Embed Size (px)
Citation preview

A Survey of the Role of Noncovalent Sulfur Interactions in DrugDesignBrett R. Beno,†,∥ Kap-Sun Yeung,‡,∥ Michael D. Bartberger,§ Lewis D. Pennington,§
and Nicholas A. Meanwell*,‡
†Department of Computer-Assisted Drug Design, Bristol-Myers Squibb Research and Development, 5 Research Parkway WallingfordConnecticut 06492, United States‡Department of Discovery Chemistry, Bristol-Myers Squibb Research and Development, 5 Research Parkway WallingfordConnecticut 06492, United States§Department of Therapeutic Discovery, Amgen Inc., One Amgen Center Drive Thousand Oaks California 91320, United States
ABSTRACT: Electron deficient, bivalent sulfur atoms havetwo areas of positive electrostatic potential, a consequence ofthe low-lying σ* orbitals of the C−S bond that are available forinteraction with electron donors including oxygen and nitro-gen atoms and, possibly, π-systems. Intramolecular interactionsare by far the most common manifestation of this effect, whichoffers a means of modulating the conformational preferencesof a molecule. Although a well-documented phenomenon, apriori applications in drug design are relatively sparse andthis interaction, which is often isosteric with an intramolecularhydrogen-bonding interaction, appears to be underappreciatedby the medicinal chemistry community. In this Perspective, wediscuss the theoretical basis for sulfur σ* orbital interactionsand illustrate their importance in the context of drug design and organic synthesis. The role of sulfur interactions in proteinstructure and function is discussed and although relatively rare, intermolecular interactions between ligand C−S σ* orbitals andproteins are illustrated.
■ INTRODUCTION
Sulfur is prevalent in biologically active natural products thatexploit its unique chemical attributes by deploying it in a widerange of heterocyclic arrangements. Prominent examples includethe fused ring systems associated with the penicillin and cepha-losporin β-lactam-based antibiotics and their synthetic homo-logues, trisulfide moieties that are triggers in the some of theenediyne DNA alkylating agents, disulfide-based cyclic dep-sipeptides, epothilones, sulfenylated diketopiperazines, bleomy-cin, and the thiazolyl peptide class of antibiotic (Figure 1).1−8
Sulfur is also a ubiquitous element in approved and experimentaldrugs, and although many are based on some of the naturalproducts noted above, the design of medicinally active, smallsynthetic molecules has frequently relied on the incorporation ofthis atom in a range of functionalities that take advantage of itsunique properties.9 These include sulfone and sulfonamidemoieties which can, for example, modulate overall polarity orionization state and provide convenient synthetic handles withwhich to generate analogues. Replacement of aromatic carbo-cycles or heterocycle rings with sulfur-containing heterocyclicrings provides a useful means of modulating substituenttrajectories that, depending on the regiochemistry, can be usedto optimize complementarity and fit within a ligand bindingpocket. This is most clearly illustrated by the structure−activityrelationships (SARs) associated with the P1 structural elements
of the inhibitors of the coagulation cascade enzyme factor Xathat are compiled in Table 1.10a,b In both of these series,the chlorothiophene makes close contact with Tyr228 in aninteraction that is generally regarded as hydrophobic in naturerather than a halogen bond but which may include an elec-trostatic component.10c,11 This structural element thus occupiesthe S1 recognition pocket of the enzyme that accommodatesarginine moieties in the natural substrates.10 The 40-folddifference in potency between the thiophene 1 and phenylhomologue 2 is reproduced in the 2,2′-bithiophene-biphenylmatched pair 3 and 4. The inhibitory potency of thiophene 5(rivaroxaban) is superior to both the para- and particularly themeta-chlorophenyl homologues 6 and 7, emphasizing theimportance of an accurate presentation of the chlorine atom toTyr228.
10 However, some of these compounds may benefit fromfavorable dipole−dipole interactions between elements of the S1substituents and proximal backbone amides of the enzyme thatexhibits some dependence on geometry.Rational control of the conformation of small molecules is a
cornerstone of both structure- and ligand-based moleculardesign.12 Chemical modification of a core scaffold or manipula-tion of a substituent designed to enrich the population of the
Received: December 1, 2014
Perspective
pubs.acs.org/jmc
© XXXX American Chemical Society A DOI: 10.1021/jm501853mJ. Med. Chem. XXXX, XXX, XXX−XXX

bound conformation of a molecule can lead to improved bindingaffinity independent of ligand−target contacts. Moreover, byselectively favoring the conformer recognized by a drug target,the population of unproductive conformations possessing littleor no affinity for the target of interest is reduced. This may lead toenhanced selectivity while mitigating off-target toxicity and/ormetabolic modification. Several strategies to influence conforma-tional preferences have been established and widely applied indrug design.12,13 Among these are introduction of intramolecularH-bonds, formation of both small and macromolecular ringsystems, utilization of attractive or repulsive nonbondingcontacts, exploitation of intramolecular dipole−dipole inter-actions, and the judicious deployment of fluorine atoms thattakes advantage of the gauche effect between this element and arange of functionalities.12−18 Certain nonoxidized sulfur atomsthat can be incorporated into drug-like molecules using a widerange of functionality, most commonly sulfur-containing hetero-cycles, can participate in attractive nonbonding interactions thatare proving to be useful in the control of molecular conformationbut which are probably underappreciated by the medicinalchemistry community.12a,19−22 This effect is based on thepresence of low lying C−S σ* orbitals on S atoms, giving rise tothe phenomenon referred to as σ-holes that possess positiveelectrostatic potential and are available for interaction withelectron donating atoms, particularly N and O.22 However, themajority of the examples of this kind of sulfur interaction havebeen noted in post facto analyses of crystallographic or otherstructural information, and there are relatively few examplesreported in the literature where this interaction has beenexploited in a prospective fashion.23,24 Indeed, in many examples
in which this effect can be clearly observed in X-ray cocrystalstructures, the phenomenon has failed to engender com-ment. The theoretical bases of the conformation-influencingeffects of these interactions have been well characterizedand widely demonstrated with model systems. However,these studies are largely restricted to the computational,crystallographic, spectroscopic, and materials science liter-ature.25
In this Perspective, we attempt to bridge the gap betweentheoretical, crystallographic, and spectroscopic studies of thesenonbonded interactions and their practical applications in drugdesign. This effect is analogous to halogen bonding althoughthe distinct geometry associated with sulfur σ* orbitals typicallyfavors an intramolecular interaction rather than the intermo-lecular effect that is more frequently associated with halogenbonding.10,11 However, examples of intermolecular sulfurinteractions between ligands and proteins that augment morecommon drug−target interactions are beginning to be docu-mented. Included among these are sulfur−aromatic interactionsand halogen bonds involving sulfur as the electron pair donor.These are discussed in the context of both sulfur-containingligands and peptide-based methionine, cysteine, and cystinemoieties in which associations extend beyond that of simplehydrophobic interactions. Both intra- and intermolecular inter-actions involving low-lying sulfur σ* orbitals have also beenimplicated in chemical reactivity, with electronic characteristicsof chemical systems responsible, in part, for specific kinetic,regiochemical, or even stereochemical outcomes. These will becommented on briefly as part of the individual sections which areorganized around a discussion of the theoretical background and
Figure 1. Sulfur-containing natural products.
Journal of Medicinal Chemistry Perspective
DOI: 10.1021/jm501853mJ. Med. Chem. XXXX, XXX, XXX−XXX
B

topology of sulfur interactions. This is followed by examplesof the application of O···S interactions and N···S associations indrug design where the range of functionalities capable ofinteracting with low-lying sulfur σ* orbitals is illustrated. In thefinal section, sulfur interactions with aromatic rings and halogenatoms are summarized and examples of intermolecularinteractions involving sulfur are described.
■ BACKGROUND THEORYThis section captures the theoretical basis for the examples whichfollow and utilizes calculations on model systems along withrelevant literature precedent to highlight key aspects of sulfurinteractions. One of the earliest examples of an intramolecularN···S interaction that stabilized a specific conformation wasreported in 1976 and involved N-phenylguanidino-substitutedthiazole adducts arising from the reaction of 4-aryl-3-arylimino-5-imino-1,2,4-thiadiazolidines with aryl cyanamides.26 Thesmall molecule single-crystal X-ray structure of 8 revealed asyn, coplanar arrangement of the electron-donating guanidinoN atom and the acceptor S atom of the thiadiazole ring whichwere separated by a distance of ∼2.5 Å. This is well within thesum of the van der Waals radii of the respective atoms whichis 3.35 Å (Table 2.).26a Theoretical calculations (vide infra)characterized the interaction of the nitrogen lone pair ofelectrons with the S−X σ* orbital as a conformationalstabilizing element and, subsequently, this interaction has
been identified in numerous examples encompassing a widevariety of chemotypes.
This syn orienting effect extends to biaryl systems in which theinteracting elements are oriented in a 2,2′-arrangement. TheX-ray structure of 2-(2′-thienyl)pyridine (9, Figure 2) provides
an archetype for the nearly coplanarizing effect of this interactionin biaryl systems (φSCCN = −3.0°; φCCCC = −4.5°).27 Thevast majority of examples in the literature involving an aromaticS···lone pair interaction that demonstrate this “s-cis-locked”conformational preference in the bound or free states can bethought of as variations on these two themes.28
■ THEORETICAL AND SPECTROSCOPIC STUDIESA combined spectroscopic and theoretical study of a family of2-acetylthiophenes demonstrates a 0.8 kcal/mol (B3LYP/6-311++G(3df,3p)) preference for the syn form of 2-acetylthiophene(10).29 A natural bond order (NBO) analysis demonstrates thatone of the two principal interactions stabilizing the syn forminvolves delocalization of the in-plane lone pair of the exocycliccarbonyl O atom into the C−S σ* orbital (Figure 3a).30 This synpreference is analogous to that determined for 2-acetylpyrrole(11) (ΔE = 5 kcal/mol) using identical methodology and, whilediminished compared to the latter (ΔE = 0.8 kcal/mol),contrasts sharply with the 1.45 kcal/mol preference for the antiform exhibited by 2-acetylfuran (12).31,32 The biaryl 2-(2′-thienyl)pyridine archetype 9 has also been studied by combinedspectroscopic and theoretical studies which have determined apreference for the syn form. This is predicted to be more stablethan the anti conformation by 0.7 kcal/mol (B3LYP/6-311++G(d,p)), in accord with the X-ray structure of 9 depicted inFigure 2.33 The lone pair interactions with the C−S σ* orbital
Table 1. Enzyme Inhibitory Activity Associated with TwoSeries of Factor Xa Inhibitor that Illustrates the Importance ofa Thiophene Ring to Correctly Orient the P1Moiety in the S1Pocket of the Enzyme
Table 2. van der Waals Radii of Sulfur, Oxygen, and Nitrogenand Their Respective Sums
vdW radius (Å) ∑ X + S (Å)
S 1.80O 1.52 O + S: 3.32N 1.55 N + S: 3.35
Figure 2. Small molecule single-crystal X-ray structure of2-(2′-thienyl)pyridine (9) illustrating an overall planar topographyfavored by an attractive N···S interaction, d1 = 2.9 Å, φSCCN = −3.0°;φCCCC= −4.5°.27
Journal of Medicinal Chemistry Perspective
DOI: 10.1021/jm501853mJ. Med. Chem. XXXX, XXX, XXX−XXX
C
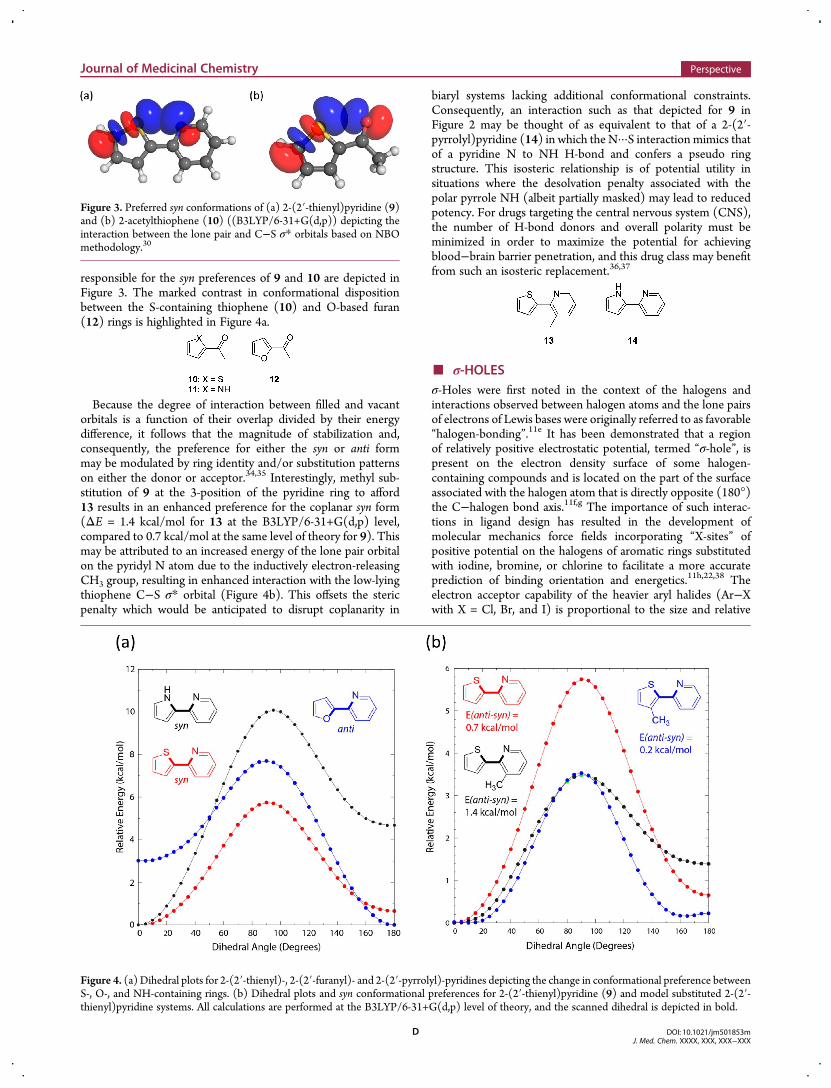
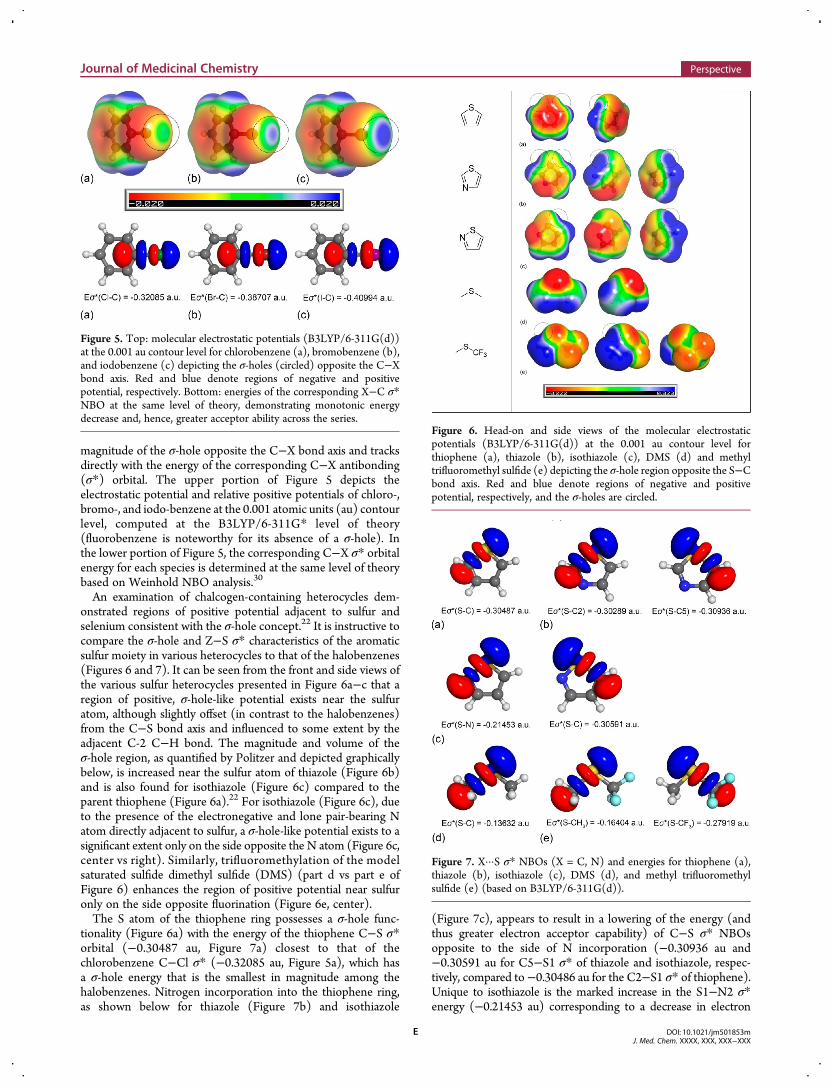
responsible for the syn preferences of 9 and 10 are depicted inFigure 3. The marked contrast in conformational dispositionbetween the S-containing thiophene (10) and O-based furan(12) rings is highlighted in Figure 4a.
Because the degree of interaction between filled and vacantorbitals is a function of their overlap divided by their energydifference, it follows that the magnitude of stabilization and,consequently, the preference for either the syn or anti formmay be modulated by ring identity and/or substitution patternson either the donor or acceptor.34,35 Interestingly, methyl sub-stitution of 9 at the 3-position of the pyridine ring to afford13 results in an enhanced preference for the coplanar syn form(ΔE = 1.4 kcal/mol for 13 at the B3LYP/6-31+G(d,p) level,compared to 0.7 kcal/mol at the same level of theory for 9). Thismay be attributed to an increased energy of the lone pair orbitalon the pyridyl N atom due to the inductively electron-releasingCH3 group, resulting in enhanced interaction with the low-lyingthiophene C−S σ* orbital (Figure 4b). This offsets the stericpenalty which would be anticipated to disrupt coplanarity in
biaryl systems lacking additional conformational constraints.Consequently, an interaction such as that depicted for 9 inFigure 2 may be thought of as equivalent to that of a 2-(2′-pyrrolyl)pyridine (14) in which the N···S interaction mimics thatof a pyridine N to NH H-bond and confers a pseudo ringstructure. This isosteric relationship is of potential utility insituations where the desolvation penalty associated with thepolar pyrrole NH (albeit partially masked) may lead to reducedpotency. For drugs targeting the central nervous system (CNS),the number of H-bond donors and overall polarity must beminimized in order to maximize the potential for achievingblood−brain barrier penetration, and this drug class may benefitfrom such an isosteric replacement.36,37
■ σ-HOLESσ-Holes were first noted in the context of the halogens andinteractions observed between halogen atoms and the lone pairsof electrons of Lewis bases were originally referred to as favorable“halogen-bonding”.11e It has been demonstrated that a regionof relatively positive electrostatic potential, termed “σ-hole”, ispresent on the electron density surface of some halogen-containing compounds and is located on the part of the surfaceassociated with the halogen atom that is directly opposite (180°)the C−halogen bond axis.11f,g The importance of such interac-tions in ligand design has resulted in the development ofmolecular mechanics force fields incorporating “X-sites” ofpositive potential on the halogens of aromatic rings substitutedwith iodine, bromine, or chlorine to facilitate a more accurateprediction of binding orientation and energetics.11h,22,38 Theelectron acceptor capability of the heavier aryl halides (Ar−Xwith X = Cl, Br, and I) is proportional to the size and relative
Figure 3. Preferred syn conformations of (a) 2-(2′-thienyl)pyridine (9)and (b) 2-acetylthiophene (10) ((B3LYP/6-31+G(d,p)) depicting theinteraction between the lone pair and C−S σ* orbitals based on NBOmethodology.30
Figure 4. (a) Dihedral plots for 2-(2′-thienyl)-, 2-(2′-furanyl)- and 2-(2′-pyrrolyl)-pyridines depicting the change in conformational preference betweenS-, O-, and NH-containing rings. (b) Dihedral plots and syn conformational preferences for 2-(2′-thienyl)pyridine (9) and model substituted 2-(2′-thienyl)pyridine systems. All calculations are performed at the B3LYP/6-31+G(d,p) level of theory, and the scanned dihedral is depicted in bold.
Journal of Medicinal Chemistry Perspective
DOI: 10.1021/jm501853mJ. Med. Chem. XXXX, XXX, XXX−XXX
D
magnitude of the σ-hole opposite the C−X bond axis and tracksdirectly with the energy of the corresponding C−X antibonding(σ*) orbital. The upper portion of Figure 5 depicts theelectrostatic potential and relative positive potentials of chloro-,bromo-, and iodo-benzene at the 0.001 atomic units (au) contourlevel, computed at the B3LYP/6-311G* level of theory(fluorobenzene is noteworthy for its absence of a σ-hole). Inthe lower portion of Figure 5, the corresponding C−X σ* orbitalenergy for each species is determined at the same level of theorybased on Weinhold NBO analysis.30
An examination of chalcogen-containing heterocycles dem-onstrated regions of positive potential adjacent to sulfur andselenium consistent with the σ-hole concept.22 It is instructive tocompare the σ-hole and Z−S σ* characteristics of the aromaticsulfur moiety in various heterocycles to that of the halobenzenes(Figures 6 and 7). It can be seen from the front and side views ofthe various sulfur heterocycles presented in Figure 6a−c that aregion of positive, σ-hole-like potential exists near the sulfuratom, although slightly offset (in contrast to the halobenzenes)from the C−S bond axis and influenced to some extent by theadjacent C-2 C−H bond. The magnitude and volume of theσ-hole region, as quantified by Politzer and depicted graphicallybelow, is increased near the sulfur atom of thiazole (Figure 6b)and is also found for isothiazole (Figure 6c) compared to theparent thiophene (Figure 6a).22 For isothiazole (Figure 6c), dueto the presence of the electronegative and lone pair-bearing Natom directly adjacent to sulfur, a σ-hole-like potential exists to asignificant extent only on the side opposite the N atom (Figure 6c,center vs right). Similarly, trifluoromethylation of the modelsaturated sulfide dimethyl sulfide (DMS) (part d vs part e ofFigure 6) enhances the region of positive potential near sulfuronly on the side opposite fluorination (Figure 6e, center).The S atom of the thiophene ring possesses a σ-hole func-
tionality (Figure 6a) with the energy of the thiophene C−S σ*orbital (−0.30487 au, Figure 7a) closest to that of thechlorobenzene C−Cl σ* (−0.32085 au, Figure 5a), which hasa σ-hole energy that is the smallest in magnitude among thehalobenzenes. Nitrogen incorporation into the thiophene ring,as shown below for thiazole (Figure 7b) and isothiazole
(Figure 7c), appears to result in a lowering of the energy (andthus greater electron acceptor capability) of C−S σ* NBOsopposite to the side of N incorporation (−0.30936 au and−0.30591 au for C5−S1 σ* of thiazole and isothiazole, respec-tively, compared to−0.30486 au for the C2−S1 σ* of thiophene).Unique to isothiazole is the marked increase in the S1−N2 σ*energy (−0.21453 au) corresponding to a decrease in electron
Figure 5. Top: molecular electrostatic potentials (B3LYP/6-311G(d))at the 0.001 au contour level for chlorobenzene (a), bromobenzene (b),and iodobenzene (c) depicting the σ-holes (circled) opposite the C−Xbond axis. Red and blue denote regions of negative and positivepotential, respectively. Bottom: energies of the corresponding X−C σ*NBO at the same level of theory, demonstrating monotonic energydecrease and, hence, greater acceptor ability across the series.
Figure 6. Head-on and side views of the molecular electrostaticpotentials (B3LYP/6-311G(d)) at the 0.001 au contour level forthiophene (a), thiazole (b), isothiazole (c), DMS (d) and methyltrifluoromethyl sulfide (e) depicting the σ-hole region opposite the S−Cbond axis. Red and blue denote regions of negative and positivepotential, respectively, and the σ-holes are circled.
Figure 7. X···S σ* NBOs (X = C, N) and energies for thiophene (a),thiazole (b), isothiazole (c), DMS (d), and methyl trifluoromethylsulfide (e) (based on B3LYP/6-311G(d)).
Journal of Medicinal Chemistry Perspective
DOI: 10.1021/jm501853mJ. Med. Chem. XXXX, XXX, XXX−XXX
E

acceptor cabability into the N−S σ* antibonding orbital(Figure 7c), yet an increase in the σ-hole potential opposite thisbond axis due to the electron-withdrawing nature of the ring Nsubstituent (Figure 6c, right.)The effects of substituents on the calculatedmaximum positive
potential (VS,max) at the σ-hole of thiazole, which is more electrondeficient than thiophene, are presented in Figure 8.22,39−41 The
electrostatic potential of tetrahydrothiophene is calculated to bezero. The calculated electrostatic potentials of the C−S σ-holeregions of thiazole are modulated by ring substituents in a fashionthat reflects their electron withdrawing/donating properties andcan enhance or reduce the asymmetry of the σ-holes.22 Thus,electron withdrawing chlorine (σp = 0.23), carboxamide (σp =0.36), and nitrile (σp = 0.66) substituents enhance the positivepotential of the C−S σ* orbitals in a differential fashion thatdepends on the site of installation while the electron donatingamino moiety (σp = −0.66) negates the positive potential ofthiazole when installed at C-4 or C-5 but not at C-2 (Figure 8).22
The C−S σ* characteristics mirror these observations, with asignificant increase of the orbital energy that corresponds witha loss in acceptor ability observed with DMS (−0.13632 au,Figure 7f). However, substitution of atoms directly involved in theantibonding orbital or on adjacent atoms with electronegativegroups can strongly modulate the acceptor characteristics. Asdepicted in Figures 6e and 7e, substitution of a CH3 moiety inDMS with highly electronegative fluorine atoms gives rise to anobservable σ-hole.Moreover, the orthogonal relationship of C−S σ* orbitals with
the lone pairs of electrons on sulfur provides an element ofstereoelectronic control over the interaction with an electro-negative donor which must navigate between the lone pairs foroptimal effect, as illustrated in Figure 9.In addition to the geometrical constraints placed on access to
these low lying σ* orbitals by the sulfur lone pairs, for intra-molecular interactions the spatial relationship between theelectron donor X and the S atom also modulates the strength ofthe effect. The strength of the interaction is reflected in the X to S
distance, with the energy inversely proportional to the extentof separation.42 The electron donating atom X and S are mostproximal in a 1,4 relationship which has the fewest number ofintervening bonds. These can be further constrained if the bondsare rotatable by incorporating the S atom into a ring and/or thedeploying the donating atom as part of an sp2 system as, forexample, in an amide where the carbonyl O atom is the donoratom. However, because of geometric constraints that preventeffective orbital overlap, these 1,4 interactions are generally con-sidered to be electrostatic in nature.43 The geometry associatedwith a 1,5- and a 1,6- X···S interaction allows the orbital con-taining the nonbonded lone pair of electrons on the donor atomto overlap more effectively with the σ* orbital of the C−S bond.These motifs have been explored quite broadly and contribute toconformational constraint in a number of circumstancesalthough, as with 1,4-relationships, repulsive forces may play anadditional important role in biasing conformation toward anattractive X···S interaction. Thus, it is important to consider anyapparent X···S interaction in the context of other intramolecularinteractions which may reinforce the effect. Likewise, in systemsin which an X···S interaction might be expected, its absence mayresult from competing interactions. For example, intramolecularsulfur interactions can be overridden by competing intra-molecular H-bonds, protein−ligand interactions, unfavorablesteric, dipole−dipole, and, possibly, n → π* interactions.
■ O···S INTERACTIONS IN MEDICINAL CHEMISTRY1,4 O···S Interactions. 1,4 O···S interactions are prevalent in
medicinal chemistry with examples including O atom donorsderived from alcohols and ethers (sp3 systems) and the carbonylmoieties of amides, esters, or ketones which also offergeometrical constraint by virtue of the π-bonded system.20
One of the earliest appreciations of the role of this phenomenonon conformational preference, and its extension to an effect onbiological properties emerged from studies of a series ofcompounds related to the nucleoside analogue ribavirin (15).44
Tiazofurin (16) and its homologues 17−22 are nucleosideanalogues, and 16 demonstrates antiviral properties in vitro andantitumor activity in vivo. The mode of action of 16 has beenattributed to inhibition of inosine monophosphate dehydrogen-ase (IMPDH), a critical enzyme in the synthesis of guanosinetriphosphate, by the metabolite tiazofurin adenine dinucleotide(TAD, 24), which binds to the nicotine adenine dinucleotide(NAD, 23) cofactor site of the enzyme.45 The similar topologicaldisposition of the carboxamide moieties of 23 and 24 contributesto structural mimicry, but, in contrast to the pyridinium ring of23, the thiazole ring of 24 is stable toward reduction by hydride, acritical aspect of the function of IMPDH. The formation of24 is envisaged to occur by phosphorylation of the C-5 hydroxylof 16 by a cellular kinase to afford the 5′-monophosphate withsubsequent reaction with adenosine triphosphate (ATP)catalyzed by NAD phosphorylase. As part of an effort tounderstand SARs for this series of IMPDH inhibitor, the solidstate structures of 16, its 2′-deoxy analogue 25, and the α-anomer26were analyzed.46,47 Themost striking observation with each of
Figure 8. Calculated electrostatic potentials (VS,max) of C−S σ* orbitalsfor sulfur-containing heterocycles and the effects of electron with-drawing and electron donating substituents.22
Figure 9. Stereochemistry of substituents, σ* orbitals, and lone pairs ofelectrons on sulfur atoms.
Journal of Medicinal Chemistry Perspective
DOI: 10.1021/jm501853mJ. Med. Chem. XXXX, XXX, XXX−XXX
F

these molecules was that the S atom of the thiazole moiety wasproximal to the O-4′ atom of the ribose ring, with contactdistances of 3.0 Å for 16 and the deoxy analogue 25 and 2.8 Å forthe α-anomer 26, indicative of an attractive electrostatic inter-action between these atoms.47 Quantummechanical calculationspredict that the electron withdrawing properties of the C-4carboxamide substituent increases the energy of the σ-hole on Sin an asymmetric fashion, with the lobe projecting toward theriboside O-4′ atom having the larger electrostatic potential.Although this electrostatic interaction is favorable, the overallgeometry of these compounds may also benefit from reducedsteric interactions between the thiazole ring and the C-2′ H andring O-4′ atom in the case of 16 and 25 and the thiazole and OHmoiety in the α-anomer 26.22,47,48
Of the analogues of 16, selenazofurin (19) is 5-fold morecytotoxic toward P388 and L1210 cells in vitro and similarlyeffective in a Lewis lung carcinoma mouse model.44c,d The con-formational relationship between the heterocycle ring and theribose moiety at the glycosyl bond is similar to that of 16, with atorsion angle that reflects close proximity between the Se an O-4′atoms and a distance of 3.0 Å, which is shorter than the 3.40 Åsum of the van der Waals radii of the two atoms.44d Computa-tional studies of derivatives of 16 and 19 indicate that theattractive O···S or O···Se interaction combined with a repulsiveO···N effect contribute to a rotational barrier of ∼4 kcal/mol forthe C-glycoside bond. Cocrystal studies of 24 and the seleniumanalogue derived from 19 revealed that both bound to the samesite as 23 in horse liver alcohol dehydrogenase by adopting similarconformations that resembled 23 but with the heterocyclesdisplaced ∼4 Å away from the catalytic Zn2+.49 Notably, the
torsion angles associated with the C-glycoside bond maintainedthe close O-4′ to S/Se interactions observed in the small moleculesingle-crystal structures of these nucleoside analogues.49
Further insight was gleaned by a systematic study ofheterocycle analogues of 16 in which it was observed thatoxazofurin (17) does not exhibit cytotoxic activity toward P338and L1210 cells in vitro.50−54 Analysis of small molecule single-crystal X-ray structures of 17 revealed an average C-glycosidebond torsion angle of 70°, which is considerably larger than theaverage 24° observed for the thiazole-based nucleosideanalogues, although the carboxamide moiety of 17 was disposedsimilarly to that of 16.50 Ab initio calculations suggested that theoxazole and ribose O-4′ atoms experience electrostatic repulsiondue to their electronegativity. This phenomenon biases theconformation of the heterocycle toward an arrangement that isnot recognized by IMPDH or the enzymes that are responsiblefor metabolizing the nucleoside into an analogue of NAD.Thiophenfurin (20) retained the cytotoxic activity of 16
toward a panel of immortalized cell lines, but furanfurin (21) wasinactive.51 In the small molecule single-crystal X-ray structure of27, a precursor of 20, the torsion angle of the C-glycosidic bondwas 46.5°, which resulted in a O···S contact distance of 3.0 Å,both of which are larger values than observed for 16. Ab initiostudies were consistent with stabilization of this conformer by anelectrostatic interaction between the O and S atoms. Thealternate conformer was ∼1 kcal/mol higher in energy but wasstabilized by a productive interaction between the C-3 protonand the furanose O-4′ atom and the barrier to rotation betweenthese two local mimina was 5−6 kcal/mol. For 21, the lowestenergy conformation had a C-glycosidic torsion angle of 70−80°,similar to 17, and this local minimum energy was 1−2 kcal/molhigher than that of the corresponding local minimum for 20.51a
In adenosine-labeled K562 cells, 20 gave 2-fold higher levels ofthe NAD analogue than 16 while metabolic conversion of 21occurred with only 10% efficiency, suggestive of conformationalpreferences also affecting metabolism.
Comparative evaluation of the NAD analogues synthesizedfrom 16 and 19−22 as inhibitors of IMPDH type I and II indi-cated relative potencies of 19 > 22 = 20 = 16≫ 21, a hierarchythat correlates with the antitumor activity of the parent nucleo-sides. These data highlight that the poor cytotoxic activity associatedwith 21 is a function of a combination of limited metabolic conver-sion and low affinity of the NAD analogue for IMPDH.Imidazofurin (18) is inactive toward cell growth, leading to the
postulate that conformation may play a role based on thehypothesis that the preferred conformation of the NAD analoguederived from this molecule is not accessible and therefore notable to efficiently bind to IMPDH.52 However, poor phos-phorylation of the 5′ OH and/or an inability to be converted tothe dinucleotide analogue of NAD are potential contributors thatwere not explicitly explored.Replacing the ribose O atomwith S was also explored as part of
the examination of this class of IMPDH inhibitor. Furanthiofurin(28) is nontoxic toward myelogenous leukemia K562 cells, whilethiophenthiofurin (29) exhibits cytotoxicity only at concen-trations markedly higher than for 20.54 In these molecules, the
Journal of Medicinal Chemistry Perspective
DOI: 10.1021/jm501853mJ. Med. Chem. XXXX, XXX, XXX−XXX
G

conformation is anticipated to be governed more by stericinteractions because calculations indicate that simple dialkylsulfides possess either a much smaller or no σ-hole on the Satom.22,48,54
Although not explicitly recognized, an attractive ribose-baseO···S interaction underlies the design of a substrate for adenosinedeaminase that provides the basis for a practical biochemicalassay with which to screen for inhibitors of this enzyme.55 Thefluorescent, isomorphic analogue of adenosine (30), thA (32),emits light at 410 nm and is a substrate for adenosinedeaminase with a Km that is just 13-fold higher than the naturalsubstrate. The product of this enzymatic deamination process,thI (33), exhibits a different fluorescence spectrum emitting amaximum at 391 nm, providing a practical and sensitive markerof enzymatic activity. An examination of the conformationalpreferences of 32 yielded an understanding of the observedbiochemical pharmacology. The solid state structures of 32, 33,and 35, the synthetic precursor of thG (34), an analogue ofguanosine (31), reveal close contacts between the thiopheneS and ribose O-4′ atoms. In each case, the distance between theO and S atoms is less than the sum of their van der Waals radii,reflective of an attractive electrostatic interaction. For 35, thereare two molecules in the unit cell with different conformationsbut both show O to S distances below the sum of the van derWaals radii. In the case of 32, the O···S interaction stabilizes anoverall conformation that is very similar to that preferred by 30,which favors an anti relationship between the heterocycle andC-5′ of the ribose ring, with the latter adopting a 3′-endo puckerconformation. Consequently, the conformations of the twomolecules overlay quite closely.55
A 1,4-O···S interaction plays a role in orienting thethiazolopyridine heterocycle of the factor Xa inhibitor edoxaban(36), which is marketed in Japan for the prevention ofvenous thromboembolism following lower-limb surgery.56 Thisphenomenon was explored in some detail in earlier studies ofthe chemotype in which a chloronaphthalene was employed asthe P1 moiety to project into the S1 subsite of the enzyme andestablish contact with Tyr288 via a hydrophobic interactionwith the π-cloud of the aryl ring.57 In the cocrystal structure of
factor Xa with 38, the carboxamide analogue of 36, the N−Meof the fused piperidine projected toward the so-called “cationhole” of the S4 subsite of the enzyme. However, the N−Memoiety appeared not to be protonated and there was noevidence of an ionic interaction with the carboxylate of theproximal Glu97.
57a Nevertheless, the SARs for the matchedpair of tetrahydrothiazolo[4,5-c]pyridines 37 and 40 indicate aclear dependence of potency on the topological disposition ofthe N−Me relative to the thiazole ring. The 10-fold differencein potency was attributed to a steric clash between the N−Me in 40and residues that define the boundary of the S4 subsite of theenzyme. This results from a projection of the N−Me toward the S4subsite rather than the “cation hole”. It was also noted that the O toS distance in 38 was 2.9 Å, closer than the van der Waals contactdistances and indicative of a productive interaction that leads to acoplanar arrangement of the amide and the thiazole ring. For thethiophene analogues 39 and 41, there is only a 2-fold difference inpotency that favors the same topology between the sulfur andN−Me as in 37. A detailed ab initio analysis of the energeticparameters associated with the two topological isomers ofthiophene-2-carboxamide and thiazole-2-carboxamide revealed astronger topological preference for the thiazole compared to thethiophene. This was attributed to a combination of an attractiveinteraction between the thiazole S and the carboxamide O atomsand a repulsive interaction between the thiazole N and carboxamideO atoms acting synergistically to favor the conformer topologyobserved in the cocrystal structure of 38. The conformationalpreference for the thiophene homologue was similarly attributed toan attractive O···S interaction. However, for the thiophene, theenergetic penalty for adopting the alternate planar conformationwas considerably lower. This is in part a function of the lowerelectrostatic potential of the S atom in thiophene compared to thatof thiazole, which explains the modest 2-fold difference in potencybetween 39 and 41.22
In the cocrystal structure of the structurally related factorXa inhibitor 42 (IC50 = 9.5 nM), the close contact between thethiazole S and adjacent amide carbonyl O atoms (3.1 Å) was con-sidered to contribute to the correct alignment of the S4 element,with a potential interaction between the thiazole N and the amideNH atoms functioning cooperatively (Figure 10).58
Journal of Medicinal Chemistry Perspective
DOI: 10.1021/jm501853mJ. Med. Chem. XXXX, XXX, XXX−XXX
H

