Embed Size (px)
Citation preview

Pcar
TSU
ARRAA
KPHTmcBE
1
tttccstch[
0h
Journal of Chromatography A, 1291 (2013) 1– 9
Contents lists available at SciVerse ScienceDirect
Journal of Chromatography A
jou rn al hom epage: www.elsev ier .com/ locate /chroma
reparation of poly(trimethyl-2-methacroyloxyethylammoniumhloride-co-ethylene glycol dimethacrylate) monolith and itspplication in solid phase microextraction of brominated flameetardants
ing-ting Yang, Lin-feng Zhou, Jun-qin Qiao, Hong-zhen Lian ∗, Xin Ge, Hong-yuan Chentate Key Laboratory of Analytical Chemistry for Life Science, School of Chemistry & Chemical Engineering and Center of Materials Analysis, Nanjingniversity, 22 Hankou Road, Nanjing 210093, China
a r t i c l e i n f o
rticle history:eceived 19 December 2012eceived in revised form 17 March 2013ccepted 18 March 2013vailable online 26 March 2013
eywords:olymer monolith microextractionigh performance liquid chromatographyrimethyl-2-ethacroyloxyethylammonium
hloriderominated flame retardantnvironmental water
a b s t r a c t
A capillary poly(trimethyl-2-methacroyloxyethylammonium chloride-co-ethylene glycol dimethacry-late) monolith was in situ synthesized by thermally initiated free radical co-polymerization usingtrimethyl-2-methacroyloxyethylammonium chloride (MATE) and ethylene glycol dimethacrylate(EGDMA) as functional monomer and cross-linker, respectively. N,N-dimethylformamide and polyethyl-ene glycol 6000 were used as solvent and porogen, respectively. The morphology and porous structureof the resulting monoliths were assessed by scanning electron microscope. In order to prepare practi-cally useful poly(MATE-co-EGDMA) monoliths with low flow resistance and good mechanical strength,some parameters such as PEG-6000 to DMF ratio, total monomer to porogen ratio, and crosslinker tomonomer ratio were optimized systematically. Moreover, the extraction mechanism was evaluatedusing two series of compounds, alkylbenzenes and weak acids, as model compounds on poly(MATE-co-EGDMA) monoliths as liquid chromatographic stationary phase. Finally, the monoliths were appliedas the solid phase microextraction medium, and a simple off-line method for simultaneous determina-
tion of three brominated flame retardants, 2,4,6-tribromophenol (TBP), tetrabromobisphenol A (TBBPA)and 4,4′-dibrominated diphenyl ether (DBDPE), in environmental waters was developed by coupling thepolymer monolith microextraction to HPLC with UV detection. The regression equations for these threebrominated flame retardants showed good linearity from their limit of quantification to 5000 ng/mL. Thelimits of detection were 0.20, 0.15 and 0.10 ng/mL for TBP, TBBPA and DBDPE, respectively. The recoveryof the proposed method was 78.7–106.1% with intra-day relative standard deviation of 1.3–4.4%.. Introduction
Monolithic stationary phases have grown in interest because ofheir characteristic enhanced mass transfer and simple prepara-ion. Polymeric monoliths, in particular, prepared by optimizinghe polymerization conditions including temperature as well asomposition of monomers and porogenic solvents offer effi-iency approaching the feature of silica-based monoliths. Recently,ome other new approaches have been proposed to improvehe performance of monolith, such as preparing monoliths underonditions of incomplete conversion, use of single crosslinkers,
ypercrosslinking of monoliths, and utilization of nanostructures1].∗ Corresponding author. Tel.: +86 25 83686075; fax: +86 25 83325180.E-mail address: [email protected] (H.-z. Lian).
021-9673/$ – see front matter © 2013 Elsevier B.V. All rights reserved.ttp://dx.doi.org/10.1016/j.chroma.2013.03.043
© 2013 Elsevier B.V. All rights reserved.
There are a number of ways in which ion exchange function-ality can be introduced in a polymeric monolithic column, suchas functionalization by co-polymerization or post-polymerizationmodification. Typical examples of post-polymerization modifi-cation employed for polymeric monoliths include reaction ofdifferent reagents with functional groups on the monolith sur-face [2], grafting of monomer to or from the surface [3,4],and coating procedures including the use of latex particles[5]. Methacrylic acid, sulfopropyl methacrylate, 2-acrylamido-2-methyl-1-propanesulfonic acid, 2-(diethylamino)ethyl methacry-late, and 2-(acryloyloxy)ethyl trimethylammonium chloride wereall reported as the functional monomer which introduced chargesto materials [6,7]. Polymeric ion exchange monoliths have beenwidely used for the separation and preconcentration of various
analytes from large molecular compounds to small ionic species[8].During the last decade, sample preparation technologieshave gained widespread attention. Methods such as single-drop

2 mato
mee(ipibsrPFtH
orflmpspr
framm(dahtbiP
tpoHpoauNIimp
2
2
fmAmA(fr
T.-t. Yang et al. / J. Chro
icroextraction, liquid-phase microextraction, stir-bar sorptivextraction and thin film microextraction have been introducedxtensively [9]. Among them, in-tube solid-phase microextractonin-tube SPME) integrates sample extraction, concentration andnjection into one step. For this reason, it became a suitable sam-le preparation technique prior to HPLC and HPLC–MS [10]. For
n-tube SPME, increasing the extraction efficiency can be achievedy either increasing the coating thickness [11] or increasing theurface area of the sorption material [12]. However, the formerequires long equilibrium time, so the latter method is more ideal.olymer monolith microextraction (PMME) was first introduced byeng’s group, now has been successfully applied to the preconcen-ration step when combined with CE [13], ICP–MS [14], HPLC [15],PLC–MS [16] and GC–MS [17].
Flame retardants are a diverse group of chemicals, mainly basedn bromine, chlorine or phosphorus, which are added to a broadange of commercial products to provide fire protection. Someame retardants may be covalently bound into materials, butost of them are simple additives. Recently, concerns over the
ersistence, ability to bioaccumulate and potential for toxicity ofome of the most widely used brominated flame retardants (BFRs),olybrominated diphenyl ethers (PBDEs), have led to increasingegulation and restriction on their production and use [18].
In this work, a functional monomer containing anion-exchangeunctionality, trimethyl-2-methacroyloxyethylammonium chlo-ide (MATE), was utilized. By direct co-polymerization of MATEnd ethylene glycol dimethacrylate (EGDMA), the anion exchangeonolithic material was prepared in a fused silica capillary. Theonolith then was used to enrich three BFRs, 2,4,6-tribromophenol
TBP), tetrabromobisphenol A (TBBPA) and 4,4′-dibrominatediphenyl ether (DBDPE), in aqueous samples, followed by HPLCnalysis. Although many polymeric monoliths mentioned aboveave been widely used as extraction medium in analytical field, upo date, no application for separation and preconcentration of BFRsy polymeric monoliths has been reported. To our knowledge, this
s the first attempt to extract BFRs in environmental waters usingMME.
The retention mechanism study of monolithic materials toarget analytes is very important to develop their applicationotential. Some researchers have reported the characterizationf different kinds of ion exchange monoliths. For example,uang et al. [19] and Abbood et al. [20] used anilines andeptides, respectively, to characterize cation-exchange abilityf poly(4-vinylphenylboronic acid-co-pentaerythritol triacrylate)nd hexylacrylate-based monoliths, while Zhang et al. [21]sed aromatic acids to characterize anion-exchange ability of-methylimidazolium-functionalized monolithic silica column.
n this present work, the hybrid hydrophobic interaction andon-exchange extraction mechanism of poly(MATE-co-EGDMA)
onolith for BFRs was elucidated by using respective model com-ounds for the first time.
