Embed Size (px)
Citation preview

Available online at www.sciencedirect.com
ScienceDirect
Journal of Nutritional Biochemistry 29 (2016) 56–63
Liver-specific deletion of the signal transducer and activator of transcription 5 geneaggravates fatty liver in response to a high-fat diet in mice☆
Myunggi Baika,b,⁎, 1, Yoon Seok Namc,1, Min Yu Piaoa, Hyeok Joong Kanga, Seung Ju Parka, Jae-Hyuk Leed
aDepartment of Agricultural Biotechnology and Research Institute of Agriculture and Life Sciences, College of Agriculture and Life Sciences, Seoul National University, 1 Gwanak-ro, Gwanak-gu,Seoul 151-921, Republic of Korea
bInstitute of GreenBio Science Technology, Pyeungchang-gun, Gangwon-do 232-916, Republic of KoreacDepartment of Pharmacology and Medical Research Center for Gene Regulation, Chonnam National University Medical School, Gwangju 501-746, Republic of Korea
dDepartment of Pathology, Chonnam National University Medical School, Gwangju 501-746, Republic of Korea
Received 28 July 2015; received in revised form 19 October 2015; accepted 29 October 2015
Abstract
Growth hormone (GH) signal is mediated by signal transducer and activator of transcription 5 (STAT5), which controls hepatic lipid metabolism.Nonalcoholic fatty liver disease (NAFLD) is clinically associated with a deficiency in GH. This study was performed to understand the role of local STAT5 signalingon hepatic lipid and glucose metabolism utilizing liver-specific STAT5 gene deletion (STAT5 LKO) mice under both normal diet and high-fat diet (HFD) feedingconditions. STAT5 LKO induced hepatic steatosis under HFD feeding, while this change was not observed in mice on normal diet. STAT5 LKO causedhyperglycemia, hyperinsulinemia, hyperleptinemia and elevated free fatty acid and cholesterol concentrations under HFD feeding but induced onlyhyperglycemia on normal diet. At the molecular level, STAT5 LKO up-regulated the expression of genes involved in lipid uptake (CD36), very low-densitylipoprotein receptor (VLDLR), lipogenic stearoyl-CoA desaturase and adipogenic peroxisome proliferator-activated receptor gamma, in both diet groups. Inresponse to HFD feeding, further increases in CD36 and VLDLR expression were found in STAT5 LKO mice. In conclusion, our study suggests that low STAT5signaling on normal diet predisposes STAT5 LKO mice to early development of fatty liver by hyperglycemia and activation of lipid uptake and adipogenesis. Adeficiency in STAT5 signaling under HFD feeding deregulates hepatic and body glucose and lipid metabolism, leading to the development of hepatic steatosis. Ourstudy indicates that low STAT5 signaling, due to low GH secretion, may increase a chance for NAFLD development in elderly people.© 2015 Elsevier Inc. All rights reserved.
Keywords: Fatty liver; Growth hormone; STAT5; High fat diet; Lipid metabolism
1. Introduction
Growth hormone (GH) signals in the liver are mediated bysignal transducer and activator of transcription 5 (STAT5). STAT5activates transcription of the insulin-like growth factor-1 (IGF-1) gene.
Abbreviations: BUN, blood urea nitrogen; FFA, free fatty acid; GH, growthhormone; HFD, high-fat diet; IGF-1, insulin-like growth factor-1; IL-6, interleukin-6;NAFLD, nonalcoholic fatty liver disease; STAT5, signal transducer and activator oftranscription 5; STAT5 LKO, liver-specific STAT5 gene deletion; TG, triglyceride;VLDLR, very low-density lipoprotein receptor.☆ Thisworkwas supported by grants from the National Research Laboratory
Program (ROA-2007-0056702) through the National Research Foundationfunded by the Ministry of Education, Science and Technology, and the Next-Generation BioGreen 21 Program (No. PJ01114001), Rural DevelopmentAdministration, Republic of Korea.⁎ Corresponding author. Department of Agricultural Biotechnology, Seoul
National University, 1 Gwanak-ro, Gwanak-gu, Seoul 151-921, Republic ofKorea. Tel.: +82-2-880-4809; fax: +82-2-873-2271.
E-mail addresses: [email protected], [email protected] (M. Baik).1 Equal contribution.
http://dx.doi.org/10.1016/j.jnutbio.2015.10.0180955-2863/© 2015 Elsevier Inc. All rights reserved.
TheGH-STAT5-IGF-1 axis regulates bodygrowth. In addition, a previousstudy showed that GH-STAT5 signaling affects both lipid and glucosemetabolism [1]. Several studies have revealed that a lack of GH-STAT5signaling induces hepatic steatosis inmice [2,3] and humans [4,5]. BloodGH and IGF-1 levels decline as age increases [6], and GH treatment inolder males improves lean bodymass and increases IGF-1 mRNA levelsin skeletalmuscle [7]. Nonalcoholic fatty liver disease (NAFLD) has beeninduced in a high-fat diet (HFD) model, as evidenced by several studies[8]. HFD feeding aggravated bodymetabolism andmetabolic disease asage increased [9]. Therefore, older people consuming an HFD mayaggravate a low GH signaling-induced metabolic disease. Littleinformation is available on the changes in lipid and glucosemetabolismand the development of metabolic diseases in low STAT5 signalingstatus under HFD feeding conditions.
Therefore, the role of local STAT5 signaling on lipid and glucosemetabolism in the liver was addressed in the current study throughuse of liver-specific STAT5 gene deletion (STAT5 LKO) mice underboth normal and HFD feeding conditions. We have hypothesizedthat STAT5 LKO may deregulate expression of genes for fatty acidmetabolism and inflammation, contributing to development ofhepatic steatosis.