In the cocrystal of rivaroxaban (5) with factor Xa, the con-formation of the thiophene carboxamide moiety reflects anattractive interaction between the heterocyclic S and the carbonylO atoms (distance = 3.0 Å) that presumably plays a role inoptimally orienting the Cl atom toward the π cloud of Tyr228which forms part of the wall of the S1 subsite.10b A productive Oto S interaction also appears to stabilize the bound conformationof the factor Xa inhibitor 43 because the cocrystal structureshows a conformational alignment in which the thiophene S andadjacent CO oxygen atoms are in close proximity (distance =2.9 Å). This topology projects the oxazolidine moiety correctlyinto the S4 pocket.59
A series of thiazole-based NPY5 receptor antagonists wereexamined as a potential treatment for obesity and hyperphagia(excessive hunger) based on the observations that NPY is apowerful stimulant of food intake in rats and that activation ofNPY5 receptors results in hyperphagia and reduced thermo-genesis.60 A design and optimization exercise that originatedwith a virtual screening approach utilizing prototype inhibitorsidentified 44 as a lead molecule that exhibited high affinity for themouse receptor used for screening purposes, IC50 = 19 nM. SARstudies evaluated the importance of the topological relationshipbetween the thiazole ring and the pendent ketone carbonyl onbinding affinity to the mouse receptor and matched pairs ofcompounds from both series were prepared by varying thesulfonamide substitution pattern (45/46 and 47/48).60 The5-substituted series represented by 45 and 46 demonstratedconsistently high affinity for the mNPY5 receptor with IC50sranging from 0.71−9.9 nM, while the 4-substituted isomersrepresented by 47 and 48were typically 10-fold weaker, althoughthe trends in SAR around the sulfonamide moiety were similar.The affinity differences between the matched pair of 4-Fsulfonamides 46 and 48 are particularly striking, withmore than a100-fold variation in potency. Molecular modeling studies on thecore scaffolds provided insight into the observation showing thatthe ketone CO of 45−48 was twisted 58° out of the plane ofthe phenyl ring due to steric interference with the ortho-Me
substituent. However, key differences were noted between thepreferred topology of the CO moiety with respect to thethiazole ring. In the 5-substituted isomers 45 and 46, the Oatom of the CO is proximal to the S atom with their spatialseparation less than the sum of the van der Waals radii of thetwo atoms, indicative of a favorable stabilizing interaction. Incontrast, in the 4-substituted isomers 47 and 48, the carbonyl Oand thiazole N atoms experience repulsion. This influences thepreferred conformation to one which aligns the dipoles of theCO and thiazole ring more favorably and projects the phenylring in the angled topology depicted for 47 and 48 rather thanthe more linear arrangement favored by 45 and 46.60−62
Calculations show that the energy penalty for the 4-substitutedisomers 47 and 48 to adopt the conformation favored by the5-substituted analogues 45 and 46 is between 7 and 10 kJ/mol(1.7−2.4 kcal/mol), which is consistent with the 5.7 kJ/mol(1.4 kcal/mol) energy difference associated with an order ofmagnitude variation in binding affinity.60 These results wereinterpreted in the context of a preferred extended conformationof the molecule, as depicted for the 5-subtitued thiazoles 45 and46. This example provides a particularly interesting illustrationof the influence of heterocycle topology and electronic propertieson conformational preferences and its manifestation as a significantbiological effect.60,62
1,4 O···S interactions appear to play a contributory role ininfluencing the conformation of oligomeric thiazole-basedγ-amino acid derivatives that adopt a helical topography.63 Themonomeric 4-aminomethyl-1,3-thiazole-5-carboxylic acid(ATC, 49) was designed to limit the conformational mobilityof the embedded amino acid moiety. Oligomers derived from 49adopted a helical arrangement in solution based on CD andNMR spectroscopy. The helicity was confirmed with a smallmolecule single-crystal X-ray structure of the tetramer 50, whichrevealed close 1,4-O···S contacts between each amide CO andthe adjacent thiazole S atom with measured distances of 3.0, 3.0,and 3.1 Å. Interestingly, it is the sp3 O atom of the ester moiety atthe terminus rather than the sp2 O atom that is closest to theadjacent S of the thiazole ring, distance = 2.9 Å, an interactionalso observed in the X-ray of an ester derived from the monomer49. The O to S atom distances of 3.0−3.1 Å reflect less than idealtorsion angles of ∼35°, suggesting that these interactions are notof optimal energy. The carbonyl O atoms of the central amide
Figure 10. Intramolecular and intermolecular interactions associatedwith the factor Xa inhibitor 42 in the cocrystal.
Journal of Medicinal Chemistry Perspective
DOI: 10.1021/jm501853mJ. Med. Chem. XXXX, XXX, XXX−XXX
I

moieties engage the adjacent NH in an H-bonding interac-tion based on a 1,9 CO(i)···HN(i + 2) relationship withdistances of 2.0 Å. This proximity is reflected in the markedlydownfield resonances of the NHs in the 1H NMR spectra,which appeared between 9.18 and 10.67 ppm, dependingon the length of the oligomer. These interactions clearlycontribute to the overall shape and stability of the molecule.Two ATC moieties were subsequently incorporated as turnmimetics into an analogue of the hemolytic cyclic decapeptidegramicidin S replacing the natural proline residues. TheATC-containing macrocyclic peptide retained similar anti-bacterial activity to the progenitor but exhibited 6-foldreduced hemolytic activity.
An attractive interaction between the thiazole S and the C-5amide carbonyl O atoms in the p38α mitogen activated protein(MAP) kinase inhibitor 51 was identified as a contributorto the potent enzyme inhibitory activity, IC50 = 3.5 nM.64 Thistopology, which is also favored by optimal alignment of theheterocycle and CO dipoles, orients the thiazole N and2-amino NH to accept and donate H-bonds from and to thebackbone of Met109, respectively, interactions that are observedin an X-ray cocrystal structure.64 Recognition of the O···S inter-action was instrumental in the design of related p38α MAPkinase inhibitors including those that anticipated an N···Sinteraction (vide infra).24,65,66 In the crystal structure of 51, thethiazole S and amide carbonyl O atom are 2.9 Å apart and theO−C−C−S dihedral angle is −1.1°.
The Src/Abl kinase inhibitor dasatinib (52, BMS-354825) alsobenefits from an intramolecular 1,4-O···S interaction thatassists in orienting the molecule for optimal presentation tokinases.66a,b In the cocrystal structure of 52 with Abl kinase, the2-amino NH and the thiazole N atom are presented to thebackbone carbonyl and amide NH of Met318, respectively, toestablish complementary H-bonding interactions.66c In theAbl-bound conformation, the amide O and thiazole S atoms of52 are 2.8 Å apart. This close O···S contact is also observed inX-ray cocrystals of 52 with several other kinases including theThr338Met form of cSrc, erythropoietin-producing hepatocel-lular tyrosine kinase A4 (EphA4), Lyn tyrosine kinase, bone
marrow kinase, and Bruton’s tyrosine kinase, with distancesranging from 2.9 to 3.1 Å.66d−h
The thieno-pyrimidine derivative 53 is a potent inhibitorof the sirtuins (SIRT) 1, 2, and 3, NAD+-dependentdeacetylases, with IC50 values of 15, 10, and 33 nM,respectively, while the analogous furopyrimidine 54 is15−40-fold weaker.67 These data are consistent with thecocrystal structure of SIRT3 with an analogue of 53 in whichthe pivalic acid moiety has been elaborated. The orientation ofthe 2-carboxamide is coplanar with the thienyl ring such thatthe O atom lies proximal to the S atom to facilitate a 1,4electrostatic interaction. This topology facilitates fourH-bonding interactions between the amide moiety andelements of the protein and a structural bridging watermolecule in a fashion that mimics that of the nicotinamidemoiety of NAD+, as depicted in Figure 11.68
A series of thiazolylamino mannosides were designedas antagonists of FimH adhesin, which is found at the tipof the type 1 pili of Escherichia coli and mediates adhesion tooligomannosides displayed on the surface of intestinalepithelial cells.68 In Crohn’s disease, there is an abnormallyhigh expression of carcinoembryonic antigen-related celladhesion molecules 5 and 6 (CEACAM5 and CEACAM6)which act as receptors for adherent invasive E. coli. An X-raycocrystal of the lead molecule 55 in the mannose bindingsite of FimH indicated that the ketone O and thiazoleS atoms were proximal, adopting a coplanar arrangement.This was shown by quantum chemical calculations to be the
Figure 11. Key interactions between the core heterocycle and ananalogue of 53 and SIRT3 in the cocrystal structure illustrating the closeO···S contact.
Journal of Medicinal Chemistry Perspective
DOI: 10.1021/jm501853mJ. Med. Chem. XXXX, XXX, XXX−XXX
J

most stable conformation, indicative of the O···S interac-tion favoring the bound conformation of the drug.Optimization afforded the bis-thiazole derivative 56 asthe most potent antagonist that prevented oligomannoseglycans from binding to FimH at concentrations of 5 nM.68
This compound also potently blocked the attachmentof adherent invasive E. coli to guinea pig erythrocytes toT84 intestinal epithelial cells in vitro in a dose-dependentfashion.
Human inflammatory protein complement C3a is a 77 residuehelical protein that binds to the G-protein coupled receptor(GPCR) C3aR, stimulating the chemotaxis of immune cells tosites of infection and the release of bactericidal agent andinflammatory cytokines based on intracellular Ca2+ mobiliza-tion.62 The C-terminal residues LGLAR are critical for receptoractivating activity and a homology model of the C3a receptor towhich the terminal arginine was docked facilitated the design oftripeptide-based inhibitors. Of these, the imidazole 57 emergedas a high affinity ligand, IC50 = 51 nM, that acted as an agonistwith an EC50 = 120 nM.62a However, the thiazole homologue58 demonstrated weaker binding affinity, IC50 = 375 nM, andexhibited only partial agonism at concentrations above 1 μM.Further optimization afforded the potent indole-based agonist59 with an IC50 = 12 nM and an EC50 of 15 nM for stimulatingintracellular Ca2+ release. The biochemical and pharmaco-logical differences between 57 and 58 have been explained bydistinct conformational preferences attributed to stereo-electronic interactions between the heterocycles and theamide moiety.Further evolution of the series focused on the benzhydryl
imidazole 60, which is characterized as an agonist of intracellularCa2+ release, EC50 = 24 nM.62b However, the thiazole analogue61 is a partial agonist with an IC50 of ∼100 nM, while the isomer62 is a full antagonist that blocks hC3a-mediated Ca2+ releasewith an IC50 of 1 μM. This was rationalized a priori based on thepreferred conformation of the amide CO with respect to theheterocycle, a topology favored by dipole arrangements. For theimidazole 60, dipole alignment and the avoidance of electrostaticrepulsion between the ring CN and the CO favors theamide topology as drawn, a circumstance mimicked by 61.However, the isomeric thiazole analogue 62 would be expectedto adopt a complementary topology based on alignment of thering and amide dipoles that is further stabilized by a productive1,4-electrostatic interaction between the CO oxygen andthiazole S atoms. The activity of 60 was mimicked by theimidazopyridine 63, which conforms to the preferred con-formation of 60 and functions as an agonist that inducesintracellular Ca2+ release with an EC50 = 15 nM. The fused lactam64, designed to recapitulate the topology of 62, is an antagonist,IC50 = 320 nM.
Although useful in conformational control, CO···S electro-static and dipole interactions can be overridden by more power-ful intramolecular interactions as evidenced by the cyclin-dependent
Figure 12. Conformation and drug−target interactions of 65 bound toCdk5 illustrating an intramolecular H-bond that stabilizes aconformation not favoring a close O···S contact.
Journal of Medicinal Chemistry Perspective
DOI: 10.1021/jm501853mJ. Med. Chem. XXXX, XXX, XXX−XXX
K

kinase 5 (Cdk5)/p25 inhibitor 65, which has a primary aminesubstituent at C-4.69 One of the NH moieties engages in aH-bond with the C-5 amide carbonyl, preorganizing the topologyof the molecule to that which complements the binding pose inthe ATP binding pocket of the kinase (Figure 12). The secondNH donates a H-bond to the backbone CO of Glu81, and thethiazole N atom accepts a H-bond from the backbone amide ofCys83. In this context, the amide CO···H2N interactionfunctions as an isostere of a ring.
In a similar series involving the weak screening hit 66,optimization was guided by X-ray cocrystal structure determi-nation, leading to 67 which was characterized as a potentinhibitor of Cdk2/cyclin A (IC50 = 1.1 nM) and Cdk5/p25(IC50 = 1.5 nM) with somewhat weaker activity toward Cdk1/cyclin A, Cdk4/cyclinD1, Cdk6/cyclin D1, and Cdk9/cyclin K(IC50 values = 7.6, 4.0, 6.6, and 1 nM, respectively), while theIC50 value for Cdk7/cyclin H was >1000 nM.70 X-ray cocrystalstructures of 66 and 67 bound to Cdk2 at the ATP siterevealed a topology similar to that observed for 65. Theenhanced potency of 67may be attributed to the sulfonamidemoiety which engages in three H-bonding interactions withthe protein that are not available to the simple allyl moietyof 66.
Interpretation of the structure−activity relationships for aseries of penem antibiotics focused on the influence of a 1,4-O···Sinteraction between the C-2 substituents and the dihydrothiazolering S atom that conferred stereoelectronic control of moleculartopography. Insights were provided by observations fromsmall molecule single-crystal X-ray data.71 The (R)-and (S)-THF isomers 68 and 69 exhibited comparable antimicrobialactivity that was similar to the C-2 CH2OCH3 derivative 70.However, 71, the methylated derivative of 70 which acts as aring opened version of 68/69, and the furan 72 exhibit muchreduced activity while antimicrobial properties were restoredwith the 2-phenyl analogue 73. The ethers 68−70 and 72crystallized in conformations in which the ring S atom and theO atom of the C-2 substituent were proximal, with the distancebetween them considerably shorter (2.8−3.0 Å) than the 3.3 Åthat defines the van der Waals radii of the two elements. Thefuran ring of 72 was coplanar with the olefin of thedihydrothiazole ring, while the plane of the phenyl ring of 73was twisted by almost 50°. The inference from these data was thatthe precise presentation of the C-2 substituent to the penicillinbinding protein was important for antimicrobial activity. This isoptimal when the C atom β- to the dihydrothiazole heterocycleorients out of the plane of the ring and is relatively small in size.71
The O atom of an alcohol can also interact closely with athiazole S atom as illustrated by two examples from the recentliterature.72,73 ZK203278 (74) is a vitamin D3 analogue withpotent immunomodulatory activity in vitro and in vivo at a con-centration that did not cause hypercalcemia, a profile thatdifferentiates the molecule from other vitamin D3 receptorligands.72 In the X-ray cocrystal structure of 74 with the ligandbinding domain of the human vitamin D nuclear receptor,the hydroxyl O atom is proximal to the thiazole S, with aninteratomic distance of 2.8 Å, a conformation also compatiblewith a favorable alignment of the dipoles associated with theC−OH moiety and the thiazole ring (Figure 13). The OH is
believed to donate a H-bond to His305 while the O atom accepts aH-bond from the NH of His397, interactions that are analogous tothose observed in the 1α,25(OH)2D3 complex. Unlike thenatural ligand and related structures with smaller moietiesattached to the alcohol, the thiazole ring makes van der Waalscontacts with residues in helixes 3, 11, and 12, stabilizing theagonist conformation of H12 while not perturbing its position ormoving H11. As a consequence, 74 acts as a full agonist of the D3receptor with the drug−target interactions thought to allosteri-cally modulate coregulator interactions, providing a potentialexplanation for the differentiated biological profile.
Crizotinib (75) was originally designed as a mesenchymal−epithelial transition factor (c-MET) kinase inhibitor but ex-presses clinical effects via inhibition of anaplastic lymphomakinase (Alk).73 In an effort to improve the potency of 75 towardboth the wild-type kinase and enzyme harboring resistantmutants, removal of the Cl atom ortho- to the fluorine of the
Figure 13. Key interactions of 74 with the ligand binding domain of thehuman vitamin D nuclear receptor from the cocrystal structureillustrating a close O···S contact between the thiazole S and hydroxylO atoms.
Journal of Medicinal Chemistry Perspective
DOI: 10.1021/jm501853mJ. Med. Chem. XXXX, XXX, XXX−XXX
L

triahalogenated phenyl ring led to some improvement in potencyas did replacing the other Cl atom with a 1,2,3-triazole ring.Further optimization focused on the pyrazole heterocycle whichoccupied a lipophilic pocket near the solvent exposed region andprojected the piperidine moiety into solvent. The pyrazole wasreplaced with a thiazole, and an exocyclic OH was introducedwith the latter designed to engage the side chain of Asp1203 as aH-bond donor. The resulting compound, 76, exhibited a Ki of0.4 nM toward the Leu1196Met Alk mutant enzyme and an EC50of 27 nM in a cell-based assay. In the X-ray cocrystal of 76 withAlk, the OH donated a H-bond to the carboxylate of Asp1203, asanticipated, while, in addition, one of themethyl groups was closeto the NHof Asp1203, which was not interacting with the inhibitor(Figure 14a). Further optimization gave the diol 77 whichexpressed a Ki of 0.2 nM, with cell-based activity improved to anEC50 of 6.6 nM, while the enantiomer demonstrated aKi of 1.1 nM.In the cocrystal structures of both 76 and 77, there is a close contactbetween the thiazole S atom and the O of the pendent tertiaryalcohol with interatomic distances between O and S measured as2.9 Å for 76 and 3.0 Å for the diol 77.73 In this circumstance, theremay be mutually reinforcing effects in which the O···S interactionenhances the capacity of the OH to donate a H-bond to Asp1203while the H-bonding interaction with Asp1203 will enhance theability of the alcohol O atom to interact with the thiazole S atom.
■ 1,4 O···S INTERACTIONS AND CONFORMATIONALPREFERENCES IN ORGANIC SYNTHESIS
The reaction of the hydroxy epoxides 78 and 79 with NaSPh inTHF afforded a mixture of the syn−syn and syn−antiγ-butyrolactones 80 and 81 in a 31:69 ratio with none of theanti−syn or anti−anti isomers observed.74 The equilibration ofsyn−syn sulfenylated γ-butyrolactones gave a similar mixture,
rationalized to be occurring via a retroaldol/aldol reaction, asdepicted in Figure 15. A smallmolecule single-crystal X-ray structureof the syn−syn lactone 80 revealed a close contact of 2.9 Å betweenthe hydroxyl O and the S atoms with the S−phenyl ring orienteddiametrically away from the OH. This stereochemical arrangementwould permit an optimal interaction between the hydroxyl O atomand the σ* of the C−S bond and allow one of the sulfur lone pairs ofelectrons to donate into the π* orbital of the adjacent carbonyl.74b
These stabilizing interactions were considered to be an importantfactor in favoring the observed stereochemical outcome of theequilibration process. A similar phenomenon is thought to underliethe epimerization of 82, which affords a 7:90 mixture at equilibriumthat favors the syn 83 isomer.75
In the small molecule single-crystal X-ray of the sulfenylatedpiperazine dione 84, the thiophenyl moiety adopts an axial con-figuration with the distance between the ester carbonyl O and theS atoms measured at 2.8 Å, indicative of a favorable interactionbetween these atoms (Figure 16).76 This biases the stereo-orientation of the phenyl ring, which as a result projects under theheterocyclic ring away from the ester carbonyl, allowing it to engagein a π-stacking arrangement with the distal amide carbonyl.
The powerful electron withdrawing properties associated witha CF3 moiety when attached to S influence the energy level of theF3C−S σ* orbital, providing an opportunity for a productiveinteraction with a proximal lone pair of electrons (vide supraFigure 7e). In the small molecule single-crystal X-ray structureof the CF3S derivative 85, prepared from the β-keto esterusing a quinidine-mediated asymmetric CF3S transfer from
Figure 14. Key ligand−protein interactions between the alcohol 76 (a) and the diol 77 (b) and the Alk enzyme illustrating close O···S contacts betweenthe thiazole S and pendent hydroxyl O atoms.
Journal of Medicinal Chemistry Perspective
DOI: 10.1021/jm501853mJ. Med. Chem. XXXX, XXX, XXX−XXX
M

N-trifluoromethylthiophthalimide, the distance between the estercarbonyl O and the S atom is very short, measured as 2.7 Å(Figure 17a).77a Further support for the existence of a productiveO···S interaction is the observation that the CF3 moiety isoriented away from the ester carbonyl O atom. This con-formation maximizes interaction of one of the CO lone pairsof electrons with the σ-hole of the electron-deficient S atom. In arelated example, a similarly short 2.8 Å distance is observedbetween the ester carbonyl O and the SCF3 S atoms of 86, acircumstance in which the O···S interaction provides stereo-electronic influence over the geometry of the CF3 with a C−S−Oangle of 161.2°.77b
■ 1,5 O···S INTERACTIONS
Intramolecular 1,5 O···S interactions are anticipated to beenergetically more favorable than 1,4-interactions because theexpanded geometry allows for increased overlap between the lonepair of electrons on the donor atom and the C−S σ* orbitals. A 1,5O···S interaction appears to be an important influence on thebiological activity and conformation of the (acylimino)-thiadiazoline moiety that is a structural hallmark of the potentangiotensin II antagonist KRH-594 (87).78 In the small moleculesingle-crystal X-ray structure of the dipotassium salt of 87, there isa close contact between the carbonyl O atom and the thiadiazole Satom (2.5 Å) that stabilizes the syn conformation between theseelements, conferring an overall planar arrangement that mimics afused bicyclic ring system. Similarly, short contacts were seen inthe simplified structure 89 (2.7 Å) as well as the acylated 2-aminothiazole 90 (2.7 Å). The oxadiazoline analogue of 87, compound88, exhibits close to an order of magnitude lower affinity for thereceptor. However, somewhat surprisingly, the small moleculesingle-crystal X-ray structure of the oxadiazoline 91 also revealedan overall planar topography and a close O···O contact of 2.7 Åthat is considerably shorter than twice the van derWaals radii of O,which amounts to 3.0 Å. Theoretical calculations on these systemsconfirmed that the conformations observed in the X-ray structuresreflected energeticminima. Consequently, the origin of the 10-folddifference in potency between 87 and 88 remains obscure,although there may be differences betweenmore electron rich acylgroups of the prototypes and the trifluoroacetyl moiety selectedfor the substructure studies 89−91.79
An O···S interaction appears to be functionally operative inthe acyliminothiazole PS-028 (92), which is a potent competitiveinhibitor of fibrinogen binding to the blood platelet glycoproteinIIb/IIIa receptor. The planar acyliminothiazoline amplifiespotency by acting as a rigid scaffold to deploy the amidine andcarboxylic acid pharmacophoric elements in a topographicalrelationship that complements receptor architecture.80
Figure 16. Small molecule single-crystal X-ray structure of 84 taken fromthe Cambridge Crystallographic Database (CSD) illustrating the closecontact between the ester CO oxygen and the S atoms, d1 = 2.8 Å.(CCDC accession number is 721744).
Figure 17. Small molecule single-crystal X-ray structures of 85 (a) and86 (b) illustrating the close contact between the carbonyl O and Satoms, d1 = 2.7 Å; d2 = 2.8 Å, ϑ = 161.2°.
Figure 15. Equilibration of syn−syn sulfenylated γ-butyrolactones via aretro-aldol reaction.
Journal of Medicinal Chemistry Perspective
DOI: 10.1021/jm501853mJ. Med. Chem. XXXX, XXX, XXX−XXX
N

Acyl iminothiazolidinines are prominent in potent andselective neonicotinoid insecticides that act as agonists ofnicotinic acetylcholine receptors, a chemotype that relies uponan overall planar topography as an important structural feature.81
This is exemplified by 93, where an intramolecular O···S inter-action favors a trans orientation between the amide carbonyland the pyridinylmethyl substituent. This appears to be ofconsiderable importance because the analogous trifluoromethylsulfonamide 94, which does not adopt a planar arrangement,is over 500-fold weaker and not toxic toward houseflies at aconcentration of 0.1 μg/g.81a,b A similar effect is observed inthe carbapenem side chain of 95, which was designed withthe intramolecular O···S interaction anticipated, and a smallmolecule single-crystal X-ray of the thiadiazole side chainelement showed the expected close O···S contact.82
An intramolecular O···S interaction was examined as anapproach to improving the profile of a series of analogues ofnocodazole (96) that inhibited both wild-type and Thr315Ilemutant Abl kinases by binding to the ATP site of the enzymeand engaging key H-bond donor and acceptor interactionswith the backbone carbonyl and NH of Met318.
83 In the synopsisof the three matched pairs represented by 97−103 compiled inTable 3, the benzothiazoles 98, 100, and 103 are considerably
more potent than the matching benzimidazoles 97, 99, and102 or the benzoxazole 101 with a 50−600-fold variation.83b,c
Interestingly, although modeling recognized a productiveO···S interaction in the benzothiazole series in the bound form(Figure 18), calculations suggested that the difference in potencybetween the benzimidazole and benzothiazole derivatives wasmore a function of desolvation costs, which are higher for themore basic benzimidazole heterocycle.83b,c In this example, theS atom very effectively replaces an NH, which would be expectedto engage in an intramolecular H-bond based on analogywith observations with the anthelmintic agent mebendazole(104), with the advantage in desolvation costs favoring sulfurover NH.12,84
An example that illustrates the advantageous incorporationa benzothiazole rather than a benizimidazole is provided by aseries of dual RAF/vascular endothelial growth factor receptor2 (VEGFR2) inhibitors 105−109 that bound to the kinasewith the DFG loop out (Table 4).85 The benzothiazole was
designed based on the notion that the amide moieties of 105 and108 were coplanar with the fused bicycle core and stabilized byan intramolecular H-bond between the NH of the imidazoleand the amide CO analogous to that observed with 104. Thelead benzimidazole 105 demonstrated potent inhibition ofboth enzymes although the effect in cells was less thanimpressive, a profile largely replicated by the aza-analogue 108.The topographical hypothesis was supported by the effects ofN-methylation (106 and 109), which distorted the amides fromcoplanarity with the fused ring system by 23−24° based onenergy minimization calculations. This resulted in a >20-foldreduction in potency for 106 and 109 toward the BRAF V600Emutant although there was no effect on VEGFR2 inhibition(Table 4). The benzothiazole ring was selected for synthesisbased on the premise of favorable O···S interactions that wouldfavor a planar topography, and 107 and 110 are of comparablepotency as kinase inhibitors to their matched benzimidazoleanalogues 105 and 108. However, the benzothiazoles 107 and,particularly, 110 exhibited enhanced potency in the cell-basedassay, which was attributed to increased membrane permeabilitycompared to 105 and 108. In the cocrystal structure of theoptimized analogue 111 in both the BRAF and VEGFR2 kinases,the amide and bicyclic heterocycle are coplanar and reveal closeCO O···S contacts of 2.8 Å. In BRAF, the molecule binds inthe ATP pocket with the exocyclic NH engaging the CO ofCys532 in a H-bond interaction complemented by the thiazole Nengaging the NH of Cys532 in the kinase hinge region. Similarinteractions were observed in VEGFR2 but with Cys919 engagingthe aminothiazole moiety with the S atom acting as the func-tional equivalent of an NH mediating a pseudo ring structure.
Table 3. Comparative SARs Associated with Abl KinaseInhibitors Based on Benzimidazole, Benzothiazole, andBenzoxazole Scaffolds
Figure 18.Modeled binding modes of 102 (a) and 103 (b) in Abl kinaseillustrating the key H-bond donor−acceptor interactions with Met318 ofthe enzyme.
Table 4. SARs Associated with the Series of BRAF andVEGFR2 Kinase Inhibitors 105−110
Journal of Medicinal Chemistry Perspective
DOI: 10.1021/jm501853mJ. Med. Chem. XXXX, XXX, XXX−XXX
O

In BRAF, Trp531 appears to coordinate with the amide COof the inhibitor in a π−π interaction arrangement whichdistinguishes this molecule from sorafenib (112).85
Checkpoint 1 kinase (CHK1) inhibitors are anticipated tosensitize tumor cells to DNA damage by inhibiting an enzymethat induces cell cycle arrest and allows for DNA repair and cellsurvival throughmodulation of the transition through the G2 andS phases of cell replication. An X-ray cocrystal structure of 113with CHK1 revealed the key drug−target interactions and theshort (2.8 Å) distance between the urea O and the thiopheneS atoms, reflective of a favorable interaction. The structure alsohighlighted the importance of an intramolecular H-bondbetween the amide CO and urea NH that formed a pseudoring, an observation that inspired the synthesis of 114−116 aspotent inhibitors of CHK1.86 An X-ray cocrystal structure of thepyridine 114 confirmed that the key enzyme−inhibitorinteractions were preserved as was the topology of thecarboxamide moiety that mimics the urea of 113, favored by aclose O···S association of 2.8 Å. The importance of this interactionon biological activity was underscored by the striking difference inpotency that was observed between the matched pair of thiophenetopological isomers 114 and 117, with the latter 1500-fold weakerthan the former. This was attributed to distortion of thecarboxamide moiety of 117 from planarity, which resulted in apoor alignment for the important H-bonding interactions with thebackbone atoms of Glu85 and Cys87 of the enzyme.
A similar ring system is present in 118, a modestly potentinhibitor of Enterococcus faecalis and Haemophilus influenzae
NAD+-dependent deoxyribonucleic acid (DNA) ligase thatwas cocrystallized with both enzymes (Figure 19).87 The X-raystructures revealed binding in the same pocket of the enzyme asthe adenine base of the NAD+ cofactor rather than at the ATPsite as well as a close O···S contact (2.9 Å in the E. faecalis DNAligase) that presumably contributes to orienting the amidemoiety in a conformation that allows it to fully engage Glu118 andLys291 in H-bonding interactions.
The cyano-substituted pyridinone 119 was identified as ascreening lead that exhibits modest affinity for the γ-aminobutyric acidA (GABAA) α1β3γ2, α2β3γ2, and α3β3γ2 receptorsubtypes with Ki values of 7, 1.8, and 0.86 μM, respectively,binding to the benzodiazepine site of the ion channel.88
However, this compound exhibited complex pharmacology,acting as an antagonist at the α1 and α3 subtypes and a partialinverse agonist at the α2 receptor. Because full GABAA agonistswith increased affinity and selectivity for α2 and α3 over the α1subtype were targeted as potential anxiolytic agents, furtheroptimization was necessary. Initial studies replaced the nitrilewith CO2Me, which increased intrinsic affinity and introducedagonist activity toward all three receptors while replacingthe dimethyl aniline with a 4-substituted pyridine improvedaqueous solubility. To enhance intrinsically poor pharmacoki-netic properties, the ester was replaced with a series of azoleheterocycles as potentially metabolically stable isosteres.Remarkably, the oxadiazole 120 proved to be essentially inactiveat all three receptors, with Ki values of >0.67 μM; however, theanalogous thiadiazole 121 exhibited potent affinity for all threereceptors, with Ki values of 90, 42, and 25 nM for the α1, α2, andα3 subtypes, respectively, where it acted as a full agonist. The4-methyl thiazole 123 performed similarly with Ki values of 45,37, and 62 nM for the α1, α2, and α3 receptors, respectively.However, this heterocycle differentially modulated the pharma-cological profile, with 123 acting as an antagonist at the α1subtype and a partial agonist at the α2 and α3 receptors, while theC-5−methyl isomer 122 was a considerably weaker ligand for allthree receptors with Ki values of 330, 380, and 250 nM for α1, α2,and α3, respectively. A small molecule single-crystal X-ray studyof 123 revealed that while the methoxyphenyl and pyridylrings were twisted out of the plane of the pyridone heterocycle
Figure 19. Key drug−target interactions between 118 and E. faecalisDNA ligase illustrating the close intramolecular O···S contact.
Journal of Medicinal Chemistry Perspective
DOI: 10.1021/jm501853mJ. Med. Chem. XXXX, XXX, XXX−XXX
P

(by 58 and 63°, respectively), the thiazole adopted an almostcoplanar arrangement that is stabilized by a close and attractive1,5-O···S contact measured at 2.8 Å. This was interpreted asestablishing a pseudo ring system that conferred topographicalmimicry with the more common bicyclic and tricyclicbenzodiazepine ligands. These data also provided an explanationfor the SARs with respect to the site of the methyl group on thethiazole ring, which was presumed to result from the induction ofsteric interference when incorrectly presented to the receptor.
In the small molecule single-crystal X-ray structure ofcocrystals of nitazoxanide (124) with carboxylic acids, thethiazole N and exocyclic NH interact with the carboxylic acidmoieties of the cocrystal formers and there are close contactsbetween the thiazole S and amide O at distances of 2.6−2.7 Å.The observed short separations presumably reflect the effect of thepowerful electron withdrawing NO2 substituent on increasingthe size of the σ-hole on the thiazole S atom.89 In each case, thereare also close contacts between theO atom of the nitro moiety andthe thiazole S atom (2.9 Å) which are less than the sum of the vander Waals radii.
Acylated 2-aminothiazoles have emerged as a prominent motifin allosteric glucokinase (GK) activators that bind to a site 20 Åaway from the glucose binding site.90 An early lead, the acyl urea125, showed good potency as a GK activator, with the N−Mehomologue 126 being 4-fold more potent. Resolution of 126revealed that activity resided entirely in the R-enantiomer,SC1.5 = 0.41 μM.91 An examination of urea substitution patternssuggested that the conformation depicted, which is stabilized byan intramolecular H-bond, is important for recognition by GK.This observation focused modeling studies on the urea element,which was subsequently shown to engage the backbone NH andCO of Arg63 via a H-bond donor/acceptor array, as depicted
in Figure 20. Replacing the urea moiety with a heterocyclicamine led to a series of thiazole derivatives, with 127representative, which provided an effective mimic of the pseudoring. In this context, the S atom functions as an isostere ofthe NH interacting with the CO and favoring the cis-amidetopology that facilitates the H-bond donor/acceptor interactionwith Arg63 while the alternative topology would experiencerepulsive O···N effects. The methylsulfonyl derivative 128 wassubsequently selected as a clinical candidate and advanced intophase 1 trials.92
The 2-amino-benzamide 129 is a potent GK activator forwhich the X-ray cocrystal structure revealed similar interactionsbetween the protein and the 2-acylamino thiazole motif to thosethat were described for 125.93 It was also noted that one of the Hatoms of the aniline moiety of 129 forms an intramolecularH-bond with the adjacent CO while the second H atomestablishes a H-bond with the hydroxyl moiety of Tyr215 of GK.However, the latter interaction was not critical because themethylated homologue 130 was similarly potent. The origin ofthis observation was revealed by an X-ray cocrystal structure inwhich Tyr215 had rotated away from 130 to expose a lipophilicpocket that was viewed as an opportunity for further structuraloptimization. The core phenyl ring was replaced with a pyridineheterocycle, deployed in a fashion to favor the boundconformation by allowing the pyridine N atom to engage theproximal amide NH while avoiding an unfavorable interactionbetween the CO and CN moieties.93b SAR studies aroundthe aniline moiety focused on probing the lipophilic pocket ofGK by replacing this element with an alkoxy or aryloxy moiety,which was, unfortunately, unsuccessful because it resulted inmarkedly less potent inhibitors. However, the 4-fluorophenylthioderivative 131 expressed good potency, EC50 = 0.25 μM, with thephenyl ring of considerable importance because alkylthioanalogues were weak GK activators. These SAR observationsare consistent with an intramolecular O···S interaction thatcorrectly orients the fluorophenyl ring for interaction with thelipophilic pocket and which would be much weaker in thealkylthio analogue based on C−S σ* orbital energetics.22 Indeed,an X-ray cocrystal structure of 131 and GK revealed theanticipated binding mode with dual close O···S contacts betweenthe amide CO and the two proximal S atoms that stabilized thebound conformation (Figure 21).
Figure 20. Key drug−target interactions between the acylated ureamoiety of 125 and Arg63 which resides in an allosteric pocket of the GKenzyme.
Journal of Medicinal Chemistry Perspective
DOI: 10.1021/jm501853mJ. Med. Chem. XXXX, XXX, XXX−XXX
Q

Intramolecular H-bonding interactions were considered toplay an important role in preorganizing the conformation of twoseries of triazolopyridazinone-based cMet kinase inhibitors132−134 that engaged the enzyme via a H-bond between thequinoline heterocycle and Met1160. Thiophene 133 was 15-foldmore potent than the furan 132 as a result of a preference for anoverall planar conformation that was mimicked by the isothiazole134 in an homologous series.94 However, these compoundssuffered from poor aqueous solubility, attributed to the highsp2 atom count and attendant planarity, a deficiency addressedby exploring a dihydro-triazolopyridinone template. However,the first iteration of this concept afforded 135, a compoundwith poor intrinsic potency which was remedied by replacingthe N−phenyl with an isothiazole ring to afford 136 with 30-foldimproved potency. The increased potency was attributed to a1,5-CO···S interaction favoring a planar topography. How-ever, solubility was compromised, necessitating further opti-mization that ultimately afforded 137 which preserved theisothiazole moiety.
A series of antagonists of inhibitor of apoptosis (IAP) withpotential in the treatment of cancer was designed from asystematic study of the 4 residue N-terminus of secondmitochondria-derived activator of caspases (Smac), with theanalysis focused on optimization of the individual residues withinthe AVPI tetrapeptide sequence recognized by X-linked IAP(XIAP).95 AVPI binds to the BIR3 domain of XIAP with an IC50
of 500 nM and also binds to the high affinity site of the BIR3domains of cIAP1/2 and the single-BIR domain ofML-IAP while
binding with lower affinity to the BIR2 domain of XIAP. Thethiadiazole GDC-0152 (138) was discovered as part of a broadsurvey of P4 in an inhibitor in which the P1, P2, and P3 siteshad been optimized and bound to XIAP-BIR3 with a Ki of 28 nMand to MLX-BIR3SG with a Ki of 14 nM. X-ray cocrystal datarevealed the key binding interactions with a cIAOP/XIAPchimera and identified a close contact between the secondaryamide O atom and the thiadiazole S atom (2.9 Å) with somedistortion from planarity. In theML-IAP/XIAP chimeric protein,this distance was shorter at 2.7 Å and associated with a moreplanar arrangement between the amide and thiadiazole ring, aconformation in which the thiadiazole ring projected the boundphenyl ring into the S4 pocket of the protein. Interestingly, inthe ML-IAP/XIAP chimera, there was a close contact (3.0 Å)between the carbonyl O atom of the proline amide O atom andthe carbonyl C atom of the secondary amide, suggestive of astabilizing n → π* interaction because the sum of the van derWaals radii of O and C is 3.2 Å.96 The angle of this interaction is96.6°, less than the 99° ≤ Φ ≤ 119° range considered to bepreferred for a n→ π* effect. However, many Xaa−Pro dipeptidemoieties in proteins exhibit smaller angles with an average of95.6° ± 8.7°, suggesting that the constraints originally describedas preferred may have been too restrictive. Compound 138but not its enantiomer was shown to be an effective killer ofMDA-MB-231 breast cancer cells in a 3 day assay while normalhuman mammary epithelial cells were spared. This effect wasassociated with a dose-dependent activation of caspases 3 and 7.In a mouse xenograft model implanted with MDA-MB-231breast cancer cells, 138 dosed once weekly at 10, 50, or 100 mpkresulted in a reduction in tumor volume and the compound wassubsequently advanced into clinical trials.
Intramolecular 1,5 O···S interactions have been assessed in thesmall molecule single-crystal X-ray structures of the carbonicanhydrase inhibitor acetazolamide (139) and the stromelysin(matrix metalloproteinase 3) inhibitors 140−142, and these havebeen compared with their conformations in cocrystal structureswith the respective enzymes.97 In the enzyme-bound structures,the O···S distances marked in the structures below are allless than the sum of the van der Waals radii for O and S, with
Figure 21. Key drug−target interactions between 131 and the GKenzyme illustrating two close intramolecular O···S contacts that stabilizethe bound conformation of the activator.
Journal of Medicinal Chemistry Perspective
DOI: 10.1021/jm501853mJ. Med. Chem. XXXX, XXX, XXX−XXX
R

the shortest distance of 2.4 Å observed for 139. This compoundincorporates a powerful electron withdrawing moiety on thethiadiazole ring, which presumably enhances the O···S interactionbecause the distances measured for 140−142 are markedly longerat 2.8, 3.0, and 2.7 Å, respectively. Similar O···S interactions wereobserved in the small molecule single-crystal X-ray structures of 139and 140, and theoretical analyses of the conformers available to thekey substructures indicated that those able to establish an O···Sinteraction were the more stable.
A family of VEGF kinase inhibitors provides a particularlyinteresting example of effective bioisosteric relationships betweenthe phthalazine ring found in PTK787 (143), the intramolecularlyH-bonded analogues AAL-993 (144) and 145, and the thioether146.98 The thioether 146 is equipotent with 145, and in thisexample of matched pairs, the S atom functions as an effectivemimic of an NH. In the small molecule single-crystal X-raystructure of 146, there is a close contact between the S and amideOof 2.8 Å, which stabilizes an overall relatively planar conformation(the torsion angle between the amide moiety and the attachedpyridine ring is 35°), mimicking the heterocycle ring in 143 and theH-bonded pseudo rings in 144 and 145. The importance of the Satom is underscored by the inactivity associated with the etheranalogue 147 and the poorer activity of the methylene derivative148. Reversing theO and S in 146 also gave an inactive compound,149, rationalized by the small molecule single-crystal X-ray whichshowed a markedly different topography in which the thioamidemoiety adopted an almost orthogonal disposition relative to thedisubstituted pyridine ring (torsion angle 108°) that results in aseparation of the O and S atoms by 3.8 Å.98
This design principle was also used in inhibitors of SIRT1,the best studied of a series of NAD+-dependent proteindeacetylases.99 NCS-7 (150) is a modestly potent SIRT1inhibitor (IC50 = 56 μM), while the sulfur analogue 151performs similarly (IC50 = 78 μM). However, the ether analogue152 is much weaker, with only 4% inhibition observed at aconcentration of 100 μM. These results were attributed to thepreferred conformation of 150 enforced by an intramolecularH-bond between the amide NH and the ether O atom. ThisH-bond is inverted relative to 152, which creates an energeticallyunfavorable mismatch with the enzyme functionality, as depictedby the proposed binding modes captured in Figure 22.99a
The acrylic acid 153 was identified as a modestly potentcytosolic phospholipase A2α inhibitor lead from a libraryscreening campaign, but preliminary SAR studies were un-productive with respect to improving the potency.100 Analysis ofthe small molecule single-crystal X-ray structure of 153 revealed aclose contact between one of the nitro O atoms and the ortho Satom which was attended by coplanarity, thereby modulating theconformation of the chlorophenyl ring. This observation inspiredthe design of an indole ring as an isostere to give 154, whichexhibited 10-fold reduced potency but was optimized to thesignificantly more potent homologue 155.
A 1,5-O···S interaction contributes to the pharmacologicalactivity of inhibitors of the serine/threonine kinase transform-ing growth factor β receptor-associated kinase 1 (TAK1 orMAP3K7), which is part of the immune and inflammatorysignaling pathway that mediates signals from several cytokinereceptors.101 The lead inhibitor 156 half-maximally inhibitedTAK1 at a concentration of 1.2 μM, and an X-ray structure ofthe molecule bound to a TAK1−TAB1 kinase fusion protein
Figure 22. Structures of the SIRT1 inhibitors 150 and 151 and theirproposed interactions with Ala227 and Tyr229 of the enzyme, illustratingstabilization of the bound conformation via an intramolecular H-bond in150 and an O···S interaction in 151. An intramolecular H-bond in theether analogue 152 favors a conformation that would poorlycomplement the key elements of the enzyme.
Journal of Medicinal Chemistry Perspective
DOI: 10.1021/jm501853mJ. Med. Chem. XXXX, XXX, XXX−XXX
S

revealed that the benzothiophene ring adopts an almost coplanararrangement with the furopyridine core (dihedral angle 7°)stabilized by a close O···S contact that measures 2.8 Å.101b
Paring back the benzothiophene ring to a phenyl moietyfacilitated a survey of substitution patterns fromwhich the 2-SMederivative 158 was found to exhibit slightly increased potencycompared to 157 (Table 5). However, ortho substituents were
generally not well tolerated while 3-substitution was morebeneficial although the 3-SMe analogue 159 was 10-fold lesspotent than 158. H-bond acceptors installed at the 3-positionafforded the best potency, with an interaction with Lys63 of theenzyme postulated to be the complex-stabilizing influence. Thisinteraction can also be accessed by the CH2−CN moiety fromC-2 (160) and the NO2 from C-3 (161). Nevertheless, the gainsin potency were modest when compared to the progenitor. Theconsideration that both the 2-SMe substituent and the H-bondacceptor at C-3 were coplanar with the phenyl ring led to theconcept of fusing either a thiazole or thiadiazole ring onto theC-2, C-3 sites of the phenyl moiety in a fashion that elegantlycombined both pharmacophoric elements.This approach was successful, with the thiadiazole 162
displaying an IC50 = 0.004 μM vs TAK1 and an EC50 value of0.012 μM in the cellular assay while the 1,3-thiazole analogue163 exhibited comparable potency, IC50 = 0.013 μM vs TAK1 andEC50 = 0.010 in the cellular assay.
101b AnX-ray cocrystal structure of162 bound to a TAK1−TAB1 fusion protein revealed that thethiadiazole ring is almost coplanar with the furan ring (dihedralangle 10°), with the conformation influenced by a short intra-molecular O···S contact of 2.7 Å. Lys63 did not appear to establish adirect H-bond with either of the benzothiadiazole N atoms of 162,although it was suggested that they may engage the enzyme via anetwork ofH2Omolecules. An interesting observationwas the closeproximity of the S atom of Cys174 of TAK1 to the C atom at thebenzothiadiazole ring junction (marked with *), which has a partialpositive charge based on polarization. The measured intermole-cular S···C distance is 3.8 Å, and calculations suggested that this
interaction may contribute 1.68 kcal/mol of binding energy to thestability of the complex.
A series of benzoxazinone derivatives that were readily pre-pared via a Knoevenagel reaction between 1,4-benzoxazin-3-oneand an aldehyde were explored as tyrosine kinase inhibitors.102
The benzoxazinone 164, in which the configuration of thetrisubstituted olefin was assigned as Z based on the small JC−Hcoupling constant (∼3 Hz) observed between the vinylic H andthe CO carbon atom, is a representative inhibitor of the Abland Kdr tyrosine kinases, expressing IC50 values of 1.9 and 1.48μM, respectively. A small molecule single-crystal X-ray structureconfirmed the olefin geometry and also revealed a close contactbetween the thiophene S and benzoxazinone O atoms of 2.8 Å.The importance of the identity of the heterocyclic ring wasunderscored by the weaker potency associated with the furanseries based on 165 that is consistent with a positive role for theO···S interaction in the expression of biological activity.102
■ 1,5 O···S INTERACTIONS IN ORGANIC SYNTHESISThe organization of the transition states in isothiourea-catalyzedreactions appears to depend upon productive intramolecular1,5 O···S interactions.103 THTP (166) proved to be a moreeffective catalyst for the acylation of alcohols by acetic anhy-dride than the analogous carbon analogue DBN (167), with thedifference in reactivity ascribed to a stabilization of theN-acylated transition state intermediate by an O···S interaction.The activity of 166 and 167 as catalysts is dependent on the sizeof the ring, with the homologues 168 and DBU (169) being lesseffective.103a Fusing an aromatic ring onto the thiazolidine providedmore active catalysts, and this was attributed to the additionalassistance conferred by a productive π−π interaction betweencatalyst and substrate in the transition state. The process wasrendered asymmetric by introducing chirality into the bicyclic ringsystem, leading to the development of a practical procedure for thekinetic resolution of alcohols and carboxylic acids. This is bestexemplified by the commercially available tetramisole (170) andrelated compounds 171−173 in which the phenyl ring facilitatesπ−π interactions in the transition state while also providingasymmetric guidance based on sterics. An O···S interaction in thetransition state was considered to contribute to preorganizationof the enolate structure such that the pendent phenyl ring is ableto induce asymmetry by influencing the stereochemical approachof the electrophile toward the nucleophilic enolate C atom
Table 5. SARs Associated with TAK1 Inhibitors 157−161 inBiochemical and Cellular Assays
Journal of Medicinal Chemistry Perspective
DOI: 10.1021/jm501853mJ. Med. Chem. XXXX, XXX, XXX−XXX
T

