of m-cresol with
isopropyl chloride: CH
3
OH OH OH
Cl CHCH
3
CH3
[H]
Properties Prismatic or acicular, colourless, shiny crystals, practically
insoluble in water, very soluble in alcohol, in ether and in light petroleum, freely
soluble in fatty oils and in liquid paraffin, very slightly soluble in glycerol. It melts
about 43C.
Identification
1. Optical rotation from -48 to -51C.
2. Thin-layer chromatography (detector - anisaldehyde).
3. Gas-chromatography.
4. The substance interacts with dinitrobenzoyl chloride in the medium of
anhydrous pyridine and heated on the water-bath for 10 min. The obtained precipitate
is crystallised from acetone, melting point of crystals is 154-157C:
OH
CO
ClNO
2
NO2
C6H
5N
O2N
NO2
O COC
6H
5N HCl+ +
The non-Pharmacopoeial reaction - by action of 1% solution of vanillin in
conc. H2SO4, the yellow colouring developed gets red after adding of water:
OH
CO
H
OCH3
O
OH
CH
CH3
[H ]+
-H2O ++-H2
+H2O
products of polymerisation
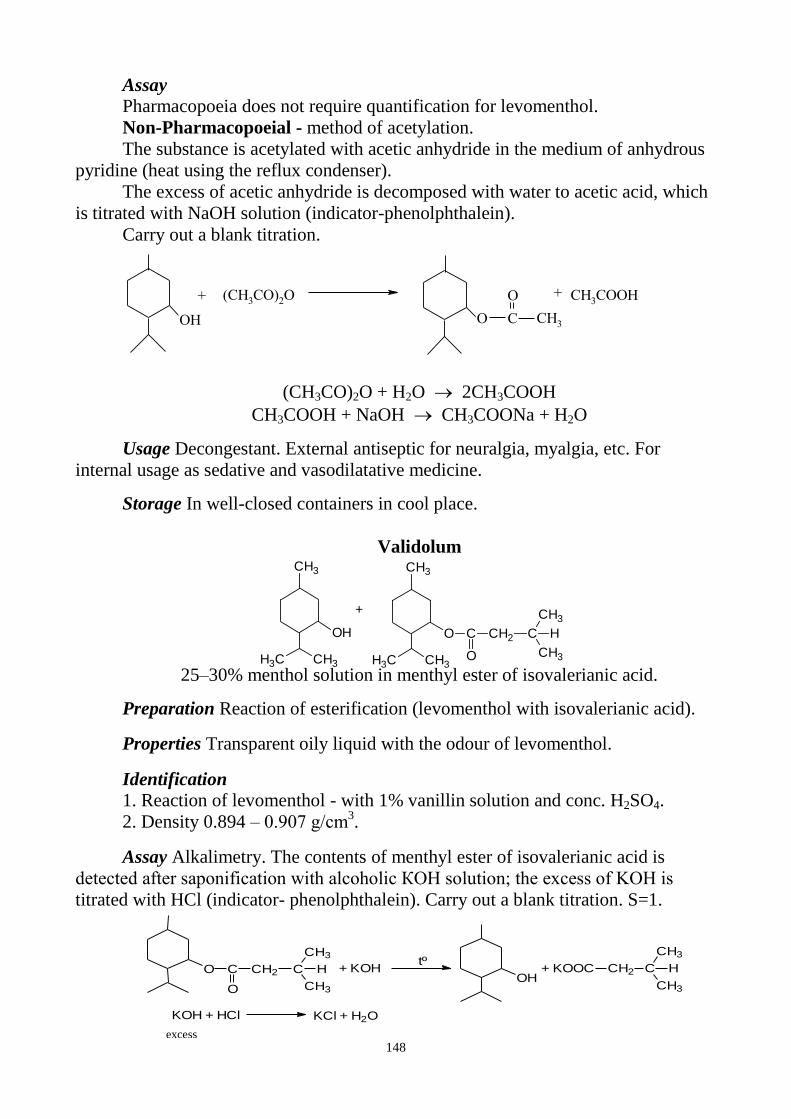
148
Assay
Pharmacopoeia does not require quantification for levomenthol.
Non-Pharmacopoeial - method of acetylation.
The substance is acetylated with acetic anhydride in the medium of anhydrous
pyridine (heat using the reflux condenser).
The excess of acetic anhydride is decomposed with water to acetic acid, which
is titrated with NaOH solution (indicator-phenolphthalein).
Carry out a blank titration.
(CH3CO)2O + H2O 2CH3COOH
CH3COOH + NaOH CH3COONa + H2O
Usage Decongestant. External antiseptic for neuralgia, myalgia, etc. For
internal usage as sedative and vasodilatative medicine.
Storage In well-closed containers in cool place.
Validolum
25–30% menthol solution in menthyl ester of isovalerianic acid.
Preparation Reaction of esterification (levomenthol with isovalerianic acid).
Properties Transparent oily liquid with the odour of levomenthol.
Identification 1. Reaction of levomenthol - with 1% vanillin solution and conc. H2SO4.
2. Density 0.894 – 0.907 g/сm3.
Assay Alkalimetry. The contents of menthyl ester of isovalerianic acid is
detected after saponification with alcoholic КОН solution; the excess of KOH is
titrated with HCl (indicator- phenolphthalein). Carry out a blank titration. S=1.
CH3
OH
CH3H3C
+
CH3
CH3H3C
O C CH2 C H
CH3
CH3O
OH
+ (CH3CO)2O O
O C CH3
+ CH3COOH
K O H + H C l
+ K O O C C H 2 C H
C H 3
C H 3 O H
t º + K O H O C C H 2 C H
C H 3
C H 3 O
K C l + H 2 O
excess
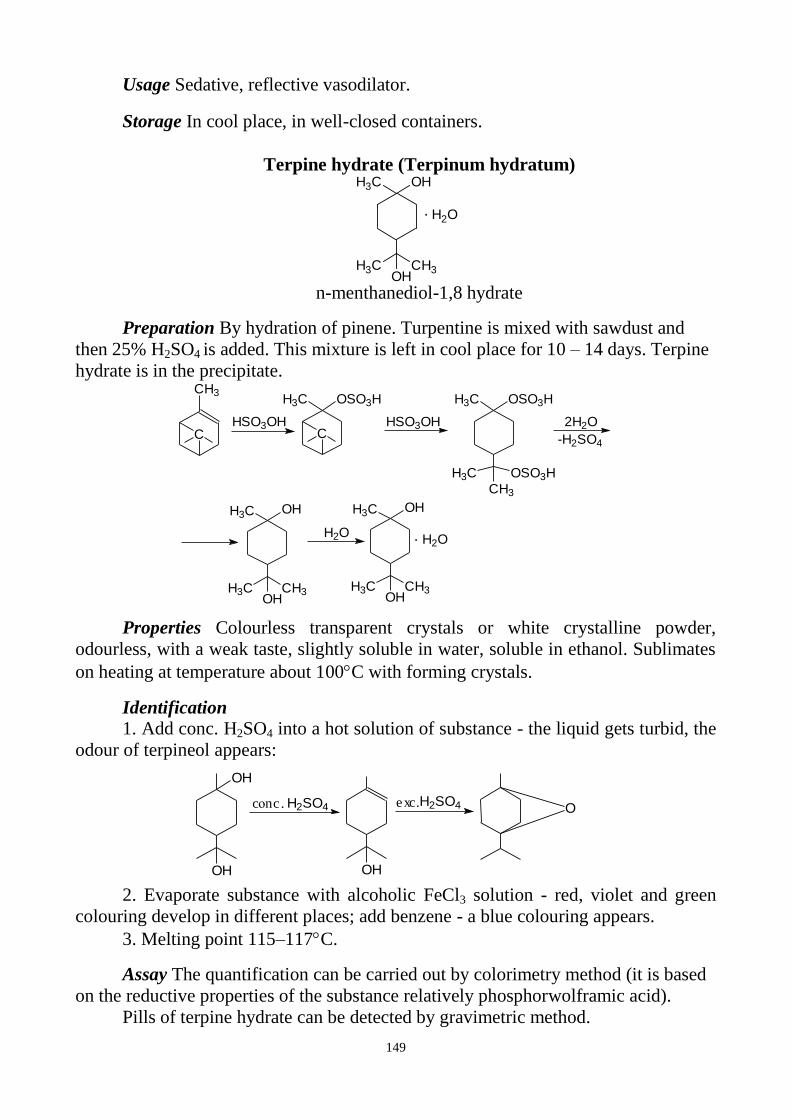
149
Usage Sedative, reflective vasodilator.
Storage In cool place, in well-closed containers.
Terpine hydrate (Terpinum hydratum)
n-menthanediol-1,8 hydrate
Preparation By hydration of pinene. Turpentine is mixed with sawdust and
then 25% H2SO4 is added. This mixture is left in cool place for 10 – 14 days. Terpine
hydrate is in the precipitate.
Properties Colourless transparent crystals or white crystalline powder,
odourless, with a weak taste, slightly soluble in water, soluble in ethanol. Sublimates
on heating at temperature about 100C with forming crystals.
Identification
1. Add conc. H2SO4 into a hot solution of substance - the liquid gets turbid, the
odour of terpineol appears:
2. Evaporate substance with alcoholic FeCl3 solution - red, violet and green
colouring develop in different places; add benzene - a blue colouring appears.
3. Melting point 115–117C.
Assay The quantification can be carried out by colorimetry method (it is based
on the reductive properties of the substance relatively phosphorwolframic acid).
Pills of terpine hydrate can be detected by gravimetric method.
OHH3C
OHCH3H3C
. H2O
. H 2 O
O H
O H C H 3 H 3 C
H 3 C
H 2 O
O H
O H C H 3 H 3 C
H 3 C
- H 2 S O 4
2 H 2 O
O S O 3 H H 3 C
O S O 3 H H 3 C
C H 3
H S O 3 O H C
O S O 3 H H 3 C
H S O 3 O H
C H 3
C
O H 2 S O 4
O H
H 2 S O 4
e x c . c о n c .
O H
O H
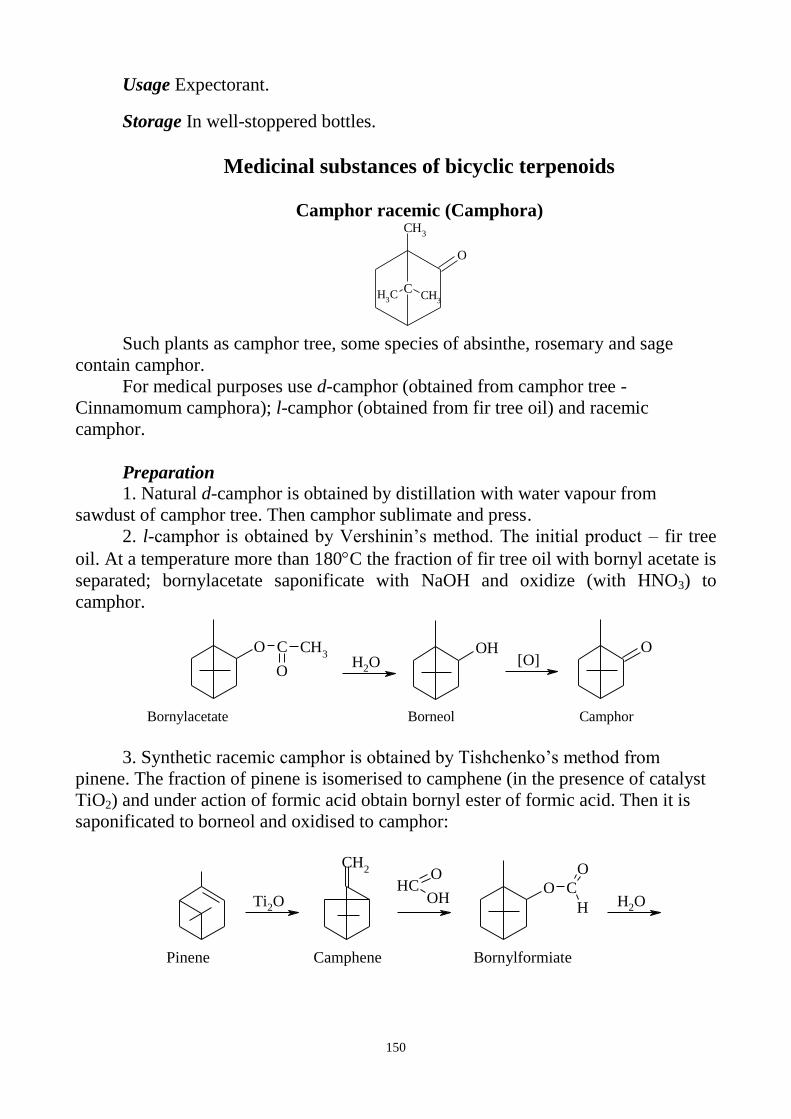
150
Usage Expectorant.
Storage In well-stoppered bottles.
Medicinal substances of bicyclic terpenoids
Camphor racemic (Camphora) CH
3
O
CCH
3CH
3
Such plants as camphor tree, some species of absinthe, rosemary and sage
contain camphor.
For medical purposes use d-camphor (obtained from camphor tree -
Cinnamomum camphora); l-camphor (obtained from fir tree oil) and racemic
camphor.
Preparation
1. Natural d-camphor is obtained by distillation with water vapour from
sawdust of camphor tree. Then camphor sublimate and press.
2. l-camphor is obtained by Vershinin’s method. The initial product – fir tree
oil. At a temperature more than 180C the fraction of fir tree oil with bornyl acetate is
separated; bornylacetate saponificate with NaOH and oxidize (with HNO3) to
camphor.
O C CH3
O
OH OH2O [O]
Bornylacetate Borneol Camphor
3. Synthetic racemic camphor is obtained by Tishchenko’s method from
pinene. The fraction of pinene is isomerised to camphene (in the presence of catalyst
TiO2) and under action of formic acid obtain bornyl ester of formic acid. Then it is
saponificated to borneol and oxidised to camphor:
O C
H
OCH
O
OH
CH2
Ti2O H2O
Pinene Camphene Bornylformiate
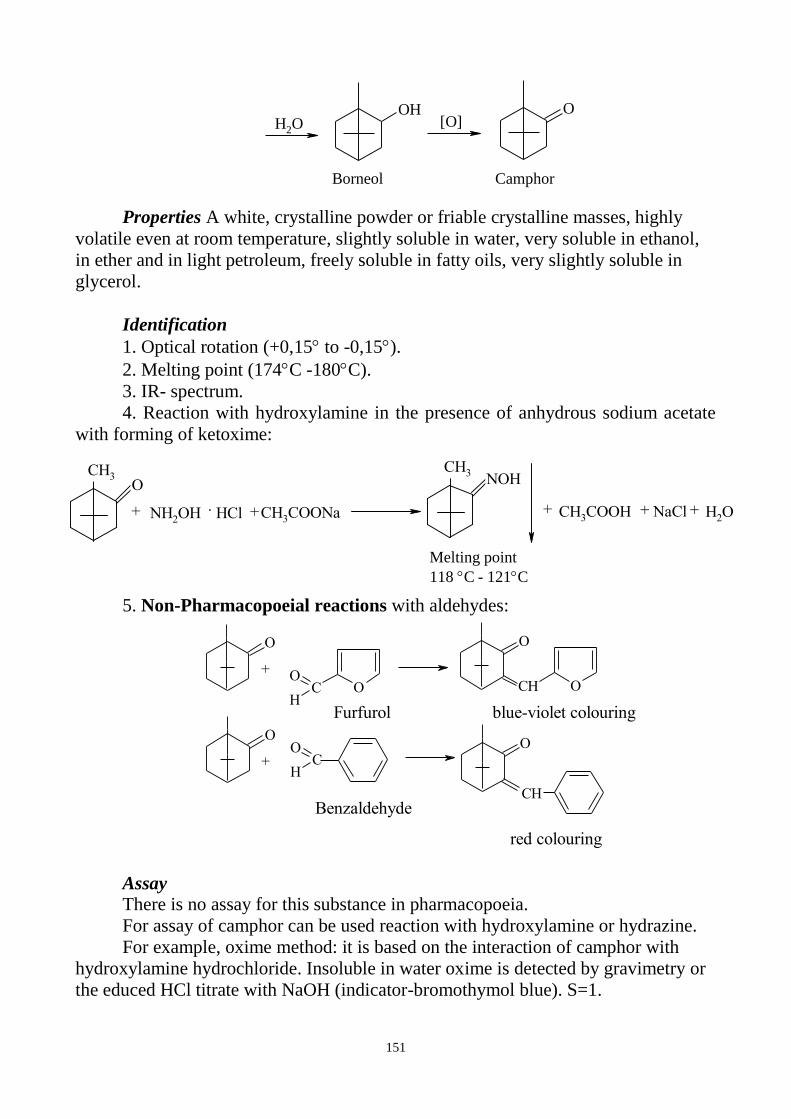
151
OH OH2O [O]
Borneol Camphor
Properties A white, crystalline powder or friable crystalline masses, highly
volatile even at room temperature, slightly soluble in water, very soluble in ethanol,
in ether and in light petroleum, freely soluble in fatty oils, very slightly soluble in
glycerol.
Identification
1. Optical rotation (+0,15 to -0,15).
2. Melting point (174C -180C).
3. IR- spectrum.
4. Reaction with hydroxylamine in the presence of anhydrous sodium acetate
with forming of ketoxime:
5. Non-Pharmacopoeial reactions with aldehydes:
O
OCO
H
O
OCH
O
CO
H
O
CH
+
+
Furfurol
Benzaldehyde
blue-violet colouring
red colouring
Assay There is no assay for this substance in pharmacopoeia.
For assay of camphor can be used reaction with hydroxylamine or hydrazine.
For example, oxime method: it is based on the interaction of camphor with
hydroxylamine hydrochloride. Insoluble in water oxime is detected by gravimetry or
the educed HCl titrate with NaOH (indicator-bromothymol blue). S=1.
Melting point
118 C - 121C
NH2OH+ .HCl +CH3COONa
CH3O
CH3NOH
+ CH3COOH NaCl H2O+ +
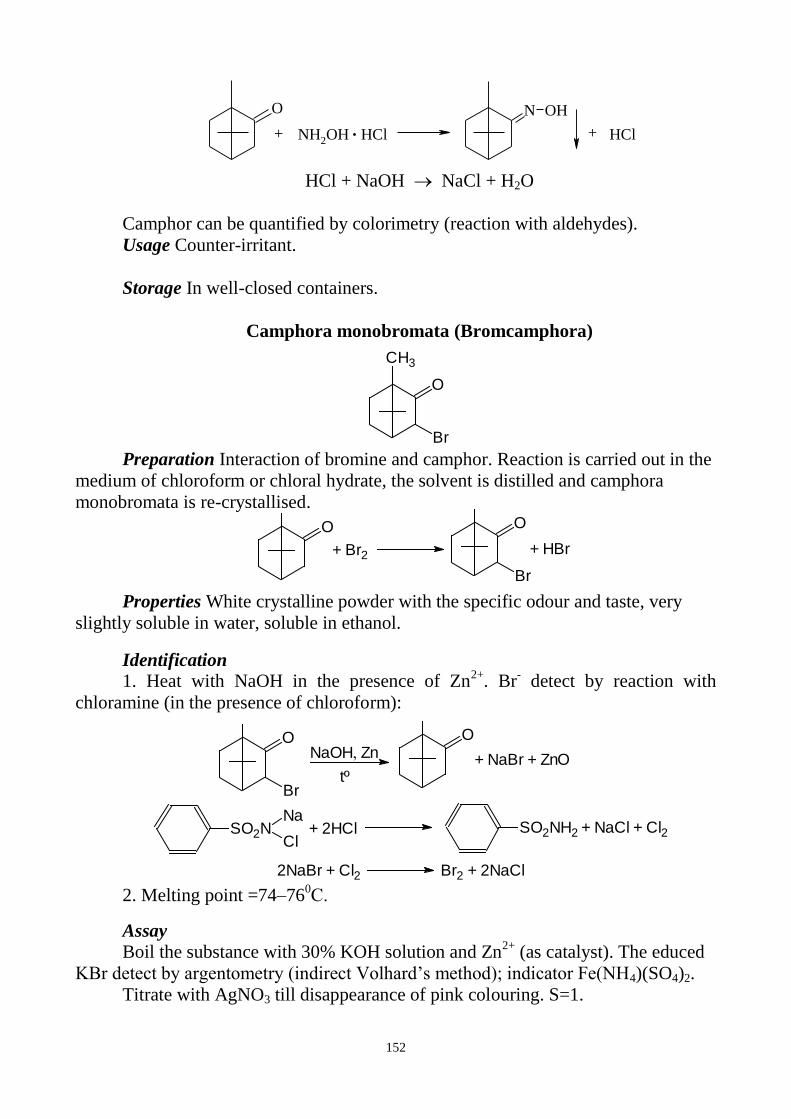
152
O N OH
+ NH2OH HCl. + HCl
HCl + NaOH NaCl + H2O
Camphor can be quantified by colorimetry (reaction with aldehydes).
Usage Counter-irritant.
Storage In well-closed containers.
Camphora monobromata (Bromcamphora)
Preparation Interaction of bromine and camphor. Reaction is carried out in the
medium of chloroform or chloral hydrate, the solvent is distilled and camphora
monobromata is re-crystallised.
Properties White crystalline powder with the specific odour and taste, very
slightly soluble in water, soluble in ethanol.
Identification
1. Heat with NaOH in the presence of Zn2+
. Br- detect by reaction with
chloramine (in the presence of chloroform):
2. Melting point =74–760С.
Assay
Boil the substance with 30% KOH solution and Zn2+
(as catalyst). The educed
KBr detect by argentometry (indirect Volhard’s method); indicator Fe(NH4)(SO4)2.
Titrate with AgNO3 till disappearance of pink colouring. S=1.
O
CH3
Br
O
+ Br2
O
Br
+ HBr
O
Br
NaOH, Zn
tº
O
+ NaBr + ZnO
SO2NNa
Cl+ 2HCl SO2NH2 + NaCl + Cl2
2NaBr + Cl2 Br2 + 2NaCl
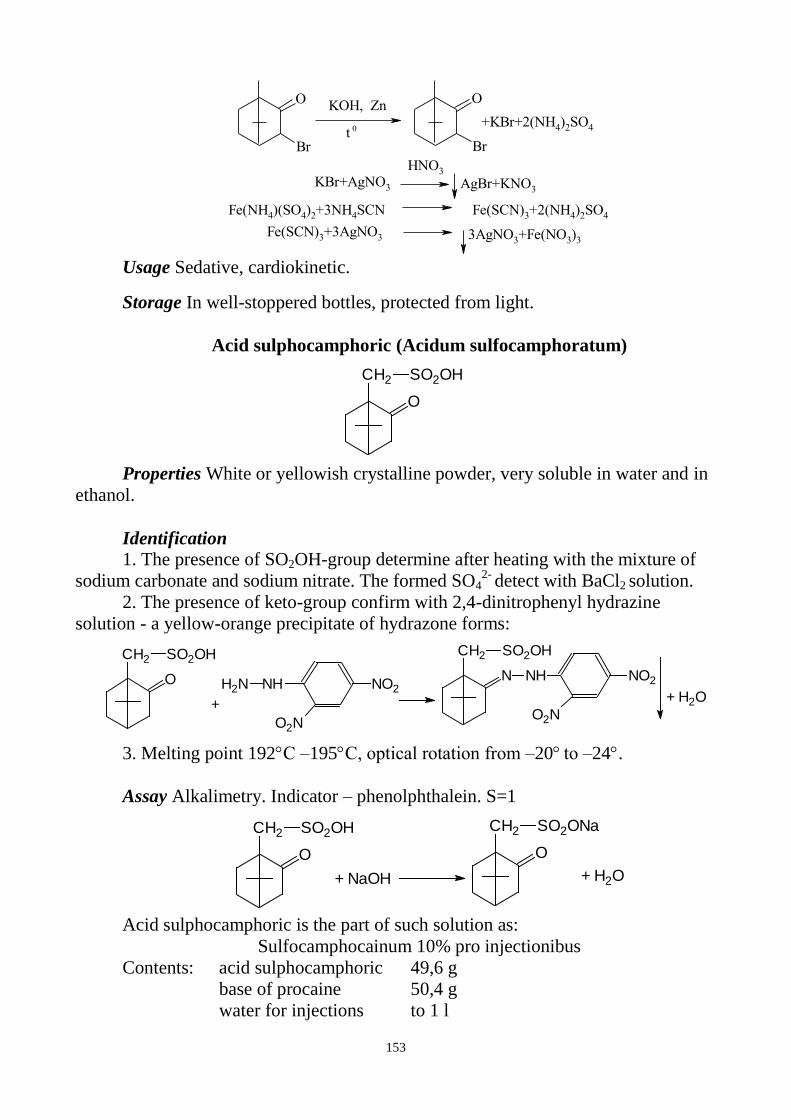
153
Br
O
Br
OKOH, Zn
t 0+KBr+2(NH4)2SO4
KBr+AgNO3
HNO3
AgBr+KNO3
Fe(NH4)(SO4)2+3NH4SCN Fe(SCN)3+2(NH4)2SO4
Fe(SCN)3+3AgNO3 3AgNO3+Fe(NO3)3
Usage Sedative, cardiokinetic.
Storage In well-stoppered bottles, protected from light.
Acid sulphocamphoric (Acidum sulfocamphoratum)
Properties White or yellowish crystalline powder, very soluble in water and in
ethanol.
Identification
1. The presence of SO2OH-group determine after heating with the mixture of
sodium carbonate and sodium nitrate. The formed SO42-
detect with BaCl2 solution.
2. The presence of keto-group confirm with 2,4-dinitrophenyl hydrazine
solution - a yellow-orange precipitate of hydrazone forms:
3. Melting point 192С –195С, optical rotation from –20 tо –24.
Assay Alkalimetry. Indicator – phenolphthalein. S=1
Acid sulphocamphoric is the part of such solution as:
Sulfocamphocainum 10% pro injectionibus
Contents: acid sulphocamphoric 49,6 g
base of procaine 50,4 g
water for injections to 1 l
O
CH2 SO2OH
+ H2O
CH2 SO2OH
N NH NO2
O2N
NO2NHH2N
O2N
+
O
CH2 SO2OH
O
CH2 SO2OH
+ NaOH
O
CH2 SO2ONa
+ H2O

154
The obtained colourless or slightly yellowish transparent liquid has pH 4,2 –
5,8. It gives reaction for -SO2OH and keto-groups of camphor. With NaOH solution
the oily precipitate of procaine-base is educed, which gives reaction for the primary
aromatic nitrogen after extraction with chloroform.
The content of sulphocamphoric acid is determined by alkalimetry, and the
content of procaine-base is determined by nitritometry.
Usage See camphor (but due to solubility in water, sulphocamphoric acid is
quickly absorbed).

