Embed Size (px)
Citation preview

Article
KDEL Receptors Assist Dengue Virus Exit from theEndoplasmic Reticulum
Graphical Abstract
Highlightsd Depletion of KDELR by siRNA reduces egress of DENV
progeny
d DENV1-3 structural protein prM interacts with KDELR
in the ER
d KDELR/prM interaction requires three positively charged
amino acids at N terminus of prM
d Disrupting this interaction inhibits DENV1 RSPs transport
from the ER to the Golgi
Authors
Ming Yuan Li, Marc Grandadam, ...,
Roberto Bruzzone, Pei Gang Wang
[email protected] (R.B.),[email protected] (P.G.W.)
In BriefViral receptors are key host factors for
virion entry; however, it is not known
whether trafficking and secretion of
progeny virus also requires host
intracellular receptors. Li et al. show that
dengue virus (DENV) interacts with host
KDEL receptors (KDELR) in the ER.
Depleting KDELR, disrupting DENV/
KDELR interaction or blocking KDELR
cycling between the ER and Golgi reduce
virus release, resulting in virus
accumulation in the ER. The authors
propose that KDELR functions as
intracellular receptors to assist in DENV
exit from the ER.
Li et al., 2015, Cell Reports 10, 1496–1507March 10, 2015 ª2015 The Authorshttp://dx.doi.org/10.1016/j.celrep.2015.02.021

Cell Reports
Article
KDEL Receptors Assist Dengue VirusExit from the Endoplasmic ReticulumMing Yuan Li,1 Marc Grandadam,2 Kevin Kwok,1 Thibault Lagache,3,4 Yu Lam Siu,1 Jing Shu Zhang,1 Kouxiong Sayteng,2
Mateusz Kudelko,1,5 Cheng Feng Qin,6 Jean-Christophe Olivo-Marin,3,4 Roberto Bruzzone,1,4,* and Pei Gang Wang1,7,*1HKU-Pasteur Research Pole and Centre of Influenza Research, School of Public Health, LKS Faculty of Medicine, The University of HongKong, Hong Kong SAR, China2Institut Pasteur du Laos, Vientiane, Lao PDR3Unite d’Analyse d’Images Biologiques, CNRS URA 2582, Department of Cell Biology and Infection, Institut Pasteur, 75015 Paris Cedex,France4Department of Cell Biology and Infection, Institut Pasteur, 75015 Paris Cedex, France5Department of Biochemistry, LKS Faculty of Medicine, The University of Hong Kong, Hong Kong SAR, China6Department of Virology, Beijing Institute of Microbiology and Epidemiology, Beijing 100071, PR China7Key Laboratory of Protein and Peptide Pharmaceuticals, Institute of Biophysics, Chinese Academy of Sciences, Beijing 100101, PR China*Correspondence: [email protected] (R.B.), [email protected] (P.G.W.)http://dx.doi.org/10.1016/j.celrep.2015.02.021This is an open access article under the CC BY-NC-ND license (http://creativecommons.org/licenses/by-nc-nd/4.0/).
SUMMARY
Membrane receptors at the surface of target cells arekey host factors for virion entry; however, it is un-known whether trafficking and secretion of progenyvirus requires host intracellular receptors. In thisstudy, we demonstrate that dengue virus (DENV) in-teracts with KDEL receptors (KDELR), which cyclebetween the ER and Golgi apparatus, for vesiculartransport from ER to Golgi. Depletion of KDELR bysiRNA reduced egress of both DENV progeny and re-combinant subviral particles (RSPs). Coimmunopre-cipitation of KDELR with dengue structural proteinprM required three positively charged residues atthe N terminus, whose mutation disrupted proteininteraction and inhibited RSP transport from theER to the Golgi. Finally, siRNA depletion of class IIArfs, which results in KDELR accumulation in theGolgi, phenocopied results obtained with muta-genized prME and KDELR knockdown. Our resultshave uncovered a function for KDELR as an internalreceptor involved in DENV trafficking.
INTRODUCTION
Dengue, a mosquito-borne viral infection endemic in over100 countries, is caused by four serotypes of dengue virus(DENV1–4). In addition to a febrile, influenza-like illness, severedengue represents a public health concern in Asia and SouthAmerica where it is a major cause of death across all ages (Guz-man et al., 2010; Messina et al., 2014). Despite the global burdenof disease, there is no specific treatment and, therefore, amolec-ular understanding of host-pathogen interactions during thecellular life cycle is needed to guide the development of effectivedrugs (Guzman et al., 2010).
DENV has two structural glycoproteins: pre-membrane (prM)and envelope (E) (Kuhn et al., 2002); E mediates interactionwith cellular receptor(s) for viral attachment and entry (Chenet al., 1997), whereas prM assists E in its correct folding (Coura-geot et al., 2000) and protects it from pre-fusion in the acidicenvironment of the secretory pathway (Zhang et al., 2003). As-sembly of DENV occurs at the ER and requires interaction ofprM and E (Mukhopadhyay et al., 2005; Pryor et al., 2004).Nascent virions bud into the lumen of the ER, accumulating indilated cisternae oriented toward the cis-Golgi, and are translo-cated to the Golgi via trafficking vesicles (Welsch et al., 2009). Inthe trans-Golgi network (TGN), prM protein is cleaved by thecellular protease furin, resulting in the release of the pr peptideand formation of infectious DENV (Li et al., 2008; Yu et al.,2008). Besides mature virions, non-infectious recombinant sub-viral particles (RSP) can be produced by cells expressing DENVprME proteins (Mukhopadhyay et al., 2005). Dengue RSP trafficalong the same compartments as infectious DENV, and repre-sent a safe and convenient tool for the study of virus-host inter-actions during secretion (Wang et al., 2009).Although DENV egress has been studied for many years, most
cellular targets identified in high-throughput screens have notbeen mapped to the secretory pathway (Sessions et al., 2009).We recently identified two cellular factors, ADP-ribosylationfactor 4 and 5 (Arf4 and Arf5), which are involved in secretionof DENV progeny (Kudelko et al., 2012). Because Arfs play animportant role in the recruitment of coat proteins necessary forthe formation of trafficking vesicles (D’Souza-Schorey andChavrier, 2006), our results indicate that Arf4+5 are acting atan early step of DENV secretion (Kudelko et al., 2012). The spe-cific involvement of Arfs, which are dispensable factors for theconstitutive pathway, in DENV trafficking suggested that thevirus uses amore complex machinery and that other cellular fac-tors besides Arf4+5 might also assist to exit the infected cell.Sorting of cargo is dependent on molecular recognition, a
process equivalent to receptor-ligand interactions; however, itis not known whether newly formed DENV exploits host factors
1496 Cell Reports 10, 1496–1507, March 10, 2015 ª2015 The Authors

