Embed Size (px)
Citation preview

1
Local blockade of epithelial PDL-1 in the airways enhances T cell function and 1
viral clearance during influenza virus infection 2
3
Beth McNally1,*, Fang Ye1, Meredith Willette1, and Emilio Flaño1,2 4
5
1 Center for Vaccines and Immunity, The Research Institute at Nationwide Children’s 6
Hospital, Columbus, OH 43205 7
2 The Ohio State University College of Medicine, Columbus, OH 43210 8
9
* Corresponding author. Mailing address: Center for Vaccines and Immunity, The 10
Research Institute at Nationwide Children’s Hospital, 700 Children’s Dr., Columbus, OH 11
43205. [email protected] 12
13
Running title: PD-1 blockade in the airways 14
15
Abbreviations: bronchoalveolar lavage (BAL), Human airway epithelial (HAE), 16
interferon-α receptor (IFNAR), mediastinal lymph node (MLN), programmed death 17
receptor-1 (PD-1), programmed death receptor 1-ligand (PDL-1), tracheal epithelial cell 18
(TEC) 19
Word count abstract: 205 20
Word count text: 4,538 21
22
JVI Accepts, published online ahead of print on 25 September 2013J. Virol. doi:10.1128/JVI.02423-13Copyright © 2013, American Society for Microbiology. All Rights Reserved.
on April 4, 2019 by guest
http://jvi.asm.org/
Dow
nloaded from

2
ABSTRACT 23
In order to maintain the gas exchange function of the lung following influenza virus 24
infection, a delicate orchestration of positive and negative regulatory pathways must be 25
maintained to attain viral eradication while minimizing local inflammation. The 26
programmed death receptor 1-ligand /programmed death receptor-1 (PDL-1/PD-1) 27
pathway plays an important immunoregulatory role particularly in the context of T cell 28
function. Here, we show that influenza virus infection of primary airway epithelial cells 29
strongly enhances PDL-1 expression, and does so in an interferon-α receptor (IFNAR) 30
signaling dependent manner. PD-1 is primarily expressed on effector T cells in the lung 31
compared to effector memory and central memory cells, and shortly after influenza virus 32
infection an increased number of PD-1+ T cells are recruited to the airways. Using in 33
vitro co-cultures of airway epithelial cells and T cells, and in vivo models of influenza 34
virus infection, we demonstrate that blockade of airway epithelial PDL-1 improves CD8 35
T cell function defined by increased production of IFNγ, granzyme B, and expression of 36
CD107ab. Furthermore, PDL-1 blockade in the airways served to accelerate influenza 37
virus clearance and enhance infection recovery. Our findings suggest that local 38
manipulation of the PDL-1/PD-1 axis in the airways may represent a therapeutic 39
alternative during acute influenza virus infection. 40
41
42
on April 4, 2019 by guest
http://jvi.asm.org/
Dow
nloaded from

3
INTRODUCTION 43
The respiratory tract is constantly exposed to a broad range of foreign antigens due to 44
respiration. The airway epithelium lining the respiratory tract not only represents the first 45
barrier of defense against respiratory pathogens it is often the primary target of infection 46
(1). Consequently, apart from a critical role in gas exchange function, airway epithelial 47
cells additionally participate in host defense mechanisms by producing cytokines and 48
chemokines (2-8). As exacerbated responses by airway epithelial cells in response to 49
innocuous antigens or pathogens can prove detrimental to lung function, airway 50
epithelial cells have also evolved to regulate pulmonary homeostasis by multiple 51
mechanisms including, among others, expression of CD200 (9), MUC-1 (10), surfactant 52
proteins (11), and reduced expression of adaptor molecules (12-14). 53
Interaction of programmed cell death-ligand 1 (PDL-1) with its receptor, programmed 54
death 1 (PD-1), a member of the B7 family of signaling molecules, has been specifically 55
linked to negative regulation of T cell immune responses (15, 16). Upon activation, PD-1 56
inhibits T cell receptor signaling by blocking PI3k/Akt activation (17). During chronic viral 57
infection and cancer, PD-1 expression leads to T cell dysfunction and exhaustion (15, 58
16, 18). Conversely, blockade of the PD-1/PDL-1 axis has been shown to either 59
enhance T cell function or reduce viral burden in a number of chronic viral infections 60
(18-21). Our understanding of the role of the PD-1/PDL-1 pathway during acute viral 61
infection, however, is just beginning to unfold. PD-1 expression is rapidly down-62
regulated during acute LCMV infection (22) but is expressed on functional CD8 T cells 63
in other acute viral infections (23, 24). PD-1 has also been shown to regulate the 64
on April 4, 2019 by guest
http://jvi.asm.org/
Dow
nloaded from

4
development of T cell memory during vaccinia virus infection (25). Fairly recently, it has 65
been shown that a number of acute respiratory viral infections including human 66
metapneumovirus, respiratory syncytial virus and influenza virus impair CD8 T cell 67
function through PD-1 (26)(27). Although previous studies have shown that lack of PD-68
1 function due to genetic mutation or to systemic blockade prevented CD8 T cell 69
inhibition and enhanced viral control, the specific role that the airway epithelium plays 70
during PDL-1/PD-1 regulation of pulmonary T cell function has not been fully elucidated. 71
Our study builds on the observations by Erickson et al. (27) to further show that PDL-1 72
is constitutively expressed in airway epithelial cells, and its expression is up-regulated 73
by influenza virus and dependent on interferon-α receptor (IFNAR) signaling. Co-culture 74
studies with airway epithelial cells and T cells in the presence of PDL-1 blocking 75
antibody revealed that PDL-1 expression on airway epithelial cells repressed T cell 76
function. Additionally, intranasal blockade of PDL-1 in influenza virus infected mice 77
resulted in increased levels of cytotoxic proteins and cytokines in the airways; enhanced 78
CD8 T cell function; and reduced viral load. Our findings suggest that PDL-1 expression 79
by airway epithelial cells during the early phase of the recall response to influenza virus 80
infection represents a mechanism for regulating lung immune homeostasis, and reveal 81
a promising strategy to modulate local epithelial-T cell interaction in the airways. 82
83
MATERIALS AND METHODS 84
85
on April 4, 2019 by guest
http://jvi.asm.org/
Dow
nloaded from

5
Mice. C57BL/6J, Balb/c, and IFNAR−/− mice (Balb/c background) were obtained from 86
The Jackson Laboratory (Bar Harbor, ME) or Harlan (Indianapolis, IN), or were bred at 87
The Research Institute at Nationwide Children’s Hospital. Mice were housed in BL2 88
containment under pathogen-free conditions. The Institutional Animal Care and Use 89
Committee at The Research Institute at Nationwide Children’s Hospital approved all of 90
the animal studies described in this study. 91
Cell culture. Primary mouse tracheal epithelial cell (TEC) cultures were obtained from 92
tracheas resected from 3-10 week old wild-type Balb/c and IFNAR-/- mice. Tracheal cell 93
isolation and culture was performed as previously described (28). In short, tracheas 94
were collected and incubated with 1.5 mg/ml pronase and 0.5 mg/ml crude pancreatic 95
DNase I. Non-adherent cells were collected and seeded onto 24 mm coated 96
membranes with 50 µg/ml type I rat tail collagen solution. Cells were incubated for 10-14 97
days post-harvest then allowed to differentiate by incubating under air-liquid interface 98
for an additional 14 days before use. 99
Virus infection. Influenza A viruses Udorn, WSN, PR8 and X31 were grown in eggs, 100
and virus titers were determined by an immunofluorescence foci assay. Male mice (6-101
12wks of age) were anesthetized with 2,2,2,-tribromoethanol and intranasally inoculated 102
with 3000 EID50 influenza virus X31 and challenged with 1x102 PFU/mouse PR8, in a 30 103
µl volume. Organs were harvested on the indicated days post treatment. Groups of 104
three to six animals were used for each data point. For the analysis of PDL-1 transcript, 105
TEC cultures derived from Balb/c and IFNAR-/- mice were exposed to 2x105 WSN for 106
2h or mock inoculated and harvested 24h post infection. Human airway epithelial (HAE) 107
on April 4, 2019 by guest
http://jvi.asm.org/
Dow
nloaded from

