Embed Size (px)
Citation preview

Lezione 6
Annotazione del genoma

• Assegnare ad ogni nucleotide del genoma un possibile ruolo. • Principalmente (ma non solo) riguarda l'annotazione dei geni. • Geni
– Codificanti proteine – Geni per RNA – Retrogeni
• Elementi regolatori – Promotori – Enhancers – siRNA
• Elementi repetitivi – LINES – SINES – Simple repeats
Annotazione del genoma

• Metodi Dire* – Mediante ricerca di match perfetti o quasi perfetti con EST, cDNA o
sequenze proteiche dello stesso organismo (allineamento cis)
Metodi Indiretti – Mediante ricerca di somiglianze con un gene noto (allineamento trans);
– Mediante ricerca di strutture simili a un gene ideale (ab initio o de novo)
– Metodi basati sul contesto (contenuto in GC, frequenze dei codoni, etc.)
– Metodi basati su segnali (siti accettori e donatori di splicing, promotori, codoni di start/stop, segnali di poliadenilazione, etc.)
– Modelli basati su contesto e segnali
Metodi Ibridi
1. Mediante l' unione di tecniche per omologia, ab initio e metodi diretti
Identificazione dei geni

Metodi ab initio
Metodi di predizione di geni ab initio si basano su: " Identificazione di segnali che permettono l'identificazione di un gene e della sua struttura (splicing, inizio e fine traduzione, etc.) " Modelli statistici che incorporano questi segnali " I segnali sono calcolati su un dataset di riferimento, cioè geni possibilmente dello stesso organismo già noti " Possono includere considerazioni evolutive (nell'ipotesi che sequenze genomiche corrispondenti a geni siano piu' conservate, e/o mostrino patterns di conservazione caratteristici).

ATG TGA
coding segment complete mRNA
ATG GT AG GT AG . . . . . . . . . start codon stop codon donor site donor site acceptor
site acceptor
site
exon exon exon intron intron
TGA
I segmenti codificanti (CDS) di un gene sono delimitati da 4 tipi di segnale: codone di inizio (ATG negli eucarioti), codone di stop (TAG, TGA, o TAA), siti donatori di splicing (solitamente GT), e siti accettori di splicing (AG)
Modelli di un gene

ATG GT AG GT AG . . . . . . . . . start codon stop codon donor site donor site acceptor
site acceptor
site
exon exon exon intron intron
TGA
Il problema dell'identificazione di geni in una sequenza genomica può essere ricondotto all'identificazione di intervalli nella sequenza genomica, delimitando gli esoni putativi e le altre regioni della struttura del gene:
gene finder
TATTCCGATCGATCGATCTCTCTAGCGTCTACGCTATCATCGCTCTCTATTATCGCGCGATCGTCGATCGCGCGAGAGTATGCTACGTCGATCGAATTG
(6,39), (107-250), (1089-1167), ...
Modelli di un gene

