Embed Size (px)
Citation preview

Kinetics of the ThermalIsomerization of 1,1,2,2-TetramethylcyclopropaneDAVID K. LEWIS,1 TIMOTHY GRAY,1 VLAD KATSVA,1 KYLE PARCELLA,1 JESSICA SCHLIER,1
BANSI L. KALRA,2 JANET CHO,2 DEBRA MISH2
1Department of Chemistry, Connecticut College, New London, CT 063202Department of Chemistry, Hollins University, Roanoke, VA 24020
Received 1 July 2003; revised 8 September 2005; accepted 21 September 2005
DOI 10.1002/kin.20149Published online in Wiley InterScience (www.interscience.wiley.com).
ABSTRACT: Reaction rates for the structural isomerization of 1,1,2,2-tetramethylcyclopropaneto 2,4-dimethyl-2-pentene have been measured over a wide temperature range, 672–750 K in astatic reactor and 1000–1120 K in a single-pulse shock tube. The combined data from the twotemperature regions give Arrhenius parameters Ea = 64.7 (±0.5) kcal/mol and log10(A, s−1) =15.47 (±0.13). These values lie at the upper end of the ranges of Ea and log A values (62.2–64.7 kcal/mol and 14.82–15.55, respectively) obtained from three previous experimental studies,each of which covered a narrower temperature range. The previously noted trend toward lowerEa values for structural isomerization of methylcyclopropanes as methyl substitution increasesextends only through the dimethylcyclopropanes (1,1- and 1,2-); Ea then appears to increasewith further methyl substitution. In contrast, the pre-exponential factors for isomerization ofcyclopropane and all of the methylcyclopropanes through tetramethylcyclopropane lie within±0.3 of log10(A, s−1) = 15.2 and show no particular trend with increasing substitution. C© 2006Wiley Periodicals, Inc. Int J Chem Kinet 38: 483–488, 2006
INTRODUCTION
Experimental kinetic studies of structural and geomet-ric isomerizations of cyclopropane (CP) and variousalkyl-substituted cyclopropanes have been carried outby a number of research groups over the past half cen-tury. The wealth of experimental data available on re-
Jessica Schlier is a visiting student from Wesleyan University,Middletown, CT 06459.
Correspondence to: David K. Lewis; e-mail: [email protected]; B. L. Kalra; e-mail: [email protected].
Contract grant sponsor: NSF.Contract grant numbers: CHE 9714356 and CHE 0213374.Contract grant sponsor: Cottrell College Science grant of
Research Corporation.Contract grant sponsor: Connecticut College.
c© 2006 Wiley Periodicals, Inc.
action rates and activation parameters, coupled withthe relative simplicity and high symmetry of the cyclo-propane ring, have made reactions of CP and its alkylderivatives convenient test cases for the developmentof computational models which examine the energiesof transition states and the dynamics of intramolecularmotions in the approach to those transition states.
Previous kinetic studies have shown that as methylgroups are substituted for H atoms on the cyclopropanering, activation energies for structural isomerizationdecrease, at least through the series cyclopropane,Ea = 65.0 kcal/mol [1]; methylcyclopropane (MCP)64.4 kcal/mol [2]; 1,1-dimethylcyclopropane (di-MCP), 61.8 kcal/mol [3]; and 1,2-dimethylcyclo-propane, 61.2–62.3 kcal/mol [4]. This trend has beenattributed to a weakening of the C C bonds in thehighly strained cyclopropane ring, leading to a lower

484 LEWIS ET AL.
energy barrier for formation of a diradical intermediate.An earlier study of 1,1,2-trimethylcyclopropane (tri-MCP) isomerization gave a value of Ea, 61.1 kcal/mol[5]; however, in our systematic re-examination of thesereactions, that isomerization has yielded a somewhathigher Ea value, 63.7 kcal/mol [6]. This suggests thatfactors other than just the strength of the C C bondbeing broken become important as more hydrogenatoms on the cyclopropane ring are replaced by methylgroups. The added steric interactions may distort thetransition state or block some easy pathways to it.
