Embed Size (px)
Citation preview

Limb gird autosomal
.le d
muscula ominant
r dystrophy with inheritance
Marconi G, Pizzi A, Arimondi CG, Vannelli B. Limb girdle muscular dys- trophy with autosomal dominant inheritance. Acta Neurol Scand 1991: 83: 234-238.
We describe a patient suffering from limb-girdle muscular dystrophy with autosomal dominant inheritance proved by the presence of other similar cases in both sexes scattered over 4 generations of his family tree. In all patients the symptoms appeared in adult age and pelvi-femoral preceded scapulo-humeral involvement. Clinical expressivity has been variable, but rather benign without any reduction in life expectancy. Myopathic changes with vacuoles were present in muscle on light and electron mi- croscopic examination. In the literature we found at least another 5 genealogies with autosomal dominant LGMD which had similar clinical and pathological features to those of our patient.
Limb girdle muscular dystrophy (LGMD) has typi- cally autosomal recessive inheritance, but some autosomal dominant cases have been reported in the literature in recent years (1-11).
In this paper we also report the clinical and pa- thological features relating to a patient suffering from slowly progressive LGMD. His pedigree shows other similar cases of both sexes scattered in four generations, which constitutes a definite proof of a dominant autosomal inheritance.
Case report
C.V. is a 48-year-old worker with no signficant per- sonal history up to the age of about 38 years, when he began to complain of weakness of the pelvi- femoral muscles, which slowly increased and was followed by progressive wasting of the thgh muscles. Afterwards the weakness and wasting spread also to the shoulder girdle muscles. Now, at age 48, he is still able to stand up and to walk with the aid of a stick. On neurological examination he presents lumbar hy- perlordosis and a waddling gait due to weakness of the gluteal muscles. He is able to rise from a chair by leaning h s hands on the thghs and by considerable bending of his trunk, but he is unable to get up from the ground on his own or to climb stairs without leaning to on the handrail, or to keep his hands raised over his head for more than a few seconds. He shows severe wasting of the girdle and the proximal limb muscles. The ocular, facial and oro-pharingeal muscles appear not to be affected and there are no muscular contractures or skeletal deformities. His tendon reflexes are normal, and no other neurologi- cal alterations or internal diseases are present. His
G. Marconi’, A. Pizzi’, C. G. Arimondi’, B. Vannelli’ Department of ’ Neurological and Psychiatric- Sciences, Human Anatomy, University of Florence, Italy
Key words: limb girdle muscolar dystrophy; autosomal dominant inheritance
G. Marconi, Department of Neurology, Univer- sity of Florence, V. le Morgagni 85, 50134 - Florence, Italy
Accepted for publication September 26, 1990
mental level has been examined and found to be normal.
Over the years the patient has frequently been hospitalised and submitted to frequent instrumental and laboratory tests. Blood tests have always been normal, with the exception of slightly increased CK values (twice or three times normal values). The echocardiogram, the ECG, the EEG and the brain CT were normal. EMG of the limb girdle muscles has always revealed small and short motor unit po- tentials and rapid recruitment up to the full interfer- ence pattern as typical of myopathy. Motor and sen- sory conduction velocities were normal.
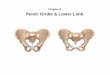
Eight successive CTs of the lower limb muscles have also been performed and have shown the pro- gressive evolution of the muscle wasting (Fig. 1,2). The first, dating back to 1982, revealed severe muscle changes at the thigh, with almost complete dis- appearance of the adductor muscles images and a lesser involvement of the quadriceps. At the calves, the soleus muscle image had also disappeared and a dyshomogeneous hypodensity of the gastrocnemius was also present. In the last CT in 1989, the picture at the thghs had worsened and almost all the posterior muscles had disappeared; the quadriceps also showed more marked hypodensity. At the legs there had been a worsening of the degenerative pattern of the gastrocnemius and, to a lesser degree, of the anterior leg muscles.
A biopsy of the quadriceps muscle performed four years after onset of symptoms revealed that the nor- mal archtecture of the muscular bundles was well preserved, but there was marked variability in fibre size because of the presence of hypotrophic ones with round or, less frequently, angular shape,
234
Autosomal Dominant LGMD
Fig. I . CT of the thigh muscles 2 (A) and 7 years (B) after the onset of symptoms.
Fig. 2. CT of the leg muscles 2 (A) and 7 years (B) after the onset of symptoms.
235

