Embed Size (px)
Citation preview

Proc. Natl. Acad. Sci. USAVol. 81, pp. 71-75, January 1984Biochemistry
Isolation of transforming sequences of two human lung carcinomas:Structural and functional analysis of the activatedc-K-ras oncogenes
(DNA transfection assays/molecular cloning/restriction endonuclease mapping/DNA sequences)
HIROFUMI NAKANO, FUMIICHIRO YAMAMOTO, CRAIG NEVILLE, DOUGLAS EVANS, TAKESHI MIZUNO*,AND MANUEL PERUCHOtDepartment of Biochemistry, State University of New York, Stony Brook, NY 11794
Communicated by Sarah Ratner, September 12, 1983
ABSTRACT Human lung tumors PR310 and PR371maintained in nude mice contain activated c-K-ras oncogenesdetectable by the ability of their DNAs to induce the morpho-logical transformation of NIH 3T3 mouse fibroblasts. Usingphage libraries constructed with DNA from NIH 3T3 mousefibroblast transformants, we have isolated human sequencesthat span >40 kilobase pairs of the c-K-ras oncogene. Based onthe conservation of these human sequences in mouse fibroblasttransformants, we conclude that the transforming ability ofthe oncogene activated in these tumors resides within a 43- to46-kilobase-pair DNA region. No clear differences were ob-served between the structures of the PR310 and PR371 clonedoncogene sequences. Nucleotide sequence analysis in concertwith DNA transfection experiments suggests that the PR371oncogene has been activated by a single base change in the firstexon, which results in the substitution of cysteine for glycine inposition 12 of the predicted amino acid sequence. The geneticalteration responsible for the transforming activity of thePR310 oncogene, however, does not reside in the first exon.These results indicate that the activation of the c-K-ras onco-gene in human lung cancer can occur by different mutationalevents.
Several oncogenes of human tumors have been detected bytheir ability to induce morphological transformation of thegrowth-controlled mouse fibroblast cell line NIH 3T3 (1, 2).We found an oncogene common to two human lung carcino-ma cell lines (Calu-1 and SK-LU-1) and one human coloncarcinoma (SK-CO-1) cell line (3). This oncogene has beenfound activated in several other human tumors (4) and tumorcell lines (4, 5) and has been identified as the human homo-logue (human c-K-ras) (6) of the Kirsten murine sarcoma vi-rus oncogene (v-K-ras) (4, 7, 8).Other human oncogenes active in the NIH 3T3 assay also
belong to the ras gene family. The oncogene of bladder carci-noma (T24-EJ) and lung carcinoma (Hs242) cell lines is thehuman homologue (human c-H-ras) (6) of the Harvey murinesarcoma virus oncogene (v-H-ras) (7, 9, 10). A third mem-ber, N-ras, has been found in other human tumor cell lines(8, 11, 12).The human c-H-ras oncogene has been cloned from the
T24-EJ and Hs242 cell lines, and the molecular nature of itsactivation has been established as a single point mutation inthe protein-encoding region of the oncogene (13-17). In con-trast with the c-H-ras and N-ras oncogenes, the size of thehuman c-K-ras oncogene appeared to be much larger andhas been estimated to be >30 kilobase pairs (kbp) (3). Thislarge size has hampered the analysis of the structure and ofthe nature of the molecular alteration undergone by the c-K-ras oncogene in some human tumors.
We report here the isolation by molecular cloning of over40 kbp of DNA of the human c-K-ras oncogene from twohuman lung tumors propagated in nude mice and the com-parative analysis of the nature of the mutation responsiblefor their activation in these tumors.
MATERIALS AND METHODSCell Cultures and Gene Transfer Experiments. Calu-1 and
SK-LU-1 are human tumor cell lines, and their origin hasbeen described (3). PR310 and PR371 (passages 8 and 2, re-spectively) are human lung adenocarcinomas (18) that havebeen maintained in nude mice and were kindly provided byJ. Fogh. DNAs from these tumors and tumor cell lines wereintroduced into NIH 3T3 cells by the calcium phosphatetransfection method as described (3).DNA Preparation and Purification of DNA Fragments.
High molecular weight DNA was prepared from NIH 3T3transformants as described (3). To prepare DNA from thehuman tumors maintained in nude mice, frozen tumor tis-sues were ground in a steel Waring blender in the presence ofliquid nitrogen. After the liquid nitrogen evaporated, the tu-mor powder was lysed with a hot (65°C) lysis buffer contain-ing 0.5% NaDodSO4, 20 mM EDTA, 10 mM Tris-HCl (pH7.5), 0.15 M NaCl, and 200 ,ug of proteinase K (Beckman)per ml. DNA was then purified by extraction with phe-nol/chloroform as described (3).