Tratto di poli-pirimidine
Modelli di un gene
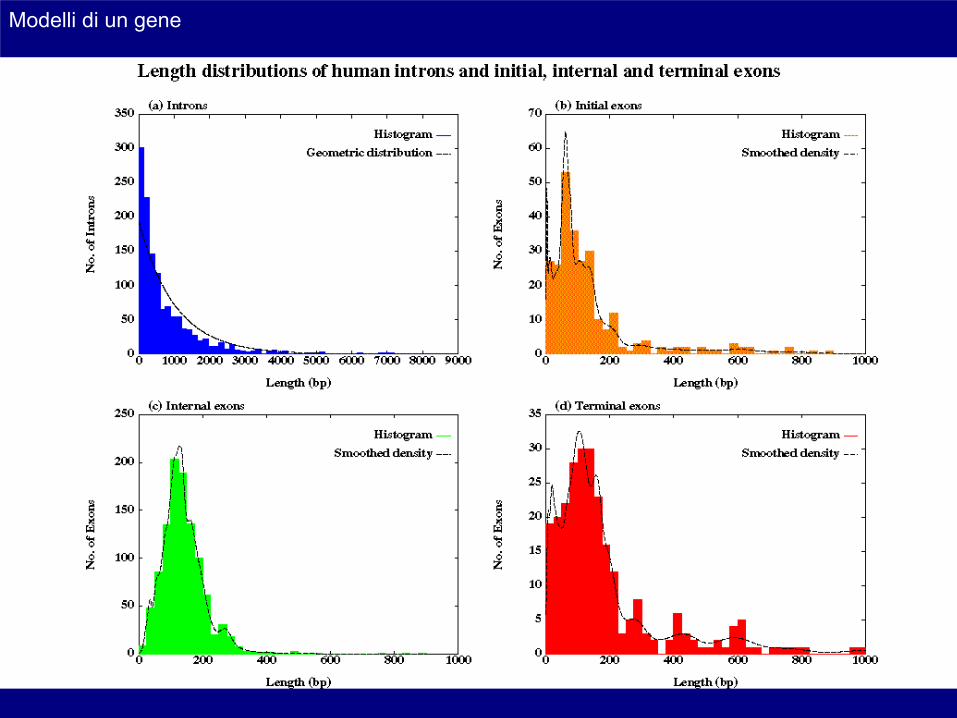
Modelli di un gene
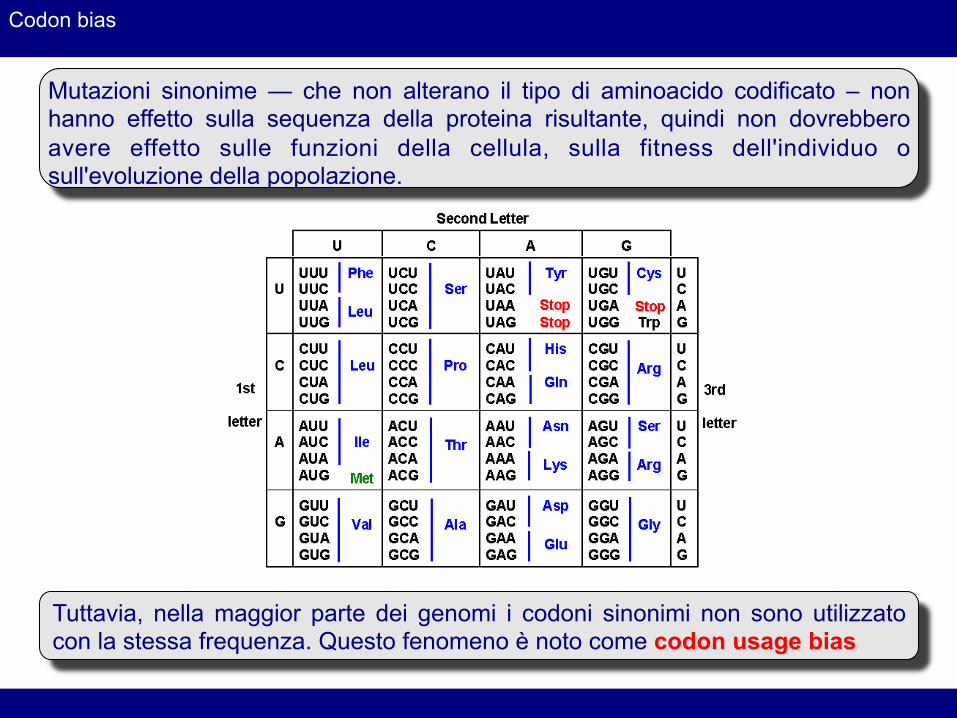
Codon bias
Mutazioni sinonime — che non alterano il tipo di aminoacido codificato – non hanno effetto sulla sequenza della proteina risultante, quindi non dovrebbero avere effetto sulle funzioni della cellula, sulla fitness dell'individuo o sull'evoluzione della popolazione.
Tuttavia, nella maggior parte dei genomi i codoni sinonimi non sono utilizzato con la stessa frequenza. Questo fenomeno è noto come codon usage bias

Altri segnali
Contenuto in G+C

• 0th-‐order • 1st-‐order • 2nd-‐order
( ) ( ) ( ) ( ) ( ) ( )∏=
−⋅=⋅⋅=N
iii sssssssssP
211231211 |pp|p|pp
( ) ( ) ( ) ( ) ( ) ( )∏=
−−⋅=⋅⋅=N
iiii ssssssssssssssP
31221324213212 |pp|p|pp
( ) ( ) ( ) ( ) ( )∏=
=⋅⋅=N
iisssssP
13210 pppp
( ) ( ) ( ) ( ) ( ) ( ) ( )∏=
−⋅=⋅⋅⋅=N
iii sssactatttsP
2111 |pp|p|p|pp
( ) ( ) ( ) ( ) ( ) ( ) ( )∏=
−−⋅=⋅⋅⋅=N
iiii sssssacgtacttattsP
312212 |pp|p|p|pp
( ) ( ) ( ) ( ) ( ) ( ) ( )∏=
=⋅⋅⋅⋅=N
iisgcattsP
10 pppppp
4321 sssss = ttacggts =Catene di Markov

