Embed Size (px)
Citation preview

The first generation DNA Sequencing
Slides 3‐17 are modified fromfaperta.ugm.ac.id/newbie/download/pak_tar/.../Instrument20072.pptslides 18‐43 are from Chengxiang Zhai at UIUC.
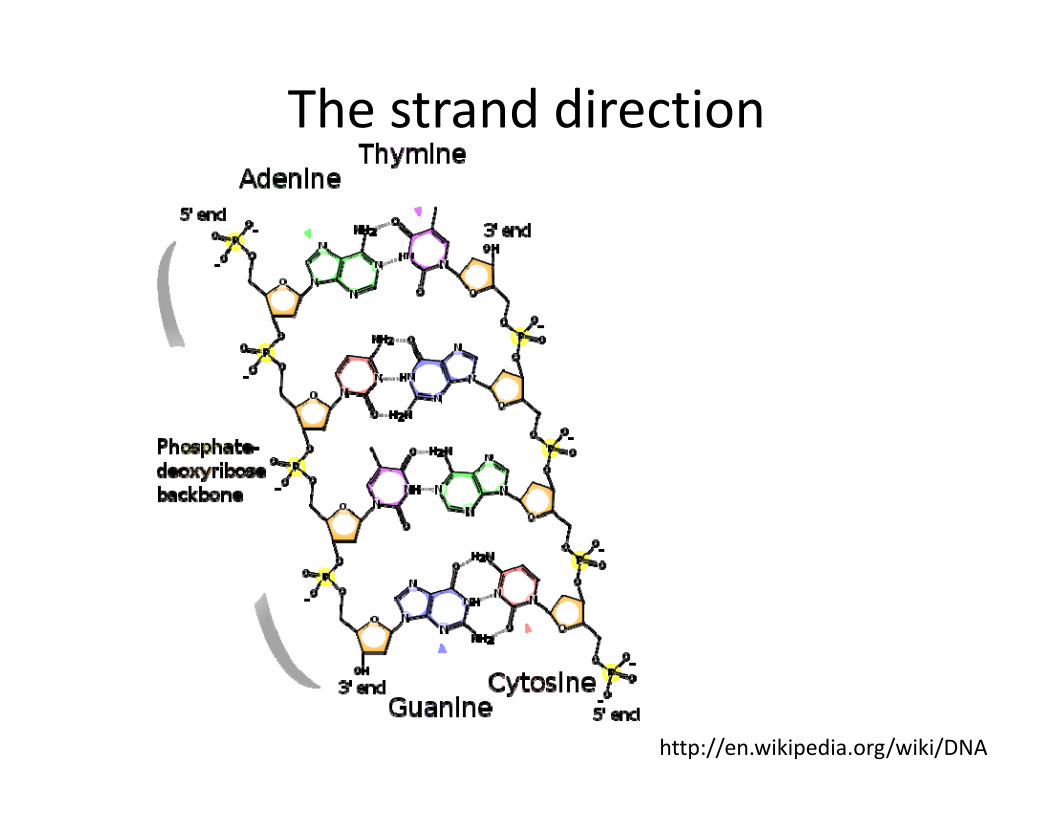
The strand direction
http://en.wikipedia.org/wiki/DNA

DNA sequencing Determination of nucleotide sequence
the determination of the precise sequence of nucleotides in a sample of DNA
Two similar methods:1. Maxam and Gilbert method
2. Sanger method
They depend on the production of a mixture of oligonucleotides labeled either radioactively or fluorescein, with one common end and differing in
length by a single nucleotide at the other end This mixture of oligonucleotides is separated by high resolution electrophoresis on polyacrilamide gels and the position of the bands
determined

Maxam-Gilbert
• Walter Gilbert– Harvard physicist– Knew James Watson– Became intrigued with
the biological side– Became a biophysicist
• Allan Maxam
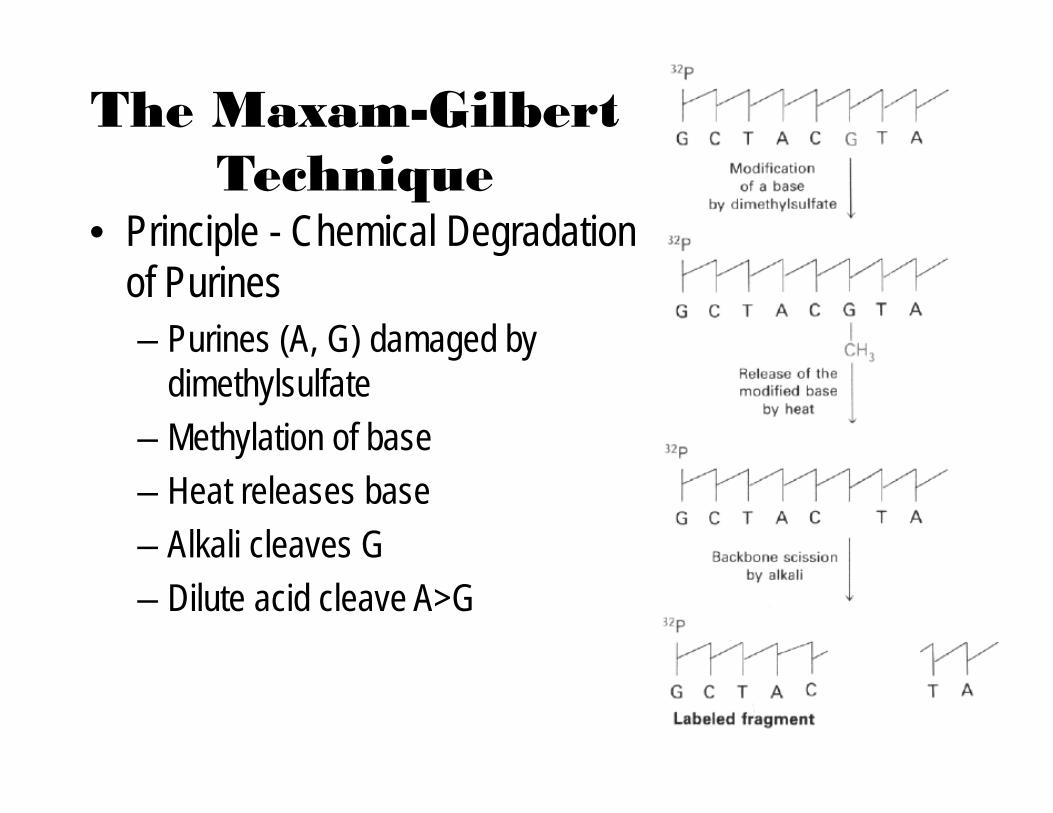
The Maxam-Gilbert Technique
• Principle - Chemical Degradation of Purines– Purines (A, G) damaged by
dimethylsulfate– Methylation of base– Heat releases base– Alkali cleaves G– Dilute acid cleave A>G
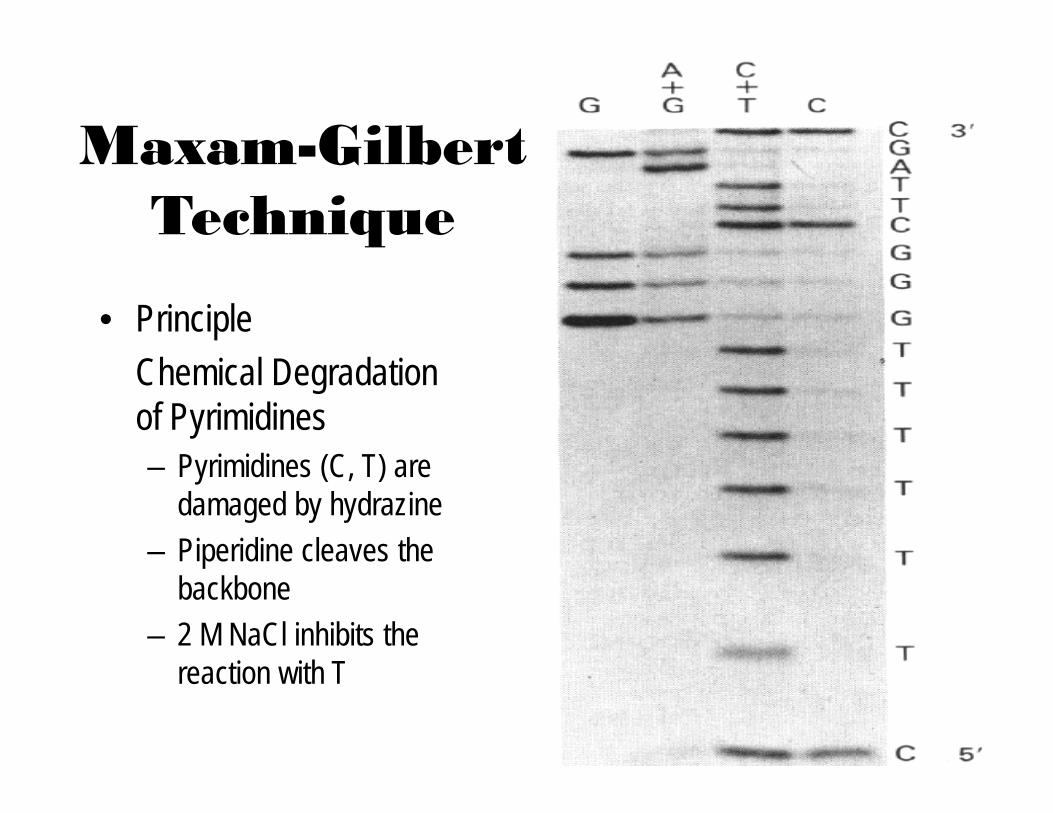
Maxam-Gilbert Technique
• PrincipleChemical Degradation of Pyrimidines– Pyrimidines (C, T) are
damaged by hydrazine– Piperidine cleaves the
backbone– 2 M NaCl inhibits the
reaction with T

