Embed Size (px)
Citation preview

Translational Cancer Mechanisms and Therapy
Local Phototherapy Synergizes withImmunoadjuvant for Treatment of PancreaticCancer through Induced Immunogenic TumorVaccineFeifan Zhou1,2, Jingxuan Yang3, Yuqing Zhang3, Mingyang Liu3, Mark L. Lang4,Min Li3, and Wei R. Chen1,2
Abstract
Purpose: To develop a synergistic combination therapyfor advanced pancreatic cancer, using local phototherapy andimmunotherapy, and to determine the efficacy and mecha-nism of the novel combination therapy using a highly met-astatic pancreatic tumor model in mice.
Experimental Design: Mice bearing Panc02-H7 pancreatictumors (both subcutaneous and orthotopic) were treatedwithnoninvasive or interventional photothermal therapy, fol-lowed by local application of an immunoadjuvant. Tumorgrowth and animal survival were assessed. Immune cell popu-lations within spleen and tumors were evaluated by FACS andIHC, and cytokine levels were determined by ELISA.
Results: Up to 75% of mice bearing subcutaneous tumorstreated with combination therapy had complete tumor regres-sion. Local photothermal therapy exposed/released damage-associated molecular patterns, which initiated an immuno-
genic tumor cell death, resulting in infiltration of antigen-presenting cells and Th1 immunity. Concomitant applicationof immunoadjuvant amplified Th1 immunity, especially thetumor-specific cytotoxic T lymphocyte response, withincreased quantity andquality of T cells. Combination therapyalso induced tumor-specific immunememory, as demonstrat-ed by resistance to tumor rechallenge and production ofmemory T cells. For the treatment of orthotopic tumor, thecombination therapy significantly reduced theprimary tumorsand metastases, and prolonged the animal survival time.
Conclusions: This study indicated that combination oflocal phototherapy and immunotherapy induced a systemicimmunity against established tumors and metastases in anaggressive, preclinical pancreatic tumor model, leading to apotential clinical method for patients with advanced pancre-atic cancer. Clin Cancer Res; 24(21); 5335–46. �2018 AACR.
IntroductionPancreatic cancer is one of the deadliest human malignancies
with an 8% 5-year survival rate for all stages (1). Currently,radical surgery is the first treatment option for pancreaticcancer. However, approximately 80% of patients, when diag-
nosed of pancreatic cancer, are at a stage not manageable withcurative surgery (2). More than 50% of patients have distalmetastases at the time of diagnosis, with a poor 5-year survivalrate of 3%. Therefore, the challenge for pancreatic cancertreatment is the control of metastasis. Although some cancertreatment modalities, such as chemotherapy, radiotherapy, andother targeted therapies, are commonly used, little progress hasbeen made in the control of tumor metastasis. To achievesuperior therapeutic effects against pancreatic cancer, new ther-apies have recently been developed, such as photothermaltherapy (PTT) and immunotherapies (3–5).
Immunotherapy has been considered as a promising treat-ment approach for advanced cancers, and several strategieshave been employed in clinical studies, including mAbs, check-point inhibitors, whole-cell vaccine, dendritic cell (DC)–basedvaccine, and chimeric antigen receptor (CAR) T cells (6–9).With the possible exception of mAb-mediated direct tumorablation, these promising treatment strategies have in commonthe requirement to break immunologic tolerance to the tumorand amplify what is often the weak tumor immunogenicity andits associated antigens (10). Furthermore, for successful cancerimmunotherapy, systemic immunity is required to fight againstmetastases and prevent recurrence (11).
Tumor cells undergoing immunogenic cell death (ICD)exhibit superior immunogenic potential by expression/releaseof damage-associated molecular patterns (DAMP), including
1Key Laboratory of Optoelectronic Devices and Systems of Ministry ofEducation and Guangdong Province, College of Optoelectronic Engineering,Shenzhen University, Shenzhen, China. 2Biophotonics Research Laboratory,Center for Interdisciplinary Biomedical Education and Research, College ofMathematics and Science, University of Central Oklahoma, Edmond,Oklahoma. 3Department of Medicine, Department of Surgery, University ofOklahoma Health Sciences Center, Oklahoma City, Oklahoma. 4Departmentof Microbiology and Immunology, University of Oklahoma Health SciencesCenter, Oklahoma City, Oklahoma.
Note: Supplementary data for this article are available at Clinical CancerResearch Online (http://clincancerres.aacrjournals.org/).
F. Zhou and J. Yang contributed equally to this article.
Corresponding Authors: Wei R. Chen, University of Central Oklahoma, 100 N.University Drive, Edmond, OK 73034. Phone: 405-974-5147; Fax: 405-974-3812;E-mail: [email protected]; and Min Li, Stanton L. Young Biomedical ResearchCenter, BRC1262A, 975 NE 10th Street, Oklahoma City, OK 73104. Phone: 405-271-1796; Fax: 405-271-1766; E-mail: [email protected]
doi: 10.1158/1078-0432.CCR-18-1126
�2018 American Association for Cancer Research.
ClinicalCancerResearch
www.aacrjournals.org 5335
on April 16, 2020. © 2018 American Association for Cancer Research. clincancerres.aacrjournals.org Downloaded from
Published OnlineFirst August 1, 2018; DOI: 10.1158/1078-0432.CCR-18-1126

heat shock proteins (HSPs), calreticulin (CRT), high-mobilitygroup box 1 (HMGB1) protein, and adenosine triphosphate(ATP) (12, 13). Tumor-derived DAMPs act as potent dangersignals to operate a series of receptors expressed by DCs,stimulating a Th1-biased immune response (14, 15). PTT isa targeted, minimally invasive treatment strategy. Absorption ofPTT-associated photon energy generates significant thermaldamage in targeted cells (16). Cancer cells generally are moresensitive to heat-induced damage than normal cells. Elevatingthe temperature in tumor tissue can cause cell stress or death,leading to the release of tumor antigens and DAMPs to breakimmunologic tolerance (17).
Immunoadjuvants are often used to modify or augment theeffects of cancer vaccine by stimulating the immune system torespond to the vaccine more vigorously, and thus enhancingadaptive antitumor immune response (18). The commonlyused aluminum salts (alum), a potent immunoadjuvant withwide clinical applications, promotes humoral immunity andTh 2 cell responses, with limited ability to efficiently drive Th1and cytotoxic T lymphocyte (CTL) responses (19, 20). Somenew adjuvants have been developed to enhance antitumor Th1-skewed CTL response. Glycated chitosan (GC), a unique immu-noadjuvant, has been used, in combination with PTT, to treatpatients with late-stage, metastatic breast cancer (21). Thecombination treatment reduced and eliminated treated prima-ry tumors and untreated distal metastases in the lungs, andsignificantly prolonged patient survival.
In this study, we investigated the effects of the combination ofPTT and GC on the treatment of pancreatic cancer using a highlymetastatic tumormodel inmice. We also investigated PTTþGC–induced long-term, tumor-specific immunity, through the induc-tion of immunogenic tumor vaccination and increased T-cellinfiltration. This study proved that synergistic photothermal–immunologic effects of PTT þ GC provided a feasible treatmentmodality for metastatic pancreatic cancer, by creating an in situvaccine through a local treatment of primary tumors.
Materials and MethodsCell culture
The mouse pancreatic cancer cell line Panc02 was establishedby implanting 3-methylcholanthrene into the pancreas of aC57BL/6 mouse (22). The Panc02-H7 cell line was derivedfrom Panc02 and displays a more aggressive tissue invasionthan other Panc02 clonal sublines (23). The mouse melanomacell line B16-F10 was purchased from ATCC. All cell lines weretested and verified to be free of Mycoplasma and used aftertwo passages from thawing. All the cells were cultured withDMEM (Gibco), supplemented with 10% FBS (ATCC), peni-cillin (100 U/mL), and streptomycin (100 mg/mL; Sigma), in ahumidified incubator with 5% CO2, 95% air at 37�C (NuAire).
Pancreatic carcinoma mouse modelC57BL/6 mice ages 6 to 8 weeks were purchased from Harlan
Sprague Dawley Co. Mice were housed in the animal facility ofthe Department of Comparative Medicine at the Universityof Oklahoma Health Sciences Center (OUHSC, Oklahoma, OK).All experiments were conducted in compliance with the Guidefor the Care and Use of Laboratory Animals published by theNIH and approved by the OUHSC Institutional Animal Care andUse Committee.
Subcutaneous tumor model. For therapeutic efficacy studies,Panc02-H7 cells (2 � 105 cells in 50 mL PBS) were injected intothe right flank of female C57BL/6 mice. Mice were treated 7 to10 days after tumor cell inoculation, when the tumors reached asize of approximately 300 mm3. To study the long-term tumor-specific immunity and cross-relativities, cured mice were rechal-lenged with Panc02-H7 (2 � 105) or B16-F10 (2 � 105) tumorcells, injected to the untreated flank of the mice. To study theeffects of PTT þ GC on untreated distal tumors, tumor cells(2 � 105) were injected to both right and left flanks of the mice.Then, the mice were treated when the first tumor reached a size ofapproximately 300 mm3, whereas the second tumor was consid-ered as a surrogate of an untreated distal metastasis.
Orthotopic tumor model. An orthotopic model of pancreatic can-cer was established using a surgical procedure (24–26). Briefly,Panc02-H7 cells (5 � 104 in 50 mL medium) were injected intothe tail of pancreas of female or male C57BL/6 mice. The ani-mals were ready for experiments in approximately 7 to 10 dayswhen the pancreatic tumors reached a size of 8 mm (approxi-mately 300 mm3) in diameter.
PTT and GC administrationFor in vitro cell treatment, cultured tumor cells were exposed
to light at a power intensity of 1.0 W/cm2 for 5 minutes from a980-nm semiconductor laser (AngioDynamics, Inc.). The lightwas delivered to the tumor cells using an optical fiber deliverysystem (Pioneer Optics), with a diffusion microlens to deliveruniform light distribution over the cell culture area.
Tumor-bearing mice were anesthetized with 4% isoflurane/96% oxygen using a precision vaporizer during the treatment. Fortumor treatments with noninvasive PTT, subcutaneous tumorswere directly irradiated by laser, whereas orthotopic tumorswere first exposed by surgical and then irradiated by laser. Thepower density at the treatment area was 0.85 W/cm2 and thetreatment duration was 10 minutes. The laser spot encompassedthe tumor plus a 0.5-cm margin surrounding the tumor.