to move along the secretory pathway. Intriguingly, depletion ofArf4+5 has also been reported to inhibit the retrograde traf-ficking of KDEL receptor (KDELR) from Golgi to ER (Volpicelli-Daley et al., 2005). The three KDELR members (KDELR1–3)identified (Hsu et al., 1992; Lewis and Pelham, 1990, 1992b;Raykhel et al., 2007) are transmembrane proteins cycling be-tween ER and Golgi apparatus to prevent leakage of ER-resi-dent proteins, such as chaperones, and retrieve them back tothe ER (Lewis and Pelham, 1990). As KDELR binding to cargothrough a C terminus KDEL motif occurs only in the Golgi appa-ratus, we investigated their possible involvement in transloca-tion of DENV from assembly and budding sites in the ER tothe Golgi.We show here that KDELR1 and KDELR2 play crucial roles for
DENV1–3, but not DENV4 secretion. KDELR interacted withDENV through three positively charged amino acids at the N ter-minus of prM. DENV secretion could be blocked either by de-pletion of KDELR, arrest of KDELR cycle, or disruption of prM/KDELR interaction. Under these conditions, progeny DENVaccumulated in the ER and did not reach the Golgi apparatus.Our results demonstrate that KDELRs function as luminal recep-tors for DENV transport along the secretory pathway.
RESULTS
KDELR Interact with prM of DENV1We previously demonstrated that depletion of Arf4+5 inhibitedDENV1 and RSP release without disrupting constitutive secre-tion (Kudelko et al., 2012). To gain insight into the underlying mo-lecular mechanism, we investigated the role of KDELRs, whichaccumulate in a peri-nuclear region and are not recycled backto the ER following Arf4+5 depletion (Volpicelli-Daley et al.,2005). All three KDELR identified thus far were detected byRT-PCR in our cellular models (Figure 1A). We observed a redis-tribution of KDELR in cells stably transfected with prME (HeLa-prME-DENV1), with an apparent reduction of co-staining withthe cis-Golgi marker GM130 in comparison to parental HeLa(Figure 1B). Moreover, partial co-localization of E and KDELRwas observed in either HeLa-prME-DENV1 or Vero E6 cells in-fected with DENV1 (Figure 1C). Although a prominent aggrega-tion of E protein was seen in HeLa-prME-DENV1 (Figure 1C),this did not reflect different distribution, as prME co-localizedwith ER marker in both cell lines (data not shown). Similar resultswere obtained in cells co-transfected with DENV1 prME andKDELR1-RFP (Figure S1A). These observations suggested theparticipation of KDELRs in DENV1 life cycle.We next performed co-immunoprecipitation (coIP) using either
dengue patient serum (DPS), containing antibodies recognizingprME (Kudelko et al., 2012), or normal human serum (NHS) ascontrol. Pellets of coIPs were analyzed with western blotting(WB) with antibodies recognizing either prM and E, or the threeKDELR. prME glycoprotein was specifically pulled down byDPS, but not NHS (Figure 1D). Although expression levels werecomparable, KDELR were detected only in pellets from HeLa-prME-DENV1, but not parental cells (Figure 1D). Similarly,when coIP was performed with replication-competent DENV1,a strong signal for KDELRwas visible only in pellets from infectedVero E6 cells (Figure 1E), confirming a biochemical interaction
between KDELR and prME. KDELR were also precipitatedfrom lysates of stable cell lines producing RSP of DENV2 andDENV3, but not DENV4 (Figure 1F), suggesting a certain degreeof specificity between serotypes.To investigate which portion of the envelope glycoprotein, prM
or E, was responsible for interaction with KDELR, the 4G2mono-clonal antibody, recognizing E but not prM, was used in coIPassays. KDELR were not present in pellets obtained following in-cubation with the 4G2 monoclonal (Figure 1G), indicating that Ewas not responsible for interaction with KDELR. These observa-tions were corroborated by detecting KDELR in coIP pellets withthe prM-6.1 monoclonal antibody, which recognizes prM but notE protein (Figure 1G). The biochemical interaction between prMand KDELR was further validated by coIP in 293T cells co-trans-fected with prME and KDELR1-myc (Figure S1B). To conclu-sively define the role of prM and E in the interaction with KDELR,we used two complementary approaches. First, glutathioneS-transferase (GST)-fusion proteins with truncated prM frag-ments were incubated with lysates of cells stably transfectedwith KDELR1-myc. Pull-down assays showed that prM-DTM,the full-length protein without the transmembrane domain(130 amino acids), pr fragment (91 amino acids) and the first40 amino acids of prM sequence (pr40) could all interact withKDELR (Figure 1H), revealing that the amino-terminal domainof prM was sufficient to mediate interaction with KDELR.Second, mature and immature RSP were used as baits to pulldown KDELR from cell lysates. Immature RSP were producedin the presence of NH4Cl, an inhibitor of furin, and thus containedfull-length prM and E (Wang et al., 2009). Mature RSP were pro-duced from immature RSP after in vitro cleavage by furin, whichreleased the pr fragment and, therefore, contained only E andM.KDELR could only be detected when immature, but not matureRSP were incubated with HeLa cell lysates and then subjectedto immune-precipitation (Figure 1I). These results demonstratethat interaction with KDELR was dependent on the N-terminalpr fragment of prM, as its release from immature RSP preventedpull-down of KDELR.
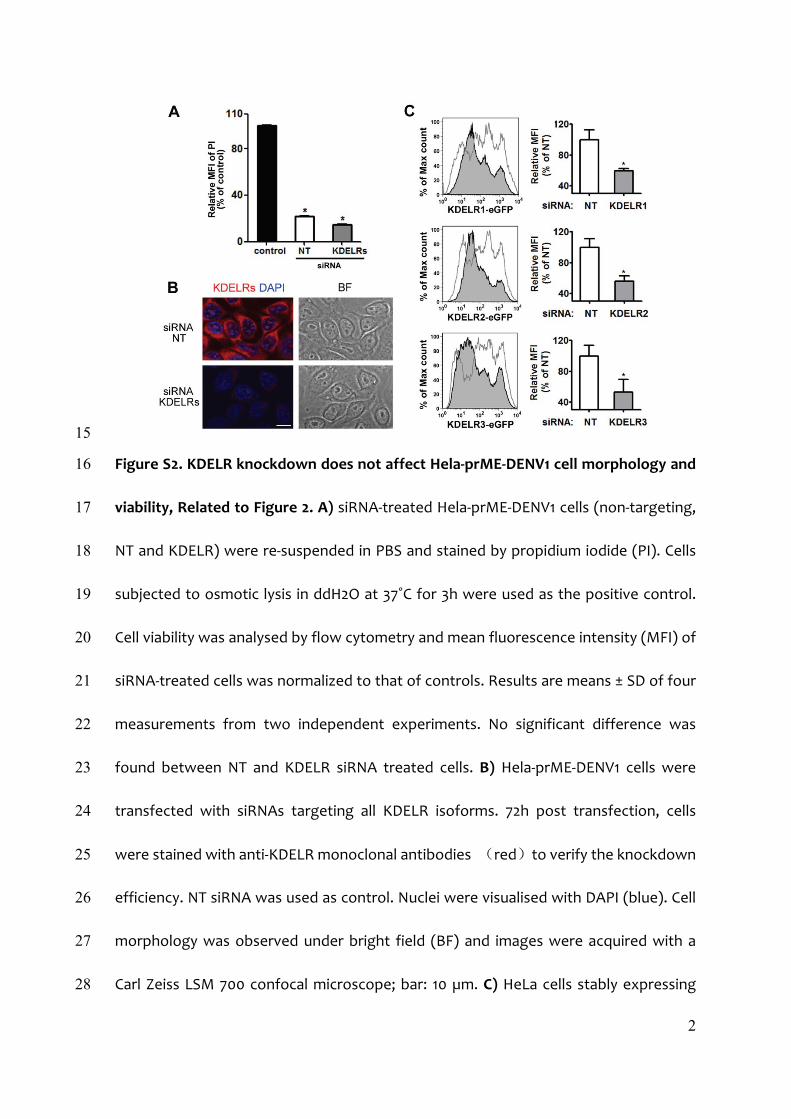
KDELR Knockdown Reduces Secretion of DENV RSPTo investigate the impact of the interaction between KDELR andprM on DENV1 life cycle, we transfected HeLa-prME-DENV1cells that constitutively secrete RSP with siRNAs targeting allthree KDELR; this resulted in an 81% ± 4% (n = 3) reduction ofKDELR protein (Figure 2A). Silencing of KDELR did not affectcell viability, as determined by propidium iodide staining (Fig-ure S2A), or morphology (Figure S2B). Our results show thatdepletion of KDELR had no effect on intracellular E proteinexpression, but significantly reduced RSP secretion (Figure 2A).To test whether the effect of KDELR on RSP release was part
of a general mechanism that would interfere with the constitutivesecretory pathway, we analyzed ssHRP release (Bard et al.,2006; Kudelko et al., 2012). No difference was observed insecreted ssHRPor intracellular HRP activity after downregulationof KDELR or Arf4+5, when compared to controls (Figure 2B). Incontrast, secretion of ssHRP-KDEL occurred only in cells treatedwith KDELR or Arf4+5 siRNAs, confirming that both manoeuvershad interfered with retrieval of KDELR to the ER, resulting in aparallel reduction of HRP activity in cell lysates (Figure 2B). These
Cell Reports 10, 1496–1507, March 10, 2015 ª2015 The Authors 1497
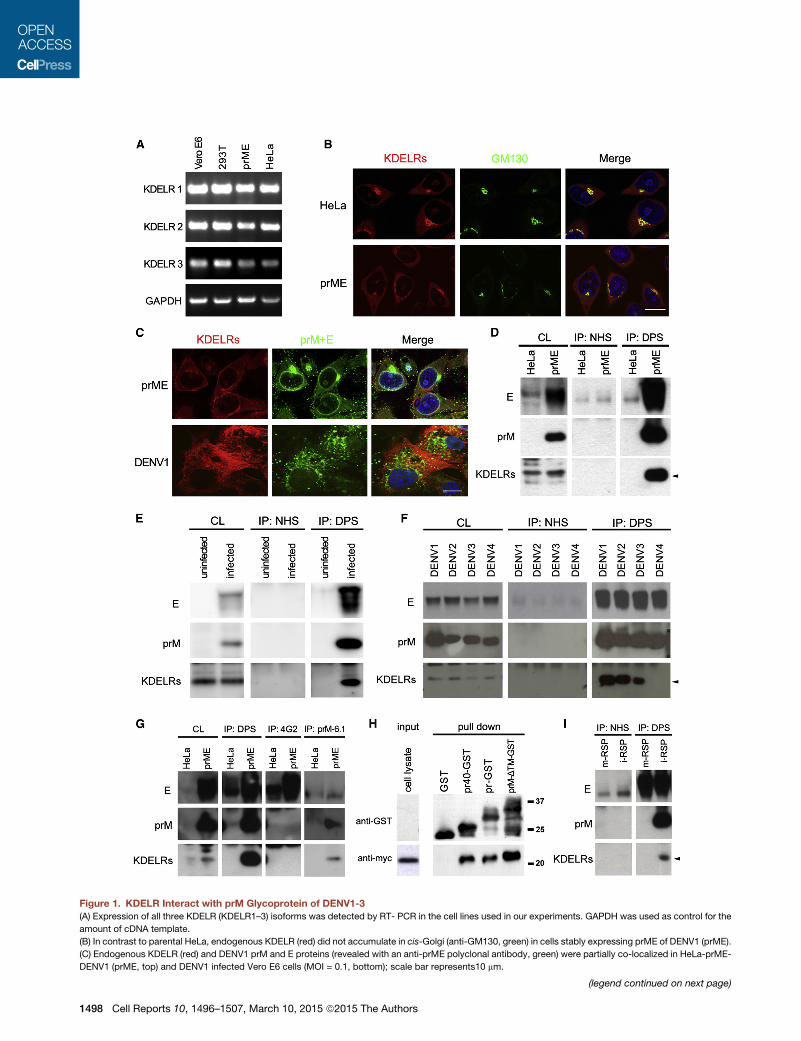
Figure 1. KDELR Interact with prM Glycoprotein of DENV1-3(A) Expression of all three KDELR (KDELR1–3) isoforms was detected by RT- PCR in the cell lines used in our experiments. GAPDH was used as control for the
amount of cDNA template.
(B) In contrast to parental HeLa, endogenous KDELR (red) did not accumulate in cis-Golgi (anti-GM130, green) in cells stably expressing prME of DENV1 (prME).
(C) Endogenous KDELR (red) and DENV1 prM and E proteins (revealed with an anti-prME polyclonal antibody, green) were partially co-localized in HeLa-prME-
DENV1 (prME, top) and DENV1 infected Vero E6 cells (MOI = 0.1, bottom); scale bar represents10 mm.
(legend continued on next page)
1498 Cell Reports 10, 1496–1507, March 10, 2015 ª2015 The Authors
results demonstrate that KDELR specifically assisted RSPrelease and their involvement was independent of perturbationof the constitutive pathway.To study the role of individual KDELR on DENV1 secretion,
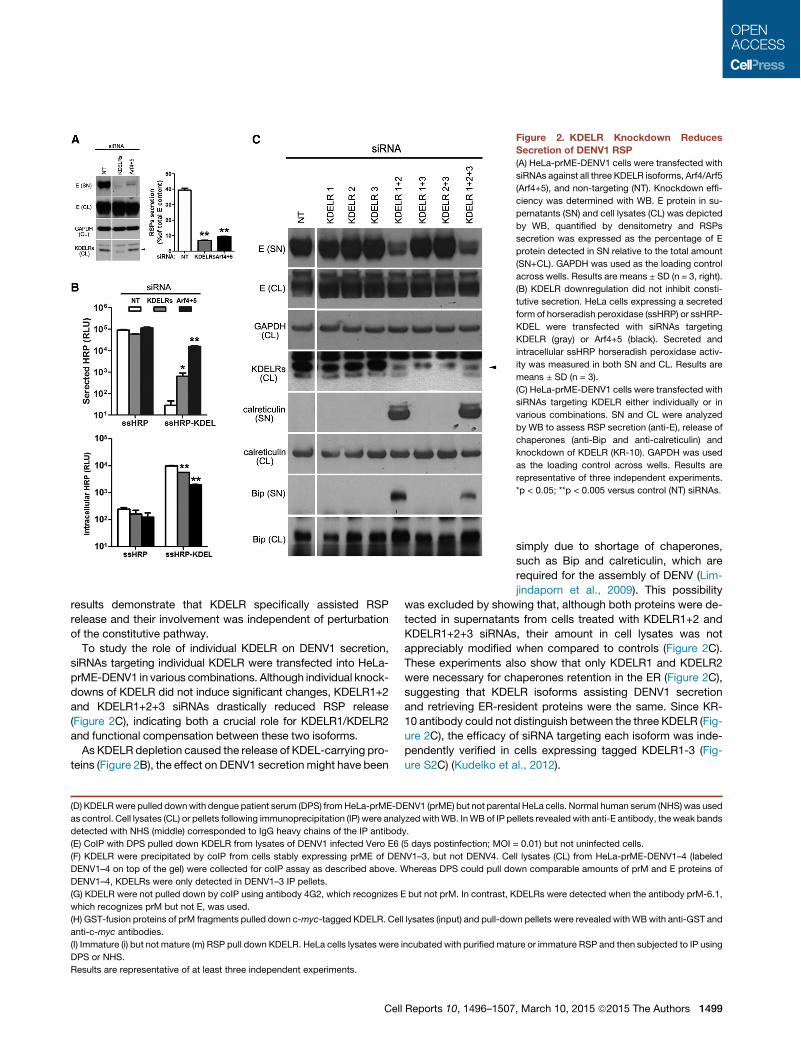
siRNAs targeting individual KDELR were transfected into HeLa-prME-DENV1 in various combinations. Although individual knock-downs of KDELR did not induce significant changes, KDELR1+2and KDELR1+2+3 siRNAs drastically reduced RSP release(Figure 2C), indicating both a crucial role for KDELR1/KDELR2and functional compensation between these two isoforms.As KDELR depletion caused the release of KDEL-carrying pro-
teins (Figure 2B), the effect on DENV1 secretion might have been
(D) KDELRwere pulled downwith dengue patient serum (DPS) fromHeLa-prME-DENV1 (prME) but not parental HeLa cells. Normal human serum (NHS) was used
as control. Cell lysates (CL) or pellets following immunoprecipitation (IP) were analyzed withWB. InWB of IP pellets revealed with anti-E antibody, theweak bands
detected with NHS (middle) corresponded to IgG heavy chains of the IP antibody.
(E) CoIP with DPS pulled down KDELR from lysates of DENV1 infected Vero E6 (5 days postinfection; MOI = 0.01) but not uninfected cells.
(F) KDELR were precipitated by coIP from cells stably expressing prME of DENV1–3, but not DENV4. Cell lysates (CL) from HeLa-prME-DENV1–4 (labeled
DENV1–4 on top of the gel) were collected for coIP assay as described above. Whereas DPS could pull down comparable amounts of prM and E proteins of
DENV1–4, KDELRs were only detected in DENV1–3 IP pellets.
(G) KDELR were not pulled down by coIP using antibody 4G2, which recognizes E but not prM. In contrast, KDELRs were detected when the antibody prM-6.1,
which recognizes prM but not E, was used.
(H) GST-fusion proteins of prM fragments pulled down c-myc-tagged KDELR. Cell lysates (input) and pull-down pellets were revealed withWBwith anti-GST and
anti-c-myc antibodies.
(I) Immature (i) but not mature (m) RSP pull down KDELR. HeLa cells lysates were incubated with purified mature or immature RSP and then subjected to IP using
DPS or NHS.
Results are representative of at least three independent experiments.
Figure 2. KDELR Knockdown ReducesSecretion of DENV1 RSP(A) HeLa-prME-DENV1 cells were transfected with
siRNAs against all three KDELR isoforms, Arf4/Arf5
(Arf4+5), and non-targeting (NT). Knockdown effi-
ciency was determined with WB. E protein in su-
pernatants (SN) and cell lysates (CL) was depicted
by WB, quantified by densitometry and RSPs
secretion was expressed as the percentage of E
protein detected in SN relative to the total amount
(SN+CL). GAPDH was used as the loading control
across wells. Results are means ± SD (n = 3, right).
(B) KDELR downregulation did not inhibit consti-
tutive secretion. HeLa cells expressing a secreted
form of horseradish peroxidase (ssHRP) or ssHRP-
KDEL were transfected with siRNAs targeting
KDELR (gray) or Arf4+5 (black). Secreted and
intracellular ssHRP horseradish peroxidase activ-
ity was measured in both SN and CL. Results are
means ± SD (n = 3).
(C) HeLa-prME-DENV1 cells were transfected with
siRNAs targeting KDELR either individually or in
various combinations. SN and CL were analyzed
by WB to assess RSP secretion (anti-E), release of
chaperones (anti-Bip and anti-calreticulin) and
knockdown of KDELR (KR-10). GAPDH was used
as the loading control across wells. Results are
representative of three independent experiments.
*p < 0.05; **p < 0.005 versus control (NT) siRNAs.
simply due to shortage of chaperones,such as Bip and calreticulin, which arerequired for the assembly of DENV (Lim-jindaporn et al., 2009). This possibility
was excluded by showing that, although both proteins were de-tected in supernatants from cells treated with KDELR1+2 andKDELR1+2+3 siRNAs, their amount in cell lysates was notappreciably modified when compared to controls (Figure 2C).These experiments also show that only KDELR1 and KDELR2were necessary for chaperones retention in the ER (Figure 2C),suggesting that KDELR isoforms assisting DENV1 secretionand retrieving ER-resident proteins were the same. Since KR-10 antibody could not distinguish between the three KDELR (Fig-ure 2C), the efficacy of siRNA targeting each isoform was inde-pendently verified in cells expressing tagged KDELR1-3 (Fig-ure S2C) (Kudelko et al., 2012).
Cell Reports 10, 1496–1507, March 10, 2015 ª2015 The Authors 1499
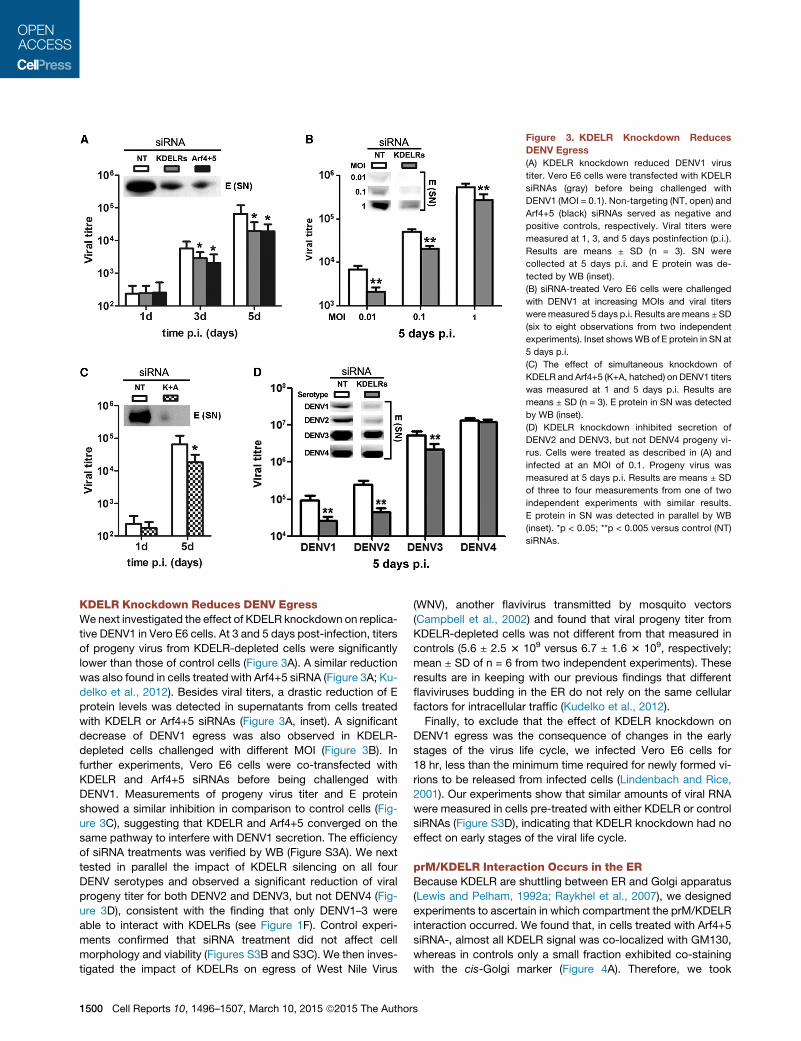
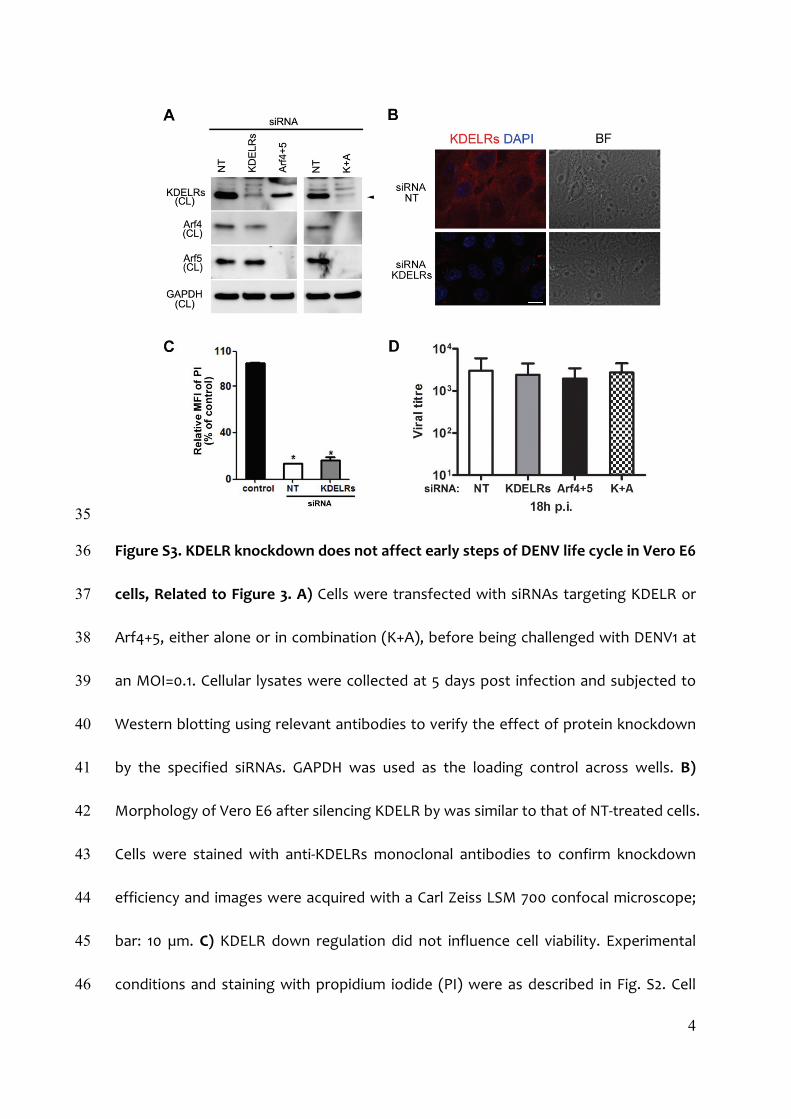
KDELR Knockdown Reduces DENV EgressWenext investigated the effect of KDELR knockdown on replica-tive DENV1 in Vero E6 cells. At 3 and 5 days post-infection, titersof progeny virus from KDELR-depleted cells were significantlylower than those of control cells (Figure 3A). A similar reductionwas also found in cells treated with Arf4+5 siRNA (Figure 3A; Ku-delko et al., 2012). Besides viral titers, a drastic reduction of Eprotein levels was detected in supernatants from cells treatedwith KDELR or Arf4+5 siRNAs (Figure 3A, inset). A significantdecrease of DENV1 egress was also observed in KDELR-depleted cells challenged with different MOI (Figure 3B). Infurther experiments, Vero E6 cells were co-transfected withKDELR and Arf4+5 siRNAs before being challenged withDENV1. Measurements of progeny virus titer and E proteinshowed a similar inhibition in comparison to control cells (Fig-ure 3C), suggesting that KDELR and Arf4+5 converged on thesame pathway to interfere with DENV1 secretion. The efficiencyof siRNA treatments was verified by WB (Figure S3A). We nexttested in parallel the impact of KDELR silencing on all fourDENV serotypes and observed a significant reduction of viralprogeny titer for both DENV2 and DENV3, but not DENV4 (Fig-ure 3D), consistent with the finding that only DENV1–3 wereable to interact with KDELRs (see Figure 1F). Control experi-ments confirmed that siRNA treatment did not affect cellmorphology and viability (Figures S3B and S3C). We then inves-tigated the impact of KDELRs on egress of West Nile Virus
(WNV), another flavivirus transmitted by mosquito vectors(Campbell et al., 2002) and found that viral progeny titer fromKDELR-depleted cells was not different from that measured incontrols (5.6 ± 2.5 3 109 versus 6.7 ± 1.6 3 109, respectively;mean ± SD of n = 6 from two independent experiments). Theseresults are in keeping with our previous findings that differentflaviviruses budding in the ER do not rely on the same cellularfactors for intracellular traffic (Kudelko et al., 2012).Finally, to exclude that the effect of KDELR knockdown on
DENV1 egress was the consequence of changes in the earlystages of the virus life cycle, we infected Vero E6 cells for18 hr, less than the minimum time required for newly formed vi-rions to be released from infected cells (Lindenbach and Rice,2001). Our experiments show that similar amounts of viral RNAwere measured in cells pre-treated with either KDELR or controlsiRNAs (Figure S3D), indicating that KDELR knockdown had noeffect on early stages of the viral life cycle.
prM/KDELR Interaction Occurs in the ERBecause KDELR are shuttling between ER and Golgi apparatus(Lewis and Pelham, 1992a; Raykhel et al., 2007), we designedexperiments to ascertain in which compartment the prM/KDELRinteraction occurred. We found that, in cells treated with Arf4+5siRNA-, almost all KDELR signal was co-localized with GM130,whereas in controls only a small fraction exhibited co-stainingwith the cis-Golgi marker (Figure 4A). Therefore, we took
Figure 3. KDELR Knockdown ReducesDENV Egress(A) KDELR knockdown reduced DENV1 virus
titer. Vero E6 cells were transfected with KDELR
siRNAs (gray) before being challenged with
DENV1 (MOI = 0.1). Non-targeting (NT, open) and
Arf4+5 (black) siRNAs served as negative and
positive controls, respectively. Viral titers were
measured at 1, 3, and 5 days postinfection (p.i.).
Results are means ± SD (n = 3). SN were
collected at 5 days p.i. and E protein was de-
tected by WB (inset).
(B) siRNA-treated Vero E6 cells were challenged
with DENV1 at increasing MOIs and viral titers
were measured 5 days p.i. Results are means ±SD
(six to eight observations from two independent
experiments). Inset showsWB of E protein in SN at
5 days p.i.
(C) The effect of simultaneous knockdown of
KDELR and Arf4+5 (K+A, hatched) on DENV1 titers
was measured at 1 and 5 days p.i. Results are
means ± SD (n = 3). E protein in SN was detected
by WB (inset).
(D) KDELR knockdown inhibited secretion of
DENV2 and DENV3, but not DENV4 progeny vi-
rus. Cells were treated as described in (A) and
infected at an MOI of 0.1. Progeny virus was
measured at 5 days p.i. Results are means ± SD
of three to four measurements from one of two
independent experiments with similar results.
E protein in SN was detected in parallel by WB
(inset). *p < 0.05; **p < 0.005 versus control (NT)
siRNAs.
1500 Cell Reports 10, 1496–1507, March 10, 2015 ª2015 The Authors