Advantages/disadvantagesMaxam-Gilbert sequencing
Requires lots of purified DNA, and many intermediate purification steps Relatively short readings
Automation not available (sequencers) Remaining use for ‘footprinting’ (partial protection against DNA modification when proteins bind to specific regions, and that produce
‘holes’ in the sequence ladder)
In contrast, the Sanger sequencing methodology requires little if any DNA purification, no restriction digests, and no labeling of
the DNA sequencing template

Sanger Method
Fred Sanger, 1958– Was originally a protein
chemist– Made his first mark in
sequencing proteins– Made his second mark in
sequencing RNA 1980 dideoxy
sequencing

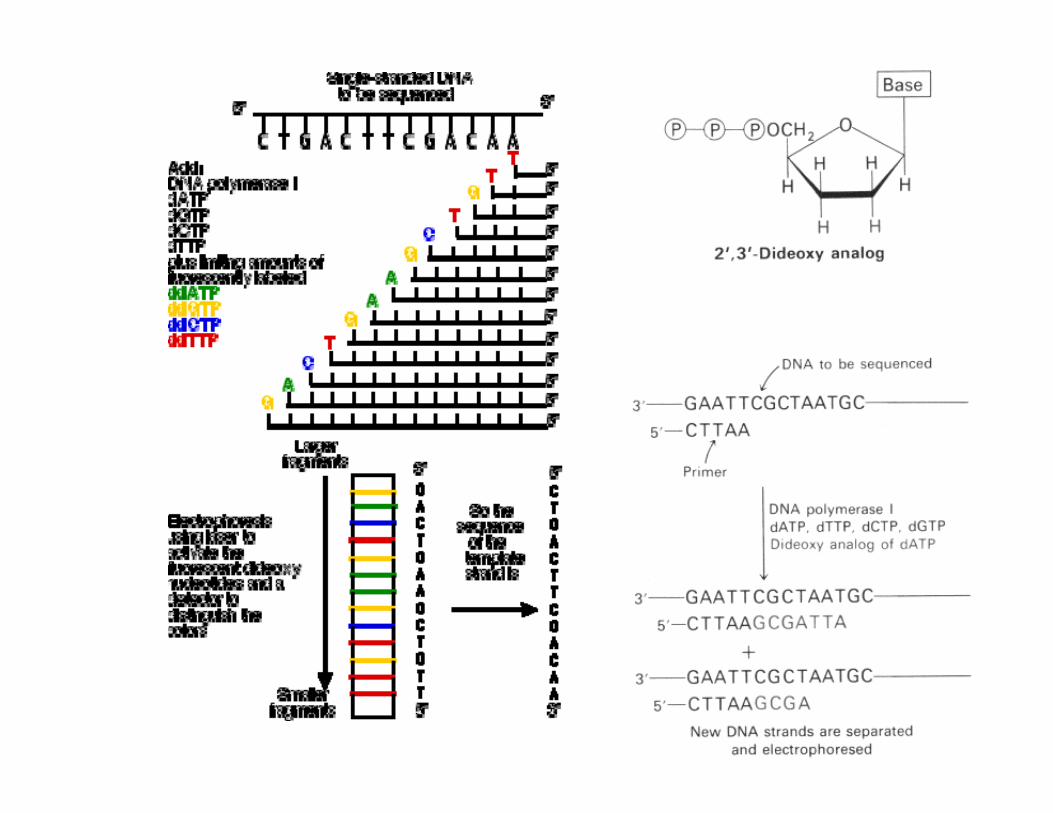
Sanger Method
in-vitro DNA synthesis using ‘terminators’, use of dideoxy-nucleotides that do not permit chain elongation after their
