Embed Size (px)
Citation preview

Lecture 20: Binding Free Energy Calculations
Dr. Ronald M. Levy [email protected]
Statistical Thermodynamics

focus on the binding problem of protein-ligand systems.
• Statistical theory of non-covalent binding equilibria • Computer models for computing free energy of binding • Binding energy distribution analysis method • Examples from our research group Center for Biophysics and Computational Biology, https://cb2.cst.temple.edu/
Outline

General chemical reaction: !!ν AA+νBB !!νCC+νDD
Very important type of “reaction”: bimolecular non-covalent binding
R(sol) + L(sol) RL(sol)
l Small molecule dimerization/association l Supramolecular complexes l Protein-ligand binding l Protein-protein binding/dimerization l Protein-nucleic acids interactions l ...
*Note*: We are implicitly assuming above that we can describe the system as being composed of 3 distinct chemical “species”, R, L, and RL (quasi-chemical description). If interactions between R and L are weak/non-specific then it would be more appropriate to treat the system as a non-ideal solution of R and L.
Sta$s$cal Theory of Non-‐Covalent Binding Equilibria

From general theory of chemical reactions, for receptor-ligand system:
( )( )
( ) ( )RL( )/ eq.
R Leq. eq.
/Binding
/ /constant
ΔA T KTbb
C CK T = = e =
C C C C−
oo
o o
( ) ( )3 3
3
1 βΨ un iui unu nu
ujj
xT = dx e
Λϕ
−−∫∏
( )ui xΨ Effective potential energy of solute i in solution.
011 =ν;=ν;=ν=ν DCBA
( ) ( ) ( ) ( )( ) ( )
( )( ) ( )RL
R L
ν νC DC DD C A B
b ν νA BA B
ν +ν ν ν T T TK T = V =C
T TT T
ϕ ϕ ϕϕ ϕϕ ϕ
− −o o

If the solution is isotropic ( invariant upon rotation of solute),
( ) ( ) 223 62
3 3
888βΨ un i iu
i θi un nu un nu uuj uj
j j
x π Z'πT = π = dx e =Λ Λ
ϕ ϕ−−∫
∏ ∏
( )i uΨ x
integrate analytically over rotational degrees of freedom (ignoring roto-vibarational couplings, OK at physiological temperatures).
Internal coordinates
When inserting into the expression for Kb(T), Λ's cancel because LRRL n+n=nWe get:
( ) RL2
R L8bZ'CK T =
π Z' Z'
o
( )RRβΨRnR
xexd=Z' −−∫ 63R
( )LLβΨLnL
xexd=Z' −−∫ 63L
!!Z'RL = ???[Gilson et al. Biophysical Journal 72, 1047-1069 (1997)]

In the complex, RL, the “external” coordinates (translations, rotations) of the ligand become internal coordinates of the complex.
( )LLRRLβΨL
LnL
RnR
ζ,x,xedζdxxd=Z' −−−∫ 66363RL
R
L
( ) ( ) 3216 ,,,, ωωω,θr=ζ L φ
Position of the ligand relative to receptor frame
Orientation of the ligand relative to receptor frame
External coordinates of the ligand relative to receptor
It is up to us to come up with a reasonable definition of “BOUND”. That is we need to define the RL species before we can compute its partition function. The binding constant will necessarily depend on this definition. Must match experimental reporting. If the binding is strong and specific the exact definition of the complexed state is often not significant.

It is convenient to introduce an “indicator” function for the complex:
( )⎪⎩
⎪⎨⎧
site binding outside is ligand0
site bindingin is ligand1=ζI L
!!Z'RL = …∫ = d∫ xR3nR−6dxL
3nL−6dζ L6I ζ L( )e
−βψRL xR ,xL ,ζ L( )then:
Next, define “binding energy” of a conformation of the complex:
( ) ( ) ( ) ( )LRRRLLRRLLLR xΨxΨζ,x,xΨ=ζ,x,xu=u −−
basically, change in effective energy for bringing ligand and receptor together at fixed internal conformation:
+

In terms of binding energy:
then:
( ) ( ) ( ) ( )LLRLRRRLLRRL ζ,x,xu+xΨ+xΨ=ζ,x,xΨ
!!
Kb T( )= C !
8π2Z'RLZ'RZ'L
=
C !
8π2
d∫ xR3nR−6dxL
3nL−6dζ L6I ζ L( )e
−βψR xR( )e−βψL xL( )
e−βu xR ,xL ,ζL( )
d∫ xR3nR−6dxL
3nL−6e−βψR xR( )
e−βψL xL( )
Now: we are not very good at computing partition functions. We are much better at computing ensemble averages:
( ) ( )
( ) ( ) ( )xρxOdx=edx
exOdx=O
xβU
xβU
∫∫∫
−
−
><