Hidden Markov Models

Hidden Markov Models

Hidden Markov Models
Stati: {S1, S2,…,SN}
Matrice delle transizioni Aij = P(qt+1 = Si | qt = Sj)
Stati iniziali πi = P(q1 = Si)
Osservazioni: {O1, O2,…,OM}
Probabilità delle osservazioni: Bj(k) = P(vt = Ok | qt = Sj)

• Algoritmo forward: Dati i parametri del modello, qual' è la probabilità di una particolare sequenza osservata?
• Algoritmo di Viterbi: Dati i parametri del modello, qual' è la sequenza di stati che più verosimilmente ha condotto alla sequenza di osservazioni?
• Baum-Welch: dato un insieme di osservazioni, e le corrispondenti sequenze di stati, quali sono i parametri del modello?
Modelli di un gene

Hidden Markov Models
Un HMM può essere costruito come un generatore di regioni genomiche:
Osservazioni: sequenza dei nucleotidi;
Stati: ruolo svolto dal nucleotide (ad es. se è il secondo nucleotide di una giunzione di splicing, se è nel mezzo della sequenza di un introne, se è nella prima posizione di un codone, etc.);
Data una sequenza nucleotidica genomica (un cromosoma, un contig), si può usare l'algoritmo di Viterbi per ottenere la più probabile sequenza di stati che l'ha prodotta -> identificare la struttura di eventuali geni presenti nella sequenza.

⎥⎥⎥⎥
⎦
⎤
⎢⎢⎢⎢
⎣
⎡
=Φ
0002.0001.000996.0001.05.00002.0998.05.00000
⎥⎥⎥⎥
⎦
⎤
⎢⎢⎢⎢
⎣
⎡
=
32.018.018.032.0
25.025.022.028.0
H
xm(i) = probabilità di essere nello stato m al momento i;
H(m,yi) = probabilità di emettere un carattere yi nello stato m;
Φmk = probabilità della transizione dallo stato k allo stato m.
Un HMM è completamente definito da: l Matrice delle transizioni fra stati (Φ) l Matrice delle emissioni (H) l Vettore di stato (x)
Hidden Markov Models

Hidden Markov Models
[Brent, 2008]

Hidden Markov Models
[Brent, 2008] Donatore
Accettore
Sequenza dell'introne

Un GHMM (detto anche explicit state duration HMM) è una variante degli HMM per il quale le osservazioni non sono singoli nucleotidi, ma interi segmenti:
Osservazioni: sequenza di vari segmenti;
Stati: ruolo svolto dal segmento (ad es. se è un sito donatore di una giunzione di splicing, se è la regione centrale di un introne);
Ogni stato è definito da un modello che definisce la probabilità di ogni osservazione. Una variante dell'algoritmo di Viterbi può essere usata per ottenere la più probabile segmentazione che l'ha prodotta -> identificare la struttura di eventuali geni presenti nella sequenza.
Generalized Hidden Markov Models (GHMM)

Generalized Hidden Markov Models (GHMM)
Esempi di modello di stato (segmento): Siti di poliadenilazione:
PSSM di sei posizioni compilata su un dataset di training
Accettore di splicing:
Catena di Markov del 1-ordine Sequenza nel mezzo di un introne:
Catena di Markov del 5-ordine (la probabilità di un segmento è il prodotto delle probabilità di ogni suo nucleotide, dati i 5 nucleotidi precedenti; ad es. la probabilità dell'ultima A dell'esapeptide TGCATA è data dalla frequenza con cui i pentapetidi TGCAT terminano in A nel dataset di training)