155
Chapter 14. ANALYSIS OF MEDICINAL SUBSTANCES DERIVATIVES
OF PHENOLS
Plan
1. General characteristic of phenols.
2. Medicinal substances derivatives of phenols: phenol, thymol, resorcinol,
phenolphthalein, xeroformium. Preparation, properties, purity, assay, usage and
storage.
Phenols are derived from aromatic hydrocarbons by substituting one or more -
OH groups for hydrogen atom of the nucleus.
According to the contents of –OH groups they are divided in one-, two-, and
three-atomic:
Chemical properties of phenols are caused by the presence of –OH group
with the mobile hydrogen atom and by aromatic properties of benzol nucleus.
Acidic properties of phenols are stronger than alcohols due to interaction of
lone-pair electrons of oxygen atom in –OH group with π-electrons of phenol
aromatic nucleus.
By simple dissolving in alkali phenolates are formed:
Solutions of phenolates in water are very hydrolysed and can be neutralised
even with СО2 – that’s why carbonates of alkaline metals do not form phenolates.
In medicinal practice are used such medical substances as: phenol, thymol,
resorcinol, phenolphthalein, xeroformium, oxolinum.
Phenol
(Phenolum purum. Acidum carbolicum crystallisatum)
OH
Hydroxybenzene
Preparation Benzene interacts with conc. Н2SO4 with forming of benzene
sulphonic acid, the excess of acid with Ca(OH)2. Then Na2CO3 is added and sodium
OH
OHHO OH
OH
HO
фенол резорцин пирогаллолphenol resorcinol pyrogallol
OH
+ NaOH
ONa
+ H2O
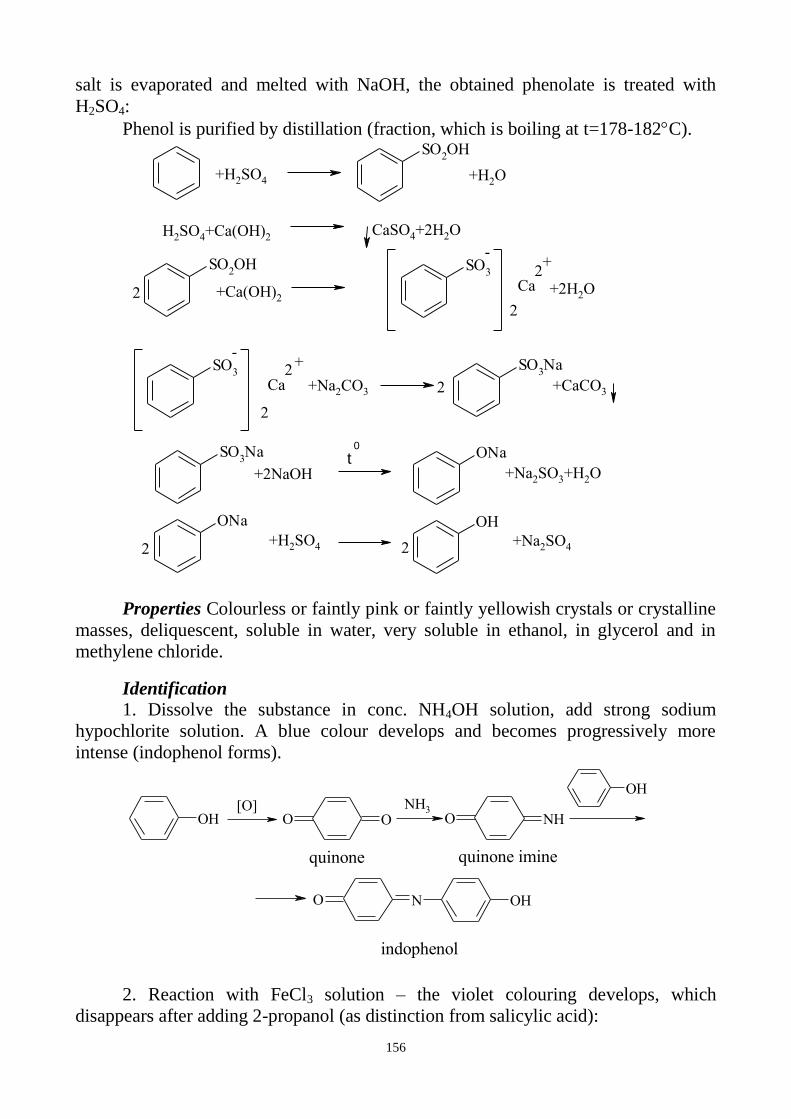
156
salt is evaporated and melted with NaOH, the obtained phenolate is treated with
H2SO4:
Phenol is purified by distillation (fraction, which is boiling at t=178-182C). SO
2OH
SO3
SO3
SO3Na
SO3Na ONa
ONa OH
SO2OH
+H2SO4 +H2O
H2SO4+Ca(OH)2CaSO4+2H2O
2 +Ca(OH)2
2
Ca2
+
+2H2O
-
2
Ca2
+-
+Na2CO3 +CaCO32
+2NaOH +Na2SO3+H2O
2+H2SO4 2 +Na2SO4
t0
Properties Colourless or faintly pink or faintly yellowish crystals or crystalline
masses, deliquescent, soluble in water, very soluble in ethanol, in glycerol and in
methylene chloride.
Identification 1. Dissolve the substance in conc. NH4OH solution, add strong sodium
hypochlorite solution. A blue colour develops and becomes progressively more
intense (indophenol forms).
OH O O O NH
OH
OHO N
[O] NH3
quinone quinone imine
indophenol
2. Reaction with FeCl3 solution – the violet colouring develops, which
disappears after adding 2-propanol (as distinction from salicylic acid):
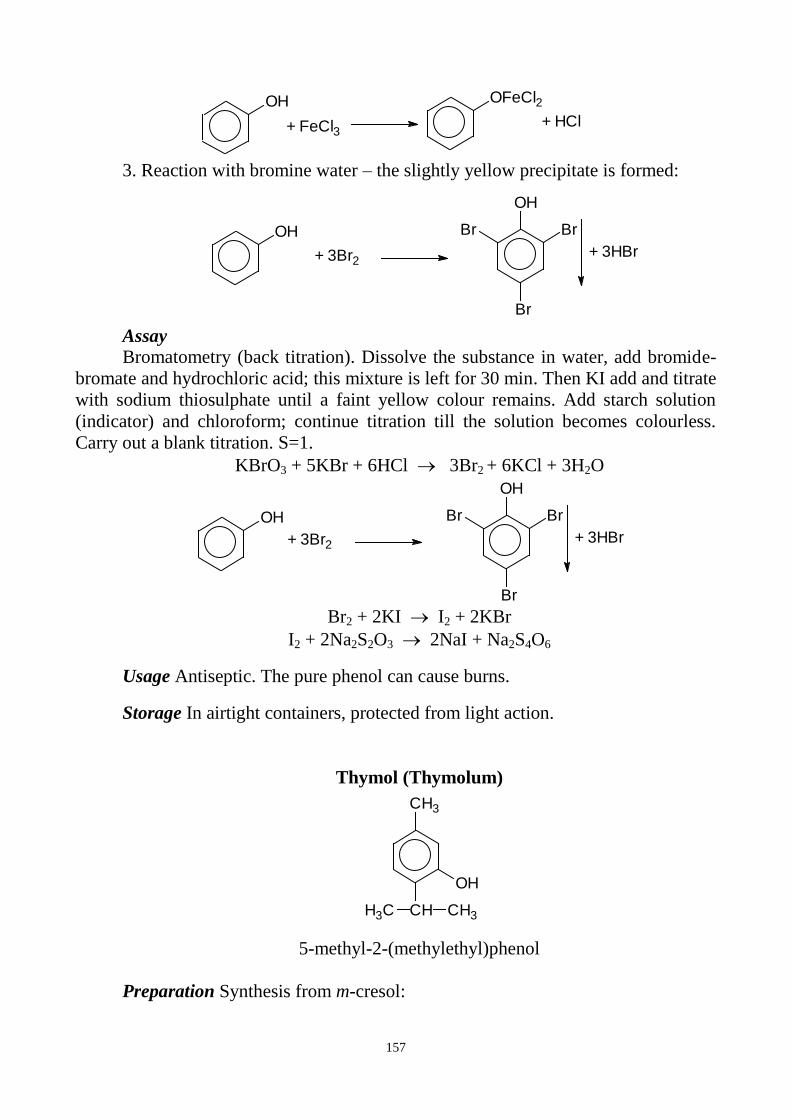
157
3. Reaction with bromine water – the slightly yellow precipitate is formed:
Assay
Bromatometry (back titration). Dissolve the substance in water, add bromide-
bromate and hydrochloric acid; this mixture is left for 30 min. Then KI add and titrate
with sodium thiosulphate until a faint yellow colour remains. Add starch solution
(indicator) and chloroform; continue titration till the solution becomes colourless.
Carry out a blank titration. S=1.
KBrO3 + 5KBr + 6HCl 3Br2 + 6KCl + 3H2O
Br2 + 2KI I2 + 2KBr
I2 + 2Na2S2O3 2NaI + Na2S4O6
Usage Antiseptic. The pure phenol can cause burns.
Storage In airtight containers, protected from light action.
Thymol (Thymolum)
5-methyl-2-(methylethyl)phenol
Preparation Synthesis from m-cresol:
OH
+ FeCl3
OFeCl2
+ HCl
OH
OH
BrBr
Br
+ 3HBr+ 3Br2
CH
CH3
CH3
OH
H3C
+ 3HBr
OH
BrBr
Br
OH
+ 3Br2
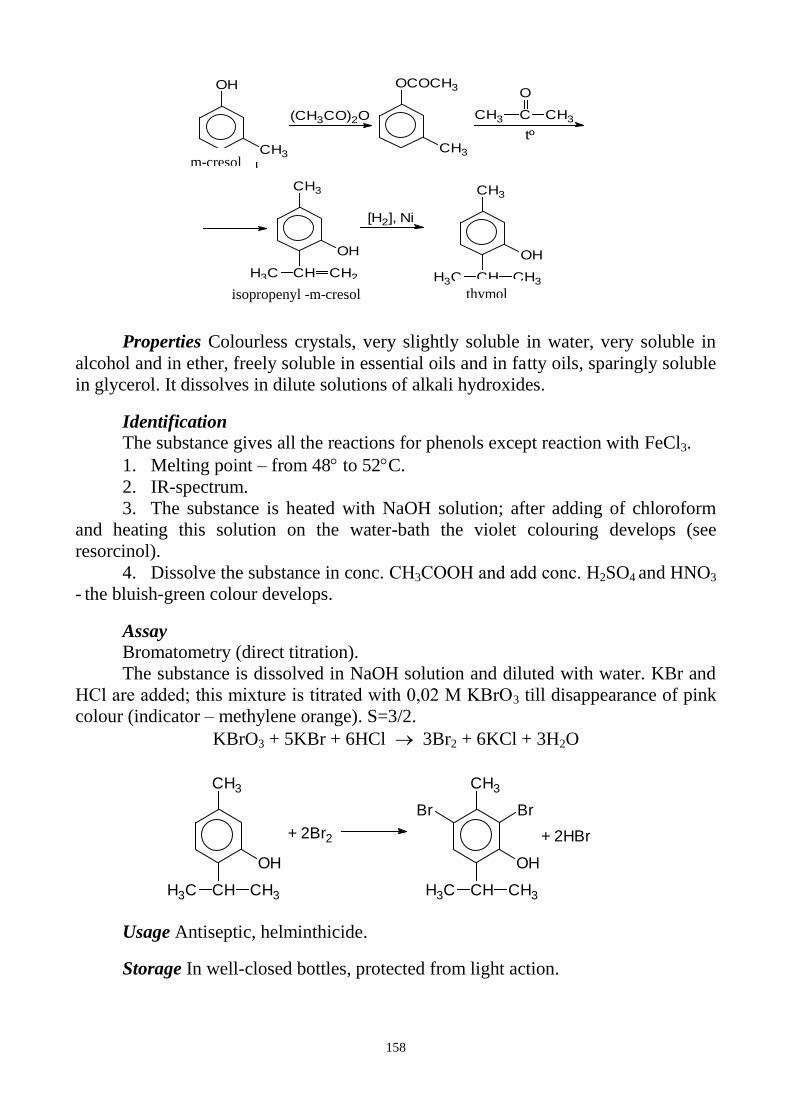
158
Properties Colourless crystals, very slightly soluble in water, very soluble in
alcohol and in ether, freely soluble in essential oils and in fatty oils, sparingly soluble
in glycerol. It dissolves in dilute solutions of alkali hydroxides.
Identification
The substance gives all the reactions for phenols except reaction with FeCl3.
1. Melting point – from 48 to 52C.
2. IR-spectrum.
3. The substance is heated with NaOH solution; after adding of chloroform
and heating this solution on the water-bath the violet colouring develops (see
resorcinol).
4. Dissolve the substance in conc. СН3СООН and add conc. H2SO4 and HNO3
- the bluish-green colour develops.
Assay
Bromatometry (direct titration).
The substance is dissolved in NaOH solution and diluted with water. KBr and
HCl are added; this mixture is titrated with 0,02 М KBrO3 till disappearance of pink
colour (indicator – methylene orange). S=3/2.
KBrO3 + 5KBr + 6HCl 3Br2 + 6KCl + 3H2O
Usage Antiseptic, helminthicide.
Storage In well-closed bottles, protected from light action.
OH
CH3
CHH3C CH3
+ 2Br2
CH3
BrBr
OH
CH CH3H3C
+ 2HBr
[H2], Ni
tº
CH3 C CH3
O
(CH3CO)2O
тимолизопропенил-м-крезол
м-крезол
OH
CH3
CHH3C CH3
OH
CH3
CH CH2H3C
CH3
OCOCH3OH
CH3 m-cresol
isopropenyl -m-cresol thymol
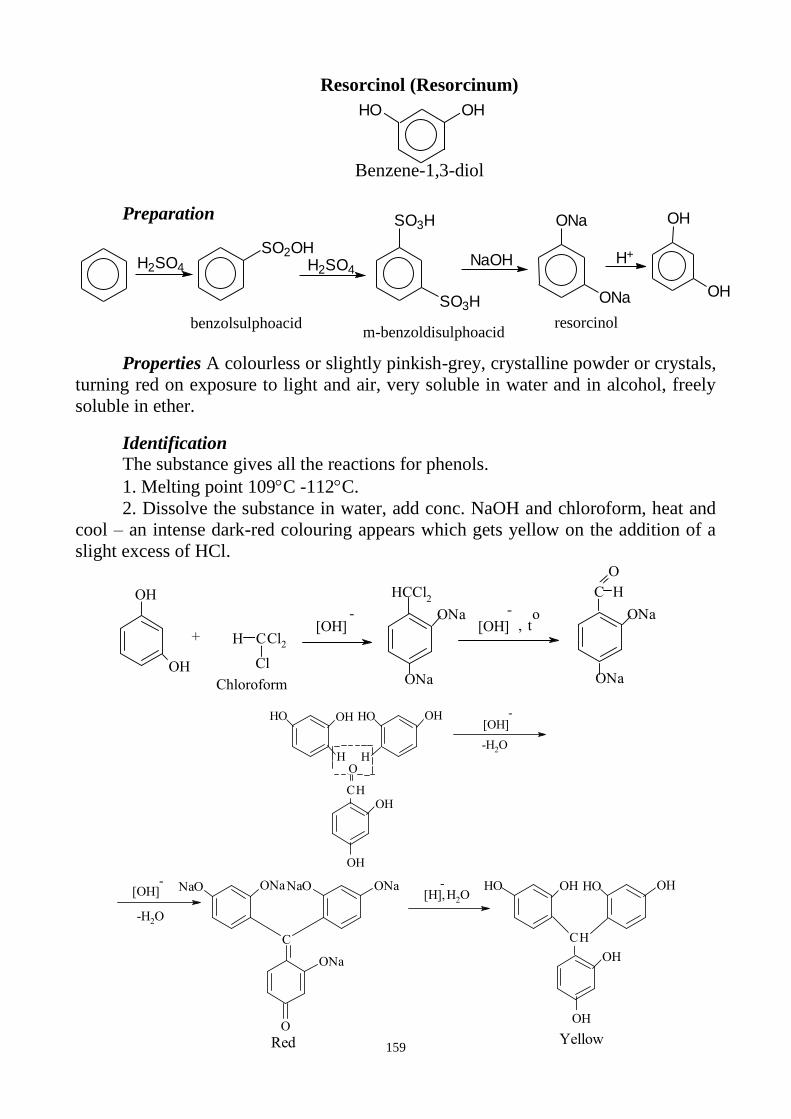
159
Resorcinol (Resorcinum)
Benzene-1,3-diol
Preparation
Properties A colourless or slightly pinkish-grey, crystalline powder or crystals,
turning red on exposure to light and air, very soluble in water and in alcohol, freely
soluble in ether.
Identification
The substance gives all the reactions for phenols.
1. Melting point 109C -112C.
2. Dissolve the substance in water, add conc. NaOH and chloroform, heat and
cool – an intense dark-red colouring appears which gets yellow on the addition of a
slight excess of HCl.
HO OH
H+
OH
OH
SO2OH
SO3H
SO3H ONa
ONa
H2SO4 H2SO4NaOH
ензолсуль окислотам- ензолдисуль окислота
резорцинbenzolsulphoacid
m-benzoldisulphoacid resorcinol
HCCl2
H
Cl
ONa
ONa
ONa
ONa
OH
OH
+
C
O
tH CCl2
[OH]-
[OH]-
,o
Chloroform
OH
C
O
[OH]-
-H2O
OH
OH
H
HO HOOH
H
H
NaO ONaNaO
C
ONa
O
ONa
,[OH]-
-H2O
[H]-H2O
OH
C
OH
OHHO HOOH
H
Red Yellow
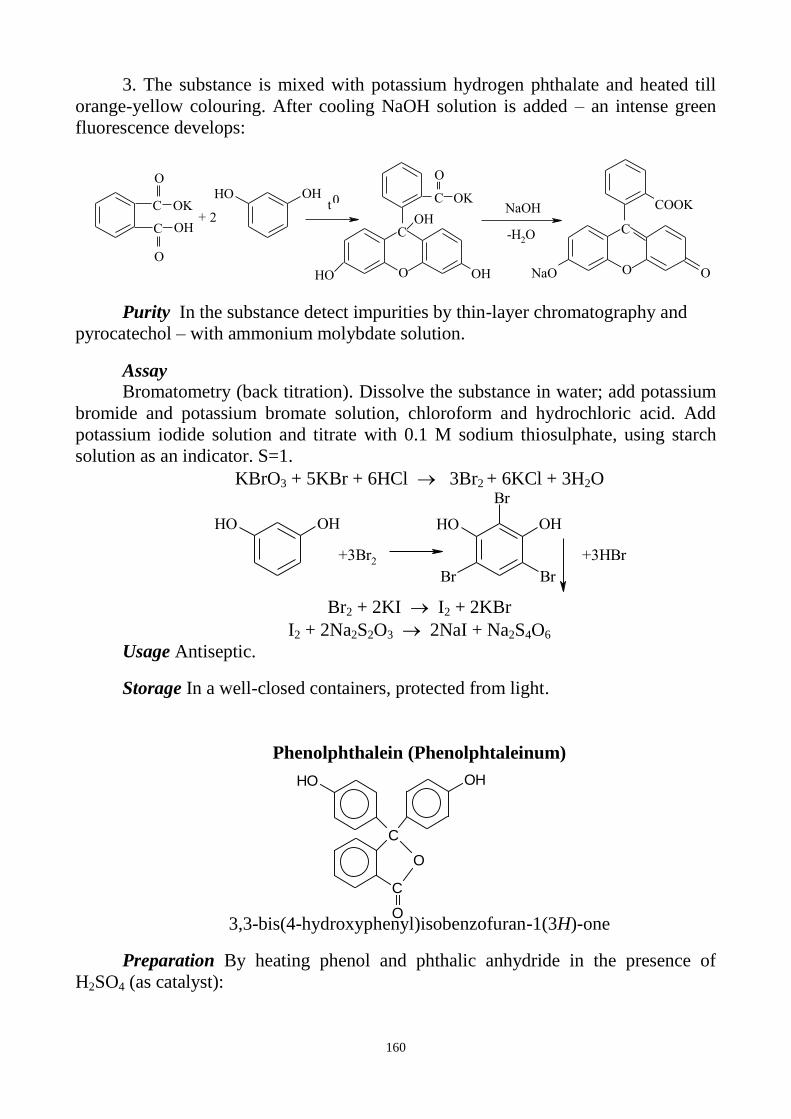
160
3. The substance is mixed with potassium hydrogen phthalate and heated till
orange-yellow colouring. After cooling NaOH solution is added – an intense green
fluorescence develops:
Purity In the substance detect impurities by thin-layer chromatography and
pyrocatechol – with ammonium molybdate solution.
Assay
Bromatometry (back titration). Dissolve the substance in water; add potassium
bromide and potassium bromate solution, chloroform and hydrochloric acid. Add
potassium iodide solution and titrate with 0.1 M sodium thiosulphate, using starch
solution as an indicator. S=1.
KBrO3 + 5KBr + 6HCl 3Br2 + 6KCl + 3H2O
OH OH OH OH
Br
Br Br
+3Br2 +3HBr
Br2 + 2KI I2 + 2KBr
I2 + 2Na2S2O3 2NaI + Na2S4O6
Usage Antiseptic.
Storage In a well-closed containers, protected from light.
Phenolphthalein (Phenolphtaleinum)
3,3-bis(4-hydroxyphenyl)isobenzofuran-1(3H)-one
Preparation By heating phenol and phthalic anhydride in the presence of
H2SO4 (as catalyst):
C
C
O
O
HO OH
OHOH
HO
C
C
O
OH
OK
O
2+t
C OK
O
OH
OHC
O
C
O
NaOH
-H2O
COOK
NaO O
0
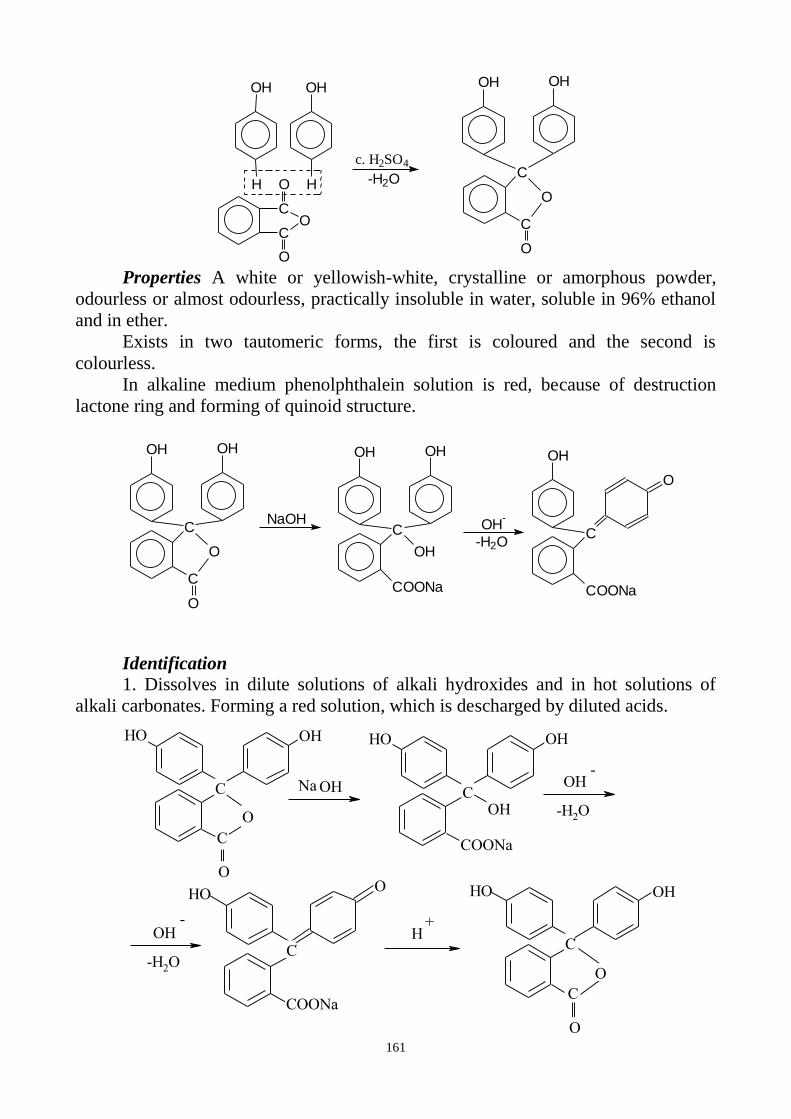
161
Properties A white or yellowish-white, crystalline or amorphous powder,
odourless or almost odourless, practically insoluble in water, soluble in 96% ethanol
and in ether.
Exists in two tautomeric forms, the first is coloured and the second is
colourless.
In alkaline medium phenolphthalein solution is red, because of destruction
lactone ring and forming of quinoid structure.
Identification
1. Dissolves in dilute solutions of alkali hydroxides and in hot solutions of
alkali carbonates. Forming a red solution, which is descharged by diluted acids.
OH
O
O
C
C
OH
-H2O
c. H2SO4
C
C
O
O
O
OH
H
OH
H
OH
O
O
C
C
OH
NaOH
OH
COONa
OH
C
OH
OH-
-H2O
OH
COONa
C
O
C
HO
C
HOO
COONa
H+
OH
O
C
O
OH-
-H2O
C
HO OH
O
C
O
OH C
HO OH
COONa
OH
Na OH-
-H2O
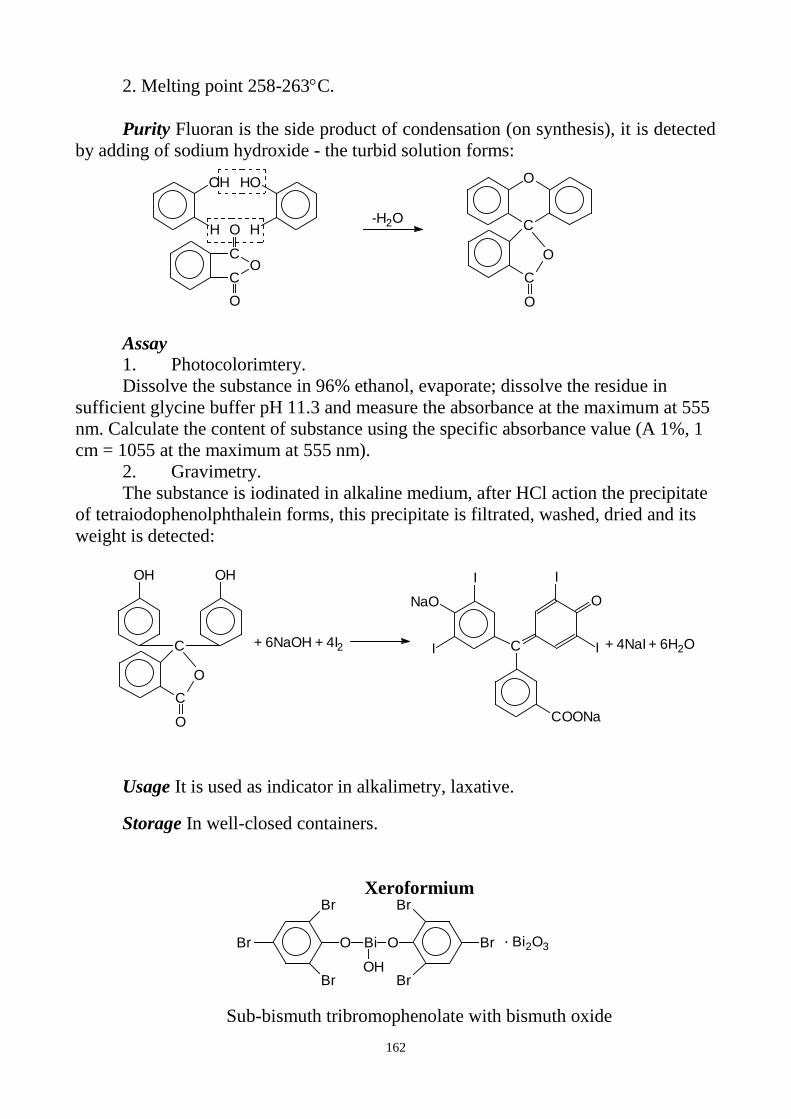
162
2. Melting point 258-263C.
Purity Fluoran is the side product of condensation (on synthesis), it is detected
by adding of sodium hydroxide - the turbid solution forms:
Assay
1. Photocolorimtery.
Dissolve the substance in 96% ethanol, evaporate; dissolve the residue in
sufficient glycine buffer pH 11.3 and measure the absorbance at the maximum at 555
nm. Calculate the content of substance using the specific absorbance value (A 1%, 1
cm = 1055 at the maximum at 555 nm).
2. Gravimetry.
The substance is iodinated in alkaline medium, after HCl action the precipitate
of tetraiodophenolphthalein forms, this precipitate is filtrated, washed, dried and its
weight is detected:
Usage It is used as indicator in alkalimetry, laxative.
Storage In well-closed containers.
Xeroformium
Sub-bismuth tribromophenolate with bismuth oxide
OH
H
C
C
O
O
O
O
C
C
O
O
-H2O
HO
H
Br
Br
Br O Bi O
OH
Br
Br
Br . Bi2O3
+ 4NaI + 6H2O
NaO
I
I C
I
I
O
COONa
+ 6NaOH + 4I2
OH
O
O
C
C
OH
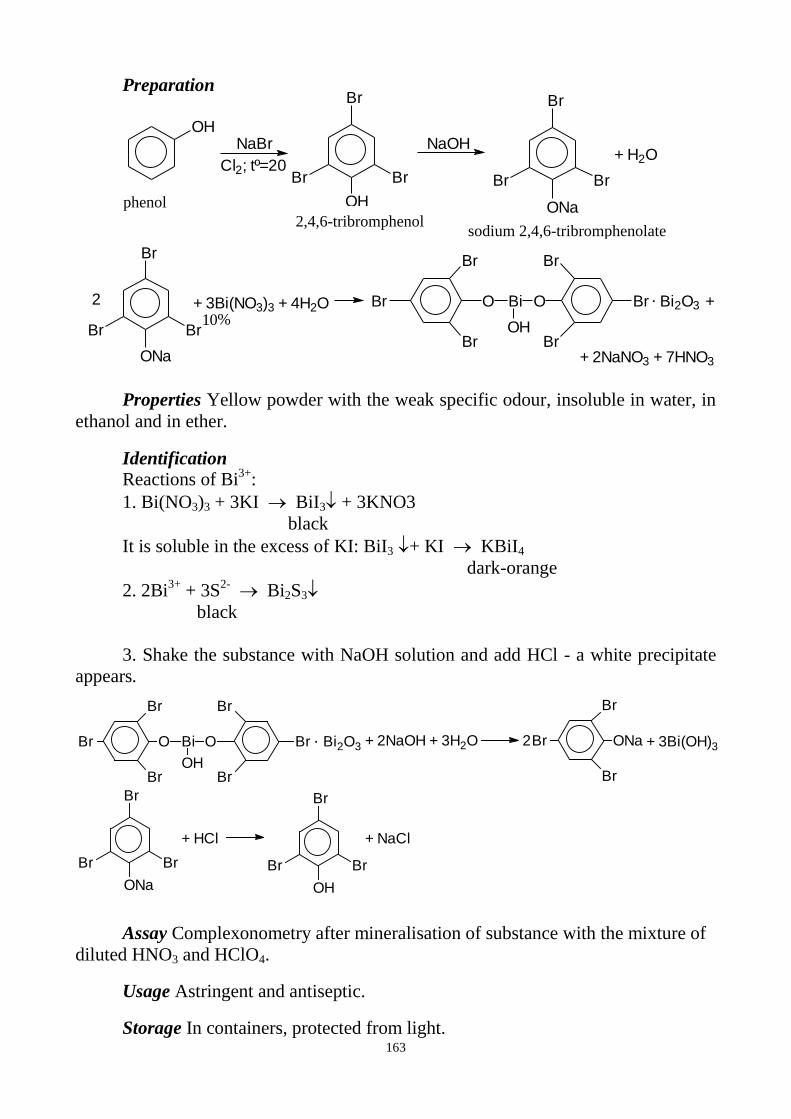
163
Preparation
Properties Yellow powder with the weak specific odour, insoluble in water, in
ethanol and in ether.
Identification
Reactions of Bi3+
:
1. Bi(NO3)3 + 3KI BiI3 + 3KNO3
black
It is soluble in the excess of KI: BiI3 + KI KBiI4
dark-orange
2. 2Bi3+
+ 3S2-
Bi2S3
black
3. Shake the substance with NaOH solution and add HCl - a white precipitate
appears.
Assay Complexonometry after mineralisation of substance with the mixture of
diluted HNO3 and HClO4.
Usage Astringent and antiseptic.
Storage In containers, protected from light.
Br
Br
Br
O Bi
OH
O Br
Br
Br
. Bi2O3 + 2NaOH + 3H2O Br
Br
Br
ONa2 + 3Bi(OH)3
Br Br
Br
ONa
+ HCl
Br Br
Br
OH
+ NaCl
10%
o o
o
o
+
+ 2NaNO3 + 7HNO3
Br
Br
Br O Bi O
OH
Br
Br
Br . Bi2O3+ 3Bi(NO3)3 + 4H2O
Br
Br Br
ONa
2
+ H2ONaOH
Cl2; tº=20
NaBr
Br
Br Br
ONa
Br
Br Br
OH
OH
phenol
2,4,6-tribromphenol sodium 2,4,6-tribromphenolate

