Embed Size (px)
Citation preview

Long non-coding RNA-ROR aggravates myocardialischemia/reperfusion injury
Weiwei Zhang1, Ying Li2 and Peng Wang1
1Department of Cardiology, Dezhou People’s Hospital, Dezhou, China2Interventional Center, Dezhou People’s Hospital, Dezhou, China
Abstract
Long non-coding RNAs (lncRNAs) play an important role in the pathogenesis of cardiovascular diseases, especially in myo-cardial infarction and ischemia/reperfusion (I/R). However, the underlying molecular mechanism remains unclear. In this study,we determined the role and the possible underlying molecular mechanism of lncRNA-ROR in myocardial I/R injury. H9c2 cellsand human cardiomyocytes (HCM) were subjected to either hypoxia/reoxygenation (H/R), I/R or normal conditions (normoxia).The expression levels of lncRNA-ROR were detected in serum of myocardial I/R injury patients, H9c2 cells, and HCM by qRT-PCR. Then, levels of lactate dehydrogenase (LDH), malondialdehyde (MDA), superoxide dismutase (SOD), and glutathioneperoxidase (GSH-PX) were measured by kits. Cell viability, apoptosis, apoptosis-associated factors, and p38/MAPK pathwaywere examined by MTT, flow cytometry, and western blot assays. Furthermore, reactive oxygen species (ROS) productionwas determined by H2DCF-DA and MitoSOX Red probes with flow cytometry. NADPH oxidase activity and NOX2 proteinlevels were measured by lucigenin chemiluminescence and western blot. Results showed that lncRNA-ROR expression wasincreased in I/R patients and in H/R treatment of H9c2 cells and HCM. Moreover, lncRNA-ROR significantly promoted H/R-induced myocardial injury via stimulating release of LDH, MDA, SOD, and GSH-PX. Furthermore, lncRNA-ROR decreased cellviability, increased apoptosis, and regulated expression of apoptosis-associated factors. Additionally, lncRNA-ROR increasedphosphorylation of p38 and ERK1/2 expression and inhibition of p38/MAPK, and rescued lncRNA-ROR-induced cell injury inH9c2 cells and HCM. ROS production, NADPH oxidase activity, and NOX2 protein levels were promoted by lncRNA-ROR.These data suggested that lncRNA-ROR acted as a therapeutic agent for the treatment of myocardial I/R injury.
Key words: lncRNA; Ischemia/reperfusion (I/R); Hypoxia/reoxygenation (H/R); Cell viability; Apoptosis
Introduction
Myocardial ischemia/reperfusion (I/R) injury leads toadverse cardiovascular outcomes following myocardialischemia, cardiac surgery or circulatory arrest and is oneof the major causes of morbidity and mortality in humanswith coronary heart disease (1). The pathology of the diseasesuggests that myocardial infarction and angina pectorisare accompanied by changes in gene expression (2). Theunderlying molecular mechanisms of myocardial I/R injuryare complex and include oxidative stress, intracellular Ca2+
overload, rapid restoration of physiological pH upon reper-fusion, mitochondrial permeability transition pore, and exagger-ated inflammation (3). Rapid alterations in ion flux andrenormalization of pH following reperfusion causes severecytotoxicity and I/R injury, characterized by cell death andfunctional deterioration because of restoration of bloodflow (4). I/R injury causes local myocardial inflammationand apoptosis, which in turn leads to irreversible damageto the myocardium. However, early restoration of blood flowthrough the occluded coronary artery might reduce mortality
by limiting the infarct size and preserving cardiac function (5,6).Despite restoration of blood flow, reperfusion alone seemsnot to be enough to save the myocardium because of thecomplications that arise from the loss of viability (7).