[Zhang, Nature 2002]
CTA GGT AAT CGT CGT
CTA GGT AAT CGT C CTA GGT AAT CGT CG
AAA CTC AGT A CTC AGT
AA CTC AGT
.... ...........
...........
Fase 0 Fase 1 Fase 2
Generalized Hidden Markov Models (GHMM)
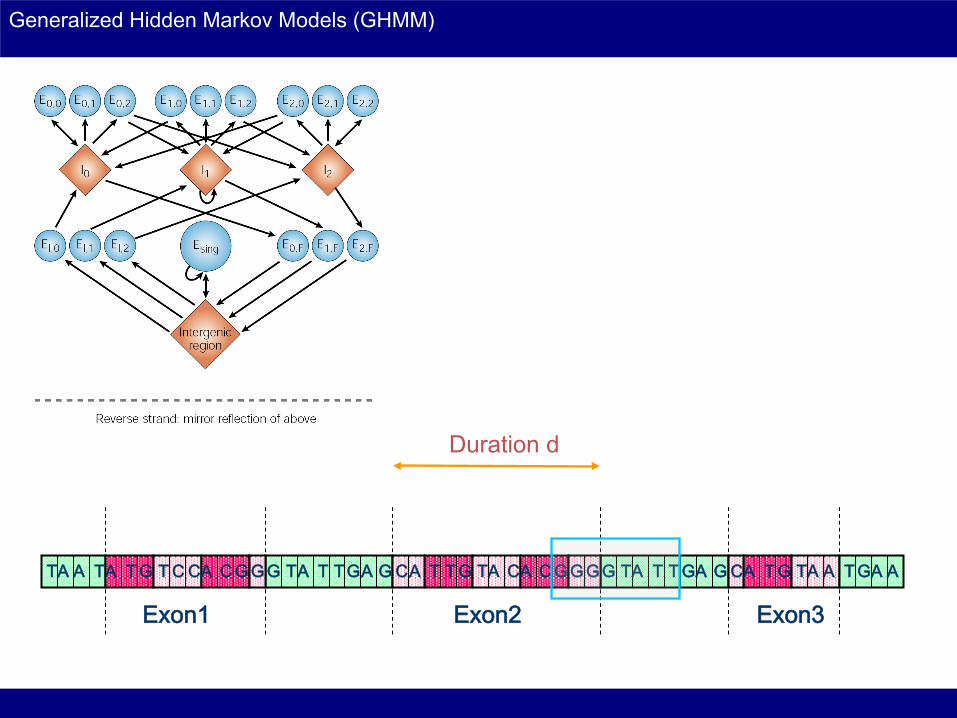
T A A A A A A A A A A A A A A A A T T T T T T T T T T T T T T T G G G G G G G G G G G G G G G C C C C C C C
Exon1 Exon2 Exon3
Duration d
Generalized Hidden Markov Models (GHMM)

" Disegnato per predire la struttura completa di un gene:
Introni, esoni, promotori, siti di poliadenilazione;
" Include: Descrizioni di segnali di inizio e fine della trascrizione, e splicing; Distribuzione delle lunghezze delle varie sottoregioni; Frequenze di composizione di esoni, introni, regioni intergeniche, regioni
C+G;
" Può predire Geni interi o frammenti; Geni multipli separati da regioni intergeniche; Geni su entrambi i filamenti della sequenza;
" Basato su un modello generale probabilistico della struttura e composizione di un gene (Explicit State Duration HMMs).
Genscan (Burge, 1997)

Genscan (Burge, 1997)
[Zhang, Nature 2002]
N – regione intergenica P - promotore F - 5’ UTR T – 3'UTR
Esngl - esone singolo (codone di inizio -> codone di stop)
Einit - esone iniziale (codone di inizio -> sito donatore di splicing)
Ek - esone interno con fase k (sito accettore di splicing -> sito donatore)
Eterm - esone terminale (sito accettore -> codone di stop)
Ik - introne con fase k: 0 – fra due codoni; 1 – dopo la prima base di un codone; 2 – dopo la seconda base di un codone

