Embed Size (px)
Citation preview

Link between Photoelectron Circular Dichroism and FragmentationChannel in Strong Field IonizationKilian Fehre,* Sebastian Eckart, Maksim Kunitski, Christian Janke, Daniel Trabert, Jonas Rist,Miriam Weller, Alexander Hartung, Lothar Ph. H. Schmidt, Till Jahnke, Reinhard Dorner,*and Markus Schoffler*
Institut fur Kernphysik, Goethe-Universitat Frankfurt, Max-von-Laue-Straße 1, 60438 Frankfurt, Germany
*S Supporting Information
ABSTRACT: The investigation of the photoelectron circular dichro-ism (PECD) in the strong field regime (800 nm, 6.9 × 1013 W/cm2)on methyloxirane (MOX) reveals a flip of the sign of PECD betweendifferent fragmentation channels. This finding is of great importancefor future experiments and applications in chemistry or pharmacy usingPECD in the strong field regime as analysis method. We suggest thatthe observed sign change of PECD is not caused by ionization fromdifferent orbitals but by effectively selecting differently orientednonisotropic subsamples of molecules via the fragmentation channel.
■ INTRODUCTION
The determination of the handedness of a chiral molecule is ofgreat importance in analytical chemistry and pharmacy. Thus,in the recent past, various approaches for the determination ofabsolute handedness were presented.1−3 In practice, however,it is sufficient to be able to distinguish between the twoenantiomers.4,5 Photoelectron circular dichroism (PECD) hasshown its potential to become a sensitive analysis tool toaccomplish this task.6,7 With a signal strength in the range of afew percent, the PECD is 3−4 orders of magnitude larger thanconventional circular dichroism (CD), allowing even thesmallest sample quantities to be examined.6 The PECDmanifests itself as a strong forward/backward asymmetry ofthe photoelectron momentum distribution with respect to thelight propagation axis, which inverts upon switching the lighthelicity or the enantiomer.8
Most previous studies on the PECD focused on single andresonance enhanced multiphoton ionization processes.9−15 Inthese experiments it could be shown that PECD can be appliedto determine enantiomeric excess.6,16 Other studies showedthat PECD is sensitive to the molecular orientation,14 thevibrational state,17,18 molecular conformation,19,20 and theinitial orbital21 from which the electrons were ejected.Three studies are devoted to the dependence of the PECD
on the fragmentation channel of the molecule. One of them21
is in the single photon ionization regime showing a richdependency on the fragmentation channel. The other two22,23
are in the (resonant) multiphoton regime revealing nodependency on the fragmentation channel. Here we expandthese studies to the strong field regime.
Recently Beaulieu et al.24 have shown PECD to be presentacross a wide range of the wavelengths and to be nonzero alsofor nonresonant strong field and tunnel ionization. In thepresent work, we show, in addition, that the sign of the PECDin the strong field regime, obtained employing the samemolecule (methyloxirane, MOX), the identical laser pulse, andthe same photon energy, depends on the fragmentationchannel.
■ EXPERIMENTAL SECTION
To facilitate the coincident detection of the fragment ions andthe electron momentum vector, we have used a symmetricCOLTRIMS (cold target recoil ion momentum spectrosco-py)25 spectrometer consisting of two identical arms (21 cmacceleration length and E = 119 V/cm electric field). Thislength and field were chosen in the way to achieve highacceleration of ions, which is needed for maximizing theefficiency of the ion detector. This spectrometer designallowed us to avoid meshes in the ion and electron arms,which improved the detection efficiency. Both sides of thespectrometer were equipped with a detector consisting of achevron stack of microchannel plates (MCP) and a hexagonaldelay-line anode.26 The electron detector used a PhotonisMCP (open area ratio, OAR, specified 60%), while the iondetector was equipped with a Hamamatsu “funnel type” MCP(OAR specified 90%).27 The main chamber was baked for 1
Received: May 3, 2019Revised: June 28, 2019Published: July 22, 2019
Article
pubs.acs.org/JPCACite This: J. Phys. Chem. A 2019, 123, 6491−6495
© 2019 American Chemical Society 6491 DOI: 10.1021/acs.jpca.9b04210J. Phys. Chem. A 2019, 123, 6491−6495
Dow
nloa
ded
via
SAL
VE
RE
GIN
A U
NIV
on
Aug
ust 1
5, 2
019
at 0
2:55
:33
(UT
C).