57M. Baik et al. / Journal of Nutritional Biochemistry 29 (2016) 56–63
2. Materials and methods
2.1. Mice and diets
All experimental procedures involving mice were approved by the ChonnamNational University Institutional Animal Use and Care Committee. STAT5 flox/flox (fl/fl)albumin-Cre mice and STAT5 fl/fl mice were kindly provided by Dr. LotharHennighausen at the National Institutes of Health (Bethesda, USA). STAT5 LKO micewere generated by breeding STAT5 fl/fl albumin-Cremicewith STAT5 fl/flmice. DNA forgenotyping was extracted from ear punches using the hot sodium hydroxide and Tris(HotSHOT) method of DNA extraction [10]. The following primers were used: STAT5wild-type allele, forward primer 5′-A AGC ATG AAA GGG TTG GAG-3′ and reverseprimer 5′-AGC AGC AAC CAG AGG ACT AC-3′; STAT5 floxed allele, forward primer 5′-AGCAGCAACCAGAGGACTAC-3′ and reverse primer5′-TACCCGCTT CCA TTGCTCAG-3′; andalbumin-Cre transgene, forward primer 5′-GGA CAA AGT CTT GTG CAT GG-3′ and reverseprimer 5′-CCA GGC TAA GTG CCT TCT CTA CA-3′.
Micewerehoused individually incarbonate cages andmaintainedunderpathogen-freeconditions at ChonnamNational University in a room that is controlled by temperature andhumidityona12-h light:12-hdark cycle,withaccess to conventional rodentnormal (chow)or HFD and water ad libitum. AIN-93G and HFD (Research Diets, Inc., New Brunswick, NJ,USA) were purchased through the Central Laboratory of Animals Corporation (Seoul,Republic of Korea). Diet composition is shown in Supporting Table 1.Malemicewere givenAIN-93G for adaptation for 1 week. At 9 weeks of age, STAT5 LKOandSTAT5fl/flmicewererandomly assigned to either thenormal diet or HFD group, and experimental dietswere fedad libitum for 12 weeks. During the experimental period, food intake and bodyweightwererecorded twice per week.
2.2. Collection and analysis of blood and tissue
At the end of the experimental period, bloodwas collected by cardiac puncturewitha 1-ml syringe,maintained for 30 min at room temperature and centrifuged at 1000g for20 min at 4°C to separate the serum. Animalswere sacrificed by decapitation after beinganesthetized with CO2. Tissue samples were weighed, rapidly frozen in liquid nitrogenand stored at −80°C.
All serum parameters were analyzed by the Green Cross Reference Laboratory(Yongin, Republic of Korea). Total cholesterol content was analyzed by enzymaticcolorimetry using CHOL (Roche, Germany). Serum triglyceride (TG) levels wereanalyzed by enzymatic colorimetry using a TG kit (Roche, Germany). Serum bloodurea nitrogen (BUN) levels were evaluated by a kinetic ultraviolet (UV) assay usingUREA/BUN (Roche, Germany). Serum free fatty acid (FFA) levels were analyzed byenzymatic colorimetry using NEFA HR.II (Wako, Japan). Serum insulin levels weredetermined with an insulin-rat/mouse enzyme-linked immunosorbent assay (ELISA)kit (Linco, USA) using an antibody raised against mouse. Serum interleukin-6 (IL-6)levels were analyzed using a mouse IL-6 ELISA kit (Raybiotech, USA). Serum GH levelswere assayed using a rodent GH ELISA kit (Endocrine, USA). Serum IGF-1 levels weredetermined using OCTEIA rat/mouse IGF-1 (IDS, USA). Serum leptin levels weredetermined using a mouse leptin ELISA kit (Millipore, USA). Levels of insulin, IL-6, GH,IGF-1 and leptinweremeasured using an ELISA reader (Molecular, USA). The coefficientvariance of the intraassay and interassay of the IGF-1, GH, glucose, insulin, TG, FFA,leptin, cholesterol, IL-6 and BUN was 7.3% and 8.8%, 8.8% and 7.7%, 1.1% and 1.9%, 1.9%and 7.6%, 1.5% and 2.4%, 1.5% and 1.5%, 1.8% and 4.6%, 1.0% and 2.7%, b10% and b12% and1.9% and 3.4%, respectively.
Tissue lipid contents were measured using a chloroform:methanol (2:1, v/v)extraction method [11].
2.3. Liver histology
Liver specimens were fixed in 10% buffered formalin (Sigma-Aldrich, St. Louis, MO,USA), equilibrated in a 70–100% ethanol series and xylene and were embedded inparaffin. Paraffin sections were stained with hematoxylin and eosin (H&E) andexamined for fat depots and inflammatory cells under a light microscope as describedbelow.
Liver specimens fixed in 10% buffered formalin were also equilibrated in 20%sucrose with gum Arabic from the acacia tree (Sigma-Aldrich, G9752) and embeddedusing optimal cutting temperature compound. Tissues were sectioned at −20°C into7-μm-thick sections using a cryostat. Cryosections were mounted by touching theglass slide to the tissue section. To detect fat deposition in the liver, frozen sectionswere fixed for 10 min in neutral-buffered 10% formalin and then rinsed with distilledwater, stained with 0.5% Oil-Red O (ORO; Sigma-Aldrich, O0625) and 60% 2-propanol(Sigma-Aldrich, 190764) for 10 min and rinsed with distilled water. Stained sections wereexamined and photographed using an Olympus 1X70 light microscope (OlympusInstruments Inc., Tokyo, Japan). The degree of ORO staining was determined by referenceORO staining (from none to the highest grade: 0, 1, 2, 3, 4 and 5).
2.4. RNA extraction, cDNA synthesis and real-time PCR
Total RNA was isolated from tissue samples using TRIzol Reagent (Invitrogen,Carlsbad, CA, USA) according to the manufacturer’s instructions. RNA quantity andquality were assessed using a NanoDrop ND-1000 UV-Vis Spectrophotometer
(NanoDrop Technologies, Wilmington, DE, USA). Total RNA (3 μg) was reverse-transcribed into cDNA using Accupower RT premix (Bioneer, Daejeon, Republicof Korea).