Continuing this exploration of the effects of suc-cessive methyl-for-hydrogen substitutions on the ki-netics and energetics of cyclopropane isomerizations,we have now looked at the next more highly substitutedmember of the series, 1,1,2,2-tetramethylcyclopropane(tetra-MCP). This should be an ideal molecule forkinetic-mechanistic studies, both experimental and the-oretical, because isomerization via the expected dirad-ical intermediate should form only a single product,2,4-dimethyl-2-pentene (2,4-DMP).
There have been three previous reports of experi-mental rate studies of tetra-MCP isomerization. Freyand Marshall [7] performed a static reactor study overthe range 708–757 K and found the reaction to be ho-mogeneous and unimolecular, with Ea = 64.4 kcal/mol and log (A, s−1) = 15.83 (later adjusted slightly toEa = 64.7 kcal/mol, log A = 15.55) [8]. Believing thework of Frey and Marshall basically sound but possiblyaffected by a systematic error due to the small temper-ature range covered (50◦C), Benson and O’Neal [9]combined a calculated pre-exponential factor based ona diradical transition state with the experimental rateconstants in the center of Frey and Marshall’s temper-ature range and concluded that Ea = 59.7 kcal/mol,log A = 14.4 best described the rate of this isomer-ization, thus continuing the trend of lower Ea with in-creasing methyl substitution reported for other methyl-substituted cyclopropanes. At that time, no other alkyl-cyclopropane isomerization showed such a large dis-crepancy between measured [7] and predicted [9] Ar-rhenius parameters.
Shortly thereafter, two other experimental studieswere carried out to resolve this discrepancy. Blumsteinet al. [8] repeated the work in a static reactor at699–759 K, and reported Ea = 63.93 kcal/mol, logA = 15.27, within experimental error of the findingsof Frey and Marshall [7]. However, they believed thisto be the result of two parallel channels for loss oftetra-MCP, the expected isomerization to 2,4-DMP(with Ea = 62.3 kcal/mol) plus a minor but signifi-cant decomposition pathway via loss of methyl radi-cal (with Ea = 77 kcal/mol). Tsang [10] studied tetra-MCP isomerization in a single-pulse shock tube at
1077–1151 K. Under the extremely homogeneous con-ditions in the shock tube, he found evidence of only onechannel for tetra-MCP depletion, the isomerization to2,4-DMP; at rates given by Ea = 62.2 kcal/mol, logA = 14.82. However, Tsang did note considerable in-stability of the product at these temperatures and hadto correct for its loss in calculating rate constants forthe tetra-MCP isomerization [10].
Given the similarity in absolute rates reported in thetwo experimental static reactor studies [7,8], and thecare with which all three previous experimental studiesappear to have been carried out [7,8,10], it is unlikelythat there are large errors in the reported experiment-based Arrhenius parameters. Still, in each of those stud-ies, rate constant corrections were necessary for theappearance of side products in experiments that con-sumed substantial fractions of the reactant. Consideringthe importance of cyclopropane isomerizations to thedevelopment and verification of computational meth-ods, activation parameters of the highest accuracy nowpossible should be made available. That is the purposeof this study.
In the work described here, the isomerization oftetra-MCP to 2,4-DMP was studied over 672–750 K inthree glass static reactor cells, and at 1000–1120 K in aglass single pulse shock tube, using two alternate ref-erence reactions for temperature determination. Com-bining the two experimental methods, we were able tocover a wide temperature range, 448◦ in this study. Wewere also able to restrict the rate data we used to runsin which extents of depletion of the reactant tetra-MCPwere relatively small (generally <25%), eliminatingthe need to know whether any side products observedwere produced directly from the reactant or from sub-sequent reactions of the primary isomerization product2,4-DMP.
