Embed Size (px)
Citation preview

Journal of Laboratory and Clinical Medicine
Volume 149, Issue 5, Pages.237-291 (May 2007)
1. Contents Page IFC
2. Masthead Page A1
3. Editorial Advisory Board Page A2
4. Author guidelines Pages A3-A4
Featured New Investigator
5. Transcriptional regulation of podocyte disease Pages 237-242 Sumant S. Chugh
Original Articles
6. Immunopathogenesis of hypersensitivity syndrome reactions to sulfonamides Pages 243-253 Manuela G. Neuman, Neil H. Shear, Izabella M. Malkiewicz, Masud Taeri, Lori E. Shapiro, Norberto Krivoy, Julia Haber, Manuel Gomez, Joel Fish, Robert Cartotto, et al.
7. Apoptosis in ibuprofen-induced Stevens–Johnson syndrome Pages 254-259 Manuela Neuman and Michael Nicar
8. Antitumor and antiinflammatory effects of tetrathiotungstate in comparison with tetrathiomolybdate Pages 260-264 Guoqing Hou, Robert Dick, Chunhua Zeng and George J. Brewer

9. Expression of angiopoietin-1 in osteoblasts and its inhibition by tumor necrosis factor-alpha and interferon-gamma Pages 265-273 Tsuyoshi Kasama, Takeo Isozaki, Tsuyoshi Odai, Mizuho Matsunawa, Kuninobu Wakabayashi, Hiroko T. Takeuchi, Satoshi Matsukura, Mitsuru Adachi, Masakazu Tezuka and Kazuo Kobayashi
10. Advanced glycation end-product-induced mitogenesis is dependent on Janus kinase 2-induced heat shock protein 70 in normal rat kidney interstitial fibroblast cells Pages 274-281 San-Cher Chen, Jinn-Yuh Guh, Hung-Chun Chen, Yu-Lin Yang, Jau-Shyang Huang and Lea-Yea Chuang
11. Impaired integration of endothelial progenitor cells in capillaries of diabetic wounds is reversible with vascular endothelial growth factor infusion Pages 282-291 Ashok K. Singh, Krishnamurthy P. Gudehithlu, Shreya Patri, Natalia O. Litbarg, Perianna Sethupathi, Jose A.L. Arruda and George Dunea
12. Information for readers Page IBC
13. Contents Page OBC

FEATURED NEW INVESTIGATOR237 Transcriptional regulation of podocyte disease
Sumant S. Chugh, Chicago, Ill
ORIGINAL ARTICLES243 Immunopathogenesis of hypersensitivity
syndrome reactions to sulfonamidesManuela G. Neuman, Neil H. Shear,Izabella M. Malkiewicz, Masud Taeri, LoriE. Shapiro, Norberto Krivoy, Julia Haber,Manuel Gomez, Joel Fish, Robert Cartotto,and Lawrence Cohen, Toronto, Ontario,Canada and Haifa, Israel
254 Apoptosis in ibuprofen-induced Stevens–Johnson syndromeManuela Neuman and Michael Nicar,Toronto, Ontario, Canada and Dallas, Tex
260 Antitumor and antiinflammatory effects oftetrathiotungstate in comparison withtetrathiomolybdateGuoqing Hou, Robert Dick, Chunhua Zeng,and George J. Brewer, Ann Arbor, Mich
265 Expression of angiopoietin-1 in osteoblastsand its inhibition by tumor necrosis factor-alpha and interferon-gammaTsuyoshi Kasama, Takeo Isozaki, TsuyoshiOdai, Mizuho Matsunawa, KuninobuWakabayashi, Hiroko T. Takeuchi, SatoshiMatsukura, Mitsuru Adachi, MasakazuTezuka, and Kazuo Kobayashi, Tokyo, Japan
274 Advanced glycation end-product-inducedmitogenesis is dependent on Janus kinase2-induced heat shock protein 70 in normalrat kidney interstitial fibroblast cellsSan-Cher Chen, Jinn-Yuh Guh, Hung-ChunChen, Yu-Lin Yang, Jau-Shyang Huang, andLea-Yea Chuang, Kaohsiung and Tainan,Taiwan
282 Impaired integration of endothelialprogenitor cells in capillaries of diabeticwounds is reversible with vascularendothelial growth factor infusionAshok K. Singh, Krishnamurthy P.Gudehithlu, Shreya Patri, Natalia O.Litbarg, Perianna Sethupathi, Jose A. L.Arruda, and George Dunea, Chicago andMaywood, Ill
READER SERVICES
3A Author Guidelines
C3 Information for Readers
Visit our website at www.elsevier.com/transres
TRANSLATIONAL RESEARCHThe Journal of Laboratory and Clinical Medicine
Volume 149, Number 5, May 2007
Contents
Translational Research: The Journal of Laboratory and Clinical Medicine (ISSN 1931-5244) is published monthly byElsevier, Inc., 360 Park Avenue South, New York, NY 10010-1710. Business and Editorial Offices: 1600 John F. KennedyBlvd., Suite 1800, Philadelphia, PA 19103-2899. Customer Service Office: 6277 Sea Harbor Drive, Orlando, FL 32887-4800. Periodicals postage paid at New York, NY and additional mailing offices.
POSTMASTER: Send address changes to Translational Research, Elsevier Periodicals Customer Service, 6277 Sea HarborDr., Orlando, FL 32887-4800.

TRANSLATIONAL RESEARCHThe Journal of Laboratory and Clinical Medicine
Editor-in-ChiefJEFFREY LAURENCE, MDProfessor of MedicineDivision of Hematology-OncologyWeill Medical College of Cornell UniversityNew York, NY
Associate Editors
Rose S. Fife, MDAssociate Dean for ResearchBarbara F. Kampen Professor of Women’s HealthProfessor of Medicine and Biochemistry andMolecular BiologyIndiana University School of MedicineIndianapolis, [email protected]
Lawrence A. Frohman, MDSection of EndocrinologyDepartment of MedicineUniversity of Illinois at ChicagoChicago, [email protected]
Joe G.N. Garcia, MDChairman, Department of Medicine (Pulmonary)University of Chicago Pritzker School of MedicineChicago, [email protected]
David W. Kamp, MDDivisions of Pulmonary and Critical Care MedicineNorthwestern UniversityChicago, [email protected]
Robert F. Todd, III, MD, PhDChief of Hematology/OncologyUniversity of Michigan Medical CenterAnn Arbor, [email protected]
Managing EditorMichael J. [email protected]
Issue ManagerAndrew P. O’Brien
Central Society for Clinical Research
David W. Kamp, PresidentRose S. Fife, Past PresidentJames B. Martins, President ElectBrian D. Hoit, Vice PresidentJordan P. Metcalf, Secretary-Treasurer
Anne N. Rushing, Executive Director555 East Wells StreetSuite 1100Milwaukee, Wis 53202-3823Fax: [email protected]
Translational ResearchPage 1AMay 2007

TRANSLATIONAL RESEARCHThe Journal of Laboratory and Clinical Medicine
Editorial Advisory Board
Morton F. Arnsdorf, MDDivision of CardiologyUniversity of Chicago HospitalsChicago, [email protected]
John P. Atkinson, MDDivision of RheumatologyWashington University School of MedicineSt. Louis, [email protected]
Robert J. Bache, MDDivision of CardiologyUniversity of Minnesota School of MedicineMinneapolis, [email protected]
George L. Bakris, MDDepartment of Preventative MedicineRush University Medical CenterChicago, [email protected]
Margo P. Cohen, MDEndocrinology and MetabolismUniversity City Science CenterPhiladelphia, [email protected]
Nicholas O. Davidson, MDDepartment of Medicine (Gastroenterology)Washington University School of MedicineSt. Louis, [email protected]
Brooks S. Edwards, MDDivision of CardiologyMayo ClinicRochester, [email protected]
Gary S. Francis, MDDept of Cardiovascular MedicineCleveland Clinic FoundationCleveland, [email protected]
Robert P. Hebbel, MDDivision of Hematology/Oncology/TransplantationUniversity of Minnesota School of MedicineMinneapolis, [email protected]
Edward N. Janoff, MDDivision of Infectious DiseaseUniversity of Colorado, Health Sciences CenterDenver, [email protected]
Julianne Imperato-McGinley, MDDivision of EndocrinologyWeill Medical College of Cornell UniversityNew York, [email protected]
Jeffrey A. Kern, MDPulmonary and Critical Care DivisionCase Western Reserve University School of MedicineCleveland, [email protected]
Alvin I. Mushlin, MDChairman, Dept. of Public HealthWeill Medical College of Cornell UniversityNew York, [email protected]
Dennis E. Niewoehner, MDPulmonary SectionMinneapolis Veterans Affairs Medical CenterMinneapolis, [email protected]
Mark M. Rasenick, Ph.DDepartments of Physiology and Biophysics andPsychiatryUniversity of Illinois at ChicagoChicago, [email protected]
Andrew Talal, MD, MPHDivision of Gastroenterology and HepatologyWeill Medical College of Cornell UniversityNew York, [email protected]
E. Kenneth Weir, MDChief of CardiologyVeteran Affairs Medical CenterMinneapolis, [email protected]
Translational ResearchPage 2A May 2007

JEFFREY LAURENCE, M.D.Translational ResearchDepartment of MedicineWeill Medical College of Cornell University411 East 69th StreetNew York, NY 10021
AUTHOR GUIDELINESEditorial Scope and Policy
Translational Research publishes original investigations in thebroad fields of laboratory and clinical medicine. It aims to expe-dite the translation of scientific discovery into new or improvedstandards of care by promoting a wide-ranging exchange betweenbasic, preclinical, clinical, epidemiologic, and health outcomesresearch. Reports of purely laboratory or animal investigationsshould have the potential for application to human disease, andreports of preliminary human investigations should have the po-tential for advancing our understanding of the biology of humandisease. Reports of public health research should have the poten-tial for application to the clinic, disease prevention, or healthcarepolicy. Case reports/series are encouraged, especially if theyprovide important mechanistic insight or illuminate a novel ther-apeutic principle. Manuscripts that are primarily methodologicwill only be considered if the method is novel, if its developmentposes or answers important biologic questions, or if its descriptionincludes data applying it to a study of potential interest to ourreadership. Papers describing refinements of routine clinical lab-oratory tests are not encouraged.
Review manuscripts are welcomed for both state-of-the-art com-prehensive reviews, directed at research scientists in specificfields, and more general informative reviews, directed at thebroader community of clinical investigators. Originality is criticalin order to contribute to the medical literature, and the perspectiveshould be fresh and the synthesis unique. Authors of reviewsshould realize that the Journal is multidisciplinary and that reviewarticles for such a journal require appropriate interpretive mate-rial. Clarity of presentation is a major criterion for acceptance.
Scientific commentary about published articles may be submittedas a Letter to the Editor. These comments should be directed atconfirming the results (from a different approach), extending theoriginal report, or refuting results or the authors’ interpretation.Reports describing preliminary findings that offer hypothesis-generating insight into a recognized problem may also be sub-mitted as a Letter to the Editor. Maximum length is 1000 words;maximum number of references is 15. The Editor reserves theright to decide on publication of letters, shorten them, removeobjectionable comments, and make other changes in accord withthe style of Translational Research.
Original research of limited scope may be submitted as a BriefReport, and should contain concise accounts of the purpose of thestudy, methods used, results, and discussion. Maximum length is1500 words; maximum number of references is 15.
Papers involving studies in human subjects must be accompanied bystatements that the research was carried out according to the princi-ples of the Declaration of Helsinki, that informed consent wasobtained, and that the author’s institutional review board has ap-proved the study. This statement must be included in the Methods
section. The Journal encourages authors to discuss the ethical con-cerns in research that involves significant risk to participants.
The publisher and editors of Translational Research subscribe tothe definitions of authorship as set forth in the Uniform Require-ments for Manuscripts Submitted to Biomedical Journals; accord-ingly, we expect each listed author to accept full responsibility forthe paper. Manuscripts submitted to Translational Research arereviewed (and ultimately published) with the understanding thatall potential copyright conflicts have been addressed by the au-thor(s) and that all overlap with other publications — by theauthors or by others — have been disclosed. Moreover, in theevent that fraud or other irregularity is alleged within 5 years ofthe appearance of a paper in Translational Research, it is ourexpectation that the authors will at our request produce both theactual data on which the paper was based and documentation ofadequate resources to have carried out the work in question.
It is understood that statements and opinions expressed in articlesand communications are those of the author(s) and not necessarilythose of the editor(s) or publisher, and the editor(s) and publisherdisclaim any responsibility or liability for such material. If themanuscript receives favorable consideration, a form transferringcopyright and confirming authorship will be sent to the corre-sponding author. It must be signed by all authors. If US Govern-ment jurisdiction precludes copyright transfer, provide a specificstatement of exemption.
Review and selectionAll articles are evaluated by the Editor for suitability for consider-ation for publication in Translational Research. Potentially accept-able submissions will also be reviewed in detail by an additionalreferee with expertise in the specific area. All revised manuscripts arecarefully re-examined with no guarantee of acceptance, and authorswill only have two opportunities to make revisions to the samemanuscript. Final acceptance is based on originality, significance,documentation of conclusions, and form of presentation.
Manuscript preparation and organizationManuscripts should be submitted online at http://ees.elsevier.com/tran-sres. The website guides authors stepwise through the submission pro-cess. Submission items include a cover letter (save as a separate file forupload), the manuscript (including title page, abstract, main text, refer-ences, and figure legends), tables, and figures. Revised manuscriptsshould also be accompanied by a unique file (separate from the coverletter) with responses to reviewers’ comments. The preferred order offiles is as follows: cover letter, response to reviews (revised manuscriptsonly), manuscript file(s), table(s), figure(s). Text, tables and figures areuploaded separately. Do not import figures or tables into the text of thearticle. Original source files (not PDF files) are required for onlinesubmission. Files should be labeled with appropriate and descriptive filenames (e.g., SmithText.doc, Fig1.tiff, Table3.doc). The manuscript mustbe written in English and typed double-spaced.
Please send queries concerning the submission or review processto the Managing Editor, Michael Franklin, at [email protected].
Authors who are unable to use the online submission system mustcontact the Managing Editor prior to submission to discuss alter-nate options.
The main sections of all manuscripts should be indicated withcapitalized head set flush with the left margin. The organization ofreview articles should be appropriate to the content of the review.The following organization is expected for manuscripts describ-ing original investigations.
Translational ResearchPage 3AMay 2007

Title page. This should include the affiliations of the author(s)and the sources of support for the investigation, when appropriate.Also indicate the address of the author to whom correspondenceand reprint requests should be directed; include business and faxnumbers.
Abstract. An abstract of 250 words or less should orient thereader to the problem, describe the major observations, and statethe principal conclusions, all in one paragraph without subhead-ings. It should be easily understood without reference to the text.
Running head and abbreviations. Include an abbreviated title(45 characters or less) and a list of definitions of any abbreviationsused in the manuscript. Because this is a multidisciplinary journal,abbreviations should be kept to a minimum. It is preferable to useonly universally understood abbreviations. Only standard chem-ical or nonproprietary pharmaceutical nomenclature should beused. All abbreviations must be defined separately in the title,abstract, and text of the manuscript.
Introduction. This should be organized and expressed in a way thatwill introduce and orient the general scientific reader to the topic.
Methods. The Methods section should include a description ofthe statistical methods used.
Results. These may be presented in tables or figures that shouldnot duplicate the text. All tables and figures must be numbered inthe order of their mention in the text.
Tables should be typed double-spaced as separate documentsfrom the text of the manuscript. Do not use ditto marks. Center thetable number at the top of the page and the title of the tablebeneath it.
Black-and-white illustrations are permitted without extra chargeto the author. Figures must be of suitable quality for publication.Resolution for halftone images should be 300 dots per inch (dpi)at the size it will appear in print. Resolution for line art should be1200 dpi at the size it will appear in print. All images should beat least 5 inches wide. Preferably, format for digital files should beTIFF (Tagged image file format). JPEG (Joint Photographic Ex-perts Group) format can be used for halftone photographs if theimage is saved with minimum compression. Do not save digitalfiles in GIF (Graphics Interchange Format) format. Do not embedfigures in the manuscript’s word processing file. Images submittedin software-specific proprietary formats (e.g., PowerPoint, Har-vard Graphics, Visio, etc.) must meet specific conditions. Instruc-tions for preparing artwork for online submission can be found atwww.ees.elsevier.com/transres.
Consistency in size of illustrations within the article is stronglypreferred. Any special instructions regarding sizing should beclearly noted. Arrangements for the use of figures requiring spe-cial handling may be made with the Editor at an additional charge.
Legends for figures should be typed double-spaced on a separatepage after the References.
Avoid duplicating previously published material. If it is necessaryto use a copyrighted table, figure, or data, the figure legend ortable footnote should give full credit to the original source andshould state that the material is reprinted with permission.
Discussion. The discussion should set the results in context andset forth the major conclusions of the authors. Information fromthe Introduction or Results section should not be repeated unlessnecessary for clarity. The authors’ speculations concerning thepossible implications of the findings may be presented in thissection but should be clearly separated from the direct inferences.
As the Journal and its audience are multidisciplinary, we encour-age the inclusion of a short concluding paragraph, under thesubheading “Speculations,” which would point out and clearlydenote such broader possibilities for the general readership.
Acknowledgment(s)In addition to the customary recognition of nonauthors who havebeen helpful to the work described, this section must disclose anysubstantive conflicts of interest.
References. All references must be cited in the text. These should benumbered serially in the text and listed, in the order cited, after anypersonal acknowledgments. Reference format should follow the styleoutlined in “Uniform Requirements for Manuscripts Submitted toBiomedical Journals“ (Vancouver style). Journal abbreviationsshould conform to the style of the Cumulated Index Medicus. If notlisted in the CIM, journal titles should not be abbreviated. Authorsare responsible for the accuracy of their references. Note: Unpub-lished results and personal communications do not belong in thereference list; they should be cited parenthetically in the text.
EXAMPLES (if six or fewer authors, list all; if seven or more, listfirst six and add et al.):
Journal articles:You CH, Lee KY, Chey WY, Menguy R. Electrogastrographicstudy of patients with unexplained nausea, bloating and vomiting.Gastroenterology 1980;79:311-4.
Books:Langer M, Chiandussi L, Chopra IJ, Martini L, editors. Theendocrines and the liver. London: Academic Press, 1984:9-34.
Chapters in books:Gustafsson JA, Eneroth P, Hokfelt T, Mode A, Norstedt G.Studies on the hypothalamo-pituitary axis: a novel concept inregulation of steroid and drug metabolism. In: Langer M, Chian-dussi L, Chapra IJ, Martini L, editors. The endocrines and theliver. London: Academic Press, 1984:9-34.
Permissions and patient consent formsDirect quotations, tables, or illustrations that have appeared in copy-righted material must be accompanied by written permission for theiruse from the copyright owner and original author, along with com-plete information as to source. Photographs of identifiable personsmust be accompanied by signed releases showing informed consent.
Required special itemsAt the time of submission, the Journal requires an explicit state-ment by the senior corresponding author warranting that themanuscript, as submitted, has been reviewed by and approved byall named authors; that the corresponding author is empowered byall of the authors to act on their behalf with respect to thesubmission of the manuscript; that the article is original; that thearticle does not infringe upon any copyright or other proprietaryright of any third party; that neither the text nor the data reportedhave been published previously (abstracts excepted); and that thearticle or a substantially similar article is not under considerationby another journal at this time. Include this author agreement inthe cover letter to be submitted online.
Patricia L. HoganPublisher, US Health Sciences JournalsElsevier360 Park Ave. SouthNew York, NY [email protected]
Translational ResearchPage 4A May 2007

FT
S
C
Wo
Fw
SJ
SSAYND
R4S
1
©
d
EATURED NEW INVESTIGATORranscriptional regulation of podocyte disease
UMANT S. CHUGH
HICAGO, ILL
The podocyte is a highly specialized visceral epithelial cell that forms the outermostlayer of the glomerular capillary loop and plays a critical role in the maintenanceof the glomerular filtration barrier. Several transcriptional factors regulate the podo-cyte function under normal and disease conditions. In this review, the role of Wilmstumor 1 (WT1), LIM homeobox transcription factor 1, beta (Lmx1b), pod1, pax-2,kreisler, nuclear factor-kappa B (NF-�B), smad7, and zinc fingers and homeoboxes(ZHX) proteins in the development of podocyte disease is outlined. The regulation ofseveral important podocyte genes, including transcriptional factors, by ZHX pro-teins, their predominant non-nuclear localization in the normal in vivo podocyte,and changes in ZHX expression related to the development of minimal changedisease and focal and segmental glomerulosclerosis are discussed. Finally, somefuture therapeutic strategies for glomerular disease are proposed. (TranslationalResearch 2007;149:237–242)
Abbreviations: BASP1 � brain acid soluble protein 1; CD2AP � CD2-associated protein; FGS �focal glomerulosclerosis; GEC � glomerular epithelial cell; Lmx1b � LIM homeobox transcrip-tion factor 1, beta; MCD � minimal change disease; NF-�B � nuclear factor-kappa B; PAN �puromycin aminonucleoside nephrosis; WT1 � Wilms tumor 1; WTIP � WT1-interacting protein;
ZHX � zinc fingers and homeoboxesttOjiotp
W
arsWiIae
ith an ever increasing number of podocyteexpressed genes being identified, there isgrowing interest in their role in the devel-
pment of disease and in their transcriptional regula-
rom the Division of Nephrology, Department of Medicine, North-estern University, Feinberg School of Medicine, Chicago, Ill.
ubmitted for publication December 29, 2006; revision submittedanuary 8, 2007; accepted for publication January 8, 2007.
upported by the following research grants: Norman S. Coplonatellite Research Grant, Carl W. Gottschalk Research Scholarward of the American Society of Nephrology, the Amgen, Inc. -oung Investigator Grant from the National Kidney Foundation, andational Institutes of Health grants DK61275, DK068203, andK077073-01.
eprint requests: Sumant S. Chugh, MD, Division of Nephrology, Tarry-753 Northwestern University, Feinberg School of Medicine, 320 Eastuperior, Chicago Ill 60611; e-mail: [email protected].
931-5244/$ – see front matter
2007 Mosby, Inc. All rights reserved.
toi:10.1016/j.trsl.2007.01.002
ion. Previous reviews on podocyte transcriptional fac-ors have largely focused on kidney development.1,2
ver the past few years, several new members haveoined the ranks, and substantial progress is being maden clarifying mechanisms of common glomerular dis-rders. This review focuses on transcriptional factorshat have shown a promising role in the development ofodocyte or glomerular disease.
ILMS TUMOR-1 (WT1)
WT1, a zinc finger protein, is the most complex ofll podocyte expressed transcriptional factors. As aesult of alternative splicing, alternative translationaltart sites, and RNA editing, at least 24 different
T1 isoforms exist, although very few of thesesoforms are likely to be expressed in the podocyte.3
soforms that lack the KTS sequence (–KTS isoform)re potent transcriptional activators and bind prefer-ntially to DNA, whereas the �KTS isoform pro-
eins may also play a role in RNA binding.4,5 Various237

dmmitlBnnlnhDFnmmmeslmdniaclfiwtldsDtwcdoKtiaaaeontdm
L(
dcadwtvcrbqsLrmgLy
P
hcacialgpccPieb1mmi
P
tueptt
Translational Research238 Chugh May 2007
egrees of loss of podocyte WT1 content6 or geneutations7 are noted in specific forms of focal glo-erulosclerosis (FGS). The most dramatic reduction
n WT1 expression in human disease is observed inhe collapsing variant of FGS. By contrast, WT1evels are unchanged in minimal change disease.orderline reduction in WT1 mRNA expressionoted in the proteinuric phase of passive Heymannephritis.8 Three genes known to be directly regu-ated by WT1 in the podocyte include podocalyxin,9
ephrin,5 and pax-2.10 In addition, WT1 mutationsave been associated with the development of Denysrash syndrome (diffuse mesangial sclerosis)11 andrasier syndrome (steroid-resistant FGS).12 A largeumber of different types of WT1 mutant or deficientice are available in literature.13 WT1 knockoutice die at midgestation because of cardiac abnor-ality before the formation of kidneys. WT1 het-
rozygous mice seemed to be healthy on initial as-essment and have up to 95% of normal WT1 mRNAevels,14 but about 40% of the same mice bred on aixed genetic background seem to die from severe
iffuse glomerulosclerosis within 13 months.15 WT1ull mice with a 470-Kb human YAC clone contain-ng the full-length WT1 gene survive beyond birthnd develop dose-dependent renal disease.14 With 1opy of the YAC clone (62% of normal WT1 mRNAevels), mice develop albuminuria at birth and dif-use mesangial sclerosis by day 10, which evolvesnto a crescentic glomerulonephritis and die by age 3eeks. WT1 null mice transgenic with 2 copies of
he YAC clone (70% normal wild-type WT1 mRNAevels) develop albuminuria at 3 weeks, followed byiffuse mesangial sclerosis, and 26% die of end-tage renal disease by 5 months of age. In a Denysrash syndrome mutant mouse (WT1tmT396), the mu-
ant protein (5% of total WT1) couples efficientlyith the normal WT1 protein and is sufficient to
ause development of sclerotic glomeruli. Two ad-itional transgenic mice were developed to expressnly the KTS� WT1 isoform (the KTS mouse) or theTS� isoform (Frasier mouse) are available. Data from
hese mice suggest that the WT1 (� KTS) isoform ismportant for the development of podocyte architecturend the integrity of the glomerular tuft.13 Two WT1-ssociated proteins, WT1-interacting protein (WTIP)nd brain acid soluble protein 1 (BASP1), are alsoxpressed in the podocyte and function as corepressorsf WT1 transcriptional factor activity. Whereas BASP1ormally associates with WT1 in the nucleus,16 WTIPranslocates from its normal site of expression in the slitiaphragm complex into the nucleus during develop-
ent of disease.17 sIM HOMEOBOX TRANSCRIPTION FACTOR 1, BETALmx1b)
Lmx1b is mutated in patients with Nail–Patella syn-rome.18–20 In the kidney, Lmx1b is expressed in podo-ytes. Lmx1b protein has 2 zinc-binding LIM domainst the amino terminus and a homeodomain in the mid-le. The LIM domains interact with other proteins,hereas the homeodomain binds to DNA. Mutations in
he LMX1B gene mostly lead to the absence or inacti-ation of the homeodomain, so that the mutated proteinannot recognize its target genes. Lmx1b in podocytesegulates the function of COL4A3 and COL4A4 genesy binding to their common regulatory site. Conse-uently, the level of expression of these 2 genes isignificantly reduced in Lmx1b �/� glomeruli.21,22
mx1b also binds to the nephrosis 2, idiopathic, steroidesistant and CD2-associated protein (CD2AP) pro-oters, and significantly reduced expression of these
enes is noted in Lmx1b �/� glomeruli. The role ofmx1b in other forms of human glomerular disease haset to be clarified.
od1
Pod1 (capsulin, epicardin) is a basic–helix–loop–elix protein expressed in developing, and adult, podo-ytes and in several other organs.23 Pod1 null mice diet birth as a result of heart and lung defects and have aomplex kidney phenotype.24 The number of glomerulis reduced because of reduced ureteric bud branches,nd glomerular differentiation is arrested at the capil-ary loop stage, with a single capillary loop in manylomeruli. Columnar podocytes with rudimentary footrocesses are noted. The precise role of Pod1 in podo-yte development and any potential role in adult podo-yte disease has yet to be clarified. The expression ofod1 may play an important role in vascular remodel-
ng during glomerular development at a stage whenndothelial cells are undergoing differentiation andranching. Interestingly, zinc fingers and homeoboxes(ZHX1) is marginally downregulated in Pod1 �/�ice.24 A single study shows downregulation of Pod1RNA expression in a new model of rapidly develop-
ng glomerular capillary loop collapse.8
ax-2
Pax-2 is an early regulator of kidney developmenthat is first expressed in the developing kidney in thereteric bud and the metanephric mesenchyme, whichventually differentiates into tubular epithelium and theodocytes.25 Following the expression of WT1 inhe s-shaped body, podocyte pax-2 is repressed and inhe fully developed normal glomerulus, pax-2 expres-
ion is noted in parietal epithelium only. WT1 seems to
dwelbeagnoswpricpssedontsw
K
dmpeokpml
N
ai
tssfqcvasrp
Z
tclpth(p(bookrfwsdcZngtcnhid
To
2
Translational ResearchVolume 149, Number 5 Chugh 239
ownregulate pax-2 expression in the podocyte,hereas in mesenchymal cells, modulation of WT1
xpression by pax-2 has also been noted.26 The regu-ation of WT1 by pax-2 is, however, more complex,ecause pax-2 represses WT1 expression in the pres-nce of groucho/transducin-like enhancer proteins andctivates it in the absence of these proteins.27 Trans-enic expression of pax-2 results in the development ofephrotic syndrome at birth and death within 1–3 daysf life.28 Podocyte-specific transgenic expression re-ults in glomerular collapse soon after birth and deathithin a few days.27 By contrast, controlled transgenicodocyte-specific expression of pax-2 in adult miceesults in the development of FGS 3 months after thenduction of pax-2 expression. In patients with theollapsing variant of FGS, increased expression ofax-2 is noted in the cells that tend to crowd the urinarypace around the glomerular tuft.29,30 It is equally pos-ible that these cells are parietal cells that normallyxpress pax-2 or that there is re-expression of pax-2 ine-differentated and proliferating podocytes from lossf WT1 expression, because these cells tend to be WT1egative. An interesting middle ground for this conten-ious issue may actually be a class of cells that expressome podocyte proteins but are normally interspersedith the parietal epithelium.31
REISLER
Kreisler (maf-1 or mafb) is expressed in podocytes ineveloping and newborn mice.32 Homoyzygous mutantice die within 24 hours of birth. The mice seem to be
roteinuric at birth, and the podocyte foot processes areffaced in the capillary loop stage of glomerular devel-pment.33 A paucity of data investigates the role ofreisler in the fully developed kidney. Gene expressionrofiling in the diabetic KK/Ta mouse reveals kreisler/af-1 to be present in the vicinity of a quantitative trait
ocus for the development of albuminuria.34
UCLEAR FACTOR-KAPPA B (NF-�B) AND Smad7
NF-�B and Smad7 are 2 transcriptional factors thatre widely expressed and seem to play a significant role
able I. List of changes in podocyte gene expressioverexpression of individual ZHX proteins for 72 hou
ZHX1
ZO-12FAT12COL4A32aminopeptidase A2dystroglycan2 PAX2
1nephrin2n2entactin22podocaly1angiopoie
n selected animal models of glomerular disease. TGF� m
ransgenic mice develop progressive glomerulosclero-is preceded by podocyte apoptosis. Increased expres-ion of Smad7 in damaged podocytes in these mice iselt to inhibit the nuclear translocation and conse-uently the anti-apoptotic properties of NF-�B.35 Re-ent evidence from HIV-transgenic mice, which de-elop severe focal sclerosis, suggests that NF-�B maylso have pro-apoptotic properties, because a persistenttate of NF-�B activation in the podocytes and otherenal epithelia induces apoptosis by increasing the ex-ression of both Fas and Fas-ligand.36
HX FAMILY
Only a limited number of target genes have been iden-ified for the transcriptional factors discussed above. Byontrast, the ZHX family of transcriptional factors regu-ates a large number of structurally and functionally im-ortant podocyte-expressed genes.37 ZHX proteins con-ain 2 C2H2-type zinc finger domains and 5 Hox-likeomeobox domains. All 3 known ZHX family membersZHX1, ZHX2, and ZHX3) are expressed in the in vivoodocyte. Of the ZHX target genes identified to dateTable I), 70% are regulated by only 1 of 3 family mem-ers. About 30% of the genes are regulated by 2 membersf the family (sometimes in the opposite direction), andnly 1 published gene (ENPEP or aminopeptidase A) isnown to be regulated by all 3 ZHX proteins. Full-lengthat ZHX3 was first cloned from a downregulated generagment noted in proteinuric glomeruli from rats injectedith �2-nephrotoxic serum.37,38 ZHX proteins have
ome unique properties that give them a key role in theevelopment of glomerular disease. Only a small per-entage of ZHX proteins (5% to 10% for ZHX2 andHX3; 20% for ZHX1) are located in normal podocyteuclei. This predominant non-nuclear localization sug-ests that most of the protein is transcriptionally inac-ive at baseline. All ZHX proteins have 2 nuclear lo-alization signals, but they remain sequestered in theon-nuclear compartment predominantly because ofeterodimer formation. Therefore, loss of heterodimer-zation, as would be observed during an increase orecrease in the expression of a single ZHX family
ltured glomerular epithelial cells (GECs) afterved from Liu et al37)
ZHX3
D2AP2COL4A4eptidase Aa 1 integrinT12Lmx1b
2nephrin2ZO-12CD2AP2P Cadherin2cathepsin L2alpha actinin 42COL4A32COL4A42aminopeptidase A2dystroglycan2B7.12angiopoietin 22VEGF A
n in curs (deri
ZHX2
eph12Caminop
xin2bettin 22W
ember, or from altered protein–protein interactions,

rpZpbstotpqgefpwn
pth
ldtonorZosZo
f individ
Translational Research240 Chugh May 2007
esults in the nuclear migration of the upregulated ZHXrotein or the binding partner of the downregulatedHX protein (Fig 1). The effect of ZHX protein on theromoters of their target genes is likely to be influencedy the DNA binding motif (eg, CdxA for ZHX3), theequence around this motif, and the concentration ofhe ZHX protein in the nucleus. In vitro overexpressionf a ZHX protein generates a higher nuclear concen-ration of that protein than knockdown of its bindingartner, and this may result in a qualitative and/oruantitative difference in the expression of targetenes. Also, ZHX proteins have a negative feedbackffect on their own promoters, but not that of otheramily members, and this absence of cross-regulationermits free ZHX proteins to regulate target genesithout altering the expression of their binding part-
Fig 1. Migration of ZHX proteins into the podocythave 2 nuclear localization signals. ZHX proteins e(90–95% for ZHX2 and ZHX3; 80% for ZHX1) idisease, loss of heterodimerization resulting from anmember caused by changes in gene expression, or asprotein–protein interaction, leads to the migration o
ers. Finally, it is possible that the ZHX region of these Z
roteins may regulate different genes, because post-ranslational cleavage of ZHX proteins into 2 fragmentsas been documented at the very least for ZHX3.The expression of ZHX proteins has been studied
ongitudinally in animal models of human glomerularisease8,37 and in human kidney biopsies.37 ZHX3 isransiently downregulated before the development ofvert proteinuria in young rats with puromycin amino-ucleoside nephrosis (PAN, single intravenous dosef 15 mg/100 g puromycin aminonucleoside). As aesult of loss of heterodimerization, small amounts ofHX1 or ZHX2 (most likely, ZHX1 � ZHX2, in viewf lack of WT1 involvement) enter the nucleus at thistage. This process is followed by a gradual recovery ofHX3 expression that coincides with the developmentf proteinuria. The mRNA expression of ZHX1 and
during development of disease. All ZHX proteinsy as heterodimers in the non-nuclear compartmentmal in vivo podocyte. During the development ofor decrease in the cellular content of a single familyof cleavage of ZHX heterodimers caused by alteredual ZHX proteins into the podocyte nucleus.
e nucleusxist mostln the norincreasea result
HX2 remains unchanged. Increased podocyte nuclear