Real-time polymerase chain reaction (PCR) was performed using QuantiTect SYBRgreen RT-PCR Master Mix (Qiagen, Valencia, CA, USA) and an Opticon sequencedetection system (Rotorgen Qiagen). Briefly, the PCR was performed in a total reactionvolume of 25 μl containing 200 ng cDNA, 12.5 μl SYBR Green RT-PCR Master Mix(Qiagen) and 1.25 μl primers (10 μM) (Bioneer). Thermal cycling parameters were asfollows: 95°C for 15 min, followed by 40 cycles at 94°C for 15 s, 60°C for 30 s and 72°Cfor 30 s. All primers were designed using Integrated DNA Technology and Primer 3software based on published sequences in GenBank and synthesized by Bioneer. Primerinformation is shown in Supporting Table 4. The ΔΔCT method was used to determinerelative fold changes, and all data were normalized to the housekeeping gene, β-actin.
2.5. Statistical analyses
Data are presented as the mean±standard error of the mean (S.E.M.). Data wereanalyzed using the general linear model procedure of SAS (SAS Institute Inc., Cary, NC,USA). The model included genotype (STAT5 fl/fl or STAT5 LKO), diet (normal or HFD)and genotype×diet interaction. Number of animals per group was six for most of data,and this statistical power isnot high in this study. The PDIFF option of LSMEANS inSASwasused to separate means. A P value b.05 was considered to indicate statistical significance.
3. Results
3.1. Expression of STAT5 signaling genes, body growth, organ weightsand blood metabolites
STAT5 LKO reduced (Pb.05) hepaticmRNA levels of STAT5a/b genesby approximately 73–86% of STAT5 fl/fl mice (Fig. 1). It also down-regulated (Pb.05) hepaticmRNA levels of the STAT5 target gene, IGF-1,by 41–59%, regardless of diet. Both fasted and fed serum GH levelswere not altered by STAT5 LKO (Table 1). However, circulating IGF-1levels in both fasted and fed states were lower (Pb.05) by 42–51% inSTAT5 LKO mice compared to STAT5 fl/fl mice (Table 1). Neithergenotypenor diet affectedmRNA levels of GH receptor or STAT3genes.
STAT5 LKO mice showed a higher (6–7%) trend in average foodintake on both normal (3.33 g and 3.56 g/day for STAT5 fl/fl vs. STAT5LKO) and HFD (2.60 g and 2.74 g/day for STAT5 fl/fl vs. STAT5 LKO)over the 12 weeks of feeding. STAT5 LKOmice (13.6 and 14.4 kcal/dayfor normal diet and HFD) ate more energy (Pb.001), which wascalculated based on food intake and energy content, compared to STATfl/fl mice (13.3 and 14.2 kcal/day for normal diet and HFD). Mice fedan HFD (2.67 g/day) ate less (Pb.001) food compared to animals fed anormal diet (3.44 g/day) for 12 weeks but consumed a similar (PN.05)amount of energy (13.8 kcal/day) as the animals fed normal diet(14.0 kcal/day). STAT5 fl/fl and LKO mice ate average 12.9% of bodyweight per day, showing normal range of control diet consumption.
Initial bodyweight of STAT5fl/flmice at 9 weeks of agewas similarto that of STAT5 LKOmice (Table 1). At 21 weeks of age, after 12 weeksof feeding, numeric values for body weight in the STAT5 LKO micewere higher compared to those of the STAT5 fl/fl animals, but thedifference did not reach statistical significance. The average bodyweight difference at 21 weeks of age between theHFDandnormal dietgroups in STAT5MKOmice (4.9 g/mouse)wasmuch higher comparedto that in the STAT5fl/flmice (1.7 g/mouse: Fig. S1). Therefore, the STAT5LKOmicehadhigher accumulatedweight gain in response toHFD feedingcompared to control STAT5fl/flmice. HFD feeding increased (Pb.05) bodyweight at 21 weeks of age.
