Upload
others
View
8
Download
0
Embed Size (px)
Citation preview
SC I ENCE TRANS LAT IONAL MED I C I N E | R E S EARCH ART I C L E
MUSCULAR DYSTROPHY
1Biozentrum, University of Basel, 4056 Basel, Switzerland. 2Robert Wood JohnsonMedical School, Rutgers University, Piscataway, NJ 08854, USA.*Corresponding author. Email: [email protected]
Reinhard et al., Sci. Transl. Med. 9, eaal4649 (2017) 28 June 2017
Copyright © 2017
The Authors, some
rights reserved;
exclusive licensee
American Association
for the Advancement
of Science. No claim
to original U.S.
Government Works.
Dow
nload
Linker proteins restore basement membrane andcorrect LAMA2-related muscular dystrophy in miceJudith R. Reinhard,1 Shuo Lin,1 Karen K. McKee,2 Sarina Meinen,1 Stephanie C. Crosson,2
Maurizio Sury,1 Samantha Hobbs,2 Geraldine Maier,1 Peter D. Yurchenco,2 Markus A. Rüegg1*
LAMA2-related muscular dystrophy (LAMA2MD or MDC1A) is the most frequent form of early-onset, fatal congenitalmuscular dystrophies. It is caused by mutations in LAMA2, the gene encoding laminin-a2, the long arm of theheterotrimeric (a2, b1, and g1) basement membrane protein laminin-211 (Lm-211). We establish that despitecompensatory expression of laminin-a4, giving rise to Lm-411 (a4,b1, and g1),muscle basementmembrane is labile inLAMA2MD biopsies. Consistent with this deficit, recombinant Lm-411 polymerized and bound to cultured myotubesonly weakly. Polymerization and cell binding of Lm-411 were enhanced by addition of two specifically designed linkerproteins. One, called aLNNd, consists of the N-terminal part of laminin-a1 and the laminin-binding site of nidogen-1.The second, called mini-agrin (mag), contains binding sites for laminins and a-dystroglycan. Transgenic expression ofmag and aLNNd in a mouse model for LAMA2MD fully restored basement membrane stability, recovered muscleforce and size, increased overall body weight, and extended life spanmore than five times to amaximum survivalbeyond 2 years. These findings provide amechanistic understanding of LAMA2MDand establish a strong basis fora potential treatment.
ed
by guest on A
pril 3, 2021http://stm
.sciencemag.org/
from
INTRODUCTIONSkeletal muscle is the largest organ in the human body accountingfor up to 50% of its mass. The functional units of skeletal muscle aresingly innervatedmuscle fibers that are formed by the fusion of myo-blasts to multinucleated cells. To withstand the mechanical forcegenerated during contraction, muscle fibers are surrounded by base-ment membrane (BM), a highly structured assembly of extracellularmatrix proteins. The bond between the muscle plasma membrane(called sarcolemma) and BM is crucial for muscle fiber stability andsignal transduction. Mutations in BM proteins, their receptors, or cyto-solic adaptors can causemuscular dystrophies of different severity andtime of onset (1).
Congenital muscular dystrophy (CMD) is a group of musculardystrophies characterized by an early onset. LAMA2-related musculardystrophy (LAMA2MD), also calledmuscular dystrophy congenital type1A (MDC1A or merosin-deficient CMD), is among the most frequentCMDs in Europe (2, 3). It is caused by mutations in the LAMA2 geneencoding the a2 subunit of laminin-211 (Lm-211). Most LAMA2MDpatients show complete absence of laminin-a2, are hypotonic (floppy)at birth, fail to ambulate, and succumb to respiratory complication. Pa-tients with less severe symptoms often express low amounts of laminin-a2 or a truncated form (4). Several mousemodels for LAMA2MDexist(5). Most widely used are the dyW/dyWmice (6) that show a very early-onset muscular dystrophy and a markedly shortened life span. A lesssevere model, dy2J/dy2J mice, expresses an N-terminally truncated mu-tant of laminin-a2 and shows a mild dystrophic pathology (7, 8).
Lm-211, a heterotrimer of the a2, b1, and g1 chains, is the prevalentlaminin found in both the endomysial BM of mature skeletal muscleand the Schwann cells of peripheral nerves (9). During embryonicdevelopment, muscle BM is composed of Lm-411 (a4, b1, and g1),Lm-511 (a5, b1, and g1), and Lm-211 (10). In rodent muscle, laminin-a4 and laminin-a5 disappear postnatally from the BM except at theneuromuscular junctions, with laminin-a2 becoming themajor subunit
in adults (9). In LAMA2MD mouse models, laminin-a4 expressionremains elevated into adulthood (9, 11, 12). Laminin-a4 lacks thelaminin N-terminal (LN) globule. In vitro BM assembly assays in com-bination withmutational analysis of Lm-111 (a1, b1, and g1) suggestedthat LN globules are important for Lm-111 self-polymerization (13, 14).Moreover, the C-terminal laminin globular (LG) domains of laminin-a4 bind only weakly to the laminin-a2 receptors a-dystroglycan (aDG)and a7b1 integrin (15–17).
We have previously demonstrated that transgenic expression of aminiaturized form of the protein agrin, called mini-agrin (mag), cansubstantially ameliorate muscular dystrophy in LAMA2 MD mice(12, 18). This function is based on the binding of mag to the coiled-coildomain of laminins at one end and to aDG at the other, thereby im-proving BM stability (19). Although this strategy confers strong benefits,muscular dystrophy still progresses in mag-expressing mice and lifeexpectancy remains markedly shorter than in wild-type controls (12, 19).Here, we tested whether additional restoration of laminin’s polymeriza-tion activity could further improve muscle structure and function in se-verely dystrophic dyW/dyW mice by introducing an additional fusionprotein, called aLNNd (laminin-a1 LN-domain nidogen-1) (20). Recentwork showed that expression of aLNNd substantially ameliorates diseasein dy2J/dy2Jmice (21).We now establish that combined expression ofaLNNd andmag in dyW/dyWmice fully restoresmuscle BMand extendsmaximum survival beyond 2 years. These data demonstrate that defectsin BM assembly and its proper connection to the sarcolemma are theprimary cause of LAMA2MD. The use of mag and aLNNd may opennew possibilities to treat LAMA2 MD.
RESULTSMuscle BM is deficient in LAMA2 MD patientsLama2-deficient mice show prominent increases in laminin-a4 and, toa lesser extent, laminin-a5 expression (9, 11, 12, 18). To examine lamininexpression in human patients, we stainedmuscle biopsies frommus-cular dystrophy patients, selected to be negative for any laminin-a2–like immunoreactivity (fig. S1A), with antibodies against laminin-a4.We observed strong laminin-a4 immunoreactivity colocalizing with
1 of 13
http://stm.sciencemag.org/
SC I ENCE TRANS LAT IONAL MED I C I N E | R E S EARCH ART I C L E
laminin-b1g1 in LAMA2 MD patients (Fig. 1A). In biopsies of age-matched controls, laminin-a4 immunoreactivity was either very lowor not detected in muscle BM (Fig. 1A), except at the neuromuscularjunction and in blood vessel BM (fig. S1B). Western blot analysis re-vealed an increase in laminin-a4 as compared to controls (Fig. 1B).Because the amount of laminin-b1g1was not different betweenLAMA2
Reinhard et al., Sci. Transl. Med. 9, eaal4649 (2017) 28 June 2017
MD patients and controls (Fig. 1B), the loss of laminin-a2 seems com-pensated for by the increase in laminin-a4. In dyW/dyWmice (6), thedistribution of laminin-a4 in 8-week-old triceps muscle was very simi-lar to that observed in human biopsies (Fig. 1C). Laminin-a4 expres-sion, asmeasured byWestern blot analysis, was higher than inwild-typemice, whereas laminin-b1g1 expression was unchanged (Fig. 1D).
by guest on April 3, 2021
http://stm.sciencem
ag.org/D
ownloaded from
E
α-Actinin
250
150
100
Laminin-α4
250kDa
Contr
ol
Laminin-α2300 kDa
LAM
A2 M
D
250
100
Laminin-β1γ1
α-Actinin
B
Contr
ol
LAM
A2 M
D
Rel
ativ
e la
min
in-α
4
Contr
ol
LAM
A2 M
D
Rel
ativ
e la
min
in-β
1γ1
*
n.s.
250
150
100
Wild type dyW/dyW
Laminin-α4kDa
α-Actinin
250
150
100
Laminin-β1γ1
α-Actinin
D
Rel
ativ
e la
min
in-α
4
Wild
type
Wild
type
Wild
type
dyW /d
yW
**
0
2
4
6
0
1
2
3
Contr
ol
**
Muscle homogenate in TBS
100,000g
100,000g
100,000g
100,000g
S1 soluble proteins P1 + 1% NP-40
P2 + collagenase
P3 + EDTA
S2membrane proteins
S4BM proteins
S3«soluble» BM proteins
Lam
inin
-β1γ
1 S
3/S
4250
kDa Laminin-α2
250 Laminin-β1γ1
250 Laminin-α4
S3 S4 S3 S4
Control LAMA2 MD
150
37
100
Laminin-β1γ1
GAPDH
Na+/K+ATPase
S1 S2 S3 S4
Control biopsy
kDaR
elat
ive
La
ma
4
dyW /d
yW
**
0
2
4
6
8
Rel
ativ
e la
min
in-β
1γ1
dyW /d
yW
n.s.
0
0.5
1.0
1.5
F G
LAM
A2 M
D
0
1
2
3
0
2
4
6
8
Laminin-α4 Laminin-β1γ1 Merge
Con
trol
LA
MA
2 M
D
A
Laminin-α4 Laminin-β1γ1 Merge
Wild
type
dy
W/d
yW
C
Fig. 1. Muscles of LAMA2 MD patients and dyW/dyW mice contain high amounts of laminin-a4 and show deficits in BM. (A) Representative immunofluorescenceimages (magnified images in insets) of human muscle biopsy cross sections stained for laminin-a4 and laminin-b1g1. n = 3 LAMA2MD patients or healthy controls. (B) Westernblot analysis and quantification of laminin chains in human muscle biopsies. a-Actinin was used as loading control. n = 4 controls, n = 3 LAMA2MD patients. (C) Representativeimmunofluorescence images (magnified images in insets) of triceps muscle from 8-week-old mice stained for laminin-a4 and laminin-b1g1. n = 4 mice per genotype.(D) Western blot analysis and quantification of laminin-a4 (left graph), laminin-b1g1 (middle graph), and Lama4 transcripts (right graph) from triceps muscle of 8-week-oldmice. n = 3 mice per genotype (Western blot) and n = 4 mice per genotype (quantitative reverse transcription polymerase chain reaction). (E) Schematic of subcellularfractionation. (F) Western blot analysis of S1 to S4 of a muscle biopsy from a control patient. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) and Na+/K+-ATPase(sodium-potassium adenosine triphosphatase) were used as markers for cytosolic and membrane proteins, respectively. (G) Western blot analysis of S3 and S4 fractionsfrom control and LAMA2 MD patient muscle biopsies probed for the indicated proteins. Right: Ratio of the laminin-b1g1 intensity in S3 and S4 fraction. n = 4 healthycontrols, n = 3 LAMA2 MD patients. Data are means ± SEM. *P < 0.05; **P < 0.01; n.s. (not significant), P > 0.05 (for exact P values, see table S3), Student’s t test. Scale bars,100 mm (A and C).