ZudcimcctrgeZgii(Zdpsli
tnFidItpdictpZtsAirpZictZst
cW
rdam
mdpeogmtaiptdIcosdaIe
C
btittqod
p
R
Translational ResearchVolume 149, Number 5 Chugh 241
HX3 staining is noted in the early stages of protein-ria, which suggests that ZHX3 protein synthesizeduring recovery of ZHX3 expression enters the nu-leus. Increased podocyte nuclear expression of ZHX3s also noted in biopsies from patients with humaninimal change disease. In a study of about 28 podo-
yte expressed genes using quantitative polymerasehain reaction,37 transient knockdown of ZHX3 in cul-ured GECs produced a gene expression profile in theecovery phase that was qualitatively identical to theene expression profile in proteinuric rat glomeruli inarly PAN. By contrast, sustained severe knockdown ofHX3 in these cells mostly alters a different set ofenes, largely as a result of entry of ZHX1 and ZHX2nto the nucleus after loss of heterodimerization. Ofnterest, 3 other transcriptional factors, WT1, Lmx1bboth ZHX2 target genes), and Pax-2 (regulated byHX1), are downregulated after severe ZHX3 knock-own. The recognition of WT1 regulation by ZHX2,ending confirmation using promoter–reporter con-tructs, is a key advance in WT1 biology, because veryittle is known about the regulation of WT1 expressionn podocytes.
Enormous potential exists in studying ZHX proteins inhe near future. The precise subcellular localization in theon-nuclear compartment is currently under investigation.urther studies are required to identify additional changes
n gene expression induced by ZHX proteins during theevelopment of minimal change disease (MCD) and FGS.n vitro changes in gene expression during recovery fromransient ZHX3 knockdown mimic the early proteinurichase of experimental MCD, whereas sustained knock-own of ZHX3 results in downregulation of WT1, whichs consistently observed in severe forms of FGS, includingollapsing glomerulopathy. This result would suggest thatransient downregulation of ZHX3 may be a critical com-onent of MCD, whereas sustained downregulation ofHX3 may contribute to the development of FGS. Addi-
ional support for these observations comes from previoustudies in rats injected with puromycin aminonucleoside.
single injection of puromycin aminonucleoside resultsn the development of MCD, whereas repeated injectionsesult in FGS.39 It is possible that repeated injection ofuromycin aminonucleoside may prevent the recovery ofHX3 expression that is normally noted after a single
ntravenous injection and may effectively convert the re-overy phase into a sustained downregulation and, hence,he FGS gene expression profile. Therefore, whereas allHX proteins are likely to contribute to the MCD–FGSpectrum, the gatekeeper role seems to have been assignedo ZHX3.
The importance of studying ZHX2 in greater detailomes from its ability to influence the expression of
T1 and Lmx1b. Whereas the established diseaseselated to these 2 transcriptional factors have beeniscussed, more studies on the effect of ZHX2 on WT1nd Lmx1b expression in other forms of human glo-erular disease are required.The importance of studying ZHX1 comes from theultitude of protein–protein interactions that have been
ocumented (mostly by yeast 2-hybrid studies) for thisrotein.40 Whereas most of these proteins may not bexpressed in the podocyte, the potential exists for somef these proteins to be actively involved in the patho-enesis of podocyte disease. Basic helix-loop-helix do-ain containing, class B, 2 (Stra13) is known to alter
he cellular redox state in cultured GECs.41 The inter-ction of ZHX1 with vimentin, a critical component ofntermediate filaments and microtubules present in theodocyte cell body and major foot processes, suggestshat ZHX proteins may be displaced toward the nucleusuring structural changes in the diseased podocyte.nteraction with P53 could also influence a variety ofellular functions. As podocytes share a large numberf specialized proteins with neurons, it is likely thatome of these other proteins may eventually also beescribed in the podocyte. Downregulation of Pax-2fter overexpression of ZHX1 in cultured GECs (Table) has the potential of explaining some changes in Pax-2xpression noted in human glomerular disease.
ONCLUSION
With continuing advancement in microarray array-ased technology, large-scale identification of additionalarget genes of podocyte expressed transcriptional factorss on the horizon. Finally, blockage of the nuclear migra-ion of ZHX proteins is an attractive future therapeuticarget for human glomerular disease, especially if subse-uent studies show a significant contribution to the devel-pment of other forms of glomerular disease, includingiabetic nephropathy and lupus nephritis.
The author wishes to thank Lionel Clement PhD for assistance inreparing Fig 1.
EFERENCES
1. Quaggin SE. Transcriptional regulation of podocyte specificationand differentiation. Microsc Res Tech 2002;57:208–11.
2. Morello R, Lee B. Insight into podocyte differentiation from thestudy of human genetic disease: nail-patella syndrome and tran-scriptional regulation in podocytes. Pediatr Res 2002;51:551–8.
3. Hohenstein P, Hastie ND. The many facets of the Wilms’ tumourgene, WT1. Hum Mol Genet 2006;15:R196–201.
4. Hammes A, Guo JK, Lutsch G, Leheste JR, Landrock D, Ziegler U,et al. Two splice variants of the Wilms’ tumor 1 gene have distinctfunctions during sex determination and nephron formation. Cell2001;106:319–29.
5. Wagner N, Wagner KD, Xing Y, Scholz H, Schedl A. The majorpodocyte protein nephrin is transcriptionally activated by the Wilms’
tumor suppressor WT1. J Am Soc Nephrol 2004;15:3044–51.
1
1
1
1
1
1
1
1
1
1
2
2
2
2
2
2
2
2
2
2
3
3
3
3
3
3
3
3
3
3
4
4
Translational Research242 Chugh May 2007
6. Barisoni L, Kriz W, Mundel P, D=Agati V. The dysregulated podo-cyte phenotype: a novel concept in the pathogenesis of collapsingidiopathic focal segmental glomerulosclerosis and HIV-associatednephropathy. J Am Soc Nephrol. 1999;10:51–61.
7. Orloff MS, Iyengar SK, Winkler CA, Goddard KA, Dart RA,Ahuja TS, et al. Variants in the Wilms’ tumor gene are associatedwith focal segmental glomerulosclerosis in the African Americanpopulation. Physiol Genomics 2005;21:212–21.
8. Clement LC, Liu G, Kanwar YS, Avila-Casado C, Chugh SS.Early changes in gene expression that influence the course ofprimary glomerular disease. Kidney Int. In press.
9. Palmer RE, Kotsianti A, Cadman B, Boyd T, Gerald W, Haber DA.WT1 regulates the expression of the major glomerular podocytemembrane protein Podocalyxin. Curr Biol. 2001;11:1805–9.
0. Ryan G, Steele-Perkins V, Morris JF, Rauscher FJ 3rd, DresslerGR. Repression of Pax-2 by WT1 during normal kidney devel-opment. Development 1995;121:867–75.
1. Pelletier J, Bruening W, Kashtan CE, Mauer SM, Manivel JC,Striegel JE, et al. Cell 1991;67:437–47.
2. Barbaux S, Niaudet P, Gubler MC, Grunfeld JP, Jaubert F,Kuttenn F, et al. Donor splice-site mutations in WT1 are respon-sible for Frasier syndrome. Nat Genet 1997;17:467–70.
3. Discenza MT, Pelletier J. Insights into the physiological role ofWT1 from studies of genetically modified mice. Physiol Genom2004;16:287–300.
4. Guo JK, Menke AL, Gubler MC, Clarke AR, Harrison D,Hammes A, et al. WT1 is a key regulator of podocyte function:reduced expression levels cause crescentic glomerulonephritisand mesangial sclerosis. Hum Mol Genet 2002;11:651–9.
5. Menke AL, IJpenberg A, Fleming S, Ross A, Medine CN, PatekCE, et al. The wt1-heterozygous mouse; a model to study thedevelopment of glomerular sclerosis. J Pathol 2003;200:667–74.
6. Carpenter B, Hill KJ, Charalambous M, Wagner KJ, Lahiri D,James DI, et al. BASP1 is a transcriptional cosuppressor for theWilms’ tumor suppressor protein WT1. Mol Cell Biol 2004;24:537–49.
7. Srichai MB, Konieczkowski M, Padiyar A, Konieczkowski DJ,Mukherjee A, Hayden PS, et al. A WT1 co-regulator controlspodocyte phenotype by shuttling between adhesion structuresand nucleus. J Biol Chem 2004;279:14398–408.
8. Dreyer SD, Zhou G, Baldini A, Winterpacht A, Zabel B, Cole W,et al. Mutations in LMX1B cause abnormal skeletal patterningand renal dysplasia in nail patella syndrome.Nat Genet 1998;19:47–50.
9. Chen H, Lun Y, Ovchinnikov D, Kokubo H, Oberg KC, PepicelliCV, et al. Limb and kidney defects in Lmx1b mutant micesuggest an involvement of LMX1B in human nail patella syn-drome. Nat Genet 1998;19:51–5.
0. Vollrath D, Jaramillo-Babb VL, Clough MV, McIntosh I, ScottKM, Lichter PR, et al. Loss-of-function mutations in theLIM-homeodomain gene, LMX1B, in nail-patella syndrome.Hum Mol Genet 1998;7:1091–8.
1. Rohr C, Prestel J, Heidet L, Hosser H, Kriz W, Johnson RL, et al.The LIM-homeodomain transcription factor Lmx1b plays a crucialrole in podocytes. J Clin Invest 2002;109:1073–82.
2. Miner JH, Morello R, Andrews KL, Li C, Antignac C, Shaw AS,et al. Transcriptional induction of slit diaphragm genes byLmx1b is required in podocyte differentiation. J Clin Invest2002;109:1065–72.
3. Quaggin SE, Schwartz L, Cui S, Igarashi P, Deimling J, Post M,et al. The basic-helix-loop-helix protein pod1 is critically impor-tant for kidney and lung organogenesis. Development 1999;126:
5771–83.4. Cui S, Li C, Ema M, Weinstein J, Quaggin SE. Rapid isolationof glomeruli coupled with gene expression profiling identifiesdownstream targets in Pod1 knockout mice. J Am Soc Nephrol2005;16:3247–55.
5. Dressler GR, Douglass EC. Pax-2 is a DNA-binding proteinexpressed in embryonic kidney and Wilms tumor. Proc NatlAcad Sci U S A 1992;89:1179–83.
6. Dehbi M, Ghahremani M, Lechner M, Dressler G, Pelletier J.The paired-box transcription factor, PAX2, positively modulatesexpression of the Wilms’ tumor suppressor gene (WT1). Onco-gene 1996;13:447–53.
7. Wagner KD, Wagner N, Guo JK, Elger M, Dallman MJ, BugeonL, et al. An inducible mouse model for PAX2-dependent glo-merular disease: insights into a complex pathogenesis. Curr Biol2006;16:793–800.
8. Dressler GR, Wilkinson JE, Rothenpieler UW, Patterson LT,Williams-Simons L, Westphal H. Deregulation of Pax-2 expres-sion in transgenic mice generates severe kidney abnormalities.Nature 1993;362:65–7.
9. Yang Y, Gubler MC, Beaufils H. Dysregulation of podocytephenotype in idiopathic collapsing glomerulopathy and HIV-associated nephropathy. Nephron 2002;91:416–23.
0. Dijkman HB, Weening JJ, Smeets B, Verrijp KC, van Kuppe-velt TH, Assmann KK, et al. Proliferating cells in HIV andpamidronate-associated collapsing focal segmental glomeru-losclerosis are parietal epithelial cells. Kidney Int 2006;70:338 – 44.
1. Bariety J, Mandet C, Hill GS, Bruneval P. Parietal podocytes innormal human glomeruli. J Am Soc Nephrol 2006;17:2770–80.
2. Imaki J, Onodera H, Tsuchiya K, Imaki T, Mochizuki T,Mishima T, et al. Developmental expression of maf-1 messengerribonucleic acids in rat kidney by in situ hybridization histo-chemistry. Biochem Biophys Res Commun 2000;272:777–82.
3. Sadl V, Jin F, Yu J, Cui S, Holmyard D, Quaggin S, et al. Themouse Kreisler (Krml1/MafB) segmentation gene is required fordifferentiation of glomerular visceral epithelial cells. Dev Biol2002;249:16–29.
4. Fan Q, Shike T, Shigihara T, Tanimoto M, Gohda T, Makita Y,et al. Gene expression profile in diabetic KK/Ta mice. Kidney Int2003;64:1978–85.
5. Schiffer M, Bitzer M, Roberts IS, Kopp JB, ten Dijke P, Mundel P,et al. Apoptosis in podocytes induced by TGF-beta and Smad7.J Clin Invest 2001;108:807–16.
6. Ross MJ, Martinka S, D=Agati VD, Bruggeman LA. NF-kappaBregulates Fas-mediated apoptosis in HIV-associated nephropa-thy. J Am Soc Nephrol 2005;16:2403–11.
7. Liu G, Clement L, Kanwar YS, Avila-Casado C, Chugh SS. ZHXproteins regulate podocyte gene expression during the develop-ment of nephrotic syndrome. J Biol Chem 2006;281;39681–92.
8. Chugh S, Yuan H, Topham PS, Haydar SA, Mittal V, Taylor GA,et al. Aminopeptidase A: a nephritogenic target antigen of neph-rotoxic serum. Kidney Int 2001;59:601–13.
9. Glasser RJ, Velosa JA, Michael AF. Experimental model of focalsclerosis. I. Relationship to protein excretion in aminonucleosidenephrosis. Lab Invest 1977 May;36:519–26.
0. Lim J, Hao T, Shaw C, Patel AJ, Szabo G, Rual JF, et al. Aprotein-protein interaction network for human inherited ataxias anddisorders of Purkinje cell degeneration. Cell 2006;125:801–14.
1. Bek MJ, Wahle S, Muller B, Benzing T, Huber TB, Kretzler M,et al. Stra13, a prostaglandin E2-induced gene, regulates the
cellular redox state of podocytes. FASEB J 2003;17:682–4.
OIr
MLR
T
FDvmDTMDbT
SCo
RIGINAL ARTICLESmmunopathogenesis of hypersensitivity syndromeeactions to sulfonamides
ANUELA G. NEUMAN, NEIL H. SHEAR, IZABELLA M. MALKIEWICZ, MASUD TAERI,ORI E. SHAPIRO, NORBERTO KRIVOY, JULIA HABER, MANUEL GOMEZ, JOEL FISH,OBERT CARTOTTO, and LAWRENCE COHEN
ORONTO, ONTARIO, CANADA AND HAIFA, ISRAEL
Cytokines play a role in the immunopathological and molecular mechanisms of sul-fonamide-induced hypersensitivity reactions (HSRs). The objective of this study was toanalyze the reliability and correlation between the clinical symptoms observed inaffected patients (n � 86) because of a sulfonamide-induced HSR and their lympho-cyte toxicity assay (LTA) values. Another goal was to determine the cytokine secretionin the patient’s sera and their expression in the peripheral blood mononuclear cells(PBMCs) and to explore whether a correlation exists among positive LTA score, cyto-kine levels, and HSR occurrence. The final goal is to determine whether these measurescould be used to predict the likelihood of a patient to experience an HSR duringsulfonamide treatment. Such a predictive ability would be valuable to the clinician toknow whether the patient would tolerate sulfonamides or whether an alternative anti-biotic should be prescribed. The LTA showed a good correlation with the clinicalinvolvement of patients with hypersensitivity syndromes. In addition, the pro-inflamma-tory cytokines presented significant differences in patients that had rash, fever, andorgan involvement than in control patients or any of the other patient groups. Expres-sion of tumor necrosis factor alpha (TNF-�) is significantly higher in patients presentingrash, fever, and organ involvement versus all other groups. It is concluded that apositive LTA is a predictor for sulfonamide-induced true HSR. In addition, T-helper cell 1cytokines [TNF-�, interleukins (ILs) 1 and 2] as well as the chemokine regulated uponactivation, normal T-cell expressed and secreted (RANTES) control the pathogenesis ofsulfonamide-induced HSR and may be used in early diagnosis of the syndrome.(Translational Research 2007;149:243–253)
Abbreviations: ELISA � enzyme-linked immunosorbent assay; G3PDH � glyceraldehyde-3-phosphate dehydrogenase; GSH � mitochondrial gluthione; GST � glutathione S transferase;HSR � hypersensitivity syndrome reaction; IL � interleukin; IFN-� � interferon-gamma; LTA �lymphocyte toxicity assay; MT � mitochondrial toxicity; NADP � nicotinamide adenine dinu-cleotide phosphate; PBMC � peripheral blood mononuclear cell; PCR � polymerase chainreaction; RANTES � regulated upon activation, normal T-cell expressed and secreted; ROS �reactive oxygen species; RT-PCR � reverse transcriptase polymerase chain reaction; SDH �succinic dehydrogenase; SJS � Stevens–Johnson syndrome; SMX � sulfamethoxazole; TEN �toxic epidermal necrolysis; TNF-� � tumor necrosis factor-alpha
rom the In Vitro Drug Safety and Biotechnology Laboratory and theepartment of Pharmacology, The Institute of Drug Research, Uni-ersity of Toronto, Toronto, Ontario, Canada; the Division of Der-atology and Sunnybrook Drug Safety Clinic, the Burn Unit, and theivision of Gastroenterology, Sunnybrook Health Science Centre,oronto, Ontario, Canada; the Department of Medicine, Faculty ofedicine, University of Toronto, Toronto, Ontario, Canada; and theepartment of Medicine B and Clinical Pharmacology Unit, Ram-am Medical Center and Bruce Rappaport Faculty of Medicine,echnion, Haifa, Israel.
upported in part by the Institute of Infection and Immunity ofanadian Institutes of Health Research and by the National Institute
USA. Supported in part by grants received by Dr. Neuman, Dr.Shapiro, and Dr. Shear from the Canadian Dermatology Foundationand the Incorporated Foundation Physician’s Services.
Submitted for publication July 23, 2006; revision submitted Decem-ber 22, 2006; accepted for publication December 22, 2006.
Reprint requests: Dr. Manuela Neuman, Director In Vitro Toxicologyand Biotechnology, MaRS Discovery District, 101 College Street,Suite 300, Lab 351, Toronto, Ontario, Canada, M5G 1L7; e-mail:[email protected].
1931-5244/$ – see front matter
© 2007 Mosby, Inc. All rights reserved.
n Alcohol Abuse and Alcoholism, National Institutes of Health, doi:10.1016/j.trsl.2006.12.001
243

Aatcpdcmtspthd
tsibrrfpshefpf
sptidwm
itsrdctc1itamHHr
ramSl(io
Hskpos
matsfaTviwvtcit
lmtmbotpnmci
ddoCNbp
Translational Research244 Neuman et al May 2007
dverse drug reactions account for 5% of all hospitaldmissions and occur in approximately 15% of hospi-alized patients.1 The majority of these reactions areommon and predictable, being primarily based on theharmacological properties of the drug given. Unpre-ictable or idiosyncratic, type B reactions pose a majoroncern in clinical practice and drug development pri-arily because they are not dose-dependent.2 However,
ype B reactions are known to occur at certain doses inusceptible individuals.3 One of the most common andotentially fatal adverse drug reactions are hypersensi-ivity syndromes associated with sulfonamides.1,4 Theypersensitivity syndrome reaction (HSR) is a host-ependent drug reaction that is idiosyncratic in nature.The true HSR is a systemic disease defined by the
riad of fever, rash, and internal organ involvement thattarts 8–12 weeks after the initiation of therapy.5 Anncidence of 1/1000 to 1/10,000 exposures is inaccurateecause many reactions are common yet under-eported. Clinical conditions range from simple skineactions and general manifestations, such as morbili-orm rash, urticaria, angioedema, fever, malaise, ana-hylaxis, bronchospasm, and erythema multiforme, tokin complications with the highest mortality rates ex-ibited by Stevens-Johnson syndrome (SJS) and toxicpidermal necrolysis (TEN), which involve multiorganunction impairment5 such as lymphadenopathy, he-atic dysfunction, hematologic dysfunction, renal dys-unction, myocarditis, or myosistis.6-8
Sulfonamides are commonly used antibiotic drugs. Theulfonamide family includes a wide spectrum of differentroducts, including sulfacetamide, sulfadiazine, sulfame-hoxazole (SMX), sulfisoxazole, sulfamethizole, sulfadox-ne/pyrimethamine (Fansidar), sulfapyridine, silver sulfa-iazine, sulfamerazine, sulfamethazine, and the mostidely used sulfa preparation sulfamethoxazole/tri-ethoprim (Septra, Bactrim).4,9,10
As a high incidence of HSRs to sulfonamides exists,n approximately 5% of the general population and 10imes greater in HIV-infected patients, significant re-earch has been conducted into finding a safe andeliable method to predict these reactions.4,11-15 Therug challenge can be complicated and dangerous be-ause of the potential for cross-reactivity or life-hreatening adverse reactions. The use of the lympho-yte toxicity assay (LTA) has been in practice since9886,16-18 and has recently been validated for SMX inmmuno-competent and immuno-compromised pa-ients.18,19 The test is based on reactive metabolites thatre generated using murine18 or canine16,17 hepaticicrosomes as a source of cytochrome P450 (P450).uman lymphocytes from patients with suspectedSRs are used as surrogate target cells for safe in vitro
e-challenge.18 g
Lymphocytes are chosen as target cells for 2 maineasons. First, they do not produce reactive metabolitesnd do not contain enzymes, which may interfere withicrosomal bioactivation of drugs added to culture.econd, they possess various detoxification enzymes,
ike epoxide hydroxylase and glutathione S transferasesGSTs), and phenotypically express individual variabil-ty in these enzymes, thus modeling the potential sourcef hypersensitivity observed in vivo.A clear understanding of the processes involved inSRs will greatly aid in its diagnosis; however, factors
uch as an unclear immunological mechanism, an un-nown epitope that causes a reaction, and whether theresence of an immunological recognition is predictivef a clinical reaction hinders understanding of hyper-ensitivity reactions.5,10,20-23
Depending on the time course of the reaction, theechanism of sulfonamide-mediated adverse drug re-
ctions falls into 1 of 2 possible pathways. Immediate-ype immune-mediated reactions are presented withymptoms of urticarial rash that is typically withoutever, occurring approximately 3 days into therapy. IgEntibodies raised against the drug are usually present.he 5-methyl-3-isoxazolyl group on SMX seem to beery involved in this immune response.4 Delayed-typemmune-mediated reactions are typically presentedith rash and fever, and they may include organ in-olvement that manifests 1–2 weeks after sulfonamideherapy is initiated.24 As drug-specific activated T-celllones have been found in these patients; this pathways considered an immune-mediated HSR. These reac-ions have limited the therapeutic use of sulfonamides.
HSRs caused by sulfonamide antimicrobials areikely a result of a combination of metabolic and im-unologic factors.10,20,25,26 The dose and duration of
herapy may play a role, as well as defects in theetabolic pathways, the degree of immunodeficiency,
acterial and viral infections, the degree of reactivexygen species (ROS) produced, if adduct forma-ion occurs, and mitochondrial toxicity (MT) isresent.1,19,23,27 Cellular immune-mediated compo-ents, such as the T cells and cytokine/chemokineediation, which can exacerbate cellular responses,
reate complex pathways that lead to a variety of clin-cal manifestations.
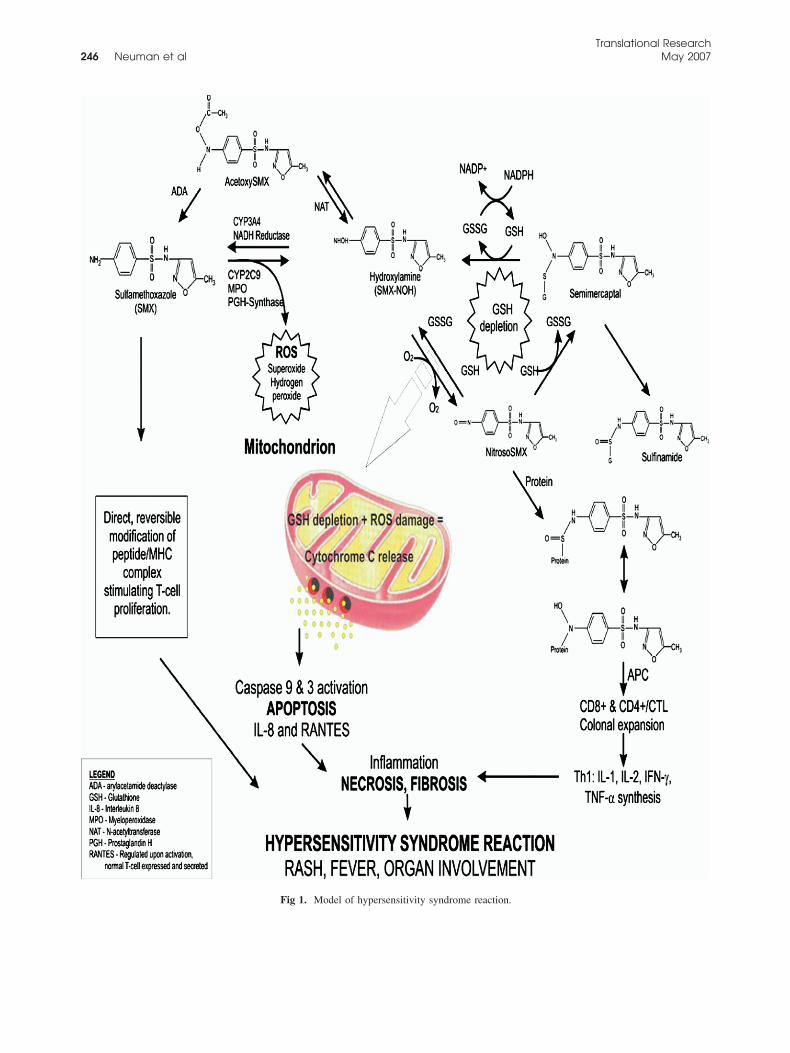
Significant evidence exists implicating the hapten andanger hypotheses in the occurrence of sulfonamide-in-uced HSRs.23 Only a small fraction of SMX undergoesxidation to a reactive metabolite, which occurs throughYP2C9, generating a hydroxylamine derivative (SMX-OH) capable of being acetylated or reduced (CYP3A4)ack to SMX (Fig 1). Further oxidation of SMX-NOHroduces a nitroso-metabolite (nitroso-SMX) capable of
enerating ROS through a redox cycle and binding cellu-
ltciinescitoaS
cl1pt(bnwopwpa
mnaehcc
tt
tu(mH
M
HGd(ActvbglccstSCpLw
●
●
sh
T
R
R
R
A1 alue; NPN ts that p
Translational ResearchVolume 149, Number 5 Neuman et al 245
ar proteins, thereby completing the process of hapteniza-ion.4 Redox cycling alone can be detrimental not only toellular proteins but to the protective balance of detoxify-ng molecules as well. Acetylation of SMX-NOH, primar-ly with N-acetyltransferase 2, helps decrease the availableitroso-SMX for covalent binding.4 In a study on ratsxposed to nitroso-SMX metabolite, Naisbitt et al22 as-essed the cytokine synthesis by responding to T lympho-ytes through a quantification of interleukin-4 (IL-4) andnterferon-gamma (IFN-�) levels. In a previous study withhe same rat model, they demonstrated that administrationf nitroso-SMX resulted in the formation of anti-drugntibodies.21 Therefore, the haptenization of the nitroso-MX might lead to a cellular immune response.The authors believe that the mitochondrion is criti-
ally damaged when it is exposed to ROS, whereas theevels of mitochondrial gluthione (GSH) are low (Fig). When SMX is metabolized via the cytochrome P450athway, the production of ROS is maintained by de-oxifying agents such as superoxide dismutaseE.C.1.15.1.1) and GST (E.C.2.5.1.18). While ROS areeing generated, the redox cycling of SMX-NOH anditroso-SMX depletes the levels of cytosolic GSH,hich would lead to a drop in the detoxifying capabilityf the mitochondrion and damage from ROS. If apo-tosis ensues, then the recruitment of chemokinesould also follow. This effect adds to the inflammatoryrocess and accelerates the cellular necrosis, leading ton HSR.The objective of this study is to understand the im-unopathological mechanism and molecular mecha-
ism of HSRs. The specific aims of the study were tonalyze the reliability and correlation between clinicalvidence in immune-competent patients who developedypersensitivity locally with systemic symptomsaused by SMX/TMP and the results of their lympho-yte toxicity assays.A secondary objective of this study is an analysis of
he cytokine secretion in the sera and their expression in
able I. Mean and standard deviation for the posit
Patient LTA result (n)LTA for SMmean (SD
(27) Positive (4) 18.40 (8.45Negative (23) 6.63 (3.26
� F (43) Positive (24) 20.31 (4.93Negative (19) 7.84 (3.36
� F � OI (16) Positive (15) 22.30 (8.34Negative (1) 3.07 (0.00
bbreviations: n, number of patients in the group; R, rash; F, fever;2.5%; SMX, value indicates cytotoxicity; PPV, positive predictive vote: LTA has an excellent positive predictive value for the patien
he peripheral blood mononuclear cell (PBMC) of pa- p
ients that presented sulfonamide-induced HSRs. Thetility of these cytokines [tumor necrosis factor-alphaTNF-�), IL, and IFN-�] as potential noninvasivearkers for susceptibility to sulfonamide-inducedSRs has also been evaluated.
ATERIALS AND METHODS
Patients. In the study, 86 individuals presented anSR to sulfonamides. The patients were referred to thelaxo Welcome-Sunnybrook Drug Safety Clinic foriagnosis by drug safety clinicians and dermatologistsL.E.S., N.H.S.) 0.5–4 years after presentation of HSR.
clinical pharmacologist and internal medicine spe-ialist (N.K.) reviewed all clinical data and its correla-ion with the laboratory data. Some patients who de-eloped SJS or TEN were hospitalized and treated byurn surgeons (J.F., R.C., J.H., and M.G.). A hepatolo-ist (L.C.) was a consultant for the cases that presentediver involvement. The clinical biochemist and pharma-ology specialist (M.G.N.) and the laboratory techni-ian (I.M.) were responsible for the laboratory diagno-is. Ethical approval for the study was obtained fromhe Scientific and Ethics Review Committees of theunnybrook and Women’s College Health Sciencesentre, and all the patients signed the inform consent toarticipate. Participants in this study underwent in vitroTA testing with SMX (Table I). The LTA test resultsere correlated with the patient’s clinical data.The subjects were divided in 2 subgroups as follows:
Patients: 86 patients who had developed a clinical HSR to a
sulfonamide.
Controls: 220 immunocompetent individuals who received sulfon-
amides without developing HSR and voluntarily participated in the
study.
Patients who were hospitalized during an acute epi-ode of HSR (SJS or TEN) in the burn unit of theospital visited the Drug Safety Clinic after they recu-
negative LTA values
PPV NPV Sensitivity Specificity
44.60% 53.40% 40.70% 78.20%
70.50% 81.50% 62.20% 79.10%
86.70% 99.50% 91.90% 99.10%
n involvement; LTA values: positive, MTT � 12.5%; negative, MTT �V, negative predictive value.resented with rash, fever, and organ involvement.
ive and
X)
))))))
OI, orga
erated from the active HSR and they agreed to partic-
Translational Research246 Neuman et al May 2007
Fig 1. Model of hypersensitivity syndrome reaction.