A C G C G A C T A G G C G C A G G T .. T A T G A T Exoninit Intron0 Exon0 Intron0 Exonterm 3’UTR
⎟⎟⎟⎟⎟⎟
⎠
⎞
⎜⎜⎜⎜⎜⎜
⎝
⎛
=
031.041.028.039.0033.028.001000010
A
⎟⎟⎟⎟⎟⎟
⎠
⎞
⎜⎜⎜⎜⎜⎜
⎝
⎛
=Π
12.060.004.006.0
Sequenza di stati assunti dal sistema: q = {q1, q2, q3 ,.., qn}
Sequenza di durate della permanenza in ogni stato: d = {d1, d2, d3 ,.., dn}
stato iniziale
durata dello stato iniziale
P(Фi,S) = πq1(d1)Pq1{s1|q1,d1} * Aq1,q2(d2)Pq2{s2|q2,d2} * ..… * Aqn-1,qn(dn)Pqn{sn|qn,dn}
segmento di sequenza probabilità di transizione
Genscan (Burge, 1997)

S), jP(S), iP(
P(S)S)i,P( S)| iP(
j ΦΣ
Φ=
Φ=Φ
ΦΦ ∈ L
P(Фi,S) = πq1(d1)Pq1{s1|q1,d1} * Aq1,q2(d2)Pq2{s2|q2,d2} * ..… * Aqn-1,qn(dn)Pqn{sn|qn,dn}
Probabilità di una sequenza S usando un particolare percorso:
Probabilità di un percorso data una sequenza S?
Teorema di Bayes
Genscan (Burge, 1997)

" Basato su Genscan, cui aggiunge un modello di conservazione evolutiva " Dato un genoma target, e un genoma di supporto (informant sequences), BLAST è utilizzato per identificare regioni di similarità locale. " Ogni base allineata nella sequenza target è segnata come gap (.), mismatch (:), o match (|). Ad esempio: Uomo: ACGGCGA-GUGCACGU Topo: ACUGUGACGUGCACUU Allineamento: ||:|:||.||||||:| " Si definisce un nuovo alfabeto di 12 lettere = { A., A:, A|, C., C:, C|, G., G:, G|, U., U:, U| } " Sequenze di questi simboli sono modellati come catene di Markov del quinto ordine. " Si usa lo stesso modello di Genscan, ma che calcola anche la probabilità della stringa di conservazione. Una variante dell'algoritmo di Viterbi è applicata per calcolare la probabilità di osservare la data sequenza di simboli.
Twinscan – Dual genome Gene Predictor
Twinscan (Korf, 2001)

[Brent, Nature Biotechnology 2007]
?
Twinscan (Korf, 2001)

?
Twinscan (Korf, 2001)
[Brent, Nature Biotechnology 2007]
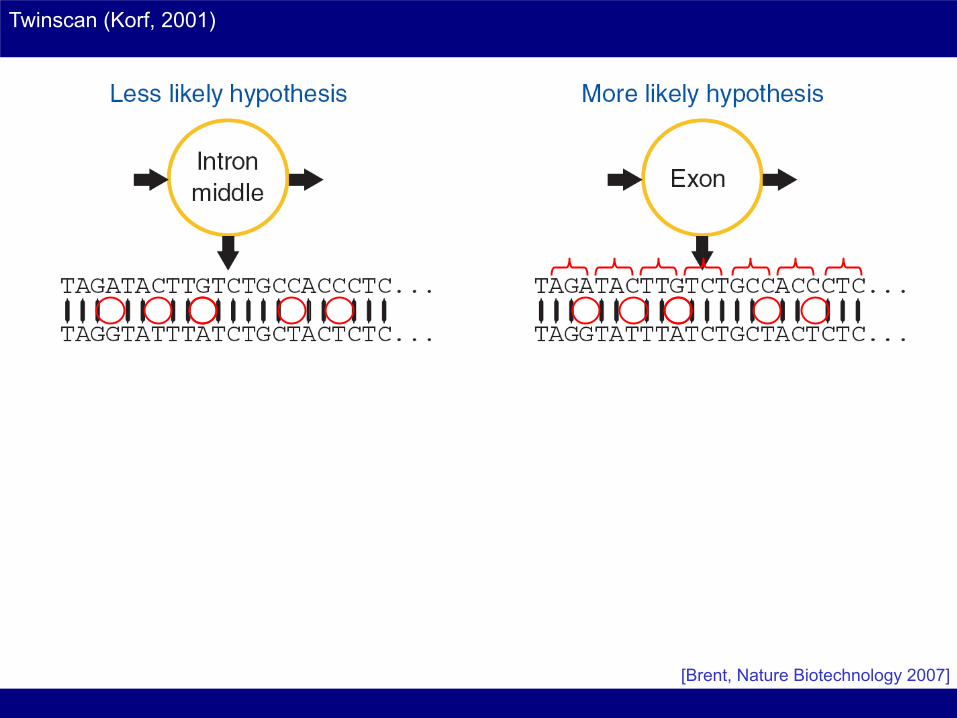
Twinscan (Korf, 2001)
[Brent, Nature Biotechnology 2007]