Following myocardial I/R injury, there is a sudden increasein cytokines and chemokines and influx of leukocytes intothe endangered myocardial region (8). Cell survival andextracellular matrix integrity by activation of pro-apoptoticsignaling pathways (including mitogen-activated proteinkinases and p38) are hampered by inflammatory responsesafter myocardial I/R injury (9). Studies indicate that celldeath is a key factor in the pathogenesis of various cardiacdiseases such as heart failure, myocardial infarction, andI/R injury (1). During heart disease, myocytes are lostdue to both apoptosis and necrosis (10). It suggests thatnecrosis plays a critical role in the pathogenesis of thecardiac disease (11). However, the underlying mechanismof cardiomyocyte death is still not clear. Thus, I/R injury is stilla major problem in the treatment of myocardial ischemia.
Correspondence: Weiwei Zhang: <[email protected]>
Received August 15, 2017 | Accepted January 25, 2018
Braz J Med Biol Res | doi: 10.1590/1414-431X20186555
Brazilian Journal of Medical and Biological Research (2018) 51(6): e6555, http://dx.doi.org/10.1590/1414-431X20186555ISSN 1414-431X Research Article
1/10

Long non-coding RNAs (lncRNAs) belong to a newlydiscovered class of genes in the human genome that havebeen proposed to be key regulators of biological process-es (12). lncRNAs consist of more than 200 nucleotides (13).Recent evidence shows that lncRNAs play an importantrole in the physiological processes such as differentiation,proliferation, apoptosis, and inflammation (14). It is alsoobserved that lncRNAs are highly regulated and specific (15).However, the role of lncRNA-ROR in myocardial I/R injuryremains unclear.
The objective of this study was to investigate the roleand the possible underlying molecular mechanism of lncRNA-ROR in myocardial I/R injury. This study will provide a newinsight for the treatment of cardiomyocytes injury.
Material and Methods
Serum samplesSerum samples of 20 normal individuals and 20 patients
with myocardial I/R injury were obtained from DezhouPeople’s Hospital. The study was approved by the ResearchEthics Committee of Dezhou People’s Hospital, and writteninformed consent was obtained from all participants. Thesamples were collected and frozen in liquid nitrogen, andstored at –80°C.
Cell culture and H/R exposureEmbryonic rat myocardium-derived cells H9c2 and
human cardiomyocytes (HCM) were purchased from theAmerican Type Culture Collection (ATCC, USA) and cul-tured in Dulbecco’s Modified Eagle’s Medium (DMEM;Sigma, USA) containing 10% fetal calf serum (FCS;Invitrogen, USA) (16). In brief, H9c2 cells and HCM inserum-free DMEM were placed in a humidified chamberequilibrated with 5% CO2 and 95% N2 for 4 h, followed byreoxygenation with 5% CO2 and 95% air for 3 h in DMEMwith 10% FCS. Hypoxia/reoxygenation (H/R) treatmentof H9c2 cells and HCM were performed as describedpreviously. Gene expression and apoptotic changes weremeasured at 24 h after re-oxygenation. Cells culturedunder normoxic conditions were used as control.
Cell transfectionSmall interfering RNAs (siRNAs) targeting mRNA
(si-lncRNA-ROR) and pcDNA3.1-lncRNA-ROR (lncRNA-ROR) were synthesized by RiboBio (China). Transfectionswere performed in 6-, 24-, or 96-well plates after seededcells were cultured for 24 h. All transfections were done withHiPerFect transfection reagent (QIAGEN, Germany) accord-ing to the manufacturer’s protocol. Briefly, 5� 103 cells/cm2
were seeded on 200 mL/cm2 culture. The siRNAs or pcDNAswere pre-incubated with HiPerFect transfection reagentat room temperature for 10 min. The complex was thentransfected into the cardiomyocytes cells at a final concen-tration of 50 nM. The transfected cells were incubatedunder normal growth conditions for 48 h.
Detection of LDH, MDA, SOD, and GSH-PXLactate dehydrogenase (LDH), malondialdehyde (MDA),
superoxide dismutase (SOD), and glutathione peroxidase(GSH-PX) commercial kits were purchased from SangonBiotech (China). The release levels of LDH, MDA, SOD,and GSH-PX were measured according to the manufac-turer’s instructions.