See
http
s://p
ubs.
acs.
org/
shar
ingg
uide
lines
for
opt
ions
on
how
to le
gitim
atel
y sh
are
publ
ishe
d ar
ticle
s.

week at 90 °C, resulting in a residual gas pressure of 1 × 10−10
mbar when the gas jet was switched off. The ionization of themethyloxirane molecules was induced by focusing a short,intense, laser pulse ( f = 60 mm, 40 fs, central wavelength 800nm, 0.3 W, 100 kHz), generated by a Ti:sapphire regenerativeamplifier (KMLabs Wyvern 500), resulting in a focal intensityof 6.9 × 1013 W/cm2 onto the supersonic gas jet. With theionization potential of 10.25 eV 28 (methyloxirane, HOMO),this results in a Keldysh parameter γ of 1.1. Switching thehelicity of the light with a motorized stage every 3 min ensuredsame experimental conditions for left-hand circular polarizedlight (LCP) and right-hand circular polarized light (RCP). Thegas jet was produced by expanding methyloxirane with itsvapor pressure at room temperature (approximately 588 mbar)through a nozzle of 30 μm diameter into vacuum. Theexperiment was run for 130 h at a count rate of 11 kHz on theion and 14 kHz on the electron detector.
■ RESULTS AND DISCUSSIONFigure 1 shows the electron momentum distribution fromionization of R-methyloxirane by a circularly polarized 40 fs
laser pulse with a central wavelength of 800 nm and anintensity of 6.9 × 1013 W/cm2. The three-dimensionalmomentum distribution has the well-known donut shapewhere the radius of the donut is approximately given by thevector potential.29 The left panel of Figure 1 shows thisdistribution in cylindrical coordinates, i.e., with the momentumcomponent parallel to the light propagation pex on thehorizontal axis, and the transverse momentum component in
the plane of polarization = +p p pet ey2
ez2 on the vertical
axis. We obtain the PECD as a function of pex and pet as
=−+
−−+
p pR p p S p p
R p p S p p
R p p S p p
R p p S p p
PECD( , ) 0.5( , ) ( , )
( , ) ( , )
0.5( , ) ( , )
( , ) ( , )
ex etLCP ex et LCP ex et
LCP ex et LCP ex et
RCP ex et RCP ex et
RCP ex et RCP ex et
Here, RLCP(pex,pet) is the number of counts received in theexperiment with left handed circularly polarized light (LCP)and R-methyloxirane, SRCP(pex,pet) indicates the number ofcounts received for right handed circularly polarized light andS-methyloxirane. We calculate the PECD in this manuscript as
an average of the PECD measurements for the two helicities.For a discussion of the sources of errors, please refer to theSupporting Information. The observed PECD ranges up to0.5% which is in line with the observation in ref 24.Expanding the mechanism that explains the final electron
momentum asymmetries in the regime of inner shell singlephoton ionization to the strong field regime, the small PECD isnot surprising. For inner shell single photon ionization, theobserved asymmetries could be successfully explained by thescattering of the electron wave packet at the positively chargedatomic cores of the chiral molecule.14 For tunnel ionization,the electron wave packet is born at rather large distances fromthe parent ion and the circularly polarized laser field quicklydrives it away from it. Here, the “scattering” processcontributing to the electron momentum asymmetries canonly take place at the chiral potential, e.g., the nonsphericalCoulomb potential of the left behind chiral molecule.In the remainder of this paper, we discuss if there is a
