Embed Size (px)
Citation preview

A Hybrid Approach to Assemble and Annotate the Brassica rapa
Transcriptome in the Cloud through the iPlant Collaborative and XSEDE
Upendra Kumar Devisetty
Postdoctoral Researcher
Maloof Lab, UC Davis
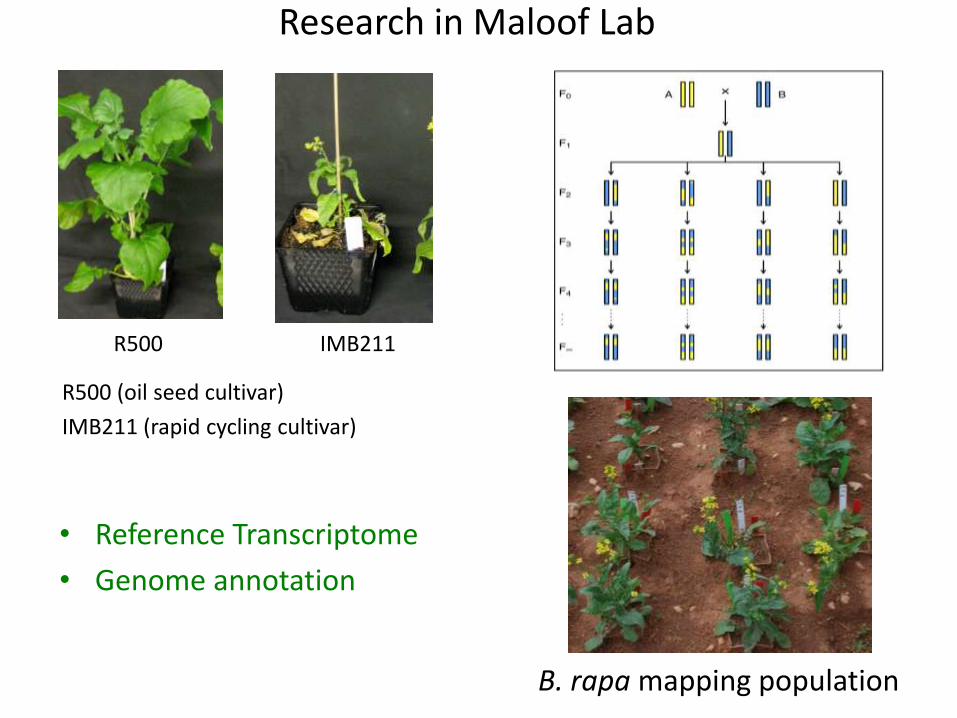
R500 IMB211
• Reference Transcriptome
• Genome annotation
R500 (oil seed cultivar)
IMB211 (rapid cycling cultivar)
B. rapa mapping population
Research in Maloof Lab
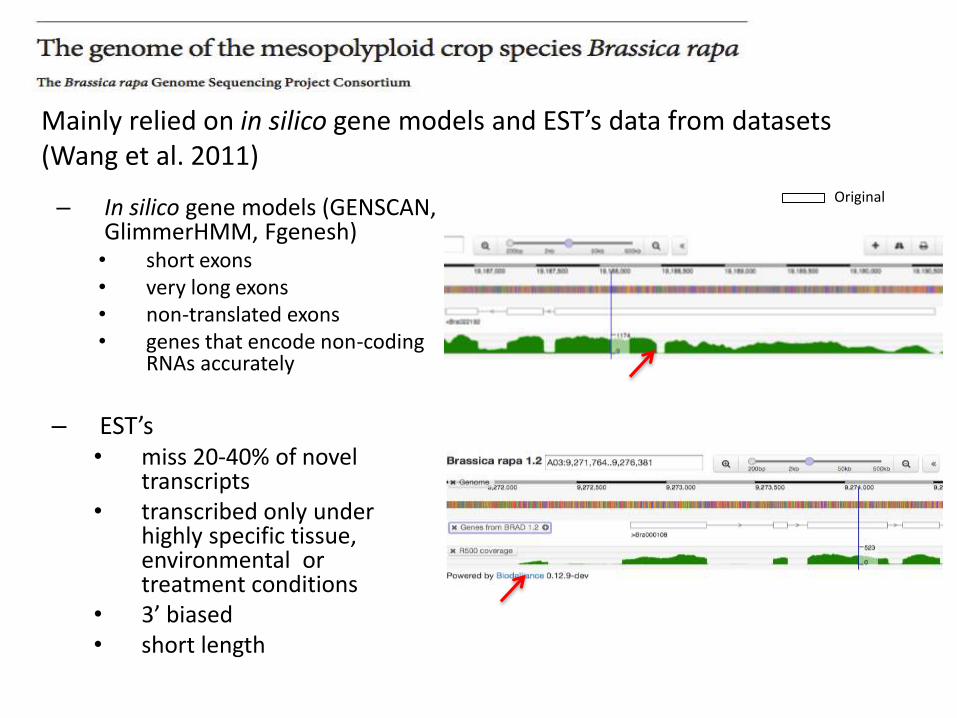
Mainly relied on in silico gene models and EST’s data from datasets (Wang et al. 2011)
– In silico gene models (GENSCAN, GlimmerHMM, Fgenesh)• short exons • very long exons• non-translated exons• genes that encode non-coding
RNAs accurately
– EST’s• miss 20-40% of novel
transcripts• transcribed only under
highly specific tissue, environmental or treatment conditions
• 3’ biased• short length
Original

Why there is a need for accurate genome annotation?