6
cultures were exposed to 2x105 influenza A Udorn strain, or mock treated for 2 hours 108
and harvested after 24 hours of incubation as indicated (28). 109
Immunofluorescence. Influenza infected HAE cultures maintained on Snapwell inserts 110
were fixed in 10% paraformaldehyde then washed and stored in 70% ethanol until 111
paraffin embedded. Six-micrometer sections of HAE the cultures or human trachea 112
were deparaffinized for immunofluorescence. Following 30’ antigen retrieval in sodium 113
citrate (pH 6), tissues were incubated in 0.2% Triton X-100 for 15’ and blocked with 5% 114
goat serum for 1 hour. PDL-1 was visualized using rabbit polyclonal to CD274, (Abcam, 115
Cambridge, MA) and Texas-red goat anti rabbit (Vector Laboratories, Burlingame, CA). 116
Control sections were stained with rabbit immunoglobulin or with secondary antibody 117
only. Tissues were mounted with Prolong Gold anti-fade reagent containing DAPI 118
(Invitrogen/Molecular Probes). Immunofluorescent images were viewed on an Olympus 119
BX61 using a 40x objective and captured using a Hamamatsu ORCA-GR digital camera 120
in concert with Slidebook version 4.2 software (Intelligent Imaging Innovations, Inc., 121
Denver, CO). 122
RNA extraction, reverse transcription, and real-time PCR. RNA isolation from 123
infected HAE cultures and TEC cultures was achieved by direct lysis of cells with TRIzol 124
(Invitrogen) and application of the resulting aqueous phase to Qiagen RNeasy Mini 125
Columns. Total RNA harvested from triplicate (TEC) or quadruplicate (HAE) cultures 126
was processed and analyzed as described in (28). 1 たg RNA extracted from infected 127
lungs or cell cultures using a combination of Trizol (Invitrogen) and/or the RNeasy Mini 128
Kit, (Qiagen), was also used for cDNA synthesis (High capacity RT Kit, Applied 129
Biosytems) followed by quantitative PCR performed using Cyber Green (Applied 130
on April 4, 2019 by guest
http://jvi.asm.org/
Dow
nloaded from

7
Biosytems) and primers for human or mouse PDL-1, the Flu NP gene and human or 131
mouse GAPDH. Data were analyzed using Prism 7500 SDS software. Mock infected 132
groups served as the calibrator sample and all samples were normalized to the 133
endogenous control, GAPDH. 134
TEC/T lymphocyte co-cultures. TEC cultures derived from Balb/c and IFNAR-/- mice 135
were co-cultured with splenic T lymphocytes at a 1:1 ratio (TECs:T cells). T 136
lymphocytes were isolated from spleens of naïve Balb/c mice by negative selection. 137
TECs seeded in 24-well plates were exposed to 1 PFU/cell influenza virus WSN for 2 138
hours. Fresh media was added after the removal of inoculum, and panned T 139
lymphocytes (purity, >95%) were added 24h post infection. Blocking antibodies to PDL-140
1 (clone MIH5, eBioscience) or control antibody (ChromePure Rat IgG, Jackson 141
Immuno Research) were added 3 hrs before the addition of T lymphocytes such that 10 142
たg blocking antibody was added to 2.5�×�105 TECs. 143
In vivo antibody blockade. Mice initially infected with influenza virus X31 were 144
challenged 30-40 days later with influenza virus PR8 as described above, and either 145
intraperitoneally injected with 200 ug of functional grade anti-mouse PDL-1, 200 ug IgG 146
or HBSS; or were intranasally treated with 150 ug anti-PDL-1 or control antibody 147
administered successively over a three day time period in 50 ug (50 ul) doses. Sham 148
mice received the same volume of HBSS in lieu of antibody. 149
Plaque assay. Lung homogenates collected in 0.5 ml DMEM-0.1% were used for viral 150
titer determination. Plaque assays were performed by exposing 80-90% confluent 151
MDCK cells grown in 12-well plates to 100 µl quantities of 10-fold serial dilutions of lung 152
homogenates for 2 hours at 37°C. The inoculated monolayers were covered with DMEM 153
on April 4, 2019 by guest
http://jvi.asm.org/
Dow
nloaded from

8
containing 0.8% methylcellulose, L-glutamine, 0.1% BSA, antibiotics and 1 たg/ml TPCK 154
trypsin (Sigma, St. Louis, MO) and incubated for 3 days at 37°C. After incubation, the 155
monolayers were permeablized with 0.2% Triton X-100. Virus infected cells were 156
immuno-stained using a monoclonal antibody that reacts with the influenza A 157
nucleoprotein (Cal Bioreagents, San Mateo, CA) followed by a 1 hr incubation with anti-158
mouse IgG peroxidase-congjugate (Sigma, St. Louis, MO) and a 10-30 min incubation 159
with True Blue Substrate (KPL, Gathersburg, MD). Quantification was performed by 160
counting plaques using a dissecting microscope and counts were expressed as log10 161
PFU per milliliter. 162
Flow cytometry 163
Epithelial cells. TECs were trypsinized to obtain a cell suspension then stained and 164
fixed with paraformaldehyde. Analysis of cell surface expression of PDL-1 on TECs was 165
achieved using PE-Rat anti-mouse CD274 (BD Pharmingen, Franklin Lakes, NJ) and 166
the BD LSRII flow cytometer with FlowJo software (TreeStar Inc., Ashland, OR). Results 167
were expressed as median fluorescence intensity (MFI) after subtracting the MFI of 168
control cells stained with the appropriate isotype control antibodies (BD Pharmingen). 169
T lymphocytes. Single-cell suspensions were obtained from the bronchoalveolar lavage, 170
lung, MLN and spleen. RBCs were lysed, and the number of cells per organ was 171
determined. Cells were blocked with Fc-block (CD16/32) and washed and stained with a 172
combination of NP366–374/Db tetramer and antibodies against CD3, CD4, CD8, CD44, 173
CD62L, CD43-activation associated glycoform, CD107ab, and PD-1 or combined with 174
intracellular staining for cytokines/cytotoxic proteins: IFN-け, IL-2, perforin and granzyme 175
B (eBioscience). Flow cytometry data were acquired on a BD LSR II (BD Biosciences) 176
on April 4, 2019 by guest
http://jvi.asm.org/
Dow
nloaded from

9
and analyzed using FlowJo software. Gates were set using negative controls and 177
isotype controls. 178
ELISA for IFN-γ and granzyme B. Concentrations of gamma interferon (IFN-γ) and 179
Granzyme B, were measured in BAL sample supernatants and co-cultutres with 180
commercially available ELISA kits (Ready-SET-Go! Kit, eBioscience). All assays were 181
carried out according to the manufacturer's instructions, and 100 µl from each sample 182
was assayed in duplicate. The lower limits of detection for these assays were 15 pg/ml 183
for IFN-け, and 40 pg/mL (Granzyme B). For statistical analysis, samples with optical 184
density readings below the limit of the standard curve of the assay were assigned a 185
value half that of the detection level. 186
Statistical analysis. T tests were used to compare both the percentage of cells positive 187
for a given antigen of interest as well as the mean relative fluorescence intensity (MRFI) 188
determined by flow cytometry analysis. Two sample homoscedastic student t-tests were 189
used for single comparisons in which a P value less than 0.05 was considered 190
statistically significant. Results are representative of at least 2-3 separate experiments 191
using 3-6 mice per group and are expressed as means�±�SD. One-way analysis of 192
variance (ANOVA) was used to determine statistical significance when comparing three 193
or more independent groups (particularly in cases in which antibody treatment was 194
compared to mock, Flu and IgG control treatments). When ANOVA demonstrated that 195
the differences among means were statistically significant (P < 0.05), the Tukey post-196
test was used to correct for multiple comparisons, and to generate multiplicity adjusted 197
P values (Prism GraphPad, version 6). 198
199
on April 4, 2019 by guest
http://jvi.asm.org/
Dow
nloaded from

10
RESULTS 200
PDL-1 is constitutively expressed in airway epithelial cells, and influenza virus 201
infection and interferon- signaling regulate its expression 202
The epithelial surfaces of the lung play an important role in regulating local immunity 203
and homeostasis. In an initial attempt to analyze the role of regulatory factors critical to 204
infection of the respiratory tract, we established polarized, well-differentiated human 205
primary airway epithelial cell cultures (HAEs) and exposed them to influenza A virus. 206
HEA cultures differentiate to form a pseudostratified mucociliary cell layer and 207
effectively mimic the airway epithelial environment. A comparison of cluster of 208
differentiation molecules following gene expression microarray-based screening 209
revealed that one gene in particular, PDL-1, was drastically upregulated in HAE cultures 210
24 h post exposure to influenza virus (S1). Our analysis showed that PDL-1 expression 211
was enhanced 83.9-fold 24h post exposure to Influenza virus compared to mock-treated 212
HAEs (Figure 1A). qRT-PCR analysis corroborated the above findings showing a 54.3-213
fold increase in PDL-1 expression in HAEs following influenza virus exposure (Figure 214
1B). Additionally, immunoflourescent staining of human trachea sections showed that 215
PDL-1 was not only constitutively expressed in airway epithelial cells, but, perhaps more 216
importantly, that the staining was prominent on the apical surface (Figure 1C). To 217
determine if influenza virus infection affected PDL-1 expression at the protein level, we 218
performed immunofluorescent analysis of PDL-1 expression on HAE cultures infected or 219
not with influenza virus. As we observed in human trachea epithelium, PDL-1 was 220
constitutively expressed in control HAEs (Figure 1D, upper panel). However, when 221
on April 4, 2019 by guest
http://jvi.asm.org/
Dow
nloaded from