Translational Relevance
Metastasis remains the biggest challenge in cancerresearch, especially with pancreatic cancer. New strategy totrigger systemic antitumor effects to control distal metastaseswill improve outcome for patients with cancer. In this study,we demonstrate that combination of local photothermaltherapy and local application of immunoadjuvant canachieve a high cure rate and a long-term tumor-specificimmunity on metastatic pancreatic tumors in mice, byincreasing the quantity and quality of splenic and tumor-infiltrating antitumor T cells. We also developed interven-tional photothermal therapy, combined with immunoadju-vant, for aggressive orthotopic pancreatic tumors in mice.The one-time treatment successfully removed the primarytumors, reduced the metastases, and prolonged animal sur-vival time. Because this novel combination has been appliedin the clinical trials for patients with late-stage, metastaticmelanoma and breast cancer, the outcomes of this study canbe readily translated to the treatment of patients with pan-creatic cancer, who face severely limited options.
Zhou et al.
Clin Cancer Res; 24(21) November 1, 2018 Clinical Cancer Research5336
on April 16, 2020. © 2018 American Association for Cancer Research. clincancerres.aacrjournals.org Downloaded from
Published OnlineFirst August 1, 2018; DOI: 10.1158/1078-0432.CCR-18-1126

For tumor treatment with interventional PTT, orthotopic tu-mors were first exposed surgically. Then, an optical fiber (PioneerOptics) with a cylindrical active lens (10 mm) was inserted intothe center of the tumor through an 18-gauge percutaneous trans-hepatic cholangiography needle. The tumor was irradiated bythe laser with a power of 1.5 W and a duration of 10 minutes.Sham controls were performed under the same conditions,without laser irradiation.
GC (50 mg/kg; Immunophotonics, Inc.) was injected intra-tumorally immediately after PTT. Tumor volume was estimatedby length (mm) � width (mm)2/2. The mice were euthanizedwhen their tumor volume exceeded 2,000 mm3.
Immunofluorescence assayIndividual tumors were fixed in 4%paraformaldehyde, and cut
into 5-mm-thick sections using a paraffin slicer before stainingwith H&E, TUNEL fluorescence dye (Sigma), or Alexa Fluor 647–conjugated anti-mouse CD8a antibody (BioLegend). Sampleswere analyzed by fluorescence microscopy (Olympus model).
DAMPs expression analysisTo detect DAMP expression in tumor tissue, protein samples
were subjected to SDS-PAGE and transferred to nitrocellulosemembranes using standard methods and commercial reagents(Bio-Rad). To detect DAMP expression on tumor cell surface, cellswere fixed and stained with antibodies and analyzed by Strate-digm S1200Ex flow cytometer (Stratedigm). The FlowJo softwarewas used for data analysis. Extracellular ATP and HMGB1 in theconditioned (serum-free) media that secreted from treated cellswere measured via ATP Bioluminescent Assay Kit (Sigma) andHMGB1 ELISA Kit (R&D systems), following the manufacturer'sinstructions.
Maturation of DCsBone marrow DCs were harvested from C57BL/6 mice using
standard methods and cultured with granulocyte-macrophagecolony-stimulating factor (GM-CSF) (27). DCs were seeded in24-well plates at 5� 105 cells/well, incubatedwith treated Panc02or Panc02-H7 cells (5 � 105) for 24 hours. Supernatants werecollected after coculture for cytokine detection with interferongamma (IFNg) ELISA Kits (R&D Systems).
Detection of immune cell infiltration into tumor tissueTumor tissues were isolated and single-cell suspensions were
prepared by passaging through a cell strainer. Single tumorcells were stained with Alexa Fluor 647 anti-mouse CD11c,PE-CD86, APC anti-mouse CD68, APC/CY7 anti-mouse CD3e,Alexa Fluor 647 anti-mouse CD8a, PE anti-mouse CD4, AlexaFluor 488 anti-mouse IFNg , or Alexa Fluor 647 anti-mouseFoxp3. Alexa Fluor 647 IgG, APC IgG, PE IgG, APC/CY7 IgG,and Alexa Fluor 488 IgG (all from BioLegend) were used ascontrol antibodies.
One day after treatment, infiltrating DCs in treated tumortissue were stained with PE anti-mouse CD11c and sorted byFACSJazz (BD Biosciences). Sorted DCs were cultured with cellsharvested from lymph node at a ratio of 1:5 for 6 days, with orwithout additional GC (50 mg/mL). The cells were tested for thepresence of tumor-specific T cells by staining with Alexa Fluor647 anti-mouse CD8a and Alexa Fluor 488 anti-mouse CD69.Data acquisition was performed on a flow cytometer, and theFlowJo software was used for data analysis.
Splenocyte analysisSpleens and serum were harvested from mice 7 days after
different treatments. For analysis of immune cell population insplenocytes, staining was performed with indicated fluorescenttagged antibodies (all from BioLegend). For analysis of memoryeffects, surface staining was performedwith APC/CY7 anti-mouseCD3e, Alexa Fluor 647 anti-mouse CD8a, PE anti-mouse CD44,and Fluor 488 anti-mouse 62L. Data acquisition was performedon a flow cytometer (Stratedigm) and the FlowJo software wasused for data analysis. For cytotoxic analysis of serum and sple-nocytes on target tumor cells, cell viability was analyzed usingthe CellTiter 96 AQ Assay (Promega).
Statistical analysisValues are expressed as mean � SEM. All the data were
analyzed with a one-way ANOVA followed by Bonferroni post-test. A P value of <0.05 was considered statistically significant.
ResultsEfficacy of PTT þ GC therapy against pancreatic tumors
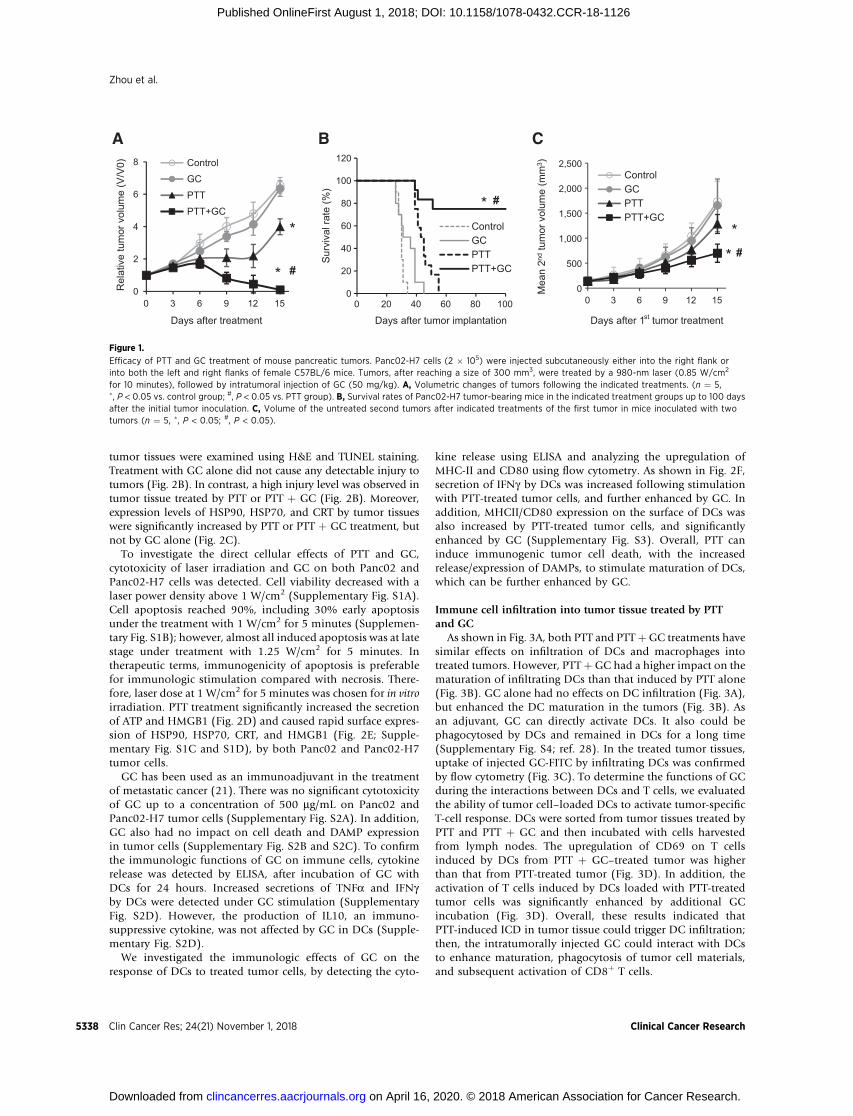
The highly tumorigenic Panc02-H7murine tumor model wasused to evaluate the effects of PTT þGC. After tumors reached asize of approximately 300 mm3, the mice were divided intofour treatment groups: untreated control, GC only, PTT only,and PTT þ GC. Mice treated with GC alone had an averagetumor burden similar to that of untreated control mice, where-as the mice treated with PTT alone had a significant smallertumor burden than that of the control mice (Fig. 1A). However,in the PTT þ GC group, complete eradication of the establishedprimary tumors was achieved within 15 days of treatment(Fig. 1A); in this group, 6 of 12 mice showed a later recurrenceof the primary tumor, which spontaneously regressed in 3 ofthose animals. Survival of the mice was monitored over a100-day period. All untreated mice died within 36 days(Fig. 1B). Mice treated with GC alone or PTT alone died within45 and 55 days, respectively (Fig. 1B). In contrast, 9 of 12 micein the PTT þ GC group survived and remained tumor free forthe 100-day monitoring period (Fig. 1B).
To determine whether PTT þ GC treatment could triggerspecific antitumor responses against untreated metastases, miceinoculated with Panc02-H7 cells on both right and left flankswere used, with one tumor being selected for treatment and thesecond being considered as a untreated surrogate metastasis.Treatment with GC alone did not slow the growth of the secondtumors. Administration of PTT alone to the first tumor signif-icantly slowed the growth of the second tumor. Treatmentwith PTT þ GC also significantly slowed the growth of thesecond tumors, to a greater extent than PTT alone (Fig. 1C).These results suggested that the PTTþGC treatment induced anantitumor immunity that controlled the treated primarytumors as well as untreated distal tumors.