advantage of this observation, which identifies the cis-Golgi asthe peri-nuclear region where KDELRwas sequestered followingArf4+5 depletion (Volpicelli-Daley et al., 2005), to perform coIP inArf4+5 knockdown cells. Despite similar expression levels in allexperimental conditions, KDELR could be precipitated onlyfrom control, but not Arf4+5 depleted cells (Figure 4B), indicatingthat prM/KDELR interaction occurred in the ER.
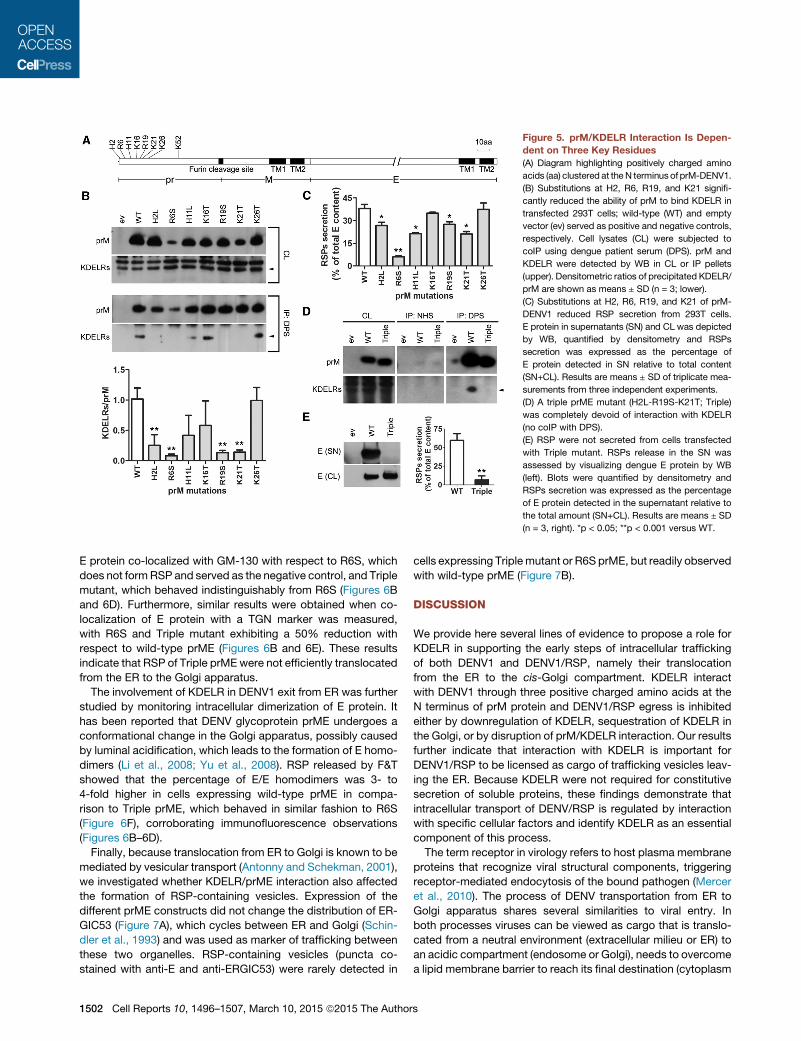
H2, R19, and K21 Are Key Residues for prM/KDELRInteractionTo identify the putative region of prM interacting with KDELR, weanalyzed theN-terminal sequence of the pr fragment and noted ahigh proportion (seven of 26 residues) of positively chargedamino acids (Figure 5A) that were conserved in DENV1–3,whereas substitutions at residues H2 and H11 were present inDENV4 (Figure S4A). To test the role of this cluster of basic res-idues we generated prME-DENV1 constructs with neutral aminoacids substitutions (Figure 5A). Mutations did not affect expres-sion of KDELR, prM, and E, with the exception of R6S, whichreduced the levels of both viral proteins when compared towild-type prME (Figure 5B, upper; and Figure S4B). Cell lysateswere subjected to coIP and the ability of mutant prM to interactwith KDELR was determined by calculating the ratio betweenprecipitated KDELR and prM, which was then normalized tothat measured for wild-type prM (Figure 5B, middle). H2L, R6S,R19S, and K21T significantly reduced prM binding to KDELR(Figure 5B, lower) and release of RSP (Figure 5C), demonstratinga positive correlation between prM/KDELR interaction and RSPsecretion.prM/E interaction is critical for the formation of DENV (Coura-
geot et al., 2000): prM functions as the chaperone of E and its R6residue is predicted to be important for both interaction and viralassembly (Li et al., 2008). To test the effect of the mutated con-structs on prM/E interaction, we performed coIP assays on ly-sates of 293T cells and found that similar levels of prM proteincould be precipitated for all mutants with the exception of R6S(Figure S4B). These observations, while confirming the predictedinvolvement of R6 (Li et al., 2008), demonstrate that none of theother mutated residues was involved in the interaction betweenprM and E.Based on the results of coIP (Figure 5B) and RSP secretion
(Figure 5C), we generated a triple mutant (H2L/R19S/K21T)designated hereinafter as ‘‘Triple.’’ The Triple mutation did not
alter the ability to bind Arf4 and Arf5 (Figure S5), but abrogatedinteraction with KDELRs (Figure 5D) and blocked the secretionof RSP by 90% (Figure 5E), demonstrating the crucial role ofH2, R19, and K21 residues for prM interaction with KDELR andDENV1 secretion.
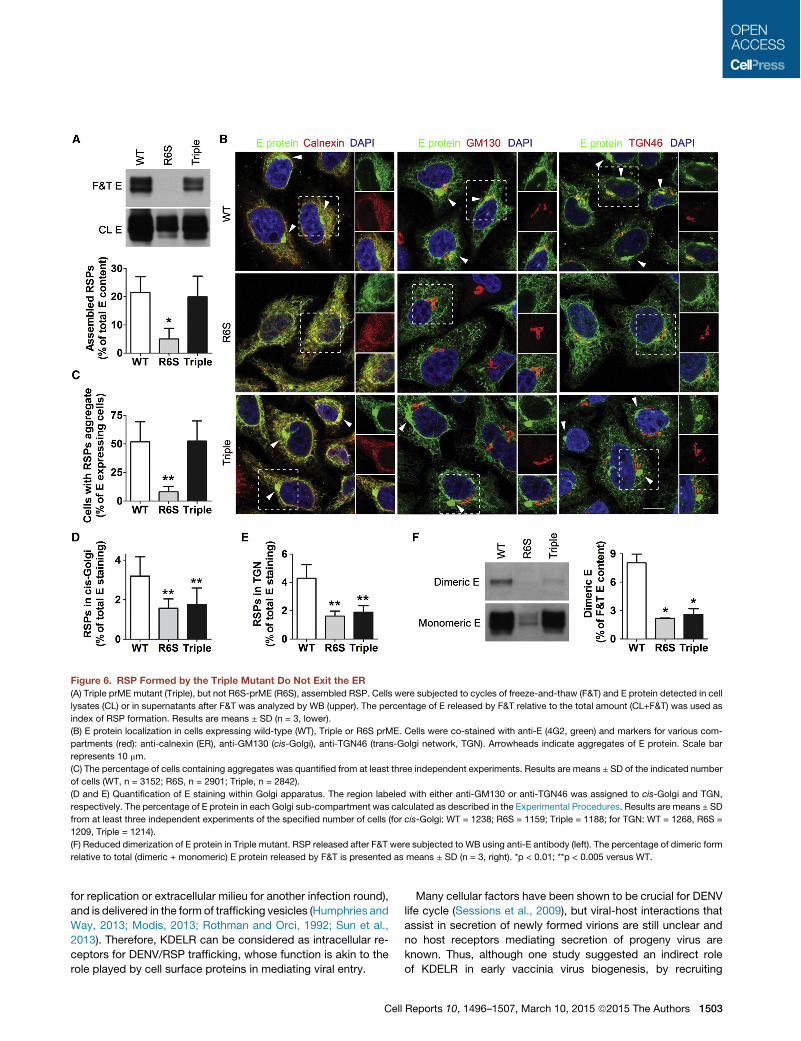
Triple Mutant prME Forms RSP and Is Translocatedwithin the ERTo exclude the possibility that disruption of prM/KDELR inter-action had compromised the assembly and formation of viralparticles, we performed freeze-and-thaw (F&T) experiments,which have been shown to release intracellular viruses fromhost cells (Burleson et al., 1992). Upon repeated cycles of F&T,cells expressing R6S released barely detectable levels of RSP(Figure 6A), confirming that disruption of prM/E interactioninhibited RSP formation. In contrast, similar amounts of RSPwere released from cells expressing either wild-type or TripleprME (Figure 6A), indicating that prM/KDELR interaction wasdispensable for the formation of viral particles and further sug-gesting that inhibition of DENV1 and RSP release was the conse-quence of a trafficking defect.Next, we studied the role of prM/KDELR interaction in RSP
traffic within the ER by immunofluorescence microscopy of cellsco-stained with anti-E as well as antibodies labeling ER, cis-Golgi and TGN. It has been observed that newly assembledDENV translocate within the ER to accumulate in dilatedcisternae oriented toward the cis-Golgi (Welsch et al., 2009)and, therefore, we used E protein aggregation as an index ofRSP trafficking within the ER. Aggregates were found next tothe Golgi apparatus in 50% of cells expressing either wild-typeor Triple mutant prME, but were hardly observed in cells stablytransfected with R6S (Figures 6B and 6C), suggesting that trans-port of newly assembled RSP inside ER lumen did not requireinteraction with KDELR. Similar aggregates were observed inDENV1-infected Vero E6 cells using either a polyclonal antibodyagainst prME or the anti-E monoclonal antibody 4G2 (Figure S6).
Triple Mutant RSP Cannot Exit from ERTo investigate whether interaction with KDELR was requiredfor DENV1 exit from ER, translocation of RSP to the cis-Golgiwas determined by measuring the percentage of total E proteinco-localized with GM130. We found that in cells expressingwild-type prME there was a significantly higher percentage of
Figure 4. prM/KDELR Interaction Occurs inthe ER(A) Depletion of Arf4+5 by siRNA blocked retrieval
of KDELR from Golgi. HeLa cells expressing
KDELR1-RFP (red) were transfected with Arf4+5
siRNA and stained with anti-GM130 antibody
(green). Non-targeting (NT) siRNA was used as
control. Scale bar represents 10 mm.
(B) KDELR sequestered in Golgi were not precipi-
tated by dengue patient serum (DPS). HeLa-prME-
DENV1 cells transfected with Arf4+5 or NT siRNAs
were subjected to coIP using DPS or normal human
serum (NHS). Cell lysates (CL) and pellets from
immunoprecipitation (IP) were analyzed by WB.
Results are representative of three independent
experiments.
Cell Reports 10, 1496–1507, March 10, 2015 ª2015 The Authors 1501
E protein co-localized with GM-130 with respect to R6S, whichdoes not formRSP and served as the negative control, and Triplemutant, which behaved indistinguishably from R6S (Figures 6Band 6D). Furthermore, similar results were obtained when co-localization of E protein with a TGN marker was measured,with R6S and Triple mutant exhibiting a 50% reduction withrespect to wild-type prME (Figures 6B and 6E). These resultsindicate that RSP of Triple prMEwere not efficiently translocatedfrom the ER to the Golgi apparatus.
The involvement of KDELR in DENV1 exit from ER was furtherstudied by monitoring intracellular dimerization of E protein. Ithas been reported that DENV glycoprotein prME undergoes aconformational change in the Golgi apparatus, possibly causedby luminal acidification, which leads to the formation of E homo-dimers (Li et al., 2008; Yu et al., 2008). RSP released by F&Tshowed that the percentage of E/E homodimers was 3- to4-fold higher in cells expressing wild-type prME in compa-rison to Triple prME, which behaved in similar fashion to R6S(Figure 6F), corroborating immunofluorescence observations(Figures 6B–6D).
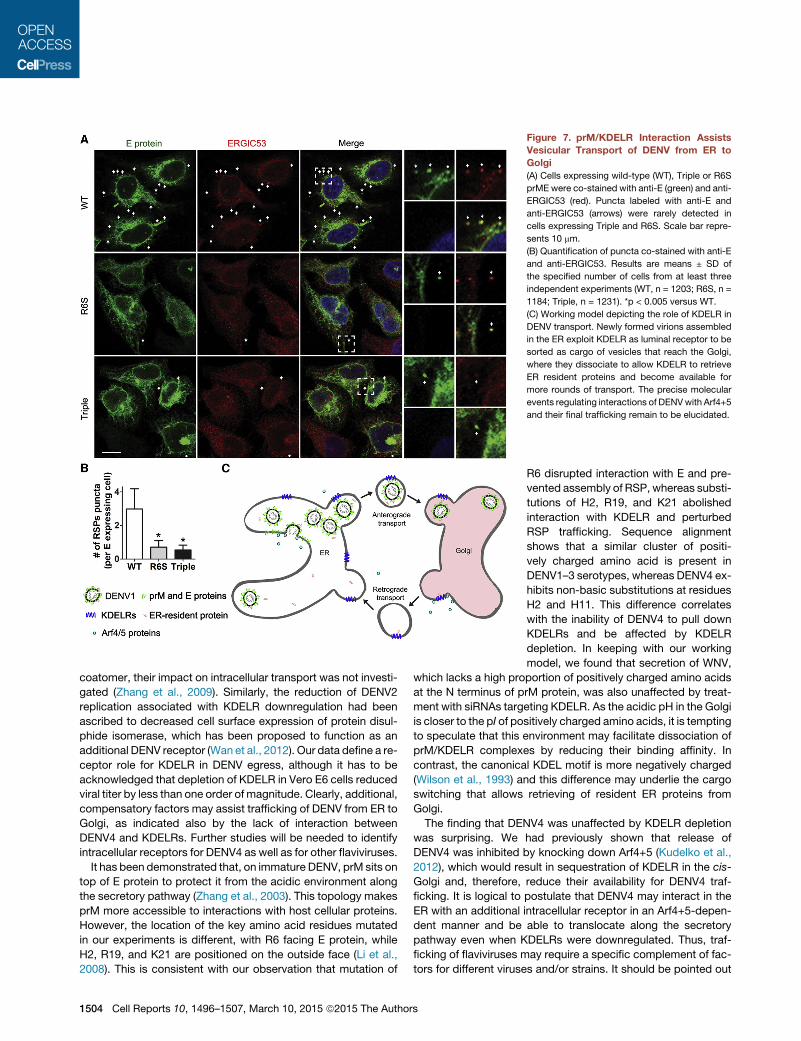
Finally, because translocation from ER to Golgi is known to bemediated by vesicular transport (Antonny and Schekman, 2001),we investigated whether KDELR/prME interaction also affectedthe formation of RSP-containing vesicles. Expression of thedifferent prME constructs did not change the distribution of ER-GIC53 (Figure 7A), which cycles between ER and Golgi (Schin-dler et al., 1993) and was used as marker of trafficking betweenthese two organelles. RSP-containing vesicles (puncta co-stained with anti-E and anti-ERGIC53) were rarely detected in
cells expressing Triplemutant or R6S prME, but readily observedwith wild-type prME (Figure 7B).
DISCUSSION
We provide here several lines of evidence to propose a role forKDELR in supporting the early steps of intracellular traffickingof both DENV1 and DENV1/RSP, namely their translocationfrom the ER to the cis-Golgi compartment. KDELR interactwith DENV1 through three positive charged amino acids at theN terminus of prM protein and DENV1/RSP egress is inhibitedeither by downregulation of KDELR, sequestration of KDELR inthe Golgi, or by disruption of prM/KDELR interaction. Our resultsfurther indicate that interaction with KDELR is important forDENV1/RSP to be licensed as cargo of trafficking vesicles leav-ing the ER. Because KDELR were not required for constitutivesecretion of soluble proteins, these findings demonstrate thatintracellular transport of DENV/RSP is regulated by interactionwith specific cellular factors and identify KDELR as an essentialcomponent of this process.The term receptor in virology refers to host plasma membrane
proteins that recognize viral structural components, triggeringreceptor-mediated endocytosis of the bound pathogen (Merceret al., 2010). The process of DENV transportation from ER toGolgi apparatus shares several similarities to viral entry. Inboth processes viruses can be viewed as cargo that is translo-cated from a neutral environment (extracellular milieu or ER) toan acidic compartment (endosome or Golgi), needs to overcomea lipid membrane barrier to reach its final destination (cytoplasm
Figure 5. prM/KDELR Interaction Is Depen-dent on Three Key Residues(A) Diagram highlighting positively charged amino
acids (aa) clustered at theN terminus of prM-DENV1.
(B) Substitutions at H2, R6, R19, and K21 signifi-
cantly reduced the ability of prM to bind KDELR in
transfected 293T cells; wild-type (WT) and empty
vector (ev) served as positive and negative controls,
respectively. Cell lysates (CL) were subjected to
coIP using dengue patient serum (DPS). prM and
KDELR were detected by WB in CL or IP pellets
(upper). Densitometric ratios of precipitated KDELR/
prM are shown as means ± SD (n = 3; lower).
(C) Substitutions at H2, R6, R19, and K21 of prM-
DENV1 reduced RSP secretion from 293T cells.
E protein in supernatants (SN) and CL was depicted
by WB, quantified by densitometry and RSPs
secretion was expressed as the percentage of
E protein detected in SN relative to total content
(SN+CL). Results are means ± SD of triplicate mea-
surements from three independent experiments.
(D) A triple prME mutant (H2L-R19S-K21T; Triple)
was completely devoid of interaction with KDELR
(no coIP with DPS).
(E) RSP were not secreted from cells transfected
with Triple mutant. RSPs release in the SN was
assessed by visualizing dengue E protein by WB
(left). Blots were quantified by densitometry and
RSPs secretion was expressed as the percentage
of E protein detected in the supernatant relative to
the total amount (SN+CL). Results are means ± SD
(n = 3, right). *p < 0.05; **p < 0.001 versus WT.
1502 Cell Reports 10, 1496–1507, March 10, 2015 ª2015 The Authors
for replication or extracellular milieu for another infection round),and is delivered in the form of trafficking vesicles (Humphries andWay, 2013; Modis, 2013; Rothman and Orci, 1992; Sun et al.,2013). Therefore, KDELR can be considered as intracellular re-ceptors for DENV/RSP trafficking, whose function is akin to therole played by cell surface proteins in mediating viral entry.
Many cellular factors have been shown to be crucial for DENVlife cycle (Sessions et al., 2009), but viral-host interactions thatassist in secretion of newly formed virions are still unclear andno host receptors mediating secretion of progeny virus areknown. Thus, although one study suggested an indirect roleof KDELR in early vaccinia virus biogenesis, by recruiting
Figure 6. RSP Formed by the Triple Mutant Do Not Exit the ER(A) Triple prME mutant (Triple), but not R6S-prME (R6S), assembled RSP. Cells were subjected to cycles of freeze-and-thaw (F&T) and E protein detected in cell
lysates (CL) or in supernatants after F&T was analyzed by WB (upper). The percentage of E released by F&T relative to the total amount (CL+F&T) was used as
index of RSP formation. Results are means ± SD (n = 3, lower).
(B) E protein localization in cells expressing wild-type (WT), Triple or R6S prME. Cells were co-stained with anti-E (4G2, green) and markers for various com-
partments (red): anti-calnexin (ER), anti-GM130 (cis-Golgi), anti-TGN46 (trans-Golgi network, TGN). Arrowheads indicate aggregates of E protein. Scale bar
represents 10 mm.
(C) The percentage of cells containing aggregates was quantified from at least three independent experiments. Results are means ± SD of the indicated number
of cells (WT, n = 3152; R6S, n = 2901; Triple, n = 2842).
(D and E) Quantification of E staining within Golgi apparatus. The region labeled with either anti-GM130 or anti-TGN46 was assigned to cis-Golgi and TGN,
respectively. The percentage of E protein in each Golgi sub-compartment was calculated as described in the Experimental Procedures. Results are means ± SD
from at least three independent experiments of the specified number of cells (for cis-Golgi: WT = 1238; R6S = 1159; Triple = 1188; for TGN: WT = 1268, R6S =
1209, Triple = 1214).
(F) Reduced dimerization of E protein in Triple mutant. RSP released after F&T were subjected to WB using anti-E antibody (left). The percentage of dimeric form
relative to total (dimeric + monomeric) E protein released by F&T is presented as means ± SD (n = 3, right). *p < 0.01; **p < 0.005 versus WT.
Cell Reports 10, 1496–1507, March 10, 2015 ª2015 The Authors 1503
coatomer, their impact on intracellular transport was not investi-gated (Zhang et al., 2009). Similarly, the reduction of DENV2replication associated with KDELR downregulation had beenascribed to decreased cell surface expression of protein disul-phide isomerase, which has been proposed to function as anadditional DENV receptor (Wan et al., 2012). Our data define a re-ceptor role for KDELR in DENV egress, although it has to beacknowledged that depletion of KDELR in Vero E6 cells reducedviral titer by less than one order of magnitude. Clearly, additional,compensatory factors may assist trafficking of DENV from ER toGolgi, as indicated also by the lack of interaction betweenDENV4 and KDELRs. Further studies will be needed to identifyintracellular receptors for DENV4 as well as for other flaviviruses.
It has been demonstrated that, on immature DENV, prM sits ontop of E protein to protect it from the acidic environment alongthe secretory pathway (Zhang et al., 2003). This topology makesprM more accessible to interactions with host cellular proteins.However, the location of the key amino acid residues mutatedin our experiments is different, with R6 facing E protein, whileH2, R19, and K21 are positioned on the outside face (Li et al.,2008). This is consistent with our observation that mutation of
Figure 7. prM/KDELR Interaction AssistsVesicular Transport of DENV from ER toGolgi(A) Cells expressing wild-type (WT), Triple or R6S
prME were co-stained with anti-E (green) and anti-
ERGIC53 (red). Puncta labeled with anti-E and
anti-ERGIC53 (arrows) were rarely detected in
cells expressing Triple and R6S. Scale bar repre-
sents 10 mm.
(B) Quantification of puncta co-stained with anti-E
and anti-ERGIC53. Results are means ± SD of
the specified number of cells from at least three
independent experiments (WT, n = 1203; R6S, n =
1184; Triple, n = 1231). *p < 0.005 versus WT.
(C) Working model depicting the role of KDELR in
DENV transport. Newly formed virions assembled
in the ER exploit KDELR as luminal receptor to be
sorted as cargo of vesicles that reach the Golgi,
where they dissociate to allow KDELR to retrieve
ER resident proteins and become available for
more rounds of transport. The precise molecular
events regulating interactions of DENVwith Arf4+5
and their final trafficking remain to be elucidated.
R6 disrupted interaction with E and pre-vented assembly of RSP, whereas substi-tutions of H2, R19, and K21 abolishedinteraction with KDELR and perturbedRSP trafficking. Sequence alignmentshows that a similar cluster of positi-vely charged amino acid is present inDENV1–3 serotypes, whereas DENV4 ex-hibits non-basic substitutions at residuesH2 and H11. This difference correlateswith the inability of DENV4 to pull downKDELRs and be affected by KDELRdepletion. In keeping with our workingmodel, we found that secretion of WNV,
which lacks a high proportion of positively charged amino acidsat the N terminus of prM protein, was also unaffected by treat-ment with siRNAs targeting KDELR. As the acidic pH in the Golgiis closer to the pI of positively charged amino acids, it is temptingto speculate that this environment may facilitate dissociation ofprM/KDELR complexes by reducing their binding affinity. Incontrast, the canonical KDEL motif is more negatively charged(Wilson et al., 1993) and this difference may underlie the cargoswitching that allows retrieving of resident ER proteins fromGolgi.The finding that DENV4 was unaffected by KDELR depletion
was surprising. We had previously shown that release ofDENV4 was inhibited by knocking down Arf4+5 (Kudelko et al.,2012), which would result in sequestration of KDELR in the cis-Golgi and, therefore, reduce their availability for DENV4 traf-ficking. It is logical to postulate that DENV4 may interact in theER with an additional intracellular receptor in an Arf4+5-depen-dent manner and be able to translocate along the secretorypathway even when KDELRs were downregulated. Thus, traf-ficking of flaviviruses may require a specific complement of fac-tors for different viruses and/or strains. It should be pointed out
1504 Cell Reports 10, 1496–1507, March 10, 2015 ª2015 The Authors