Marconi et al.
scattered among normal and hypertrophic fibres, some of the latter showing splitting. There were no necrotic fibres nor phagocytosis nor inflammatory reaction. Fibres with internal nuclei were more fre- quent than normally. Perimysial and endomisyal connective tissue was slightly increased. Some of the fibres, especially the hypotrophic ones, showed the presence of one, or sometimes several, vacuoles of variable size and shape with irregular margin surrounded by a rim of granular or filamentous material which stained red with Gomori Thricrome and was basophilic in HE preparations (Fig. 3). Fibres with ragged-red appearance in the Gomori trichrome were not uncommon. Glycogen and lipid content were normal. In histochemical preparations the changes in size appeared to involve all fibre types. Fibre type groupings were not present but there was a slight type 1 predominance. With NADH-TR reaction some fibres had a lobulated appearance and other corresponding to ragged-red ones showed much darker stained sub-sarcolemmal regions. On electron microscopic examination the variability in fibre size was evident, and some fibres contained in subsarcolemmal regions a large amount of mitochondria which were normal in shape and structure. The vacuoles contained filamentous debris, glycogen and mitochondria. No other specific changes were found.
Genetic study
It has been possible to obtain available data con- cerning the siblings offive generations (Fig. 3). There was no consanguinity and the people marrying into the family had no neuromuscular disease. The pater- nal grandfather of the proband (P), who died at 84
Fig. 3. Muscle biopsy. Hypotrophic fibre with vacuoles (HE x400).
years, had gait disorders which have been reported as similar to the P’s, but of milder extent. The grand- father’s sister and three brothers and their children had no symptoms or signs of myopathy.
The P’s father, who died at 74 years, was affected with a pelvi-femoral myopathy with onset at age 40 years. Two of his brothers and their children and grandchildren are healthy. One brother, who died at 40, had myopathic symptoms, and another who lives in Argentine is paraplegic, but has not actually been examined by the authors personally. These two brother have also had several children and grand- children who have been described as healthy.
Besides the P, three other relatives who are undoubtedly myopathic have been found. One of them, one of the P’s brothers who is now 39 years old, was examined by the authors at the age of 34 when he showed pelvi-femoral myopathy with slight gait disorders and difficulty in getting up from the ground. His EMG showed myopathic evidence and the CK was slightly increased (302 U/L as compared with the 160U/L of the norm). The onset of symptoms had been at age 22 years, and now he has two healthy children of 8 and 10 years.
The second relative is the P’s 31-year-old sister, who has LGMD which is more severe in the pelvi- femoral muscles. She was examined at age 25 years when symptoms were very mild. Her EMG was myopathic and her CK slightly increased. She has a daughter of 8 years who is clinically healthy.
The third case concerns the P’s 24-year-old daughter, who is affected with a myopathy which had its onset at about age 18. This myopathy resembles her father’s, with myopathic EMG and slight increase in serum CK. She has a healthy 8-month- old son. The P has also a brother with two children who have been described as normal and two daughters and a son who have been examined by the AA personally and have not shown any myopathic signs, their EMGs and serum CK being normal.
Discussion
The P presents clinical, EMG and muscle biopsy findings typical of a LGMD. In his pedigree there are another 5 certain, and 3 probable, cases with a similar type of myopathy, scattered over 4 gener- ations and belonging to both sexes. These data indi- cate an autosomal dominant inheritance. Other muscular dystrophies with this type of inheritance can easily be ruled out. In particular the lack of facial muscle involvement rules out a facio-humeral dys- trophy, while the non-prevalent involvement of the anterior tibial and peroneal muscles and the lack of calf hypertrophy, as well as the onset in adulthood, rule out scapulo-peroneal and childhood pseudo- hypertrophic muscular dystrophy, respectively.
236

Autosomal Dominant LGMD
The disease revealed itself clinically in the 2nd decade in the patient’s daughter, in the 3rd and 4th in the other patients, with onset in the pelvi-femoral muscles and slow spreading to the shoulder girdle where it has remained of a milder degree with partial involvement also of the leg muscles. On the whole the myopathy has been rather benign, with differences in severity, mild in the patient’s father and very severe in the patient himself.
The clinical picture, the lab tests and the patholo- gical feature of the biopsy specimens of the patient are very similar to those of the LGMDs with autoso- mal recessive inheritance. The CT findings also correspond with the latter forms as regards both the hypodensity patterns and the most prevalently affected muscles; also as regards the early involve- ment of the soleus muscle, which is very frequent in recessive cases (12).
The literature reports only a few cases of LGMD with autosomal dominant inheritance. Some of these
differ from our cases because of involvement of males ( 5 ) or females (3) only or by onset in childhood (6), or by almost exclusive affection of the quadri- ceps (4).
At the end, only 5 pedigrees remain with features corresponding to those of our patients which are summarized in Table 1 (2,7,9-11). As may be seen in all patients the onset of symptoms has been between the 2nd and 6th decades, with more severe pelvi-femoral involvement, slow course and variable, but very rarely very severe, degrees of muscular dam- age. Life expectancy has never been reduced. Gilchrist et al. (9) found also asymptomatic patients with increased serum CK. The clinical variability is reflected in the different expressions of the disease where dominant inheritance is involved.
The CK values were also mainly normal or just slightly higher than the norm, and the muscular biopsy constantly gave myopathic results, though with varying aspects (7) and without any specific
Fig. 4. Pedigree of family with autosomal dominant limb-girdle muscular dystrophy. P Proband 0 Normal
Affected 0 Probable
Table 1. Clinical and pathological data of patients of 6 pedigrees suffering from autosomal dominant LGMD
Onset age A.A. (decades) Location Course Severity CK EMG Biopsy
Schneiderman 3-4 LG Slow Mod N M M, RV Chulkow 2-6 LG Slow ModlSvr SI NIM M, RV Gilchrist 2-3 PF Slow Mod/Svr I M M, RV Serratrice 1-4 LG Slow Mod SI M M Somer 3 LG/DM Slow Mod SI M M Marconi 2-5 LG Slow Mod/Svr SI M M, RV
LG: limbgirdle; PF: pelvi-femoral; DM: distal muscles; Mod: moderate: Svr: severe; N: normal; I: increased; SI: slightly increased: M: myopathic; RV: rimmed vacuoles.
231