connection of the observed PECD with the fate of the left-behind molecular ion. Pioneering work on resonant multi-photon ionization of MOX at 420 nm has found that theelectron energy changes with the fragment channel, but for agiven electron energy the PECD is independent of thefragmention.22 As this study was in the resonance enhancedmultiphoton ionization (REMPI) regime, only the first abovethreshold ionization peak was studied, which corresponds toelectron energies below 2.5 eV.This is very different from the present situation at 800 nm
where the electron energy results mainly from acceleration ofthe electron by the laser field. Figure 2 shows our resultshighlighting that PECD in the strong field regime is indeedvery different from the multiphoton case.In order to obtain the information on the associated
fragmentation channel, the time-of-flight (TOF) of thecorresponding ion was measured in coincidence with thephotoelectron. The TOF spectrum is shown in Figure 2A andreveals that the electron distribution is composed ofcontributions from several fragmentation channels. We foundalmost no ion−ion coincidences, confirming that multi-ionization events play only a minor role (less than 1%). Theparent ion C3H6O
+ (C3H6O → C3H6O+ + e−) with a TOF of
4640 ns shows a very narrow TOF distribution, while all othermasses show a much broader TOF distribution. This can beeasily explained by the momentum, which the chargedfragment receives in the dissociation from the unchargedfragment. An ion emitted in the direction of the ion detectorresults in a shorter TOF, and in the opposite direction, theTOF is correspondingly longer. The observed fragment massdistribution is qualitatively similar to what has been observedin the multiphoton regime.22 A black arrow connects the TOFpeak with the associated electron PECD, which displays asignal strength of about 8% (Figure 2M). This signal is muchstronger than the PECD integrated over all ion TOFs as shownin Figure 1. The transparent blue highlight of the TOF peakindicates the negative sign of the slope of the PECD in theTOF spectrum in the region around 0.2 < pet < 0.6 au. ThePECD spectrum in this pet region associated with the fragmentCH3
+ (C3H6O → C2H3O + CH3+ + e−) has a positive slope
and a signal strength of about 2% (Figure 2 B). The positiveslope of the PECD is reflected in the red transparent highlightof the corresponding peak in the TOF distribution in Figure2A.
Figure 1. Left: Final electron momenta from ionization by laser pulsesof 800 nm, 6.9 × 1013 W/cm2 in cylindrical coordinates for R-methyloxirane and LCP without gate on the ion time-of-flight (TOF).pex is the electron momentum parallel to the laser propagationdirection. pet is the component perpendicular to it. The thirddimension φ, the angle in the polarization plane, is integrated over.Right: For methyloxirane without gate on the ion TOF,
=−+p pPECD( , ) 0.5
R p p S p p
R p p S p pex et( , ) ( , )
