Embed Size (px)
Citation preview

1
Linux & Shell ScriptingSmall Group
Lecture 3
How to Learn to Code Workshop
http://onish.web.unc.edu/how-to-learn-how-to-code-linux-small-group/
Erin Osborne Nishimura
Where are we?
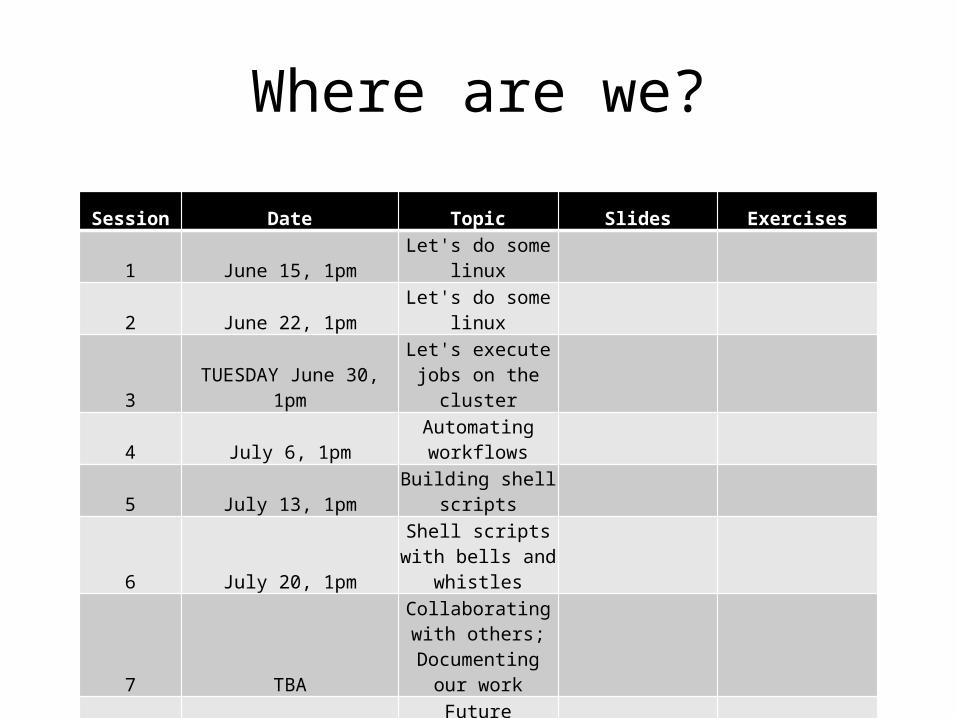
Session Date Topic Slides Exercises
1 June 15, 1pm Let's do some linux
2 June 22, 1pm Let's do some linux
3 TUESDAY June 30, 1pmLet's execute jobs
on the cluster
4 July 6, 1pmAutomating workflows
5 July 13, 1pm Building shell scripts
6 July 20, 1pmShell scripts with bells and whistles
7 TBA
Collaborating with others;
Documenting our work
8 Aug 3, pmFuture directions for users and the course

Questions & Comments

Group pop quizWhat does this do?$ ls
1_cdc1.txt2_cdc2.txt3_cdc3.txt4_cdc4.txt5_cdc5.txtmy_cyclins_README.logmy_cyclins_downloaddata.txt
$ wc *.txt | sort > captured.txt
How would you unpack this?project_update.tar.gz
You have a file that contains a list of Arabidopsis transcription factor names:
"ID","Name","Species","GeneID","Family","Evidence""T000676_1.01","ANT","Arabidopsis_thaliana","AT4G37750-TAIR-G","AP2","D""T000588_1.01","AT1G22985","Arabidopsis_thaliana","AT1G22985-TAIR-G","AP2","D""T000614_1.01","AT1G75490","Arabidopsis_thaliana","AT1G75490-TAIR-G","AP2","D"
What all the IDs of transcription factors that contain the bHLH domain?How many are there?

Learning objectives week 2
• Move files to and from kure
• Decompress and unpackage a tarball
• Read files
• Start to use the wild card *
• Start to chain commands together using |