Immunogenic tumor cell death induced by PTTTo investigate the immunologic mechanism of PTT þ GC
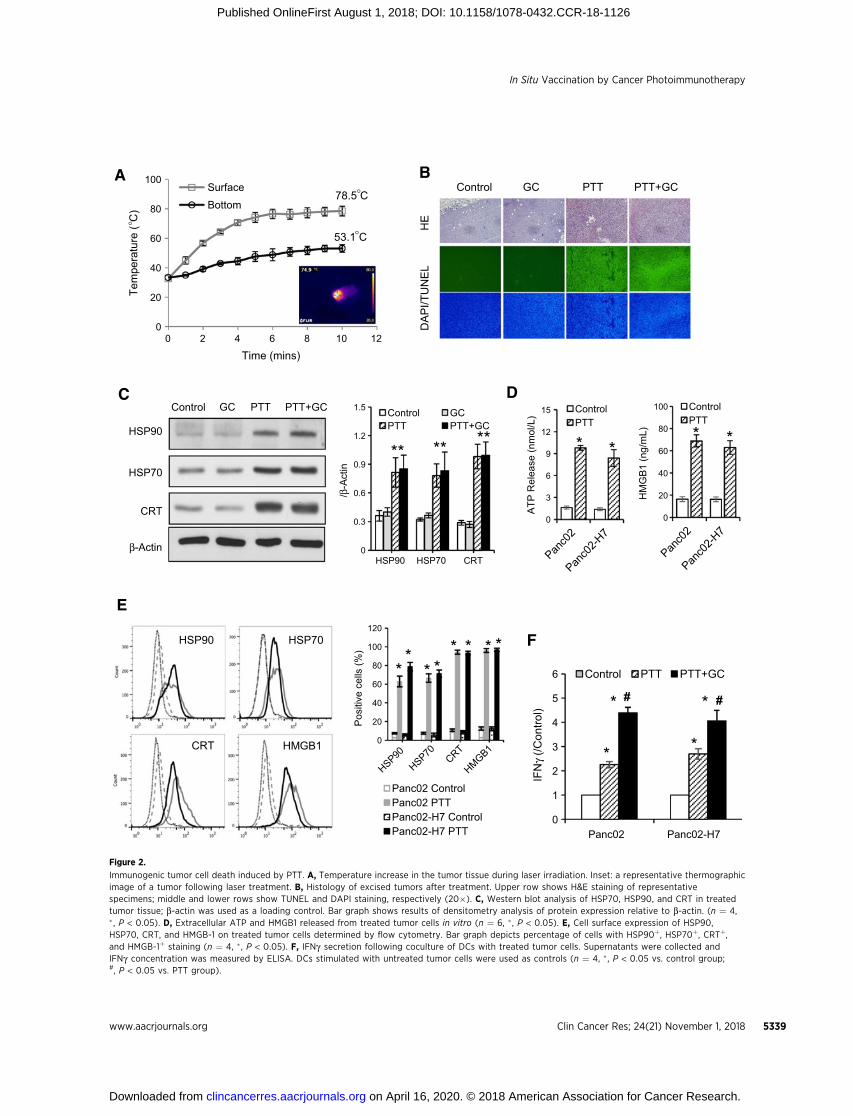
treatment, the direct effects of PTT and GC on tumor cells weredetermined. During PTT treatment, the tumor proximal surfacetemperature increased rapidly and remained stable after 5 min-utes at 75�C; the temperature at the bottom of tumor tissueincreased steadily and reached a stable temperature of 53�C(Fig. 2A). To confirm photothermal cytotoxicity, injury levels in
In Situ Vaccination by Cancer Photoimmunotherapy
www.aacrjournals.org Clin Cancer Res; 24(21) November 1, 2018 5337
on April 16, 2020. © 2018 American Association for Cancer Research. clincancerres.aacrjournals.org Downloaded from
Published OnlineFirst August 1, 2018; DOI: 10.1158/1078-0432.CCR-18-1126
tumor tissues were examined using H&E and TUNEL staining.Treatment with GC alone did not cause any detectable injury totumors (Fig. 2B). In contrast, a high injury level was observed intumor tissue treated by PTT or PTT þ GC (Fig. 2B). Moreover,expression levels of HSP90, HSP70, and CRT by tumor tissueswere significantly increased by PTT or PTT þ GC treatment, butnot by GC alone (Fig. 2C).
To investigate the direct cellular effects of PTT and GC,cytotoxicity of laser irradiation and GC on both Panc02 andPanc02-H7 cells was detected. Cell viability decreased with alaser power density above 1 W/cm2 (Supplementary Fig. S1A).Cell apoptosis reached 90%, including 30% early apoptosisunder the treatment with 1 W/cm2 for 5 minutes (Supplemen-tary Fig. S1B); however, almost all induced apoptosis was at latestage under treatment with 1.25 W/cm2 for 5 minutes. Intherapeutic terms, immunogenicity of apoptosis is preferablefor immunologic stimulation compared with necrosis. There-fore, laser dose at 1 W/cm2 for 5 minutes was chosen for in vitroirradiation. PTT treatment significantly increased the secretionof ATP and HMGB1 (Fig. 2D) and caused rapid surface expres-sion of HSP90, HSP70, CRT, and HMGB1 (Fig. 2E; Supple-mentary Fig. S1C and S1D), by both Panc02 and Panc02-H7tumor cells.
GC has been used as an immunoadjuvant in the treatmentof metastatic cancer (21). There was no significant cytotoxicityof GC up to a concentration of 500 mg/mL on Panc02 andPanc02-H7 tumor cells (Supplementary Fig. S2A). In addition,GC also had no impact on cell death and DAMP expressionin tumor cells (Supplementary Fig. S2B and S2C). To confirmthe immunologic functions of GC on immune cells, cytokinerelease was detected by ELISA, after incubation of GC withDCs for 24 hours. Increased secretions of TNFa and IFNgby DCs were detected under GC stimulation (SupplementaryFig. S2D). However, the production of IL10, an immuno-suppressive cytokine, was not affected by GC in DCs (Supple-mentary Fig. S2D).
We investigated the immunologic effects of GC on theresponse of DCs to treated tumor cells, by detecting the cyto-
kine release using ELISA and analyzing the upregulation ofMHC-II and CD80 using flow cytometry. As shown in Fig. 2F,secretion of IFNg by DCs was increased following stimulationwith PTT-treated tumor cells, and further enhanced by GC. Inaddition, MHCII/CD80 expression on the surface of DCs wasalso increased by PTT-treated tumor cells, and significantlyenhanced by GC (Supplementary Fig. S3). Overall, PTT caninduce immunogenic tumor cell death, with the increasedrelease/expression of DAMPs, to stimulate maturation of DCs,which can be further enhanced by GC.
Immune cell infiltration into tumor tissue treated by PTTand GC
As shown in Fig. 3A, both PTT and PTT þGC treatments havesimilar effects on infiltration of DCs and macrophages intotreated tumors. However, PTT þ GC had a higher impact on thematuration of infiltrating DCs than that induced by PTT alone(Fig. 3B). GC alone had no effects on DC infiltration (Fig. 3A),but enhanced the DC maturation in the tumors (Fig. 3B). Asan adjuvant, GC can directly activate DCs. It also could bephagocytosed by DCs and remained in DCs for a long time(Supplementary Fig. S4; ref. 28). In the treated tumor tissues,uptake of injected GC-FITC by infiltrating DCs was confirmedby flow cytometry (Fig. 3C). To determine the functions of GCduring the interactions between DCs and T cells, we evaluatedthe ability of tumor cell–loaded DCs to activate tumor-specificT-cell response. DCs were sorted from tumor tissues treated byPTT and PTT þ GC and then incubated with cells harvestedfrom lymph nodes. The upregulation of CD69 on T cellsinduced by DCs from PTT þ GC–treated tumor was higherthan that from PTT-treated tumor (Fig. 3D). In addition, theactivation of T cells induced by DCs loaded with PTT-treatedtumor cells was significantly enhanced by additional GCincubation (Fig. 3D). Overall, these results indicated thatPTT-induced ICD in tumor tissue could trigger DC infiltration;then, the intratumorally injected GC could interact with DCsto enhance maturation, phagocytosis of tumor cell materials,and subsequent activation of CD8þ T cells.
A B
0
2
4
6
8
15129630
Rel
ativ
e tu
mor
vol
ume
(V/V
0)
Days after treatment
ControlGC
PTT
PTT+GC
*
#***#
0
500
1,000
1,500
2,000
2,500
15129630
Mea
n 2n
dtu
mor
vol
ume
(mm
3 )
Days after 1st tumor treatment
ControlGCPTTPTT+GC
C
0
20
40
60
80
100
120
100806040200
Sur
viva
l rat
e (%
)Days after tumor implantation
ControlGCPTTPTT+GC
* #
Figure 1.
Efficacy of PTT and GC treatment of mouse pancreatic tumors. Panc02-H7 cells (2 � 105) were injected subcutaneously either into the right flank orinto both the left and right flanks of female C57BL/6 mice. Tumors, after reaching a size of 300 mm3, were treated by a 980-nm laser (0.85 W/cm2
for 10 minutes), followed by intratumoral injection of GC (50 mg/kg). A, Volumetric changes of tumors following the indicated treatments. (n ¼ 5,�, P < 0.05 vs. control group; #, P < 0.05 vs. PTT group). B, Survival rates of Panc02-H7 tumor-bearing mice in the indicated treatment groups up to 100 daysafter the initial tumor inoculation. C, Volume of the untreated second tumors after indicated treatments of the first tumor in mice inoculated with twotumors (n ¼ 5, �, P < 0.05; #, P < 0.05).
Zhou et al.
Clin Cancer Res; 24(21) November 1, 2018 Clinical Cancer Research5338
on April 16, 2020. © 2018 American Association for Cancer Research. clincancerres.aacrjournals.org Downloaded from
Published OnlineFirst August 1, 2018; DOI: 10.1158/1078-0432.CCR-18-1126
A B
C
0
20
40
60
80
100
0 2 4 6 8 10 12
Tem
pera
ture
(°C
)
Time (mins)
SurfaceBottom
53.1 °C
78.5 °CControl GC PTT PTT+GC
DA
PI/T
UN
EL
HE
HSP90
HSP70
CRT
β-Actin
Control GC PTT PTT+GC
0
0.3
0.6
0.9
1.2
1.5
HSP90 HSP70 CRT
/β-A
ctin
Control GCPTT PTT+GC
** ** **
D
E
HSP90 HSP70
CRT HMGB1
0
3
6
9
12
15
ATP
Rel
ease
(nm
ol/L
)
ControlPTT
* *
F
0
1
2
3
4
5
6
Panc02 Panc02-H7
IFN
γ (/C
ontro
l)
Control PTT PTT+GC
#
* *
* *#
0
20
40
60
80
100
120
Pos
itive
cel
ls (%
)
Panc02 ControlPanc02 PTTPanc02-H7 ControlPanc02-H7 PTT
**
* ** * * *
0
20
40
60
80
100
HM
GB
1 (n
g/m
L)
ControlPTT* *
Figure 2.