To transform the expression for Kb so that it looks like an average:
multiply and divide by: !! d∫ ζ L6I ζ L( )=VsiteΩsite
!!
Kb T( )= C !
8π2Z'RLZ'RZ'L
=
C !
8π2
d∫ xR3nR−6dxL
3nL−6dζ L6I ζ L( )e
−βψR xR( )e−βψL xL( )
e−βu xR ,xL ,ζL( )
d∫ xR3nR−6dxL
3nL−6e−βψR xR( )
e−βψL xL( ) =
VsiteV !
Ωsite8π2
d∫ xR3nR−6dxL
3nL−6dζ L6I ζ L( )e
−βψR xR( )e−βψL xL( )
e−βu xR ,xL ,ζL( )
d∫ xR3nR−6dxL
3nL−6dζ L6I ζ L( )e
−βψR xR( )e−βψL xL( )
then:
or:
!! Kb T( )= Vsite
V !Ωsite8π2 < e
−βu xR ,xL ,ζL( ) >R+L+I( )

We can see that binding constant can be expressed in terms of an average of the exponential of the binding energy over the ensemble of conformations of the complex in which the ligand and the receptor are not interacting while the ligand is placed in the binding site.
( ) ( )( )
site siteRL2 2
R L8 8βu xR L L
b R+L+I
,x ,ζV ΩZ'CK T = eπ Z' Z' V π
−= < >
o
o
Standard free energy of binding: ( ) ( )TKkT=TΔA bb ln−!
( ) ( )( )
site site2ln ln ln
8βu xR L L
b R+L+I
,x ,ζV ΩΔA T = kT kT kT eV π
−− − − < >o
o
( )naltranslatio!bΔA
(analytic formulas) (numerical computation)
( ) ( ) ( ) ( )LRRRLLRRLLLR xΨxΨζ,x,xΨ=ζ,x,xu=u −−
Summary of Binding Free Energy Theory
Binding energy:
Binding Constant:

Interpretation in terms of binding thermodynamic cycle:
RL R + L
R(L) “Virtual” state in which ligand is in binding site without interacting with receptor
Ligand and receptor in solution at concentration Cº
Ligand bound to receptor in solution at concentration Cº
Loss of translational, rotational freedom (to fit binding site definition)
Binding while in receptor site (independent of concentration)
( )rt+ΔAb!
!!ΔAb exc.( )
!bΔA
( ) ( )exc.rt bbb ΔA++ΔA=ΔA !!
BEDAM method and computer exercise will focus on the computation of by computer simulations. !!ΔAb exc.( )

Binding Free Energy Models [Gallicchio and Levy, Adv. Prot. Chem (2012)]
K AB=C ∘
8π2Z ABZ AZ B
Double Decoupling Method (DDM)
Relative Binding Free Energies (FEP)
Potential of Mean Force/ Pathway Methods
MM/PBSA Mining Minima (M2)
Exhaustive docking
Docking & Scoring
BEDAM (Implicit solvation)
λ-dynamics
Statistical mechanics theory
Binding Energy Distribution Analysis Method

Free Energy Perturbation (FEP/TI) Double Decoupling (DDM) Jorgensen, Kollman, McCammon
(1980’s – present) Jorgensen, Gilson, Roux, . . .
(2000’s – to present)
: Challenges:
• Dissimilar ligand sets • Numerical instability
• Dependence on starting conformations • Multiple bound poses
• Slow convergence
Statistical mechanics based, in principle account for: • Total binding free energy • Entropic costs • Ligand/receptor reorganization
Free Energy Perturba$on and Double Decoupling Methods
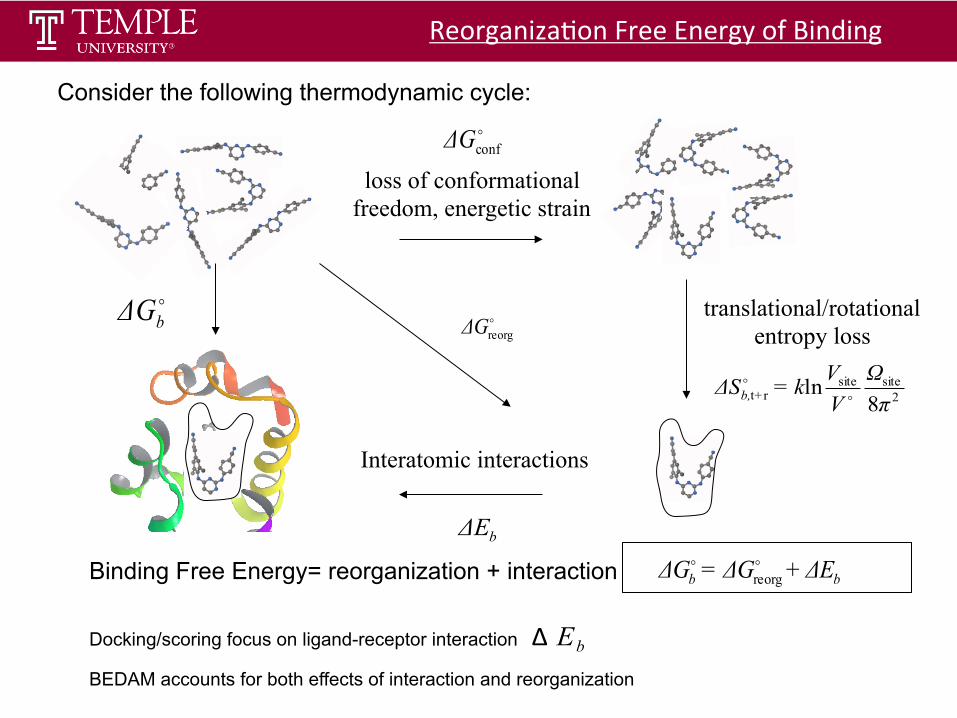
loss of conformational freedom, energetic strain
translational/rotational entropy loss
!reorgΔG
bΔE
!bΔG
Interatomic interactions
2sitesite
rt 8ln
πΩ
VVk=ΔS +b, !
!
Reorganiza$on Free Energy of Binding
Consider the following thermodynamic cycle: !confΔG
Binding Free Energy= reorganization + interaction bb ΔE+ΔG=ΔG !!reorg
Docking/scoring focus on ligand-receptor interaction Δ EbBEDAM accounts for both effects of interaction and reorganization