Quantitative real-time PCR (qRT-PCR)Total RNA was isolated using TRIzol reagent (Invitrogen),
and complementary DNA (cDNA) was synthesized withPrimeScript reverse transcriptase (TaKaRa, China) andoligo-dT (20 bp) following the manufacturer’s instructions.Reverse transcription PCR (RT-PCR) or real-time PCRwas performed to analyze mRNA expression. The RT-PCRprogram was as follows: 94°C for 5 min, followed by35 cycles of 94°C for 30 s, 50°C annealing for 30 s, and72°C for 30 s. Real-time PCR was performed using SYBRPremix Ex TaqTM II kit (TaKaRa) as follows: 94°C for 10 s,followed by 40 cycles of 94°C for 5 s, 52°C for 30 s toanneal, and 72°C for 15 s. The relative level of lncRNA-ROR was determined using the 2-DDCt analysis method.
Western blot analysisProteins were extracted from the primary cardiomyo-
cytes in RIPA buffer (1% Triton X-100, 150 mmol/L NaCl,5 mmol/L EDTA, and 10 mmol/L Tris-HCl, pH 7.0; Solarbio,China) supplemented with a protease inhibitor cocktail(Cat: I3786-1ML, Sigma). The cell lysates were separatedby 10% sodium dodecyl sulfate-polyacrylamide gel electro-phoresis (SDS-PAGE) and transferred electrophoreticallyto a PVDF membrane (Millipore Corporation, USA). Afterblocking with 8% milk in PBS, pH 7.5, the membraneswere incubated with the following specific primary anti-bodies of Bax (ab32503), Bcl-2 (ab59348), cytochrome C(ab13575), Smac/Diablo (ab32023), cleaved-capase-3(ab13847), cleaved-capase-9 (ab2324), p-p38 (ab47363),p38 (ab31828), p-ERK (ab214362), and ERK1/2 (ab196883;all at a dilution of 1:1000, Abcam, UK). After overnightincubation, the appropriate HRP-conjugated anti-rabbitIgG secondary antibody (ab205781, Abcam, all at a dilu-tion of 1:5000) was subsequently applied and immunode-tection was achieved using the ECL Plus detection system(Millipore Corporation) according to the manufacturer’sinstructions. Band intensity was quantified using ImageLabt Software (Bio-Rad, China). GAPDH (ab8245, Abcam)was used as an internal control.
Cell viability assayTo explore the effect of lncRNA on cell viability, 5000 cells
per well in a 100 mL medium were seeded in 96-well plates.Every 24 h after transfection, 20 mL of the 3-(4,5-dimethyl-thiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) reagent(Solarbio) was added to wells and incubated with thesecells for 4 h. After removing the medium, blue formazan wasdissolved with 200 mL dimethyl sulfoxide (DMSO; Sigma),
Braz J Med Biol Res | doi: 10.1590/1414-431X20186555
Role of lncRNA-ROR in myocardial I/R injury 2/10
and absorbance was measured at 570 nm. Wells contain-ing only cardiomyocyte cells served as blanks.
Cell apoptosis assayTo quantify apoptotic cells, flow cytometry was per-
formed with an Annexin V-fluorescein-5-isothiocyanate apop-tosis detection kit (Bio-vision, USA). After transfection for48 h, cells were harvested in a 5-mL tube. Then, the cellswere washed with cold PBS and re-suspended in 1�binding buffer (10 mM HEPES, 140 mM NaCl, 2.5 mMCaCl2, pH 7.4) at a final concentration of 1�106 cells/mL.FITC-AnnexinV (5 mL) and propidium iodide (PI, 5 mL)were gently mixed and incubated with the cells for 15 minat room temperature. After incubation, the samples wereanalyzed by flow cytometry within 1 h.