Twinscan (Korf, 2001)
[Brent, Nature Biotechnology 2007]

• GENSCAN
• TWINSCAN
• N-SCAN
Target GGTGAGGTGACCAAGAACGTGTTGACAGTA
Target GGTGAGGTGACCAAGAACGTGTTGACAGTA Conservation |||:||:||:|||||:||||||||...... sequence
Target GGTGAGGTGACCAAGAACGTGTTGACAGTA Informant1 GGTCAGC___CCAAGAACGTGTAG...... Informant2 GATCAGC___CCAAGAACGTGTAG...... Informant3 GGTGAGCTGACCAAGATCGTGTTGACACAA
Emette una sequenza
Emette una sequenza e la sua conservazione
Emette colonne di un allineamento multiplo
NSCAN - Multiple Species Gene Predictor
N-scan (Gross, 2006)

Basato sui Conditional Random Fields (CRF); I CRF sono modelli discriminativi, per l'etichettatura di una sequenza di input (mentre gli HMM sono generativi); E' rappresentato da un modello grafico, in cui i vertici sono le variabili, e gli archi descrivono un rapporto di dipendenza fra variabili; L'input è un allinamento multiplo fra la sequenza target e una serie di genomi (informants); Può includere anche dati di espressione (ad es. ESTs);
Contast (Gross, 2007)
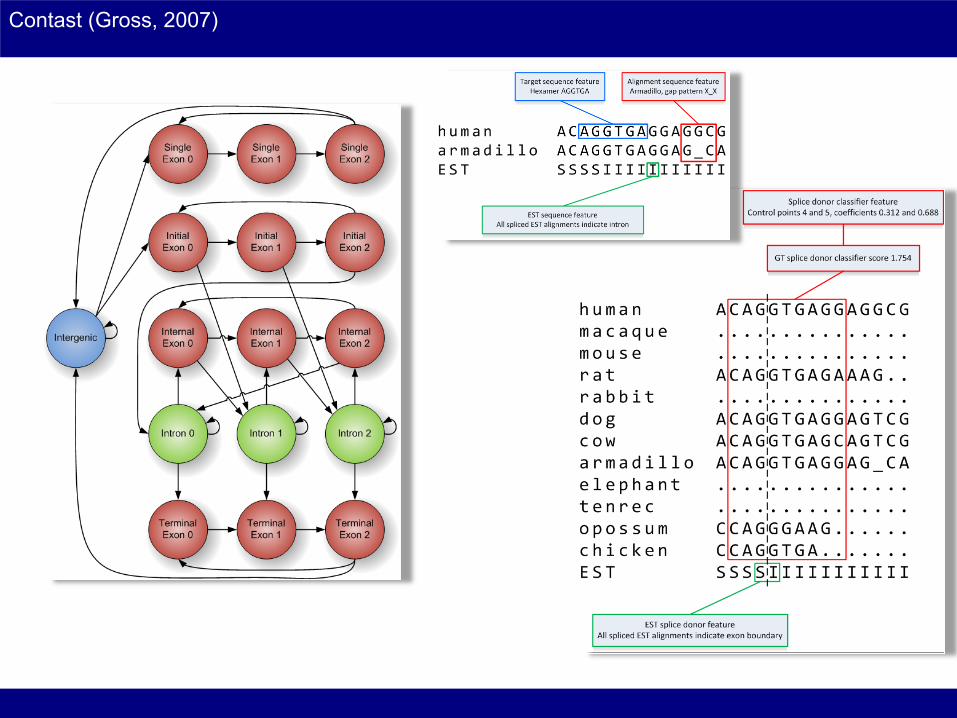
Contast (Gross, 2007)
TN FP FN TN TN TP FN TP FN
Realtà
Predizione
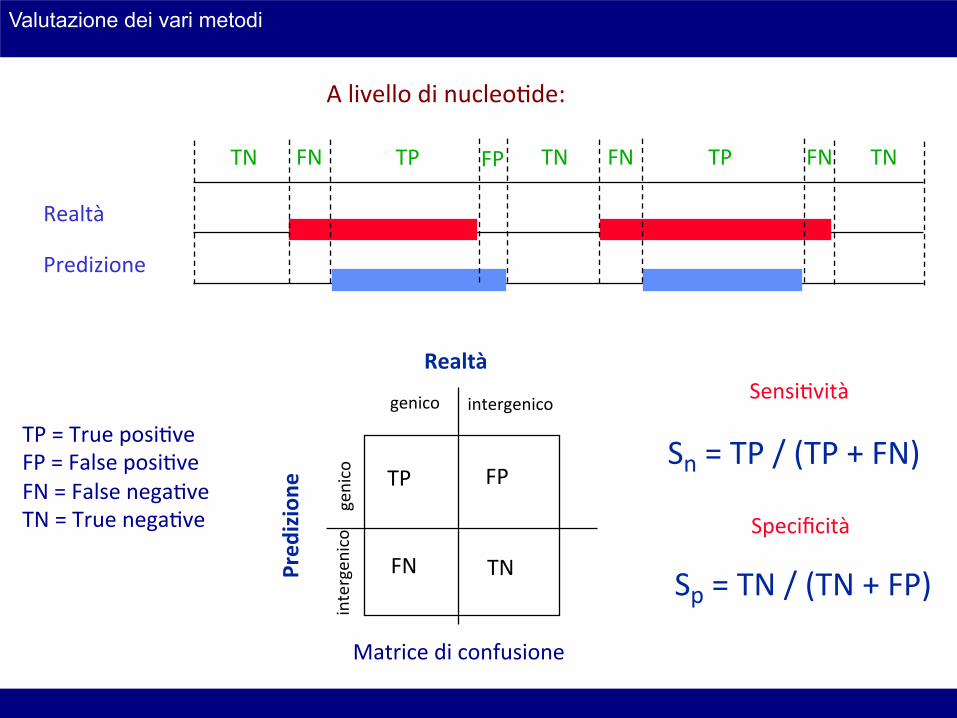
Sn = TP / (TP + FN)
Sp = TN / (TN + FP)
SensiHvità
Specificità
A livello di nucleoHde:
Valutazione dei vari metodi
TP = True posiHve FP = False posiHve FN = False negaHve TN = True negaHve
Pred
izione
Realtà
TP
FN TN
FP
genico
genico
intergen
ico
intergenico
Matrice di confusione