( , ) ( , )LCP ex et LCP ex et
LCP ex et LCP ex et−
−+0.5
R p p S p p
R p p S p p
( , ) ( , )
( , ) ( , )RCP ex et RCP ex et
RCP ex et RCP ex et.
The Journal of Physical Chemistry A Article
DOI: 10.1021/acs.jpca.9b04210J. Phys. Chem. A 2019, 123, 6491−6495
6492
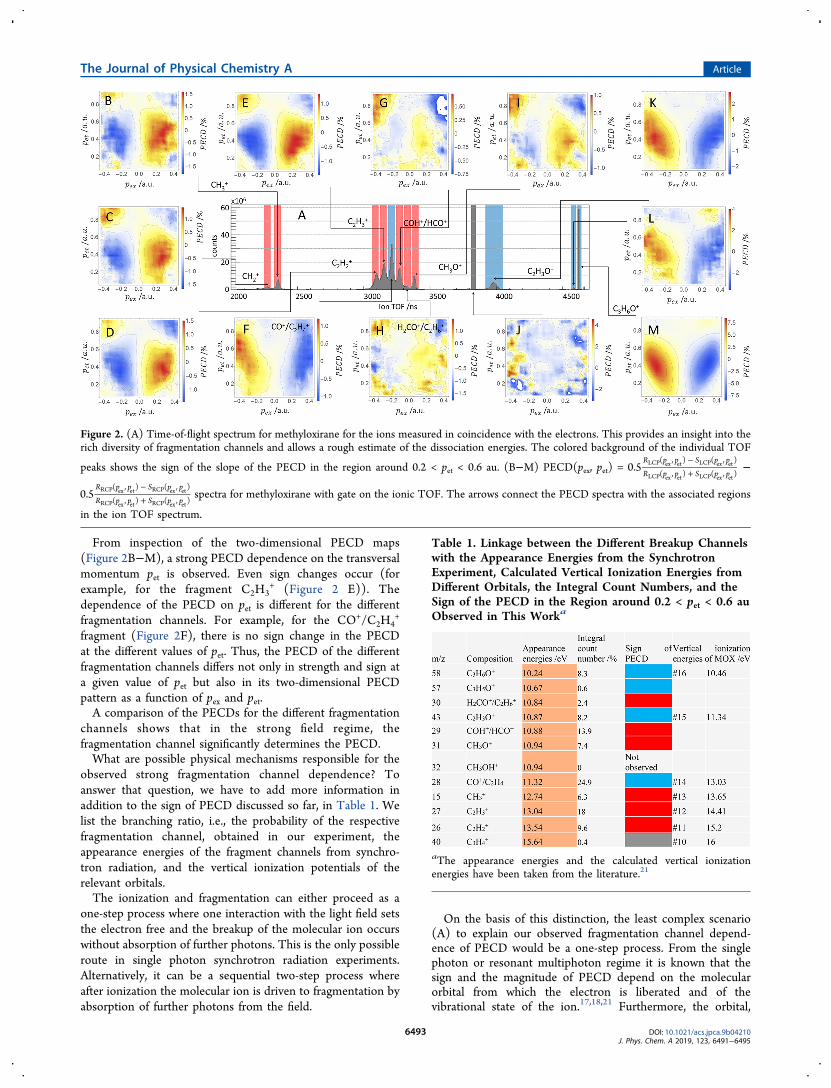
From inspection of the two-dimensional PECD maps(Figure 2B−M), a strong PECD dependence on the transversalmomentum pet is observed. Even sign changes occur (forexample, for the fragment C2H3
+ (Figure 2 E)). Thedependence of the PECD on pet is different for the differentfragmentation channels. For example, for the CO+/C2H4
+
fragment (Figure 2F), there is no sign change in the PECDat the different values of pet. Thus, the PECD of the differentfragmentation channels differs not only in strength and sign ata given value of pet but also in its two-dimensional PECDpattern as a function of pex and pet.A comparison of the PECDs for the different fragmentation
channels shows that in the strong field regime, thefragmentation channel significantly determines the PECD.What are possible physical mechanisms responsible for the
observed strong fragmentation channel dependence? Toanswer that question, we have to add more information inaddition to the sign of PECD discussed so far, in Table 1. Welist the branching ratio, i.e., the probability of the respectivefragmentation channel, obtained in our experiment, theappearance energies of the fragment channels from synchro-tron radiation, and the vertical ionization potentials of therelevant orbitals.The ionization and fragmentation can either proceed as a
one-step process where one interaction with the light field setsthe electron free and the breakup of the molecular ion occurswithout absorption of further photons. This is the only possibleroute in single photon synchrotron radiation experiments.Alternatively, it can be a sequential two-step process whereafter ionization the molecular ion is driven to fragmentation byabsorption of further photons from the field.
On the basis of this distinction, the least complex scenario(A) to explain our observed fragmentation channel depend-ence of PECD would be a one-step process. From the singlephoton or resonant multiphoton regime it is known that thesign and the magnitude of PECD depend on the molecularorbital from which the electron is liberated and of thevibrational state of the ion.17,18,21 Furthermore, the orbital,
Figure 2. (A) Time-of-flight spectrum for methyloxirane for the ions measured in coincidence with the electrons. This provides an insight into therich diversity of fragmentation channels and allows a rough estimate of the dissociation energies. The colored background of the individual TOF
peaks shows the sign of the slope of the PECD in the region around 0.2 < pet < 0.6 au. (B−M) PECD(pex, pet) =−+0.5
R p p S p p
R p p S p p
( , ) ( , )
( , ) ( , )LCP ex et LCP ex et
LCP ex et LCP ex et−
−+0.5
R p p S p p
R p p S p p
( , ) ( , )
( , ) ( , )RCP ex et RCP ex et
RCP ex et RCP ex etspectra for methyloxirane with gate on the ionic TOF. The arrows connect the PECD spectra with the associated regions
in the ion TOF spectrum.