Measurement of reactive oxygen species (ROS)production
For examining the accumulation of intracellular ROS inH9c2 cells, the ROS assay kit purchased from BeyotimeInstitute of Biotechnology (Haimen, China) was used accord-ing to the manufacturer’s instructions. Briefly, after treat-ment, cells were grown in a 96-well plate and incubatedwith 10 mmol/L of H2DCF-DA at 37°C for 1 h. The fluores-cence intensity was measured using the fluorescenceplate reader (BD Falcon, USA) at Ex./Em. = 488/525 nm.
Measurement of mitochondrial ROS by MitoSOXRedFor detection of mitochondrial superoxide generation,
MitoSOXRed assay (Invitrogen/Molecular Probes, USA)was performed. In brief, the treated H9c2 cells were incu-bated with 5 mM of MitoSOX Red for 30 min at 37°C.MitoSOX Red fluorescent intensity was determined at510 nm excitation and 580 nm emission. After incubation,these cells were washed twice with PBS, trypsinized,resuspended, and immediately submitted to flow cytome-try analysis.
Measurement of NADPH oxidase activityNADPH oxidase activity was detected by using the
lucigenin-enhanced chemiluminescence method as pre-viously described (17). Briefly, treated cells were washed
in PBS and re-suspended in cold Krebs-HEPES buffer.Then, 300 mL cell suspensions were homogenized with100 strokes in a Dounce homogenizer on ice, and aliquotsof the homogenates were used immediately. Subsequently,100 mL of homogenates were added to 900 mL of phos-phate buffer, pH 7.0, containing 1 mM EGTA, 150 mMsucrose, 5 mM lucigenin, and 100 mM NADPH to start thereaction. Chemiluminescence was measured every 15 sfor 10 min in a luminometer. A buffer blank (less than 5%of the cell signal) was subtracted from each reading. Thedifferences between the values obtained before and afteradding NADPH were calculated, and these data repre-sented the activity of NADPH oxidase.