Restriction endonuclease fragments from cellular, plas-mid, or bacteriophage DNA were fractionated by agarose gelelectrophoresis and purified by the KI/glass powder method(8) with slight modifications. Agarose strips containing theDNA fragments were dissolved in saturated KI, and the so-lution was centrifuged at 1,000 x g through 0.1 ml of glasspowder packed in a pipette tip. The column was washedtwice with 1 ml of 70%o KI and once with a buffer containing0.1 M NaCl, 5 mM Tris (pH 7.5), and 5 mM EDTA in 50%ethanol. The DNA was then eluted with 50-100 ,ul of 1 mMTris'HCl, pH 7.5/1 mM EDTA. DNA fragments purified inthis way were used in nick translations, in vitro ligations,restriction endonuclease digestions, and gene transfer ex-periments.
Construction of Partial Phage Libraries. DNA from NIH3T3 secondary and tertiary transformants, derived from lungtumor DNA, was digested to completion with the restrictionendonucleases HindIll, BamHI, or Xba I, and fragments ofdifferent sizes were fractionated by agarose gel electrophore-sis and purified by the KI/glass powder method. DNA of theX47.1 (19) or Xgt-wes XB (20) phage vectors was digested alsowith these enzymes, and the annealed vector arms were pu-rified as above.
Abbreviation: kbp, kilobase pair(s).*Present address: Mitsubishi-Kasei Institute of Life Sciences, 11Minamiooya, Machida-shi, Tokyo, Japan.tTo whom reprint requests should be addressed.
71
The publication costs of this article were defrayed in part by page chargepayment. This article must therefore be hereby marked "advertisement"in accordance with 18 U.S.C. §1734 solely to indicate this fact.
Dow
nloa
ded
by g
uest
on
Apr
il 2,
202
0

72 Biochemistry: Nakano et al.
The purified DNA inserts and phage arms were incubated,at a molar ratio of 1:2 and a concentration of 100-300 ;Lg/ml,with T4 ligase at 90C for 24-40 hr. Partial phage librarieswere constructed by in vitro packaging of the ligated DNA asdescribed by Hohn (21). The phage libraries were screenedfor the presence of human oncogene sequences by the meth-od of Benton and Davis (22), with different DNA fragmentsof the oncogene previously subcloned in plasmid vectors(Fig. 1) as radioactive probes.DNA, Sequence Analysis. The nucleotide sequence of the
first exon of the PR310 and PR371 cloned oncogenes wasdetermined by the procedure ofMaxam and Gilbert (24). Thesequence of the first exon of the PR310 transforming genewas determined by reading both strands (i) from the HinfIsite towards the 5' end after labeling with the Klenow en-zyme and (ii) from the Stu I site towards the 3' end afterlabeling with the T4 polynucleotide kinase. The sequence ofthe first exon of the PR371 oncogene was determined afterthe Stu I-Xba I fragment was subcloned into the Sma I-XbaI site of pchtk5. pchtk5 is the 3-kbp HindIII fragment ofXchtk-1, a Charon 4A derivative containing the chicken thy-midine kinase gene (25) cloned into the HindIII site ofpBR322. The HindIII site of the recombinant plasmid locat-ed at 56 bp from the Sma I-Stu I site was end-labeled withthe Klenow enzyme and the sequence was determined to-wards the 3' end of the oncogene.
RESULTSStrategy for Cloning the Oncogenes of PR310 and PR371.
PR371 and PR310 are human lung tumors that have beenmaintained by successive passages in nude mice. DNA fromthese tumors was able to induce foci of morphologicallytransformed NIH 3T3 cells with efficiencies of 0.4 and 0.05foci per ;kg, respectively. NIH 3T3 secondary and tertiarytransformants were derived by successive transfection cy-
cles as described (3). The Alu blot pattern indicated that theoncogene activated in these human lung tumors was thesame as that previously identified in the Calu-1, SK-LU-1,and SK-CO-1 cell lines (3) (Fig. 2A).A fragment of the oncogene of Calu-1 had been cloned pre-
viously by constructing complete genomic libraries in Char-on 4A vectors by partially digesting DNA of an NIH 3T3secondary transformant with EcoRI and screening with theBLUR8 clone containing human repetitive sequences. Over-lapping fragments of the Calu-1 oncogene were isolated fur-ther by screening the libraries with restriction fragmentscontained in the first isolated phage, XL2-3.4 (8).We used a different approach for cloning the oncogenes
from PR310 and PR371. DNA fragments of XL2-3.4 wereused to determine the position of contiguous restriction en-donuclease sites located outside the cloned region of the on-cogene by genomic blots of NIH 3T3 transformant DNAs.Once a restriction endonuclease fragment of a convenientsize was found, it was cloned in appropriate phage vectorsby complete digestion and size fractionation of mouse trans-formant DNAs.