164
Oxolinum
1,2,3,4-tetraoxo-1,2,3,4-tetrahydronaphthaline dihydrate
Properties White crystalline powder. Freely soluble in water. Aqueous
solutions are unstable, in alkaline medium they get dark.
Usage For treatment of virous diseases of eyes, skin and influenza.
Storage In well-closed containers, protected from light.
O
O
O
O
. 2H2O
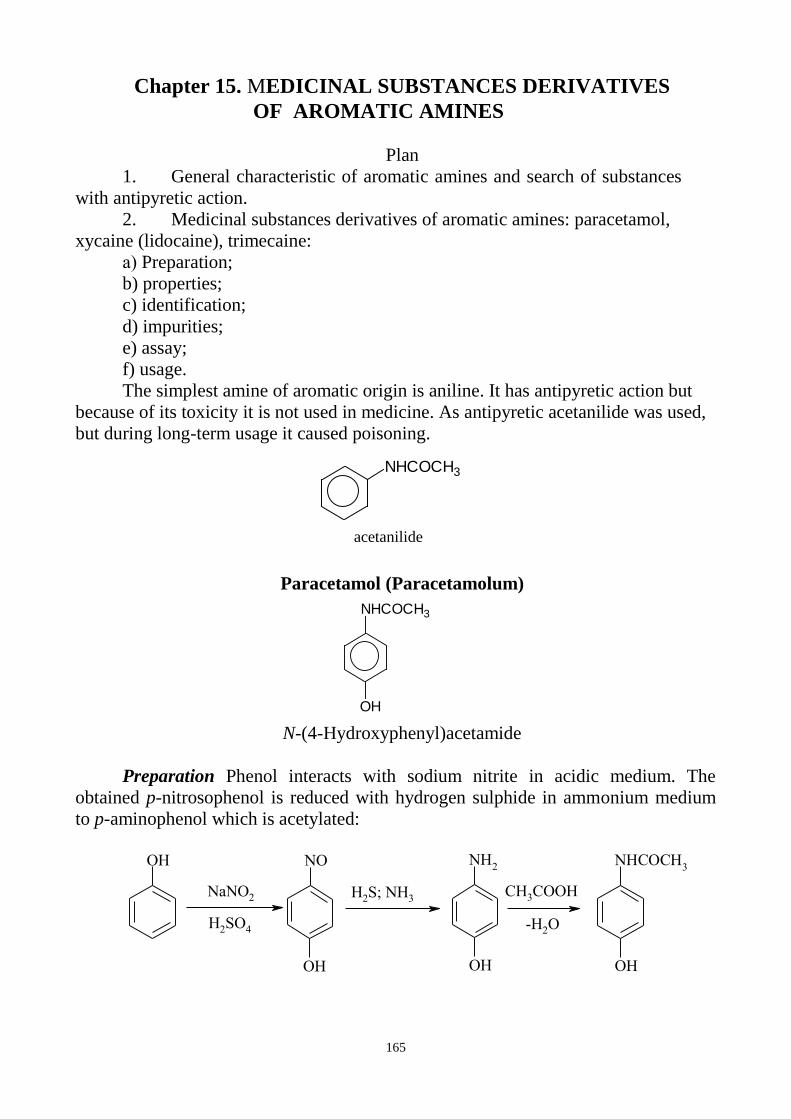
165
Chapter 15. MEDICINAL SUBSTANCES DERIVATIVES
OF AROMATIC AMINES
Plan
1. General characteristic of aromatic amines and search of substances
with antipyretic action.
2. Medicinal substances derivatives of aromatic amines: paracetamol,
xycaine (lidocaine), trimecaine:
а) Preparation;
b) properties;
c) identification;
d) impurities;
e) assay;
f) usage.
The simplest amine of aromatic origin is aniline. It has antipyretic action but
because of its toxicity it is not used in medicine. As antipyretic acetanilide was used,
but during long-term usage it caused poisoning.
Paracetamol (Paracetamolum)
N-(4-Hydroxyphenyl)acetamide
Preparation Phenol interacts with sodium nitrite in acidic medium. The
obtained p-nitrosophenol is reduced with hydrogen sulphide in ammonium medium
to p-aminophenol which is acetylated:
NHCOCH
3
OH
NH2
OH
OH
OH
NO
CH3COOHNaNO2
H2SO4
H2S; NH3
-H2O
NHCOCH3
OH
NHCOCH3
ацетанилидacetanilide
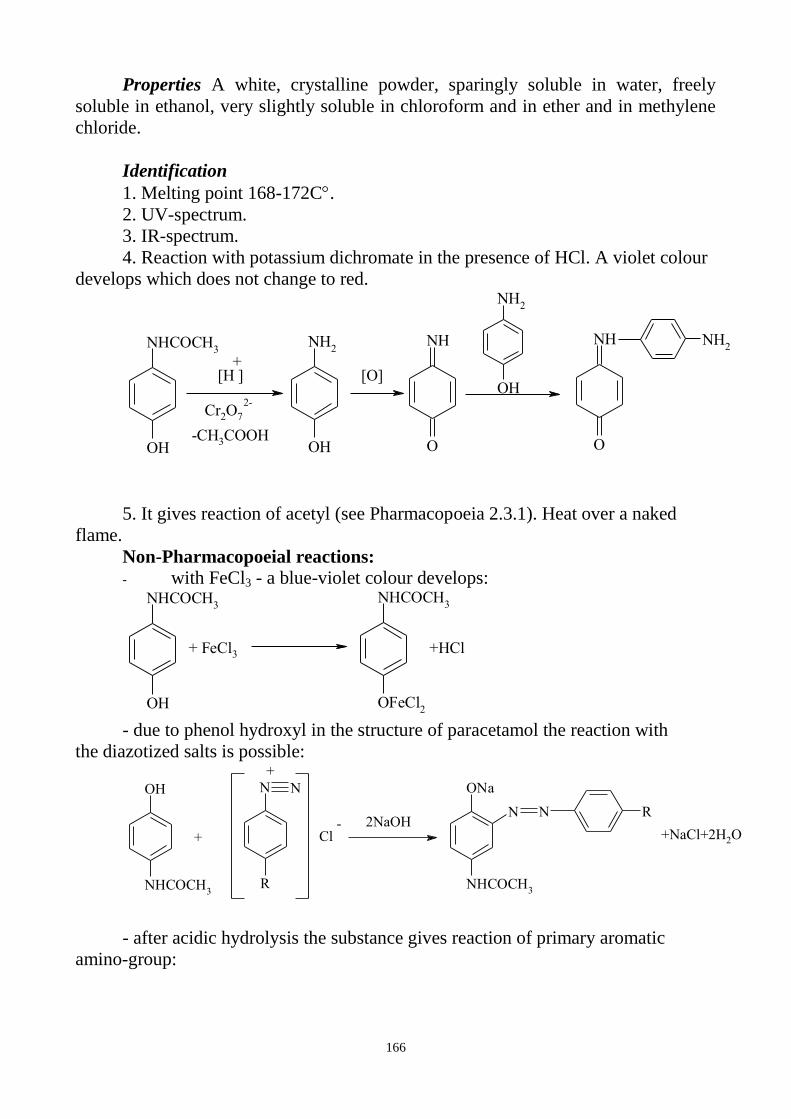
166
Properties A white, crystalline powder, sparingly soluble in water, freely
soluble in ethanol, very slightly soluble in chloroform and in ether and in methylene
chloride.
Identification
1. Melting point 168-172C.
2. UV-spectrum.
3. IR-spectrum.
4. Reaction with potassium dichromate in the presence of HCl. A violet colour
develops which does not change to red.
NHCOCH3
OH
NH2
OH
NH
O
NH2
OH
NH
O
NH2
[H ]
Cr2O7
-CH3COOH
+
2-
[O]
5. It gives reaction of acetyl (see Pharmacopoeia 2.3.1). Heat over a naked
flame.
Non-Pharmacopoeial reactions:
- with FeCl3 - a blue-violet colour develops: NHCOCH
3
OH
NHCOCH3
OFeCl2
+ FeCl3 +HCl
- due to phenol hydroxyl in the structure of paracetamol the reaction with
the diazotized salts is possible:
OH
NHCOCH3
N
R
N
ONa
NHCOCH3
N N R
+ Cl
+
- 2NaOH+NaCl+2H2O
- after acidic hydrolysis the substance gives reaction of primary aromatic
amino-group:
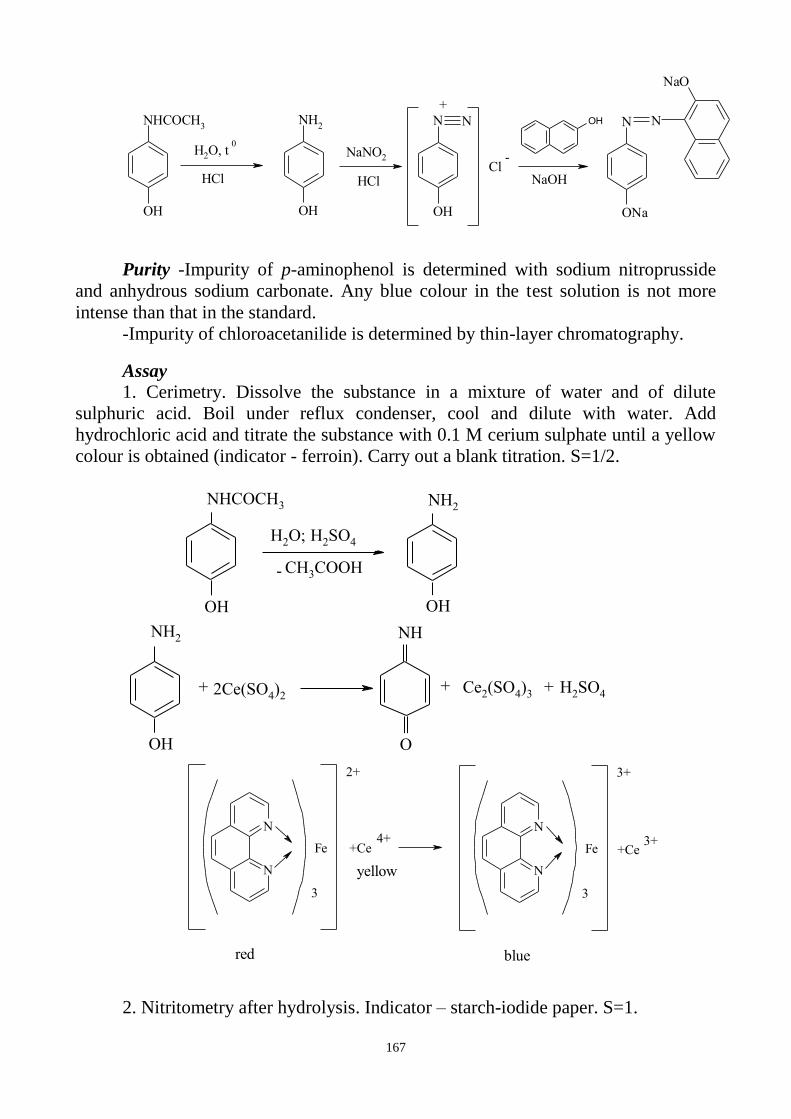
167
NHCOCH3
OH
N
OH
N
N
ONa
N
NaO
NH2
OH
OH
Cl
+
-
NaOH
H2O, t
HCl
0NaNO2
HCl
Purity -Impurity of p-aminophenol is determined with sodium nitroprusside
and anhydrous sodium carbonate. Any blue colour in the test solution is not more
intense than that in the standard.
-Impurity of chloroacetanilide is determined by thin-layer chromatography.
Assay
1. Cerimetry. Dissolve the substance in a mixture of water and of dilute
sulphuric acid. Boil under reflux condenser, cool and dilute with water. Add
hydrochloric acid and titrate the substance with 0.1 M cerium sulphate until a yellow
colour is obtained (indicator - ferroin). Carry out a blank titration. S=1/2.
OH
OH
H2SO4H2O;
NH2
+
CH3COOH
OH
NH2 NH
+ +Ce2(SO4)3 H2SO4
NHCOCH3
O
2Ce(SO4)2
-
N
N
N
N
3
Fe
2+
+Ce4+
3
Fe
3+
+Ce3+
red
yellow
blue
2. Nitritometry after hydrolysis. Indicator – starch-iodide paper. S=1.
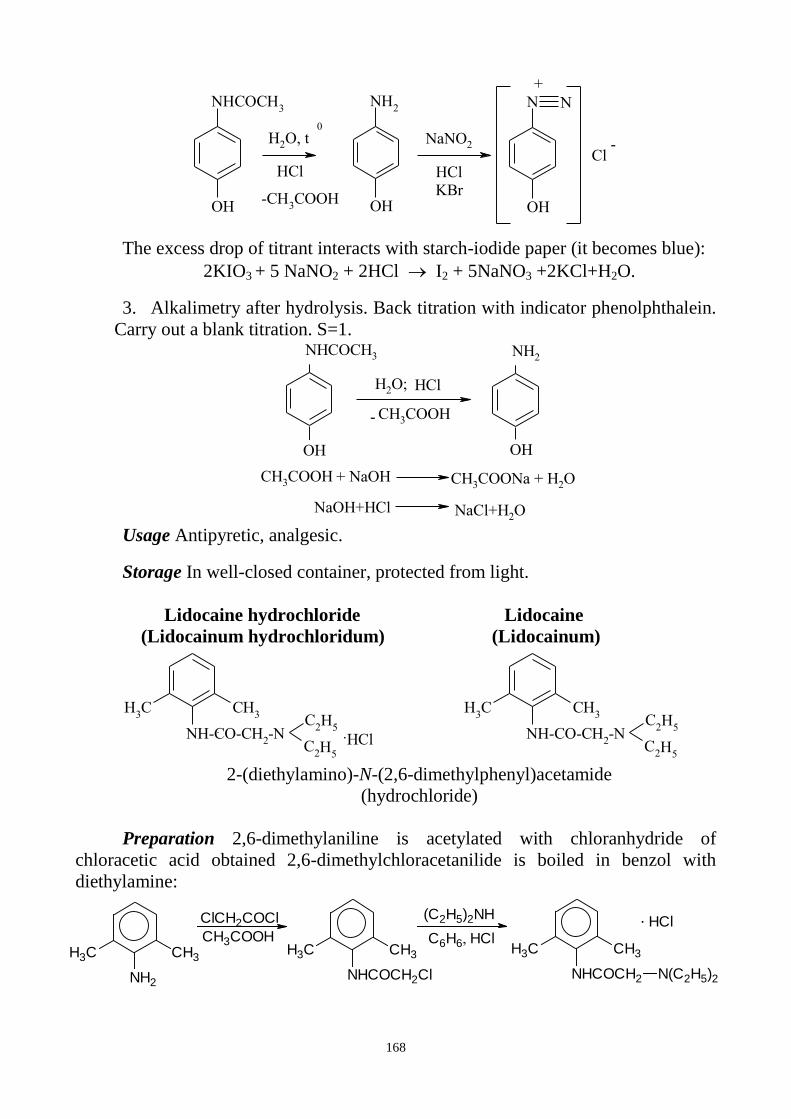
168
NHCOCH3
OH
N
OH
N
NH2
OH-CH
3COOH
Cl
+
-H2O, t
HCl
0
NaNO2
HClKBr
The excess drop of titrant interacts with starch-iodide paper (it becomes blue):
2KIO3 + 5 NaNO2 + 2HCl I2 + 5NaNO3 +2KCl+H2O.
3. Alkalimetry after hydrolysis. Back titration with indicator phenolphthalein.
Carry out a blank titration. S=1.
OH
H2O;
NH2
CH3COOH
OH
NHCOCH3
-
CH3COOH + NaOH CH3COONa + H2O
NaOH+HCl NaCl+H2O
HCl
Usage Antipyretic, analgesic.
Storage In well-closed container, protected from light.
Lidocaine hydrochloride Lidocaine
(Lidocainum hydrochloridum) (Lidocainum)
CH3
CH3
NH-CO-CH2-N
C2
C2H5
H5
.HCl
CH3
CH3
NH-CO-CH2-N
C2
C2H5
H5
2-(diethylamino)-N-(2,6-dimethylphenyl)acetamide
(hydrochloride)
Preparation 2,6-dimethylaniline is acetylated with chloranhydride of
chloracetic acid obtained 2,6-dimethylchloracetanilide is boiled in benzol with
diethylamine:
CH3H3C
NH2
ClCH2COCl
CH3COOHCH3H3C
NHCOCH2Cl
(C2H5)2NH
C6H6, HCl . HCl
CH3H3C
NHCOCH2 N(C2H5)2
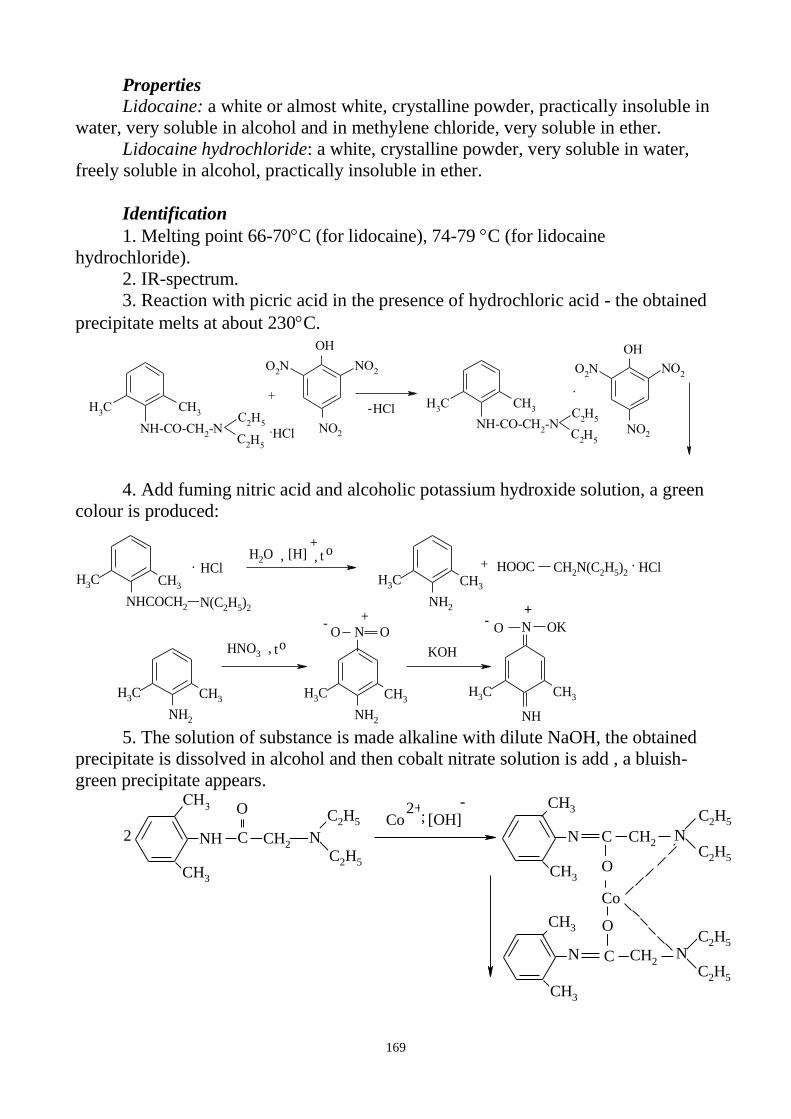
169
Properties
Lidocaine: a white or almost white, crystalline powder, practically insoluble in
water, very soluble in alcohol and in methylene chloride, very soluble in ether.
Lidocaine hydrochloride: a white, crystalline powder, very soluble in water,
freely soluble in alcohol, practically insoluble in ether.
Identification
1. Melting point 66-70C (for lidocaine), 74-79 C (for lidocaine
hydrochloride).
2. IR-spectrum.
3. Reaction with picric acid in the presence of hydrochloric acid - the obtained
precipitate melts at about 230C.
4. Add fuming nitric acid and alcoholic potassium hydroxide solution, a green
colour is produced:
5. The solution of substance is made alkaline with dilute NaOH, the obtained
precipitate is dissolved in alcohol and then cobalt nitrate solution is add , a bluish-
green precipitate appears. CH
3
2 NH C CH2N
C2H5
C2H5
O
CH3
Co2+
; [OH]-
CH3
N
N
N
CH3
CH3
C
C
O
O
Co
N
CH3
CH2
CH2
C2H5
C2H5
C2H5
C2H5
CH3H3C
NHCOCH2 N(C2H5)2
. HClH2O , [H]
+, t o
CH3H3C
NH2
+ HOOC CH2N(C2H5)2. HCl
CH3H3C
NH2
HNO3 , to
CH3H3C
NH2
NO O+
-
KOH
NH
CH3H3C
OKNO-
+
CH3
CH3
NH-CO-CH2-N
O2N NO
2
C2
C2H5
H5
CH3
CH3
NH-CO-CH2-N
O2N NO
2
C2
C2H5
H5
.
+
OH
NO2
HCl
HCl
OH
NO2
-
.
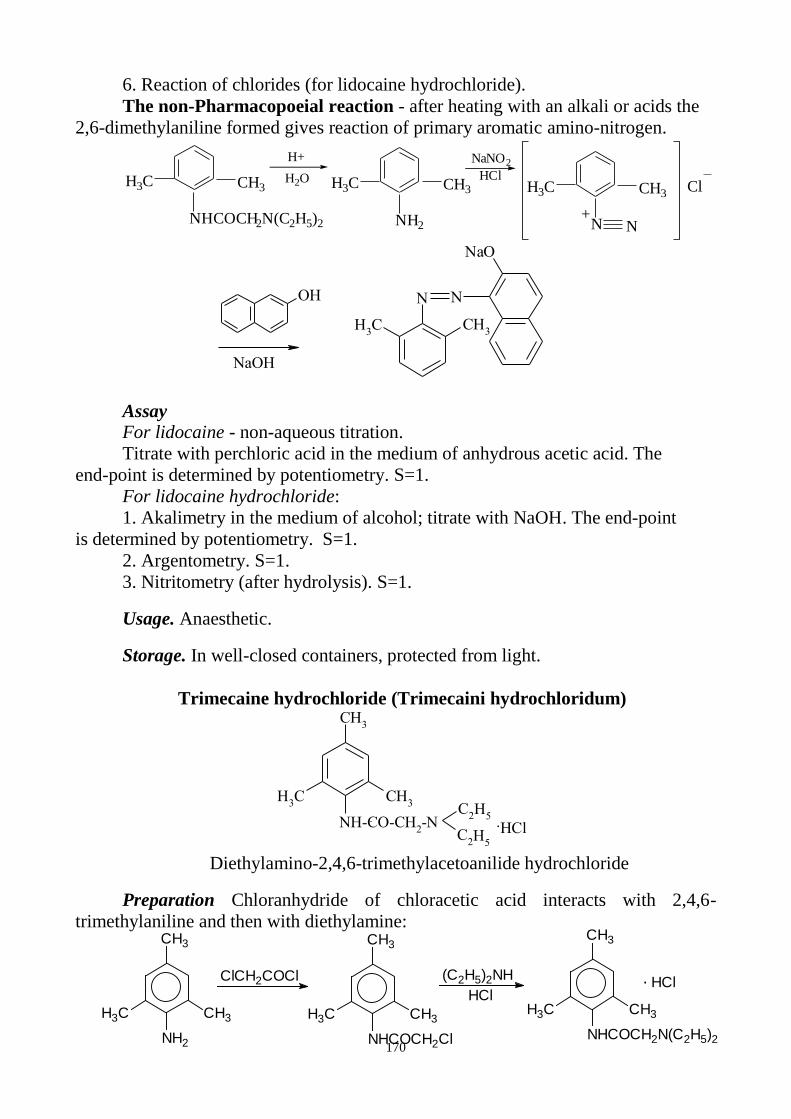
170
6. Reaction of chlorides (for lidocaine hydrochloride).
The non-Pharmacopoeial reaction - after heating with an alkali or acids the
2,6-dimethylaniline formed gives reaction of primary aromatic amino-nitrogen.
H3C
NHCOCH2N(C2H5)2
CH3
H+
H2O H3C
NH2
CH3 H3C
N
CH3
NaNO2
HCl
N+
Cl
N
CH3
CH3
N
NaO
OH
NaOH
Assay
For lidocaine - non-aqueous titration.
Titrate with perchloric acid in the medium of anhydrous acetic acid. The
end-point is determined by potentiometry. S=1.
For lidocaine hydrochloride:
1. Akalimetry in the medium of alcohol; titrate with NaOH. The end-point
is determined by potentiometry. S=1.
2. Argentometry. S=1.
3. Nitritometry (after hydrolysis). S=1.
Usage. Anaesthetic.
Storage. In well-closed containers, protected from light.
Trimecaine hydrochloride (Trimecaini hydrochloridum)
CH3
CH3
NH-CO-CH2-N
CH3
C2
C2H5
H5
.HCl
Diethylamino-2,4,6-trimethylacetoanilide hydrochloride
Preparation Chloranhydride of chloracetic acid interacts with 2,4,6-
trimethylaniline and then with diethylamine:
CH3H3C
CH3
NH2
ClCH2COCl
CH3H3C
CH3
NHCOCH2Cl
(C2H5)2NH
HClCH3H3C
CH3
NHCOCH2N(C2H5)2
. HCl
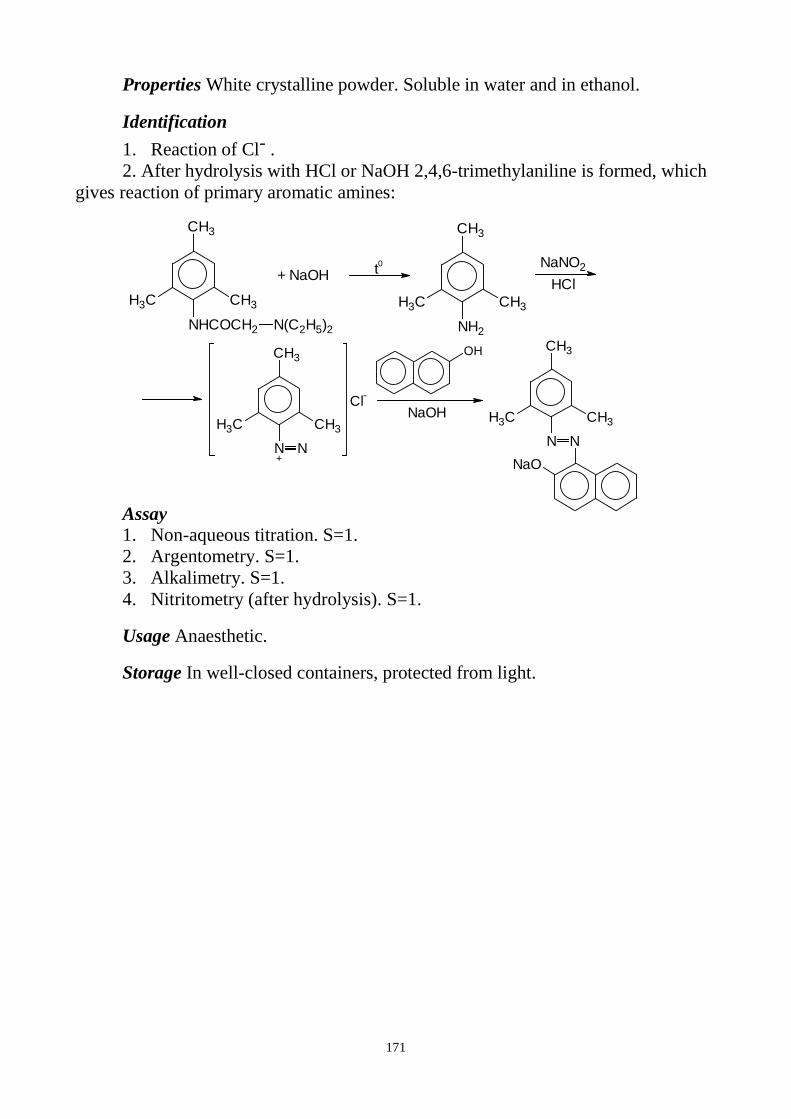
171
Properties White crystalline powder. Soluble in water and in ethanol.
Identification
1. Reaction of Cl- .
2. After hydrolysis with HCl or NaOH 2,4,6-trimethylaniline is formed, which
gives reaction of primary aromatic amines:
Assay
1. Non-aqueous titration. S=1.
2. Argentometry. S=1.
3. Alkalimetry. S=1.
4. Nitritometry (after hydrolysis). S=1.
Usage Anaesthetic.
Storage In well-closed containers, protected from light.
HCl
NaNO2
CH3H3C
CH3
NH2
t0
+ NaOH
CH3H3C
NHCOCH2 N(C2H5)2
CH3
CH3H3C
CH3
N N+
Cl-
NaOH
OH
CH3H3C
CH3
N N
NaO