Binding Energy Distribu$on Analysis Method (Sta$s$cal Theory)
[Gilson, McCammon et al., (1997)]
BAABβΔF
AB ZZZC=e=K b
28π
!!−
ΔW+ΔU=ΔE Binding “energy” of a fixed conformation of the complex. W(): solvent PMF (implicit solvation model)
0siteβΔE°
AB eVC=K −
Entropically favored
:... 0 Ligand in binding site in absence of ligand-receptor interactions
"" confSTEFAB Δ−Δ=Δ !
!ABF
AB eK Δ−== β Area

Binding Energy Distribu$on Analysis Method (Compu$ng Method)
P0 (ΔE): encodes all enthalpic and entropic effects
• Solution: 1) treat binding energy as biasing
potential = λ ΔE λ=0: uncoupled/unbound state, weakly coupled states λ=1: full coupled /bound state 2) Hamiltonian Replica Exchange
+WHAM
ΔE [kcal/mol]
P0(Δ
E ) [
kcal
/mol
-1] e− βΔE
P0(ΔE)
( ) ( )ΔEPeΔEdVCeVC=K βΔEsite
βΔE°AB 00site
−− ∫= !
Integration problem: region at favorable ΔE’s is seriously undersampled.
Main contribution to integral
• Ideal for HPC cluster computing and distributed grid network
ΔW+ΔUEE=ΔE =− unboundbound
] Gallicchio, Lapelosa, & Levy, 2010; Xia, Flynn, Gallichio & et al, 2015
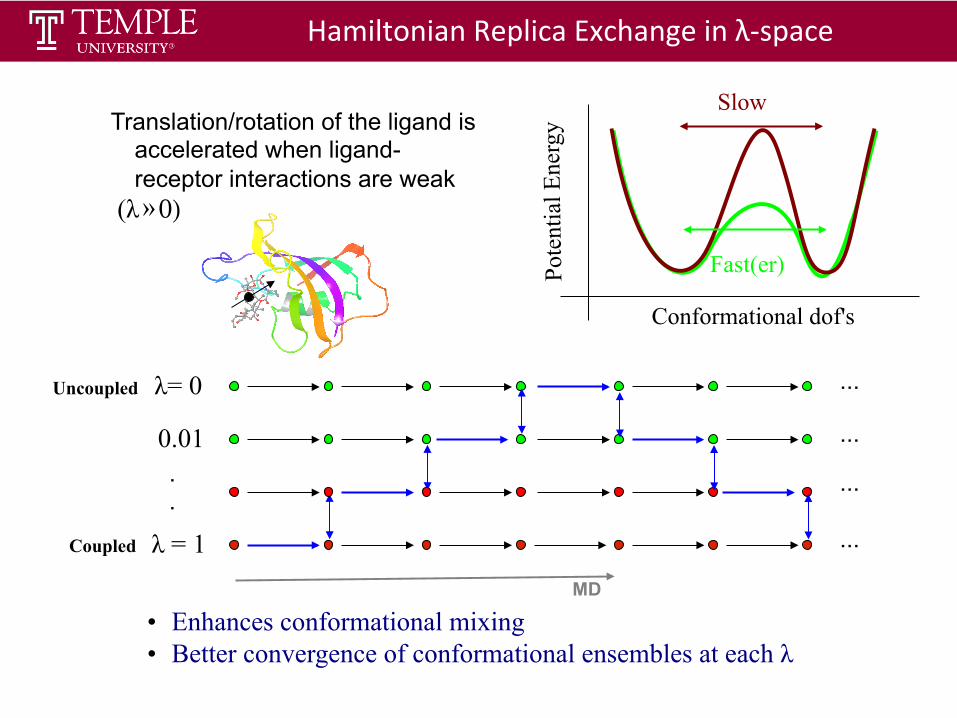
Hamiltonian Replica Exchange in λ-‐space
• Enhances conformational mixing • Better convergence of conformational ensembles at each λ
... λ= 0
λ = 1 ...
.
.
0.01 ...
...
Translation/rotation of the ligand is
accelerated when ligand-receptor interactions are weak
(λ » 0)
Slow
Conformational dof's
Fast(er) Pote
ntia
l Ene
rgy
MD
Coupled
Uncoupled