Statistical analysisData are reported as means±SE. Differences between
groups were compared by a one-way ANOVA usingGraphpad prism software 6.0 (GraphPad Software, SanUSA). Po0.05 was considered to be statistically signifi-cant difference.
Results
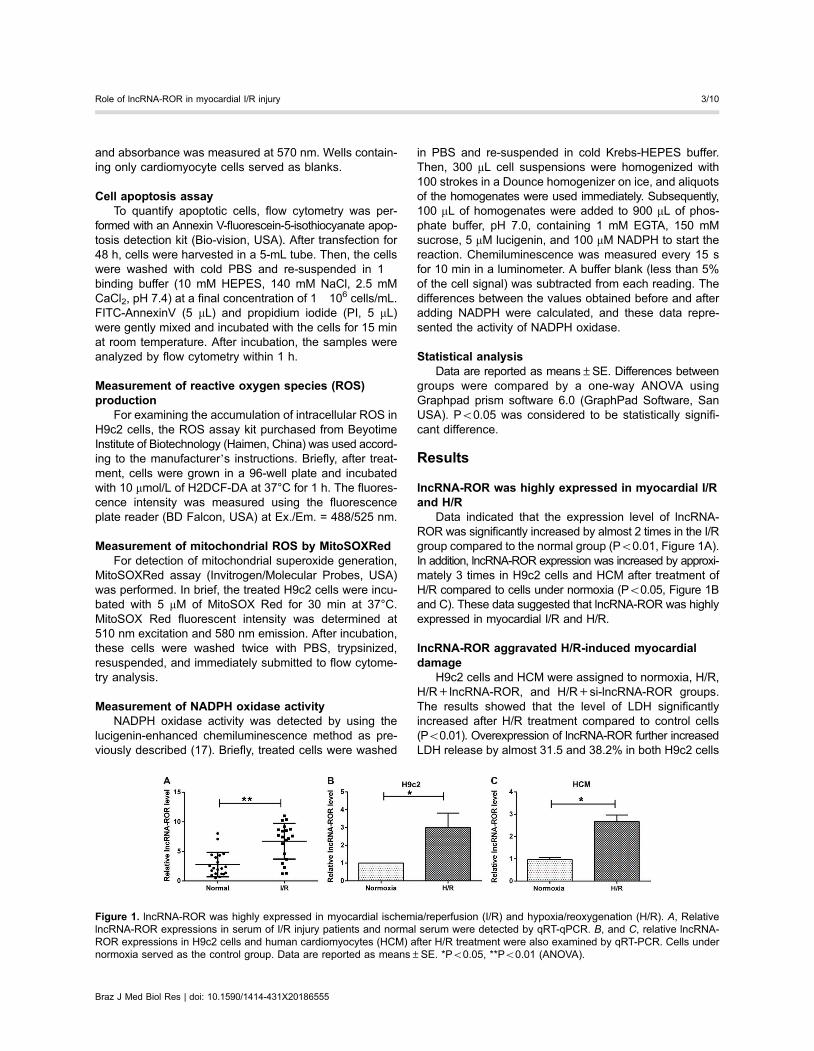
lncRNA-ROR was highly expressed in myocardial I/Rand H/R
Data indicated that the expression level of lncRNA-ROR was significantly increased by almost 2 times in the I/Rgroup compared to the normal group (Po0.01, Figure 1A).In addition, lncRNA-ROR expression was increased by approxi-mately 3 times in H9c2 cells and HCM after treatment ofH/R compared to cells under normoxia (Po0.05, Figure 1Band C). These data suggested that lncRNA-ROR was highlyexpressed in myocardial I/R and H/R.
lncRNA-ROR aggravated H/R-induced myocardialdamage
H9c2 cells and HCM were assigned to normoxia, H/R,H/R+lncRNA-ROR, and H/R+si-lncRNA-ROR groups.The results showed that the level of LDH significantlyincreased after H/R treatment compared to control cells(Po0.01). Overexpression of lncRNA-ROR further increasedLDH release by almost 31.5 and 38.2% in both H9c2 cells
Figure 1. lncRNA-ROR was highly expressed in myocardial ischemia/reperfusion (I/R) and hypoxia/reoxygenation (H/R). A, RelativelncRNA-ROR expressions in serum of I/R injury patients and normal serum were detected by qRT-qPCR. B, and C, relative lncRNA-ROR expressions in H9c2 cells and human cardiomyocytes (HCM) after H/R treatment were also examined by qRT-PCR. Cells undernormoxia served as the control group. Data are reported as means±SE. *Po0.05, **Po0.01 (ANOVA).
Braz J Med Biol Res | doi: 10.1590/1414-431X20186555
Role of lncRNA-ROR in myocardial I/R injury 3/10
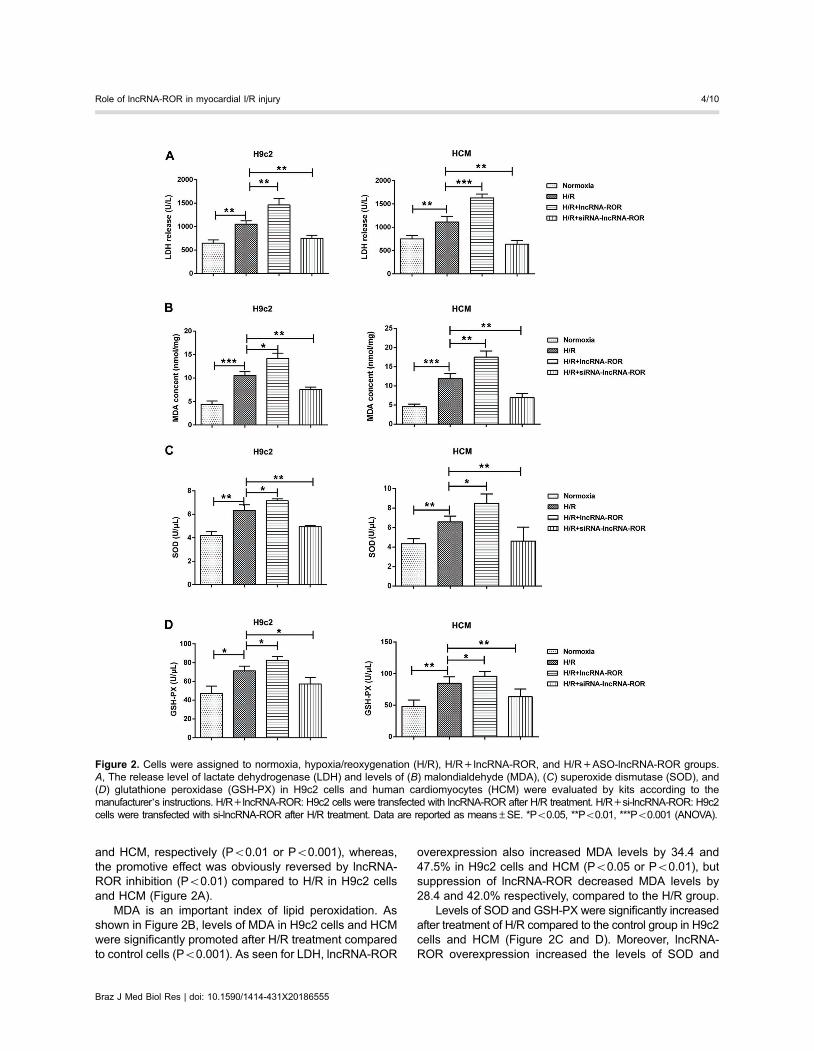