Table 1. Linkage between the Different Breakup Channelswith the Appearance Energies from the SynchrotronExperiment, Calculated Vertical Ionization Energies fromDifferent Orbitals, the Integral Count Numbers, and theSign of the PECD in the Region around 0.2 < pet < 0.6 auObserved in This Worka
aThe appearance energies and the calculated vertical ionizationenergies have been taken from the literature.21
The Journal of Physical Chemistry A Article
DOI: 10.1021/acs.jpca.9b04210J. Phys. Chem. A 2019, 123, 6491−6495
6493

which carries the vacancy together with the vibrational state ofthe molecular ion, predetermines the dissociation pathway.Thus, in a one-step process, selection of the fragmentationchannel can influence PECD because it selects a definiteorbital from which the electron is freed.While a one-step scenario can explain the channel
dependence of PECD, it cannot be reconciled with theadditional facts listed in Table 1. Most strikingly, theprobability of the individual fragment channels has nocorrelation with the ionization potential for the correspondingorbital. For example, the ionization potential (IP) of the orbitalassigned to the fragment C3H5O
+ with the mass 57 u is about 2eV lower than that of the fragment CH3
+ with the mass of 15 u,but the fragment CH3
+ occurs 10 times more often than thefragment C3H5O
+. In the tunneling regime studied here, theionization probability falls exponentially with increasingbinding energy in clear contradiction to the branching ratioslisted in Table 1. Another observation that contradicts the one-step scenario A is that fragmentation channels assigned to thesame orbitals (see Table 1, for instance, C2H3O
+, COH+/HCO+, and CH3O
+ fragmentation channels of orbital no. 15)show different PECD signals (in sign as well as in magnitude).This rules out any one-step scenario to explain the channeldependence of PECD in the strong field case.The next more complex scenario (B) is to combine the
assumption that it is the orbital, which determines the sign ofPECD with the absorption of a second photon, whichdetermines the final ionic channel. In this case, the tunnelionization probability of the orbital would determine theprobability for each sign of the PECD.Again, the exponential binding energy dependence of the
tunnel ionization probability has to be taken into account andthe overall electron signal would be dominated by theionization of orbital no. 16. The next one, no. 15, alreadyrequires about 1 eV more of energy and would be suppressed.The other orbitals would not contribute significantly to theoverall signal. We find, however, that channels for both signs ofPECD have similar overall yield (which is reflected by thecomparatively small TOF integrated PECD). From this weconclude that within the two-step process, orbital dependenceof the PECD is not sufficient to explain our observation.Furthermore, the two-step process decouples the orbital
dependence of the PECD and the fragmentation channel. Theelectron is set free and driven by the laser field on a very fasttime scale. Later during the laser pulse, the molecular ionabsorbs further photons and is driven by the strong field intofragmentation. As by that time the electrons are already faraway, this slow dissociation would not influence the electronanymore (compare to literature30). Thus, within scenario B,one cannot explain the observed inversion of PECD betweenfragmentation channels.The third more complex scenario (C) then is to consider
not only the orbital dependence of PECD but additionally thatPECD depends strongly on molecular orientation with respectto the polarization plane, even for a given orbital. This issimply because PECD is caused by the asymmetry of thepotential and changing the orientation of that potentiallandscape with respect to the direction to the light that isdriving the electron, thus altering the PECD. For single photonionization this has been demonstrated recently by Tia et al.14
For photon induced fragmentation, it is also known that itstrongly depends on the orientation of the molecule withrespect to the polarization of the lasers electric field.31−33
In this scenario, selecting a fragment ion channel effectivelyselects a molecular subsample of specific orientation in space.The corresponding PECD then would be that of the orientedsample of molecules and hence depend on the fragmentchannel. This scenario accounts for all of the compiledinformation. It can explain the channel dependence even if theelectron signal is dominated by ionization from the HOMO, asit is common in strong field ionization.
■ CONCLUSIONIn this article, we investigated the PECD in the strong fieldregime and report on a so far unobserved and strongdependence of the forward/backward asymmetry of theelectron distribution on the fragmentation channel. We findthat the mechanism responsible for this phenomenon isdifferent from the one in the corresponding case for singlephoton ionization. We attribute our finding to the selection ofa specific subset of molecular orientations for eachfragmentation channel, which is enforced by the differenttransition dipole moments linked to that channel.