EXPERIMENTAL SECTION
Materials
Tetra-MCP (99.96%)1 was obtained from the API Stan-dard Reference Materials project at Carnegie MellonUniversity. Either Matheson CP grade (99%) cyclo-propane (CP) or Aldrich reagent grade (99%) cyclo-hexene (CH) was added to each reactant mixtureas an internal thermometer. These reagents, in ap-propriate ratios, were diluted with Matheson reagentgrade (99.9999%) argon to prepare reaction mixtures.
1Reagent purities listed here are as specified by the vendors fromwhom the samples were obtained. GC analyses confirmed thesepercentages.

KINETICS OF THE THERMAL ISOMERIZATION OF 1,1,2,2-TETRAMETHYLCYCLOPROPANE 485
Samples of 2,4-DMP (99.6%) and a variety of otherreagents from ChemSampCo (formerly Wiley Organ-ics, Inc.) were diluted with argon and used for theidentification of products and as calibration mixtures.All reagents were degassed before use through mul-tiple freeze–pump–thaw cycles, then distilled fromthe liquid phase into a gas storage bulb leaving be-hind any less volatile impurities. High purity gradehelium was used as the driver gas in the shock-tuberuns.
Apparatus
The static reactor used in this study is a well-insulatedaluminum-block tube furnace into which glass reac-tor cells are inserted and removed to initiate and ter-minate the reaction. Three 100 cm3 cells were usedrandomly in this study; they were all well seasonedthrough many hundreds of hours of heating while filledwith a variety of hydrocarbon gases. The cells wereevacuated and filled on an oil- and mercury-free high-vacuum line. A fourth glass reactor cell was used ini-tially, but then sidelined because of its curious abilityto produce an unexpected competing product at lowtemperatures—see Results and Discussion. A 2.54-cmdiameter single-pulse shock tube was used for the1000–1120 K experiment; its operating characteristicshave been described previously [11]. Reactant andproduct samples were analyzed on a Varian 1440-20gas chromatograph with hydrogen flame ionization de-tector. A 3-m column containing 20% polypropyleneglycol saturated with AgNO3 on 80–100 meshChromosorb W was used at room temperature.
Sample Preparation
Table I lists reaction mixtures containing different per-centages of tetra-MCP and either CP or CH in argon;these were prepared in 1- or 2-L glass storage flaskswith greaseless stopcocks and thoroughly mixed be-fore use.
Table I Sample Reaction Mixtures
Reactor %tetra-MCP %CP (or CH∗)
Single-pulse shock tube 0.67 0.67
2.00 2.00
3.00 3.00
1.02 0.99∗Static reactor 1.18 2.26
2.00 2.00
Kinetic Runs
For the runs at 672–750 K carried out in the static reac-tor, the reactor cells were filled at room temperature topressures ranging from 89 to 328 Torr, then inserted intothe hot oven for times ranging from 45 to 150 min. Theheating at constant volume produced pressures (calcu-lated from measured initial pressure and �T ) rangingfrom 200 to 800 Torr during reaction. The cells wereremoved from the reactor and cooled rapidly to roomtemperature, then product gas was introduced into theGC directly from the cells. Tests with an enclosed ther-mocouple showed that it took somewhat longer for thecells to reach reaction temperature when inserted intothe oven than to cool below reaction temperature whenremoved; this thermal lag was corrected for in calcu-lating the rate constants in Table II. Once a cell andits contents reached thermal equilibrium with the ovencore, movement of the thermocouple along the cell’swall and within the heated gas produced temperaturevariations of less than a degree.