(Figure 23).103i An O−···S interaction was also considered tostabilize the transition state in a fashion that led to asymmetric induc-tion in an intramolecular reaction producing β-lactones and in aninter- and intramolecular Michael addition−lactonization process.103j
■ 1,6 O···S INTERACTIONSA 1,6 O···S interaction was identified in the pyrimidinone-basedangiotensin II (AII) antagonist milfasaran (174, LR-B/081),which was selected as a candidate for clinical study.104 A smallmolecule single-crystal X-ray structure of 174 at 17K allowed detailedstudy of the electronic properties of this molecule and revealed aninteraction between the thiophene S and amide O atoms, with thedistancemeasured as 3.2 Å, just less than the sumof the van derWaalsradii. In addition, theOatomof the ester carbonylmoietymakes closecontact with one of the C-5 H atoms (2.2 Å) and may stabilize therelative conformation between the thiophene and pyrimidinone rings.Interestingly, it was also noted that there is a close association (3.3 Å)between the thiophene S atom and one of the tetrazole N atoms.
An X-ray cocrystal structure of the benzo[4,5]thieno[3,2-b]-furan-based IKKβ inhibitor 175, IC50 = 45 nM, with the structurallyrelated JNK3 kinase identified the key ligand−protein interactionsdepicted in Figure 24 and noted a close contact of 2.8 Å between the1,6-disposed urea O and thienyl S atoms.105 SAR studies revealedthat replacement of the thiophene ring with alternatives led to a 3−4-fold reduction in potency, suggesting a role for the O···S asso-ciation in modulating the conformation of the urea moiety.
Another interesting motif that relies upon a 1,6 O···Sinteraction can be seen in the small molecule single-crystalX-ray structure of the xanthydrol 176, a derivative of theantibiotic disulfide xenorhabdin 1, which was isolated fromXenorhabdus spp. usingMicrococcus luteus as the test organism inan antibacterial-guided bioassay.106 The O···S distances in thetwo molecules in the unit cell were 2.6 Å.
■ N···S INTERACTIONS1,4-N···S Interactions. An early survey of the CSD identified
78 structures that contained 1,4-N···S structural motifs.107 Ofthese, 69% had an N···S separation of less than 3.3 Å, within thesum of the van der Waals radii of N and S, that were associatedwith torsional angles of less than 20° (Figure 25).107
An interesting example of a functional role for a 1,4 N···Seffect is related to the studies of amide hydrolysis catalyzedby the cysteine protease papain.108 The structures of thethioacyl-enzyme model compounds N-benzoylglycine thio-ester 177 and N-benzoylglycine dithioester 178 weredetermined by X-ray crystallography. Short interatomicdistances between the N and the S atoms of 2.9 Å wererevealed based on the conformation depicted. Thisconformation has a N−Cα−C−S dihedral angle of between5° and 20°, with the three key atoms N−S−C1 aligned in alinear fashion, which maximizes the N−S nonbonding orbitalinteractions. The relative population of this conformer insolution, as measured by resonance Raman spectroscopy,was enhanced by increasing the basicity of the N atomthrough the introduction of electron-donating para sub-stituents on the phenyl ring, thereby strengthening theN···S interaction.109 It was suggested that the N···Sinteraction may function as a thermodynamic sink to favorthe collapse of the tetrahedral intermediate in the oxyanionhole to generate the thioacyl-enzyme intermediate.109b
Figure 23. Proposed structure of enolate intermediates derived from theisothiourea catalysts based on 170 and 173 illustrating an interactionbetween the anionic O atom and the proximal S atom that stabilizes thetopology shown, thereby maximizing the influence of the phenyl ring onthe steric approach of an electrophile.
Figure 24. Key drug−target interactions between 175 and JNK3 kinaseillustrating the close intramolecular O···S association.
Figure 25. 1,4-N···S structural motif searched in the CSD.
Journal of Medicinal Chemistry Perspective
DOI: 10.1021/jm501853mJ. Med. Chem. XXXX, XXX, XXX−XXX
U

Kinetic measurements on the effect of para substitution alsosuggested that the rate-limiting step during the sub-sequent deacylation process of the thioacyl-enzyme inter-mediate may involve overcoming the stabilizing N···Sinteraction.
In the small molecule single-crystal X-ray structures ofN-substituted derivatives of the antiulcer drug rabeprazole(179), an inhibitor of the gastric H+/K+-ATPase proton pump,the sulfoxide S atom and the N atom of the pyridine ring of180−182 are in close, sub-van der Waals radii contact, withN···S interatomic distances of 2.8, 2.8, and 2.7 Å, respectively.110
The pseudo four-membered ring comprised of S1−C15−C16−N4 that is associated with this conformation is nearlycoplanar, with dihedral angles between 0.6° and 12.8°.Interestingly, in an analysis by cold-spray ionization massspectrometry of a mixture of ethyl methyl sulfoxide andpyridine in MeOH at room temperature, a molecular ion peakat m/z 172 was observed. This corresponds to an ethyl methylsulfoxide/pyridine complex, suggestive of an intermolecularN···S interaction. These results provided evidence that theinhibition mechanism of 179 involves a 1,4-N···S nonbondinginteraction that draws the pyridine N atom into closeproximity with the benzimidazole iminium ion in theprotonated form of the drug. This accelerates the reactioncascade that generates a highly reactive sulfenamide inter-mediate in gastric parietal cells, conferring a use-dependentaspect to the pharmacology. The proton pump enzyme isinactivated via reaction of a cysteine residue with the activatedelectrophilic sulfenamide to afford a disulfide derivative, asdepicted in Figure 26.110
A transannular interaction between a N atom and a sulfoxide Swas observed in the crystal structures of the 1,5-thiazoninederivatives 183 and 184, with both structures exhibiting a nearlinear alignment of the N−S−O atoms and a N···S contactdistance of approximately 2.8 Å.111
In an example taken from the material sciences arena, anintramolecular N···S nonbonding interaction was designed intothe photochromic 2,3-dithiazolylbenzothiophene 185,which, along with a ArC−H···N H-bond, predisposes themolecule for photocyclization to the cyclohexadienederivative 186 (Figure 27).112 Because these contacts favor
coplanarity of the three heterocyclic rings, the two 2,5-disubstituted thiazole rings were only slightly tilted despitethe steric interference between the two methyl groups. Acrystal structure of 185 confirmed the molecular arrange-ment in which both the N···S and C-4−H···N distances,measured at 3.0 and 2.7 Å, respectively, are less than the sumof the van der Waals radii (Figure 28). The structure also
revealed a close proximity of the thiazole methyl group to theplane of the adjacent thiazole ring (3.1 and 3.2 Å), suggesting
Figure 26. Inhibitory mechanism associated with the gastric H+/K+-ATPase proton pump inhibitor 179 illustrating activation of thesulfoxide moiety into a sulfenic acid that reacts with a cysteine in theprotein.
Figure 27. Photocyclization of 2,3-dithiazolylbenzothiophene 185 andthe dithiophene analogue 187.
Figure 28. X-ray crystal structure of compound 185 showingthe intramolecular 1,4 N···S interaction and H···N close contact,d1 = 3.0 Å, d2 = 2.7 Å, φ = 30.1° (CCDC accession number796946).112
Journal of Medicinal Chemistry Perspective
DOI: 10.1021/jm501853mJ. Med. Chem. XXXX, XXX, XXX−XXX
V

that C−H to π−face interactions may also play a role in thestabilization of the photoreactive structure. Indeed, a smallmolecule single crystal of 185 turned blue upon irradiationwith UV light. In contrast, a single crystal of the dithiopheneanalogue 187, which is devoid of N atoms, resulted in aparallel conformation that is nonphotoreactive and themolecule did not exhibit photochromism. Remarkably,irradiation of a solution of 185 in hexane produced 186with a quantum yield of 0.98, among the highest syntheticphotochromic molecules, while photocyclization in MeOHdecreased the quantum yield to 0.54. The photocyclization of187 to 188 provided a lower quantum yield, but the values inthe two different solvents were similar (0.49 and 0.38,respectively). These results indicate that the attractive N···Sinteraction and the C-4−H···N H-bonding in 185 werecompromised in the polar protic solvent, leading to a reducedstabilization of the photoreactive state and a lower quantumyield.An intramolecular no···σ* nonbonding interaction between
N and S atoms was anticipated to stabilize the bioactiveconformation of the 5-(pyrimidin-4-yl)thiazole-based p38αMAP kinase inhibitor 190.24,64 This compound wasdesigned to replace the close O···S interaction observed inthe bound conformation of the thiazole-5-carboxamideanalogue 189 in the p38α kinase active site. An X-raycocrystal structure of 190 bound to the active site of theunphosphorylated p38α enzyme revealed coplanaritybetween the thiazole and pyrimidine rings, with the thiazoleS atom and the pyrimidine N1 atom positioned toward eachother separated by a distance of 3.0 Å, supporting theproposed stabilizing interaction (Figure 29). Further
indication of the importance of this kind of intramolecularN···S contact came from the small molecule X-ray structuresof the isomeric analogues 191 and 192, which showed a5-fold difference in kinase inhibitory potency. The moreactive analogue 191 crystallized in the bioactive conforma-tion depicted by 190, maintaining a 2.9 Å N···S distance andprojecting the 4-chlorophenyl ring into the kinase selectivitypocket. The less active 2,4-disubstituted pyrimidine isomer192 also showed a similar short N···S contact in the smallmolecule single-crystal X-ray structure, but this favored aconformation not preferred for binding to the p38α kinaseactive site.
The 5-(pyrimidin-4-yl)thiazole motif is also an importantstructural element of the potent and selective B-Raf V600Ekinase inhibitor dabrafenib (GSK2118436, 193) which wasapproved by the FDA in May 2013 for the treatment ofmelanoma associated with mutations in the B-Raf V600E geneproduct.113
NMR experiments using NOE methodology applied tothe series of phosphodiesterase (PDE) IV inhibitors194−196 that incorporate five-membered heterocycles atthe C-4 position of the phthalazine template suggested thatin solution the triazole of 195 and the imidazole of 196were rotated out of the plane of the phthalazine ring in bothapolar and polar solvents of different dielectric constants.114
Molecular dynamics simulations in the gas phase alsoindicated that the azole and phthalazine rings were almostorthogonal to each other. Interestingly, these results were incontrast to the planar conformation observed with 195 inthe solid state by X-ray crystallography in which N-2 of thetriazole ring projected toward the C-5 proton of thephthalazine carbocyclic ring. This coplanar conformation ispresumably stabilized by two complementary but weakintramolecular Ar−C−H···N interactions and is likely thepreferred one for binding to the PDE IV enzyme. However,similar experimental analyses performed on 194 revealedan almost coplanar arrangement between the thiazole andphthalazine rings, with a dihedral angle of about 16° insolution and in the gas phase. Presumably, this conformationis stabilized by a productive N···S effect. The conformationalpreference of these molecules appears to be related to PDE IVinhibitory potency because 194, the most active inhibitor,paid a much lower energy cost than 195 and 196 to adopt thebioactive conformation.
Figure 29. X-ray cocrystal structure of the complex of p38α kinaseand 190 illustrating the close intramolecular 1,4 N···S interaction,d = 3.0 Å, φ = 14.6° (PDB ID: 3NWW).24 For clarity, only thegatekeeper residue Thr106 and residues forming direct H-bonds to190 are shown.
Journal of Medicinal Chemistry Perspective
DOI: 10.1021/jm501853mJ. Med. Chem. XXXX, XXX, XXX−XXX
W

A similar kind of thiazole-2-N-heteroaryl motif is incorporatedinto the P2*moiety of the hepatitis C virus (HCV) NS3/4A pro-tease inhibitor simeprevir (TMC435, 197), which was approvedby the FDA as add-on therapy to pegylated-interferon-α andribavirin in November 2013. The quinoline and thiazole ringsof the P2* moiety are coplanar due to an attractive N···Sinteraction, as revealed by a cocrystal of 197 and the NS3/NS4Aprotease complex structure, and are positioned over the catalyticresiduesHis57 and Asp81.
115 This fragment, together with the fusedphenyl element of the quinoline heterocycle and the cyclopentylring, effectively shields the catalytic region of the enzyme frombulksolvent. Urea-basedmacrocyclicHCVNS3/4Aprotease inhibitorsutilizing the same 2-(quinolin-2-yl)thiazole P2* moiety have alsobeen described.116
A thiazole-2-N-heteroaryl ring arrangement is conspicuous innatural products including the marine alkaloid lodopyridone(198), isolated from the actinomycete Saccharomonospora sp.,which demonstrates cytotoxicity against HCT-116 human coloncancer cells (IC50 = 3.6 μM) and inhibits human quinonereductase 2 (NQO2, QR2).117 The structure of 198, asdetermined by X-ray crystallography, contains a 2-(quinolin-2-yl)thiazole ring which exhibits a preference for the N andS orientation depicted. A small molecule single-crystal X-raystructure of an advanced intermediate in a total synthesisof lodopyridone confirmed the preferred topology of the2-(quinolin-2-yl)thiazole ring, and this preference was alsoobserved in the benzothiazole derivative 199 in the solid state,with the molecule adopting a nearly planar conformation.118
While the phenolic proton forms an intramolecular H-bond tothe quinoline N atom, potentially weakening the intramolecularN···S interaction to some extent, the observed conformationalpreference is assisted by an intramolecular aryl C−H···Ninteraction between the quinoline C-3H and the benzothiazole.119
■ 1,4- N···S INTERACTIONS IN BITHIAZOLE ANDRELATED RING SYSTEMS
Bithiazoles are naturally occurring ring systems, with early examplesidentified in the thiopeptide antibiotic natural productsmicrococcinP (200) and thiomuracin A (201).120 It can be envisioned that thepreferred conformations of these molecules will be influenced by aninterplay between multiple attractive N···S and O···S interactionsand repulsive N···N and N···O interactions. An X-ray crystallo-graphic structure of 202, the bis-4-bromoanilide derivative of 200,revealed an interesting coplanar arrangement of the thiazole−thiazole−pyridine ring system that displays two neighboringN···S interactions.121 The thiazole at C-3 of the pyridine ringof 202 is in an almost orthogonal orientation relative to theheterocycle at C-2, presumably due to steric buttressingeffects. However, an N···S contact between the two thiazolerings could be envisioned if they were both to adopt a coplanartopography with the pyridine ring. Of note is the con-formation of the two amides at the C-4 position of the twothiazole rings in which the carbonyl oxygen atom is trans to theN atom of the thiazole. This presumably reflects the potentialfor a productive H-bond interaction between the NH andthiazole N and favorable alignment of the amide and hetero-cycle dipoles.62
Journal of Medicinal Chemistry Perspective
DOI: 10.1021/jm501853mJ. Med. Chem. XXXX, XXX, XXX−XXX
X

Cystothiazole A (203), isolated from the myxobacteriumCystobacter fuscus, is representative of another class of naturalproduct that contains a bithiazole core.122 This compoundexhibits potent antifungal activity against the phytopatho-genic fungus Phytophthora capsici at 2 μg/disk as well ascytotoxic activity against human colon carcinoma HCT-116and human leukemia K562 cells. During the chemicalsynthesis of metabolites of cystothiazole A, the crystalstructure of the intermediate (S)-204 was determinedwhich confirmed the close proximity of the N and Satoms of the bithiazole system and the attendantcoplanarity.122 Interestingly, a potential 1,4- or 1,5- O···S inter-action in the (S)-2-(thiazol-2-yl)propane-1,2-diol moiety was notobserved.
The light emitting chemical oxyluciferin (205) that is the basisof firefly bioluminescence incorporates a novel benzothiazole−thiazolidinone structure. A small molecule single-crystal X-raystructure revealed that the 5-methoxy analogue 206 exists in theenol form, with the benzothiazole and thiazole rings in an almostcoplanar arrangement (dihedral angle ≤6°) that aligns both setsof N and S atoms proximal in space.123 These structural featureswere also observed in the bis-mesylate derivative 207, and it is areasonable assumption that attractive N···S and repulsive N···Ninteractions are operative in these molecules and may have aneffect on the fascinating bioluminescence properties of thefirefly.
The [4,5′-bithiazole]-2′-carboxamide 208 is a corrector of theΔF508 cystic fibrosis trans-membrane conductance regulator(CFTR) with potential for the treatment of cystic fibrosis.28,124
This compound provides a novel example of conformationalcontrol by a combination of N···S and O···S interactions.Compound 208 exhibited a Kd of 0.7 μM in an assay usingepithelial cells expressing humanΔF508-CFTR. Replacement ofeither of the thiazole rings of 208with moieties that did not favoran overall planar conformation were inactive, indicating therelevance of the bis-heterocycle core conformation for biologicalactivity.28,124 The 4′-methyl-4,5′-bithiazole core of 208 canadopt two conformations, designated s-trans and s-cis as depicted
in the simplified model analogue 211, that present differentorientations of the aniline moiety relative to the amide group anddifferent directionality of the H-bond donors and acceptors.Density functional theory calculations at the B3LYP/6-31+G-(d,p) level showed that the minimum energy conformer for s-cis-211 was that with the bithiazole ring nearly coplanar. This wasmore stable than the s-trans isomer 211 set at a dihedral angle of151° by 0.79 and 1.26 kcal/mol in the gas phase and in H2O,respectively. The coplanar conformation of s-trans-211 wasabout 0.6 kcal/mol higher in energy than the noncoplanarconformer of s-trans-211. This preference was enhanced by anattractive N to S interaction, which apparently overrides thedestabilizing effect of allylic 1,3-like strain between the C-4′ CH3and the C-5 H atom. Interestingly, the C-2′-amide conformationis locked by a favorable O···S interaction together with a weakintramolecular H-bond between the amide NH and the N of thethiazole ring. The alternative amide conformation obtained by a180° rotation of the C-2′-CO bond was disfavored by about9 kcal/mol based on gas phase calculations, probably due to theabsence of the two favorable interactions and the presence of anunfavorable lone pair/lone pair repulsion between the carbonylO and the thiazole N atoms. Modeling studies suggested that theC-5-methyl-C-4′-ethyl analogue 209 was less likely to adopta s-cis coplanar conformation due to a 7 kcal/mol energy penaltyresulting from steric repulsion between the methyl and ethylgroups. However, the s-cis energy minimum of 209, whichshowed a distortion of 55° from planarity, was still slightly lowerin energy than the minimum calculated for the s-trans isomer,thereby providing an explanation for the reduced CFTRcorrector activity of 209 relative to 208. These results, alongwith further analyses, led to the design of the conformationallyconstrained bridged bithiazole analogue 210 (Figure 30), which
Figure 30. Small molecule single-crystal X-ray crystal of the CFTRcorrector 210 showing an intramolecular 1,4 N···S interaction, d = 3.1 Å,φ = 18.3° (CCDC accession number 687310).
Journal of Medicinal Chemistry Perspective
DOI: 10.1021/jm501853mJ. Med. Chem. XXXX, XXX, XXX−XXX
Y

was indeed a more active corrector than 208. It was shown that aN···S interaction was more favorable in seven-membered ringbridged constrained bithiazole analogues like 210 than in thecorresponding eight-membered ring bridged analogues basedon the calculated N···S interatomic distance and S−C−C−Ndihedral angle obtained from computer modeling.124c FurtherSARs derived by replacement of one of the thiazole rings withoxazole, oxadiazole, and thiadiazole coupled with computa-tional conformational analysis suggested that the alignment ofO, S, and N atoms on the same face with a dual O···S/N···Sinteraction was more likely to ensure access to the putativeU-shaped bioactive conformation, as depicted in the represen-tations of 208 and 210 and displayed in the crystal structure of210 (Figure 30)124c
The bithiazole derivative 212 emerged from screening of alibrary of aminothiazoles as a dual inhibitor of sphingosinekinases SphK1 and SphK2, which are enzymes that phosphor-ylate sphingosine, to afford sphingosine-1-phosphate.125 Thismolecule can potentially form simultaneous 1,4- and 1,5- N···Sinteractions that favor a conformation exhibiting an interestinglinear, planar array of the three thiazole rings. However, molec-ular modeling studies placed the 4-methylthiazole N atom awayfrom the S atom of the neighboring thiazole ring in the dockedpose of 212, suggesting the influence of protein/ligandinteractions in the kinase binding site.
The 4′-methyl-4,5′-bithiazole derivative 213 was identified asan inhibitor of phosphoinositide 3-kinase γ (PI3Kγ), a potentialtarget for the treatment of autoimmune and inflammatorydiseases, through high-throughput screening.126a Although noX-ray crystallographic information was reported, the boundconformation of 213 in the ATP-binding site of PI3Kγ as proposedby docking experiments displayed an interesting alignment of theN-3···S-1′···O (of the carbonyl) atoms. This may result from dual1,4-N···S and 1,5-O···S interactions. The N-3′ and amide NHserved as the typical kinase inhibitor donor−acceptor pair formingtwoH-bonds with the backbone of Val882 in the hinge region of theenzyme. Replacing the central thiazole ring with an oxazole (214)or an oxadiazole (215) maintained the kinase inhibitory activity,presumably due to preservation of the N···S···O and O···S···Oalignments in these two analogues.126b
Bioactivation of the antituberculosis drug ethionamide (216)is performed by the flavin adenine dinucleotide-containingmonooxygenase EthA in Mycobacterium tuberculosis to afford an
ethionamide−NAD adduct which inhibits the mycobacterialenoyl-ACP reductase InhA that is critically involved in cell wallbiosynthesis. Because gene transcription of EthA is suppressedby the transcriptional regulator EthR, inhibition of this proteinhas been proposed as a means of increasing the concentration ofthe bioactive metabolite of 216 in an effort to relieve the dose-limiting toxicity exhibited by this drug. A series of 3-(thiophen-2-yl)-1,2,4-oxadiazoles exemplified by 217−219 were shown to beinhibitors of EthR, with 217 exhibiting ∼10-fold better activityat inhibiting the interaction of EthR with its DNA promoterthan 218.127a This was attributed to a stabilization of the con-formation of the (thiophen-2-yl)acetamide moiety of 217 thatwas more compatible with the shape of the hydrophobic bindingpocket in that region of EthR by an O to S interaction. In theX-ray structure of the EthR/217 complex, the O and S atoms inthe (thiophen-2-yl)acetamide moiety are 3.1 Å apart. The SARfor a N···S attraction within the 3-(thiophen-2-yl)-1,2,4-oxadiazole series was not completely clear, and the orientationof the S atom, whether adjacent to N-2 or N-4, appeared not tobe critical. However, the N···S distances for both 217 and 218 intheir complexes with EthR are less than the 3.35 Å sum of the Nand S van der Waals radii and the two heterocyclic rings adopt anear coplanar geometry (dihedral angle ∼17°). Interestingly,consistent with the difference in partial negative charge that isreflective of the H-bonding potential differences between theN-2 and N-4 atoms of the 1,2,4-oxadiazole ring, the N-2···Sdistance in 218 is longer than the N-4···S distance in 217. This issuggestive of a weaker N···S attraction in 218. More remarkable isthe conformation of the 3-(thiazol-2-yl)-1,2,4-oxadiazole ringsystem exhibited by 219, the most potent analogue of the series, incomplex with EthR that is captured in Figure 31.127a The two ringsare in a perfectly coplanar orientation (dihedral angle = 0.1°) witha N-4···S interatomic distance of 3.1 Å, a nonbonding interactionthat appears to overcome the repulsion apparent between N-2 andN. Remarkably, the 2.9 Å distance between N-2 and N is shorterthan the 3.10 Å sum of van der Waals radii of two N atoms.
This kind of thiazole-oxazole/oxadiazole motif is alsofound in naturally occurring compounds which are synthe-sized by the post-translational heterocyclization of cysteine, serine,and threonine residues of ribosomally synthesized linear precursorpeptides.120 Recently, a widely conserved biosynthetic gene clusterof heterocycle synthetases responsible for the synthesis of thiazole
Journal of Medicinal Chemistry Perspective
DOI: 10.1021/jm501853mJ. Med. Chem. XXXX, XXX, XXX−XXX
Z

and oxazole rings on ribosomally produced peptides wasidentified.128 Plantazolicin (220) and its N-terminal didesmethylanalogue were identified in select strains of Bacillus amylolique-faciens and Bacillus pumilus and belong to a family of thiazole/oxazole-modified microcin natural products that have demon-strated selective antibacterial activity against the causative agent ofanthrax,Bacillus anthracis.129b The opportunities for attractiveN···S interactions within 5-methyl-4-(thiazol-2-yl)oxazole ring sys-tems and multiple N···O repulsive, nonbonding interactionsbetween the oxazole rings likely contributes to the unique three-dimensional organization of this molecule, in particular withregard to the substrate binding interaction with S-adenosyl-L-methionine-dependent methyltransferase (BamL), which re-mains to be fully characterized.129c
A series of 2-(1H-pyrazol-1-yl)thiazole-based antagonists ofthe EP1 receptor that recognizes prostaglandin E2 offer potentialto treat overactive bladder symptoms.130a In this series,replacement of the thiazole ring of the lead compound 221with an oxazole heterocycle (222) resulted in a dramatic decreasein potency. The thiophene analogue 223 was weakly potentcompared to 221 but was more active than either the furan (224)or pyridine (225) analogues. Although direct structural evidenceis lacking, these data suggest that the conformation of the5,5-biheterocycle moiety is influenced by an attractive N···Sinteraction or repulsive N···N and N···O interactions that mayplay a role in the observed SARs. Intriguingly, conversion of thepyrazole ring of 221 to an imidazole (226) led to a 10-folderosion of potency. This could be a function of the more polarand basic nature of the imidazole in 226 that may incur a higherdesolvation penalty for binding to the receptor.130b The sharpdecline in potency of the isomer 227 is presumably also due to anunfavorable repulsiveN···N interaction between the thiazoleN atomand the two imidazole N atoms, which gives rise to a conformationwith a dihedral angle between the two heterocycles that is sub-optimal for binding when compared to that of the prototype 221.
(Pyrazol-3-yl)thiazole 228 was identified as an inhibitor ofbacterial DNA gyrase B (GyrB) from a high-throughput screen-ing campaign directed toward the discovery of compoundsinhibiting the ATPase activity of the GyrB subunit.131 Thismolecule presents an interesting array of the three heterocycleswith an almost coplanar geometry in the bound state (the twodihedral angles are about −18.5°), as shown in the structure ofthe complex with Staphylococcus aureus (S. aureus) GyrBdetermined by X-ray crystallography (Figure 32). The pyrazole
ring of 228 exists in the tautomeric form depicted, favoring aH-bonding interaction between its N-1H and Asp81 of the enzymewhile the N-2 atom engages in a second H-bonding interactionwith a conserved H2O molecule. The indicated N···S distancebetween the pyrazole and the thiazole rings as well as the S···Sdistance between the thiazole and the thiophene rings (3.3 and3.5 Å, respectively) are slightly less than the sum of the van derWaals radii of 3.4 and 3.6 Å, respectively, suggestive of attractiveinteractions. Development of SARs demonstrated that an ethylester at the 5-position of the pyrazole provided equal potency totheMeSmoiety. The modest activity of 228 could be increased by10-fold by replacing the thiophene ringwith a 3-pyridyl ring (229),an improvement attributed to an additional H-bondinginteraction between a C−H of the pyridine ring and Arg136.
Figure 31. X-ray cocrystal structure of the EthR/219 complex showingan intramolecular 1,4 N···S interaction and a close N···N contact, d1 =3.1 Å, d2 = 2.9 Å, φ = 0.1° (PDB ID: 3SFI).127a
Figure 32. X-ray cocrystal structure of the S. aureus GyrB/228 complexshowing an intramolecular 1,4 N···S interaction and a close S···S contact,d1 = 3.3 Å, d2 = 3.5 Å, φ = −18.9° (PDB ID: 3G75).131
Journal of Medicinal Chemistry Perspective
DOI: 10.1021/jm501853mJ. Med. Chem. XXXX, XXX, XXX−XXX
AA

Interestingly, larger and more lipophilic substituents suchas cyclohexyl (230) and phenyl (231) were favored at the4-position of the thiazole ring when compared to a methylgroup, with a several-fold enhancement of potency relative to229. These sterically larger substituents would increase thedihedral angle between the pyrazole and the thiazole rings,suggesting that close proximity between the N and S atoms ofthese two rings is not a prerequisite for the observed gyraseinhibitory activity and can be compensated for by other inter-actions, for example, favorable van der Waals contacts.131
The highly potent bacterial dihydrofolate reductase (DHFR)inhibitor 232 adopts a rather unexpected conformation in thecocrystal structure of its S. aureus DHFR complex, with themolecule binding in a pocket adjacent to the NADPHcofactor.132 The thiazole and the benzimidazole rings arealmost coplanar (dihedral angle = 11°), with the thiazole Satom adjacent to the benzimidazole N-2 nitrogen atom andseparated by 3.1 Å. This is indicative of an attractive N···Sinteraction that appears to override the potential nonbondingrepulsion between the thiazole N atom and the benzimidazoleN-1 nitrogen atom. In the cocrystal structure, the thiazole C−Sσ* exhibited a preference for the CN lone pair electrons of thebenzimidazole moiety over those of the exocyclic N−C(aryl) σbond. Unlike in the structure of 219 described above, the 3.1 Åinter-ring distance between the two N atoms in 232 is longer andapproximates the sum of the van der Waals radii of two N atoms(3.1 Å). The SARs for this series of compounds suggestedthat the N···S interaction was beneficial to the inhibitory activity(compare matched pairs 232 with 233 and 234 with 235),although it may not be an absolute requirement, as demonstratedby the good potency of the furan analogues 236 and 237.Compound 232was highly selective toward the bacterial enzymebecause the size of the analogous pocket in human DHFR isnot sufficiently large to accommodate the thiazole ring. Thiscompound demonstrated antibacterial potency, with an MIC of0.0125 μg/mL toward wild-type S. aureus, was active againsttrimethoprim-resistant strains in vitro and exhibited in vivoefficacy in a mouse model of survival after a lethal S. aureuschallenge.132
Thiazolo[5,4-d]thiazole is an interesting, planar fused-ringvariant of bithiazole that offers a C2 symmetrical projection ofsubstituents from a ring system that has rarely been exploredin medicinal chemistry. The small molecule single-crystal
X-ray structure of 238 showed that the two pyridine ringsare almost coplanar with the central thiazolothiazole ringand the pyridine N atoms are proximal to the thiazole S atoms,suggestive of an influence of an N···S interaction on the solidstate conformation.133a Interestingly, the 1H NMR spectrumof the unsymmetrical 2,5-disubstituted thiazolo[5,4-d]-thiazole 240 showed that the proton signals of the quinolinering matched well with those of the symmetrical compound239, while those of the pyridine ring were much broader thanobserved in 238. These results suggest that the pyridinemoiety of 240 is rotamerically mobile presumably due to aweaker N···S interaction with the thiazolo[5,4-d]thiazole ringrelative to that between the quinoline and the thiazolo[5,4-d]-thiazole ring in the same molecule.133b
Another interesting thiazolo[5,4-d]thiazole system is 2,5-thienythiazolothiazole which was employed as a monomer unitfor poly(2,5-thienythiazolothiazole) derivatives that weredeveloped as organic electronic materials.134 A N···S contactdistance of 3.2 Å and a coplanar arrangement of the threeheterocycles was observed in the crystalline monomer 241. Thedistance between the N and S atoms is within the sum of van derWaals radii of the two heteroatoms (3.35 Å), consistent with anattractive intramolecular interaction. These structural featurescontribute to the properties of this type of polymer that exhibitshighly ordered structures in thin films and strong fluorescence insolution.
The two 2,3-benzodiazepines 242 and 243 were found to befar more potent inhibitors of both the closed and openconformations of all four homomeric α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptor channelsthan the unsubstituted 2,3-benzodiazepine 244 as well as the N-3amide and urea derivatives 245 and 246.135 Although theincreased activity can be partially explained by the filling of theside pocket in the “M” site of the AMPA receptors by thethiadiazole moieties of 242 and 243, the influence of an apparentstabilizing conformational bias induced by a N-2···S interac-tion between the benzodiazepine and the thiadiazole rings (asdrawn for 242 and 243) cannot be excluded. Interestingly, theamide 245 was the least potent among the N-3-substitutedanalogues, with activity similar to the unsubstituted 2,3-benzodiazepine 244. The amide carbonyl of 245 is likely to berotated away from the benzodiazepine N-2 atom to avoid arepulsive nonbonding interaction, while the urea analogue 246could adopt a planar conformation favored by an intramolecularH-bond between N-2 and the urea NH, with the topology asshown. In essence, the thiadiazole rings of 242 and 243 not onlyoccupied the side pocket more effectively but also appeared to be
Journal of Medicinal Chemistry Perspective
DOI: 10.1021/jm501853mJ. Med. Chem. XXXX, XXX, XXX−XXX
AB

functionally equivalent to a H-bond donor in which the S atomreplaces the more conventional NH of 246, leading to theimproved activity.
A crystal structure of the tankyrase 1 inhibitor 247 bound tothe poly(ADPribose) polymerase revealed that the SCH2 moietywas in plane with the 1,2,4-oxadiazole ring (the S−C−C−Ndihedral angle = 0.1°), with the S atom proximal to the oxadiazoleN-4 atom, a conformation that is indicative of a N···Sinteraction (Figure 33).136 Although this effect appears to besomewhat weak because the distance between the twoheteroatoms is 3.1 Å, it may help orient the oxadiazole C-34-methoxyphenyl substituent to project into the adenosinebinding groove of the tankyrase 1 enzyme. This compoundexhibited a preference for an N···S rather than an O···S contactbetween the S atom and the oxadiazole ring, consistent with amore partially negative charge at the oxadiazole N-4 atom.However, protein−ligand interactions associated with thisregion of 247, especially a favorable π-stacking interactionbetween the C-3 4-methoxyphenyl group and the imidazole ofHis1201 of tankyrase 1, may contribute to the observed con-formational disposition.
■ 1,5-N···S INTERACTIONS
In a series of 2-aminothiazole-derived kinase insert domainreceptor (KDR) inhibitors, it was observed that compounds witha basic nitrogen atom in heteroaryl rings ortho to the 2-aminogroup were more active, exemplified by the 34-fold increase inpotency of the 2-pyridinyl analogue 248 compared to its3-pyridinyl isomer 249.137a More dramatically, the analogous2-aminooxazole 250was >250-fold less potent than thiazole 248.Quantum mechanical calculations revealed that the twoheterocycles of the molecule could adopt two isoenergeticcoplanar conformations, A and B, which were the two lowestenergy conformers identified for the thiazole series (Figure 34).Conformer A was presumably stabilized in part by anintramolecular N···S interaction, although this was not strong
enough to result in an energetic preference for A relative to B.Conformer A was more likely to represent the bioactiveconformation because substitution at the C-3 of the pyridinering did not significantly affect the potency. The moleculararrangement in conformer A would also place the thiazole Natom and its 2-amino group in a position to form H-bonds withthe backbone amide proton and carbonyl oxygen of Cys919,respectively, in the kinase active site. Interestingly, for the oxazoleseries, the corresponding conformer A′ was energetically lessfavorable than the lowest energy conformer B′ by 5.8 kcal/mol,probably as a result of the destabilization by the repulsionbetween the lone pairs of the O and N atoms. Theconformational bias induced by these intramolecular interactionsprovides an explanation for the observed variations in KDRinhibitory activity for this series of inhibitor. Activity in cell-basedassays and in vivo animal pharmacokinetic parameters could bemodulated by varying the substituent at C-4 of the pyridinering of 248, with hERG ion channel inhibition attenuated byreplacing the phenyl ring with a cyanomoiety.137b The optimizedanalogue 251 demonstrated a dose-dependent pharmacody-namic effect in a mouse model that evaluated the inhibition ofVEGF-induced tyrosine autophosphorylation of mouse lungKDR.
An early example of the successful application of a favorable1,5- N···S stabilizing interaction utilized the (pyridinyl-2-ylamino) or (pyrimidin-4-ylamino)thiazole structural motif indual Src/Abl kinase inhibitors to provide a planar template forH-bonding to the enzyme target. In the X-ray cocrystal structure
Figure 33. X-ray crystal cocrystal structure of 247 and poly(ADPribose)polymerase showing an intramolecular 1,4 N···S interaction, d1 = 3.1 Å,φ = 0.1° (PDB ID: 3UDD).136
Figure 34. Conformational energy analysis of the 2-aminothiazole and2-aminooxazole cores.
Journal of Medicinal Chemistry Perspective
DOI: 10.1021/jm501853mJ. Med. Chem. XXXX, XXX, XXX−XXX
AC

of the Abl kinase in complex with 52, the thiazole and pyrimidinerings are coplanar, with the O atom of the 5-carboxamidecarbonyl group adopting a conformation that placed it proximalto the S atom, distance = 3.0 Å.66c Dual 1,4- and 1,5-sulfurinteractions promote a linear array of the O, S, and N atoms.H-bonds were formed between the NH of the 4-amino groupof 52 and the carbonyl O of Met318 and between the N-3 ofthe thiazole ring and the amide NH of Met318 in the hingeregion of the ATP-binding site of Abl kinase. In a series ofcocrystal structures of 52with a several kinases, the distancesbetween the pyrimidine N and thiazole S atoms range from2.7 to 2.8 Å.66 Pyridine and pyrimidine rings were initiallydesigned as urea or amide replacements to mimic theconformation of the carbonyl group in 252, as shown foranalogues 253−257. The SARs indicated that the corre-sponding isomeric pyridine or pyrimidine analogues inwhich the N atom was distal to the S atom of the thiazolewere several fold to >20-fold less active.66b,138 The isostericreplacement of amide/urea by a N-heterocycle essentiallyswitched the conformational influence of an O···S inter-action with that of a N···S interaction which played the samerole. This kind of (N-heteroaryl-amino)thiazole motif wassubsequently investigated through SAR studies, computermodeling, and X-ray crystallography in the several series ofkinase inhibitors discussed below.
X-ray cocrystal structures of the bound conformation of theCHK1 inhibitor 258 and its more potent analogue 259 with thekinase demonstrated that the pyridine−thiazole structuralelement in each adopted conformation A depicted in Figure 34and functioned as a H-bond acceptor−donor engaging Cys87(Figure 35).19,139,140 The N···S distances in 258 and 259 are2.9 Å, with N−C−N−C−S dihedral angles of 0.7° and −7.1°,respectively.
Isosteric replacement of a urea group with a pyridine ring wasfurther illustrated in a thiophene carboxamide-based series ofselective Janus kinase 2 (JAK2) inhibitors.141 The design strategyfocused on reducing the number of H-bond donors and polarsurface area in an effort to improve the oral drug-like propertiesof the high throughput screening hit 260 (Table 6). This
approach maintained the bioactive conformation of 260, which ismodulated by an O···S interaction, to one similarly favored by aN···S contact in the 2-pyridinyl analogue 261. As depicted by theSARs, analogues 262−264 that are devoid of these nonbondinginteractions are less active against JAK2 compared to 260 and261, which are similarly potent. Medicinal chemistry effortsdirected toward solving chemical and metabolic stabilityissues led to the identification of 265, which exhibited goodkinase selectivity, activity in cell-based assays, 100% oralbioavailability in rats, and in vivo activity in a mouse PK/PDmodel of myeloproliferative neoplasms. Computer-baseddocking of 265 into the JAK2 binding site to gain insight onthe binding interactions was performed and included an intra-molecular 1,5- N ···S interaction via an N−C−N−C−S dihedralangle constraint between the thiophene and pyridine rings thatenforced a planar heterocyclic core.
Figure 35.X-ray cocrystal structure of the CHK1/259 complex showingan intramolecular 1,5 N···S interaction, d1 = 2.9 Å,φ1 = 0.7°,φ2 =−7.1°(PDB ID: 3TKI).139 For clarity, only gatekeeper residue Leu84 andresidues forming direct H-bonds to 259 are shown.
Table 6. Structures of the Selective JAK2 Inhibitors 260−265and Their JAK2 and JAK3 Inhibitory Potencies
Journal of Medicinal Chemistry Perspective
DOI: 10.1021/jm501853mJ. Med. Chem. XXXX, XXX, XXX−XXX
AD