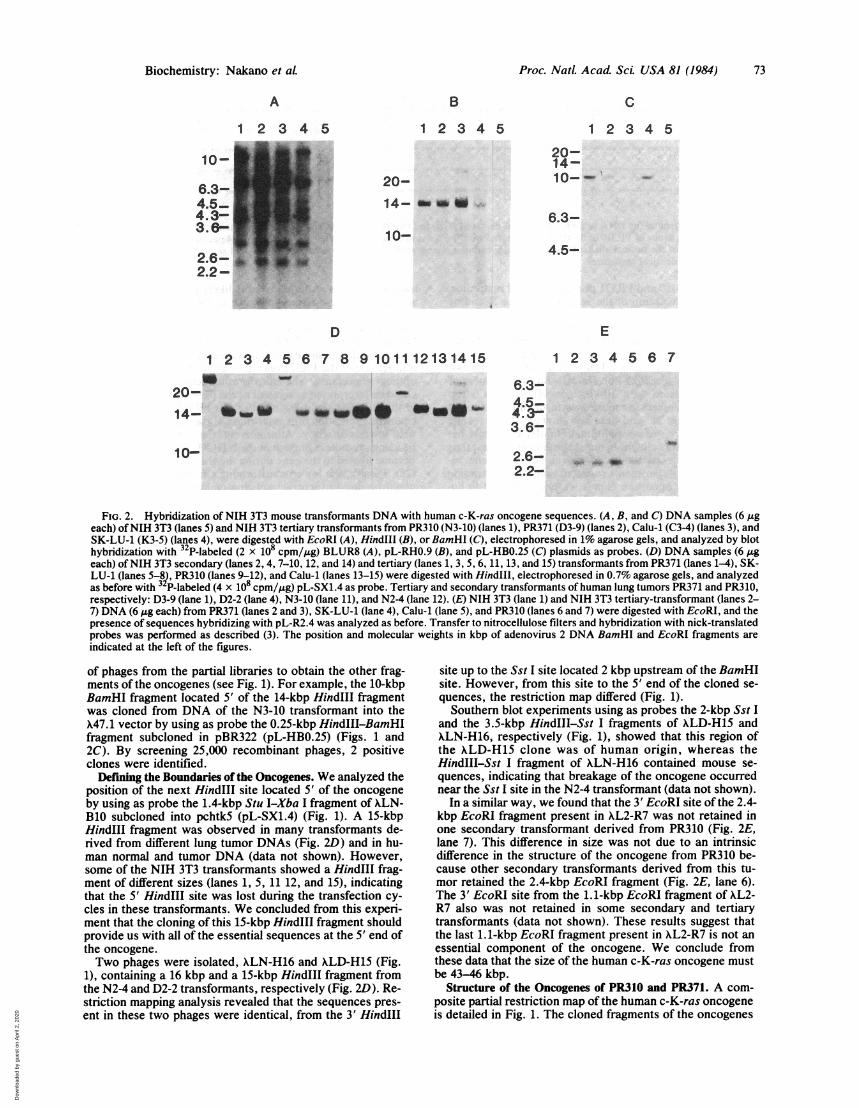
Thus, the 0.93-kbp EcoRI-HindIII fragment of XL2-3.4subcloned in pBR322 (pL-RH0.9) (Fig. 1) was used in ourinitial cloning step, to determine the position of the nextHindIII site in the oncogene. We analyzed several NIH 3T3transformants derived from DNAs of different human lungtumors. In all cases a 14-kbp HindIII fragment was foundthat hybridized with the probe (Fig. 2B). The 14-kbp HindIIIfragment of NIH 3T3 tertiary transformant N3-10 (Fig. 2B),derived from DNA of PR310, was cloned into the X47.1 vec-tor by screening the library with pL-RH0.9 as described.From =150,000 recombinant phages constructed by in vitropackaging of the enriched 14-kbp HindIII fragment, 88 posi-tive plaques were detected. The cloning procedure was thussimplified because it was sufficient to screen a lower number
pL-SX1 .4
BRR l
pL-HBO.25
R HB RI 11
pL-R3.0 pL-RHO.9
R R RR H11
pL-R3.1 pL-R2.4
R R RH R H RH RH RI/ If
_----IL I -_II _i
PR3lO '______________'-HRS~g )LN-BlO ,___________________________
*~~~~~~~~I11 -4 gx m PP gs mBg S X B
~kLD-H14XLN-H16 XLN-HIO
PR3710LD-H15 A, __==._____. _.. _l_L -3
)LC - H14 XL2-R7Calu -1
-5 Kbp 0 10 20 30 40
FIG. 1. Partial restriction map of the human c-K-ras oncogene. A composite restriction map of the c-K-ras oncogene activated in PR310,PR371, and Calu-1 human lung carcinomas was derived from the restriction maps of the DNA fragments cloned in overlapping or contiguousphage vectors. XLN-H16 and XLD-H15 are X47.1 Hind.ll derivatives constructed with DNA of NIH 3T3 secondary transformants N2-4 and D2-2 (see Fig. 2) derived from PR310 and PR371 tumor DNA, respectively. XLN-B10 is a X47.1 BamHI recombinant from N3-10 tertiary transfor-mant of PR310. XLN-H14, XLD-H14, and XLC-H14 contain the 14-kbp HindIII fragments of N3-10, D3-9, and C3-4 tertiary transformants ofPR310, PR371, and Calu-1 (Fig. 2) cloned into X47.1. XLN-H1O and XLD-H10 are recombinant X47.1 phages containing a 10.5-kbp HindIIIfragment from N3-10 and D3-9 transformants, respectively. XLN-X8 is a Xgt-wes XB derivative containing an 8.5-kbp Xba I fragment from N3-10 transformant. XL2-3.4 and XL2-R7 are Charon 4A clones containing sequences of a Calu-1-derived NIH 3T3 secondary transformant (8). Thetop of the figure shows maps of recombinant plasmids containing fragments of the oncogene sequences: pL-SX1.4 is the 1.4-kbp Stu I-Xba Ifragment of XLD-H15 cloned into the Sma I-Xba I sites of pchtk5; pL-HBO.25 is a pBR322 derivative containing the 0.25-kbp HindIII-BamHIfragment of XLN-H14; pL-R3.0, pL-R3.1, and pL-R2.4 are the 3.0-, 3.1-, and 2.4-kbp EcoRI fragments of XLN-H14, XL2-3.4, and XL2-R7,respectively, cloned into the EcoRI site of pBR322; and pL-RHO.9 is the 0.93-kbp EcoRI-HindIII fragment of XL2-3.4 cloned into the EcoRI-Hindill site of pBR322. These plasmids were used as radioactive probes to screen the partial genomic libraries and to analyze the boundaries ofthe oncogene. Restriction endonuclease cleavage sites: B, BamHI; Bg, BgI II; R, EcoRI; H, HindIII; P, Pvu II; Sm, Sma I; S, Sst I; X, Xba I;