. Experimental
.1. Chemicals
Ethylene glycol dimethacrylate (EGDMA) was purchasedrom Alfa Aesar (Tianjin, China), trimethyl-2-
ethacroyloxyethylammonium chloride (MATE) was fromdamas Reagent (Shanghai, China), and �-methacryloxypropyltri-ethoxysilane (�-MAPS) was from Yaohua (Shanghai, China). 2,2′-
zobis(2-methylpropionitrile) (AIBN), N,N-dimethylformamideDMF) and polyethylene glycol-6000 (PEG-6000) were purchasedrom Shanghai Chemical Reagent (Shanghai, China). All of theseeagents were of analytical reagent grade.
gr. A 1291 (2013) 1– 9
Toluene, ethylbenzene, propylbenzene, n-butylbenzene,m-toluic acid (mTA), 3,5-dihydroxybenzoic acid (DHBA), 3-hydroxybenzoic acid (HBA) and phenylacetic acid (PAA) werepurchased from J&K Chemical (Beijing, China).
2,4,6-Tribromophenol (TBP) was purchased from TCI (Tokyo,Japan), and tetrabromobisphenol A (TBBPA) and 4,4′-dibrominateddiphenyl ether (DBDPE) were from Accustandard (New Haven,USA). The analytes were prepared as 1.0 mg/mL stock solution inmethanol and stored at 4 ◦C.
Dipotassium hydrogen phosphate, hydrochloric acid and phos-phoric acid were purchased from Nanjing Chemical Reagent(Nanjing, China), and tris(hydroxymethyl)aminomethane (Tris)was from Biosharp (Seoul, Korea).
Methanol (MeOH) was HPLC grade (Merck, Darmstadt,Germany). Double distilled water (DDW) was used for all exper-iments.
2.2. Equipment
The scanning electron microscopy (SEM) images of poly(MATE-co-EGDMA) monolith were obtained using a S-3400N scanningelectron microscope (Hitachi, Tokyo, Japan). The pH values ofmobile phase were measured with a Seven Multi electrochemi-cal analytical meter (Metter-Toledo, Schwerzenbach, Switzerland).The electrode system was standardized with ordinary aqueousbuffers of pH 2.00, 4.01 and 7.01 at 25 ◦C (Mettler-Toledo, Shanghai,China).
HPLC measurements were performed on three liquid chromato-graph (LC) systems according to different purposes: LC 1 – Agilent1200 separations module equipped with a quaternary pump, athermostatted column compartment, a diode array detector (DAD)and a chemstation (Agilent, Santa Clara, USA); LC 2 – Lab Tech600 liquid chromatograph pump (Lab Tech, Beijing, China); LC 3– Waters Alliance 2695 separations module equipped with a vac-uum degasser, a quaternary pump, an auto-sampler, a 996 UV–visphotodiode-array detector (PDA) and an Empower chromatogra-phy manager system (Milford, MA, USA).
2.3. Preparation of poly(MATE-co-EGDMA) monolith
Prior to polymerization, the fused silica capillary (530 �m i.d.,Reafine Chromatography Ltd., Hengshui, China) was pretreatedaccording to the following procedure [22]: first, the capillarycolumn was rinsed with 0.1 M NaOH, water and 0.1 M HCl, respec-tively, for 1 h, and then dried by passage of nitrogen gas. �-MAPSsolution by its dilution with DMF at a volume ratio of 1:1 wasinjected into the capillary. The capillary was sealed with rubberstoppers at both ends and put into an oven at 60 ◦C for 12 h, andfinally, rinsed with methanol to flush out residual reagent and onceagain dried by nitrogen gas.
The monolith was prepared from polymerization reaction ofthe mixture consisted of monomer MATE and crosslinker EGDMA,porogens DMF and PEG-6000, and initiator AIBN (Fig. 1). At first,the polymerization mixture was sonicated to obtain homogeneoussolution, and then the solution was filled into the capillary. Aftersealed at both ends with rubber stoppers, the capillary was putinto an oven at 60 ◦C for 17 h and then washed with methanol at0.3 mL/min for 30 min to remove unreacted monomer, crosslinkerand porogen before use.
2.4. PMME procedure
The PMME device shown in Fig. 1 was referred to the work ofFeng and co-workers [13]. The brief description of it is that theindispensable part of the PMME device is an interface capable ofconnecting the monolithic capillary and a syringe seamlessly. In

T.-t. Yang et al. / J. Chromatogr. A 1291 (2013) 1– 9 3
MME
tsTwidp5cictd2l1ao5c0w
2
lXap(tsw
3
3
3
ptD
Fig. 1. P
he following extraction experiments the metallic needle of theyringe was replaced by a 5 cm long prepared monolithic capillary.he whole extraction procedure contains precondition, sampling,ashing and desorption. A LSP04-1A syringe infusion pump (Baod-
ng, China) was employed for delivery of sample, washing andesorption solutions. For preconditioning, 0.8 mL 18 mM phos-hate buffer solution (PBS) containing 40% (v/v, %) methanol (pH.6, after mixing with methanol) was injected via the monolithicapillary at 0.1 mL/min. The sorption of analytes was realized bynjecting sample solution at 0.1 mL/min for 24 min, and then 0.3 mLlean air was injected via the monolithic capillary at 0.1 mL/mino remove the residual sample matrix in the capillary. For theesorption step, 0.5 mL 80% methanol–20% water mixture (pH.3, with phosphoric acid) was injected via the monolithic capil-
ary at 0.05 mL/min and the whole effluent was collected into a.5 mL vial for the subsequent analysis by HPLC on LC 1. The sep-ration was controlled with Agilent chemstation and was carriedut on an Agela Venusil XBP-C18 column (150 mm × 2.1 mm i.d.,
�m) (Agela Technology Inc., Tianjin, China). The mobile phase wasonsisted of 20% water and 80% methanol (v:v) with the flow rate of.2 mL/min. Injection volume was 10 �L and detection wavelengthas 235 nm.
.5. Water sample preparation
Tap water was collected in our laboratory. Rain water was col-ected in the campus of our university, and lake water was fromuanwu Lake in Nanjing. Each water sample was filtered through
0.45 �m cellulose acetate membrane. Sample solution was pre-ared by adding methanol until final methanol content reached 40%v/v, %), and adding 0.9 M dipotassium hydrogen phosphate solu-ion to keep PBS concentration being 18 mM. After adjusting theolution pH to 5.6 with phosphoric acid the flask was filled withater to the volume for PMME of BFRs.
. Results and discussion
.1. Optimization of poly(MATE-co-EGDMA) monolith
.1.1. Selection of solvent
In order to obtain transparent and homogeneous pre-olymerization mixture liquid, several common solvents includingoluene, ethanol, ethyl acetate, isopropanol, acetonitrile, methanol,MF and dimethylsulfoxide were tested. The choice of solvent
device.
was subjected to the restrictions of polymerization temperature,the type of initiator, and the solubility of monomer and poro-gen. Toluene, ethanol, ethyl acetate isopropanol, acetonitrile andmethanol were not adopted for their limited solubility for PEG-6000. At last, DMF was selected as the solvent, as well as theco-porogen, for the polymerization reaction.
3.1.2. Permeability of the monolithic materialThe permeability (K) of a porous medium is a measure of its
capacity to transmit a fluid driven by an imposed pressure dropacross the column. According to Darcy’s law, permeability (K) canbe expressed as follows [16]:
K = (�L/�P) × � (1)
where � (Pa s) is the dynamic viscosity of eluent, � (m/s) the linearvelocity of mobile phase, L (m) the effective column length and �P(Pa) the pressure drop.