2 of 13
http://stm.sciencemag.org/
SC I ENCE TRANS LAT IONAL MED I C I N E | R E S EARCH ART I C L E
by guest on April 3, 2021
http://stm.sciencem
ag.org/D
ownloaded from
Transcriptional changes in Lama4 coincidedwith increased laminin-a4in dyW/dyW muscle (Fig. 1D). Both wild-type and dyW/dyW miceexpressed a similar amount of laminin-a4 at birth (fig. S2, A and B).Although expression of laminin-a4 decreased over the next 8 weeksinwild-typemice, it remained high in dyW/dyWmice (fig. S2B), suggest-ing that dyW/dyW mice continue to express the embryonic laminin-a4,whereas this isoform is progressively replaced by laminin-a2 in wild-type mice. We also observed a similar increase, although less strongthan laminin-a4, of laminin-a5 in both LAMA2 MD patients anddyW/dyWmice (fig. S3). In summary, these results show that expressionof the embryonic laminin-a4 and laminin-a5 isoforms remains highinmuscle BM in bothLAMA2MDpatients and dyW/dyWmice, withoutalterations in either laminin-b1 or laminin-g1 expression. These resultsconfirm previous immunostainings of biopsies from LAMA2 MDpatients (22, 23) and support the notion that dyW/dyW mice closely re-semble the human disease.
To investigate why Lm-411 cannot functionally compensate for theloss of Lm-211, muscle BM in LAMA2 MD biopsies was analyzed bysubcellular fractionation using a combination of differential extraction,enzymatic digestion, and ultracentrifugation (Fig. 1E). Similar methodshave been used to determine BM stability in dy2J/dy2Jmice, the mousemodel expressing a truncated form of laminin-a2 (8). After removal ofsoluble and membrane proteins, the second pellet (P2) was treatedwith collagenase to release loosely attached BM proteins into the thirdsupernatant (S3) (Fig. 1E). Finally, stably bound components of theBM fromP3were solubilized by adding high concentrations of EDTAand appeared in S4. In control biopsies, soluble proteinswere enrichedin S1, membrane proteins were enriched in S2, and laminin-b1g1appeared in S4 (Fig. 1F). Analysis of S3 and S4 fractions from controlpatients showed that laminin-a2 predominately appeared in S4 (Fig.1G, left lanes, top). Laminin-a4, however, was recovered from fractionS3 in LAMA2 MD patient samples (Fig. 1G, right lanes, middle). Tocompare the distribution of all laminin isoforms in S3 and S4, we alsoprobed for laminin-b1g1 in controls andLAMA2MDpatients. Laminin-b1g1 appeared predominately in S4 for controls butwasmainly detectedin S3 for LAMA2MDpatients (Fig. 1G, bottom and right). These resultsshow that Lm-211 is strongly attached to the BM in controls, whereasthe compensatory Lm-411 in LAMA2MD patients is weakly attached.
aLNNd and mag induce accumulation of Lm-411 onC2C12 myotubesReasons for theweak BMattachment of Lm-411 could be lack of the LNglobule, which is important for Lm-111 self-polymerization in vitro(24), or lack of binding to aDG and a7b1 integrin, as observed inbinding assays using laminin-a4 fragments (Fig. 2A) (15–17). In pre-vious work, we showed that mag binds to laminins via high-affinitybinding to the coiled-coil domain (25, 26) and to aDG (27) (Fig. 2A andfig. S4A) but is unlikely to bind to a7b1 integrins (28). Other work hasshown that a chimeric protein, called aLNNd, restores polymerizationof LN-truncated Lm-111 (DLN–Lm-111) and improves binding ofDLN–Lm-111 to cultured Schwann cells (20). aLNNd comprises theLN domain of laminin-a1 and the laminin/collagen IV binding siteof nidogen-1 (Fig. 2A and fig. S4B). Because previous experiments usedonly fragments of Lm-411 tomeasure binding affinities to a7b1 integ-rins and to aDG, we compared binding of recombinant, full-lengthLm-411 and Lm-111. In contrast to the Lm-111 control, no or very littlebinding of Lm-411 to integrin a7X2b1 (Fig. 2B) or to aDG (Fig. 2C) wasdetected. Adding an equimolar concentration of purifiedmag increasedbinding of Lm-411 to aDG (Fig. 2D). Mag also significantly improved
Reinhard et al., Sci. Transl. Med. 9, eaal4649 (2017) 28 June 2017
binding of recombinant, full-length Lm-511 to aDG (fig. S4C). Self-polymerization of recombinant Lm-411wasmeasured using a sedimen-tation technique previously usedwith truncated Lm-111 (20).WhereasLm-111 polymerized with increasing concentrations, Lm-411 did notpolymerize (Fig. 2E). Lm-411 combinedwith equimolar concentrationsof aLNNd, however, dose-dependently formed polymers similar toLm-111 (Fig. 2E).
Finally, we assessed the ability of mag and aLNNd to promote bind-ing of Lm-411 to cultured C2C12 myotubes grown to confluence. Incontrast to Lm-211, Lm-411 did not bind to C2C12myotubes (Fig. 2F).Whereas an equimolar concentration of mag improved binding, no sig-nificant increase was observed in the presence of aLNNd alone (Fig.2F).Combinedadditionofmag andaLNNd,however, enhancedbindingof Lm-411, reaching a similar staining intensity as for Lm-211 (Fig. 2F).These experiments suggest that aLNNd promotes Lm-411 polymer-ization, whereas mag mediates high-affinity binding to aDG, andtogether, they may be sufficient to compensate for the loss of Lm-211in LAMA2 MD.
Muscle BMs are fully restored in double-transgenicdyW/dyW miceTo test this hypothesis, we generated double-transgenic dyW/dyW micethat expressed aLNNd andmag. To allow comparison with previouswork using mag (12, 18, 19, 29), expression of aLNNd was driven bythe same muscle creatine kinase (MCK) promoter. Two stable aLNNdtransgenic mouse lines were established, which were independentlymated to obtain mice of the following genotypes: dyW/dyW mice,dyW/dyWmice expressing mag (dyW/dyWmag), dyW/dyWmice expres-sing aLNNd (dyW/dyW aLNNd), and dyW/dyWmice expressing bothtransgenes (dyW/dyWDT).Western blot analysis of total lysates from8-week-old triceps muscle demonstrated the presence of the transgenes(Fig. 3A). The two independent dyW/dyW aLNNd lines A and Bexpressed similar amounts of the transgene (fig. S5A). Because therewas also no difference in the phenotypes between dyW/dyWDTmiceof each line (fig. S5, B and C), we focused our analysis on line A. Boththe mag and the aLNNd transgenic proteins were already present atbirth (P0), and their amount seemed to increase over the first three post-natal weeks (fig. S5, D and E). As noted previously (12), the mag trans-gene appeared on Western blots as two bands with an apparent Mr(relative molecular mass) of 80 and 110 kDa, respectively (Fig. 3A andfig. S5E). The upper band corresponds to the full-length mag protein,whereas the lower band is the result of proteolytic cleavage by the agrin-specific protease neurotrypsin (30), as shown by immunoprecipitation/SDS-PAGE followed by tandem mass spectrometry (fig. S5F) and magexpression in neurotrypsin-deficient mice (fig. S5G). At 8 weeks of age,aLNNd and mag proteins colocalized perfectly with laminin-g1 indyW/dyW DT mice, indicating that both transgenes were incorporatedinto the muscle BM (Fig. 3B). Both transgenic proteins were boundto Lm-411, as shown by coimmunoprecipitation (Fig. 3C). The expres-sion of the transgenes did not affect the total amount of laminin-a4 or oflaminin-b1g1 (Fig. 3,D andE) compared to nontransgenic dyW/dyWmice.
To test whether the transgenes changed attachment of laminin-a4 tomuscle BM, differential extractionwas used (Fig. 1E). In dyW/dyWmice,most laminin-a4 appeared in fraction S1, which contains secreted andcytosolic proteins. Themajority of the remaining laminin-a4was recov-ered in S3, and only aminor fraction appeared in S4 (Fig. 4A).Whereasmag did not change the distribution of laminin-a4 in the differentfractions, aLNNd increased the proportion in S4 (Fig. 4A). In dyW/dyW
DTmice, the proportion of laminin-a4was reduced in S1 but increased
3 of 13
http://stm.sciencemag.org/
SC I ENCE TRANS LAT IONAL MED I C I N E | R E S EARCH ART I C L E
by guest on April 3, 2021
http://stm.sciencem
ag.org/D
ownloaded from
in S4 compared to dyW/dyWmice, demonstrating a striking improvementof laminin-a4 incorporation into BM (Fig. 4A). To examine the associ-ationof all laminin isoformswith theBM,we compared the proportionoflaminin-b1g1 present in each fraction (Fig. 4B). The proportion oflaminin-b1g1 in the S4 fraction of single transgenicmice (dyW/dyWmagor dyW/dyWaLNNd)was improved compared todyW/dyWmice butwasstill lower than that in wild-type mice. In contrast, the proportion oflaminin-b1g1 in the S4 fractionofdyW/dyWDTmicewas indistinguishablefrom that seen in wild-type mice and higher than either single trans-genic mouse (Fig. 4B). Furthermore, in dyW/dyWDTmice, the amountof mag and aLNNd was higher in the S4 fraction relative to the S3 frac-tion (fig. S5H), showing cofractionation of the transgenic proteins withLm-411. Finally, BM structure, judged by electron density and continu-
Reinhard et al., Sci. Transl. Med. 9, eaal4649 (2017) 28 June 2017
ity, was indistinguishable between dyW/dyW DT mice and wild-typelittermate controls (Fig. 4C). In contrast, BMs in dyW/dyW miceappeared patchy and of low electron density (Fig. 4C), whereas singletransgenic mice showed an intermediate improvement in BM structure(Fig. 4C). These results demonstrate that simultaneous expression ofmag and aLNNd in dyW/dyW mice mediates incorporation of Lm-411into the BM and restores BM structure to an extent that reaches thequality of wild-type controls.
Transgenes greatly ameliorate muscular dystrophyTo test whether the restoration in BM structure also improved function,we next examined histology of skeletal muscle by staining cross sectionswith hematoxylin and eosin (H&E). Triceps from 8-week-old dyW/dyW
mag
αLNNd
A Lm-111/211 Lm-411
α4
β1 γ1β1 γ1
α2/1
Integrin IntegrinDystroglycan Dystroglycan Laminin (nM)
Bin
ding
(A45
0)
Bin
ding
(A45
0)
Integrin α7X2β1 (nM)
D
B
No Lm Lm-211 Lm-411
Lm-411+ αLNNd
Lm-411+ mag
Lm-411+ mag + αLNNd
Lam
inin
-γ1
Lam
inin
-γ1
F
Rel
ativ
e la
min
in-γ
1 IF
0 20 40 600
0.5
1.0
1.5
Lm-111
Lm-411
0 20 40 600
1
2
3
Lm-111
Lm-411
C
0 20 40 600
0.1
0.2
0.3
0.4
0.5
Bin
ding
(A45
0)
Lm-411
Lm-411 + mag
Laminin (nM)
***
***
0
0.5
1.0
1.5
Lm-211
Lm-411
Lm-411 + αLNNdLm-411 + αLNNd + mag
Lm-411 + mag
0 0.2 0.4 0.60
0.1
0.2
0.3
Pol
ymer
(mg/
ml)
Laminin (mg/ml)
Lm-411
Lm-111Lm-411+ αLNNd
E
n.s.