• Accurate and comprehensive genome annotation (e.g. gene models) is imperative for functional studies
• Useful for accurate mRNA abundance and detection of eQTLs (expression QTLs) in mapping populations
Objectives
• To detect transcripts that are not present in the existing genome reference of B. rapa (novel transcripts)
• To update the existing gene models of B. rapa genome
UK Devisetty et al. 2014 G3: Genes|Genomes|Genetics
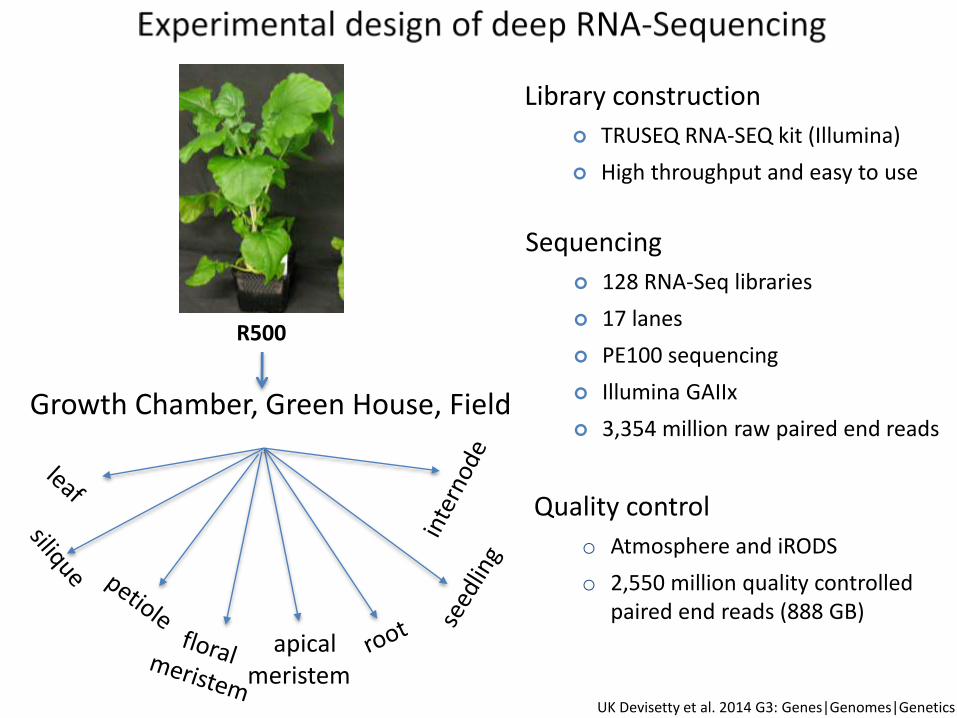
Growth Chamber, Green House, Field
apical meristem
R500
Library construction
TRUSEQ RNA-SEQ kit (Illumina)
High throughput and easy to use
Sequencing
128 RNA-Seq libraries
17 lanes
PE100 sequencing
Illumina GAIIx
3,354 million raw paired end reads
Quality control
o Atmosphere and iRODS
o 2,550 million quality controlled paired end reads (888 GB)
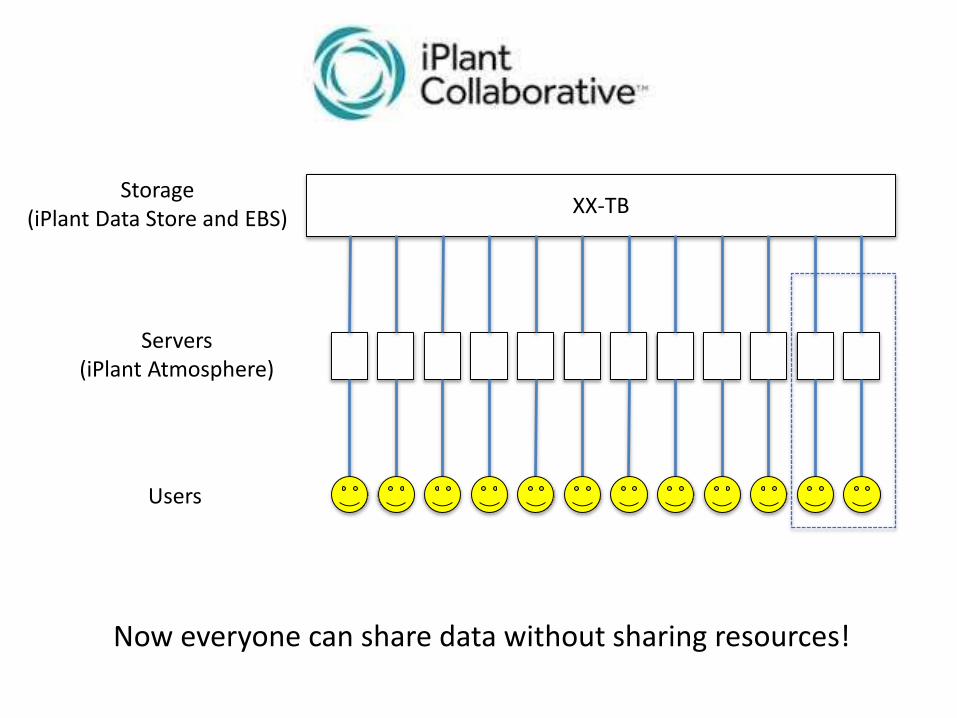
Servers(iPlant Atmosphere)
XX-TBStorage
(iPlant Data Store and EBS)
Users
Now everyone can share data without sharing resources!