11
compared with mock treated cultures, PDL-1 staining increased in intensity in HAEs 24 222
h post exposure to influenza (Figure 1D, lower panel). Altogether, these data indicate 223
that PDL-1 is constitutively expressed by the human airway epithelium, and that 224
influenza virus infection increases the expression of PDL-1 in airway epithelial cells 225
within 24 hours of infection. 226
To further analyze regulation of PDL-1 expression on airway epithelial cells we 227
generated primary murine tracheal epithelial cell cultures (TECs). As we observed with 228
the HAEs, exposure of TECs to influenza virus resulted in a significant increase (27.6-229
fold) in PDL-1 mRNA expression by 24 h (Figure 2A). As interferon-α receptor (IFNAR) 230
signaling is critical for the anti-viral response of the airway epithelium (28), we also 231
analyzed the role of IFNAR on the regulation of PD-1L expression in airway epithelial 232
cells during the response to influenza virus. The data show that there was a 21.1-fold 233
difference in PDL-1 mRNA expression when comparing influenza virus infected IFNAR-234
/- TECs to wild-type TECs, indicating that PDL-1 mRNA expression is dependent on 235
signaling. We next corroborated these findings at the protein level by FACS analysis of 236
PDL-1 cell surface expression. The data show that influenza virus infection enhanced 237
the cell surface expression of PDL-1 on TECs, but that PDL-1 expression on IFNAR-238
deficient TEC cultures was not different from isotype control staining (Figure 2B-C). 239
Altogether, these data indicate that influenza virus-induced increase of PDL-1 240
expression on epithelial cells is IFNAR signaling dependent. 241
PD-1+ T cells are present in the lung early during influenza virus infection 242
on April 4, 2019 by guest
http://jvi.asm.org/
Dow
nloaded from

12
In order for PDL-1 to be functional on airway epithelial cells, contact with cells 243
expressing its receptor is necessary. Therefore, we investigated whether or not a 244
significant population of PD-1 expressing cells was available in the respiratory tract. In 245
order to do this, cohorts of mice were intranasally infected with influenza A virus, and 246
cell populations were analyzed in the bronchoalveolar lavage (BAL) and lung 247
parenchyma at different times points after infection. The data shows that PD-1 was 248
expressed in 10-40% of the CD4 and CD8 T cells present in the airways and lung 249
parenchyma of naïve mice, and that there were not significant changes in the frequency 250
of PD-1-expressing T cells during the first week after influenza virus infection (Figures 251
3A-B). The analysis of T cell numbers shows that a small population of PD-1-expressing 252
CD4 and CD8 T cells is present in the lung parenchyma (~1000 cells) while the number 253
of PD-1+ T cells in the airways was very low (10 CD8 T cells, 100 CD4 T cells, Figure 254
3C-D). However, we observed a significant increase in the absolute number of PD-1+ 255
CD4 and CD8 T cells present in the airways by day 3 after influenza virus infection 256
(Figure 3C). Additionally, the number of PD-1+ T cells in the lung parenchyma also 257
increased over time though the differences were not statistically significant (Figure 3D). 258
Although the number of PD1-expressing T cells returned to baseline levels in the lung 259
parenchyma by day 12 post-infection, greater numbers of PD-1+ CD4 and CD8 T cells 260
remained in the airways of influenza infected mice up to one month after infection. To 261
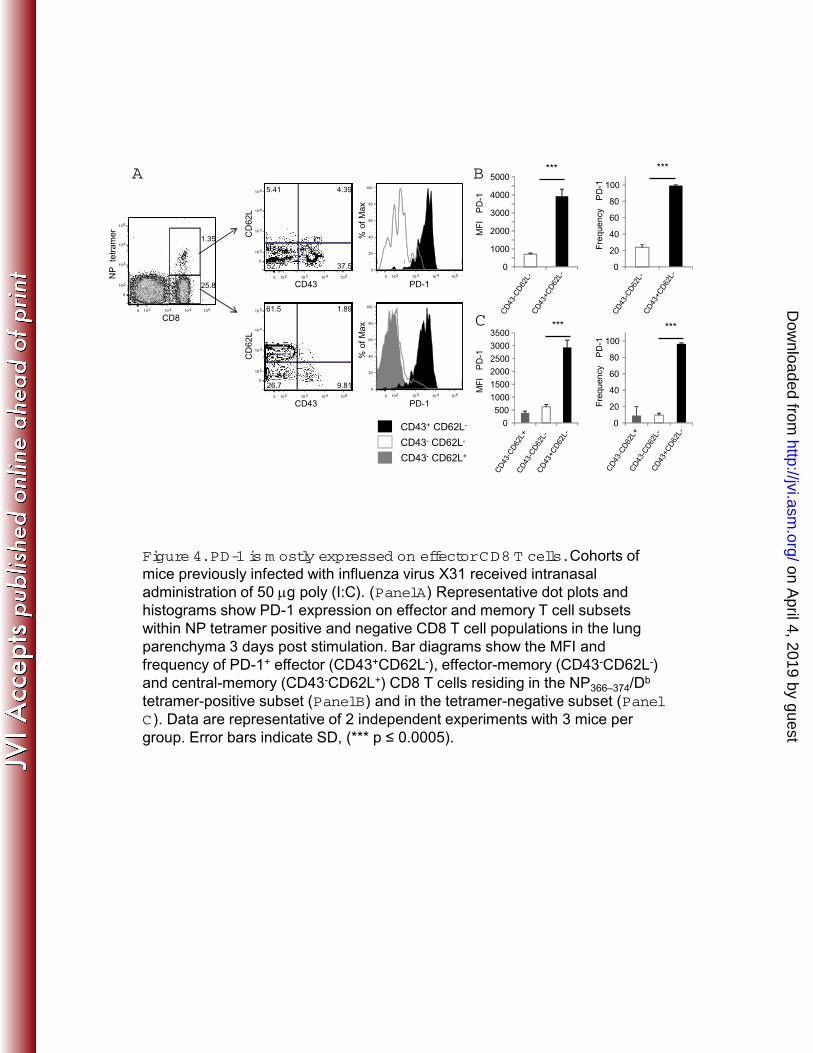
more precisely determine the expression pattern of PD-1 on different populations of 262
influenza virus-specific effector and memory T cells, influenza virus memory mice were 263
intranasally stimulated with poly (I:C) to induce non-specific activation of T cells in the 264
respiratory tract. We found that PD-1 was expressed on effector CD8 T cells but was 265
on April 4, 2019 by guest
http://jvi.asm.org/
Dow
nloaded from

13
absent from the cell surface of effector-memory and central-memory CD8 T cells as 266
defined by the expression of L-selectin and CD43-activation associated glycoform 267
(Figure 4). This pattern of expression was found on influenza virus NP366–374-specific 268
CD8 T cells as well as NP tetramer-negative CD8 T cells. Altogether, these data 269
indicate that the majority of influenza virus-specific effector CD8 T cells present in the 270
lung express PD-1. 271
PDL-1 expression on airway epithelial cells controls CD8 T cell function 272
To determine if PDL-1 expression on the airway epithelium modulated the function of T 273
cells we used a memory system that allowed us to analyze the function of virus-specific 274
T cells. We incubated T cells isolated from the spleens of day 30-40 influenza virus-275
infected mice with primary, polarized TECs in the presence or absence of PDL-1-276
blocking antibodies. TECs were first exposed to influenza virus for 24 h to induce the 277
up-regulation of PDL-1 on the epithelial cell surface, and then co-cultured with T cells 278
for an additional 36 h. T cell function was assessed by measuring the levels of 279
granzyme B secreted into the culture medium. As shown in Fig. 5, granzyme B was 280
undetectable in uninfected co-cultures regardless of the presence or not of anti-PDL-1 281
antibodies. However, in the presence of influenza virus, granzyme B was released into 282
the culture media, and PDL-1 blockade resulted in significantly higher levels of 283
granzyme B in culture supernatants. These data indicate that PDL-1 expressed on 284
airway epithelial cells can inhibit the cytotoxic activity of T cells through its interaction 285
with PD-1, and that this process can be reversed in vitro by blockade of PDL-1/PD-1 286
interaction. To address the functional role of PDL-1/PD-1 interactions in vivo during 287
on April 4, 2019 by guest
http://jvi.asm.org/
Dow
nloaded from

14
influenza virus infection, we performed PDL-1 blockade studies and monitored 288
parameters of T cell function in the airways. For these studies, we used influenza virus 289
memory mice to ensure that an elevated number of virus-specific T cells were present in 290
the respiratory tract very early after challenge. The memory mice were intranasally 291
challenged with a heterosubtypic influenza virus (PR8) infection, injected with PDL-1 292
blocking antibody or with isotype control antibody, and the levels of granzyme B and of 293
IFNγ were assayed in BAL fluid 3 days after viral challenge. As shown in Figure 6, 294
intraperitoneal administration of anti-PDL-1 blocking antibody resulted in a significant 295
increase in the concentration of granzyme B and IFNγ in the airways when compared to 296
mice treated with control IgG. 297
Although these in vivo experiments demonstrated that PDL-1/PD-1 interaction regulates 298
the effector functions during influenza virus infection, they did not specifically address 299
the role of the airway epithelium in this process. This is important because the PDL-300
1/PD-1 pathway has been shown to impact the priming of T cell responses (29-34). 301
Thus, in order to elucidate the role of the PDL-1/PD-1 axis in regulating intraepithelial T 302
cell function we administered anti-PDL-1 blocking antibodies intranasally to induce 303
blockade in the airways. Preliminary studies demonstrated that intranasal administration 304
of antibodies had a specific local effect in the airways but not in the lung parenchyma, 305
draining lymph nodes, or spleen (S2). The results show that intranasal administration of 306
anti-PDL-1 blocking antibodies resulted in significantly higher concentrations of 307
granzyme B and IFNγ in the airways 3 days after influenza virus infection (Figure 7 A-B). 308
By day 5 after influenza virus infection the levels of granzyme B and IFNγ in mice 309
on April 4, 2019 by guest
http://jvi.asm.org/
Dow
nloaded from