Fig. 2. Binding of recombinant full-length Lm-411 to muscle receptors, myotubes, and self-polymerization is enhanced by mag and aLNNd. (A) Schematic ofLm-111, Lm-211, and Lm-411 and the linker molecules mag and aLNNd. Binding interactions of mag, aLNNd, laminins, integrins, and dystroglycan are indicated. (B) Bindingcurves of integrin a7X2b1 to Lm-111 and Lm-411. (C) Binding curves of Lm-111 and Lm-411 to purified aDG. (D) Binding curves of Lm-411 and Lm-411 plus an equimolarconcentration of mag to purified aDG. Data in (B) to (D) are means ± SEM with n = 3 per condition. Curve fitting used the equation Y = Bmax*X/(Kd + X). (E) Polym-erization assay for Lm-111, Lm-411, and an equimolar mixture of Lm-411 and aLNNd. Polymer formation was monitored by SDS–polyacrylamide gel electrophoresis(PAGE) and Coomassie blue staining after centrifugation. r2 = 0.998 (Lm-111), r2 = 0.941 (Lm-411 and aLNNd), and r2 = 0.0013 (Lm-411) using linear regression. (F) Confluentlayer of cultured C2C12 myotubes after incubation with no Lm, recombinant Lm-211, Lm-411, or Lm-411 in the presence of an equimolar concentration of aLNNd and/or mag.C2C12-bound Lm-211 or Lm-411 was visualized by staining with antibodies to laminin-g1. Representative immunofluorescence images (left) and quantification (right) oflaminin-g1 immunofluorescence (IF). Scale bar, 200 mm. n = 9 cultures (Lm-411 and mag), n = 6 cultures (Lm-411, Lm-411 + aLNNd, Lm-411 + aLNNd + mag, and Lm-211). Dataare means ± SEM. ***P < 0.001; n.s., P > 0.05 (for exact P values, see table S3), one-way analysis of variance (ANOVA) with Bonferroni post hoc test.
4 of 13
http://stm.sciencemag.org/
SC I ENCE TRANS LAT IONAL MED I C I N E | R E S EARCH ART I C L E
by guest on April 3, 2021
http://stm.sciencem
ag.org/D
ownloaded from
mice showed signs of muscular dystrophy, including fibrotic regions,centralizedmyonuclei indicatingmuscle fiber degeneration/regeneration,and differences in fiber size (Fig. 5A). Expression of a single transgeneimproved but did not completely overcome these pathological indicators,whereas dyW/dyW DT histology was comparable to wild-type controlswith the exception of centralized myonuclei (Fig. 5A). Qualitative as-sessment of fibrosis by collagen stainingwith SiriusRed dye also showedthat the transgenes reduced fibrosis (Fig. 5B). The collagen content, asquantified by the relative amount of hydroxyproline, was reduced toamounts similar to those in wild-type mice (Fig. 5C). Quantitative as-sessment of histological changes, such as fiber size variation, medianfiber size, and total fiber number, revealed that the single transgenesameliorated the muscular dystrophy and that the double-transgenicdyW/dyWDTmice reached values similar to those of wild-type controls
Reinhard et al., Sci. Transl. Med. 9, eaal4649 (2017) 28 June 2017
(Fig. 5D and table S1). Fibrosis in dyW/dyW correlated with increasedinflammation (macrophages stained with anti-F4/80 antibodies) incross sections of 8-week-old triceps muscle (fig. S6) and an almost10-fold increase in the F4/80-encoding transcriptAdgre1 (Fig. 5E).Mac-rophage number and levels of Adgre1 were decreased by the transgenes(fig. S6 and Fig. 5E). A very similar normalization by the transgenes wasobserved forTnc expression, encoding thematricellular protein tenascin-C, shown to be increased in dyW/dyW mice (31, 32).
Quantification of the percentage of muscle fibers with centralizedmyonuclei (CNFs) indicated that single- and double-transgenicdyW/dyWmice had the same or an even higher number of such fibers(Fig. 5G). Centralizedmyonuclei inmuscular dystrophies are indicativeof recent muscle regeneration as the consequence of previous muscledegeneration. Although the number of centralized myonuclei in dyW/dyW
250
150
100
Laminin-α4kDa
α-Actinin
250
150
100
Laminin-β1γ1
α-Actinin
D
Wild
type
dy
W/d
yW
DT
mag αLNNd
150
250
37
αLNNd
GAPDH
75
150
mag100
Wild type dyW/dyW dy
W/dyW αLNNd
dyW/dyW
mag dy
W/dyW DT
A
Rel
ativ
e la
min
in-α
4
Rel
ativ
e la
min
in-β
1γ1
Wild type
dyW/dyW
dyW/dyW mag
dyW/dyW αLNNd
dyW/dyW DT
Laminin-γ1
0
2
4
6
0
0.5
1.0
1.5
n.s.
n.s. ***
kDa
MergeB
E
C
100
150kDa
250
250
αLNNd
mag
Laminin-α4
Laminin-β1γ1
IP Ig
Y
IP la
min
in-α
4
IP Ig
Y
dyW/dyWDT dy
W/dyW
IP la
min
in-α
4
dy
W/d
yW
DT
dy
W/d
yW
Input
Wild type dyW/dyWdy
W/dyWαLNNd
dyW/dyW
mag dy
W/dyWDT
Fig. 3. Transgenic expression of mag and aLNNd in dyW/dyW mice. (A) Western blot analysis to detect aLNNd and mag in lysates from triceps muscle from 8-week-old mice. GAPDH was used as loading control. The aLNNd-specific band is indicated by an arrow because the antibodies also detect nonspecific bands at higher Mr;mag runs on SDS-PAGE as two bands because of its cleavage by neurotrypsin (see fig. S5, F and G, for details). (B) Representative immunofluorescence (magnifiedimages in insets) images of triceps cross sections from 8-week-old mice stained for mag, aLNNd, and laminin-g1. n = 4 mice per genotype. Scale bar, 100 mm.(C) Western blot analysis of immunoprecipitates using anti–laminin-a4 antibodies or immunoglobulin Y (IgY) (as control) and lysates of triceps muscle from 8-week-olddyW/dyW DT or dyW/dyW mice. (D) Western blot analysis of laminin-a4 and laminin-b1g1 in lysates of triceps muscles from 8-week-old mice. a-Actinin was used asloading control. (E) Protein quantification in lysates from the different mice. n = 3 mice per genotype. Data are means ± SEM. ***P < 0.001; n.s., P > 0.05 (for exactP values, see table S3), one-way ANOVA with Bonferroni post hoc test.
5 of 13
http://stm.sciencemag.org/
SC I ENCE TRANS LAT IONAL MED I C I N E | R E S EARCH ART I C L E
by guest on April 3, 2021
http://stm.sciencem
ag.org/D
ownloaded from
mice is increased, there is also evidence that regenerating fibers undergoapoptosis (29, 33, 34), thus hampering successful regeneration and low-ering the number of centralized myonuclei. Consequently, treatmentsthat improve survival of regenerating fibers, such as expression ofBcl2, result in a higher number of fibers with centralized myonuclei(29). On the other hand, treatments that preventmuscle degenerationwill lower the percentage of muscle fibers with centralized myonuclei.In previous work, we have shown that mag both lowers muscle de-generation and improves muscle regeneration (19). This dual functionresults in an increased number of fibers with centralized myonuclei indyW/dyW mag mice compared to dyW/dyW mice (29), which isconsistent with our data in 8-week-old mice (Fig. 5G). To explore pos-sible reasons for the high number of centralized myonuclei in dyW/dyW
DTmice, we examined cross sections of tricepsmuscle from3-week-oldmice using H&E (fig. S7A) and Sirius Red staining (fig. S7B). At thisearly time point, the triceps of dyW/dyW DTmice showed similar signsof muscular dystrophy as did those of dyW/dyWmice (fig. S7, A and B).In addition, analysis of 3-week-old muscle for the presence of the em-bryonic form of myosin heavy chain (eMHC) indicated that dyW/dyW
DT mice show a high degree of regeneration (fig. S7, C and D). At theage of 8 weeks, dyW/dyW DT mice contained only few eMHC-positivefibers compared to dyW/dyWmice (fig. S7E), and the amount of eMHCdetected in lysates was not different from that in wild-type controls (fig.S7F). These data suggest that because of the low expression of mag andaLNNd in the first postnatal weeks (fig. S5, D and E), stabilization ofmuscle BMmight only be achieved later, leaving themyonuclei still at acentral position at 8 weeks of age. Consistent with this idea, the number
Reinhard et al., Sci. Transl. Med. 9, eaal4649 (2017) 28 June 2017
of fibers with centralized myonuclei in the triceps of dyW/dyWDTmicewas significantly higher at 3 weeks than at 8 weeks (fig. S7G).
Expression of the transgenes had a very similar ameliorating effectin 8-week-old tibialis anterior (TA) muscle (fig. S8, A and B) and thediaphragm (fig. S8C) as in triceps muscle. Quantification of fibrosis byeithermeasuring hydroxyproline content (Fig. 5H for TA) or determiningthe Sirius Red–positive area (Fig. 5I for diaphragm) confirmed the sub-stantial improvement by single transgenes and the reduction close towild-type by the simultaneous action of both transgenes. Finally, wemeasured creatine kinase activity in the blood as an estimate for damageof all skeletal muscles. Again, dyW/dyW DT mice were improved com-pared to dyW/dyW and not different from wild-type controls (Fig. 5J).
Next, we assessed disease progression in 16-week-old mice. We ex-cluded dyW/dyWmice in this analysis because of their low survival rate.Gross muscle architecture and fibrosis remainedmarkedly improved inthe double-transgenic compared to single-transgenic dyW/dyWmice, asquantified by the variance coefficient of fiber diameter (fig. S9A) and therelative content of hydroxyproline in triceps (fig. S9B) and TA (fig. S9C).In summary, the overall histological phenotype of dyW/dyWDTmusclebecame similar to that inwild-typemice, with the notable exception thatthe number of centralized myonuclei was higher than that in wild-typecontrols.
Transgenes improve muscle function and markedly prolonglife spanAt the age of 8 weeks, dyW/dyW mice were easily distinguishable fromcontrol littermates by their small size and kyphosis (Fig. 6A). dyW/dyW
A
Rel
ativ
e la
min
in-β
1γ1
[% o
f (S
1 + S
2 + S
3 + S
4)]
Wild type
dyW/dyW
dyW/dyW mag
dyW/dyW αLNNd
dyW/dyW DT
S1 S2 S3 S4
n.s.
*
****
n.s.
*n.s.
*n.s.
*
*
**
n.s.n.s.
n.s.
n.s.S
1S
2S
3S
4
S1
S2
S3
S4
S1
S2
S3
S4
S1
S2
S3
S4
100
37
250150
dyW/dyW
dyW/dyW
αLNNd dy
W/dyW mag
dyW/dyW DT
Laminin-α4
GAPDH
Na+/K+-ATPase
kDa
**
37
250
dyW/dyW
dyW/dyW
αLNNd dy
W/dyW mag
dyW/dyW DT
Laminin-β1γ1
GAPDH
kDa
Wild typeB
Wild typedyW/dyW mag dyW/dyW dyW/dyW αLNNd dyW/dyW DT C
Rel
ativ
e la
min
in-α
4[%
of (
S1
+ S2
+ S3
+ S4)
]
S1 S2 S4S30
20
40
60
80
0
20
40
60
80
S1
S2
S3
S4
S1
S2
S3
S4
S1
S2
S3
S4
S1
S2
S3
S4
S1
S2
S3
S4
Fig. 4. Expression of mag and aLNNd in dyW/dyW mice improves muscle BM. (A and B) Western blot analysis (top) and quantification (bottom) of laminin-a4 (A) andlaminin-b1g1 (B) in the fractions obtained by differential extraction of triceps muscles of 8-week-old mice (see scheme in Fig. 1E). GAPDH and Na+/K+-ATPase were usedas markers for the soluble (S1) and the membrane proteins (S2), respectively. n = 4 mice (dyW/dyW), n = 6 mice (dyW/dyW mag, dyW/dyW aLNNd, and dyW/dyW DT), n = 5mice (wild type). Data are means ± SEM. *P < 0.05; **P < 0.01; n.s., P > 0.05 (for exact P values, see table S3), one-way ANOVA with Bonferroni post hoc test.(C) Transmission electron micrographs of triceps muscle from 8-week-old mice. White arrow, sarcolemma; white arrowhead, BM. Scale bar, 100 nm.
6 of 13
http://stm.sciencemag.org/
SC I ENCE TRANS LAT IONAL MED I C I N E | R E S EARCH ART I C L E
by guest on April 3, 2021
http://stm.sciencem
ag.org/D
ownloaded from
ControldyW/dyW mag dyW/dyW dyW/dyW αLNNd dyW/dyW DT
ControldyW/dyW mag dyW/dyW
H&
E
dyW/dyW αLNNd dyW/dyW DT A
B
Rel
ativ
e hy
drox
ypro
line
n.s.*n.s.
n.s.