The N-(pyridin-2-yl)thiazol-2-amine 266 was designed as aGK activator using a structure-based approach in which thisstructural element functioned as a replacement of the thiazoleamide moiety present in an earlier series represented by267.142,143 As revealed by a cocrystal structure with GK, theN-(pyridin-2-yl)thiazol-2-amine group of 266 maintained aplanar geometry with a N···S distance of 2.9 Å, enabling theamino group and the thiadiazole N to act as donor−acceptorpartners and form dual H-bonds with Arg63 in the allostericbinding pocket of the enzyme.142 In this context, the N···Sinteraction in 266 effectively mimics the N···HN H-bondinteraction between the urea NH and pyridine N atom of 268.144
As observed in a GK/268 cocrystal structure, the intramolecularH-bond locked the pyridinyl NH and the urea carbonyl in thepreferred s-cis conformation that projected these two functionalgroups in a fashion that established complementary donor−acceptor H-bonds with Arg63.
The N-(pyrazol-3-yl)-1,3,4-thiadiazol-2-amine (S)-269 is aninhibitor of the enoyl−acyl carrier protein reductase (InhA) ofMycobacterium tuberculosis that interferes with mycolic acidbiosynthesis by binding to the substrate binding site of InhA.145
In an X-ray cocrystal structure of (S)-269 bound to InhA, theN-(pyrazol-3-yl)-1,3,4-thiadiazol-2-amine moiety exhibited thesignature of an N···S nonbonding interaction with a planararray of the two heterocycles and a N···S distance of 2.8 Å(Figure 36).145 This flat scaffold provided a H-bond donor−
acceptor pair comprised of the thiadiazole N and the amine NHthat complemented the backbone amide NH and carbonyl ofMet98 in the binding site. This arrangement ideally positioned thedifluorobenzyl group to project into a hydrophobic pocket that isnormally occupied by the alkyl side chain of fatty acid substrates.Also noteworthy is the observation that the 4-methylthiazolering of (S)-269, which interacted with the NAD cofactor,appeared to form an intramolecular O···S interaction with thependent hydroxyl group with a distance of 2.9 Å, although theS−C−C−O dihedral angle is approximately 23°. The structuralfeatures conferred by the N···S interaction weremanifested in theSARs because replacing the pyrazole ring of rac-269 withheterocycles capable of forming a favorable N···S interaction(270−272) maintained or improved the potency in the InhAenzymatic assay. In contrast, the isomeric pyrazole analogue 273,which is devoid of a potential N···S interaction, showed a 59-folddecline in activity. The amide analogue 274, which was also50-fold less potent, presents an interesting example because theX-ray cocrystal structure with InhA revealed a close contactbetween the thiadiazole S atom and the amide carbonylO (distance between O and S = 2.9 Å, dihedral angle 25.2°),thereby mimicking the N···S interaction in 269 (Figure 37). The
thiadiazole amide-4-methylthiazole moiety of 274 bound toInhA in a similar fashion to 269, adopting an almost identicalconformation. However, the position of the difluorobenzylgroup of 274 was restricted by the conformation of the amidemoiety, which projected the aromatic ring in the oppositedirection to that of 269, presumably accounting for the reducedpotency.
Figure 36. X-ray cocrystal structure of the M. tuberculosis enoyl-acylcarrier protein reductase (InhA)/(S)-269 complex showing intra-molecular 1,5 N···S and 1,4 O···S interactions, d1 = 2.8 Å, d2 = 2.9 Å,φ1 = 2.2°, φ2 = −8.0°, φ3 = 23.1° (PDB ID: 4BQP).145
Figure 37. X-ray cocrystal structure of the M. tuberculosis enoyl−acylcarrier protein reductase (InhA)/274 complex showing intramolecular1,5 N···S and 1,4 O···S interactions, d1 = 2.9 Å, d2 = 2.9 Å, φ1 = −0.3°,φ2 = −5.9°, φ3 = 25.2° (PDB ID: 4BQR).145
Journal of Medicinal Chemistry Perspective
DOI: 10.1021/jm501853mJ. Med. Chem. XXXX, XXX, XXX−XXX
AE

The N-(pyridin-2-yl)-5-(pyrimidin-4-yl)thiazol-2-amine 275is an inhibitor of the cell cycle regulatory kinase Cdk4 thatexhibits >300-fold selectivity against Cdk1 and Cdk2 as wellas the non-cell cycle-related Cdk5, Cdk7, and Cdk9.146 Theselectivity was achieved by exploiting the limited space betweenthe gate keeper residue Phe93 and the pyrimidine ring of 275 viathe introduction of a methyl group at the C-6 position of theheterocyclic ring. Inhibitor 275 represents an interestingmolecular scaffold in that both attractive 1,4- and 1,5- N···Sinteractions between the thiazole S atom and the N atoms of thepyrimidine and pyridine rings, respectively, can exist simulta-neously. These interactions influence the orientation of thethree rings to adopt a coplanar arrangement. Although notunambiguously determined, it may be hypothesized that thecoplanarity would project the C-6 CH3 group in an optimalmanner into the Cdk4 gatekeeper binding pocket, conferringthe high selectivity observed for Cdk4 relative to other Cdkenzymes.
The N-(pyridin-2-yl)thiazol-2-amine moiety was also identi-fied as a useful scaffold for inhibitors of prion diseases.147 Inprion-infected neuroblastoma ScN2a-cl3 cell lines, it appeared thatthe antiprion activity of 276−279was dependent upon the presenceand position of the N atom of the pyridine ring. The 2-pyridylanalogue 277, which can potentially form an N···S interaction, wasmore potent although this aspect of the physical chemistry was notexplicitly characterized.148a,b Density functional theory calculationson the related prion inhibitor 280 indicated that at neutral pH thelowest energy conformation of the N-(pyridin-2-yl)thiazol-2-aminemoiety is coplanar. At a more acidic pH, the aminopyridine (pKa∼5.6) is protonated, which results in an out-of-plane arrangementbetween the thiazole and the pyridine rings, results that are consistentwith the disruption of a N···S interaction.148c
In inhibitors of glycogen synthase kinase 3 (GSK3)exemplified by 281, an atypical aryl C−H to N interactionbetween the positively polarized C-4 proton of the pyrazole ringand N-3 of the quinazoline was recognized as being crucial tostabilizing the bound planar conformation of the molecule.23
This topography enabled the appropriate projection of theH-bond donor−acceptor−donor motif of the 5-aminopyrazole,allowing formation of a key H-bonding triplet interaction withthe hinge region of the kinase. The conformation depicted in
281a that is observed in the cocrystal structure of the compoundbound to the ATP-binding site of GSK3 incorporates an Ar−C−H···N interaction. This conformer is 0.4 kcal/mol more stablethan conformer 281b, in which a conventional H-bonding inter-action exists between the pyrazole NH and the quinazoline Natom. A conformation anchored by an Ar−C−H···N interactionalso appears be operative in 282, which rendered the moleculeunable to form two of the three essential H-bonds to the hingeregion of the enzyme, resulting in an approximate 100-fold re-duction in activity compared to compound 281. More inter-estingly, the 2-aminothiazole analogue 283 exhibited a strongpreference for a planar conformation, attributed to the partiallypositive S atom and partially negative quinazoline N-3 atomengaging via a polar interaction. This conferred 9 kcal/mol ofstabilization to the molecule based on ab initio calculations atthe RHF/6-31G* level. Indeed, this preferred conformationwas observed in the cocrystal structure of 283 complexed toGSK3, which also unveiled an unusual H-bond between thethiazole C-4 H atom and a backbone carbonyl O atom,mimicking the role of the pyrazole N-2 H atom of 281. The6-fold decrease in potency of 283 relative to 281 may beattributed to the weaker Ar−C-3−H···O interaction com-pared to a more conventional H-bond. Notably, this was thefirst demonstration of an intermolecular C−HH-bond betweena ligand and protein playing a role in stabilization of thecomplex.
The tactic of employing a 2-aminothiazole as an isostere of a3-aminopyrazole to maintain a planar conformation and a donorcapability was investigated in the JAK2 inhibitor 284.149 Thethiazole analogue 285 appeared to be almost as potent as 284 ina JAK2 enzymatic assay at the Km of ATP; however, 285 was16-fold less potent at a high concentration of ATP (5 mM),suggestive of weaker binding affinity. In a TEL-JAK2proliferation assay, 285 exhibited only moderate inhibitoryactivity, with a GI50 of about 0.37 μM, and this compound wasalso more than 10-fold less stable in rat liver microsomes whencompared to 284. Cellular activity could be improved by theincorporation of a morpholine ring in the solvent-exposedregion of the molecule, as demonstrated by the relatedpyrimidine 286; however, the intrinsic JAK2 kinase inhibitoryactivity of 286 was still about 7-fold less than that of thepyrazole analogue.150
Journal of Medicinal Chemistry Perspective
DOI: 10.1021/jm501853mJ. Med. Chem. XXXX, XXX, XXX−XXX
AF

Conformational stabilization by a N···S interaction wassuccessfully used as a design principle to optimize the kinaseinhibition profile of a structurally related, quinazoline-basedinhibitor of Aurora A and B kinases derived from the leadcompound ZM447439 (287).151a The SAR survey gave rise tothe potent thiazole analogue 288, with a >100-fold increase inactivity in the Aurora A and B enzymatic assays compared to 287and significantly enhanced inhibition of phosphorylation ofhistone H3 in SW620 cells.151b Modeling of the heterocyclic coreof 288 using ab initio calculations at the RHF/6-31G* levelsuggested that the two heterocyclic rings adopted a coplanarconformation in which the thiazole S and the quinazoline N-3atoms were oriented proximal to each other. This conformationwas favored by 17.2 kcal/mol relative to the alternate con-formation in which the thiazole N and the quinazoline N-3 atomswere proximal. This topology optimally positioned the3-fluorophenylacetamide side chain on C-5 of the thiazole ringto occupy the hydrophobic selectivity pocket. Modeling studiespredicted that the C-5 side chain could bind to the DFG-outconformation of Aurora kinase, leading to the enhanced cellularpotency of 288. The corresponding C-4-substituted analogueshowed reduced kinase inhibitory activity and >100-fold loss inpotency in SW620 cells, consistent with the conformationalanalyses. Interestingly, the oxazole analogue 289 was a weakinhibitor compared to 287 in both the enzymatic and cellularassays, probably because the molecule preferred a 28° torsionalangle between the oxazole and the quinazoline rings, whichwould inflict a significant energy penalty in order to assume thebioactive coplanar conformation. Replacement of the morpho-line ring of 288 with a 4-(hydroxymethyl)piperidine ringprovided a compound that as its phosphate prodrug demon-strated pharmacodynamic effects consistent with Aurora kinaseinhibition in nude mice implanted with subcutaneous SW620human colorectal carcinoma xenografts. Not surprisingly, thebioisosteric quinazolin-4-yl-pyrazol-3-yl-amine analogues, whichcan adopt the same bioactive conformation as 288 for the reasonsdiscussed above for the GSK3 examples 281 and 283, were alsovery potent Aurora kinase inhibitors.
Another series of potent Aurora kinase inhibitors includes290 and 291, which feature an imidazo[1,2-a]pyrazine core withan aminoisothiazole ring appended at the C-8 position.152 Ingeneral, isothiazole- and thiophene-containing analogues weremore potent, although these heterocycles were not absolutelyrequired. X-ray cocrystal structures of compounds 290 and 291in complex with Aurora kinase A revealed that the inhibitorsbound in the ATP pocket, with the kinase in the catalyticallyactive DFG-in conformation.152a,c The NH of the pyrazolemoiety formed a H-bond with the carboxylate of Asp274, aninteraction critical for enhancing inhibitory potency. Thebioactive conformations of both compounds are very likelystabilized by intramolecular N···S interactions, as indicated bythe close contact between the N and S atoms (3.0 and 2.9 Å).This planar conformation positions theN-1 and C-8NHH-bonddonor−acceptor pair for H-bonding to Ala213 in the hinge regionwhile projecting the isothiazole methyl group toward the solvent-exposed region. Elaboration of this methyl group with basicamines and polar groups, exemplified by 291, was essential forimproving cellular activity toward inhibition of phosphorylationof histone H3 in HCT-116 cells while also serving as an avenuefor increasing solubility.152d,e
Interesting SARs highlighting the potential of a S atom to serveas an isostere for a NH was observed in a series of pyrrolopyrazine-based antimalarial agents.153 The benzimidazole derivative 292exhibited potent growth inhibition against asexual blood stages ofthe drug-sensitive Plasmodium falciparum NF54 strain, with anEC50 value of 50 nM. N-Methylation of the benzimidazole N atom(293) decreased potency by 7-fold, suggesting that a productiveH-bond between the benzimidazole NH and the pyrrolopyrazineN-1 atom was important for activity. It can be envisioned that thisH-bond together with an additional H-bond between theNHof theC-6 amino group and the benzimidazole N atom would support acoplanar conformation of the tetracyclic ring system. Notably,replacement of the benzimidazolewith a benzothiazole ring restored
Journal of Medicinal Chemistry Perspective
DOI: 10.1021/jm501853mJ. Med. Chem. XXXX, XXX, XXX−XXX
AG

the potency of 292, as shown by 294, establishing the potential of aN···S interaction to mimic a N···NH H-bond in this context.
A series of pyrido[2,3-d]pyrimidine-4-amine derivativesrepresented by 295 were identified as potent inhibitors of theHCV NS5A replicase complex.154 The corresponding Oanalogue 296, however, exhibited dramatically reduced potency,against genotypes 1a and 1b virus, 51-fold and 113-fold, respec-tively, when compared to the S-containing prototype 295. It wassuggested that “The sulfur atom’s larger size, greater polarizability,longer bond length, and lower bond angle (C−S−C) compared tooxygen may result in signif icantly dif ferent topologies for 295 and296, and may account for the potency dif ference.”154 However, a 1,6-N···S interaction between the sulfide S and the pyrimidineN atoms in295would be expected to influence the conformationof themoleculeto stabilize an overall tetracyclic array. Thismay result in orientation ofthe aniline and amide substituents in a fashion that is suitable forinteraction with the NS5A protein which could contribute to thesuperior activity of 295 compared to 296. Structural evidence forrelated intramolecular 1,5-N···S interactions involving phenylsulfidederivatives has been provided by X-ray crystallographic studies ofthe oxazolinyl-phenyl benzyl sulfide 297 and its analogues.155 The2-oxazolidine ring and phenylsulfide moiety in 297 are in a planargeometrical arrangement with the distance between the S and Natoms measured at 2.8 Å, which is shorter than the sum of van derWaals radii, 3.35 Å, of these two atoms.
■ SULFUR···AROMATIC INTERACTIONS
The possibility that in proteins the interactions between thesulfur atoms in cysteine andmethionine and the aromatic rings inphenylalanine, tyrosine, tryptophan, and histidine or cofactors(e.g., heme) could provide a larger amount of stabilization thanexpected for simple van der Waals contacts was proposed in1978.156 An analysis of the X-ray crystal structure data providedin the Atlas of Macromolecular Structure on Microf iche, a pre-decessor of the Protein Data Bank, revealed numerous contactsbetween sulfur atoms and π-bonded atoms in proteins where theinteratomic distances were ≤5.0 Å.157,158 This distance cutoffwas derived from the sum of the van der Waals radii for S andπ-bonded atoms, with 0.75 Å added to account for uncertaintiesin atomic coordinates. Alternating chains of these S···π inter-actions (e.g., Cys−Tyr−Cys−Phe) were also identified in severalprotein structures. An earlier solution phase study demonstratedthat disulfide and aromatic ring-containing compounds can form1:1 complexes, realizing approximately 1 kcal/mol in enthalpic
stabilization, which exceeds the estimate for van der Waalsinteractions by several fold.159,160 On the basis of the foregoing, itwas concluded that individual S···π interactions in proteinsprovide additional stability beyond van der Waals contacts and itwas proposed that alternating chains of S···π contacts facilitatefolding and electron transfer within proteins.156 The formationof charge transfer complexes, unusually strong van der Waalsinteractions, and π−π bonding between S atoms and aromaticrings were suggested to rationalize the S···π association. Sub-sequent efforts to characterize the geometric and energeticaspects of sulfur···aromatic close contacts have focused in threemain categories including statistical analyses of X-ray crystalstructure data, computational studies of model systems, andexperimental spectroscopic investigations.159,161−164 In the fol-lowing sections, the findings from these studies are summarizedand several reports in which sulfur···aromatic interactions maycontribute significantly to protein···protein and protein···ligandassociation are highlighted, although only a subset of illustrativeexamples are discussed.164
■ CRYSTALLOGRAPHIC DATABASE ANALYSESOF SULFUR···AROMATIC INTERACTIONS
Subsequent to the initial report, S···π interactions in the X-raycrystal structures of 22 proteins were evaluated and an empiricalequation for predicting their frequency was derived(eq 1).156,161a The correlation coefficient between the predictedand actual number of S···π interactions in the 22 protein set was0.86, a high level of predictivity despite the inclusion of counts ofonly three different amino acids (Met, Arg, Tyr) in the equation.An inverse correlation between the number of S···π and π−πinteractions was interpreted to imply competition between S···πand π−π interactions in proteins. It was concluded that thefrequency of S···π interactions observed differed significantlyfrom what would be expected for random contacts and wasstrongly correlated with the presence of the basic amino acidarginine.
= +× + × − ×
YN
2.54(4 Met 2 Arg Tyr) 83
(1)
Y = predicted number of sulfur ··· aromatic interactions;N = totalnumber of amino acids in protein.There are multiple possible configurations in which aromatic
rings in amino acid side chains may interact with the sulfur atomof methionine or cysteine residues in the context of proteins, andmany of the structural database analyses have focused onidentifying the geometric parameters of these and the frequencywith which they occur (Figure 38).
An attempt was made to define the preferred geometry forS···π interactions based on analysis of a larger set of 36 proteinX-ray crystal structures with ≤2 Å resolution.161b For each S···π
Figure 38. Possible interactions geometries for sulfur···aromatic inter-actions. In A and B, the S atom is located above the ring plane, while in C,the S atom interacts with the edge of the ring. The latter configuration isunlikely to represent a S···π interaction.
Journal of Medicinal Chemistry Perspective
DOI: 10.1021/jm501853mJ. Med. Chem. XXXX, XXX, XXX−XXX
AH

contact, three geometric parameters were evaluated, the first ofwhich was the distance (d) from Phe, Tyr, or Trp aromatic ringcentroids to the Cys or Met sulfur atoms (Figure 39a). Proximalsulfur atoms and aromatic rings for which d was determined tobe ≤6 Å were identified as S···π interactions. For these, theelevation angle (φ1) between the centroid to S atom vector andthe ring plane was determined. A second angle parameter, φ2,spanning the vector connecting the ring centroid and a ringcarbon atom and the projection of the ring centroid to sulfuratom vector onto the ring plane, was also evaluated. Analysisof normalized distributions indicated a maximum in d atapproximately 5.3 Å separation for S···π contacts, while theelevation angle, φ1, for S···π interactions had a maximum in the0−10° range, andφ2 showed a broad distribution. The maximumof 5.3 Å for d is consistent with sulfur−carbon van der Waalscontacts. The observed small values of φ1 correspond to S atomslocated approximately in the plane of the aromatic ring ratherthan above the ring face. On the basis of this, it was suggestedthat referring to these sulfur/aromatic ring contacts as S···πinteractions was misleading. It was also noted that almost 50% ofthe S atoms in methionine, cysteine, and cystine residues in the36 protein crystal structures were within the 6 Å cutoff distanceused to define S···π interactions. This is in accord with the earlierstudy which reported a higher than expected number of S···πinteractions.161a
Sulfur···aromatic interactions were evaluated as part of a moreextensive analysis of the intraprotein contacts made by cysteinesulfhydryl groups in 49 X-ray crystal structures.161c Theparameter utilized in this study measured the distance fromthe relevant cysteine sulfur atom to the closest ring atom in theproximal phenylalanine, tyrosine, tryptophan, or histidine ring(Figure 39b). The angle, ϑ, between the aromatic ring planenormal vector at the ring atom closest to the cysteine sulfur andthe ring atom to sulfur vector was evaluated to discriminatebetween interactions of S with the ring face or edge. Ring−faceinteractions were defined as those with angles less than 45°, andthose with angles greater than 45° were deemed ring edge inter-actions. Of the 28 cysteine sulfur···aromatic contacts identified,19 were ring−face interactions and the remaining nine were edgecontacts. This suggests a preference for cysteine sulfhydrylgroups to interact with the faces of aromatic rings. A study ofsulfur···aromatic interactions involving methionine that em-ployed similar geometric criteria found comparable numbers ofsulfur to ring−face and ring−edge interactions.161f It was noted
that on the basis of a random distribution and the larger areaavailable for ring edge interactions, a substantially larger fractionof edge contacts would be expected. Shorter sulfur···ring atomcontact distances when the sulfur atoms were located moredirectly above the aromatic ring planes were interpreted to implylarger favorable interaction energies for sulfur contacts withring faces. A study that focused on only the interaction ofphenylalanine with cysteine residues not involved in disulfidebonds found that for 682 cysteine residues in 609 X-ray crystalstructures, there were 268 interactions with phenylalaninerings.161e Of these, 207 were in geometries indicative of attractiveinteractions between sulfur lone pairs of electrons and a region ofpositive electrostatic potential at short distances (0−1.3 Å) aboveand below the plane of the phenyl ring (Figure 40A). Only four
were consistent withH-bonding between the π-face of the phenylring and the cysteine S−H moiety (Figure 40B). The authorsnoted several possibilities to rationalize the infrequentoccurrence of S−H···π H-bonds. Repulsion between lone-pairelectrons on the cysteine sulfur atoms and the π-electronsin the phenyl ring of phenylalanine could enforce stringentgeometric requirements on S−H···π H-bonds. Phenylalanineand cysteine residues are distributed differentially within proteins(hydrophobic core vs surface). Finally, comparatively weak S−H···π H-bonds may compete with other stronger interactionsinvolving phenylalanine.Another study of 753 protein crystal structures examined
interactions of cysteine residues with the aryl rings ofphenylalanine, tyrosine, and tryptophan.161h The geometriescorresponding to the maxima in a normalized two-dimensionalhistogram comparing sulfur to ring centroid distance, and theangle between the centroid−sulfur vector and the ring plane wereanalyzed and are shown in Figure 41. These corresponded to
three minimum energy configurations identified in a high-level,gas phase, ab initio quantum mechanical study of the benzene−H2S complex.The analysis of interactions of SSC (cystine) and CSC
(methionine) moieties with phenylalanine and tyrosine aromaticrings in a set of 1085 nonredundant X-ray crystal structures noteda preference for both SSC and CSC groups to interact with thering edges, although contacts between the sulfur atoms and theπ-faces of the aromatic rings were also observed.161g Recently, ananalysis of the entire PDB (>80K structures) focused on theinteraction of methionine sulfur atoms with the aryl rings of
Figure 39. (a) Geometric parameters for S···π interactions defined byReid et al.161b Figure adapted from ref 161b based on the Met160/Phe149S···π interaction in the 1.8 Å X-ray crystal structure of domain VI andLE1 of human netrin-G2 (PDB ID: 3TBD).165 (b) Polar angle analyzedin a study of S···π interactions between cysteine and aromatic residues inproteins.161c The S···π interaction between Cys59 and Phe66 in the 1.8 ÅX-ray crystal structure of chloride intracellular channel 1 (CLIC1)complex with glutathione (PDB ID: 1KON) is shown.166
Figure 40. Schematic representations of phenylalanine/cysteineinteractions identified in protein structures.161e
Figure 41. Schematic representations of predominant Phe/Tyr/Trpinteractions with cysteine identified in an analysis of 753 proteinstructures.161h
Journal of Medicinal Chemistry Perspective
DOI: 10.1021/jm501853mJ. Med. Chem. XXXX, XXX, XXX−XXX
AI

phenylalanine, tyrosine, and tryptophan.161i This study revealedthat methionine sulfur atoms are nearly twice as likely to belocated within 7 Å of an aromatic ring center of mass than atomsin a control set including all non-hydrogen atoms. As with theearlier studies that evaluated smaller numbers of crystal struc-tures, a maximum in the probability distribution was observedat an aromatic ring center of mass/sulfur atom distance ofapproximately 5 Å. Another interesting observation was thatmethionine−aromatic interactions (7 Å distance cutoff) occur inapproximately one-third of all protein structures. Furthermore,multiple methionine−aromatic interactions were present in themajority of the protein structures analyzed. This large-scale studycorroborates the earlier reports described above that indicated apreference for S···π distances of∼5 Å and larger numbers of S···πinteractions than expected based on chance. An examination ofα-helical FXXXC, FXXXM, CXXXF, and MXXXF motifs in thePDB identified a preference for cysteine and methionine sulfuratoms to be proximal to the meta and para carbons of thephenylalanine ring above the ring edge.163b While these studiesutilized X-ray crystal structures of proteins to evaluate S···πinteractions, a more recent report focused on the small moleculestructures available in the CSD.161d,167 From a set of 2266 crystalstructures containing one or more C−S−C moieties and one ormore aromatic rings, 240310 sulfur···aromatic interactions (ringcentroid to S atom distance ≤10 Å) were identified. Forcomparison, reference distributions of contacts between thecentral methylene carbon in C−CH2−C substructures andaromatic rings were also evaluated. In accord with findings forS···π contacts in proteins, a maximum in the S···ring centroiddistance histogram was observed at approximately 5 Å. Severalangle parameters were also evaluated and used along with thedistance preference described above to define an ideal geometryfor sulfur···aromatic interactions involving divalent sulfur. Thesulfur···aromatic interaction present in the small molecule single-crystal X-ray structure of 298 was highlighted as an example ofthe ideal geometry (Figure 42).168 The sulfur atom is located at
the periphery of the aromatic ring, approximately within the ringplane, with the sulfur substituents directed away from the ring
and the sulfur lone pairs of electrons proximal to ring hydrogenatoms.
Overall, there appears to be a consensus from these studies ofprotein and small molecule crystal structures that sulfur···aromatic contacts occur more frequently than expected based onchance, with preferred sulfur to ring centroid distances ofapproximately 5 Å. However, no such consensus exists withregard to a preference for the sulfur atom to interact with theπ-face or the edge of the aromatic ring and other atoms in themethionine and cysteine side chains may contribute tointeractions with the aryl rings of aromatic amino acids.While this was not explicitly addressed in several of the reportsoutlined above, S−H···π H-bonds appear to occur some-what infrequently in proteins. The high frequency withwhich sulfur···aromatic interactions are present in proteinssuggests that they may play a stabilizing role in foldingand intermolecular interactions, although it does not indicatethe magnitude of this stabilization. This may be estimatedfrom computational and experimental studies on modelsystems.
■ COMPUTATIONAL STUDIES OFSULFUR···AROMATIC MODEL SYSTEMS
Quantification of the stabilization energy that can be realizedfrom the association of divalent sulfur with aromatic rings, aswell as determination of the ideal geometries and underlyingfactors contributing to the favorable energetics, have been theobjectives of several computational studies. These utilizedmolecular mechanics force-field based methods, semiempirical,or ab initio calculations and many of them employed modelsystems (e.g., benzene plus H2S, methanethiol, DMS, etc.) assurrogates for aromatic amino acids, cysteine, andmethionine. Ina comparison of complexes of benzene and cyclohexane withdimethyl disulfide (DMDS) using empirical conformationalenergy program for peptides (ECEPP) molecular mechanicscalculations, the lowest energy minimum found on the benzene/DMDS potential energy surface positioned one of the sulfuratoms of DMDS within van der Waals contact distance ofbenzene carbon atoms above the face of the ring.162a Theinteraction energy was −3.3 kcal/mol, and this complex wasapproximately 0.8 kcal/mol more stable than the lowest energyminimum identified for cyclohexane/DMDS. The greaterstabilization of the benzene/DMDS complex was attributedprimarily to favorable nonbonded interactions between a sulfuratom and aromatic carbon atoms, and electrostatic interactionswere not considered to be a dominant factor.The potential energy surface for the interaction of benzene
with methanethiol was examined using Hartree−Fock ab initiocalculations with the effects of electron correlation estimatedwith second-order Møller−Plesset (MP2) single-point energycalculations.162b Previously reported geometries for benzeneand methanethiol optimized at the RHF/3-21G(*) level oftheory were kept fixed during the subsequent computations, andgeometry optimization of the methanethiol and benzenecomplex was performed starting from 10 initial configurations,also using RHF/3-21G(*) calculations. This resulted in the
Figure 42. X-ray crystal structure of 298 highlighting a representativeS···aromatic interaction identified in a study of small molecule crystalstructures, d1 = 4.9 Å.161d,168 Image created with ConQuest.169
Journal of Medicinal Chemistry Perspective
DOI: 10.1021/jm501853mJ. Med. Chem. XXXX, XXX, XXX−XXX
AJ

identification of five local minima, two of which correspondedto S−H···π H-bonding geometries with the S−H bond vectordirected toward the center of the face of the benzene ring(Figure 43A). The other three directed a C−H bond vector from
the methanethiol group toward the face of the benzene ring andplaced the sulfur atom above the edge of the benzene ring. Thesewere termed S−C−H···π configurations (Figure 43B). For all ofthese minima, the ring centroid to sulfur atom distance fell in therange of 4.0−4.8 Å, in rough agreement with the results from thecrystal structure analyses described above. No minimum energystructure in which the sulfur atom of methanethiol was located atthe periphery of the benzene ring within the plane could belocated. At the highest level of theory utilized, MP2(FC)/6-31G*//HF/3-21G(*), the binding energy for the differentconfigurations ranged from −2.5 to −3.0 kcal/mol, with theS−C−H···π structures slightly more stable. In an attempt toprobe the significance of positively charged arginine residuesnoted previously, the impact of a positive point charge on theenergy of the benzene/methanethiol complex was alsoevaluated.161a Both stabilization and destabilization wereobserved depending on the location of the point charge relativeto the complex.The benzene/DMS complex, a model system more
representative of the methionine−aromatic interactions ob-served in proteins, was studied using molecular mechanics,HF/6-31G*, B3LYP/6-31G*, and MP2/6-31G* calcula-tions.162c Several configurations of this complex were optimizedat each of the three ab initio levels. At all three levels of theory,the complex geometry with the DMS located above the face ofthe ring, with the sulfur distal to the ring, was found to be moststable (Figure 44A). The MP2/6-31G* energy reported for one
of the configurations was determined based on the B3LYP/6-31G* optimized geometry because a corresponding local
minimum could not be identified with MP2/6-31G* calcu-lations. At all ab initio levels of theory, complex structures inwhich the sulfur lies in the plane of the benzene ring along theperiphery (Figure 44C) were slightly less stable than thosecomplexes in which the DMS was located above the plane ofthe ring, with both methyl groups interacting with the π-face(Figure 44A). In all cases, the ring centroid to sulfur atomdistances were between 4.9 and 5.9 Å and molecular mechanicscalculations using various force-fields were in reasonable accordwith the MP2/6-31G* results.The interactions between cysteine and aromatic amino acid
side chains were modeled as the complex of benzene withmethanethiol.161e Starting with bond lengths and anglesconstrained to experimental values during subsequent calcu-lations, three different configurations for the benzene/methanethiol complex were optimized at the MP2/6-31G**level. MP2 interactions energies using several basis sets up to6-311+G(2d,p) were computed and corrected for basis setsuperposition error (BSSE). At the MP2/6-311+G(2d,p)//MP2/6-31G** level of theory, the configuration in which theS−H bond vector in the methanethiol was directed towardthe center of the π-face of the benzene ring had an interactionenergy of −3.7 kcal/mol (Figure 45A). This was approximately
2 kcal/mol more favorable than the corresponding values cal-culated for the other two configurations. The distance from thering centroid to the sulfur atom varied from 3.7 to 4.9 Å for thethree complex geometries studied. Interaction energies werecalculated at the MP2/6-311G(2d,p) level for four S−H···π H-bonding contacts identified in protein crystal structures. Thesecalculations used geometries extracted from the crystal structureatomic coordinates, and stabilization energies were estimated tobe −2.3 to −2.6 kcal/mol.Very high level ab initio calculations were utilized to examine
the benzene/H2S dimer as a model for S···π interactions.162d Acomplex with the H2S located above the benzene plane,hydrogen atoms directed toward the π-face of the ring, and asulfur atom to ring centroid distance of 3.8 Å was identified asthe most favorable configuration (Figure 46A). The interactionenergy was −2.64 kcal/mol based on CCSD(T)/aug-cc-pVTZ
Figure 43. Schematic representation of the S−H···π (A) and S−C−H···πminima identified by an ab initio quantummechanical study of thebenzene/methanethiol complex.162b Interaction energies are for themost stable minimum of each type calculated at the MP2(FC)/6-31G*//HF/3-21G(*) level.
Figure 44. Schematic representation of the minima identified for thebenzene/DMS complex.162c The interaction energies for A and C weredetermined at the MP2/6-31G* level and at the MP2/6-31G*//B3LYP/6-31G* level for B.
Figure 45. Schematic representation of the minima identified for thebenzene/methanethiol complex.161e Reported interaction energies werecalculated at the MP2/6-311+G(2d,p)//MP2/6-31G** level andcorrected for BSSE.
Figure 46. Minima identified for the benzene/H2S complex.161h,162d
Reported interaction energies were calculated at the CCSD(T)/aug-cc-pVTZ level.
Journal of Medicinal Chemistry Perspective
DOI: 10.1021/jm501853mJ. Med. Chem. XXXX, XXX, XXX−XXX
AK

calculations corrected for BSSE, and the configuration of thecomplex with the sulfur atom of H2S directed toward the π-faceof the benzene ring was less stable. On the basis of an energydecomposition analysis, electrostatic attraction between thehydrogen atoms of H2S and the π-electrons of benzene wasproposed to best describe the nature of the benzene/H2Sinteraction. In another study of the benzene/H2S system, twoother minima were identified on the MP2/aug-cc-pVDZpotential energy surface that were further characterized withCCSD(T)/aug-cc-pVTZ calculations (Figure 46B,C).161h Inone of these, the sulfur atom of H2S was proximal to the π-face ofbenzene, while in the other, the sulfur atom was in the plane ofthe benzene ring along one of the C−H bond axes. These twominima were 1.5−1.9 kcal/mol less stable than the “hydrogendown” configuration.162d
The primary focus of a study of the complexes of benzene withH2S andDMSwas density functional and semiempirical methodsincorporating corrections for the dispersion energy that can bean important component of weak intermolecular interac-tions.162f,170 Two configurations for the benzene/DMS complexwere examined using MP2/aug-cc-pVDZ calculations. Theconfiguration in which the DMS is above the plane of thebenzene ring with the sulfur directed away from the ringface (Figure 47A) was about 1.8 kcal/mol more stable than the
second complex structure. In the latter, the DMS was located atthe periphery of the benzene ring, with the sulfur atom orientedalong a C−H bond axis (Figure 47B). For the more stable of thetwo, based on comparison of BLYP-D* (dispersion-correctedDFT) and BLYP calculations, dispersion was found to play acritical role in stabilizing the complex.In the most comprehensive computational study related to
S···π interactions reported to date, high-level quantum chemicalcalculations were used to examine the potential energy surfacesfor complexes of benzene, phenol, and indole with DMS andpropane.161i The complex of benzene with dimethyl ether wasalso studied. Configurations with the central non-hydrogen atomin DMS, propane, and dimethyl ether oriented up, away from theπ-face of the aromatic ring, or down, toward the π-face, wereevaluated using a range of theory up to CCSD(T)/6-311+G-(d,p)//MP2/6-31G(d) for the benzene/DMS, benzene/pro-pane, phenol/DMS, and phenol/propane systems. The othercomplexes were studied using M06-2X/6-311+G(d,p) andMP2/6-311+G(d,p) calculations. At all levels of theory, the“sulfur up” configuration for DMS interacting with benzene,phenol, or indole was more stable than the “sulfur down”configuration by 1.4−2.9 kcal/mol (Figure 48). For the “sulfurup” configuration, the sulfur to ring centroid distance wasapproximately 4.8 Å, in good agreement with the statisticalstudies of crystal structures described above. Dispersion energywas noted as a critical component of the overall binding energiesfor the complexes. It also lead to a greater stabilization of about
1 kcal/mol for the DMS “sulfur up” complex relative to thepropane “up” complex. This suggests a differentiation betweenthe interactions of methionine with aromatic rings and purelyhydrophobic contacts.To further probe the energetics of methionine/aromatic inter-
actions in a biological context, molecular dynamics simulationswere performed on two protein−protein complexes.161i TheTRAIL-DR5 (methionine/tyrosine) and LTα-TNFR1 (methionine/tryptophan) complexes included methionine/aromatic inter-actions that the authors demonstrated with point mutationswere critical for function and binding (Figure 49).171,172 Both
simulations showed a bias toward 5 Å separation betweenthe sulfur atom of the methionine residue and the aromaticring centroid of either phenol or tryptophan. Interactionenergies were calculated at the M06-2X/6-311+G(d,p) andMP2/6-311+G(d,p) levels for the methionine/tyrosine andmethionine/tryptophan residue pairs in geometries from 128different structures extracted from the molecular dynamicstrajectories. These varied from −1.4 to −1.9 kcal/mol for themethionine/tyrosine interaction and −1.9 to −2.9 kcal/mol forthe methionine−tryptophan interaction. Furthermore, when anearby arginine residue was included in the quantum calculationsfor themethionine−tyrosinemodel, the stabilization increased toapproximately −5 kcal/mol. This is in accord with earlier ob-servations regarding the correlation between proximal arginineresidues and S···π interactions as well as the computationalresults.161a,162b
The computational studies described above generally concurthat at least for simple models systems, complexes betweenaryl rings and small sulfur-containing compounds (H2S, DMS)adopt preferred geometries with the sulfur atom approximately5 Å from the ring centroid with stabilizing interaction energies inthe range of ∼1−3 kcal/mol. In those examples where it wasevaluated, dispersion was found to be a significant component inthe overall interaction energy. As with the database studies, thereis less accord with regard to the placement of the sulfur atomrelative to the ring face or edge. However, recent high level
Figure 47. Schematic representation of the configurations of thebenzene/DMS complex.162f Reported interaction energies weredetermined at the MP2/aug-cc-pVDZ level.
Figure 48. Sulfur-up (A) and sulfur-down (B) configurations for thecomplexes of DMS with aryl rings.161i Interaction energies weredetermined at the CCSD(T)/6-311+G(d,p)//MP2/6-31G(d) level.
Figure 49. (a): the X-ray crystal structure of the TRAIL/DR5 complexshowing a close contact between TRAIL Tyr237 and DR5Met99, d1=5.2 Å (PDB ID: 1D0G).171 (b): The X-ray crystal structure of the LTα/TNFR1 complex with a close contact between LTαMet120 and TNFR1Trp107 highlighted, d2 = 5.4 Å (PDB ID: 1TNR).172
Journal of Medicinal Chemistry Perspective
DOI: 10.1021/jm501853mJ. Med. Chem. XXXX, XXX, XXX−XXX
AL