Week 2
• Killdevil • module• Bsub

7
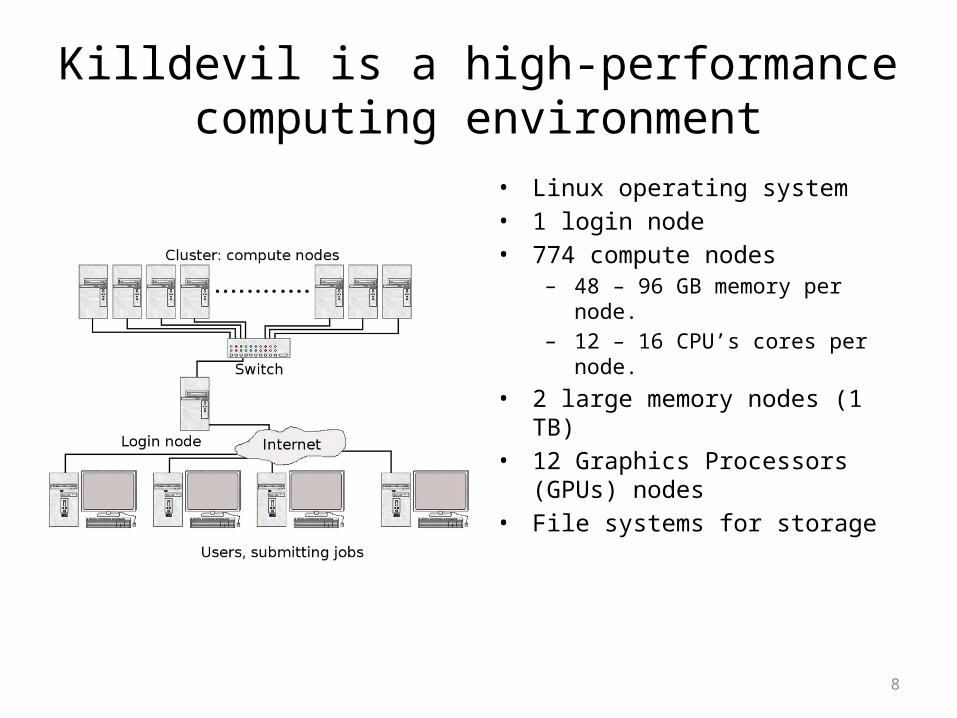
No seriously, what is Killdevil?
8
Killdevil is a high-performance computing environment
• Linux operating system• 1 login node• 774 compute nodes
– 48 – 96 GB memory per node.– 12 – 16 CPU’s cores per node.
• 2 large memory nodes (1 TB)• 12 Graphics Processors (GPUs) nodes• File systems for storage

If a job takes more than 3 seconds to complete, cancel it!
Use bsub instead!

Load File Sharing (LSF)
• LSF -- allocates nodes to job submissions -- max 64 on kure, 1024 on killdevil -- fair scheduling -- takes into consideration your status, queue, your
job, recent activity, node requirements
$ bqueues #This will show you all the ‘queues’
$ bjobs -u all #This will show all the jobs right now!

Execute big jobs on kure using bsub
• Execute jobs on kure with bsub to get off the head node$ bsub -q week [-n 1] [-M <number>] [-o %J.log] “<command line>”
Use the queue called ‘week’;I want one node; Give me more memory!output anything about how this job runs to a log file.
$ bjobs # check all running jobs$ bpeek [NUMBER] # see the screen output of a job$ bkill <NUMBER> # terminate a job

Big jobs – exercise #1• The directory /proj/seq/data/ contains lots of whole genome sequencing resources. These are big files.
– Go to /proj/seq/data/
• Now navigate to the directory:– /proj/seq/data/ce10_NCBI/Sequence/WholeGenomeFasta– This directory contains the C. elegans genome (genome.fa).
• Try to count the lines in this genome. If it takes longer than 5 seconds, cancel out.• Next, re-submit the command using bsub:
• /proj/seq/data/ce10_NCBI/Sequence/WholeGenomeFasta/genome.fa
$ bsub -q week “wc genome.fa”
• Check your job with “bpeek” and “bjobs”. When your job is finished, check your e-mail.• Try to save the output of your job to a logfile in your home directory:
$ bsub –q week –o ~/bjobs_output_%J.log “wc genome.fa”
• Watch your job with “bpeek” and “bjobs”. Check your e-mail. Anything there? Go to your home directory. Anything there?

Parallel processing $ bsub -q week -n <numberGreaterThanOne> -R “span[hosts=1]” [-M <number>] [-o %J.log] “<command line>”
You must use –n and –R together when submitting parallel jobs
-n <number> #This is the number of nodes (also called jobs) to use. Maximum of 12.
-R “span[hosts=1]” #This tells LSF to put all of those nodes/jobs on one host.

Modules on killdevil• $ module avail
– View available modules on killdevil
• $ module list- List the modules you have loaded up and ready to use
• $ module add <modulename>• $ module load <modulename>
– These are the same. They both load an available module into your list of usable modules. They will load for your ssh session and be removed when you log out.
• $ module initadd <modulename>– Load this module every time I log in, as soon as I log in
• $ module initrm<modulename>– Remove this module from the list of all modules that load as soon as I log in.
• $ module• $ module –H• $ module –help
– These are the same. They all take you to the help page

Exercise 2• Browse the list of available modules• Do you recognize any of these applications?• Which have you used before?• Load the module bedtools to your list of available modules• Read a little bit about bedtools using its manual page• Read about bedtools getfasta• What does bedtools getfasta do?• Load another module, maybe something you recognize.• Can you find the manual or help message for this utility? (Hint, the point of this is
that some of these are really hard to find manuals for and that you need to go to the internet for help; Sometimes that doesn’t even work).