For the shock tube experiments at 1000–1120 K,sample mixture pressures introduced into the samplesection of the shock tube ranged from 35 to 100 Torr.The reaction mixtures were heated for 800 ± 50 μs attotal reaction pressures of 2–3 bar (p again calculatedfrom measured initial pressure and �T ). Reaction timewas determined by measuring the duration of the highpressure plateau (about 700 μs) and adding 100 μs,or 14%. That percentage corresponds to the additionalproduct expected to be formed during the finite cool-ing process assuming (1) an initial cooling rate (fromrecorded pressure traces) of 5 × 105 K/s, (2) a first-order irreversible reaction with Ea = 65 kcal/mol, and(3) small extents of conversion of reactant to productduring the 700 μs plateau. As extent of conversion in-creases, more reactant is consumed before the coolingwave so less remains to react and the cooling correctiondecreases. The percentage of product formed during thecooling is only moderately dependent on activation en-ergy; for example, it should increase from 14% to 18%if Ea dropped from 65 to 45 kcal/mol. Since the reac-tions chosen as reference reactions have rate constantsand activation energies similar to the subject reaction,there should be no significant error introduced by treat-ing the experiment as an ideal “square wave” thermalpulse of 800 μs duration rather than integrating downthe cooling curve to calculate the correction for eachproduct.
Postshock samples extracted for the GC analysiswere taken by expansion into an evacuated 25 cm3 bulbattached to the shock tube end plate, immediately af-ter the shock. This is about 3% of the volume of theshock tube sample section; therefore, it was assumed
486 LEWIS ET AL.
Table II Reaction Temperatures, Times, and RateConstants Displayed in Fig. 1
Temp (K) Time k (s−1)
685.9 120 min 8.83 × 10−6
689.9 90 min 1.21 × 10−5
697.0 90 min 1.50 × 10−5
694.1 120 min 1.28 × 10−5
672.5 90 min 4.67 × 10−6
674.4 120 min 5.41 × 10−6
712.0 90 min 3.41 × 10−5
749.7 45 min 3.65 × 10−4
741.2 60 min 1.64 × 10−4
718.1 90 min 4.01 × 10−5
740.8 60 min 1.23 × 10−4
729.7 60 min 8.25 × 10−5
730.7 60 min 8.27 × 10−5
723.3 60 min 7.90 × 10−5
705.8 60 min 2.11 × 10−5
711.8 60 min 3.32 × 10−5
1081 8.0 × 10−4 s 2.77 × 102
1120 8.0 × 10−4 s 7.55 × 102
1105 8.0 × 10−4 s 5.15 × 101
1091 8.0 × 10−4 s 4.03 × 102
1090 8.0 × 10−4 s 3.96 × 102
1006 8.0 × 10−4 s 3.62 × 101
1017 8.0 × 10−4 s 3.75 × 101
1087 8.0 × 10−4 s 2.92 × 102
1062 8.0 × 10−4 s 1.53 × 102
1015 8.0 × 10−4 s 3.95 × 101
1098 8.0 × 10−4 s 2.14 × 102
1023 8.0 × 10−4 s 4.79 × 101
1004 8.0 × 10−4 s 2.95 × 102
1000 8.0 × 10−4 s 2.82 × 101
that the sample taken for analysis (compressed morethan five-fold during the reaction) resided very close tothe end plate where reaction time was measured, whilereacting.
Calculations
Rate constants for the thermal isomerization of tetra-MCP were calculated from the extent of its conversionto 2,4-DMP, as determined from the GC peak heights,corrected for measured sensitivity differences, of thehydrocarbons in the product samples. Rate constantswere calculated from the integrated rate expressionfor an irreversible first-order reaction. Reaction tem-peratures were determined from the extent of conver-sion of CP to propene or of CH to 1,3-butadiene plusethene, using the well-established Arrhenius param-eters for these irreversible first-order isomerizations:Ea = 65.0 kcal/mol, log (A, s−1) = 15.2 for CP [1]
and Ea = 66.6 kcal/mol, log (A, s−1) = 15.15 for CH[10]. For ln[(CP)t/(CP)0], we used GC peak heightsof CP and its isomerization product propene in heatedsamples, again corrected for differences in GC detec-tor response: ln[(CP)/(CP + propene)]. Similarly, GCresponses to 1,3-butadiene and CH were used for tem-perature calculations for the samples to which CHhad been added. This assumes that all losses of CPor CH were accounted for through product propeneor 1,3-butadiene, respectively. We believe that this issupported by the absence of other product peaks at-tributable to CP or CH decomposition, other than theethene co-produced from CH.