15
treated with anti-PDL1 antibodies were comparable to those of non-challenged mice 310
(Figure 7 C-D). These results indicate that intranasal PDL-1 blockade can enhance and 311
accelerate the cytotoxic function of immune cells in the airways. 312
We next analyzed the effect of intranasal PDL-1 blockade on CD8 T cell function. 313
Influenza virus-challenged mice treated with anti-PDL-1 antibodies had a statistically 314
significant higher frequency and absolute number of IFNγ CD107ab-double positive 315
cells than mice treated with control antibody or mock treated (Figure 7E-F). The 316
percentage of IFNγ and of CD107ab-single positive CD8 T cells was also higher in mice 317
treated with anti-PDL-1 antibodies but the differences were not statistically significant. 318
We did not observe any differences in CD8 T cell function in the draining lymph nodes 319
or the spleen between mice treated or not with anti-PDL-1 antibodies (data not shown). 320
Altogether, our data indicate that preventing the PDL-1/PD-1 interaction between the 321
airway epithelia and T cells results in enhanced IFNγ production and cytolytic function 322
by influenza virus-specific CD8 T cells. 323
PDL-1 blockade in the airways reduces influenza virus load and ameliorates 324
disease 325
We next analyzed the consequences of intranasal PDL-1 blockade on influenza virus 326
control on days 3 and 5 following a secondary viral challenge. The viral loads were 327
measured in lung homogenates using both qRT-PCR and infectious plaque assays. 328
The data show that PDL-1 blockade resulted in a slight reduction of influenza virus 329
nucleoprotein (NP) transcripts and of infectious viral titers by day three post infection 330
(Figure 8A). However, by day 5 post infection NP transcript levels and infectious virus 331
on April 4, 2019 by guest
http://jvi.asm.org/
Dow
nloaded from

16
titers were below the limit of detection in the anti-PDL-1 treated group (Figure 8B). 332
Conversely, untreated or control IgG-treated mice had significantly greater levels of 333
expression of NP RNA in the lungs as measured by qRT-PCR, and several orders of 334
magnitude (102-105) higher viral titers measured by plaque assay. We also subjected 335
each mouse from each group to whole body plethysmography on day 5 post infection 336
with Influenza. Enhanced pause (Penh) values in Influenza memory mice exposed to a 337
heterosubtypic influenza challenge but treated with α-PDL-1 registered values 338
equivalent to Influenza memory mice that did not receive the secondary influenza 339
challenge. Conversely, Penh values were significantly elevated in influenza memory 340
mice that were exposed to a heterosubtypic influenza challenge but were left untreated 341
or treated with control α-IgG (S3). Lastly, to determine the effect of intranasal anti-PDL-342
1 treatment on acute respiratory disease we monitored weight loss. We observed that 343
mice treated with anti-PDL-1 antibodies during influenza virus challenge did not present 344
any significant weight loss between day 0 and day 5 of treatment, while untreated or 345
control IgG-treated mice had a significant decrease in weight loss (Figure 9) suggesting 346
that intranasal anti-PDL-1 treatment can influence acute respiratory disease severity. 347
Altogether, our data indicate that blockade of PDL-1/PD-1 interaction in the airways 348
during influenza virus infection can result in enhanced viral control and disease 349
recovery. 350
351
DISCUSSION 352
on April 4, 2019 by guest
http://jvi.asm.org/
Dow
nloaded from

17
In this report, we showed that influenza virus infection of primary airway epithelial cells 353
strongly enhances PDL-1 expression, and does so in an IFNAR signaling dependent 354
manner. Most importantly, we observed that intranasal administration of anti-PDL-1 355
blocking antibodies to induce local blockade of PDL-1 in the airways improved T cell 356
function in the acute phase of infection as defined by increased production of IFNγ, 357
granzyme B, and cell surface mobilization of CD107ab. PDL-1 blockade in the airways 358
resulted in accelerated influenza virus clearance and enhanced infection recovery as 359
measured by weight loss. These data indicate that at the onset of influenza virus 360
infection the PD-1/PDL-1 axis is available to limit effector T cell function in the airways. 361
Our findings showing that PDL-1 was constitutively expressed on airway epithelial cells 362
prompted us to hypothesize that the PDL-1/PD-1 axis could be regulating local lung 363
immune responses as a means to maintain the integrity of the epithelial lining of the 364
respiratory tract during infection. The epithelial surfaces of the lung have been shown to 365
express molecules such as CD200 among others to regulate the function of immune 366
cells and maintain lung homeostasis (9). The observation that expression of PDL-1 in 367
airway epithelial cells is controlled by viral infection and IFNAR signaling suggests that 368
the PDL-1/PD-1 pathway plays an important role during inflammatory responses in the 369
lung. Innate interferons are critical for the response of airway epithelial cells to 370
respiratory viral infection (28). These observations indicate that interferon signaling also 371
contributes to the regulation of inflammatory homeostasis in the lung by enhancing the 372
expression of PDL-1 in airway epithelial cells, adding another layer of complexity to the 373
regulatory pathways operated by interferon stimulated genes. Our findings with airway 374
epithelial cells are in accordance with observations in other systems. Mice lacking TLR3 375
on April 4, 2019 by guest
http://jvi.asm.org/
Dow
nloaded from

18
had reduced PDL-1 expression (35); and a major downstream product of TLR3 376
signaling, IFNβ has been shown to enhance PDL-1 expression on monocytes, dendritic 377
cells and hippocampal cultures (35, 36). Furthermore, synthetic dsRNA has been shown 378
to trigger PDL-1 expression on epithelial cells (37, 38). Taken together, the above 379
findings suggest that PDL-1 expression in the periphery during the effector phase of the 380
response to virus infection likely represents a generalized host regulatory response to 381
viral infection rather than a virus-specific immunoevasion strategy. 382
The PD-1/PDL-1 signaling cascade is a prominent mechanism by which T cell non-383
responsiveness occurs in the face of chronic viral infections and cancer. The role of PD-384
1 expressing T cells during acute infection has remained controversial (39), although 385
recent observations report increased primary T cell responses in the absence of PD-1 386
signaling (25, 27, 40). Additionally, a recent study showed that the majority of PD-1 387
expressing T cells in healthy human adults do not exhibit an exhausted phenotype (41). 388
Our data show that PD-1 is primarily expressed on effector T cells in the lung compared 389
to effector memory and central memory cells, and that shortly after influenza virus 390
infection an increased number of PD-1+ T cells are recruited to the airways. Zelinskyy et 391
al. (23) also demonstrated that during acute Friend retrovirus infection, the PD-1-392
expressing CD8 T cells were terminally differentiated cytotoxic effectors. These studies 393
indicate that PD-1 is not a marker of T cell exhaustion per se, and suggest a likely 394
scenario during acute infection in which the PD-1/PDL-1 axis modulates inflammation 395
and ultimately serves to prevent tissue damage from activated effector T cells. 396
Using co-cultures of primary airway epithelial cells and T cells, and a mouse model of 397
influenza virus infection, we determined that blockade of airway epithelial PDL-1 398
on April 4, 2019 by guest
http://jvi.asm.org/
Dow
nloaded from

19
improves CD8 T cell function defined by increased production of IFNγ, granzyme B, and 399
expression of CD107ab. Importantly, therapeutic PDL-1 blockade in the airways 400
accelerated influenza virus clearance and enhanced infection recovery as measured by 401
weight loss. Our results are in accordance with recent findings by Erickson (11) showing 402
that viral acute respiratory infections impair CD8 T cells through PD-1, and that 403
systemic inhibition of PD-1 prevented CD8 T cell impairment. Our study adds that PD-404
1/PDL-1 regulation of CD8 T cell effector function is mediated locally by the epithelial 405
cells of the lung, it is regulated by IFNAR signaling, and can be therapeutically 406
manipulated in the airways by intranasal administration of anti-PDL-1 blocking 407
antibodies. As PD-1 signaling has been shown to impact dendritic cell maturation, 408
antigen presentation, and T cell priming and expansion (34, 42-44), it is important to 409
differentiate the systemic role of PD-1/PDL-1 during the generation of adaptive immune 410
responses from the its specific role at the mucosal site of infection during epithelial-T 411
cell interaction cross-talk. 412
Our findings are in line with several studies demonstrating that loss of PDL-1 during 413
acute viral infection can promote viral clearance (45, 46) and that PDL-1 blockade at the 414
onset of infection may serve as a potential therapeutic (27, 35). Clearly, further work is 415
needed to optimize the timing of therapy and to address role of other cell populations 416
expressing PD-1/PDL-1 in the lung. Nevertheless, our findings indicate that local 417
manipulation of the PDL-1/PD-1 pathway in the airways may represent a therapeutic 418
alternative during acute influenza virus infection. 419
420
on April 4, 2019 by guest
http://jvi.asm.org/
Dow
nloaded from