and Xh, Xho I. m, Location of the first four regions of the oncogenes hybridizing with the v-k-ras oncogene (clone HiHi3) (23); m, position inthe 4.8-kbp and 2.4-kbp EcoRI fragments located at the 5' and 3' ends of the oncogene, respectively, of two other regions hybridizing with theviral clone pKBE-2 (23) but not with the clone HiHi3; S, location of DNA fragments that contain Alu repeat sequences. The direction oftranscription was determined by hybridization with specific restriction fragments of the v-K-ras clones.
H R
Proc. NatL Acad Sci. USA 81 (1984)
Dow
nloa
ded
by g
uest
on
Apr
il 2,
202
0
Proc. Natl. Acad Sci. USA 81 (1984) 73
A
1 2 3 4 5
B
1 2 3 4 5
C
1 2 3 4 5
10-
6.3-4.5-.4.3-3.6-'
2.6-2.2-
D
20-14- swm 0
10-
1 2 3 4 5 6 7 8 9101112131415
a~~~20- _
14- vto _ot o Wa_
10-
1 2 3 4 5 6 7
6.3-
4.5-4.3-3.6-
2.6-2.2-
FIG. 2. Hybridization of NIH 3T3 mouse transformants DNA with human c-K-ras oncogene sequences. (A, B, and C) DNA samples (6 ,ugeach) ofNIH 3T3 (lanes 5) and NIH 3T3 tertiary transformants from PR310 (N3-10) (lanes 1), PR371 (D3-9) (lanes 2), Calu-1 (C3-4) (lanes 3), andSK-LU-1 (K3-5) (lanes 4), were digested with EcoRI (A), HindIII (B), or BamHI (C), electrophoresed in 1% agarose gels, and analyzed by blothybridization with 32 P-labeled (2 x 108 cpm/,ug) BLUR8 (A), pL-RHO.9 (B), and pL-HBO.25 (C) plasmids as probes. (D) DNA samples (6 ,geach) ofNIH 3T3 secondary (lanes 2, 4, 7-10, 12, and 14) and tertiary (lanes 1, 3, 5, 6, 11, 13, and 15) transformants from PR371 (lanes 1-4), SK-LU-1 (lanes 5-8), PR310 (lanes 9-12), and Calu-1 (lanes 13-15) were digested with HindIlI, electrophoresed in 0.7% agarose gels, and analyzedas before with 3 P-labeled (4 x 108 cpm/,ug) pL-SX1.4 as probe. Tertiary and secondary transformants of human lung tumors PR371 and PR310,respectively: D3-9 (lane 1), D2-2 (lane 4), N3-10 (lane 11), and N24 (lane 12). (E) NIH 3T3 (lane 1) and NIH 3T3 tertiary-transformant (lanes 2-7) DNA (6 Ag each) from PR371 (lanes 2 and 3), SK-LU-1 (lane 4), Calu-1 (lane 5), and PR310 (lanes 6 and 7) were digested with EcoRI, and thepresence of sequences hybridizing with pL-R2.4 was analyzed as before. Transfer to nitrocellulose filters and hybridization with nick-translatedprobes was performed as described (3). The position and molecular weights in kbp of adenovirus 2 DNA BamHI and EcoRI fragments are
indicated at the left of the figures.