quantum mechanical studies suggest a preference for aconfiguration with the sulfur above the ring plane with either ahydrogen atom (e.g., H2S, cysteine) or methyl groups (DMS,methionine) more proximal to the π-face and the sulfur atommore distal. In the following section, we review the experimentalstudies that help elucidate the specifics of the S···π interaction.
■ EXPERIMENTAL STUDIES OF SULFUR···AROMATICMODEL SYSTEMS
Relatively few experimental studies have focused on the S···πinteraction and, like the computational analyses described above,many of these have utilized small model systems. A few have alsoexamined the interaction in peptides. An early report notedupfield shifts for the methyl protons in the NMR spectrum ofdimethyl disulfide upon addition of benzene to the sample.159a
The relationship between benzene mole fraction and chemicalshift revealed the formation of a 1:1 complex between thecomponents in which the DMS protons were shielded by thefield of the benzene ring. This work was later extended to includecomplexes of DMS and DMDS with benzene, toluene, p-cresol,and 1-methylnaphthalene in CCl4 solution, and many of thesecombinations also formed 1:1 complexes.159b For the DMS/1-methylnaphthalene and DMDS/1-methylnaphthalene com-plexes, the dependence of proton chemical shift on temperaturewas measured to determine their heats of formation. The ΔHof complex formation was −0.79 kcal/mol for the DMS/1-methylnaphthalene complex and −0.96 kcal/mol for theDMDS/1-methylnaphthalene complex. These values were 3−5-fold larger than estimates of the van der Waals energy for sulfur···aromatic side chain interactions.160
A subsequent NMR study of a disulfide-bridged oxytocinpeptide and a series of analogues highlighted a chemical shiftdifference for Tyr2 2′, and 6′ protons in the desamino-oxytocinpeptide 299 compared to the analogue 300 in which both sulfuratoms of the disulfide bridge were replaced with methylenecarbons.163a The resonance for the Tyr2 2′ and 6′ protons in thepeptide disulfide 299 was shifted downfield by 0.08 ppm relativeto the corresponding resonance in the 1HNMR spectrum of 300,in which the disulfide was replaced with an ethyl linker. This wasattributed to interactions between the Cys6 sulfur atom and Tyr2.In support of this observation, oxidation of the sulfur of Cys6yielded a peptide lacking the more pronounced downfieldchemical shift for the Tyr2 2′ and 6′ protons, as did inversion ofthe chirality of Tyr2. Replacement of the Cys1 sulfur atom with amethylene moiety yielded a peptide which also demonstrated thedownfield shift effect for the Tyr2 2′ and 6′ protons, furtherimplicating Cys6 as a component of the interaction.
Both 1H NMR and circular dichroism (CD) spectroscopywere utilized to study the interactions of phenylalanine withcysteine and methionine located at the i and i + 4 positions ofsmall α-helical peptides.163b In α-helices, residues at i and i + 4positions (three intervening residues) are located on the sameface of the helix (Figure 50). The side chains may contact eachother depending on the types of residues at these positions.When phenylalanine was included at the ith position and cysteineormethionine at the i + 4th position of model 15 residue peptides,
nuclear Overhauser effect (NOE) cross-peaks were observedbetween their side chains.173 These NOE cross-peaks, indicativeof spatial proximity, were also noted in model peptides in whichthe positions of the phenylalanine and cysteine/methionine wereswapped. For the peptides incorporating cysteine, upfield shiftswere also observed for the cysteine Cα protons. This suggeststhat the phenylalanine rings were close to the cysteine Cα atomsin these peptides. CD spectra were measured for peptidescontaining combinations of phenylalanine with cysteine andmethionine located at the ith and i + 4th positions. The percentageof α-helix content for each peptide was determined from themeasured ellipticity. The free energy for the interaction ofphenylalanine with cysteine and methionine was estimated bydetermining the ΔGint required to reproduce the experimentallydetermined helical content with a previously developedalgorithm (AGADIR).174 This predicts helical content inpeptides based on helix/coil transition theory. For a peptidewith phenylalanine at the ith position andmethionine at the i + 4th
position, the ΔGint was estimated to be −0.65 kcal/mol, whichagrees closely with an earlier estimate of −0.75 kcal/mol thatwas also based on helix−coil transition theory.175 Thephenylalanine−cysteine (i/i + 4) interaction was estimated tobe significantly stronger at −2.0 kcal/mol. These results suggestthat S···π interactions can stabilize α-helices when aromaticresidues and cysteine or methionine are incorporated at spatiallyproximal positions. The ΔGint values determined in this studyare in rough accord with the results of computational studiesdescribed earlier.The interaction of methionine with aromatic residues has also
been examined in the context of analogues of a peptide knownto adopt a β-hairpin conformation.163c In these peptides,tryptophan or phenylalanine were positioned diagonally acrossfrom a methionine residue, with the side chains of the aryl andsulfur-containing residues on the same face of the β-hairpin.1H NMR spectroscopy was used to confirm that β-hairpinconformations were maintained. Interactions between thearomatic residues and methionine were evaluated based onchanges in chemical shifts for diagnostic H atoms, leading to thesuggestion that interactions between methionine and phenyl-alanine or tryptophan were similar to interactions of norleucinewith these aromatic residues. Thermal denaturation studies alsoindicated similarities in the interactions of methionine andnorleucine with tryptophan. To determine the magnitude of themethionine interactions with phenylalanine and tryptophan,double mutant cycles were employed with the phenylalanine−methionine and tryptophan−methionine interaction stabiliza-tion energies both determined to be −0.3 kcal/mol.176 Forcomparison, the cyclohexylalanine−methionine, phenylalanine−norleucine, and tryptophan−norleucine stabilization energieswere−0.5,−0.1, and−0.3 kcal/mol, respectively. On the basis ofthe NMR and double mutant cycle results, it was concluded thatcontacts between methionine and the aryl rings of phenylalanine
Figure 50. Model of canonical α-helix with Phe at ith position and Cysat i + 4.
Journal of Medicinal Chemistry Perspective
DOI: 10.1021/jm501853mJ. Med. Chem. XXXX, XXX, XXX−XXX
AM

and tryptophan are largely classical nonspecific hydrophobicinteractions. However, there may be some differences in themethionine−tryptophan and methionine−phenylalanine inter-actions, with a possible small thermodynamic contribution tothe latter. S···π interactions have also been studied in the contextof molecular torsion balances, with a 3:1 preference for theconformation of 301 in which the S atom is directed toward theface of a phenyl ring determined from NMR experiments inCDCl3 solution at 298 K.177
Overall, these somewhat limited experimental studies supportthe idea that small molecules containing divalent sulfur canassociate with aromatic compounds in solution and that, at leastin the context of peptides, a similar interaction is possiblebetween cysteine and methionine and amino acids containingaryl rings. Similar to the computational studies described earlier,these experiments suggest that the S···π interaction is relativelyweak. A maximum of approximately 2 kcal/mol of stabilizationis likely for cysteine−aromatic interactions (which may involveS−H···π H-bonding) and ∼1 kcal/mol for methionine···aromatic interactions. It should also be noted that the term“S···π” interaction may be somewhat of a misnomer, at leastfor the interactions of cysteine and methionine with aromaticamino acids.161b Both the computational and experimental worksummarized above suggest that interactions between aryl ringsand other atoms bound to sulfur may contribute to the overallstabilization achieved. In the following, examples from theliterature where S···π interactions are either demonstrated orpostulated to play a significant role are summarized.
■ SELECTED EXAMPLES OF SULFUR···AROMATICINTERACTIONS IN BIOLOGICAL SYSTEMS
To probe the importance of a specific S···π interaction, theimpact on oxidation−reduction potentials of mutating Met56 toall of the other natural amino acids in the Clostridium beijerinckiiflavodoxin protein was evaluated.178 Flavodoxins are electrontransferases that utilize a flavin mononucleotide as a cofactor inthe oxidation−reduction process. In the crystal structure ofC. beijerinckii flavodoxin, the side chain of Met56 makes directcontact with edge of the face of the isoalloxazine ring of thecofactor 302 in a geometry consistent with some of the earlierdescriptions of S···π interactions (Figure 51).179 Significantchanges in the oxidation−reduction potentials measured for themutant proteins led to the conclusion that Met56 was central tothe oxidation reduction characteristics of Clostridium beijerinckiiflavodoxin protein and that the specific S···π interaction betweenthe methionine and flavin cofactor contributed to the observedproperties.
In a report describing the design, synthesis, and biologicalactivity of EGFR tyrosine kinase inhibitors, it was proposed thata chlorophenyl ring in potent isoflavone compounds formssulfur···aromatic interactions with Cys773 of EGFR tyrosinekinase.180 In a homology model of the EGFR kinase, Cys773 waspredicted to be in the sugar pocket of the ATP binding site andmodeling of compound 303 into the ATP binding site suggesteda potential sulfur···aromatic interaction. This sulfur···aromaticinteraction was later specifically incorporated into a kinasepseudoreceptor model derived from a 3D-QSAR method byanother group.181 Extending previous work that showed thatmutation of residue 33 in the human equilibrative nucleosidetransporters 1 and 2 (hENT1, hENT2) resulted in modifiedsensitivity to two inhibitors, dilazep (304) and dipyridamole(305), several mutants of hENT2 were examined includingIle33Cys and Ile33Met.182,183 In an assay that evaluated[3H]-uridine transport by wild-type (WT)-hENT2, hENT2-Ile33Met, and hENT2-Ile33Cys, 14- and 18-fold improvementsin the Ki values for inhibition by 305 were observed with thehENT2-Ile33Met and hENT2-Ile33Cys proteins, respectively,relative to WT-hENT2. This increased sensitivity in the mutantswas attributed to S···π interactions between Cys33 and Met33 and305. It was also noted that the difference inΔG derived from themeasured Ki values for the mutant proteins relative to WTcorresponded approximately to the amount of interaction energypredicted for S···π interactions.
A sulfur···aromatic interaction was identified between ligandand protein in the cocrystal structure of the juvenile hormoneesterase (JHE) of the Manduca sexta tobacco hornworm incomplex with the transition state inhibitor 3-octylthio-1,1,1-trifluoropropan-2-one (OTFP, 306).184 In the cocrystalstructure, determined at 2.7 Å resolution, the sulfur atom of306 is located 4.0 Å from the centroid of the Phe259 phenyl ring(Figure 52). Aromatic amino acids frequently occur at thisposition in JHEs. This corroborated an earlier suggestion thatS···π interactions might play a significant role in the SAR aroundtrifluoromethyl ketone inhibitors of the esterase.185 To probe theimportance of the inhibitor S···π interaction with Phe259 inenzyme inhibition by 306, a JHE Phe259Ile mutant protein wasgenerated. The IC50 values for 306 and its analogue TFDK
Figure 51. X-ray cocrystal structure of the C. beijerinckii flavodoxin withbound flavin mononucleotide cofactor 302 showing an intermolecularS···π interaction between the sulfur atom in Met56 and the cofactor,d1 = 4.1 Å (PDB ID: 5NLL).179
Journal of Medicinal Chemistry Perspective
DOI: 10.1021/jm501853mJ. Med. Chem. XXXX, XXX, XXX−XXX
AN
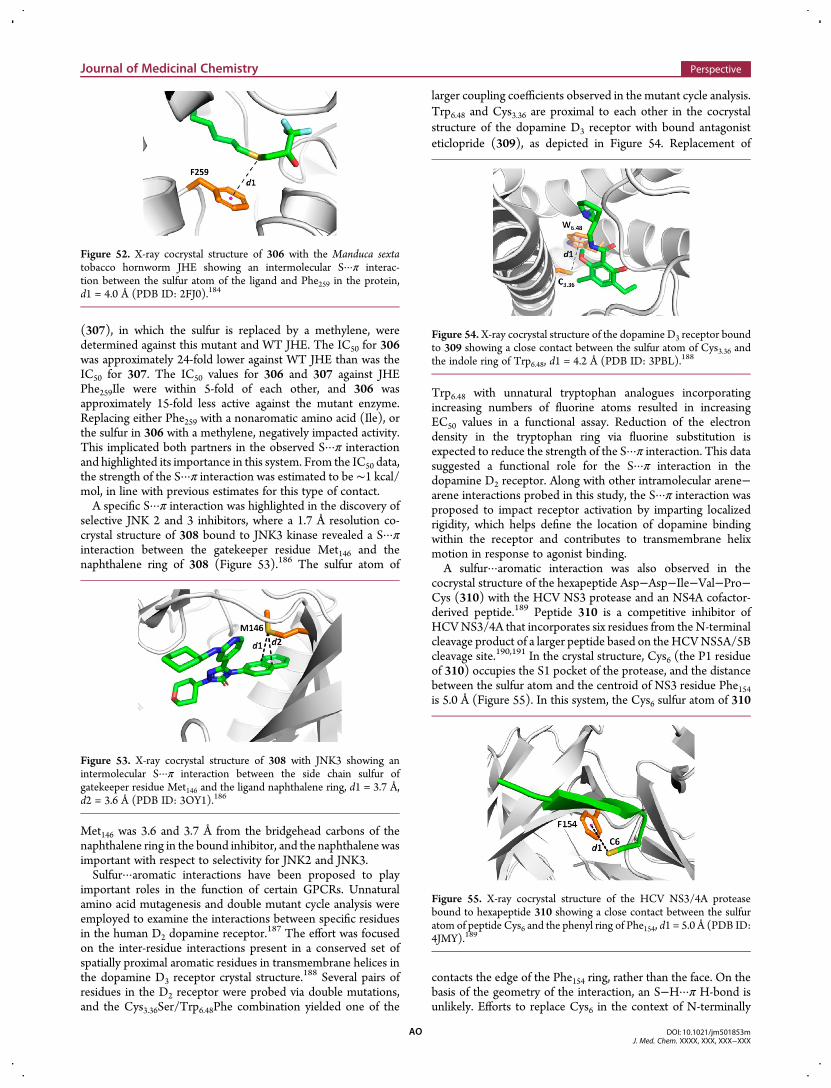
(307), in which the sulfur is replaced by a methylene, weredetermined against this mutant and WT JHE. The IC50 for 306was approximately 24-fold lower against WT JHE than was theIC50 for 307. The IC50 values for 306 and 307 against JHEPhe259Ile were within 5-fold of each other, and 306 wasapproximately 15-fold less active against the mutant enzyme.Replacing either Phe259 with a nonaromatic amino acid (Ile), orthe sulfur in 306 with a methylene, negatively impacted activity.This implicated both partners in the observed S···π interactionand highlighted its importance in this system. From the IC50 data,the strength of the S···π interaction was estimated to be ∼1 kcal/mol, in line with previous estimates for this type of contact.A specific S···π interaction was highlighted in the discovery of
selective JNK 2 and 3 inhibitors, where a 1.7 Å resolution co-crystal structure of 308 bound to JNK3 kinase revealed a S···πinteraction between the gatekeeper residue Met146 and thenaphthalene ring of 308 (Figure 53).186 The sulfur atom of
Met146 was 3.6 and 3.7 Å from the bridgehead carbons of thenaphthalene ring in the bound inhibitor, and the naphthalene wasimportant with respect to selectivity for JNK2 and JNK3.Sulfur···aromatic interactions have been proposed to play
important roles in the function of certain GPCRs. Unnaturalamino acid mutagenesis and double mutant cycle analysis wereemployed to examine the interactions between specific residuesin the human D2 dopamine receptor.187 The effort was focusedon the inter-residue interactions present in a conserved set ofspatially proximal aromatic residues in transmembrane helices inthe dopamine D3 receptor crystal structure.
188 Several pairs ofresidues in the D2 receptor were probed via double mutations,and the Cys3.36Ser/Trp6.48Phe combination yielded one of the
larger coupling coefficients observed in the mutant cycle analysis.Trp6.48 and Cys3.36 are proximal to each other in the cocrystalstructure of the dopamine D3 receptor with bound antagonisteticlopride (309), as depicted in Figure 54. Replacement of
Trp6.48 with unnatural tryptophan analogues incorporatingincreasing numbers of fluorine atoms resulted in increasingEC50 values in a functional assay. Reduction of the electrondensity in the tryptophan ring via fluorine substitution isexpected to reduce the strength of the S···π interaction. This datasuggested a functional role for the S···π interaction in thedopamine D2 receptor. Along with other intramolecular arene−arene interactions probed in this study, the S···π interaction wasproposed to impact receptor activation by imparting localizedrigidity, which helps define the location of dopamine bindingwithin the receptor and contributes to transmembrane helixmotion in response to agonist binding.A sulfur···aromatic interaction was also observed in the
cocrystal structure of the hexapeptide Asp−Asp−Ile−Val−Pro−Cys (310) with the HCV NS3 protease and an NS4A cofactor-derived peptide.189 Peptide 310 is a competitive inhibitor ofHCVNS3/4A that incorporates six residues from the N-terminalcleavage product of a larger peptide based on the HCVNS5A/5Bcleavage site.190,191 In the crystal structure, Cys6 (the P1 residueof 310) occupies the S1 pocket of the protease, and the distancebetween the sulfur atom and the centroid of NS3 residue Phe154is 5.0 Å (Figure 55). In this system, the Cys6 sulfur atom of 310
contacts the edge of the Phe154 ring, rather than the face. On thebasis of the geometry of the interaction, an S−H···π H-bond isunlikely. Efforts to replace Cys6 in the context of N-terminally
Figure 52. X-ray cocrystal structure of 306 with the Manduca sextatobacco hornworm JHE showing an intermolecular S···π interac-tion between the sulfur atom of the ligand and Phe259 in the protein,d1 = 4.0 Å (PDB ID: 2FJ0).184
Figure 53. X-ray cocrystal structure of 308 with JNK3 showing anintermolecular S···π interaction between the side chain sulfur ofgatekeeper residue Met146 and the ligand naphthalene ring, d1 = 3.7 Å,d2 = 3.6 Å (PDB ID: 3OY1).186
Figure 54. X-ray cocrystal structure of the dopamine D3 receptor boundto 309 showing a close contact between the sulfur atom of Cys3.36 andthe indole ring of Trp6.48, d1 = 4.2 Å (PDB ID: 3PBL).188
Figure 55. X-ray cocrystal structure of the HCV NS3/4A proteasebound to hexapeptide 310 showing a close contact between the sulfuratom of peptide Cys6 and the phenyl ring of Phe154, d1 = 5.0 Å (PDB ID:4JMY).189
Journal of Medicinal Chemistry Perspective
DOI: 10.1021/jm501853mJ. Med. Chem. XXXX, XXX, XXX−XXX
AO

acetylated analogues of 310 resulted in uniformly less activepeptides in an enzymatic assay based on NS3/4A inhibition. Of12 natural and non-natural amino acids containing hydrophobic,polar, or charged side chains tested as Cys replacements, onlynorvaline, with an n-propyl side chain, imparted roughly similaractivity (5-fold increase in IC50).
190
As detailed above, intra- and intermolecular stabilizinginteractions between sulfur (and/or bound atoms) and aromaticrings have been observed in the context of peptides, proteins,protein/protein, and protein/ligand complexes. Examples ofS···π interactions have been seen in the context of medicinalchemistry, and both computational and experimental studies onmodel systems have provided estimates of the strengths of theseinteractions and favored interaction geometries. However, mostof the published work has focused on surrogates for inter- andintraprotein interactions. Computational and experimentalstudies directly relevant to complexes of drug-like moleculeswith proteins and DNA/RNA would more clearly define theimportance of S···π interactions, potentially benefiting medicinalchemists as they strive to design compounds with improvedproperties and potency.
■ SULFUR···HALOGEN INTERACTIONS
The halogen atoms chlorine, bromine, and iodine possessσ-holes of varying magnitude depending on the electrondonating or withdrawing properties of the moieties to whichthey are bound.11g,192 Interaction of these halogens with Lewisbases (e.g., a lone pair of electrons on a carbonyl oxygen atom)may result in stabilizing interactions referred to as halogen bonds.Computational results suggest that individual halogen bonds canprovide up to several kcal/mol of stabilization energy, and themagnitude of the interactions increases with the size andpolarizability of the halogens involved (Cl < Br < I).11b,193 It isplausible that sulfur atoms with nonbonded pairs of electronscould act as Lewis bases and form halogen bonds with halogenσ-hole donors.194 This interaction, which is somewhat sparselyrepresented in the literature, is explored below.As with sulfur···aromatic interactions, computational and
experimental studies of sulfur−halogen interactions have beenprimarily limited to small model systems. The potential forcomplex formation between CF3Cl, CF3Br, CF3I, and DMS inliquid krypton solution was assessed using both FTIR and Ramanspectroscopy augmented by computational analyses at the abinitio MP2/aug-cc-pVDZ(-PP) level of theory.195 CF3Cl/DMScomplex formation was not observed spectroscopically, althoughboth FTIR and Raman spectra indicated the formation of CF3Br/DMS and CF3I/DMS 1:1 complexes and a CF3I/DMS 2:1 com-plex. For the CF3Br/DMS and CF3I/DMS 1:1 complexes, thecomplexation enthalpies determined by Van’t Hoff analyses overa range of temperatures from 118 to 163 K in liquid krypton weredetermined to be −2.3 and −4.2 kcal/mol, respectively. Thecorresponding values determined based on the ab initio com-putational results incorporating zero-point vibrational, thermal,and solvation energy corrections were −2.3 and −4.4 kcal/mol,respectively, in excellent agreement with experiment. Thecalculations predict a less favorable complexation energy for aCF3Cl/DMS complex of about −1 kcal/mol. It was noted thatthe experimentally determined enthalpies of complexation forDMS and dimethyl ether with CF3Br and CF3I are comparable,suggesting that S and O are similar in their abilities to participate
in halogen bonds. The geometries of the three complexesdetermined at the MP2/aug-cc-pVDZ(-PP) level were similar tothe Cl−S distance in the CF3Cl/DMS complex measured as 3.4Å, while both the CF3Br/DMS and CF3I/DMS complexes hadC−X distances of 3.3 Å. The C−X−S angle increasedfrom 165.9° for the CF3Cl/DMS complex to 172.2° for theCF3I/DMS complex, while the angle between the X atom and theC−S−C plane of DMS ranged from 83.6° to 94.3° and increasedwith the size of the halogen atom X. It should be noted that theenthalpies of complexation were determined at low temperaturesusing halogen bond donors activated by the electron withdrawingeffect of the CF3 group. Thus, the overall stabilization resulting fromthe sulfur···halogen interactions in this work is likely larger thanwhatcan be expected for sulfur···halogen interactions in the typicalmedicinal chemistry context.Another recent study focused on the potential to exploit
halogen bonds between ligand molecules and the sulfur atoms ofmethionine residues in the binding sites of proteins. Theinteractions of halobenzenes with DMS, methyl propyl sulfide,and N- and C-terminally capped methionine were examinedusing ab initio calculations.194 At theMP2/QZVPP level of theory,the interaction energies for the complexes of chlorobenzene,bromobenzene, and iodobenzene with DMS were −2.4, −3.1,and −4.6 kcal/mol, respectively. At the same level, benzene ispredicted to form a complex with DMS having an interactionenergy of −2.6 kcal/mol while phenol forms a H-bondedcomplex with DMS that is stabilized by 7.0 kcal/mol. Theseresults suggest that halogen bonds to methionine with arylchlorides or bromides acting as acceptors do not providesubstantial stabilization compared to unsubstituted aryl C−H tomethionine side chain sulfur interactions while aryl iodines couldbe expected to provide ∼2 kcal/mol of additional stabilizationenergy. Key geometric parameters for the halobenzene/DMSmodel complexes calculated at the MP2/QZVPP level areprovided in Figure 56. These halobenzene−DMS interactions
are also predicted to be ∼2.4 kcal/mol weaker than a H-bondfrom phenol to a methionine S atom. However, a simpleapproximation of ligand desolvation in which the H2Omoleculesin iodobenzene:H2O, benzene:H2O, and phenol:H2O complexesare replaced with DMS dramatically reduces the stabilization inthe case of phenol, with less significant impacts on theiodobenzene and benzene systems.The energies for the interactions of DMS with the
iodobenzene, benzene, and phenol complexes with H2O are
Figure 56. Geometric parameters for S···X halogen bonds based onhigh-level ab initio calculations.194
Journal of Medicinal Chemistry Perspective
DOI: 10.1021/jm501853mJ. Med. Chem. XXXX, XXX, XXX−XXX
AP

−2.7,−1.4, and−0.3 kcal/mol, respectively. This model suggeststhat desolvation effects result in a sulfur−iodine halogen bondthat is stronger than a H-bond with a methionine sulfur acting asthe acceptor. A comparison of the interactions of each of thehalobenzenes with DMS, methyl propyl sulfide, and N- andC-terminally capped methionine model systems showed littledependency on the sulfur-containing moiety used, withinteraction energies varying by about 0.5 kcal/mol within eachseries, as calculated at the MP2/TZVPP level of theory. Distanceand spherical energy scans exploring the geometric dependenciesfor the halobenzene/DMS complexes were also reported in thiswork. Distance tolerances for the halobenzene−DMS inter-actions increased with halogen size, and the maximumstabilization is achieved when the C−X bond is approximatelyperpendicular to the DMS C−S−C plane with the halogen atomdirectly above or below the sulfur atom. Directional require-ments for the chlorobenzene and bromobenzene interactionswith DMS were shown to be more stringent than those foriodobenzene.Visual inspection of small molecule crystal structures from the
CSD containing intramolecular sulfur−halogen contacts atdistances less than or equal to the sum of the van der Waals radiifor sulfur and the relevant halogen did not reveal any compounds inwhich the sulfur atom and the halogen were positioned for thehalogen atom to act as a halogen bond donor to sulfur. However,there are examples of intermolecular protein−ligand halogenbonds to sulfur in the PDB, and these include complexes wherechlorine, bromine, and iodine act as the halogen bond donor.A comprehensive X-ray crystallographic study of the
complexes of halogenated benzenes with the T4 lysozymeLeu99Ala mutant protein involved the successful determinationof structures of C6BrF5, C6ClF5, C6F5I, and C6H5I bound to ahydrophobic cavity at resolutions of 1.7−1.8 Å.196 Of these, onlythe T4 lysozyme (L99A)/C6F5I complex X-ray data showed clearelectron density indicative of a single bound orientation for theligand in which the iodine atom forms a halogen bond to thesulfur atom ofMet102 in the binding site (Figure 57). C6H5I binds
to the protein in two different orientations, of which only one isconsistent with a halogen bond toMet102. In this binding orienta-tion, the S···I distance is 3.3 Å compared to the correspondingdistance of 2.9 Å for the C6F5I complex. This difference and theobservation of only a single binding orientation suggests thatC6F5I forms a stronger halogen bond to the Met102 sulfur atomthan does C6H5I. This is in accord with expectations based on theelectron withdrawing effect of the fluorine atoms.
Several examples of S···X halogen bonds in the context ofmedicinal chemistry studies have been reported. These includethe cocrystal structure of the bacterial ZipA protein with 311,which was identified as an inhibitor of the ZipA−FtsZ protein−protein interaction, Ki = 12 μM, via high throughput screen-ing.197 The cocrystal structure of 311 bound to ZipA wasdetermined at 2.0 Å resolution and includes a close contactbetween the chlorine atom in 311 and the sulfur of ZipA residueMet42, with the geometry appropriate for a halogen bond. TheS−Cl distance is 3.1 Å, the S−Cl−C angle is 166°, and the anglebetween the S−Cl vector and the C−S−C plane in Met42 is 59°,however, this interaction was not specifically addressed indeference to a primary focus on the application of shape-basedcomputational scaffold hopping methodology (Figure 58).
An example of an intermolecular S···Br halogen bond comesfrom a study comparing the results of high throughput screeningand computational docking experiments based on the cocrystalstructure of 312 with the Chagas’ disease-related thiol proteasecruzain which was determined at 1.3 Å resolution.198
Examination of this structure revealed a potential halogenbond between the Br atom of 312 and the S atom of Met68, withan S···Br distance of 3.3 Å and a C−Br−S angle of 172°, while theangle between the S−Br vector and the C−S−C plane in Met68is 21° (Figure 59). In this example, although the interatomicdistance and C−Br−S angle are consistent with a halogen bond,the C−Br bond vector is not directed toward the Met68 sulfuratom in a fashion that would result in maximum stabilization.A potential sulfur···iodine halogen bond is observed in the
1.8 Å X-ray cocrystal structure of 313 with the urokinase typeplasminogen activator (uPA) protein.199 In this structure, the
Figure 57. X-ray cocrystal structure of C6F5I bound to T4 lysozymeLeu99Ala mutant showing an intermolecular S···I halogen bond betweenthe side chain sulfur ofMet102 and the ligand iodine atom, d1 = 2.9 Å, ϑ =166.3° (PDB ID: 3DN3).196
Figure 58. X-ray cocrystal structure of 311 with the ZipA proteinshowing an intermolecular S···Cl halogen bond between the side chainsulfur of Met42 and a ligand chlorine atom, d1 = 3.1 Å, ϑ = 165.9° (PDBID: 1Y2F).197 For the purpose of clarity, only ZipA residue Met42 isshown.
Journal of Medicinal Chemistry Perspective
DOI: 10.1021/jm501853mJ. Med. Chem. XXXX, XXX, XXX−XXX
AQ

iodine in 313 is 3.7 Å from the sulfur atom of Cys42, the C−I−Sangle is 143°, and the angle between the S···I vector and theSCys58−SCys42−CCys42 plane is 28°, as depicted in Figure 60.This geometry is nonideal for halogen bonds and is consistentwith the description of this specific interaction as secondaryrelative to the interaction between the iodine in 313 and thebackbone carbonyl oxygen atom of Val41.
11b
Although the above examples highlight potential protein−ligand S···X halogen bonds or, at a minimum, close contacts,none suggest whether the specific interaction positively con-tributes to the binding energy for the protein−ligand com-plex. However, the limited SAR associated with 314, an ATP-competitive inhibitor of JNKs 1−3, suggests a positive effect for aS···Cl halogen bond.200 In the 2.4 Å resolution cocrystal structureof 314 bound to JNK3, there is a clear S···Cl halogen bondinginteraction between the chlorine in 314 and gatekeeper residueMet46 (Figure 61). The S···Cl distance is 3.3 Å, the C−Cl−Sangle is 160°, and the C−Cl bond vector is approximatelyperpendicular to the Met42 C−S−C plane. The IC50 of 314 in anin vitro JNK3 assay is 57 nM, while compound 315, in which thechlorine is replaced by a methyl group, has an approximately7-fold higher IC50 of 410 nM. Given the roughly comparable sizeof methyl groups and chlorine atoms, it is reasonable to infer thatthe improved IC50 observed for 314 may be due to a specificS···Cl halogen bond. JNK1 and JNK2 are kinases with highhomology to JNK3 and 314 is more active than 315 or the des-chloro analogue 316 toward these enzymes, which also containmethionine gatekeeper residues, and it appears likely that S···Clhalogen bonds are present in these complexes.The limited number of reports focused on S···X halogen bonds
suggests that they are infrequently exploited in the context of drug
design. As medicinal chemists and molecular modelers becomemore aware of this type of interaction and its potential utility in thequest for improved potency and molecular properties, it isanticipated that more examples are likely to be forthcoming.
■ INTERMOLECULAR O···C−S σ* INTERACTIONSIntermolecular interactions between the low lying C−S σ*orbital of a sulfur-containing ligand and a protein target arerelatively rare, in part a function of the geometric requirementsfor this interaction which is predisposed to an intramoleculareffect. However, there are two recent examples describing a closeassociation between backbone amide oxygen atoms and sulfur-containing ligands that are suggestive of a productiveinteraction.201,202 The CHK1 inhibitor 317 was identifiedusing an affinity selection-mass spectrometry-based automatedligand identification system to survey mixtures of compoundsprepared in combinatorial libraries.201a The affinity of 317 forCHK1 kinase was determined to be <100 nM in the screen, andkinase inhibitory activity was confirmed in a biochemical assay,IC50 = 75 nM, while affinity competition studies suggested thatthe compound bound to the ATP binding site. This wasconfirmed by an X-ray cocrystal solved at 1.9 Å resolution,which indicated binding to the hinge region of the enzyme andrevealed an interesting constellation of interactions with theprotein (Figure 62a). TheC-6 hydrogen of the dihydrobenzofuranis close to the carbonyl of Cys87 (3.3 Å), while the C-5 H of thethiazole is 3.1 Å away from the backbone amide of Glu85 at anangle of 161.2°, geometries suggestive of partial H-bondinginteractions. The Glu85 carbonyl is also proximal to the thiazole Satom at a distance of 3.3 Å, just below the sum of the van derWaals radii, with a CO···S angle of 141.0°. The thiazoleH-bond donor effect is unusual in that this H atom is adjacent tosulfur rather than nitrogen, which is energetically favored andmore common.203 The bound conformer is also stabilized byintramolecular H-bonding interactions between the amide NHand both the thiazole and piperazine N atoms. In addition, thering and amide dipoles are favorably aligned, all of which maycontribute to preorganization of the molecule for binding.Optimization of 317 focused on improving CHK1 inhibitorypotency and selectivity and took full advantage of the insightsgleaned from the cocrystal structure to introduce additionalintermolecular H-bonding interactions. The isoindolone 318emerged as a refined homologue with potent CHK1 inhibition,IC50 = 1 nM, and 300-fold improved activity in a cell-basedassay.201b In the cocrystal structure of 318 with CHK1, theisoindolone O atom is closely associated with the thiazole S
Figure 59. X-ray cocrystal structure of 312 with the cruzain proteinshowing an intermolecular S···Br close contact between the side chainsulfur of Met68 and the ligand bromine atom, d1 = 3.3 Å, ϑ = 172.4°(PDB ID: 3KKU).198 For clarity, only cruzain residue Met68 is shown.
Figure 60. X-ray cocrystal structure of 313 with the uPA proteinshowing an intermolecular S···I close contact between the side chainsulfur of Cys42 and the ligand iodine atom, d1 = 3.7 Å, ϑ = 143.3° (PDBID: 1GJD).199 For clarity, only uPA residues Cys42 and Cys58 are shown.
Figure 61. X-ray cocrystal structure of the JNK3/314 complex showingan intermolecular S···Cl halogen bonding interaction between the sidechain sulfur of Met146 and the ligand chlorine atom, d1 = 3.4 Å, ϑ =160.4° (PDB ID: 2P33).200 For clarity, only gatekeeper residue Met146and hinge residue Met149 are shown.
Journal of Medicinal Chemistry Perspective
DOI: 10.1021/jm501853mJ. Med. Chem. XXXX, XXX, XXX−XXX
AR
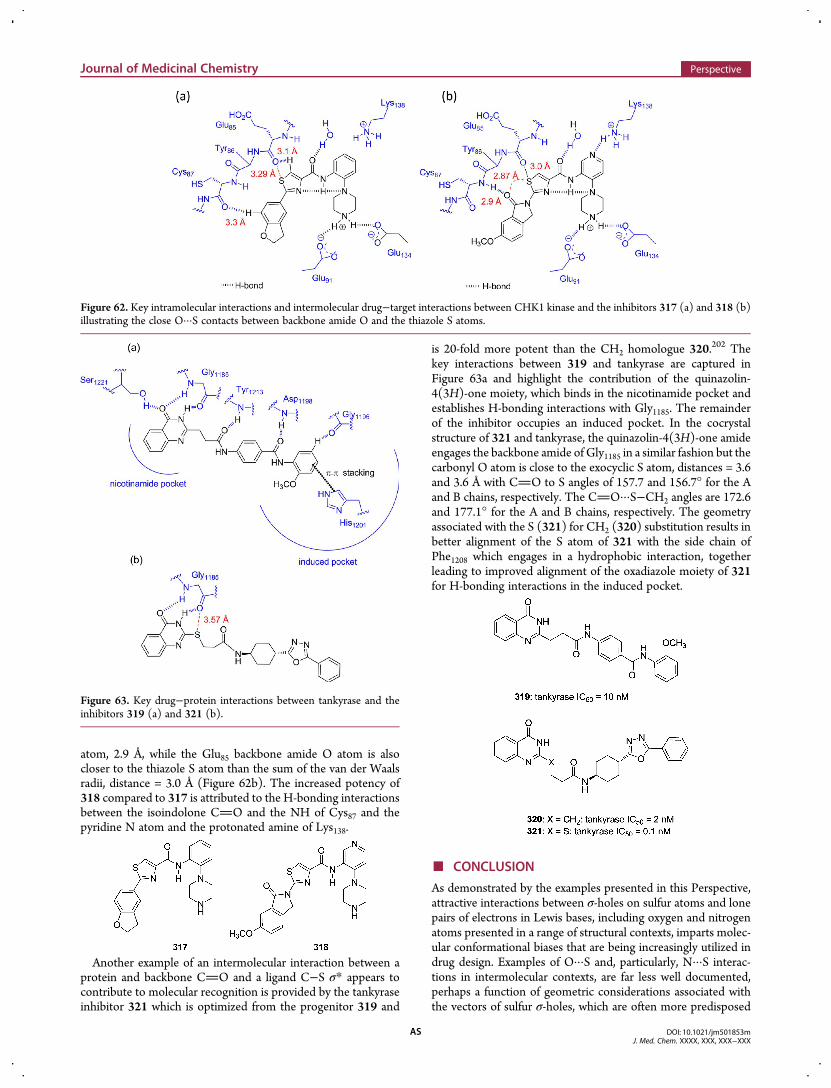
atom, 2.9 Å, while the Glu85 backbone amide O atom is alsocloser to the thiazole S atom than the sum of the van der Waalsradii, distance = 3.0 Å (Figure 62b). The increased potency of318 compared to 317 is attributed to the H-bonding interactionsbetween the isoindolone CO and the NH of Cys87 and thepyridine N atom and the protonated amine of Lys138.
Another example of an intermolecular interaction between aprotein and backbone CO and a ligand C−S σ* appears tocontribute to molecular recognition is provided by the tankyraseinhibitor 321 which is optimized from the progenitor 319 and
is 20-fold more potent than the CH2 homologue 320.202 Thekey interactions between 319 and tankyrase are captured inFigure 63a and highlight the contribution of the quinazolin-4(3H)-one moiety, which binds in the nicotinamide pocket andestablishes H-bonding interactions with Gly1185. The remainderof the inhibitor occupies an induced pocket. In the cocrystalstructure of 321 and tankyrase, the quinazolin-4(3H)-one amideengages the backbone amide of Gly1185 in a similar fashion but thecarbonyl O atom is close to the exocyclic S atom, distances = 3.6and 3.6 Å with CO to S angles of 157.7 and 156.7° for the Aand B chains, respectively. The CO···S−CH2 angles are 172.6and 177.1° for the A and B chains, respectively. The geometryassociated with the S (321) for CH2 (320) substitution results inbetter alignment of the S atom of 321 with the side chain ofPhe1208 which engages in a hydrophobic interaction, togetherleading to improved alignment of the oxadiazole moiety of 321for H-bonding interactions in the induced pocket.
■ CONCLUSION
As demonstrated by the examples presented in this Perspective,attractive interactions between σ-holes on sulfur atoms and lonepairs of electrons in Lewis bases, including oxygen and nitrogenatoms presented in a range of structural contexts, imparts molec-ular conformational biases that are being increasingly utilized indrug design. Examples of O···S and, particularly, N···S interac-tions in intermolecular contexts, are far less well documented,perhaps a function of geometric considerations associated withthe vectors of sulfur σ-holes, which are often more predisposed
Figure 62. Key intramolecular interactions and intermolecular drug−target interactions between CHK1 kinase and the inhibitors 317 (a) and 318 (b)illustrating the close O···S contacts between backbone amide O and the thiazole S atoms.
Figure 63. Key drug−protein interactions between tankyrase and theinhibitors 319 (a) and 321 (b).
Journal of Medicinal Chemistry Perspective
DOI: 10.1021/jm501853mJ. Med. Chem. XXXX, XXX, XXX−XXX
AS