that, although the Triple mutant was mainly localized in the ER, itwas still able to pull down both Arf4 and Arf5, confirming thatbinding to Arf4+5 and KDELR in the ER are independent events.Arf4+5 are localized at both Golgi and ER (Duijsings et al., 2009)and may play two crucial roles for DENV secretion, by beinginvolved in KDELR recycling and interacting with prM protein.Further experiments will be required to ascertain the preciselocation and role of class II Arf/prM interaction in DENVtrafficking.The function of prM in DENV biology is attracting more atten-
tion. Thus, prM has been recently shown to interact with the lightchain Tctex-1 of dynein and play a role in late stages of virusreplication (Brault et al., 2011). We demonstrate here that prMinteracts with KDELR during virus secretion. Our working hy-pothesis is that DENV1–3 use unoccupied KDELR, which arerecognized by a binding motif in the N terminus of prM, to exitthe ER as cargo of vesicles en route to the Golgi apparatus (Fig-ure 7C). We had previously characterized the function of class IIArf proteins in DENV/RSP egress (Kudelko et al., 2012). Simulta-neous depletion of Arf4+5 efficiently sequester intracellularKDELR in the Golgi and, therefore, it is logical to postulate thatboth factors converge on the same pathway to inhibit DENV/RSP secretion (Figure 7C). However, results with DENV4 andWNV suggest that additional host proteins are specificallyinvolved in sorting flaviviruses through late secretory compart-ments and assisting their release from infected cells. In recentyears, evidence has accumulated to suggest that, besides theirwell-established function in retrieving chaperones, KDELR canbe activated by cargo to trigger signaling pathways that regulateanterograde and retrograde traffic (Giannotta et al., 2012; Pulvir-enti et al., 2008). Specifically, it has been proposed that KDELRsrecognize chaperones that are carried by ER vesicles en route toGolgi (Cancino et al., 2013). It is tempting to speculate, therefore,that during DENV1–3 biogenesis, newly formed virions bind toKDELR to activate cell signaling pathways that facilitate theirtranslocation to the Golgi.
EXPERIMENTAL PROCEDURES
Cells, viruses, antibodies, and siRNA experiments are described in the Supple-
mental Experimental Procedures. Primers used for RT-PCR, GST pull-down,
and site-directed mutagenesis are shown in Tables S1, S2, and S3,
respectively.
Protein Analysis and RSP QuantificationGel electrophoresis and WB analysis were carried out as previously described
(Kudelko et al., 2012) To quantify RSP secretion, the area and mean lumines-
cence signals detected by WB in supernatants (SN) and cellular lysates (CL)
were measured by densitometry using Image Quant TL (Thermo Fisher). For
each condition, the relative amount of secreted RSP (E signal in SN) was calcu-
lated as the percentage of total signal (ESN/ESN+ECL).
Virus Infection ExperimentsViral stocks of DENV1-4 andWNV were titrated by determining the tissue cul-
ture infective dose 50% (TCID50/ml) in Vero E6 cells challenged with 10-fold
serial dilutions of infectious supernatants for 90 min at 37!C. Cells were
subsequently incubated in DMEM with 2.5% fetal calf serum. At 5–7 days
postinfection for DENV1–3 and 3–5 days postinfection for DENV4 and
WNV, culture supernatant was removed and cell monolayers were fixed in
4% formaldehyde. The percentage of cytopathic effects was used to calcu-
late the viral titer.
For measurements of progeny virus production, viral RNA was extracted
from culture supernatants and quantified by real-time RT-PCR (see the Sup-
plemental Experimental Procedures). The amount of viral RNA transcripts
was then calculated by generating a standard curve with 10-fold dilutions of
RNA isolated from a known amount of DENV1 stock and expressed as
TCID50/ml, as described above.
GST Pull-Down AssayFragments of the prM sequence of DENV1 were amplified by PCR (Table S2).
Ampliconswere subcloned in frame into the bacterial expression vector pGEX-
4T-1 to produce N-terminal tagged GST constructs (see the Supplemental
Experimental Procedures). Twenty micrograms of each purified protein bound
to sepharose 4B-glutathione beads was mixed with lysates of HeLa cells sta-
bly expressing cMyc-KDELR, incubated overnight at 4!C, and extensively
washed before eluting bound proteins, according to the manufacturer’s in-
structions, for WB analysis.
CoimmunoprecipitationSub-confluent monolayers of HeLa-prME-DENV1 or 293T cells transfected
with the specified constructs were lysed on ice for 30min with 1ml RIPA buffer,
supplemented with freshly added 1 mM PMSF and protease inhibitors cock-
tail. Cell debris were removed by centrifugation at 13,000 rpm for 15 min at
4!C and lysates were pre-cleared by incubation with 30 ml of 50% protein G
sepharose beads (Amersham Pharmacia) for 1 hr. Pre-cleared lysates
(400 ul) were then incubated for 2 hr at 4!Cwith additional 30 ml of 50% protein
G sepharose beads previously treatedwith either specific antibodies or control
IgGs. Beads were then pelleted by centrifugation at 13,000 rpm for 30 s at 4!C
and bound proteins were eluted by boiling in gel loading buffer, separated by
electrophoresis and analyzed with WB.
Freeze-and-Thaw AssayFor subcellular fractionation (Xu et al., 1997), sub-confluent HeLa cells stably
expressing either wild-type prME-DENV1 or the specified mutants were first
detached in PBS plus 5 mM EDTA at 37!C for 5 min and washed three times
on icewith PBS supplementedwith 1mMEGTA. Cells were then re-suspended
in a buffer containing 10% wieght/vol sucrose, 20 mM Tris HCl, 150 mM NaCl,
10 mM magnesium acetate, 1 mM EGTA (pH 7.6) supplemented with freshly
added 1 mM PMSF and protease inhibitors cocktail, and then subjected to
eight cycles of freeze (dry ice) and thaw (37!C water bath), 1 min each step.
Nuclei and cellular debris were removed by a short (5 s) spin atmaximumspeed
in a bench-top centrifuge at 4!C. Supernatants were collected and centrifuged
for 30 min at maximum speed at 4!C to pellet the membrane fraction. The final
supernatants, containing newly formed RSP, were analyzed with WB.
Fluorescence MicroscopyFor fluorescence microscopy, cells grown on glass coverslips were fixed, per-
meabilized, and incubated with primary antibodies (see the Supplemental
Experimental Procedures). Samples were then probed with appropriate sec-
ondary antibodies conjugated with fluorescein isothiocyanate or Texas Red
(both from Life Technologies). Nuclei were stained with DAPI and coverslips
were mounted on glass slides for image acquisition using either an Axio
Observer Z1 inverted microscope or an LSM 700 confocal microscope (Carl
Zeiss).
Quantitative Analysis of Fluorescent ImagesTo extract and quantify cells stained with the viral E protein, we developed
a specific protocol, ‘‘Stained cells,’’ in the ICY software (http://icy.
bioimageanalysis.org) (de Chaumont et al., 2012). To extract and quantify cells
that containedRSP aggregates, we developed a separate protocol, ‘‘Cells with
aggregates,’’ in the ICY software. Details of these protocols are provided in the
Supplemental Experimental Procedures.
To determine RSP localization in the Golgi apparatus, weighted co-localiza-
tion coefficients of E with Golgi markers were computed using the ZEN2011
co-localization coefficient software (Carl Zeiss). The sums of intensities of
pixels corresponding to anti-E (So) and to co-staining with anti-E and either
cis-Golgi or TGN marker (Sc) were computed and then weighted co-localiza-
tion coefficients, which are equal to the ratio of Sc to So, were used to
Cell Reports 10, 1496–1507, March 10, 2015 ª2015 The Authors 1505