STAT5LKO increased(P≤.01) liverweight and relative liverweight/bodyweight. Numeric values of epididymal fat weight, relative epididymalfat/body weight, abdominal fat, relative abdominal fat/body weight,total fat and relative total fat/body weight in STAT5 LKO mice werehigher compared to those in STAT5 fl/fl animals without statisticalsignificance in theHFDgroup, but not in normal diet group.HFD feedingincreased (P≤.02) body fat accumulation, including the weightsof epididymal fat, relative epididymal fat/body weight, subcutaneousfat, relative subcutaneous fat/body weight, abdominal fat, relative
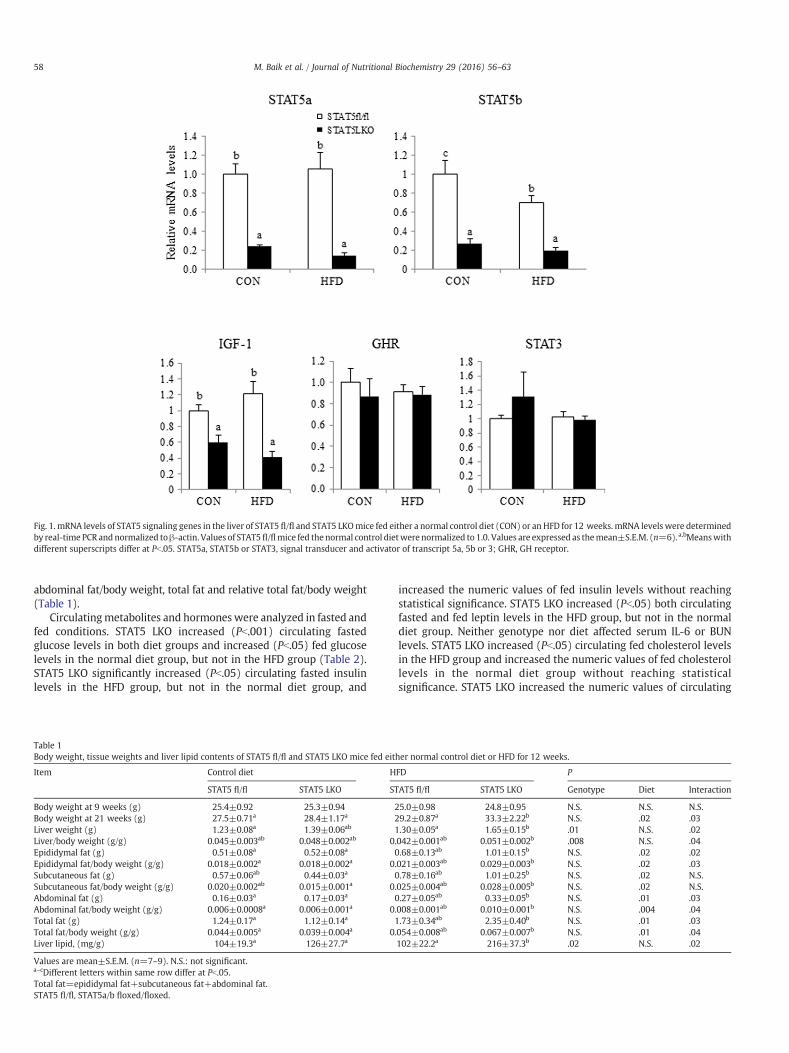
Fig. 1. mRNA levels of STAT5 signaling genes in the liver of STAT5 fl/fl and STAT5 LKOmice fed either a normal control diet (CON) or an HFD for 12 weeks. mRNA levels were determinedby real-time PCR andnormalized toβ-actin. Values of STAT5fl/flmice fed the normal control dietwere normalized to 1.0. Values are expressed as themean±S.E.M. (n=6).a,bMeanswithdifferent superscripts differ at Pb.05. STAT5a, STAT5b or STAT3, signal transducer and activator of transcript 5a, 5b or 3; GHR, GH receptor.
58 M. Baik et al. / Journal of Nutritional Biochemistry 29 (2016) 56–63
abdominal fat/body weight, total fat and relative total fat/body weight(Table 1).
Circulatingmetabolites and hormoneswere analyzed in fasted andfed conditions. STAT5 LKO increased (Pb.001) circulating fastedglucose levels in both diet groups and increased (Pb.05) fed glucoselevels in the normal diet group, but not in the HFD group (Table 2).STAT5 LKO significantly increased (Pb.05) circulating fasted insulinlevels in the HFD group, but not in the normal diet group, and
Table 1Body weight, tissue weights and liver lipid contents of STAT5 fl/fl and STAT5 LKO mice fed eit
Item Control diet H
STAT5 fl/fl STAT5 LKO ST
Body weight at 9 weeks (g) 25.4±0.92 25.3±0.94 2Body weight at 21 weeks (g) 27.5±0.71a 28.4±1.17a 2Liver weight (g) 1.23±0.08a 1.39±0.06ab 1Liver/body weight (g/g) 0.045±0.003ab 0.048±0.002ab 0.Epididymal fat (g) 0.51±0.08a 0.52±0.08a 0Epididymal fat/body weight (g/g) 0.018±0.002a 0.018±0.002a 0.Subcutaneous fat (g) 0.57±0.06ab 0.44±0.03a 0Subcutaneous fat/body weight (g/g) 0.020±0.002ab 0.015±0.001a 0.Abdominal fat (g) 0.16±0.03a 0.17±0.03a 0Abdominal fat/body weight (g/g) 0.006±0.0008a 0.006±0.001a 0.Total fat (g) 1.24±0.17a 1.12±0.14a 1Total fat/body weight (g/g) 0.044±0.005a 0.039±0.004a 0.Liver lipid, (mg/g) 104±19.3a 126±27.7a
Values are mean±S.E.M. (n=7–9). N.S.: not significant.a–cDifferent letters within same row differ at Pb.05.Total fat=epididymal fat+subcutaneous fat+abdominal fat.STAT5 fl/fl, STAT5a/b floxed/floxed.
increased the numeric values of fed insulin levels without reachingstatistical significance. STAT5 LKO increased (Pb.05) both circulatingfasted and fed leptin levels in the HFD group, but not in the normaldiet group. Neither genotype nor diet affected serum IL-6 or BUNlevels. STAT5 LKO increased (Pb.05) circulating fed cholesterol levelsin the HFD group and increased the numeric values of fed cholesterollevels in the normal diet group without reaching statisticalsignificance. STAT5 LKO increased the numeric values of circulating
her normal control diet or HFD for 12 weeks.
FD P
AT5 fl/fl STAT5 LKO Genotype Diet Interaction
5.0±0.98 24.8±0.95 N.S. N.S. N.S.9.2±0.87a 33.3±2.22b N.S. .02 .03.30±0.05a 1.65±0.15b .01 N.S. .02042±0.001ab 0.051±0.002b .008 N.S. .04.68±0.13ab 1.01±0.15b N.S. .02 .02021±0.003ab 0.029±0.003b N.S. .02 .03.78±0.16ab 1.01±0.25b N.S. .02 N.S.025±0.004ab 0.028±0.005b N.S. .02 N.S..27±0.05ab 0.33±0.05b N.S. .01 .03008±0.001ab 0.010±0.001b N.S. .004 .04.73±0.34ab 2.35±0.40b N.S. .01 .03054±0.008ab 0.067±0.007b N.S. .01 .04102±22.2a 216±37.3b .02 N.S. .02

Table 2Serum parameters in STAT5 fl/fl and STAT5 LKO mice either control normal diet or HFD for 12 weeks in 8 h fasted or fed states.