toward an intramolecular engagement. However, emergingevidence suggests that this type of protein−ligand contactcould contribute favorably to the augmentation of drug−targetbinding energies, offering opportunity for further exploitation.Sulfur···aromatic interactions are common in proteins and havealso been observed in protein−ligand complexes. However, whilethe available examples do not clearly demonstrate improvedligand affinity as a result of specific intermolecular sulfur···aromatic interactions, studies of the association of HCVNS3/4Aprotease with small peptides suggests that they can impart asignificant favorable impact on recognition and binding.189,190
Sulfur may also interact with chlorine, bromine, and iodine,adopting the role of a Lewis base and donating electrons into theσ-holes found in organic halides. As with the sulfur···aromaticinteractions, there are relatively few examples of sulfur halogenbonds, but in at least one case, a sulfur halogen bond potentiallycontributes to the improved potency of a kinase inhibitor.200
Computational studies have elucidated the origins of the sulfurinteractions described in this Perspective and, along withexperimental studies on models systems, support the premisethat they can afford stabilization in both intermolecular andintramolecular contexts. However, additional computational andexperimental studies utilizing surrogates more representative ofdrug-like molecules would provide valuable insights regardingthe maximum potential of sulfur···aromatic and sulfur···halogeninteractions to enhance drug−target binding affinity.Because the role of noncovalent interactions involving sulfur in
compound conformation and ligand−protein interactions maybe underappreciated, this phenomenon may have been over-looked in many drug design campaigns. By fostering a greaterawareness of the presence of low-lying σ* orbitals associated withelectron deficient sulfur atoms, we hope to encourage broaderapplication by the medicinal chemistry community who maybe able to take advantage of this phenomenon to modulateproperties of interest in drug design. This, in turn, will lead to adeepening of the understanding of the role of sulfur σ-holeinteractions as a mediator of both intra- and intermolecularcontacts, furthering exploitation of the effect and enhancingpractical utility. A successful drug represents the confluenceof a range of properties and the strategic exploitation of sulfurinteractions offers one approach to address some of the manychallenges encountered in contemporary discovery campaigns.
■ AUTHOR INFORMATIONCorresponding Author*Phone: 203-677-6679. Fax: 203-677-7884. E-mail: [email protected] Contributions∥B.R.B. and K.S.Y. contributed equally to this Perspective.NotesThe authors declare no competing financial interest.The images in Figures 2, 16, 17, 28−33, 35−37, 39, 49−55, and57−61, were generated with the PyMOL Molecular GraphicsSystem, Version 1.7 (Schrodinger, LLC).Biographies
Brett R. Beno received his Ph.D. degree in 1997 from the University ofCalifornia, Los Angeles, under the supervision of Professor K. N. Houk.He remained in Professor Houk’s group as a postdoctoral scholar untiljoining the Computer Assisted Drug Design department at Bristol-Myers Squibb in 1998. At BMS, he has contributed to many drugdiscovery programs in the areas of neuroscience and virology and iscurrently a coleader of a drug discovery team focused on HIV.
Kap-Sun Yeung is a Principal Scientist in the Discovery ChemistryDivision at Bristol-Myers Squibb. He has expertise in antiviral drugdiscovery and key contributions to the HIV-1 attachment inhibitor andHCVNS5B polymerase inhibitor projects at Bristol-Myers Squibb, bothof which have progressed compounds into phase II clinical studies. Heobtained his Ph.D. in organic synthesis from Cambridge University inthe United Kingdom and performed postdoctoral research at the ScrippsResearch Institute in La Jolla before joining Bristol-Myers Squibb.
Michael D. Bartberger received his undergraduate degree in Chemistry atthe University of Central Florida in 1992. After obtaining his Ph.D. inExperimental and Theoretical Physical Organic Chemistry at the Universityof Florida with Professor W. R. Dolbier, Jr., he moved to the University ofCalifornia, Los Angeles, as a National Institutes ofHealth postdoctoral fellow,furthering his studies in theoretical organic chemistry and chemical biology inthe research group of Professor K.N.Houk. In 2001, he joinedAmgenwherehe is currently a Principal Scientist in the Computational Chemistry divisionof Therapeutic Discovery and contributor to research efforts ranging fromoncology and metabolic disorders to inflammation and neuroscience.
Lewis D. Pennington is a Principal Scientist in the Medicinal Chemistrygroup at Amgen in Thousand Oaks, California. Prior to joining Amgen in2003, heworked at Array BioPharma in Boulder, CO, after receiving a Ph.D.in Chemistry under the mentorship of Professor Larry E. Overman at theUniversity of California, Irvine, in 2002. Preceding his graduate studies, hewas employed for three years in theMedicinal Chemistry department at EliLilly & Co. in Indianapolis, IN, after earning a B.S. in Chemistry (withHighest Honors in Chemistry) from the University of Michigan, AnnArbor, in 1993 under the guidance of Professor Masato Koreeda.
Nicholas A. Meanwell received his Ph.D. degree from the University ofSheffield, Sheffield, England, under the supervision of Dr. D. NevilleJones and conducted postdoctoral studies at Wayne State University,Detroit, MI, in collaboration with Professor Carl R. Johnson. He joinedBristol-Myers Squibb in 1982, where he has supervised teams that haveadvanced clinical candidates in several areas of antiviral drug discovery,including BMY-433771, an inhibitor of respiratory syncytial virus fusion,the HIV-1 attachment inhibitor BMS-626529 that is being developed asthe prodrug BMS-663068, the HCVNS3 protease inhibitor asunaprevir,the HCV NS5A inhibitor daclatasvir, and the HCV NS5B RNA-dependent RNA polymerase inhibitor beclabuvir.
■ ACKNOWLEDGMENTS
The numerous stimulating discussions and camaraderie betweenM.D.B. and Prof. Dean J. Tantillo (Department of Chemistry,University of California, Davis) are acknowledged with gratitude.M.D.B. and L.D.P. also extend their appreciation to Dr. David St.Jean, Jr. (Therapeutic Discovery, Amgen, Inc.) for useful discussions.
■ ABBREVIATIONS USED
AII, angiotensin II; Alk, anaplastic lymphoma kinase; AMPA,α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid; ATC,4-aminomethyl-1,3-thiazole-5-carboxylic acid; ATP, adenosinetriphosphate; au, atomic units; BSSE, basis set superpositionerror; CD, circular dichroism; Cdk, cyclin-dependent kinase;CEACAM, carcinoembryonic antigen-related cell adhesionmolecule; CHK1, checkpoint 1 kinase; c-MET, mesenchymal−epithelial transition factor; CSD, Cambridge CrystallographicDatabase; CFTR, cystic fibrosis trans-membrane conductanceregulator; CHK1, checkpoint kinase 1; d, distance; DHFR,dihydrofolate reductase; DMDS, dimethyl disulfide; DMS,dimethyl sulfide; DNA, deoxyribonucleic acid; EphA4,erythropoietin-producing hepatocellular tyrosine kinase A4;GABA, γ-amino butyric acid; GK, glucokinase; GSK3, glycogen
Journal of Medicinal Chemistry Perspective
DOI: 10.1021/jm501853mJ. Med. Chem. XXXX, XXX, XXX−XXX
AT

synthase kinase 3; GPCR, G-protein coupled receptor; GyrB,DNA gyrase B; hC3a, human inflammatory protein complementC3a; HCV, hepatitis C virus; IAP, inhibitor of apoptosis;IMPDH, inosine monophosphate dehydrogenase; JAK, Januskinase; JHE, juvenile hormone esterase; JNK, c-Jun N-terminalkinase; KDR, kinase insert domain receptor; MAP kinase,mitogen-activated protein kinase; NAD, nicotine adeninedinucleotide; NBO, natural bond order; NOE, nuclear Over-hauser effect; PDE, phosphodiesterase; PI3Kγ, phosphoinositide3-kinase γ; SAR, structure−activity relationship; SIRT, sirtuin;Smac, second mitochondria-derived activator of caspases; S. aureus,Staphylococcus aureus; TAD, tiazofurin adenine dinucleotide;TAK1, transforming growth factor β receptor-associated kinase 1;uPA, urokinase type plasminogen activator; VEGFR2, vascularendothelial growth factor receptor 2;WT,wild-type; XIAP, X-linkedinhibitor of apoptosis
■ REFERENCES(1) (a) Nicolaou, K. C.; Chen, J. S.; Dalby, S. M. From nature to thelaboratory and into the clinic. Bioorg. Med. Chem. 2009, 17, 2290−2303.(b) Prinsep, M. R. Sulfur-containing natural products from marineinvertebrates. Stud. Nat. Prod. Chem. 2003, 28, 617−751. (c) Parry, R. J.Biosynthesis of some sulfur-containing natural products investigationsof the mechanism of carbon−sulfur bond formation. Tetrahedron 1983,39, 1215−1238.(2) Aoki, H.; Okuhara, M. Natural β-lactam antibiotics. Annu. Rev.Microbiol. 1980, 34, 159−181.(3) (a) Basak, A.; Mandal, S.; Bag, S. S. Chelation-controlled Bergmancyclization: synthesis and reactivity of enediynyl ligands. Chem. Rev.2003, 103, 4077−4094. (b) Shen, B.; Hindra; Yan, X.; Huang, T.; Ge,H.; Yang, D.; Teng, Q.; Rudolf, J. D.; Lohman, J. R. Enediynes:exploration ofmicrobial genomics to discover new anticancer drug leads.Bioorg. Med. Chem. Lett. 2015, 25, 9−15.(4) (a) VanderMolen, K.M.;McCulloch,W.; Pearce, C. J.; Oberlies, N.H. Romidepsin (istodax, NSC 630176, FR901228, FK228, depsipep-tide): a natural product recently approved for cutaneous T-celllymphoma. J. Antibiot. 2011, 64, 525−531. (b) Bertino, E. M.;Otterson, G. A. Romidepsin: a novel histone deacetylase inhibitor forcancer. Expert Opin. Invest. Drugs 2011, 20, 1151−1158.(5) Borthwick, A. D. 2,5-Diketopiperazines: synthesis, reactions,medicinal chemistry, and bioactive natural products. Chem. Rev. 2012,112, 3641−3716.(6) (a) Feyen, F.; Cachoux, F.; Gertsch, J.; Wartmann, M.; Altmann, K.H. Epothilones as lead structures for the synthesis-based discovery ofnew chemotypes for microtubule stabilization. Acc. Chem. Res. 2008, 41,21−31. (b) Borzilleri, R. M.; Vite, G. D. Discovery of ixabepilone(IXEMPRA), a first-in-class epothilone analog for treatment ofmetastatic breast cancer. Annu. Rep. Med. Chem. 2009, 44, 301−322.(c) Borzilleri, R. M.; Vite, G. D. Epothilones: new tubulin polymer-ization agents in preclinical and clinical development. Drugs Future2002, 27, 1149−1163.(7) (a) Hecht, S. M. Bleomycin: new perspectives on the mechanism ofaction. J. Nat. Prod. 2000, 63, 158−168. (b) Hecht, S. M. The chemistryof activated bleomycin. Acc. Chem. Res. 1986, 19, 383−391.(8) Bagley, M. C.; Dale, J. W.; Merritt, E. A.; Xiong, X. Thiopeptideantibiotics. Chem. Rev. 2005, 105, 685−714.(9) Ilardi, E. A.; Vitaku, E.; Njardarson, J. T. Data-mining for sulfur andfluorine: an evaluation of pharmaceuticals to reveal opportunities fordrug design and discovery. J. Med. Chem. 2014, 57, 2832−2842.(10) (a) Choi-Sledeski, Y. M.; Kearney, R.; Poli, G.; Pauls, H.; Gardner,C.; Gong, Y.; Becker, M.; Davis, R.; Spada, A.; Liang, G.; Chu, V.; Brown,K.; Collussi, D.; Leadley, R., Jr.; Rebello, S.; Moxey, P.; Morgan, S.;Bentley, R.; Kasiewski, C.; Maignan, S.; Guilloteau, J.-P.; Mikol, V.Discovery of an orally efficacious inhibitor of coagulation factor Xawhich incorporates a neutral P1 ligand. J. Med. Chem. 2003, 46, 681−684. (b) Roehrig, S.; Straub, A.; Pohlmann, J.; Lampe, T.; Pernerstorfer,J.; Schlemmer, K.-H.; Reinemer, P.; Perzborn, E. Discovery of the novel
antithrombotic agent 5-chloro-N-({(5S)-2-oxo-3-[4-(3-oxomorpholin-4-yl)phenyl]-1,3-oxazolidin-5-yl}methyl)thiophene-2-carboxamide(BAY 59-7939): an oral, direct factor Xa inhibitor. J. Med. Chem. 2005,48, 5900−5908. (c) Perzborn, E.; Roehrig, S.; Straub, A.; Kubitza, D.;Misselwitz, F. The discovery and development of rivaroxaban, an oral,direct factor Xa inhibitor. Nature Rev. Drug Discovery 2011, 10, 61−75.(d) Chan, C.; Borthwick, A. D.; Brown, D.; Burns-Kurtis, C. L.;Campbell, M.; Chaudry, L.; Chung, C.-w.; Convery, M. A.; Hamblin, J.N.; Johnstone, L.; Kelly, H. A.; Kleanthous, S.; Patikis, A.; Patel, C.;Pateman, A. J.; Senger, S.; Shah, G. P.; Toomey, J. R.; Watson, N. S.;Weston, H. E.; Whitworth, C.; Young, R. J.; Zhou, P. Factor Xainhibitors: S1 binding interactions of a series of N-{(3S)-1-[(1S)-1-methyl-2-morpholin-4-yl-2-oxoethyl]-2-oxopyrrolidin-3-yl}-sulfonamides. J. Med. Chem. 2007, 50, 1546−1557.(11) (a) Lu, Y.; Shi, T.; Wang, Y.; Yang, H.; Yan, X.; Luo, X.; Jiang, H.;Zhu,W. Halogen bondinga novel interaction for rational drug design?J. Med. Chem. 2009, 52, 2854−2862. (b) Wilcken, R.; Zimmermann, M.O.; Lange, A.; Joerger, A. C.; Boeckler, F. M. Principles and applicationsof halogen bonding in medicinal chemistry and chemical biology. J. Med.Chem. 2013, 56, 1363−1388. (c) Sirimulla, S.; Bailey, J. B.; Vegesna, R.;Narayan, M. Halogen interactions in protein−ligand complexes:implications of halogen bonding for rational drug design. J. Chem. Inf.Model. 2013, 53, 2781−2791. (d) Hardegger, L. A.; Kuhn, B.; Spinnler,B.; Anselm, L.; Ecabert, R.; Stihle, M.; Gsell, B.; Thoma, R.; Diez, J.;Benz, J.; Plancher, J.-M.; Hartmann, G.; Banner, D. W.; Haap, W.;Diederich, F. Systematic investigation of halogen bonding in protein−ligand interactions. Angew. Chem., Int. Ed. 2011, 50, 314−318.(e) Scholfield, M. R.; Zanden, C. M. V.; Carter, M.; Ho, P. S. Halogenbonding (X-bonding): a biological perspective. Protein Sci. 2013, 22,139−152. (f) Clark, T.; Hennemann, M.; Murray, J. S.; Politzer, P.Halogen bonding: the σ-hole. J. Mol. Model. 2007, 13, 291−296.(g) Politzer, P.; Murray, J. S.; Clark, T. Halogen bonding and otherσ-hole interactions: a perspective. Phys. Chem. Chem. Phys. 2013, 15,11178−11189. (h) Ibrahaim, M. A. A. Molecular mechanical study ofhalogen bonding in drug discovery. J. Comput. Chem. 2011, 32, 2564−2574. (i) Desiraju, G. R.; Ho, P. S.; Kloo, L.; Anthony C. Legon, A. C.;Marquardt, R.; Metrangolo, P.; Politzer, P.; Resnati, G.; Rissanen, K.Definition of the halogen bond (IUPAC Recommendations 2013). PureAppl. Chem. 2013, 85, 1711−1713. (j) Matter, H.; Nazare, M.;Gussregen, S.; Will, D. W.; Schreuder, H.; Bauer, A.; Urmann, M.;Ritter, K.; Wagner, M.; Wehner, V. Evidence for C−Cl/C−Br···πinteractions as an important contribution of protein−ligand bindingaffinity. Angew. Chem., Int. Ed. 2009, 48, 2911−2916.(12) (a) Brameld, K. A.; Kuhn, B.; Reuter, D. C.; Stahl, M. Smallmolecule conformational preferences derived from crystal structuredata. A medicinal chemistry focused analysis. J. Chem. Inf. Model. 2008,48, 1−24. (b) Scharfer, C.; Schulz-Gasch, T.; Ehrlich, H.-C.; Guba, W.;Rarey, M.; Stahl, M. Torsion angle preferences in druglike chemicalspace: a comprehensive guide. J. Med. Chem. 2013, 56, 2016−2028.(13) Mann, A. Conformational restriction and/or steric hindrance inmedicinal chemistry. In The Practice of Medicinal Chemistry, 2nd ed.;Wermuth, C. G., Ed.; Academic Press: London, 1996; pp 233−250.(14) (a) Kuhn, B.; Mohr, P.; Stahl, M. Intramolecular hydrogenbonding in medicinal chemistry. J. Med. Chem. 2010, 53, 2601−2611.(b) Shalaeva, M.; Caron, G.; Abramov, Y. A.; O’Connell, T. N.;Plummer,M. S.; Yalamanchi, G.; Farley, K. A.; Goetz, G. H.; Philippe, L.;Shapiro, M. J. Integrating intramolecular hydrogen bonding (IMHB)considerations in drug discovery using ΔlogP as a tool. J. Med. Chem.2013, 56, 4870−4879.(15) (a) Yu, K. L.; Johnson, R. L. Synthesis and chemical properties oftetrazole peptide analogues. J. Org. Chem. 1987, 52, 2051−2059.(b) Zabrocki, J.; Marshall, G. R. The 1,5-disubstituted tetrazole ring as acis-amide bond surrogate. Methods Mol. Med. 1998, 23, 417−436.(16) (a) Venkatraman, S.; Njoroge, F. G. Macrocyclic inhibitors ofHCV NS3 protease. Exp. Opin. Ther. Pat. 2009, 19, 1277−1303.(b) Giordanetto, F.; Kihlberg, J. Macrocyclic drugs and clinicalcandidates: what can medicinal chemists learn from their properties?J. Med. Chem. 2014, 57, 278−295. (c) Lam, P. Y. S.; Jadhav, P. K.;Eyermann, C. J.; Hodge, C. N.; Ru, Y.; Bacheler, L. T.; Meek, J. L.; Otto,
Journal of Medicinal Chemistry Perspective
DOI: 10.1021/jm501853mJ. Med. Chem. XXXX, XXX, XXX−XXX
AU

M. J.; Rayner, M. M.; Wong, Y. N.; Chang, C.-H.; Weber, P. C.; Jackson,D. A.; Sharpe, T. R.; Erikson-Viitanen, S. Rational design of potent,bioavailable, nonpeptide cyclic ureas as HIV protease inhibitors. Science1994, 263, 380−384. (d) Lam, P. Y. S.; Ru, Y.; Jadhav, P. K.; Aldrich, P.E.; DeLucca, G. V.; Eyermann, C. J.; Chang, C.-H.; Emmett, G.; Holler,E. R.; Daneker, W. F.; Li, L.; Confalone, P. N.; McHugh, R. J.; Han, Q.;Li, R.; Markwalder, J. A.; Seitz, S. P.; Sharpe, T. R.; Bacheler, L. T.;Rayner, M. M.; Klabe, R. M.; Shum, L.; Winslow, D. L.; Kornhauser, D.M.; Jackson, D. A.; Erickson-Viitanen, S.; Hodge, C. N. Cyclic HIVprotease inhibitors: synthesis, conformational analysis, P2/P2′structure−activity relationship, and molecular recognition of cyclicureas. J. Med. Chem. 1996, 39, 3514−3525.(17) (a) Bryan, M. C.; Falsey, J. R.; Frohn, M.; Reichelt, A.; Yao, G.;Bartberger, M. D.; Bailis, J. M.; Zalameda, L.; SanMiguel, T.; Doherty, E.M.; Allen, J. G. N-Substituted azaindoles as potent inhibitors of Cdc7kinase. Bioorg. Med. Chem. Lett. 2013, 23, 2056−2060. (b) Chien, R. J.;Corey, E. J. Strong conformational preferences of heteroaromatic ethersand electron pair repulsion. Org. Lett. 2010, 12, 132−135.(18) (a) Zimmer, L. E.; Sparr, C.; Gilmour, R. Fluorine conformationaleffects in organocatalysis: an emerging strategy for molecular design.Angew. Chem., Int. Ed. 2011, 50, 11860−11871. (b) Schuler, M.;O’Hagan, D.; Slawin, A. M. Z. The vicinal F−C−C−F moiety as a toolfor influencing peptide conformation. Chem. Commun. 2005, 4324−4326.(19) Dudkin, V. Y. Bioisosteric equivalence of five-memberedheterocycles. Chem. Heterocycl. Compd. 2012, 48, 27−32.(20) Nagao, Y. Chemical pharma-sciences that incorporate non-covalent bonded interactions. Heterocycles 2013, 87, 1−29.(21) Iwaoka, M.; Isozumi, N. Hypervalent non-bonded interactions ofa divalent sulfur atom. Implications in protein architecture and thefunctions. Molecules 2012, 17, 7266−7283.(22) Murray, J. S.; Lane, P.; Politzer, P. Simultaneous σ-hole andhydrogen bonding by sulfur- and selenium-containing heterocycles. Int.J. Quantum Chem. 2008, 108, 2770−2781.(23) Pierce, A. C.; ter Haar, E.; Binch, H. M.; Kay, D. P.; Patel, S. R.; Li,P. CH···O and CH···N hydrogen bonds in ligand design: a novelquinazolin-4-ylthiazol-2-ylamine protein kinase inhibitor. J. Med. Chem.2005, 48, 1278−1281.(24) Lin, S.; Wrobleski, S. T.; Hynes, J., Jr.; Pitt, S.; Zhang, R.; Fan, Y.;Doweyko, A. M.; Kish, K. F.; Sack, J. S.; Malley, M. F.; Kiefer, S. E.;Newitt, J. A.; McKinnon, M.; Trzaskos, J.; Barrish, J. C.; Dodd, J. H.;Schieven, G. L.; Leftheris, K. Utilization of a nitrogen−sulfurnonbonding interaction in the design of new 2-aminothiazol-5-yl-pyrimidines as p38α MAP kinase inhibitors. Bioorg. Med. Chem. Lett.2010, 20, 5864−5868.(25) Jackson, N. E.; Savoie, B. M.; Kohlstedt, K. L.; Olvera de la Cruz,M.; Schatz, G. C.; Chen, L. X.; Ratner, M. A. Controlling conformationsof conjugated polymers and small molecules: the role of nonbondinginteractions. J. Am. Chem. Soc. 2013, 135, 10475−10483.(26) (a) Akiba, K.; Tsuchiya, T.; Inamoto, N.; Onuma, K.; Nagashima,N.; Nakamura, A. Chemistry of hypervalent sulfur. IV. The structure ofthe 1:1 adduct of “Hector’s base” with arylcyanamides. Bond switch onhypervalent sulfur. Chem. Lett. 1976, 723−726. (b) Akiba, K.; Tsuchiya,Y.; Inamoto, N. Chemistry of hypervalent sulfur. V. A 13C-NMR study ofthe 1:1 adduct of “Hector’s base” with arylcyanamides. Evidence forintramolecular S···N interaction. Tetrahedron Lett. 1976, 17, 3819−3820.(27) Ghosh, R.; Simonsen, S. H. Structure of 2-(2′-thienyl)pyridine at193 K. Acta Crystallogr., Sect. C: Cryst. Struct. Commun. 1993, C49,1031−1032.(28) Yu, J. Y.; Yoo, C. L.; Yang, B.; Lodewyk, M. W.; Meng, L.; El-Idreesy, T. T.; Fettinger, J. C.; Tantillo, D. J.; Verkman, A. S.; Kurth, M.J. Potent s-cis-locked bithiazole correctors of ΔF508 cystic fibrosistransmembrane conductance regulator cellular processing for cysticfibrosis therapy. J. Med. Chem. 2008, 51, 6044−6054.(29) Rittner, R.; Ducati, L. C.; Tormena, C. F.; Fiorin, B. C.; Braga, C.B. Conformational preferences for some 5-substituted 2-acetylthio-phenes through infrared spectroscopy and theoretical calculations.Spectrochim. Acta, Part A 2011, 79, 1071−1076.
(30) Weinhold, F.; Landis, C. R. Natural bond orbitals and extensionsof localized bonding concepts. Chem. Educ. Res. Pract. Eur. 2001, 2, 91−104.(31) Ducati, L. C.; Braga, C. B.; Rittner, R.; Tormena, C. F. A criticalevaluation of the s-cis−trans isomerism of 2-acetylpyrrole and its N-methyl derivative through infrared and NMR spectroscopies andtheoretical calculations. Spectrochim. Acta, Part A 2013, 116, 196−203.(32) Rittner, R.; Ducati, L. C.; Tormena, C. F.; Cormanich, R. A.;Fiorin, B. C.; Braga, C. B.; Abraham, R. J. Studies on the s-cis−transisomerism for some furan derivatives through IR and NMR spectros-copies and theoretical calculations. Spectrochim. Acta, Part A 2013, 103,84−89.(33) Gokce, H.; Bahceli, S. Analysis of molecular structure andvibrational spectra of 2-(2′-thienyl)pyridine. J. Mol. Struct. 2011, 1005,100−106.(34) Fleming, I. Frontier Orbitals and Organic Chemical Reactions;Wiley & Sons: New York, 1976.(35) Bartberger, M. D.; Tantillo, D. J. Work in progressthe effect ofsubstituents and ring composition on such donor−acceptor interactionsis a current area of study in our two groups and will be reported in duecourse.(36) Sims, P. A.; Wong, C. F.; Vuga, D.; McCammon, J. A.; Sefton, B.M. Relative contributions of desolvation, inter- and intra-molecularinteractions to binding affinity in protein kinase systems. J. Comput.Chem. 2005, 26, 668−681.(37) Hitchcock, S. A.; Pennington, L. D. Structure−brain exposurerelationships. J. Med. Chem. 2006, 49, 7559−7583.(38) Jorgensen, W. L.; Schyman, P. Treatment of halogen bonding inthe OPLS-AA force field: application to potent anti-HIV agents. J. Chem.Theory Comput. 2012, 8, 3895−3901.(39) St. Jean, D. J., Jr.; Fotsch, C. Mitigating heterocycle metabolism indrug discovery. J. Med. Chem. 2012, 55, 6002−6020.(40) Edwards, P. D.; Wolanin, D. J.; Andisik, D. W.; Davis, M. W.Peptidyl α-ketoheterocyclic inhibitors of human neutrophil elastase. 2.Effect of varying the heterocyclic ring on in vitro potency. J. Med. Chem.1995, 38, 76−85.(41) Maryanoff, B. E.; Costanzo, M. J. Inhibitors of proteases andamide hydrolases that employ an α-ketoheterocycle as a key enablingfunctionality. Bioorg. Med. Chem. 2008, 16, 1562−1595.(42) Angyan, J. G.; Poirer, R. A.; Kucsman, A.; Csizmadia, I. G.Bonding between nonbonded sulfur and oxygen atoms in selectedorganic molecules (a quantum chemical study). J. Am. Chem. Soc. 1987,109, 2237−2245.(43) Markham, G. D.; Bock, C. W. Intramolecular sulfur−oxygeninteractions: an ab initio molecular orbital and density functional theoryinvestigation. J. Mol. Struct.: THEOCHEM 1997, 418, 139−154.(44) (a) Srivastava, P. C.; Pickering, M. V.; Allen, L. B.; Streeter, D. G.;Campbell, M. T.; Witkowski, J. T.; Sidwell, R. W.; Robins, R. K.Synthesis and antiviral activity of certain thiazole C-nucleosides. J. Med.Chem. 1977, 20, 256−262. (b) Goldstein, B. M.; Takusagawa, F.;Berman, H. M.; Srivastava, P. C.; Robins, R. K. Structural studies of anew antitumor agent: tiazofurin and its inactive analogues. J. Am. Chem.Soc. 1983, 105, 7416−7422. (c) Gebeyehu, G.; Marquez, V. E.; VanCott, A.; Cooney, D. A.; Kelley, J. A.; Jayaram, H. N.; Ahluwalia, G. S.;Dion, R. L.; Wilson, Y. A.; Johns, D. G. Ribavirin, tiazofurin, andselenazofurin: mononucleotides and nicotinamide adenine dinucleotideanalogs. Synthesis, structure, and interactions with IMP dehydrogenase.J. Med. Chem. 1985, 28, 99−105. (d) Goldstein, B. M.; Takusagawa, F.;Berman, H. M.; Srivastava, P. C.; Robins, R. K. Structural studies of anew antitumor and antiviral agent: selenazofurin and its α anomer. J. Am.Chem. Soc. 1985, 107, 1394−1400. (e) Streeter, D. G.; Robins, R. K.Comparative in vitro studies of tiazofurin and a selenazole analog.Biochem. Biophys. Res. Commun. 1983, 115, 544−550. (f) Goldstein, B.M.; Mao, D. T.; Marquez, V. E. Ara-tiazofurin: conservation of structuralfeatures in an unusual thiazole nucleoside. J. Med. Chem. 1988, 31,1026−1031. (g) Goldstein, B. M.; Colby, T. D. Conformationalconstraints in NAD analogs: implications for dehydrogenase bindingand specificity. Adv. Enzyme Regul. 2000, 40, 405−426.
Journal of Medicinal Chemistry Perspective
DOI: 10.1021/jm501853mJ. Med. Chem. XXXX, XXX, XXX−XXX
AV

(45) (a) Jayaram, H. N.; Dion, R. L.; Glazer, R. I.; Johns, D. G.; Robins,R. K.; Srivastava, P. C.; Cooney, D. A. Initial studies on the mechanismof action of a new oncolytic thiazole nucleoside, 2-β-D-ribofuranosylth-iazole-4-carboxamide (NSC 286193). Biochem. Pharmacol. 1982, 31,2371−2380. (b) Tiazofurin: molecular and clinical action. Weber, G.;Prajda, N.; Abonyi, M.; Look, K. Y.; Tricot, G. Anticancer Res. 1996, 16,3313−3322. (c) Goldstein, B. M.; Bell, J. E.; Marquez, V. E.Dehydrogenase binding by tiazofurin anabolites. J. Med. Chem. 1990,33, 1123−1127.(46) Li, H.; Goldstein, B. M. Carboxamide group conformation in thenicotinamide and thiazole-4-carboxamide rings: implications for enzymebinding. J. Med. Chem. 1992, 35, 3560−3567.(47) Burling, F. T.; Goldstein, B. M. Computational studies ofnonbonded sulfur−oxygen and selenium−oxygen interactions in thethiazole and selenazole nucleosides. J. Am. Chem. Soc. 1992, 114, 2313−2320.(48) Makara, G. M.; Keseru, G. M. On the conformation of tiazofurinanalogues. J. Med. Chem. 1997, 40, 4154−4159.(49) (a) Li, H.; Hallows, W. H.; Punzi, J. S.; Marquez, V. E.; Carrell, H.L.; Pankiewicz, K. W.; Watanabe, K. A.; Goldstein, B. M. Crystallo-graphic studies of two alcohol dehydrogenase-bound analogues ofthiazole-4-carboxamide adenine dinucleotide (TAD), the activeanabolite of the antitumor agent tiazofurin. Biochemistry 1994, 33,23−32. (b) Jayaram, H. N.; Ahluwalia, G. S.; Dion, R. L.; Gebeyehu, G.;Marquez, V. E.; Kelley, J. A.; Robins, R. K.; Cooney, D. A.; Johns, D. G.Conversion of 2-β-D-ribofuranosylselenazole-4-carboxamide to ananalog of NAD with potent IMP dehydrogenase-inhibitory properties.Biochem. Pharmacol. 1983, 32, 2633−2636. (c) Li, M.; Dyda, F.;Benhart, I.; Pastant, I.; Davies, D. R. Crystal structure of the catalyticdomain of Pseudomonas exotoxin A complexed with a nicotinamideadenine dinucleotide analog: implications for the activation process andfor ADP ribosylation. Proc. Natl. Acad. Sci. U. S. A. 1996, 93, 6902−6906.(d) Colby, T. D.; Vanderveen, K.; Strickler, M. D.; Markham, G. D.;Goldstein, B. M. Crystal structure of human type II inosinemonophosphate dehydrogenase: implications for ligand binding anddrug design. Proc. Natl. Acad. Sci. U. S. A. 1999, 96, 3531−3536.(50) (a) Franchetti, P.; Cristalli, G.; Grifantini, M.; Cappellacci, L.;Vittori, S.; Nocentini, G. Synthesis and antitumor activity of 2-β-D-ribofuranosyloxazole-4-carboxamide (oxazofurin). J. Med. Chem. 1990,33, 2849−2852. (b) Goldstein, B. M.; Li, H.; Hallows, W. H.; Langs, D.A.; Franchetti, P.; Cappellacci, L.; Grifantini, M. C-Glycosyl bondconformation in oxazofurin: crystallographic and computational studiesof the oxazoles analogue of tiazofurin. J. Med. Chem. 1994, 37, 1684−1688.(51) (a) Franchetti, P.; Cappellacci, L.; Grifantini, M.; Barzi, A.;Nocentini, G.; Yang, H.; O’Connor, A.; Jayaram, H. N.; Carrell, C.;Goldstein, B. M. Furanfurin and thiophenfurin: two novel tiazofurinanalogues. Synthesis, structure, antitumor activity, and interactions withinosine monophosphate dehydrogenase. J. Med. Chem. 1995, 38, 3829−3837. (b) Franchetti, P.; Cappellacci, L.; Perlini, P.; Jayaram, H. N.;Buler, A.; Schneider, B. P.; Collart, F. R.; Huberman, E.; Grifantini, M.Isosteric analogues of nicotinamide adenine dinucleotide derived fromfuranfurin, thiophenfurin, and selenophenfurin as mammalian inosinemonophosphate dehydrogenase (type I and II) inhibitors. J. Med. Chem.1998, 41, 1702−1707.(52) Franchetti, P.; Marchetti, S.; Cappellacci, L.; Yalowitz, J. A.;Jayaram, H. N.; Goldstein, B. M.; Grifantini, M. A new C-nucleosideanalogue of tiazofurin: synthesis and biological evaluation of 2-β-D-ribofuranosylimidazole-4-carboxamide (imidazofurin). Bioorg. Med.Chem. Lett. 2001, 11, 67−69.(53) Popsavin, M.; Spaic, S.; Svircev, M.; Kojic, V.; Bogdanovic, G.;Popsavin, V. Synthesis and in vitro antitumour screening of 2-(β-D-xylofuranosyl)thiazole-4-carboxamide and two novel tiazofurin ana-logues with substituted tetrahydrofurodioxole moiety as a sugar mimic.Bioorg. Med. Chem. Lett. 2012, 22, 6700−6704. (b) Zatorski, A.;Watanabe, K. A.; Carr, S. F.; Goldstein, B. M.; Pankiewicz, K. W.Chemical synthesis of benzamide adenine dinucleotide: inhibition ofinosine monophosphate dehydrogenase (types I and II). J. Med. Chem.1996, 39, 2422−2426.
(54) Franchetti, P.; Marchetti, S.; Cappellacci, L.; Jayaram, H. N.;Yalowitz, J. A.; Goldstein, B. M.; Barascut, J.-L.; Dukhan, D.; Imbach, J.-L.; Grifantini, M. Synthesis, conformational analysis, and biologicalactivity of C-thioribonucleosides related to tiazofurin. J. Med. Chem.2000, 43, 1264−1270.(55) (a) Shin, D.; Sinkeldam, R. W.; Tor, Y. Emissive RNA alphabet. J.Am. Chem. Soc. 2011, 133, 14912−14915. (b) Sinkeldam, R. W.;McCoy, L. S.; Shin, D.; Tor, Y. Enzymatic interconversion of isomorphicfluorescent nucleosides: adenosine deaminase transforms an adenosineanalogue into an inosine analogue. Angew. Chem., Int. Ed. 2013, 52,14026−14030.(56) Camm, A. J.; Bounameaux, H. Edoxaban: a new oral direct factorXa inhibitor. Drugs 2011, 71, 1503−1526. (b) Escolar, G.; Diaz-Ricart,M. Edoxaban tosilate: direct factor Xa inhibitor prevention of post-operative venous thromboembolism treatment of atrial fibrillation.Drugs Future 2009, 34, 861−872.(57) (a) Haginoya, N.; Kobayashhi, S.; Komoriya, S.; Yoshino, T.;Suzuki, M.; Shimada, T.; Watanabe, K.; Hirokawa, Y.; Furugori, T.;Nagahara, T. Synthesis and conformational analysis of a non-amidinefactor Xa inhibitor that incorporates 5-methyl-4 ,5,6,7-tetrahydrothiazolo[5,4-c]pyridine as S4 binding element. J. Med.Chem. 2004, 47, 5167−5182. (b) Komoriya, S.; Kobayashi, S.; Osanai,K.; Yoshino, T.; Nagata, T.; Haginoya, N.; Nakamoto, Y.; Mochizuki, A.;Nagahara, T.; Suzuki, M.; Shimada, T.; Watanabe, K.; Isobe, Y.;Furugoori, T. Design, synthesis, and biological activity of novel factor Xainhibitors: Improving metabolic stability by S1 and S4 ligandmodification. Bioorg. Med. Chem. 2006, 14, 1309−1330. (c) Nagata,T.; Yoshino, T.; Haginoya, N.; Yoshikawa, K.; Nagamochi, M.;Kobayashi, S.; Komoriya, S.; Yokomizo, A.; Muto, R.; Yamaguchi, M.;Osanai, K.; Suzuki, M.; Kanno, H. Discovery of N-[(1R,2S,5S)-2-{[(5-chloroindol-2-yl)carbonyl]amino}-5-(dimethylcarbamoyl)cyclohexyl]-5-methyl-4,5,6,7-tetrahydrothiazolo[5,4-c]pyridine-2-carboxamide hy-drochloride: a novel, potent and orally active direct inhibitor of factorXa. Bioorg. Med. Chem. 2009, 17, 1193−1206.(58) Yoshikawa, K.; Kobayashi, S.; Nakamoto, Y.; Haginoya, N.;Komoriya, S.; Yoshino, T.; Nagata, T.; Mochizuki, A.; Watanabe, K.;Suzuki, M.; Kanno, H.; Ohta, T. Design, synthesis, and SAR of cis-1,2-diaminocyclohexane derivatives as potent factor Xa inhibitors. Part II:exploration of 6−6 fused rings as alternative S1 moieties. Bioorg. Med.Chem. 2009, 17, 8221−8233.(59) Adler, M.; Kochanny, M. J.; Ye, B.; Rumennik, G.; Light, D. R.;Biancalana, S.;Whitlow,M. Crystal structures of two potent nonamidineinhibitors bound to factor Xa. Biochemistry 2002, 41, 15514−15523.(60) (a) Guba, W.; Neidhart, W.; Nettekoven, M. Novel and potentNPY5 receptor antagonists derived from virtual screening and iterativeparallel chemistry design. Bioorg. Med. Chem. Lett. 2005, 15, 1599−1603.(b) Nettekoven, M.; Guba, W.; Neidhart, W.; Mattei, P.; Pflieger, P.;Roche, O.; Taylor, S. Isomeric thiazole derivative as ligands for theneuropeptide Y5 receptor. Bioorg. Med. Chem. Lett. 2005, 15, 3446−3449. (c) Nettekoven, M.; Guba, W.; Neidhart, W.; Mattei, P.; Pflieger,P.; Plancher, J.-M.; Taylor, S. Aminothiazole derivatives as neuropeptideY5 receptor ligands: finding the balance between affinity andphysicochemical properties. ChemMedChem 2006, 1, 45−48.(61) (a) Gibson, S.; McGuire, R.; Rees, D. C. Principal componentsdescribing biological activities and molecular diversity of heterocyclicaromatic ring fragments. J. Med. Chem. 1996, 39, 4065−4072.(b) Breitung, E. M.; Shu, C.-F.; McMahon, R. J. Thiazole and thiopheneanalogues of donor−acceptor stilbenes: molecular hyperpolarizabilitiesand structure−property relationships. J. Am. Chem. Soc. 2000, 122,1154−1160.(62) (a) Reid, R. C.; Yau, M.-K.; Singh, R.; Hamidon, J. K.; Reed, A. N.;Chu, P.; Suen, J. Y.; Stoermer, M. J.; Blakeney, J. S.; Lim, J.; Faber, J. M.;Fairlie, D. P. Downsizing a human inflammatory protein to a smallmolecule with equal potency and functionality. Nature Commun. 2013,4, No. 2802, DOI: 10.1038/ncomms3802. (b) Reid, R. C.; Yau, M.-K.;Singh, R.; Lim, J.; Fairlie, D. P. Stereoelectronic effects dictate molecularconformation and biological function of heterocyclic amides. J. Am.Chem. Soc. 2014, 136, 11914−11917.
Journal of Medicinal Chemistry Perspective
DOI: 10.1021/jm501853mJ. Med. Chem. XXXX, XXX, XXX−XXX
AW