Immunogenic tumor cell death induced by PTT. A, Temperature increase in the tumor tissue during laser irradiation. Inset: a representative thermographicimage of a tumor following laser treatment. B, Histology of excised tumors after treatment. Upper row shows H&E staining of representativespecimens; middle and lower rows show TUNEL and DAPI staining, respectively (20�). C, Western blot analysis of HSP70, HSP90, and CRT in treatedtumor tissue; b-actin was used as a loading control. Bar graph shows results of densitometry analysis of protein expression relative to b-actin. (n ¼ 4,� , P < 0.05). D, Extracellular ATP and HMGB1 released from treated tumor cells in vitro (n ¼ 6, �, P < 0.05). E, Cell surface expression of HSP90,HSP70, CRT, and HMGB-1 on treated tumor cells determined by flow cytometry. Bar graph depicts percentage of cells with HSP90þ, HSP70þ, CRTþ,and HMGB-1þ staining (n ¼ 4, � , P < 0.05). F, IFNg secretion following coculture of DCs with treated tumor cells. Supernatants were collected andIFNg concentration was measured by ELISA. DCs stimulated with untreated tumor cells were used as controls (n ¼ 4, � , P < 0.05 vs. control group;#, P < 0.05 vs. PTT group).
In Situ Vaccination by Cancer Photoimmunotherapy
www.aacrjournals.org Clin Cancer Res; 24(21) November 1, 2018 5339
on April 16, 2020. © 2018 American Association for Cancer Research. clincancerres.aacrjournals.org Downloaded from
Published OnlineFirst August 1, 2018; DOI: 10.1158/1078-0432.CCR-18-1126
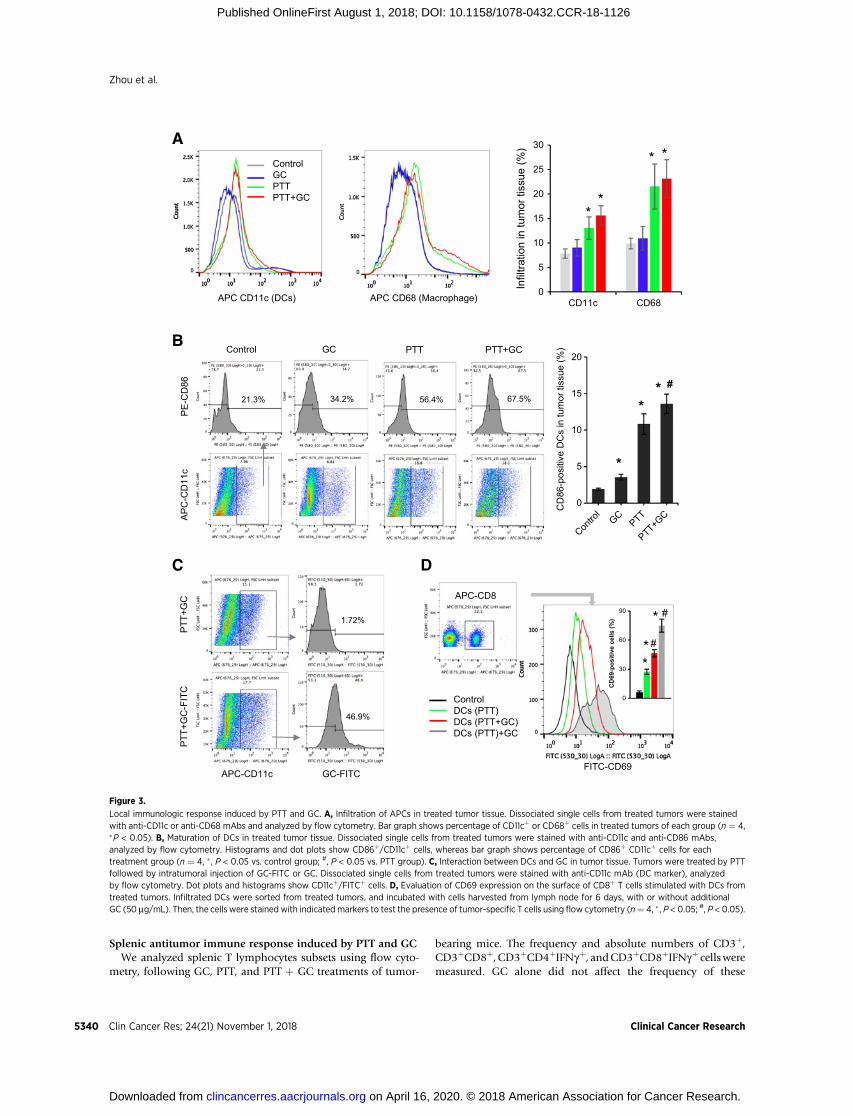
Splenic antitumor immune response induced by PTT and GCWe analyzed splenic T lymphocytes subsets using flow cyto-
metry, following GC, PTT, and PTT þ GC treatments of tumor-
bearing mice. The frequency and absolute numbers of CD3þ,CD3þCD8þ, CD3þCD4þIFNgþ, andCD3þCD8þIFNgþ cellsweremeasured. GC alone did not affect the frequency of these
B
C
PTT+
GC
PTT+
GC
-FIT
C
APC-CD11c GC-FITC
1.72%
46.9%
Control GC PTT PTT+GC
PE
-CD
86A
PC
-CD
11c
21.3% 34.2% 56.4% 67.5%
D
0
5
10
15
20
CD
86-p
ositi
ve D
Cs
in tu
mor
tiss
ue (%
)
*
** #
ControlDCs (PTT)DCs (PTT+GC)DCs (PTT)+GC
FITC-CD69
APC-CD8
0
30
60
90C
D69
-pos
itive
cel
ls (%
)
**
*
#
#
APC CD11c (DCs) APC CD68 (Macrophage)
ControlGCPTTPTT+GC
0
5
10
15
20
25
30
CD11c CD68
Infil
tratio
n in
tum
or ti
ssue
(%)
**
* *A
Figure 3.
Local immunologic response induced by PTT and GC. A, Infiltration of APCs in treated tumor tissue. Dissociated single cells from treated tumors were stainedwith anti-CD11c or anti-CD68 mAbs and analyzed by flow cytometry. Bar graph shows percentage of CD11cþ or CD68þ cells in treated tumors of each group (n ¼ 4,�P < 0.05). B, Maturation of DCs in treated tumor tissue. Dissociated single cells from treated tumors were stained with anti-CD11c and anti-CD86 mAbs,analyzed by flow cytometry. Histograms and dot plots show CD86þ/CD11cþ cells, whereas bar graph shows percentage of CD86þ CD11cþ cells for eachtreatment group (n ¼ 4, � , P < 0.05 vs. control group; #, P < 0.05 vs. PTT group). C, Interaction between DCs and GC in tumor tissue. Tumors were treated by PTTfollowed by intratumoral injection of GC-FITC or GC. Dissociated single cells from treated tumors were stained with anti-CD11c mAb (DC marker), analyzedby flow cytometry. Dot plots and histograms show CD11cþ/FITCþ cells. D, Evaluation of CD69 expression on the surface of CD8þ T cells stimulated with DCs fromtreated tumors. Infiltrated DCs were sorted from treated tumors, and incubated with cells harvested from lymph node for 6 days, with or without additionalGC (50 mg/mL). Then, the cells were stained with indicatedmarkers to test the presence of tumor-specific T cells using flow cytometry (n¼ 4, � , P < 0.05; #, P < 0.05).
Zhou et al.
Clin Cancer Res; 24(21) November 1, 2018 Clinical Cancer Research5340
on April 16, 2020. © 2018 American Association for Cancer Research. clincancerres.aacrjournals.org Downloaded from
Published OnlineFirst August 1, 2018; DOI: 10.1158/1078-0432.CCR-18-1126
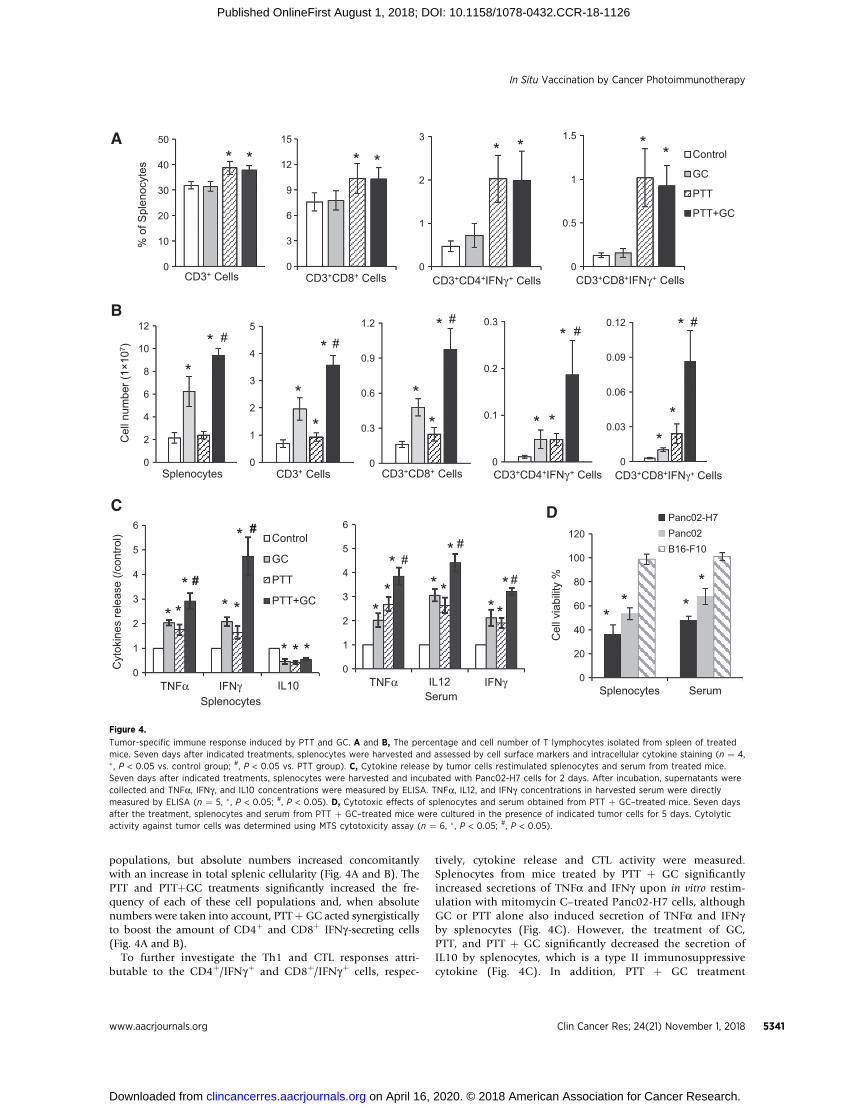
populations, but absolute numbers increased concomitantlywith an increase in total splenic cellularity (Fig. 4A and B). ThePTT and PTTþGC treatments significantly increased the fre-quency of each of these cell populations and, when absolutenumbers were taken into account, PTTþGC acted synergisticallyto boost the amount of CD4þ and CD8þ IFNg-secreting cells(Fig. 4A and B).