Siri
us R
ed
Con
trol
dy
W/d
yW
dy
W/d
yW
mag
dy
W/d
yW
αLN
Nd
dy
W/d
yW
DT
Mea
n of
med
ian
fiber
di
amet
er (µ
m)
D* n.s.
Con
trol
dy
W/d
yW
dy
W/d
yW
mag
dy
W/d
yW
αLN
Nd
dy
W/d
yW
DT
Fibe
r num
ber
0–5
5–10
10–1
515
–20
20–2
525
–30
30–3
535
–40
40–4
545
–50
50–5
555
–60
60–6
565
–70
70–7
575
–80
80–8
585
–90
>90
Fiber diameter class (µm)
0
1
2
3
0
10
20
30
40
50
0
2000
4000
6000
GE
0
10
20
30
40
Fibr
otic
are
a (%
)
Con
trol
dy
W/d
yW
mag
dy
W/d
yW
αLN
Nd
dy
W/d
yW
DT
dy
W/d
yW
*n.s.****
n.s.
0
5
10
15
Rel
ativ
e A
dg
re
1 (F
4/80
)
Con
trol
dy
W/d
yW
mag
dy
W/d
yW
αLN
Nd
dy
W/d
yW
DT
dy
W/d
yW
n.s.
***
*n.s.
C
Con
trol
dy
W/d
yW
dy
W/d
yW
mag
dy
W/d
yW
αLN
Nd
dy
W/d
yW
DT
n.s.
*****
Rel
ativ
e hy
drox
ypro
line
n.s.
0
1
2
3
4
5
Con
trol
dy
W/d
yW
mag
dy
W/d
yW
αLN
Nd
dy
W/d
yW
DT
dy
W/d
yW
Con
trol
dy
W/d
yW
mag
dy
W/d
yW
αLN
Nd
dy
W/d
yW
DT
dy
W/d
yW
H I J
0
5
10
15
20
25
0
1
2
3
4
5
Rel
ativ
e C
K
CN
F (%
)
Con
trol
dy
W/d
yW
DT
dy
W/d
yW
****
n.s.
*
0
5
10
15
20
Rel
ativ
e T
nc
F**
n.s.
n.s.
***
Con
trol
dy
W/d
yW
mag
dy
W/d
yW
αLN
Nd
dy
W/d
yW
DT
dy
W/d
yW
n.s.
**n.s.*
n.s.n.s.
Num
ber o
f fib
ers
(%)
0
5
10
15dy
W/dyW
Control
dyW/dyW mag
dyW/dyW αLNNd
dyW/dyW DT
Fig. 5. Transgenic expressionofaLNNdandmag indyW/dyWmice improvesmusclehistology in8-week-oldmice. (A) Representative images (magnified images in insets)of H&E-stained cross sections from tricepsmuscle. Anexample of amuscle fiberwith centralizedmyonuclei is indicatedwith an arrow. n=5mice per genotype. (B) Representative
images (magnified images in insets) of Sirius Red–stained cross sections (collagen in red) from triceps muscle. n = 3 mice per genotype. (C) Relative hydroxyproline content intriceps indicative of collagen content. Values are normalized to controls. n=4mice (dyW/dyWmag, dyW/dyW aLNNd, and dyW/dyWDT),n= 5mice (dyW/dyW), n= 6mice (control).(D) Distribution of themuscle fiber diameters (left), the mean of median fiber diameter (middle), and the total fiber number (right) in 8-week-old triceps muscle. n = 4malemice (dyW/dyW and control), n= 5malemice (dyW/dyW aLNNd and dyW/dyWDT), n= 6malemice (dyW/dyWmag). Statistical evaluation of fiber size distribution is shown in table S1.(E) Expression of the inflammatory marker Adgre1 (encoding F4/80). n = 4mice (dyW/dyW, dyW/dyWmag, dyW/dyW aLNNd, and control), n = 5mice (dyW/dyWDT). (F) Expressionof transcripts encoding thematricellular protein tenascin-C in tricepsmuscle. n = 4mice (dyW/dyWmag, dyW/dyW aLNNd, and control), n = 5mice (dyW/dyW and dyW/dyWDT).(G) Quantification of CNFs in tricepsmuscle.n=3mice per genotype. (H) Quantificationof relativehydroxyproline content in TA from8-week-oldmice. n=3mice (dyW/dyWDT),n= 4mice (dyW/dyWmag and dyW/dyW aLNNd), n = 5mice (dyW/dyW), n= 6mice (control). (I) Quantification of fibrotic area (visualized by Sirius Red staining) in diaphragm crosssections. n = 4mice (dyW/dyW and dyW/dyWmag), n = 7mice (dyW/dyW aLNNd, dyW/dyW DT, and control). (J) Creatine kinase activity in blood. n = 7mice (dyW/dyW and dyW/dyW
DT), n = 15mice (control). Data aremeans ± SEM. *P< 0.05; **P < 0.01; ***P < 0.001; n.s., P > 0.05 (for exact P values, see table S3), one-way ANOVAwith Bonferroni post hoc test.Controls are wild-type or dyW/+ littermates. Scale bars, 100 mm.
Reinhard et al., Sci. Transl. Med. 9, eaal4649 (2017) 28 June 2017 7 of 13
http://stm.sciencemag.org/
SC I ENCE TRANS LAT IONAL MED I C I N E | R E S EARCH ART I C L E
by guest on April 3, 2021
http://stm.sciencem
ag.org/D
ownloaded from
DTmice looked similar to control littermates, with the exception of awaddling gait, slight hindlimb paralysis, and a leaner appearance (Fig.6A). Peripheral neuropathy is a characteristic feature of dyW/dyW mice(35), and expression of the transgenes using the MCK promoter doesnot restore this deficit. Despite this neuropathy, in a vertical grid hangtest, dyW/dyWDT passed the 3-min test, whereas dyW/dyWwere largelyunable to hold themselves on the grid (Fig. 6B). The expression of magalone improved hang time, whereas little effect was seen in miceexpressing aLNNd (Fig. 6B). Consistent with the improved hang time,peak tetanic force measured in isolated extensor digitorum longus(EDL) at 8 weeks was improved in single- and double-transgenic com-pared to dyW/dyWmice (Fig. 6C). At 16 weeks of age, the effect of thecombined expression of mag and aLNNd was superior to mice expres-sing only one transgene (Fig. 6D). Similar results were obtained forthe peak twitch force andmeasurements in soleus muscles (table S2).Notably, these improvements in force were evident in spite of themuscle atrophy resulting from hindlimb paralysis.
To further assess the therapeutic potential of combined mag andaLNNd expression, we measured body mass and life span. Both body
Reinhard et al., Sci. Transl. Med. 9, eaal4649 (2017) 28 June 2017
mass and life span were drastically reduced in dyW/dyW mice, with apeak bodymass of about 9 g and amedian survival of just 15.5 weeks.The expression of mag or aLNNd resulted in an about 40% increase inbodymass and in extended life span.Median life span reached 50weeksin dyW/dyW mag mice, as reported before (19). The survival curve ofdyW/dyW aLNNd mice was biphasic, extending the survival of longsurvivors (9 of 16) only. The expression of both transgenes togetherincreased body mass by more than 90% and increased life span, withamedian life span of 81 weeks and 5 of 16mice reaching an age beyond2 years. Two-year-old dyW/dyWDTmice were quite active in spite ofthe complete hindlimb paralysis (fig. S10A). Examination of musclehistology in the triceps and the diaphragm showed preservation ofmuscle tissue (fig. S10, B and C). Finally, staining triceps muscles ofthe 2-year-old mice for laminin-a4 and laminin-a5 showed that thesarcolemmal BM of dyW/dyW DT mice still contained high amountsof both proteins, whereas the two laminin-a chains were confined toblood vessels in wild-type controls (fig. S10, D and E). In summary,these data show that expression of a single transgene leads to a modestincrease in body mass and a robust extension of life span. Combined
Fdy
W/dyW
dyW/dyW mag
dyW/dyW αLNNd
dyW/dyW DT
Sur
viva
l (%
)
Age (weeks)
Bod
y w
eigh
t (g)
Age (weeks)
E
dyW/dyW
dyW/dyW mag
dyW/dyW αLNNd
dyW/dyW DT
Control
0
50
100
150
200
dy
W/d
yW
dy
W/d
yW
αLN
Nd
dy
W/d
yW
mag
dy
W/d
yW
DT
Tim
e on
grid
(s)
B
**
**
n.s.
***n.s.
C
Pea
k te
tani
c fo
rce
(mN
)
0
100
200
300
**
Wild
type
dy
W/d
yW
dy
W/d
yW
mag
dy
W/d
yW
αLN
Nd
dy
W/d
yW
DT
Wild
type
dy
W/d
yW
mag
dy
W/d
yW
αLN
Nd
dy
W/d
yW
DT
**
**
A
dyW/dyW
dyW/dyW DT
Control
***
0
10
20
30
105 15
0
100
200
300
400
500D
Pea
k te
tani
c fo
rce
(mN
)
0 20 40 60 80 100 1200
50
100
Fig. 6. Transgenic expression of aLNNd and mag in dyW/dyW mice improves muscle function, increases body weight, and prolongs life span. (A) Picture of arepresentative dyW/dyW, dyW/dyW DT, and control mouse (8 weeks old). Note that dyW/dyW mice appear small because of the severe kyphosis, and dyW/dyW DT mice areleaner than controls. n = 10 mice per genotype. (B) Quantification of grip strength of 8-week-old mice. Grip strength was measured by hang time on a vertical grid. Testwas stopped after 180 s (dotted line). n = 4 mice (dyW/dyW), n = 6 mice (dyW/dyW mag), n = 7 mice (dyW/dyW aLNNd), n = 9 mice (dyW/dyW DT). (C) Peak tetanic force ofEDL muscle from 8-week-old mice. n = 9 mice (dyW/dyW), n = 10 mice (dyW/dyW mag), n = 12 mice (dyW/dyW aLNNd), n = 13 mice (dyW/dyW DT), n = 14 mice (control).(D) Peak tetanic force of EDL from 16-week-old mice. n = 4 mice (dyW/dyW aLNNd), n = 5 mice (dyW/dyW mag and control), n = 6 mice (dyW/dyW DT). Data in (B) and (C) aremeans ± SEM. *P < 0.05; **P < 0.01; ***P < 0.001; n.s., P > 0.05 (for exact P values, see table S3), one-way ANOVA with Bonferroni post hoc test. (E) Body weight of 5- to15-week-old male mice. Mice that died between 5 and 15 weeks of age were excluded from analysis. n = 7 mice (dyW/dyW), n = 6 (dyW/dyW mag and dyW/dyW aLNNd),n = 11 mice (dyW/dyW DT), n = 8 mice (control). Data are means ± SEM. *P < 0.05, **P < 0.01 (for exact P values, see table S3), two-way ANOVA with Bonferroni posthoc test. (F) Survival curves for dyW/dyW (n = 19), dyW/dyW mag (n = 16), dyW/dyW aLNNd (n = 16), and dyW/dyW DT (n = 17) mice. Marks indicate mice that were stillalive at the end of the study. **P < 0.01 (dyW/dyW mag versus dyW/dyW DT), ***P < 0.001 (dyW/dyW versus dyW/dyW DT or dyW/dyW versus dyW/dyW mag) (for exact P values,see table S3), log-rank test. Controls are wild-type or dyW/+ littermates.