Predizione
A livello di esone:
Esone sbagliato
Esone correPo
Esone mancante
Sn = SensiHvità Numero di esoni correPamente prede*
Numero di esoni nel dataset
Sp = Specificità Numero di esoni correPamente prede*
Numero di esoni prede*
Realtà
Valutazione dei vari metodi

A livello di gene:
Sn = SensiHvità numero di geni correPamente prede* numbero di geni nel dataset
Sp = Specificità numero di geni correPamente prede*
numero di geni prede*
Si considera una predizione corretta a livello di gene se tutti i suoi esoni sono stati correttamnete predetti.
Valutazione dei vari metodi
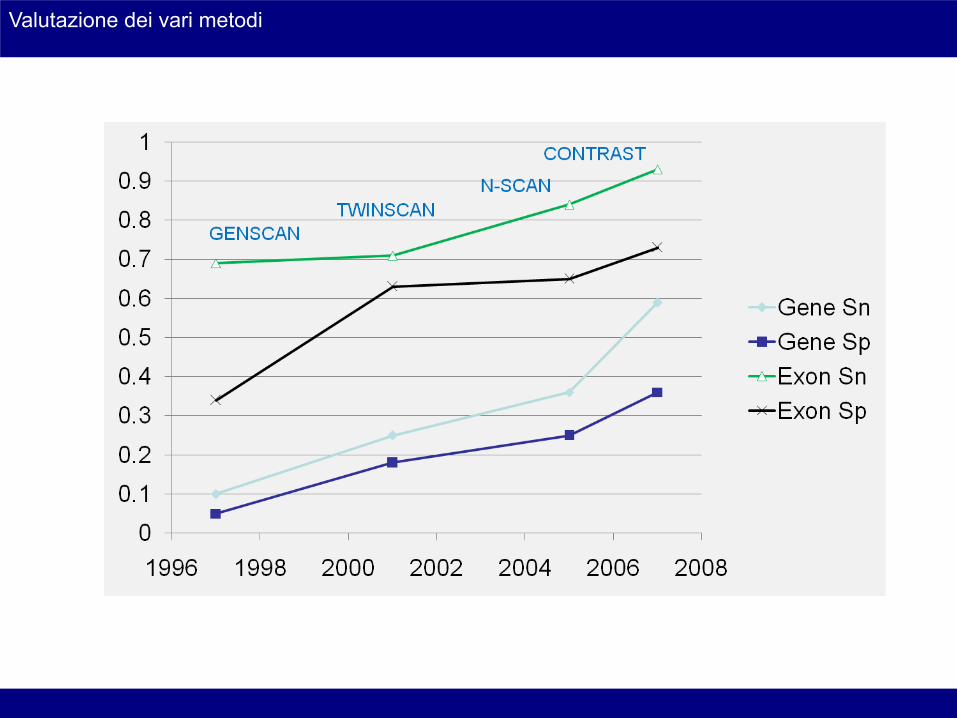
Valutazione dei vari metodi