RESULTS AND DISCUSSION
At temperatures below 1200 K, structural isomeriza-tion of cyclopropanes is generally believed to occurmainly via cleavage of a C C bond to form a 1,3-diradical, followed by 1,2-hydrogen transfer to forma product alkene. As expected from this mechanism,2,4-DMP was the major product, and in most experi-ments the only product, formed from tetra-MCP iso-merization. Conditions that produced other detectableproducts are described below.
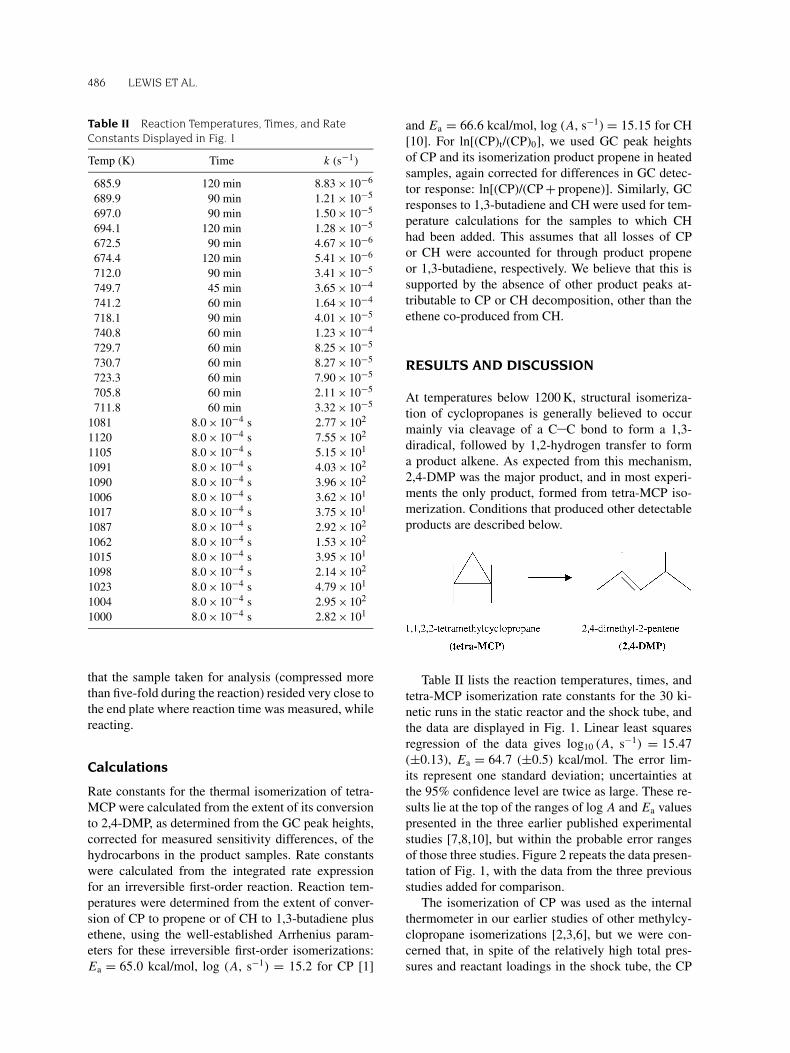
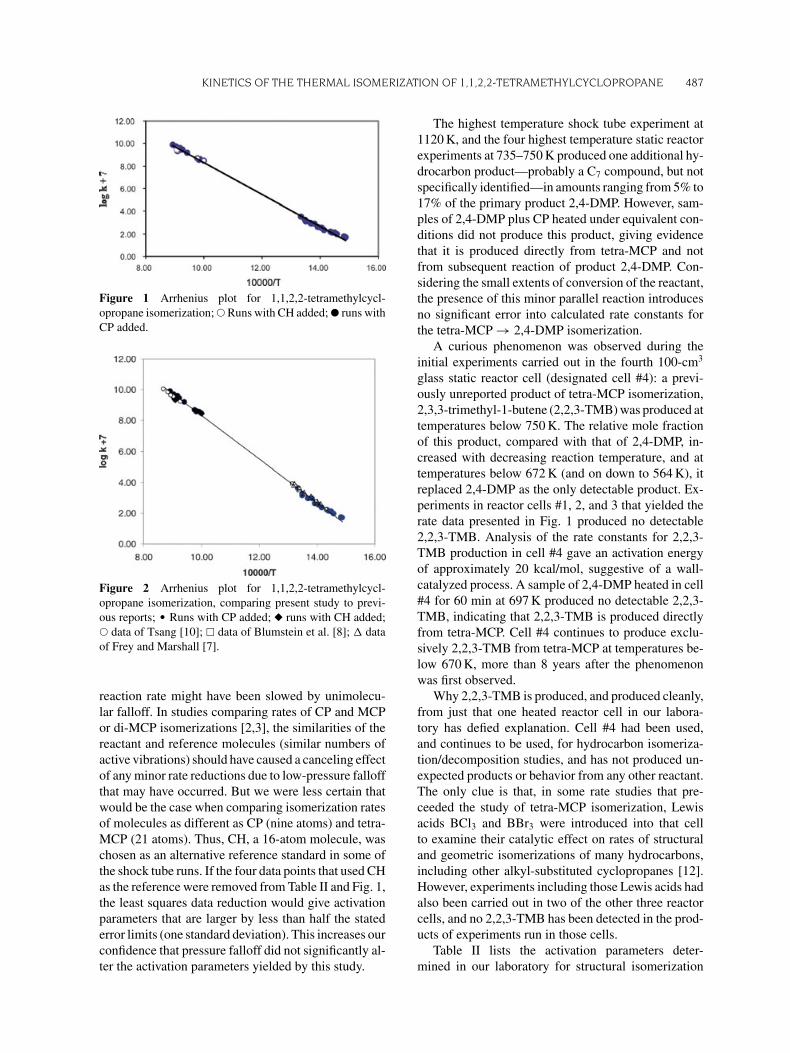
Table II lists the reaction temperatures, times, andtetra-MCP isomerization rate constants for the 30 ki-netic runs in the static reactor and the shock tube, andthe data are displayed in Fig. 1. Linear least squaresregression of the data gives log10 (A, s−1) = 15.47(±0.13), Ea = 64.7 (±0.5) kcal/mol. The error lim-its represent one standard deviation; uncertainties atthe 95% confidence level are twice as large. These re-sults lie at the top of the ranges of log A and Ea valuespresented in the three earlier published experimentalstudies [7,8,10], but within the probable error rangesof those three studies. Figure 2 repeats the data presen-tation of Fig. 1, with the data from the three previousstudies added for comparison.
The isomerization of CP was used as the internalthermometer in our earlier studies of other methylcy-clopropane isomerizations [2,3,6], but we were con-cerned that, in spite of the relatively high total pres-sures and reactant loadings in the shock tube, the CP
KINETICS OF THE THERMAL ISOMERIZATION OF 1,1,2,2-TETRAMETHYLCYCLOPROPANE 487
Figure 1 Arrhenius plot for 1,1,2,2-tetramethylcycl-
opropane isomerization; � Runs with CH added; � runs with
CP added.