To further investigate the Th1 and CTL responses attri-butable to the CD4þ/IFNgþ and CD8þ/IFNgþ cells, respec-
tively, cytokine release and CTL activity were measured.Splenocytes from mice treated by PTT þ GC significantlyincreased secretions of TNFa and IFNg upon in vitro restim-ulation with mitomycin C–treated Panc02-H7 cells, althoughGC or PTT alone also induced secretion of TNFa and IFNgby splenocytes (Fig. 4C). However, the treatment of GC,PTT, and PTT þ GC significantly decreased the secretion ofIL10 by splenocytes, which is a type II immunosuppressivecytokine (Fig. 4C). In addition, PTT þ GC treatment
B
A
0
10
20
30
40
50%
of S
plen
ocyt
es
CD3+ Cells
* *
0
3
6
9
12
15
CD3+CD8+ Cells
* *
0
1
2
3
CD3+CD4+IFNγ+ Cells
* *
0
0.5
1
1.5
CD3+CD8+IFNγ+ Cells
Control
GC
PTT
PTT+GC
* *
0
2
4
6
8
10
12
Cel
l num
ber (
1×10
7 )
Splenocytes
*
#*
0
1
2
3
4
5
CD3+ Cells
*
*
* #
0
0.3
0.6
0.9
1.2
CD3+CD8+ Cells
*
*
* #
0
0.1
0.2
0.3
CD3+CD4+IFNγ+ Cells
* *
* #
0
0.03
0.06
0.09
0.12
CD3+CD8+IFNγ+ Cells
**
* #
C D
0
20
40
60
80
100
120
Splenocytes Serum
Cel
l via
bilit
y %
Panc02-H7Panc02B16-F10
** *
*
0
1
2
3
4
5
6
TNFα IFNγ IL10
Cyt
okin
es re
leas
e (/c
ontro
l)
Splenocytes
Control
GC
PTT
PTT+GC
#
#
* * * *
* * *
*
*
0
1
2
3
4
5
6
TNFα IL12 IFNγSerum
***
* *
*#
#
* *
* #
Figure 4.
Tumor-specific immune response induced by PTT and GC. A and B, The percentage and cell number of T lymphocytes isolated from spleen of treatedmice. Seven days after indicated treatments, splenocytes were harvested and assessed by cell surface markers and intracellular cytokine staining (n ¼ 4,� , P < 0.05 vs. control group; #, P < 0.05 vs. PTT group). C, Cytokine release by tumor cells restimulated splenocytes and serum from treated mice.Seven days after indicated treatments, splenocytes were harvested and incubated with Panc02-H7 cells for 2 days. After incubation, supernatants werecollected and TNFa, IFNg , and IL10 concentrations were measured by ELISA. TNFa, IL12, and IFNg concentrations in harvested serum were directlymeasured by ELISA (n ¼ 5, � , P < 0.05; #, P < 0.05). D, Cytotoxic effects of splenocytes and serum obtained from PTT þ GC–treated mice. Seven daysafter the treatment, splenocytes and serum from PTT þ GC–treated mice were cultured in the presence of indicated tumor cells for 5 days. Cytolyticactivity against tumor cells was determined using MTS cytotoxicity assay (n ¼ 6, � , P < 0.05; #, P < 0.05).
In Situ Vaccination by Cancer Photoimmunotherapy
www.aacrjournals.org Clin Cancer Res; 24(21) November 1, 2018 5341
on April 16, 2020. © 2018 American Association for Cancer Research. clincancerres.aacrjournals.org Downloaded from
Published OnlineFirst August 1, 2018; DOI: 10.1158/1078-0432.CCR-18-1126

also resulted in high levels of TNFa, IL12, and IFNg in serum(Fig. 4C).
To confirm the tumor-specific CTLs generated by PTT þ GCtreatment, cytotoxic effects of splenocytes and serum fromtreated mice were tested against different target tumor cells(Panc02-H7, Panc02, or B16-F10). Splenocytes and serumobtained from PTT þ GC–treated mice markedly reduced theviability of Panc02-H7 cells and parental Panc02 cells(Fig. 4D), whereas the viability of B16-F10 cells was notaffected (Fig. 4D). These results suggested that PTT couldinduce a Th1 cell response to trigger CTLs. Apparently, PTT þGC treatment significantly amplified the antitumor immuneresponse, finally triggering an effective tumor-specific CTL.
T-cell infiltration into secondary tumor induced byPTT þ GC treatment
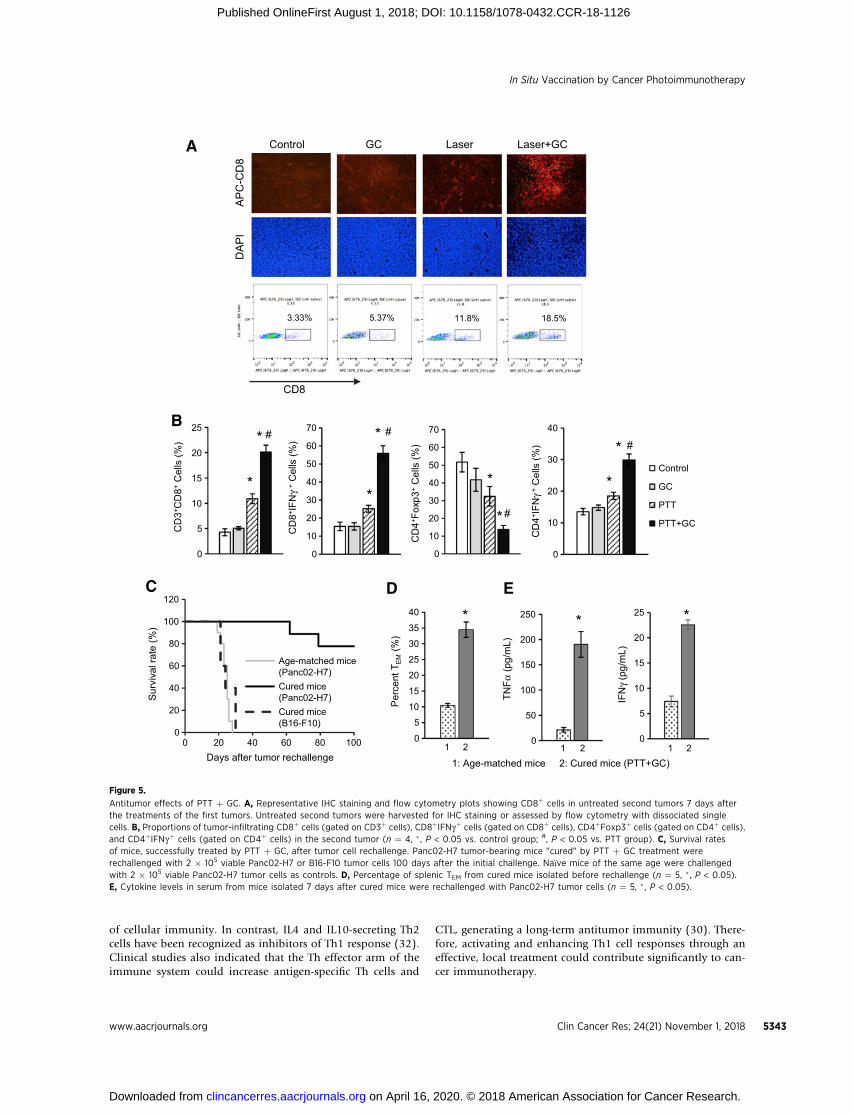
To further confirm the synergistic antitumor effects triggeredby PTT and GC, immune cells in the untreated second tumor,implanted at the same time as the treated first tumor on thesamemouse, were analyzed on day 7. As shown in Fig. 5A and B,IHC and flow cytometry analysis of tumors showed that treat-ment with GC on the first tumors failed to promote CD8þ
infiltration into the second tumors. In contrast, for mice withPTT þ GC–treated first tumors, the percentage of CD8þ in thesecond tumors significantly increased (�18.5%), whichappeared to be higher than that in the mice with PTT-treatedfirst tumors (�11.8%). Finally, the percentage of CD8þCTL(CD8þIFNgþ) infiltration was greatly increased in the mice withPTT þ GC–treated first tumors (Fig. 5B). In addition, helperT cells that are important for regulating adaptive immunitieswere also analyzed by flow cytometer. GC treatment of the firsttumors had no effect on the infiltration of either regulatory Tcells (Treg; CD4þFoxp3þ) or Th1 cells (CD4þIFNgþ) into thesecond tumors. However, PTT treatment of first tumors slightlyreduced Tregs and increased Th1 cells in the second tumors. Incontrast, PTT þ GC greatly reduced the percentage of Tregs, butsignificantly increased Th1 cells, in the untreated second tumors(Fig. 5B). These results further confirmed that PTT þ GC trig-gered a synergistic antitumor response that effectively sup-pressed the untreated distal tumors.
Long-term immune effects of PTT þ GC therapyTo evaluate whether the long-term immunity can be induced
by PTT þ GC treatment, the cured tumor-bearing mice wererechallenged with Panc02-H7 tumor cells. It was observed that7 of 9 rechallenged mice survived for 100 days after tumor cellrechallenge and did not develop visible or palpable tumors(Fig. 5C). In contrast, age-matched control mice bearingPanc02-H7 tumors all developed tumors (10/10) and diedwithin 30 days of tumor inoculation (Fig. 5C). To demonstratespecificity of Panc02-H7 tumor rejection, PTT þ GC curedtumor-bearing mice were challenged with B16-F10 tumor cells.All mice in this group (5/5) succumbed to B16-F10 tumorgrowth and died within 30 days (Fig. 5C). The increasedeffector memory T cells (TEM cells, CD3þCD8þCD44þCD62Lþ)in splenocytes of cured mice before rechallenge also confirmedthe antitumor immune memory effects, triggered by PTT þ GC(Fig. 5D). One week after tumor rechallenge, cytokines inserum were analyzed to confirm the specific antitumor immu-nity. The serum levels of TNFa and IFNg were significantlyincreased in the mice treated with PTT þ GC (Fig. 5E). These
results further confirmed that PTT þ GC could induce a tumor-specific T-cell response, a major cause of elimination of treatedtumors and resistance to tumor rechallenge, through genera-tion of CTLs and TEM.