Items Control diet HFD P
STAT5 fl/fl STAT5 LKO STAT5 fl/fl STAT5 LKO Genotype Diet Interaction
FastedIGF-1 (ng/ml) 350±35.0b 152±29.6a 372±64.5b 189±32.4a b.001 N.S. .004GH (ng/ml) 1.13±0.03 1.02±0.03 1.04±0.03 1.05±0.08 N.S. N.S. N.S.Glucose (mg/dl) 121±9.10a 180±23.4bc 139±7.30ab 206±23.8c b.001 N.S. .03Insulin (ng/ml) 0.40±0.10a 1.00±0.30ab 0.30±0.10a 2.30±0.80b .003 N.S. .001Leptin (ng/ml) 1.20±0.40a 3.80±1.00a 2.20±0.50a 19.0±5.60b .017 N.S. .001IL-6 (pg/ml) 27.6±5.88 43.6±12.5 38.7±9.85 25.4±3.14 N.S. N.S. N.S.BUN (mg/dl) 30.6±2.70 28.4±3.80 24.6±1.80 27.8±2.60 N.S. N.S. N.S.
FedIGF-1 (mg/dl) 402±28.6b 192±28.2a 456±20.4b 193±50.2a b.001 N.S. b.001GH (ng/ml) 0.97±0.04 0.94±0.02 0.96±0.03 0.90±0.01 N.S. N.S. N.S.Glucose (mg/dl) 222±19.2a 293±30.7b 266±15.6ab 274±14.6ab .07 N.S. N.S.Insulin (ng/ml) 1.84±0.37 3.22±0.30 4.48±1.07 9.34±3.49 N.S. .04 .05TG (mg/dl) 148±21.7 172±37.1 125±18.8 254±101 N.S. N.S. N.S.FFA (μEq/L) 1533±155ab 1544±120ab 1477±68.0a 1988±211b N.S. N.S. N.S.Leptin (ng/ml) 4.36±0.42a 10.7±4.61a 10.5±4.48a 25.1±4.03b .03 .03 .01Total cholesterol (mg/dl) 134±7.50a 184±18.6a 170±7.10a 290±28.4b .003 .02 b.001IL-6 (pg/ml) 22.9±3.85 19.9±2.90 21.3±2.37 15.0±1.24 N.S. N.S. N.S.BUN (mg/dl) 29.3±1.39 30.6±2.13 31.5±1.78 32.4±4.10 N.S. N.S. N.S.
Values are mean±S.E.M. N.S.: not significant.a–cDifferent letters within same row differ at Pb.05.STAT5 fl/fl, STAT5a/b floxed/floxed.
59M. Baik et al. / Journal of Nutritional Biochemistry 29 (2016) 56–63
fed TG levels without reaching statistical significance. STAT5 LKOincreased (Pb.05) circulating fedFFA levels in theHFDgroup,butnot in thenormal diet group.
3.2. STAT5 LKO aggravates hepatic fat accumulation in the HFDgroup and deregulates the expression of lipid metabolism genesin the liver
STAT5 LKO increased (Pb.05) liver lipid content in the HFD group,but not in the normal diet group (Table 2). We observed a yellowishappearance of the liver in STAT5 LKOmice fed theHFD,whereas that ofthe other groupswas normal (Fig. 2). FollowingH&E and ORO stainingof liver sections, a higher degree of visible lipid droplets was observedin STAT5 LKO mice fed the HFD compared to the three other groups(Fig. 2). Quantification of ORO staining data revealed that STAT LKOmice had increased hepatic fat accumulation in the HFD group but ithad increased trend of hepatic fat accumulation without statisticalsignificance (Fig. 2).
We examined hepatic mRNA levels of lipid metabolism genes,including those involved in lipid uptake and lipogenesis, and low-density lipoprotein receptor (LDLR). STAT5 LKO up-regulated (Pb.05)expression of the lipid uptake gene, CD36, in the normal diet groupand further induced CD36 expression in the HFD group (Fig. 3). STAT5LKO also up-regulated (Pb.05) expression of the peroxisome pro-liferator-activated receptor gamma (PPARγ) gene in both the normaldiet and HFD groups. STAT5 LKO increased (Pb.05) expression of thelipogenic sterol regulatory element bindingprotein 1c (SREBP1c) genewhen data from the normal diet and HFD groups were combined.STAT5 LKO did not affect expression of the lipogenic fatty acidsynthase (FASN) gene in either the normal diet orHFDgroups, butHFDfeeding decreased FASN expression (Fig. 3). STAT5 LKOmice showed astrong (5- to 6-fold) up-regulation (Pb.05) of hepatic stearoyl-CoAdesaturase 2 (SCD2) gene expression under both diet conditions.STAT5 LKO decreased (PN.05) cholesterol uptake LDLRmRNA levels inthe normal diet group, and it showed decreased tendency of LDLRexpression in the HFD group (Fig. 3). STAT5 LKO increased (Pb.05)expression of the very low-density lipoprotein receptor (VLDLR) genein the HFD group, but not in the normal diet group.
3.3. STAT5 LKO induces hepatic inflammation and deregulates hepaticexpression of inflammatory genes
Wenext examinedhepatic infiltration of inflammatory cells inH&Esections and found that the frequency of inflammation occurrencewasincreased in STAT5 LKO mice (Fig. 4A); in the normal diet group, nooccurrence was found, but inflammatory cells were detected in 14.3%of STAT5 LKO mice fed a normal diet (Fig. 4B). In the HFD group,inflammatory cells were detected in 28.6% of STAT5 fl/flmice, and theoccurrence of inflammatory cells was increased by up to 50% of STAT5LKO mice.
STAT5 LKO profoundly induced (39- to 41-fold) hepatic lipocalin 2(LCN2) gene expression in both diet groups (Fig. 4C). STAT5 LKOalso increased hepatic tumor necrosis factor alpha (TNFα) geneexpressiononnormal diet and further inducedTNFα expressionunderHFD feeding.
4. Discussion
In this study, liver-specific deletion of both STAT5a and STAT5bgene expression and the corresponding transcriptional down-regulationof the STAT5 target gene, IGF-1, were confirmed. Circulating IGF-1 levels,under both fasted and fed states, decreased by 42–51% in STAT5 LKOmice, indicating that STAT5 signaling was reduced by ~50% by STAT5LKO, consistent with a previous report [3].