integration DNA synthesis using deoxy- and dideoxynucleotides that
results in termination of synthesis at specific nucleotidesRequires a primer, DNA polymerase, a template, a mixture of
nucleotides, and detection system Incorporation of di-deoxynucleotides into growing strand
terminates synthesis Synthesized strand sizes are determined for each di-deoxynucleotide by using gel or capillary electrophoresis
Enzymatic methods
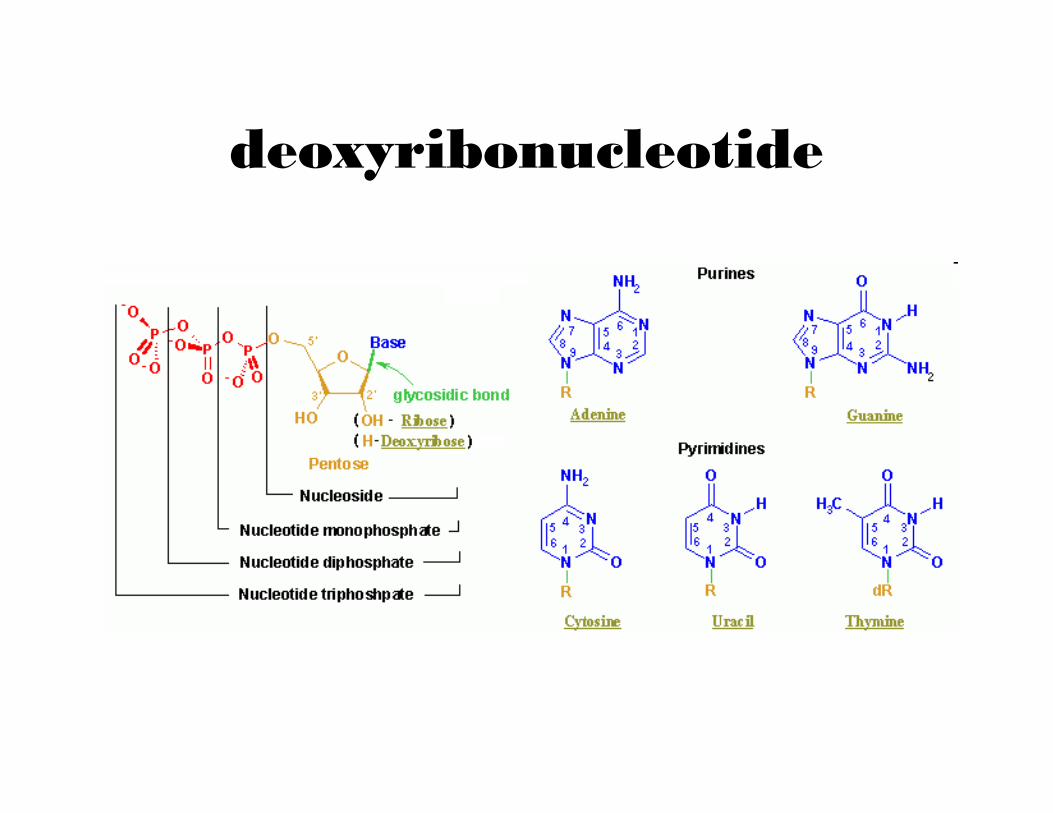
deoxyribonucleotide
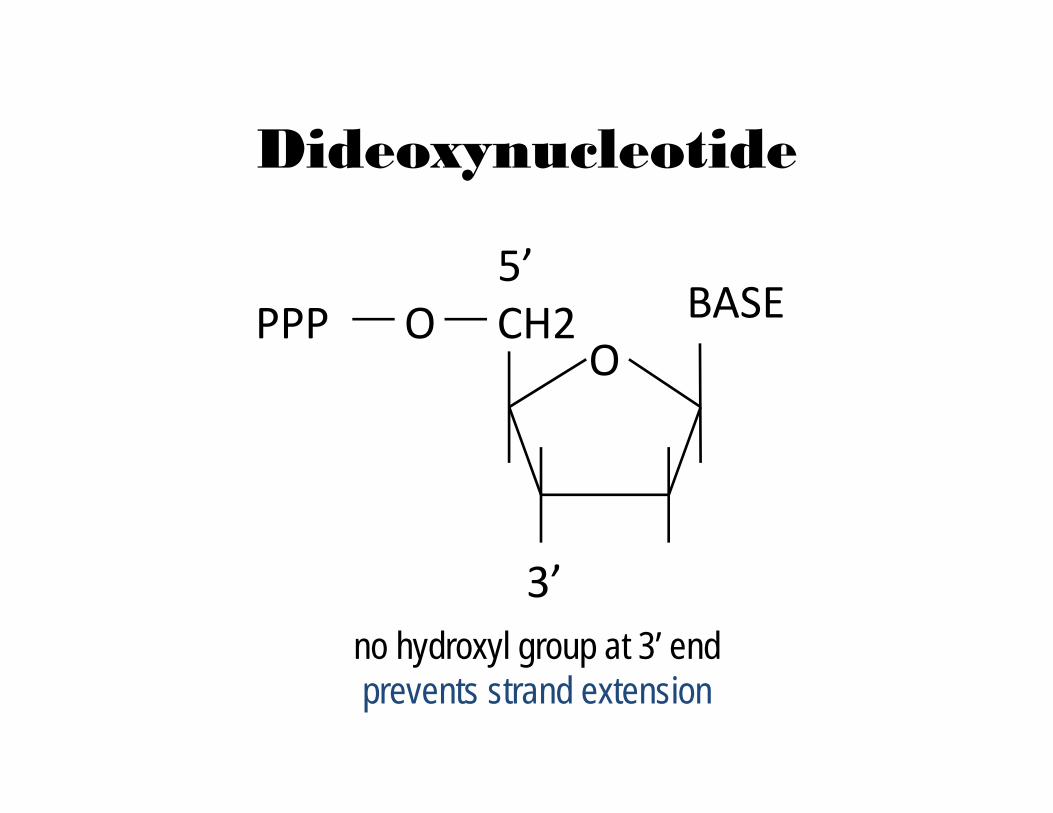
Dideoxynucleotide
no hydroxyl group at 3’ endprevents strand extension
CH2O
OPPP5’
3’
BASE
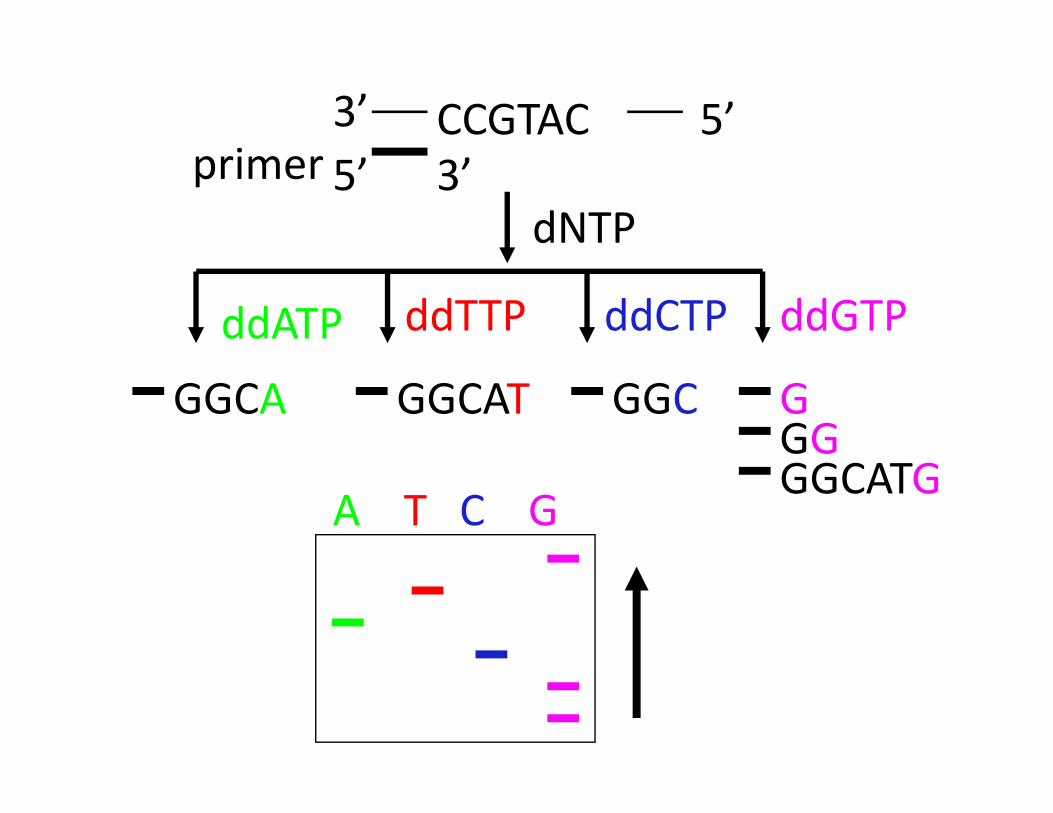
CCGTAC3’ 5’5’ 3’primer
dNTP
ddATP
GGCA
ddTTP
GGCAT
ddCTP
GGC G
ddGTP
GGGGCATG
A T C G
Sample Output
1 lane
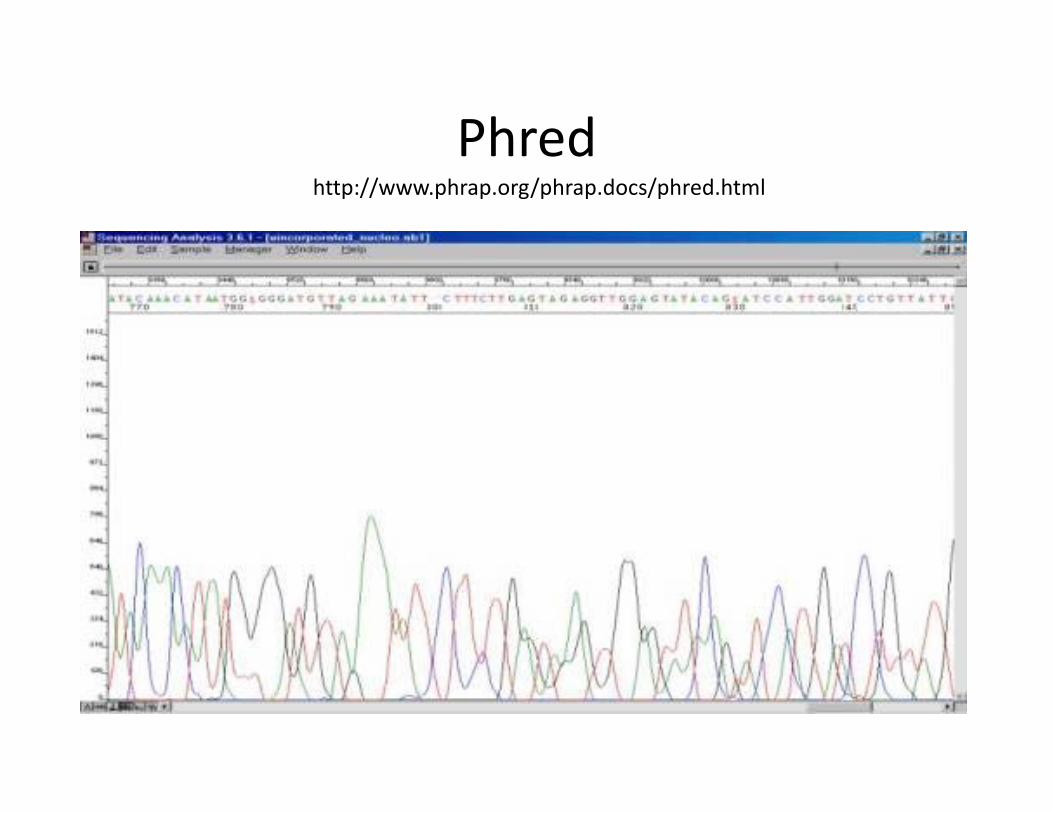
Phredhttp://www.phrap.org/phrap.docs/phred.html