(63) (a) Mathieu, L.; Legrand, B.; Dang, C.; Vezenkov, L.; Wenger, E.;Didierjean, C.; Amblard, M.; Avrlant-Petit, M.-C.; Masurier, N.;Lisowski, V.; Martinez, J.; Maillard, L. T. Helical oligomers ofthiazole-based γ-amino acids: synthesis and structural studies. Angew.Chem., Int. Ed. 2013, 52, 6006−6010. (b) Legrand, B.; Mathieu, L.;Lebrun, A.; Andriamanarivo, S.; Lisowski, V.; Masurier, N.; Zirah, S.;Kamg, Y. K.; Martinez, J.; Maillard, L. T. Thiazole-based γ-buildingblocks as reverse-turn mimetic to design a gramicidin S analogue:conformational and biological evaluation. Chem.Eur. J. 2014, 20,6713−6720.(64) Hynes, J., Jr.; Wu, H.; Pitt, S.; Shen, D. R.; Zhang, R.; Schieven, G.L.; Gillooly, K. M.; Shuster, D. J.; Taylor, T. L.; Yang, X.-X.; McIntyre, K.W.; McKinnon, M.; Zhang, H.; Marathe, P. H.; Doweyko, A. M.; Kish,K.; Kiefer, S. E.; Sack, J. S.; Newitt, J. A.; Barrish, J. C.; Dodd, J.; Leftheris,K. The discovery of (R)-2-(sec-butylamino)-N-(2-methyl-5-(methylcarbamoyl)phenyl)-thiazole-5-carboxamide (BMS-640994)a potent and efficacious p38α MAP kinase inhibitor. Bioorg. Med.Chem. Lett. 2008, 18, 1762−1767.(65) Wrobleski, S. T.; Lin, S.; Dhar, T. G. M.; Dyckman, A. J.; Li, T.;Pitt, S.; Zhang, R.; Fan, Y.; Doweyko, A. M.; Tokarski, J. S.; Kish, K. F.;Kiefer, S. E.; Sack, J. S.; Newitt, J. A.; Witmer, M. R.; McKinnon, M.;Barrish, J. C.; Dodd, J. H.; Schieven, G. L.; Leftheris, K. Theidentification of novel p38α isoform selective kinase inhibitors havingan unprecedented p38α binding mode. Bioorg. Med. Chem. Lett. 2013,23, 4120−4126.(66) (a) Lombardo, L. J.; Lee, F. Y.; Chen, P.; Norris, D.; Barrish, J. C.;Behnia, K.; Castaneda, S.; Cornelius, L. A. M.; Das, J.; Doweyko, A. M.;Fairchild, C.; Hunt, J. T.; Inigo, I.; Johnston, K.; Kamath, A.; Kan, D.;Klei, H.; Marathe, P.; Pang, S.; Peterson, R.; Pitt, S.; Schieven, G. L.;Schmidt, R. J.; Tokarski, J.; Wen, M.-L.; Wityak, J.; Borzilleri, R. M.Discovery of N-(2-chloro-6-methyl-phenyl)-2-(6-(4-(2-hydroxyethyl)-piperazin-1-yl)-2-methylpyrimidin-4-ylamino)thiazole-5-carboxamide(BMS-354825), a dual Src/Abl kinase inhibitor with potent antitumoractivity in preclinical assays. J. Med. Chem. 2004, 47, 6658−6661.(b) Das, J.; Chen, P.; Norris, D.; Padmanabha, R.; Lin, J.; Moquin, R. V.;Shen, Z.; Cook, L. S.; Doweyko, A. M.; Pitt, S.; Pang, S.; Ren Shen, D.;Fang, Q.; Fex, H. F.; McIntyre, K. W.; Shuster, D. J.; Gillooly, K. M.;Behnia, K.; Schieven, G. L.;Wityak, J.; Barrish, J. C. 2-Aminothiazole as anovel kinase inhibitor template. Structure−activity relationship studiestoward the discovery of N-(2-chloro-6-methylphenyl)-2-[[6-[4-(2-hydroxyethyl)-1-piperazinyl)]-2-methyl-4-pyrimidinyl]amino)]-1,3-thiazole-5-carboxamide (dasatinib, BMS-354825) as a potent pan-Srckinase inhibitor. J. Med. Chem. 2006, 49, 6819−6832. (c) Tokarski, J. S.;Newitt, J.; Chang, C. Y. J.; Cheng, J. D.; Wittekind, M.; Kiefer, S. E.;Kish, K.; Lee, F. Y. F.; Borzilerri, R.; Lombardo, L. J.; Xie, D.; Zhang, Y.;Klei, H. E. The structure of dasatinib (BMS-354825) bound to activatedABL kinase domain elucidates its inhibitory activity against imatinib-resistant ABL mutants. Cancer Res. 2006, 66, 5790−5797. (d) Getlik,M.; Grutter, C.; Simard, J. R.; Kluter, S.; Rabiller, M.; Rode, H. B.;Robubi, A.; Rauh, D. Hybrid compound design to overcome thegatekeeper T338M mutation in cSrc. J. Med. Chem. 2009, 52, 3915−3926. (e) Farenc, C. J. A.; Hameetman, L.; Zoutman, W.; Tensen, C. P.;Siegal, G. Crystal structure of the EphA4 protein tyrosine kinase domainin the apo- and dasatinib-bound state. FEBS Lett. 2011, 585, 3593−3599. (f)Williams, N. K.; Lucet, I. S.; Klinken, S. P.; Ingley, E.; Rossjohn,J. Crystal structures of the Lyn protein tyrosine kinase domain in its apo-and inhibitor bound state. J. Biol. Chem. 2009, 284, 284−291.(g) Muckelbauer, J.; Sack, J. S.; Ahmed, N.; Burke, J.; Chang, C. Y.;Gao, M.; Tino, J.; Xie, D.; Tebben, A. J. X-ray crystal structure of bonemarrow kinase in the X chromosome: a Tec family kinase. Chem. Biol.Drug Des. 2011, 78, 739−748. (h) Marcotte, D. J.; Liu, Y. T.; Arduini, R.M.; Hession, C. A.; Miatkowski, K.; Wildes, C. P.; Cullen, P. F.; Hong,V.; Hopkins, B. T.; Mertsching, E.; Jenkins, T. J.; Romanowski, M. J.;Baker, D. P.; Silvian, L. F. Structures of human Bruton’s tyrosine kinasein active and inactive conformations suggest a mechanism of activationfor TEC family kinases. Protein Sci. 2010, 19, 429−439.(67) Disch, J. S.; Evindar, G.; Chiu, C. H.; Blum, C. A.; Dai, H.; Jin, L.;Schuman, E.; Lind, K. E.; Belyanskaya, S. L.; Deng, J.; Coppo, F.;Aquilani, L.; Graybill, T. L.; Cuozzo, J. W.; Lavu, S.; Mao, C.; Vlasuk, G.
P.; Perni, R. B. Discovery of thieno[3,2-d]pyrimidine-6-carboxamides aspotent inhibitors of SIRT1, SIRT2, and SIRT3. J. Med. Chem. 2013, 56,3666−3679.(68) Brument, S.; Sivignon, A.; Dumych, T. I.; Moreau, N.; Roos, G.;Guerardel, Y.; Chalopin, T.; Deniaud, D.; Bilyy, R. O.; Darfeuille-Michaud, A.; Bouckaert, J.; Gouin, S. G. Thiazolylaminomannosides aspotent antiadhesives of type 1 piliated Escherichia coli isolated fromCrohn’s disease patients. J. Med. Chem. 2013, 56, 5395−5406.(69) (a) Laha, J. K.; Zhang, X.; Qiao, L.; Liu, M.; Chatterjee, S.;Robinson, S.; Kosik, K. S.; Cuny, G. D. Structure−activity relationshipstudy of 2,4-diaminothiazoles as Cdk5/p25 kinase inhibitors. Bioorg.Med. Chem. Lett. 2011, 21, 2098−2101. (b) Ahn, J. S.; Radhakrishnan,M. L.; Mapelli, M.; Choi, S.; Tidor, B.; Cuny, G. D.; Musacchio, A.; Yeh,L.-A.; Kosik, K. S. Defining Cdk5 ligand chemical space with smallmolecule inhibitors of tau phosphorylation. Chem. Biol. 2005, 12, 811−823.(70) Schonbrunn, E.; Betzi, S.; Alam, R.; Martin, M. P.; Becker, A.;Han, H.; Francis, R.; Chakrasali, R.; Jakkaraj, S.; Kazi, A.; Sebti, S. M.;Cubitt, C. L.; Gebhard, A. W.; Hazlehurst, L. A.; Tash, J. S.; Georg, G. I.Development of highly potent and selective diaminothiazole inhibitorsof cyclin-dependent kinases. J. Med. Chem. 2013, 56, 3768−3782.(71) Tanaka, R.; Oyama, Y.; Imajo, S.; Matsuki, S.; Ishiguro, M.Structure−activity relationships of penem antibiotics: crystallographicstructures and implications for their antimicrobial activities. Bioorg. Med.Chem. 1997, 5, 1389−1399.(72) Rochel, N.; Moras, D. Crystal structure of a vitamin D3 analogue,ZK203278, showing dissociated profile. Anticancer Res. 2012, 32, 335−340.(73) Huang, Q.; Johnson, T. W.; Bailey, S.; Brooun, A.; Bunker, K. D.;Burke, B. J.; Collins, M. R.; Cook, A. S.; J. Cui, J.; Dack, K. N.; Deal, J. G.;Deng, Y.-L.; Dinh, D.; Engstrom, L. D.; He, M.; Hoffman, J.; Hoffman,R. L.; Johnson, P. S.; Kania, R. S.; Lam, H.; Lam, J. L.; Le, P. T.; Li, Q.;Lingardo, L.; Liu, W.; West Lu, M.; McTigue, M.; Palmer, C. L.;Richardson, P. F.; Sach, N. W.; Shen, H.; Smeal, T.; Smith, G. L.;Stewart, A. E.; Timofeevski, S.; Tsaparikos, K.; Wang, H.; Zhu, H.; Zhu,J.; Zou, H. Y.; Edwards, M. P. Design of potent and selective inhibitors toovercome clinical anaplastic lymphoma kinase mutations resistant tocrizotinib. J. Med. Chem. 2014, 57, 1170−1187.(74) (a) Rodríguez, S.; Kneeteman, M.; Izquierdo, J.; Lopez, I.;Gonzalez, F. V.; Peris, G. Diastereoselective synthesis of γ-hydroxy α,β-epoxyesters and their conversion into β-hydroxy α-sulfenyl γ-butyrolactones. Tetrahedron 2006, 62, 11112−11123. (b) Gonzalez, F.V.; Jain, A.; Rodríguez, S.; Saez, J. A.; Vicent, C.; Peris, G.Stereoisomerization of β-hydroxy-α-sulfenyl-γ-butyrolactones con-trolled by two concomitant 1,4-type nonbonded sulfur−oxygeninteractions as analyzed by X-ray crystallography. J. Org. Chem. 2010,75, 5888−5894.(75) Silverman, R. B. Mechanism of isomerization of a β-keto sulfide. J.Org. Chem. 1981, 46, 4789−4791.(76) Dubey, R.; Polaske, N. W.; Nichol, G. S.; Olenyuk, B. Efficientorganocatalytic α-sulfenylation of substituted piperazine-2,5-diones.Tetrahedron Lett. 2009, 50, 4310−4313.(77) (a) Wang, X.; Yang, T.; Cheng, X.; Shen, Q. Enantioselectiveelectrophilic trifluoromethylthiolation of β-ketoesters: a case ofreactivity and selectivity bias for organocatalysis. Angew. Chem., Int.Ed. 2013, 52, 12860−12864. (b) Bootwicha, T.; Liu, X.; Pluta, R.;Atodiresei, I.; Rueping, M. N-Trifluoromethylthiophthalimide: a stableelectrophilic SCF3 reagent and its application in the catalytic asymmetrictrifluoromethylsulfenylation. Angew. Chem., Int. Ed. 2013, 52, 12856−12859.(78) (a) Hiram, T.; Nomiyama, J.; Sakae, N.; Nishimura, K.;Yokomoto, M.; Inoue, S.; Tamura, K.; Okuhira, M.; Amano, H.;Nagao, Y. Acyliminothiadiazoline derivatives: new, highly potent, andorally active angiotensin II receptor antagonists. Bioorg. Med. Chem. Lett.1996, 6, 1469−1474. (b) Hirata, T.; Goto, S.; Tamura, K.; Okuhira, M.;Nagao, Y. Quantitative structure−activity relationships of benzoylimi-nothiadiazoline derivatives as angiotensin II receptor antagonists. Bioorg.Med. Chem. Lett. 1997, 7, 385−388. (c) Nagao, Y.; Hirata, T.; Goto, S.;Sano, S.; Kakehi, A.; Iizuka, K.; Shiro, M. Intramolecular nonbonded S···
Journal of Medicinal Chemistry Perspective
DOI: 10.1021/jm501853mJ. Med. Chem. XXXX, XXX, XXX−XXX
AX

O interaction recognized in (acylimino)thiadiazoline derivatives asangiotensin II receptor antagonists and related compounds. J. Am. Chem.Soc. 1998, 120, 3104−3110.(79) (a) Meyer, E.; Joussef, A. C.; Gallardo, H.; Bortoluzzi, A. J.;Longo, R. L. 1,5-Type nonbonded O···S and S···S interactions in(acylimino) and (thioacylimino)benzothiazoline systems. Crystalstructures and theoretical calculations. Tetrahedron 2003, 59, 10187−10193. (b) Nagao, Y.; Iimori, H.; Goto, S.; Hirata, T.; Sano, S.; Chuman,H.; Shiro, M. Remarkable discrepancy in the predominant structures ofacyl (or thioacyl)iminothiadiazoles, acyl (or thioacyl)aminooxadiazolesand related compounds having the potential for rotational, geometricaland tautomeric isomerism. Tetrahedron Lett. 2002, 43, 1709−1712.(80) Manaka, A.; Sato, M.; Aoki, M.; Tanaka, M.; Ikeda, T.; Toda, Y.;Yamane, Y.; Nakaike, S. 2-Acylimino-3H-thiazoline derivatives: a noveltemplate for platelet gpIIb/IIIa receptor antagonists. Bioorg. Med. Chem.Lett. 2001, 11, 1031−1035.(81) (a) Jeschke, P.; Nauen, R.; Beck, M. E. Nicotinic acetylcholinereceptor agonists: a milestone for modern crop protection. Angew.Chem., Int. Ed. 2013, 52, 9464−9485. (b) Tomizawa, M.; Zhang, N.;Durkin, K. A.; Olmstead, M. M.; Casida, J. E. The neonicotinoidelectronegative pharmacophore plays the crucial role in the high affinityand selectivity for the Drosophila nicotinic receptor: an anomaly for thenicotinoid cation−π interaction model. Biochemistry 2003, 42, 7819−7827. (c) Tomizawa, M.; Kagabu, S.; Ohno, I.; Durkin, K. A.; Casida, J.E. Potency and selectivity of trifluoroacetylimino and pyrazinoyliminonicotinic insecticides and their fit at a unique binding site niche. J. Med.Chem. 2008, 51, 4213−4218. (d) Ohno, I.; Tomizawa, M.; Aoshima, A.;Kumuzawa, S.; Kagabu, S. Trifluoroacetyl neonicotinoid insecticideswith enhanced hydrophobicity and effectiveness. J. Agric. Food Chem.2010, 58, 4999−5003. (e) Tomizawa, M.; Kagabu, S.; Casida, J. E.Receptor structure-guided neonicotinoid design. J. Agric. Food Chem.2011, 59, 2918−2922. (f) Tomizawa, M.; Durkin, K. A.; Ohno, I.;Nagura, K.; Manabe, M.; Kumuzawa, S.; Kagabu, S. N-Haloacetyliminoneonicotinoids: potency and molecular recognition at the insectnicotinic receptor. Bioorg. Med. Chem. Lett. 2011, 21, 3583−3586.(82) Nagao, Y.; Iimori, H.; Nam, K. H.; Sano, S.; Shiro, M. Synthesisand antibacterial activity of new 1β-methylcarbapenems having thepotential for an intramolecular nonbonded S···O interactions. Chem.Pharm. Bull. 2001, 49, 1660−1661.(83) (a) Park, H.; Hong, S.; Hong, S. Nocodazole is a high-affinityligand for the cancer-related kinases ABL, cKIT, BRAF, and MEK.ChemMedChem 2012, 7, 53−56. (b) Park, H.; Hong, S.; Kim, J.; Hong,S. Discovery of picomolar ABL kinase inhibitors equipotent for wild typeand T315I mutant via structure-based de novo design. J. Am. Chem. Soc.2013, 135, 8227−8237. (c) Hong, S.; Kim, J.; Yun, S.-M.; Lee, H.; Park,Y.; Hong, S.-S.; Hong, S. Discovery of new benzothiazole-basedinhibitors of breakpoint cluster region-Abelson kinase including theT315I mutant. J. Med. Chem. 2013, 56, 3531−3545.(84) Martins, F. T.; Neves, P. P.; Ellena, J.; Cami, G. E.; Brusau, E. V.;Narda, G. E. Intermolecular contacts influencing the conformational andgeometric features of the pharmaceutically preferred mebendazolepolymorph C. J. Pharm. Sci. 2009, 98, 2336−2344.(85) Okaniwa, M.; Hirose, M.; Imada, T.; Ohaashi, T.; Hayashai, Y.;Miyazaki, T.; Arita, T.; Yabuki, M.; Kakoi, K.; Kato, J.; Takagi, T.;Kawamoto, T.; Yao, S.; Sumita, A.; Tsutsumi, S.; Tottori, T.; Oki, H.;Sang, B.-C.; Yano, J.; Aertgeerts, K.; Yoshida, S.; Ishikawa, T. Design andsynthesis of novel DFG-out RAF/vascular endothelial growth factorreceptor 2 inhibitors. 1. Exploration of [5,6]-fused bicyclic scaffolds. J.Med. Chem. 2012, 55, 3452−3478.(86) Zhao, L.; Zhang, Y.; Dai, C.; Guzi, T.; Wiswell, D.; Seghezzi, W.;Parry, D.; Fischmann, T.; Siddiqui, M. A. Design, synthesis and SAR ofthienopyridines as potent CHK1 inhibitors. Bioorg. Med. Chem. Lett.2010, 20, 7216−7221.(87) Murphy-Benenato, K.; Wang, H.; McGuire, H. M.; Davis, H. E.;Gao, N.; Prince, D. B.; Jahic, H.; Stokes, S. S.; Boriack-Sjodin, P. A.Identification through structure-based methods of a baceterial NAD+-dependent DNA ligase inhibitor that avoids known resistancemutations. Bioorg. Med. Chem. Lett. 2014, 24, 360−366.
(88) Collins, I.; Moyes, C.; Davey, W. B.; Rowley, M.; Bromidge, F. A.;Quirk, K.; Atack, J. R.; McKernan, R. M.; Thompson, S.-A.; Wafford, K.;Dawson, G. R.; Pike, A.; Sohal, B.; Tsou, N. N.; Ball, R. G.; Castro, J. L. 3-Heteroaryl-2-pyridones: benzodiazepine site ligands with functionalselectivity for α2/α3-subtypes of human GABAA receptor-ion channels.J. Med. Chem. 2002, 45, 1887−1900.(89) Corazon Felix-Sonda, B.; Rivera-Islas, J.; Herrera-Ruiz, D.;Morales-Rojas, H.; Hopfl, H. Nitazoxanide cocrystals in combinationwith succinic, glutaric and 2,5-dihydroxybenzoic acid. Cryst. Growth Des.2014, 14, 1086−1102.(90) Grewal, A. S.; Sekhon, B. S.; Lather, V. Recent updates onglucokinase activators for the treatment of type 2 diabetes mellitus.Mini-Rev. Med. Chem. 2014, 14, 585−602.(91) Haynes, N.-E.; Corbett, W. L.; Bizzarro, F. T.; Guertine, K. R.;Hilliard, D. W.; Holland, G. W.; Kester, R. F.; Mahaney, P. E.; Qi, L.;Spence, C. L.; Tengi, J.; Dvorozniak, M. T.; Railkar, A.; Matschinsky, F.M.; Grippo, J. F.; Grimsby, J.; Sarabu, R. Discovery, structure−activityrelationships, pharmacokinetics, and efficacy of glucokinase activator(2R)-3-cyclopentyl-2-(4-methanesulfonylphenyl)-N-thiazole-2-yl-pro-pionamide (RO0281675). J. Med. Chem. 2010, 53, 3618−3625.(92) Cheruvallath, Z. S.; Gwaltney, S. L., II; Sabat, M.; Tang, M.; Feng,J.; Wang, H.; Miura, J.; Guntupalli, P.; Jennings, A.; Hosfield, D.; Lee, B.;Wu, Y. Design, synthesis and SAR of novel glucokinase activators.Bioorg. Med. Chem. Lett. 2013, 23, 2166−2171.(93) (a) Nishimura, T.; Iino, T.; Mitsuya, M.; Bamba, M.; Watanabe,H.; Tsukahara, D.; Kamata, K.; Sasaki, K.; Ohyama, S.; Hosaka, H.;Futamura, M.; Nagata, Y.; Eiki, J.-i. Identification of novel and potent 2-amino benzamide derivatives as allosteric glucokinase activators. Bioorg.Med. Chem. Lett. 2009, 19, 1357−1360. (b) Mitsuya, M.; Kamata, K.;Bamba, M.; Watanabe, H.; Sasaki, Y.; Sasaki, K.; Ohyama, S.; Hosaka,H.; Nagata, Y.; Eiki, J.-i.; Nishimura, T. Discovery of novel 3,6-disubstituted 2-pyridinecarboxamide derivatives as GK activators.Bioorg. Med. Chem. Lett. 2009, 19, 2718−2712.(94) (a) Bode, C.M.; Boezio, A. A.; Albrecht, B. K.; Bellon, S. F.; Berry,L.; Broome, M. A.; Choquette, D.; Dussault, I.; Lewis, R. T.; Lin, M.-H.J.; Rex, K.; Whittington, D. A.; Yang, Y.; Harmange, J.-C. Discovery andoptimization of a potent and selective triazolopyridinone series of c-Metinhibitors. Bioorg. Med. Chem. Lett. 2012, 22, 4089−4093. (b) Albrecht,B. K.; Harmange, J.-C.; Bauer, D.; Berry, L.; Bode, C.; Boezio, A. A.;Chen, A.; Choquette, D.; Dussault, I.; Fridrich, C.; Hirai, S.; Hoffman,D.; Larrow, J. F.; Kaplan-Lefko, P.; Lin, J.; Lohman, J.; Long, A. M.;Moriguchi, J.; O’Connor, A.; Potashman, M. H.; Reese, M.; Rex, K.;Siegmund, A.; Shah, K.; Shimanovich, R.; Springer, S. K.; Teffera, Y.;Yang, Y.; Zhang, Y.; Bellon, S. F. Discovery and optimization oftriazolopyridazines as potent and selective inhibitors of the c-Met kinase.J. Med. Chem. 2008, 51, 2879−2882. (c) Boezio, A. A.; Berry, L.;Albrecht, B. K.; Bauer, D.; Bellon, S. F.; Bode, C.; Chen, A.; Choquette,D.; Dussault, I.; Hirai, S.; Kaplan-Lefko, P.; Larrow, J. F.; Lin, M.-H. J.;Lohman, J.; Potashman, M. H.; Rex, K.; Santostefano, M.; Shah, K.;Shimanovich, R.; Springer, S. K.; Teffera, Y.; Yang, Y.; Zhang, Y.;Harmange, J. C. Discovery and optimization of potent and selectivetriazolopyridazine series of c-Met inhibitors. Bioorg. Med. Chem. Lett.2009, 19, 6307−6312.(95) Flygare, J. A.; Beresini, M.; Budha, N.; Chan, H.; Chan, I. T.;Cheeti, S.; Cohen, F.; Deshayes, K.; Doerner, K.; Eckhardt, S. G.; Elliott,L. O.; Feng, B.; Franklin, M. C.; Frankovitz Reisner, S.; Gazzard, L.;Halladay, J.; Hymowitz, S. G.; La, H.; LoRusso, P.; Maurer, B.; Murray,L.; Plise, E.; Quan, C.; Stephan, J.-P.; Young, S. G.; Tom, J.; Tsui, V.;Um, J.; Varfolomeev, E.; Vucic, D.; Wagner, A. J.; Wallweber, H. J. A.;Wang, L.; Ware, J.; Wen, Z.; Wong, H.; Wong, J. M.; Wong, M.; Wong,S.; Yu, R.; Zobel, K.; Fairbrother, W. J. Discovery of a potent small-molecule antagonist of inhibitor of apoptosis (IAP) proteins and clinicalcandidate for the treatment of cancer (GDC-0152). J. Med. Chem. 2012,55, 4101−4113.(96) (a) Bartlett, G. J.; Choudhary, A.; Raines, R. T.; Woolfson, D. N.n→π* Interactions in proteins. Nature Chem. Biol. 2010, 6, 615−620.(b) Newberry, R. W.; Van Veller, B.; Guzei, I. A.; Raines, R. T. n→π*Interactions of amides and thioamides: implications for protein stability.J. Am. Chem. Soc. 2013, 135, 7843−7846. (c) Hodges, J. A.; Raines, R. T.
Journal of Medicinal Chemistry Perspective
DOI: 10.1021/jm501853mJ. Med. Chem. XXXX, XXX, XXX−XXX
AY

Energetics of an n→π* interaction that impacts protein structure. Org.Lett. 2006, 8, 4695−4697. (d) Choudhoury, A.; Gandla, D.; Krow, G. R.;Raines, R. T. Nature of amide carbonyl−carbonyl interactions inproteins. J. Am. Chem. Soc. 2009, 131, 7244−7246.(97) Nagao, Y.; Honjo, T.; Iimori, H.; Goto, S.; Sano, S.; Shiro, M.;Yamaguchi, K.; Sei, Y. Intramolecular nonbonded S···O interaction inacetazolamide and thiadiazolinethione molecules in their dimericcrystalline structures and complex crystalline structures with enzymes.Tetrahedron Lett. 2004, 45, 8757−8761.(98) (a) Honda, T.; Tajima, H.; Kaneko, Y.; Ban, M.; Inaba, T.;Takeno, Y.; Okamoto, K.; Aono, H. Conformation−activity relationshipon novel 4-pyridylmethylthio derivatives with antiangiogenic activity.Bioorg. Med. Chem. Lett. 2008, 18, 2939−2943. (b) Tajima, H.; Honda,T.; Kawashima, K.; Sasabuchi, Y.; Yamamoto, M.; Ban, M.; Okamoto,K.; Inoue, K.; Inaba, T.; Takeno, Y.; Aono, H. Pyridylmethylthioderivatives as VEGF inhibitors. Part 1. Bioorg. Med. Chem. Lett. 2010, 20,7234−7238. (c) Tajima, H.; Honda, T.; Kawashima, K.; Sasabuchi, Y.;Yamamoto, M.; Ban, M.; Okamoto, K.; Inoue, K.; Inaba, T.; Takeno, Y.;Tsuboi, T.; Tonouchi, A.; Aono, H. Pyridylmethylthio derivatives asVEGF inhibitors. Part 2. Bioorg. Med. Chem. Lett. 2011, 21, 1232−1235.(99) (a) Suzuki, T.; Imai, K.; Nakagawa, H.; Miyata, N. 2-Anilinobenzamides as SIRT inhibitors. ChemMedChem 2006, 1,1059−1062. (b) Suzuki, T.; Imai, K.; Imai, E.; Iida, S.; Ueda, R.;Tsumoto, H.; Nakagawa, H.; Miyata, N. Design, synthesis, enzymeinhibition, and tumor cell growth inhibition of 2-anilinobenzamidederivatives as SIRT1 inhibitors. Bioorg. Med. Chem. 2009, 17, 5900−5905.(100) Tomoo, T.; Nakatsuka, T.; Katayama, T.; Hayashi, Y.; Fujieda,Y.; Terakawa, M.; Nagahira, K. Design, synthesis and biologicalevaluation of 2-(1-aryl-1H-indol-5-yl)propanoic acids as new indole-based cytosolic phospholipase A2α inhibitors. J. Med. Chem. 2014, 57,7244−7262.(101) (a) Hornberger, K. R.; Chen, X.; Crew, A. P.; Kleinberg, A.; Ma,L.; Mulvihill, M. J.; Wang, J.; Wilde, V. L.; Albertella, M.; Bittner, M.;Cooke, A.; Kadhim, S.; Kahler, J.; Maresca, P.; May, E.; Meyn, P.;Romashko, D.; Tokar, B.; Turton, R. Discovery of 7-aminofuro[2,3-c]pyridine inhibitors of TAK1: optimization of kinase selectivity andpharmacokinetics. Bioorg. Med. Chem. Lett. 2013, 23, 4511−4516.(b) Hornberger, K. R.; Berger, D. M.; Crew, A. P.; Dong, H.; Kleinberg,A.; Li, A.-H.; Medeiros, M. R.; Mulvihill, M. J.; Siu, K.; Tarrant, J.; Wang,J.; Weng, F.; Wilde, V. L.; Albertella, M.; Bittner, M.; Cooke, A.; Gray,M. J.; Maresca, P.; May, E.; Meyn, P.; Peick, W., Jr.; Romashko, D.;Tanowitz, M.; Tokar, B. Discovery and optimization of 7-aminofuro-[2,3-c]pyridine inhibitors of TAK1. Bioorg. Med. Chem. Lett. 2013, 23,4517−4522.(102) Honda, T.; Terao, T.; Aono, H.; Ban, M. Synthesis of novel 1,4-benzoxazin-3-one derivatives as inhibitors against tyrosine kinases.Bioorg. Med. Chem. 2009, 17, 699−708.(103) (a) Birman, V. B.; Li, X.; Han, Z. Nonaromatic amidinederivatives as acylation catalysts. Org. Lett. 2007, 9, 37−40.(b) Kobayashi, M.; Okamoto, S. Unexpected reactivity of annulated3H-benzothiazol-2-ylideneamines as an acyl transfer catalyst. Tetrahe-dron Lett. 2006, 47, 4347−4350. (c) Birman, V. B.; Li, X.Benzotetramisole: a remarkably enantioselective acyl transfer catalyst.Org. Lett. 2006, 8, 1351−1354. (d) Birman, V. B.; Jiang, H.; Li, X.; Guo,L.; Uffman, E. W. Kinetic resolution of 2-oxazoldinones via catalytic,enantioselective N-acylation. J. Am. Chem. Soc. 2006, 128, 6536−6537.(e) Birman, V. B.; Guo, L. Kinetic resolution of propargylic alcoholscatalyzed by benzotetramisole. Org. Lett. 2006, 8, 4859−4861.(f) Birman, V. B.; Jiang, H.; Li, X. Enantioselective synthesis of lobelinevia nonenzymatic desymmetrization. Org. Lett. 2007, 9, 3237−3240.(g) Birman, V. B.; Li, X. Homobenzotetramisole: an effective catalyst forkinetic resolution of aryl-cycloalkanols. Org. Lett. 2008, 10, 1115−1118.(h) Yang, X.; Lu, G.; Birman, V. B. Benzotetramisole-catalyzed dynamickinetic resolution of azlactones.Org. Lett. 2010, 12, 892−895. (i) Shiina,I.; Nakata, K.; Ono, K.; Onda, Y.; Itagaki, M. Kinetic resolution ofracemic α-arylalkanoic acids with achiral alcohols via the asymmetricesterification using carboxylic anhydrides and acyl-transfer catalysts. J.Am. Chem. Soc. 2010, 132, 11629−11641. (j) Leverett, C. A.; Purohit, V.
C.; Romo, D. Enantioselective, organocatalyzed, intramolecular aldollactonizations with keto acids leading to bi- and tricyclic β-lactones andtopology-morphing transformations. Angew. Chem., Int. Ed. 2010, 49,9479−9483. (k) Belmessieri, D.; Morrill, L. C.; Simal, C.; Slawin, A. M.Z.; Smith, A. D. Organocatalytic functionalization of carboxylic acids:isothiourea-catalyzed asymmetric intra- and intermolecular Michaeladdition−lactonizations. J. Am. Chem. Soc. 2011, 133, 2714−2720.(l) Morrill, L. C.; Stark, D. G.; Taylor, J. E.; Smith, S. R.; Squires, J. A.;D’Hollander, A. C. A.; Simal, C.; Shapland, P.; O’Riordan, T. J. C.;Smith, A. D. Organocatalytic Michael addition−lactonisation ofcarboxylic acids using α,β-unsaturated trichloromethyl ketones as α,β-unsaturated ester equivalents.Org. Biomol. Chem. 2014, 12, 9016−9027.(104) (a) Salimbeni, A.; Canevotti, R.; Paleari, F.; Poma, D.; Caliari, S.;Fici, F.; Cirillo, R.; Renzetti, A. R.; Subissi, A.; Belvisi, L.; Bravi, G.;Scolastico, C.; Giachetti, A. N-3-Substituted pyrimidinones as potent,orally active, AT1 selective angiotensin II receptor antagonists. J. Med.Chem. 1995, 38, 4806−4820. (b) Destro, R.; Soave, R. A new non-peptide angiotensin II receptor antagonist. Acta Crystallogr., Sect. C:Cryst. Struct. Commun. 1995, 51, 1383−1385. (c) Destro, R.; Soave, R.;Barzaghi, M.; Lo Presti, L. Progress in understanding of drug−receptorinteractions, Part 1: experimental charge-density study of an angiotensinII receptor antagonist (C30H30N6O3S) atT = 17 K.Chem.Eur. J. 2005,11, 4621−4634. (d) Soave, R.; Barzaghi, M.; Destro, R. Progress inunderstanding of drug−receptor interactions, part 2: experimental andtheoretical electrostatic moments and interaction energies of anangiotensin II receptor antagonist (C30H30N6O3S). Chem.Eur. J.2007, 13, 6942−6956.(105) Sugiyama, H.; Yoshida, M.; Mori, K.; Kawamoto, T.; Sogabe, S.;Takagi, T.; Oki, H.; Tanaka, T.; Kimura, H.; Ikeura, Y. Synthesis andstructure−activity relationship studies of benzothieno[3,2-b]furanderivatives as a novel class of IKKβ inhibitors. Chem. Pharm. Bull.2007, 55, 613−624.(106) McInerney, B. V.; Gregson, R. P.; Lacey, M. J.; Akhurst, R. J.;Lyons, G. R.; Rhodes, S. H.; Smith, D. R. J.; Engelhardt, L. M.; White, A.H. Biologically active metabolites from Xenorhabdus spp. Part 1.Dithiolopyrrolone derivatives with antibiotic activity. J. Nat. Products1991, 54, 774−784.(107) Burling, F. T.; Goldstein, B. M. A database study of nonbondedintramolecular sulfur−nucleophile contacts. Acta Crystallogr., Part B:Struct. Sci. 1993, B49, 738−744.(108) (a) Huber, C. P.; Carey, P. R.; Hsi, S.-C.; Lee, H.; Storer, A. C.Conformational study ofN-acyl amino acid esters and thiol esters by FT-IR and X-ray crystallography: evidence for a N···S interaction in thiolesters. J. Am. Chem. Soc. 1984, 106, 8263−8268. (b) Varughese, K. I.;Storer, A. C.; Carey, P. R. Directional preference for a catalyticallyimportant N−S contact seen in acyl-thiolproteases. J. Am. Chem. Soc.1984, 106, 8252−8257.(109) (a) Carey, P. R.; Lee, H.; Ozaki, Y.; Storer, A. C. Rate−structurecorrelation for enzyme−substrate intermediates: resonance Raman andkinetic studies on some N-benzoylglycine (dithioacyl)papains. J. Am.Chem. Soc. 1984, 106, 8258−8262. (b) Carey, P. R.; Storer, A. C.Molecular details of enzyme-substrate transients by resonance Ramanspectroscopy. Acc. Chem. Res. 1983, 16, 455−460.(110) Hayashi, K.; Ogawa, S.; Sano, S.; Shiro, M.; Yamaguchi, K.; Sei,Y.; Nagao, Y. Intramolecular nonbonded S···N interaction inrabeprazole. Chem. Pharm. Bull. 2008, 56, 802−806.(111) (a) Kuti, M.; Ratbai, J.; Kapovits, I.; Kucsman, A.; Parkanyi, L.;Argay, Gy.; Kalman, A. Transannular sulfur−nitrogen interaction in 1,5-thiazonine derivatives: an X-ray study. J. Mol. Struct. 1994, 318, 161−169. (b) Kuti, M.; Ratbai, J.; Kapovits, I.; Jalsovszky, I.; Argay, Gy.;Kalman, A.; Parkanyi, L. Transannular sulfur−nitrogen interaction in1,5-thiazocine derivatives: an X-ray study. J. Mol. Struct. 1996, 382, 1−11.(112) Fukumoto, S.; Nakashima, T.; Kawai, T. Photon-quantitativereaction of a dithiazolylarylene in solution. Angew. Chem., Int. Ed. 2011,50, 1565−1568.(113) Rheault, T. R.; Stellwagen, J. C.; Adjabeng, G. M.; Hornberger,K. R.; Petrov, K. G.; Waterson, A. G.; Dickerson, S. H.; Mook, R. A., Jr.;Laquerre, S. G.; King, A. J.; Rossanese, O. W.; Arnone, M. R.;
Journal of Medicinal Chemistry Perspective
DOI: 10.1021/jm501853mJ. Med. Chem. XXXX, XXX, XXX−XXX
AZ

Smitheman, K. N.; Kane-Carson, L. S.; Han, C.; Moorthy, G. S.; Moss,K. G.; Uehling, D. E. Discovery of dabrafenib: a selective inhibitor of Rafkinases with antitumor activity against B-Raf-driven tumors. ACS Med.Chem. Lett. 2013, 4, 358−362.(114) Haack, T.; Fattori, R.; Napoletano, M.; Pellacini, F.; Fronza, G.;Raffainic, G.; Ganazzolic, F. Phthalazine PDE IV inhibitors: conforma-tional study of some 6-methoxy-1,4-disubstituted derivatives. Bioorg.Med. Chem. 2005, 13, 4425−4433.(115) Cummings, M. D.; Lindberg, J.; Lin, T.-I.; de Kock, H.; Lenz, O.;Lilja, E.; Fellander, S.; Baraznenok, V.; Nystrom, S.; Nilsson, M.; Vrang,L.; Edlund, M.; Rosenquist, Å.; Samuelsson, B.; Raboisson, P.; Simmen,K. Induced-fit binding of the macrocyclic noncovalent inhibitorTMS435 to its HCV NS3/NS4A protease target. Angew. Chem., Int.Ed. 2010, 49, 1652−1655.(116) Kazmierski, W. M.; Hamatake, R.; Duan, M.; Wright, L. L.;Smith, G. K.; Jarvest, R. L.; Ji, J.-J.; Cooper, J. P.; Tallant, M. D.; Crosby,R. M.; Creech, K.; Wang, A.; Li, X.; Zhang, S.; Zhang, Y.-K.; Liu, Y.;Ding, C. Z.; Zhou, Y.; Plattner, J. J.; Baker, S. J.; Bu,W.; Liu, L. Discoveryof novel urea-based hepatitis C protease inhibitors with high potencyagainst protease-inhibitor-resistant mutants. J. Med. Chem. 2012, 55,3021−3026.(117) Maloney, K. N.; MacMillan, J. B.; Kauffman, C. A.; Jensen, P. R.;DiPasquale, A. G.; Rheingold, A. L.; Fenical, W. Lodopyridone, astructurally unprecedented alkaloid from a marine actinomycete. Org.Lett. 2009, 11, 5422−5424.(118) George, I. R.; Lewis, W.; Moody, C. J. Synthesis of lodopyridone.Tetrahedron 2013, 69, 8209−8215.(119) da Silva, L. E.; Joussef, A. C.; Forob, S.; Schmidt, B. 2-(1,3-Benzothiazol-2-yl)quinolin-8-ol. Acta Crystallogr., Sect. E: Struct. Rep.Online 2006, E62, o880−o881.(120) Arnison, P. G.; Bibb, M. J.; Bierbaum, G.; Bowers, A. A.; Bugni,T. S.; Bulaj, G.; Camarero, J. A.; Campopiano, D.; Challis, J. G. L.;Clardy, J.; Cotter, P. D.; Craik, D. J.; Dawson, M.; Dittmann, E.;Donadio, S.; Dorrestein, P. C.; Entian, K.-D.; Fischbach, M. A.;Garavelli, J. S.; Goransson, U.; Gruber, C. W.; Haft, D. H.; Hemscheidt,T. K.; Hertweck, C.; Hill, C.; Horswill, A. R.; Jaspars, M.; Kelly, W. L.;Klinman, J. P.; Kuipers, O. P.; Link, A. J.; Liu, W.; Marahiel, M. A.;Mitchell, D. A.; Moll, G. N.; Moore, B. S.; Muller, R.; Nair, S. K.; Nes, I.F.; Norris, G. E.; Olivera, B. M.; Onaka, H.; Patchett, M. L.; Piel, J.;Reaney, M. J. T.; Rebuffat, S.; Ross, R. P.; Sahl, H.-G.; Schmidt, E. W.;Selsted, M. E.; Severinov, K.; Shen, B.; Sivonen, K.; Smith, L.; Stein, T.;Sussmuth, R. D.; Tagg, J. R.; Tang, G.-L.; Truman, A. W.; Vederas, J. C.;Walsh, C. T.; Walton, J. D.; Wenzel, S. C.; Willey, J. M.; van der Donk,W. A. Ribosomally synthesized and post-translationally modifiedpeptide natural products: overview and recommendations for a universalnomenclature. Nat. Prod. Rep. 2013, 30, 108−160.(121) James, M. N. G.; Watson, K. J. Chemistry of micrococcin P. PartIX. The crystal and molecular structure of micrococcinic acid bis-4-bromoanilide. J. Chem. Soc. C 1966, 1361−1371.(122) Iwaki, Y.; Yamamura, S.; Akita, H. Asymmetric syntheses andstructure elucidation of cystothiazole A metabolites of the myxobacte-rium Cystobacter fuscus. Tetrahedron: Asymmetry 2008, 19, 2192−2200.(123) (a) Naumov, P.; Ozawa, Y.; Ohkubo, K.; Fukuzumi, S. Structureand spectroscopy of oxyluciferin, the light emitter of the fireflybioluminescence. J. Am. Chem. Soc. 2009, 131, 11590−11605.(b) Maltsev, O. V.; Nath, N. K.; Naumov, P.; Hintermann, L. Why isfirefly oxyluciferin a notoriously labile substance. Angew. Chem., Int. Ed.2014, 53, 847−850.(124) (a) Yoo, C. L.; Yu, G. J.; Yang, B.; Robins, L. I.; Verkmanb, A. S.;Kurth, M. J. 4′-Methyl-4,5′-bithiazole-based correctors of defectiveΔF508-CFTR cellular processing. Bioorg. Med. Chem. Lett. 2008, 18,2610−2614. (b) Coffman, K. C.; Nguyen, H. H.; Phuan, P.-W.; Hudson,B. M.; Yu, G.; Bagdazarian, A. L.; Montgomery, D.; Lodewyk, M. W.;Yang, B.; Yoo, C. L.; Verkman, A. S.; Tantillo, D. J.; Kurth, M. J.Constrained bithiazoles: small molecule correctors of defective ΔF508-CFTR protein trafficking. J. Med. Chem. 2014, 57, 6729−6738. (c) Ye,L.; Hu, B.; El-Badri, F.; Hudson, B. M.; Phuan, P.-W.; Verkman, A. S.;Tantillo, D. J.; Kurth, M. J. ΔF508-CFTR correctors: synthesis and
evaluation of thiazole-tethered imidazolones, oxazoles, oxadiazoles, andthiadiazoles. Bioorg. Med. Chem. Lett. 2014, 24, 5840−5844.(125) Vogt, D.; Weber, J.; Ihlefeld, K.; Bruggerhoff, A.; Proschak, E.;Stark, H. Design, synthesis and evaluation of 2-aminothiazole derivativesas sphingosine kinase inhibitors. Bioorg. Med. Chem. 2014, 22, 5354−5367.(126) (a) Oka, Y.; Yabuuchi, T.; Fujii, Y.; Ohtake, H.; Wakahara, S.;Matsumoto, K.; Endo, M.; Tamura, Y.; Sekiguchi, Y. Discovery andoptimization of a series of 2-aminothiazole-oxazoles as potentphosphoinositide 3-kinase γ inhibitors. Bioorg. Med. Chem. Lett. 2012,22, 7534−7538. (b) Oka, Y.; Yabuuchi, T.; Oi, T.; Kuroda, S.; Fujii, Y.;Ohtake, H.; Inoue, T.; Wakahara, S.; Kimura, K.; Fujita, K.; Endo, M.;Taguchi, K.; Sekiguchi, Y. Discovery of N-{5-[3-(3-hydroxypiperidin-1-yl)-1,2,4-oxadiazol-5-yl]-4-methyl-1,3-thiazol-2-yl}acetamide(TASP0415914) as an orally potent phosphoinositide 3-kinase γinhibitor for the treatment of inflammatory diseases. Bioorg. Med. Chem.2013, 21, 7578−7583.(127) (a) Flipo, M.; Desroses, M.; Lecat-Guillet, N.; Dirie, B.; Carette,X.; Leroux, F.; Piveteau, C.; Demirkaya, F.; Lens, Z.; Rucktooa, P.;Villeret, V.; Christophe, T.; Jeon, H. K.; Locht, C.; Brodin, P.; Deprez,B.; Baulard, A. R.; Willand, N. Ethionamide boosters: synthesis,biological activity, and structure−activity relationships of a series of1,2,4-oxadiazole EthR inhibitors. J. Med. Chem. 2011, 54, 2994−3010.(b) Flipo, M.; Desroses, M.; Lecat-Guillet, N.; Villemagne, B.;Blondiaux, N.; Leroux, F.; Piveteau, C.; Mathys, V.; Flament, M.-P.;Siepmann, J.; Villeret, V.; Wohlkonig, A.; Wintjens, R.; Soror, S. H.;Christophe, T.; Jeon, H. K.; Locht, C.; Brodin, P.; Deprez, B.; Baulard, A.R.; Willand, N. Ethionamide boosters. 2. Combining bioisostericreplacement and structure-based drug design to solve pharmacokineticissues in a series of potent 1,2,4-oxadiazole EthR inhibitors. J. Med.Chem. 2012, 55, 68−83.(128) Lee, S. W.; Mitchell, D. A.; Markley, A. L.; Hensler, M. E.;Gonzalez, D.; Wohlrab, A.; Dorrestein, P. C.; Nizet, V.; Dixon, J. E.Discovery of a widely distributed toxin biosynthetic gene cluster. Proc.Natl. Acad. Sci. U. S. A. 2008, 105, 5879−5884.(129) (a) Kalyon, B.; Helaly, S. E.; Scholz, R.; Nachtigall, J.; Vater, J.;Borriss, R.; Sussmuth, R. D. Plantazolicin A and B: Structure elucidationof ribosomally synthesized thiazole/oxazole peptides from Bacillusamyloliquefaciens FZB42.Org. Lett. 2011, 13, 2996−2999. (b) Molohon,K. J.; Melby, J. O.; Lee, J.; Evans, B. S.; Dunbar, K. L.; Bumpus, S. B.;Kelleher, N. L.; Mitchell, D. A. Structure determination and interceptionof biosynthetic intermediates for the plantazolicin class of highlydiscriminating antibiotics. ACS Chem. Biol. 2011, 6, 1307−1313.(c) Sharma, A.; Blair, P. M.; Mitchell, D. A. Synthesis of plantazolicinanalogues enables dissection of ligand binding interactions of a highlyselective methyltransferase. Org. Lett. 2013, 15, 5076−5079.(130) (a) Atobe, M.; Naganuma, K.; Kawanishi, M.; Morimoto, A.;Kasahara, K.-i.; Ohashi, S.; Suzuki, H.; Hayashi, T.; Miyoshi, S. SAR-based optimization of 2-(1H-pyrazol-1-yl)-thiazole derivatives as highlypotent EP1 receptor antagonists. Bioorg. Med. Chem. Lett. 2013, 23,6569−6576. (b) Atobe, M.; Naganuma, K.; Kawanishi, M.; Morimoto,A.; Kasahara, K.-i.; Ohashi, S.; Suzuki, H.; Hayashi, T.; Miyoshi, S. Hit-to-lead optimization of 2-(1H-pyrazol-1-yl)-thiazole derivatives as anovel class of EP1 receptor antagonists. Bioorg. Med. Chem. Lett. 2013,23, 6064−6067.(131) Ronkin, S. M.; Badia, M.; Bellon, S.; Grillot, A.-L.; Gross, C. H.;Grossman, T. H.; Mani, N.; Parsons, J. D.; Stamos, D.; Trudeau, M.;Wei, Y.; Charifson, P. S. Discovery of pyrazolothiazoles as novel andpotent inhibitors of bacterial gyrase. Bioorg. Med. Chem. Lett. 2010, 20,2828−2831.(132) Lam, T.; Hilgers, M. T.; Cunningham, M. L.; Kwan, B. P.;Nelson, K. J.; Brown-Driver, V.; Ong, V.; Trzoss, M.; Hough, G.; Shaw,K. J.; Finn, J. Structure-based design of new dihydrofolate reductaseantibacterial agents: 7-(benzimidazol-1-yl)-2,4-diaminoquinazolines. J.Med. Chem. 2014, 57, 651−668.(133) (a) Zampese, J. A.; Keene, F. R.; Steel, P. J. Diastereoisomericdinuclear ruthenium complexes of 2,5-di(2-pyridyl)thiazolo[5,4-d]-thiazole. Dalton Trans. 2004, 4124−4129. (b) Knighton, R. C.; Hallett,A. J.; Kariuki, B. M.; Pope, S. J. A. A one-step synthesis towards new
Journal of Medicinal Chemistry Perspective
DOI: 10.1021/jm501853mJ. Med. Chem. XXXX, XXX, XXX−XXX
BA