Reweighting techniques are necessary to recover the unbiased true observables (results) from more advanced sampling methods:
• Weighted Histogram Analysis Method (WHAM) Ferrenberg & Swendsen (1989) Kumar, Kollman et al. (1992) Bartels & Karplus (1997) Gallicchio, Levy et al. (2005) • Unbinned Weighted Histogram Analysis Method (UWHAM) equivalent to Multistate Bennett Acceptance Ratio (MBAR) Shirts & Chodera J. Chem. Phys. (2008). Tan, Gallicchio, Lapelosa, Levy JCTC (2012).
Reweigh$ng Techniques in Free Energy Calcula$ons

Results for Binding to Mutants of T4 Lysozyme
L99A Hydrophobic cavity
L99A/M102Q Polar cavity
Brian Matthews Brian Shoichet Benoit Roux David Mobley Ken Dill John Chodera Graves, Brenk and Shoichet, JMC (2005)
BEDAM: 20ns HREM, 12 replicas λ=10-6, 10-5, 10-4, 10-3, 10-2, 0.1, 0.15, 0.25, 0.5, 0.75, 1, 1.2
IMPACT + OPLS-AA/AGBNP2

Binders vs. Non-‐Binders
L99A T4 Lysozyme, Apolar Cavity
L99A/M102Q T4 Lysozyme, Polar Cavity

Rutgers/Temple – E. Gallicchio, N. Deng, P. He, R. Levy
Scripps - A. Perryman, S. Forli, D. Santiago, A. Olson,
SAMPL4

Large-Scale Screening by Binding Free energy Calculations: HIV-Integrase LEDGF Inhibitors
. . . . .
. . . . .
450 SAMPL4 Ligand Candidates ~350 scored with BEDAM
Docking + BEDAM Screening IN/LEDGF Binding Site
• HIV-IN is responsible for the integration of viral genome into host genome. • The human LEDGF protein links HIV-IN to the chromosome • Development of LEDGF binding inhibitors for novel HIV therapies
• SAMPL4 blind challenge: computational prediction of undisclosed experimental screens. • Docking provides little screening discrimination: “everything binds”! • Much more selectivity from absolute binding free energies • BEDAM predictions ranked first among 25 computational groups in SAMPL4, • 2.5 x fold enrichment factor in top 10% of focused library
-5
-5

Asynchronous Replica Exchange for Computa$onal Grids
• Separate local file-based asynchronous exchanges and remote MD simulations
Limited to large MD period (> 1 ps) but robust to failures of individual MD processes because no synchronizing process is required.
• Metroplis independence sampling approaching the infinite swapping limit (100s to 1000s swaps/cycle)
Exchanges between all pairs of neighbors can be performed in a local CPU independent of MD jobs running remotely.
Current Grid Computing Network: Temple University 450 CPUs Brooklyn College@CUNY 2000 CPUs World Community Grid at IBM 600,000+ CPUs
Xia, Flynn, Gallicchio, Zhang, He, Tan, & Levy, J. Comput. Chem., 2015. Gallicchio, Xia, Flynn, Zhang, Samlalsingh, Mentes, &Levy, Comput. Phys. Comm., 2015.
https://github.com/ComputationalBiophysicsCollaborative/AsyncRE
MD running remotely
Exchange locally

Async REMD for β-‐cyclodextrin-heptanoate Host-Guest System
β-‐cyclodextrin-heptanoate complex
MD running remotely
Exchange locally )
Converged binding energy distributions of λ=1 from1D Sync REMD (72ns x 16 replica)

2D Async RE Results for β-‐cyclodextrin-heptanoate Complex
Binding energy distributions of λ=1 from different REMD simulations at T=300K
MD running remotely
Exchange locally )
Binding free energy as a function of λ from different REMD simulations at T=300K





