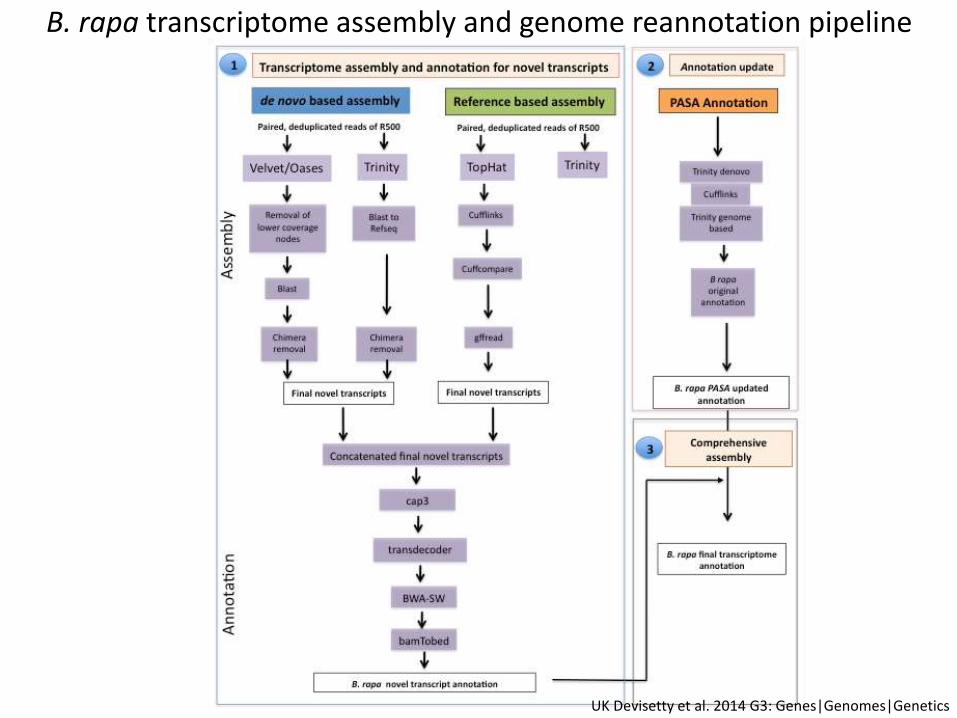
B. rapa transcriptome assembly and genome reannotation pipeline
UK Devisetty et al. 2014 G3: Genes|Genomes|Genetics
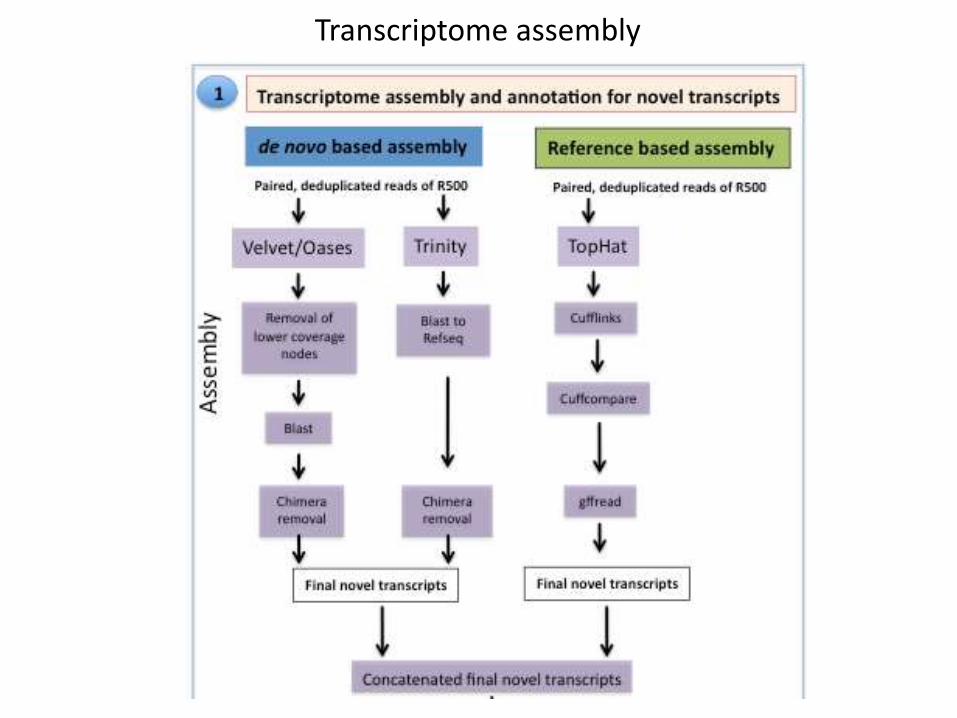
Transcriptome assembly
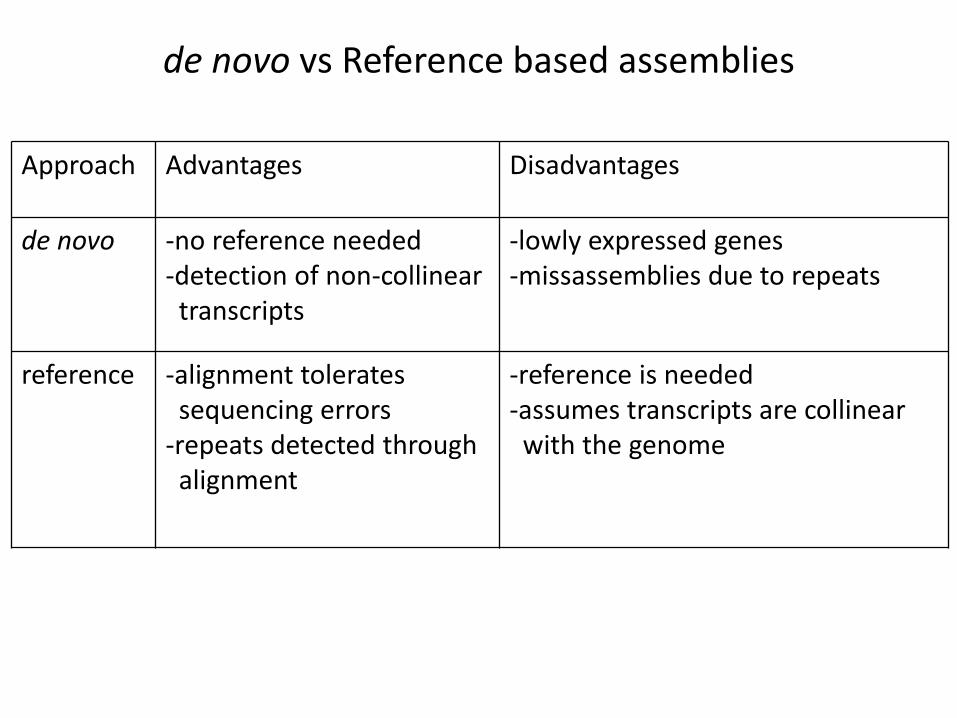
de novo vs Reference based assemblies
Approach Advantages Disadvantages
de novo -no reference needed-detection of non-collinear
transcripts
-lowly expressed genes-missassemblies due to repeats
reference -alignment tolerates sequencing errors
-repeats detected through alignment
-reference is needed-assumes transcripts are collinear
with the genome
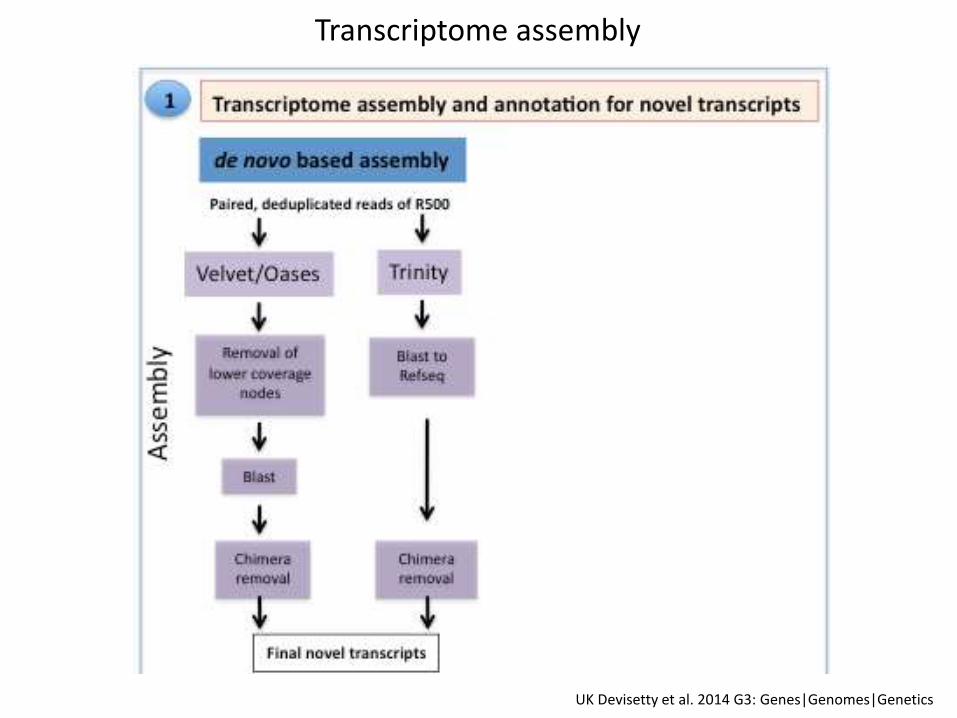
Transcriptome assembly
UK Devisetty et al. 2014 G3: Genes|Genomes|Genetics