of phages from the partial libraries to obtain the other frag-ments of the oncogenes (see Fig. 1). For example, the 10-kbpBamHI fragment located 5' of the 14-kbp HindIII fragmentwas cloned from DNA of the N3-10 transformant into theX47.1 vector by using as probe the 0.25-kbp HindIII-BamHIfragment subcloned in pBR322 (pL-HBO.25) (Figs. 1 and2C). By screening 25,000 recombinant phages, 2 positiveclones were identified.
Defining the Boundaries of the Oncogenes. We analyzed theposition of the next HindIII site located 5' of the oncogeneby using as probe the 1.4-kbp Stu I-Xba I fragment of XLN-B10 subcloned into pchtk5 (pL-SX1.4) (Fig. 1). A 15-kbpHindIII fragment was observed in many transformants de-rived from different lung tumor DNAs (Fig. 2D) and in hu-man normal and tumor DNA (data not shown). However,some of the NIH 3T3 transformants showed a HindIII frag-ment of different sizes (lanes 1, 5, 11 12, and 15), indicatingthat the 5' HindIII site was lost during the transfection cy-cles in these transformants. We concluded from this experi-ment that the cloning of this 15-kbp HindIII fragment shouldprovide us with all of the essential sequences at the 5' end ofthe oncogene.Two phages were isolated, XLN-H16 and XLD-H15 (Fig.
1), containing a 16 kbp and a 15-kbp HindIl fragment fromthe N2-4 and D2-2 transformants, respectively (Fig. 2D). Re-striction mapping analysis revealed that the sequences pres-ent in these two phages were identical, from the 3' HindIII
site up to the Sst I site located 2 kbp upstream of the BamHIsite. However, from this site to the 5' end of the cloned se-quences, the restriction map differed (Fig. 1).Southern blot experiments using as probes the 2-kbp Sst I
and the 3.5-kbp HindIII-Sst I fragments of XLD-H15 andXLN-H16, respectively (Fig. 1), showed that this region ofthe XLD-H15 clone was of human origin, whereas theHindIII-Sst I fragment of XLN-H16 contained mouse se-
quences, indicating that breakage of the oncogene occurrednear the Sst I site in the N2-4 transformant (data not shown).
In a similar way, we found that the 3' EcoRI site of the 2.4-kbp EcoRI fragment present in XL2-R7 was not retained inone secondary transformant derived from PR310 (Fig. 2E,lane 7). This difference in size was not due to an intrinsicdifference in the structure of the oncogene from PR310 be-cause other secondary transformants derived from this tu-mor retained the 2.4-kbp EcoRI fragment (Fig. 2E, lane 6).The 3' EcoRI site from the 1.1-kbp EcoRI fragment of XL2-R7 also was not retained in some secondary and tertiarytransformants (data not shown). These results suggest thatthe last 1.1-kbp EcoRI fragment present in XL2-R7 is not an
essential component of the oncogene. We conclude fromthese data that the size of the human c-K-ras oncogene mustbe 43-46 kbp.
Structure of the Oncogenes of PR310 and PR371. A com-posite partial restriction map of the human c-K-ras oncogeneis detailed in Fig. 1. The cloned fragments of the oncogenes
20-14-10-r
6.3-
4.5-
E
Biochemistry: Nakano et aL
Dow
nloa
ded
by g
uest
on
Apr
il 2,
202
0

74 Biochemistry: Nakano etaLP
1 10 20 30 37c-K-As MET TH GLU TYR LYS LEU VAL VAL VAL GLY ALA GLY GLY VAL GLY LYS SER ALA LEU THR ILE GLN LEU ILE GIN ASN HIS PHE VAL ASP GW TYR ASP PRO THR ILE GLUM310 A GGC CTG CTG AAA ATG ACT GMA TAT AMACTT GTG GTA GTT GGW GCT GGI IGGC GTA GGC AAG ACT GCC TTG ACG ATA CAG CTA ATT CAG MAT CATm GTG 6AC GMA TAT GAT CC4 ACA ATA GAG GTA MAT CT GTT
STw-1 1 50 HlINF-1.c-K-RAS CYS SPLICER671 ***** ******
SER GLN
v-K-HAS* A
GA** ** C *T T HG C
c-H-w CCCT TGA GlGCGG G "G 'G *G *C **C T C G C C C G C C C C C T G *G G GHU1A
FIG. 3. Comparison of the DNA sequence of the first exon of PR310 and PR371 oncogenes. The nucleotide sequence of the first exon regionof the transforming genes of PR310 and PR371 is compared with the previously reported sequences of the first 124 nucleotides of the v-K-rasoncogene (26) and of the normal allele of the T24 bladder carcinoma oncogene (13-16). The predicted amino acid sequence is also indicated.Asterisks indicate identical sequences.