Efficacy of PTT þ GC therapy on orthotopic pancreatictumors
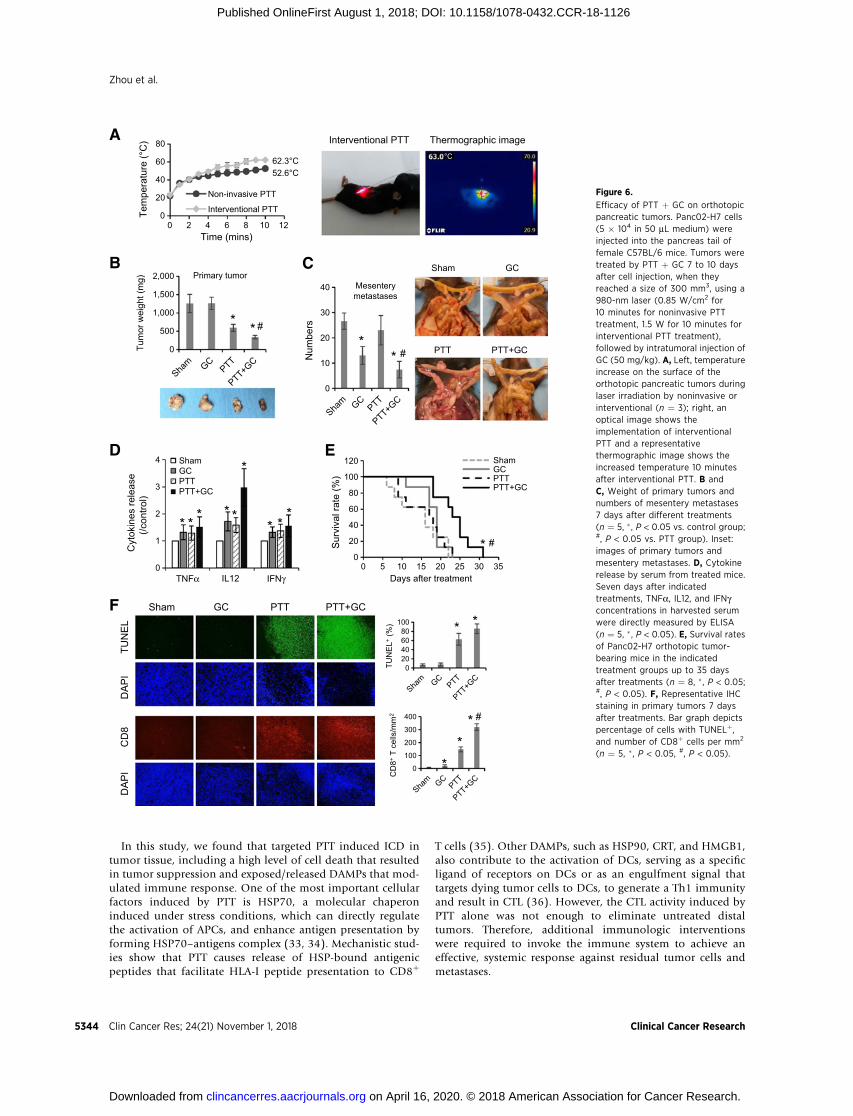
On the basis of the strong effects of PTT þ GC therapy on thesubcutaneous Panc02-H7 tumors, we investigate its effect onorthotopic pancreatic tumors and metastases. Mice bearingPanc02-H7 orthotopic tumors were exposed by surgery andtreated by noninvasive laser irradiation, with the sameparametersused on subcutaneous tumors. However, the tumor proximalsurface temperature only increased to 52.6�C under the laserirradiation (Fig. 6A).
To enhance the local thermal effects and improve the ther-apeutic efficacy for deep-buried pancreatic cancer, intervention-al PTT was developed using the fiber with a cylindrical activetip, which was inserted into the tumor, as shown in Fig. 6A.Under the treatment of interventional PTT, the tumor surfacetemperature increased to 62.3�C (Fig. 6A). Treated mice weresacrificed for anatomical analysis 7 days later. For the animals inthe Sham group, the abdomen was distended due to the tumorsand ascites, both in female and male mice (SupplementaryFig. S5). However, for the animals in the PTT þ GC group,there was no obvious ascites (Supplementary Fig. S5). As shownin Fig. 6B and Supplementary Fig. S6A, the interventional PTTsuccessfully damaged the primary tumors; however, there wasno noticeable impact of GC on primary tumors. Combined withGC, interventional PTT significantly reduced the metastases(Fig. 6C; Supplementary Fig. S6B), and resulted in the produc-tion of high-level TNFa, IL12, and IFNg in serum (Fig. 6D).Finally, as shown in Fig. 6E, the average survival time of themice in the PTT þ GC group was significantly higher than thatof other groups. The median survival time for animals in theSham, GC, PTT, and PTT þ GC treatment groups were 15, 19,17.5, and 24 days, respectively. In addition, IHC analysis usingTUNEL dye and CD8 antibody staining further confirmed thatPTT could damage the tumor cells to shrink the tumors and pro-vide antigen source; GC treatment could enhance the immuneresponse to attack remain tumor cells (Fig. 6F; SupplementaryFig. S6C). These results showed the efficacy of interventionalPTT, in combination with GC, in the treatment of metastaticpancreatic tumors, removing the primary tumor by local ther-mal effects and controlling the metastases by induced antitumorimmunity.
DiscussionIn this study, we demonstrated that a combination of
PTT and GC adjuvant induced a synergistic photothermal–immunologic response with local intervention to control thetreated primary tumors and untreated metastases, by increasingthe quantity and quality of antitumor T cells, leading to a long-term antitumor immunity against metastatic pancreatic tumorsin mice.
CD4þ Th1 and Th2 responses play an important role incancer immunotherapy (29, 30). Th1 cells have been recog-nized as promoters of cell-mediated antitumor response byregulating the survival and persistence of CD8þ CTLs (31). Th1cytokines, including TNFa, IFNg , and IL12, are typical markers
Zhou et al.
Clin Cancer Res; 24(21) November 1, 2018 Clinical Cancer Research5342
on April 16, 2020. © 2018 American Association for Cancer Research. clincancerres.aacrjournals.org Downloaded from
Published OnlineFirst August 1, 2018; DOI: 10.1158/1078-0432.CCR-18-1126
of cellular immunity. In contrast, IL4 and IL10-secreting Th2cells have been recognized as inhibitors of Th1 response (32).Clinical studies also indicated that the Th effector arm of theimmune system could increase antigen-specific Th cells and
CTL, generating a long-term antitumor immunity (30). There-fore, activating and enhancing Th1 cell responses through aneffective, local treatment could contribute significantly to can-cer immunotherapy.
C
A
B
0
10
20
30
40
CD
4+ IFN
γ+C
ells
(%)
Control
GC
PTT
PTT+GC
*
* #
CD8
Control GC Laser Laser+GC
AP
C-C
D8
DAP
I
3.33% 5.37% 11.8% 18.5%
0
10
20
30
40
50
60
70
CD
8+ IFN
γ+
Cel
ls (%
)
*
* #
0
10
20
30
40
50
60
70
CD
4+ Fox
p3+
Cel
ls (%
)
* #
*
D
0
50
100
150
200
250
TNFα
(pg/
mL)
1 20
5
10
15
20
25IF
Nγ
(pg/
mL)
* *
1 20
5
10
15
20
25
30
35
40
Per
cent
TE
M(%
)
*
1 2
1: Age-matched mice 2: Cured mice (PTT+GC)
E
0
20
40
60
80
100
120
0 20 40 60 80 100
Sur
viva
l rat
e (%
)
Days after tumor rechallenge
Age-matched mice(Panc02-H7)Cured mice(Panc02-H7)Cured mice(B16-F10)
0
5
10
15
20
25
CD
3+ CD
8+C
ells
(%)
*
* #
Figure 5.
Antitumor effects of PTT þ GC. A, Representative IHC staining and flow cytometry plots showing CD8þ cells in untreated second tumors 7 days afterthe treatments of the first tumors. Untreated second tumors were harvested for IHC staining or assessed by flow cytometry with dissociated singlecells. B, Proportions of tumor-infiltrating CD8þ cells (gated on CD3þ cells), CD8þIFNgþ cells (gated on CD8þ cells), CD4þFoxp3þ cells (gated on CD4þ cells),and CD4þIFNgþ cells (gated on CD4þ cells) in the second tumor (n ¼ 4, � , P < 0.05 vs. control group; #, P < 0.05 vs. PTT group). C, Survival ratesof mice, successfully treated by PTT þ GC, after tumor cell rechallenge. Panc02-H7 tumor-bearing mice "cured" by PTT þ GC treatment wererechallenged with 2 � 105 viable Panc02-H7 or B16-F10 tumor cells 100 days after the initial challenge. Na€�ve mice of the same age were challengedwith 2 � 105 viable Panc02-H7 tumor cells as controls. D, Percentage of splenic TEM from cured mice isolated before rechallenge (n ¼ 5, � , P < 0.05).E, Cytokine levels in serum from mice isolated 7 days after cured mice were rechallenged with Panc02-H7 tumor cells (n ¼ 5, � , P < 0.05).
In Situ Vaccination by Cancer Photoimmunotherapy
www.aacrjournals.org Clin Cancer Res; 24(21) November 1, 2018 5343
on April 16, 2020. © 2018 American Association for Cancer Research. clincancerres.aacrjournals.org Downloaded from
Published OnlineFirst August 1, 2018; DOI: 10.1158/1078-0432.CCR-18-1126
In this study, we found that targeted PTT induced ICD intumor tissue, including a high level of cell death that resultedin tumor suppression and exposed/released DAMPs that mod-ulated immune response. One of the most important cellularfactors induced by PTT is HSP70, a molecular chaperoninduced under stress conditions, which can directly regulatethe activation of APCs, and enhance antigen presentation byforming HSP70–antigens complex (33, 34). Mechanistic stud-ies show that PTT causes release of HSP-bound antigenicpeptides that facilitate HLA-I peptide presentation to CD8þ
T cells (35). Other DAMPs, such as HSP90, CRT, and HMGB1,also contribute to the activation of DCs, serving as a specificligand of receptors on DCs or as an engulfment signal thattargets dying tumor cells to DCs, to generate a Th1 immunityand result in CTL (36). However, the CTL activity induced byPTT alone was not enough to eliminate untreated distaltumors. Therefore, additional immunologic interventionswere required to invoke the immune system to achieve aneffective, systemic response against residual tumor cells andmetastases.