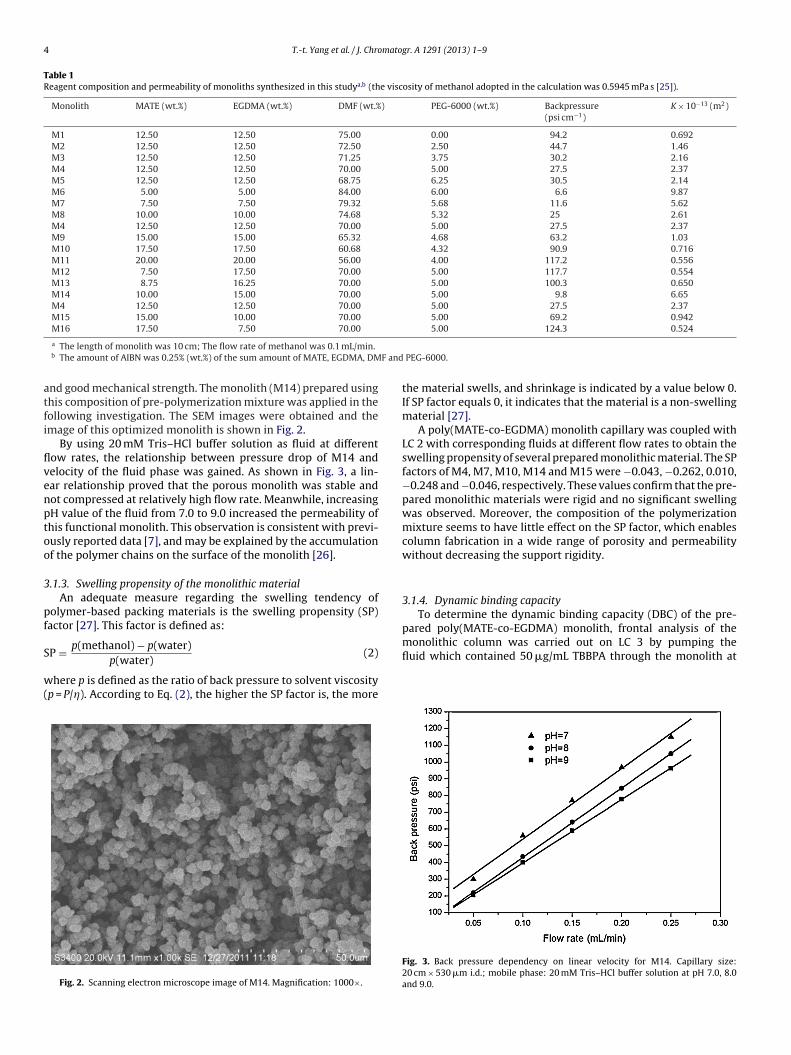
It is known that the composition of pre-polymerization mixturehas great influence on the monolithic structure [23]. Insufficientaddition of porogen would result in uniform monolithic structureand sample solution would have to pass through the column underlarge backpressure. On the opposite, more solvent is required ifthe amount of porogen is large. High crosslinker to monomer ratiocan lead to the monolithic material with a larger specific surfacearea. However, low monomer content will lead to little reactivesites and poor permeability of the monolithic material due to exces-sive crosslinking. On the other hand, low crosslinker to monomerratio will lead to weak rigidity of the material [24]. To reduce thebackpressure, several parameters including PEG-6000 to DMF ratio,total monomer to porogen ratio, and crosslinker to monomer ratiowere optimized carefully to prepare the monolith with appropriateporosity (Table 1). First, the monomer and the crosslinker amountswere kept constant while the content of the porogen was changed.Next, the ratio of crosslinker to monomer and the ratio of PEG-6000to DMF were kept as constants while the ratio of total monomerto total porogen was changed. Last, the amount of PEG-6000 andthe amount of DMF were remained unchanged while the ratio ofcrosslinker to monomer was changed.
A poly(MATE-co-EGDMA) monolith capillary (10 cm in length)was coupled with LC 2. The column pressure with methanol as the
fluid at 0.1 mL/min was recorded. The K values of each monolithprepared in this study are listed in Table 1.After optimization, the mixture of 10% MATE, 15% EGDMA, 70%DMF and 5% PEG-6000 (wt.%) can provide suitable permeability
4 T.-t. Yang et al. / J. Chromatogr. A 1291 (2013) 1– 9
Table 1Reagent composition and permeability of monoliths synthesized in this studya,b (the viscosity of methanol adopted in the calculation was 0.5945 mPa s [25]).
Monolith MATE (wt.%) EGDMA (wt.%) DMF (wt.%) PEG-6000 (wt.%) Backpressure(psi cm−1)
K × 10−13 (m2)
M1 12.50 12.50 75.00 0.00 94.2 0.692M2 12.50 12.50 72.50 2.50 44.7 1.46M3 12.50 12.50 71.25 3.75 30.2 2.16M4 12.50 12.50 70.00 5.00 27.5 2.37M5 12.50 12.50 68.75 6.25 30.5 2.14M6 5.00 5.00 84.00 6.00 6.6 9.87M7 7.50 7.50 79.32 5.68 11.6 5.62M8 10.00 10.00 74.68 5.32 25 2.61M4 12.50 12.50 70.00 5.00 27.5 2.37M9 15.00 15.00 65.32 4.68 63.2 1.03M10 17.50 17.50 60.68 4.32 90.9 0.716M11 20.00 20.00 56.00 4.00 117.2 0.556M12 7.50 17.50 70.00 5.00 117.7 0.554M13 8.75 16.25 70.00 5.00 100.3 0.650M14 10.00 15.00 70.00 5.00 9.8 6.65M4 12.50 12.50 70.00 5.00 27.5 2.37M15 15.00 10.00 70.00 5.00 69.2 0.942M16 17.50 7.50 70.00 5.00 124.3 0.524
F and
atfi
flvenptoo
3
pf
S
w(
monolithic column was carried out on LC 3 by pumping the
a The length of monolith was 10 cm; The flow rate of methanol was 0.1 mL/min.b The amount of AIBN was 0.25% (wt.%) of the sum amount of MATE, EGDMA, DM
nd good mechanical strength. The monolith (M14) prepared usinghis composition of pre-polymerization mixture was applied in theollowing investigation. The SEM images were obtained and themage of this optimized monolith is shown in Fig. 2.
By using 20 mM Tris–HCl buffer solution as fluid at differentow rates, the relationship between pressure drop of M14 andelocity of the fluid phase was gained. As shown in Fig. 3, a lin-ar relationship proved that the porous monolith was stable andot compressed at relatively high flow rate. Meanwhile, increasingH value of the fluid from 7.0 to 9.0 increased the permeability ofhis functional monolith. This observation is consistent with previ-usly reported data [7], and may be explained by the accumulationf the polymer chains on the surface of the monolith [26].
.1.3. Swelling propensity of the monolithic materialAn adequate measure regarding the swelling tendency of
olymer-based packing materials is the swelling propensity (SP)actor [27]. This factor is defined as:
p(methanol) − p(water)
P =p(water)(2)
here p is defined as the ratio of back pressure to solvent viscosityp = P/�). According to Eq. (2), the higher the SP factor is, the more
Fig. 2. Scanning electron microscope image of M14. Magnification: 1000×.
PEG-6000.
the material swells, and shrinkage is indicated by a value below 0.If SP factor equals 0, it indicates that the material is a non-swellingmaterial [27].