20
ACKNOWLEDGMENTS 421
We thank the Biomedical Genomics Core and Flow Cytometry Core Laboratory at 422
the Research Institute, and Dr. Mark Peeples and Sara Mertz for help with the HAE 423
cultures. This work was supported in part by NIH grant AI082962 and by The Research 424
Institute. 425
426
on April 4, 2019 by guest
http://jvi.asm.org/
Dow
nloaded from

21
REFERENCES 427
1. Taubenberger JK, Morens DM. 2008. The pathology of influenza virus 428
infections. Annu Rev Pathol 3:499-522. 429
2. Diamond G, Legarda D, Ryan LK. 2000. The innate immune response of the 430
respiratory epithelium. Immunol Rev 173:27-38. 431
3. Bals R, Hiemstra PS. 2004. Innate immunity in the lung: how epithelial cells fight 432
against respiratory pathogens. Eur Respir J 23:327-333. 433
4. Kato A, Schleimer RP. 2007. Beyond inflammation: airway epithelial cells are at 434
the interface of innate and adaptive immunity. Curr Opin Immunol 19:711-720. 435
5. Schleimer RP, Kato A, Kern R, Kuperman D, Avila PC. 2007. Epithelium: at 436
the interface of innate and adaptive immune responses. J Allergy Clin Immunol 437
120:1279-1284. 438
6. Shaykhiev R, Bals R. 2007. Interactions between epithelial cells and leukocytes 439
in immunity and tissue homeostasis. J Leukoc Biol 82:1-15. 440
7. Shornick LP, Wells AG, Zhang Y, Patel AC, Huang G, Takami K, Sosa M, 441
Shukla NA, Agapov E, Holtzman MJ. 2008. Airway epithelial versus immune 442
cell Stat1 Function for Innate Defense against Respiratory Viral Infection. J 443
Immunol 180:3319-3328. 444
8. Humlicek AL, Manzel LJ, Chin CL, Shi L, Excoffon KJ, Winter MC, Shasby 445
DM, Look DC. 2007. Paracellular permeability restricts airway epithelial 446
responses to selectively allow activation by mediators at the basolateral surface. 447
J Immunol 178:6395-6403. 448
9. Snelgrove RJ, Goulding J, Didierlaurent AM, Lyonga D, Vekaria S, Edwards 449
L, Gwyer E, Sedgwick JD, Barclay AN, Hussell T. 2008. A critical function for 450
CD200 in lung immune homeostasis and the severity of influenza infection. Nat 451
Immunol 9:1074-1083. 452
10. Ueno K, Koga T, Kato K, Golenbock DT, Gendler SJ, Kai H, Kim KC. 2008. 453
MUC1 mucin is a negative regulator of toll-like receptor signaling. Am J Respir 454
Cell Mol Biol 38:263-268. 455
11. Yamada C, Sano H, Shimizu T, Mitsuzawa H, Nishitani C, Himi T, Kuroki Y. 456
2006. Surfactant protein A directly interacts with TLR4 and MD-2 and regulates 457
inflammatory cellular response. Importance of supratrimeric oligomerization. J 458
Biol Chem 281:21771-21780. 459
12. Jia HP, Kline JN, Penisten A, Apicella MA, Gioannini TL, Weiss J, McCray 460
PB, Jr. 2004. Endotoxin responsiveness of human airway epithelia is limited by 461
low expression of MD-2. Am J Physiol Lung Cell Mol Physiol 287:L428-437. 462
13. Muir A, Soong G, Sokol S, Reddy B, Gomez MI, Van Heeckeren A, Prince A. 463
2004. Toll-like receptors in normal and cystic fibrosis airway epithelial cells. Am J 464
Respir Cell Mol Biol 30:777-783. 465
14. Greene CM, Carroll TP, Smith SG, Taggart CC, Devaney J, Griffin S, O'Neill 466
S J, McElvaney NG. 2005. TLR-induced inflammation in cystic fibrosis and non-467
cystic fibrosis airway epithelial cells. J Immunol 174:1638-1646. 468
on April 4, 2019 by guest
http://jvi.asm.org/
Dow
nloaded from

22
15. Butte MJ, Keir ME, Phamduy TB, Sharpe AH, Freeman GJ. 2007. 469
Programmed death-1 ligand 1 interacts specifically with the B7-1 costimulatory 470
molecule to inhibit T cell responses. Immunity 27:111-122. 471
16. Freeman GJ, Long AJ, Iwai Y, Bourque K, Chernova T, Nishimura H, Fitz LJ, 472
Malenkovich N, Okazaki T, Byrne MC, Horton HF, Fouser L, Carter L, Ling V, 473
Bowman MR, Carreno BM, Collins M, Wood CR, Honjo T. 2000. Engagement 474
of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to 475
negative regulation of lymphocyte activation. J Exp Med 192:1027-1034. 476
17. Francisco LM, Sage PT, Sharpe AH. 2010. The PD-1 pathway in tolerance and 477
autoimmunity. Immunol Rev 236:219-242. 478
18. Barber DL, Wherry EJ, Masopust D, Zhu B, Allison JP, Sharpe AH, Freeman 479
GJ, Ahmed R. 2006. Restoring function in exhausted CD8 T cells during chronic 480
viral infection. Nature 439:682-687. 481
19. Day CL, Kaufmann DE, Kiepiela P, Brown JA, Moodley ES, Reddy S, 482
Mackey EW, Miller JD, Leslie AJ, DePierres C, Mncube Z, Duraiswamy J, 483
Zhu B, Eichbaum Q, Altfeld M, Wherry EJ, Coovadia HM, Goulder PJ, 484
Klenerman P, Ahmed R, Freeman GJ, Walker BD. 2006. PD-1 expression on 485
HIV-specific T cells is associated with T-cell exhaustion and disease progression. 486
Nature 443:350-354. 487
20. Trautmann L, Janbazian L, Chomont N, Said EA, Wang G, Gimmig S, 488
Bessette B, Boulassel MR, Delwart E, Sepulveda H, Balderas RS, Routy JP, 489
Haddad EK, Sekaly RP. 2006. Upregulation of PD-1 expression on HIV-specific 490
CD8 + T cells leads to reversible immune dysfunction. Nat Med. 491
21. Urbani S, Amadei B, Tola D, Massari M, Schivazappa S, Missale G, Ferrari 492
C. 2006. PD-1 expression in acute hepatitis C virus (HCV) infection is associated 493
with HCV-specific CD8 exhaustion. J Virol 80:11398-11403. 494
22. Wherry EJ, Blattman JN, Murali-Krishna K, van der Most R, Ahmed R. 2003. 495
Viral persistence alters CD8 T-cell immunodominance and tissue distribution and 496
results in distinct stages of functional impairment. J Virol 77:4911-4927. 497
23. Zelinskyy G, Myers L, Dietze KK, Gibbert K, Roggendorf M, Liu J, Lu M, 498
Kraft AR, Teichgraber V, Hasenkrug KJ, Dittmer U. 2011. Virus-specific CD8+ 499
T cells upregulate programmed death-1 expression during acute friend retrovirus 500
infection but are highly cytotoxic and control virus replication. J Immunol 501
187:3730-3737. 502
24. Bowen DG, Shoukry NH, Grakoui A, Fuller MJ, Cawthon AG, Dong C, 503
Hasselschwert DL, Brasky KM, Freeman GJ, Seth NP, Wucherpfennig KW, 504
Houghton M, Walker CM. 2008. Variable patterns of programmed death-1 505
expression on fully functional memory T cells after spontaneous resolution of 506
hepatitis C virus infection. Journal of virology 82:5109-5114. 507
25. Allie SR, Zhang W, Fuse S, Usherwood EJ. 2011. Programmed death 1 508
regulates development of central memory CD8 T cells after acute viral infection. 509
J Immunol 186:6280-6286. 510
26. Telcian AG, Laza-Stanca V, Edwards MR, Harker JA, Wang H, Bartlett NW, 511
Mallia P, Zdrenghea MT, Kebadze T, Coyle AJ, Openshaw PJ, Stanciu LA, 512
on April 4, 2019 by guest
http://jvi.asm.org/
Dow
nloaded from