8 of 13
http://stm.sciencemag.org/
SC I ENCE TRANS LAT IONAL MED I C I N E | R E S EARCH ART I C L E
expression ofmag and aLNNd has an evenmore profound effect thatincludes stabilization of body weight and prolonged preservation ofmuscle histology, which together allows some of these transgenicmice to survive as long as control littermates in spite of the completehindlimb paralysis.
by guest on April 3, 2021
http://stm.sciencem
ag.org/D
ownloaded from
DISCUSSIONOur work provides a framework for the mechanistic understandingof LAMA2 MD and presents an efficacious preclinical treatment.The approach is based on the observation that muscle BM of LAMA2MD patients is unstable despite the compensatory increase of laminin-a4. This phenomenon was also observed in dyW/dyWmice. Thus, thedisease pathology in dyW/dyWmice mirrors that seen in LAMA2MDpatients not only phenotypically but also mechanistically. Wild-typeand dyW/dyW mice express high levels of laminin-a4 at birth. Whereaslaminin-a2 replaces laminin-a4 in muscle BM postnatally in wild-typemice, laminin-a4 remains high throughout the life span of dyW/dyW
mice. Such sustained expression of laminin-a4 is a unique feature ofLAMA2MD. The slight increase of laminin-a5 in LAMA2MDpatientsand mouse models, in contrast, may be the consequence of ongoingmuscle degeneration/regeneration, because such increase has beenobserved during muscle regeneration after injury (36) and in severalothermuscular dystrophies (37). Laminin-a4 and laminin-a5 persistin the muscle BM of LAMA2MD patients at an age of at least 14 years(the oldest donor in our cohort), suggesting that the therapeutictranslation of this approach into clinics should be possible well afterthe onset of the disease.
The therapeutic approach presented here uses the specific increaseof laminin-a4 in LAMA2MD as the binding scaffold for the two linkerproteins, mag and aLNNd. A cornerstone of this study was our findingthat combined expression of mag and aLNNd is sufficient to enhancethe cell binding and polymerization capacity of Lm-411 in culturedC2C12 myotubes. These findings were corroborated in vivo, with thecombination of mag and aLNNd restoring BM structure and functionin dystrophic dyW/dyWmice. In contrast, expression of either transgenesingly only partially shifted extractability of Lm-411 and partially im-proved BM structure. These results are thus the direct in vivo supportfor the model (14) that the formation and maintenance of the primarylaminin network require both cell surface binding (restored in thedyW/dyW DT mice by mag) and laminin self-assembly (restored byaLNNd). The therapeutic effect of a single transgene in dyW/dyW micemay be based on the increased amount of laminin-a5. In the dyW/dyW
mag mice, Lm-511 could provide the necessary LN domains to formlaminin hetero-oligomers with Lm-411, whereas binding to muscle fi-bers would be mediated by mag (12). Similar hetero-oligomers betweenLm-411 and Lm-511 could also be formed in dyW/dyWaLNNd, with thelaminin-a5 mediating some binding to muscle receptors. The lamininself-assemblymodel (14) is also supported by the finding that transgenicexpression of aLNNd alone is sufficient to ameliorate muscle functionand BM structure in dy2J/dy2Jmice (21). Muscular dystrophy in dy2J/dy2J
mice is due to an N-terminal truncation of laminin-a2, representing amild form of LAMA2 MD. In these mice, laminin-a2 still contains allthe C-terminal LG domains that bind to the sarcolemma, and thus, res-toration of laminin self-assembly by aLNNd is sufficient [see also com-mentary (38)].
Lack of Lm-211 causes structural instability to contracting musclefibers, leading to their degeneration. This then initiates a cascade ofsecondary events, which all accelerate disease progression. The secondary
Reinhard et al., Sci. Transl. Med. 9, eaal4649 (2017) 28 June 2017
events, such as apoptosis/necrosis of regeneratingmuscle fibers, inflam-mation, and fibrosis, are often common to severalmuscular dystrophies(39). Although approaches to inhibit such downstream events are ap-plicable to several diseases, their efficacy is limited. For example, inhi-bition of fibrosis by the angiotensin II type 1 receptor blocker losartanameliorates the disease in mouse models for Marfan’s syndrome,Duchenne muscular dystrophy, and LAMA2MD (40–42). In dyW/dyW
mice, losartan treatment dampens the expression of matricellularproteins, a class of matrix proteins that does not contribute directly toBM structure but rather serves as regulators of cell function (43). In-creased expression of matricellular proteins is an early disease markerin dyW/dyWmice (32), and there is evidence that normalization of thosemarkers, in conjunction with the improvement of muscle regeneration,ameliorates disease progression (44). We examined the expression of thematricellular protein tenascin-C and found that its expression was thesame as that in wild-type controls upon expression of both linker pro-teins. Together with our data demonstrating reduction of fibrosis andinflammation, these data indicate that prevention of the primary deficitof muscle BM instability in LAMA2MD prevents secondary events.
Forced expression of Lama1, encoding laminin-a1, also tackles thestructural deficit in LAMA2 MD. In these experiments, ubiquitoustransgenic expression fully restored the functionality of the BM andprolonged survival of dystrophicmice for up to 2 years (45, 46). Becausethe transgene was also expressed in the peripheral nerve, the mice didnot showhindlimbparalysis. Although this is proof that laminin-a1 canreplace laminin-a2, translation of this approach into clinics is fraughtwith challenges. LAMA1 is too large to insert into suitable gene therapyvectors; production of Lm-111, with a size of about 850 kDa, ischallenging; and the efficacy of injected protein remains low (47, 48).The strategy presented here, in contrast, has potential for translation intoclinics because the complementary DNAs (cDNAs) encoding aLNNd ormag are small and can be incorporated into adeno-associated virus(AAV) vectors. First, systemic application of muscle-targeted AAV thatexpresses mag ameliorated the disease phenotype in dyW/dyWmice to asimilar extent as transgenic expression (49). Second, AAV-mediatedgene transfer into skeletal muscle allows expression of the gene of inter-est for at least 10 years in human patients (50). Third, both linker pro-teins are secreted and can also be incorporated into the BM in adjacent,noninfected muscle fibers. Fourth, mag and aLNNd are derived fromproteins (agrin, laminin-a1, and nidogen-1) that are expressed inLAMA2MDpatients. Thus, possible immune responses to the expressedlinker proteins might be restricted to neoepitopes generated by thefusion of domains from different proteins. Infection efficacy and im-mune responses are likely less of a challenge than in gene transferstrategies for Duchenne muscular dystrophy, where the transferredmicrodystrophin can only act in the infected muscle fibers and patientsmay generate antibodies against the expressed protein (51). Despitethese advantages of our approach, successful translation into patientswill require the targeting of all muscles by systemic delivery of AAV.Restoration of whole-body muscle function for at least 9 months hasrecently been achieved in dog models for X-linked myotubular myop-athy by one-time peripheral venous infusion of myotubularin-expressing AAV (52). However, systemic delivery of AAV has not beenreported for muscular dystrophy patients. Moreover, high-efficacytreatment of LAMA2MD patients would require delivery of two dif-ferent AAVvectors and at young age. Injection of recombinant proteinsmight be another option for treatment, but success might be hamperedbecause mag and aLNNd strongly bind to extracellular matrix proteinsthat are not specific for muscle BM.
9 of 13
http://stm.sciencemag.org/
SC I ENCE TRANS LAT IONAL MED I C I N E | R E S EARCH ART I C L E
Dow
nloa
On the basis of the data obtained in mice, the combined treatmentwith mag and aLNNd should greatly improve quality of life measures,such as muscle function and bodymass, and most importantly prolonglife span. In the mice, median survival increased from 16 weeks for thedyW/dyWmice to more than 81 weeks in the dyW/dyWDTmice. In thedyW/dyWDTmice cohort, one-third of themice reached an age ofmorethan 2 years, an age that only one-half of female and two-thirds of malewild-type C57/BL6 mice reach (53). The life span extension wasachieved despite hindlimb paralysis resulting from defective peripheralnerve myelination, which is not corrected in the transgenic mice due tothe muscle-specific expression of the transgenes. The observation thatlife span extension is possible in spite of hindlimb paralysis also showsthat the lower efficacy of the single transgenes is likely based on anincomplete restoration of muscle function and not on phenotypescaused in nonmuscle tissue. Whereas the peripheral neuropathy is aprominent feature of all mouse models of LAMA2MD (5), the musculardystrophy is the predominant phenotype in human patients (54). Thereis only one case report in whichmutations in LAMA2 have been shownto result in a limb-girdle type of muscular dystrophy with prevalentperipheral nerve involvement (55).
by guest on April 3, 2021
http://stm.sciencem
ag.org/ded from
MATERIALS AND METHODSStudy designThe objective of this study was to test the use of two designed smalllinker proteins for the potential treatment of LAMA2MD.We reasonedthat addition of these proteins could restore the muscle BM despiteLm-211 deficiency. We tested the capacity of the linker proteins invitro and by transgenic expression in a mouse model for LAMA2MD. No statistical methods were used to predetermine sample sizes,but sample sizes are similar to those reported in the field. We did notuse any method of randomization and did not exclude mice from thestudy except those that died before the planned age of analysis. Evalua-tions of immunohistochemistry and muscle histology were performedby investigators blinded to the specific sample.
MiceDyW/dyW mice [B6.129S1(Cg)-Lama2tm1Eeng/J; available from theJacksonLaboratory] containing aLacZ insertion in theLama2 (6) servedas mouse model for LAMA2 MD. Mice expressing the mag transgeneunder the MCK promoter were described previously (12). The aLNNdtransgene is a chimeric protein consisting of the laminin-a1 LN-LEadomains fused to the C-terminal part of nidogen-1 (G2-G3 domain)(20). Its corresponding cDNA was subcloned into a vector containingthe MCK promoter sequence (12). This construct was linearized withPac I and injected into the pronuclei of fertilized mouse oocytes. Weused two lines that expressed aLNNd at high levels. Neurotrypsin-deficient mice were described previously (30). Genotyping of dyW/dyW
andmag was performed as described previously (6, 12, 19). Genotypingof aLNNd was performed with the following primers: 5′-CTCATCT-CAGAAGAGGATCTG-3′ and 5′-GAATAATACGAGGTGCAGAT-GACTTC-3′. The transgenicmouse lines weremaintained on aC57BL/6J background. All mice analyzed were from breedings of dyW/+ micethat were hemizygous for aLNNd or mag, which allowed to receive allgenotypes from the samebreeding.Controlmicewerewild type or dyW/+from the same litters. Unless otherwise indicated, female andmalemicewere used. To ensure optimal access of the dystrophicmice towater andfood, all cages were supplied with long-necked water bottles and wetfood. All mouse experiments were performed according to the federal
Reinhard et al., Sci. Transl. Med. 9, eaal4649 (2017) 28 June 2017
guidelines for animal experimentation and approved by the authoritiesof the Canton of Basel-Stadt.
Human samplesMuscle biopsies analyzed were from donors of both genders (ages 5 to16 years). They were collected as extra tissue that was removed duringa diagnostic biopsy or as remaining or extra tissue that was removedduring amedical procedure after patient consent. Biopsieswere receivedfrom the Institute of Pathology, University Hospital, Basel, Switzerland,and three institutes associated with EuroBioBank (www.eurobiobank.org) and TREAT-NMD (www.treat-nmd.eu): the “Myobank AFM”hosted by the Institute de Myologie, Paris, France; the “Cells, tissuesandDNA frompatients with neuromuscular diseases” hosted byC. BestaNeurological Institute, Milano, Italy (funded by the Telethon Networkof Genetic Biobanks, project no. GTB12001); and “The Muscle TissueCulture Collection” hosted by the Friedrich-Baur-Institute, Munich,Germany [associated with MD-NET and mito-NET, which are bothfunded by Federal Ministry of Education and Research (BMBF)].