A poly(MATE-co-EGDMA) monolith capillary was coupled withLC 2 with corresponding fluids at different flow rates to obtain theswelling propensity of several prepared monolithic material. The SPfactors of M4, M7, M10, M14 and M15 were −0.043, −0.262, 0.010,−0.248 and −0.046, respectively. These values confirm that the pre-pared monolithic materials were rigid and no significant swellingwas observed. Moreover, the composition of the polymerizationmixture seems to have little effect on the SP factor, which enablescolumn fabrication in a wide range of porosity and permeabilitywithout decreasing the support rigidity.
3.1.4. Dynamic binding capacityTo determine the dynamic binding capacity (DBC) of the pre-
pared poly(MATE-co-EGDMA) monolith, frontal analysis of the
fluid which contained 50 �g/mL TBBPA through the monolith at
Fig. 3. Back pressure dependency on linear velocity for M14. Capillary size:20 cm × 530 �m i.d.; mobile phase: 20 mM Tris–HCl buffer solution at pH 7.0, 8.0and 9.0.
T.-t. Yang et al. / J. Chromatogr. A 1291 (2013) 1– 9 5
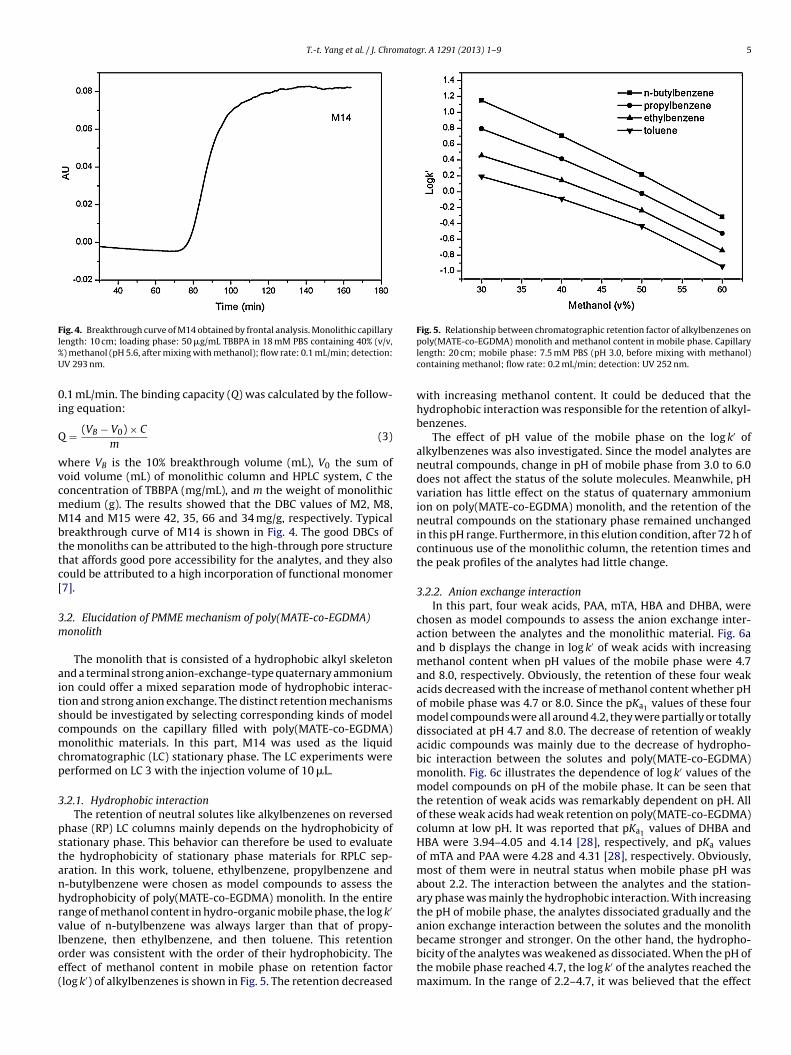
Fig. 4. Breakthrough curve of M14 obtained by frontal analysis. Monolithic capillaryl%U
0i
Q
wvcmMbttc[
3m
aitscmcp
3
pstanhrvloe(
Fig. 5. Relationship between chromatographic retention factor of alkylbenzenes on
ength: 10 cm; loading phase: 50 �g/mL TBBPA in 18 mM PBS containing 40% (v/v,) methanol (pH 5.6, after mixing with methanol); flow rate: 0.1 mL/min; detection:V 293 nm..1 mL/min. The binding capacity (Q) was calculated by the follow-ng equation:
= (VB − V0) × C
m(3)
here VB is the 10% breakthrough volume (mL), V0 the sum ofoid volume (mL) of monolithic column and HPLC system, C theoncentration of TBBPA (mg/mL), and m the weight of monolithicedium (g). The results showed that the DBC values of M2, M8,14 and M15 were 42, 35, 66 and 34 mg/g, respectively. Typical
reakthrough curve of M14 is shown in Fig. 4. The good DBCs ofhe monoliths can be attributed to the high-through pore structurehat affords good pore accessibility for the analytes, and they alsoould be attributed to a high incorporation of functional monomer7].
.2. Elucidation of PMME mechanism of poly(MATE-co-EGDMA)onolith
The monolith that is consisted of a hydrophobic alkyl skeletonnd a terminal strong anion-exchange-type quaternary ammoniumon could offer a mixed separation mode of hydrophobic interac-ion and strong anion exchange. The distinct retention mechanismshould be investigated by selecting corresponding kinds of modelompounds on the capillary filled with poly(MATE-co-EGDMA)onolithic materials. In this part, M14 was used as the liquid
hromatographic (LC) stationary phase. The LC experiments wereerformed on LC 3 with the injection volume of 10 �L.
.2.1. Hydrophobic interactionThe retention of neutral solutes like alkylbenzenes on reversed
hase (RP) LC columns mainly depends on the hydrophobicity oftationary phase. This behavior can therefore be used to evaluatehe hydrophobicity of stationary phase materials for RPLC sep-ration. In this work, toluene, ethylbenzene, propylbenzene and-butylbenzene were chosen as model compounds to assess theydrophobicity of poly(MATE-co-EGDMA) monolith. In the entireange of methanol content in hydro-organic mobile phase, the log k′
alue of n-butylbenzene was always larger than that of propy-
benzene, then ethylbenzene, and then toluene. This retentionrder was consistent with the order of their hydrophobicity. Theffect of methanol content in mobile phase on retention factorlog k′) of alkylbenzenes is shown in Fig. 5. The retention decreasedpoly(MATE-co-EGDMA) monolith and methanol content in mobile phase. Capillarylength: 20 cm; mobile phase: 7.5 mM PBS (pH 3.0, before mixing with methanol)containing methanol; flow rate: 0.2 mL/min; detection: UV 252 nm.
with increasing methanol content. It could be deduced that thehydrophobic interaction was responsible for the retention of alkyl-benzenes.
The effect of pH value of the mobile phase on the log k′ ofalkylbenzenes was also investigated. Since the model analytes areneutral compounds, change in pH of mobile phase from 3.0 to 6.0does not affect the status of the solute molecules. Meanwhile, pHvariation has little effect on the status of quaternary ammoniumion on poly(MATE-co-EGDMA) monolith, and the retention of theneutral compounds on the stationary phase remained unchangedin this pH range. Furthermore, in this elution condition, after 72 h ofcontinuous use of the monolithic column, the retention times andthe peak profiles of the analytes had little change.