■ ASSOCIATED CONTENT*S Supporting InformationThe Supporting Information is available free of charge on theACS Publications website at DOI: 10.1021/acs.jpca.9b04210.
Discussion of systematic errors in the calculation of thePECD (PDF)
■ AUTHOR INFORMATIONCorresponding Authors*K.F.: e-mail, [email protected].*R.D.: e-mail, [email protected].*M.S.: e-mail, [email protected] Fehre: 0000-0003-2519-5564NotesThe authors declare no competing financial interest.
■ ACKNOWLEDGMENTSWe acknowledge support from Deutsche Forschungs-gemeinschaft via Sonderforschungsbereich 1319 (ELCH).K.F. and A.H. acknowledge support by the German NationalMerit Foundation. M.S. thanks the Adolf-Messer Foundationfor financial support.
■ REFERENCES(1) Pitzer, M.; Kunitski, M.; Johnson, A. S.; Jahnke, T.; Sann, H.;Sturm, F.; Schmidt, L. P. H.; Schmidt-Bocking, H.; Dorner, R.;Stohner, J.; et al. Direct determination of absolute molecularstereochemistry in gas phase by Coulomb explosion imaging. Science(Washington, DC, U. S.) 2013, 341, 1096−1100.(2) Patterson, D.; Schnell, M.; Doyle, J. M. Enantiomer-specificdetection of chiral molecules via microwave spectroscopy. Nature2013, 497, 475−477.(3) Herwig, P.; Zawatzky, K.; Grieser, M.; Heber, O.; Jordon-Thaden, B.; Krantz, C.; Novotny, O.; Repnow, R.; Schurig, V.;Schwalm, D.; et al. Imaging the absolute configuration of a chiralepoxide in the gas phase. Science (Washington, DC, U. S.) 2013, 342,1084−1086.(4) Horsch, P.; Urbasch, G.; Weitzel, K.-M. Circular Dichroism inIon Yields in Multiphoton Ionization of (R)-Propylene OxideEmploying Femtosecond Laser Pulses. Z. Phys. Chem. 2011, 225,587−594.
The Journal of Physical Chemistry A Article
DOI: 10.1021/acs.jpca.9b04210J. Phys. Chem. A 2019, 123, 6491−6495
6494

(5) Breunig, H. G.; Urbasch, G.; Horsch, P.; Cordes, J.; Koert, U.;Weitzel, K.-M. Circular dichroism in ion yields of femtosecond-lasermass spectrometry. ChemPhysChem 2009, 10, 1199−1202.(6) Kastner, A.; Lux, C.; Ring, T.; Zullighoven, S.; Sarpe, C.;Senftleben, A.; Baumert, T. Enantiomeric Excess Sensitivity to BelowOne Percent by Using Femtosecond Photoelectron CircularDichroism. ChemPhysChem 2016, 17, 1119−1122.(7) Comby, A.; Bloch, E.; Bond, C. M. M.; Descamps, D.; Miles, J.;Petit, S.; Rozen, S.; Greenwood, J. B.; Blanchet, V.; Mairesse, Y. Real-time determination of enantiomeric and isomeric content usingphotoelectron elliptical dichroism. Nat. Commun. 2018, 9, 143.(8) Lux, C.; Wollenhaupt, M.; Bolze, T.; Liang, Q.; Kohler, J.; Sarpe,C.; Baumert, T. Circular dichroism in the photoelectron angulardistributions of camphor and fenchone from multiphoton ionizationwith femtosecond laser pulses. Angew. Chem., Int. Ed. 2012, 51, 5001−5005.(9) Beaulieu, S.; Comby, A.; Descamps, D.; Petit, S.; Legare, F.;Fabre, B.; Blanchet, V.; Mairesse, Y. Multiphoton photoelectroncircular dichroism of limonene with independent polarization statecontrol of the bound-bound and bound-continuum transitions. J.Chem. Phys. 2018, 149, 134301.(10) Beaulieu, S.; Comby, A.; Fabre, B.; Descamps, D.; Ferre, A.;Garcia, G.; Geneaux, R.; Legare, F.; Nahon, L.; Petit, S.; Ruchon, T.;Pons, B.; Blanchet, V.; Mairesse, Y. Probing ultrafast dynamics ofchiral molecules using time-resolved photoelectron circular dichroism.Faraday Discuss. 