• XSEDE is the most powerful integrated advanced digital resources and services in the world funded by NSF
• Scientists and Engineers around the world use XSEDE resources and services: supercomputers, collections of data, help services
• Consists of supercomputers, high-end visualization, data analysis and storage around the country
xsede.org
xsede.org
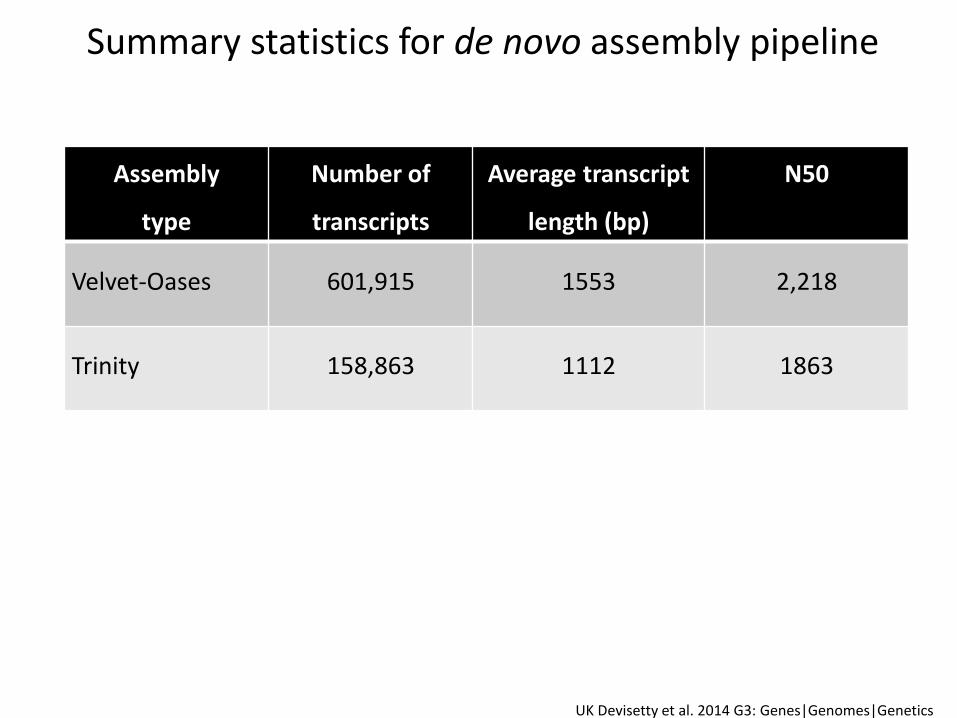
Summary statistics for de novo assembly pipeline
UK Devisetty et al. 2014 G3: Genes|Genomes|Genetics
Assembly
type
Number of
transcripts
Average transcript
length (bp)
N50
Velvet-Oases 601,915 1553 2,218
Trinity 158,863 1112 1863
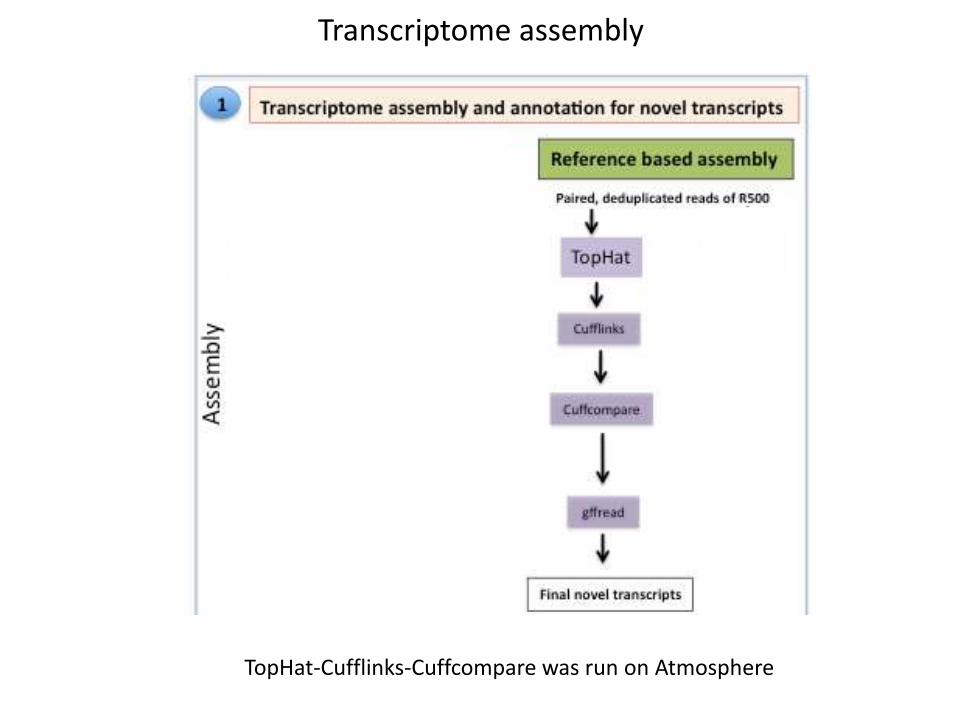
Transcriptome assembly
TopHat-Cufflinks-Cuffcompare was run on Atmosphere
Summary statistics for Reference assembly pipeline