represent the percentage of RSP translocated to either cis-Golgi or TGN.
To determine the number of RSP-containing vesicles, we manually counted
(in blind) puncta co-labeled with anti-E and anti-ERGIC that were adjacent to
perinuclear E-staining. The total number of double-labeled puncta per field
was then calculated, divided by the number of cells expressing E protein
and displayed as the number of puncta per cell. Data sets for quantitative anal-
ysis were acquired from an average of 40–50 fields from four to five indepen-
dent experiments for each condition.
Statistical AnalysisResults are shown asmeans ±SD. Statistical significancewas analyzed by the
Student’s unpaired t test, with a confidence limit for significance set at 0.05 or
less.
SUPPLEMENTAL INFORMATION
Supplemental Information includes Supplemental Experimental Procedures,
six figures, and three tables and can be found with this article online at
http://dx.doi.org/10.1016/j.celrep.2015.02.021.
AUTHOR CONTRIBUTIONS
M.Y.L., K.K., Y.L.S., and M.K. performed the experiments on prM/KDELR
interaction. M.Y.L., J.S.Z., and K.S. performed the experiments with replicable
dengue viruses. T.L. and J.-C.O.-M. designed the software for co-localization
analysis. C.F.Q. produced and purified the monoclonal antibody. M.G., J.-C.
O.-M., R.B., and P.G.W. analyzed the results. R.B. and P.G.W. prepared and
revised the manuscript.
ACKNOWLEDGMENTS
HKU-Pasteur Research Pole is a member of the Institut Pasteur International
Network. We thank V. Malhotra (Center for Genomic Regulation, Barcelona)
for the gift of soluble horseradish peroxidase construct; R. Gijsbers (Katholieke
Universiteit, Leuven) for the retroviral vector; P. Despres (Institut Pasteur,
Paris) for the anti-E antibody 4E11; P. Buchy (Institut Pasteur du Cambodge,
Phnom Penh) and M. Dupont-Rouzeyrol (Institut Pasteur de Nouvelle-Caledo-
nie, Noumea) formouse anti-prME antibody and dengue patients sera; S. Nop-
porn (Chiang Mai University, Thailand) for anti-prM antibody prM-6.1; P.H.M.
Leung (The Hong Kong Polytechnic University, Hong Kong SAR) for the anti-
prME polyclonal antibody; J. Guo and the Faculty Core Facility of the LKS
Faculty of Medicine of the University of Hong Kong for support with confocal
microscopy; P. Chavrier (Institut Curie, Paris), S. Sanyal (HKU-Pasteur
Research Pole), and C. Zurzolo (Institut Pasteur, Paris) for critical comments;
and members of HKU-Pasteur for useful discussion and technical advice.
This work was funded by RFCID (grant #10091312 to P.G.W.), Institut Pasteur
International Network (ACIP A-12-10 to P.G.W. andM.G.), BNP Paribas CIB (to
R.B.), and ANR-10-INBS-04-06 FranceBioImaging and ANR-10-LABX-62-
IBEID (to T.L. and J.-C.O.-M.). M.Y.L. was supported in part by The University
of Hong Kong through a postgraduate scholarship and by the LKS Faculty of
Medicine through a Research Postgraduate Student Exchange Scheme. T.L.
was funded by a Roux fellowship from Institut Pasteur.
Received: April 23, 2014
Revised: October 16, 2014
Accepted: February 4, 2015
Published: March 5, 2015
REFERENCES
Antonny, B., and Schekman, R. (2001). ER export: public transportation by the
COPII coach. Curr. Opin. Cell Biol. 13, 438–443.
Bard, F., Casano, L., Mallabiabarrena, A., Wallace, E., Saito, K., Kitayama, H.,
Guizzunti, G., Hu, Y., Wendler, F., Dasgupta, R., et al. (2006). Functional geno-
mics reveals genes involved in protein secretion and Golgi organization.
Nature 439, 604–607.
Brault, J.B., Kudelko, M., Vidalain, P.O., Tangy, F., Despres, P., and Pardigon,
N. (2011). The interaction of flavivirus M protein with light chain Tctex-1 of
human dynein plays a role in late stages of virus replication. Virology 417,
369–378.
Burleson, P.G., Chambers, T.M., and Wiedbrauk, D.L. (1992). Virology: A Lab-
oratory Manual (San Diego, CA: Academic Press).
Campbell, G.L., Marfin, A.A., Lanciotti, R.S., and Gubler, D.J. (2002). West Nile
virus. Lancet Infect. Dis. 2, 519–529.
Cancino, J., Jung, J.E., and Luini, A. (2013). Regulation of Golgi signaling and
trafficking by the KDEL receptor. Histochem. Cell Biol. 140, 395–405.
Chen, Y., Maguire, T., Hileman, R.E., Fromm, J.R., Esko, J.D., Linhardt, R.J.,
and Marks, R.M. (1997). Dengue virus infectivity depends on envelope protein
binding to target cell heparan sulfate. Nat. Med. 3, 866–871.
Courageot, M.P., Frenkiel, M.P., Dos Santos, C.D., Deubel, V., andDespres, P.
(2000). Alpha-glucosidase inhibitors reduce dengue virus production by
affecting the initial steps of virion morphogenesis in the endoplasmic reticu-
lum. J. Virol. 74, 564–572.
D’Souza-Schorey, C., and Chavrier, P. (2006). ARF proteins: roles in mem-
brane traffic and beyond. Nat. Rev. Mol. Cell Biol. 7, 347–358.
de Chaumont, F., Dallongeville, S., Chenouard, N., Herve, N., Pop, S., Pro-
voost, T., Meas-Yedid, V., Pankajakshan, P., Lecomte, T., Le Montagner, Y.,
et al. (2012). Icy: an open bioimage informatics platform for extended repro-
ducible research. Nat. Methods 9, 690–696.
Duijsings, D., Lanke, K.H.W., van Dooren, S.H.J., van Dommelen, M.M.T.,
Wetzels, R., de Mattia, F., Wessels, E., and van Kuppeveld, F.J.M. (2009). Dif-
ferential membrane association properties and regulation of class I and class II
Arfs. Traffic 10, 316–323.
Giannotta, M., Ruggiero, C., Grossi, M., Cancino, J., Capitani, M., Pulvirenti,
T., Consoli, G.M., Geraci, C., Fanelli, F., Luini, A., and Sallese, M. (2012).
The KDEL receptor couples to Gaq/11 to activate Src kinases and regulate
transport through the Golgi. EMBO J. 31, 2869–2881.
Guzman, M.G., Halstead, S.B., Artsob, H., Buchy, P., Farrar, J., Gubler, D.J.,
Hunsperger, E., Kroeger, A., Margolis, H.S., Martınez, E., et al. (2010). Dengue:
a continuing global threat. Nat. Rev. Microbiol. 8 (12, Suppl), S7–S16.
Hsu, V.W., Shah, N., and Klausner, R.D. (1992). A brefeldin A-like phenotype is
induced by the overexpression of a human ERD-2-like protein, ELP-1. Cell 69,
625–635.
Humphries, A.C., and Way, M. (2013). The non-canonical roles of clathrin and
actin in pathogen internalization, egress and spread. Nat. Rev. Microbiol. 11,
551–560.
Kudelko, M., Brault, J.B., Kwok, K., Li, M.Y., Pardigon, N., Peiris, J.S., Bruz-
zone, R., Despres, P., Nal, B., andWang, P.G. (2012). Class II ADP-ribosylation
factors are required for efficient secretion of dengue viruses. J. Biol. Chem.
287, 767–777.
Kuhn, R.J., Zhang,W., Rossmann, M.G., Pletnev, S.V., Corver, J., Lenches, E.,
Jones, C.T., Mukhopadhyay, S., Chipman, P.R., Strauss, E.G., et al. (2002).
Structure of dengue virus: implications for flavivirus organization, maturation,
and fusion. Cell 108, 717–725.
Lewis, M.J., and Pelham, H.R. (1990). A human homologue of the yeast HDEL
receptor. Nature 348, 162–163.
Lewis, M.J., and Pelham, H.R. (1992a). Ligand-induced redistribution of a hu-
man KDEL receptor from the Golgi complex to the endoplasmic reticulum. Cell
68, 353–364.
Lewis, M.J., and Pelham, H.R. (1992b). Sequence of a second human KDEL
receptor. J. Mol. Biol. 226, 913–916.
Li, L., Lok, S.M., Yu, I.M., Zhang, Y., Kuhn, R.J., Chen, J., and Rossmann, M.G.
(2008). The flavivirus precursor membrane-envelope protein complex: struc-
ture and maturation. Science 319, 1830–1834.
Limjindaporn, T., Wongwiwat, W., Noisakran, S., Srisawat, C., Netsawang, J.,
Puttikhunt, C., Kasinrerk, W., Avirutnan, P., Thiemmeca, S., Sriburi, R., et al.
(2009). Interaction of dengue virus envelope protein with endoplasmic reticu-
lum-resident chaperones facilitates dengue virus production. Biochem. Bio-
phys. Res. Commun. 379, 196–200.
1506 Cell Reports 10, 1496–1507, March 10, 2015 ª2015 The Authors