itdp
5mpas
u
fiwepevsfwrcftIsh
wwMwHcio
ba2ptpmdtNgp
p
cpatytpdtbafpAa
�dcmecjtamphtt
Ctc2aae
KrsItdhlt
iCaT
Translational ResearchVolume 149, Number 5 Neuman et al 247
pate in the study. They accepted to have their labora-ory diagnostic performed at 2 different times: (1)uring the acute phase and (2) when they no longerresented symptoms.Drugs and chemicals. The tetrazolium salt 3-(4,
-dimethylthiazol-2-yl) 2, 5 diohenyl-tetrazolium bro-ide (MTT) was obtained from Sigma Chemical Com-
any (St. Louis, Mo). All remaining reagents were ofnalytical grade obtained from the usual commercialources.
Laboratory methods. Cytotoxicity assay. The studysed an in vitro LTA test.18
Microsome preparation. Swiss mice were obtainedrom the National Institutes of Health and were treatedntraperitoneally with phenobarbital, 10 mg/kg of bodyeight per day for 3 days, to induce cytochrome P450
nzymes. Animals were sacrificed in conformity withrotocol approved by the animal ethics committee. Liv-rs were removed aseptically and homogenized in 3olumes (w/v) of 0.15-M KCl. The homogenate waspun for 10 min at 500 � g. The supernatant was spunor 15 min at 9000 � g, and the resulting supernatantas then spun for 1 h at 100,000 � g. The pellet was
esuspended in EDTA/KCl buffer at pH 7.4. The cyto-hrome P450 activity was verified in the microsomalraction. Minimal concentration of cytochrome P450 inhe induced microsomes was 1-mol P450/mg protein.nduced microsomes from phenobarbital-treated micehowed greater P450 activity than those from mice thatad not been pretreated with the drug.Lymphocyte preparation. Fresh, heparinized bloodas diluted 3-fold with minimal essential medium andas layered on a Ficoll–Paque density gradient. PB-Cs containing 22% monocytes and 78% lymphocytesere collected by centrifugation and suspended in aEPES-buffered medium. Lymphocytes were then
ounted using a microscope by a technician specializedn hematological analysis and resuspended at a densityf 2 � 106 cells/mL.Incubation of lymphocytes. Lymphocytes were incu-
ated at 37°C for 24 h, along with 0.6-mM nicotin-mide adenine dinucleotide phosphate (NADP),.24-mM glucose-6-phosphate, and 2-�M glucose-6-hosphate dehydrogenase as a control (primary con-rol). The treatments were as follows: lymphocytes plusarent drug only (secondary control), lymphocytes plusicrosomes only, and finally lymphocytes plus parent
rug plus microsomes (test group). Microsomal activa-ion of the parent drug has been previously shown to beADP-dependent; thus, the addition of NADP andlucose-6-phosphate to the culture is crucial to theroduction of the reactive metabolites.Each reaction involves the incubation of patient lym-
hocytes (viability �95%) at 37°C with 0.28-mg mi- d
rosomal protein, 0.6-mM NADP, 2.24-mM glucose-6-hosphate, 2 U of glucose-6-phosphate dehydrogenase,nd the causative drug. Cytotoxicity was assessed usinghe tetrazolium dye MTT (3-{4,5-dimethyl thiazol-2-l}2,5 diphenyl-tetrazolium bromide). Viable, intact cellsake up the yellow dye MTT and can convert it to a purpleroduct using the mitochondrial enzyme succinic dehy-rogenase (SDH) (E.C. 1.3.99.1). The principle behindhe MTT dye conversion test is that lymphocytes harmedy reactive drug metabolites undergo mitochondrial dam-ge and lose the ability to convert the dye from MTT toormazan. This purple product can be quantitated spectro-hotometrically at an endpoint mode using 2 wavelengths:bsorbance is measured at 560 nm, whereas absorbance
t 690 nm is used as a reference.At the end of the 24-h incubation period, MTT (100L) was added for 1 h. The cells were lysed, and theye was dissolved with 100-�L isopropyl alcohol. Theytotoxicity was measured using an enzyme-linked im-unosorbent assay (ELISA) reader. An LTA result
qual to or higher than 12.5% of cytotoxicity wasonsidered to be positive for immunocompetent sub-ects.18 Each experimental condition using the conven-ional methodology was studied in triplicate sample,nd the mean percent of dead lymphocytes was deter-ined. The change from baseline values (calculated as
ercent dead cells in the presence of drug and murineepatic microsomes minus the percent of dead cells inhe absence of drug) was used as the measure ofoxicity.
The Maxline Microplate Reader (Molecular Deviceorporation, Menlo Park, Calif) was used to determine
he release of cytokines in media. The reader wasonnected to a computer using SOFT MAX software.3 for Windows to program the template, the temper-ture, and the kinetic or endpoint-mode of readingccording to specific experimental needs for eachndpoint.
Cytokine measurement. Cytoscreen, Immunoassayits, Human IL-1, IL-2, IL-4, IL-5, IL-6, IL-8, IL-10,
egulated upon activation, normal T-cell expressed andecreted (RANTES), and TNF-�, ELISA (Biosourcenternational, Camarillo, Calif) were used for the quan-itative determination of cytokines in cell culture me-ia. The assay is designed to recognize both naturaluman and recombinant human cytokines. The corre-ation coefficient was linear (r � 0.989) in a concen-ration range between 2 and 500 pg/mL.
Cytokine expression in PBMC. Total cellular RNA wassolated from PBMC by a modification of the method ofhomczynski and Sacchi.17 Taq DNA Polymerase andmplifier sets for human IL-1, human IL-6, and humanNF-� as well as glyceraldehyde-3-phosphate dehy-
rogenase (G3PDH) control were purchased from
CPcg5rpnCCCITAtAGsPorPokiau�fGadctCrspfipcp(weDlDpus1
S
oc
(ial
R
ttasat(rftFtitt1
aasctv(dtwsavi9erb5nrLp
o
Translational Research248 Neuman et al May 2007
LONTECH Laboratories, Inc. (Palo Alto, Calif).olymerase chain reaction (PCR) was performed ac-ording to the method of Kawarski.18 The cytokineene expression was evaluated by RT-PCR. Briefly,0–400-ng total RNA was reverse transcribed, theesulting cDNA was amplified with each gene-specificrimer, and the PCR reaction was optimized for cycleumber. The IL-1 sequence was as follows: (5=) 5=AA GGA GAG CAT GGT GGT AGT AGC AACAA CG 3= and (3=) 5= TAG TGC CGT GAG TTTCC AGA AGA AGA GGA GG 3=. The primers for
L-6 were as follows: upstream: 5= ATG AAC TCCTC TCC ACA AGC GC 3= and downstream (3=): 5=GAG AGC CCT CAG GCT GGA CTG 3=. For TNF-�,
he primers were as follows: 5= primer- 5=ATG AGCCT GAA AGC ATG ATC CGG3= and 3= primer: 5=CA ATG ATC CCA AAG TAG ACC TGC CC 3=. In
ubsequent experiments, the RNA concentration andCR cycle number were chosen from the linear portionf the curve for each cytokine. From the product ofeverse transcriptase polymerase chain reaction (RT-CR), 5 �L were used for electrophoresis. The amountf DNA was normalized for the amount of the house-eeping G3PDH, which did not change under conditionn these experiments. In each sample, both the cytokinend the housekeeping gene, G3PDH, were quantifiednder the same conditions. G3PDH is preferred to-actin, because levels of the �-actin mRNA have been
ound to be regulated by a variety of agents, whereas3PDH was reported to be refractory to regulatory
gents.28 The same RT-PCR technique under the con-itions requested by each cytokine was used to measurehanges in different samples, which received differentreatments. For the PCR amplification, a Perkin-Elmeretus DNA Thermal Cycler model 480 (Cetus Corpo-
ation, Emeryville, Calif) was used. PCR products wereeparated in 2% agarose gel. In addition to being theredicted size, the nature of PCR products was con-rmed by automated sequencing. For the gel-electro-horesis, a Gel apparatus (Bio-Rad Laboratories, Her-ules, Calif) was used. A cDNA synthesis kit wasurchased from Bethesda Research Laboratories, Inc.Bethesda, Md). The electrophoresis was visualizedith ethidium bromide, and the image analysis of the
xpression was quantitated with a Phophoimager: SI/ensitometer (model PST-486) connected to an ana-
ytic, automatic program ImageQantNewT (Molecularynamics, Sunnyvale, Calif). Each determination waserformed in triplicate in at least 3 different cell pop-lations. The results of the cytokines cellular expres-ion were presented in percentage with controls at00% value.Data analysis. Results are expressed as mean � SD.
tatistical analysis was performed using 1-way analysis o
f variance between the groups. A P � 0.05 wasonsidered as significant.All statistical analysis was done using SPSS 11.5
SPSS Incorporated, Chicago, Ill). Sensitivity, specific-ty, positive predictive and negative predictive values,nd positive and negative likelihood ratios were calcu-ated using the SPSS 11.5 program.
ESULTS
Patient population. In this study, the following pa-ients were used: 220 controls (160 women and 60 men)hat did not present any symptoms of HSRs to sulfon-mides and 86 patients that had a previous history ofulfonamide use. The patients with a history of andverse reaction and symptoms were placed in one ofhe following categories: (1) rash, (2) rash and fever, or3) rash, fever, and organ involvement. Patients withash had a wide range of symptoms: eczema, non-ollicular papules, blisters, conjunctivitis, desquama-ion of skin, pruritic, erythematous, and skin vasiculitis.ever was considered any indication of increased body
emperature. Organ involvement included the follow-ng: thrombocytopenia, raised liver enzymes, hepatocy-otoxicity, leukocytosis, eosnophilia, lymphadenopa-hy, and pancytopenia. The clinical syndrome appears–56 days after the initial therapy.5
Of the 86 exposed patients, 14 were men with a meange of 46.64 � 28.94 and 72 were women with a meange of 52.07 � 14.40. Of the total 86 patients, 43 hadymptoms and had positive LTA results and 43 hadlinical symptoms and had negative LTA results. Con-rol population (n � 220) was as follows: 60 maleolunteers (36.42 � 8.64) and 160 female volunteers49.44 � 21.40). Overall, 2 of 220 control individualsid not present any symptoms but were positive forheir LTA, and 218 did not present any symptoms andere negative for their LTA values. The statistical
ignificance is shown in the Table I.29 The statisticalnalysis for patients with rash, fever, and organ in-olvement indicated a sensitivity of 92.90%, suggest-ng a strong true-positive rate, and a specificity of9.1%, which nicely identifies patients without the dis-ase. Of the group of 16 individuals that had presentedash, fever, and organ, and organ involvement, 12 hadeen treated in the burn unit for SJS or TEN. Overall,of these 12 patients had LTA performed as a diag-
ostic test during the SJS/TEN episode, and it wasepeated in this study 2–4 years after the episode. TheTA remained positive to the drug even after 2–4 yearsost-SMX-induced SJS.Cytokine results. Cytokines were measured in the sera
f patients 2–4 years after the HSR occurred. The level
f TNF-� for women with positive LTA values was
sudAL
srTIgeaHIwpwfo
Iavtvliiptwvs�
hooh�Epv
T
IIIIIIIR
Np*†
‡
§ ver, and�
FtFwcFfptr
Translational ResearchVolume 149, Number 5 Neuman et al 249
tatistically higher than in men with positive LTA val-es. The level of TNF-� in HSR-LTA-negative patientsid not differ from the normal population (60 pg/mL).
significant correlation of 0.001 was noted for HSR-TA-positive and higher TNF-�.Regarding the distribution of cytokine levels in the
era of different groups (controls, rash, rash � fever,ash � fever � organ involvement) summarized inable II, the population with only rash presented higher
L-5 levels than controls (P � 0.05) and the otherroups. In patients presenting rash � fever, IL-18 lev-ls were significantly higher (P � 0.05) than controlsnd subjects with rash only. The patients with trueSRs presented significantly higher values for IL-1,
L-2, and IL-12 (P � 0.001) than all other groups. IL-6as found to be less in the sera of control patients andatients presenting only rash. In addition, IL-18 levelsere significantly higher than patients with rash �
ever (P � 0.05) and patients with just rash (P � 0.001)r controls (P � 0.001).Figure 2 presents the values of TNF-�, Fas, and
L-8 in control volunteers, patients presenting rash,nd patients presenting rash � fever � organ in-olvement. TNF-� is 6 times more elevated in pa-ients presenting rash � fever � organ involvementersus the controls and patients presenting the sameevels in controls and those with only rash (Fig 2). Its also 3 times more elevated in individuals present-ng rash with fever. Fas levels in the individualsresenting rash � fever � organ involvement are 3imes more elevated than in patients presenting rashith fever pointing to the apoptotic processes in-olved in the immunopathogenesis of true hypersen-itivity syndromes. The individuals presenting rash
able II. The distribution of cytokine levels among th
Interleukines pg/mL(mean � SD) Control (220) Rash
L-1 50 � 15 55 �L-2 65 � 15 75 �L-4 78 � 10� 46 �L-5 25 � 15 66 �L-6 38 � 10 36 �L-12 69 � 10 58 �L-18 130 � 5 110 �ANTES 50 � 10 45 �
ote: Controls were considered volunteer individuals that took suatients that took sulfonamide developed only a rash.P � 0.05 higher than levels in patients with rash and controls.P � 0.05 higher than the levels in patients with rash and fever anP � 0.05 higher than controls.P � 0.001 higher than the levels in patients with rash, rash and feP � 0.001 higher than the levels of HSRs patients.
fever � organ involvement had IL-8 levels 5 times (
igher than the levels in the individuals presentingnly rash or rash with fever or controls. Expressionsf ILs other than TNF-� were not significantlyigher between the patients presenting rash � fever
organ involvement and those presenting only rash.xpression of TNF-� was significantly elevated inatients with rash � fever � organ involvementersus controls or patients with rash or rash � fever
rent groups studied
Rash � fever (43)Rash � fever � organ
involvement (16)
80 � 8 180 � 20§
90 � 15 260 � 10§
42 � 4 39 � 455 � 10‡ 50 � 1552 � 8 69 � 6*72 � 12 195 � 15§
190 � 5* 250 � 10†
55 � 15 150 � 15§
es and did not develop any reactions to the drug; the group of
01 higher than patients with rash and controls.
controls.
ig 2. Correlation among serum levels of TNF-�, Fas, and IL-8 andhe clinical symptoms in patients with HSRs. The levels of TNF-�,as, and IL-8 were significantly (P � 0.0001) elevated in patientsith rash � fever � organ involvement (RashFO) as compared with
ontrols and patients that presented only rash or rash � fever (Rash-ever). TNF-� levels and Fas levels in patients presenting rash �ever were significantly higher (P � 0.001) versus the control and theatients presenting only rash. No changes have been observed be-ween the levels of IL-8 in control and in patients presenting rash orash � fever.
e diffe
(27)
10151010‡
10101515
lfonamid
d P � 0.0
Fig 3).

D
pcrcGtdwcfect
svpmdaa
mvmet(pts
RpdGcatvsso
pDcLpcSfsccwricvcss
dctasialaisntseame
it
Fsparaprfv
Translational Research250 Neuman et al May 2007
ISCUSSION
It is critical to have an understanding of the biochemicalrocesses that occur during HSR. It allows for a betteromprehension of predisposing factors, potential cross-eactivity, and the duration of treatment, and it aids clini-ians in their assessment of further or continued therapy.4
ender is thought to be a predisposing factor; however, inhis study, no significant difference existed between gen-er-related positive or negative LTA results, and yetomen were the majority (83.7%) of subjects. This result
ould be, in part, because of the higher percentage ofemale patients referred to the clinic. However, it has beenstablished that gender differences can be based on cyto-hrome P450 expression, hormonal influence, drug me-abolism, or obesity.30,31
Current in vitro investigations contribute to under-tanding of the fundamental biology underlying ad-erse drug reactions, providing clues to their immuno-athogenesis. A positive predictive value of 92.3% hasade the in vitro LTA test a reliable tool for the
iagnosis of the true HSRs caused by sulfonamidentibiotics. It is in this respect that an LTA test is mostcknowledged as being a predictor test for true HSR.Clinical symptoms of rash, fever, and organ involve-ent that occur in HSRs may be because of the in-
olvement of cells such as neutrophils, monocytes, andacrophages, as well as specialized cells such as Lang-
rhans cells within keratinocytes.23,32,33 In this study,he chemoattractant substances such as chemokinesIL-8 and RANTES) were higher in the HSR thatresented the organ involvement when compared withhe individuals that presented less important symptoms
ig 3. Expression of TNF-� in the PBMCs of patients with hyper-ensitivity syndromes to sulfonamides (Line A). Line B is the ex-ression of glucose-6-phosphate dehydrogenase, which was taken toct as a housekeeping enzyme/cytokine. Column 1: patient presentingash � fever � organ involvement after treatment with sulfonamidentibiotics has significant (P 0.001) expression of TNF-� (686ics) versus control volunteers (0 pics). Column 2: patient presentingash � fever (88 pics). Column 3 and 4: patient presenting rash �ever � organ involvement (550 pics). Columns 5 and 6: contrololunteers. Columns 7 and 8: patients presenting only rash.
uch as fever. In their review, Rubin and Kretz- g
ommel32 suggest that metabolites of SMX may com-ete with chloride ions for this pathway producingrug-free radicals, which are promptly detoxified withSH conjugation or acetylation. The system requires
ontrolled levels of GSH to maintain low levels of toxicgents. Depletion of GSH by its use in phase II reac-ions may leave organelles such as the mitochondriaulnerable to ROS (Fig 1). Based on this idea, thepecific mitochondrial enzyme SDH activity (data nothown) that correlates directly to mitochondrial toxicityccurring in HSRs was measured.Mitochondrial damage may be only part of the com-
lications involved in HSRs, but it is a critical one.efects in any of the 5 complexes in the respiratory
hain would greatly add to mitochondrial toxicity. TheTA is a marker of one such disorder because it em-loys the use of SDH (E.C. 1.3.99.1), an enzyme part ofomplex II in the mitochondrial respiratory chain.34 AsDH is very specific to mitochondrial activity, its dys-unction would indicate cell death when the cell istressed. As is the case with the LTA, cells from sus-eptible individuals have shown high degrees of mito-hondrial apoptosis when exposed to drug metabolites,hich would indicate a defective pathway in the respi-
atory chain that most likely involves SDH. Deficiencyn glutathione synthetase (E.C.6.3.2.3) showed in-reased toxicity to sulfadiazine metabolites generatedia the hepatic microsomal system. An inherited defi-iency in GST activity within the target cells is respon-ible for the development of sulfonamide toxicity inusceptible individuals.
Cytokines are regulatory peptides that can be pro-uced by virtually every cell in the body.35-38 Theytokine family consists of several subfamilies: the ILs;he TNF family; IL-6 and related cytokines; interferons;nd chemokines such as IL-8, RANTES, colony-timulating factors, and others. In most tissues, includ-ng the liver, constitutive production of cytokines isbsent or minimal. However, as physiologic and patho-ogic stimuli activate cells, the production of theseutocrine, paracrine, and endocrine effector moleculesncrease, and they, in turn, orchestrate the tissue’s re-ponse to stimuli such as inflammation, apoptosis, andecrosis,39-43 but paradoxically they can also mediatehe regeneration of liver tissue after injury.44-46 In thistudy, the levels of IL-5, a Th2-type response, waslevated in patients with only a rash, pointing to anllergic type of reaction in these individuals. Further-ore, patients that had presented a true HSR 2–4 years
arlier had significantly elevated levels Fas and TNF-�.Cytokines involved in immune responses play a myr-
ad of roles. The T-cell response generated because ofhe presence of haptenized SMX metabolites has been
iven much attention.47,48 It has been found that SMX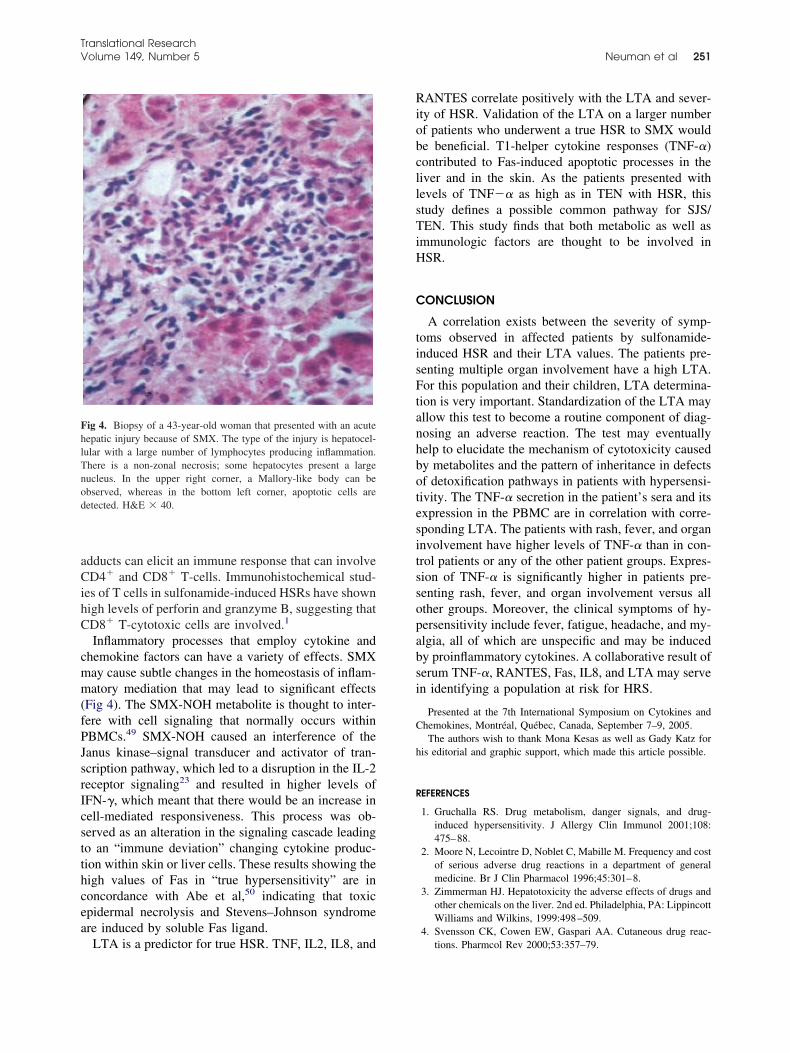
aCihC
cmm(fPJsrIcstthcea
RiobcllsTiH
C
tisFtanhbotesitssopabsi
C
h
R
FhlTnod
Translational ResearchVolume 149, Number 5 Neuman et al 251
dducts can elicit an immune response that can involveD4� and CD8� T-cells. Immunohistochemical stud-
es of T cells in sulfonamide-induced HSRs have shownigh levels of perforin and granzyme B, suggesting thatD8� T-cytotoxic cells are involved.1
Inflammatory processes that employ cytokine andhemokine factors can have a variety of effects. SMXay cause subtle changes in the homeostasis of inflam-atory mediation that may lead to significant effects
Fig 4). The SMX-NOH metabolite is thought to inter-ere with cell signaling that normally occurs withinBMCs.49 SMX-NOH caused an interference of theanus kinase–signal transducer and activator of tran-cription pathway, which led to a disruption in the IL-2eceptor signaling23 and resulted in higher levels ofFN-�, which meant that there would be an increase inell-mediated responsiveness. This process was ob-erved as an alteration in the signaling cascade leadingo an “immune deviation” changing cytokine produc-ion within skin or liver cells. These results showing theigh values of Fas in “true hypersensitivity” are inoncordance with Abe et al,50 indicating that toxicpidermal necrolysis and Stevens–Johnson syndromere induced by soluble Fas ligand.
ig 4. Biopsy of a 43-year-old woman that presented with an acuteepatic injury because of SMX. The type of the injury is hepatocel-ular with a large number of lymphocytes producing inflammation.here is a non-zonal necrosis; some hepatocytes present a largeucleus. In the upper right corner, a Mallory-like body can bebserved, whereas in the bottom left corner, apoptotic cells areetected. H&E � 40.
LTA is a predictor for true HSR. TNF, IL2, IL8, and
ANTES correlate positively with the LTA and sever-ty of HSR. Validation of the LTA on a larger numberf patients who underwent a true HSR to SMX woulde beneficial. T1-helper cytokine responses (TNF-�)ontributed to Fas-induced apoptotic processes in theiver and in the skin. As the patients presented withevels of TNF� as high as in TEN with HSR, thistudy defines a possible common pathway for SJS/EN. This study finds that both metabolic as well as
mmunologic factors are thought to be involved inSR.
ONCLUSION
A correlation exists between the severity of symp-oms observed in affected patients by sulfonamide-nduced HSR and their LTA values. The patients pre-enting multiple organ involvement have a high LTA.or this population and their children, LTA determina-
ion is very important. Standardization of the LTA mayllow this test to become a routine component of diag-osing an adverse reaction. The test may eventuallyelp to elucidate the mechanism of cytotoxicity causedy metabolites and the pattern of inheritance in defectsf detoxification pathways in patients with hypersensi-ivity. The TNF-� secretion in the patient’s sera and itsxpression in the PBMC are in correlation with corre-ponding LTA. The patients with rash, fever, and organnvolvement have higher levels of TNF-� than in con-rol patients or any of the other patient groups. Expres-ion of TNF-� is significantly higher in patients pre-enting rash, fever, and organ involvement versus allther groups. Moreover, the clinical symptoms of hy-ersensitivity include fever, fatigue, headache, and my-lgia, all of which are unspecific and may be inducedy proinflammatory cytokines. A collaborative result oferum TNF-�, RANTES, Fas, IL8, and LTA may serven identifying a population at risk for HRS.
Presented at the 7th International Symposium on Cytokines andhemokines, Montréal, Québec, Canada, September 7–9, 2005.The authors wish to thank Mona Kesas as well as Gady Katz for
is editorial and graphic support, which made this article possible.
EFERENCES
1. Gruchalla RS. Drug metabolism, danger signals, and drug-induced hypersensitivity. J Allergy Clin Immunol 2001;108:475–88.
2. Moore N, Lecointre D, Noblet C, Mabille M. Frequency and costof serious adverse drug reactions in a department of generalmedicine. Br J Clin Pharmacol 1996;45:301–8.
3. Zimmerman HJ. Hepatotoxicity the adverse effects of drugs andother chemicals on the liver. 2nd ed. Philadelphia, PA: LippincottWilliams and Wilkins, 1999:498–509.
4. Svensson CK, Cowen EW, Gaspari AA. Cutaneous drug reac-
tions. Pharmcol Rev 2000;53:357–79.
1
1
1
1
1
1
1
1
1
1
2
2
2
2
2
2
2
2
2
2
3
3
3
3
3
3
3
3
3
3
4
4
4
4
4
Translational Research252 Neuman et al May 2007
5. Knowles SR, Uetrecht J, Shear NH. Idiosyncratic drug reac-tions: the reactive metabolite syndromes. Lancet 2000;356:1587–91.
6. Shear NH, Spielberg S. Anticonvulsant hypersensitivity syn-drome. J Clin Invest 1988;82:1826–32.
7. Jones D, Chiap V, Resor S, Grossman M. Phenytoin-like hyper-sensitivity associated with lamotrigine. J Acad Dermatol 1997;36:1016–8.
8. Svensson CK, Cowen E, Gaspari AA. Cutaneous drug reactions.Pharmacol Rev 2000;53:357–79.
9. Masters PA, O’Bryan TA, Zurlo J, Miller DQ, Joshi N. Tri-methoprim-sulfamethoxazole revisited. Arch Intern Med 2003;163:402–10.
0. Cribb A, Lee B, Trepanier L, Spielberg S. Adverse reactions tosulfonamide and sulfonamide-trimethoprim antimicrobials: clin-ical syndromes and pathogenesis. Adverse Drug React ToxicolRev 1996;15:9–50.
1. Lazarou J, Pomeranz B, Corey P. Incidence of adverse drugreactions in hospitalized patients. JAMA 1998;279:1200–5.
2. Jick J. A possible explanation based on patterns of immunedysregulation seen in HIV-1 disease. Clin Exp Dermatol 1982;22:118–23.
3. Schnyder B, Burkhart C, Shnyder-Frutig K, von Greyerz S,Naisbitt DJ, Pirmohamed M, et al. Recognition of sulfamethox-azole and its reactive metabolites by drug-specific CD4� T cellsfrom allergic individuals. J Immunol 2000;164:6647–54.
4. Smith KJ, Skelton HG, Yeager J, Ledsky R, Hg TH, Wager KF.Increased drug reactions in HIV-1-positive patients: a possibleexplanation based on patterns of immune dysregulation seen inHIV-1 disease. The Military Medical Consortium for the Ad-vancement of Retroviral Research (MMCARR). Clin Exp Der-matol 1997;22:118–23.
5. Coopman SA, Johnson RA, Platt R, Stern RS. Cutaneous diseaseand drug reactions in HIV infection. N Engl J Med 1993;328:1670–4.
6. Krivoy N, Struminger L, Bendersky R, Avivi I, Neuman MG,Pollack S. Rifampin-induced thrombocytopenia: diagnosis by anovel in vitro lymphocyte toxicity assay. Isr Med Assoc J 2001;3:536.
7. Sbeit W, Krivoy N, Shiller M, Furah R, Cohen HI, Struminger L,et al. Nimesulide-induced acute hepatitis. Ann Pharmacother2001;35:1049–52.
8. Neuman MG, Malkiewicz IM, Shear NH. A novel lymphocytetoxicity assay to assess drug hypersensitivity syndromes. ClinBiochem 2000;33:517–24.
9. Neuman MG, Malkiewicz IM, Phillips EJ, Rachlis AR, Ong D,Yeung E, et al. Monitoring adverse drug reactions to sulfonamideantibiotics in human immunodeficiency virus-infected individu-als. Ther Drug Mon 2002;24:728–36.
0. Cribb A, Miller M, Leeder J, Hill H, Spielberg S. Reactions ofthe nitroso and hydroxylamine metabolites of sulfamethoxazolewith reduced glutathione. Drug Metab Dispos 1991;19:900–6.
1. Gill HJ, Hough SJ, Naisbitt DJ, Maggs JL, Kitteringham NR,Pirmohamed M, et al. The relationship between the dispositionand immunogenicity of sulphamethozazole in the rat. J Pharma-col Exp Ther 1997;282:795–801.
2. Naisbitt DJ, Gordon SF, Pirmohamed M, Brurkhart C, Cribb AE,Pichler WJ, et al. Antigenicity and immunogenicity of sulpha-methoxazole: demonstration of metabolism-dependent hapteni-zation and T-cell proliferaton in vivo. Br J Pharmacol 2001;133:295–305.
3. Reilly TP, Ju C. Mechanistic perspectives on sulfonamide-in-duced cutaneous drug reactions. Curr Opin Allergy Clin Immu-
nol 2002;2:307–15.4. Reilly TP, Lash LH, Doll MA, Hein DW, Woster PM, SvessonCK. A role for bioactivation and covalent binding within epider-mal keratinocytes in sulfonamide-induced cutaneous drug reac-tions. J Invest Dermatol 2000;114:1164–73.
5. Rieder M, Uetrecht J, Shear NH, Cannon M, Miller M. Diagnosisof sulfonamide hypersensitivity reactions by in vitro “rechal-lenge” with hydroxylamine metabolites. Ann Inter Med 1989;110:286–9.
6. Cribb A, Spielberg S. Hepatic microsomal metabolism of sulfa-methoxazole to the hydroxylamine. Drug Metab Dispos 1990;18:784–7.
7. Riley RJ, Leeder JS. In vitro anaylsis of metabolic predisposition todrug hypersensitivity reactions. Clin Exp Immunol 1995;99:1–6.
8. Neuman MG, Shear NH, Bellentani S, Tiribelli C. Role ofcytokines in ethanol-induced hepatotoxicity in Hep G2 cells.Gastroenterol 1998;114:157–69.
9. Mark DB. Decision making in clinical medicine. In: BraunwaldE, Fauci AS, Kasper DL, Hauser SL, Longo DL, Jameson JL,eds. Principles of internal medicine—Harrison’s 15th ed. NewYork: McGraw-Hill, 2001:8–14.
0. Thurmann PA, Hompesch BC. Influence of gender on the phar-macokinetics and pharmacodynamics of drugs. Int J Clin Phar-macol Ther 1998;36:586–90.
1. Kotlyar M. Effects of obesity on the cytochrome P450 enzymesystem. Int J Clin Pharmacol Ther 1999;37:8–19.
2. Rubin RL, Kretz-Rommel A. Phagocyte-mediated oxidation inidiosyncratic adverse drug reactions. Curr Opin Hematol 2001;8:34–40.
3. Burner U, Furtmuller PG, Kettle AJ, Koppenol WH, Obinger C.Mechanism of reaction of myeloperoxidase with nitrite. J BiolChem 2000;275:20597–601.
4. Turnbull DM, Bindoff LA. Muscle disease. In: Marshall WJ,Bangert SK, eds. Clincal biochemistry metabolic and clinicalaspects. New York: Churchill Livingstone, 1995:551–2.
5. Dinarello C. Biologic basis for interleukin-1 in disease. Blood1996;87:2095–147.
6. Tracey K, Cerami A. Tumor necrosis factor, other cytokines anddisease. Annu Rev Cell Biol 1993;9:317–43.
7. Neuman MG, Cameron RG, Shear NH, Feuer G. Drug-inducedapoptosis of skin cells and liver. In: Cameron RG, Feuer G, eds.Handbook of experimental pharmacology: apoptosis modulatonby drugs. Heidelberg, Germany: Springer Verlag Publishers,1999:344–55.
8. Neuman MG. Apoptosis in disease of the liver. Crit Rev Clin LabSci 2001;38:109–66.
9. Neuman MG. Apoptosis in liver disease. Rom J Gastroenterol2002;11:3–7.
0. Mizuhara H, O’Neill E, Seki N, Ogawa T, Kusunoki C, OtsukaK, et al. T cell activation-associated hepatic injury: mediation bytumor necrosis factors and protection by interleukin 6. J Exp Med1994;179:1529–37.
1. Orange J, Salazar-Mather T, Opal S, Biron C. Mechanism forvirus-induced liver disease: tumor necrosis factor-mediated pa-thology independent of natural killer and T cells during murinecytomegalovirus infection. J Virol 1997;71:9248–58.
2. Trauner M, Meier P, Boyer J. Molecular pathogenesis of cho-lestasis. N Engl J Med 1998;339:1217–27.
3. Friedman S. Molecular regulation of hepatic fibrosis, and inte-grated cellular response to tissue injury. J Biol Chem 2000;275:2247–50.
4. Akerman P, Cote PYS, McClain C, Nelson S, Bagby GJ, White HS.Antibodes to tumor necrosis factor-alpha inhibit liver regeneration
after partial hepatectomy. Am J Physiol 1992;263:G579–85.
4
4
4
4
4
5
Translational ResearchVolume 149, Number 5 Neuman et al 253
5. Yamada Y, Kirillova I, Peschon J, Fausto N. Initiation of livergrowth by tumor necrosis factor: deficient liver regeneration inmice lacking type I tumor necrosis factor receptor. Proc NatlAcad Sci USA 1997;94:1441–6.
6. Cressman D, Greenbaum L, DeAngelis R, Ciliberto G, Furth E,Poli V, et al. Liver failure and defective hepatocyte regenerationin interleukin-6-defecient mice. Science 1996;274:1379–83.
7. Ring J, Brockow K. Adverse drug reactions: mechanisms andassessment. Eur Surg Res 2002;34:170–5.
8. Neuman MG, Shear NH, Malkiewicz IM, Abbot F. P450’s 2E1
inducers trigger valproic acid-induced apoptosis in vitro [ab-stract]. Clin Invest Med 1998;21:582.
9. Hess DA, O’Leary EF, Lee JT, Almawi WY, Madrenas J, RiederMJ. Inhibition of cytokine production and interference in IL-2receptor-mediated Jak-Stat signaling by the hydroxylamine me-tabolite of sulfamethoxazole. FASEB J 2001;15:1855–7.
0. Abe R, Shimizu T, Shibaki A, Nakamura H, Watanabe H,Shimizu H. Toxic epidermal necrolysis and Stevens-Johnsonsyndrome are induced by soluble Fas ligand. Am J Pathol.
2003;162:1515–20.
As
M
T
Atmdap
FDTC
Sb
RBSe
1
©
d
2
poptosis in ibuprofen-induced Stevens–Johnsonyndrome
ANUELA NEUMAN and MICHAEL NICAR
ORONTO, ONTARIO, CANADA AND DALLAS, TEX
Cytokines play a role in the immunopathological and molecular mechanisms ofdrug-induced hypersensitivity reactions (HSR). The objective of the current reportwas to analyze the reliability and correlation between the clinical symptoms ob-served in a patient that presented an ibuprofen-induced Stevens–Johnson Syn-drome (SJS), her lymphocyte toxicity assay to the incriminated drug, and thecytokine secretion in the patient’s sera. In her skin biopsy, the apoptotic keratino-cytes is shown. Clinically, the patient presented a triad that characterizes “true” HSR(rash, fever, and liver involvement). The pro-inflammatory cytokines were signifi-cantly higher in sera from the patient than in sera from control patients (analyzedpreviously in the authors’ laboratory). More specifically, the high level of tumornecrosis factor alpha (TNF-�) is as high as in patients found to have toxic epidermalnecrolysis (TEN) and presenting “true” HSR, eg, rash, fever, and organ involvement.The same is the case with the apoptotic markers Fas, caspase activity, and M30.T-cell cytokines control the pathogenesis of SJS/TEN contributing to apoptotic pro-cesses in the liver and in the skin. (Translational Research 2007;149:254–259)
Abbreviations: ELISA � enzyme-linked immunosorbent assay; HSR � hypersensitivity syndromereaction; IL � interleukin; LTA � lymphocyte toxicity assay; RANTES � regulated upon activa-tion, normal T cell expressed and secreted; SJS � Stevens–Johnson syndrome; TEN � toxic
epidermal necrolysis; TNF-� � tumor necrosis factor-alpha.edmad
iaa1ttmgats(va
dverse drug reactions account for 5% of allhospital admissions and occur in approxi-mately 15% of hospitalized patients.1 Most of
hese reactions are common and predictable, being pri-arily based on the pharmacological properties of the
rug given. Unpredictable or idiosyncratic, type B re-ctions pose a major concern in clinical practice andrimarily because they are not dose-dependent.2 How-
rom the In Vitro Drug Safety and Biotechnology Laboratory, MaRSiscovery Centre, and Department of Pharmacology, University oforonto, Toronto, Ontario, Canada, and 2Baylor University Medicalenter, Dallas, Tex.
ubmitted for publication July 26, 2006; revision submitted Novem-er 19, 2006; accepted for publication December 19, 2006.
eprint requests: Manuela Neuman, PhD, In Vitro Drug Safety andiotechnology Laboratory, MaRS Discovery District, 101 Collegetreet, Suite 300, Lab 351, Toronto, Ontario, Canada M5G 1L7;-mail: [email protected].
931-5244/$ – see front matter
2007 Mosby, Inc. All rights reserved.
toi:10.1016/j.trsl.2006.12.005
54
ver, type B reactions are known to occur at certainoses in susceptible individuals.3 One of the most com-on and potentially fatal adverse drug reactions is
ssociated with antimicrobials and anti-inflammatoryrugs.4
The “true” hypersensitivity syndrome reaction (HSR)s a systemic disease defined by the triad of fever, rash,nd internal organ involvement that starts 8 to 12 weeksfter the initiation of therapy. An incidence of 1/1000 to/10,000 exposures is inaccurate because many reac-ions are common, yet under-reported.5 Clinical condi-ions range from simple skin reactions and generalanifestations such as morbiliform rash, urticaria, an-
ioedema, fever, malaise, anaphylaxis, bronchospasm,nd erythema multiforme, to skin complications withhe highest mortality rates exhibited by Stevens–John-on syndrome (SJS) and toxic epidermal necrolysisTEN) also known as Lyell’s syndrome.6,7 These in-olve multi-organ function impairment, such as lymph-denopathy, hepatic dysfunction (liver function 3 times
he upper normal limit), hematologic dysfunction
(roa
ctawpftotn
M
htVTactShhsd
dafit
bvopmtcteFSm
eaio
[(shlamctotiShftrdskTbyeft
n
5mpas
tnbLpcEcp0gspwaMMM
Translational ResearchVolume 149, Number 5 Neuman and Nicar 255
white blood count �2000, platelets �100,000/mL),enal dysfunction (hematuria, nephritis), myocarditis,r myositis.8-13 These reactions have limited the ther-peutic use of many drugs including ibuprofen.HSRs due to drugs of use are likely caused by a
ombination of metabolic and immunologic fac-ors.14,15 Cellular immune-mediated components, suchs the T cells and cytokine/chemokine mediation,hich can exacerbate cellular responses, create com-lex pathways that lead to a variety of clinical mani-estations. A drop in the detoxifying capability will leado cell death. If apoptosis ensues, then the recruitmentf chemokines would also follow. This effect adds tohe inflammatory process and accelerates the cellularecrosis, leading to an HSR.16-19
ATERIALS AND METHODS
Case report. A 7-year-old African-American girl wasospitalized with rash, pruritus, and fever. Previously,he girl was given ibuprofen, as medication for fever.iral determinations showed no signs of viral infection.he vesicular eruptions became bullous, coalescent,nd led to desquamation of the skin. Based on thelinical manifestation, the hospital dermatologist madehe diagnosis of SJS. A biopsy was taken during theJS episode. During the hospitalization she developedigh alanine aminotransferase � 1.5 than normal andigh aspartate aminotransferase � 8 than normal. Con-idering the high liver enzymes and the bilirubin founduring this period, liver involvement was confirmed.The biopsy material was analyzed that was obtained
uring the acute SJS/TEN episode. Two years after thecute episode of SJS, the blood was analyzed to con-rm the hypersensitivity to the specific drug. Moreover,
he cytokine profile of the patient was determined.By light microscopy the differential diagnosis made
y the hospital pathologist revealed “a subepidermalesicle formed by ballooning degeneration and necrosisf keratinocytes at the basal layer with edema of theapillary dermis, peri-vascular lympho-histiocytic der-atitis. Scatter necrotic keratinocytes and intact stra-
um corneum. No viral inclusion is identified.” Theonstellation for these findings is consistent with ery-hema multiform/SJS.20 Abe et al21 showed that toxicpidermal necrolysis and SJS are induced by solubleas ligand leading to cell death. Because a landmark ofJS–TEN is the skin cell death, it was decided to use 1arker of apoptosis.The levels of different cytokines and cell death mark-
rs in the blood were analyzed to determine the reli-bility and correlation between clinical evidence of SJSn the patient and the immuno-histochemistry markers
f inflammation and cell death. The utility of cytokines ptumor necrosis factor-alpha (TNF-�), interleukinsILs), and markers of cell death] as potential noninva-ive markers for susceptibility to drug-induced HSRsas also been evaluated. The results obtained by ana-yzing these specimen(s) with data accumulated in theuthors’ laboratory over the last 15 years were deter-ined. The control specimens used for correlation
ame from healthy individuals that volunteer. The pa-ients were referred to in the In Vitro Toxicology lab-ratory by drug safety clinicians, dermatologists, andhe burn unit surgeons that treated SJS and TEN. Eth-cal approval for the studies was obtained from thecientific and Ethics Review Committees of the sameospital. The subjects were divided into 2 subgroups asollows: 6 patients who developed a clinical SJS/TENo ibuprofen and 60 immunocompetent individuals thateceived the same drug as the SJS/TEN patients withouteveloping HSR and voluntarily participated in thetudy. The lymphocyte toxicity assay (LTA) and cyto-ine analysis was performed 2–3 years after the SJS orEN. The 2 individuals that have the LTA performedoth during the clinical presentation of SJS and 2 or 3ears after the episode presented similar LTA values atach point in time. In 3 control individuals, the ibupro-en–LTA was performed also at 2 consecutive points inime and presented the same negative value.
The cytokine measurements were performed only inonactive clinical SJS.Drugs and chemicals. The tetrazolium salt 3-(4,
-dimethylthiazol-2-yl) 2, 5 diohenyl-tetrazolium bro-ide (MTT) was obtained from Sigma Chemical Com-
any (St. Louis, Mo). All remaining reagents were ofnalytical grade obtained from the usual commercialources.
Laboratory methods. Cytotoxicity assay–lymphocyteoxicity assay. The method was first described for sulpho-amide and anticonvulsant-induced toxicity.14 Wholelood was layered on a Ficoll–Paque density gradient.ymphocytes were collected by centrifugation and sus-ended in a HEPES-buffered medium. Cells were thenounted and resuspended at a density of 2 � 106 cells/mL.ach reaction involves the incubation of patient lympho-ytes (viability �95%) at 37°C with 0.28-mg microsomalrotein (liver microsomes from mice induced-P450),.6-mM NADP, 2.24-mM glucose-6-phosphate, 2 U oflucose-6-phosphate dehydrogenase, and ibuprofen at thepecific concentration. At the end of the 24-h incubationeriod, MTT (100 �L) was added for 1 h. The reactionas stopped, and the cells were lysated with isopropyl
lcohol. Cytotoxicity was measured using the Maxlineicroplate Reader (Molecular Device Corporation,enlo Park, Calif) connected to a computer using SOFTAX software 2.3 for Windows (Molecular Device Cor-
oration) allowing for the template to be programmed.