172
Chapter 16. MEDICINAL SUBSTANCES DERIVATIVES OF
AROMATIC ACIDS
Plan
1. Medicinal substances derivatives of benzoic acid: benzoic acid, sodium
benzoate – Preparation, properties, identification and assay, usage and storage.
2. Medicinal substances derivatives of salicylic acid: salicylic acid, sodium
salicylate, acetylsalicylic acid, phenyl salicylate, methyl salicylate. Preparation,
properties, identification and assay, usage and storage.
Aromatic acids – are derivatives of aromatic hydrocarbons, which contain one
or some hydrogen atoms of benzene nucleus substituted by carboxyl group:
CO
OH
The presence of carboxyl group in aromatic hydrocarbon decreases toxicity.
Benzoic acid (Ка=6,27.10-5
) is stronger than acetic acid (Ка=1,82.10-5
).
Benzoic acid (Acidum benzoicum)
CO
OH
Benzenecarboxylic acid
Benzoic acid occurs as its benzyl ester in benzoic resins.
Preparation
1. By oxidising of toluene:
CH3
COOH
Toluene
+3MnO2+3H2SO4 +3MnSO4+4H2O
2. By chlorination of toluene with further oxidising of the formed
benzenetrichloride:
CH3
C Cl
Cl
Cl
C
OH
OH
OH
COOHCl2
3H2O -H2O
Toluene Benzenetrichloride Trihydroxyphenylmethane
CH3 C
O
OH
C6H5 C
O
OH
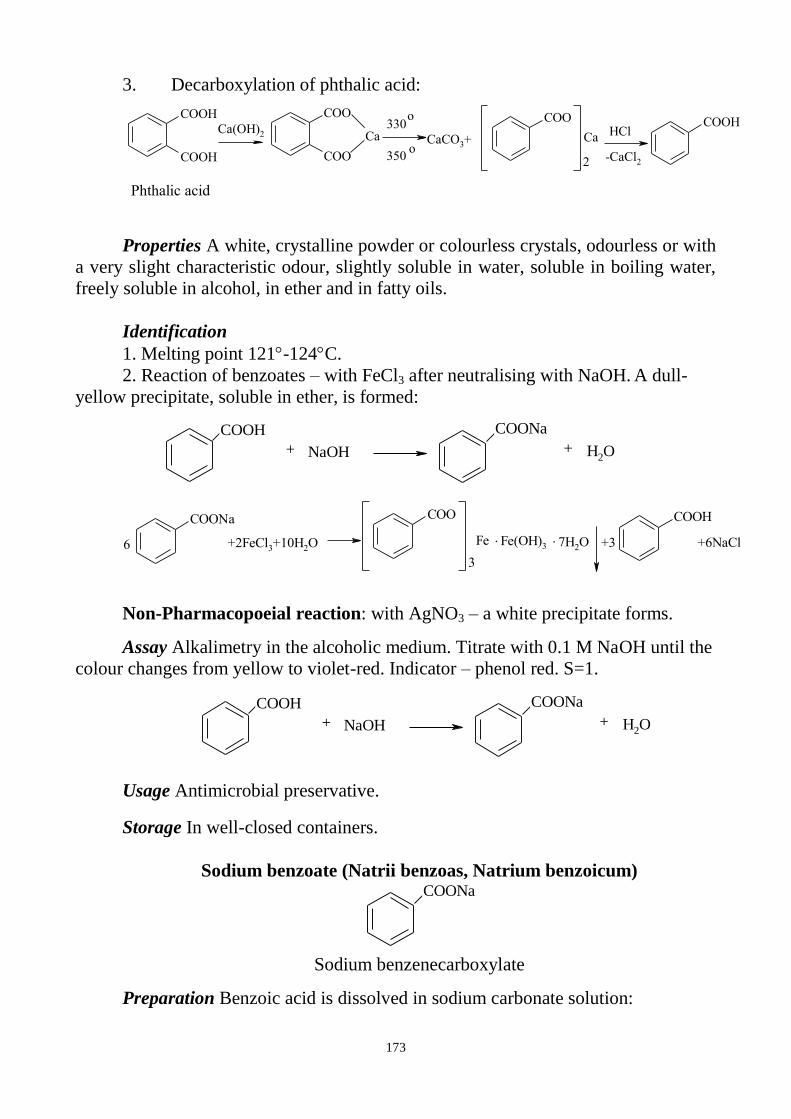
173
3. Decarboxylation of phthalic acid:
COOH
COOH
COO
COO
COO
COOH
Phthalic acid
Ca(OH)2 Ca330
350
o
oCaCO3+
2
CaHCl
-CaCl2
Properties A white, crystalline powder or colourless crystals, odourless or with
a very slight characteristic odour, slightly soluble in water, soluble in boiling water,
freely soluble in alcohol, in ether and in fatty oils.
Identification
1. Melting point 121-124C.
2. Reaction of benzoates – with FeCl3 after neutralising with NaOH. A dull-
yellow precipitate, soluble in ether, is formed:
COONa COO
COOH
6 +2FeCl3+10H2O
3
Fe . Fe(OH)3. 7H2O +3 +6NaCl
Non-Pharmacopoeial reaction: with AgNO3 – a white precipitate forms.
Assay Alkalimetry in the alcoholic medium. Titrate with 0.1 M NaOH until the
colour changes from yellow to violet-red. Indicator – phenol red. S=1.
Usage Antimicrobial preservative.
Storage In well-closed containers.
Sodium benzoate (Natrii benzoas, Natrium benzoicum) COONa
Sodium benzenecarboxylate
Preparation Benzoic acid is dissolved in sodium carbonate solution:
COOH
+ NaOH
COONa
+ H2O
COOH
+ NaOH
COONa
+ H2O
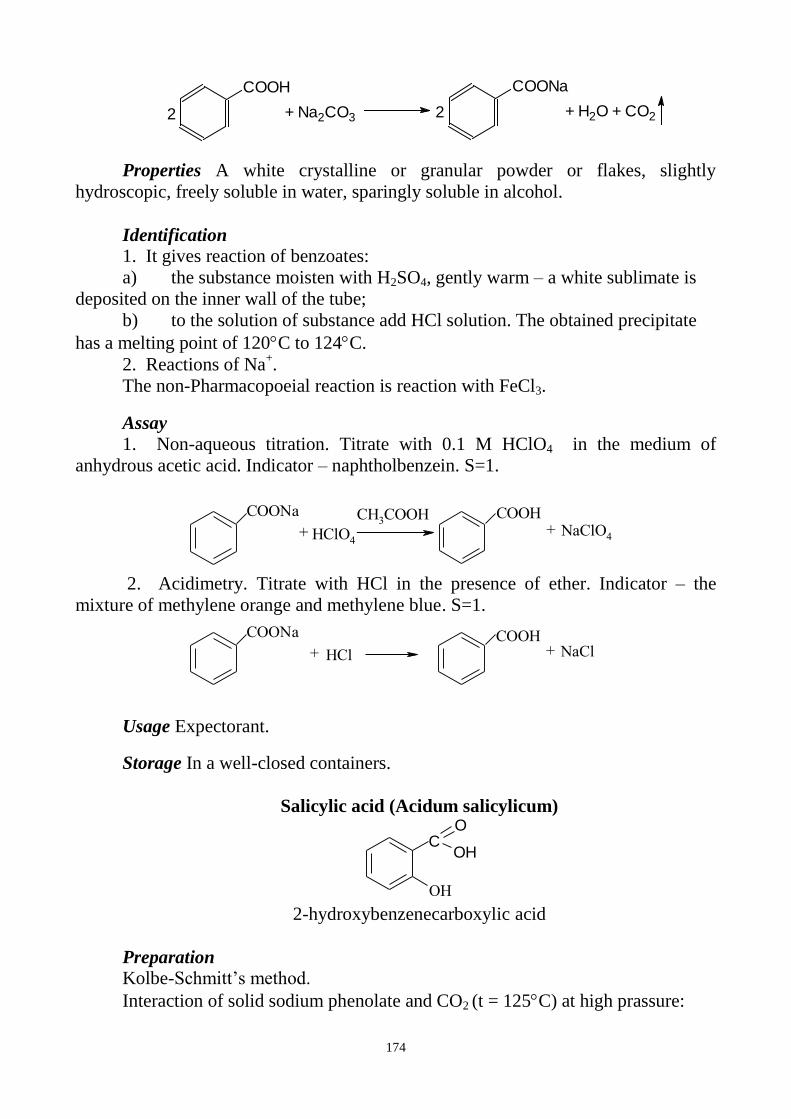
174
Properties A white crystalline or granular powder or flakes, slightly
hydroscopic, freely soluble in water, sparingly soluble in alcohol.
Identification
1. It gives reaction of benzoates:
a) the substance moisten with H2SO4, gently warm – a white sublimate is
deposited on the inner wall of the tube;
b) to the solution of substance add HCl solution. The obtained precipitate
has a melting point of 120C to 124C.
2. Reactions of Na+.
The non-Pharmacopoeial reaction is reaction with FeCl3.
Assay
1. Non-aqueous titration. Titrate with 0.1 M HClO4 in the medium of
anhydrous acetic acid. Indicator – naphtholbenzein. S=1.
2. Acidimetry. Titrate with HCl in the presence of ether. Indicator – the
mixture of methylene orange and methylene blue. S=1.
Usage Expectorant.
Storage In a well-closed containers.
Salicylic acid (Acidum salicylicum)
OH
CO
OH
2-hydroxybenzenecarboxylic acid
Preparation
Kolbe-Schmitt’s method.
Interaction of solid sodium phenolate and CO2 (t = 125C) at high prassure:
COOH
2 + Na2CO3 2
COONa
+ H2O + CO2
COONa COOH+ +HCl NaCl
COONa COOH
+ +HClO4NaClO4
CH3COOH
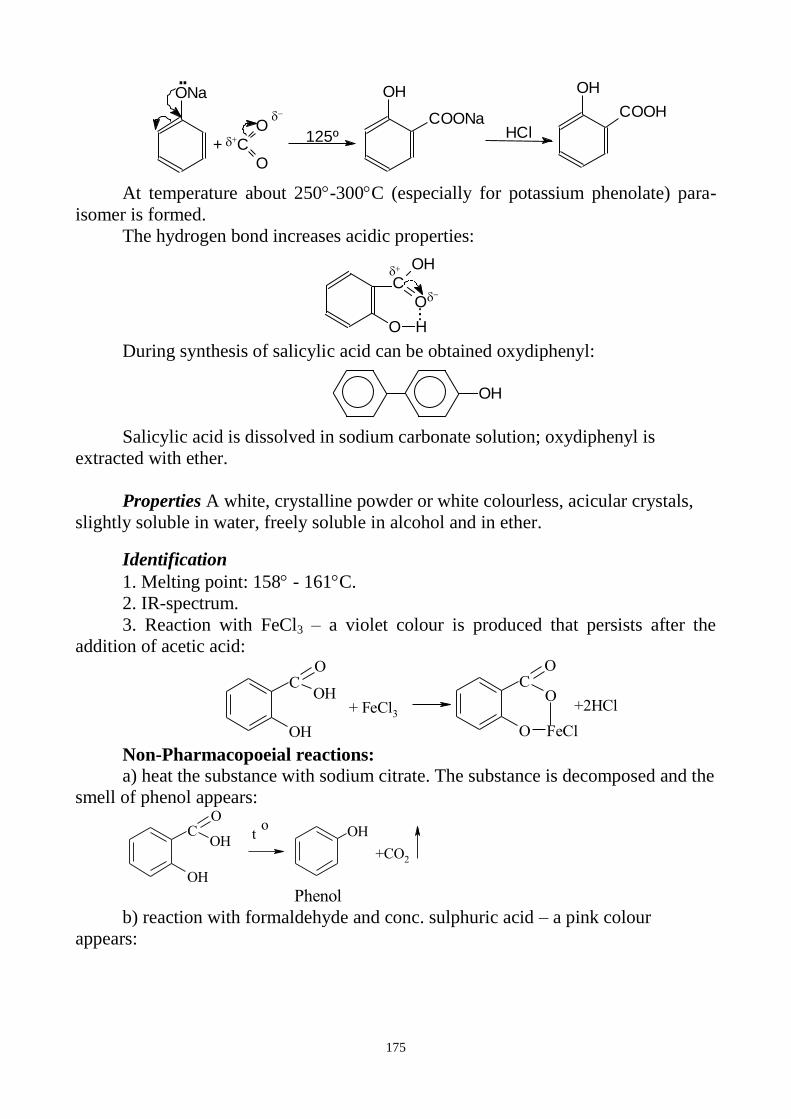
175
At temperature about 250-300C (especially for potassium phenolate) para-
isomer is formed.
The hydrogen bond increases acidic properties:
During synthesis of salicylic acid can be obtained oxydiphenyl:
Salicylic acid is dissolved in sodium carbonate solution; oxydiphenyl is
extracted with ether.
Properties A white, crystalline powder or white colourless, acicular crystals,
slightly soluble in water, freely soluble in alcohol and in ether.
Identification
1. Melting point: 158 - 161C.
2. IR-spectrum.
3. Reaction with FeCl3 – a violet colour is produced that persists after the
addition of acetic acid:
OH
CO
OH
O FeCl
CO
O+ FeCl3
+2HCl
Non-Pharmacopoeial reactions:
a) heat the substance with sodium citrate. The substance is decomposed and the
smell of phenol appears:
OH
CO
OHOHt
o
+CO2
Phenol b) reaction with formaldehyde and conc. sulphuric acid – a pink colour
appears:
C
OH
O
O H
....
d
d
OH
ONa..
+ dC
O
O
d
125º
OH
COONaHCl
OH
COOH
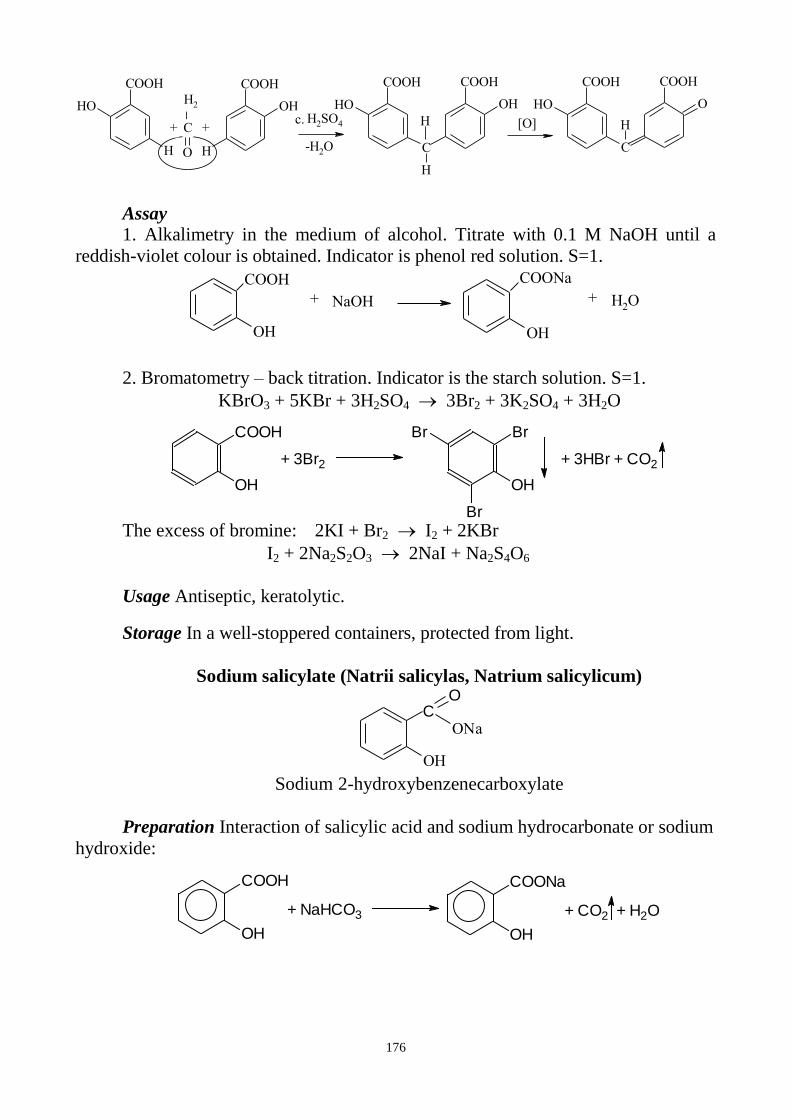
176
OH
H
COOH
O
COOH
H
OH OH
COOH
C
H
COOH
OH
H
OH
COOH
C
H
COOH
O
+ C
H2
+H2SO4c.
-H2O
[O]
Assay
1. Alkalimetry in the medium of alcohol. Titrate with 0.1 M NaOH until a
reddish-violet colour is obtained. Indicator is phenol red solution. S=1.
2. Bromatometry – back titration. Indicator is the starch solution. S=1.
KBrO3 + 5KBr + 3H2SO4 3Br2 + 3K2SO4 + 3H2O
The excess of bromine: 2KI + Br2 I2 + 2KBr
I2 + 2Na2S2O3 2NaI + Na2S4O6
Usage Antiseptic, keratolytic.
Storage In a well-stoppered containers, protected from light.
Sodium salicylate (Natrii salicylas, Natrium salicylicum)
OH
CO
ONa
Sodium 2-hydroxybenzenecarboxylate
Preparation Interaction of salicylic acid and sodium hydrocarbonate or sodium
hydroxide:
COOH
OH
+ NaHCO3
COONa
OH
+ CO2 + H2O
+ 3HBr + CO2
Br Br
Br
OH
+ 3Br2
OH
COOH
OH OH
COOH
+ NaOH
COONa
+ H2O
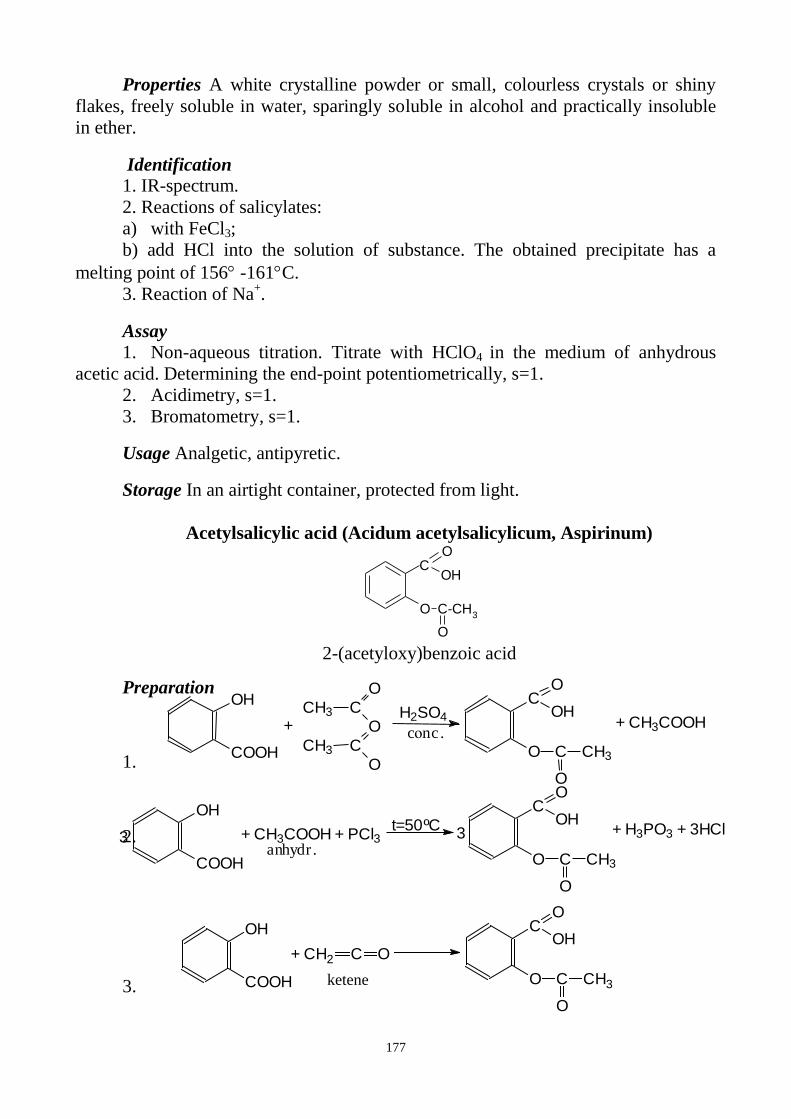
177
Properties A white crystalline powder or small, colourless crystals or shiny
flakes, freely soluble in water, sparingly soluble in alcohol and practically insoluble
in ether.
Identification
1. IR-spectrum.
2. Reactions of salicylates:
a) with FeCl3;
b) add HCl into the solution of substance. The obtained precipitate has a
melting point of 156 -161C.
3. Reaction of Na+.
Assay 1. Non-aqueous titration. Titrate with HClO4 in the medium of anhydrous
acetic acid. Determining the end-point potentiometrically, s=1.
2. Acidimetry, s=1.
3. Bromatometry, s=1.
Usage Analgetic, antipyretic.
Storage In an airtight container, protected from light.
Acetylsalicylic acid (Acidum acetylsalicylicum, Aspirinum)
O C-CH3
O
CO
OH
2-(acetyloxy)benzoic acid
Preparation
1.
2.
3.
O
C
C
O
O
OH
CH3
OH
COOH
+C
O
O
C
O
CH3
CH3
H2SO4
cоnc.+ CH3COOH
+ H3PO3 + 3HCl3
O
C
C
O
O
OH
CH3
t=50+ CH3COOH + PCl33
OH
COOHanhydr.
ºC
кетен со спиртами и эфирами дает соответствующий эфир
OH
COOH
+ CH2 C O
O
C
C
O
O
OH
CH3
ketene
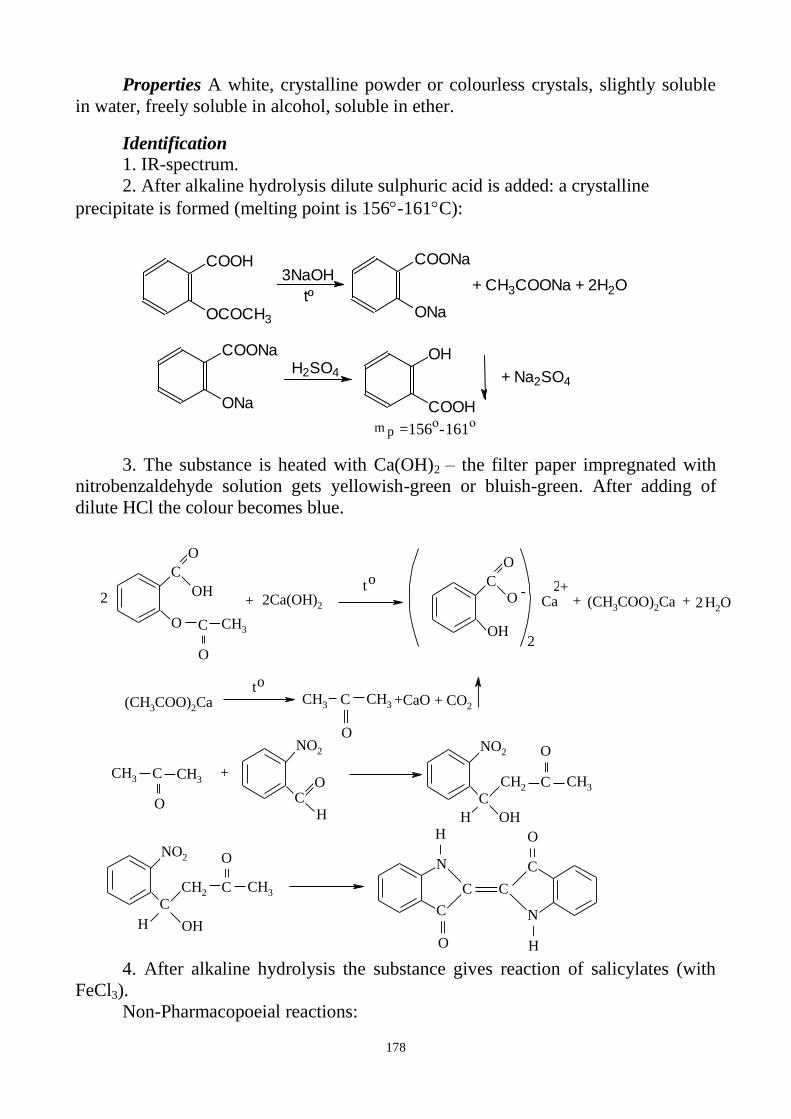
178
Properties A white, crystalline powder or colourless crystals, slightly soluble
in water, freely soluble in alcohol, soluble in ether.
Identification 1. IR-spectrum.
2. After alkaline hydrolysis dilute sulphuric acid is added: a crystalline
precipitate is formed (melting point is 156-161C):
3. The substance is heated with Ca(OH)2 – the filter paper impregnated with
nitrobenzaldehyde solution gets yellowish-green or bluish-green. After adding of
dilute HCl the colour becomes blue.
4. After alkaline hydrolysis the substance gives reaction of salicylates (with
FeCl3).
Non-Pharmacopoeial reactions:
COOH
OCOCH3
3NaOH
tº
COONa
ONa
+ CH3COONa + 2H2O
C O O N a
O N a
H 2 S O 4
O H
C O O H
+ N a 2 S O 4
m p = 1 5 6
о - 1 6 1
о
2
2
C
O C
O
CH3
OH+ 2Ca(OH)2
t C
O
C
O
CH3
OH
O -Ca
2++ (CH3COO)2Ca + H2O
(CH3COO)2Cat
CH3 CH3 + +CaO CO2
O
CCH3 CH3+
C
O
CO
OHH
NO2
C
O
H
NO2
CH2
H
CH3C
OH
C
ONO2
CH2
HC
N
C C
N
C
H O
O
o
o
2
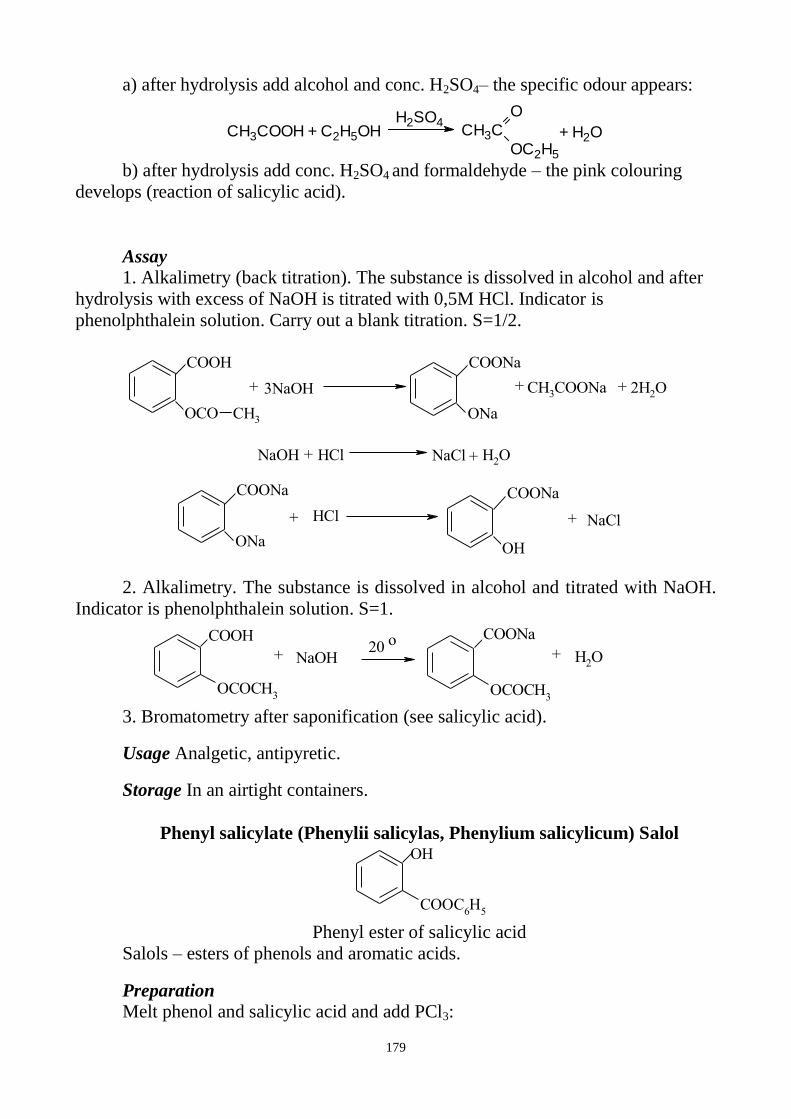
179
a) after hydrolysis add alcohol and conc. H2SO4– the specific odour appears:
b) after hydrolysis add conc. H2SO4 and formaldehyde – the pink colouring
develops (reaction of salicylic acid).
Assay
1. Alkalimetry (back titration). The substance is dissolved in alcohol and after
hydrolysis with excess of NaOH is titrated with 0,5M HCl. Indicator is
phenolphthalein solution. Carry out a blank titration. S=1/2.
COOH
OCO CH3
+ 3NaOH
COONa
ONa
+ CH3COONa + 2H2O
2. Alkalimetry. The substance is dissolved in alcohol and titrated with NaOH.
Indicator is phenolphthalein solution. S=1.
3. Bromatometry after saponification (see salicylic acid).
Usage Analgetic, antipyretic.
Storage In an airtight containers.
Phenyl salicylate (Phenylii salicylas, Phenylium salicylicum) Salol
COOC6H
5
OH
Phenyl ester of salicylic acid
Salols – esters of phenols and aromatic acids.
Preparation
Melt phenol and salicylic acid and add PCl3:
CH3COOH + C2H5OHH2SO4
CH3C
O
OC2H5
+ H2O
NaOH + HCl NaCl + H2O
COONa
ONa
+ HCl
COONa
OH
+ NaCl
OCOCH3 OCOCH
3
COOH
+ NaOH
COONa
+ H2O20 o