Sanger sequencing• Laser excitation of fluorescent labels as fragments of discreet lengths
exit the capillary, coupled to four‐color detection of emission spectra, provides the readout that is represented in a Sanger sequencing ‘trace’. Software translates these traces into DNA sequence, while also generating error probabilities for each base‐call.
• Simultaneous electrophoresis in 96 or 384 independent capillaries provides a limited level of parallelization.
• After three decades of gradual improvement, the Sanger biochemistry can be applied to achieve read‐lengths of up to ~1,000 bp, and per‐base ‘raw’ accuracies as high as 99.999%. In the context of highthroughput shotgun genomic sequencing, Sanger sequencing costs on the order of $0.50 per kilobase.

Comparison
• Sanger Method– Enzymatic– Requires DNA synthesis– Termination of chain
elongation
• Maxam Gilbert Method– Chemical– Requires DNA– Requires long stretches of
DNA– Breaks DNA at different
nucleotides

How to obtain the human genome sequence
• The Sanger sequencing can only generate 1kb long DNA segments.
• How to obtain the human genome that are 3 billion letters? The answer is to get pieces of DNA segments and assemble them into the genome.

Challenges with Fragment Assembly
• Sequencing errors
~1‐2% of bases are wrong
•Repeats
false overlap due to repeat
Bacterial genomes:5%Mammals: 50%

Repeat Types• Low-Complexity DNA (e.g. ATATATATACATA…)
• Microsatellite repeats (a1…ak)N where k ~ 3‐6(e.g. CAGCAGTAGCAGCACCAG)
• Transposons/retrotransposons– SINE Short Interspersed Nuclear Elements
(e.g., Alu: ~300 bp long, 106 copies)
– LINE Long Interspersed Nuclear Elements~500 ‐ 5,000 bp long, 200,000 copies
– LTR retroposons Long Terminal Repeats (~700 bp) at each end
• Gene Families genes duplicate & then diverge
• Segmental duplications ~very long, very similar copies

Strategies for whole‐genome sequencing
1. Hierarchical – Clone‐by‐clone yeast, worm, humani. Break genome into many long fragmentsii. Map each long fragment onto the genomeiii. Sequence each fragment with shotgun
2. Online version of (1) – Walking rice genomei. Break genome into many long fragmentsii. Start sequencing each fragment with shotguniii. Construct map as you go
3. Whole Genome Shotgun fly, human, mouse, rat, fugu
One large shotgun pass on the whole genome