Lindenbach, B.D., and Rice, C.M. (2001). Flaviviridae: the viruses and their
replication. In Fields virology (Philadelphia, PA: Lippincott Williams & Wilkins),
pp. 991–1041.
Mercer, J., Schelhaas, M., and Helenius, A. (2010). Virus entry by endocytosis.
Annu. Rev. Biochem. 79, 803–833.
Messina, J.P., Brady, O.J., Scott, T.W., Zou, C., Pigott, D.M., Duda, K.A.,
Bhatt, S., Katzelnick, L., Howes, R.E., Battle, K.E., et al. (2014). Global spread
of dengue virus types: mapping the 70 year history. Trends Microbiol. 22,
138–146.
Modis, Y. (2013). Class II fusion proteins. Adv. Exp. Med. Biol. 790, 150–166.
Mukhopadhyay, S., Kuhn, R.J., and Rossmann, M.G. (2005). A structural
perspective of the flavivirus life cycle. Nat. Rev. Microbiol. 3, 13–22.
Pryor, M.J., Azzola, L., Wright, P.J., and Davidson, A.D. (2004). Histidine 39 in
the dengue virus type 2 M protein has an important role in virus assembly.
J. Gen. Virol. 85, 3627–3636.
Pulvirenti, T., Giannotta, M., Capestrano, M., Capitani, M., Pisanu, A., Polish-
chuk, R.S., San Pietro, E., Beznoussenko, G.V., Mironov, A.A., Turacchio, G.,
et al. (2008). A traffic-activated Golgi-based signalling circuit coordinates the
secretory pathway. Nat. Cell Biol. 10, 912–922.
Raykhel, I., Alanen, H., Salo, K., Jurvansuu, J., Nguyen, V.D., Latva-Ranta, M.,
and Ruddock, L. (2007). A molecular specificity code for the three mammalian
KDEL receptors. J. Cell Biol. 179, 1193–1204.
Rothman, J.E., and Orci, L. (1992). Molecular dissection of the secretory
pathway. Nature 355, 409–415.
Schindler, R., Itin, C., Zerial, M., Lottspeich, F., and Hauri, H.P. (1993). ERGIC-
53, a membrane protein of the ER-Golgi intermediate compartment, carries an
ER retention motif. Eur. J. Cell Biol. 61, 1–9.
Sessions, O.M., Barrows, N.J., Souza-Neto, J.A., Robinson, T.J., Hershey,
C.L., Rodgers, M.A., Ramirez, J.L., Dimopoulos, G., Yang, P.L., Pearson,
J.L., and Garcia-Blanco, M.A. (2009). Discovery of insect and human dengue
virus host factors. Nature 458, 1047–1050.
Sun, E., He, J., and Zhuang, X. (2013). Live cell imaging of viral entry. Curr.
Opin. Virol. 3, 34–43.
Volpicelli-Daley, L.A., Li, Y., Zhang, C.J., and Kahn, R.A. (2005). Isoform-selec-
tive effects of the depletion of ADP-ribosylation factors 1-5 on membrane
traffic. Mol. Biol. Cell 16, 4495–4508.
Wan, S.W., Lin, C.F., Lu, Y.T., Lei, H.Y., Anderson, R., and Lin, Y.S. (2012).
Endothelial cell surface expression of protein disulfide isomerase activates
b1 and b3 integrins and facilitates dengue virus infection. J. Cell. Biochem.
113, 1681–1691.
Wang, P.G., Kudelko, M., Lo, J., Siu, L.Y., Kwok, K.T., Sachse, M., Nicholls,
J.M., Bruzzone, R., Altmeyer, R.M., and Nal, B. (2009). Efficient assembly
and secretion of recombinant subviral particles of the four dengue serotypes
using native prM and E proteins. PLoS ONE 4, e8325.
Welsch, S., Miller, S., Romero-Brey, I., Merz, A., Bleck, C.K., Walther, P., Ful-
ler, S.D., Antony, C., Krijnse-Locker, J., and Bartenschlager, R. (2009).
Composition and three-dimensional architecture of the dengue virus replica-
tion and assembly sites. Cell Host Microbe 5, 365–375.
Wilson, D.W., Lewis, M.J., and Pelham, H.R. (1993). pH-dependent binding of
KDEL to its receptor in vitro. J. Biol. Chem. 268, 7465–7468.
Xu, Z., Bruss, V., and Yen, T.S. (1997). Formation of intracellular particles by
hepatitis B virus large surface protein. J. Virol. 71, 5487–5494.
Yu, I.M., Zhang, W., Holdaway, H.A., Li, L., Kostyuchenko, V.A., Chipman,
P.R., Kuhn, R.J., Rossmann, M.G., and Chen, J. (2008). Structure of the imma-
ture dengue virus at low pH primes proteolytic maturation. Science 319, 1834–
1837.
Zhang, Y., Corver, J., Chipman, P.R., Zhang, W., Pletnev, S.V., Sedlak, D.,
Baker, T.S., Strauss, J.H., Kuhn, R.J., and Rossmann, M.G. (2003). Structures
of immature flavivirus particles. EMBO J. 22, 2604–2613.
Zhang, L., Lee, S.Y., Beznoussenko, G.V., Peters, P.J., Yang, J.S., Gilbert,
H.Y., Brass, A.L., Elledge, S.J., Isaacs, S.N., Moss, B., et al. (2009). A role
for the host coatomer and KDEL receptor in early vaccinia biogenesis. Proc.
Natl. Acad. Sci. USA 106, 163–168.
Cell Reports 10, 1496–1507, March 10, 2015 ª2015 The Authors 1507