aIcestEdMia
wcssc6ttiiaufpsap
aNctea
R
itrinLphitwvr
sr
dTaTpvtatda
pFFtcecnole
aecdl
Ts
IIIIIIIRTFMCC
*†
Translational Research256 Neuman and Nicar May 2007
Cytokine measurement in sera. Cytoscreen, immuno-ssay kits, human Fas, IL-1, IL-2, IL-4, IL-5, IL-6,L-8, IL-10, IL-18 regulated upon activation, normal Tell expressed and secreted (RANTES), and TNF-�nzyme-linked immunosorbent assay (ELISA; Bio-ource International, Camarillo, Calif) were used forhe quantitative determination. M30-ApoptosenseLISA (Bender MedSystems, Vienna, Austria). Stan-ards and reference reagents available from BenderedSystems were used. Each specimen was analyzed
n triplicate with a sensitivity and specificity of 95%nd 90%, respectively.
Immunohistochemistry. The slide arrived intact, and itas perfectly suitable for further analysis. The specimen
ontained a peri-lesional and lesional tissue (involvedkin). The 5-�m formalin fixed, paraffin embedded tissueection was previously mounted on positive charged mi-roscope slide. Tissue section was then baked overnight at0°C, de-waxed in xylene, and hydrated to distilled waterhrough decreasing concentrations of alcohol. Immunohis-ochemical procedure for Cleaved Caspase 3 (Cell Signal-ng, Beverly, Mass) was performed on the NEXES auto-mmunostainer (Ventana Medical Systems, Tuscon, Ariz),ll at a dilution of 1:25. Immunodetection was carried outsing the Ventana iView DAB detection kit and blockedor endogenous biotin (Ventana Avidin/Biotin Block) asart of the automated staining protocol. Before immuno-taining, the slide for cleaved caspase 3 was heat-inducedntigen retrieved, with citrate buffer. The counter-stain ofreference was hematoxylin, for nuclear detail.For reading, an Olympus microscope equipped with
n Olympus camera (Olympus America, Inc., Melville,Y) was used. The image was transmitted from the
amera to the computer that captured the images withhe help of a “Ropper” software. The images of differ-nt fields from the slide were taken and compared usingmorphometry program.
ESULTS
In controls, LTA shows normal ranges of toxicity forbuprofen. This patient presented a high level of cyto-oxicity to ibuprofen as compared with controls with aesult of 33%. The normal level or cut-off for thisbuprofen-LTA on patients is 15% (10 � 5). In immu-ocompetent individuals found to have SJS or TEN, theTA ranges between 24% and 35%. The immunocom-etent person is defined as not presenting acquired orereditary immunodeficiency, and as not being undermmunosupressive drugs or radiotherapy. The valida-ion for the LTA in ibuprofen-induced hypersensitivityas performed, and the test had a positive predictivealue of 91%. The statistical analysis for patients with
ash, fever, and organ involvement indicated an LTA iensitivity of 92.90%, suggesting a strong true-positiveate and a specificity of 99.1%.
The distribution of cytokine levels in the sera ofifferent groups (controls and TEN) is summarized inable I. It is clear that the cytokine values of the patientre in the range of the levels of the patients presentingEN. Fas (�3) levels and IL8 (�5) in the individualsresenting rash � fever � organ involvement are ele-ated, pointing to the apoptotic processes involved inhe immunopathogenesis of TEN. The values of Fasnd IL8 in the case presented are at the same level ashe TEN patients (see Table I). There is a statisticalifference between the levels of cytokines in controlnd in individuals presenting SJS/TEN.Immunohistochemistry results are presented in the
ictures taken from the same slides on different fields.igure 1 contains both normal and abnormal skin cells.igure 2 shows the presence of caspase antibodies and
herefore activation of death signal and desquamatingells forming sheets. In Fig 2, there are normal-lookingpidermal cells, but at the basal layer, one can see theoloration of caspase 3. Figure 3 shows only a fewormal-looking epidermal cells. The entire architecturef the skin tissue is changed. Many ballooned cells areinked one to the other producing desquamation of thepithelia, compatible with the SJS.The imunohistochemistry result confirms that there
re skin cells with a high caspase (the name of thenzyme) activity compatible with cell death (apoptoticells). The overall picture is consistent with toxic epi-ermal necrolysis. In the same line, the high serumevels of proinflammatory cytokines reveal the similar-
able I. Serum levels of cytokines and capases inera of normal controls and TEN patients
Cytokines(mean � SD) Control (60) Patient
Rash � fever �
organinvolvement (6)
L-1 pg/mL 50 � 15 165 180 � 20*L-2 pg/mL 65 � 15 140 260 � 10*L-4 pg/mL 78 � 10 46 39 � 4†
L-5 pg/mL 25 � 15 66 50 � 15L-6 pg/mL 38 � 10 36 69 � 6†
L-8 pg/mL 69 � 10 158 195 � 15*L-10 pg/mL 130 � 5 170 250 � 10†
ANTES pg/mL 50 � 10 165 150 � 15*NF-� pg/mL 50 � 10 380 350 � 10*as ng/mL 2.0 � 1.0 28 25.0 � 10.0*30 (%) 20 � 10 38 60.0 � 10.0*aspase 8 IU/mL 5.0 � 1.0 18 25.0 � 15.0*aspase 3 IU/mL 4.0 � 1.0 33 35.0 � 10.0*
P � 0.001 higher vs control;P � 0.05 higher vs control.
ty with the level of cytokines observed in the persons

wi
D
oS
mTnsc
al cells (
Translational ResearchVolume 149, Number 5 Neuman and Nicar 257
ith rash, fever, and organ involvement due to a drug-nduced SJS or TEN.
ISCUSSION
Current investigations contribute to the understandingf the underlying adverse drug reaction that caused the
Fig 1. Immuno-histochemistry slide in the left bottobodies (activation of death signal). Epidermal and dermsheets. Foci of cleaved caspase 3 can be seen in derm
Fig 2. Presents with normal looking epidermal cell3, which is the signal to the cell death. There is acell are desquamating (Magnification x 40).
JS/TEN, providing clues to their immunopathogenesis. s
Clinical symptoms of rash, fever, and organ involve-ent that occur in HSRs and specifically in a patient’sEN may be due to the involvement of cells such aseutrophils, monocytes, and macrophages, as well as topecialized cells such as keratinocytes. However, ex-luding neutrophils, these cells are also antigen-pre-
many keratinocytes show presence of caspase anti-ith pale nuclei. There are desquamating cells formingupper left corner) (Magnification x 60).
e basal layer one can see the coloration of caspasein between the cells showing clear that sheets of
m corneral cells w
s, but at thdisruption
enting cells and thus are capable of biotransformation,

ccRmkttpt
sapITwishtesvcwha
d
toi
tti
C7INHra
R
Translational Research258 Neuman and Nicar May 2007
onjugation, and immune cell stimulation. Afterhemo-attractant, substances such as chemokines (IL-8,ANTES, MCP) activate neutrophils.22-25 Proinflam-atory cytokines such as IL-1�, and TNF-� are cyto-
ines that stimulate the synthesis of acute-phase pro-eins. IL-18 is proinflammatory at a very early step inhe immune response. IL-6 stimulates most acute-phaseroteins, whereas IL-10 is a prototype anti-inflamma-ory cytokine that regulates the B-cell function.26
Urticaria induced by beta-lactam antibiotics, analge-ics/antipyretics, and nonsteroidal anti-inflammatorygents was found to have highly significant meanlasma levels of cytokines and proteins (IL-2, IL-6,L-10, TNF-�) during inflammation and significantNF-�, C-reactive protein, IL-10, soluble IL-2 receptoreeks after treatment.27 High IL-10 was found in var-
ous drug-induced skin reactions.28 Although thesetudies may suggest protein patterns between differentypersensitivity reactions, these studies do not explainhe maximum length of time that cytokine levels arelevated. Higher T-cell numbers may be present forpecific hypersensitivity to drugs many years after ad-erse reactions occurred, and they can markedly in-rease cytokine levels.29 This is also in concordanceith the current case in which the patient maintainedigh levels of proinflammatory cytokines 2–3 yearsfter the SJS.The results of serum and immunohistochemistry
Fig 3. Skin biopsy treated with cleaved -caspase3entire architecture of the skin tissue is changed. Thproducing desquamation of the epithelia. There iscontinuation of the basal layer. In this slide where yis clear. (Magnification x 40).
emonstrate that the genetic background, environmen-
al factors, or immune-compromise system had previ-usly been activated to develop cytotoxicity towardbuprofen characterized by apoptotic processes.
This article summarizes and demonstrates for the firstime the link between the systemic reaction involvinghe liver-induced damage and the skin-induced damagen ibuprofen-induced SJS.
This work was presented at the 7th International Symposium onytokines and Chemokines, Montréal, Québec, Canada, September-9, 2005, and was supported in part by the Institute of Infection andmmunity of Canadian Institutes of Health Research and by theational Institute on Alcohol and Alcoholism, National Institutes ofealth, USA. The laboratory work was supported in parts by grants
eceived by Dr. Neuman from the Canadian Dermatology Foundationnd the Incorporated Foundation Physician’s Services.
EFERENCES
1. Gruchalla RS. Drug metabolism, danger signals, and drug-inducedhypersensitivity. J Allergy Clin Immunol 2001;108:475–88.
2. Moore N, Lecointre D, Noblet C, Mabille M. Frequency and costof serious adverse drug reactions in a department of generalmedicine. Br J Clin Pharmacol 1996;45:301–8.
3. Zimmerman HJ. Hepatotoxicity the adverse effects of drugs andother chemicals on the liver. 2nd ed. Philadelphia, Pa: LippincottWilliams and Wilkins; 1999:498–509.
4. Svensson CK, Cowen EW, Gaspari AA. Cutaneous drug reac-tions. Pharmcol Rev 2000;53:357–79.
5. Lazarou J, Pomeranz B, Corey P. Incidence of adverse drugsreactions in hospitalized patients. JAMA 1998;279:1200–5.
6. Lyell A. Toxic epidermal necrolysis: an eruption resembling
only few of normal looking epidermal cells. Theany ballooned cells that are linked one to the othermpty space representing a clear disruption of thee basal layer the coloration of caspase-3 antibodies
presentsere are ma large eou see th
scalding of the skin. Br J Dermatol 1956;68:355–61.

1
1
1
1
1
1
1
1
1
1
2
2
2
2
2
2
2
2
2
2
Translational ResearchVolume 149, Number 5 Neuman and Nicar 259
7. Lyell A. Toxic epidermal necrolysis (an eruption resemblingscalding of the skin) a repraisal. Br J Dermatol 1979;100:69–86.
8. Bastuji-Garin S, Rzany B, Stern R, Shear NH, Naldi L, RoujeauJC. Clinical classification of cases of toxic epidermal necrolysis,Stevens-Johnson syndrome, and erythema multiformae. ArchDermatol 1993;129:912–96.
9. Knowles SR, Uetrecht J, Shear NH. Idiosyncratic drug reactions:the reactive metabolite syndromes. Lancet 2000;356:1587–91.
0. Park BK, Pirmohamed M, Kitteringham NR. Idiosyncratic drugreactions: a mechanistic evaluation of risk factors. Br J ClinPharmacol 1992;34:377–95.
1. Roujeau JC, Stern RS. Severe cutaneous drug reactions to drugs.N Engl J Med 1995;33:1600–7.
2. Ring J, Brockow K. Adverse drug reactions: mechanisms andassessment. Eur Surg Res 2002;34:170–5.
3. Sharma VK, Sethuraman G, Kumar B. Cutaneous drug reactions:clinical pattern and causative agents: a 6 years series fromChandigarh, India. J Postgrad Med 2001;47:95–9.
4. Neuman MG, Malkiewicz IM, Shear NH. A novel lymphocytetoxicity assay to assess drug hypersensitivity syndromes. ClinBiochem 2000;33:517–24.
5. Schnyder B, Burkhart C, Shnyder-Frutig K, von Greyerz S,Naisbitt DJ, Pirmohamed M, et al. Recognition of sulfame-thoxazole and its reactive metabolites by drug-specific CD4�T cells from allergic individuals. J Immunol 2000;164:6647–54.
6. Mizuhara H, O’Neill E, Seki N, Ogawa T, Kusunoki C, OtsukaK, et al. T cell activation-associated hepatic injury: mediation bytumor necrosis factors and protection by interleukin 6. J Exp Med1994;179:1529–37.
7. Neuman MG. Apoptosis in disease of the liver. Crit Rev Clin LabSci 2001;38:109–66.
8. Neuman MG, Malkiewicz IM, Phillips EJ, Rachlis AR, Ong D,Yeung E, Shear NH. Monitoring adverse drug reactions to sul-fonamide antibiotics in human immunodeficiency virus-infected
individuals. Ther Drug Mon 2002;24:728–36.9. Neuman MG, Shear NH, Bellentani S, Tiribelli C. Role ofcytokines in ethanol-induced hepatotoxicity in Hep G2 cells.Gastroenterology 1998;114:157–69.
0. Neuman MG. Apoptosis in liver disease. Rom J Gastroenterol2002;11:3–7.
1. Neuman MG, Cohen L, Gomez M, Fish J, Shear N, Nicar M.Cytokine network in ibuprofen-induced hepatocytotoxicity [ab-stract]. Dig Disease Sci 2005;50:10.
2. Jules E. Skin biopsy. In: Jules E, ed. Immunopathology—apractical approach to diagnosis. 2nd ed. Chicago, Ill: AmericanSociety for Clinical Pathology Press, 2003:187–223.
3. Abe R, Shimizu T, Shibaki A, Nakamura H, Watanabe H,Shimizu H. Toxic epidermal necrolysis and Stevens-Johnsonsyndrome are induced by soluble Fas ligand. Am J Pathol 2003;162:1515–20.
4. Dinarello C. Biologic basis for interleukin-1 in disease. Blood1996;87:2095–2147.
5. Tracey K, Cerami A. Tumor necrosis factor, other cytokines anddisease. Annu Rev Cell Biol 1993;9:317–43.
6. Neuman MG, Shear NH, Malkiewicz IM, Abbot F. P450’s 2E1inducers trigger valproic acid-induced apoptosis in vitro. ClinInvest Med 1998;21:582.
7. Neuman MG, Cameron RG, Shear NH, Feuer G. Drug-in-duced apoptosis of skin cells and liver. In: Cameron RG,Fauer G, eds. Handbook of experimental pharmacology: ap-optosis modulation by drugs, vol. 142. Heidelberg: SpringerVerlag; 1999:344 –55.
8. Caproni M, Antiga E, Parodi A, Schena D, Marzano A,Quaglino P, et al. Elevated circulating CD40 ligand in patientswith erythema multiforme and Stevens-Johnson syndrome/toxic epidermal necrolysis spectrum. Br J Dermatol 2006;154:1006 –7.
9. Pestka S, Krause CD, Sarkar D, Walter MR, Shi Y, Fisher PB.Interleukin-10 and related cytokines and receptors. Annu Rev
Immunol 2004;22:929–79.
Att
G
A
Tsa
FIA
SUAbAte
Sc
RSb
1
©
d
2
ntitumor and antiinflammatory effects ofetrathiotungstate in comparison withetrathiomolybdate
UOQING HOU, ROBERT DICK, CHUNHUA ZENG, and GEORGE J. BREWER
NN ARBOR, MICH
Tetrathiomolybdate (TM) is an anticopper drug under development for treatingWilson’s disease. Its mechanism of action involves forming a tight tripartite complexin the blood with serum albumin and available copper. When available copperlevels are lowered in animals with TM, strong antiangiogenic and antitumor effectsare observed. Similarly, TM has excellent efficacy in animal models of fibrotic,inflammatory, and autoimmune diseases, and it protects against heart damagefrom doxorubicin (DXR) and liver damage from acetaminophen, carbon tetrachlo-ride, and concanavalin A. Tetrathiotungstate (TT) also forms a similar tripartitecomplex in the blood and has similar effects to TM on copper. In this article, whetherTT had similar antitumor effects, and similar effects in protecting the heart againstDXR toxicity, as TM was evaluated. It was found that the 2 drugs were comparablein their effects when doses were used that lowered copper availability to the sameextent. (Translational Research 2007;149:260–264)
Abbreviations: Cp � ceruloplasmin; CK-MB � creatine kinase-MB; DXR � doxorubicin; LDH �lactic dehydrogenase; LLHM � Lewis lung high metastatic; TM � tetrathiomolybdate; TT �
tetrathiotungstate.icbiaihino
oostaac
Tm
etrathiomolybdate (TM) is an anticopper drugbeing developed for Wilson’s disease.1–5 In ad-dition to efficacy in Wilson’s disease, it has been
hown that lowering copper levels with TM producesn antiangiogenic, anticancer effect, probably due to
rom the Department of Human Genetics, and the Department ofnternal Medicine, University of Michigan Medical School, Annrbor, Mich.
upported in part by Pipex Therapeutics, Inc., Ann Arbor, Mich. Theniversity of Michigan has licensed the antiangiogenic uses of TM tottenuon LLC, San Diego, Calif, and recently licensed the antifi-rotic and antiinflammatory uses of TM to Pipex Therapeutics, Inc.,nn Arbor, Mich. Dr. Brewer has equity in and is a paid consultant
o both Attenuon LLC and Pipex Therapeutics, Inc. Mr. Dick hasquity in Attenuon LLC.
ubmitted for publication August 18, 2006; revision submitted De-ember 7, 2006; accepted for publication December 9, 2006.
eprint requests: George J. Brewer, University of Michigan Medicalchool, 5024 Kresge Bldg. II, Ann Arbor, MI 48109-0534; e-mail:[email protected].
931-5244/$ – see front matter
2007 Mosby, Inc. All rights reserved.
toi:10.1016/j.trsl.2006.12.003
60
nhibition of many copper-dependent proangiogenicytokines.6–12 An antifibrotic effect of TM has alsoeen shown, possibly from inhibition of the transform-ng growth factor beta pathway.13–15 More recently,ntiinflammatory effects of TM have been shown,15,16
ncluding inhibition of doxorubicin (DXR)-inducedeart damage in a mouse model.17 In these studies theres strong inhibition of inflammatory cytokines tumorecrosis factor-alpha, interleukin-1-beta, and inhibitionf the immune regulatory cytokine, interleukin-2.16,17
In the literature some papers indicate that the effectsf tetrathiotungstate (TT) on copper are similar to thosef TM.18,19 Thus, absorbed TT seems to form the sametable 3-way complex with albumin, copper, and itselfhat TM forms. McQuaid et al18 conclude that TT “has
genuine decoppering effect and could be considereds an alternative to thiomolybdate in the treatment ofopper storage diseases.”In this study, TT was compared and evaluated with
M in some tumor and inflammatory disease animalodels in which TM has shown efficacy. In this article,
he antitumor effect of TT in comparison with TM in a

mm
M
tJ�chAcAfor1
j(hUwawvt
odlMl
otgcfidswmtTd
TSsTLeolbof
wf
TDtmo
oaa2wgsm
o�0
R
tCesfitFgt
Fswcet0
Translational ResearchVolume 149, Number 5 Hou et al 261
ouse xenograft tumor model, and in the DXR mouseodel of cardiac damage, is examined.
ATERIALS AND METHODS
Mice and mouse food. All mice were 7–8 weeks old athe start of the various studies, were C57BL/6 purchased fromackson Laboratory (Bar Harbor, Me), and were housed at 21
2°C on a 12-h light/dark cycle in polycarbonate cagesontaining corn cob bedding. Experimental animals wereoused in the University of Michigan Unit for Laboratorynimal Medicine facility and treated in accord with a proto-
ol approved by the University of Michigan Institutionalnimal Care and Use Committee. They were fed a mouse diet
rom Harlan/Teklad (Madison, Wisc) that had been depletedf copper and zinc. Copper acetate and zinc acetate wereeadded such that the final contents were 2-mg copper and0-mg zinc/kg of food.
Materials. DXR was purchased from Pharmacia and Up-ohn Co (Kalamazoo, Mich). The Lewis lung high metastaticLLHM) cancer cells were originally a gift from Dr. Mo-amed K. Khan, of the Department of Radiation Oncology,niversity of Michigan, whom the authors thank; these cellsere used in Khan et al9 and have subsequently been prop-
gated in the authors’ laboratory. TM and TT for this studyere a gift from Professor Dimitri Coucouvanis, of the Uni-ersity of Michigan Chemistry Department, whom the au-hors thank.
Assay methods. Ceruloplasmin (Cp) was measured by thexidase method, using 3,3=-dimethoxybenzidine (0-dianisi-ine) as substrate, as previously published.17 Troponin I,actic dehydrogenase (LDH), and creatine kinase-MB (CK-
B) were measured by kit methods as previously pub-ished.17
Mouse tumor study. In an earlier study of TT using a dosef TT by oral gavage that was equimolar to TM, it was foundhat TT was much less effective than TM in inhibiting tumorrowth (Merajver S. and Brewer G, unpublished). In theurrent study, preliminary work was performed in mice tond a dose of TT that would be equivalent to TM in terms ofecreasing levels of blood Cp, the surrogate marker of coppertatus on which the authors have published extensively.6–15 Itas found that it takes a dose of a little more than twice asuch TT as TM, on a molar basis, to decrease Cp levels into
he effective range. Therefore, in this study, 0.036 mg/mL ofT and 0.015 mg/mL of TM were used in the drinking wateruring the study.Twenty-five mice were divided into 3 groups, 10 to receive
M, 10 to receive TT, and 5 to serve as no treatment controls.even days before tumor cell injection, the first 2 groups weretarted on TM and TT, respectively, in their drinking water.hen all 25 mice were injected in the flank with 1 � 106
LHM cells by subcutaneous injection. Tumor volume wasstimated at various time points by measuring the dimensionsf the tumor with calipers and using the formula 0.52 �ength � width squared.9 This was done in a blinded fashion,y 2 observers, and the data from the 2 averaged. At the endf the study, the mice were sacrificed, the tumors dissected
ree, and weighed. Body weight, Cp levels, and hematocrit were measured periodically during the study, using bloodrom a tail vein.
Mouse DXR study. In this study, the same drinking waterM and TT dose regimens were used during the week beforeXR injection as were used in the tumor study above. During
he 4 days of study after DXR, because the mice often drinkuch less water, an oral gavage of 0.48 mg of TT and 0.2 mg
f TM daily were used to control Cp levels.Twenty mice were divided into 4 groups, 5 to receive DXR
nly, 5 to receive TM plus DXR, 5 to receive TT plus DXR,nd 5 to receive saline only and serve as controls. Seven daysfter TM and TT were started in the drinking water of groups
and 3, respectively, DXR in a dose of 20-mg/kg bodyeight was given as a single dose intraperitoneally to mice inroups 1, 2, and 3. Four days after DXR, the mice wereacrificed, and serum Cp, troponin I, CK-MB, and LDH wereeasured according to previously published methods.17
Statistical analysis. For the comparison of means, analysisf variance was used. All data in the figures are given as mean
the standard error. Significantly different values at P �.05 or less are marked with asterisks in the figures.
ESULTS
Mouse tumor study. As can be observed from Fig 1,he dose of TT used produced an equivalent lowering ofp levels as TM, and perhaps even a little more low-ring during the latter part of the study. There waslightly more effect on the hematocrit (anemia is therst indication of overtreatment copper deficiency) in
he TT treated groups than in the TM group (Table I).or example, at 14 days, the hematocrits of the TMroup (52%) and control group (51%) averaged abouthe same, whereas the TT group averaged 43%. There
ig 1. The means and standard errors of the blood Cp levels arehown over time in the 3 groups of mice in the tumor study. Thereere 10 mice each in the TM and TT treatment groups and 5 in the
ontrol group. TT treatment with the dose selected reduced Cp levelsquivalent to that of TM treatment. The asterisks indicate that thereatment groups indicated had a significantly lower mean Cp (P �.05) than the control mean at that point in time.
as no significant effect of either TT or TM on body