An intro to Exercise 3• Fasta files
– Genome sequences are kept as fasta files.– http://genetics.bwh.harvard.edu/pph/FASTA.html
• Bed files– Gives the intervals of a genome– https://genome.ucsc.edu/FAQ/FAQformat.html#format1
• Gff files– Gives intervals of a genome– https://genome.ucsc.edu/FAQ/FAQformat.html#format3

Exercise 3A
• Let’s use bedtools getfasta. • The point of this exercise will be to make a fasta file
containing the sequences of all the genes that are located on the left hand of chromosome I in the yeast genome.
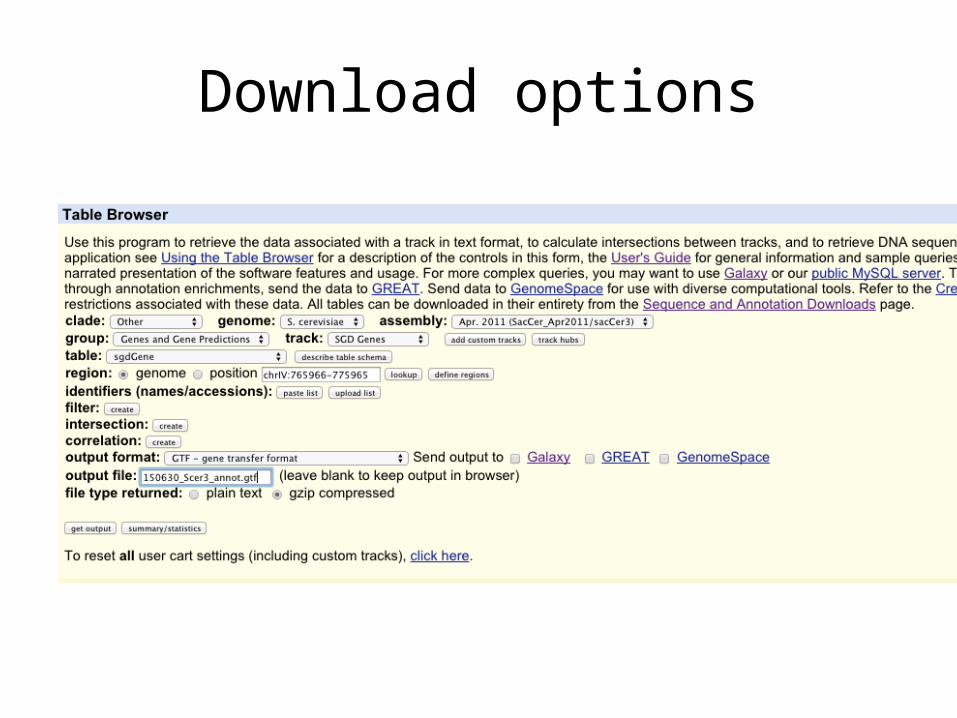
• Start a README.txt file for this project• Obtain a gtf/gff file of the entire yeast genome. This is the
full annotation of every gene in the yeast genome– Go to UCSC genome browser’s table page:– https://genome.ucsc.edu/cgi-bin/hgTables?command=start– Download a gtf file by filling out the form to exactly match the
next slide…
Download options

Exercise 3B (cont)
• Download the .gtf.gz file to your local computer• Upload it to killdevil.• Unzip it using
$ gunzip <filename>• Look inside of it to see what a .gtf file looks like.• Compare the file with the description of a gtf
file on UCSC genome browser (slide 16)

Exercise3C• Make a smaller annotation file that contains just the coding sequences
(“CDS”) of genes that are on the left side of chromosome 1. These genes will all start with the letters “YAL”.– Filter for “YAL”; Filter for “CDS”; save the results in the file chrI_left_CDS.gtf– How many genes are in there?
• Execute bedtools. Learn how to use bedtools. – Input – The yeast genome. We downloaded this in week2; exercise2. I think
it’s called S_cerevisiae.fa. You will need to know the FULL PATH to this file.– Input – The gtf file called chrI_left_CDS.gtf– Input – The NAME you want to give to your output fasta file.
• Use the bedtools getfasta help pages to figure out how to use this code.– Hint: you will need the arguments (-fi, -bed, and -fo, and one other option)
• Check that all your sequences start with an “ATG”.• Check that you have the correct number of fasta sequence entries (“>”).

Exercise 3D
• Complete your README.txt file





