180
The temperature is increased to 100C and then the mixture is cooled to 50-
55оС.
Properties A white crystalline powder with weak odour. Practically insoluble
in water, soluble in alcohol and in alkali solution, freely soluble in chloroform, very
soluble in ether.
Identification
1. To alcoholic solution of the substance add FeCl3 solution - a violet colouring
develops. After hydrolysis add an acid, the smell of phenol appears and the
precipitate of salicylic acid develops (melting point 158-161C).
C6H4OHCOOC6H5 + 3NaOH C6H5ONa + C6H4ONaCOONa + 2H2O
2. Reaction with formaldehyde on heating in the presence of conc. sulphuric
acid – a pink colouring develops (see salicylic acid).
Assay
1. Alkalimetry (back titration) after alkaline hydrolysis:
The excess of NaOH and of products of hydrolysis are titrated with HCl in the
presence of bromocresol purple:
NaOH + HCl NaCl + H2O
The products of hydrolysis are neutralised to sodium salicylate, which is
neutral relatively phenolphthalein and bromocresol purple.
One molecule of the substance is hydrolysed with one molecule of NaOH, S=1.
Usage For treatment of enterocolitis and colitis, cystitis, pyelitis.
Storage In well-closed containers, protected from light.
OH
COOH
+ 3
OH
+ PCl360º
3 3
OH
COC6H5
O
+ H3PO4 + 3HCl
OH
COOC6H5
+ 3NaOH
ONa
COONa
+
ONa
+ 2H2O
ONa
COONa
+ HCl
COONa
OH
+ NaCl
+ NaCl
OH
+ HCl
ONa
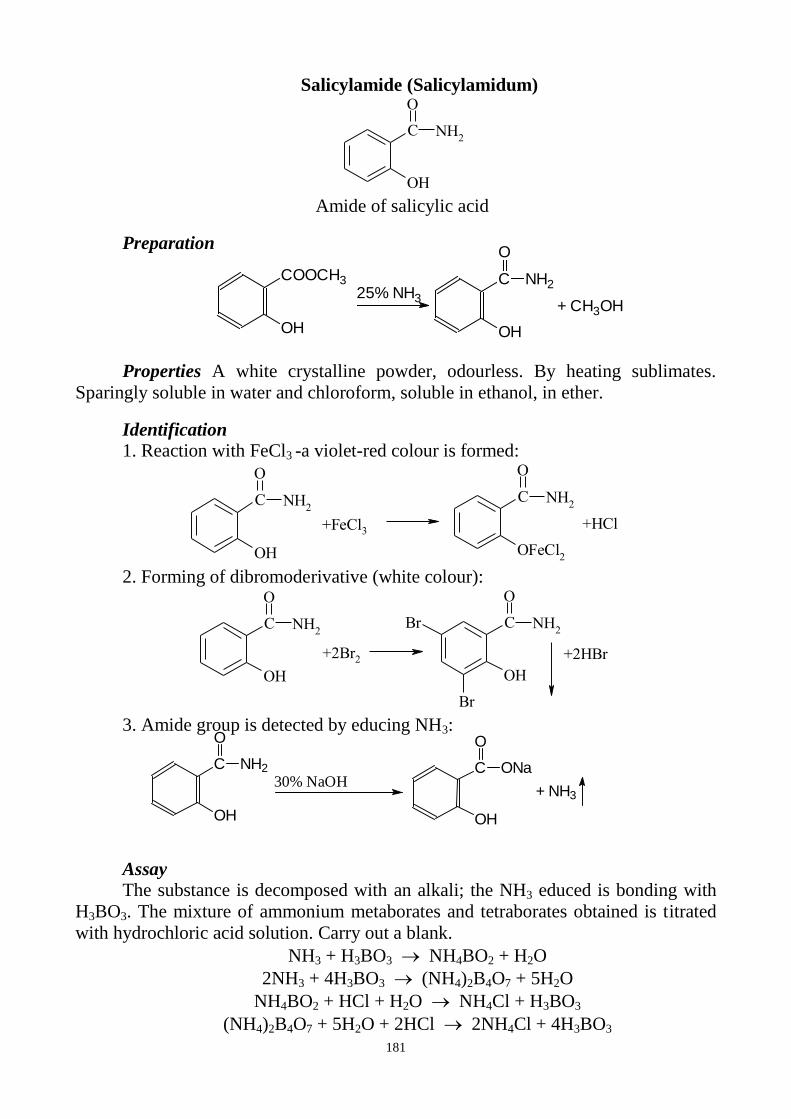
181
Salicylamide (Salicylamidum)
C
OH
O
NH2
Amide of salicylic acid
Preparation
Properties A white crystalline powder, odourless. By heating sublimates.
Sparingly soluble in water and chloroform, soluble in ethanol, in ether.
Identification
1. Reaction with FeCl3 -a violet-red colour is formed:
C
OH
O
NH2
C
O
OFeCl2
NH2
+FeCl3+HCl
2. Forming of dibromoderivative (white colour):
C
OH
O
NH2
C
OH
O
Br
Br NH2
+2Br2 +2HBr
3. Amide group is detected by educing NH3:
Assay
The substance is decomposed with an alkali; the NH3 educed is bonding with
H3BO3. The mixture of ammonium metaborates and tetraborates obtained is titrated
with hydrochloric acid solution. Carry out a blank.
NH3 + H3BO3 NH4BO2 + H2O
2NH3 + 4H3BO3 (NH4)2B4O7 + 5H2O
NH4BO2 + HCl + H2O NH4Cl + H3BO3
(NH4)2B4O7 + 5H2O + 2HCl 2NH4Cl + 4H3BO3
C NH2
O
OHOH
COOCH325% NH3
+ CH3OH
C NH2
O
OH
30% NaOHC
O
OH
ONa
+ NH3
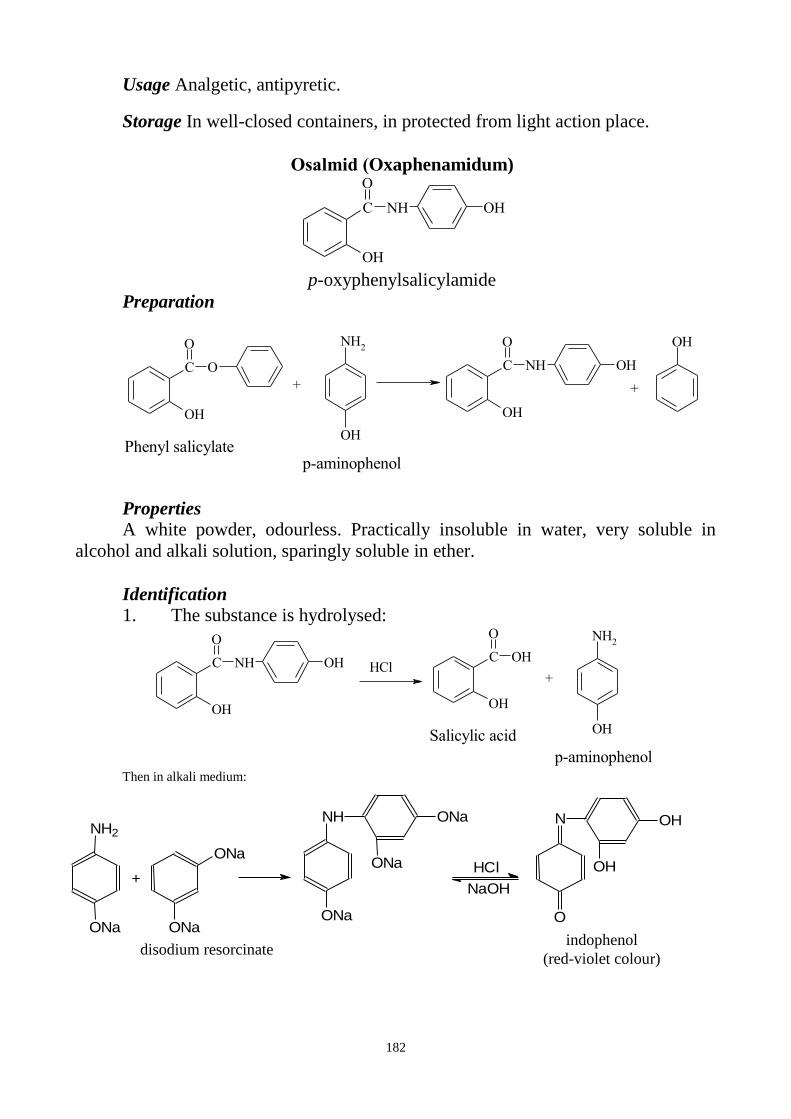
182
Usage Analgetic, antipyretic.
Storage In well-closed containers, in protected from light action place.
Osalmid (Оxaphenamidum)
C
OH
O
NH OH
p-oxyphenylsalicylamide
Preparation
C
OH
O
O
NH2
OH
C
OH
O
NH OH
OH
Phenyl salicylate
+
p-aminophenol
+
Properties
A white powder, odourless. Practically insoluble in water, very soluble in
alcohol and alkali solution, sparingly soluble in ether.
Identification
1. The substance is hydrolysed:
C
OH
O
OH
NH2
OH
C
OH
O
NH OH
Salicylic acid
+
p-aminophenol
HCl
Then in alkali medium:
NH2
ONa
+
ONa
ONa
HCl
NaOH
NH
ONa
ONa
ONa
OH
OHN
O
резорцинат aиндо енол
(красновато-фиолетового цвета)indophenol
(red-violet colour) disodium resorcinate

183
Assay Determination of nitrogen by sulphuric acid digestion (Kjeldahl
method).
Usage Choleretic.
Storage In well-closed containers, protected from light.
Bismuth subgallate (Dermatolum, Bismuthum subgallicum)
Complex of bismuth and gallic acid
Preparation Dissolve bismuth nitrate in acetic acid and add water. Heat the
solution to 30–40С and add the solution of gallic acid.
Properties A yellow powder, practically insoluble in water, in alcohol. It
dissolves in mineral acids with decomposition, and in solutions of alkali hydroxides,
producing a reddish-brown liquid.
Identification 1. Mix the substance with water and add phosphoric acid. Heat to boiling, cool
and filter. Bismuth subphosphate is in precipitate. Filtrate the solution and add FeCl3
solution into filtrate - a black-blue colouring develops (reaction of gallic acid).
2. The precipitate obtained in the test (1) dissolve in nitric acid and carry
out reaction of bismuth with thiourea:
Assay
Complexometry. Heat to boiling with HNO3. Add potassium chlorate, heat to
boiling, add water and heat until the solution becomes colourless (mineralization).
Titrate with 0.1 M sodium edetate until yellow colour is obtained using xylenol
orange triturate as indicator. S=1.
Bi(NO3)3 +
HO
HO
HO
COOH + 2H2O
HO
HO
HO
COOBi
OH
OH
+ 3HNO3
HO
HO
HO
COO
_
Bi(OH)2
yellow-orange colour
Bi3+ + [Bi((NH2)2CS)3]
3+3(NH2)2CS

184
.Bi3+
Ind[ ] +
CH2
CH2CH2 N
N
CH2COONa
CH2COONa
CH2COOH
CH2COOH
CH2
N
N
CH2COO
CH2COO
CH2COO
CH2COOBi Na
-
HNO3
+ H3Ind + NaNO3
+
Usage Antiseptic, astringent.
Storage In well- closed containers, protected from light.
Bismuth subsalicylate (Bismuthi subsalicylas)
Complex of bismuth and salicylic acid
Properties A white powder, practically insoluble in water and in alcohol. It
dissolves in mineral acids with decomposition.
Identification
1. Add HCl , heat and boil for 5 min. Retain the filtrate for identification (2).
Wash the residue with dil. HCl, dissolve the residue in NaOH solution, neutralise
with dil. HCl. The solution gives reaction of salicylates (with FeCl3 - see
Pharmacopoeia 2.3.1).
2. The obtained filtrate (identification 1) gives reaction of bismuth (see bismuth
subgallate).
Assay Complexometry. Dissolve the substance in hot dilute perchloric acid
and titrate with sodium edetate until a yellow colour is obtained using xylenol orange
triturate as indicator (see bismuth subgallate). S=1.
Usage Antiseptic, astringent.
Storage In well-closed containers, protected from light.