wb
siptmfdotm0d
tTuitc
TC
caaTi0
oDt
Td
��712
Fsivu2g0
FsTttpDcat0
Fssaw�
Translational Research262 Hou et al May 2007
eight, compared with controls, during the study (Ta-le I).The effect of TM and TT on tumor volume during the
tudy is shown in Fig 2. Both drugs had a very signif-cant effect on inhibiting tumor growth, with TM ap-earing to be somewhat more effective, particularly inhe latter stages of the experiment. Tumor weight data,easured after sacrifice, confirmed the significant ef-
ects of both drugs on inhibiting tumor growth, but itid not confirm that there was a difference in the effectsf the 2 drugs. The control tumors averaged 0.66 g, TMumors 0.22 g, and TT tumors 0.26 g. Both TM and TTeans were significantly different from controls at P �
.003 and 0.04, respectively, but were not significantlyifferent from each other.Mouse DXR study. The Cp data at day 0 and day 4 for
he 4 groups of mice are shown in Fig 3. At day 0, theM and TT groups had equivalently lowered Cp val-es. At day 4, the DXR only mice had a spike in Cp thats always observed, because Cp is an acute-phase reac-ant. The increase in Cp at day 4 indicates that DXR is
able I. Mean body weight and hematocrit valuesuring the tumor study
Time (Days)
Body weight (g) Hematocrit (%)
TM TT Control TM TT Control
7 26 26 26 54 53 542 27 26 27 53 49 51
26 25 27 46 42 474 26 25 28 52 43 512 27 25 28 38 31 41
050
100150200250300350400450
7 10 13 16 19 22
Days After Tumor Injection
Tum
or V
olum
e (m
m3 )
TMTTControl
**
*******
ig 2. The means and standard errors of the tumor volumes arehown over time in the 3 groups of mice. There were 10 mice eachn the TM and TT treatment groups and 5 in the control group. Tumorolumes were determined by 2 blinded observers using calipers andsing the formula 0.52 � length � width squared.9 The results of theobservers were averaged. The asterisks indicate that the treatment
roups indicated had a significantly lower mean tumor volume (P �.05) than the control mean at that point in time.
ausing an inflammatory reaction. The Cp at day 4 in H
M-treated animals remained well controlled, but thep in TT-treated animals spiked to some degree.Troponin I in the serum is a specific measure of
ardiac damage and is shown in Fig 4. The day 4 levelsre markedly elevated in DXR only controls, but theyre kept at almost control levels by both TM and TT.he means of the TM and TT groups were both signif-
cantly different from the DXR-only mean at P �.001.The serum levels of CK-MB, another specific marker
f cardiac damage, are shown in Fig 5. Again, levels inXR-only animals are markedly elevated. Again, TM
herapy significantly inhibits this increase (P � .02).
ig 3. The means and standard errors of the blood Cp levels arehown at days 0 and 4 in the 4 groups of mice in the DXR study.here were 5 mice in each of the 4 groups. At day 0, the TM and TT
reatment groups had equivalently lower mean Cp levels. At day 4,he DXR-only group had a spike in mean Cp, caused by an acute-hase response, probably mediated from inflammation caused byXR. The mean Cp at day 4 in TM-treated animals remained well
ontrolled (kept at low values), but the mean Cp in TT-treatednimals spiked up to some degree. The asterisks indicate that thereated groups, at the indicated time, had a significantly lower (P �.05) mean Cp than the DXR-only group.
ig 4. The means and standard errors of the blood troponin I, apecific measure of heart damage, in the 4 groups of mice in the DXRtudy at day 4. There were 5 mice in each of the 4 groups. Thesterisks indicate that the means of the TM- and TT-treated groupsere significantly different than the mean of the DXR-only group (P0.05).
owever, in TT animals, although the mean is lower
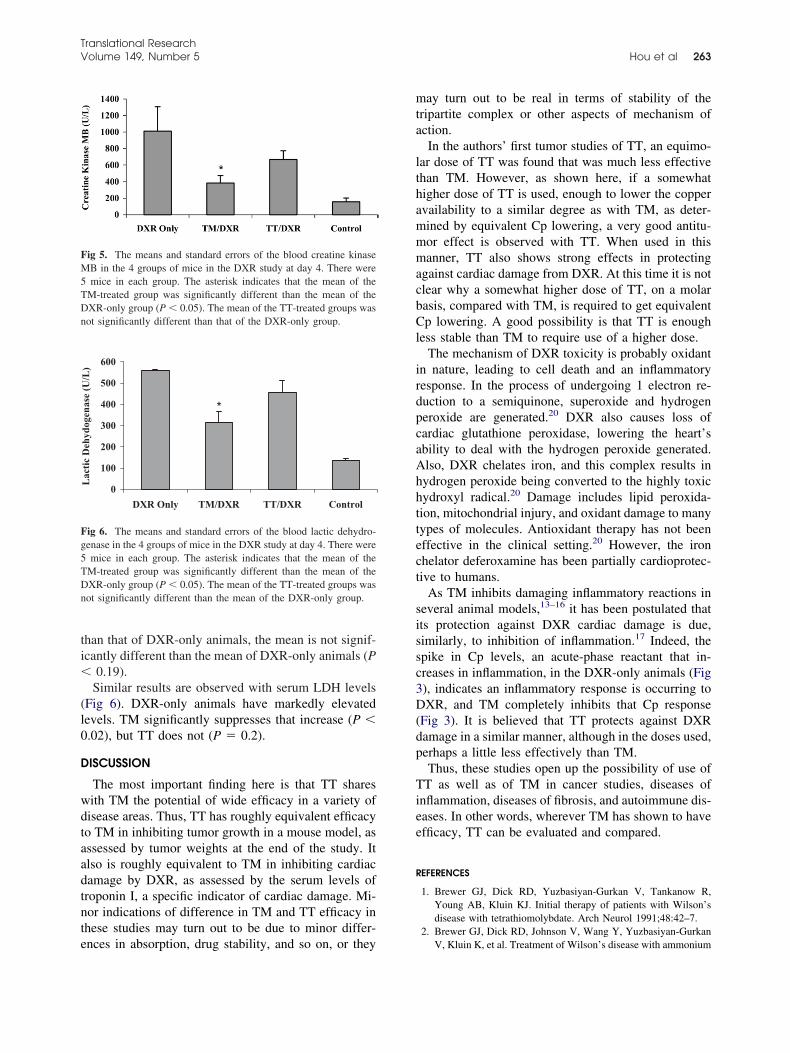
ti�
(l0
D
wdtaadtnte
mta
lthammmacbCl
irdpcaAhhttect
sissc3D(dp
Tiee
R
FM5TDn
Fg5TDn
Translational ResearchVolume 149, Number 5 Hou et al 263
han that of DXR-only animals, the mean is not signif-cantly different than the mean of DXR-only animals (P
0.19).Similar results are observed with serum LDH levels
Fig 6). DXR-only animals have markedly elevatedevels. TM significantly suppresses that increase (P �.02), but TT does not (P � 0.2).
ISCUSSION
The most important finding here is that TT sharesith TM the potential of wide efficacy in a variety ofisease areas. Thus, TT has roughly equivalent efficacyo TM in inhibiting tumor growth in a mouse model, asssessed by tumor weights at the end of the study. Itlso is roughly equivalent to TM in inhibiting cardiacamage by DXR, as assessed by the serum levels ofroponin I, a specific indicator of cardiac damage. Mi-or indications of difference in TM and TT efficacy inhese studies may turn out to be due to minor differ-
ig 5. The means and standard errors of the blood creatine kinaseB in the 4 groups of mice in the DXR study at day 4. There weremice in each group. The asterisk indicates that the mean of the
M-treated group was significantly different than the mean of theXR-only group (P � 0.05). The mean of the TT-treated groups wasot significantly different than that of the DXR-only group.
0
100
200
300
400
500
600
DXR Only TM/DXR TT/DXR Control
Lac
tic
Deh
ydog
enas
e (U
/L)
*
ig 6. The means and standard errors of the blood lactic dehydro-enase in the 4 groups of mice in the DXR study at day 4. There weremice in each group. The asterisk indicates that the mean of the
M-treated group was significantly different than the mean of theXR-only group (P � 0.05). The mean of the TT-treated groups wasot significantly different than the mean of the DXR-only group.
nces in absorption, drug stability, and so on, or they
ay turn out to be real in terms of stability of theripartite complex or other aspects of mechanism ofction.In the authors’ first tumor studies of TT, an equimo-
ar dose of TT was found that was much less effectivehan TM. However, as shown here, if a somewhatigher dose of TT is used, enough to lower the coppervailability to a similar degree as with TM, as deter-ined by equivalent Cp lowering, a very good antitu-or effect is observed with TT. When used in thisanner, TT also shows strong effects in protecting
gainst cardiac damage from DXR. At this time it is notlear why a somewhat higher dose of TT, on a molarasis, compared with TM, is required to get equivalentp lowering. A good possibility is that TT is enough
ess stable than TM to require use of a higher dose.The mechanism of DXR toxicity is probably oxidant
n nature, leading to cell death and an inflammatoryesponse. In the process of undergoing 1 electron re-uction to a semiquinone, superoxide and hydrogeneroxide are generated.20 DXR also causes loss ofardiac glutathione peroxidase, lowering the heart’sbility to deal with the hydrogen peroxide generated.lso, DXR chelates iron, and this complex results inydrogen peroxide being converted to the highly toxicydroxyl radical.20 Damage includes lipid peroxida-ion, mitochondrial injury, and oxidant damage to manyypes of molecules. Antioxidant therapy has not beenffective in the clinical setting.20 However, the ironhelator deferoxamine has been partially cardioprotec-ive to humans.
As TM inhibits damaging inflammatory reactions ineveral animal models,13–16 it has been postulated thatts protection against DXR cardiac damage is due,imilarly, to inhibition of inflammation.17 Indeed, thepike in Cp levels, an acute-phase reactant that in-reases in inflammation, in the DXR-only animals (Fig), indicates an inflammatory response is occurring toXR, and TM completely inhibits that Cp response
Fig 3). It is believed that TT protects against DXRamage in a similar manner, although in the doses used,erhaps a little less effectively than TM.Thus, these studies open up the possibility of use of
T as well as of TM in cancer studies, diseases ofnflammation, diseases of fibrosis, and autoimmune dis-ases. In other words, wherever TM has shown to havefficacy, TT can be evaluated and compared.
EFERENCES
1. Brewer GJ, Dick RD, Yuzbasiyan-Gurkan V, Tankanow R,Young AB, Kluin KJ. Initial therapy of patients with Wilson’sdisease with tetrathiomolybdate. Arch Neurol 1991;48:42–7.
2. Brewer GJ, Dick RD, Johnson V, Wang Y, Yuzbasiyan-Gurkan
V, Kluin K, et al. Treatment of Wilson’s disease with ammonium
1
1
1
1
1
1
1
1
1
1
2
Translational Research264 Hou et al May 2007
tetrathiomolybdate. I. Initial therapy in 17 neurologicallyaffected patients. Arch Neurol 1994;51:545–54.
3. Brewer GJ, Johnson V, Dick RD, Kluin KJ, Fink JK, BrunbergJA. Treatment of Wilson disease with ammonium tetrathiomo-lybdate. II. Initial therapy in 33 neurologically affected patientsand follow-up with zinc therapy. Arch Neurol 1996;53:1017–25.
4. Brewer GJ, Hedera P, Kluin KJ, Carlson M, Askari F, Dick RB,et al. Treatment of Wilson disease with ammonium tetrathiomo-lybdate. III. Initial therapy in a total of 55 neurologically affectedpatients and follow-up with zinc therapy. Arch Neurol 2003;60:379–85.
5. Brewer GJ, Askari F, Lorincz MT, Carlson M, Schilsky M, KluinKJ, et al. Treatment of Wilson disease with ammonium tetrathio-molybdate. IV. Comparison of tetrathiomolybdate and trientinein a double-blind study of treatment of the neurologic presenta-tion of Wilson disease. Arch Neurol 2006;63:521–7.
6. Cox C, Merajver SD, Yoo S, Dick RD, Brewer GJ, Lee JS, et al.Inhibition of the growth of squamous cell carcinoma by tetra-thiomolybdate-induced copper suppression in a murine model.Arch Otolaryngol Head Neck Surg 2003;129:781–5.
7. Cox CD, Teknos TN, Barrios M, Brewer GJ, Dick RD, MerajverSD. The role of copper suppression as an antiangiogenic strategyin head and neck squamous cell carcinoma. Laryngoscope 2001;111:696–701.
8. Kent MS, Madewell BR, Dank G, Dick RB, Merajver SD,Brewer GJ. An anticopper antiangiogenic approach for advancedcancer in spontaneously occurring tumors, using tetrathiomolyb-date: a pilot study in a canine animal mode. J Trace Elem ExpMed 2004;17:9–20.
9. Khan MK, Miller MW, Taylor J, Gill NK, Dick RD, Van GolenKL, et al. Radiotherapy and antiangiogenic TM in lung cancer.Neoplasia 2002;4:164–70.
0. Pan Q, Bao LW, Kleer CG, Brewer GJ, Merajver SD. Antian-giogenic tetrathiomolybdate enhances the efficacy of doxorubi-
cin against breast carcinoma. Mol Cancer Ther 2003;2:617–22.1. Pan Q, Kleer CG, Van Golen KL, Irani J, Bottema KM, Bias C,et al. Copper deficiency induced by tetrathiomolybdate sup-presses tumor growth and angiogenesis. Cancer Res 2002;62:4854–9.
2. Van Golen KL, Bao L, Brewer GJ, Pienta KJ, Kamradt JM,Livant DL, et al. Suppression of tumor recurrence and metastasisby a combination of the PHSCN sequence and the antiangiogeniccompound tetrathiomolybdate in prostate carcinoma. Neoplasia2002;4:373–9.
3. Brewer GJ, Ullenbruch MR, Dick RB, Olivarez L, Phan SH.Tetrathiomolybdate therapy protects against bleomycin-in-duced pulmonary fibrosis in mice. J Lab Clin Med 2003;141:210 – 6.
4. Brewer GJ, Dick R, Ullenbruch MR, Jin H, Phan SH. Inhibitionof key cytokines by tetrathiomolybdate in the bleomycin modelof pulmonary fibrosis. J Inorg Biochem 2004;98:2160–7.
5. Askari FK, Dick RB, Mao M, Brewer GJ. Tetrathiomolybdatetherapy protects against concanavalin A and carbon tetrachloridehepatic damage in mice. Exp Biol Med 2004;229:857–63.
6. Ma S, Hou G, Dick RD, Brewer GJ. Tetrathiomolybdate protectsagainst liver injury from acetaminophen in mice. J Appl Res ClinExp Ther 2004;4:419–26.
7. Hou G, Dick R, Abrams GD, Brewer GJ. Tetrathiomolybdateprotects against cardiac damage by doxorubicin in mice. J LabClin Med 2005;146:299–303.
8. McQuaid A, Lamand M, Mason J. Thiotungstate-copper inter-actions II. The effects of tetrathiotungstate on systemic coppermetabolism in normal and copper-treated rats. J Inorg Biochem1994;53:205–18.
9. Young BW, Bremner I, Mills CF. Effects of tetrathiotungstateand dithiotungstate on copper metabolism in rats. J Inorg Bio-chem 1982;16:121–34.
0. Myers C. The role of iron in doxorubicin-induced cardiomyop-
athy. Semin Oncol 1998;25:10–4.
Eii
TKM
T
FDiCN
SD
xpression of angiopoietin-1 in osteoblasts and itsnhibition by tumor necrosis factor-alpha andnterferon-gamma
SUYOSHI KASAMA, TAKEO ISOZAKI, TSUYOSHI ODAI, MIZUHO MATSUNAWA,UNINOBU WAKABAYASHI, HIROKO T. TAKEUCHI, SATOSHI MATSUKURA, MITSURU ADACHI,ASAKAZU TEZUKA, and KAZUO KOBAYASHI
OKYO, JAPAN
Angiogenesis is a crucial component of bone remodeling under both normal andpathophysiological conditions. Among the various mediators that regulate the angio-genic process is the angiopoietin (Ang) family of growth factors. Ang-1 stabilizes newblood vessels by recruiting surrounding mesenchymal cells and promoting their differ-entiation into vascular smooth muscle cells, whereas Ang-2 is a natural antagonist ofAng-1 and can inhibit angiogenesis. The expression of Ang-1 and Ang-2 in humanosteoblasts (hOBs) isolated from rheumatoid arthritis (RA) and osteoarthritis (OA) pa-tients and from healthy individuals has been examined. After incubation in the pres-ence or absence of tumor necrosis factor-alpha (TNF-�) and/or interferon-gamma(IFN-�), the culture supernatants were assayed for Ang using an enzyme-linked immu-nosorbent assay. In addition, expression of Ang protein and mRNA was examined usingimmunohistochemical techniques and quantitative real-time polymerase chain reac-tion, respectively. It was found that hOBs expressed Ang-1 but not Ang-2 protein, andcultured hOBs from RA and OA patients and from healthy individuals all spontaneouslysecreted significant amounts of Ang-1 in the absence of any stimulation. Althoughstimulation with TNF-� or IFN-� had little or no effect on Ang-1 secretion, costimulationwith IFN-� plus TNF-� dose- and time-dependently diminished secretion of Ang-1 fromhOBs. This inhibitory effect was mediated in part by nuclear factor-kappa B via up-regulated expression of inducible nitric oxide synthase and enhanced synthesis of nitricoxide. Taken together, these findings suggest that OBs are an important cellular sourceof Ang-1 and may modulate bone remodeling through regulation of angiogenesis.(Translational Research 2007;149:265–273)
Abbreviations: Ang � angiopoietin; DMEM � Dulbecco’s modified eagle’s medium; ELISA �enzyme-linked immunosorbent assay; FBS � fetal bovine serum; hOB � human osteoblast;IFN-� � interferon-gamma; IL � interleukin; iNOS � inducible NO synthase; IP-10 � interferon-inducible protein-10; LDH � lactate dehydrogenase; L-NAME � L-nitro arginine methyl ester;NO � nitric oxide; NF-�B � nuclear factor-kappa B; OA � osteoarthritis; OB � osteoblast; PCR� polymerase chain reaction; RA � rheumatoid arthritis; rRNA � ribosomal RNA; SEM �standard error of the mean; siRNA � small interfering RNA; SNP � sodium nitroprusside; TNF-�� tumor necrosis factor-alpha; VEGF � vascular endothelial growth factor.
rom the Division of Rheumatology and Clinical Immunology, Firstepartment of Internal Medicine, Showa University School of Med-
cine, Tokyo, Japan; the Department of Orthopedics, Denencyofuentral Hospital, Tokyo, Japan; and the Department of Immunology,ational Institute of Infectious Diseases, Tokyo, Japan.
ubmitted for publication September 7, 2006; revision submitted
Reprint requests: Tsuyoshi Kasama, MD, PhD, Division of Rheumatol-ogy and Clinical Immunology, First Department of Internal Medicine,Showa University School of Medicine, 1-5-8 Hatanodai, Shinagawa-ku,
1931-5244/$ – see front matter
© 2007 Mosby, Inc. All rights reserved.
Tokyo 142-8666, Japan; e-mail: [email protected].
ecember 5, 2006; accepted for publication December 9, 2006. doi:10.1016/j.trsl.2006.12.007
265

TpooccntrssObmne
gAavctbcvslitci
ArnV(TnikaMkiluO
M
Dm
LaLaAGns(ss
if3bhBtscrihedisaet
wCws
alomt(cp�op
iocuio(a
Translational Research266 Kasama et al May 2007
he maintenance of skeletal homeostasis is a dynamicrocess driven by the coordinated cellular activities ofsteoblasts (OBs), osteocytes, and osteoclasts. More-ver, angiogenesis in bone, which requires communi-ation between endothelial cells and OBs, is a crucialomponent of bone remodeling occurring under bothormal conditions and the pathophysiological condi-ions that result from injury (eg, fracture) or disease [eg,heumatoid arthritis (RA)].1–3 But although the expres-ion of a variety of angiogenic factors is strongly as-ociated with the cells involved in bone formation (eg,Bs), and the importance of angiogenic factors forlood vessel invasion of hyaline cartilage, growth plateorphogenesis, and bone remodeling is well recog-
ized,4 the molecular mechanisms governing angiogen-sis in bone remain unclear.Among the various mediators that regulate angio-
enic activity are the angiopoietins (Angs). Ang-1,ng-2, and their specific receptors, Tie1 and Tie2, are
ll involved in the angiogenic processes triggered byascular endothelial growth factor (VEGF) via its re-eptors.5,6 Once VEGF-mediated formation of imma-ure vessels has occurred, Ang-1 stabilizes the newlood vessels by recruiting surrounding mesenchymalells to the site and promoting their differentiation intoascular smooth muscle cells. On the other hand, Ang-2eems to act primarily as an angiostatic factor, modu-ating Ang-1-mediated vessel stabilization by compet-tively inhibiting the binding of Ang-1 to Tie2, al-hough recent studies have shown that, under certainircumstances, Ang-2 also may induce angiogenesis vianteraction with VEGF-dependent pathways.5,7,8
It was recently reported that expression of Ang-1 andng-2/Tie is upregulated in the RA synovium and in
heumatoid fibroblasts, and that their expression in sy-ovial cells and other cells is modulated by hypoxia,EGF, and proinflammatory cytokines [interleukin
IL)-1 and tumor necrosis factor-alpha (TNF-�)].9–12
his suggests that a variety of mediators make a sig-ificant contribution to the angiogenic response in bothschemic and inflamed tissues. In that regard, it isnown that OBs are an important cellular source ofngiogenic factors such as VEGF13,14 and Ang-1.15,16
oreover, evidence now suggests that nuclear factor-appa B (NF-�B) and nitric oxide (NO) may playmportant roles in the regulation of OB metabo-ism.17,18 Therefore, the aim of this study was to betternderstand the regulation of angiopoietin expression inBs.
ATERIALS AND METHODS
Reagent preparation. Completed medium consisted ofulbecco’s modified eagle’s medium (DMEM; Nissui Phar-
aceutical Co., Tokyo, Japan) supplemented with 2-mM o-glutamine, 100-U/mL penicillin, 100-�g/mL streptomycin,nd 10% heat-inactivated fetal bovine serum (FBS; Gibcoaboratories, Grand Island, NY). Monoclonal and biotinyl-ted polyclonal antibodies (Abs) against human Ang-1 andng-2 and TNF-� and interferon-gamma (IFN-�) were fromenzyme/Techne (Cambridge, Mass). The NO scavenger L-itro arginine methyl ester (L-NAME) and the NO donorodium nitroprusside (SNP) were from Sigma Chemical Co.St Louis, Mo). The NF-�B p65 siRNA (Silencer NF-�B p65iRNA) and nonsilencing siRNA (Silencer Negative ControliRNA) were from Ambion (Austin, Tex).
Preparation of human osteoblasts (hOBs). hOBs weresolated from metaphyseal trabecular bone from the proximalemur of RA (N � 3) and osteoarthritis (OA) patients (N �) during total hip arthroplasty. In addition, OBs from healthyone were obtained during total hip arthroplasty in otherwiseealthy individuals (N � 2) with femoral neck fracture.riefly, after removing pieces of cortical bone, articular car-
ilage, and soft connective tissue, the fragments were cut intomall pieces and incubated for 30 min at 37°C in DMEMontaining 1% collagenase and then extensively washed. Theesultant bone explants were cultured on tissue culture platesn DMEM containing 10% FBS. Once the cell monolayersad reached confluence (ie, after 3–5 weeks in culture), thexplants were removed, and the cells were replated to a cellensity 1 � 105/mL and incubated for 3 days before exper-mentation. The cells obtained showed a flattened polygonalhape with multiple spindle-shaped legs and exhibited char-cteristics of osteoblast-like phenotype, including osteocalcinxpression and positive staining for bone alkaline phospha-ase and mineralization (von Kossa stain) (data not shown).
All human experiments were carried out in accordanceith protocols approved by the Human Subjects Researchommittee at the authors’ institution, and informed consentas obtained from all patients before their enrollment in the
tudy.Measurement of antigenic Ang-1 and Ang-2. Ang-1
nd Ang-2 were quantified using a double ligand enzyme-inked immunosorbent assay (ELISA) that was a modificationf an assay described previously.19 For Ang-1 measurements,onoclonal murine anti-human Ang-1 (0.5 �g/mL) served as
he primary Ab and biotinylated polyclonal goat anti-Ang-10.1 �g/mL) served as the secondary Ab. For Ang-2, mono-lonal murine anti-human Ang-2 (1 �g/mL) served as therimary Ab, and biotinylated polyclonal goat anti-Ang-2 (2g/mL) served as the secondary Ab. The limits of sensitivityf the Ang-1 and Ang-2 ELISAs were �150 pg/mL and �50g/mL, respectively.
Immunohistochemistry. Cell-associated Ang-1 was visual-zed immunohistochemically using a modification of a previ-usly published assay.19 Briefly, human OBs were grown to nearonfluence in an 8-well LabTech chamber slide and then stim-lated with cytokines for 24 h, after which they were thenncubated with polyclonal rabbit anti-Ang-1 Ab (1:500 dilution)r pre-immune rabbit IgG. Biotinylated goat anti-rabbit IgGNichirei, Japan), and peroxidase-conjugated streptavidin serveds second and third reagents, respectively. The color was devel-
ped using a 3,3-diaminobenzidine tetrahydrochloride detection
kd
ru1wCRsmAdrDtm1mrdfip
�ciFfi(mcrwc
mtdua
OdnuJuk
sPaGwtf�
R
ohoi(cgEoamald(
asaAhdsEtAtliafthdns
Tu
nOR
N3asi
Translational ResearchVolume 149, Number 5 Kasama et al 267
it (Nichirei, Japan), after which the slides were rinsed withistilled water and counterstained with Mayer’s hematoxylin.
Isolation of total RNA and real-time polymerase chaineaction (PCR). Total RNA was extracted from human OBssing TRIzol reagent (Invitrogen, San Diego, Calif), after which-�g samples were reverse transcribed into cDNA by incubationith TaqMan RT reagents (Applied Biosystems, Foster City,alif) first for 120 min at 37°C and then for 10 min at 25°C.eal-time PCR was then carried out using an ABI Prism 7900
equence detection system (Applied Biosystems). The reactionixture included 40 ng of cDNA, which was amplified usingmpliTaq Gold DNA polymerase (Applied Biosystems). Foretection of Ang-1, Ang-2, inducible NO synthase (iNOS), andibosomal RNA (rRNA) expression, appropriate Assays-on-emand primers and probes (Applied Biosystems) were used in
he PCR. The amplification protocol entailed incubation for 2in at 50°C and then 10 min at 95°C, followed by 40 cycles of
5 s at 95°C and 1 min at 60°C. For quantification, data wereanipulated to calculate the number of target mRNA copies per
RNA copy and expressed as fold increases over control (me-ium alone). In some experiments, the DNA fragments ampli-ed by PCR were subjected to 2% agarose gel electrophoresis asreviously described.20
siRNA transfection. Cells were seeded to a density of 1.5105 cells per well in 6-well plates and then incubated in
ompleted medium without antibiotics for 24 h before exper-mentation, at which time they were 60% to 70% confluent.ifty nM siRNAs and Lipofectamine 2000 (Invitrogen) at anal concentration of 10 �g/mL in 500 �L of Opti-MEMInvitrogen) were mixed gently at room temperature for 20in to allow complexes to form. Thereafter, 1500 �L of
ompleted medium without antibiotics was added, and theesultant solution was mixed and overlaid on the cells, whichere then incubated with the siRNA/Lipofectamine 2000
omplexes for 24 h at 37°C before isolation of the total RNA.Assessment of NO production. NO release was deter-ined by evaluating the accumulation of its oxidation reac-
ion product, nitrate, in the culture medium. Nitrate wasetected spectrophotometrically based on the Griess reactionsing a commercially available kit (Dojin, Tokyo, Japan)ccording to the manufacturer’s recommendations.
Evaluation of cytotoxicity. To evaluate cytotoxicity toBs, cell viability was evaluated in part based on trypan blueye exclusion. In addition, the amount of lactate dehydroge-ase (LDH) released into the culture medium was analyzedsing an automatic autoanalyzer system (Hitachi, Tokyo,apan), and the incidence of apoptosis was determined in situsing an ApopTag plus peroxidase in situ apoptosis detectionit (Chemicon International, Temecula, Calif).
Statistical analysis. Data are expressed as the means �tandard error of the mean (SEM) and were analyzed on aower Macintosh computer using a statistics software pack-ge (Statview 4.5; Abacus Concept, Inc., Berkeley, Calif).roup data were compared by analysis of variance, afterhich the means of groups whose variances were determined
o significantly differ were compared using the Student t-testor comparison of the means of multiple groups. Values of P
0.05 were considered significant. p
ESULTS
Secretion of OB-derived angiopoietins and the effectsf TNF-� and IFN-� on levels of hOB-derived Ang-1. It wasypothesized that OBs are an important cellular sourcef the angiogenic factors needed during bone remodel-ng. To test this idea, hOBs were first isolated from RARA hOB) and OA patients (OA hOB) and healthyontrols, after which secreted and cell-associated an-iopoietins (Ang-1 and Ang-2) were assayed byLISA. Significant amounts of Ang-1 were spontane-usly secreted into the culture supernatants by RA, OA,nd healthy hOBs (mean: 6206.7 pg/mL, 6349.9 pg/L, and 5990.4 pg/mL, respectively) over the course of24-h incubation period in the absence of any stimu-
ation (Table I). By contrast, little or no Ang-2 wereetected in the culture supernatants of any cells testedTable I).
Next the effects of the inflammatory cytokine TNF-�nd the immunoregulatory cytokine IFN-� on Ang-1ecretion were examined. It was found that, individu-lly, neither TNF-� nor IFN-� significantly affectedng-1 secretion from hOBs (Fig 1, A). On the otherand, when applied together, TNF-� and IFN-� in-uced a significant dose-dependent reduction in Ang-1ecretion from RA, OA, and healthy hOBs (Fig 1, A).xamination of the time course of this effect revealed
hat, under control conditions, spontaneous secretion ofng-1 from unstimulated RA hOBs was detectable in
he culture supernatants within 2 h, after which theevels gradually increased over the course of the 24-hncubation period (Fig 1, B). In the presence of TNF-�nd IFN-�, however, secretion of Ang-1 was attenuatedrom the 8th to the 24th h of incubation. In contrast toheir effect on Ang-1 secretion, TNF-� and/or IFN-�ad no effect on the levels of cell-associated Ang-1uring the 24-h incubation period; moreover, they hado significant stimulatory or inhibitory effect on Ang-2ecretion (data not shown).
Consistent with the spontaneous expression of Ang-1
able I. Secretion of Ang-1 and Ang-2 fromnstimulated hOBs
Cells Ang-1 (pg/mL) Ang-2 (pg/mL)
ormal hOBs 5990.4 � 206.8 87.0 � 18.1A hOBs 6349.9 � 1365.5 154.2 � 33.9A hOBs 6206.7 � 415.5 113.3 � 23.6
otes: Cultured hOBs from 2 normal individuals, 3 OA patients, andRA patients were incubated for 24 h with medium alone. Ang-1nd Ang-2 levels in the culture supernatants were assayed using
pecific ELISAs. Data are expressed as the means � SEM of 3ndependent experiments.
rotein, RT-PCR analysis revealed spontaneous steady-

sioctle
i
tatAita1Au��Tl
ichtstfiNT8wtoNiNwana
ANleepmpcamws
tNw
FTOw(cclTmomc
Translational Research268 Kasama et al May 2007
tate expression of Ang-1 mRNA in cultured RA hOBsncubated in control medium (Fig 2, A and B). More-ver, exposing the cells to TNF-� plus IFN-� signifi-antly diminished levels of Ang-1 mRNA to 32.8% ofhat observed in control medium (Fig 2, B). Costimu-ation with TNF-� and IFN-� exerted similar inhibitoryffects on both OA and healthy hOBs (data not shown).Costimulation with TNF-� and IFN-� had no signif-
ig 1. Spontaneous secretion of Ang-1 from hOBs and the effects ofNF-� and IFN-�. (A) Cultured hOBs from RA patients (RA hOBs),A patients (OA hOBs), and healthy individuals (healthy hOBs)ere incubated for 24 h with the indicated concentrations of TNF-�
0.2-20 ng/mL) and/or IFN-� (1000 U/mL). Ang-1 levels in theulture supernatants were assayed using specific ELISAs. (B) Timeourse of the secretion of hOB-derived Ang-1. Cultured hOBs iso-ated from RA patients were stimulated at time 0 with 20-ng/mLNF-� (�), 1000-U/mL IFN-� (□), TNF-� plus IFN-� (�), oredium alone (Œ). Supernatants were collected after 2, 4, 8, and 24 h
f stimulation and assayed by ELISA. Data are expressed as theeans � SEM of 3 independent experiments; *P � 0.05 versus
ontrol medium.
cant effect on LDH release, cell viability assessed by F
rypan blue dye exclusion, or the incidence of apoptosismong RA hOBs (data not shown). This suggests thatheir inhibitory effects on the expression and secretionng-1 are specific and do not merely reflect cytokine-
nduced cytotoxicity. Consistent with that idea, whenhe effects of TNF-� and/or IFN-� on the secretion ofnother cytokine, interferon-inducible protein-10 (IP-0), were examined, it was found that, in contrast tong-1, IP-10 secretion from OBs was significantlypregulated by either TNF-� or IFN-� (medium � 11.9
2.9 pg/mL, TNF-� � 166.2 � 3.7, IFN-� � 3577.151.4, TNF-� plus IFN-� � 3619.8 � 71.0). Thus,
NF-� and IFN-� seem to exert specific effects thatead to the downregulation of Ang-1 secretion.
Expression of NF-�B and its role in cytokine-mediatednhibition of Ang-1 secretion from hOBs. Given that theytokine-mediated inhibition of Ang-1 secretion fromOBs took about 8 h to develop, it was hypothesized thathe delay reflected the time needed to express and/orynthesize mediators involved in the inhibition. To testhis idea, the expression of NF-�B mRNA in hOBs wasrst assayed. It was found that although expression ofF-�B mRNA was induced by stimulation with eitherNF-� or IFN-�, together the 2 cytokines elicited an.4-fold increase over control in the levels of NF-�Bithin about 4 h (Fig 3, A); moreover, significant induc-
ion of NF-�B mRNA also was detected after 2 h or 8 hf stimulation (Fig 3, B). Then the potential role played byF-�B in the downregulation of Ang-1 expression was
nvestigated by evaluating the effect of knocking downF-�B expression using siRNA. It was found that,hereas addition of control siRNA had no specific effect,
ddition of siRNA targeted to NF-�B completely elimi-ated the downregulation of Ang-1 expression by TNF-�nd IFN-� (Fig 3, C).
Involvement of NO synthesis in the downregulation ofng-1 expression in hOBs. Evidence suggests thatF-�B and NO may play important roles in the regu-
ation of OB metabolism.17,18 With that in mind, theffect of NO synthesis mediated via NF-�B on Ang-1xpression was examined. It was found that, in theresence of TNF-� and IFN-�, expression of iNOSRNA was significantly upregulated in hOBs, as com-
ared with the levels observed in the presence of eitherytokine alone or in control medium (Fig 4, A). Inddition, evaluation of the nitrate levels in the cultureedium revealed that the increased iNOS expressionas accompanied by a corresponding increase in the
ynthesis of NO (Fig 4, B).To confirm the role played by NO/iNOS in the cy-
okine-mediated inhibition of Ang-1, the effects of L-AME (an NO scavenger) and SNP (an NO donor)ere tested on Ang-1 inhibition in hOBs. As shown in
ig 4, C, the inhibitory effects of TNF-� and IFN-� on
tpL2Ap6(
D
Rep
rTadrasTeTaav
tained us
Translational ResearchVolume 149, Number 5 Kasama et al 269
he expression of Ang-1 mRNA and secretion of Ang-1rotein was partially reversed by exposing the cells to-NAME: Expression of Ang-1 mRNA increased from2.7% to 61.5% of control, whereas levels of secretedng-1 protein increased from 3231.5 pg/mL to 4205.0g/mL. Conversely, SNP reduced Ang-1 secretion from206.7 pg/mL (control medium) to 3544.7 pg/mLSNP) (Fig 4, D).
ISCUSSION
It has been shown here that OBs from patients withA or OA as well as healthy hOBs constitutivelyxpress and secrete Ang-1. Furthermore, both the ex-
Fig 2. PCR analysis of Ang-1 mRNA expression ahOBs. (A) Electrophoresis gel showing a represeGAPDH mRNA under the indicated conditions in eaRA, and the PCR was performed (Lane 3). (B) CuTNF-� (20 ng/mL) and IFN-� (1000 U/mL), after wwas carried out. Data are means � SEM of 3 indepLeft panel: representative photomicrographs showin(arrowheads) within RA hOBs. Right panel: cells s
ression and the secretion of Ang-1 were negatively I
egulated by the combination of TNF-� and IFN-�.his downregulation of OB-derived Ang-1 was medi-ted via an NF-�B-dependent pathway leading to in-uction of NO synthesis. Notably, it was previouslyeported that Ang-1 is upregulated in endothelial cellsnd RA synovial fibroblasts by TNF-� via an NF-�Bignaling pathway.10–12 In this study, by contrast,NF-� signaling had little or no effect on constitutivexpression of Ang-1 in OBs. It is well known thatNF-� and NF-�B are both important for the inductionnd expression of a variety of cytokines, chemokines,nd growth factors,21–23 but their effects on OBs canary. For instance, TNF-� upregulates expression of
nohistochemical localization of antigenic Ang-1 inpression pattern of RA hOB-derived Ang-1 andNA was isolated from bone tissue of a patient withBs from RA patients were incubated for 4 h withl RNA was isolated and quantitative real-time PCRperiments; * P � 0.05 versus control medium. (C)
unohistochemical localization of antigenic Ang-1ing control IgG (original magnification, 400�).
nd immuntative exch lane. Rltured hOhich tota
endent exg the imm
L-6, colony stimulating factor-1, and ICAM-1,24–26

bpRs(
(cho
05 versus
Translational Research270 Kasama et al May 2007
ut it downregulates expression of osteocalcin, alkalinehosphatase, insulin-like growth factor-1, andunx2.27–30 In addition, previous studies also have
hown that exposing OBs to inflammatory cytokines
Fig 3. Expression of NF-�B in hOBs and its role inRA patients were incubated for 4 h with TNF-� (20isolated and real-time PCR was carried out to assemRNA was observed in hOBs incubated for 4 h withof OB-derived NF-�B. (C) Effects of NF-�B-speceither NF-�B-specific siRNA or negative control siwhich total RNA was isolated and real-time PCR w� SEM from 3 independent experiments; *P � 0.
TNF-� and IL-1) and IFN-� for prolonged periods T
48–72 h) reduces cell viability and numbers and in-reases the incidence of apoptosis.17,31 In this study,owever, no significant cell damage or apoptosis wasbserved in cells exposed to TNF-� and IFN-� for 24 h.
bition of hOB-derived Ang-1. Cultured hOBs fromnd IFN-� (1000 U/mL), after which total RNA was
expression. (A) Enhanced expression of NF-�Bplus IFN-�. (B) Time course of enhanced inductionA on Ang-1 expression. RA-OBs transfected withre incubated for 4 h with TNF-� plus IFN-�, afterd out to assess Ang-1 expression. Data are meansTNF-�/IFN-� with control siRNA.
the inhing/mL) ass NF-�BTNF-�
ific siRNRNA weas carrie
hus, the effects of TNF-� signaling may differ de-