Strategia per l'annotazione di un genoma
[Brent, Nature Biotechnology 2007]

" Non possono identificare: Geni sovrapposti; Geni annidati; Frame-shifts o errori di sequenziamento; Codoni di inizio e stop alternativi;
Giunzioni di splicing non canoniche; Splicing alternativo; Salto del codone di stop (TGA) causato da selenocisteine;
" Sono in genere organismo-specifici; " Identificano bene geni simili a qualcosa visto in precedenza; " Sono disegnati per identificare solo geni codificanti per proteine.
Limiti degli algoritmi per identificazione di geni

Genomics Session
Geni per RNA non codificanti

RNA non codificante
l Di tutto l'RNA trascritto negli eucarioti superiori, il 98% non è mai tradotto in proteine;
l Di questo 98%, circa il 50-70% è costituito da introni; l Il resto origina da geni non codificanti proteine, fra cui geni
per rRNA, tRNA e una vasta serie di altri geni per RNA non codificante (non-coding RNA, ncRNAs);
l Anche alcuni introni sono stati visti contenere ncRNAs, ad
exsempio gli snoRNA; l Il numero di ncRNA diversi nei genomi di mammifero è
sconosciuto (secondo stime recenti > 15000).

RNA non codificante

Funzioni dell’RNA: 1. Immagazzinamento/trasferimento dell'informazione genetica: ñ RNA genomico ñ Molti virus hanno genomi composti da RNA è Singolo filamento (ssRNA) [ad es. HIV] è Doppio filamento (dsRNA) [ad es. Rotavirus]
ñ RNA messaggero
2. Strutturale 3. Catalitico 4. Regolatorio
RNA non codificante

Gli ncRNA si possono genericamente classificare in due gruppi in base alla loro funzione:
l NcRNA housekeeping, i quali sono espressi sempre e sono necessari per le funzioni normali e la sopravvivenza della cellula;
l NcRNA regolatori o modulatori, i quali sono espressi per rispondere a particolari esigenze;
l NcRNA regolatori possono influire sull'espressione di altri geni
modulando la loro trascrizione o traduzione
RNA non codificante

Esempi di ncRNA housekeeping: • Apparato per la sintesi proteica:
l Transfer RNA (tRNA); l RNA Ribosomiale (rRNA); l snRNA: RNA dello spliceosoma; l snoRNA (small nucleolar RNA) : ruolo accessorio agli rRNA;
• tmRNA (tRNA like mRNA): degradazione delle proteine; • gRNA: editing dell'RNA; • RNA della telomerasi: primer per la sintesi del DNA dei telomeri;
RNA non codificante

Esempi di ncRNA modulatori: • Micro RNA (miRNA): regolatori della traduzione; • Small interfering RNAs (siRNA): silenziamento di geni; • Riboswitch RNA: controllo dell'espressione genica; • ncRNA modulatori delle funzioni di proteine; • ncRNA regolatori della localizzazione di RNA e proteine.
RNA non codificante

I ncRNA svolgono la loro funzione: " In maniera sequenza-specifica (es. per appaiamento di basi con un target); " In maniera struttura-specifica (es. per interazione con ligandi proteici); " In maniera sia sequenza- che struttura-specifica.
RNA non codificante





