Hierarchical Sequencing vs. Whole Genome Shotgun
• Hierarchical Sequencing– Advantages: Easy assembly– Disadvantages:
• Build library & physical map; • Redundant sequencing
• Whole Genome Shotgun (WGS)– Advantages: No mapping, no redundant sequencing– Disadvantages: Difficult to assemble and resolve repeats
Whole Genome Shotgun appears to get more popular…
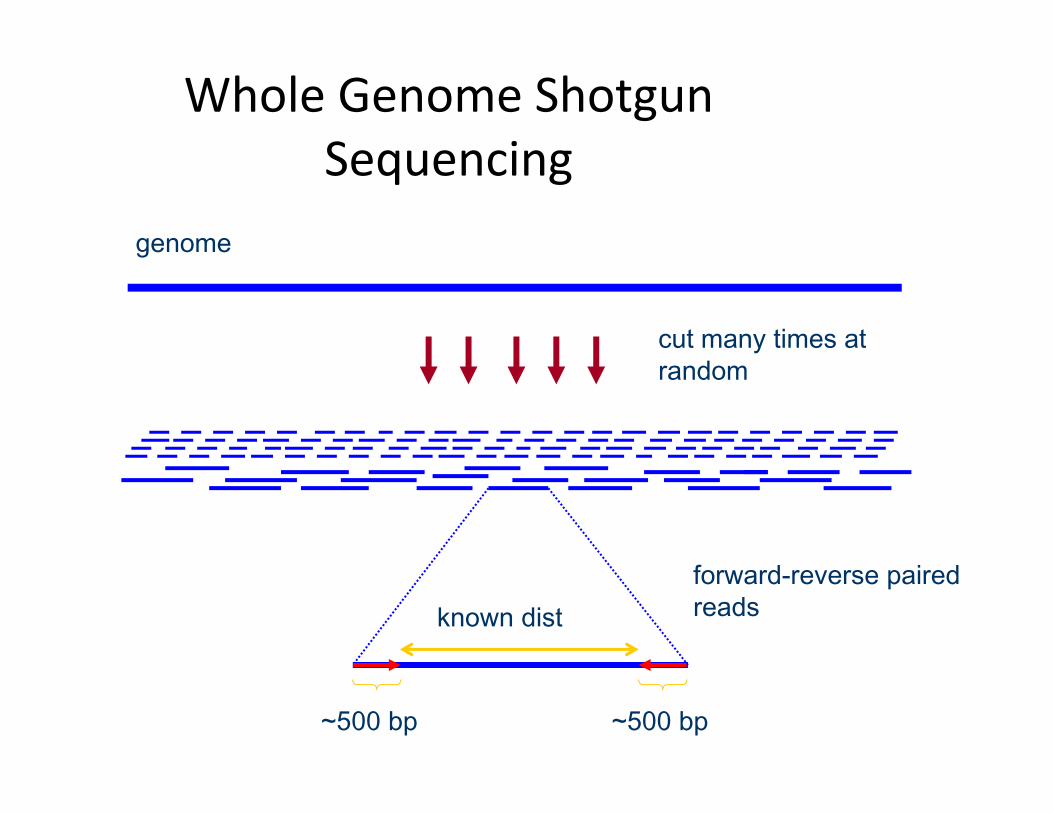
Whole Genome Shotgun Sequencing
cut many times at random
genome
forward-reverse paired readsknown dist
~500 bp~500 bp
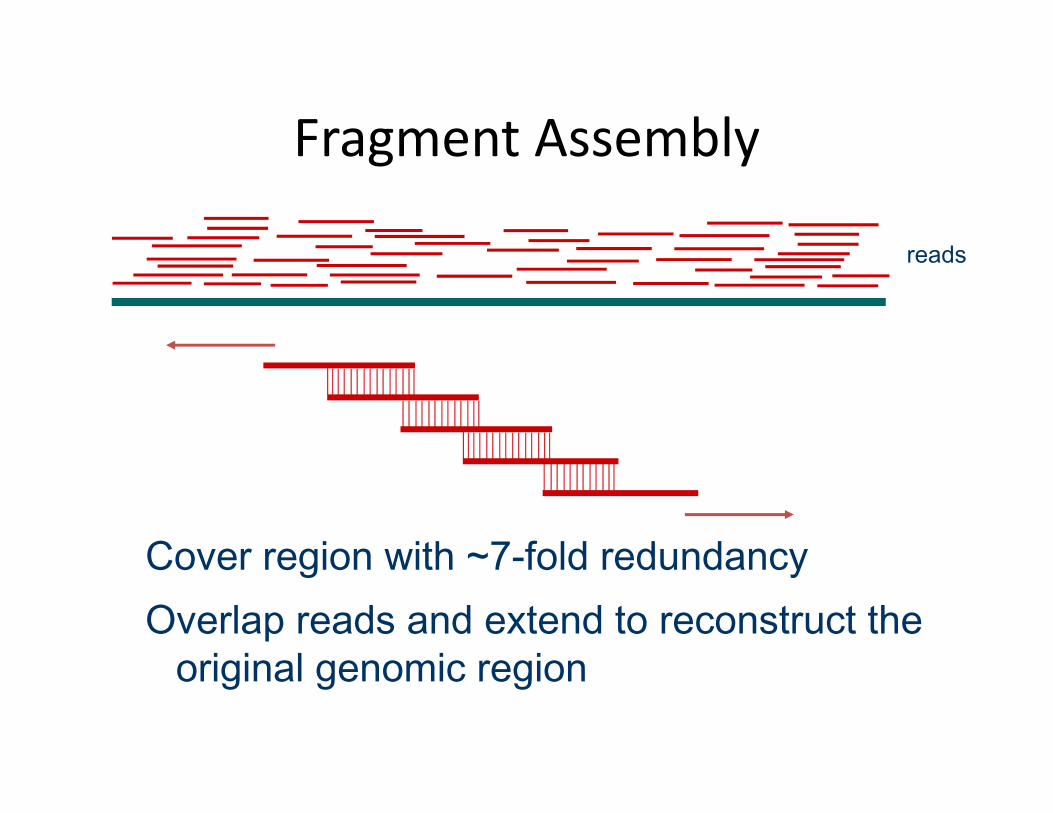
Fragment Assembly
Cover region with ~7-fold redundancyOverlap reads and extend to reconstruct the
original genomic region
reads

Read Coverage
Length of genomic segment: GNumber of reads: NLength of each read: L
Definition: Coverage C = NL/ G
C

Enough Coverage
How much coverage is enough?
According to the Lander‐Waterman model:
Assuming uniform distribution of reads, C=7 results in 1 gap per 1,000 nucleotides