from Calu-1, PR310, and PR371 present identical restrictionmaps (with the exception of the sequences at the 5' end ofthe oncogene). The 0.9-kbp EcoRI-HindIII fragment of XL2-3.4 hybridized to a 14-kbp HindIII fragment and to a 6.8-kbpEcoRI fragment present in NIH 3T3 transformant DNAs de-rived from Calu-1, SK-LU-1, PR310, and PR371 DNAs aswell as in the original tumor DNAs (Fig. 2B and data notshown). Similarly, the 0.25-kbp HindIII-BamHI fragment ofXLN-B10 hybridized to a 10-kbp BamHI and a 4.6-kbpEcoRI fragment present in mouse transformant DNAs and inhuman DNA (Fig. 2C and data not shown). These resultsindicate that there are no additional HindIII fragments be-tween the fragments cloned in our recombinant phages, thusdemonstrating the continuity of the cloned oncogene se-quences. Similar experiments revealed that other restrictionfragments of our oncogene clones were also present in hu-man normal and tumor DNAs. These results suggest thatthere are no gross alterations in the structure of the onco-
genes relative to the sequences of the normal proto-onco-gene and also that there are no clear differences in the struc-ture of the oncogenes activated in different tumors.
Sequence of the First Exon of the PR310 and PR371 Onco-genes. We determined the sequence of the region of thecloned oncogenes containing the putative first exon (see Fig.1). Comparison of the sequence of this region revealed thatthe two human oncogenes differed by a single base at posi-tion 34 (Fig. 3), which corresponds to the first base of codon12 of the predicted protein sequence. The oncogene ofPR310 contains deoxyguanosine, while this residue in thePR371 oncogene is thymidine. This difference results in theincorporation of glycine and cysteine in the predicted pro-teins of PR310 and PR371 oncogenes, respectively. Fig. 3also shows that the predicted amino acid sequence of thefirst exon of the PR310 oncogene is identical to that of thenormal allele of the T24-EJ bladder carcinoma oncogene.Transforming Activity of c-K-ras and c-H-ras Chimeric
Genes. We designed a functional assay to test if the presenceof cysteine at codon 12 of the PR371 oncogene was sufficientfor the acquisition of transforming activity of this gene. Weconstructed chimeric genes by substituting the first mutantexon of the bladder carcinoma oncogene (13-16) with thoseof the two lung carcinomas. Recombinant plasmids contain-ing the first exon of the PR310 or PR371 oncogenes, ligatedto the last three exons of the T24 oncogene (Table 1), were
analyzed for their ability to induce oncogenic transformationof NIH 3T3 cells. DNA of pLDBXP, which contains the firstexon of the PR371 oncogene, was able to induce foci of mor-phologically transformed NIH 3T3 cells compared with pTB-1, which contains the intact T24 bladder carcinoma trans-forming gene (Table 1).Because the chimeric gene containing the first exon of the
PR310 oncogene, like the normal T24 proto-oncogene (13-16), was not able to induce oncogenic transformation in NIH3T3 cells (Table 1), we conclude that the presence of thecysteine encoded in the first exon of the PR371 oncogenemust be the result of a mutational event. These results also
indicate that this mutation is most likely responsible for theactivation of the c-K-ras oncogene in PR371 tumor cells.The chimeric gene contained in pLDBXP showed a trans-
forming efficiency 20- to 50-fold lower than that of pTB-1(Table 1). The reason for this lower efficiency is not clear atpresent. One possibility is that sequences upstream of theBamHI site of the PR371 oncogene could be required for itsoptimal expression. The transforming activity of pLDBXPcould be interpreted as a result of the presence of sequencesin the BamHI-Xba I fragment of the PR371 oncogene thatenable the initiation of transcription of the chimeric gene butwith lower efficiency. Alternatively, the hybrid gene couldbe using promoters present in the NIH 3T3 carrier DNA thatbecame ligated to the gene during the transfection procedure(23). Finally, the lower transforming efficiency of pLDBXPcould be due to incorrect splicing or lower stability of themRNA caused by the presence of an abnormal intron be-tween the first and second exons of the chimeric gene. Addi-tional experiments will be necessary to decide between thesepossibilities.
DISCUSSIONThe ras genes are a family of well-conserved genes in highereukaryotes encoding immunologically related proteins of Mr21,000 (p21), whose physiological function is unknown (28).