3.2.2. Anion exchange interactionIn this part, four weak acids, PAA, mTA, HBA and DHBA, were
chosen as model compounds to assess the anion exchange inter-action between the analytes and the monolithic material. Fig. 6aand b displays the change in log k′ of weak acids with increasingmethanol content when pH values of the mobile phase were 4.7and 8.0, respectively. Obviously, the retention of these four weakacids decreased with the increase of methanol content whether pHof mobile phase was 4.7 or 8.0. Since the pKa1 values of these fourmodel compounds were all around 4.2, they were partially or totallydissociated at pH 4.7 and 8.0. The decrease of retention of weaklyacidic compounds was mainly due to the decrease of hydropho-bic interaction between the solutes and poly(MATE-co-EGDMA)monolith. Fig. 6c illustrates the dependence of log k′ values of themodel compounds on pH of the mobile phase. It can be seen thatthe retention of weak acids was remarkably dependent on pH. Allof these weak acids had weak retention on poly(MATE-co-EGDMA)column at low pH. It was reported that pKa1 values of DHBA andHBA were 3.94–4.05 and 4.14 [28], respectively, and pKa valuesof mTA and PAA were 4.28 and 4.31 [28], respectively. Obviously,most of them were in neutral status when mobile phase pH wasabout 2.2. The interaction between the analytes and the station-ary phase was mainly the hydrophobic interaction. With increasingthe pH of mobile phase, the analytes dissociated gradually and theanion exchange interaction between the solutes and the monolith
became stronger and stronger. On the other hand, the hydropho-bicity of the analytes was weakened as dissociated. When the pH ofthe mobile phase reached 4.7, the log k′ of the analytes reached themaximum. In the range of 2.2–4.7, it was believed that the effect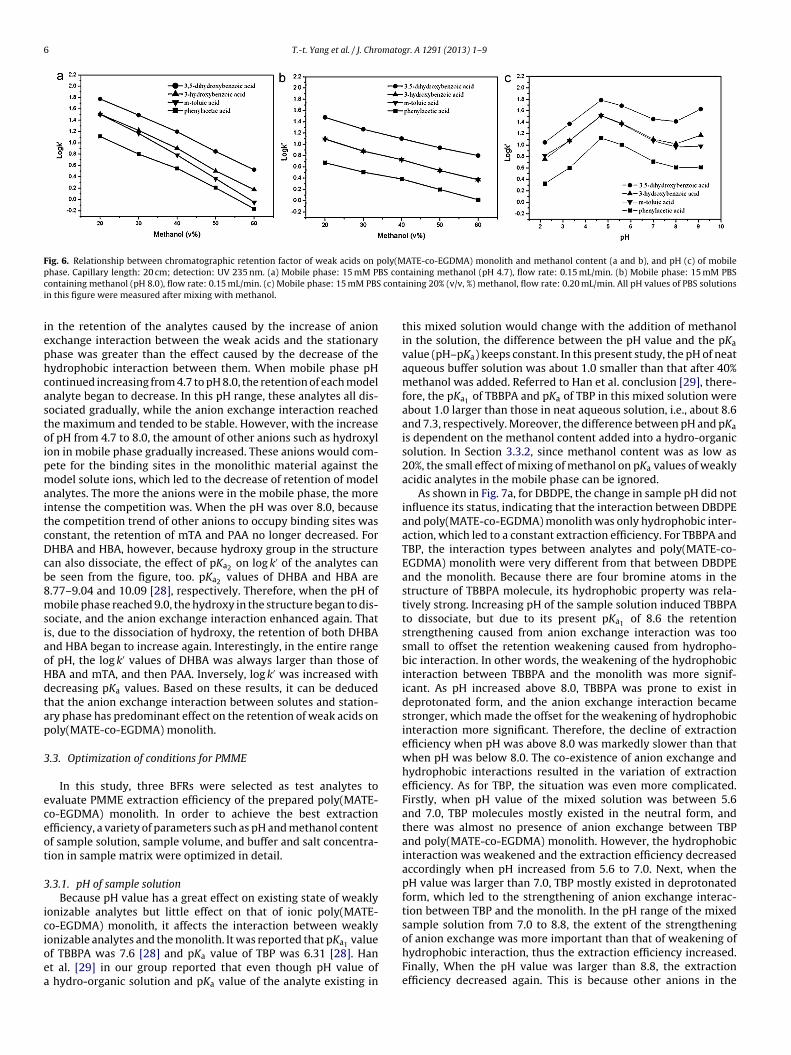
6 T.-t. Yang et al. / J. Chromatogr. A 1291 (2013) 1– 9
Fig. 6. Relationship between chromatographic retention factor of weak acids on poly(MATE-co-EGDMA) monolith and methanol content (a and b), and pH (c) of mobilep BS conc contai
iephcastoipmaitcDcb8msiaoHdtap
3
eceot
3
icioea
hase. Capillary length: 20 cm; detection: UV 235 nm. (a) Mobile phase: 15 mM Pontaining methanol (pH 8.0), flow rate: 0.15 mL/min. (c) Mobile phase: 15 mM PBSn this figure were measured after mixing with methanol.
n the retention of the analytes caused by the increase of anionxchange interaction between the weak acids and the stationaryhase was greater than the effect caused by the decrease of theydrophobic interaction between them. When mobile phase pHontinued increasing from 4.7 to pH 8.0, the retention of each modelnalyte began to decrease. In this pH range, these analytes all dis-ociated gradually, while the anion exchange interaction reachedhe maximum and tended to be stable. However, with the increasef pH from 4.7 to 8.0, the amount of other anions such as hydroxylon in mobile phase gradually increased. These anions would com-ete for the binding sites in the monolithic material against theodel solute ions, which led to the decrease of retention of model
nalytes. The more the anions were in the mobile phase, the morentense the competition was. When the pH was over 8.0, becausehe competition trend of other anions to occupy binding sites wasonstant, the retention of mTA and PAA no longer decreased. ForHBA and HBA, however, because hydroxy group in the structurean also dissociate, the effect of pKa2 on log k′ of the analytes cane seen from the figure, too. pKa2 values of DHBA and HBA are.77–9.04 and 10.09 [28], respectively. Therefore, when the pH ofobile phase reached 9.0, the hydroxy in the structure began to dis-
ociate, and the anion exchange interaction enhanced again. Thats, due to the dissociation of hydroxy, the retention of both DHBAnd HBA began to increase again. Interestingly, in the entire rangef pH, the log k′ values of DHBA was always larger than those ofBA and mTA, and then PAA. Inversely, log k′ was increased withecreasing pKa values. Based on these results, it can be deducedhat the anion exchange interaction between solutes and station-ry phase has predominant effect on the retention of weak acids onoly(MATE-co-EGDMA) monolith.
.3. Optimization of conditions for PMME
In this study, three BFRs were selected as test analytes tovaluate PMME extraction efficiency of the prepared poly(MATE-o-EGDMA) monolith. In order to achieve the best extractionfficiency, a variety of parameters such as pH and methanol contentf sample solution, sample volume, and buffer and salt concentra-ion in sample matrix were optimized in detail.
.3.1. pH of sample solutionBecause pH value has a great effect on existing state of weakly
onizable analytes but little effect on that of ionic poly(MATE-o-EGDMA) monolith, it affects the interaction between weakly
onizable analytes and the monolith. It was reported that pKa1 valuef TBBPA was 7.6 [28] and pKa value of TBP was 6.31 [28]. Hant al. [29] in our group reported that even though pH value ofhydro-organic solution and pKa value of the analyte existing in
taining methanol (pH 4.7), flow rate: 0.15 mL/min. (b) Mobile phase: 15 mM PBSining 20% (v/v, %) methanol, flow rate: 0.20 mL/min. All pH values of PBS solutions
this mixed solution would change with the addition of methanolin the solution, the difference between the pH value and the pKa
value (pH–pKa) keeps constant. In this present study, the pH of neataqueous buffer solution was about 1.0 smaller than that after 40%methanol was added. Referred to Han et al. conclusion [29], there-fore, the pKa1 of TBBPA and pKa of TBP in this mixed solution wereabout 1.0 larger than those in neat aqueous solution, i.e., about 8.6and 7.3, respectively. Moreover, the difference between pH and pKa
is dependent on the methanol content added into a hydro-organicsolution. In Section 3.3.2, since methanol content was as low as20%, the small effect of mixing of methanol on pKa values of weaklyacidic analytes in the mobile phase can be ignored.