AntibodiesFor immunostaining and Western blot analysis, the following anti-bodies were used: a-actinin (1:5000; Sigma, catalog no. A7732), agrin[chicken, for detection of mag, produced in-house (56), 1:1000 forWestern blots, 1:200 for immunostainings], eMHC (1:100 for Westernblot, 1:1200 for immunostainings; developed by H. Blau, obtained fromDevelopmental Studies Hybridoma Bank, catalog no. F1.652), F4/80(1:100; Abcam, catalog no. ab6640), laminin-a1 (for detection ofaLNNd by Western blot, 1:2000; R&D Systems, catalog no. AF4187),aLNNd [for detection of aLNNd by immunostainings, 1:100 (21)],laminin-a2 [for Western blot analysis (57), 1:500], laminin-a2 (N-terminal, for immunostainings of human samples, clone 4H8-2,1:400; Sigma, catalog no. L0663), laminin-a2 (C-terminal, for immu-nostainings of human samples, clone 5H2, 1:5000; Merck Millipore,catalog no. MAB1922), laminin-a4 (21) (1:1000 for Western blots,1:200 for immunostainings), laminin-a5 [clone 504, gift fromL. Sorokin(58), 1:1000], laminin-b1g1 (1:1000 forWestern blots, 1:100 for immu-nostainings; Sigma, catalog no. L9393), laminin-g1 (1:200; Millipore,catalog no. MAB1914), Na+/K+-ATPase (1:1000; Cell Signaling, catalogno. 3010), GAPDH (1:1000; Cell Signaling, catalog no. 2118), andCD31(1:100; Abcam, catalog no. ab9498).
Recombinant and native protein preparation forbinding assaysRecombinant Lm-111, nidogen-1, and aLNNd were purified byhemagglutinin–affinity chromatography from stably transfected humanembryonic kidney (HEK) 293 cells, as described (59). Mag was purifiedby metal-chelating chromatography, as described (20, 59, 60). Recom-binant Lm-211 was purified by heparin affinity chromatography fromcells grown in the presence 5 mMfurin inhibitor-1 (Calbiochem, catalogno. 344930) to reduce LG domain cleavage (61). Recombinant Lm-511containing Lma5–C-terminal FLAG tag was purified fromHEK293 byanti–FLAG-M2 affinity chromatography (60). Recombinant mousenidogen-1 was purified from conditioned medium by HisPur-cobaltchelating chromatography (Thermo Scientific, catalog no. 89965). TypeIV collagen was extracted and purified from lathyritic mouse EHStumor, as described (62). Integrin constructs coding for soluble integrina7X2 and integrin b1 were stably transfected into 293 cells (J. Takagi,Osaka University, Japan) and purified onHisPur-cobalt chelating chro-matography followed by FLAG-agarose affinity chromatography. aDG
10 of 13
http://www.eurobiobank.orghttp://www.eurobiobank.orghttp://www.treat-nmd.euhttp://stm.sciencemag.org/
SC I ENCE TRANS LAT IONAL MED I C I N E | R E S EARCH ART I C L E
by guest on April 3, 2021
http://stm.sciencem
ag.org/D
ownloaded from
was extracted from rabbitmuscles usingwheat germagglutinin–agarosebeads (Vector Laboratories, catalog no. al-1023) as described (63) or asrecombinant protein as described previously (64).
Polymerization assaysFor polymerization assays, a 1:1 molar mixture of purified Lm-411 +aLNNd or Lm-411 and Lm-111 alone were incubated overnight onice in 50 mM tris-HCl (pH 7.4), 90 mMNaCl, and 1 mMCaCl2. Sub-sequently, aliquots of various concentrations were incubated at 37°Cfor 3 hours in polymerization buffer [50mM tris-HCl (pH 7.4), 90mMNaCl, 1 mMCaCl2, 0.15% Triton X-100, bovine serum albumin (BSA)(30 ng/ml)]. Polymerized laminins were separated from nonpolymer-ized forms by centrifugation at 11,000g. Supernatant and pellet wereloaded on SDS-PAGE followed by Coomassie blue staining and quan-titation, as described (57, 60).
In vitro binding assaysTo determine laminin binding to aDG, crude aDG was coated ontohigh-binding 96-well plates (Costar, catalog no. 3691) in bicarbonatebuffer overnight at 4°C. Plates were incubated for 1 hour at room tem-peraturewith blocking buffer [50mM tris-HCl (pH7.4), 150mMNaCl,3% BSA, 1mMMgCl2, 1 mMCaCl2], washed with phosphate-bufferedsaline (PBS), and incubated with various amounts of recombinantlaminins (in blocking buffer) for 2 hours at room temperature. Lamininbinding was detected with aE4 (10 mg/ml; anti–Lm-111) antibodies andsecondary anti-rabbit horseradish peroxidase (HRP), developed withUltra-TMB (Thermo Scientific 34028), and absorbance was measuredat 450 nm. To determine laminin binding to integrin a7X2b1, recombi-nant Lm-111 or Lm-411 (in bicarbonate buffer) was coated to high-binding 96-well plates overnight at 4°C. Plates were incubated for1 hour at room temperature with blocking buffer [50 mM tris-HCl(pH 7.4), 90 mM NaCl, 1% BSA, 1 mM MnCl2, 0.1% Tween-20],washed with PBS, and incubated with various concentrations of in-tegrin a7X2b1 (in blocking buffer) for 2 hours at RT. Bound integrinwas detected with anti-VELCRO antibody (1:1000) and streptavidin-HRP antibodies and detected with Ultra-TMB, as described above.
Histology and histological quantificationsMuscles were mounted on 7% gum tragacanth (Sigma), and rapidlyfrozen 2-methylbutane was cooled in liquid nitrogen (−150°C). Crosssections of 12 mm thickness were cut on a cryostat. General histologywas assessed after tissue fixation with 4% paraformaldehyde by H&Estaining (Merck) or Picro Sirius Red stain [Direct Red 80 (Sigma) inpicric acid solution]. Images were acquired with an Olympus iX81microscope using cellSens software (Olympus). To assess the severityof muscular dystrophy, the H&E-stained cross sections were blindlyexamined and scored by two to three observers. The basis of the judg-ment was a qualitative extent of muscle degeneration, cell infiltration,fibrosis, the shape ofmuscle fibers and their number, size and size var-iation, and the number of fibers containing centralized myonuclei.Each mouse was given a relative value termed “dystrophic severityindex” from 0 (no muscular dystrophy) to 4 (severe muscular dystro-phy). For fiber number and fiber size analysis, laminin-g1–stainedmid-belly cross sections were evaluated using cellSens software (OlympusSoft Imaging System). The muscle fiber size was quantified using theminimum distance of parallel tangents at opposing particle borders(minimal “Feret’s diameter”), as described elsewhere (65). The fibroticarea in diaphragm was quantified as the percentage of Sirius Red–positive area per cross-sectional area.
Reinhard et al., Sci. Transl. Med. 9, eaal4649 (2017) 28 June 2017
Hydroxyproline assayFibrosis in muscles was determined by measuring the amount ofcollagen-specific amino acid hydroxyproline. Methods for isolation,freeze-drying, and amino acid analysis were as described (19). Aminoacid analysis was done by the Analytical Research and Services, Univer-sity of Bern, Bern, Switzerland.
Grip strength and in vitro muscle force measurementGrip strength was assessed by placing the mice on a vertical grid andmeasuring its hang time. The experiment was stopped after 3 min. Invitro muscle force measurements were performed on isolated EDL andsoleus muscle using the 1200A Isolated Muscle System (Aurora Scien-tific). By stimulationwith a single electrical pulse (2.5 kHz, 15V, 0.2ms),muscles were adjusted to the optimum muscle length (Lo) achieved atpeak twitch force (Pt). Peak tetanic force (Po) was recordedwith 500-msstimulation (150 Hz for EDL, 120 Hz for soleus). Specific peak andtwitch forces were calculated by normalization to the cross-sectional ar-ea (CSA) by using the following formula: CSA (mm2) = muscle wetweight (mg)/[fiber length (lf, mm) × 1.06 mg/mm
3], with lf = lo ×0.44 for EDL or lf = lo × 0.71 for soleus (66).
Statistical analysisStatistical analysis was performed using unpaired, two-tailed Student’s ttest for comparisons of two groups. Welch’s correction was applied ifunequal variance between groupswas detected byF test.One- or two-wayANOVA followed by Bonferroni post hoc test was used for the compar-isons of more than two groups.We assumed normal distribution of thevariables analyzed. Survival analyses were performed using the Kaplan-Meier method, and the significance of differences between curves wascalculated using the log-rank test. All statistical tests were performedusing Prism version 6 (GraphPad Software). Statistical significancewas set at P < 0.05. All data are presented as means ± SEM, unless in-dicated otherwise in the figure legend. Individual-level data and exactP values are shown in table S3.
SUPPLEMENTARY MATERIALSwww.sciencetranslationalmedicine.org/cgi/content/full/9/396/eaal4649/DC1Materials and MethodsFig. S1. Expression of laminin-a2 and laminin-a4 in muscles of LAMA2 MD and healthy controls.Fig. S2. Expression of laminin-a4 at different ages in wild-type and dyW/dyW mice.Fig. S3. Expression of laminin-a5 in control and diseased muscles of humans and mice.Fig. S4. Schematic representation of transgenic proteins and effect of mag on aDG binding ofLm-511.Fig. S5. Expression and processing of transgenes.Fig. S6. Inflammation is reduced by expression of aLNNd and mag in dyW/dyW mice.Fig. S7. Muscle degeneration and regeneration in 3- and 8-week-old dyW/dyW and dyW/dyWDTmice.Fig. S8. Transgenic expression of aLNNd and mag in dyW/dyW mice improves muscle histologyof TA and diaphragm muscle in 8-week-old mice.Fig. S9. Expression of mag and aLNNd in dyW/dyW mice improves variance of fiber diameterand collagen content in 16-week-old mice.Fig. S10. Phenotype and continued presence of laminin-a4 and laminin-a5 in 2-year-olddyW/dyW DT mice.Table S1. Statistical analysis of fiber size classes shown in Fig. 5D.Table S2. Muscle force in isolated EDL or soleus muscle from 8- or 16-week-old mice.Table S3. Single data points and exact P values.
REFERENCES AND NOTES1. E. Mercuri, F. Muntoni, Muscular dystrophies. Lancet 381, 845–860 (2013).2. F. L. M. Norwood, C. Harling, P. F. Chinnery, M. Eagle, K. Bushby, V. Straub, Prevalence of
genetic muscle disease in Northern England: In-depth analysis of a muscle clinicpopulation. Brain 132, 3175–3186 (2009).
11 of 13
http://www.sciencetranslationalmedicine.org/cgi/content/full/9/396/eaal4649/DC1http://stm.sciencemag.org/
SC I ENCE TRANS LAT IONAL MED I C I N E | R E S EARCH ART I C L E
by guest on April 3, 2021
http://stm.sciencem
ag.org/D
ownloaded from
3. E. M. Clement, L. Feng, R. Mein, C. A. Sewry, S. A. Robb, A. Y. Manzur, E. Mercuri, C. Godfrey,T. Cullup, S. Abbs, F. Muntoni, Relative frequency of congenital muscular dystrophysubtypes: Analysis of the UK diagnostic service 2001–2008. Neuromuscul. Disord. 22,522–527 (2012).
4. F. Geranmayeh, E. Clement, L. H. Feng, C. Sewry, J. Pagan, R. Mein, S. Abbs, L. Brueton,A.-M. Childs, H. Jungbluth, C. G. De Goede, B. Lynch, J.-P. Lin, G. Chow, C. Sousa,O. O’Mahony, A. Majumdar, V. Straub, K. Bushby, F. Muntoni, Genotype–phenotypecorrelation in a large population of muscular dystrophy patients with LAMA2 mutations.Neuromuscul. Disord. 20, 241–250 (2010).
5. L. T. Guo, X. U. Zhang, W. Kuang, H. Xu, L. A. Liu, J.-T. Vilquin, Y. Miyagoe-Suzuki, S. Takeda,M. A. Ruegg, U. M. Wewer, E. Engvall, Laminin a2 deficiency and muscular dystrophy;genotype-phenotype correlation in mutant mice. Neuromuscul. Disord. 13, 207–215(2003).