UK Devisetty et al. 2014 G3: Genes|Genomes|Genetics
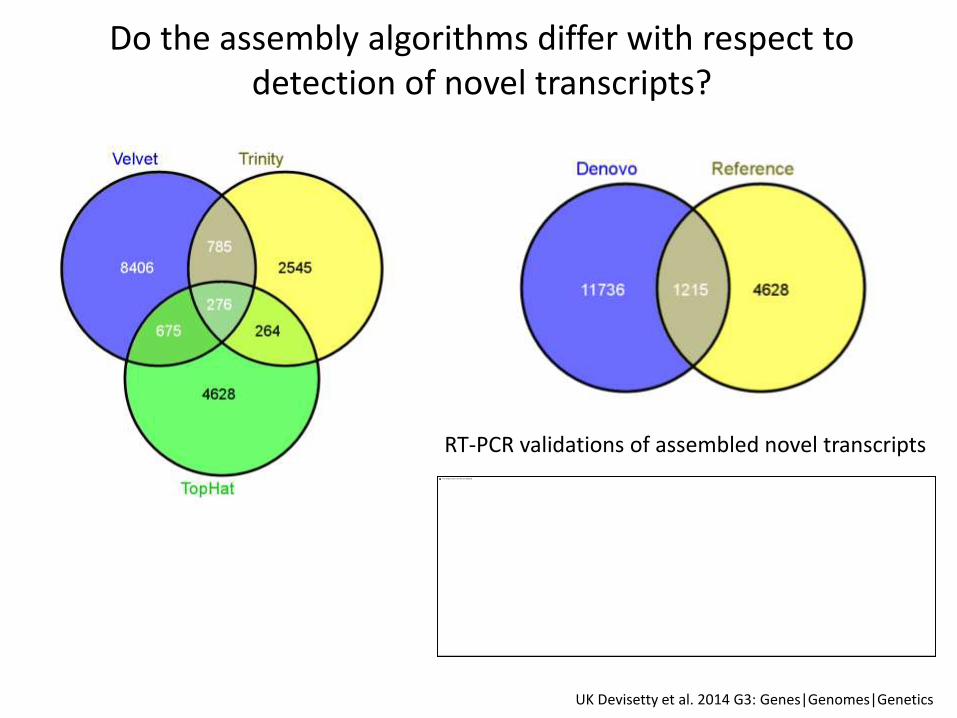
Do the assembly algorithms differ with respect to detection of novel transcripts?
UK Devisetty et al. 2014 G3: Genes|Genomes|Genetics
RT-PCR validations of assembled novel transcripts
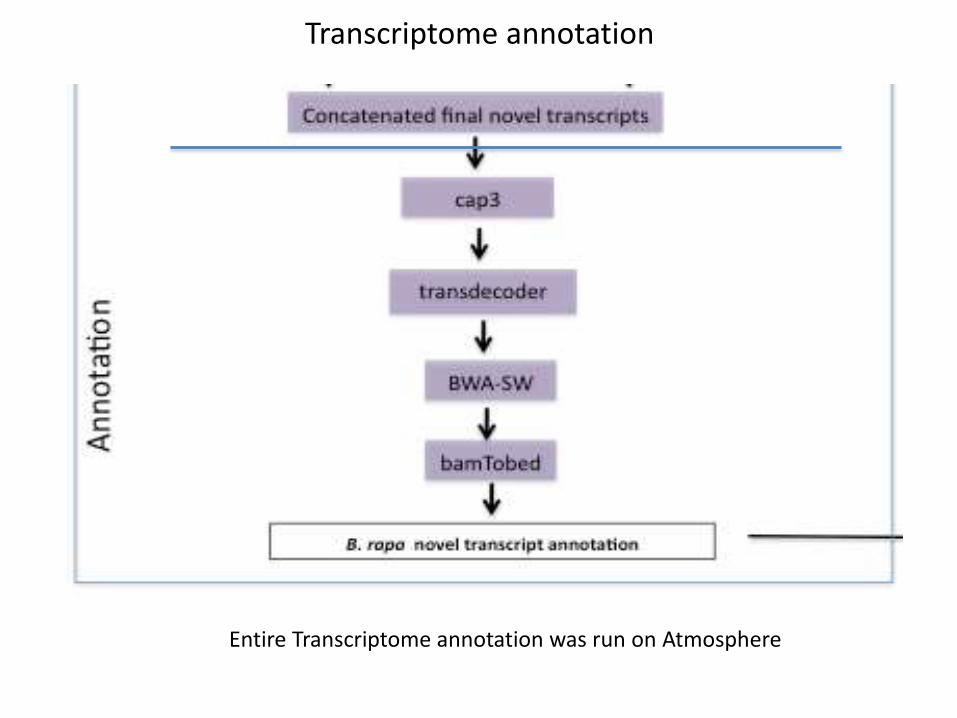
Transcriptome annotation
Entire Transcriptome annotation was run on Atmosphere
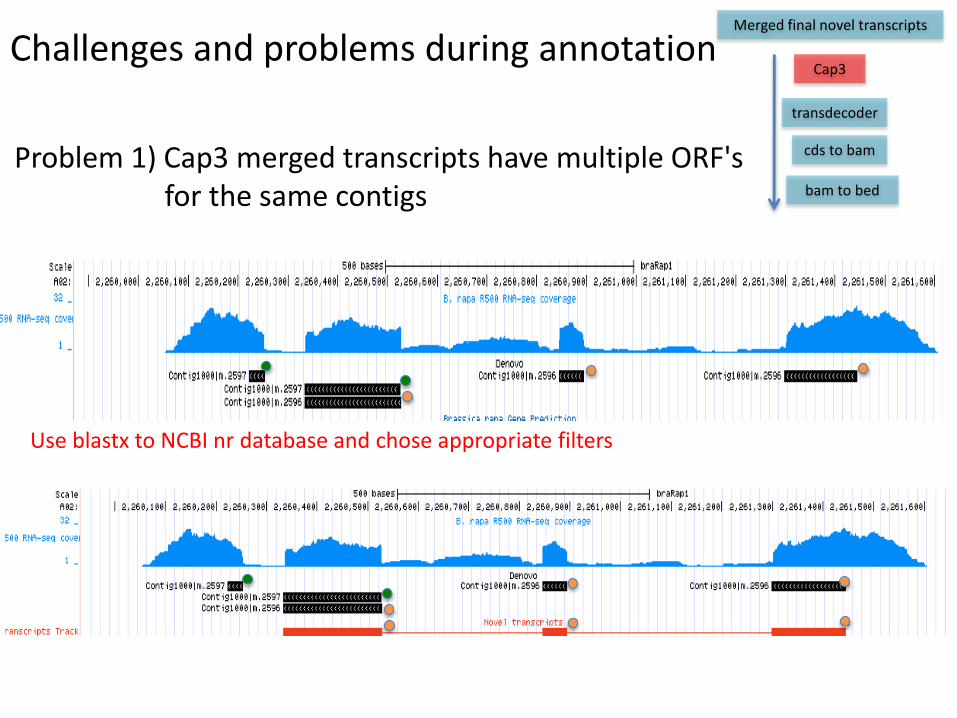
Problem 1) Cap3 merged transcripts have multiple ORF'sfor the same contigs
Challenges and problems during annotationCap3
transdecoder
cds to bam
Merged final novel transcripts
bam to bed
Use blastx to NCBI nr database and chose appropriate filters
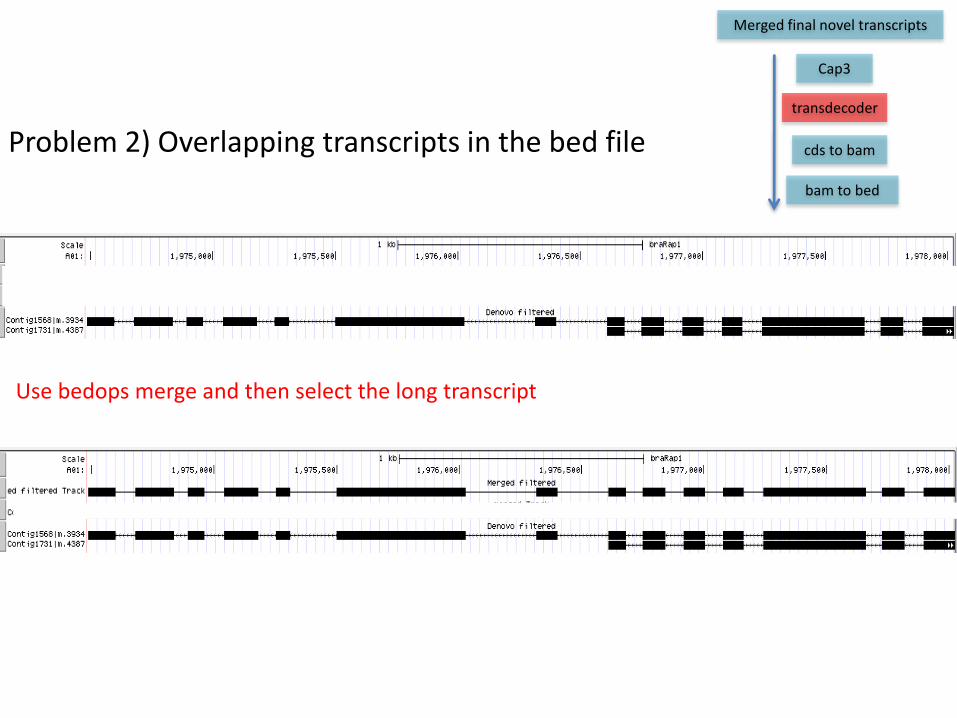
Problem 2) Overlapping transcripts in the bed file