Cell Reports Supplemental Information
KDEL Receptors Assist Dengue Virus Exit from the Endoplasmic Reticulum Ming Yuan Li, Marc Grandadam, Kevin Kwok, Thibault Lagache, Yu Lam Siu, Jing Shu Zhang, Kouxiong Sayteng, Mateusz Kudelko, Cheng Feng Qin, Jean-Christophe Olivo-Marin, Roberto Bruzzone, and Pei Gang Wang

1
Supplemental Figures and Legends 1
2
3
Figure S1. KDELR interact with DENV1 prM glycoprotein, Related to Figure 1. A) 4
KDELR1 and DENV1‐E protein were partially co‐localised in HeLa cells co‐transfected 5
with prME and KDELR1‐RFP (red). Cells were stained with anti‐E 4G2 antibody (green) 6
and nuclei were visualised with DAPI (blue). Images were acquired with a Zeiss Axio 7
Observer Z1 inverted microscope; bar: 10 µm. B) Co‐immunoprecipitation of prM with 8
KDELR1‐myc. Lysates from 293T cells transfected with empty vector (ev), prME or 9
prME and KDELR1‐myc were subjected to immunoprecipitation using rabbit either an 10
anti‐c‐myc antibody or normal rabbit IgG. Cell lysates (CL) or pellets were analysed by 11
Western blotting using anti‐myc or anti‐prME antibodies. prM/KDELR interaction was 12
observed also when KDELR1 was used as the bait (data not shown). These results are 13
representative of two independent experiments. 14
2
15
Figure S2. KDELR knockdown does not affect Hela‐prME‐DENV1 cell morphology and 16
viability, Related to Figure 2. A) siRNA‐treated Hela‐prME‐DENV1 cells (non‐targeting, 17
NT and KDELR) were re‐suspended in PBS and stained by propidium iodide (PI). Cells 18
subjected to osmotic lysis in ddH2O at 37˚C for 3h were used as the positive control. 19
Cell viability was analysed by flow cytometry and mean fluorescence intensity (MFI) of 20
siRNA‐treated cells was normalized to that of controls. Results are means ± SD of four 21
measurements from two independent experiments. No significant difference was 22
found between NT and KDELR siRNA treated cells. B) Hela‐prME‐DENV1 cells were 23
transfected with siRNAs targeting all KDELR isoforms. 72h post transfection, cells 24
were stained with anti‐KDELR monoclonal antibodies (red)to verify the knockdown 25
efficiency. NT siRNA was used as control. Nuclei were visualised with DAPI (blue). Cell 26
morphology was observed under bright field (BF) and images were acquired with a 27
Carl Zeiss LSM 700 confocal microscope; bar: 10 µm. C) HeLa cells stably expressing 28

3
eGFP‐tagged KDELR1‐3 were transfected with either NT (open symbols) or the 29
indicated siRNAs specific for individual receptor isoforms (grey‐tinted symbols). Cells 30
were fixed 72h post‐transfection and analysed by flow cytometry. MFI of cells treated 31
with siRNAs for KDELR was normalized to that of NT controls (right‐hand panels). 32
Results are means ± SD of nine observations from three independent experiments. 33
*P<0.005 vs lysed control cells (B) or NT (C) by the unpaired Student’s t‐test. 34
4
35
Figure S3. KDELR knockdown does not affect early steps of DENV life cycle in Vero E6 36
cells, Related to Figure 3. A) Cells were transfected with siRNAs targeting KDELR or 37
Arf4+5, either alone or in combination (K+A), before being challenged with DENV1 at 38
an MOI=0.1. Cellular lysates were collected at 5 days post infection and subjected to 39
Western blotting using relevant antibodies to verify the effect of protein knockdown 40
by the specified siRNAs. GAPDH was used as the loading control across wells. B) 41
Morphology of Vero E6 after silencing KDELR by was similar to that of NT‐treated cells. 42
Cells were stained with anti‐KDELRs monoclonal antibodies to confirm knockdown 43
efficiency and images were acquired with a Carl Zeiss LSM 700 confocal microscope; 44
bar: 10 µm. C) KDELR down regulation did not influence cell viability. Experimental 45
conditions and staining with propidium iodide (PI) were as described in Fig. S2. Cell 46

5
viability was analysed by flow cytometry and MFI of siRNA‐treated cells was 47
normalized to that of controls. Results are means ± SD of four measurements from 48
two independent experiments. No significant difference was found between cells 49
treated with either NT or KDELR siRNA. D) siRNA‐treated Vero E6 cells were 50
challenged with DENV1 at an MOI=1. Viral titres, measured by real‐time RT‐PCR at 18h 51
post infection (p.i.) were similar to those of control cells treated with non‐targeting 52
(NT) siRNA. Results are means ± SD of three independent experiments. *P<0.005 vs 53
control cells by the unpaired Student’s t‐test. 54

6
55
Figure S4. The R6S point mutation disrupts the interaction between prM and E 56
proteins, Related to Figure 5. A) Sequence alignment of the N‐terminal 40 amino acids 57
of DENV1‐4 prM. Positively charged residues are showed in red. The three key amino 58
acids for prM/KDELRs interaction are indicated by asterisks. B) Lysates from cells 59
transfected with either wild‐type or mutant prME (see Fig. 4) were subjected to 60
immunoprecipitation using the anti‐E monoclonal antibody 4E11. prM and E proteins in 61
cell lysates (CL) or pellets (IP) were then analysed by Western blotting using the 62
indicated antibodies. These experiments showed that a drastic reduction of the prM/E 63
in the IP was observed only for the R6S mutation. These results are representative of 64
two independent experiments. 65

7
66
Figure S5. A triple prME mutant (Triple) does not affect the interaction between prM 67
and Arf4/Arf5, Related to Figure 5. Lysates from cells transfected with either empty 68
vector (ev), wild‐type (WT), or Triple (see also Experimental Procedures and Fig. 5) 69
were subjected to immunoprecipitation using dengue patient serum (DPS). Normal 70
human serum (NHS) was used the negative control. Arf4, Arf5 and prM proteins in cell 71
lysates (CL) or pellets (IP) were then analysed by Western blotting using the indicated 72
antibodies. Similar amounts of Arf4 and Arf5 could be precipitated from cells 73
expressing either WT or the Triple prME mutant. These results are representative of 74
two independent experiments. 75

8
76
Figure S6. Aggregates of E protein are present in DENV1 infected Vero E6 cells, 77
Related to Figure 6. Cells were infected at an MOI=0.1 and stained 5 days later with 78
either a polyclonal antibody against both prM and E (left panel), or a monoclonal 79
antibody recognizing only E (right panel). Arrowheads indicate aggregates of E 80
protein, similarly to what observed in HeLa‐prME‐DENV1 cells (see Fig. 6B, WT panels). 81
Images were acquired with a Carl Zeiss LSM 700 confocal microscope; bar: 10 µm. 82

9
Supplemental Tables 83
Table S1. Primers used to identify KDELR by RT‐PCR, Related to Figure 1. 84
KDELR1‐f 5’TGTGGTGTTCACTGCCCGA3’
KDELR1‐r 5’CGGTAAACGCCTAGCGCAA3’
KDELR2‐f 5’AACATTTTCCGGCTGACT3’
KDELR2‐r 5’GACAAGATACAAAGCACGA3’
KDELR3‐f 5’AAGTGCTGCAAGGGCATCT3’
KDELR3‐r 5’CCAATGACTGGGACCAGAA3’

10
Table S2. Primers used to produce prM fragments used as baits for GST pull‐downs, 85
Related to Figure 1. 86
prM‐GST‐f 5’CGCGGATCCTTCCACCTGACCACCAGAG3’
pr‐GST‐r 5’CCGCTCGAGTCATCATCTCTTGTCTCTCCTGTGCT3’
prM‐ΔTM‐GST‐r 5’CCG CTCGAG TCA TCA CAG GGC CCA GGT CTC CAC CT3’
pr40‐GST‐r 5’CCG CTCGAG TCA TCA ATC CAT GGC GAT CAG GGT AC3’

11
Table S3. Primers used in overlapping PCR for site‐directed mutagenesis, Related to 87
Figure 5. 88
P1 5’CCATTGACGTCAATGGGAGT3’
P2‐H2L 5’GGTCAGGAGGAAGCAGTTCA3’
P3‐H2L 5’TGAACTGCTTCCTCCTGACC3’
P2‐R6S 5’TCGCCGCCGCTGGTGGTCA3’
P3‐R6S 5’TGACCACCAGCGGCGGCGA3’
P2‐H11L 5’ACACGATCATGAGGGGCTC3’
P3‐H11L 5’GAGCCCCTCATGATCGTGT3’
P2‐K16T 5’TCCTGCGTGGACACGATCA3’
P3‐K16T 5’TGATCGTGTCCACGCAGGA3’
P2‐R19S 5’AGGCTCTTGCCGCTCTCCT 3’
P3‐R19S 5’AGGAGAGCGGCAAGAGCCT3’
P2‐K21T 5’TGAACAGCAGGCTCGTGCC3’
P3‐K21T 5’GGCACGAGCCTGCTGTTCA3’
P2‐K26T 5’GCGCTGGTCGTGAACAGCA 3’
P3‐K26T 5’TGCTGTTCACGACCAGCGC 3’
P4 5’TCTAGACTCGAGCTAGCTAG3’
Bold‐faced letters designate the mutated base. 89

12
Supplemental Experimental Procedures, Related to Experimental Procedures. 90
Cells, Viruses and Antibodies 91
HeLa and human embryonic kidney (293T) cells were maintained in DMEM 92
supplemented with 10% foetal bovine serum (FBS) and 1% penicillin/streptomycin at 93
37°C, with 5% CO2. The stable cell lines expressing prME‐DENV1 or its mutants were 94
established using the retroviral vector pCHMWS‐IRES‐Hygromycin (kindly provided by 95
Dr. Rik Gijsbers, Division of Molecular Medicine, Katholieke Universiteit, Leuven, 96
Belgium), selected following a 2‐week period in the presence of 500 µg/mL 97
hygromycin and maintained thereafter the same medium, as previously described 98
(Wang et al., 2009). Vero E6 cells, which were derived from African green monkey 99
kidney, were maintained in DMEM supplemented with 5% foetal calf serum (FCS) at 100
37°C, with 5% CO2. All work with infectious DENV1 strain (DENV1 Hawaii), DENV2 strain 101
(Dengue 2 New Guinea), DENV3 strain (Dengue 3 strain H87), DENV 4 strain (Dengue 4 102
Jamaique 8343) and WNV strain (West Nile B956, lineage II) was performed in a 103
biosafety level 2 plus laboratory (Institut Pasteur du Laos, Vientiane, Lao PDR) with 104
Vero E6 cells. For biochemistry, the following antibodies were used: mouse anti‐E 105
monoclonal antibodies (mAb) 4E11 (a gift of Dr. Philippe Despres, Institut Pasteur, 106
Paris, France); mouse anti‐E mAb 4G2 was prepared using hybridoma cells D1‐4G2‐4‐15 107
from ATCC (Manassas, VA, USA); mouse anti‐prME antibody and dengue patients sera 108
(kindly provided by Dr. Philippe Buchy, Institut Pasteur, Cambodia and Myrielle 109
Dupont‐Rouzeyrol, Institut Pasteur de Nouvelle‐Calédonie, Nouméa, New‐Caledonia) 110