Lander‐Waterman Model• Major Assumptions
– Reads are randomly distributed in the genome– The number of times a base is sequenced follows a Poisson
distribution
• Implications– G= genome length, L=read length, N = # reads– Mean of Poisson: =LN/G (coverage)– % bases not sequenced: p(X=0) =0.0009 = 0.09%– Total gap length: p(X=0)*G– Total number of gaps: p(X=0)*N
( )!
xep X xx
Average times
This model was used to plan the Human Genome Project…
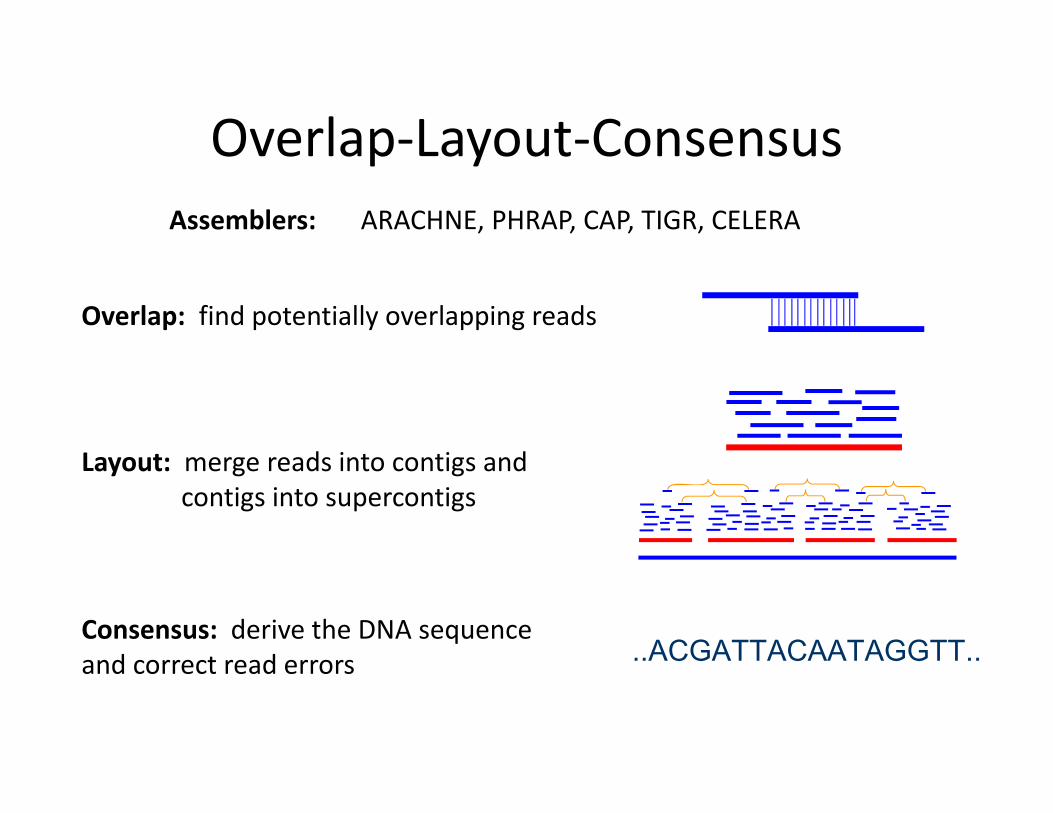
Overlap‐Layout‐Consensus Assemblers: ARACHNE, PHRAP, CAP, TIGR, CELERA
Overlap: find potentially overlapping reads
Layout: merge reads into contigs and contigs into supercontigs
Consensus: derive the DNA sequence and correct read errors ..ACGATTACAATAGGTT..

Overlap
• Find the best match between the suffix of one read and the prefix of another
• Due to sequencing errors, need to use dynamic programming to find the optimal overlap alignment
• Apply a filtration method to filter out pairs of fragments that do not share a significantly long common substring

Overlapping Reads
TAGATTACACAGATTAC
TAGATTACACAGATTAC|||||||||||||||||
• Sort all k‐mers in reads (k ~ 24)
• Find pairs of reads sharing a k-mer
• Extend to full alignment – throw away if not >95% similar
T GA
TAGA| ||
TACA
TAGT||