185
Chapter 17. MEDICINAL SUBSTANCES DERIVATIVES OF
AROMATIC AMINOACIDS
Plan
1. General characteristic of p-aminobenzoic acid derivatives.
2. Medicinal substances, derivatives of p-aminobenzoic acid (benzocaine,
procaine, procainamide, tetracaine). Preparation, properties, identification, assay,
usage and storage.
3. Medicinal substances, derivatives of p-aminosalicylic acid (sodium p-
aminosalicylate, calcium benzamidosalicylate). Preparation, properties, identification,
assay, usage and storage.
4. Derivatives of o-aminobenzoic (anthranilic) acid (mefenamic acid,
mefenamate sodium).
5. Medicinal substances, derivatives of phenylacetic acid (diclofenac sodium).
As fatty aminoacids the aromatic aminoacids have acidic and basic properties
(acidic properties are more strong):
It is a part of fermentative complex, which is necessary for the growth of
bacteria. Esters of p-aminobenzoic acid have an anaesthetic activity and they are
synthetic substances, which are used instead of cocaine (the first anaesthetic), which
causes drug dependence.
Anaesthetic activity of cocaine is caused by the presence of benzoic acid in its
structure. In general the structure of anaesthetic group is:
Derivatives of p-aminobenzoic acid Benzocaine (Anaesthesinum)
NH2
COOC2H5 Ethyl 4-aminobenzoate
CH2 CH CH C
O
OCH3NH3C
CH CH2CH2C6H5
O
OC
CH
NH2
COOH
N ( C )n , Х=O, S, NH.CO ArХ
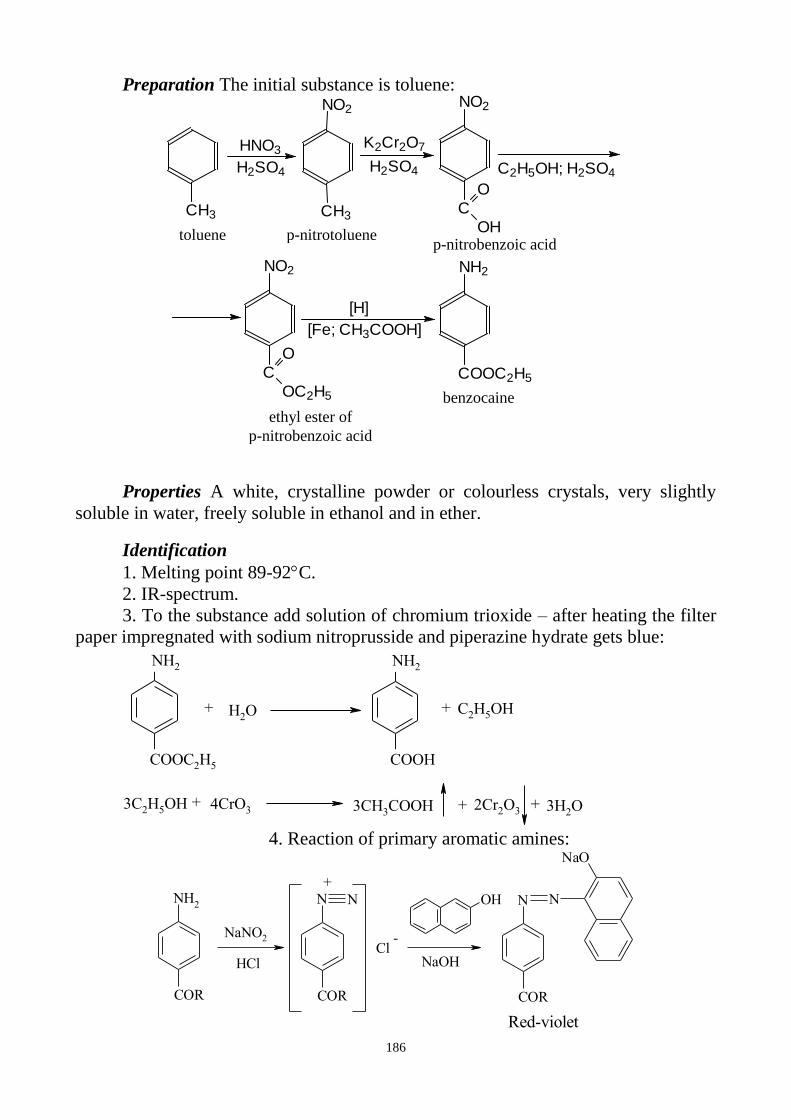
186
Preparation The initial substance is toluene:
Properties A white, crystalline powder or colourless crystals, very slightly
soluble in water, freely soluble in ethanol and in ether.
Identification
1. Melting point 89-92C.
2. IR-spectrum.
3. To the substance add solution of chromium trioxide – after heating the filter
paper impregnated with sodium nitroprusside and piperazine hydrate gets blue:
NH2
COOC2H5
+ H2O
NH2
COOH
+ C2H5OH
3C2H5OH + 4CrO3 3CH3COOH + 2Cr2O3+ 3H2O
4. Reaction of primary aromatic amines:
N
COR
N
N
COR
N
NaO
NH2
COR
OH
Cl
+
-
NaOH
NaNO2
HCl
Red-violet
benzocaine
p-nitrobenzoic acid
CH3
HNO3
H2SO4
CH3
NO2
K2Cr2O7
H2SO4
NO2
C
O
OH
C2H5OH; H2SO4
NO2
C
O
OC2H5
[H]
[Fe; CH3COOH]
NH2
COOC2H5
толуол п-нитротолуол
этиловый эфир п-нитробензойнойкислоты
toluene p-nitrotoluene
ethyl ester of
p-nitrobenzoic acid
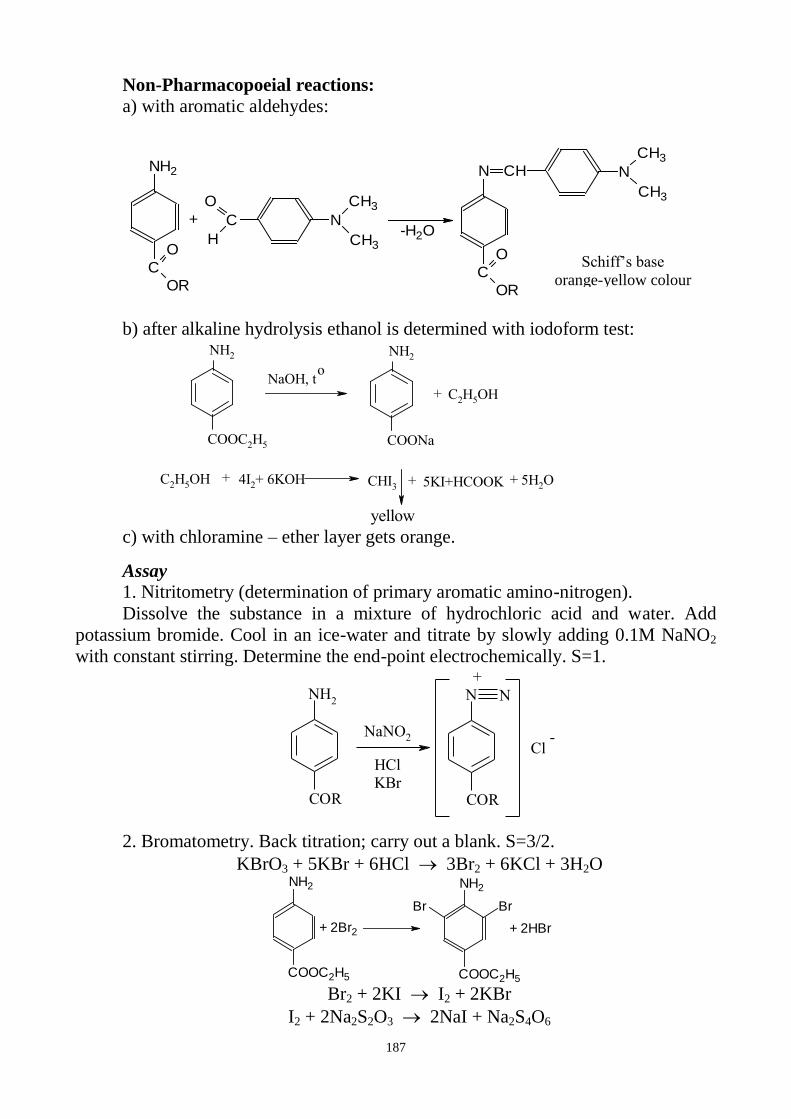
187
Non-Pharmacopoeial reactions:
a) with aromatic aldehydes:
b) after alkaline hydrolysis ethanol is determined with iodoform test:
c) with chloramine – ether layer gets orange.
Assay
1. Nitritometry (determination of primary aromatic amino-nitrogen).
Dissolve the substance in a mixture of hydrochloric acid and water. Add
potassium bromide. Cool in an ice-water and titrate by slowly adding 0.1M NaNO2
with constant stirring. Determine the end-point electrochemically. S=1.
N
COR
N
NH2
COR
Cl
+
-NaNO2
HClKBr
2. Bromatometry. Back titration; carry out a blank. S=3/2.
KBrO3 + 5KBr + 6HCl 3Br2 + 6KCl + 3H2O
Br2 + 2KI I2 + 2KBr
I2 + 2Na2S2O3 2NaI + Na2S4O6
желто-оранжевого цветаоснование Шиффа
N
CH3
CH3
C
O
OR
N CH
-H2ON
CH3
CH3
C
O
H
+
C
O
OR
NH2
Schiff’s base
orange-yellow colour
NH2
COOC2H5
+ 2Br2
BrBr
NH2
COOC2H5
+ 2HBr
NH2
COOC2H5
NH2
COONa
+ C2H5OH
C2H5OH + 4I2+ 6KOH CHI3+ + 5H2O
NaOH, to
5KI+HCOOK
yellow
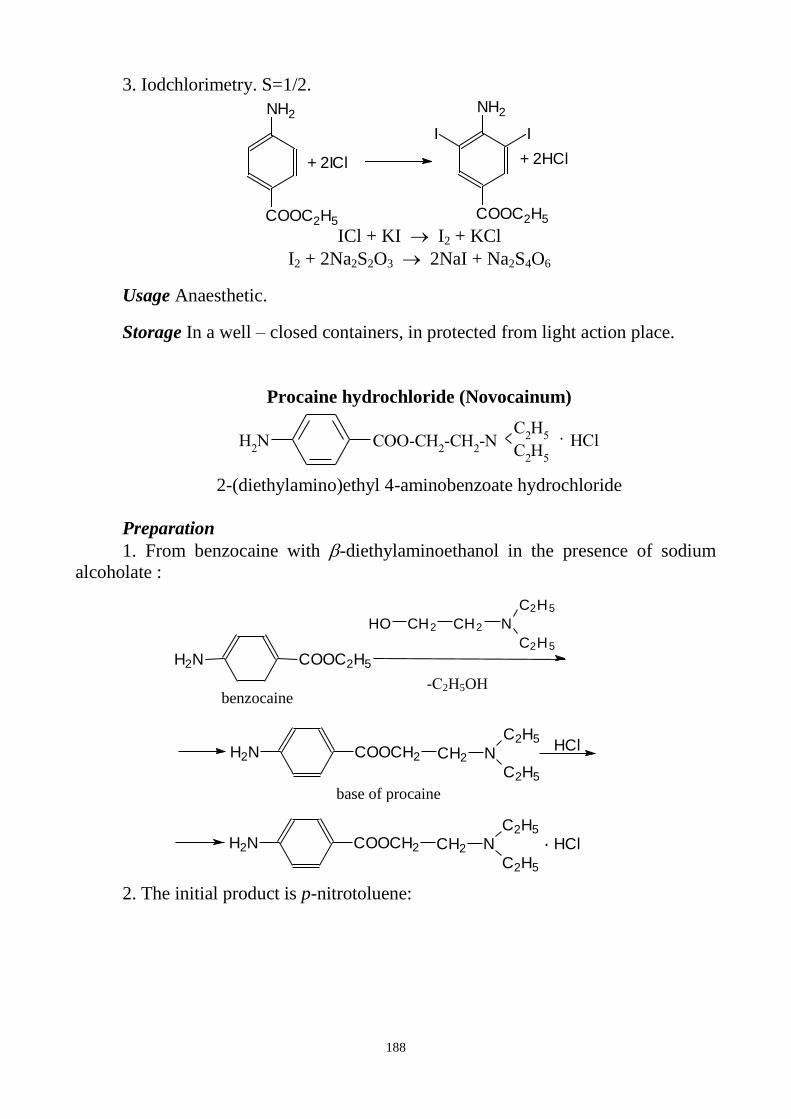
188
3. Iodchlorimetry. S=1/2.
ICl + KI I2 + KCl
I2 + 2Na2S2O3 2NaI + Na2S4O6
Usage Anaesthetic.
Storage In a well – closed containers, in protected from light action place.
Procaine hydrochloride (Novocainum)
NH2
COO-CH2-CH
2-N
C2H
5
C2H
5
. HCl
2-(diethylamino)ethyl 4-aminobenzoate hydrochloride
Preparation
1. From benzocaine with -diethylaminoethanol in the presence of sodium
alcoholate :
2. The initial product is p-nitrotoluene:
NH2
COOC2H5
+ 2ICl
NH2
COOC2H5
II
+ 2HCl
HCl
H2N COOC2H5
HO CH2 CH2 N
C2H5
C2H5
H2N COOCH2 CH2 N
C2H5
C2H5
CH2 N
C2H5
C2H5
H2N COOCH2 . HCl
анестезин
снование новокаина
benzocaine
base of procaine
-С2H5OH
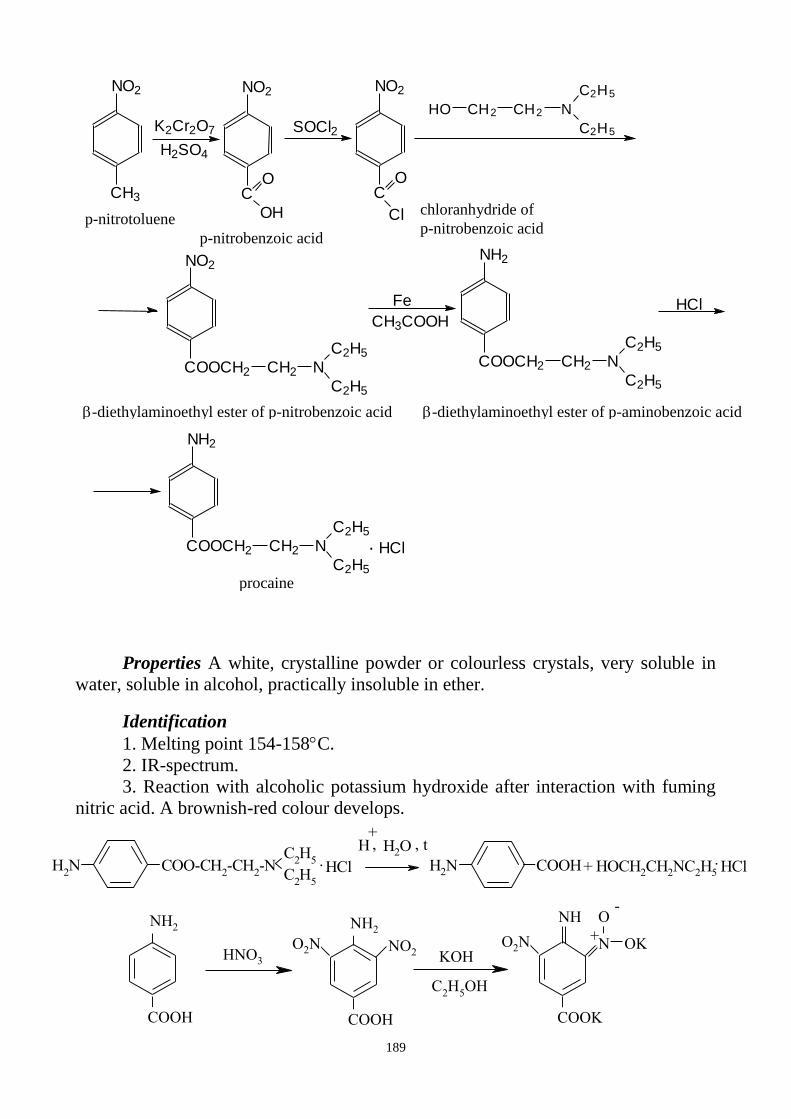
189
Properties A white, crystalline powder or colourless crystals, very soluble in
water, soluble in alcohol, practically insoluble in ether.
Identification
1. Melting point 154-158C.
2. IR-spectrum.
3. Reaction with alcoholic potassium hydroxide after interaction with fuming
nitric acid. A brownish-red colour develops.
C2H
5OH
NH2
COOH
HNO3
NH2
COOH
O2N NO2 KOH
COOK
O2N N+
-
OK
NH O
новокаин
п-аминобензойнойензойной
нитробензойной кислотыкислота
п-нитро ензо наяп-нитротолуол
. HClCOOCH2 CH2 N
C2H5
C2H5
NH2
HCl
COOCH2 CH2 N
C2H5
C2H5
NH2
CH3COOH
Fe
NO2
COOCH2 CH2 N
C2H5
C2H5
HO CH2 CH2 N
C2H5
C2H5
NO2
CO
Cl
SOCl2
NO2
CO
OH
H2SO4
K2Cr2O7
NO2
CH3
p-nitrotoluene
procaine
-diethylaminoethyl ester of p-nitrobenzoic acid
chloranhydride of
p-nitrobenzoic acid p-nitrobenzoic acid
-diethylaminoethyl ester of p-aminobenzoic acid
NH2
COO-CH2-CH
2-N
C2H
5
C2H
5
H H2O, , t
H2N COOH HOCH2CH2NC2H5+ .HCl
+
. HCl
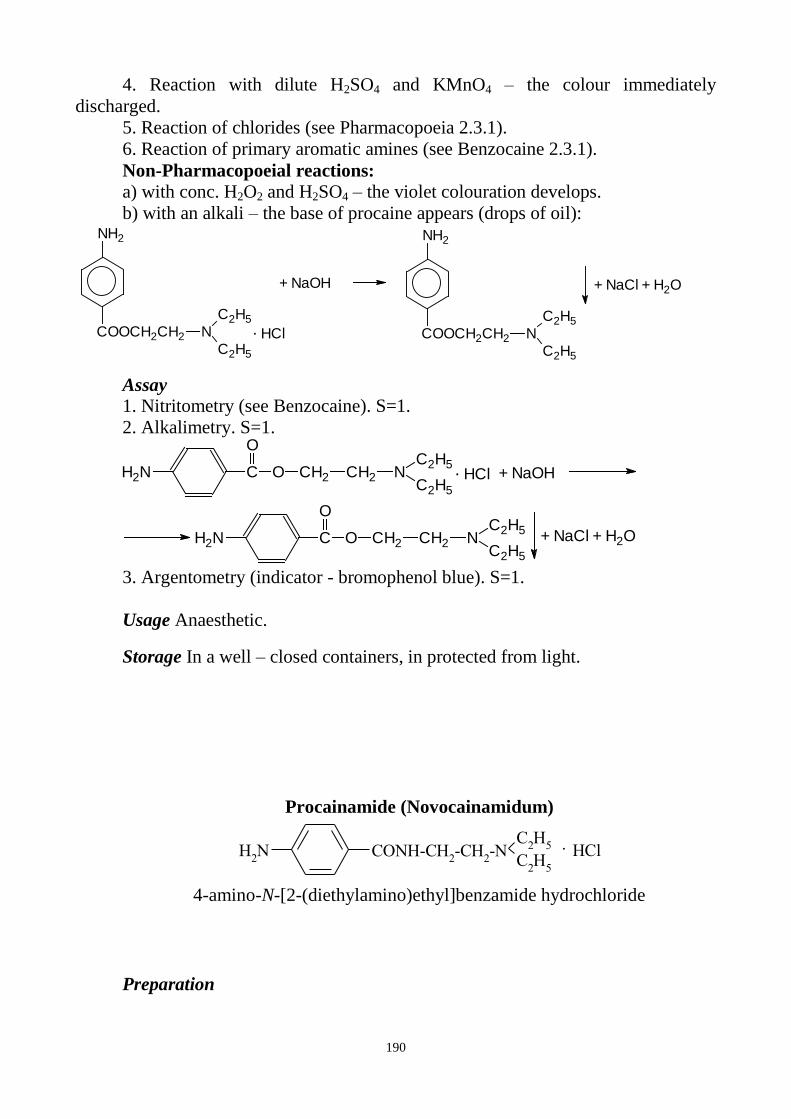
190
4. Reaction with dilute H2SO4 and KMnO4 – the colour immediately
discharged.
5. Reaction of chlorides (see Pharmacopoeia 2.3.1).
6. Reaction of primary aromatic amines (see Benzocaine 2.3.1).
Non-Pharmacopoeial reactions:
a) with conc. H2O2 and H2SO4 – the violet colouration develops.
b) with an alkali – the base of procaine appears (drops of oil):
Assay
1. Nitritometry (see Benzocaine). S=1.
2. Alkalimetry. S=1.
3. Argentometry (indicator - bromophenol blue). S=1.
Usage Anaesthetic.
Storage In a well – closed containers, in protected from light.
Procainamide (Novocainamidum)
NH2
CONH-CH2-CH
2-N
C2H
5
C2H
5
. HCl
4-amino-N-[2-(diethylamino)ethyl]benzamide hydrochloride
Preparation
NH2
COOCH2CH2 N
C2H5
C2H5
NH2
COOCH2CH2 N
C2H5
C2H5
+ NaCl + H2O
. HCl
+ NaOH
+ NaCl + H2OH2N C O CH2 CH2 NC2H5
C2H5
O
. HClH2N C O CH2 CH2 NC2H5
C2H5
O
+ NaOH
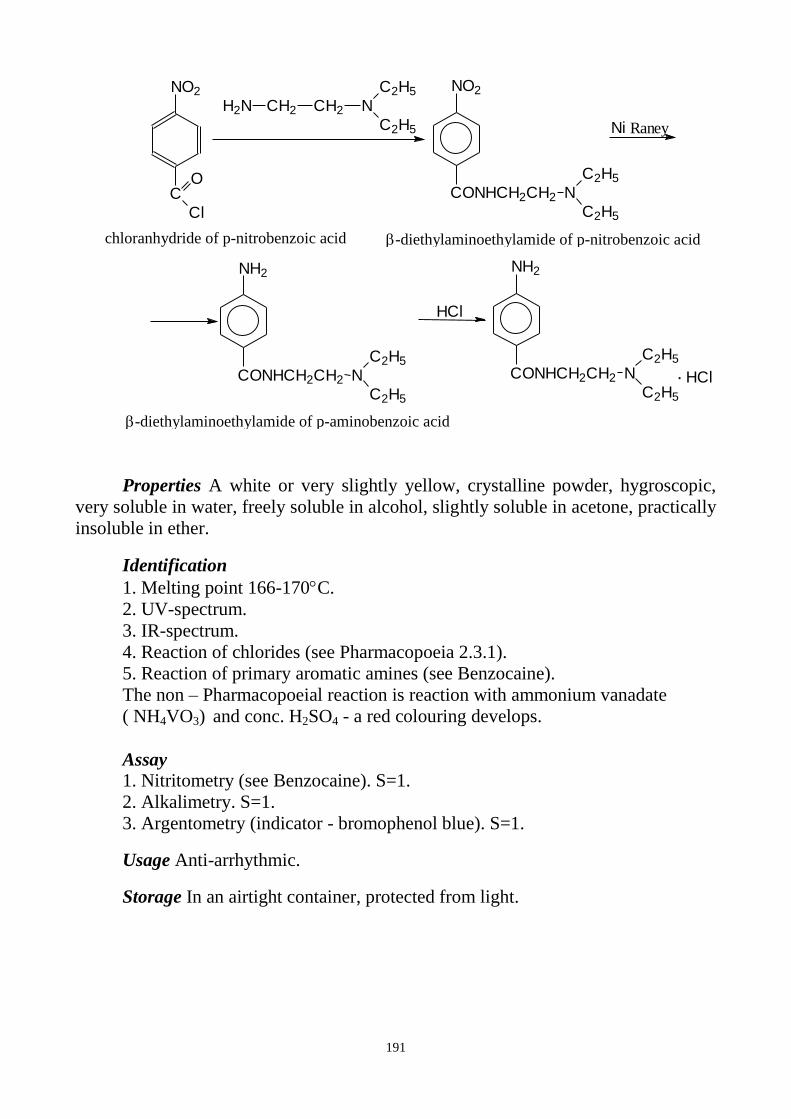
191
Properties A white or very slightly yellow, crystalline powder, hygroscopic,
very soluble in water, freely soluble in alcohol, slightly soluble in acetone, practically
insoluble in ether.
Identification
1. Melting point 166-170C.
2. UV-spectrum.
3. IR-spectrum.
4. Reaction of chlorides (see Pharmacopoeia 2.3.1).
5. Reaction of primary aromatic amines (see Benzocaine).
The non – Pharmacopoeial reaction is reaction with ammonium vanadate
( NH4VO3) and conc. H2SO4 - a red colouring develops.
Assay
1. Nitritometry (see Benzocaine). S=1.
2. Alkalimetry. S=1.
3. Argentometry (indicator - bromophenol blue). S=1.
Usage Anti-arrhythmic.
Storage In an airtight container, protected from light.
-диэтиламиноэтиламид
-диэтиламиноэтиламид
п-аминобензойной кислоты
-нит обензойной кислотып-нитробензойной кислоты
CONHCH2CH2
C2H5
C2H5
N
NH2
HCl
CONHCH2CH2
C2H5
C2H5
N
NH2
Ni
NO2
CONHCH2CH2
C2H5
C2H5
N
H2N CH2 CH2 N
C2H5
C2H5
NO2
CO
Cl
. HCl
Raney
-diethylaminoethylamide of p-aminobenzoic acid
-diethylaminoethylamide of p-nitrobenzoic acid chloranhydride of p-nitrobenzoic acid
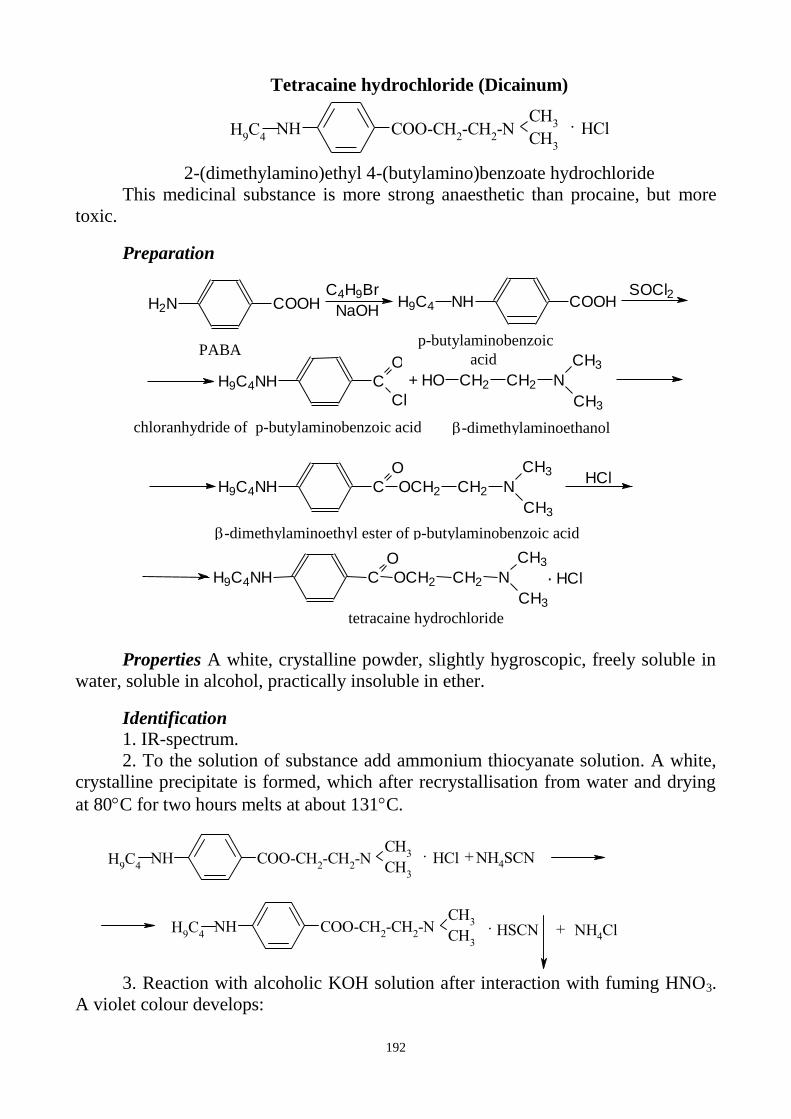
192
Tetracaine hydrochloride (Dicainum)
COO-CH2-CH
2-NNH
CH3
CH3
H9C
4. HCl
2-(dimethylamino)ethyl 4-(butylamino)benzoate hydrochloride
This medicinal substance is more strong anaesthetic than procaine, but more
toxic.
Preparation
Properties A white, crystalline powder, slightly hygroscopic, freely soluble in
water, soluble in alcohol, practically insoluble in ether.
Identification
1. IR-spectrum.
2. To the solution of substance add ammonium thiocyanate solution. A white,
crystalline precipitate is formed, which after recrystallisation from water and drying
at 80C for two hours melts at about 131C.
3. Reaction with alcoholic KOH solution after interaction with fuming HNO3.
A violet colour develops:
H2N COOH NaOH
C4H9BrCOOHNHH9C4
SOCl2
C
O
Cl
H9C4NH + HO CH2 CH2 N
CH3
CH3
CH9C4NH OCH2 CH2
O
N
CH3
CH3
HCl
N
CH3
CH3
CH9C4NH OCH2 CH2
O. HCl
ПАБК ензо
диметиламиноэтиловый эфир п-бутиламинобензойной кислоты
п-бутиламинобензойной кислотыхлорангидрид диметиламиноэтанол
дикаин
-dimethylaminoethyl ester of p-butylaminobenzoic acid
chloranhydride of p-butylaminobenzoic acid -dimethylaminoethanol
p-butylaminobenzoic
acid PABA
tetracaine hydrochloride
COO-CH2-CH
2-NNH
CH3
CH3
H9C
4
COO-CH2-CH
2-NNH
CH3
CH3
H9C
4
+ NH4SCN
. HSCN + NH4Cl
. HCl
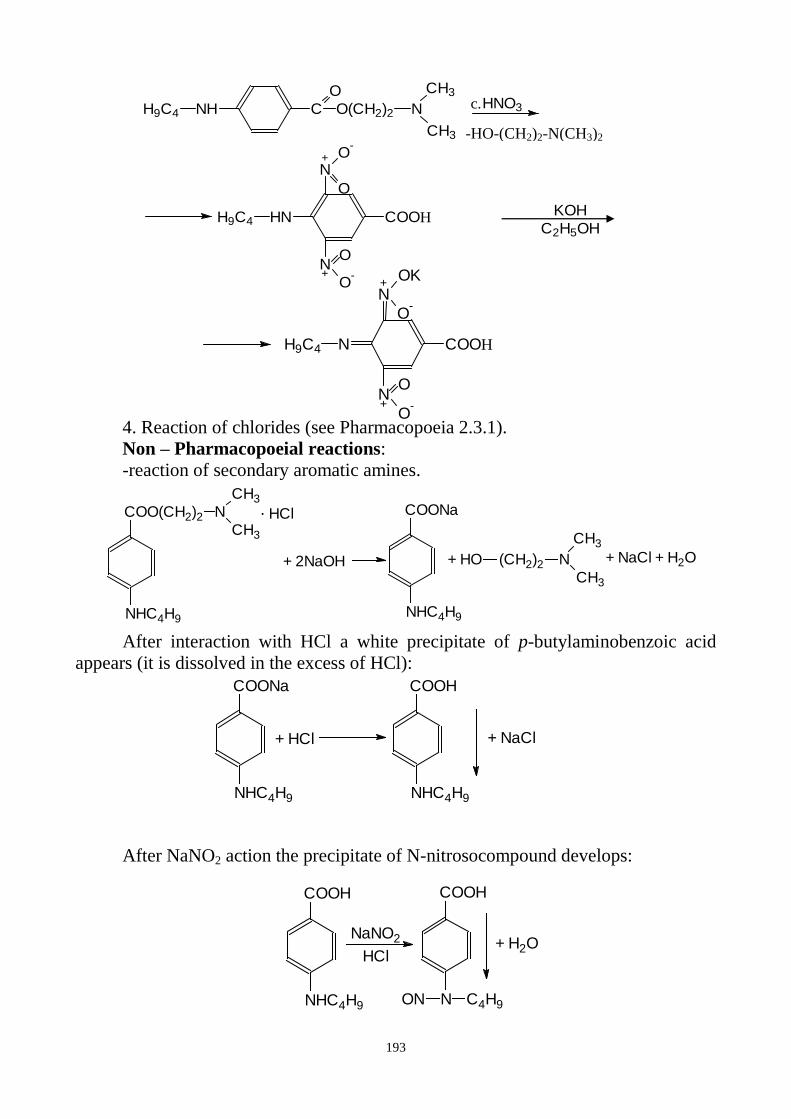
193
4. Reaction of chlorides (see Pharmacopoeia 2.3.1).
Non – Pharmacopoeial reactions:
-reaction of secondary aromatic amines.
After interaction with HCl a white precipitate of p-butylaminobenzoic acid
appears (it is dissolved in the excess of HCl):
After NaNO2 action the precipitate of N-nitrosocompound develops:
NHC4H9
COO(CH2)2 . HCl
+ 2NaOH + NaCl + H2O
NHC4H9
COONa
+ HO (CH2)2 N
CH3
CH3
N
CH3
CH3
NHC4H9
COOH
NaNO2
HCl
COOH
NON C4H9
+ H2O
+ NaCl
NHC4H9
COOH
+ HCl
NHC4H9
COONa
-HO-(CH2)2-N(CH3)2
NHH9C4 C O(CH2)2 N
CH3
CH3
OHNO3
HNH9C4
N
N
O
O-
O-
O
COOH
+
+
KOH
C2H5OH
c.
N
N
O -
O
C O O H
O K
O -
N H 9 C 4
+
+
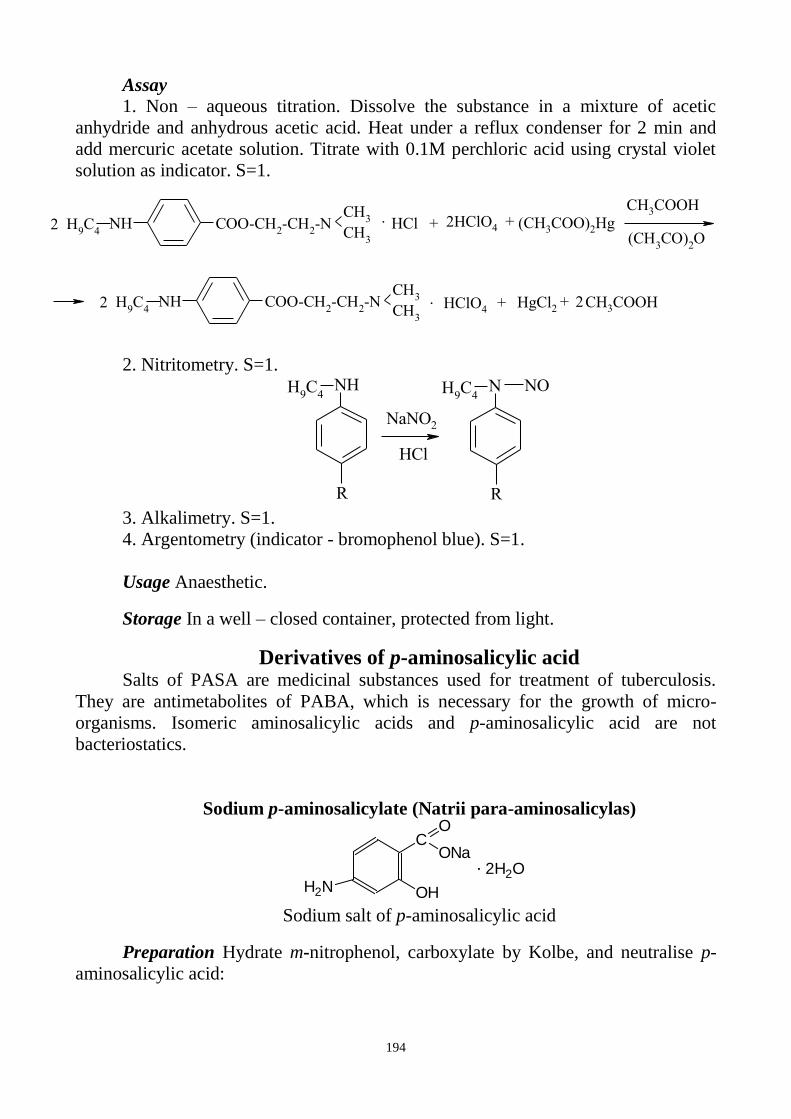
194
Assay
1. Non – aqueous titration. Dissolve the substance in a mixture of acetic
anhydride and anhydrous acetic acid. Heat under a reflux condenser for 2 min and
add mercuric acetate solution. Titrate with 0.1M perchloric acid using crystal violet
solution as indicator. S=1.
2. Nitritometry. S=1. NH
R
H9C
4N
R
H9C
4NO
NaNO2
HCl
3. Alkalimetry. S=1.
4. Argentometry (indicator - bromophenol blue). S=1.
Usage Anaesthetic.
Storage In a well – closed container, protected from light.
Derivatives of p-aminosalicylic acid Salts of PASA are medicinal substances used for treatment of tuberculosis.
They are antimetabolites of PABA, which is necessary for the growth of micro-
organisms. Isomeric aminosalicylic acids and p-aminosalicylic acid are not
bacteriostatics.
Sodium p-aminosalicylate (Natrii para-aminosalicylas)
Sodium salt of p-aminosalicylic acid
Preparation Hydrate m-nitrophenol, carboxylate by Kolbe, and neutralise p-
aminosalicylic acid:
H2N
CO
ONa
OH
. 2H2O
COO-CH2-CH
2-NNH
CH3
CH3
H9C
4
COO-CH2-CH
2-NNH
CH3
CH3
H9C
4
(CH3CO)
2O
+ 2HClO4 + (CH3COO)2Hg
. HClO4+ HgCl2
+ CH3COOH2
2
CH3COOH. HCl
2
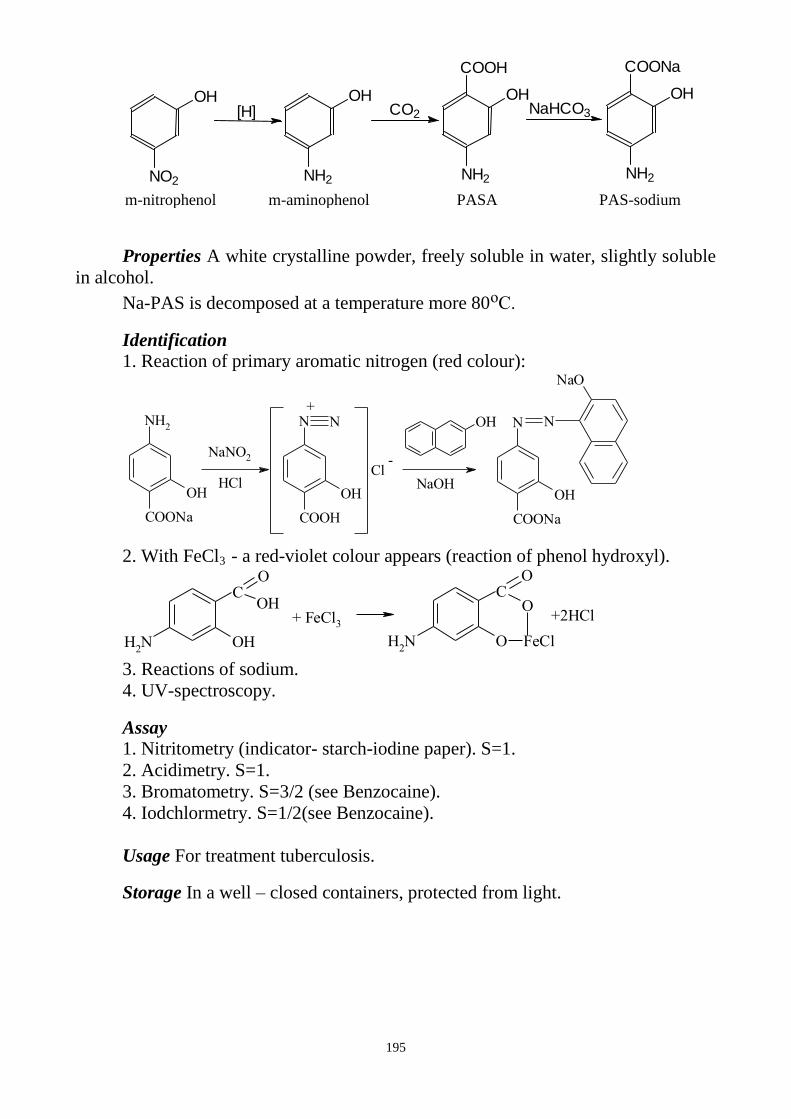
195
Properties A white crystalline powder, freely soluble in water, slightly soluble
in alcohol.
Na-PAS is decomposed at a temperature more 80оС.
Identification
1. Reaction of primary aromatic nitrogen (red colour):
N
OH
COOH
N
N
COONa
OH
N
NaO
NH2
COONa
OH
OH
Cl
+
-
NaOH
NaNO2
HCl
2. With FeCl3 - a red-violet colour appears (reaction of phenol hydroxyl).
OHNH2
CO
OH
O FeClNH2
CO
O+ FeCl3
+2HCl
3. Reactions of sodium.
4. UV-spectroscopy. Assay
1. Nitritometry (indicator- starch-iodine paper). S=1.
2. Acidimetry. S=1.
3. Bromatometry. S=3/2 (see Benzocaine).
4. Iodchlormetry. S=1/2(see Benzocaine).
Usage For treatment tuberculosis.
Storage In a well – closed containers, protected from light.
m-nitrophenol PASA PAS-sodium m-aminophenol
OH
NO2
[H]
NH2
OHCO2
NH2
OH
COOH
NaHCO3
NH2
OH
COONa
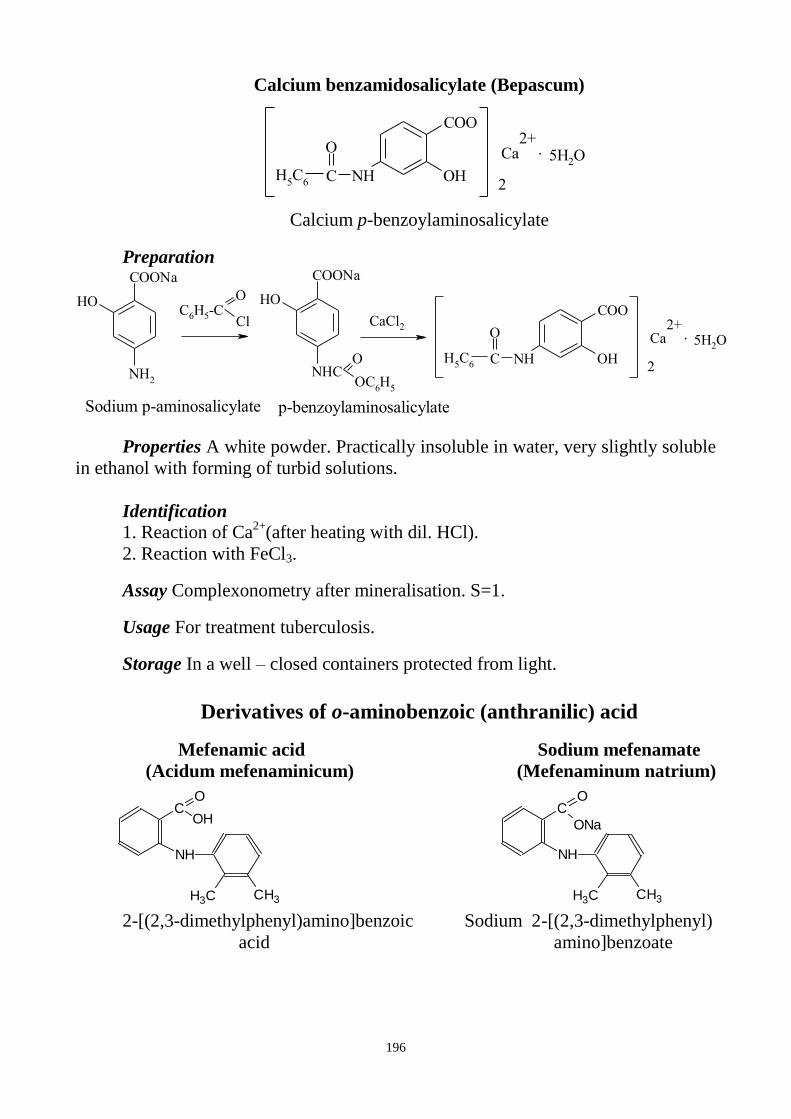
196
Calcium benzamidosalicylate (Bepascum)
COO
OHNHC
O
H5C
6 2
Ca2+
. 5H2O
Calcium p-benzoylaminosalicylate
Preparation
NH2
COONa
OH O
Cl
NHC
COONa
OH
O
OC6H
5
COO
OHNHC
O
H5C
6
C6H5-C
Sodium p-aminosalicylate p-benzoylaminosalicylate
CaCl2
2
Ca2+
. 5H2O
Properties A white powder. Practically insoluble in water, very slightly soluble
in ethanol with forming of turbid solutions.
Identification
1. Reaction of Ca2+
(after heating with dil. HCl).
2. Reaction with FeCl3.
Assay Complexonometry after mineralisation. S=1.
Usage For treatment tuberculosis.
Storage In a well – closed containers protected from light.
Derivatives of o-aminobenzoic (anthranilic) acid
Mefenamic acid Sodium mefenamate
(Acidum mefenaminicum) (Mefenaminum natrium)
2-[(2,3-dimethylphenyl)amino]benzoic Sodium 2-[(2,3-dimethylphenyl)
acid amino]benzoate
CO
NH
CH3H3C
ONa
CO
NH
CH3H3C
OH
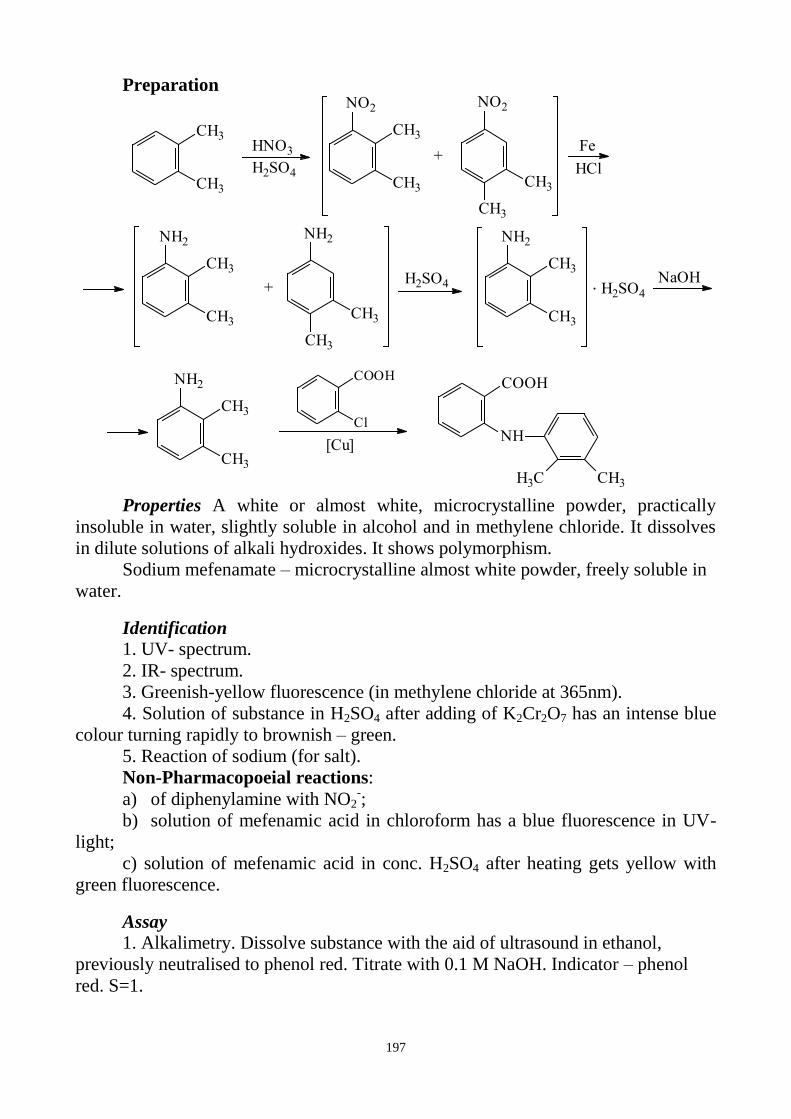
197
Preparation
Properties A white or almost white, microcrystalline powder, practically
insoluble in water, slightly soluble in alcohol and in methylene chloride. It dissolves
in dilute solutions of alkali hydroxides. It shows polymorphism.
Sodium mefenamate – microcrystalline almost white powder, freely soluble in
water.
Identification
1. UV- spectrum.
2. IR- spectrum.
3. Greenish-yellow fluorescence (in methylene chloride at 365nm).
4. Solution of substance in H2SO4 after adding of K2Cr2O7 has an intense blue
colour turning rapidly to brownish – green.
5. Reaction of sodium (for salt).
Non-Pharmacopoeial reactions:
a) of diphenylamine with NO2-;
b) solution of mefenamic acid in chloroform has a blue fluorescence in UV-
light;
c) solution of mefenamic acid in conc. H2SO4 after heating gets yellow with
green fluorescence.
Assay
1. Alkalimetry. Dissolve substance with the aid of ultrasound in ethanol,
previously neutralised to phenol red. Titrate with 0.1 M NaOH. Indicator – phenol
red. S=1.
NaOH. H2SO4
CH3
CH3
NH2
H2SO4+
CH3
NH2
CH3
CH3
CH3
NH2
COOH
NH
CH3H3C
CH3
CH3
NH2COOH
Cl
[Cu]
HCl
Fe
CH3
NO2
CH3
+
CH3
CH3
NO2
H2SO4
HNO3
CH3
CH3