Marconi et al. Autosomal Dominant LGMD
features except for the frequent presence of vacuoles which are in any case also occasionally observed in other myopathies including autosomic recessive LGMD and typically in oculopharingeal myopathies and have no clear significance. Probably they are lysosomal in origin (13,7). The presence of sub-sar- colemmal accumulations of normal mitochondria found in many of our patient's fibres is a common finding in many myopathies and has no specific sig- nificance.
In conclusion we have identified a form of LGMD which is inherited in a dominant autosomal fashion and which have uniform clinical, electromyographic and histologic features similar to those found in the more common recessive inherited type of LGMD.
References
1.
2.
3.
4.
5.
HENSON TE, MULLER J, DE MYER WE. Hereditary myopathy limited to females. Arch Neurol 1967: 17:
SCHNEIDERMAN IJ, SAMPSON WI, SCHOENE WC, HAYDON GB. Genetic studies of a family with two unusual autosomal dominant conditions muscular dystrophy and Pelger-Willi anomaly. Am J Med 1969: 46: 380-393. HEYCK H, LANDAHN G. Die progressive dystrophischen myopathien. Berlin: Springer-Verlag 1969: 58-60. BACON PA, SMITH B. Familial muscular dystrophy of late onset. J Neurol Neurosurg Psychiatry 1971: 34: 93-97. DE COSTER w , DE RENCK I, THIERY E. A late autosomal
238-247.
dominant form of limb-girdle muscular dystrophy: a clinical genetic, and morphological study. Eur Neurol 1974: 12:
6. BETHLEM I, VAN VIJNGAARDEN GK. Benign myopathy, with autosomal dominant inheritance: a report on three pe- digrees. Brain 1976: 99: 91-100.
7. CHUTKOW JG, HEFFNER RR, KRAMER AA, EDWARDS IA. Adult onset autosomal dominant limb-girdle muscular dys- trophy. Ann Neurol 1986: 20: 240-248.
8. GRAHAM IM, RAWNSLEY ES, NORDGREN R, FRATKIN I. Autosomal dominant limb-girdle muscular dystrophy with progressive cardiomyopathy: report of large family and delineation of natural history. Am J Hum Genet 1986: 25:
9. GILCHRIST JM, PERICAK-VANCE M, SILVERMAN L, ROSES AD. Clinical and genetic investigation in autosomal dominant limb-girdle muscular dystrophy. Neurology 1988:
10. SERRATRICE G, PELLIONIER JF. Deux familles de myopathies benignes predominant sur les cintures d'hkrtdite autosomique dominante. Rev Neurol 1988: 144: 43-46.
11. SOMMER H, LANLUMAR V, PALJARVI L, PARTANEU J, HALTRA M. Adult onset limb-girdle muscular dystrophy with autosomal dominant inheritance. In: BARTSOCAS CS, ed. Genetics of neuromuscular disorders. New York: Alan R. Liss, 1989: 69-71.
12. MARCONI G, VILLARI N, SBRILLI C , PIZZI A, FERRARI A. L'uso della TC e della RMN nelle malattie neuromuscolari. Rieducazione motoria del cerebroleso adulto. Ed. Liviana Editrice Italy, 1985: 389-396.
13. NONAKA I, SUNAHARA N, SATOYOSHI E et al. Autosomal recessive distal muscular dystrophy: a comparative study with distal myopathy with rimmed vacuole formation. Ann Neurol 1985: 17: 51-59.
159-172.
720-721.
38: 5-9.
238














![Genetic Testing and Treatment: Part 1, …...• 40‐50% for limb‐girdle muscular dystrophy on exome sequencing [Ghaoui et al, JAMA Neurol 2015;72:1424‐1432] [Reddy et al, J Hum](https://img.pdfslide.net/doc/110x75/5f3730805b1a5c148c428f1e/genetic-testing-and-treatment-part-1-a-40a50-for-limbagirdle-muscular.jpg)