pu
ptkwtmpi
tcryveamNc
Translational ResearchVolume 149, Number 5 Kasama et al 271
ending on the cell type and the inducible mediatornder study.Synthesis of both NO and iNOS has been observed
reviously in OBs.32,33 In this study, it was found thathe NO inhibitor L-NAME partially reversed the cyto-ine-induced downregulation of Ang-1 expression,hereas the NO donor SNP could inhibit Ang-1 secre-
ion from OBs. This suggests that NO is an importantediator of cytokine-induced inhibition of Ang-1 ex-
ression and secretion in hOBs. Consistent with that
Fig 4. Role of NO in the downregulation of Ang-1patients were incubated for 4 h or 24 h with TNF-RNA was isolated and real-time PCR was carriedculture supernatant were measured as an index ofpatients were incubated 4 h or 24 h with TNF-� pL-NAME (1 mM) or the NO donor SNP (1 mM),carried to assess Ang-1 expression (C), or culture sby ELISA (D). Data are means � SEM from 3 indor control medium (D).
dea, NO seems to regulate the activities by several cell s
ypes during neovascularization, including endothelialells and monocytes,34 although earlier studies of theegulatory function of NO during angiogenesis haveielded contradictory results. For instance, NO acts initro as an autocrine regulator of the microvascularvents necessary for neovascularization and mediatesngiogenesis.35 By contrast, in an in vivo angiogenesisodel (chick embryo chorioallantoic membrane), theO synthase inhibitor NG-monomethyl-L-arginine in-
reased both collagen biosynthesis and vascular den-
on in hOBs. (A) and (B) Cultured hOBs from RAmL) and/or IFN-� (1000 U/mL), after which totalsess iNOS expression (A), or nitrate levels in theuction (B). (C) and (D) Cultured hOBs from RA
in the presence or absence of the NO scavengerh total RNA was isolated and real-time PCR wasts were collected and Ang-1 levels were measuredexperiments; *P � 0.05 versus TNF-�/IFN-� (C)
expressi� (20 ng/out to asNO prodlus IFN-�after whicupernatanependent
ity, which suggests NO inhibits angiogenesis.36 In

aMla
prvbmi
sfptlg
R
1
1
1
1
1
1
1
1
1
1
2
2
2
2
2
2
2
2
Translational Research272 Kasama et al May 2007
ddition, NO may induce VEGF expression in OBs.37
ost likely the function of NO depends on a variety ofocal factors, including the cell type and the stage ofngiogenesis.The Ang-1 receptor Tie2 also is constitutively ex-
ressed in bone tissues,38 which in the context of theseesults suggests that regulation of angiogenesis in boneia an Ang-1/Tie2 pathway could be a key mechanismy which bone remodeling is modulated. The specificolecular events triggered by Ang-1 signaling via Tie2
n OBs remain to be determined, however.In conclusion, OBs seem to be an important cellular
ource of Ang-1. A more complete understanding of theunctions of OB-derived Ang-1 under both normalhysiological and pathological conditions should con-ribute significantly to the understanding of the modu-ation of bone remodeling through regulation of angio-enesis.
EFERENCES
1. Ducy P, Schinke T, Karsenty G. The osteoblast: a sophisticatedfibroblast under central surveillance. Science 2000;289:1501–4.
2. Tanaka Y, Nakayamada S, Okada Y. Osteoblasts and osteoclastsin bone remodeling and inflammation. Curr Drug Targets In-flamm Allergy 2005;4:325–8.
3. Kasama T, Yajima N, Shiozawa F, Hanaoka R. Immunobiologyand regulatory mechanisms of the expression of angiogenic andangiostatic factors in arthritis inflammation. In: Zubar, ed. RVNew angiogenesis research. New York, NY: Nova Science Pub-lishers, 2005:257–77.
4. Zelzer E, Olsen BR. Multiple roles of vascular endothelialgrowth factor (VEGF) in skeletal development, growth, andrepair. Curr Top Dev Biol 2005;65:169–87.
5. Hayes AJ, Huang WQ, Mallah J, Yang D, Lippman ME, Li LY.Angiopoietin-1 and its receptor Tie-2 participate in the regulationof capillary-like tubule formation and survival of endothelialcells. Microvasc Res 1999;58:224–37.
6. Polverini PJ. Angiogenesis in health and disease: insights intobasic mechanisms and therapeutic opportunities. J Dent Educ2002;66:962–75.
7. Ahmad SA, Liu W, Jung YD, Fan F, Wilson M, Reinmuth N, etal. The effects of angiopoietin-1 and -2 on tumor growth andangiogenesis in human colon cancer. Cancer Res 2001;61:1255–9.
8. Hu B, Guo P, Fang Q, Tao HQ, Wang D, Nagane M, et al.Angiopoietin-2 induces human glioma invasion through the ac-tivation of matrix metalloprotease-2. Proc Natl Acad Sci U S A2003;100:8904–9.
9. Shahrara S, Volin MV, Connors MA, Haines GK, Koch AE.Differential expression of the angiogenic Tie receptor family inarthritic and normal synovial tissue. Arthritis Res 2002;4:201–8.Epub 2002 Jan 16.
0. Gravallese EM. Bone destruction in arthritis. Ann Rheum Dis2002;61(suppl 2):ii84–6.
1. DeBusk LM, Chen Y, Nishishita T, Chen J, Thomas JW, Lin PC.Tie2 receptor tyrosine kinase, a major mediator of tumor necrosisfactor alpha-induced angiogenesis in rheumatoid arthritis. Arthri-tis Rheum 2003;48:2461–71.
2. Scott BB, Zaratin PF, Gilmartin AG, Hansbury MJ, Colombo A,
Belpasso C, et al. TNF-alpha modulates angiopoietin-1 expres-sion in rheumatoid synovial fibroblasts via the NF-kappa Bsignalling pathway. Biochem Biophys Res Commun 2005;328:409–14.
3. Goad DL, Rubin J, Wang H, Tashjian AH Jr, Patterson C.Enhanced expression of vascular endothelial growth factor inhuman SaOS-2 osteoblast-like cells and murine osteoblasts in-duced by insulin-like growth factor I. Endocrinology 1996;137:2262–8.
4. Schlaeppi JM, Gutzwiller S, Finkenzeller G, Fournier B. 1,25-Dihydroxyvitamin D3 induces the expression of vascular endo-thelial growth factor in osteoblastic cells. Endocr Res 1997;23:213–29.
5. Horner A, Bord S, Kelsall AW, Coleman N, Compston JE. Tie2ligands angiopoietin-1 and angiopoietin-2 are coexpressed withvascular endothelial cell growth factor in growing human bone.Bone 2001;28:65–71.
6. Arai F, Hirao A, Ohmura M, Sato H, Matsuoka S, Takubo K, etal. Tie2/angiopoietin-1 signaling regulates hematopoietic stemcell quiescence in the bone marrow niche. Cell 2004;118:149–61.
7. Hukkanen M, Hughes FJ, Buttery LD, Gross SS, Evans TJ,Seddon S, et al. Cytokine-stimulated expression of induciblenitric oxide synthase by mouse, rat, and human osteoblast-likecells and its functional role in osteoblast metabolic activity.Endocrinology 1995;136:5445–53.
8. Van Bezooijen RL, Papapoulos SE, Lowik CW. Effect of inter-leukin-17 on nitric oxide production and osteoclastic bone re-sorption: is there dependency on nuclear factor-kappaB andreceptor activator of nuclear factor kappaB (RANK)/RANK li-gand signaling? Bone 2001;28:378–86.
9. Kasama T, Shiozawa F, Kobayashi K, Yajima N, Hanyuda M,Takeuchi TT, et al. Vascular endothelial growth factor expres-sion by activated synovial leukocytes in rheumatoid arthritis.Critical involvement of the interaction with synovial fibroblasts.Arthritis Rheum 2001;44:2512–24.
0. Lu J, Kasama T, Kobayashi K, Yoda Y, Shiozawa F, HanyudaM, et al. Vascular endothelial growth factor expression andregulation of murine collagen-induced arthritis. J Immunol 2000;164:5922–7.
1. Li Q, Verma IM. NF-kappaB regulation in the immune system.Nat Rev Immunol 2002; 2:725–34.
2. Bonizzi G, Karin M. The two NF-kappaB activation pathwaysand their role in innate and adaptive immunity. Trends Immunol2004;25:280–8.
3. Jimi E, Ghosh S. Role of nuclear factor-kappaB in the immunesystem and bone. Immunol Rev 2005;208:80–7.
4. Kurokouchi K, Kambe F, Yasukawa K, Izumi R, Ishiguro N,Iwata H, et al. TNF-alpha increases expression of IL-6 andICAM-1 genes through activation of NF-kappaB in osteoblast-like ROS17/2.8 cells. J Bone Miner Res 1998;13:1290–9.
5. Chae HJ, Chae SW, Chin HY, Bang BG, Cho SB, Han KS, et al.The p38 mitogen-activated protein kinase pathway regulatesinterleukin-6 synthesis in response to tumor necrosis factor inosteoblasts. Bone 2001;28:45–53.
6. Yao GQ, Sun BH, Insogna KL, Weir EC. Nuclear factor-kappaBp50 is required for tumor necrosis factor-alpha-induced colony-stimulating factor-1 gene expression in osteoblasts. Endocrinol-ogy 2000;141:2914–22.
7. Nanes MS, McKoy WM, Marx SJ. Inhibitory effects of tumornecrosis factor-alpha and interferon-gamma on deoxyribonu-cleic acid and collagen synthesis by rat osteosarcoma cells
(ROS 17/2.8). Endocrinology 1989;124:339 – 45.
2
2
3
3
3
3
3
3
3
3
3
Translational ResearchVolume 149, Number 5 Kasama et al 273
8. Kuroki T, Shingu M, Koshihara Y, Nobunaga M. Effects ofcytokines on alkaline phosphatase and osteocalcin production,calcification and calcium release by human osteoblastic cells.Br J Rheumatol 1994;33:224–30.
9. Gilbert L, He X, Farmer P, Boden S, Kozlowski M, Rubin J, etal. Inhibition of osteoblast differentiation by tumor necrosisfactor-alpha. Endocrinology 2000;141:3956–64.
0. Gilbert L, He X, Farmer P, Rubin J, Drissi H, van Wijnen AJ, et al.Expression of the osteoblast differentiation factor RUNX2 (Cbfa1/AML3/Pebp2alpha A) is inhibited by tumor necrosis factor-alpha.J Biol Chem 2002;277:2695–701. Epub 001 Nov 26.
1. Damoulis PD, Hauschka PV. Nitric oxide acts in conjunctionwith proinflammatory cytokines to promote cell death in osteo-blasts. J Bone Miner Res 1997;12:412–22.
2. MacPherson H, Noble BS, Ralston SH. Expression and func-tional role of nitric oxide synthase isoforms in human osteoblast-like cells. Bone 1999;24:179–85.
3. Hughes FJ, Buttery LD, Hukkanen MV, O’Donnell A, MacloufJ, Polak JM. Cytokine-induced prostaglandin E2 synthesis and
cyclooxygenase-2 activity are regulated both by a nitric oxide-dependent and -independent mechanism in rat osteoblasts invitro. J Biol Chem 1999;274:1776–82.
4. Ziche M, Morbidelli L. Nitric oxide and angiogenesis. J Neu-rooncol 2000; 50:139–48.
5. Ziche M, Morbidelli L, Masini E, Amerini S, Granger HJ, MaggiCA, et al. Nitric oxide mediates angiogenesis in vivo and endo-thelial cell growth and migration in vitro promoted by substanceP. J Clin Invest 1994;94:2036–44.
6. Pipili-Synetos E, Sakkoula E, Maragoudakis ME. Nitric oxide isinvolved in the regulation of angiogenesis. Br J Pharmacol 1993;108:855–7.
7. Wang FS, Kuo YR, Wang CJ, Yang KD, Chang PR, HuangYT, et al. Nitric oxide mediates ultrasound-induced hypoxia-inducible factor-1alpha activation and vascular endothelialgrowth factor-A expression in human osteoblasts. Bone 2004;35:114 –23.
8. Lewinson D, Maor G, Rozen N, Rabinovich I, Stahl S, Rach-miel A. Expression of vascular antigens by bone cells duringbone regeneration in a membranous bone distraction system.
Histochem Cell Biol 2001;116:381– 8. Epub 2001 Oct 18.
Amhf
SJ
K
FaKn
S9
S1
2
dvanced glycation end-product-induceditogenesis is dependent on Janus kinase 2-induced
eat shock protein 70 in normal rat kidney interstitialibroblast cells
AN-CHER CHEN, JINN-YUH GUH, HUNG-CHUN CHEN, YU-LIN YANG,AU-SHYANG HUANG, and LEA-YEA CHUANG
AOHSIUNG AND TAINAN, TAIWAN
Kidney interstitial fibroblast proliferation is important in the pathogenesis of diabeticrenal fibrosis. In this regard, advanced glycation end-product (AGE)-induced pro-liferation in normal rat kidney interstitial fibroblast (NRK-49F) cells is dependent onthe Janus kinase 2 (JAK2) signal transducers and activators of transcription (STAT)pathway. Heat shock protein (Hsp) is a molecular target of JAK/STAT. Thus, the roleof Hsp70 in AGE-induced mitogenesis in NRK-49F cells was studied. The AGE dose(100–200 �g/mL) and time (16–72 h) dependently increased Hsp70 protein expres-sion. AGE-induced Hsp70 was attenuated by AG-490 (a JAK2 inhibitor) and N-acetylcysteine. AGE also increased tyrosine phosphorylation of Hsp70, cyclin E, andcyclin D1 (to a lesser extent) while increasing Hsp70 protein interactions with STAT1,STAT3, STAT5b, cyclin D1, and cyclin E. AGE-induced tyrosine phosphorylation ofHsp70 and cyclin E (but not cyclin D1) was attenuated by AG-490. AGE-inducedmitogenesis, cyclin D1, and cyclin E were attenuated by Hsp70 antisense oligode-oxynucleotide and 2-aminopurine (an Hsp70 inhibitor). AGE-induced Hsp70 andmitogenesis were also attenuated by N-acetylcysteine. It was concluded thatAGE-induced Hsp70 protein expression and tyrosine phosphorylation are depen-dent on JAK2 in NRK-49F cells. AGE increased protein–protein interactions amongHsp70, STAT1, STAT3, STAT5b, cyclin D1, and cyclin E. Moreover, AGE-induced mi-togenesis is dependent on Hsp70 and oxidative stress. (Translational Research 2007;149:274–281)
Abbreviations: AGE � advanced glycation end-product; BSA � bovine serum albumin; DMEM� Dulbecco’s modified eagle’s medium; DMSO � dimethyl sulfoxide; DN � diabetic nephrop-athy; ECL � enhanced chemiluminescence; FCS � Fetal calf serum; Hsp � heat shock protein;iNOS � inducible nitric oxide synthase; JAK2 � Janus kinase 2; MTT � 3,[4,5-dimethylthiazolyl-2]-2,5-diphenyltetrazolium bromide; NRK-49F � normal rat kidney interstitial fibroblast; ODN �oligodeoxynucleotide; PBS � phosphate-buffered saline; RAGE � receptor for AGE; ROS �reactive oxygen species; SEM � standard errors of the mean; STAT � signal transducers andactivators of transcription
rom the Graduate Institute of Medicine, Department of Biochemistrynd Department of Internal Medicine, Kaohsiung Medical University,aohsiung, Taiwan; and the Department of Biological Science and Tech-ology, Chung Hwa College of Medical Technology, Tainan, Taiwan.
upported in part by the National Science Council of Taiwan (NSC-1-2320-B-037-044 to LYC; NSC-89-2314-B- 037-008 to JYG).
ubmitted for publication April 9, 2006; revision submitted August
Reprint requests: Lea-Yea Chuang, PhD, Department of Biochemis-try, Kaohsiung Medical University, 100 Shi-Chuan 1st Road, Taiwan807. e-mail: [email protected].
1931-5244/$ – see front matter
© 2007 Mosby, Inc. All rights reserved.
6, 2006; accepted for publication August 19, 2006. doi:10.1016/j.trsl.2006.08.005
74

Taefi(d
ahedo
tidkaoicipND
r(oiA
liwehapHm
Aeaps
M
ewtI
((SCaIaDddaIfsmsiA
twm1C(oAw(o
7g3iocAAewpAp
dl5wag
bswlt
Translational ResearchVolume 149, Number 5 Chen et al 275
he morphology of diabetic nephropathy (DN) is char-cterized by early renal hypertrophy/hyperplasia andxtracellular matrix expansion followed by late renalbrosis.1 Additionally, advanced glycation end-productAGE) is important in the pathogenesis of DN andiabetic renal fibrosis.1
Diabetic renal fibrosis consists of glomerulosclerosisnd tubulointerstitial fibrosis.1 Tubulointerstitial injuryas been postulated to be a final common pathway tond-stage renal disease in DN.2 An integral part ofiabetic renal fibrosis is the activation and proliferationf interstitial fibroblasts.3
AGE binds to many cell-surface receptors. Amonghem, receptor for AGE (RAGE) is the best character-zed AGE receptor.4 The AGE–RAGE interaction in-uces cellular oxidative stress, NF-�B and the Janusinase 2 (JAK2; a tyrosine kinase)-signal transducersnd activators of transcription (STAT) pathway, and son.1,4 It has been shown that AGE induces proliferationn normal rat kidney interstitial fibroblast (NRK-49F)ells by activating the JAK2-STAT pathway1,5 whilenhibiting the inducible nitric oxide synthase (iNOS)athway.6 Moreover, AGE-induced proliferation inRK-49F cells is dependent on redox state and cyclin1.5,7
One common molecular target of JAK/STAT andeactive oxygen species (ROS) is heat shock proteinHsp),8 a molecular chaperone.9 Moreover, inhibitionf NO also induces renal expression of Hsp.10 Interest-ngly, one Hsp (Hsp47) is increased in DN in vivo andGE-treated mesangial cells in vitro.11,12
Hsp70 is a family of cytosolic Hsps, which is trans-ocated to the nucleus upon stress.13 Angiotensin IInduces Hsp70 protein expression in the kidney14
hereas ROS induces Hsp70 protein expression bynhancing STAT binding to the Hsp70 promoter.8 Itas also found that Hsp70 protects mesangial cellsgainst oxidative stress.15 Interestingly, Hsp70 sup-resses iNOS in fibroblasts.16 However, the role ofsp70 in AGE-induced mitogenesis in renal cells re-ains unknown.Therefore, the roles of Hsp70 protein were studied inGE-induced mitogenesis in NRK-49F cells. To better
lucidate the molecular mechanisms underlying thesections, tyrosine phosphorylation of Hsp70 protein androtein–protein interactions of Hsp 70 were also as-essed with cyclin D1, cyclin E, and STAT1, 3, 5b.
ETHODS
Reagents. Fetal calf serum (FCS), Dulbecco’s modifiedagle’s medium (DMEM), antibiotics, lipofectin, moleculareight standards, trypsin-ethylene diamine tetra-acetic acid,
rypan blue, and other medium additives were obtained from
nvitrogen Corp. (Carlsbad, Calif). Anti-phosphotyrosine a4G-10) antibody was obtained from Upstate BiotechnologyLake Placid, NY). Anti-Hsp70 antibody was obtained fromtressgen Biotechnologies Corp. (Victoria, British Columbia,anada). STAT1, STAT3, STAT5b, cyclin D1, and cyclin Entibodies were obtained from Santa Cruz Biotechnology,nc. (Santa Cruz, Calif). Protein A/G-coupled agarose beadsnd AG-490 were obtained from Calbiochem Corp. (Saniego, Calif). Methylglyoxal was obtained from Sigma-Al-rich Co. (St. Louis, Mo). Horseradish peroxidase-conjugatedonkey anti-goat, goat anti-rabbit, or anti-mouse secondaryntibodies were obtained from Santa Cruz Biotechnology,nc. Enhanced chemiluminescence (ECL) kits were obtainedrom Amersham Corp. (Amersham, UK). N,N=-methylenebi-acrylamide, acrylamide, SDS, ammonium persulfate, Te-ed, Tween 20, bovine serum albumin (BSA), dimethyl
ulfoxide (DMSO), �-aminopropionitrile, N-ethyl-maleim-de, and all other chemicals were obtained from Sigma-ldrich Co.Culture conditions. NRK-49F cells were obtained from
he American Type Culture Collection (Manassas, Va). Cellsere grown in culture dishes and maintained in DMEM (5.5M D-glucose) supplemented with 100-IU/mL penicillin,
00-�g/mL streptomycin, and 5% FCS in a humidified 5%O2 incubator at 37°C. Cells were exposed to serum-free
0.1% FCS) DMEM for 24 h before timed exposure to BSAr AGE. In some experiments, cells were pretreated withG-490 for 16 h or other inhibitors for 1 h. 2-Aminopurineas dissolved in phosphate-buffered saline (PBS)/acetic acid
200:1).17 N-Acetylcysteine was dissolved in PBS, whereasther inhibitors were dissolved in DMSO.
Preparation of AGE. AGE was prepared by incubating.2-mg/mL fatty acid-free BSA with 100 mM methyl-lyoxal18 in 100 mM phosphate buffer, pH 7.4, for 50 h at7°C in sterile conditions. Control BSA was obtained afterncubation of BSA in the same conditions with the exceptionf the absence of methylglyoxal. AGE was eluted on a PD-10olumn with DMEM to remove salts and unreacted carbonyls.GE was sterilized by filtration and kept at �20°C until used.GE content was determined spectrofluorometrically with
xcitation set at 355 nm and emission set at 460 nm,19 and itas expressed as the percentage of relative fluorescence com-ared with the control BSA. The average fluorescence forGE was 200 arbitrary units compared with 1 arbitrary uniter milligram for BSA.
Measurement of intracellular ROS. Intracellular ROS wasetected using the fluorescent probe CM-H2DCF-DA (Mo-ecular Probes, Inc., Eugene, Ore).20 Briefly, cells were plated
� 103 cells/well in 96-well plates. The cells were loadedith 20-�mol/L CM-H2DCFDA, incubated for 15 min at 37°,
nd analyzed with FLUOstar galaxy BMG using FLUOstaralaxy software (ReTiSoft, Inc., Toronto, Ontario, Canada).
Immunoblotting and immunoprecipitation. Immuno-lotting and immunoprecipitation were performed as de-cribed in an earlier study.5 Briefly, serum-deprived cellsere treated with BSA or AGE as described above. Total cell
ysates were harvested, resolved by 10% SDS-PAGE, andhen transferred to 0.45 �m Protran membranes (Schieicher
nd Schuell, Keene, NH). The membranes were blocked in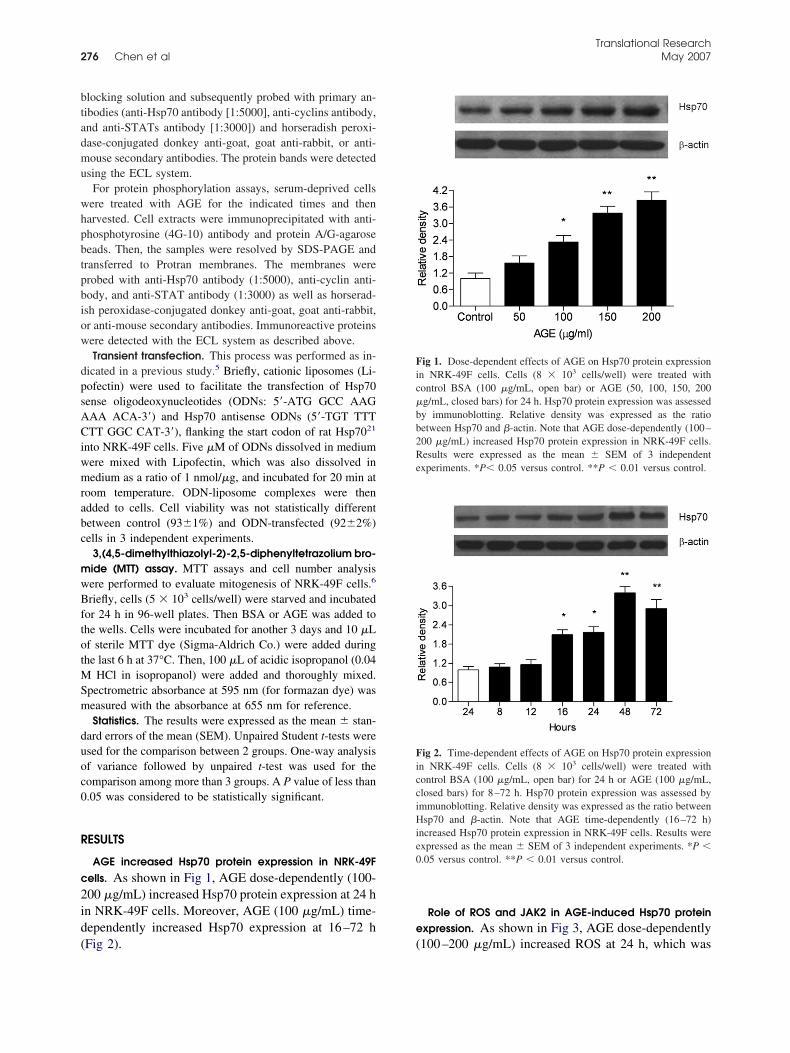
btadmu
whpbtpbiow
dpsACiwmrabc
mwBftotMSm
duoc0
R
c2id(
e
Fic�bb2Re
FicciHie0
Translational Research276 Chen et al May 2007
locking solution and subsequently probed with primary an-ibodies (anti-Hsp70 antibody [1:5000], anti-cyclins antibody,nd anti-STATs antibody [1:3000]) and horseradish peroxi-ase-conjugated donkey anti-goat, goat anti-rabbit, or anti-ouse secondary antibodies. The protein bands were detected
sing the ECL system.For protein phosphorylation assays, serum-deprived cells
ere treated with AGE for the indicated times and thenarvested. Cell extracts were immunoprecipitated with anti-hosphotyrosine (4G-10) antibody and protein A/G-agaroseeads. Then, the samples were resolved by SDS-PAGE andransferred to Protran membranes. The membranes wererobed with anti-Hsp70 antibody (1:5000), anti-cyclin anti-ody, and anti-STAT antibody (1:3000) as well as horserad-sh peroxidase-conjugated donkey anti-goat, goat anti-rabbit,r anti-mouse secondary antibodies. Immunoreactive proteinsere detected with the ECL system as described above.Transient transfection. This process was performed as in-
icated in a previous study.5 Briefly, cationic liposomes (Li-ofectin) were used to facilitate the transfection of Hsp70ense oligodeoxynucleotides (ODNs: 5=-ATG GCC AAGAA ACA-3=) and Hsp70 antisense ODNs (5=-TGT TTTTT GGC CAT-3=), flanking the start codon of rat Hsp7021
nto NRK-49F cells. Five �M of ODNs dissolved in mediumere mixed with Lipofectin, which was also dissolved inedium as a ratio of 1 nmol/�g, and incubated for 20 min at
oom temperature. ODN-liposome complexes were thendded to cells. Cell viability was not statistically differentetween control (93�1%) and ODN-transfected (92�2%)ells in 3 independent experiments.
3,(4,5-dimethylthiazolyl-2)-2,5-diphenyltetrazolium bro-ide (MTT) assay. MTT assays and cell number analysisere performed to evaluate mitogenesis of NRK-49F cells.6
riefly, cells (5 � 103 cells/well) were starved and incubatedor 24 h in 96-well plates. Then BSA or AGE was added tohe wells. Cells were incubated for another 3 days and 10 �Lf sterile MTT dye (Sigma-Aldrich Co.) were added duringhe last 6 h at 37°C. Then, 100 �L of acidic isopropanol (0.04
HCl in isopropanol) were added and thoroughly mixed.pectrometric absorbance at 595 nm (for formazan dye) waseasured with the absorbance at 655 nm for reference.Statistics. The results were expressed as the mean � stan-
ard errors of the mean (SEM). Unpaired Student t-tests weresed for the comparison between 2 groups. One-way analysisf variance followed by unpaired t-test was used for theomparison among more than 3 groups. A P value of less than.05 was considered to be statistically significant.
ESULTS
AGE increased Hsp70 protein expression in NRK-49Fells. As shown in Fig 1, AGE dose-dependently (100-00 �g/mL) increased Hsp70 protein expression at 24 hn NRK-49F cells. Moreover, AGE (100 �g/mL) time-ependently increased Hsp70 expression at 16–72 h
Fig 2). (Role of ROS and JAK2 in AGE-induced Hsp70 proteinxpression. As shown in Fig 3, AGE dose-dependently
ig 1. Dose-dependent effects of AGE on Hsp70 protein expressionn NRK-49F cells. Cells (8 � 103 cells/well) were treated withontrol BSA (100 �g/mL, open bar) or AGE (50, 100, 150, 200g/mL, closed bars) for 24 h. Hsp70 protein expression was assessedy immunoblotting. Relative density was expressed as the ratioetween Hsp70 and �-actin. Note that AGE dose-dependently (100–00 �g/mL) increased Hsp70 protein expression in NRK-49F cells.esults were expressed as the mean � SEM of 3 independentxperiments. *P� 0.05 versus control. **P � 0.01 versus control.
ig 2. Time-dependent effects of AGE on Hsp70 protein expressionn NRK-49F cells. Cells (8 � 103 cells/well) were treated withontrol BSA (100 �g/mL, open bar) for 24 h or AGE (100 �g/mL,losed bars) for 8–72 h. Hsp70 protein expression was assessed bymmunoblotting. Relative density was expressed as the ratio betweensp70 and �-actin. Note that AGE time-dependently (16–72 h)
ncreased Hsp70 protein expression in NRK-49F cells. Results werexpressed as the mean � SEM of 3 independent experiments. *P �.05 versus control. **P � 0.01 versus control.
100–200 �g/mL) increased ROS at 24 h, which was

ao((
cdcpHlcayoc
F2�2cdMAc
Fpp1(abARm
Fpac3aAicttopMcmev
Translational ResearchVolume 149, Number 5 Chen et al 277
ttenuated by N-acetylcysteine (antioxidant). More-ver, N-acetylcysteine and AG-490 attenuated AGE100 �g/mL)-induced Hsp70 protein expression at 24 h
ig 3. Dose-dependent effects of AGE on ROS in NRK-49F cells at4 h. Cells (8 � 103 cells/well) were treated with control BSA (100g/mL, open bar) or AGE (50, 100, 150, 200 �g/mL, closed bars) for4 h. H2O2 (100 �M, open bar) served as a positive control. Intra-ellular ROS was measured by CM-H2DCF-DA. Note that AGEose-dependently (100–200 �g/mL) increased intracellular ROS.oreover, N-acetylcysteine (10 mM pretreated for 1 h) attenuatedGE (100 �g/mL)-induced ROS. *P � 0.05, **P � 0.01 versus
ontrol. #P � 0.05 versus 100 �g/mL AGE.
ig 4. Effects of N-acetylcysteine and AG-490 on AGE-induced Hsp70rotein expression in NRK-49F cells. Cells (8 � 103 cells/well) wereretreated with N-acetylcysteine (10 mM) for 1 h or AG-490 (5 �M) for6 h before treatment with control BSA (100 �g/mL, open bar) or AGE100 �g/mL, closed bars) for 24 h. Hsp70 protein expression wasssessed by immunoblotting. Relative density was expressed as the ratioetween Hsp70 and �-actin. Note that N-acetylcysteine (lane 3) andG-490 (lane 4) attenuated AGE-induced Hsp70 protein expression.esults were expressed as the mean � SEM of 3 independent experi-ents. *P � 0.05 versus control.
Fig 4). y
AGE increased tyrosine phosphorylation of Hsp70, cy-lin D1, and cyclin E. AGE increases cell-cycle-ependent mitogenesis by increasing cyclin D1 andyclin E levels in NRK-49F cells.5 Protein tyrosinehosphorylation is critical in mitogenesis,22 whereassp70, cyclin D1, and cyclin E can be phosphory-
ated.23 Thus, tyrosine phosphorylation of Hsp70 andyclin E were found to be time-dependently increasedt 30–240 min (Fig 5). In contrast, tyrosine phosphor-lation of cyclin D1 increased to a lesser extent andnly at 60 min. Moreover, AGE-induced Hsp70 andyclin E (but not cyclin D1) protein tyrosine phosphor-
ig 5. Time-dependent effects of AGE (100 �g/mL) on tyrosinehosphorylation of Hsp70 (upper panel), cyclin D1 (middle panel),nd cyclin E (lower panel) proteins in NRK-49F cells. Cells (8 � 103
ells/well) were treated with control BSA (100 �g/mL, open bars) for0 min or AGE (100 �g/mL, closed bars) for 15–240 min. Cells werelso pretreated with AG-490 (5 �M) for 16 h before treatment withGE (100 �g/mL) for 30 min (lane 3). Total cell lysates were
mmunoprecipitated with anti-phosphotyrosine antibody. Immuneomplexes were separated by a polyacrylamide gel and immunoblot-ed with Hsp70, cyclin D1, or cyclin E antibodies. Note that AGEime-dependently (30–240 min) increased tyrosine phosphorylationf Hsp70 and cyclin E. In contrast, AGE increased tyrosine phos-horylation of cyclin D1 to a lesser extent and only at 60 min.oreover, AGE-induced tyrosine phosphorylation of Hsp70 and cy-
lin E (but not cyclin D1) was attenuated by AG-490 (lane 3) at 30in. Results were expressed as the mean � SEM of 3 independent
xperiments. *P � 0.05, **P � 0.01 versus control. #P � 0.05ersus lane 3, ##P � 0.01 versus lane 3.
lation was dependent on JAK2 (Fig 5).