STAT5 LKOdid not affect bodyweight and hepatic fat accumulationunder normal diet feeding. However, after HFD feeding for 25 weeks,STAT5 LKO mice exhibited increased body weight, liver weight andhepatic lipid content and developed a fatty liver. Thus, our resultsdemonstrate that STAT5 LKO induced hepatic steatosis in response toHFD feeding.
Normal diet-fed STAT5 LKO mice showed increased fasted and fedglucose levels, a trend toward increased fasted and fed insulin levelsand a trend toward increased leptin levels, compared to STAT5 fl/flmice. STAT5 LKO did not affect circulating TG, cholesterol or FFAconcentrations under normal diet feeding. However, under HFDfeeding, STAT5 LKO mice exhibited increased circulating fastedglucose, fasted insulin, fasted and fed leptin and FFA and cholesterolconcentrations. Overall, STAT5 LKO caused significant metabolic and

Fig. 2. Liver appearance, H&E and ORO staining of liver sections of STAT5 fl/fl and STAT5 LKOmice fed either a normal control diet (CON) or an HFD for 12 weeks. Representative photosfrom six animals are shown.Magnification of H&E and ORO staining was ×200 and ×400, respectively. ORO grade (0–5 grade: 0=none, 5=highest) was determined as described in thematerials and methods section. a–cMeans with different superscript letters indicate significance at Pb.05. N=6.
60 M. Baik et al. / Journal of Nutritional Biochemistry 29 (2016) 56–63
endocrinal deregulation under HFD feeding, while these changesweremoderate under normal diet feeding.
Our study suggests that STAT5 LKO may cause insulin resistanceunder HFD feeding, considering that STAT5 LKO mice had hypergly-cemia, hyperinsulinemia and elevated FFA concentrations. Previously,insulin resistance was reported in mice with STAT5 deletion [12]. Thisstudy suggests that NAFLD may result from insulin resistance and theconcomitant impairment of lipid metabolism within peripheraltissues, including the liver, skeletal muscle and adipose tissue [13].This peripheral insulin resistance may have potentially contributed tothe development of hepatic steatosis observed in our study.
In this study, STAT5 LKO mice exhibited hyperleptinemia with noincrease in food intake under HFD feeding. Leptin is known to reducefood intake and increase energy expenditure. Therefore, our resultsimply that HFD feeding under deficient STAT5 signaling may cause astate of leptin resistance. Previously, hyperleptinemia exerted no
anorexic effect in HFD-induced obesemice [14]. They also suggest thatearly-stage development of NAFLD without apparent inflammation isa result of uncontrolled hepatic lipogenesis and gluconeogenesisand that these metabolic deregulations are associated with alteredinsulin/leptin signaling in both the hypothalamus and liver. Therefore,there is a possibility that STAT5 LKO deregulates hepatic lipid andglucose metabolism, causing insulin resistance and leptin resistance,contributing to the development of NAFLD under HFD feeding.
At the molecular level, STAT5 LKO up-regulated the expressionof CD36, VLDLR, SCD2 and PPARγ genes in both normal diet-fed andHFD-fed mice. Similar increases in CD36, PPARγ, VLDLR and SCD geneexpression in the liver were reported in STAT5-deficientmice [1,3,15].SCD is required for the biosynthesis of oleate and palmitoleate [16],and increased SCD activity has been reported in humans and animalswith fatty liver [17]. Therefore, up-regulation of SCD2 gene expressionin this studymay have contributed to the development of fatty liver in

Fig. 3.mRNA levels of lipidmetabolism genes in the liver of STAT5fl/fl and STAT5 LKOmice fed either a normal control diet (CON) or anHFD for 12 weeks.mRNA levels were determinedby real-time PCR and normalized to β-actin. Values of STAT5 fl/flmice fed a normal control diet were normalized to 1.0. Values are expressed as themean±S.E.M. (n=6). a–cMeans withdifferent superscripts differ at Pb.05. CD36, cluster of differentiation 36.
61M. Baik et al. / Journal of Nutritional Biochemistry 29 (2016) 56–63
STAT5 LKO mice. Under HFD feeding, STAT5 LKO mice exhibitedfurther increased CD36 and VLDLR mRNA levels. Previously, GHsignaling repressed CD36 expression [18]. CD36 expression wasincreased when hepatic steatosis was induced [19]. In normalconditions, VLDLR is mainly expressed in adipose tissue and muscleand only at very low levels in the liver [20]. In the absence of LDLR,such as in LDLR knockoutmice, VLDLR expressionwas up-regulated inthe liver [21]. Endoplasmic reticulum stress-dependent expression ofVLDLR led to steatosis by increasing lipoprotein delivery to the liver[22]. Therefore, there is a possibility that elevated VLDLR expressionmay, in part, contribute to the development of fatty liver in STAT5 LKOmice under HFD feeding. Our findings suggest that the combinedeffects of increased lipid uptake by increased circulating FFA levels andup-regulation of CD36 and VLDLR genes may, in part, contribute toaggravated hepatic fat accumulation in STAT5 LKOmice in response toHFD feeding.
In the current study, STAT5 LKO did not affect FASN geneexpression. HFD feeding decreased FASN expression. Thus, fatty acidsynthesis may not be a reason for hepatic steatosis in STAT5 LKOmice.SREBP1c acts as a transcription factor for activation of hepaticadipogenic and lipogenic gene expression including acetyl CoAcarboxylase, FASN and SCD2 [23,24]. Increased SREBP1c expressionin STAT5 LKO may explain up-regulation of SCD2 expression in ourstudy. However, no increase of hepatic FASN transcription was
observed in this study. Liver X receptor can directly activate FASNtranscription through binding to liver X receptor element of FASNpromoter [25]. There is a possibility that other factors including liver Xreceptor rather than SREBP1c have major roles in regulatingtranscriptional activation of FASN gene in this study. PPARγ is atranscriptional regulator of adipogenesis and CD36 is a target gene ofPPARγ [26]. Thus, increased PPARγ expression may up-regulate CD36expression, leading to hepatic steatosis in STAT5 LKO. Previously,hepatic PPARγ expression was increased in animals with fatty liver,and liver-specific knockout of PPARγ could prevent fatty liver [27,28].