198
2. Non-aqueous titration. Solvent – dimethylformamide, titrate with sodium
hydroxide in a mixture of methanol and benzol. Indicator – thymol blue. S=1.
Sodium mefenamate is quantified by gravimetry.
Usage Anti-inflammatory, analgesic.
Because it is believed that aspirin and amino-pyrine owe their general purpose
analgesic efficacy to a combination of peripheral and central effects, a wide variety of
arylanthranilic acids were screened for antinociceptive (analgesic) activity if they
showed significant anti-inflammatory action. It has become evident that the
combination of both effects is a rarity among these compounds.
Storage In a well – closed container.
Derivatives of phenylacetic acid
Diclofenac sodium (Diclofenac-Natrium)
Sodium 2-[(2,6-dichlorophenyl)amino]phenyl]acetate
Preparation The initial product is bromobenzene:
Properties A white or slightly yellowish, crystalline powder, slightly
hygroscopic, sparingly soluble in water, freely soluble in methanol, soluble in
alcohol, slightly soluble in acetone.
Identification
1. IR-spectrum.
2. Thin-layer chromatography.
CH2 C
O
ONa
NH
Cl
Cl
NH
CH3H3C
COOH
NaOHNH
CH3H3C
COONa
+ H2O
COONa
NH
ClCl
Br +
ClCl
NH
CO
CH3
N
ClCl
ONaOH
tº
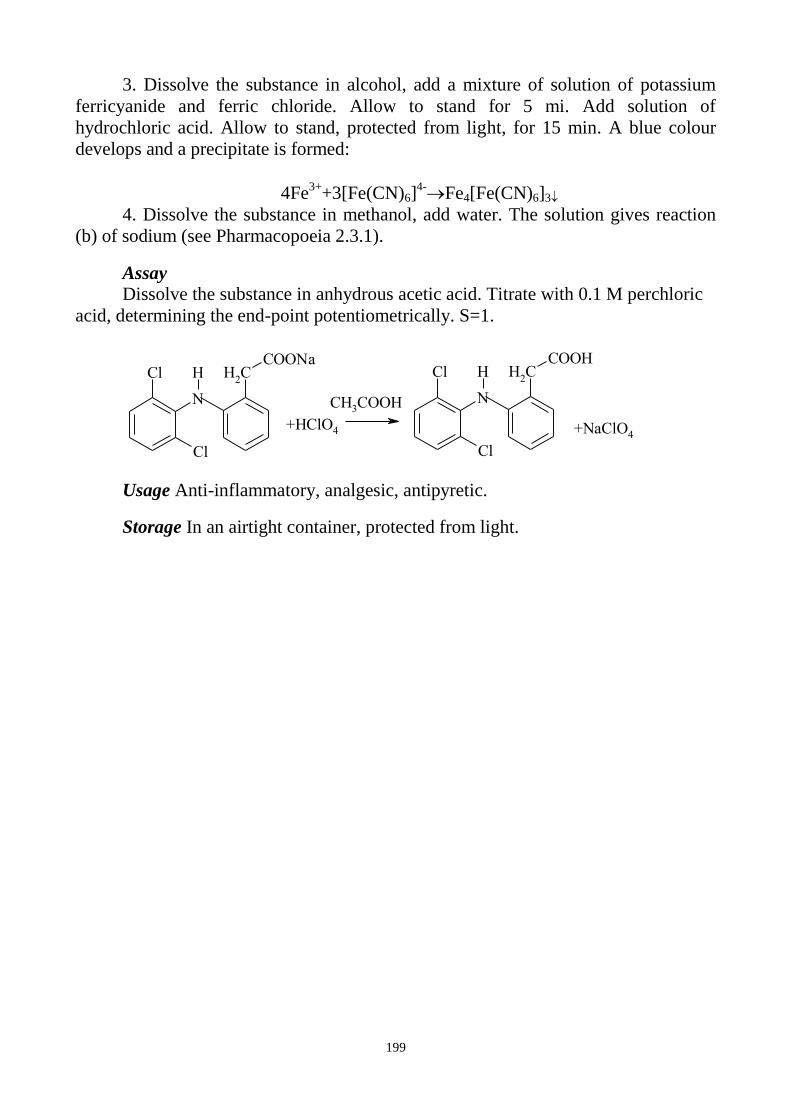
199
3. Dissolve the substance in alcohol, add a mixture of solution of potassium
ferricyanide and ferric chloride. Allow to stand for 5 mi. Add solution of
hydrochloric acid. Allow to stand, protected from light, for 15 min. A blue colour
develops and a precipitate is formed:
4Fe3+
+3[Fe(CN)6]4-Fe4[Fe(CN)6]3
4. Dissolve the substance in methanol, add water. The solution gives reaction
(b) of sodium (see Pharmacopoeia 2.3.1).
Assay
Dissolve the substance in anhydrous acetic acid. Titrate with 0.1 M perchloric
acid, determining the end-point potentiometrically. S=1.
Cl
N
Cl
H CH2
COONaCl
N
Cl
H CH2
COOH
+HClO4 +NaClO4
CH3COOH
Usage Anti-inflammatory, analgesic, antipyretic.
Storage In an airtight container, protected from light.
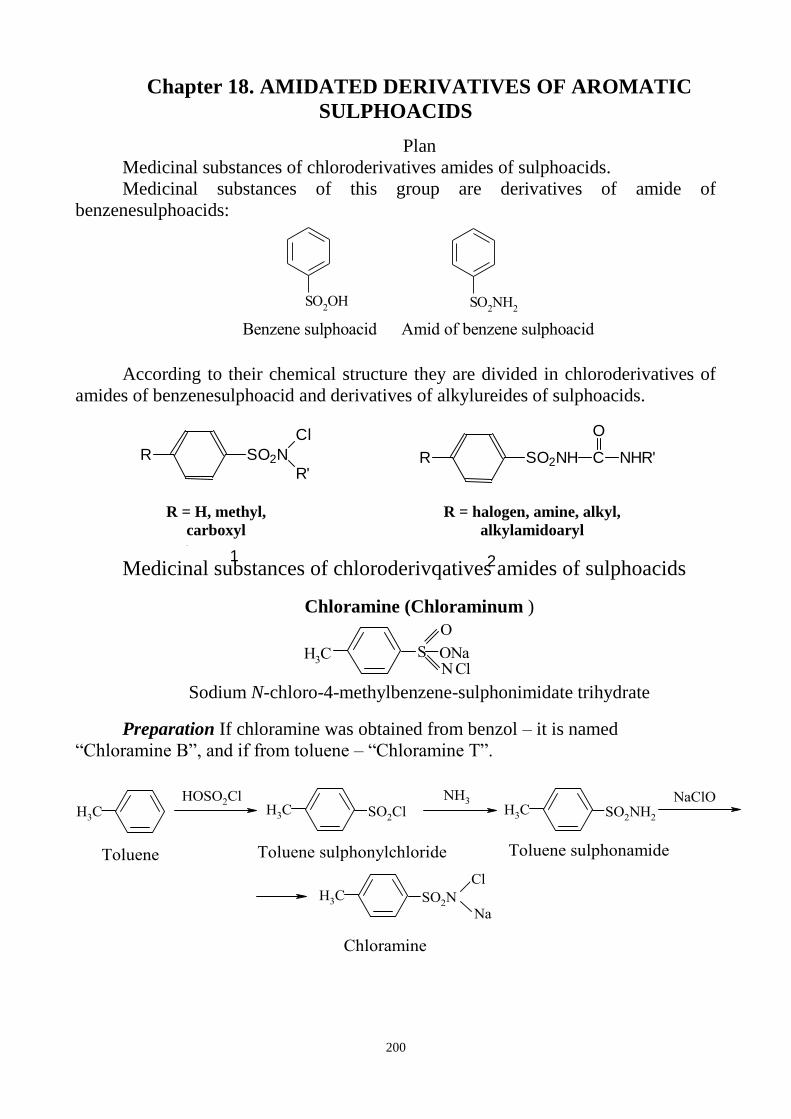
200
Chapter 18. AMIDATED DERIVATIVES OF AROMATIC
SULPHOACIDS
Plan
Medicinal substances of chloroderivatives amides of sulphoacids.
Medicinal substances of this group are derivatives of amide of
benzenesulphoacids:
SO2OH SO
2NH
2
Benzene sulphoacid Amid of benzene sulphoacid
According to their chemical structure they are divided in chloroderivatives of
amides of benzenesulphoacid and derivatives of alkylureides of sulphoacids.
Medicinal substances of chloroderivqatives amides of sulphoacids
Chloramine (Chloraminum )
Sodium N-chloro-4-methylbenzene-sulphonimidate trihydrate
Preparation If chloramine was obtained from benzol – it is named
“Chloramine B”, and if from toluene – “Chloramine T”.
H3CHOSO2Cl
H3C SO2ClNH3
H3C SO2NH2
NaClO
H3C SO2N
Cl
Na
Toluene Toluene sulphonylchloride Toluene sulphonamide
Chloramine
H3C S
O
ONaN Cl
R SO2N
Cl
R'
R'=Na, Cl
R=H, COOH R= CH3, Cl, NH2
R SO2NH C NHR'
O
R'= C4H9, C3H7
1 2
R = H, methyl,
carboxyl
R=Na, -Cl
R = halogen, amine, alkyl,
alkylamidoaryl
R = alkyl, cycloalkyl
201
During synthesis NaClO can educe NaOH, which changes direction of
reaction:
Properties A white or slightly yellow, crystalline powder. Freely soluble in
water, soluble in ethanol, practically insoluble in chloroform and in ether.
Identification
1. The solution of substance turns red litmus paper blue and then bleaches:
2. To the solution of substance add dilute H2O2. A white precipitate is formed
which dissolves on heating. White crystals melt at 137-140C.
3. Ignite the substance (cautiously, because of risk of deflagration). Dissolve
the residue in water – the solution gives reaction of chlorides (see Pharmacopoeia
2.3.1).
4. Ignite the substance (cautiously, because of risk of deflagration). Dissolve
the residue in water – the solution gives reaction of sodium (see Pharmacopoeia
2.3.1).
5. Ignite the substance (cautiously, because of risk of deflagration). Dissolve
the residue in water – the solution gives reaction of sulphates (see Pharmacopoeia
2.3.1).
Non-Pharmacopoeial reaction
In the presence of hydrochloric acid the free chlorine is educed:
Cl2 is determined with potassium iodide in the presence of chloroform
(chloroform layer gets violet):
SO2N
Cl
Na
+ HCl SO2NH2 + Cl2 + NaCl
Cl2 + 2KI I2 + 2KCl
H3C S
O
ONaNCl
+ H2O2 H3C SO2NH2O2
+NaCl +
H3C SO2NH2+ NaOH H3C SO2N
Na
H+ H2O
NaClO+H2O NaOH+HClO
H3C SO2NNa
ClH2O+ +H3C SO2NH2 NaClO
NaClO+H2O NaOH+HClO
HClO HCl+[O]
202
Assay
Iodometry. Dissolve substance in water, add KI and dilute H2SO4. Titrate with
sodium thiosulphate, using starch solution as indicator. S=1/2.
Usage Antiseptic, disinfectant.
Storage In an airtight container, protected from light at temperature of 8 to
15C.
Pantocidum
N-dichloro-p-carboxybenzolsulphamide
Properties A white crystalline powder with a weak smell of chlorine. Very
slightly soluble in water and in dilute acids, freely soluble in alkali solutions.
Identification
1. It turns red an alkaline methylene red solution, and then bleaches it.
2. By action of HCl it educes Cl2:
Free chlorine is detected with KI solution (see chloramine).
Assay
Iodometry, direct titration. The substance is dissolved in an alkali solution, KI
and the excess of H2SO4 are added. The I2 educed is titrated with sodium thiosulphate
using the starch solution as an indicator. S=1/4.
C SO2N
Cl
Cl
O
HO
C SO2N
Cl
Cl
O
HO
+ 2HCl C
O
HO
SO2NH2 + 2Cl2
H3C S
O
ONaN Cl
+ +2KI H2SO4 H3C SO2NH2+ NaClI2
K2SO4+ +
I2 + 2Na2S2O3 2NaI + Na2S4O6
I2 + 2Na2S2O3 2NaI + Na2S4O6
+ 2K2SO4SO2NH22HOOC+
+ 8KI + 3H2SO4SO2N
Cl
Cl
2NaOOC 4I2 + Na2SO4 + 4KCl +
203
The content of free chlorine – not less 50%.
Usage Antiseptic.
Storage In a well-stoppered bottles, protected from light, in a cool, dry place.
Medicinal substances derivatives of alkylureides of sulphoacids
Butamide (Butamidum)
Tolbutamide (Tolbutamidum)
N-(p-methylbenzolsulphonyl)-N`-butyl urea
Properties A white, crystalline powder, odourless. Practically insoluble in
water, soluble in alcohol, freely soluble in acetone and in chloroform.
Preparation
Identification
1. After alkaline hydrolysis and on heating it educes ammonium (specific
smell; red litmus paper gets blue). The smell of butylamine appears:
2. On long-term heating in the presence of 50% sulphuric acid (with a reflux
condenser) and neutralising the precipitate of p-toluene sulphamide (melting point of
135-138оС) appears.
H3C SO2 NH C NH C4H9
O
H3C SO2NH C
O
NHC4H9
H2O
H2SO4H3C SO2NH2
+ CO2 + C4H9NH2
H3CHOSO2Cl
H3C SO2ClNH3
H3C SO2NH2
NaOH
C2H5OH
SO2NH3C
Na
H
C4H9N C OSO2NH3C
Na
C
O
NH C4H9
HCl
толуол п-толуолсульфохлорид п-толуолсульфамид
п-толуолсульфамид-натрий
утилизоциана
натрий-бутамид
бутамид
H3C SO2NH C
O
NHC4H9
toluene p-toluenesulphochloride p-toluenesulphamide
sodium p-toluenesulphamide
butylisocyanate
sodium butamide
butamide
NH3
++ C4H9 NH2H3C SO2OK+ 3KOHH3C SO2NH C
O
NHC4H9
+ K2CO3
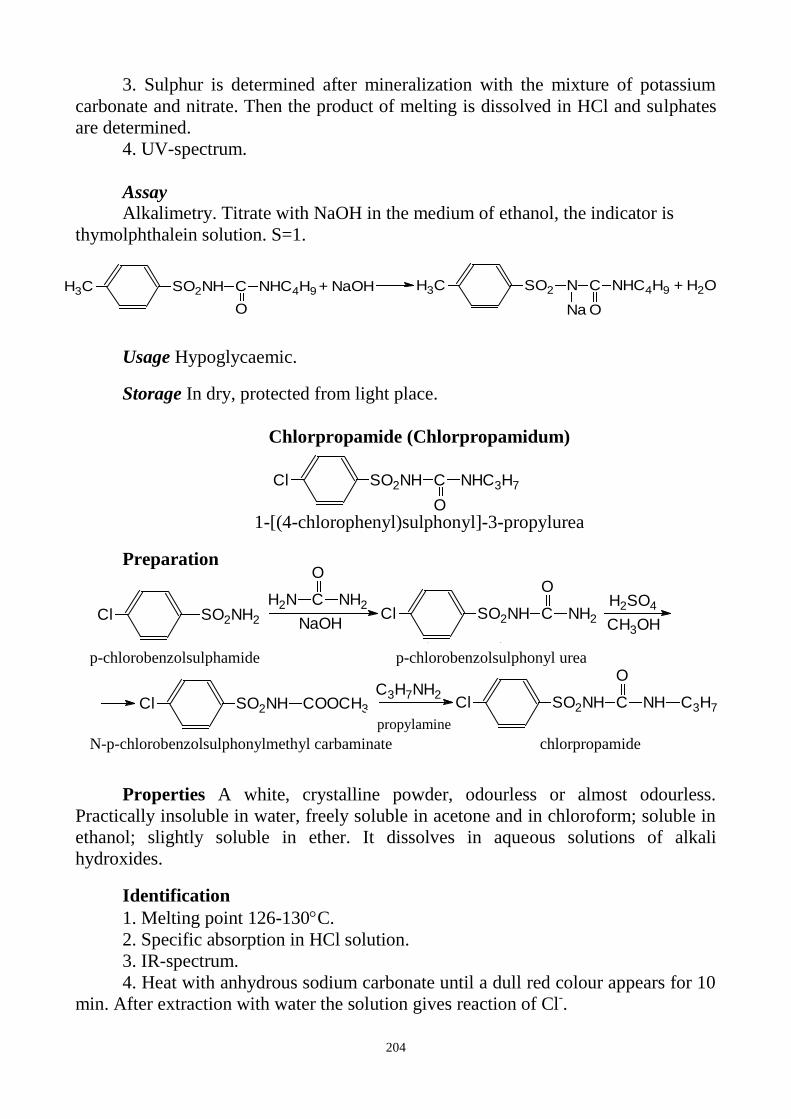
204
3. Sulphur is determined after mineralization with the mixture of potassium
carbonate and nitrate. Then the product of melting is dissolved in HCl and sulphates
are determined.
4. UV-spectrum.
Assay
Alkalimetry. Titrate with NaOH in the medium of ethanol, the indicator is
thymolphthalein solution. S=1.
Usage Hypoglycaemic.
Storage In dry, protected from light place.
Chlorpropamide (Chlorpropamidum)
1-[(4-chlorophenyl)sulphonyl]-3-propylurea
Preparation
Properties A white, crystalline powder, odourless or almost odourless.
Practically insoluble in water, freely soluble in acetone and in chloroform; soluble in
ethanol; slightly soluble in ether. It dissolves in aqueous solutions of alkali
hydroxides.
Identification
1. Melting point 126-130C.
2. Specific absorption in HCl solution.
3. IR-spectrum.
4. Heat with anhydrous sodium carbonate until a dull red colour appears for 10
min. After extraction with water the solution gives reaction of Cl-.
H3C SO2NH C
O
NHC4H9 + NaOH H3C SO2 N C NHC4H9 + H2O
Na O
SO2NH C
O
NHC3H7Cl
хлорпропамид
пропиламин
п-хлорбензолсульфонилмочевинап-хлорбензолсульфамид
SO2NH CCl
O
NH C3H7
C3H7NH2SO2NHCl COOCH3
NaOH CH3OH
H2SO4SO2NH CCl
O
NH2
H2N C NH2
O
Cl SO2NH2
N-п-хлорбензолсульфонилметилкарбаминат
p-chlorobenzolsulphamide p-chlorobenzolsulphonyl urea
N-p-chlorobenzolsulphonylmethyl carbaminate chlorpropamide
propylamine
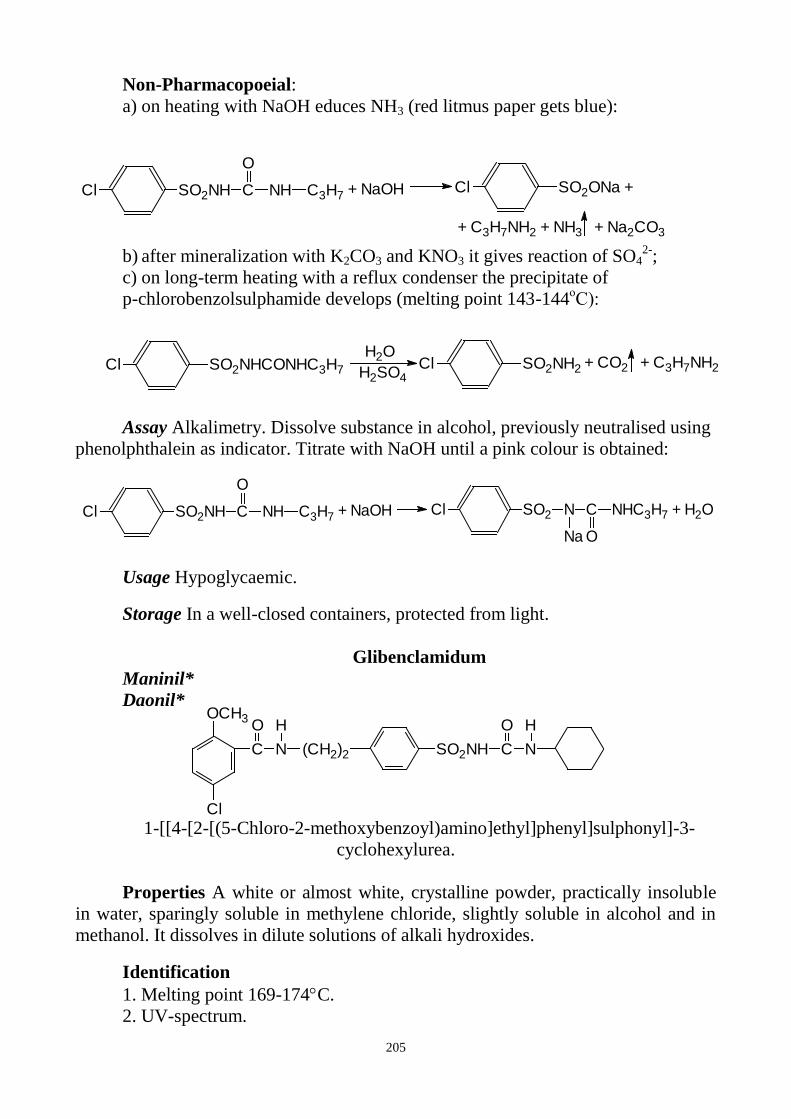
205
Non-Pharmacopoeial:
a) on heating with NaOH educes NH3 (red litmus paper gets blue):
b) after mineralization with K2CO3 and KNO3 it gives reaction of SO42-
;
c) on long-term heating with a reflux condenser the precipitate of
p-chlorobenzolsulphamide develops (melting point 143-144оС):
Assay Alkalimetry. Dissolve substance in alcohol, previously neutralised using
phenolphthalein as indicator. Titrate with NaOH until a pink colour is obtained:
Usage Hypoglycaemic.
Storage In a well-closed containers, protected from light.
Glibenclamidum
Maninil*
Daonil*
1-[[4-[2-[(5-Chloro-2-methoxybenzoyl)amino]ethyl]phenyl]sulphonyl]-3-
cyclohexylurea.
Properties A white or almost white, crystalline powder, practically insoluble
in water, sparingly soluble in methylene chloride, slightly soluble in alcohol and in
methanol. It dissolves in dilute solutions of alkali hydroxides.
Identification
1. Melting point 169-174C.
2. UV-spectrum.
SO2NH CCl
O
NH C3H7 + NaOH Cl SO2ONa
+ C3H7NH2 + NH3 + Na2CO3
+
Cl SO2NHCONHC3H7
H2O
H2SO4Cl SO2NH2 + CO2 + C3H7NH2
SO2NH CCl
O
NH C3H7 + NaOH SO2 N C
Na O
Cl NHC3H7 + H2O
Cl
OCH3
C
O
N
H
(CH2)2 SO2NH C
O
N
H
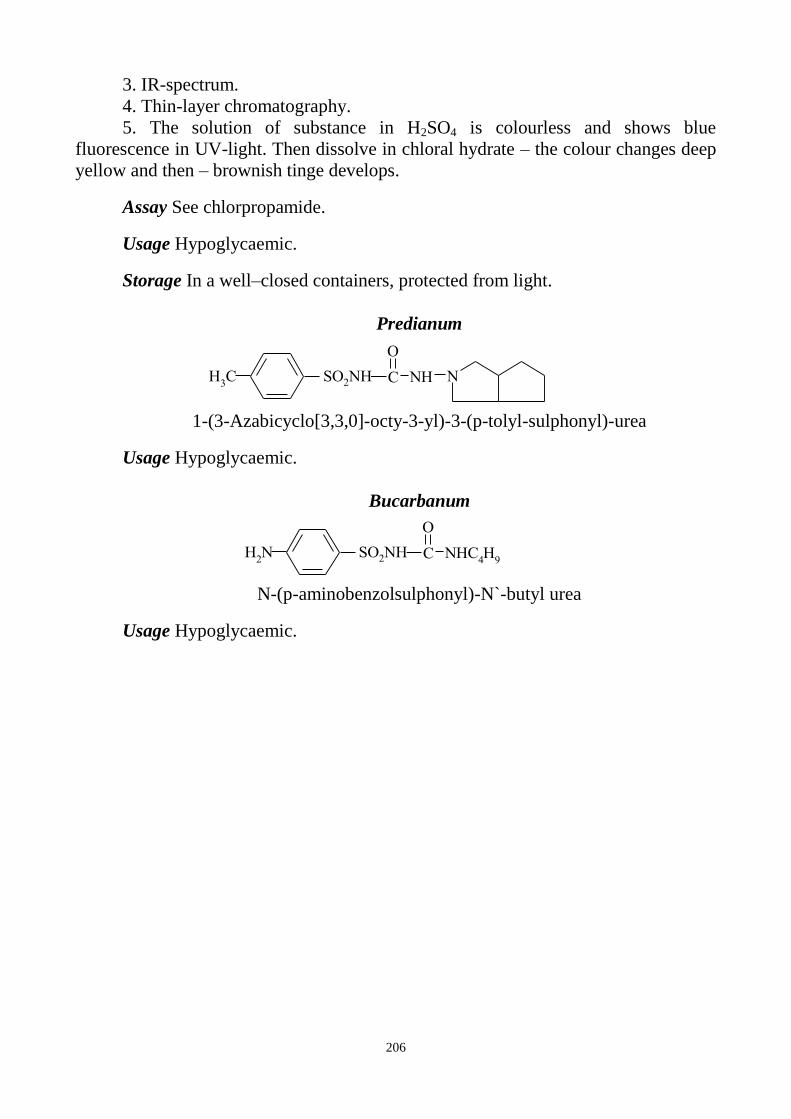
206
3. IR-spectrum.
4. Thin-layer chromatography.
5. The solution of substance in H2SO4 is colourless and shows blue
fluorescence in UV-light. Then dissolve in chloral hydrate – the colour changes deep
yellow and then – brownish tinge develops.
Assay See chlorpropamide.
Usage Hypoglycaemic.
Storage In a well–closed containers, protected from light.
Predianum
1-(3-Azabicyclo[3,3,0]-octy-3-yl)-3-(p-tolyl-sulphonyl)-urea
Usage Hypoglycaemic.
Bucarbanum
N-(p-aminobenzolsulphonyl)-N`-butyl urea
Usage Hypoglycaemic.
H2N C
O
NHC4H
9SO2NH
H3C C
O
NH NSO2NH

207
Chapter 19. MEDICINAL SUBSTANCES DERIVATIVES OF
AMIDES OF SULPHANILIC ACID (SULPHANILAMIDES)
Plan
1. Preparation of derivatives of sulphanilic acid.
2. Properties and reactions of identification for sulphanilamides.
3. Methods of quantification for sulphanilamides.
4. The basic medicinal substances, derivatives of amides of sulphanilic acid;
specific reactions for identification, methods of quantification.
Sulphanilic acid is the source for Preparation of great number of medicinal
substances, combined according to their chemical structure and pharmacological
action into the group of sulphanilamides with general formula:
Knowledge of remarkable effects of such compounds in combating bacterial
infections resulted by chance from experiments on the bacterial properties of dyes.
Work on azo-dyes containing a sulphonamide nucleus have been started about 1930.
Two years later a German patent was issued covering the preparation of a number of
these dyes, including prontosil:
The first experimental data on the treatment of streptococcal infections in
animals by means of prontosil were published in 1935. Simultaneously, there
appeared a series of clinical reports on the value of prontosil in streptococcal
infections. In the same year it was suggested that the action of prontosil involved a
metabolism of the compound in the body with liberation of sulphanilamide. This led
to a study of sulphanilamide (sulfanilamide) itself, and this simple compound, which
had been prepared in 1908, was shown to be effective against streptococci.
Sulfanilamide itself has been largely superseded in medicine by derivatives,
which are less toxic or are for individual purposes preferable. These derivatives are
widely used in treatment of streptococcal, pneumococcal, meningococcal,
gonococcal, and E. coli infections.
The derivatives of sulfanilamide, that have found most successful application
in medicine, have in place of hydrogen of the sulphonamido-group such substituents
as acetyl, amidino- (guanyl), or heterocyclic radical.
Sulfacetamide, a simple acetyl derivative of sulfanilamide, known also as
albucid, was introduced for use in injuries and infections of the eyes as its
sodium salt. Sulfadiazine is used for general purposes and so are its dimethyl
NN SO2NH2
NH2
H2N
RHN SO2NHR'