Overlapping Reads and Repeats
• A k‐mer that appears N times, initiates N2
comparisons
• For an Alu that appears 106 times 1012comparisons – too much
• Solution:Discard all k‐mers that appear more than
t Coverage, (t ~ 10)
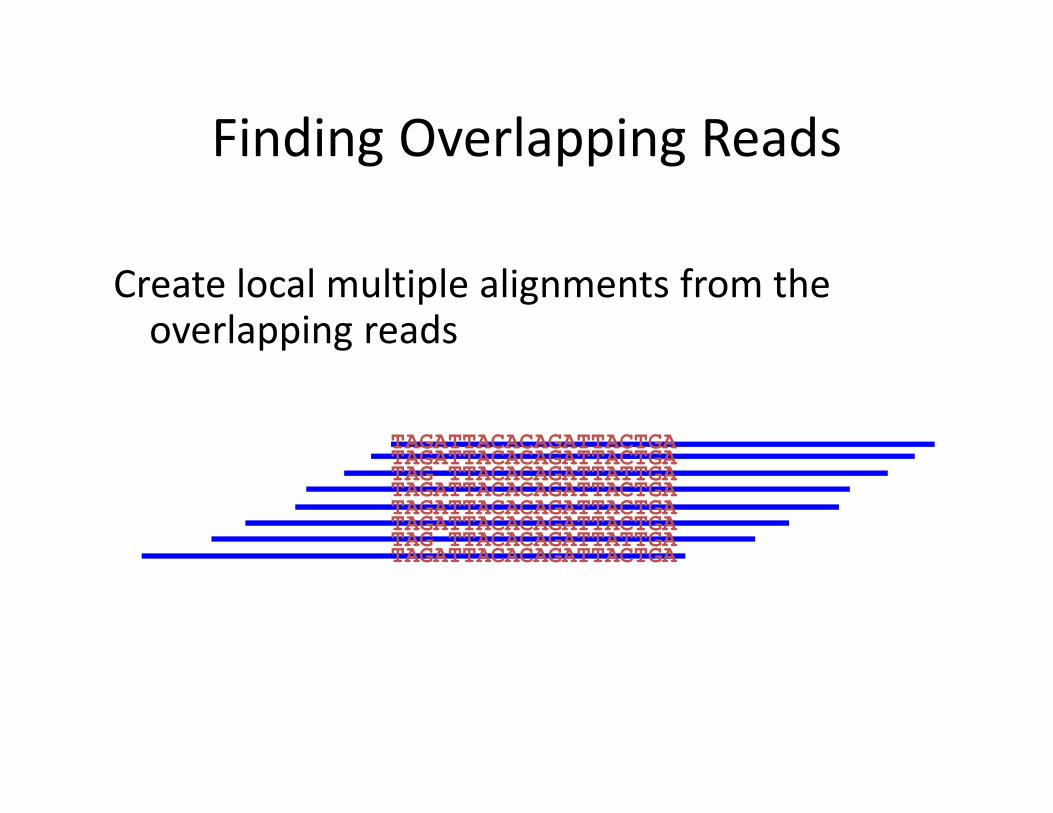
Finding Overlapping Reads
Create local multiple alignments from the overlapping reads
TAGATTACACAGATTACTGATAGATTACACAGATTACTGATAG TTACACAGATTATTGATAGATTACACAGATTACTGATAGATTACACAGATTACTGATAGATTACACAGATTACTGATAG TTACACAGATTATTGATAGATTACACAGATTACTGA
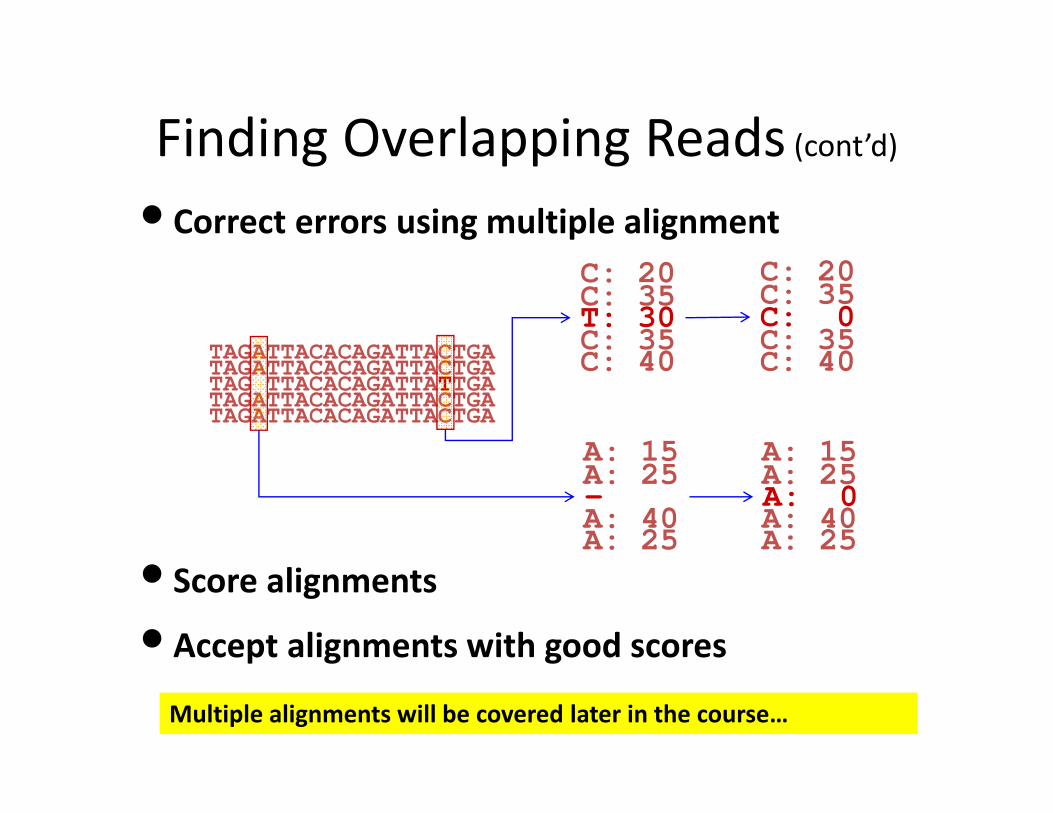
Finding Overlapping Reads (cont’d)
• Correct errors using multiple alignment
TAGATTACACAGATTACTGATAGATTACACAGATTACTGATAG TTACACAGATTATTGATAGATTACACAGATTACTGATAGATTACACAGATTACTGA
C: 20C: 35T: 30C: 35C: 40
C: 20C: 35C: 0C: 35C: 40
• Score alignments
•Accept alignments with good scores
A: 15A: 25A: 40A: 25-
A: 15A: 25A: 40A: 25A: 0
Multiple alignments will be covered later in the course…

Layout
• Repeats are a major challenge• Do two aligned fragments really overlap, or are they from two copies of a repeat?
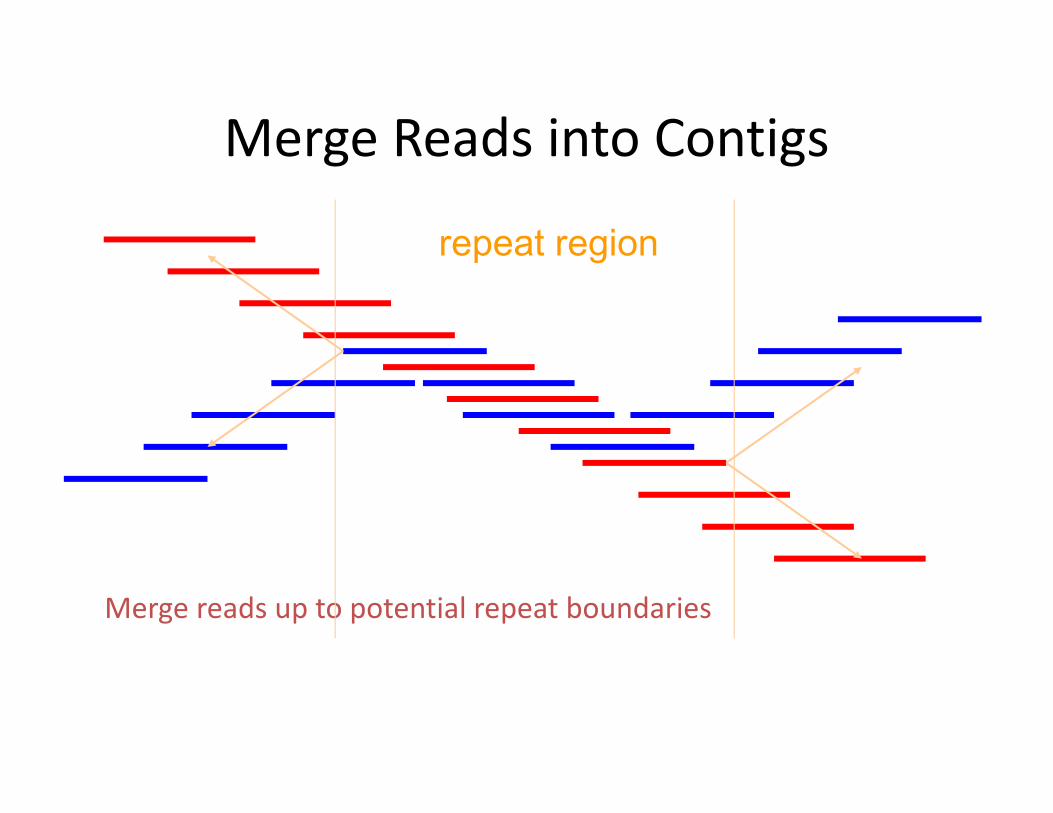
Merge Reads into Contigs
Merge reads up to potential repeat boundaries
repeat region