and HCM, respectively (Po0.01 or Po0.001), whereas,the promotive effect was obviously reversed by lncRNA-ROR inhibition (Po0.01) compared to H/R in H9c2 cellsand HCM (Figure 2A).
MDA is an important index of lipid peroxidation. Asshown in Figure 2B, levels of MDA in H9c2 cells and HCMwere significantly promoted after H/R treatment comparedto control cells (Po0.001). As seen for LDH, lncRNA-ROR
overexpression also increased MDA levels by 34.4 and47.5% in H9c2 cells and HCM (Po0.05 or Po0.01), butsuppression of lncRNA-ROR decreased MDA levels by28.4 and 42.0% respectively, compared to the H/R group.
Levels of SOD and GSH-PX were significantly increasedafter treatment of H/R compared to the control group in H9c2cells and HCM (Figure 2C and D). Moreover, lncRNA-ROR overexpression increased the levels of SOD and
Figure 2. Cells were assigned to normoxia, hypoxia/reoxygenation (H/R), H/R+lncRNA-ROR, and H/R+ASO-lncRNA-ROR groups.A, The release level of lactate dehydrogenase (LDH) and levels of (B) malondialdehyde (MDA), (C) superoxide dismutase (SOD), and(D) glutathione peroxidase (GSH-PX) in H9c2 cells and human cardiomyocytes (HCM) were evaluated by kits according to themanufacturer’s instructions. H/R+lncRNA-ROR: H9c2 cells were transfected with lncRNA-ROR after H/R treatment. H/R+si-lncRNA-ROR: H9c2cells were transfected with si-lncRNA-ROR after H/R treatment. Data are reported as means±SE. *Po0.05, **Po0.01, ***Po0.001 (ANOVA).
Braz J Med Biol Res | doi: 10.1590/1414-431X20186555
Role of lncRNA-ROR in myocardial I/R injury 4/10
GSH-PX by 13.0 or 15.6% in H9c2 cells (Po0.05) and28.6 or 13.2% in HCM, respectively (Po0.05), whereas,lncRNA-ROR suppression decreased by 21.9 or 19.7% inH9c2 cells (Po0.01) and 30.0 or 25.0% in HCM (Po0.05)compared to the H/R group.