ita5Atc
Ne(ta
Eppsisac
Fbc�lcaSi
Fb�oiscpe
Fi�ocoeetocS0
Translational Research278 Chen et al May 2007
Protein–protein interaction of STAT1, STAT3, and STAT5bn AGE-treated NRK-49F cells. Protein–protein interac-ion is important in the function of proteins.24 Thus, wessessed protein–protein interactions among STAT1, 3,b, and cyclin D1/cyclin E by co-immunoprecipitation.s shown in Fig 6, AGE (100 �g/mL) increased pro-
ein–protein interactions among STAT1, 3, 5b, and
ig 6. Effects of AGE (100 �g/mL) on protein–protein interactionetween cyclin D1/cyclin E and STAT1, 3, 5b proteins in NRK-49Fells. Cells (8 � 103 cells/well) were treated with control BSA (100g/mL, open bars) or AGE (100 �g/mL, closed bars) for 24 h. Total cell
ysates were immunoprecipitated with cyclin D1 antibody (left panels) oryclin E antibody (right panels). Immune complexes were separated by
polyacrylamide gel and immunoblotted with STAT1, STAT3, orTAT5b antibodies. Results were expressed as the mean � SEM of 3
ndependent experiments. *P � 0.05, **P � 0.01 versus control.
ig 7. Effects of AGE (100 �g/mL) on protein–protein interactionsetween Hsp70 and cyclins/STATs proteins in NRK-49F cells. Cells (8
103 cells/well) were treated with control BSA (100 �g/mL, open bars)r AGE (100 �g/mL, closed bars) for 24 h. Total cell lysates weremmunoprecipitated with Hsp70 antibody. Immune complexes wereeparated by a polyacrylamide gel and immunoblotted with cyclin D1/yclin E (left panels) or STAT1, STAT3, or STAT5b antibodies (rightanels). Results were expressed as the mean � SEM of 3 independentxperiments. *P � 0.05, **P � 0.01 versus control.
yclin D1/cyclin E at 24 h. s
Protein–protein interaction of Hsp70 in AGE-treatedRK-49F cells. Being a molecular chaperone, Hsp70 isxpected to interact with many proteins. Thus, AGE100 �g/mL) increased protein–protein interactions be-ween Hsp70 and cyclin D1, cyclin E, STAT1, STAT3,nd STAT5b at 24 h (Fig 7).
Role of Hsp70 in AGE-induced cyclin D1 and cyclin. Hsp70 antisense ODN and 2-aminopurine (an Hsp70rotein inhibitor)17 were used to knock down Hsp70rotein. As shown in Fig 8, Hsp70 antisense (but notense) ODN attenuated AGE (100 and 200 �g/mL)-nduced Hsp70, cyclin D1, and cyclin E protein expres-ion at 24 h. As shown in Fig 9, 2-aminopurine alsottenuatesd AGE (100 and 200 �g/mL)-induced Hsp70,yclin D1, and cyclin E protein expression at 24 h.
Role of Hsp70 and ROS in AGE-induced mitogenesis. As
ig 8. Effects of Hsp70 antisense oligodeoxynucleotide on AGE-nduced Hsp70, cyclin D1, and cyclin E protein expression. Cells (8
103 cells/well) were treated with control BSA (100 �g/mL, leftpen bar; 200 �g/mL, right open bar) or AGE (100 �g/mL, leftlosed bars; 200 �g/mL, right closed bars) for 24 h. Oligode-xynucleotides were transiently transfected, whereas Hsp70 proteinxpression was assessed by immunoblotting. Relative density wasxpressed as the ratio between each specific protein and �-actin. Notehat Hsp70 antisense (lane 3), but not sense (lane 4), oligode-xynucleotide attenuated AGE (lane 2)-induced Hsp70, cyclin D1, oryclin E protein expression. Results were expressed as the mean �EM of 3 independent experiments. *P � 0.05 versus BSA, **P �.01 versus BSA. #P � 0.05 versus lane 2 or 4.
hown in Fig 10, Hsp70 antisense (but not sense) ODN,

2i
D
cttctSN
efiplSc
Np
HebeontiHs
daci(tfim
FapcofRp(Re0
Fo4ocoHwH2AfwpB
Translational ResearchVolume 149, Number 5 Chen et al 279
-aminopurine, and N-acetylcysteine attenuated AGE-nduced mitogenesis at 3 days.
ISCUSSION
This demonstration is the first in which AGE in-reases Hsp70 protein expression and Hsp70 proteinyrosine phosphorylation. Moreover, Hsp70 protein in-eracts with STAT1, STAT3, STAT5b, cyclin D1, andyclin E. These findings complement studies showinghat AGE induces mitogenesis by activating the JAK2,TAT1, STAT3, and STAT5-cyclin D1 pathway inRK-49F cells.1,5
The authors found that AGE-induced Hsp70 proteinxpression was dependent on ROS and JAK2. Thisnding corroborates a study showing that hydrogeneroxide increases Hsp70 protein expression in vascu-ar smooth muscle cells, whereas JAK2-activatedTAT binds to the promoter of Hsp70 gene.8 To further
ig 9. Effects of 2-aminopurine on AGE-induced Hsp70, cyclin D1,nd cyclin E protein expression. Cells (8 � 103 cells/well) wereretreated with 2-aminopurine (5 mM) for 1 h before treatment withontrol BSA (100 �g/mL, left open bar; 200 �g/mL, right open bar)r AGE (100 �g/mL, left closed bars; 200 �g/mL, right closed bars)or 24 h. Hsp70 protein expression was assessed by immunoblotting.elative density was expressed as the ratio between each specificrotein and �-actin. Note that 2-aminopurine (lane 3) attenuated AGElane 2)-induced Hsp70, cyclin D1, or cyclin E protein expression.esults were expressed as the mean � SEM of 3 independentxperiments. *P � 0.05 versus BSA, **P � 0.01 versus BSA. #P �.05 versus lane 2, ##P � 0.01 versus lane 2.
haracterize the functions of AGE-induced Hsp70 in w
RK-49F cells, protein tyrosine phosphorylation androtein–protein interactions of Hsp70 were studied.Thus, AGE increased tyrosine phosphorylation ofsp70, cyclin E, and cyclin D1 (although to a lesser
xtent). Tyrosine phosphorylation of cyclin D1 in fi-roblasts25 and tyrosine kinase-induced Hsp70 proteinxpression26 have been found in other studies. On thether hand, tyrosine phosphorylation of cyclin E is aovel finding. The authors also found that AG-490 (ayrosine kinase and JAK2 inhibitor) attenuated AGE-nduced mitogenesis and tyrosine phosphorylation ofsp70 and cyclin E. This finding corroborates a study
howing that JAK2 interacts with Hsp70.27
Hsp70 interacts with Hsp90 and many signal trans-ucers.13,28 Thus, the authors found that Hsp70 inter-cts with STAT1, STAT3, STAT5b, cyclin D1, andyclin E. The Hsp90/Hsp70 complex has been found tonteract with STAT3 to promote nuclear translocationactivation) of STAT3,9 whereas the interaction be-ween Hsp70 and STAT1 and STAT5b is a novelnding. Interestingly, heat shock cognate protein 70 (aember of the Hsp70 protein superfamily) interacts
ig 10. Effects of Hsp70 antisense oligodeoxynucleotide, 2-amin-purine and N-acetylcysteine on AGE-induced mitogenesis in NRK-9F cells. Cells were pretreated with 2-aminopurine (3 mM, lane 4)r N-acetylcysteine (10 mM, lane 5) for 1 h before treatment withontrol BSA (100 �g/mL, left open bar; 200 �g/mL, right open bar)r AGE (100 �g/mL, closed bars; 200 �g/mL, gray bars) for 3 days.sp70 antisense oligodeoxynucleotide (lane 3) was transfected,hereas mitogenesis was determined by the MTT assay. Note thatsp70 antisense (lane 3, but not sense) oligodeoxynucleotide,-aminopurine (lane 4), and N-acetylcysteine (lane 5) attenuatedGE (lane 2)-induced mitogenesis. PBS/acetic acid: solvent control
or 2-aminopurine. PBS: solvent control for N-acetylcysteine. Resultsere expressed as the mean � SEM of 3 independent experimentserformed in triplicate. *P � 0.05 versus BSA, **P � 0.01 versusSA. #P � 0.05 versus lane 2, ##P � 0.01 versus lane 2.
ith cyclin D1 to stabilize cyclin D1 in fibroblasts.29

Mt
aEsrStb
atm2SObtm
otogtsmocH
ipirq4a
rrMitslHD
ppAS
MH
R
1
1
1
1
1
1
1
1
1
Translational Research280 Chen et al May 2007
oreover, Hsp70/Hsp90 stimulates mitogenesis by in-eracting with mitogenic signaling molecules.28
The significance of the protein–protein interactionsmong STAT1, STAT3, STAT5b, and cyclin D1/cyclinfound in this study is not known. However, a recent
tudy found that cyclin D1 interacts with STAT3 toeduce STAT3 activation (nuclear translocation).30 AsTAT3 induces the expression of cyclin D1 protein,31
his study indicates that a negative feedback loop existsetween STAT3 and cyclin D1.Hsp is involved in mitogenesis.28 In this study, the
uthors found that N-acetylcysteine concomitantly at-enuated AGE-induced Hsp70 protein expression anditogenesis. Moreover, both Hsp70 antisense ODN and
-aminopurine attenuated AGE-induced mitogenesis.imilarly, a previous study found that Hsp70 antisenseDN decreased serum-stimulated mitogenesis in fibro-lasts.16 The authors have also shown that Hsp70 pro-ein protected mesangial cells against ROS-inhibiteditogenesis.15
The roles of Hsp in the kidney include both chaper-ne functions and nonchaperone functions32, whichhereby helps uncoupling protein, signaling, membrane,rganelle, and transcriptional networks to regulate cellrowth during stress.9,28 These findings demonstratehat AGE affects the interaction between Hsp70 and theignaling network. A previous study found thatethylglyoxal-modified Hsp27 has enhanced chaper-
ne and anti-apoptotic functions.33 Additionally, gly-osylation enhances the nuclear transport functions ofsp70.34
These findings must be viewed in the context of themportance of interstitial fibroblast in DN.2,3 Thus,revious studies found that diabetic renal fibrosis isnduced by AGE35 and attenuated by AGE inhibitors inats.36 The authors have also found that RAGE is re-uired for AGE-induced collagen production in NRK-9F cells.37 Moreover, reduction of renal fibroblaststtenuates renal fibrosis.38
The role of the JAK2/STAT pathway in glomeruloscle-osis and DN in vivo has been shown previously.39,40 Theole of AGE/RAGE in DN in vivo has also been shown.4
oreover, increased Hsp70 protein level has been foundn peripheral blood mononuclear cells in diabetic pa-ients.41 In contrast, the authors found that Hsp70 anti-ense oligodeoxynucleotide attenuated AGE-induced pro-iferation in NRK-49F cells in vitro. Thus, the role ofsp70 in DN remains to be shown in model systems ofN in vivo.The authors conclude that AGE increases Hsp70
rotein expression and Hsp70 protein tyrosine phos-horylation via the JAK2 pathway in NRK-49F cells.GE also increases Hsp70 protein interactions with
TAT1, STAT3, STAT5b, cyclin D1, and cyclin E.oreover, AGE-induced mitogenesis is dependent onsp70 and oxidative stress.
EFERENCES
1. Chuang LY, Guh JY. Extracellular signals and intracellular path-ways in diabetic nephropathy. Nephrology 2001;6:165–72.
2. Eddy AA. Progression in chronic kidney disease. Adv ChronicKidney Dis 2005;12:353–65.
3. Qi W, Chen X, Poronnik P, Pollock CA. The renal corticalfibroblast in renal tubulointerstitial fibrosis. Int J Biochem CellBiol 2006;38:1–5.
4. Bohlender JM, Franke S, Stein G, Wolf G. Advanced glycationend products and the kidney. Am J Physiol Renal Physiol 2005;289:F645–F659.
5. Guh JY, Huang JS, Chen HC, Hung WC, Lai YH, Chuang LY.Advanced glycation end product-induced proliferation in NRK-49F cells is dependent on the JAK2/STAT5 pathway and cyclinD1. Am J Kidney Dis 2001;38:1096–104.
6. Huang JS, Chuang LY, Guh JY, Chen CJ, Yang YL, Chiang TA,et al. Effect of nitric oxide-cGMP-dependent protein kinaseactivation on advanced glycation end-product-induced prolifera-tion in renal fibroblasts. J Am Soc Nephrol 2005;16:2318–29.
7. Nath KA, Grande J, Croatt A, Haugen J, Kim Y, Rosenberg ME.Redox regulation of renal DNA synthesis, transforming growthfactor-beta1 and collagen gene expression. Kidney Int 1998;53:367–81.
8. Madamanchi NR, Li S, Patterson C, Runge MS. Reactive oxygenspecies regulate heat-shock protein 70 via the JAK/STAT path-way. Arterioscler Thromb Vasc Biol 2001;21:321–6.
9. Soti C, Pal C, Papp B, Csermely P. Molecular chaperones asregulatory elements of cellular networks. Curr Opin Cell Biol2005;17:210–5.
0. Bravo J, Quiroz Y, Pons H, Parra G, Herrera-Acosta J, JohnsonRJ, et al. Vimentin and heat shock protein expression are inducedin the kidney by angiotensin and by nitric oxide inhibition.Kidney Int 2003;86(Suppl):S46–S51.
1. Razzaque MS, Kumatori A, Harada T, Taguchi T. Coexpressionof collagens and collagen-binding heat shock protein 47 in hu-man diabetic nephropathy and IgA nephropathy. Nephron 1998;80:434–43.
2. Ohashi S, Abe H, Takahashi T, Yamamoto Y, Takeuchi M, AraiH, et al. Advanced glycation end products increase collagen-specific chaperone protein in mouse diabetic nephropathy. J BiolChem 2004;279:19816–23.
3. Mayer MP, Bukau B. Hsp70 chaperones: cellular functions andmolecular mechanism. Cell Mol Life Sci 2005;62:670–84.
4. Ishizaka N, Aizawa T, Ohno M, Usui SS, Mori I, Tang SS, et al.Regulation and localization of HSP70 and HSP25 in the kidneyof rats undergoing long-term administration of angiotensin II.Hypertension 2002;39:122–8.
5. Chen HC, Guh JY, Tsai JH, Lai YH. Induction of heat shockprotein 70 protects mesangial cells against oxidative injury.Kidney Int 1999;56:1270–3.
6. Renis M, Cardile V, Grasso S, Palumbo M, Scifo C. Switchingoff HSP70 and i-NOS to study their role in normal and H2O2-stressed human fibroblasts. Life Sci 2003;74:757–69.
7. Doerwald L, Onnekink C, van Genesen ST, de Jong WW,Lubsen NH. Translational thermotolerance provided by smallheat shock proteins is limited to cap-dependent initiation andinhibited by 2-aminopurine. J Biol Chem 2003;278:49743–50.
8. Geoffroy K, Wiernsperger N, Lagarde M, El BS. Bimodal effect
of advanced glycation end products on mesangial cell prolifera-
1
2
2
2
2
22
2
2
2
2
3
3
3
3
3
3
3
3
3
3
4
4
Translational ResearchVolume 149, Number 5 Chen et al 281
tion is mediated by neutral ceramidase regulation andendogenous sphingolipids. J Biol Chem 2004;279:34343–52.
9. Munch G, Keis R, Wessels A, Riederer P, Bahner U, Heidland A,et al. Determination of advanced glycation end products in serumby fluorescence spectroscopy and competitive ELISA. Eur J ClinChem Clin Biochem 1997;35:669–77.
0. Fukami K, Ueda S, Yamagishi S, Kato S, Inagaki Y, TakeuchiM, et al. AGEs activate mesangial TGF-beta-Smad signaling viaan angiotensin II type I receptor interaction. Kidney Int 2004;66:2137–47.
1. Feinstein DL, Galea E, Aquino DA, Li GC, Xu H, Reis DJ. Heatshock protein 70 suppresses astroglial-inducible nitric-oxide syn-thase expression by decreasing NFkappaB activation. J BiolChem 1996;271:17724–32.
2. Chernoff J. Protein tyrosine phosphatases as negative regulatorsof mitogenic signaling. J Cell Physiol 1999;180:173–81.
3. Knowlton AA, Grenier M, Kirchhoff SR, Salfity M. Phosphor-ylation at tyrosine-524 influences nuclear accumulation ofHSP72 with heat stress. Am J Physiol Heart Circ Physiol 2000;278:H2143–H2149.
4. Vidal M. Interactome modeling. FEBS Lett 2005;579:1834–8.5. Scovassi AI, Stivala LA, Rossi L, Bianchi L, Prosperi E. Nuclear
association of cyclin D1 in human fibroblasts: tight binding tonuclear structures and modulation by protein kinase inhibitors.Exp Cell Res 1997;237:127–34.
6. Okubo S, Bernardo NL, Elliott GT, Hess ML, Kukreja RC.Tyrosine kinase signaling in action potential shortening andexpression of HSP72 in late preconditioning. Am J Physiol HeartCirc Physiol 2000;279:H2269–H2276.
7. Sarkar S, Pollack BP, Lin KT, Kotenko SV, Cook JR, Lewis A,et al. hTid-1, a human DnaJ protein, modulates the interferonsignaling pathway. J Biol Chem 2001;276:49034–42.
8. Helmbrecht K, Zeise E, Rensing L. Chaperones in cell cycleregulation and mitogenic signal transduction: a review. CellProlif 2000;33:341–65.
9. Diehl JA, Yang W, Rimerman RA, Xiao H, Emili A. Hsc70regulates accumulation of cyclin D1 and cyclin D1-dependentprotein kinase. Mol Cell Biol 2003;23(5):1764–74.
0. Bienvenu F, Gascan H, Coqueret O. Cyclin D1 represses STAT3activation through a Cdk4-independent mechanism. J Biol Chem
2001;276:16840–7.1. Calo V, Migliavacca M, Bazan V, Macaluso M, Buscemi M,Gebbia N, et al. STAT proteins: from normal control of cellularevents to tumorigenesis. J Cell Physiol 2003;197:157–68.
2. Borkan SC, Gullans SR. Molecular chaperones in the kidney.Annu Rev Physiol 2002;64:503–27.
3. Oya-Ito T, Liu BF, Nagaraj RH. Effect of methylglyoxal modi-fication and phosphorylation on the chaperone and anti-apoptoticproperties of heat shock protein 27. J Cell Biochem 2006;99:279–91.
4. Guinez C, Morelle W, Michalski JC, Lefebvre T. O-GlcNAcglycosylation: a signal for the nuclear transport of cytosolicproteins? Int J Biochem Cell Biol 2005;37:765–74.
5. Zhou G, Li C, Cai L. Advanced glycation end-products induceconnective tissue growth factor-mediated renal fibrosis predom-inantly through transforming growth factor beta-independentpathway. Am J Pathol 2004;165:2033–43.
6. Forbes JM, Thallas V, Thomas MC, Founds HW, Burns WC,Jerums G, et al. The breakdown of preexisting advanced glyca-tion end products is associated with reduced renal fibrosis inexperimental diabetes. FASEB J 2003;17:1762–4.
7. Huang JS, Guh JY, Chen HC, Hung WC, Lai YH, Chuang LY.Role of receptor for advanced glycation end-product (RAGE)and the JAK/STAT-signaling pathway in AGE-induced collagenproduction in NRK-49F cells. J Cell Biochem 2001;81:102–13.
8. Iwano M, Fischer A, Okada H, Plieth D, Xue C, Danoff TM, etal. Conditional abatement of tissue fibrosis using nucleosideanalogs to selectively corrupt DNA replication in transgenicfibroblasts. Mol Ther 2001;3:149–59.
9. Takahashi T, Abe H, Arai H, Matsubara T, Nagai K, MatsuuraM, et al. Activation of STAT3/Smad1 is a key signaling pathwayfor progression to glomerulosclerosis in experimental glomeru-lonephritis. J Biol Chem 2005;280:7100–6.
0. Banes AK, Shaw S, Jenkins J, Redd H, Amiri F, Pollock DM,et al. Angiotensin II blockade prevents hyperglycemia-in-duced activation of JAK and STAT proteins in diabetic ratkidney glomeruli. Am J Physiol Renal Physiol 2004;286:F653–F659.
1. Yabunaka N, Ohtsuka Y, Watanabe I, Noro H, Fujisawa H,Agishi Y. Elevated levels of heat-shock protein 70 (HSP70) inthe mononuclear cells of patients with non-insulin-dependent
diabetes mellitus. Diabetes Res Clin Pract 1995;30:143–7.
Iiv
AP
C
Dtl
FISCL
S(
St
2
mpaired integration of endothelial progenitor cellsn capillaries of diabetic wounds is reversible withascular endothelial growth factor infusion
SHOK K. SINGH, KRISHNAMURTHY P. GUDEHITHLU, SHREYA PATRI, NATALIA O. LITBARG,ERIANNA SETHUPATHI, JOSE A. L. ARRUDA, and GEORGE DUNEA
HICAGO AND MAYWOOD, ILL
To understand impaired angiogenesis in diabetic wounds, polyvinyl tubes were im-planted subcutaneously in rats to form a granulation tissue for 2 weeks and the gran-ulation tissue was studied after inducing diabetes with streptozotocin. By 1 week ofdiabetes, the granulation tissue was bloody and thinner than controls, its medial layerwas depleted of microvessels, and the surviving vessels appeared dehisced. Vascularendothelial growth factor (VEGF) in the diabetic granulation tissue was reduced by 50%compared with control granulation tissue. After 3 days of diabetes, the diabetic tissueshowed a greater degree of apoptosis in the microvessels. Chemotactic factors [stro-mal cell-derived factor-1� and chemokine receptor-4 (CXCR-4)], responsible for at-tracting bone marrow cells, showed equal intensity in control and diabetic tissues. Asexpected, progenitor endothelial CD-34 cells were observed in abundance in both thecontrol and the diabetic granulation tissues. However, although the CD-34-positivecells appeared mostly to be integrated in the blood vessels of the control tissue, fewersuch cells were present in the blood vessels of the diabetic tissues, suggesting thatCD-34 failed to integrate into new blood vessels. Infusion of VEGF in the granulationtissue of diabetic rats for 1 week resulted in complete prevention of the microvasculardefect compared with the contralateral granulation tissue that showed the typicaldiabetic changes. It was concluded that diabetes causes reduction of VEGF in thewound, resulting in loss of blood vessels by apoptosis and possible failure of CD-34 cellsto integrate into the vessel structure. (Translational Research 2007;149:282–291)
Abbreviations: CXCR-4 � chemokine receptor-4; CD-34 (or CD-31) � cluster of differentiation(cell marker)-34 (or -31); FITC � fluorescein isothiocyanate; SDF-1� � stromal cell-derivedfactor-1 alpha; STZ � streptozotocin; TUNEL � terminyl deoxynucleotidyl transferase biotin-dUTP
nick end labeling; VEGF � vascular endothelial growth factorsaf
s
RCC
1
©
iabetes leads to a generalized microvasculardisease that ultimately causes extensive dam-age to the heart, eye, kidney, and nervous sys-
em. The affected vessels undergo either excessive pro-iferation, as in the eye, or regression and tissue
rom the Division of Nephrology, Cook County Hospital, Chicago,ll, the Hektoen Institute for Medical Research, Chicago, Ill, theection of Nephrology, University of Illinois at Chicago and thehicago VAMC, Chicago, Ill; and the Section of Nephrology,oyola-Hines Medical Center, Maywood Ill.
upported in part by the Juvenile Diabetes Foundation InternationalGrant JDA 1-2000-241).
ubmitted for publication September 8, 2006; accepted for publica-
ion November 8, 2006.d
82
carring, as in most other tissues.1 Wound healing islso impaired, largely because of poor new blood vesselormation (angiogenesis).2,3
In normal wound healing, angiogenesis takes place toupply blood to the newly formed healed tissue. This
eprint requests: Ashok K. Singh, PhD, Stroger Hospital of Cookounty, 637 South Wood Street, Durand Building, 2nd floor,hicago Ill 60612. e-mail: [email protected].
931-5244/$ – see front matter
2007 Mosby, Inc. All rights reserved.
oi:10.1016/j.trsl.2006.11.005

pltgfgwtcmt
swbdsvtltbawb
M
braC
dRcidbtta
t1aostcr
t
1
2
3
Tflafgd
gdidatmgcaam
(1sMd
tnwcfeotfll
wc(hi
Translational ResearchVolume 149, Number 5 Singh et al 283
rocess is regulated by systemic factors (blood circu-ation, underlying disease), regional factors (oxygenension, edema, infection), and locally generatedrowth factors.4–7 Progenitor endothelial cells recruitedrom local tissue or bone marrow take part in angio-enesis by forming an extensive tubular structure net-ork that is later remodeled by selective pruning and
rimming of redundant vessels to build the mature vas-ulature.8–10 Studies in animals have shown that bonearrow-derived progenitor cells contribute substan-
ially to the formation of new blood vessels.3,11–15
It is unclear what particular factor or factors is re-ponsible for causing the microvascular damage andhich specific step in the angiogenic process is affectedy diabetes. Also, little is known about the effect ofiabetes on the recruitment of progenitor cells to theite of injury and on their participation in new bloodessel formation. To investigate these questions, diabe-es was induced in a normally healing wound, and theevels of vascular endothelial growth factor (VEGF) inhe wound environment and the participation of CD-34one marrow cells in angiogenesis were studied at 3nd 7 days after inducing diabetes. Further experimentsere conducted to reverse the microvascular changesy infusing VEGF in the diabetic wound.
ETHODS
Wound healing model. Granulation tissue was inducedy the subcutaneous implantation of polyvinyl tubes in theat. The experiments on rats were conducted in accordancend with approval of the Institutional Animal Care and Useommittee of the Stroger Hospital of Cook County.A piece of polyvinyl chloride tubing (L � 25 mm, internal
iameter � 7 mm) (PVC 180 Nalge Nunc International,ochester, NY) was sealed at both open ends by heat appli-ation to create an enclosed chamber (inside volume approx-mately 0.7 mL). Overall, 8 holes (diameter 0.5 mm) wererilled around the chamber to allow for steady diffusionetween the tube contents and the surrounding tissue. Theubes were stored in 70% alcohol for sterility. Before implan-ation, the tubes were washed vigorously with sterile salinend were air-dried.
Sprague–Dawley rats (males, 225–250 g) were anesthe-ized by intraperitoneal injection of sodium nembutal (5 mg/00 g wt), and their backs were shaved and cleaned withlcohol and povidone. Next, two 1-cm incisions were maden either side of the lumbar region. Using blunt dissection, 2ubcutaneous pockets were made around the incision into whichhe polyvinyl tubes were inserted (2 per rat). The incisions werelosed with silk sutures, and the animals were allowed toecover and form granulation tissue around the tube.
Experimental plan. Animals implanted with polyvinylubes were sorted as follows:
. An experimental group in which the granulation tissues
were developed for 2 weeks followed by inducing diabe- ttes by injecting streptozotocin (STZ; 55 mg/kg bodyweight in 10-mM citrate buffer pH 5.5, intravenously).The rats were sacrificed at the end of 3 weeks.
. A nondiabetic control group consisting of animals inwhich the granulation tissues were developed for 2 weeksfollowed by the injection of the citrate buffer intrave-nously and sacrificed after 1 additional week.
. An insulin-treated group similar to the experimentalgroup 1, except that 12 h after the STZ injection andconfirmation of diabetes (by blood glucose being above300 mg/dL), the animals were implanted with insulinpumps (Alzet; Durect Corporation, Cupertino, Calif). Thedelivery method was 1.0 IU/day to maintain normogly-cemia for 1 week, after which the rats were sacrificed.
o study early events and the mechanism of blood vesselormation, experiments were also conducted in 9-day granu-omas in which the experimental group was injected with STZfter 6 days of tube implantation and the diabetes maintainedor 3 further days before termination (group 4). The controlroup consisted of 9-day granulomas without the induction ofiabetes (group 5).After the above study, another experiment was set up in 2
roups of rats to investigate whether VEGF treatment ofiabetic wounds could reverse the adverse changes observedn diabetic rats. This experiment included (1) a group ofiabetic rats like the experimental group 1 above in which,fter injection of STZ, the right-side granulation tissue wasreated with a continuous infusion of VEGF delivered via aini-osmotic pump for 1 week. The left-side contralateral
ranulation tissue served as an untreated diabetic control foromparison. (2) A group of non-diabetic control rats in whicht similar times one granulation tissue was treated with VEGFs above and the contralateral granulation tissue left as nor-al control.Murine VEGF was purchased from R and D Systems, Inc.
Minneapolis, Minn) and dissolved at a concentration of0 �g/mL in sterile normal saline containing 10% normal raterum and used for filling the mini-osmotic pump (Alzetodel 2001; 7 days life; Durect Corporation). The pump
elivered VEGF at the rate of 10 ng/h.VEGF measurement in granuloma fluid. The granulation
issue formed after subcutaneous implantation of the polyvi-yl tubes completely encapsulates the polyvinyl tube by 1eek. The tube is full at all times with a plasma-like fluid
alled the granuloma fluid that can be aspirated convenientlyrom the outside for analysis. The granuloma fluid is inquilibrium with the wound environment, and concentrationf VEGF (and other cytokines) in the granuloma fluid reflectshe wound VEGF. VEGF was measured in the granulomauid using a commercial murine VEGF sandwich enzyme-
inked immunosorbent assay kit from R and D Systems.Processing of granulation tissues. The granulation tissues
ere surgically resected, and their wet weights were re-orded. The extracted granulomas were fixed in Histochoicea non-formalin fixative from Amresco Inc., Solon, Oh) foristological and immunohistochemical studies. Tissues fixedn Histochoice were embedded in paraffin, and 4�-thick sec-
ions were cut by a microtome. Sections were de-paraffinized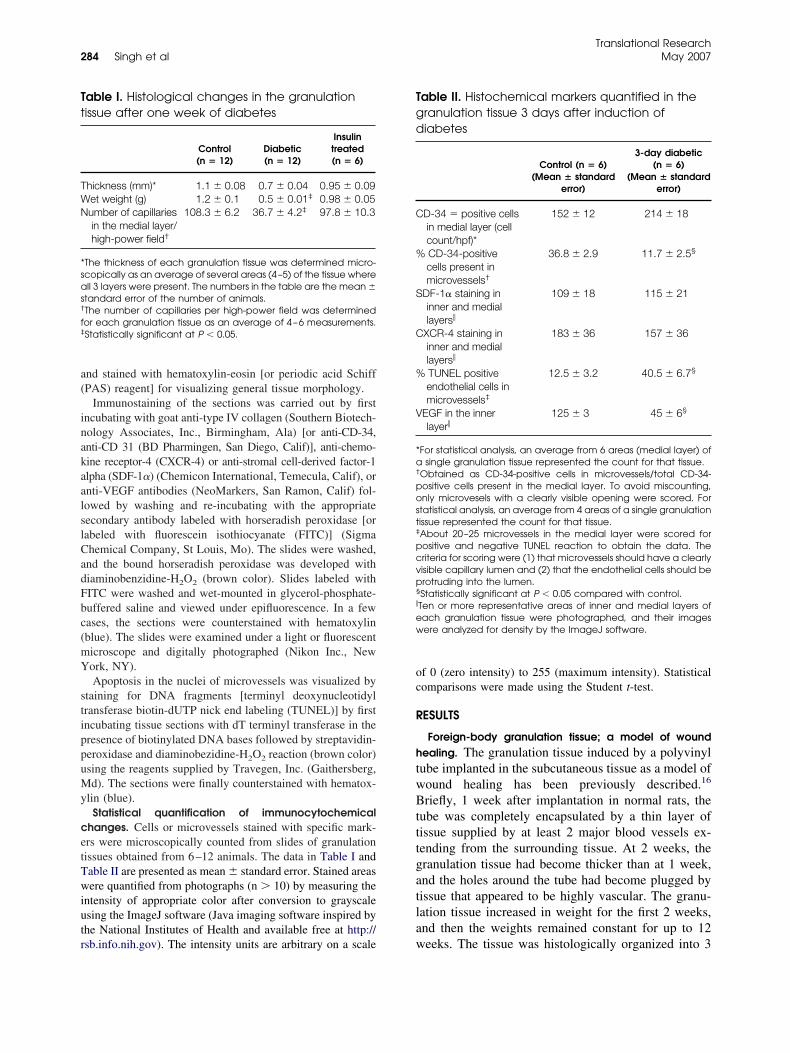
a(
inakaalslCadFbc(mY
stippuMy
cetTwiutr
oc
R
htwBtttgatla
Tt
TWN
*sas†
f‡
Tgd
C
%
S
C
%
V
*a†
post‡
pcvp§
�
ew
Translational Research284 Singh et al May 2007
nd stained with hematoxylin-eosin [or periodic acid SchiffPAS) reagent] for visualizing general tissue morphology.
Immunostaining of the sections was carried out by firstncubating with goat anti-type IV collagen (Southern Biotech-ology Associates, Inc., Birmingham, Ala) [or anti-CD-34,nti-CD 31 (BD Pharmingen, San Diego, Calif)], anti-chemo-ine receptor-4 (CXCR-4) or anti-stromal cell-derived factor-1lpha (SDF-1�) (Chemicon International, Temecula, Calif), ornti-VEGF antibodies (NeoMarkers, San Ramon, Calif) fol-owed by washing and re-incubating with the appropriateecondary antibody labeled with horseradish peroxidase [orabeled with fluorescein isothiocyanate (FITC)] (Sigmahemical Company, St Louis, Mo). The slides were washed,nd the bound horseradish peroxidase was developed withiaminobenzidine-H2O2 (brown color). Slides labeled withITC were washed and wet-mounted in glycerol-phosphate-uffered saline and viewed under epifluorescence. In a fewases, the sections were counterstained with hematoxylinblue). The slides were examined under a light or fluorescenticroscope and digitally photographed (Nikon Inc., Nework, NY).Apoptosis in the nuclei of microvessels was visualized by
taining for DNA fragments [terminyl deoxynucleotidylransferase biotin-dUTP nick end labeling (TUNEL)] by firstncubating tissue sections with dT terminyl transferase in theresence of biotinylated DNA bases followed by streptavidin-eroxidase and diaminobezidine-H2O2 reaction (brown color)sing the reagents supplied by Travegen, Inc. (Gaithersberg,d). The sections were finally counterstained with hematox-
lin (blue).Statistical quantification of immunocytochemical
hanges. Cells or microvessels stained with specific mark-rs were microscopically counted from slides of granulationissues obtained from 6–12 animals. The data in Table I andable II are presented as mean � standard error. Stained areasere quantified from photographs (n � 10) by measuring the
ntensity of appropriate color after conversion to grayscalesing the ImageJ software (Java imaging software inspired byhe National Institutes of Health and available free at http://
able I. Histological changes in the granulationissue after one week of diabetes
Control(n � 12)
Diabetic(n � 12)
Insulintreated(n � 6)
hickness (mm)* 1.1 � 0.08 0.7 � 0.04 0.95 � 0.09et weight (g) 1.2 � 0.1 0.5 � 0.01‡ 0.98 � 0.05umber of capillariesin the medial layer/high-power field†
108.3 � 6.2 36.7 � 4.2‡ 97.8 � 10.3
The thickness of each granulation tissue was determined micro-copically as an average of several areas (4–5) of the tissue wherell 3 layers were present. The numbers in the table are the mean �
tandard error of the number of animals.The number of capillaries per high-power field was determinedor each granulation tissue as an average of 4–6 measurements.Statistically significant at P � 0.05.
sb.info.nih.gov). The intensity units are arbitrary on a scale w
f 0 (zero intensity) to 255 (maximum intensity). Statisticalomparisons were made using the Student t-test.
ESULTS
Foreign-body granulation tissue; a model of woundealing. The granulation tissue induced by a polyvinylube implanted in the subcutaneous tissue as a model ofound healing has been previously described.16
riefly, 1 week after implantation in normal rats, theube was completely encapsulated by a thin layer ofissue supplied by at least 2 major blood vessels ex-ending from the surrounding tissue. At 2 weeks, theranulation tissue had become thicker than at 1 week,nd the holes around the tube had become plugged byissue that appeared to be highly vascular. The granu-ation tissue increased in weight for the first 2 weeks,nd then the weights remained constant for up to 12
able II. Histochemical markers quantified in theranulation tissue 3 days after induction ofiabetes
Control (n � 6)(Mean � standard
error)
3-day diabetic(n � 6)
(Mean � standarderror)
D-34 � positive cellsin medial layer (cellcount/hpf)*
152 � 12 214 � 18
CD-34-positivecells present inmicrovessels†
36.8 � 2.9 11.7 � 2.5§
DF-1� staining ininner and mediallayers�
109 � 18 115 � 21
XCR-4 staining ininner and mediallayers�
183 � 36 157 � 36
TUNEL positiveendothelial cells inmicrovessels‡
12.5 � 3.2 40.5 � 6.7§
EGF in the innerlayer�
125 � 3 45 � 6§
For statistical analysis, an average from 6 areas (medial layer) ofsingle granulation tissue represented the count for that tissue.
Obtained as CD-34-positive cells in microvessels/total CD-34-ositive cells present in the medial layer. To avoid miscounting,nly microvesels with a clearly visible opening were scored. Fortatistical analysis, an average from 4 areas of a single granulationissue represented the count for that tissue.About 20–25 microvessels in the medial layer were scored forositive and negative TUNEL reaction to obtain the data. Theriteria for scoring were (1) that microvessels should have a clearlyisible capillary lumen and (2) that the endothelial cells should berotruding into the lumen.Statistically significant at P � 0.05 compared with control.Ten or more representative areas of inner and medial layers ofach granulation tissue were photographed, and their imagesere analyzed for density by the ImageJ software.
eeks. The tissue was histologically organized into 3