23
Johnston SL. 2011. RSV-induced bronchial epithelial cell PD-L1 expression 513
inhibits CD8+ T cell nonspecific antiviral activity. J Infect Dis 203:85-94. 514
27. Erickson JJ, Gilchuk P, Hastings AK, Tollefson SJ, Johnson M, Downing 515
MB, Boyd KL, Johnson JE, Kim AS, Joyce S, Williams JV. 2012. Viral acute 516
lower respiratory infections impair CD8+ T cells through PD-1. J Clin Invest 517
122:2967-2982. 518
28. Ioannidis I, McNally B, Willette M, Peeples ME, Chaussabel D, Durbin JE, 519
Ramilo O, Mejias A, Flano E. 2012. Plasticity and Virus Specificity of the Airway 520
Epithelial Cell Immune Response during Respiratory Virus Infection. J Virol 521
86:5422-5436. 522
29. Probst HC, McCoy K, Okazaki T, Honjo T, van den Broek M. 2005. Resting 523
dendritic cells induce peripheral CD8+ T cell tolerance through PD-1 and CTLA-524
4. Nat Immunol 6:280-286. 525
30. Keir ME, Freeman GJ, Sharpe AH. 2007. PD-1 regulates self-reactive CD8+ T 526
cell responses to antigen in lymph nodes and tissues. J Immunol 179:5064-5070. 527
31. Martin-Orozco N, Wang YH, Yagita H, Dong C. 2006. Cutting Edge: 528
Programmed death (PD) ligand-1/PD-1 interaction is required for CD8+ T cell 529
tolerance to tissue antigens. J Immunol 177:8291-8295. 530
32. Wei S, Shreiner AB, Takeshita N, Chen L, Zou W, Chang AE. 2008. Tumor-531
induced immune suppression of in vivo effector T-cell priming is mediated by the 532
B7-H1/PD-1 axis and transforming growth factor beta. Cancer research 68:5432-533
5438. 534
33. Wahl C, Bochtler P, Chen L, Schirmbeck R, Reimann J. 2008. B7-H1 on 535
hepatocytes facilitates priming of specific CD8 T cells but limits the specific recall 536
of primed responses. Gastroenterology 135:980-988. 537
34. Goldberg MV, Maris CH, Hipkiss EL, Flies AS, Zhen L, Tuder RM, Grosso 538
JF, Harris TJ, Getnet D, Whartenby KA, Brockstedt DG, Dubensky TW, Jr., 539
Chen L, Pardoll DM, Drake CG. 2007. Role of PD-1 and its ligand, B7-H1, in 540
early fate decisions of CD8 T cells. Blood 110:186-192. 541
35. Lafon M, Megret F, Meuth SG, Simon O, Velandia Romero ML, Lafage M, 542
Chen L, Alexopoulou L, Flavell RA, Prehaud C, Wiendl H. 2008. Detrimental 543
contribution of the immuno-inhibitor B7-H1 to rabies virus encephalitis. J Immunol 544
180:7506-7515. 545
36. Schreiner B, Mitsdoerffer M, Kieseier BC, Chen L, Hartung HP, Weller M, 546
Wiendl H. 2004. Interferon-beta enhances monocyte and dendritic cell 547
expression of B7-H1 (PD-L1), a strong inhibitor of autologous T-cell activation: 548
relevance for the immune modulatory effect in multiple sclerosis. Journal of 549
neuroimmunology 155:172-182. 550
37. Heinecke L, Proud D, Sanders S, Schleimer RP, Kim J. 2008. Induction of B7-551
H1 and B7-DC expression on airway epithelial cells by the Toll-like receptor 3 552
agonist double-stranded RNA and human rhinovirus infection: In vivo and in vitro 553
studies. J Allergy Clin Immunol 121:1155-1160. 554
38. Tsuda M, Matsumoto K, Inoue H, Matsumura M, Nakano T, Mori A, Azuma 555
M, Nakanishi Y. 2005. Expression of B7-H1 and B7-DC on the airway epithelium 556
on April 4, 2019 by guest
http://jvi.asm.org/
Dow
nloaded from

24
is enhanced by double-stranded RNA. Biochem Biophys Res Commun 330:263-557
270. 558
39. Brown KE, Freeman GJ, Wherry EJ, Sharpe AH. 2010. Role of PD-1 in 559
regulating acute infections. Curr Opin Immunol 22:397-401. 560
40. Channappanavar R, Twardy BS, Suvas S. 2012. Blocking of PDL-1 interaction 561
enhances primary and secondary CD8 T cell response to herpes simplex virus-1 562
infection. PLoS ONE 7:e39757. 563
41. Duraiswamy J, Ibegbu CC, Masopust D, Miller JD, Araki K, Doho GH, Tata 564
P, Gupta S, Zilliox MJ, Nakaya HI, Pulendran B, Haining WN, Freeman GJ, 565
Ahmed R. 2011. Phenotype, function, and gene expression profiles of 566
programmed death-1(hi) CD8 T cells in healthy human adults. J Immunol 567
186:4200-4212. 568
42. Talay O, Shen CH, Chen L, Chen J. 2009. B7-H1 (PD-L1) on T cells is required 569
for T-cell-mediated conditioning of dendritic cell maturation. Proc Natl Acad Sci U 570
S A 106:2741-2746. 571
43. Yao S, Wang S, Zhu Y, Luo L, Zhu G, Flies S, Xu H, Ruff W, Broadwater M, 572
Choi IH, Tamada K, Chen L. 2009. PD-1 on dendritic cells impedes innate 573
immunity against bacterial infection. Blood 113:5811-5818. 574
44. Rowe JH, Johanns TM, Ertelt JM, Way SS. 2008. PDL-1 blockade impedes T 575
cell expansion and protective immunity primed by attenuated Listeria 576
monocytogenes. J Immunol 180:7553-7557. 577
45. Iwai Y, Terawaki S, Ikegawa M, Okazaki T, Honjo T. 2003. PD-1 inhibits 578
antiviral immunity at the effector phase in the liver. J Exp Med 198:39-50. 579
46. Kirchberger S, Majdic O, Steinberger P, Bluml S, Pfistershammer K, 580
Zlabinger G, Deszcz L, Kuechler E, Knapp W, Stockl J. 2005. Human 581
rhinoviruses inhibit the accessory function of dendritic cells by inducing 582
sialoadhesin and B7-H1 expression. J Immunol 175:1145-1152. 583
584
585
on April 4, 2019 by guest
http://jvi.asm.org/
Dow
nloaded from

25
FIGURE LEGENDS 586
Figure 1. PDL-1 is constitutively expressed in the human airway epithelium and is 587
up-regulated by influenza infection in HAEs. (Panel A) mRNA expression of PDL-1 588
in mock treated HAE cultures or in HAE cultures 24 h post exposure to 2x105 PFU 589
influenza virus (Udorn strain). (Panel B) Relative mRNA levels of PDL-1 in Influenza 590
infected HAE cells validated by qRT-PCR. (Panel C) Immunofluorescent analysis of 591
PDL-1 expression (red) in a paraffin embedded human trachea section. The box inset 592
represents a high magnification detail of the apical side of the epithelium. (Panel D) 593
Immunofluorescent analysis of PDL-1 expression (red) in paraffin embedded HAE 594
cultures either mock treated (upper panel) or infected with influenza virus (lower panel). 595
In all immunofluorescent images, the nucleus is counterstained with DAPI (blue). Error 596
bars indicate SD, (*** p ≤ 0.0005). 597
598
Figure 2. IFNAR signaling regulates PDL-1 expression in TEC cultures. (Panel A) 599
PDL-1 mRNA expression analyzed in TEC cultures derived from balb/c (wt) and IFNAR-600
/- mice 24 h after +/- exposure to 2x105 PFU influenza virus (WSN). (Panels B & C) Cell 601
surface expression of PDL-1 on wt or IFNAR-/- TEC cultures infected with 0.1 PFU/cell 602
or 1.0 PFU/cell Influenza virus 24 and 60 hour post infection. Panel B: Representative 603
histograms. Panel C: Median fluorescent intensity (MFI) in which values used for the bar 604
graph represent the MFI of PD1L minus the MFI of the isotype control. All panels are 605
representative of 2-3 separate experiments in which TEC cultures were performed in 606
triplicate. Error bars indicate SD, (*** p ≤ 0.0005). 607
608
on April 4, 2019 by guest
http://jvi.asm.org/
Dow
nloaded from

26
609
Figure 3. Influenza virus infection induces early, non-specific recruitment of PD-1+ 610
effector T cells into the lung airways. Cohorts of naïve C57BL/6 mice were infected 611
with influenza virus X31 and the T cell phenotype was assessed at the indicated time 612
points. Representative dot plots show the frequency of PD-1+ CD44+ T cells present in 613
the CD4 and CD8 subpopulations of the BAL (Panel A) and Lung parenchyma (Panel B) 614
at the indicated time points after infection. Bar diagrams show the number of antigen 615
experienced PD1+ CD4 & PD-1+ CD8 T cells present in the BAL (Panel C), and the lung 616
parenchyma (Panel D). Data are representative of 3 independent experiments with 3 617
mice per group, error bars indicate SD, (* p ≤ 0.05, ** p ≤ 0.005). 618
619
Figure 4. PD-1 is mostly expressed on effector CD8 T cells. Cohorts of mice 620
previously infected with influenza virus X31 received intranasal administration of 50 ug 621
poly (I:C). (Panel A) Representative dot plots and histograms show PD-1 expression on 622
effector and memory T cell subsets within NP tetramer positive and negative CD8 T cell 623
populations in the lung parenchyma 3 days post stimulation. Bar diagrams show the 624
MFI and frequency of PD-1+ effector (CD43+CD62L-), effector-memory (CD43-CD62L-) 625
and central-memory (CD43-CD62L+) CD8 T cells residing in the NP366–374/Db tetramer-626
positive subset (Panel B) and in the tetramer-negative subset (Panel C). Data are 627
representative of 2 independent experiments with 3 mice per group. Error bars indicate 628
SD, (*** p ≤ 0.0005). 629
630
on April 4, 2019 by guest
http://jvi.asm.org/
Dow
nloaded from