Merge Reads into Contigs (cont’d)
• Ignore non‐maximal reads• Merge only maximal reads into contigs
repeat region
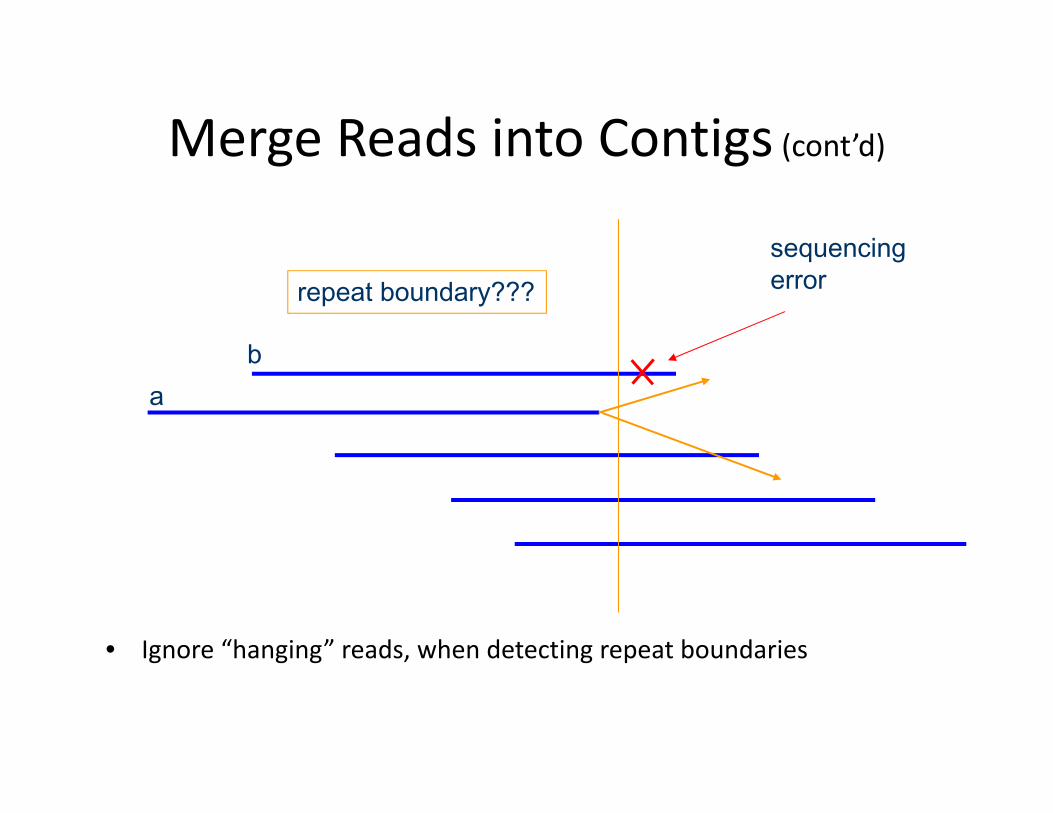
Merge Reads into Contigs (cont’d)
• Ignore “hanging” reads, when detecting repeat boundaries
sequencing errorrepeat boundary???
ba

Merge Reads into Contigs (cont’d)
?????
Unambiguous
• Insert non-maximal reads whenever unambiguous

Link Contigs into Supercontigs
Too dense: Overcollapsed?(Myers et al. 2000)
Inconsistent links: Overcollapsed?
Normal density

Link Contigs into Supercontigs (cont’d)
Find all links between unique contigs
Connect contigs incrementally, if 2 links
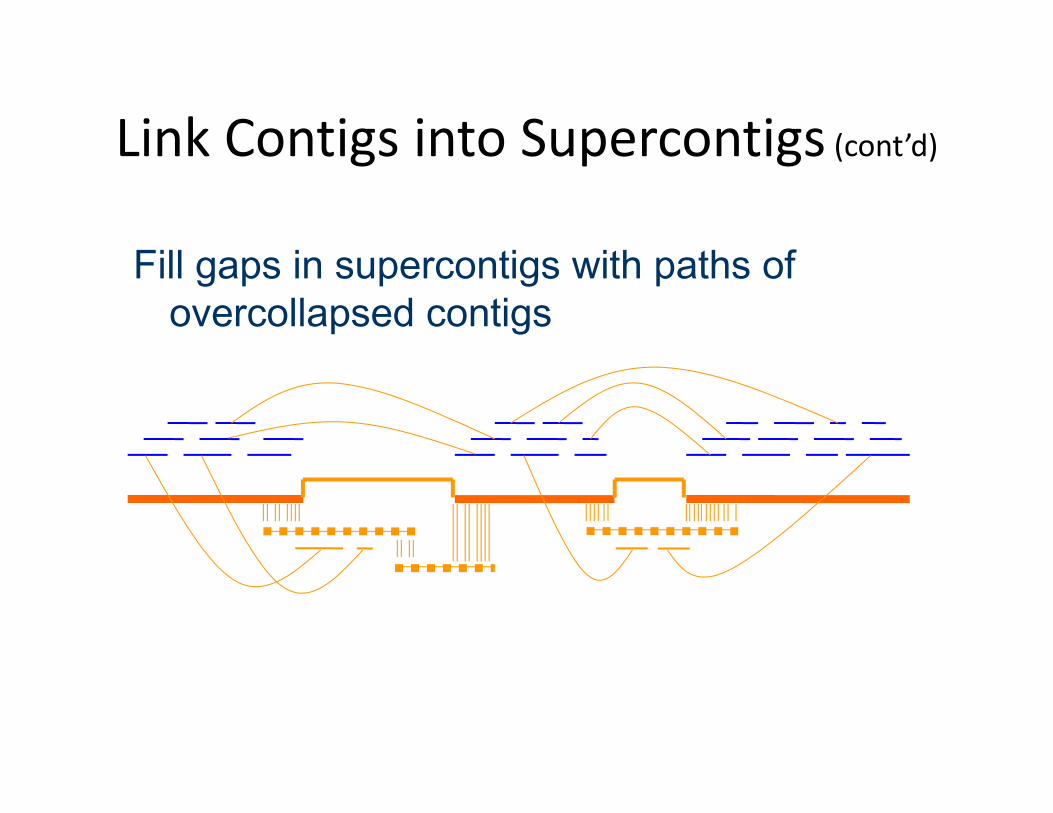
Link Contigs into Supercontigs (cont’d)
Fill gaps in supercontigs with paths of overcollapsed contigs

Consensus
• A consensus sequence is derived from a profile of the assembled fragments
• A sufficient number of reads is required to ensure a statistically significant consensus
• Reading errors are corrected

Derive Consensus Sequence
Derive multiple alignment from pairwise read alignments
TAGATTACACAGATTACTGA TTGATGGCGTAA CTATAGATTACACAGATTACTGACTTGATGGCGTAAACTATAG TTACACAGATTATTGACTTCATGGCGTAA CTATAGATTACACAGATTACTGACTTGATGGCGTAA CTATAGATTACACAGATTACTGACTTGATGGGGTAA CTA
TAGATTACACAGATTACTGACTTGATGGCGTAA CTA
Derive each consensus base by weighted voting