ltclmpaabwcttlAulwa
rntb
nS
tbc2ias3Igotw(
1psdtcscswtg
riFhrw(
bgtsofltidp
ubt
Fdwgtwncs
Translational ResearchVolume 149, Number 5 Singh et al 285
ayers. The inner layer in contact with the polyvinylube was about 200 �m thick, consisted of mononu-lear epitheloid cells and fibroblasts, had little extracel-ular matrix, and was devoid of blood vessels. Theedial layer was thicker and consisted of tightly
acked fibroblastic cells and extracellular matrix fibersrranged in parallel to length of the tube. This layer waslso richly endowed with fine blood vessels (best visi-le by microvessel staining; see Fig 2). The outer layeras more amorphous, with loose adventitious tissue
ontaining large blood vessels. The 1-week granulationissues were as histologically organized as the olderissues up to 6 weeks, except that each of the 3 histo-ogical layers was thinner than in the older tissues.fter 6 weeks, the medial vascular layer became grad-ally depleted of microvessels and, by 12 weeks, thisayer, as well as the inner layer, was completely filledith fibrous tissue. The tube was filled at all times withplasma-like fluid called the granuloma fluid.In the STZ diabetic rats, the implantation of the tube
esulted in a poorly developed granulation tissue andon-healing of the skin sutures culminating in the ex-eriorization of the tube within a few days. It, therefore,
ig 1. Morphology of the granulation tissue of control, 1-weekiabetic, and insulin-treated rats. Vacant area on the right lower sideas the location of the tube. Compared with control (A), the diabeticranulation tissue (B) was thinner and the medial layer appeared to beearing apart from the inner layer. Pools of red blood cells (Fig 3)ere observed in the inner and medial layers. Treatment with insulinormalized blood glucose levels and prevented the morphologicalhanges observed in the diabetic granulation tissue (C). Hand Etaining.
ecame necessary to develop a granulation tissue in d
ormal rats for 2 weeks and then induce diabetes withTZ.STZ-induced diabetes in rats. After a single iv injec-
ion of STZ, rats become diabetic within 24 h withlood glucose levels of 420 � 30 mg/dL (n � 20)ompared with normal levels of 102 � 10 mg/dL (n �0). If untreated, the rats remain hyperglycemic indef-nitely with progressive loss of weight (in younger rats,s used in this study, weight loss is measured as ab-ence of weight gain) becoming significant only after–4 weeks of diabetes. If treated with insulin (1.0U/day delivered via mini-osmotic pumps), the bloodlucose normalized to 110 � 14 mg/dL through the lifef the pump. In experiments wherein diabetic rats werereated with VEGF infusion in one of the bilateralound tissues, the blood glucose levels remained high
� 350 mg/dL) as expected in diabetic rats.Morphological changes in the granulation tissue afterweek of diabetes. Figure 1, A shows the typical mor-hology of the control granulation tissue after H and Etaining. After 2 weeks of normal growth and 1 week ofiabetes, grossly the granulation tissue was bloody andhinner than the control tissue (Table I). Microscopi-ally, in most areas, the medial layer appeared to beeparating from the inner layer, and pools of red bloodells were observed in the inner layer where it had noteparated from the medial layer (Fig 1, B). Treatmentith insulin normalized blood glucose levels prevented
he morphological changes observed in the diabeticranulation tissue (Fig 1, C).The medial layer of control granulation tissue was
ichly endowed with microvessels, which were signif-cantly lost after induction of diabetes (Fig 2, Table I).urthermore, many surviving vessels appeared de-isced and to be bleeding as judged by the presence ofed blood cells in the parenchyma (Fig 3). Treatmentith insulin prevented the microvascular changes
Fig 2, C).VEGF levels in the granuloma fluid in normal and dia-
etic rats. Many have reported that VEGF levels in theranuloma fluid of normal rats are typically 25–50imes higher than in serum (or plasma).16 Figure 4hows that VEGF level (ng/mL) in the granuloma fluidf diabetic rats was 50% lower than in the granulomauid of normal rats (Fig 4). As the protein concentra-
ions in the granuloma fluids of normal, diabetic, andnsulin-treated rats were similar (data not shown), theifferences in the VEGF levels expressed as ng/mgrotein remained as shown in Fig 4.Mechanism of microvessel loss in the diabetic wound. To
nderstand the mechanism of microvessel loss in dia-etic wounds, studies were conducted in granulationissue soon after inducing diabetes (3 days). Within 3
ays of diabetes, the medial layer of the granulation
ttnmshlls
ngr
gDCs(S
be like th
Fgwu
Fdonnef
Translational Research286 Singh et al May 2007
issue showed fewer microvessels than did the controlissue, although the difference was not statistically sig-ificant (data not shown), which suggested that theicrovessel loss and rarification, as was clearly ob-
erved after 1 week of diabetes (see above and Fig 2),ad begun as early as 3 days after diabetes. VEGFevels in the granuloma fluid were also not significantlyower in the diabetic tissue by this time (data not
Fig 2. Microvessels in the granulation tissue revecontrol tissue was richly endowed with microvesselinduction of diabetes (B). Many surviving vesselpresence of red blood cells in the parenchymamicrovascular changes, and the tissue appeared to
ig 3. Bleeding vessels in the medial layer of 1-week diabeticranulation tissue revealed by peroxidase reaction (arrows). Sectionas counterstained with hematoxylin. The vacant area in the leftpper corner was the location of the tube.
hown). However, by immunostaining, VEGF, which is t
ormally concentrated in the inner layer of the controlranulation tissue, as shown before,16 was significantlyeduced 3 days after diabetes (Table II).
To investigate whether the microvessels were under-oing apoptosis, tissues were stained for fragmentedNA using the TUNEL immunostaining technique.ompared with control microvessels, the diabetic tissue
howed a significantly greater degree of apoptosisFig 5, Table II). Tissues were also immunostained forDF-1� and CXCR-4 (receptor for SDF) as evidence of
ollagen type IV immunofluorescent staining. Theedial layer (A), which were significantly lost afterd dehisced and to be bleeding as judged by then Fig 3). Treatment with insulin prevented thee control tissue (C).
ig 4. VEGF levels in the granuloma fluid of normal control,iabetic, and insulin-treated rats. VEGF level in the granuloma fluidf 1-week diabetic rats was 50% lower than in the granuloma fluid oformal control rats. Insulin treatment of diabetic rats completelyormalized the VEGF to control levels. Limit bars indicate standardrror of mean (n � 10). *Denotes a statistically significant differencerom control and insulin-treated groups at P � 0.05.
aled by cs in the ms appeare(shown i
he chemotactic activity that attracts bone marrow cells

tttoafwteiftophtwqbc
tgoagwsadisptntam
gtttcstalntVsigocfVit(c
D
cgowiioibmcewftiiigsog
vait
Fi3co
Translational ResearchVolume 149, Number 5 Singh et al 287
o an injured site, and CD-34 (and CD-31) cell markero track the infiltration of progenitor endothelial cells inhe tissue. A strong chemotactic response for migrationf bone marrow cells was suggested in both the controlnd the diabetic granulation tissues by an equal stainingor SDF-1� and CXCR-4 (Table II). Also, CD-34 cellsere observed in abundance both in the control and in
he diabetic granulation tissues (Fig 6, Table II). How-ver, although the CD-34-positive cells were mostlyntegrated in the blood vessels in the control tissue,ewer such cells were present in the blood vessels ofhe diabetic tissues, suggesting a defective integrationf these cells in diabetic vessels. Instead, they wereresent freely in the inner layer in numbers significantlyigher than present in control tissues (Table II). Essen-ially similar results were obtained after tissue sectionsere stained for CD-31 antigen (not shown). Fre-uently, “pseudo-capillaries” were visible in the dia-etic tissue that appeared to be devoid of endothelialells.
Prevention of microvessel damage in the granulationissue by infusion of VEGF. In diabetic rats with bilateralranulation tissue, a continuous infusion of VEGF inne granulation tissue for 1 week resulted in healthy-ppearing granulation tissue compared with the thinranulation tissue of the contralateral side (Fig 7). Theet weight of the VEGF-infused granulation tissue was
ignificantly higher than the diabetic granulation tissuend was as much as that of the normal control (non-iabetic) rats (Table III). The gross color of the VEGF-nfused granulation tissue was pink in the normal rats,uggesting good vascularity compared with the pale,oorly vascularized contralateral diabetic granulationissue. Histologically, in contrast to the contralateralon-treated diabetic granulation tissue, the VEGF-reated granulation tissue was as thick and well-formeds the normal granulation tissue (Table III). Further-
ig 5. Apoptosis in the endothelial cells of microvessels as seen bymmune-staining for fragmented DNA (TUNEL) in control (A) and-day diabetic granulation tissue (B). Compared with control, mi-rovessels in the diabetic tissue showed a significantly greater degreef apoptosis (brown-stained nuclei) (see Table II for quantitation).
ore, the microvessel density of the VEGF-treated i
ranulation tissue was also normalized compared withhe massive loss of microvessels observed in the con-ralateral diabetic tissue (Fig 8, Table III). Predictably,he integration of CD-34 (and CD-31) cells in mi-rovessels was corrected as also apoptosis of microves-els to normal levels by treatment with VEGF, whereashe contralateral diabetic granulation tissue remainedbnormal in these aspects (Fig 9, Table III). From theevels of VEGF measured in the granuloma fluid fromormal, diabetic, and VEGF-treated diabetic granula-ion tissue, it was clear that the 50% deficiency ofEGF levels in the diabetic tissue was overcome (and
omewhat exceeded the normal levels) by the VEGFnfusion. As expected, the infusion of VEGF in oneranulation tissue did not increase the systemic levelsf VEGF and therefore could not have affected theontralateral diabetic granulation tissue (see Table IIIor serum levels). Normal control animals treated withEGF did not show any remarkable gross or histolog-
cal changes in the granulation tissue (not shown) evenhough the VEGF concentration in the granuloma fluid8.54 � 0.52 ng/mL) expectedly exceeded the normalontrol levels (see Table III for normal control levels).
ISCUSSION
Wound healing is a unique regenerative cellular pro-ess accompanied by blood vessel formation (angio-enesis) that ultimately results in the rapid constructionf new tissue. In diabetes, angiogenesis is defective andound healing is impaired, and the sequence of events
n these defects, cellular and biochemical, is best stud-ed in cutaneous wounds. In wound healing, severalrderly sequential phases exist, namely coagulation,nfiltration of the wound by cells from the blood (andone marrow), proliferation of the infiltrated cells, for-ation of new blood vessels (angiogenesis) and extra-
ellular matrix, and finally a longer process of remod-ling of the early tissue to form a more mature healedound. Implanting an inert polyvinyl tube (or other
oreign materials) under the skin to create granulationissue is a useful model for studying wound heal-ng.17–20 It is akin to the incisional skin wound modelsn that it displays all phases of wound healing, includ-ng coagulation, infiltration, proliferation, and angio-enesis. Instead of re-epithelialization, as observed inkin wounding, a pseudo-epithelial layer of cells envel-ps the foreign body that forms the inner layer of theranulation tissue.This foreign-body granulation model has several ad-
antages as follows: (1) It gives rise to a demarcatedrea of healing clearly separable from the normal un-njured area; (2) it allows for the size of the granulationissue to be controlled (by the size of the foreign body
nserted) and to yield sufficient tissue for histological
aetmotps
t
obl
shown).
F1po1s
Tqaw
TWV
V
M
C
%
%
*an
Translational Research288 Singh et al May 2007
nd biochemical analysis; (3) it generates an abundantxtractable granulation fluid that is in equilibrium withhe wound tissue and can be used for analyses andonitoring of the wound environment16; and (4) it
ffers a convenient space for injecting test substanceshat can diffuse into the wound and modify the healingrocess. For these reasons, this model was selected totudy angiogenesis in diabetic wounds.
After 1 week of diabetes, thinning of the granulation
Fig 6. CD-34-positive cells in the microvesselsCD-34-positive cells stained brown were visible intissues. However, although the CD-34-positive celltissue (red arrows), fewer such cells were present ipresent freely in the medial and inner layers. Essenwere stained for CD-31 antigen (not shown). Frequethat appeared to be devoid of endothelial cells (not
ig 7. Morphology of the granulation tissue from diabetic rats afterweek of infusion with VEGF showing well-formed, healthy ap-
earing granulation tissue compared with the thin granulation tissuef the untreated contralateral side typically seen in diabetic rats (see-week diabetic granulation tissue in Fig 1 for comparison). PAStaining.
issue and dehiscence of the medial vascular layer was d
bserved because of fewer and weaker (also leakier)lood vessels accompanied by a decrease of VEGFevels in the granuloma fluid. As early as 3 days after
l (A) and 3-day diabetic granulation tissue (B).ce in both the control and the diabetic granulationostly integrated in the blood vessels of the controld vessels of the diabetic tissue. Rather, they wereilar results were obtained when the tissue sectionseudo-capillaries” were visible in the diabetic tissue
able III. Gross and histochemical changesuantified in the granulation tissue of diabetic ratsfter treatment of one bilateral granulation tissueith VEGF for 1 week
Diabetic granulation tissue
Normalgranulation
tissue
MeasurementsUntreated
(n � 4)VEGF-treated
(n � 4)Untreated
(n � 4)
hickness (mm) 0.6 � 0.05* 1.3 � 0.1 1.1 � 0.1et weight (g) 0.5 � 0.04* 1.1 � 0.03 1.0 � 0.05
EGF level ingranuloma fluid(ng/mL)
2.9 � 0.2* 7.2 � 0.3 5.9 � 0.4
EGF level in serum(ng/mL)
0.13 � 0.04 0.12 � 0.02
icrovessel density(number of capillariesin medial layer/high-power field)
42 � 6* 123 � 8 110 � 9
D-34-positive cells inmedial layer (cellcount/hpf)
230 � 16 210 � 19 190 � 15
CD-34-positive cellspresent inmicrovessels
5 � 1* 42 � 5 38 � 3
TUNEL (positiveendothelial cells inmicrovessels)
48 � 3* 9 � 2 11 � 2
Statistically significant at P � 0.05 compared with VEGF-treatednd normal. Please consult footnotes of Tables I and II for expla-ation of the measurement parameters.
of controabundan
s were mn the blootially simntly, “ps
iabetes, extensive apoptosis of the endothelial cells

owwanVfnggsac
tf-ihtculwob
dctt“tbCtsbsit(nnsasoa
mally dev
FtTCigT
Translational ResearchVolume 149, Number 5 Singh et al 289
ccurred, forming the medial layer microvessels, asell as significantly decreased amounts of VEGF in theound tissue. Low VEGF could be responsible for
poptosis of the blood vessels as it has been shown thatewly formed vessels are much more sensitive toEGF levels.21,22 Support for this view comes also
rom the work of Howdieshell, et al17 who showed thateutralizing VEGF with an antibody in a foreign-bodyranulation model impaired angiogenesis and inhibitedranulation tissue formation. The authors likewise havehown previously in a tube granulation model thatntagonizing VEGF locally with a receptor antagonist
Fig 8. Microvessels revealed by collagen type IV immtreated for 1 week by infusion of VEGF. Left: Microwas preserved to normal level compared with the mass(not shown here, but see diabetic tissue in Fig 2 for comthe inner layer of the granulation tissue, which is nor
ig 9. CD-34-positive cells (brown) in the microvessels of granula-ion tissue of diabetic rats treated for 1 week by infusion of VEGF.reated granulation tissue shows a normal level of integration ofD-34 cells in microvessels (red arrows) compared with the lack of
ntegration of CD-34 cells in the contralateral untreated diabeticranulation tissue (compare with diabetic tissue in Fig 6, see alsoable III).
aused attrition of blood vessels and disorganization of t
he inner matrix layer.16 It may be pertinent to state thatactors other than VEGF such as angiopoietins-1 and2, tissue factor, and Cyr61, although not investigatedn this study, may also be involved in angiogenesis ofealing wounds.23–25 Nevertheless, infusing VEGF inhe diabetic wound completely prevented the microvas-ular and other accompanying changes obsered in thentreated contralateral diabetic wound, suggesting thatow VEGF was the most important factor controllingound healing. It is, however, possible that factorsther than VEGF are also involved, but they may haveeen normalized by correction of VEGF levels.Although the granulation tissue is formed by cells
erived from bone marrow and local tissue, the relativeontribution of the two is not clear.26–28 It is possiblehat, in response to the initial inflammation caused byhe wound, bone marrow-derived progenitor cellshome” to the inflamed site and together with the localissue differentiate to form new structures. Of theseone marrow-derived progenitor cells, the CD-34 (andD-31) cells seem to be the predecessor of the endo-
helial cells of the new blood vessels.14,28 A variety ofignaling chemokines influence the homing of theseone marrow-derived progenitor cells to the injuredite. Among the chemokines, SDF-1� produced by thenjured tissue attracts CD-34 cells expressing the recep-or CXCR-4.29–31 These results show that the CD-34and CD-31) cells did not effectively integrate into theew blood vessels in the diabetic wound despite theirormal mobilization to the injured site as was alsouggested by similar immune staining for the SDF-1�nd CXCR-4 in both the control and the diabetic tis-ues. Such failure of integration could be either becausef the lack of a differentiation factor such as VEGF ordefect in the CD-34 cells of the diabetic rats. Al-
scent staining in the granulation tissue of diabetic ratssity of the VEGF-treated diabetic granulation tissue
f microvessels seen in the contralateral diabetic tissueRight: Occasionally, microvessels were seen to reachoid of blood vessels.
unofluorevessel denive loss oparison).
hough CD-34 cells from long-standing diabetic pa-

tcmbmowt
adamCVfww
Vdfdsmavca
Rc
R
1
1
1
1
1
1
1
1
1
1
2
2
2
2
2
2
Translational Research290 Singh et al May 2007
ients fail to differentiate into capillaries in cultureompared with CD-34 cells from nondiabetic hu-ans,32 it is unlikely that it may be the case here
ecause of the short duration of diabetes (3 days) in thisodel. That low VEGF may be responsible for the lack
f integration of CD-34 cells in the new blood vesselsas strongly suggested by the complete correction of
his defect by infusion of VEGF.In conclusion, these results show that diabetes causesloss of blood vessels by two mechanisms: (1) cell
eath of newly formed vessels (wound vessels) bypoptosis and (2) inhibition of new blood vessel for-ation because of the inability of the CD-34 (andD-31) cells to integrate into the vessel structure. LowEGF level in the wound tissue appears to be the critical
actor impairing angiogenesis as the microvascular defectsere completely corrected by VEGF infusion in theound tissue.Speculations. Although it is shown here that lowEGF was the main factor in causing microvascularamage in diabetic wounds, it may also be responsibleor the generalized microvascular organ damage iniabetes (except in the eye, where VEGF has beenhown to be increased in diabetes). The integrity oficrovessels in general being dependent on a constant
nd steady turnover, an impaired integration of theascular progenitor cells in diabetes would graduallyause loss of microvessels with progressive scarringnd eventual organ failure.
The authors wish to thank Valentina Svoren, MS, and Levappaport, MD, provided help with the histology and immunocyto-hemistry.
EFERENCES
1. Hanahan D. Signaling vascular morphogenessis and mainte-nance. Science 1997;277:48–50.
2. Abaci A, Oguzhan A, Kahraman S, Eryol NK, Unal S, Arinc H,et al. Effect of diabetes mellitus on formation of coronary col-lateral vessels. Circulation 1999;99:2239–42.
3. Waltenberger J. Impaired collateral vessel development in dia-betes: potential cellular mechanisms and therapeutic implications(Review). Cardiovasc Res 2001;49:554–60.
4. Galeano M, Deodata B, Altavilla D, Cucinotta D, Arsic N,Marini H, et al. Adeno-associated viral vector-mediated humanvascular endothelial growth factor gene transfer stimulates an-giogenesis and wound healing in the genetically diabetic mouse.Diabetologia 2003;46:546–55.
5. Galeano M, Altavilla D, Cuciotta D, Russo GT, Calo M, Bitto A,et al. Recombinant human erythropoietin stimulates angiogenesisand wound healing in the genetically diabetic mouse. Diabetes2004;53:2509–17.
6. Keswani SG, Katz AB, Lim FY, Zoltick P, Radu A, Alaee D,et al. Adenoviral mediated gene transfer of PDGF-B enhanceswound healing in type I and type II diabetic wounds. WoundRepair Regen 2004;12:497–504.
7. Graiani G, Eamnueli C, Desortes E, Van Linthout S, Pinna A,
Figueroa CD, et al. Nerve growth factor promotes reparativeangiogenesis and inhibits endothelial apoptosis in cutaneouswounds in type I diabetic mice. Diabetologia 2004;47:1047–54.
8. Hunt TK. The physiology of wound healing. Ann Emerg Med1988;17:1265–72.
9. Waldorf H, Fewkes J. Wound healing. Adv Dermatol 1995;10:77–97.
0. Jain RK. Molecular regulation of vessel maturation. Nat Med2003;9:685–93.
1. Asahara T, Murohara T, Sullivan A, Silver M, van der Zee R,Li T, et al. Isolation of putative progenitor endothelial cellsfor angiogenesis. Science 1997;275:964 –7.
2. Dzau VJ, Gnecchi M, Pachori AS, Morello F, Melo LG. Ther-apeutic potential of endothelial progenitor cells in cardiovasculardiseases (Brief Review). Hypertension 2005;46:7–18.
3. Minamino K, Adachi Y, Okigaki M, Ito H, Togawa Y, Fujita K,et al. Macrophage colony-stimulating factor (M-CSF), as well asgranulocyte colony-stimulating factor (G-CSF), accelerates neo-vascularization. Stem Cells 2005;23:347–54.
4. Murayama T, Tepper O, Silver M, Losordo DW, Isner JM, AsaharaT, et al. Determination of bone marrow-derived endothelial cellsresponsible for post-natal vasculogenesis in physiological andpathological neovascularization. Exp Hematol 2002;30:967–73.
5. Galiano RD, Tepper OM, Pelo CR, Bhatt KA, Callaghan M,Bastidas N, et al. Topical vascular endothelial growth factoraccelerates diabetic wound healing through increased angiogen-esis and by mobilizing and recruiting bone marrow derived cells.Am J Pathol 2004;164:1935–47.
6. Gudehithlu KP, Ahmed N, Wu H, Litbarg NO, Garber SL,Arruda JAL, et al. Antagonism of vascular endothelial growthfactor results in microvessel attrition and disorganization ofwound tissue. J Lab Clin Med 2005;145:194–203.
7. Howdieshell TR, Riegner C, Gupta V, Callaway D, GrembowiczK, Sathyanarayana MD, et al. Normoxic wound fluid containshigh levels of vascular endothelial growth factor. Ann Surg 1998;228:707–15.
8. Rodriguez MC, Saiz MP, Carbonell T, Mitjavila MT. Local andsystemic responses to iron dextran injected in a granuloma pouchin the rat. J Lab Clin Med 1999;134:42–8.
9. Teixeira AS, Andrade SP. Glucose-induced inhibition of angio-genesis in the rat sponge granuloma is prevented by aminogua-nidine. Life Sci 1999;64:655–62.
0. Li Y, Metori K, Koike K, Kita F, Che QM, Sato T, et al.Granuloma maturation in the rat is advanced by the oral admin-istration of Eucommia ulmoides OLIVER leaf. Biol Pharm Bull2000;23:60–5.
1. Benjamin LE, Keshet E. Conditional switching of vascular en-dothelial growth factor (VEGF) expression in tumors: inductionof endothelial cell shedding and regression of hemangioblasto-ma-like vessels by VEGF withdrawal. Proc Natl Acad Sci USA1997;94:8761–6.
2. Benjamin LE, Golijanin D, Itin A, Pode D, Keshet E. Selectiveablation of immature blood vessels in established human tumorsfollows vascular endothelial growth factor withdrawal. J ClinInvest 1999;103:159–65.
3. Kampfer H, Pfeilschifter J, Frank S. Expressional regulation ofangiopoietin-1 and -2 and the Tie-1 and -2 receptor tyrosinekinases during cutaneous wound healing: a comparative study ofnormal and impaired repair. Lab Invest 2001;81:361–73.
4. Chen J, Kasper T, Heck T, Nakagawa K, Humpert PM, Bai L,et al. Tissue factor as a link between wounding and tissuerepair. Diabetes 2005;54:2143–54.
5. Chen CC, Mo FE, Lau LF. The angiogenic factor Cyr61 activatesa genetic program for wound healing in human skin fibroblasts.
J Biol Chem 2001;276:47329–37.
2
2
2
2
3
3
3
Translational ResearchVolume 149, Number 5 Singh et al 291
6. Bucala R, Spiegel LA, Chesney J, Hogan M, Cerami A. Circu-lating fibrocytes define a new leukocyte subpopulation that me-diates tissue repair. Mol Med 1994;1:71–81.
7. Niku M, Ilmonen L, Pessa-Morrikawa T, Iivanainen A. Limitedcontribution of circulating cells to the development and mainte-nance of nonhematopoietic bovine tissues. Stem Cells 2004;22:12–20.
8. Sivan-Loukianova E, Awad OA, Stepanovic V, Bikenbach J,Schatteman GC. CD 34� blood cells accelerate vascularizationand healing of diabetic mouse skin wounds. J Vasc Res 2003;40:368–77.
9. Gillitzer R, Goebeler M. Chemokines in cutaneous wound heal-
ing. J Leukoc Biol 2001;69:513–22.0. Kollet O, Shivtiel S, Chen YQ, Suriawinata J, Thung SN, DabevaMD, et al. HGF, SDF-1, and MMP-9 are involved in stress-induced human CD34� stem cell recruitment to the liver. J ClinInvest 2003;112:160–9.
1. Ceradini DJ, Kulkarni AR, Callaghan MJ, Tepper OM, BastidasN, Kleinman ME, et al. Progenitor cell trafficking is regulated byhypoxic gradients through HIF-1 induction of SDF-1. Nat Med2004;10:858–64.
2. Tepper OM, Galiano RD, Capla JM, Kalka C, Gagne PJ,Jacobowitz GR, et al. Human endothelial progenitor cellsfrom Type II diabetics exhibit impaired proliferation, adhe-sion, and incorporation in vascular structures. Circulation
2002;106:2781– 6.
INFORMATION FOR READERS
COMMUNICATIONS: Communications regarding articles andeditorial management should be addressed to the Managing Edi-tor, Michael Franklin, at [email protected]. Authors shouldconsult the Instructions for Authors before submitting manu-scripts to Translational Research.
CUSTOMER SERVICE (orders, claims, online, change of ad-dress): Elsevier Periodicals Customer Service, 6277 Sea HarborDrive, Orlando, FL 32887-4800. Tel: (800) 654-2452 (U.S. andCanada); (407) 345-4000 (outside U.S. and Canada). Fax: (800)225-6030 (U.S. and Canada); (407) 363-9661 (outside U.S. andCanada). E-mail: [email protected]. Address changes must besubmitted four weeks in advance.
YEARLY SUBSCRIPTION RATES: United States and posses-sions: Individual $216.00; Institution $524.00; Student/Resident$104.00. All other countries (prices include airspeed delivery):Individual $267.00; Institution $575.00; Student/Resident$137.00. Single Issues $48.00. To receive student/resident rate,orders must be accompanied by name of affiliated institution, dateof term and the signature of program/residency coordinator oninstitution letterhead. Order will be billed at the individual rateuntil proof of status is received. Current prices are in effect forback volumes and back issues.
Further information on this journal is available from the publisher,Elsevier, or from the journal’s Web site (http://www.elsevier.com/transres). Information on other Elsevier products is availablethrough Elsevier’s Web site (http://www.elsevier.com).
Advertising information: Advertising orders and inquiries canbe sent to: USA, Canada, and South America, Ariel Medina,Elsevier Inc, 360 Park Avenue South, New York, NY 10010;phone (212)633-3689; fax (212)633-3820. Classified advertisingorders and inquiries can be sent to Simone Imbert, Elsevier Inc,360 Park Avenue South, New York, NY 10010; phone (212)462-1908; fax (212)633-3820. Europe and the rest of the world,Julie Toop; phone _44 (0) 1865 843016; fax _44 (0) 1865 843976;e-mail: [email protected].
Author Inquiries: For inquiries relating to the submission ofarticles (including electronic submission), please visit Elsevier’sAuthor Gateway at http://authors.elsevier.com. The Author Gate-way also provides the facility to track accepted articles and set upe-mail alerts to inform you of when an article’s status haschanged, as well as detailed artwork guidelines, copyright infor-mation, frequently asked questions, and more. Please see AuthorGuidelines for individual journal. Contact details for questionsarising after acceptance of an article, especially those relating toproofs, are provided after registration of an article for publication.
Reprints: For queries about author offprints, e-mail:[email protected]. To order 100 or more reprints foreducational, commercial, or promotional use, contact the Com-mercial Reprints Department, Elsevier Inc, 360 Park Avenue South,New York, NY 10010-1710; fax (212)462-1935; e-mail:[email protected]. Reprints of single articles available onlinemay be obtained by purchasing Pay-Per-View access for $30 perarticle on the journal Web site, http://www.elsevier.com/transres.
© 2007 Mosby, Inc. All rights reserved. This journal and theindividual contributions contained in it are protected under copy-right by Elsevier, Inc., and the following terms and conditionsapply to their use. Photocopying: Single photocopies of singlearticles may be made for personal use as allowed by nationalcopyright laws. Permission of the Publisher and payment of a feeis required for all other photocopying, including multiple orsystematic copying, copying for advertising or promotional pur-poses, resale, and all forms of document delivery. Special ratesare available for educational institutions that wish to make pho-tocopies for non-profit educational classroom use. Permissionsmay be sought directly from Elsevier’s Rights Department inPhiladelphia, PA, USA: telephone (215)239-3804; fax (215)239-3805; e-mail: [email protected]. Requests mayalso be completed online via the Elsevier homepage (http://www.elsevier. com/locate/permissions). In the USA, users mayclear permissions and make payments through the CopyrightClearance Center, Inc., 222 Rosewood Drive, Danvers, MA01923, USA; phone: (978)750-8400, fax: (978)750-4744, and inthe UK through the Copyright Licensing Agency Rapid ClearanceService (CLARCS), 90 Tottenham Court Road, London W1P0LP, UK; phone: (44)20-7631-5555; fax: (44)20-7631-5500.Other countries may have a local reprographic rights agency forpayments. Derivative Works: Subscribers may reproduce tablesof contents or prepare lists of articles including abstracts forinternal circulation within their institutions. Permission of thePublisher is required for resale or distribution outside the institu-tion. Permission of the Publisher is required for all other deriva-tive works, including compilations and translations. ElectronicStorage or Usage: Permission of the Publisher is required to storeor use electronically any material contained in this journal, in-cluding any article or part of an article. Except as outlined above,no part of this publication may be reproduced, stored in a retrievalsystem or transmitted in any form or by any means, electronic,mechanical, photocopying, recording or otherwise, without priorwritten permission of the Publisher. Address permissions requeststo: Elsevier Rights Department, at the fax and e-mail addressesnoted above. Notice: No responsibility is assumed by the Pub-lisher for any injury and/or damage to persons or property as amatter of products liability, negligence or otherwise, or from anyuse or operation of any methods, products, instructions or ideascontained in the material herein. Because of rapid advances in themedical sciences, in particular, independent verification of diag-noses and drug dosages should be made. Although all advertisingmaterial is expected to conform to ethical (medical) standards,inclusion in this publication does not constitute a guarantee orendorsement of the quality or value of such product or of theclaims made of it by its manufacturer.
Indexed or abstracted in Index Medicus, Science Citation Index,Current Contents/Clinical Medicine, Current Contents/Life Sci-ences, and MEDLINE.
Microform edition available from ProQuest Information andLearning, 300 North Zeeb Rd, Ann Arbor, MI 48106-1346.
Available electronically from Ovid Technologies, Inc, 333 Sev-enth Ave, New York, NY 10001; telephone (800)950-2035.

FEATURED NEW INVESTIGATOR237 Transcriptional regulation of podocyte disease
Sumant S. Chugh, Chicago, Ill
ORIGINAL ARTICLES243 Immunopathogenesis of hypersensitivity
syndrome reactions to sulfonamidesManuela G. Neuman, Neil H. Shear,Izabella M. Malkiewicz, Masud Taeri, LoriE. Shapiro, Norberto Krivoy, Julia Haber,Manuel Gomez, Joel Fish, Robert Cartotto,and Lawrence Cohen, Toronto, Ontario,Canada and Haifa, Israel
254 Apoptosis in ibuprofen-induced Stevens–Johnson syndromeManuela Neuman and Michael Nicar,Toronto, Ontario, Canada and Dallas, Tex
260 Antitumor and antiinflammatory effects oftetrathiotungstate in comparison withtetrathiomolybdateGuoqing Hou, Robert Dick, Chunhua Zeng,and George J. Brewer, Ann Arbor, Mich
265 Expression of angiopoietin-1 in osteoblastsand its inhibition by tumor necrosis factor-alpha and interferon-gammaTsuyoshi Kasama, Takeo Isozaki, TsuyoshiOdai, Mizuho Matsunawa, KuninobuWakabayashi, Hiroko T. Takeuchi, SatoshiMatsukura, Mitsuru Adachi, MasakazuTezuka, and Kazuo Kobayashi, Tokyo, Japan
274 Advanced glycation end-product-inducedmitogenesis is dependent on Janus kinase2-induced heat shock protein 70 in normalrat kidney interstitial fibroblast cellsSan-Cher Chen, Jinn-Yuh Guh, Hung-ChunChen, Yu-Lin Yang, Jau-Shyang Huang, andLea-Yea Chuang, Kaohsiung and Tainan,Taiwan
282 Impaired integration of endothelialprogenitor cells in capillaries of diabeticwounds is reversible with vascularendothelial growth factor infusionAshok K. Singh, Krishnamurthy P.Gudehithlu, Shreya Patri, Natalia O.Litbarg, Perianna Sethupathi, Jose A. L.Arruda, and George Dunea, Chicago andMaywood, Ill
READER SERVICES
3A Author Guidelines
C3 Information for Readers
Visit our website at www.elsevier.com/transres
TRANSLATIONAL RESEARCHThe Journal of Laboratory and Clinical Medicine
Volume 149, Number 5, May 2007
Contents





