27
Figure 5. PDL-1 blockade on airway epithelial cells enhances CD8 T cell function. 631
T cells isolated from the spleens of influenza-infected mice were co-cultured with TEC 632
cells infected or not with influenza virus WSN in the presence of anti-PDL-1 blocking 633
antibody or control IgG antibody. 36 hours after co-culture, supernatants were 634
harvested for the determination of granzyme B secretion by ELISA. Data are combined 635
from 2 separate experiments employing triplicate wells per group. Error bars indicate 636
SD, (* p ≤ 0.05). 637
638
Figure 6. PDL-1 blockade increases the concentration of granzyme B and IFNγ in 639
the airways during recall. Influenza-memory mice were secondarily challenged with 640
heterosubtypic influenza virus PR8, and intra-peritoneally treated with anti-mouse PDL-641
1 blocking antibody or rat IgG control. 3 days post-infection, BAL samples were assayed 642
for the presence of granzyme B (Panel A) or interferon-γ (Panel B) by ELISA. Data are 643
representative of 3 independent experiments (3 mice per group). Error bars indicate SD, 644
(* p ≤ 0.05). 645
646
Figure 7. Intranasal PDL-1 blockade enhances the secretion of granzyme B and 647
IFNγ in the airways during influenza virus recall. Influenza virus memory mice were 648
challenged with influenza virus PR8 (or mock challenged with HBSS), and treated 649
intranasally with anti-mouse PDL-1 blocking antibody or IgG control or HBSS (Flu). 650
ELISA was used to determine the levels of Granzyme B or IFN� present in the BAL on 651
days 3 & 5 post PDL-1 blockade (panels A-D). One-way ANOVA was used to compare 652
on April 4, 2019 by guest
http://jvi.asm.org/
Dow
nloaded from

28
the differences between means, and when these differences were significant (p ≤ 0.05), 653
the Tukey test was used to correct for multiple comparisons and to generate the 654
multiplicity adjusted P values shown. (Panel A) Granzyme B, day 3 (ANOVA, **p<.01); 655
(Panel B) IFNγ, day 3 (ANOVA, **p<.01); (Panel C) Granzyme B, day 5 (ANOVA, 656
**p<.01); (Panel D) IFNγ, day 5 (ANOVA, ****p<.0001). (Panel E) Frequency of IFNγ 657
and CD107ab-expressing NP366–374-specific CD8 T cells in lung. (Panel F) Number of 658
IFNγ and CD107ab expressing NP366–374-specific CD8 T cells in lung. Data are 659
representative of 2-4 independent experiments (3 mice per group). Error bars indicate 660
SD, (* p ≤ 0.05). 661
662
Figure 8. Reduced viral load in anti-PDL-1 treated mice. Influenza memory mice (30-663
40 days post intranasal inoculation with the Influenza X-31 strain) were intranasally 664
challenged with 1x102 PFU/ml influenza virus PR8 (or mock challenged with HBSS) and 665
treated with intra-nasal instillations of anti-mouse PDL-1 blocking antibody, rat IgG or 666
HBSS (Flu). On days 3 and 5 after initiation of PDL-1 blockade, influenza virus load in 667
lung homogenates was determined by qRT-PCR for the NP transcript (left panels) and 668
by plaque assay (right panels). (Panel A) Day 3. (Panel B) Day 5. Values for plaque 669
assay represent the means (+/- SD) of RSV log10 PFU/ml where 1.7 log10 PFU/ml 670
represents the limit of detection. Comparisons were made by one-way ANOVA 671
(****p<0.0001 for all panels), and the Tukey post-test was used to correct for multiple 672
comparisons and to generate the multiplicity adjusted P values shown. Data are 673
on April 4, 2019 by guest
http://jvi.asm.org/
Dow
nloaded from

29
representative of at least 2 independent experiments (3 mice per group). Error bars 674
indicate SD, (* p ≤ 0.05). 675
676
Figure 9. Intranasal PDL-1 blockade prevents influenza-associated weight loss. 677
Percent weight loss in mice inoculated and treated as in Figure 8 was determined by 678
dividing the average starting weight of each group (day 0, pre inoculation) by the 679
average weight of each treatment group on day 5 post-infection. Data are 680
representative of 3 independent experiments with 3 mice per group each. Error bars 681
indicate SD, (* p ≤ 0.05, *** p ≤0.0005). 682
on April 4, 2019 by guest
http://jvi.asm.org/
Dow
nloaded from

0
10
20
30
40
50
60
Mock Flu
Mock
Flu
A
D
54.3x
***
0
2000
4000
6000
8000
10000
Mock Flu
PD
-L1 R
ela
tive e
xpre
ssio
n
B
1 PD
-L1 R
ela
tive e
xpre
ssio
n 89.9x
***
1
C
Figure 1. PDL-1 is cons titu tive ly expres s ed in the human a irway ep ithe lium and is up-regula ted by influenza infec tion in HAEs . (Pane l A)
mRNA expression of PDL-1 in mock treated HAE cultures or in HAE cultures
24 h post exposure to 2x105 PFU influenza virus (Udorn strain). (Pane l B)
Relative mRNA levels of PDL-1 in Influenza infected HAE cells validated by
qRT-PCR. (Pane l C) Immunofluorescent analysis of PDL-1 expression (red) in
a paraffin embedded human trachea section. The box inset represents a high
magnification detail of the apical side of the epithelium. (Pane l D)
Immunofluorescent analysis of PDL-1 expression (red) in paraffin embedded
HAE cultures either mock treated (upper panel) or infected with influenza virus
(lower panel). In all immunofluorescent images, the nucleus is counterstained
with DAPI (blue). Error bars indicate SD, (*** p ≤ 0.0005).
on April 4, 2019 by guest
http://jvi.asm.org/
Dow
nloaded from

0
1000
2000
3000
4000
5000
6000
27.6x
*** A
C
B
Isotype control
wt
mock
0.1 PFU / cell 24h
1 PFU / cell 24h
0.1 PFU / cell 60h
1 PFU / cell 60h
0 10 2 10 3 10 4 10 5 0
20
40
60
80
100
0 10 2 10 3 10 4 10 5 0
20
40
60
80
100
0 10 2 10 3 10 4 10 5 0
20
40
60
80
100
0 10 2 10 3 10 4 10 5 0
20
40
60
80
100
0 10 2 10 3 10 4 10 5 0
20
40
60
80
100
0 10 2 10 3 10 4 10 5 0
20
40
60
80
100
0 10 2 10 3 10 4 10 5 0
20
40
60
80
100
0 10 2 10 3 10 4 10 5 0
20
40
60
80
100
0 10 2 10 3 10 4 10 5
0
20
40
60
80
100
0 10 2 10 3 10 4 10 5 0
20
40
60
80
100
PDL-1
IFNAR -/-
-50
0
50
100
150
200
250
mock
0.1
PF
U /
cell
24h
0.1
PF
U /
cell
60h
1.0
PF
U /
cell
24h
1.0
PF
U /
cell
60h
PD
L-1
MF
I
IFNAR-/-
wt
Wt m
ock
Wt F
lu
IFN
AR
-/-
Flu
IFN
AR
-/-
mock P
D-L
1 R
ela
tive e
xpre
ssio
n
Figure 2. IFNAR signaling regulates PDL-1 expression in TEC cultures.
(Panel A) PDL-1 mRNA expression analyzed in TEC cultures derived from
balb/c (wt) and IFNAR-/- mice 24 h after +/- exposure to 2x105 PFU influenza
virus (WSN). (Panels B & C ) Cell surface expression of PDL-1 on wt or
IFNAR-/- TEC cultures infected with 0.1 PFU/cell or 1.0 PFU/cell Influenza
virus 24 and 60 hour post infection. Panel B: Representative histograms.
Panel C: Median fluorescent intensity (MFI) in which values used for the bar
graph represent the MFI of PD1L minus the MFI of the isotype control. All
panels are representative of 2-3 separate experiments in which TEC cultures
were performed in triplicate. Error bars indicate SD, (*** p ≤ 0.0005).
on April 4, 2019 by guest
http://jvi.asm.org/
Dow
nloaded from