Table 1. Transfection of NIH 3T3 cells with recombinantplasmids containing chimeric c-K-ras-c-H-ras oncogenes
Foci
Plasmid Total no.* No. per ngtpXP2.1 0, 0 <0.001pLDBXP 163, 85 0.41 ± 0.13pLNBXP 0, 0 <0.001pTB-1 508, 615 11.2 ± 1.1
Purified plasmid DNAs were digested to completion with BamHI,and 300 ng each of pXP2.1, pLDBXP, and pLNBXP and 50 ng ofpTB-1 were mixed with 60 jig of NIH 3T3 carrier DNA. Transfec-tion assays were performed by the addition of 30 pg of DNA to eachof two plates of NIH 3T3 cultures as described (3), and foci of mor-phologically transformed cells were scored after 15-20 days. pXP2.1is the 2.1-kbp Xba I-Pvu II fragment of pTB-1 (27), which containsthe last three coding exons of the T24 c-H-ras oncogene, insertedinto the Xba I-Sma I site of pchtk-2 (25). pLDBXP and pLNBXPwere constructed by insertion of the 4.6-kbp BamHI-Xba I frag-ments of XLDH15 and XLNH16, respectively (Fig. 1), into theBamHI-Xba I site of pXP2.1. Therefore, these plasmids contained achimeric gene generated by ligation in the correct orientation of thefirst exon of the c-K-ras oncogene from either PR371 (pLDBXP) orPR310 (pLNBXP), with the last three exons of the c-H-ras T24 on-cogene. pTB-1 is a pBR322 derivative containing the T24 bladdercarcinoma transforming gene (27). The Pvu II site at 2.1 kbp 3' of theXba I site of pTB-1 is located outside the essential region of the T24oncogene (16, 27).*In two independent experiments.tAverage values of foci per ng of recombinant plasmid from the twodifferent experiments.
Proc. Natl. Acad Sci. USA 81 (1984)
Dow
nloa
ded
by g
uest
on
Apr
il 2,
202
0

Proc. NatL Acada Sci USA 81 (1984) 75
In a variety of human tumors, these genes have undergonestructural alterations that confer to them oncogenic capabili-ty in the NIH 3T3 assay.The activation mechanism of the transforming gene of
T24-EJ human bladder carcinoma cell lines (c-H-ras) hasbeen established as a single point mutation in codon 12 of thefirst exon (13-16). More recently, the human c-H-ras onco-gene has been found activated in a different tumor, and themutation was localized in the second exon (17). Thus, it ap-pears that the activation of the same oncogene (c-H-ras) canoccur by single substitutions of different amino acids in thep21 protein.We molecularly cloned over 40 kbp of human DNA se-
quences from NIH 3T3 transformants derived from DNA oftwo human lung tumors, PR371 and PR310. This allowed usto perform a comparative analysis of the mechanisms bywhich the c-K-ras oncogene has been independently activat-ed in two naturally occurring human lung carcinomas.We found that the oncogene of PR371 was activated by a
single base substitution in the first exon similar to the activa-tion of T24-EJ bladder carcinoma oncogene and that the ge-netic alteration responsible for the transforming activity ofthe PR310 oncogene does not reside in the first exon. We donot know the nature of the mutation of the PR310 oncogene,although we like to speculate that this transforming genecontains a point mutation in another protein-encoding re-gion. Whatever the mechanisms, our studies demonstratethat the activation of the same proto-oncogene (c-K-ras) canoccur by different mutational events in human tumor cells.
While other types of activation of the ras oncogenes couldalso occur in other tumors, the examples studied so far indi-cate that this type of point mutation is found often. Thiscould simply be due to a selection by the NIH 3T3 systemused in these studies. Only genes able to transform the recip-ient cells in a single step and able to induce the transformedphenotype when present in a single or a few copies in thehost genome will be detected in this assay. Although thefunctional role of the p21 protein encoded by the ras proto-oncogenes is not understood, it has become clear from thesestudies that mutations altering the structure of specific do-mains in ras gene-encoded proteins could be an importantand common step in some human cancers. The cause ofthese point mutations and the role that these oncogenes playin the initiation progression or maintenance of the humantumors remain to be established.
After this manuscript was completed, similar studies onthe structure of human c-K-ras oncogenes were reported(29, 30). Our results and those of these reports are essentiallyin agreement. The reasons for the slight differences observedin the restriction maps of the c-K-ras oncogenes studied byus and the Calu-1 oncogene and the normal proto-oncogenestudied by these groups are not clear at present.
We thank C. Lama for her excellent technical assistance, J. Foghfor providing us with tumor specimens, J. Kwoh for sequence dataof the chicken thymidine kinase gene, E. M. Scolnick and R. Ellisfor providing the HiHi3 and pKBE-2 clones, M. Freundlich for read-ing the manuscript, J. Koenig for preparation of the manuscript, andM. Inouye for his generous support. This work was also supported
by grants from the New York Health Research Council (HRC 14-033) and the National Cancer Institute (CA 33021).