As shown in Fig. 7a, for DBDPE, the change in sample pH did notinfluence its status, indicating that the interaction between DBDPEand poly(MATE-co-EGDMA) monolith was only hydrophobic inter-action, which led to a constant extraction efficiency. For TBBPA andTBP, the interaction types between analytes and poly(MATE-co-EGDMA) monolith were very different from that between DBDPEand the monolith. Because there are four bromine atoms in thestructure of TBBPA molecule, its hydrophobic property was rela-tively strong. Increasing pH of the sample solution induced TBBPAto dissociate, but due to its present pKa1 of 8.6 the retentionstrengthening caused from anion exchange interaction was toosmall to offset the retention weakening caused from hydropho-bic interaction. In other words, the weakening of the hydrophobicinteraction between TBBPA and the monolith was more signif-icant. As pH increased above 8.0, TBBPA was prone to exist indeprotonated form, and the anion exchange interaction becamestronger, which made the offset for the weakening of hydrophobicinteraction more significant. Therefore, the decline of extractionefficiency when pH was above 8.0 was markedly slower than thatwhen pH was below 8.0. The co-existence of anion exchange andhydrophobic interactions resulted in the variation of extractionefficiency. As for TBP, the situation was even more complicated.Firstly, when pH value of the mixed solution was between 5.6and 7.0, TBP molecules mostly existed in the neutral form, andthere was almost no presence of anion exchange between TBPand poly(MATE-co-EGDMA) monolith. However, the hydrophobicinteraction was weakened and the extraction efficiency decreasedaccordingly when pH increased from 5.6 to 7.0. Next, when thepH value was larger than 7.0, TBP mostly existed in deprotonatedform, which led to the strengthening of anion exchange interac-tion between TBP and the monolith. In the pH range of the mixedsample solution from 7.0 to 8.8, the extent of the strengthening
of anion exchange was more important than that of weakening ofhydrophobic interaction, thus the extraction efficiency increased.Finally, When the pH value was larger than 8.8, the extractionefficiency decreased again. This is because other anions in the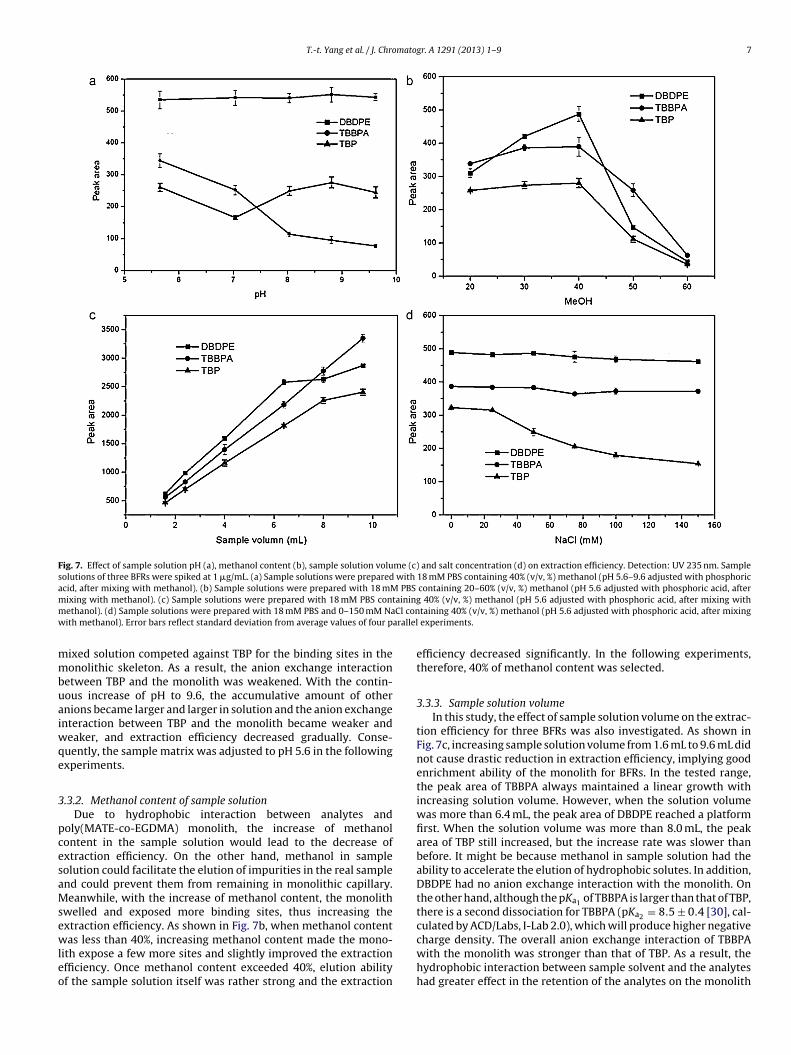
T.-t. Yang et al. / J. Chromatogr. A 1291 (2013) 1– 9 7
Fig. 7. Effect of sample solution pH (a), methanol content (b), sample solution volume (c) and salt concentration (d) on extraction efficiency. Detection: UV 235 nm. Samplesolutions of three BFRs were spiked at 1 �g/mL. (a) Sample solutions were prepared with 18 mM PBS containing 40% (v/v, %) methanol (pH 5.6–9.6 adjusted with phosphoricacid, after mixing with methanol). (b) Sample solutions were prepared with 18 mM PBS containing 20–60% (v/v, %) methanol (pH 5.6 adjusted with phosphoric acid, afterm ainingm Cl conw arallel
mmbuaiwqe
3
pcesaMsewleo
ixing with methanol). (c) Sample solutions were prepared with 18 mM PBS contethanol). (d) Sample solutions were prepared with 18 mM PBS and 0–150 mM Naith methanol). Error bars reflect standard deviation from average values of four p
ixed solution competed against TBP for the binding sites in theonolithic skeleton. As a result, the anion exchange interaction
etween TBP and the monolith was weakened. With the contin-ous increase of pH to 9.6, the accumulative amount of othernions became larger and larger in solution and the anion exchangenteraction between TBP and the monolith became weaker and
eaker, and extraction efficiency decreased gradually. Conse-uently, the sample matrix was adjusted to pH 5.6 in the followingxperiments.
.3.2. Methanol content of sample solutionDue to hydrophobic interaction between analytes and
oly(MATE-co-EGDMA) monolith, the increase of methanolontent in the sample solution would lead to the decrease ofxtraction efficiency. On the other hand, methanol in sampleolution could facilitate the elution of impurities in the real samplend could prevent them from remaining in monolithic capillary.eanwhile, with the increase of methanol content, the monolith
welled and exposed more binding sites, thus increasing thextraction efficiency. As shown in Fig. 7b, when methanol content
as less than 40%, increasing methanol content made the mono-ith expose a few more sites and slightly improved the extractionfficiency. Once methanol content exceeded 40%, elution abilityf the sample solution itself was rather strong and the extraction
40% (v/v, %) methanol (pH 5.6 adjusted with phosphoric acid, after mixing withtaining 40% (v/v, %) methanol (pH 5.6 adjusted with phosphoric acid, after mixing
experiments.
efficiency decreased significantly. In the following experiments,therefore, 40% of methanol content was selected.