2016, 194, 325−348.(11) Goetz, R. E.; Isaev, T. A.; Nikoobakht, B.; Berger, R.; Koch, C.P. Theoretical description of circular dichroism in photoelectronangular distributions of randomly oriented chiral molecules aftermulti-photon photoionization. J. Chem. Phys. 2017, 146, 024306.(12) Kastner, A.; Ring, T.; Kruger, B. C.; Park, G. B.; Schafer, T.;Senftleben, A.; Baumert, T. Intermediate state dependence of thephotoelectron circular dichroism of fenchone observed via femto-second resonance-enhanced multi-photon ionization. J. Chem. Phys.2017, 147, 013926.(13) Nahon, L.; Garcia, G. A.; Powis, I. Valence shell one-photonphotoelectron circular dichroism in chiral systems. J. ElectronSpectrosc. Relat. Phenom. 2015, 204, 322−334.(14) Tia, M.; Pitzer, M.; Kastirke, G.; Gatzke, J.; Kim, H.-K.; Trinter,F.; Rist, J.; Hartung, A.; Trabert, D.; Siebert, J.; et al. Observation ofEnhanced Chiral Asymmetries in the Inner-Shell Photoionization ofUniaxially Oriented Methyloxirane Enantiomers. J. Phys. Chem. Lett.2017, 8, 2780−2786.(15) Zhang, Y.; Rouxel, J. R.; Autschbach, J.; Govind, N.; Mukamel,S. X-ray circular dichroism signals: A unique probe of local molecularchirality. Chemical science 2017, 8, 5969−5978.(16) Nahon, L.; Nag, L.; Garcia, G. A.; Myrgorodska, I.;Meierhenrich, U.; Beaulieu, S.; Wanie, V.; Blanchet, V.; Geneaux,R.; Powis, I. Determination of accurate electron chiral asymmetries infenchone and camphor in the VUV range: Sensitivity to isomerismand enantiomeric purity. Phys. Chem. Chem. Phys. 2016, 18, 12696−12706.(17) Garcia, G. A.; Dossmann, H.; Nahon, L.; Daly, S.; Powis, I.Identifying and Understanding Strong Vibronic Interaction EffectsObserved in the Asymmetry of Chiral Molecule PhotoelectronAngular Distributions. ChemPhysChem 2017, 18, 500−512.(18) Contini, G.; Zema, N.; Turchini, S.; Catone, D.; Prosperi, T.;Carravetta, V.; Bolognesi, P.; Avaldi, L.; Feyer, V. Vibrational statedependence of beta and D asymmetry parameters: The case of thehighest occupied molecular orbital photoelectron spectrum of methyl-oxirane. J. Chem. Phys. 2007, 127, 124310.(19) Turchini, S. Conformational effects in photoelectron circulardichroism. J. Phys.: Condens. Matter 2017, 29, 503001.(20) Turchini, S.; Catone, D.; Zema, N.; Contini, G.; Prosperi, T.;Decleva, P.; Stener, M.; Rondino, F.; Piccirillo, S.; Prince, K. C.; et al.Conformational sensitivity in photoelectron circular dichroism of 3-methylcyclopentanone. ChemPhysChem 2013, 14, 1723−1732.(21) Garcia, G. A.; Dossmann, H.; Nahon, L.; Daly, S.; Powis, I.Photoelectron circular dichroism and spectroscopy of trifluoromethyl-