Figure 2 Arrhenius plot for 1,1,2,2-tetramethylcycl-
opropane isomerization, comparing present study to previ-
ous reports; � Runs with CP added; � runs with CH added;
� data of Tsang [10]; � data of Blumstein et al. [8]; � data
of Frey and Marshall [7].
reaction rate might have been slowed by unimolecu-lar falloff. In studies comparing rates of CP and MCPor di-MCP isomerizations [2,3], the similarities of thereactant and reference molecules (similar numbers ofactive vibrations) should have caused a canceling effectof any minor rate reductions due to low-pressure falloffthat may have occurred. But we were less certain thatwould be the case when comparing isomerization ratesof molecules as different as CP (nine atoms) and tetra-MCP (21 atoms). Thus, CH, a 16-atom molecule, waschosen as an alternative reference standard in some ofthe shock tube runs. If the four data points that used CHas the reference were removed from Table II and Fig. 1,the least squares data reduction would give activationparameters that are larger by less than half the statederror limits (one standard deviation). This increases ourconfidence that pressure falloff did not significantly al-ter the activation parameters yielded by this study.
The highest temperature shock tube experiment at1120 K, and the four highest temperature static reactorexperiments at 735–750 K produced one additional hy-drocarbon product—probably a C7 compound, but notspecifically identified—in amounts ranging from 5% to17% of the primary product 2,4-DMP. However, sam-ples of 2,4-DMP plus CP heated under equivalent con-ditions did not produce this product, giving evidencethat it is produced directly from tetra-MCP and notfrom subsequent reaction of product 2,4-DMP. Con-sidering the small extents of conversion of the reactant,the presence of this minor parallel reaction introducesno significant error into calculated rate constants forthe tetra-MCP → 2,4-DMP isomerization.
A curious phenomenon was observed during theinitial experiments carried out in the fourth 100-cm3
glass static reactor cell (designated cell #4): a previ-ously unreported product of tetra-MCP isomerization,2,3,3-trimethyl-1-butene (2,2,3-TMB) was produced attemperatures below 750 K. The relative mole fractionof this product, compared with that of 2,4-DMP, in-creased with decreasing reaction temperature, and attemperatures below 672 K (and on down to 564 K), itreplaced 2,4-DMP as the only detectable product. Ex-periments in reactor cells #1, 2, and 3 that yielded therate data presented in Fig. 1 produced no detectable2,2,3-TMB. Analysis of the rate constants for 2,2,3-TMB production in cell #4 gave an activation energyof approximately 20 kcal/mol, suggestive of a wall-catalyzed process. A sample of 2,4-DMP heated in cell#4 for 60 min at 697 K produced no detectable 2,2,3-TMB, indicating that 2,2,3-TMB is produced directlyfrom tetra-MCP. Cell #4 continues to produce exclu-sively 2,2,3-TMB from tetra-MCP at temperatures be-low 670 K, more than 8 years after the phenomenonwas first observed.
Why 2,2,3-TMB is produced, and produced cleanly,from just that one heated reactor cell in our labora-tory has defied explanation. Cell #4 had been used,and continues to be used, for hydrocarbon isomeriza-tion/decomposition studies, and has not produced un-expected products or behavior from any other reactant.The only clue is that, in some rate studies that pre-ceeded the study of tetra-MCP isomerization, Lewisacids BCl3 and BBr3 were introduced into that cellto examine their catalytic effect on rates of structuraland geometric isomerizations of many hydrocarbons,including other alkyl-substituted cyclopropanes [12].However, experiments including those Lewis acids hadalso been carried out in two of the other three reactorcells, and no 2,2,3-TMB has been detected in the prod-ucts of experiments run in those cells.
Table II lists the activation parameters deter-mined in our laboratory for structural isomerization

488 LEWIS ET AL.
Table III Activation Parameters for Total Structural Isomerization Reactions (to All Product Isomers) forCyclopropane and Four Methylcyclopropanes
Reactant log10 (A, s−1) Ea (kcal/mol) k700 K (s−1) k1100 K (s−1) Ea** Ref.