ligands based on aryl-functionalised thiazolo[5,4-d]thiazole chromo-phores. Tetrahedron Lett. 2010, 51, 5419−5422.(134) Naraso; Wudl, F. Two poly(2,5-thienythiazolothiazole)s:observation of spontaneous ordering in thin films. Macromolecules2008, 41, 3169−3174.(135) Wang, C.; Han, Y.; Wu, A.; Solyom, S.; Niu, L. Mechanism andsite of inhibition of AMPA receptors: pairing a thiadiazole with a 2,3-benzodiazepine scaffold. ACS Chem. Neurosci. 2014, 5, 138−147.(136) Shultz, M. D.; Kirby, C. A.; Stams, T.; Chin, D. N.; Blank, J.;Charlat, O.; Cheng, H.; Cheung, A.; Cong, F.; Feng, Y.; Fortin, P. D.;Hood, T.; Tyagi, V.; Xu, M.; Zhang, B.; Shao, W. [1,2,4]Triazol-3-ylsulfanylmethyl)-3-phenyl-[1,2,4]oxadiazoles: antagonists of the Wntpathway that inhibit tankyrases 1 and 2 via novel adenosine pocketbinding. J. Med. Chem. 2012, 55, 1127−1136.(137) (a) Bilodeau, M. T.; Rodman, L. D.; McGaughey, G. B.; Coll, K.E.; Koester, T. J.; Hoffman, W. F.; Hungate, R. W.; Kendall, R. L.;McFall, R. C.; Rickert, K. W.; Rutledge, R. Z.; Thomas, K. A. Thediscovery ofN-(1,3-thiazol-2-yl)pyridin-2-amines as potent inhibitors ofKDR kinase. Bioorg. Med. Chem. Lett. 2004, 14, 2941−2945.(b) Bilodeau, M. T.; Balitza, A. E.; Koester, T. J.; Manley, P. J.;Rodman, L. D.; Buser-Doepner, C.; Coll, K. E.; Fernandes, C.; Gibbs, J.B.; Heimbrook, D. C.; Huckle, W. R.; Kohl, N.; Lynch, J. J.; Mao, X.;McFall, R. C.;McLoughlin, D.;Miller-Stein, C.M.; Rickert, K.W.; Sepp-Lorenzino, L.; Shipman, J. M.; Subramanian, R.; Thomas, K. A.; Wong,B. K.; Yu, S.; Hartman, G. D. Potent N-(1,3-thiazol-2-yl)pyridin-2-amine vascular endothelial growth factor receptor tyrosine kinaseinhibitors with excellent pharmacokinetics and low affinity for the hERGion channel. J. Med. Chem. 2004, 47, 6363−6372.(138) Das, J.; Moquin, R. V.; Lin, J.; Liu, C.; Doweyko, A. M.; DeFex,H. F.; Fang, Q.; Pang, S.; Pitt, S.; Shen, D. R.; Schieven, G. L.; Barrish, J.C.; Wityak, J. Discovery of 2-amino-heteroaryl-benzothiazole-6-anilidesas potent p56lck inhibitors. Bioorg. Med. Chem. Lett. 2003, 13, 2587−2590.(139) Dudkin, V. Y.; Rickert, K.; Kreatsoulas, C.; Wang, C.; Arrington,K. L.; Fraley, M. E.; Hartman, G. D.; Yan, Y.; Ikuta, M.; Stirdivant, S. M.;Drakas, R. A.; Walsh, E. S.; Hamilton, K.; Buser, C. A.; Lobell, R. B.;Sepp-Lorenzino, L. Pyridyl aminothiazoles as potent inhibitors of Chk1with slow dissociation rates. Bioorg. Med. Chem. Lett. 2012, 22, 2609−2612.(140) Dudkin, V. Y.; Wang, C.; Arrington, K. L.; Fraley, M. E.;Hartman, G. D.; Stirdivant, S. M.; Drakas, R. A.; Rickert, K.;Walsh, E. S.;Hamilton, K.; Buser, C. A.; Hardwick, J.; Tao, W.; Beck, S. C.; Mao, X.;Lobell, R. B.; Sepp-Lorenzino, L. Pyridyl aminothiazoles as potent Chk1inhibitors: optimization of cellular activity. Bioorg. Med. Chem. Lett.2012, 22, 2613−2619.(141) Haidle, A. M.; Zabierek, A. A.; Childers, K. K.; Rosenstein, C.;Mathur, A.; Altman, M. D.; Chan, G.; Xu, L.; Bachman, E.; Mo, J.-R.;Bouthillette, M.; Rush, T.; Tempesta, P.; Marshall, C. G.; Young, J. R.Thiophene carboxamide inhibitors of JAK2 as potential treatments formyleoproliferative neoplasms. Bioorg. Med. Chem. Lett. 2014, 24, 1968−1973.(142) Hinklin, R. J.; Boyd, S. A.; Chicarelli, M. J.; Condroski, K. R.;DeWolf,W. E., Jr.; Lee, P. A.; Lee,W.; Singh, A.; Thomas, L.; Voegtli, W.C.;Williams, L.; Aicher, T. D. Identification of a new class of glucokinaseactivators through structure-based design. J. Med. Chem. 2013, 56,7669−7678.(143) Waring, M. J.; Johnstone, C.; McKerrecher, D.; Pike, K. G.;Robb, G. Matrix-based multiparameter optimization of glucokinaseactivators: the discovery of AZD1092. Med. Chem. Commun. 2011, 2,775−779.(144) Hinklin, R. J.; Aicher, T. D.; Anderson, D. A.; Baer, B. R.; Boyd,S. A.; Condroski, K. R.; DeWolf, W. E., Jr.; Kraser, C. F.; McVean, M.;Rhodes, S. P.; Sturgis, H. L.; Voegtli, W. C.; Williams, L.; Houze, J. B.Discovery of 2-pyridylureas as glucokinase activators. J. Med. Chem.2014, 57, 8180−8186.(145) Shirude, P. S.; Madhavapeddi, P.; Naik, M.; Murugan, K.;Shinde, V.; Nandishaiah, R.; Bhat, J.; Kumar, A.; Hameed, S.; Holdgate,G.; Davies, G.; McMiken, H.; Hegde, N.; Ambady, A.; Venkatraman, J.;Panda, M.; Bandodkar, B.; Sambandamurthy, V. K.; Read, J. A. Methyl-
thiazoles: a novel mode of inhibition with the potential to develop novelinhibitors targeting InhA in Mycobacterium tuberculosis. J. Med. Chem.2013, 56, 8533−8542.(146) Shimamura, T.; Shibata, J.; Kurihara, H.; Mita, T.; Otsuki, S.;Sagara, T.; Hirai, H.; Iwasawa, Y. Identification of potent 5-pyrimidinyl-2-aminothiazole CDK4, 6 inhibitors with significant selectivity overCDK1, 2, 5, 7, and 9. Bioorg. Med. Chem. Lett. 2006, 16, 3751−3754.(147) Ghaemmaghami, S.; Russo, M.; Renslo, A. R. Successes andchallenges in phenotype-based lead discovery for prion diseases. J. Med.Chem. 2014, 57, 6919−6929.(148) (a) Gallardo-Godoy, A.; Gever, J.; Fife, K. L.; Silber, B. M.;Prusiner, S. B.; Renslo, A. R. 2-Aminothiazoles as therapeutic leads forprion diseases. J. Med. Chem. 2011, 54, 1010−1021. (b) Silber, B. M.;Rao, S.; Fife, K. L.; Gallardo-Godoy, A.; Renslo, A. R.; Dalvie, D. K.;Giles, K.; Freyman, Y.; Elepano, M.; Gever, J. R.; Li, Z.; Jacobson, M. P.;Huang, Y.; Benet, L. Z.; Prusiner, S. B. Pharmacokinetics andmetabolism of 2-aminothiazoles with antiprion activity in mice.Pharm. Res. 2013, 30, 932−950. (c) Li, Z.; Silber, B. M.; Rao, S.;Gever, J. R.; Bryant, C.; Gallardo-Godoy, A.; Dolghih, E.; Widjaja, K.;Elepano, M.; Jacobson, M. P.; Prusiner, S. B.; Renslo, A. R. 2-Aminothiazoles with improved pharmacotherapeutic properties fortreatment of prion disease. ChemMedChem 2013, 8, 847−857.(149) Ioannidis, S.; Lamb, M. L.; Almeida, L.; Guan, H.; Peng, B.;Bebernitz, G.; Bell, K.; Alimzhanov, M.; Zinda, M. Replacement ofpyrazol-3-yl amine hinge binder with thiazol-2-yl amine: Discovery ofpotent and selective JAK2 inhibitors. Bioorg. Med. Chem. Lett. 2010, 20,1669−1673.(150) Guan, H.; Lamb, M. L.; Peng, B.; Huang, S.; DeGrace, N.; Read,J.; Hussain, S.; Wu, J.; Rivard, C.; Alimzhanov, M.; Bebernitz, G.; Bell,K.; Ye, M.; Zinda, M.; Ioannidis, S. Discovery of novel JAK2−STATpathway inhibitors with extended residence time on target. Bioorg. Med.Chem. Lett. 2013, 23, 3105−3110.(151) (a) Dietchfield, C.; Johnson, V. L.; Tighe, A.; Ellston, R.;Haworth, C.; Johnson, T.; Mortlock, A.; Keen, N.; Taylor, S. S. Aurora Bcouples chromosome alignment with anaphase by targeting BubR1,Mad2, and Cenp-E to kinetochores. J. Cell. Biol. 2003, 161, 267−280.(b) Jung, F. H.; Pasquet, G.; Lambert-van der Brempt, C.; Lohmann, J.-J.M.; Warin, N.; Renaud, F.; Germain, H.; De Savi, C.; Roberts, N.;Johnson, T.; Dousson, C.; Hill, G. B.; Mortlock, A. A.; Heron, N.;Wilkinson, R. W.; Wedge, S. R.; Heaton, S. P.; Odedra, R.; Keen, N. J.;Green, S.; Brown, E.; Thompson, K.; Brightwell, S. Discovery of noveland potent thiazoloquinazolines as selective aurora A and B kinaseinhibitors. J. Med. Chem. 2006, 49, 955−970.(152) (a) Belanger, D. B.; Curran, P. J.; Hruza, A.; Voigt, J.; Meng, Z.;Mandal, A. K.; Siddiqui, M. A.; Basso, A. D.; Gray, K. Discovery ofimidazo[1,2-a]pyrazine-based Aurora kinase inhibitors. Bioorg. Med.Chem. Lett. 2010, 20, 5170−5174. (b) Meng, Z.; Kulkarni, B. A.;Kerekes, A. D.; Mandal, A. K.; Esposite, S. J.; Belanger, D. B.; Reddy, P.A.; Basso, A. D.; Tevar, S.; Gray, K.; Jones, J.; Smith, E. B.; Doll, R. J.;Siddiqui, M. A. Bioisosteric approach to the discovery of imidazo[1,2-a]pyrazines as potent Aurora kinase inhibitors. Bioorg. Med. Chem. Lett.2011, 21, 592−598. (c) Belanger, D. B.; Williams, M. J.; Curran, P. J.;Mandal, A. K.; Meng, Z.; Rainka, M. P.; Yu, T.; Shih, N.-Y.; Siddiqui, M.A.; Liu, M.; Tevar, S.; Lee, S.; Liang, L.; Gray, K.; Yaremko, B.; Jones, J.;Smith, E. B.; Prelusky, D. B.; Basso, A. D. Discovery of orally bioavailableimidazo[1,2-a]pyrazine-based Aurora kinase inhibitors. Bioorg. Med.Chem. Lett. 2010, 20, 6739−6743. (d) Yu, T.; Tagat, J. R.; Kerekes, A.D.; Doll, R. J.; Zhang, Y.; Xiao, Y.; Esposite, S.; Belanger, D. B.; Curran,P. J.; Mandal, A. K.; Siddiqui, M. A.; Shih, N.-Y.; Basso, A. D.; Liu, M.;Gray, K.; Tevar, S.; Jones, J.; Lee, S.; Liang, L.; Ponery, S.; Smith, E. B.;Hruza, A.; Voigt, J.; Ramanathan, L.; Prosise, W.; Hu, M. Discovery of apotent, injectable inhibitor of aurora kinases based on the imidazo-[1,2-a]-pyrazine core. ACS Med. Chem. Lett. 2010, 1, 214−218. (e) Kerekes,A. D.; Esposite, S. J.; Doll, R. J.; Tagat, J. R.; Yu, T.; Xiao, Y.; Zhang, Y.;Prelusky, D. B.; Tevar, S.; Gray, K.; Terracina, G. A.; Lee, S.; Jones, J.;Liu, M.; Basso, A. D.; Smith, E. B. Aurora kinase inhibitors based on theimidazo[1,2-a]pyrazine core: fluorine and deuterium incorporationimprove oral absorption and exposure. J. Med. Chem. 2011, 54, 201−210.
Journal of Medicinal Chemistry Perspective
DOI: 10.1021/jm501853mJ. Med. Chem. XXXX, XXX, XXX−XXX
BB

(153) Reker, D.; Seet, M.; Pillong, M.; Koch, C. P.; Schneider, P.;Witschel, M. C.; Rottmann, M.; Freymond, C.; Brun, R.; Schweizer, B.;Illarionov, B.; Bacher, A.; Fischer, M.; Diederich, F.; Schneider, G.Deorphaning pyrrolopyrazines as potent multi-target antimalarialagents. Angew. Chem., Int. Ed. 2014, 53, 7079−7084.(154) (a) Krueger, A. C.; Madigan, D. L.; Beno, D. W.; Betebenner, D.A.; Carrick, R.; Green, B. E.; He, W.; Liu, D.; Maring, C. J.; McDaniel, K.F.; Mo, H.; Molla, A.; Motter, C. E.; Pilot-Matias, T. J.; Tufano, M. D.;Kempf, D. J. Novel hepatitis C virus replicon inhibitors: synthesis andstructure−activity relationships of fused pyrimidine derivatives. Bioorg.Med. Chem. Lett. 2012, 22, 2212−2215. (b) DeGoey, D. A.; Betebenner,D. A.; Grampovnik, D. J.; Liu, D.; Pratt, J. K.; Tufano, M. D.; He, W.;Krishnan, P.; Pilot-Matias, T. J.; Marsh, K. C.; Molla, A.; Kempf, D. J.;Maring, C. J. Discovery of pyrido[2,3-d]pyrimidine-based inhibitors ofHCV NS5A. Bioorg. Med. Chem. Lett. 2013, 23, 3627−3630.(155) Mugesh, G.; Singh, H. B.; Butcher, R. J. Non-bonded S···Ninteractions in organosulfur derivatives: crystal and molecular structuresof [2-(4,4-dimethyl-2-oxazolinyl)phenyl] benzyl sulfide and bis[2-(4,4-dimethyl-2-oxazolinyl)phenyl] sulfide. J. Chem. Res. 1999, 472−473.(156) Morgan, R. S.; Tatsch, C. E.; Gushard, R. H.; McAdon, J. M.;Warme, P. K. Chains of alternating sulfur and π-bonded atoms in eightsmall proteins. Int. J. Pept. Protein Res. 1978, 11, 209−217.(157) Feldman, R. J. Atlas of Macromolecular Structure on Microfiche;Tracor-Jitco, Inc.: Rockville, MD, 1976.(158) Berman, H. M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I. N.; Bourne, P. E. The Protein Data Bank.Nucleic Acids Res. 2000, 28, 235−242.(159) (a) Bodner, B. L.; Jackman, L.M.;Morgan, R. S.Abstracts AnnualMeeting Biophysical Society NMR evidence for a 1:1 complex betweensulfur-containing and aromatic molecules. Biophys. J. 1977, 17, 57a.(b) Bodner, B. L.; Jackman, L. M.; Morgan, R. S. NMR study of 1:1complexes between divalent sulfur and aromatic compounds: a modelfor interactions in globular proteins. Biochem. Biophys. Res. Commun.1980, 94, 807−813.(160) (a) Gibson, K. D.; Scheraga, H. A. Minimization of polypeptideenergy. I. Preliminary structures of bovine pancreatic ribonuclease S-peptide. Proc. Natl. Acad. Sci. U. S. A. 1967, 58, 420−427. (b) Nemethy,G.; Scheraga, H. A. Protein folding. Q. Rev. Biophys. 1977, 10, 239−352.(161) (a) Morgan, R. S.; McAdon, J. M. Predictor for sulfur−aromaticinteractions in globular proteins. Int. J. Pept. Protein Res. 1980, 15, 177−180. (b) Reid, K. S. C.; Lindley, P. F.; Thornton, J. M. Sulfur−aromaticinteractions in proteins. FEBS Lett. 1985, 190, 209−213. (c) Pal, D.;Chakrabarti, P. Different types of interactions involving cysteinesulfhydryl group in proteins. J. Biomol. Struct. Dynam. 1998, 15,1059−1072. (d) Zauhar, R. J.; Colbert, C. L.; Morgan, R. S.; Welsh, W. J.Evidence for a strong sulfur−aromatic interaction derived fromcrystallographic data. Biopolymers 2000, 53, 233−248. (e) Duan, G.;Smith, V. H., Jr.; Weaver, D. F. Characterization of aromatic−thiol π-type hydrogen bonding and phenylalanine−cysteine side chaininteractions through ab initio calculations and protein database analyses.Mol. Phys. 2001, 99, 1689−1699. (f) Pal, D.; Chakrabarti, P. Non-hydrogen bond interactions involving the methionine sulfur atom. J.Biomol. Struct. Dynam. 2001, 19, 115−128. (g) Iwaoka, M.; Takemoto,S.; Okada, M.; Tomoda, S. Weak nonbonded S···X (X=O, N, S)interactions in proteins. Statistical and theoretical studies. Bull. Chem.Soc. Jpn. 2002, 75, 1611−1625. (h) Ringer, A. L.; Senenko, A.; Sherrill,C. D. Models of S/π interactions in protein structures: comparison ofthe H2S−benzene complex with PDB data. Protein Sci. 2007, 16, 2216−2223. (i) Valley, C. C.; Cembran, A.; Perlmutter, J. D.; Lewis, A. K.;Labello, N. P.; Gao, J.; Sachs, J. N. The methionine−aromatic motifplays a unique role in stabilizing protein structure. J. Biol. Chem. 2012,287, 34979−34991. (j) Chakrabarti, P.; Bhattacharyya, R. Geometry ofnonbonded interactions involving planar groups in proteins. Prog.Biophys. Mol. Biol. 2007, 95, 83−137.(162) (a) Nemethy, G.; Scheraga, H. A. Strong interaction betweendisulfide derivatives and aromatic groups in peptides and proteins.Biochem. Biophys. Res. Commun. 1981, 98, 482−487. (b) Cheney, B. V.;Schultz, M. W.; Cheney, J. Complexes of benzene with formamide andmethanethiol as models for interactions of protein substructures.
Biochim. Biophys. Acta 1989, 996, 116−124. (c) Pranata, J. Sulfur−aromatic interactions: a computational study of the DMS−benzenecomplex. Bioorg. Chem. 1997, 25, 213−219. (d) Tauer, T. P.; Derrick, M.E.; Sherrill, C. D. Estimates of the ab initio limit for sulfur−πinteractions: the H2S−benzene dimer. J. Phys. Chem. A 2005, 109, 191−196. (e) Hermida-Ramon, J. M.; Cabaleiro-Lago; Rodriguez-Otero, J.Theoretical characterization of structures and energies of benzene−(H2S)n and (H2S)n (n = 1−4) clusters. J. Chem. Phys. 2005, 122, 204315-1−204315-5. (f) Morgado, C. A.; McNamara, J. P.; Hillier, I. A.; Burton,N. A.; Vincent, M. A. Density functional and semiempirical molecularorbital methods including dispersion corrections for the accuratedescription of noncovalent interactions involving sulfur-containingmolecules. J. Chem. Theory Comput. 2007, 3, 1656−1664.(163) (a) Lebl,M.; Sugg, E. E.; Hruby, V. J. Proton n.m.r. spectroscopicevidence for sulfur−aromatic interactions in peptides. Int. J. Pept. ProteinRes. 1987, 29, 40−45. (b) Viguera, A. R.; Serrano, L. Side chaininteractions between sulfur-containing amino acids and phenylalanine inα-helices. Biochemistry 1995, 34, 8771−8779. (c) Tatko, C. D.; Waters,M. L. Investigation of the nature of the methionine−π interaction in β-hairpin peptide model systems. Protein Sci. 2004, 13, 2515−2522.(164) For a more complete discussion of examples see: (a) Meyer, E.A.; Castellano, R. K.; Diederich, F. Interactions with aromatic rings inchemical and biological recognition. Angew. Chem., Int. Ed. 2003, 42,1210−1250. (b) Salonen, L. M.; Ellermann, M.; Diederich, F. Aromaticrings in chemical and biological recognition: energetics and structures.Angew. Chem., Int. Ed. 2011, 50, 4804−4842.(165) Brasch, J.; Harrison, O. J.; Ahlsen, G.; Liu, Q.; Shapiro, L. Crystalstructure of domain VI and LE1 of human Netrin-G2. J. Mol. Biol. 2011,414, 723−734.(166) Harrop, S. J.; DeMaere, M. Z.; Fairlie, W. D.; Reztsova, T.;Valenzuela, S. M.; Mazzanti, M.; Tonini, R.; Qiu, M. R.; Jankova, L.;Warton, K.; Bauskin, A. R.; Wu, W. M.; Pankhurst, S.; Campbell, T. J.;Breit, S. N.; Curmi, P. M. Crystal structure of a soluble form of theintracellular chloride ion channel CLIC1 (NCC27) at 1.4 Å resolution. J.Biol. Chem. 2001, 276, 44993−45000.(167) Allen, F. H. The Cambridge Structural Database: a quarter of amillion crystal structures and rising. Acta Crystallogr., Sect. B: Struct. Sci.2002, B58, 380−388.(168) Karolak-Wojciechowska, J.; Kiec-Kononowicz, K. Structure of 6-(4-chlorobenzylidene)-2,3-dihydroimidazo[2,1-b]thiazol-5(6H)-one.Conformational analysis of Z isomers of 5-benzylidenethiohydantoinderivatives. Acta Crystallogr., Sect. C: Cryst. Struct. Commun. 1991, C47,2371−2374.(169) Bruno, I. J.; Cole, J. C.; Edgington, P. R.; Kessler, M.; Macrae, C.F.; McCabe, P.; Pearson, J.; Taylor, R. New software for searching theCambridge Structural Database and visualising crystal structures. ActaCrystallogr., Sect. B: Struct. Sci. 2002, B58, 389−397.(170) (a) Gellman, S. H. On the role of methionine residues in thesequence-independent recognition of nonpolar protein surfaces.Biochemistry 1991, 30, 6634−6636. (b) Yoshida, T.; Mashima, A.;Sasahara, K.; Chuman, H. A simple and efficient dispersion correction tothe Hartree−Fock theory. Bioorg. Med. Chem. Lett. 2014, 24, 1037−1042 and references therein.(171) Hymowitz, S. G.; Christinger, H. W.; Fuh, G.; Ultsch, M.;O’Connell, M.; Kelley, R. F.; Ashkenazi, A.; deVos, A. M. Triggering celldeath: the crystal structure of Apo2L/TRAIL in a complex with deathreceptor 5. Mol. Cell 1999, 4, 563−571.(172) Banner, D. W.; D’Arcy, A.; Janes, W.; Gentz, R.; Schoenfeld, H.J.; Broger, C.; Loetscher, H.; Lesslauer, W. Crystal structure of thesoluble human 55 kD TNF receptor−human TNF beta complex:implications for TNF receptor activation. Cell 1993, 73, 431−445.(173)Williamson,M. P. Applications of theNOE inmolecular biology.Annu. Rep. NMR Spectrosc. 2009, 65, 77−109.(174)Munoz, V.; Serrano, L. Elucidating the folding problem of helicalpeptides using empirical parameters. Nature Struct. Biol. 1994, 1, 399−409.(175) Stapley, B. J.; Rohl, C. A.; Doig, A. J. Addition of side chaininteractions to modified Lifson−Roig helix−coil theory: application to
Journal of Medicinal Chemistry Perspective
DOI: 10.1021/jm501853mJ. Med. Chem. XXXX, XXX, XXX−XXX
BC

energetic of phenylalanine−methionine interactions. Protein Sci. 1995,4, 2383−2391.(176) (a) Cockroft, S. L.; Hunter, C. A. Chemical double-mutantcycles: dissecting non-covalent interactions. Chem. Soc. Rev. 2007, 36,172−188. (b) Kawai, S. H.; Bailey, M. D.; Halmos, T.; Forgione, P.;LaPlante, S. R.; Llinas-Brunet, M.; Naud, J.; Goudreau, N. The use ofchemical double-mutant cycles in biomolecular recognition studies:application to HCV NS3 protease inhibitors. ChemMedChem 2008, 3,1654−1657.(177) Motherwell, W. B.; Joelle, M.; Aliev, A. E.; Nic, M.; Coles, S. J.;Horton, P. N.; Hursthouse, M. B.; Chessari, G.; Hunter, C. A.; Vinter, J.G. Noncovalent functional-group−arene interactions. Angew. Chem., Int.Ed. 2007, 46, 7823−7826.(178) Druhan, L. J.; Swenson, R. P. Role of methionine 56 in thecontrol of the oxidation−reduction potentials of the Clostridiumbeijerinckii flavodoxin; effects of substitutions by aliphatic amino acidsand evidence for a role of sulfur−flavin interactions. Biochemistry 1998,37, 9668−9678.(179) Ludwig, M. L.; Pattridge, K. A.; Metzger, A. L.; Dixon, M. M.;Eren, M.; Feng, Y.; Swenson, R. P. Control of oxidation−reductionpotentials in flavodoxin from Clostridium beijerinckii: the role ofconformation changes. Biochemistry 1997, 36, 1259−1280.(180) Traxler, P.; Gren, J.; Mett, H.; Sequin, U.; Furet, P. Use of apharmacophore model for the design of EGFR tyrosine kinaseinhibitors: isoflavones and 3-phenyl-4(1H)-quinolones. J. Med. Chem.1999, 42, 1018−1026.(181) Peng, T.; Pei, J.; Zhou, J. 3D-QSAR and receptor modeling oftyrosine kinase inhibitors with flexible atom receptor model (FLARM).J. Chem. Inf. Comput. Sci. 2003, 43, 298−303.(182) Visser, F.; Vickers, M. F.; Ng, A. M.; Baldwin, S. A.; Young, J. D.;Cass, C. E. Mutation of residue 33 of human equilibrative nucleosidetransporters 1 and 2 alters sensitivity to inhibition of transport by dilazepand dipyridamole. J. Biol. Chem. 2002, 277, 395−401.(183) Visser, F.; Zhang, J.; Raborn, R. T.; Baldwin, S. A.; Young, J. D.;Cass, C. E. Residue 33 of human equilibrative nucleoside transporter 2 isa functionally important component of both the dipyridamole andnucleoside binding sites. Mol. Pharmacol. 2005, 67, 1291−1298.(184) Wogulis, M.; Wheelock, C. E.; Kamita, S. G.; Hinton, A. C.;Whetstone, P. A.; Hammock, B. D.; Wilson, D. K. Structural studies of apotent insect maturation inhibitor bound to the juvenile hormoneesterase of Manduca sexta. Biochemistry 2006, 45, 4045−4057.(185) Heelock, C. E.; Colvin, M. E.; Uemura, I.; Olmstead, M. M.;Sanborn, J. R.; Nakagawa, Y.; Jones, A. D.; Hammock, B. D. Use of abinitio calculations to predict the biological activity of carboxylesteraseinhibitors. J. Med. Chem. 2002, 45, 5576−5593.(186) Probst, G. D.; Bowers, S.; Sealy, J. M.; Truong, A. P.; Horn, R. K.;Galemmo, R. A., Jr.; Konradi, A.W.; Sham, H. L.; Quincy, D. A.; Pan, H.;Yao, N.; Lin, M.; Toth, G.; Artis, D. R.; Zmolek, W.; Wong, K.; Qin, A.;Lorentzen, C.; Nakamura, D. F.; Quinn, K. P.; Sauer, J.-M.; Powell, K.;Ruslim, L.; Wright, S.; Chereau, D.; Ren, Z.; Anderson, J. P.; Bard, F.;Yednock, T. A.; Griswold-Prenner, I. Highly selective c-Jun N-terminalkinase (JNK) 2 and 3 inhibitors with in vitro CNS-like pharmacokineticproperties prevent neurodegradation. Bioorg. Med. Chem. Lett. 2011, 21,315−319.(187) Daeffler, K. N.-M.; Lester, H. A.; Dougherty, D. A. Functionallyimportant aromatic−aromatic and sulfur−π interactions in the D2
dopamine receptor. J. Am. Chem. Soc. 2012, 134, 14890−14896.(188) Chien, E. Y. T.; Liu, W.; Zhao, Q.; Katritch, V.; Won Han, G.;Hanson, M. A.; Shi, L.; Newman, A. H.; Javitch, J. A.; Cherezov, V.;Stevens, R. C. Structure of the human dopamine D3 receptor in complexwith a D2/D3 selective antagonist. Science 2010, 330, 1091−1095.(189) LaPlante, S. R.; Nar, H.; Lemke, C. T.; Jakalian, A.; Aubry, N.;Kawai, S. H. Ligand bioactive conformation plays a critical role in thedesign of drugs that target the hepatitis C virus NS3 protease. J. Med.Chem. 2014, 57, 1777−1789.(190) (a) Llinas-Brunet, M.; Bailey, M.; Fazal, G.; Goulet, S.; Halmos,T.; LaPlante, S.; Maurice, R.; Poirier, M.; Poupart, M.-A.; Thibeault, D.;Wernic, D.; Lamarre, D. Peptide-based inhibitors of the hepatitis C virusserine protease. Bioorg. Med. Chem. Lett. 1998, 8, 1713−1718.
(b) Llinas-Brunet, M.; Bailey, M.; Deziel, R.; Fazal, G.; Gorys, V.;Goulet, S.; Halmos, T.; Maurice, R.; Poirier, M.; Poupart, M.-A.;Rancourt, J.; Thibeault, D.; Wernic, D.; Lamarre, D. Studies on the C-terminal of hexapeptide inhibitors of the hepatitis C virus serineprotease. Bioorg. Med. Chem. Lett. 1998, 8, 2719−2724.(191) Kakiuchi, N.; Hijikata, M.; Komoda, Y.; Tanji, Y.; Hirowatori, Y.;Shimotohno, K. Bacterial expression and analysis of cleavage activity ofHCV serine proteinase using recombinant and synthetic substrate.Biochem. Biophys. Res. Commun. 1995, 210, 1059−1065.(192) Politzer, P.; Murray, J. M.; Clark, T. Halogen bonding: anelectrostatically-driven highly directional noncovalent interaction. Phys.Chem. Chem. Phys. 2010, 12, 7748−7757.(193) Aufinger, P.; Hays, F. A.;Westhof, E.; Ho, P. S. Halogen bonds inbiological macromolecules. Proc. Natl. Acad. Sci. U. S. A. 2004, 101,16789−16794.(194)Wilcken, R.; Zimmermann, M. O.; Lange, A.; Zahn, S.; Kirchner,B.; Boeckler, F. M. Addressing methionine in molecular design throughdirected sulfur−halogen bonds. J. Chem. Theory Comput. 2011, 7, 2307−2315.(195) Hauchecorne, D.; Moiana, A.; van der Veken, B.; Herrebout, W.A. Halogen bonding to a divalent sulfur atom: an experimental study ofthe interactions of CF3X (X = Cl, Br, I) with dimethyl sulfide. Phys.Chem. Chem. Phys. 2011, 13, 10204−10213.(196) Liu, l.; Baase, W. A.; Matthews, B. W. Halogenated benzenesbound within a non-polar cavity in T4 lysozyme provide examples of I···S and I···Se halogen-bonding. J. Mol. Biol. 2009, 385, 595−605.(197) Rush, T. S., III; Grant, A.; Mosyak, L.; Nicholls, A. A shape-based3-D scaffold hopping method and its application to a bacterial protein−protein interaction. J. Med. Chem. 2005, 48, 1489−1495.(198) Ferreira, R. S.; Simeonov, A.; Jadhav, A.; Eidam, O.; Mott, B. T.;Keiser, M. J.; McKerrow, J. H.; Maloney, D. J.; Irwin, J. J.; Shoichet, B. K.Complementarity between a docking and a high-throughput screen indiscovering new cruzain inhibitors. J. Med. Chem. 2010, 53, 4891−4905.(199) Katz, B. A.; Sprengeler, P. A.; Luong, C.; Verner, E.; Elrod, K.;Kirtley, M.; Janc, J.; Spencer, J. R.; Breitenbucher, J. G.; Hui, H.; McGee,D.; Allen, D.; Martelli, A.; Mackman, R. L. Engineering inhibitors highlyselective for the S1 sites of Ser190 trypsin-like serine protease targets.Chem. Biol. 2001, 8, 1107−1121.(200) Alam,M.; Beevers, R. E.; Ceska, T.; Davenport, R. J.; Dickson, K.M.; Fortunato, M.; Gowers, L.; Haughan, A. F.; James, L. A.; Jones, M.W.; Kinsella, N.; Lowe, C.; Meissner, J. W. G.; Nicolas, A.-L.; Perry, B.G.; Philips, D. J.; Pitt, W. R.; Platt, A.; Ratcliffe, A. J.; Sharpe, A.; Tait, L. J.Synthesis and SAR of aminopyrimidines as novel c-Jun N-terminalkinase (JNK) inhibitors. Bioorg. Med. Chem. Lett. 2007, 17, 3463−3467.(201) (a) Huang, X.; Cheng, C. C.; Fischmann, T. O.; Duca, J. S.; Yang,X.; Richards, M.; Shipps, G. W., Jr. Discovery of a novel series of CHK1kinase inhibitors with a distinctive hinge binding mode. ACSMed. Chem.Lett. 2012, 3, 123−128. (b) Huang, X.; Cheng, C. C.; Fischmann, T. O.;Duca, J. S.; Richards, M.; Tadikonda, P. K.; Adulla Reddy, P.; Zhao, L.;Siddiqui, M. A.; Parry, D.; Davis, N.; Seghezzi, W.; Wiswell, D.; Shipps,G.W., Jr. Structure-based design and optimization of 2-aminothiazole-4-carboxamide as a new class of CHK1 inhibitors. Bioorg. Med. Chem. Lett.2013, 23, 2590−2594.(202) Hua, Z.; Bregman, H.; Buchanan, J. L.; Chakka, N.; Guzman-Perez, A.; Gunaydin, H.; Huang, X.; Gu, Y.; Berry, V.; Liu, J.; Teffera, Y.;Huang, L.; Egge, B.; Emkey, R.; Mullady, E. L.; Schneider, S.; Andrews,P. S.; Acquaviva, L.; Dovey, J.; Mishra, A.; Newcomb, J.; Saffran, D.;Serafino, R.; Strathdee, C. A.; Turci, S. M.; Stanton, M.; Wilson, C.;DiMauro, E. F. Development of novel dual binders as potent, selective,and orally bioavailable tankyrase inhibitors. J. Med. Chem. 2013, 56,10003−10015.(203) Pierce, A. C.; Sandretto, K. L.; Bemis, G. W. Kinase inhibitorsand the case for CH···O hydrogen bonds in protein−ligand binding.Proteins: Struct. Funct. Genet. 2002, 49, 567−576.
Journal of Medicinal Chemistry Perspective
DOI: 10.1021/jm501853mJ. Med. Chem. XXXX, XXX, XXX−XXX
BD





