6. W. Kuang, H. Xu, P. H. Vachon, L. Liu, F. Loechel, U. M. Wewer, E. Engvall, Merosin-deficientcongenital muscular dystrophy. Partial genetic correction in two mouse models.J. Clin. Invest. 102, 844–852 (1998).
7. H. Xu, X.-R. Wu, U. M. Wewer, E. Engvall, Murine muscular dystrophy caused by amutation in the laminin a2 (Lama2) gene. Nat. Genet. 8, 297–302 (1994).
8. H. Colognato, P. D. Yurchenco, The laminin a2 expressed by dystrophic dy2J mice isdefective in its ability to form polymers. Curr. Biol. 9, 1327–1330 (1999).
9. B. L. Patton, J. H. Miner, A. Y. Chiu, J. R. Sanes, Distribution and function of laminins inthe neuromuscular system of developing, adult, and mutant mice. J. Cell Biol. 139,1507–1521 (1997).
10. D. Gullberg, C. F. Tiger, T. Velling, Laminins during muscle development and in musculardystrophies. Cell. Mol. Life Sci. 56, 442–460 (1999).
11. B. Ringelmann, C. Röder, R. Hallmann, M. Maley, M. Davies, M. Grounds, L. Sorokin,Expression of laminin a1, a2, a4, and a5 chains, fibronectin, and tenascin-C in skeletalmuscle of dystrophic 129ReJdy/dy mice. Exp. Cell Res. 246, 165–182 (1999).
12. J. Moll, P. Barzaghi, S. Lin, G. Bezakova, H. Lochmüller, E. Engvall, U. Müller, M. A. Ruegg,An agrin minigene rescues dystrophic symptoms in a mouse model for congenitalmuscular dystrophy. Nature 413, 302–307 (2001).
13. S. Li, D. Harrison, S. Carbonetto, R. Fassler, N. Smyth, D. Edgar, P. D. Yurchenco,Matrix assembly, regulation, and survival functions of laminin and its receptors inembryonic stem cell differentiation. J. Cell Biol. 157, 1279–1290 (2002).
14. P. D. Yurchenco, Basement membranes: Cell scaffoldings and signaling platforms. ColdSpring Harb. Perspect. Biol. 3, a004911 (2011).
15. J. F. Talts, T. Sasaki, N. Miosge, W. Göhring, K. Mann, R. Mayne, R. Timpl, Structural andfunctional analysis of the recombinant G domain of the laminin a4 chain and itsproteolytic processing in tissues. J. Biol. Chem. 275, 35192–35199 (2000).
16. H. von der Mark, I. Williams, O. Wendler, L. Sorokin, K. von der Mark, E. Pöschl, Alternativesplice variants of a7b1 integrin selectively recognize different laminin isoforms. J. Biol.Chem. 277, 6012–6016 (2002).
17. R. Nishiuchi, J. Takagi, M. Hayashi, H. Ido, Y. Yagi, N. Sanzen, T. Tsuji, M. Yamada,K. Sekiguchi, Ligand-binding specificities of laminin-binding integrins: A comprehensivesurvey of laminin–integrin interactions using recombinant a3b1, a6b1, a7b1 and a6b4integrins. Matrix Biol. 25, 189–197 (2006).
18. C. F. Bentzinger, P. Barzaghi, S. Lin, M. A. Ruegg, Overexpression of mini-agrin in skeletalmuscle increases muscle integrity and regenerative capacity in laminin-a2-deficientmice. FASEB J. 19, 934–942 (2005).
19. S. Meinen, P. Barzaghi, S. Lin, H. Lochmüller, M. A. Ruegg, Linker molecules betweenlaminins and dystroglycan ameliorate laminin-a2–deficient muscular dystrophy atall disease stages. J. Cell Biol. 176, 979–993 (2007).
20. K. K. McKee, S. Capizzi, P. D. Yurchenco, Scaffold-forming and adhesive contributions ofsynthetic laminin-binding proteins to basement membrane assembly. J. Biol. Chem.284, 8984–8994 (2009).
21. K. K. McKee, S. C. Crosson, S. Meinen, J. R. Reinhard, M. A. Ruegg, P. D. Yurchenco, Chimericprotein repair of laminin polymerization ameliorates muscular dystrophy phenotype.J. Clin. Invest. 127, 1075–1089 (2017).
22. B. L. Patton, A. M. Connolly, P. T. Martin, J. M. Cunningham, S. Mehta, A. Pestronk,J. H. Miner, J. R. Sanes, Distribution of ten laminin chains in dystrophic and regeneratingmuscles. Neuromuscul. Disord. 9, 423–433 (1999).
23. C. A. Sewry, J. Philpot, D. Mahony, L. A. Wilson, F. Muntoni, V. Dubowitz, Expression oflaminin subunits in congenital muscular dystrophy. Neuromuscul. Disord. 5, 307–316(1995).
24. P. D. Yurchenco, B. L. Patton, Developmental and pathogenic mechanisms of basementmembrane assembly. Curr. Pharm. Des. 15, 1277–1294 (2009).
25. A. J. Denzer, R. Brandenberger, M. Gesemann, M. Chiquet, M. A. Ruegg, Agrin binds to thenerve–muscle basal lamina via laminin. J. Cell Biol. 137, 671–683 (1997).
26. R. A. Kammerer, T. Schulthess, R. Landwehr, B. Schumacher, A. Lustig, P. D. Yurchenco,M. A. Ruegg, J. Engel, A. J. Denzer, Interaction of agrin with laminin requires a coiled-coilconformation of the agrin-binding site within the laminin g1 chain. EMBO J. 18,6762–6770 (1999).
Reinhard et al., Sci. Transl. Med. 9, eaal4649 (2017) 28 June 2017
27. M. Gesemann, V. Cavalli, A. J. Denzer, A. Brancaccio, B. Schumacher, M. A. Ruegg,Alternative splicing of agrin alters its binding to heparin, dystroglycan, and the putativeagrin receptor. Neuron 16, 755–767 (1996).
28. P. T. Martin, J. R. Sanes, Integrins mediate adhesion to agrin and modulate agrinsignaling. Development 124, 3909–3917 (1997).
29. S. Meinen, S. Lin, R. Thurnherr, M. Erb, T. Meier, M. A. Rüegg, Apoptosis inhibitors andmini-agrin have additive benefits in congenital muscular dystrophy mice. EMBO Mol.Med. 3, 465–479 (2011).
30. R. Reif, S. Sales, S. Hettwer, B. Dreier, C. Gisler, J. Wölfel, D. Lüscher, A. Zurlinden,A. Stephan, S. Ahmed, A. Baici, B. Ledermann, B. Kunz, P. Sonderegger, Specific cleavageof agrin by neurotrypsin, a synaptic protease linked to mental retardation. FASEB J.21, 3468–3478 (2007).
31. J. A. Doe, R. D. Wuebbles, E. T. Allred, J. E. Rooney, M. Elorza, D. J. Burkin, Transgenicoverexpression of the a7 integrin reduces muscle pathology and improves viability in thedyW mouse model of merosin-deficient congenital muscular dystrophy type 1A.J. Cell Sci. 124, 2287–2297 (2011).
32. T. Mehuron, A. Kumar, L. Duarte, J. Yamauchi, A. Accorsi, M. Girgenrath, Dysregulation ofmatricellular proteins is an early signature of pathology in laminin-deficient musculardystrophy. Skelet. Muscle 4, 14 (2014).
33. W. Kuang, H. Xu, J. T. Vilquin, E. Engvall, Activation of the lama2 gene in muscleregeneration: Abortive regeneration in laminin alpha2-deficiency. Lab. Invest.79, 1601–1613 (1999).
34. J. Yamauchi, A. Kumar, L. Duarte, T. Mehuron, M. Girgenrath, Triggering regenerationand tackling apoptosis: A combinatorial approach to treating congenital musculardystrophy type 1 A. Hum. Mol. Genet. 22, 4306–4317 (2013).
35. K. I. Gawlik, M. Durbeej, Skeletal muscle laminin and MDC1A: Pathogenesis and treatmentstrategies. Skelet. Muscle 1, 9 (2011).
36. J. Huijbregts, J. D. White, M. D. Grounds, The absence of MyoD in regenerating skeletalmuscle affects the expression pattern of basement membrane, interstitial matrix andintegrin molecules that is consistent with delayed myotube formation. Acta Histochem.103, 379–396 (2001).
37. C.-F. Tiger, M.-F. Champliaud, F. Pedrosa-Domellof, L.-E. Thornell, P. Ekblom, D. Gullberg,Presence of laminin a5 chain and lack of laminin a1 chain during human muscledevelopment and in muscular dystrophies. J. Biol. Chem. 272, 28590–28595 (1997).
38. S. D. Funk, J. H. Miner, Muscular dystrophy meets protein biochemistry, the mother ofinvention. J. Clin. Invest. 127, 798–800 (2017).
39. E. Engvall, U. M. Wewer, The new frontier in muscular dystrophy research: Booster genes.FASEB J. 17, 1579–1584 (2003).
40. J. P. Habashi, D. P. Judge, T. M. Holm, R. D. Cohn, B. L. Loeys, T. K. Cooper, L. Myers,E. C. Klein, G. Liu, C. Calvi, M. Podowski, E. R. Neptune, M. K. Halushka, D. Bedja,K. Gabrielson, D. B. Rifkin, L. Carta, F. Ramirez, D. L. Huso, H. C. Dietz, Losartan, an AT1antagonist, prevents aortic aneurysm in a mouse model of Marfan syndrome. Science312, 117–121 (2006).
41. R. D. Cohn, C. van Erp, J. P. Habashi, A. A. Soleimani, E. C. Klein, M. T. Lisi, M. Gamradt,C. M. ap Rhys, T. M. Holm, B. L. Loeys, F. Ramirez, D. P. Judge, C. W. Ward, H. C. Dietz,Angiotensin II type 1 receptor blockade attenuates TGF-b–induced failure of muscleregeneration in multiple myopathic states. Nat. Med. 13, 204–210 (2007).
42. M. Elbaz, N. Yanay, S. Aga-Mizrachi, Z. Brunschwig, I. Kassis, K. Ettinger, V. Barak, Y. Nevo,Losartan, a therapeutic candidate in congenital muscular dystrophy: Studies in thedy2J/dy2J mouse. Ann. Neurol. 71, 699–708 (2012).
43. P. Bornstein, E. H. Sage, Matricellular proteins: Extracellular modulators of cell function.Curr. Opin. Cell Biol. 14, 608–616 (2002).
44. A. Accorsi, A. Kumar, Y. Rhee, A. Miller, M. Girgenrath, IGF-1/GH axis enhances losartantreatment in Lama2-related muscular dystrophy. Hum. Mol. Genet. 25, 4624–4634 (2016).
45. K. Gawlik, Y. Miyagoe-Suzuki, P. Ekblom, S. Takeda, M. Durbeej, Laminin a1 chainreduces muscular dystrophy in laminin a2 chain deficient mice. Hum. Mol. Genet. 13,1775–1784 (2004).
46. K. I. Gawlik, M. Durbeej, Transgenic overexpression of laminin a1 chain in laminin a2chain–deficient mice rescues the disease throughout the lifespan. Muscle Nerve 42, 30–37(2010).
47. P. M. Van Ry, P. Minogue, B. L. Hodges, D. J. Burkin, Laminin-111 improves musclerepair in a mouse model of merosin-deficient congenital muscular dystrophy.Hum. Mol. Genet. 23, 383–396 (2014).
48. J. E. Rooney, J. R. Knapp, B. L. Hodges, R. D. Wuebbles, D. J. Burkin, Laminin-111 proteintherapy reduces muscle pathology and improves viability of a mouse model ofmerosin-deficient congenital muscular dystrophy. Am. J. Pathol. 180, 1593–1602 (2012).