CP 15.20 65.0 0.8 × 10−5 1.9 × 102 65.0 [1]
MCP 15.37 64.4 1.8 × 10−5 3.8 × 102 63.8 [2]
di-MCP 15.04 61.8 5.6 × 10−5 5.8 × 102 62.3 [3]
tri-MCP 15.28 63.7 2.5 × 10−5 4.2 × 102 63.4 [6]
tetra-MCP 15.47 64.7 1.9 × 10−5 4.1 × 102 63.8 This work
Rate constants at 700 and 1100 K are taken from the least squares reduction of each data set. For the explanation of E∗∗a see text.
of cyclopropane and four methylcyclopropanes. Thereis a clear trend of decreasing activation energies withincreasing methyl substitution from CP through di-MCP, then a reversal of that trend with further methyl-for-hydrogen substitution. In comparison, the pre-exponential factors show no particular trend; in factthey are equivalent (95% confidence level) at 15.2(±0.3). Actual rate constants, at 700 K and at 1100 K,increase with methyl substitution from CP through di-MCP then decrease with further methyl substitution, instep with the trend in activation energies.
Given the statistical equivalence (within 2σ uncer-tainty ranges) of the pre-exponential factors for to-tal structural isomerization of cyclopropane and fourmethylcyclopropanes, it is interesting to compare theactivation energies that are obtained from the experi-mental rate data (using average k values at 700 K fromeach study) if log A is taken to be 15.2 for each re-action. Those values are included in Table III as E∗∗
a .This comparison produces a more even decline of 1.2–1.5 kcal/mol per methyl group added to CP from CPthrough di-MCP, followed by a gradual rise in Ea withfurther methyl substitution. We propose that this is thebest overall representation of the activation parametersfor CP and the family of methylcyclopropanes fromMCP through tetra-MCP.
B.L.K. thanks Hollins University for partial support. We also
thank Steven Hughes, Kevin Wilkinson, and Sara Wilkinson
for their assistance with portions of this study.
BIBLIOGRAPHY
1. (a). Bradley, J. N.; Frend, M. A. Trans Faraday Soc 1971,
67, 72–82; (b) Jeffers, P. M.; Lewis, D. K.; Sarr, M. S. J
Phys Chem 1973, 77, 3037–3041.
2. Kalra, B. L.; Cho, J. Y.; Lewis, D. K. J Phys Chem A
1999, 103, 362–364.
3. Kalra, B. L.; Lewis, D. K. Int J Chem Kinet 2001, 33,
853–858.
4. Flowers, M. C.; Frey, H. M. Proc R Soc (London) 1961,
A260, 424–432.
5. O’Neal, H. E.; Henfling, D. Int J Chem Kinet 1972, 4,
117–126.
6. Lewis, D. K.; Hughes, S. V.; Miller, J. D.; Schlier, J.;
Wilkinson, A. K.; Wilkinson, S. R.; Kalra, B. L. Int J
Chem Kinet 2006, 38, 475–482.
7. Frey, H. M.; Marshall, D. C. J Chem Soc 1962, 3052–
3055.
8. Blumstein, C.; Henfling, D.; Sharts, C. M.; O’Neal,
H. E. Int J Chem Kinet 1970, 2, 1–10.
9. (a). O’Neal, H. E.; Benson, S. W. J Phys Chem1968, 72,
1866–1887; (b) Benson, S. W.; O’Neal, H. E. Kinetic
Data on Gas Phase Unimolecular Reactions; NSRDS
NBS 21, U. S. Government Printing Office: Washington,
DC, 1970; p. 225.
10. Tsang, W. Int J Chem Kinet 1973, 5, 651–662.
11. Lewis, D. K.; Giesler, S. E.; Brown, M. S. Int J Chem
Kinet 1978, 10, 277–294.
12. (a) Lewis, D. K.; Bergman, J.; Manjoney, R.; Paddock,
R.; Kalra, B. L. J Phys Chem 1984, 88, 4112–4116; (b)
Kalra, B. L.; Clark, K. G.; Lewis, D. K. J Phys Chem
1988, 92, 263–264.





