0
20
40
60
80
0 2 4 6 8 10 12
Tem
pera
ture
(°C
)
Time (mins)
Non-invasive PTT
Interventional PTT
62.3°C52.6°C
A
B C
D
Interventional PTT Thermographic image
Sham GC
PTT PTT+GC
0
20
40
60
80
100
120
0 5 10 15 20 25 30 35
Sur
viva
l rat
e (%
)
Days after treatment
ShamGCPTTPTT+GC
#*
E
F Sham GC PTT PTT+GC
DA
PI
CD
8D
AP
ITU
NE
L
020406080
100
TUN
EL+
(%) * *
0
100
200
300
400
CD
8+T
cells
/mm
2
**
* #
0
1
2
3
4
TNFα IL12 IFNγ
Cyt
okin
es re
leas
e (/c
ontro
l)
ShamGCPTTPTT+GC
* * * * *
*
* **
0
500
1,000
1,500
2,000
Tum
or w
eigh
t (m
g) Primary tumor
* * #
0
10
20
30
40
Num
bers
Mesentery metastases
** #
°C
Figure 6.
Efficacy of PTT þ GC on orthotopicpancreatic tumors. Panc02-H7 cells(5 � 104 in 50 mL medium) wereinjected into the pancreas tail offemale C57BL/6 mice. Tumors weretreated by PTT þ GC 7 to 10 daysafter cell injection, when theyreached a size of 300 mm3, using a980-nm laser (0.85 W/cm2 for10 minutes for noninvasive PTTtreatment, 1.5 W for 10 minutes forinterventional PTT treatment),followed by intratumoral injection ofGC (50 mg/kg). A, Left, temperatureincrease on the surface of theorthotopic pancreatic tumors duringlaser irradiation by noninvasive orinterventional (n ¼ 3); right, anoptical image shows theimplementation of interventionalPTT and a representativethermographic image shows theincreased temperature 10 minutesafter interventional PTT. B andC, Weight of primary tumors andnumbers of mesentery metastases7 days after different treatments(n ¼ 5, � , P < 0.05 vs. control group;#, P < 0.05 vs. PTT group). Inset:images of primary tumors andmesentery metastases. D, Cytokinerelease by serum from treated mice.Seven days after indicatedtreatments, TNFa, IL12, and IFNgconcentrations in harvested serumwere directly measured by ELISA(n ¼ 5, � , P < 0.05). E, Survival ratesof Panc02-H7 orthotopic tumor-bearing mice in the indicatedtreatment groups up to 35 daysafter treatments (n ¼ 8, � , P < 0.05;#, P < 0.05). F, Representative IHCstaining in primary tumors 7 daysafter treatments. Bar graph depictspercentage of cells with TUNELþ,and number of CD8þ cells per mm2
(n ¼ 5, �, P < 0.05, #, P < 0.05).
Zhou et al.
Clin Cancer Res; 24(21) November 1, 2018 Clinical Cancer Research5344
on April 16, 2020. © 2018 American Association for Cancer Research. clincancerres.aacrjournals.org Downloaded from
Published OnlineFirst August 1, 2018; DOI: 10.1158/1078-0432.CCR-18-1126

GC is an immunoadjuvant that consists of a functionalizedchitosan, or N-dihydrogalacto-chitosan (37). As a soluble andnontoxic compound, GC possesses improved properties forimmunologic stimulation as shown in preclinical and clinicalstudies, indicating its immunologic effect for the treatment ofmetastatic cancers (21, 38). In combination with PTT, GC-treatedtumor-bearing animals had a significantly higher cure rate thananimals treated with other commonly used adjuvants, such ascomplete Freund's adjuvant, incomplete Freund's adjuvant, andCorynebacterium parvum (37). Chitosan, the precursor of GC,has been reported to promote DCmaturation by inducing a type IIFN response, which in turn enhanced antigen-specific Th1responses (39). GC was also shown to stimulate secretion ofIFNg and TNFa by DCs. On the basis of the immunogenic tumorantigen source and infiltrating DCs produced by PTT, intratu-moral injected GC could interact with and activate infiltratingDCs, to enhance antigen uptake by DCs. Furthermore, GC canbe taken up by DCs to further enhance the antigen presentationto T cells, augmenting the specific antitumor immune response,to eliminate metastatic tumors and inhibit tumor recurrence.Although GC has been shown to induce Th1 immunity andpromote proliferation of splenocytes, as a potent adjuvant incancer therapy, the immune modulation functions of GC needto be further determined in the future.
Local PTT induces immunogenic tumor cell death, whichprovides sources of tumor antigens, creating an in situ autol-ogous immunogenic cell vaccine. As an immunoadjuvant, GC,following PTT, enhances antigen uptake by DCs and presen-tation to T cells, amplifying tumor-specific T-cell response toattack residual tumor cells at the treatment site and untreatedmetastases at distant sites. As a whole tumor cell vaccine,algenpantucel-L (also known as hyperacute-PC vaccine) holdshigh promise in treating patients with pancreatic cancer (8).Algenpantucel-L consists two irradiated, live, human allogeneicpancreatic cancer cell lines that express murine a-1,3-galacto-syltransferase, responsible for the synthesis of a-galactosylatedepitopes on cell surface proteins (8). To avoid autoimmunity,antigen-specific antitumor vaccine against a single epitope hasbeen developed. However, tumors can evade recognition anddestruction through an antigen loss variance (40). DuringPTT þ GC treatment, an autologous vaccine-like approach,using patients' own whole tumor cells as the source of antigens,allows the host immune system to have access to the wholespectrum of unique and shared antigens expressed by thetargeted tumor cells in each individual patient. Furthermore,PTT þ GC does not require ex vivo selection and processing ofpatient antigens, avoiding obstacles, such as poor immunoge-nicity and different antigen expressions. Other targeted thera-pies, such as photodynamic therapy and radiotherapy, couldalso induce an in situ autologous cancer vaccine (41, 42).Especially, some chemotherapy agents, such as mitoxantrone,have been shown to trigger ICD, which stimulates an antitumorimmunity (43). On the basis of our therapeutic strategy, thecombination of GC with PDT, radiotherapy, or mitoxantronecould also be therapeutically feasible for the treatment ofmetastatic tumors, which should be investigated in the future.
Panc02-H7 cell line, derived from Panc02 by implanting 3-methylcholanthrene, shows more aggressive invasion than otherPanc02 clonal sublines (22, 23). Orthotopic implantations ofPanc02-H7 cells can quickly cause peritoneumdissemination anddistal organs metastases, hence becoming an ideal model for
immunotherapy of pancreatic cancer (44). Noninvasive laserirradiation worked well on the subcutaneous pancreatic tumors.However, the temperature increasewas limited due to lowabsorp-tion of light by the soft, almost transparent, pancreatic tissue.To achieve the optimal temperature distribution in tumor tissuethat can kill as many tumor cells while keeping tumor proteinsviable, local regional interventional PTT was developed. Theparameters of laser irradiation were determined according tothe tumor size and tissue property, based on our ex vivo modelfor simulating temperature distribution in the target tissue (45).The interventional PTT þ GC resulted in significantly reducedprimary tumors andmetastases. These data further supported themechanism of PTT þ GC: the interventional PTT damaged theprimary tumors; the combination of GC induced systemic immu-nity to control metastases. Although this pilot study showedthe potential of interventional PTTþGC for orthotopic pancreatictumors, as a new treatment modality for pancreatic cancer, itstherapeutic effects and antitumor mechanism should be furtherinvestigated in the future.
In summary, our preclinical data demonstrate the effective-ness of PTT þ GC on advanced pancreatic cancer by inducing asynergistic photothermal–immunologic response, connectingthe tumoricidal and immunologic processes and trigging aneffective CTL for tumor elimination. More importantly, weconfirmed that interventional PTT þ GC, as a one-time, localapplication, could successfully control both local tumors anddistal metastases, and prolong animal survival time. This studystrongly demonstrates the potential of this novel approach ininducing in situ ICD-based tumor vaccine through local inter-vention for patients with advanced cancers.
Disclosure of Potential Conflicts of InterestW.R. Chen is an unpaid member of the Board of Directors of Immunopho-
tonics, Inc. Nopotential conflicts of interest were disclosed by the other authors.
Authors' ContributionsConception and design: F. Zhou, J. Yang, Y. Zhang, M. Li, W.R. ChenDevelopment of methodology: F. Zhou, J. Yang, M. Liu, M. Li, W.R. ChenAcquisition of data (provided animals, acquired and managed patients,provided facilities, etc.): F. Zhou, J. Yang, Y. Zhang, W.R. ChenAnalysis and interpretation of data (e.g., statistical analysis, biostatistics,computational analysis): F. Zhou, J. Yang, Y. Zhang, M. LiWriting, review, and/or revision of themanuscript: F. Zhou, J. Yang, Y. Zhang,M. Liu, M.L. Lang, M. Li, W.R. ChenAdministrative, technical, or material support (i.e., reporting or organizingdata, constructing databases): F. Zhou, M. Li,Study supervision: F. Zhou, Y. Zhang, M. Li, W.R. Chen
AcknowledgmentsThis study was supported in part by grants from the NIH R21 EB015509,
RS20132225-106, R01 CA205348 (to W.R. Chen), R01 CA138701, R01CA186338, and R01 CA203108 (to M. Li), and HR16-085 Oklahoma Centerfor Advancement of Science and Technology (to W.R. Chen).
The costs of publication of this article were defrayed in part by thepayment of page charges. This article must therefore be hereby markedadvertisement in accordance with 18 U.S.C. Section 1734 solely to indicatethis fact.
Received April 11, 2018; revised June 24, 2018; accepted July 25, 2018;published first August 1, 2018.
In Situ Vaccination by Cancer Photoimmunotherapy
www.aacrjournals.org Clin Cancer Res; 24(21) November 1, 2018 5345
on April 16, 2020. © 2018 American Association for Cancer Research. clincancerres.aacrjournals.org Downloaded from
Published OnlineFirst August 1, 2018; DOI: 10.1158/1078-0432.CCR-18-1126

References1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2018. CA Cancer J Clin
2018;68:7–30.2. Spadi R, Brusa F, Ponzetti A, Chiappino I, Birocco N, Ciuffreda L, et al.