1. Weinberg, R. A. (1981) Biochim. Biophys. Acta 651, 25-35.2. Cooper, G. M. (1982) Science 218, 801-806.3. Perucho, M., Goldfarb, M., Shimizu, K., Lama, C., Fogh, J. &
Wigler, M. (1981) Cell 27, 467-476.4. Pulciani, S., Santos, E., Lauver, A. V., Long, L. K., Aaron-
son, S. A. & Barbacid, M. (1982) Nature (London) 300, 539-541.
5. Murray, M. J., Shilo, B. Z., Shih, C., Cowing, D., Hsu, H. W.& Weinberg, R. A. (1981) Cell 25, 355-361.
6. Chang, E. H., Gonda, M. A., Ellis, R. W., Scolnick, E. M. &Lowy, D. R. (1982) Proc. Natl. Acad. Sci. USA 79,4848-4852.
7. Der, C. J., Krontiris, T. G. & Cooper, G. M. (1982) Proc.Natl. Acad. Sci. USA 79, 3637-3640.
8. Shimizu, K., Goldfarb, M., Suard, Y., Perucho, M., Li, Y.,Kamata, T., Feramisco, J., Stavenzer, E., Fogh, J. & Wigler,M. H. (1983) Proc. Natl. Acad. Sci. USA 80, 2112-2116.
9. Parada, L., Tabin, C., Shih, C. & Weinberg, R. (1982) Nature(London) 297, 474-478.
10. Santos, E., Tronick, S. R., Aaronson, S. A., Pulciani, S. &Barbacid, M. (1982) Nature (London) 298, 343-347.
11. Murray, M. J., Cunningham, J. M., Parada, L. F., Dautry, F.,Lebowitz, P. .& Weinberg, R. A. (1983) Cell 33, 749-757.
12. Hall, A., Marshall, C. J., Spurr, N. K. & Weiss, R. A. (1983)Nature (London) 303, 396-400.
13. Tabin, C. J., Bradley, S. M., Bargmann, C. I., Weinberg,R. A., Papageorge, A. G., Scolnick, E. M., Dhar, R., Lowy,D. R. & Chang, E. H. (1982) Nature (London) 300, 143-148.
14. Reddy, E. P., Reynolds, R., Santos, E. & Barbacid, M. (1982)Nature (London) 300, 149-152.
15. Taparowsky, E., Suard, Y., Fasano, O., Shimizu, K., Gold-farb, M. & Wigler, M. (1982) Nature (London) 300, 762-765.
16. Capon, D. J., Chen, E. Y., Levinson, A. D., Seburg, P. H. &Goeddel, D. V. (1983) Nature (London) 302, 33-37.
17. Yuasa, Y., Srivastava, S. K., Dunn, C. Y., Rhim, J. S.,Reddy, E. P. & Aaronson, S. A. (1983) Nature (London) 303,775-779.
18. Fogh, J., Tiso, J., Orfeo, T., Sharkey, F. E., Daniels, W. P. &Fogh, J. M. (1980) J. Natl. Cancer Inst. 64, 745-751.
19. Loenen, W. A. & Brammar, W. J. (1980) Gene 10, 249-259.20. Leder, P., Tiemeier, D. & Enquist, L. (1977) Science 196, 175-
177.21. Hohn, B. (1979) Methods Enzymol. 68, 299-309.22. Benton, W. & Davis, R. (1977) Science 196, 180-182.23. Perucho, M., Hanahan, D. & Wigler, M. (1980) Cell 22, 309-
317.24. Maxam, A. M. & Gilbert, W. (1980) Methods Enzymol. 65,
499-560.25. Perucho, M., Hanahan, D., Lipsich, L. & Wigler, M. (1980)
Nature (London) 285, 207-210.26. Tsuchida, N., Ryder, T. & Ohtsubo, E. (1982) Science 217,
937-939.27. Goldfarb, M., Shimizu, K., Perucho, M. & Wigler, M. (1982)
Nature (London) 296, 404-409.28. Ellis, R. W., DeFeo, D., Shih, T. Y., Gonda, M. A., Young,
H. A., Tsuchida, N., Lowy, D. R. & Scolnick, E. M. (1981)Nature (London) 292, 506-511.
29. Shimizu, K., Birnbaum, D., Ruley, M. A., Fasano, O., Suard,Y., Edlund, L., Taparowsky, E., Goldfarb, M. & Wigler, M.(1983) Nature (London) 304, 497-500.
30. McGrath, J. P., Capon, D. J., Smith, D. H., Chen, E. Y., See-burg, P. H., Goeddel, D. V. & Levinson, A. D. (1983) Nature(London) 304, 501-506.
Biochemistry: Nakano et aL
Dow
nloa
ded
by g
uest
on
Apr
il 2,
202
0