and methyl-oxirane: A comparative study. Phys. Chem. Chem. Phys.2014, 16, 16214−16224.(22) Rafiee Fanood, M. M.; Powis, I.; Janssen, M. H. M. Chiralasymmetry in the multiphoton ionization of methyloxirane usingfemtosecond electron-ion coincidence imaging. J. Phys. Chem. A 2014,118, 11541−11546.(23) Lehmann, C. S.; Ram, N. B.; Powis, I.; Janssen, M. H. M.Imaging photoelectron circular dichroism of chiral molecules byfemtosecond multiphoton coincidence detection. J. Chem. Phys. 2013,139, 234307.(24) Beaulieu, S.; Ferre, A.; Geneaux, R.; Canonge, R.; Descamps,D.; Fabre, B.; Fedorov, N.; Legare, F.; Petit, S.; Ruchon, T.; et al.Universality of photoelectron circular dichroism in the photo-ionization of chiral molecules. New J. Phys. 2016, 18, 102002.(25) Dorner, R.; Mergel, V.; Jagutzki, O.; Spielberger, L.; Ullrich, J.;Moshammer, R.; Schmidt-Bocking, H. Cold Target Recoil IonMomentum Spectroscopy: A ‘momentum microscope’ to view atomiccollision dynamics. Phys. Rep. 2000, 330, 95−192.(26) Jagutzki, O.; Cerezo, A.; Czasch, A.; Dorner, R.; Hattas, M.;Huang, M.; Mergel, V.; Spillmann, U.; Ullmann-Pfleger, K.; Weber,T.; Schmidt-Bocking, H.; Smith, G. D. W.; et al. Multiple hit readoutof a microchannel plate detector with a three-layer delay-line anode.IEEE Trans. Nucl. Sci. 2002, 49, 2477−2483.(27) Fehre, K.; Trojanowskaja, D.; Gatzke, J.; Kunitski, M.; Trinter,F.; Zeller, S.; Schmidt, L. P. H.; Stohner, J.; Berger, R.; Czasch, A.;et al. Absolute ion detection efficiencies of microchannel plates andfunnel microchannel plates for multi-coincidence detection. Rev. Sci.Instrum. 2018, 89, 045112.(28) McAlduff, E. J.; Houk, K. N. Photoelectron spectra ofsubstituted oxiranes and thiiranes. Substituent effects on ionizationpotentials involving σ orbitals. Can. J. Chem. 1977, 55, 318−332.(29) Eckart, S.; Fehre, K.; Eicke, N.; Hartung, A.; Rist, J.; Trabert,D.; Strenger, N.; Pier, A.; Schmidt, L. P. H.; Jahnke, T.; et al. DirectExperimental Access to the Nonadiabatic Initial Momentum Offsetupon Tunnel Ionization. Phys. Rev. Lett. 2018, 121, 1307.(30) Waitz, M.; Asliturk, D.; Wechselberger, N.; Gill, H. K.; Rist, J.;Wiegandt, F.; Goihl, C.; Kastirke, G.; Weller, M.; Bauer, T.; et al.Electron Localization in Dissociating H2
+ by Retroaction of aPhotoelectron onto Its Source. Phys. Rev. Lett. 2016, 116, 43001.(31) Hansen, J. L.; Holmegaard, L.; Nielsen, J. H.; Stapelfeldt, H.;Dimitrovski, D.; Madsen, L. B. Orientation-dependent ionizationyields from strong-field ionization of fixed-in-space linear andasymmetric top molecules. J. Phys. B: At., Mol. Opt. Phys. 2012, 45,015101.(32) Litvinyuk, I. V.; Lee, K. F.; Dooley, P. W.; Rayner, D. M.;Villeneuve, D. M.; Corkum, P. B. Alignment-dependent strong fieldionization of molecules. Phys. Rev. Lett. 2003, 90, 233003.(33) Fehre, K.; Eckart, S.; Kunitski, M.; Pitzer, M.; Zeller, S.; Janke,C.; Trabert, D.; Rist, J.; Weller, M.; Hartung, A.; et al.Enantioselective fragmentation of an achiral molecule in a stronglaser field. Sci. Adv. 2019, 5, No. eaau7923.
The Journal of Physical Chemistry A Article
DOI: 10.1021/acs.jpca.9b04210J. Phys. Chem. A 2019, 123, 6491−6495
6495










![Circular Dichroism in the Photoelectron Angular ...€¦ · depictedinFigure 1cwheretheregionoftheB band[18] actsas the resonant intermediate state. The vertical ionization potential](https://img.pdfslide.net/doc/110x75/6060b90c231d1648f6685fa6/circular-dichroism-in-the-photoelectron-angular-depictedinfigure-1cwheretheregionoftheb.jpg)


