Current therapeutic strategies for advanced pancreatic cancer: a reviewfor clinicians. World J Clin Oncol 2016;7:27.
3. Hu Y, Chi C, Wang S, Wang L, Liang P, Liu F, et al. A comparativestudy of clinical intervention and interventional photothermal therapyfor pancreatic cancer. Adv Mater 2017;29:1700448.
4. Manji GA, Olive KP, Saenger YM, Oberstein P. Current and emergingtherapies inmetastatic pancreatic cancer. Clin Cancer Res 2017;23:1670–8.
5. Koido S, Homma S, Takahara A, Namiki Y, Tsukinaga S, Mitobe J, et al.Current immunotherapeutic approaches in pancreatic cancer. Clin DevImmunol 2011;2011:267539.
6. Couzin-Frankel J. Cancer immunotherapy. Science 2013;342:1432–3.7. Mellman I, Coukos G, Dranoff G. Cancer immunotherapy comes of age.
Nature 2011;480:480–9.8. Yadav D, deLu C, Yadav R. Vaccine therapy for pancreatic cancer a battle
against deadly cancer. J Cancer Sci Ther 2014;6:268–77.9. Curran KJ, Brentjens RJ. Chimeric antigen receptor T cells for cancer
immunotherapy. J Clin Oncol 2015;33:1703–6.10. Blankenstein T, Coulie PG, Gilboa E, Jaffee EM. The determinants of
tumour immunogenicity. Nat Rev Cancer 2012;12:307–13.11. Spitzer MH, Carmi Y, Reticker-Flynn NE, Kwek SS, Madhireddy D, Martins
MM, et al. Systemic immunity is required for effective cancer immuno-therapy. Cell 2017;168:487–502.
12. Kroemer G, Galluzzi L, Kepp O, Zitvogel L. Immunogenic cell death incancer therapy. Annu Rev Immunol 2013;31:51–72.
13. Kepp O, Tesniere A, Zitvogel L, Kroemer G. The immunogenicity of tumorcell death. Curr Opin Oncol 2009;21:71–6.
14. Ludgate CM. Optimizing cancer treatments to induce an acute immuneresponse: radiation Abscopal effects, PAMPs, and DAMPs. Clin Cancer Res2012;18:4522–5.
15. Kono H, Rock KL. How dying cells alert the immune system to danger.Nat Rev Immunol 2008;8:279–89.
16. Hamblin MR, Huang Y-Y. Handbook of photomedicine. Boca Raton:Taylor & Francis; 2013:763–70.
17. den BrokMH, Sutmuller RP, van der Voort R, Bennink EJ, Figdor CG, RuersTJ, et al. In situ tumor ablation creates an antigen source for the generationof antitumor immunity. Cancer Res 2004;64:4024–9.
18. Montomoli E, Piccirella S, Khadang B, Mennitto E, Camerini R, De Rosa A.Current adjuvants and newperspectives in vaccine formulation. Expert RevVaccines 2011;10:1053–61.
19. Brewer JM, Conacher M, Hunter CA, Mohrs M, Brombacher F, Alexander J.Aluminium hydroxide adjuvant initiates strong antigen-specific Th2responses in the absence of IL-4-or IL-13-mediated signaling. J Immunol1999;163:6448–54.
20. CoffmanRL, Sher A, Seder RA. Vaccine adjuvants: putting innate immunityto work. Immunity 2010;33:492–503.
21. Li X, Ferrel GL, Guerra MC, Hode T, Lunn JA, Adalsteinsson O, et al.Preliminary safety and efficacy results of laser immunotherapy for thetreatment of metastatic breast cancer patients. Photochem Photobiol Sci2011;10:817–21.
22. Corbett T, Roberts B, Leopold W, Peckham J, Wilkoff L, Griswold D, et al.Induction and chemotherapeutic response of two transplantable ductaladenocarcinomas of the pancreas in C57BL/6 mice. Cancer Res 1984;44:717–26.
23. Wang B, Shi Q, Abbruzzese JL, Xiong Q, Le X, Xie K. A novel, clinicallyrelevant animal model of metastatic pancreatic adenocarcinoma biologyand therapy. Int J Pancreatol 2001;29:37–46.
24. Li M, Zhang Y, Bharadwaj U, Zhai QJ, Ahern CH, Fisher WE, et al. Down-regulation of ZIP4 by RNA interference inhibits pancreatic cancer growthand increases the survival of nude mice with pancreatic cancer xenografts.Clin Cancer Res 2009;15:5993–6001.
25. Li M, Zhang Y, Liu Z, Bharadwaj U, Wang H, Wang X, et al. Aberrantexpression of zinc transporter ZIP4 (SLC39A4) significantly contributesto human pancreatic cancer pathogenesis and progression. Proc Natl AcadSci U S A 2007;104:18636–41.
26. Zhang Y, Yang J, Cui X, Chen Y, Zhu VF, Hagan JP, et al. A novelepigenetic CREB-miR-373 axis mediates ZIP4-induced pancreatic cancergrowth. EMBO Mol Med 2013;5:1322–34.
27. Helft J, B€ottcher J, Chakravarty P, Zelenay S, Huotari J, Schraml BU, et al.GM-CSF mouse bone marrow cultures comprise a heterogeneous popu-lation of CD11cþ MHCIIþ macrophages and dendritic cells. Immunity2015;42:1197–211.
28. El-Hussein A, Lam SS, Raker J, Chen WR, Hamblin MR.N-Dihydrogalactochitosan as a potent immune activator for dendriticcells. J Biomed Mater Res A 2017;105:963–72.
29. Kennedy R, Celis E. Multiple roles for CD4þ T cells in antitumor immuneresponses. Immunol Rev 2008;222:129–44.
30. Knutson KL, Disis ML. Tumor antigen-specific T helper cells in cancerimmunity and immunotherapy. Cancer Immunol Immunother 2005;54:721–8.
31. Zhu J, Paul WE. Peripheral CD4þ Tcell differentiation regulated bynetworks of cytokines and transcription factors. Immunol Rev 2010;238:247–62.
32. Johansson M, DeNardo DG, Coussens LM. Polarized immune responsesdifferentially regulate cancer development. Immunol Rev 2008;222:145–54.
33. Nishikawa M, Takemoto S, Takakura Y. Heat shock protein derivatives fordelivery of antigens to antigen presenting cells. Int J Pharm2008;354:23–7.
34. Torigoe T, Tamura Y, Sato N. Heat shock proteins and immunity: appli-cation of hyperthermia for immunomodulation. Int J Hyperther 2009;25:610–6.
35. Murshid A, Gong J, Calderwood SK. The role of heat shock proteins inantigen cross presentation. Front Immunol 2012;3:63.
36. Garg AD, Nowis D, Golab J, Vandenabeele P, Krysko DV, Agostinis P.Immunogenic cell death, DAMPs and anticancer therapeutics: anemerging amalgamation. BBA-Rev Cancer 2010;1805:53–71.
37. Chen WR, Korbelik M, Bartels KE, Liu H, Sun J, Nordquist RE. Enhance-ment of laser cancer treatment by a chitosan-derived immunoadjuvant.Photochem Photobiol 2005;81:190–5.
38. ChenWR, Singhal AK, Liu H, Nordquist RE. Antitumor immunity inducedby laser immunotherapy and its adoptive transfer. Cancer Res 2001;61:459–61.
39. Carroll EC, Jin L,Mori A,Mu~noz-Wolf N,Oleszycka E,MoranHB, et al. Thevaccine adjuvant chitosan promotes cellular immunity via DNA sensorcGAS-STING-dependent induction of type I interferons. Immunity 2016;44:597–608.
40. Schlom J. Therapeutic cancer vaccines: current status and moving forward.JNCI 2012;104:599–613.
41. Garg AD, Vandenberk L, Koks C, Verschuere T, Boon L, Van Gool SW, et al.Dendritic cell vaccines based on immunogenic cell death elicit dangersignals and T cell–driven rejection of high-grade glioma. Sci Transl Med2016;8:328ra27.
42. Formenti SC, Demaria S. Radiation therapy to convert the tumor into anin situ vaccine. Int J Radiat Oncol Biol Phys 2012;84:879–80.
43. MichaudM,Martins I, Sukkurwala AQ, Adjemian S,Ma Y, Pellegatti P, et al.Autophagy-dependent anticancer immune responses induced bychemotherapeutic agents in mice. Science 2011;334:1573–7.
44. Wang B, Wei D, Crum VE, Richardson EL, Xiong HH, Luo Y, et al. Anovel model system for studying the double-edged roles of nitric oxideproduction in pancreatic cancer growth and metastasis. Oncogene2003;22:1771–82.
45. Liu S, Doughty A, West C, Tang Z, Zhou F, Chen WR. Determination oftemperature distribution in tissue for interstitial cancer photothermaltherapy. Int J Hyperthermia 2017;6:1–8.
Clin Cancer Res; 24(21) November 1, 2018 Clinical Cancer Research5346
Zhou et al.
on April 16, 2020. © 2018 American Association for Cancer Research. clincancerres.aacrjournals.org Downloaded from
Published OnlineFirst August 1, 2018; DOI: 10.1158/1078-0432.CCR-18-1126

2018;24:5335-5346. Published OnlineFirst August 1, 2018.Clin Cancer Res Feifan Zhou, Jingxuan Yang, Yuqing Zhang, et al. Vaccineof Pancreatic Cancer through Induced Immunogenic Tumor Local Phototherapy Synergizes with Immunoadjuvant for Treatment
Updated version
10.1158/1078-0432.CCR-18-1126doi:
Access the most recent version of this article at:
Material
Supplementary
http://clincancerres.aacrjournals.org/content/suppl/2018/08/01/1078-0432.CCR-18-1126.DC1
Access the most recent supplemental material at:
Cited articles
http://clincancerres.aacrjournals.org/content/24/21/5335.full#ref-list-1
This article cites 44 articles, 13 of which you can access for free at:
E-mail alerts related to this article or journal.Sign up to receive free email-alerts
Subscriptions
Reprints and
To order reprints of this article or to subscribe to the journal, contact the AACR Publications Department at
Permissions
Rightslink site. Click on "Request Permissions" which will take you to the Copyright Clearance Center's (CCC)
.http://clincancerres.aacrjournals.org/content/24/21/5335To request permission to re-use all or part of this article, use this link
on April 16, 2020. © 2018 American Association for Cancer Research. clincancerres.aacrjournals.org Downloaded from
Published OnlineFirst August 1, 2018; DOI: 10.1158/1078-0432.CCR-18-1126





