3.3.3. Sample solution volumeIn this study, the effect of sample solution volume on the extrac-
tion efficiency for three BFRs was also investigated. As shown inFig. 7c, increasing sample solution volume from 1.6 mL to 9.6 mL didnot cause drastic reduction in extraction efficiency, implying goodenrichment ability of the monolith for BFRs. In the tested range,the peak area of TBBPA always maintained a linear growth withincreasing solution volume. However, when the solution volumewas more than 6.4 mL, the peak area of DBDPE reached a platformfirst. When the solution volume was more than 8.0 mL, the peakarea of TBP still increased, but the increase rate was slower thanbefore. It might be because methanol in sample solution had theability to accelerate the elution of hydrophobic solutes. In addition,DBDPE had no anion exchange interaction with the monolith. Onthe other hand, although the pKa1 of TBBPA is larger than that of TBP,there is a second dissociation for TBBPA (pKa2 = 8.5 ± 0.4 [30], cal-culated by ACD/Labs, I-Lab 2.0), which will produce higher negative
charge density. The overall anion exchange interaction of TBBPAwith the monolith was stronger than that of TBP. As a result, thehydrophobic interaction between sample solvent and the analyteshad greater effect in the retention of the analytes on the monolith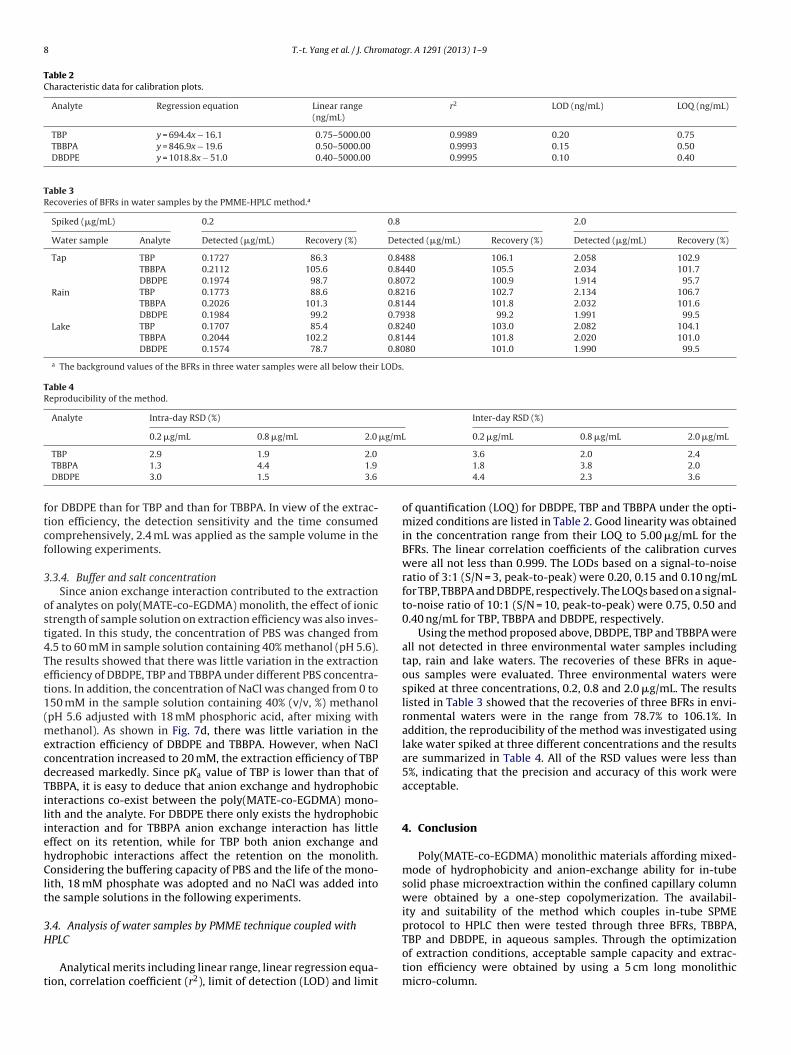
8 T.-t. Yang et al. / J. Chromatogr. A 1291 (2013) 1– 9
Table 2Characteristic data for calibration plots.
Analyte Regression equation Linear range(ng/mL)
r2 LOD (ng/mL) LOQ (ng/mL)
TBP y = 694.4x − 16.1 0.75–5000.00 0.9989 0.20 0.75TBBPA y = 846.9x − 19.6 0.50–5000.00 0.9993 0.15 0.50DBDPE y = 1018.8x − 51.0 0.40–5000.00 0.9995 0.10 0.40
Table 3Recoveries of BFRs in water samples by the PMME-HPLC method.a
Spiked (�g/mL) 0.2 0.8 2.0
Water sample Analyte Detected (�g/mL) Recovery (%) Detected (�g/mL) Recovery (%) Detected (�g/mL) Recovery (%)
Tap TBP 0.1727 86.3 0.8488 106.1 2.058 102.9TBBPA 0.2112 105.6 0.8440 105.5 2.034 101.7DBDPE 0.1974 98.7 0.8072 100.9 1.914 95.7
Rain TBP 0.1773 88.6 0.8216 102.7 2.134 106.7TBBPA 0.2026 101.3 0.8144 101.8 2.032 101.6DBDPE 0.1984 99.2 0.7938 99.2 1.991 99.5
Lake TBP 0.1707 85.4 0.8240 103.0 2.082 104.1TBBPA 0.2044 102.2 0.8144 101.8 2.020 101.0DBDPE 0.1574 78.7 0.8080 101.0 1.990 99.5
a The background values of the BFRs in three water samples were all below their LODs.
Table 4Reproducibility of the method.
Analyte Intra-day RSD (%) Inter-day RSD (%)
0.2 �g/mL 0.8 �g/mL 2.0 �g/mL 0.2 �g/mL 0.8 �g/mL 2.0 �g/mL
ftcf
3
ost4Tet1(mecdTiliehClt
3H
t
TBP 2.9 1.9 2.0
TBBPA 1.3 4.4 1.9
DBDPE 3.0 1.5 3.6
or DBDPE than for TBP and than for TBBPA. In view of the extrac-ion efficiency, the detection sensitivity and the time consumedomprehensively, 2.4 mL was applied as the sample volume in theollowing experiments.
.3.4. Buffer and salt concentrationSince anion exchange interaction contributed to the extraction
f analytes on poly(MATE-co-EGDMA) monolith, the effect of ionictrength of sample solution on extraction efficiency was also inves-igated. In this study, the concentration of PBS was changed from.5 to 60 mM in sample solution containing 40% methanol (pH 5.6).he results showed that there was little variation in the extractionfficiency of DBDPE, TBP and TBBPA under different PBS concentra-ions. In addition, the concentration of NaCl was changed from 0 to50 mM in the sample solution containing 40% (v/v, %) methanolpH 5.6 adjusted with 18 mM phosphoric acid, after mixing with
ethanol). As shown in Fig. 7d, there was little variation in thextraction efficiency of DBDPE and TBBPA. However, when NaCloncentration increased to 20 mM, the extraction efficiency of TBPecreased markedly. Since pKa value of TBP is lower than that ofBBPA, it is easy to deduce that anion exchange and hydrophobicnteractions co-exist between the poly(MATE-co-EGDMA) mono-ith and the analyte. For DBDPE there only exists the hydrophobicnteraction and for TBBPA anion exchange interaction has littleffect on its retention, while for TBP both anion exchange andydrophobic interactions affect the retention on the monolith.onsidering the buffering capacity of PBS and the life of the mono-
ith, 18 mM phosphate was adopted and no NaCl was added intohe sample solutions in the following experiments.