49. C. Qiao, J. Li, T. Zhu, R. Draviam, S. Watkins, X. Ye, C. Chen, X. Xiao, Amelioration oflaminin-a2-deficient congenital muscular dystrophy by somatic gene transfer ofminiagrin. Proc. Natl. Acad. Sci. U.S.A. 102, 11999–12004 (2005).
50. G. Buchlis, G. M. Podsakoff, A. Radu, S. M. Hawk, A. W. Flake, F. Mingozzi, K. A. High, FactorIX expression in skeletal muscle of a severe hemophilia B patient 10 years after AAV-mediated gene transfer. Blood 119, 3038–3041 (2012).
12 of 13
http://stm.sciencemag.org/
SC I ENCE TRANS LAT IONAL MED I C I N E | R E S EARCH ART I C L E
by ghttp://stm
.sciencemag.org/
Dow
nloaded from
51. J. R. Mendell, K. Campbell, L. Rodino-Klapac, Z. Sahenk, C. Shilling, S. Lewis, D. Bowles,S. Gray, C. Li, G. Galloway, V. Malik, B. Coley, K. R. Clark, J. Li, X. Xiao, J. Samulski,S. W. McPhee, R. J. Samulski, C. M. Walker, Dystrophin immunity in Duchenne’s musculardystrophy. N. Engl. J. Med. 363, 1429–1437 (2010).
52. D. L. Mack, K. Poulard, M. A. Goddard, V. Latournerie, J. M. Snyder, R. W. Grange,M. R. Elverman, J. Denard, P. Veron, L. Buscara, C. Le Bec, J.-Y. Hogrel, A. G. Brezovec,H. Meng, L. Yang, F. Liu, M. O’Callaghan, N. Gopal, V. E. Kelly, B. K. Smith, J. L. Strande,F. Mavilio, A. H. Beggs, F. Mingozzi, M. W. Lawlor, A. Buj-Bello, M. K. Childers, SystemicAAV8-mediated gene therapy drives whole-body correction of myotubular myopathy indogs. Mol. Ther. 25, 839–854 (2017).
53. A. Turturro, P. Duffy, B. Hass, R. Kodell, R. Hart, Survival characteristics and age-adjusteddisease incidences in C57BL/6 mice fed a commonly used cereal-based diet modulated bydietary restriction. J. Gerontol. A Biol. Sci. Med. Sci. 57, B379–B389 (2002).
54. C. G. Bonnemann, C. H. Wang, S. Quijano-Roy, N. Deconinck, E. Bertini, A. Ferreiro,F. Muntoni, C. Sewry, C. Beroud, K. D. Mathews, S. A. Moore, J. Bellini, A. Rutkowski,K. N. North, Diagnostic approach to the congenital muscular dystrophies. Neuromuscul.Disord. 24, 289–311 (2014).
55. S. H. S. Chan, A. R. Foley, R. Phadke, A. A. Mathew, M. Pitt, C. Sewry, F. Muntoni, Limbgirdle muscular dystrophy due to LAMA2 mutations: Diagnostic difficulties due toassociated peripheral neuropathy. Neuromuscul. Disord. 24, 677–683 (2014).
56. M. Gesemann, A. J. Denzer, M. A. Ruegg, Acetylcholine receptor-aggregatingactivity of agrin isoforms and mapping of the active site. J. Cell Biol. 128, 625–636(1995).
57. Y.-S. Cheng, M.-F. Champliaud, R. E. Burgeson, M. P. Marinkovich, P. D. Yurchenco, Self-assembly of laminin isoforms. J. Biol. Chem. 272, 31525–31532 (1997).
58. J. Song, Z. Lokmic, T. Lämmermann, J. Rolf, C. Wu, X. Zhang, R. Hallmann,M.-J. Hannocks, N. Horn, M. A. Ruegg, A. Sonnenberg, E. Georges-Labouesse,T. H. Winkler, J. F. Kearney, S. Cardell, L. Sorokin, Extracellular matrix of secondarylymphoid organs impacts on B-cell fate and survival. Proc. Natl. Acad. Sci. U.S.A. 110,E2915–E2924 (2013).
59. K. K. McKee, D.-H. Yang, R. Patel, Z.-L. Chen, S. Strickland, J. Takagi, K. Sekiguchi,P. D. Yurchenco, Schwann cell myelination requires integration of laminin activities. J. CellSci. 125, 4609–4619 (2012).
60. K. K. McKee, D. Harrison, S. Capizzi, P. D. Yurchenco, Role of laminin terminalglobular domains in basement membrane assembly. J. Biol. Chem. 282, 21437–21447(2007).
61. S. P. Smirnov, P. Barzaghi, K. K. McKee, M. A. Ruegg, P. D. Yurchenco, Conjugation of LGdomains of agrins and perlecan to polymerizing laminin-2 promotes acetylcholinereceptor clustering. J. Biol. Chem. 280, 41449–41457 (2005).
62. P. D. Yurchenco, H. Furthmayr, Self-assembly of basement membrane collagen.Biochemistry 23, 1839–1850 (1984).
Reinhard et al., Sci. Transl. Med. 9, eaal4649 (2017) 28 June 2017
63. D. E. Michele, R. Barresi, M. Kanagawa, F. Saito, R. D. Cohn, J. S. Satz, J. Dollar, I. Nishino,R. I. Kelley, H. Somer, V. Straub, K. D. Mathews, S. A. Moore, K. P. Campbell,Post-translational disruption of dystroglycan-ligand interactions in congenital musculardystrophies. Nature 418, 417–422 (2002).
64. J. H. Yoon, R. Xu, P. Martin, A method to produce and purify full-length recombinant alphadystroglycan: Analysis of N- and O-linked monosaccharide composition in CHO cellswith or without LARGE overexpression. PLOS Curr. 5 (2013).
65. A. Briguet, I. Courdier-Fruh, M. Foster, T. Meier, J. P. Magyar, Histological parameters forthe quantitative assessment of muscular dystrophy in the mdx-mouse. Neuromuscul.Disord. 14, 675–682 (2004).
66. S. V. Brooks, J. A. Faulkner, Contractile properties of skeletal muscles from young, adultand aged mice. J. Physiol. 404, 71–82 (1988).
Acknowledgments: We thank F. Oliveri for technical assistance throughout the project,U. Sauder for help in electron microscopy, P. Jenö for mass spectrometry, the animal facility formouse handling, and the Transgenic Mouse Core facility of the University of Basel for cDNAinjections into mouse embryos. We thank D. Ham for critical reading of the manuscript andN. Rion for sharing material. Neurotrypsin knockout mice were received from Neurotune Ltd. andC. Wagner (University of Zurich, Switzerland), and laminin-a5 antibodies were from L. Sorokin(University of Münster, Germany). We thank S. Frank (University Hospital Basel), S. Krause(Friedrich Baur Institute, Munich, Germany), M. Mora (C. Besta Neurological Institute, Milano,Italy), and M. Chapart (Hôpital de la Pitié-Salpêtrière, Paris, France) for providing biopsies.Funding: This work was supported by the Cantons of Basel-Stadt and Basel-Landschaft, grantsfrom the Swiss Foundation for Research on Muscle Diseases, the Association Française contreles Myopathies, and the Neuromuscular Research Association Basel. Author contributions:J.R.R., P.D.Y., and M.A.R. designed experiments. J.R.R., S.L., S.M., S.C.C., M.S., and G.M. performedexperiments and analyzed the data. K.K.M., with the help of S.H., generated and characterizedantibodies and recombinant laminins and provided all the data shown in Fig. 2. J.R.R. andM.A.R. wrote the manuscript. Competing interests: All authors declare that they haveno competing interests. Data and materials availability: Relevant data that support thefindings of this study are available from M.A.R. ([email protected]) upon request.
Submitted 23 November 2016Resubmitted 3 March 2017Accepted 4 May 2017Published 28 June 201710.1126/scitranslmed.aal4649
Citation: J. R. Reinhard, S. Lin, K. K. McKee, S. Meinen, S. C. Crosson, M. Sury, S. Hobbs, G. Maier,P. D. Yurchenco, M. A. Rüegg, Linker proteins restore basement membrane and correct LAMA2-related muscular dystrophy in mice. Sci. Transl. Med. 9, eaal4649 (2017).
ue
13 of 13
st on April 3, 2021
[email protected]://stm.sciencemag.org/
dystrophy in mice-related muscularLAMA2Linker proteins restore basement membrane and correct
Hobbs, Geraldine Maier, Peter D. Yurchenco and Markus A. RüeggJudith R. Reinhard, Shuo Lin, Karen K. McKee, Sarina Meinen, Stephanie C. Crosson, Maurizio Sury, Samantha
DOI: 10.1126/scitranslmed.aal4649, eaal4649.9Sci Transl Med
spans. These proteins could be a missing link for muscular dystrophy therapy.had stable basement membranes, improved muscle function, and prolonged life−−1 and nidogen-1αlaminin-
LNNd, composed of parts ofαa shorter form of agrin and the fusion protein −−expressing two linker proteins proteins could be used to fortify the basement membrane, using laminin-411 as a scaffold. Transgenic mice
studied whether linker et al.degeneration. Using a transgenic mouse model of muscular dystrophy, Reinhard basement membrane and lack of proper connections to the muscle plasma membrane, leading to musclechain of laminin-211, a basement membrane component. Deleterious muscle function results from the unstable
The most common form of congenital muscular dystrophy is caused by mutations in the gene encoding oneBuilding a better basement membrane
ARTICLE TOOLS http://stm.sciencemag.org/content/9/396/eaal4649
MATERIALSSUPPLEMENTARY http://stm.sciencemag.org/content/suppl/2017/06/26/9.396.eaal4649.DC1
CONTENTRELATED
http://stm.sciencemag.org/content/scitransmed/12/541/eaaz2415.fullhttp://stm.sciencemag.org/content/scitransmed/11/520/eaat6072.fullhttp://science.sciencemag.org/content/sci/366/6463/351.fullhttp://stm.sciencemag.org/content/scitransmed/4/164/164ra160.fullhttp://stm.sciencemag.org/content/scitransmed/2/42/42ra54.fullhttp://stm.sciencemag.org/content/scitransmed/4/139/139ra85.fullhttp://stm.sciencemag.org/content/scitransmed/4/144/144ra103.fullhttp://stm.sciencemag.org/content/scitransmed/8/335/335ra58.full
REFERENCES
http://stm.sciencemag.org/content/9/396/eaal4649#BIBLThis article cites 65 articles, 22 of which you can access for free
PERMISSIONS http://www.sciencemag.org/help/reprints-and-permissions
Terms of ServiceUse of this article is subject to the
registered trademark of AAAS. is aScience Translational MedicineScience, 1200 New York Avenue NW, Washington, DC 20005. The title
(ISSN 1946-6242) is published by the American Association for the Advancement ofScience Translational Medicine
of Science. No claim to original U.S. Government Works.Copyright © 2017 The Authors, some rights reserved; exclusive licensee American Association for the Advancement
by guest on April 3, 2021
http://stm.sciencem
ag.org/D
ownloaded from
http://stm.sciencemag.org/content/9/396/eaal4649http://stm


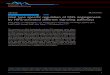



![Laminin-332 and Integrins: Signaling Platform for …Laminin-332 and Integrins 31 laminin globular (LG) subdomains (LG1-5) [2]. The latter is the major interaction sites for cell surface](https://img.pdfslide.net/doc/110x75/5f712e9e3f945d798f112220/laminin-332-and-integrins-signaling-platform-for-laminin-332-and-integrins-31-laminin.jpg)


![NTPC DLN Systems Vamsi[1]](https://img.pdfslide.net/doc/110x75/55cf9466550346f57ba1bc91/ntpc-dln-systems-vamsi1.jpg)



![Standard Combustion vs DLN[1]](https://img.pdfslide.net/doc/110x75/5461752eb1af9f7d228b494d/standard-combustion-vs-dln1.jpg)