In this study, STAT5 LKO significantly increased circulatingcholesterol concentrations in the HFD group, but not in the normaldiet group. STAT5 LKO decreased hepatic cholesterol uptake and LDLRmRNA levels in the normal diet group and showedadecreased trend inthe HFD group. LDLR is important for cholesterol uptake [29]. Thus,down-regulation of LDLR mRNA may, in part, be responsible forincreased total cholesterol concentrations in STAT5 LKO micecompared with STAT5 fl/fl mice.
In this study, the hepatic infiltration frequency of inflammatorycells was moderately increased in STAT5 LKO mice fed a normal diet,and the HFD feeding augmented the infiltration frequency ofinflammatory cells. STAT5 LKO up-regulated mRNA levels of inflam-mation-associated LCN2 and TNFα genes in both diet groups. UnderHFD feeding, STAT5 LKO further increased TNFα mRNA levels. Our

Fig. 4. H&E staining (A), inflammatory frequency (B) andmRNA levels of inflammatory genes in the liver of STAT5 fl/fl and STAT5 LKOmice fed either a normal control diet (CON) or anHFD for 12 weeks. (A) Arrows indicate inflammatory cells in the hepatic parenchyma. The area of inflammatory cells ismagnified and shown in the inset. Magnification for H&E stainingand insets was ×200 and ×400, respectively. Scale bars represent 50 μm. (B) Presence of inflammatory cells was observed in H&E stained section of six to eight mice per group.Inflammatory frequency (%)=(number of mice that had inflammatory cells per group/total mice number per group)×100. (C) mRNA levels were determined by real-time PCR andnormalized to β-actin. Values of STAT5 fl/fl mice fed the normal control diet were normalized to 1.0. Values are expressed as the mean±S.E.M. (n=6). a–cMeans with differentsuperscripts differ at Pb.05.
62 M. Baik et al. / Journal of Nutritional Biochemistry 29 (2016) 56–63
findings reveal that STAT5 LKO induces moderately hepatic inflam-mation andderegulates the expressionof inflammation genes and thatHFD feeding aggravates these changes. However, only one inflamma-tion frequency value was available for each treatment, and thus, ourfrequency data have a limitation for statistical analysis.
Overall, STAT5 LKO caused significant metabolic and endocrinalderegulation and induced hepatic steatosis upon HFD feeding, whilethese changes were minor with normal diet feeding. NAFLD mainlyaffects themiddle-aged and the elderly, andwith advancing age,morerisk factors arise [30]. CirculatingGH and IGF-1 levels generally declineas age increases [6]. Thus, the decline in GH with age may, in part, beresponsible for the progression of metabolic diseases, includingNAFLD, in the elderly. It has been reported that an HFD differentiallyinfluences body metabolism and metabolic disease with age inanimals [9]. In response to HFD feeding, older mice showed larger
increases in body weight and serum total cholesterol and glucoselevels compared with younger animals [9]. Our study suggests thatNAFLD may be more easily developed in an elderly person due tolower circulating levels of GH and thus low STAT5 signaling whenexposed to an HFD. Therefore, more cautionmay be needed against anHFD in elderly people to prevent aggravation of NAFLD.
In conclusion, our study suggests that a deficiency in STAT5-IGF-1signaling under a normal diet predisposes STAT5 LKO mice to earlydevelopment of fatty liver by activating lipid uptake and adipogenesis. Adeficiency in STAT5-IGF-1 signaling under HFD feeding conditionsaggravates hepatic and body glucose and lipid metabolism, as evidencedby hyperglycemia, hyperinsulinemia, hyperleptinemia and elevatedcirculating FFA and cholesterol concentrations and by correspondingchanges in the expression of genes involved in lipid and cholesteroluptake, leading to thedevelopmentofhepatic steatosis and inflammation.

63M. Baik et al. / Journal of Nutritional Biochemistry 29 (2016) 56–63
Authors’ disclosure
The authors have no conflicts of interest to disclose.
Appendix A. Supplementary data
Supplementary data to this article can be found online at http://dx.doi.org/10.1016/j.jnutbio.2015.10.018.
References
[1] Barclay JL, Nelson CN, Ishikawa M, Murray LA, Kerr LM, McPhee TR, et al. GH-dependent STAT5 signaling plays an important role in hepatic lipid metabolism.Endocrinology 2011;152:181–92.
[2] Fan Y, Menon RK, Cohen P, Hwang D, Clemens T, DiGirolamo DJ, et al. Liver-specific deletion of the growth hormone receptor reveals essential role of growthhormone signaling in hepatic lipid metabolism. J Biol Chem 2009;284:19937–44.
[3] Cui Y, Hosui A, Sun R, Shen K, Gavrilova O, Chen W, et al. Loss of signal transducerand activator of transcription 5 leads to hepatosteatosis and impaired liverregeneration. Hepatology 2007;46:504–13.
[4] Ichikawa T, Hamasaki K, Ishikawa H, Ejima E, Eguchi K, Nakao K. Non-alcoholicsteatohepatitis and hepatic steatosis in patients with adult onset growth hormonedeficiency. Gut 2003;52:914.
[5] Takahashi Y, Iida K, Takahashi K, Yoshioka S, Fukuoka H, Takeno R, et al. Growthhormone reverses nonalcoholic steatohepatitis in a patient with adult growthhormone deficiency. Gastroenterology 2007;132:938–43.
[6] Giustina A, Veldhuis JD. Pathophysiology of the neuroregulation of growthhormone secretion in experimental animals and the human 1. Endocr Rev 1998;19:717–97.
[7] Sattler FR, Castaneda-Sceppa C, Binder EF, Schroeder ET, Wang Y, Bhasin S, et al.Testosterone and growth hormone improve body composition and muscleperformance in older men. J Clin Endocrinol Metab 2009;94:1991–2001.