.4. Analysis of water samples by PMME technique coupled with
PLCAnalytical merits including linear range, linear regression equa-ion, correlation coefficient (r2), limit of detection (LOD) and limit
3.6 2.0 2.41.8 3.8 2.04.4 2.3 3.6
of quantification (LOQ) for DBDPE, TBP and TBBPA under the opti-mized conditions are listed in Table 2. Good linearity was obtainedin the concentration range from their LOQ to 5.00 �g/mL for theBFRs. The linear correlation coefficients of the calibration curveswere all not less than 0.999. The LODs based on a signal-to-noiseratio of 3:1 (S/N = 3, peak-to-peak) were 0.20, 0.15 and 0.10 ng/mLfor TBP, TBBPA and DBDPE, respectively. The LOQs based on a signal-to-noise ratio of 10:1 (S/N = 10, peak-to-peak) were 0.75, 0.50 and0.40 ng/mL for TBP, TBBPA and DBDPE, respectively.
Using the method proposed above, DBDPE, TBP and TBBPA wereall not detected in three environmental water samples includingtap, rain and lake waters. The recoveries of these BFRs in aque-ous samples were evaluated. Three environmental waters werespiked at three concentrations, 0.2, 0.8 and 2.0 �g/mL. The resultslisted in Table 3 showed that the recoveries of three BFRs in envi-ronmental waters were in the range from 78.7% to 106.1%. Inaddition, the reproducibility of the method was investigated usinglake water spiked at three different concentrations and the resultsare summarized in Table 4. All of the RSD values were less than5%, indicating that the precision and accuracy of this work wereacceptable.
4. Conclusion
Poly(MATE-co-EGDMA) monolithic materials affording mixed-mode of hydrophobicity and anion-exchange ability for in-tubesolid phase microextraction within the confined capillary columnwere obtained by a one-step copolymerization. The availabil-ity and suitability of the method which couples in-tube SPMEprotocol to HPLC then were tested through three BFRs, TBBPA,
TBP and DBDPE, in aqueous samples. Through the optimizationof extraction conditions, acceptable sample capacity and extrac-tion efficiency were obtained by using a 5 cm long monolithicmicro-column.
mato
A
oNNatvm
R
[[[
[[[
[
[[
[
[
[[[[[[
T.-t. Yang et al. / J. Chro
cknowledgements
This work was supported by National Basic Research Programf China (973 program, 2009CB421601, 2011CB911003), Nationalatural Science Foundation of China (21275069, 90913012),ational Science Funds for Creative Research Groups (21121091),nd Analysis and Test Fund of Nanjing University. Authors alsohank Professor Yu-qi Feng, Department of Chemistry, Wuhan Uni-ersity, for his aspiring and kind help in the preparation of PMMEonolithic materials.
eferences
[1] F. Svec, J. Chromatogr. A 1228 (2012) 250.[2] A. Nordborg, F. Lime, A. Shchukarev, K. Irgum, J. Sep. Sci. 31 (2008) 2143.[3] K. Eder, C.G. Huber, M.R. Buchmeiser, Macromol. Rapid Commun. 28 (2007)
2029.[4] S. Eeltink, E.F. Hilder, L. Geiser, F. Svec, J.M.J. Frechet, G.P. Rozing, P.J. Schoen-
makers, W.T. Kok, J. Sep. Sci. 30 (2007) 407.[5] P. Zakaria, J.P. Hutchinson, N. Avdalovic, Y. Liu, P.R. Haddad, Anal. Chem. 77
(2005) 417.[6] A. Nordborg, E.F. Hilder, Anal. Bioanal. Chem. 394 (2009) 71.[7] Y. Li, B.H. Gu, H.D. Tolley, M.L. Lee, J. Chromatogr. A 1216 (2009) 5525.[8] A. Nordborg, E.F. Hilder, P.R. Haddad, Annu. Rev. Anal. Chem. 4 (2011) 197.[9] C. Dietz, J. Sanz, C. Cámara, J. Chromatogr. A 1103 (2006) 183.
[[[
[
gr. A 1291 (2013) 1– 9 9
10] T. Kumazawa, X.P. Lee, K. Sato, O. Suzuki, Anal. Chim. Acta 494 (2003) 49.11] Y. Liu, Y. Shen, M.L. Lee, Anal. Chem. 69 (1997) 190.12] S.L. Chong, D.X. Wang, J.D. Hayes, B.W. Wilhite, A. Malik, Anal. Chem. 69 (1997)
3889.13] M. Zhang, F. Wei, Y.F. Zhang, J. Nie, Y.Q. Feng, J. Chromatogr. A 1102 (2006) 294.14] J. Yin, B. Hu, M. He, M.M. Zheng, Y.Q. Feng, J. Anal. At. Spectrom. 24 (2009) 76.15] L.F. Zhou, X.G. He, J.Q. Qiao, H.Z. Lian, X. Ge, H.Y. Chen, J. Chromatogr. A 1256
(2012) 15.16] Q.W. Yu, X. Wang, Q. Ma, B.F. Yuan, H.B. He, Y.Q. Feng, Anal. Method 4 (2012)
1538.17] D. Luo, F. Chen, K. Xiao, Y.Q. Feng, Talanta 77 (2009) 1701.18] A. Papachlimitzou, J.L. Barber, S. Losada, P. Bersuder, R.J. Law, J. Chromatogr. A
1219 (2012) 15.19] H. Huang, Z. Lin, Y. Lin, X.B. Sun, Y.Y. Xie, L. Zhang, G.N. Chen, J. Chromatogr. A
1251 (2012) 82.20] A. Abbood, C. Herrenknecht, G. Proczek, S. Descroix, J. Rodrige, M. Taverna, C.
Smadja, Anal. Bioanal. Chem. 400 (2011) 459.21] P.F. Zhang, J. Chen, L. Jia, J. Chromatogr. A 1218 (2011) 3459.22] R.A. Wu, H.F. Zou, M.L. Ye, Z.D. Lei, J.Y. Ni, Anal. Chem. 732 (2001) 4918.23] S. Xie, F. Svec, J.M.J. Frechet, Chem. Mater. 10 (1998) 4072.24] C. Viklund, F. Svec, J.M.J. Frechet, K. Irgum, Chem. Mater. 8 (1996) 744.25] Wiley, http://onlinelibrary.wiley.com/doi/10.1002/9780470423851.app1/pdf26] K. Kontturi, S. Mafe, J.A. Manzanares, B.L. Svarfvar, P. Viinikka, Macromolecules
29 (1996) 5740.
27] C.P. Bisjak, J. Chromatogr. A 1154 (2007) 269.28] Reaxys, https://www.reaxys.com/reaxys/secured/start.do29] S.Y. Han, C. Liang, K. Zou, J.Q. Qiao, H.Z. Lian, X. Ge, H.Y. Chen, Talanta 101 (2012)64.30] ACD/Labs, https://www.ilab.acdlabs.com















![Journal of Inorganic Biochemistry - or.nsfc.gov.cnor.nsfc.gov.cn/bitstream/00001903-5/439248/1/1000008889503.pdf · appreciated [21,22]. Liriodenine is a representative oxoaporphine](https://img.pdfslide.net/doc/110x75/5b63e89d7f8b9a6c178c99a3/journal-of-inorganic-biochemistry-ornsfcgovcnornsfcgovcnbitstream00001903-54392481.jpg)