208
homologue, sulfadimidine (also known as sulfamethazine and sulfamezathine),
sulfamethoxypyridazine and sulfadimethoxine. These substances are more slowly
excreted, and can be given in less frequent doses. Sulfacarbamide is used for urinary
tract infections. Sulfathiazole may cause toxic symptoms, but its derivatives
succinylsulfathiazole and phthalylsulfathiazole, are of low toxicity: these substances,
as well as sulfaguanidine, have the advantage of being only slightly absorb by
intestinal mucosa, and can therefore be used in relatively large doses in the treatment
of intestine infections. Salazodine and salazodimethoxine are products of interaction
with salicylic acid; they are given in the treatment of infections of the large intestine.
Their action is based on liberation of 5-aminosalicylic acids, which have anti-
inflammatory activity.
Preparation
1. The initial substance is sulphanilic acid:
NH2
SO2OH NH
2SO
2ONa
NHCH3CO SO
2ONa NH SO
2ClCH
3CO
R-NH2
NH SO2NHRCH
3CO SO
2NHRNH
2
NaOH (CH3CO)2O
PCl5
H2SO4
HOH
Sulpanilic acid Sodium salt of sulphanilic acid
Sodium p-acetamidebenzene sulphonate p-acetamidebenzene sulphochloride
2. The most rational is the synthesis from N-carbomethoxy aniline: R-NH
2
SO2NHRNH
2
NHCOCH3
O
NHCOCH3
SO2Cl
O
NHCOCH3
SO2NHR
O
NaOH
HOSO2Cl
N-carbomethoxy aniline p-carbomethoxyaminobenzene sulphochloride
Properties and identification
The most of sulphanilamides are amphoteric compounds. Their basic properties
are caused by the presence of aromatic aminogroup; they dissolve in acids with
forming of salts.
NH2 SO
2NHRHCl .
But these salts are easy hydrolysed in water.
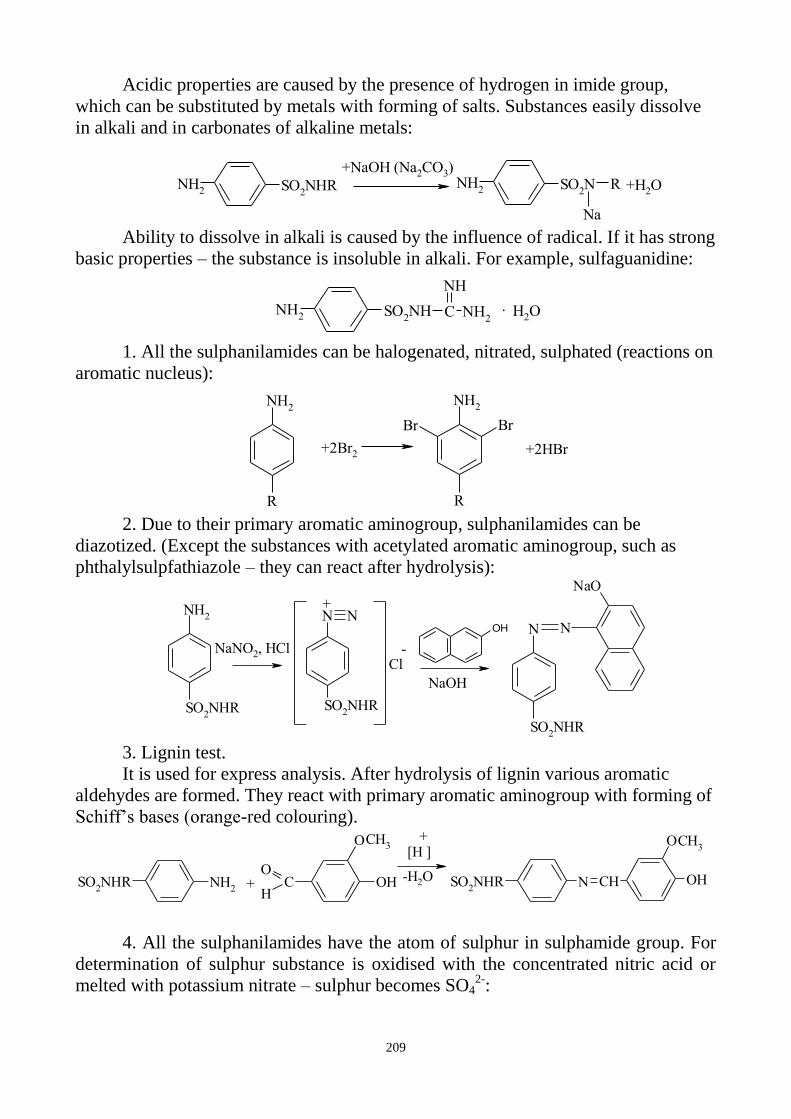
209
Acidic properties are caused by the presence of hydrogen in imide group,
which can be substituted by metals with forming of salts. Substances easily dissolve
in alkali and in carbonates of alkaline metals:
NH2 SO
2NNH
2 SO2NHR R
Na
+H2O
+NaOH (Na2CO3)
Ability to dissolve in alkali is caused by the influence of radical. If it has strong
basic properties – the substance is insoluble in alkali. For example, sulfaguanidine:
NH2 SO
2NH C
NH
NH2
. H2O
1. All the sulphanilamides can be halogenated, nitrated, sulphated (reactions on
aromatic nucleus):
NH2
R
NH2
R
Br Br
+2Br2 +2HBr
2. Due to their primary aromatic aminogroup, sulphanilamides can be
diazotized. (Except the substances with acetylated aromatic aminogroup, such as
phthalylsulpfathiazole – they can react after hydrolysis):
N
SO2NHR
N
SO
2NHR
NH2
N
NaO
OH
SO2NHR
N
NaNO2, HCl
+
Cl-
NaOH
3. Lignin test.
It is used for express analysis. After hydrolysis of lignin various aromatic
aldehydes are formed. They react with primary aromatic aminogroup with forming of
Schiff’s bases (orange-red colouring).
NH2
SO2NHR C
O
OHH
O
CH3
NSO2NHR CH
O
OH
CH3
+
[H ]
-H2O
+
4. All the sulphanilamides have the atom of sulphur in sulphamide group. For
determination of sulphur substance is oxidised with the concentrated nitric acid or
melted with potassium nitrate – sulphur becomes SO42-
:

210
NH2 SO
2NHR
conc. HNO3
H2SO4 + CO2 + NH4NO3 + NO + NO2 + H2O
H2SO4 + BaCl2 BaSO4 + 2HCl
5. Hydrogen of imide group causes ability to interact with the salts of heavy
metals (CuSO4, CoCl2, FeCl3) with forming of coloured complexes, soluble and
insoluble in water. With this reaction substances can be identified.
NH2 SO
2N R
Na
R
NH2 SO
2N R
Cu
NH2 SO
2N
2СuSO4
+Na2SO4
Substance dissolve in 0,1 М solution of alkali and add the solution of salt of
heavy metal. In the presence of excess of alkali hydroxides of heavy metals can be
precipitated.
6. With 1% solution of sodium nitroprusside in the presence of alkali and with
subsequent adding of acid a red or brown-red precipitate develops.
7. On heating in a dry test-tube these substances develop the melted products
with various colouring and educe various gases. It is a reaction for identification and
distinguishing of sulphanilamides.
8. UV-spectroscopy.
Methods of quantification
1. The main method for quantification of the most sulphanilamides is
determination of primary aromatic amino-nitrogen (nitritometry).
Titrate with 0.1 M sodium nitrite in the medium of dilute hydrochloric acid in
the presence of potassium bromide (as catalyst) after cooling in ice-water. Determine
end-point electrochemically or by use of prescribed indicator:
N SO2NHRN
NH2 SO
2NHR
NaNO2, HCl
KBr
+Cl
-
2. Acid-base titration.
It is based on the properties of hydrogen in imide group. Acidic forms are
titrated with an alkali in the presence of thymolphthalein as indicator:
NH2 SO
2NNH
2 SO2NHR R
Na
+H2O+NaOH
Substances with constant of dissociation 10-7
-10-8
(sulfathiazole) are dissolved
in aqueous-acetone solution or in alcohol. Substances with constant of dissociation
10-9
(phthalylsulfathiazole, salazodine) titrate only in non-aqueous solvents
(dimethylformamide), titrate with solution of alkali in benzol with methanol.
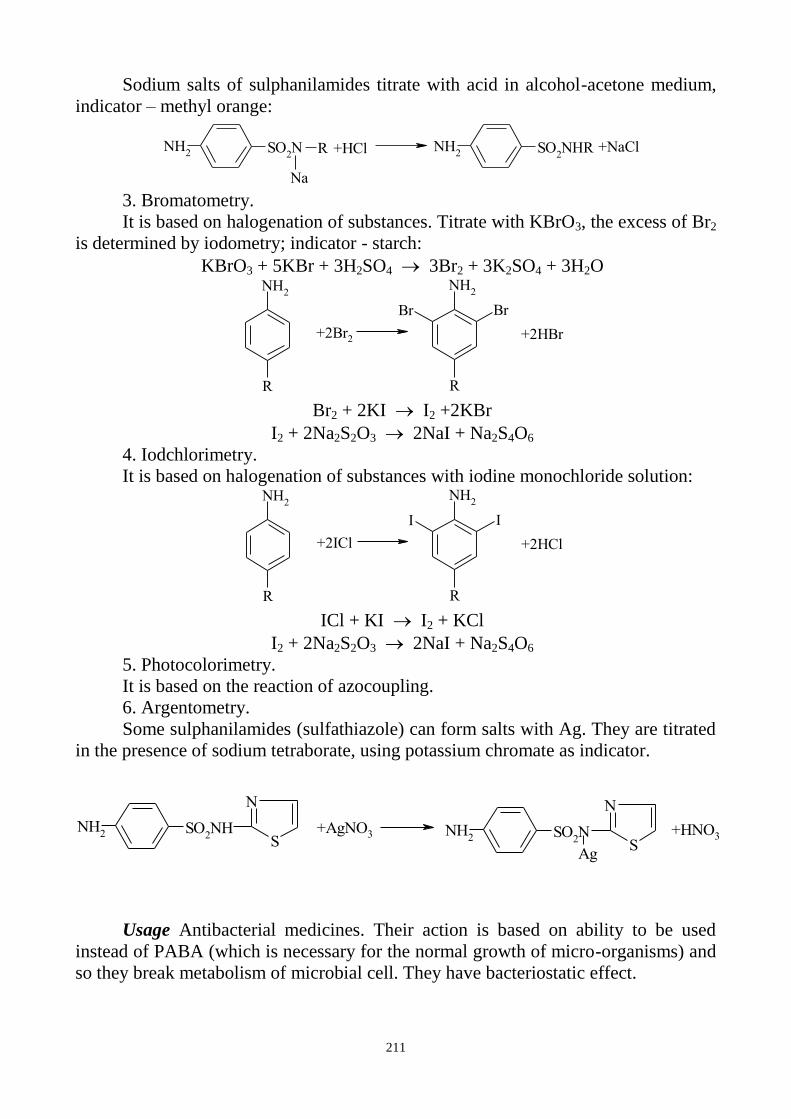
211
Sodium salts of sulphanilamides titrate with acid in alcohol-acetone medium,
indicator – methyl orange:
NH2 SO
2N NH
2 SO2NHRR
Na
+HCl +NaCl
3. Bromatometry.
It is based on halogenation of substances. Titrate with KBrO3, the excess of Br2
is determined by iodometry; indicator - starch:
KBrO3 + 5KBr + 3H2SO4 3Br2 + 3K2SO4 + 3H2O NH
2
R
NH2
R
Br Br
+2Br2 +2HBr
Br2 + 2KI I2 +2KBr
I2 + 2Na2S2O3 2NaI + Na2S4O6
4. Iodchlorimetry.
It is based on halogenation of substances with iodine monochloride solution: NH
2
R
NH2
R
I I
+2ICl +2HCl
ICl + KI I2 + KCl
I2 + 2Na2S2O3 2NaI + Na2S4O6
5. Photocolorimetry.
It is based on the reaction of azocoupling.
6. Argentometry.
Some sulphanilamides (sulfathiazole) can form salts with Ag. They are titrated
in the presence of sodium tetraborate, using potassium chromate as indicator.
NH2 SO
2NH
N
SNH
2 SO2N
N
SAg
+AgNO3 +HNO3
Usage Antibacterial medicines. Their action is based on ability to be used
instead of PABA (which is necessary for the normal growth of micro-organisms) and
so they break metabolism of microbial cell. They have bacteriostatic effect.

212
MAIN MEDICINAL SUBSTANCES
Sulfanilamide (Streptocidum)
NH2 SO
2NH
2
4-aminobenzenesulphonamide
Properties A white or yellowish-white crystals or fine powder, slightly soluble
in water, freely soluble in acetone, sparingly soluble in alcohol, practically insoluble
in methylene chloride. It dissolves in solutions of alkali hydroxides and in dilute
mineral acids.
Identification
1. Melting point: 164.5C to 166.0C.
2. IR-spectrum.
3. Thin-layer chromatography.
4. Reaction of primary aromatic amines.
Non-Pharmacopoeial reactions:
a) on heating with alkali the smell of ammonium appears:
NH2 SO
2NH
2NH
2 SO2ONa+ NaOH
to
+ NH3
b) on melting the product obtained has a blue-violet colouring; smells of
ammonium and aniline appears;
c) by action of oxidisers (H2O2 or FeCl3) a red-violet colouring appears.
Assay Determination of primary aromatic amino-nitrogen and all the methods
may be used except neutralisation.
Storage Store protected from light.
Sulfacetamide sodium (Sulfacylum-natrium)
H2N SO2 N C
ONa
CH3. H2O
Sodium N-[(4-aminophenyl)sulphonyl]acetamide
Properties A white or yellowish-white, crystalline powder, freely soluble in
water, slightly soluble in ethanol, practically insoluble in ether.
Identification
1. UV-spectrum.
2. IR-spectrum.
3. Dissolve the substance in water, add dilute acetic acid and filter. The
precipitate, washed with a small quantity of water melts at 181C to 185C.

213
4. Dissolve the precipitate obtained in the test (3) in alcohol. Add sulphuric
acid and heat. The odour of ethyl acetate is perceptible:
5. Dissolve the precipitate obtained in the test (3), with heating, in water – the
solution gives reaction of primary aromatic amines with formation of orange-red precipitate.
6. Reactions of Na+.
Non-Pharmacopoeial reactions:
a) with СuSO4 - a bluish-green precipitate appears;
b) after hydrolysis appears the smell of acetic acid:
NH2 SO
2NH
2H2N SO2 N C
ONa
CH3. H2O
to
+CH3COOH +NaCl
HCl,
Assay Determination of primary aromatic amino-nitrogen. May be applied all
the methods mentioned above.
Storage Store protected from light.
Sulfaguanidine (Sulginum)
NH2 SO
2NH C
NH
NH2
. H2O
(4-aminophenylsulphonyl)guanidine
Properties A white or almost white, fine crystalline powder, very slightly
soluble in water, slightly soluble in acetone, very slightly soluble in alcohol,
practically insoluble in methylene chloride. It dissolves in dilute solutions of mineral
acids.
Identification
1. Melting point: 189C to 193C, determined on the dried substance.
2. IR-spectrum.
3. Thin-layer chromatography.
4. Dissolve the substance in hydrochloric acid; dilute the solution with water –
the solution, without further acidification, gives the reaction of primary aromatic
amines.
5. Suspend the substance in water, add -naphthol solution and a mixture of
water and strong sodium hypochlorite solution – a red colour develops.
H2N SO2 N C
OH
CH3
, H2O to
H2N SO2 NH2+CH3COOH
CH3COOH + C2H5OH
H2SO4
H2SO4
-H2OCH3COOC2H5
,
c.

214
Non-Pharmacopoeial reactions:
a) the product obtained after melting has a violet-red colouring and a smell of
ammonium appears (distinguishing from the other sulphanilamides except
sulfacarbamide):
NH C
NHR
NH2
NH=C-NH-C=NH
NHR NHR
2 +NH3
to
b) for distinguishing from sulfacarbamide the examined substance shake with
0.1 М sodium hydroxide solution, add 2-3 drops of phenolphthalein – a red colouring
appears (the substance is insoluble in alkali);
c) on heating with alkali educes ammonium:
NH2 SO
2NH C
NH
NH2
NH2 SO
3Na+3NaOH
to
+3NH3 +Na2CO3+H2O
Assay Determination of primary aromatic amino-nitrogen and all the methods
mentioned above.
Storage Store protected from light.
Sulfacarbamide (Urosulfanum)
NH2 SO
2NH C
O
NH2
. H2O
p-aminobenzolsulphanyl urea
Properties A white crystalline powder, odourless, with a sour taste. Slightly
soluble in water, very slightly soluble in alcohol, practically insoluble in acetone, in
dilute acids and in solutions of alkali hydroxides.
Identification
1. The product after melting has a violet-red colour and a smell of ammonium
appears.
2. On heating with alkali educes ammonium:
NH2 SO
2NH C
O
NH2
NH2 SO
3Na+NaOH
to
+2NH3 +Na2CO3
3. On heating with 5% sodium nitrite solution a red colouring appears.
Assay Nitritometry and methods mentioned above.
Storage In a well-closed containers.

215
Sulfathiazole (Norsulfazolum)
NH2 SO
2NH
N
S
4-amino-N-(thiazol-2-yl)
Sulfathiazole-sodium (Norsulfazolum-natrium)
NH2 SO
2N
N
SNa
Sodium 4-amino-N-(thiazol-2-yl)
Succinylsulfathiazole (Succinylsulfathiazolum)
NH SO2NHC
O
CH2-CH
2
N
SC
O
H
4-oxo-4-[[4-(thiazol-2-ylsulphamoyl)phenyl]amino]butanoic acid
Properties
Sulfathiazole - a white or slightly yellowish, crystalline powder, practically
insoluble in water, slightly soluble in alcohol, practically insoluble in ether and in
methylene chloride. It dissolves in dilute solutions of alkali hydroxides and in dilute
mineral acids.
Sulfathiazole-sodium – a white or slightly yellowish crystals, odourless, freely
soluble in water.
Succinylsulfathiazole - a white or yellowish-white, crystalline powder, very
slightly soluble in water, slightly soluble in alcohol and in acetone, practically
insoluble in ether. It dissolves in dilute solutions of alkali hydroxides and carbonates.
Identification
1. Melting point (for sulfathiazole 200-203C; for succinylsulfathiazole after
alkaline hydrolysis 196-204C).
2. IR-spectroscopy.
3. On heating fumes are evolved which blacken lead acetate paper (for
succinylsulfathiazole).
4. Reaction with hydroquinone and sulphuric acid. After heating add toluene,
shake – the upper (toluene) layer shows an intense pink colour (for
succinylsulfathiazole).
5. Thin-layer chromatography (for sulfathiazole).
6. With CuSO4 solution – a greyish-blue or purple precipitate is formed (for
sulfathiazole).
7. Reaction of Na+.
8. Reaction of primary aromatic amines.
216
Assay Nitritometry, determining the end-point electrometrically (for
succinylsulfathiazole – after acidic hydrolysis) and methods mentioned above.
Storage Store protected from light.
Sulfaethidole (Aethazolum)
NH2 SO
2NH
NN
S C2H
5 2(p-aminobenzolsulfamide)-5-ethyl-1,3,4-thiadiazole
Sulfaethidole-sodium (Aethazolum-natrium)
NH2 SO
2N
NN
S C2H
5Na
Sodium 2(p-aminobenzolsulfamide)-5-ethyl-1,3,4-thiadiazole
Properties A white or almost white powder, odourless. Practically insoluble in
water, very slightly soluble in ether, soluble in solutions of alkali hydroxides.
Sulfaethidole-sodium – white crystalline powder. Freely soluble in water,
practically insoluble in ether.
Identification
1. With CuSO4 develops a green precipitate becomes black.
2. With СoCl3 develops a white precipitate.
3. After melting develops a brown product, and the smell of hydrogen sulphide
appears.
Assay Nitritometry and methods mentioned above.
Storage In well-stoppered bottles, protected from light.
Sulfadimidine (Sulfadimezinum)
N
N
NH2 SO
2NH
CH3
CH3
4-amino-N-(4,6-dimethylpyrimidin-2-yl)
Properties White or almost white powder or crystals, very slightly soluble in
water and in ether, soluble in acetone, slightly soluble in alcohol. It dissolves in
solutions of alkali hydroxides and in dilute mineral acids.
Identification
1. IR-spectrum.
217
2. Thin-layer chromatography.
3. On heating to about 270C the substance decomposes and a white or
yellowish-white sublimate is formed which after recrystallisation from toluene melts
at 150-154C.
4. Reaction of primary aromatic amines.
5. With CuSO4 a yellowish-green precipitate develops which quickly becomes
brown.
6. With sodium nitroprusside solution a violet colouring appears.
Assay Nitritometry and all the methods mentioned above.
Storage In a well-closed containers, protected from light.
Phthalylsulfathiazole (Phthalazolum)
NH SO2NHC
O
C
O
OH
N
S
2-[[4-(thiazol-2-ylsulphamoyl)phenyl]carbamoyl]benzoic acid
Properties A white or yellowish-white, crystalline powder, practically
insoluble in water and in ether, freely soluble in dimethylformamide, slightly
soluble in acetone and in alcohol.
Identification
1. IR-spectroscopy.
2. After boiling with dilute sodium hydroxide solution add dilute hydrochloric
acid, neutralise – the obtained crystals melt at 200 - 203C.
3. With dilute sulphuric acid and zinc powder – fumes are evolved which
produce a black stain on lead acetate paper.
4. With resorcinol and sulphuric acid after heating add dilute sodium hydroxide
solution. Dilute this brownish-red mixture with water – an intense green fluorescence
appears which disappears on acidification:
OH
OH
C
C
O
O
O
O
C
OH
CO
OH
Oconc. H2SO4
-2H2O2 +
NaOH, H2O
.H2S + (CH3COO)2Pb PbS 2CH3COOH+
218
O
C
OH OH
COONa
OH
O
C
NaO
COONa
O
-H2O
5. Reaction of primary aromatic amines (after hydrolysis).
Purity Sulfathiazole and other primary aromatic amines are detected by thin-
layer chromatography.
Assay Alkalimetry in the medium of dimethylformamide.
Titrate with sodium hydroxide until the colour becomes blue using
thymolphthalein as indicator. Carry out a blank titration. S=1/2.
NH SO2NHC
O
C
O
OH
N
SNH SO
2NC
O
C
O
ONa
N
SNa
2NaOH
HCON(CH3)2
+H2O
Storage Store protected from light.
Sulfadimethoxine (Sulfadimethoxinum)
N
N
O
O
NH2 SO
2NH
CH3
CH3
6-(p-aminobenzolsulfamide)-2,4-dimethoxypyridine
Properties A white powder, odourless.
Identification
1. With CuSO4 develops greyish-yellow amorphous precipitate.
2. Reaction of primary aromatic amines.
Assay Nitritometry and methods mentioned above.

219
Sulfalene (Sulfalenum)
NH2 SO
2NH
N
N
OH3C
2-(p-aminobenzolsulfamide)-3methoxypyrazine
Properties A white or yellowish, crystalline powder, odourless.
Identification
1. UV-spectra (in alkaline and acidic medium).
2. Reaction of primary aromatic amines.
3. Reaction with CuSO4 develops greyish-green precipitate becomes greenish-
blue.
Assay Nitritometry (calculate on dry substance).
Storage Store protected from light.
Sulfadiazine (Sulfazinum)
NH2 SO
2NH
N
N
4-amino-N-pyrimidin-2-ylbenzenesulphonamide
Properties A white or slightly yellowish, crystalline powder, odourless.
Practically insoluble in water, slightly soluble in acetone, very slightly soluble in
alcohol. It dissolves in solutions of alkali hydroxides and in dilute mineral acids.
Identification
1. IR-spectrum.
2. Thin-layer chromatography.
3. On heating at temperature about 270C the obtained sublimate after
recrystallisation melts at 123-127C.
4. Reaction of primary aromatic amines.
5. With CuSO4 solution develops yellowish-green precipitate becomes greyish-
violet.
6. With CoСl3 solution develops violet precipitate.
7. Reaction with alcoholic resorcinol solution and sulphuric acid – after heating
of substance. A red colouring appears which becomes blue or red-blue after adding of
anhydrous acetic acid and ammonia solution.
Assay Nitritometry. The end-point is stated electrometrically.

220
Sulfamethoxypyridazine (Sulfapyridazinum)
NH2 SO
2NH
NN
OCH3
4-amino-N-(6-methoxypyridazin-3-yl)benzenesulphonamide
Properties A white or slightly yellowish, crystalline powder, colouring slowly
on exposure to light, practically insoluble in water, sparingly soluble in acetone,
slightly soluble in alcohol, vary slightly soluble in methylene chloride. It dissolves in
solutions of alkali hydroxides and in dilute mineral acids.
Identification
1. IR-spectrum.
2. Thin-layer chromatography.
3. Dissolve the substance in sulphuric acid; heat gently until a crystalline
precipitate appears. Cool and add dilute sodium hydroxide solution, ether, and shake.
Separate the ether layer, dry over anhydrous sodium sulphate and filter. Evaporate the
solvent by heating in a water-bath. An oily residue is obtained which becomes
crystalline on cooling. The residue (2-amino-6-methoxypyridazine) melts at 102C to
106C:
NH2 SO
2NH
NN
OCH3
NH2 SO
3H NH
2
NN
OCH3
H2SO4, H2O
+
2-amino-6-methoxypyridazine
4. Dissolve the substance in hydrochloric acid, dilute with water. The solution,
without further acidification, gives reaction of primary aromatic amines.
Assay Carry out the assay of primary aromatic amino-nitrogen.
Usage Sulphanilamide with long-term action.
Storage Store in a well-closed containers, protected from light.
Sulfamethoxypyridazine-sodium (Sulfapyridazinum-natrium)
NH2 SO
2N
NN
OCH3
Na
. 4H2O
Sodium 4-amino-N-(6-methoxypyridazin-3-yl)benzenesulphonamide
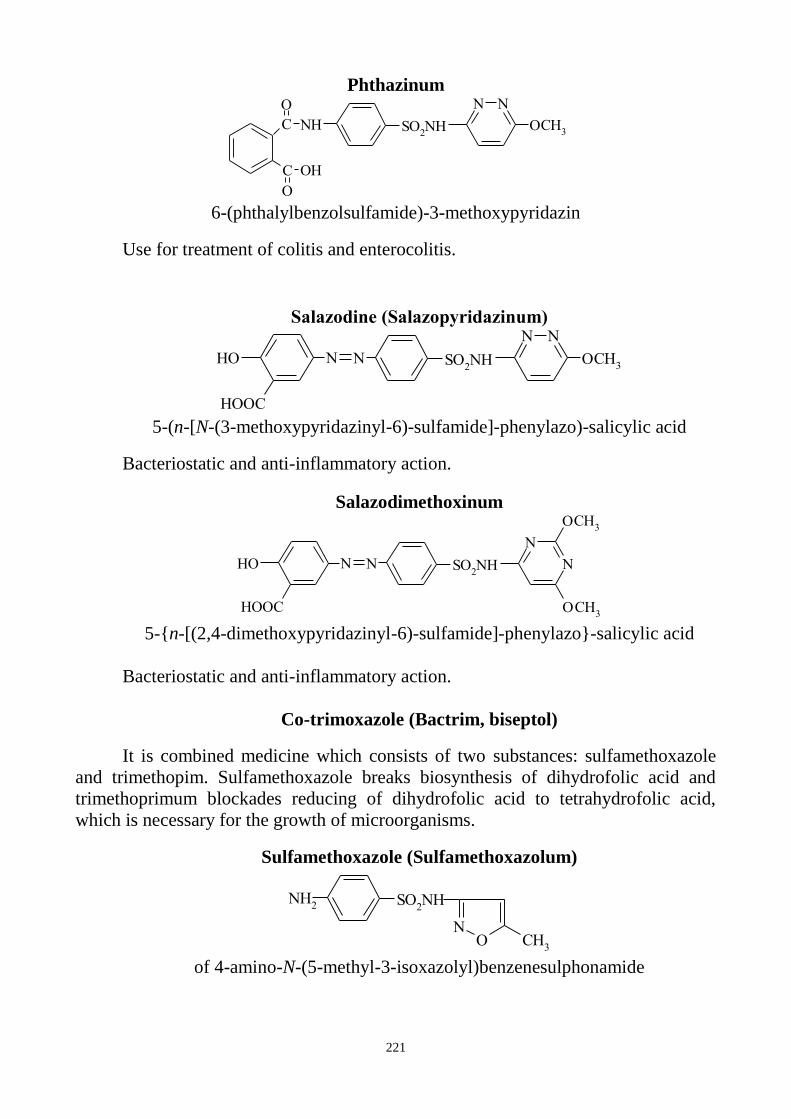
221
Phthazinum NN
OCH3
NH SO2NHC
O
C
O
OH
6-(phthalylbenzolsulfamide)-3-methoxypyridazin
Use for treatment of colitis and enterocolitis.
Salazodine (Sаlazopyridazinum) NN
OCH3
N SO2NHNOH
HOOC 5-(n-[N-(3-methoxypyridazinyl-6)-sulfamide]-phenylazo)-salicylic acid
Bacteriostatic and anti-inflammatory action.
Salazodimethoxinum
N
N
O
O
N SO2NHNOH
HOOC
CH3
CH3
5-{n-[(2,4-dimethoxypyridazinyl-6)-sulfamide]-phenylazo}-salicylic acid
Bacteriostatic and anti-inflammatory action.
Co-trimoxazole (Bactrim, biseptol)
It is combined medicine which consists of two substances: sulfamethoxazole
and trimethopim. Sulfamethoxazole breaks biosynthesis of dihydrofolic acid and
trimethoprimum blockades reducing of dihydrofolic acid to tetrahydrofolic acid,
which is necessary for the growth of microorganisms.
Sulfamethoxazole (Sulfamethoxazolum)
NH2 SO
2NH
NO CH
3 of 4-amino-N-(5-methyl-3-isoxazolyl)benzenesulphonamide

222
Properties A white or almost white crystalline powder, practically insoluble in
water, freely soluble in acetone, sparingly soluble in alcohol, slightly soluble in ether.
It dissolves in dilute solutions of sodium hydroxide.
Identification
1. Melting point 169-172C.
2. IR-spectrum.
3. Thin-layer chromatography.
4. Reaction of primary aromatic amines.
Assay Assay of primary aromatic amines (nitritometry), determining the end-
point electrometrically.
Storage In a well-closed container, protected from light.

223
References
1. Beckett A.H., Stenlake J.B. Practical Pharmaceutical Chemistry. New
Delhi: CBS Publishers and Distributors, 2002. –Fourth edition. – P. I. – 326 p., P. II.-
602 p.
2. British Pharmacopoeia. Version 11 and Supplements, 2007. – CD.
3. David G. Watson. Pharmaceutical analysis. – New York: Churchill
Livingston, 1999. – 337 p.
4. European Pharmacopoeia. Fourth edition and Supplement, 2004.
Council of Europe, Strasbourg. –CD.
5. European Pharmacopoeia. Fifth edition and Supplement, 2006. Council
of Europe, Strasbourg. –CD.
6. John H. Block, John M. Beale, Jr. Organic, Medicinal and
Pharmaceutical chemistry. – Philadelphia: Lippincott, Williams & Wikins, 2004. –
991 p.
7. Textbook of pharmaceutical chemistry / Revised by L.M. Atherden. –
Oxford: University press, 1999. – 915 p.
8. United States Pharmacopoeia, 2007. – CD.
9. Беликов В.Г. Фармацевтическая химия . – В 2 ч. Учебное пособие. –
4-е изд., перераб. и доп. М.: МЕД пресс-информ, 2007. – 624 с.
10. Государственная фармакопея СССР. Х издание. – М.: Медицина,
1968. – 1079с.
11. Державна фармакопея України.- 1-е вид.- X.: РІРЕГ, 2001.- 556 с.
12. Державна фармакопея України. - 1-е вид. Доповнення І. - X.: РІРЕГ,
2004. - 494 с.
13. Машковский М. Д. Лекарственнме средства. - 15-е изд., перераб.,
нспр. и доп. -М.: ООО "Издательство Новая Волна", 2005. - 1200 с.
14. Фармацевтична хімія. Навчальний посібник / За загальною ред.
П.О. Безуглого. – Вінниця: НОВА КНИГА, 2006. – 552 с.