Use bedops merge and then select the long transcript
transdecoder
cds to bam
Merged final novel transcripts
bam to bed
Cap3
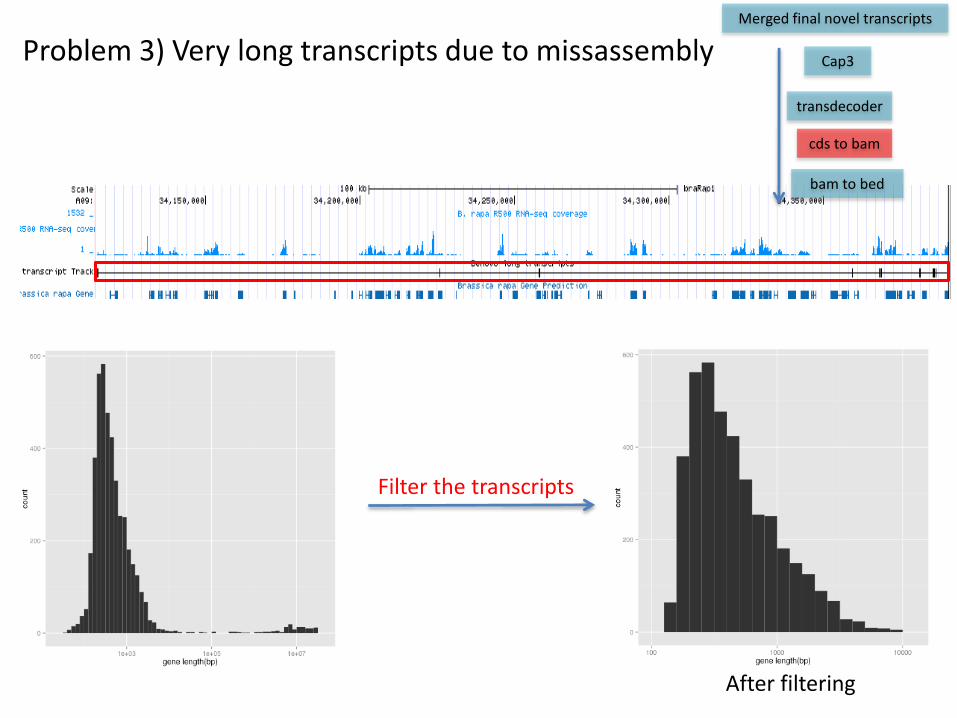
Problem 3) Very long transcripts due to missassembly
transdecoder
cds to bam
Merged final novel transcripts
bam to bed
Filter the transcripts
After filtering
Cap3

Problem 4) No lines connecting the exons in bed file
Use a custom script and join the lines. Check UCSC bed file guideline
cds to bam
Merged final novel transcripts
bam to bed
Cap3
transdecoder
UK Devisetty et al. 2014 G3: Genes|Genomes|Genetics
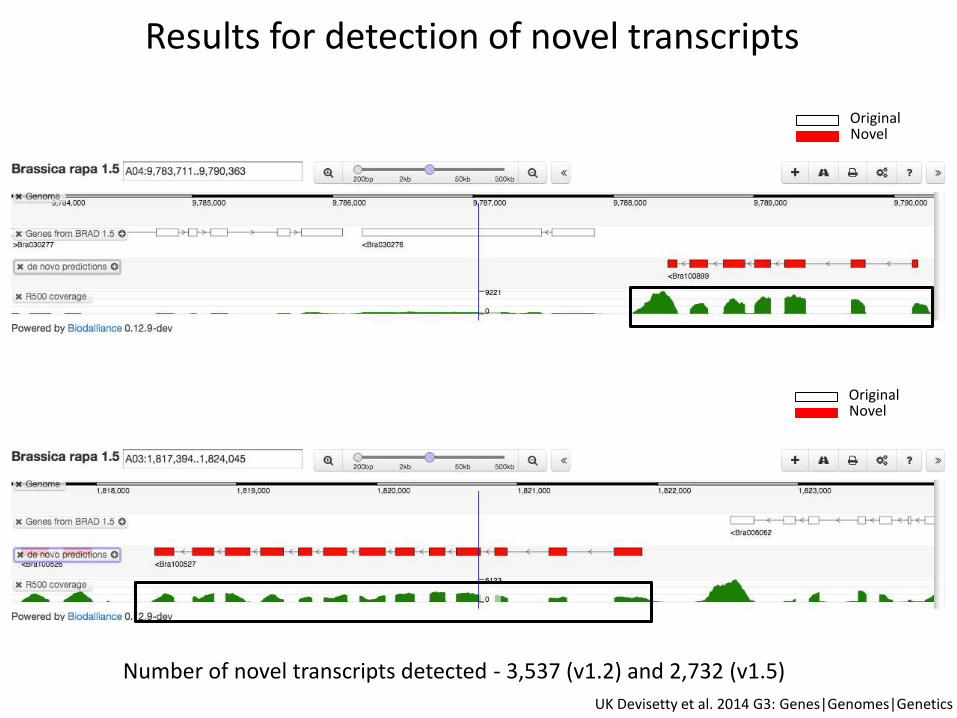
Results for detection of novel transcripts
Number of novel transcripts detected - 3,537 (v1.2) and 2,732 (v1.5)
OriginalNovel
OriginalNovel