Figure 3. Influenza virus infection induces early, non-specific
recruitm ent of PD-1+ effector T cells into the lung airw ays. Cohorts of
naïve C57BL/6 mice were infected with influenza virus X31 and the T cell
phenotype was assessed at the indicated time points. Representative dot
plots show the frequency of PD-1+ CD44+ T cells present in the CD4 and
CD8 subpopulations of the BAL (Panel A) and Lung parenchyma (Panel
B) at the indicated time points after infection. Bar diagrams show the
number of antigen experienced PD1+ CD4 & PD-1+ CD8 T cells present
in the BAL (Panel C), and the lung parenchyma (Panel D ). Data are
representative of 3 independent experiments with 3 mice per group, error
bars indicate SD, (* p ≤ 0.05, ** p ≤ 0.005).
PD
-1
CD44
PD
-1
CD44
BAL
CD8
CD4
d0 d1 d3 d7
A
Lung
CD8
CD4
d0 d1 d3 d7
B
BAL
Lung
C
D
1
10
100
1000
10000
100000
1
10
100
1000
10000
100000
1
10
100
1000
10000
100000
1000000
1
10
100
1000
10000
100000
*
* *
* ** **
*
*
PD
-1+
CD
8 T
cells
1 3 7 12 30 0 1 3 7 12 30 0
1 3 7 12 30 0 1 3 7 12 30 0
day day
PD
-1+
CD
4 T
cells
PD
-1+
CD
8 T
cells
PD
-1+
CD
4 T
cells
on April 4, 2019 by guest
http://jvi.asm.org/
Dow
nloaded from
0 10 2 10 3 10 4 10 5
CD8
0
10 2
10 3
10 4
10 5
NP
te
tram
er
25.8
1.35
0 10 2 10 3 10 4 10 5
0
10 2
10 3
10 4
10 5 5.41 4.39
37.5 52.7
0 10 2 10 3 10 4 10 5
CD43
0
10 2
10 3
10 4
10 5
CD
62L
61.5 1.89
9.81 26.7
0 10 2 10 3 10 4 10 5
0
20
40
60
80
100
0 10 2 10 3 10 4 10 5
PD-1
0
20
40
60
80
100
% o
f M
ax
CD43- CD62L+
CD43- CD62L-
CD43+ CD62L- 0
500
1000
1500
2000
2500
3000
3500
MF
I
PD
-1
0
20
40
60
80
100
Fre
quency
P
D-1
0
1000
2000
3000
4000
5000
0
20
40
60
80
100 A B
Fre
quency
P
D-1
CD
62L
MF
I
PD
-1
CD43
% o
f M
ax
PD-1
C
*** ***
*** ***
Figure 4. PD-1 is m ostly expressed on effector CD8 T cells. Cohorts of
mice previously infected with influenza virus X31 received intranasal
administration of 50 mg poly (I:C). (Panel A) Representative dot plots and
histograms show PD-1 expression on effector and memory T cell subsets
within NP tetramer positive and negative CD8 T cell populations in the lung
parenchyma 3 days post stimulation. Bar diagrams show the MFI and
frequency of PD-1+ effector (CD43+CD62L-), effector-memory (CD43-CD62L-)
and central-memory (CD43-CD62L+) CD8 T cells residing in the NP366–374/Db
tetramer-positive subset (Panel B) and in the tetramer-negative subset (Panel
C ). Data are representative of 2 independent experiments with 3 mice per
group. Error bars indicate SD, (*** p ≤ 0.0005).
on April 4, 2019 by guest
http://jvi.asm.org/
Dow
nloaded from

0
50
100
150
200
250
300
control aPDL1
Gra
nzym
e B
(p
g/m
l)
*
control aPDL1
Influenza Mock
Figure 5. PDL-1 blockade on airw ay epithelial cells enhances CD8 T cell
function. T cells isolated from the spleens of influenza-infected mice were co-
cultured with TEC cells infected or not with influenza virus WSN in the
presence of anti-PDL-1 blocking antibody or control IgG antibody. 36 hours
after co-culture, supernatants were harvested for the determination of
granzyme B secretion by ELISA. Data are combined from 2 separate
experiments employing triplicate wells per group. Error bars indicate SD, (* p ≤ 0.05).
on April 4, 2019 by guest
http://jvi.asm.org/
Dow
nloaded from

0
200
400
600
800
1000
1200
1400
1600
IgG aPDL-1
Gra
nzym
e B
(p
g/m
L)
A
B
0
500
1000
1500
2000
2500
IgG aPDL-1
IFN
g (p
g/m
L)
*
*
Figure 6. PDL-1 blockade increases the concentration of granzym e B and
IFNg in the airw ays during recall. Influenza-memory mice were secondarily
challenged with heterosubtypic influenza virus PR8, and intra-peritoneally
treated with anti-mouse PDL-1 blocking antibody or rat IgG control. 3 days
post-infection, BAL samples were assayed for the presence of granzyme B
(Panel A) or interferon-g (Panel B) by ELISA. Data are representative of 3
independent experiments (3 mice per group). Error bars indicate SD, (* p ≤ 0.05).
on April 4, 2019 by guest
http://jvi.asm.org/
Dow
nloaded from

0
400
800
1200
1600
Mock Flu IgG aPDL1
0
2000
4000
6000
8000
Mock Flu IgG aPDL1 Gra
nzym
e B
(pg/m
l) *
*
A
C
0
400
800
1200
1600
Mock Flu IgG aPDL1
IFN
g (p
g/m
L)
*
IFN
g (p
g/m
L)
0
2000
4000
6000
8000
Mock Flu IgG aPDL1 Gra
nzym
e B
(pg/m
l)
B
D
CD10
7ab
CD10
7ab/
IFNg
IFNg
0
2
4
6
IgG
PDL-1
% C
D8 T
cells
#
CD
8 T
cells
E F
*
*
0
500
1000
1500
* *
*** ****
Figure 7. Intranasal PDL-1 blockade enhances the secretion of granzym e
B and IFNg in the airw ays during influenza virus recall. Influenza virus
memory mice were challenged with influenza virus PR8 (or mock challenged
with HBSS), and treated intranasally with anti-mouse PDL-1 blocking antibody
or IgG control or HBSS (Flu). ELISA was used to determine the levels of
Granzyme B or IFNg present in the BAL on days 3 & 5 post PDL-1 blockade
(panels A-D). One-way ANOVA was used to compare the differences between
means, and when these differences were significant (p ≤ 0.05), the Tukey test
was used to correct for multiple comparisons and to generate the multiplicity
adjusted P values shown. (Panel A) Granzyme B, day 3 (ANOVA, **p<.01);
(Panel B) IFNg, day 3 (ANOVA, **p<.01); (Panel C ) Granzyme B, day 5
(ANOVA, **p<.01); (Panel D ) IFNg, day 5 (ANOVA, ****p<.0001). (Panel E)
Frequency of IFNg and CD107ab-expressing NP366–374-specific CD8 T cells in
lung. (Panel F) Number of IFNg and CD107ab expressing NP366–374-specific
CD8 T cells in lung. Data are representative of 2-4 independent experiments (3
mice per group). Error bars indicate SD, (* p ≤ 0.05).
on April 4, 2019 by guest
http://jvi.asm.org/
Dow
nloaded from

A
B
0
5000
10000
15000
20000
25000
Mock Flu IgG aPDL1
d 3
NP
RN
A
Re
lative E
xpre
ssio
n
0 200 400 600 800
1000 1200 1400 1600 1800
Mock Flu IgG aPDL1
d 5
NP
RN
A
Re
lative E
xpre
ssio
n
**** ****
1
2
4
8
Mock Flu IgG PD1L
d3 V
iral lo
ad
Log
10 P
FU
/ml
1
2
4
8
Mock Flu IgG PD1L
d5 V
iral lo
ad
Log
10 P
FU
/ml ****
****
Figure 8. Reduced viral load in anti-PDL-1 treated m ice. Influenza memory
mice (30-40 days post intranasal inoculation with the Influenza X-31 strain)
were intranasally challenged with 1x102 PFU/ml influenza virus PR8 (or mock
challenged with HBSS) and treated with intra-nasal instillations of anti-mouse
PDL-1 blocking antibody, rat IgG or HBSS (Flu). On days 3 and 5 after
initiation of PDL-1 blockade, influenza virus load in lung homogenates was
determined by qRT-PCR for the NP transcript (left panels) and by plaque
assay (right panels). (Panel A) Day 3. (Panel B) Day 5. Values for plaque
assay represent the means (+/- SD) of RSV log10 PFU/ml where 1.7 log10
PFU/ml represents the limit of detection. Comparisons were made by one-way
ANOVA (****p<0.0001 for all panels), and the Tukey post test was used to
correct for multiple comparisons and to generate the multiplicity adjusted P
values shown. Data are representative of at least 2 independent experiments
(3 mice per group). Error bars indicate SD, (* p ≤ 0.05).
on April 4, 2019 by guest
http://jvi.asm.org/
Dow
nloaded from

0
20
40
60
80
100
120
Mock Flu IgG PDL1
% s
tart
ing w
eig
ht
day 0
day 5
* ***
Figure 9. Intranasal PDL-1 blockade prevents influenza-associated
w eight loss. Percent weight loss in mice inoculated and treated as in Figure 8
was determined by dividing the average starting weight of each group (day 0,
pre inoculation) by the average weight of each treatment group on day 5 post-
infection. Data are representative of 3 independent experiments with 3 mice
per group each. Error bars indicate SD, (* p ≤ 0.05, *** p ≤0.0005).
on April 4, 2019 by guest
http://jvi.asm.org/
Dow
nloaded from





