13
and mouse anti‐prM mAb prM‐6.1 (a gift from Dr. Sittisombut Nopporn, Chiang Mai 111
University, Chiang Mai, Thailand) had been previously used (Junjhon et al., 2008; 112
Kudelko et al., 2012); mouse anti‐KDELR mAb, rabbit anti‐GM130 mAb, rabbit anti‐Myc 113
tag antibody and anti‐GAPDH mAb from Abcam (Cambridge, MA, USA); rabbit 114
anti‐Arf4 from ProteinTech Group Inc (Chicago, IL, USA); mouse anti‐Arf5 mAb, rabbit 115
anti‐Bip mAb and rabbit anti‐calreticulin antibody from Cell Signaling Technology 116
(Beverly, MA, USA). For immunofluorescence, antibodies were as follows: mouse 117
anti‐E mAb 4G2, rabbit anti‐prME polyclonal antibody (kindly provided by Dr. Polly H.M. 118
Leung, The Hong Kong Polytechnic University, Hong Kong SAR), rabbit anti‐calnexin 119
mAb from Cell Signaling Technology, rabbit anti‐ERGIC53 from Sigma (Saint Louis, MO, 120
USA), rabbit anti‐GM130 mAb from Abcam, rabbit anti‐TGN46 from Abnova 121
Corporation (Walnut, CA, USA). 122
123
siRNA experiments 124
All siRNAs used in this work, including non‐targeting (NT) siRNA (D‐001206) and 125
transfection reagents DharmaFECT 1 (T‐2001) were purchased from Dharmacon 126
Research Inc. (Lafayette, CO, USA). Arf4 siRNA (L‐011582) and Arf5 siRNA (L‐011584) 127
were provided as SMARTpool ON‐TARGET plus siRNAs, which are pools of four siRNAs 128
targeting various sites in a single gene. KDELR1 siRNA (J‐019136‐09/11), KDELR2 siRNA 129
(J‐0.12315‐05/07) and KDELR3 siRNA (J‐012316‐05/06) were provided as individual 130
ON‐TARGET plus siRNAs targeting one site in a single gene. Briefly, siRNAs mixed with 131

14
DharmaFECT 1 reagents (Dharmacon) were added to 24‐well plates in DMEM medium 132
without FBS and antibiotics. Twenty minutes later, 0.8 mL cells (75,000 cells/mL in 133
DMEM supplemented with 2.5% FCS) were added to each well so that the final siRNA 134
concentration was 100nM for Arf4+5, and 150nM for both KDELR and NT. Cells were 135
incubated at 37°C for 48 hours. For RSPs experiments, medium was replaced with 0.3 136
mL of DMEM containing 2% FBS (without antibiotics) and, 14 hours later, supernatant 137
containing secreted RSPs was collected, cleared by centrifugation at 4,000 rpm for 15 138
min and analysed by Western blotting (Wang et al., 2009). Cells were lysed in RIPA 139
buffer containing: 1% Triton X‐100, 150 mM NaCl, 50 mM Tris‐HCl, (pH 7.5), 1 mM EDTA, 140
0.5% Na‐deoxycholate, freshly added 1 mM PMSF and protease inhibitors cocktail 141
(Roche Applied Science, Mannheim, Germany), for 15 min on ice, with frequent 142
vortexing. For dengue virus 1 (DENV1) infection assay, siRNA treatment was 143
performed 48 hours before viral infection using the same conditions described above. 144
Supernatant containing secreted progeny virus was collected, cleared by 145
centrifugation at 4000 rpm for 15 min and further processed to measured viral titre by 146
Real Time RT‐PCR. Cells were lysed in RIPA buffer as detailed above. 147
148
Production, purification and quantification of GST‐fusion proteins 149
A single colony of BL21 bacteria transformed with pGEX‐4T‐1 expression vector 150
encoding GST‐fused prM fragments or GST protein alone was lifted from LB agar 151
dishes containing 100µg/ml ampicillin and grown overnight at 37°C in 5ml LB medium 152

15
supplemented with the same ampicillin concentration. To induce expression of GST 153
fusion proteins, isopropyl‐1thio‐b‐D‐galactopyanoside (IPTG) was added at a final 154
concentration of 1mM after cooling down on ice for 5min a 500 ml bacterial culture in 155
mid‐log phase (OD600 ~0.6‐0.7). After shaking for 2h at 25°C, bacteria were harvested 156
by centrifugation at 4,000 g for 10min at 4°C and re‐suspended in 10ml of ice‐cold lysis 157
buffer I [0.4M NaCL, 50 mM Tris‐HCL pH 7.5, 0.3% (v/v) Triton X‐100], supplemented 158
with freshly added 2% (v/v) N‐lauroylsarcosine, 100 μg/ml lysozyme, 0.6 mM PMSF (all 159
from Sigma‐Aldrich, St. Louis, MO) and 1x protease inhibitors cocktail (Roche Applied). 160
The bacterial cell suspension was transferred to a 50ml Falcon tube, mixed well with 161
2% N‐lauroylsarcosine, and then homogenized on ice by sonication (9x10sec pulses) 162
using a cell disruptor (Vibra CellTM, Sonics and Materials, Inc., CT, USA) at 35% 163
amplitude. Cell debris were spun down at 12,000 g for 1 hr at 4°C. To purify GST fusion 164
proteins, clarified supernatants were transferred to a clean Falcon tube containing 165
200μl of a 50% slurry of pre‐washed sepharose glutathione 4B beads (Amersham 166
Biosciences, MA, USA). Following an overnight incubation at 4°C on a rotating plate, 167
beads were extensively washed with 800μl of ice‐cold lysis buffer II (0.2 M NaCl, 168
50mM Tris‐HCl pH 7.5, 0.15% Triton X‐100,), freshly supplemented with 0.6mM PMSF, 169
1X Complete protease cocktail inhibitor and 1% N‐lauroylsarcosine. Supernatants were 170
discarded and the beads were eventually mixed with 500μl of lysis buffer II. Five 171
microlitres of this mixture were loaded on a 12% SDS‐PAGE, stained with Coomassie 172
brilliant blue (G250; Sigma‐Aldrich) and quantified by densitometry using Image Quant 173

16
TL (Thermo Fisher Scientific Inc., Rockford, IL, USA) against a standard curve of bovine 174
serum albumin samples (2‐fold dilutions ranging from 54‐1,448ng/μl) ran in parallel. 175
176
Mutagenesis 177
Site‐directed mutagenesis was performed by overlapping PCR using codon optimized 178
DENV1 prME as the template. Seven positive charged amino acids in the N‐terminal 179
portion of prM were substituted with neutral amino acid individually or in combination 180
(Table S3). prME mutants were digested with BamHI/XhoI and subcloned into 181
pcDNA3.1 (Invitrogen) or pCHMWS‐IRES‐Hygromycin vectors for further studies. 182
183
Flow Cytometry 184
Stable HeLa cell lines expressing eGFP tagged KDELR1, 2 or 3, were transfected with 185
siRNAs targeting individual isoforms as indicated. Cells were re‐suspended and fixed 186
72h post‐transfection. For cell viability assays, siRNA‐treated HeLa‐prME‐DENV1 or 187
Vero E6 cells were re‐suspended in PBS containing 10µg/ml propidium iodide (PI) and 188
incubated on ice for 30min. Cells re‐suspended in ddH2O (3h at 37˚C before PI staining 189
as above) to induce osmotic lysis served as the positive controls. For both sets of 190
experiments, after washing with cold PBS, cell suspensions were subjected to flow 191
cytometry using a FACSCalibur (BD Biosciences, San Jose, CA, USA), and more than 192
20,000 singlet living cells were collected. The post‐acquisition data analysis was 193
performed using FlowJo software (TreeStar, Ashland, OR, USA). 194

17
195
RT‐PCR 196
Total RNA from HeLa, HeLa‐prME‐DENV1, 293T and VeroE6 cells was extracted using 197
RNeasy Mini kit (QIAGEN, Valencia, CA, USA) and eluted in 50µl RNase‐free water, 198
according to the manufacturer’s instructions. cDNA synthesis was performed with 199
SuperScript III First‐Strand Synthesis System for RT‐PCR kit (Invitrogen) using 1µg of 200
total RNA in one reaction mixture of 20µl that contained dNTP and Oligo (dT)20, as 201
recommended. Specific individual primers (Table S1) designed for the three KDELR 202
isoforms and GAPDH were then used in the following amplification reactions with 203
FastStart Taq DNA Polymerase, dNTPack (Roche Applied Science). PCR cycling 204
conditions were as follows: 95°C for 4 min, followed by 35 cycles of 95°C for 30 sec, 205
54°C for 30s and 72°C for 1 min. PCR products were analysed by agarose gel 206
electrophoresis and detected by ethidium bromide staining. 207
208
Real‐time RT‐PCR (TaqMan) 209
For measurements of progeny virus production, viral RNA was extracted from culture 210
supernatants (150µl) using Nucleospin@ RNA virus kit (MACHEREY‐NAGEL, Düren, 211
Germany), following the instructions provided by the manufacturer, and eluted in 50µl 212
RNase‐free water. The same procedure was used for viral replication assay, except 213
that 20µl of RNase‐Free proteinase K (20mg/ml stock solution; Roche Applied Science) 214
were added to the lysis mixture and DNA was removed by using RNase‐Free DNase kit 215

18
(QIAGEN, Chatsworth, CA, USA) in the washing steps, as recommended. The sequence 216
of DENV universal primers to detect all four serotypes, of West Nile Virus primers and 217
their specific fluorogenic probe (FAM/BHQ‐1) have been previously reported (Linke et 218
al., 2007; Warrilow et al., 2002). Briefly, for each reaction 5µl of RNA were combined 219
with 2µl of each primer (10µM) and 1µl of probe (10µM) in a total mixture volume of 220
25µl by using EXPRESS One‐Step SuperScript qRT‐PCR Kits (Invitrogen). The cycling 221
conditions used were: 45°C for 30 min and 95°C for 2 min, followed by 40 cycles as 95°C 222
for 15 sec and 60°C for 30 sec. Real‐time PCR was performed with the MiniOpticon™ 223
Real‐time PCR Detection system (Bio‐Rad, CA, USA) and products were detected by 224
measuring the fluorescence signal from the FAM reporter. 225
226
Quantitative analysis of fluorescent images 227
In protocol “Stained cells”, we first extracted signals of nuclei, stained by DAPI, and of 228
viral E protein, stained by the anti‐E monoclonal antibody 4G2, which were 229
significantly brighter than the cell background with a K‐mean algorithm (Forgy, 1965). 230
The Regions of Interest (ROIs) defined by the extracted signals were then filled to 231
form connected regions, and the boolean intersection between nuclei ROIs and E 232
protein ROIs was computed to extract the nuclei that are surrounded by E signals. 233
Finally, by counting surrounded nuclei we obtained the number of stained cells in each 234
image. In “Cells with aggregates” protocol, we first extracted signal corresponding to 235
cell nuclei with a K‐mean algorithm and filled holes in nuclei ROIs to obtain connected 236

19
regions as described previously. In parallel, we used a wavelet‐based detection 237
method (Olivo‐Marin, 2002), implemented as a plugin named “Spot detector” in the 238
ICY software (scale=5, threshold=100), to extract the large spots corresponding to 239
RSPs aggregates, stained with the anti‐E antibody, which were significantly brighter 240
than the surrounding background. We filtered these detections with a size criterion 241
(spots>100 pixels), and a compactness criterion (4π Area/Perimeter2 >0.15; Perfect 242
disks have a compactness equal to 1, whereas the compactness of filamentous spots 243
tends to 0) to obtain round and large aggregate spots. We then defined nuclear 244
neighbourhoods by dilating nuclear ROIs by 15 pixels and filtered aggregate spots that 245
were inside these dilated nuclei ROIs to select aggregates spots that were sufficiently 246
close to cell nuclei. Finally, by counting the nuclei with neighbourhood‐containing 247
aggregates, we obtained the number of cells with RSPs aggregates. 248
249
Supplemental References 250
Forgy, E.W. (1965). Cluster analysis of multivariate data: efficiency versus 251 interpretability of classifications. Biometrics 21, 768‐769. 252
Junjhon, J., Lausumpao, M., Supasa, S., Noisakran, S., Songjaeng, A., Saraithong, P., 253 Chaichoun, K., Utaipat, U., Keelapang, P., Kanjanahaluethai, A., et al. (2008). 254 Differential modulation of prM cleavage, extracellular particle distribution, and virus 255 infectivity by conserved residues at nonfurin consensus positions of the dengue virus 256 pr‐M junction. J. Virol. 82, 10776‐10791. 257
Kudelko, M., Brault, J.B., Kwok, K., Li, M.Y., Pardigon, N., Peiris, J.S., Bruzzone, R., 258 Despres, P., Nal, B., and Wang, P.G. (2012). Class II ADP‐ribosylation Factors Are 259 Required for Efficient Secretion of Dengue Viruses. J. Biol. Chem. 287, 767‐777. 260
Linke, S., Ellerbrok, H., Niedrig, M., Nitsche, A., and Pauli, G. (2007). Detection of West 261 Nile virus lineages 1 and 2 by real‐time PCR. J.Virol. Methods 146, 355‐358. 262

20
Olivo‐Marin, J.C. (2002). Extraction of spots in biological images using multiscale 263 products. Pattern. Recogn. 35, 1989‐1996. 264
Wang, P.G., Kudelko, M., Lo, J., Siu, L.Y., Kwok, K.T., Sachse, M., Nicholls, J.M., 265 Bruzzone, R., Altmeyer, R.M., and Nal, B. (2009). Efficient assembly and secretion of 266 recombinant subviral particles of the four dengue serotypes using native prM and E 267 proteins. PLoS One 4, e8325. 268
Warrilow, D., Northill, J.A., Pyke, A., and Smith, G.A. (2002). Single rapid TaqMan 269 fluorogenic probe based PCR assay that detects all four dengue serotypes. J.Med. 270 Virol. 66, 524‐528. 271






![Dengue Fever/Severe Dengue Fever/Chikungunya Fever · Dengue fever and severe dengue (dengue hemorrhagic fever [DHF] and dengue shock syndrome [DSS]) are caused by any of four closely](https://img.pdfslide.net/doc/110x75/5e87bf3e7a86e85d3b149cd7/dengue-feversevere-dengue-feverchikungunya-dengue-fever-and-severe-dengue-dengue.jpg)






