[8] Collins S, Martin TL, Surwit RS, Robidoux J. Genetic vulnerability to diet-inducedobesity in the C57BL/6J mouse: physiological and molecular characteristics.Physiol Behav 2004;81:243–8.
[9] Korou L-MA, Doulamis IP, Tzanetakou IP, Mikhailidis DP, Perrea DN. The effect ofbiological age on the metabolic responsiveness of mice fed a high-fat diet. LabAnim 2013;47:241–4.
[10] Truett GE, Heeger P, Mynatt RL, Truett AA, Walker JA, Warman ML. Preparation ofPCR-quality mouse genomic DNA with hot sodium hydroxide and tris (HotSHOT).BioTechiniques 2000;29:52–4.
[11] Folch J, Lees M, Sloane-Stanley GH. A simple method for the isolation andpurification of total lipids from animal tissues. J Biol Chem 1957;226:497–509.
[12] Mueller KM, Kornfeld JW, Friedbichler K, Blaas L, Egger G, Esterbauer H, et al.Impairment ofhepatic growthhormone andglucocorticoid receptor signalingcausessteatosis and hepatocellular carcinoma in mice. Hepatology 2011;54:1398–409.
[13] Barshop NJ, Francis CS, Schwimmer JB, Lavine JE. Nonalcoholic fatty liver diseaseas a comorbidity of childhood obesity. Ped Health 2009;3:271–81.
[14] Guo J, Jou W, Gavrilova O, Hall KD. Persistent diet-induced obesity in male C57BL/6mice resulting from temporary obesigenic diets. PLoS One 2009;4:e5370.
[15] Sos BC, Harris C, Nordstrom SM, Tran JL, Balázs M, Caplazi P, et al. Abrogation ofgrowth hormone secretion rescues fatty liver in mice with hepatocyte-specificdeletion of JAK2. J Clin Invest 2011;121:1412–23.
[16] Attie AD, Krauss RM, Gray-Keller MP, Brownlie A, Miyazaki M, Kastelein JJ, et al.Relationship between stearoyl-CoA desaturase activity and plasma triglyceridesin human and mouse hypertriglyceridemia. J Lipid Res 2002;43:1899–907.
[17] Kotronen A, Seppänen-Laakso T, Westerbacka J, Kiviluoto T, Arola J, Ruskeepää AL,et al. Hepatic stearoyl-CoA desaturase (SCD)-1 activity and diacylglycerol but notceramide concentrations are increased in the nonalcoholic human fatty liver.Diabetes 2009;58:203–8.
[18] Cheung L, Andersen M, Gustavsson C, Odeberg J, Fernandez-Perez L, Norstedt G,et al. Hormonal and nutritional regulation of alternative CD36 transcripts in ratliver – a role for growth hormone in alternative exon usage. BMCMol Biol 2007;8:60.
[19] Zhou J, Febbraio M, Wada T, Zhai Y, Kuruba R, He J, et al. Hepatic fatty acidtransporter Cd36 is a common target of LXR, PXR, and PPARgamma in promotingsteatosis. Gastroenterology 2008;134:556–67.
[20] Oka K, Ishimura-Oka K, Chu MJ, Sullivan M, Krushkal J, Li WH, et al. Mouse very-low-density-lipoprotein receptor (VLDLR) cDNA cloning, tissue-specific expres-sion and evolutionary relationship with the low-density-lipoprotein receptor. EurJ Biochem 1994;224:975–82.
[21] Degrace P, Moindrot B, Mohamed I, Gresti J, Du ZY, Chardigny JM, et al.Upregulation of liver VLDL receptor and FAT/CD36 expression in LDLR−/−apoB100/100 mice fed trans-10,cis-12 conjugated linoleic acid. J Lipid Res 2006;47:2647–55.
[22] Jo H, Choe SS, Shin KC, Jang H, Lee JH, Seong JK, et al. Endoplasmic reticulum stressinduces hepatic steatosis via increased expression of the hepatic very low-densitylipoprotein receptor. Hepatology 2013;57:1366–77.
[23] Shimomura I, Shimano H, Korn BS, Bashmakov Y, Horton JD. Nuclear sterolregulatory element-binding proteins activate genes responsible for the entireprogram of unsaturated fatty acid biosynthesis in transgenic mouse liver. J BiolChem 1998;273:35299–306.
[24] Tabor DE, Kim JB, Spiegelman BM, Edwards PA. Identification of conserved cis-elements and transcription factors required for sterol-regulated transcription ofstearoyl-CoA desaturase 1 and 2. J Biol Chem 1999;274:20603–10.
[25] Joseph SB, Laffitte BA, Patel PH, Watson MA, Matsukuma KE, Walczak R, et al.Direct and indirect mechanisms for regulation of fatty acid synthase geneexpression by liver X receptors. J Biol Chem 2002;277:11019–25.
[26] Tontonoz P, Nagy L, Alvarez JG, Thomazy VA, Evans RM. PPARgamma promotesmonocyte/macrophage differentiation and uptake of oxidized LDL. Cell 1998;93:241–52.
[27] Gavrilova O, Haluzik M, Matsusue K, Cutson JJ, Johnson L, Dietz KR, et al. Liverperoxisome proliferator-activated receptor γ contributes to hepatic steatosis,triacylglycerol clearance, and regulation of body fat mass. J Biol Chem 2003;278:34268–76.
[28] Matsusue K, Haluzik M, Lambert G, Yim SH, Gavrilova O, Ward JM, et al. Liverspecific disruption of PPARγ in leptin-deficient mice improves fatty liver butaggravates diabetic phenotypes. J Clin Invest 2003;111:737–47.
[29] Brown MS, Goldstein JL. A receptor-mediated pathway for cholesterol homeostasis.Science 1986;232:34–47.
[30] Frith J, Day CP, Henderson E, Burt AD, Newton JL. Non-alcoholic fatty liver diseasein older people. Gerontology 2009;55:607–13.