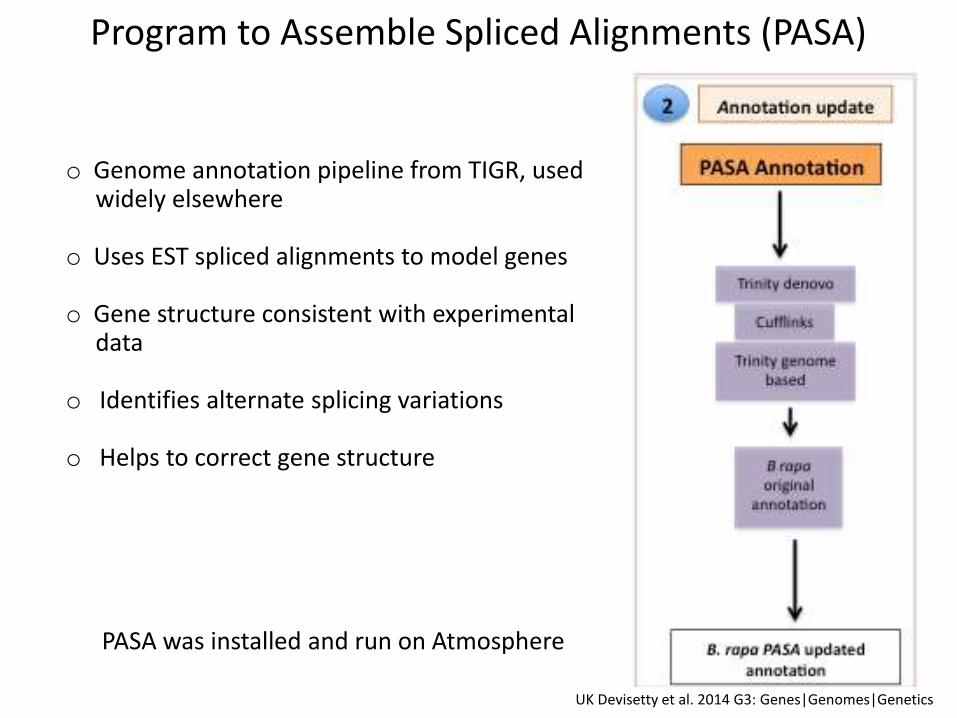
o Genome annotation pipeline from TIGR, used widely elsewhere
o Uses EST spliced alignments to model genes
o Gene structure consistent with experimental data
o Identifies alternate splicing variations
o Helps to correct gene structure
Program to Assemble Spliced Alignments (PASA)
UK Devisetty et al. 2014 G3: Genes|Genomes|Genetics
PASA was installed and run on Atmosphere
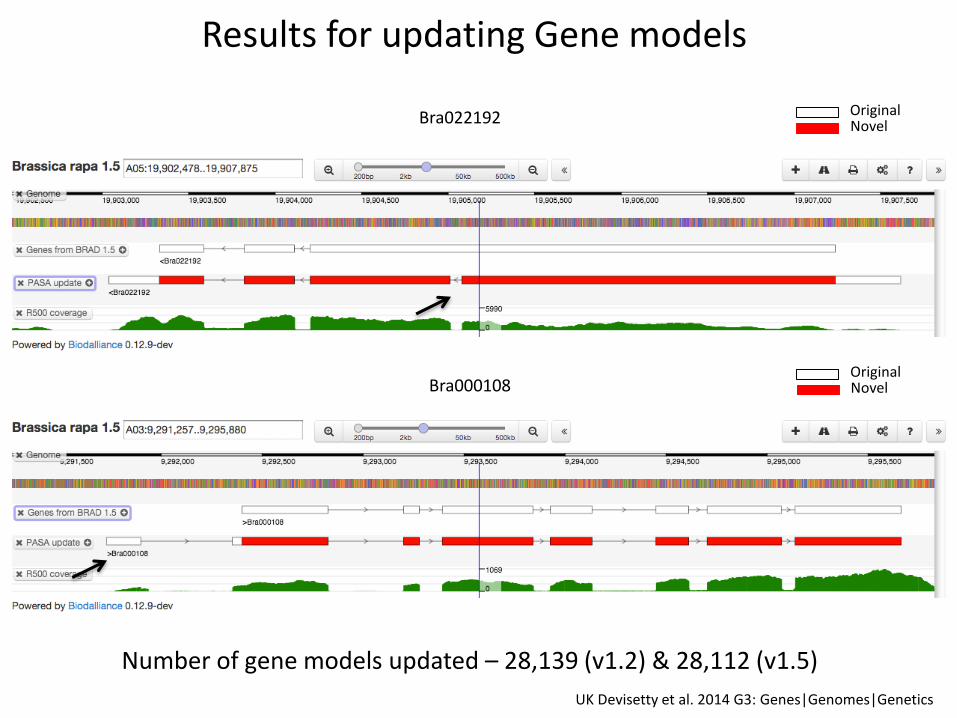
Number of gene models updated – 28,139 (v1.2) & 28,112 (v1.5)
UK Devisetty et al. 2014 G3: Genes|Genomes|Genetics
Results for updating Gene models
OriginalNovelBra000108
OriginalNovelBra022192
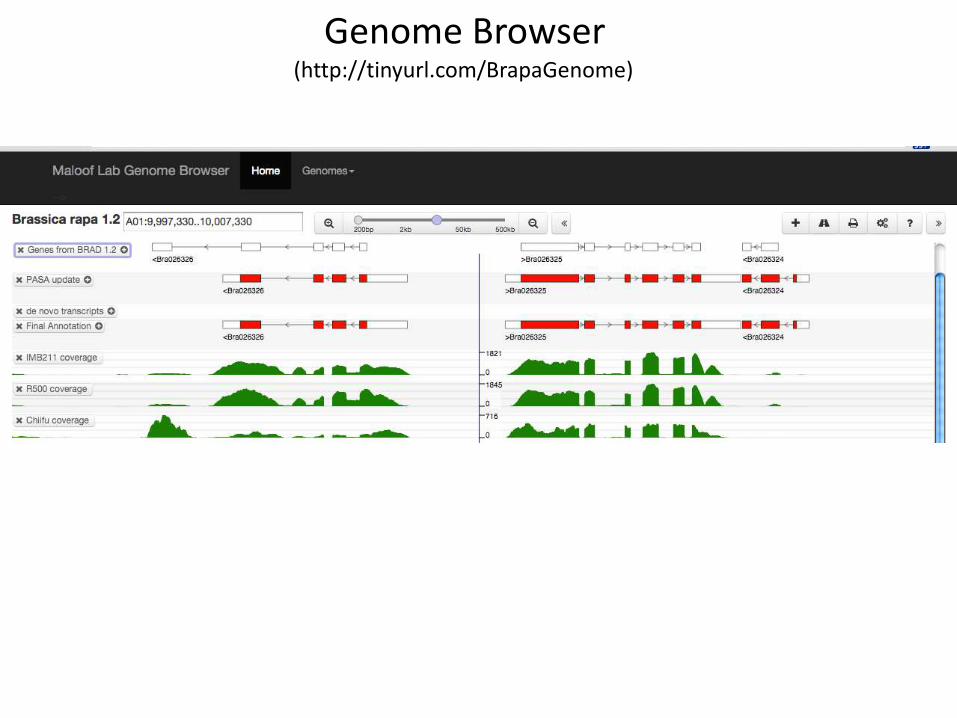
Genome Browser(http://tinyurl.com/BrapaGenome)

Conclusions
• Deep RNA-Seq provides enough coverage for the detection of a large number unknown transcripts and genome improved annotation
• Neither de novo assembly nor reference-based category is the best choice and hybrid assembly can offer more accurate assembly and annotation
• Problems during genome re-annotation needs to be addressed before a fully annotated genome is obtained
• iPlant Collaborative and XSEDE provides the systems andpeople to facilitate transcriptome assembly and genome reannotation

ACKNOWLEDGEMENTS
• Julin Maloof
• Mike Covington
• Cody Markelz
• An Tat
• Kazu Nozue
• Saradadevi Lekkala
• Maloof lab
• Harmer lab
• Cynthia Weinig
• Marc T. Brock
• Matthew Rubin
• Brian Haas
• Andy Edmonds
• Edwin Skidmore
• Sangeeta Kuchimanchi
• Matt Vaughan





