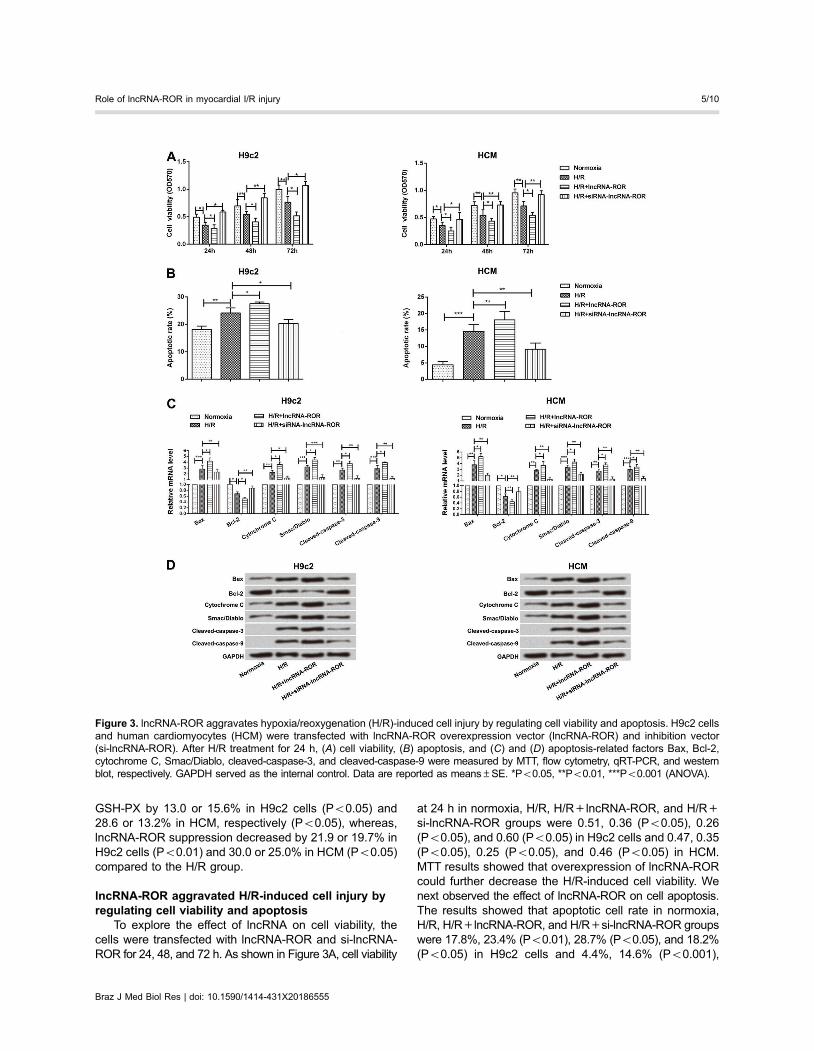
lncRNA-ROR aggravated H/R-induced cell injury byregulating cell viability and apoptosis
To explore the effect of lncRNA on cell viability, thecells were transfected with lncRNA-ROR and si-lncRNA-ROR for 24, 48, and 72 h. As shown in Figure 3A, cell viability
at 24 h in normoxia, H/R, H/R+lncRNA-ROR, and H/R+si-lncRNA-ROR groups were 0.51, 0.36 (Po0.05), 0.26(Po0.05), and 0.60 (Po0.05) in H9c2 cells and 0.47, 0.35(Po0.05), 0.25 (Po0.05), and 0.46 (Po0.05) in HCM.MTT results showed that overexpression of lncRNA-RORcould further decrease the H/R-induced cell viability. Wenext observed the effect of lncRNA-ROR on cell apoptosis.The results showed that apoptotic cell rate in normoxia,H/R, H/R+lncRNA-ROR, and H/R+si-lncRNA-ROR groupswere 17.8%, 23.4% (Po0.01), 28.7% (Po0.05), and 18.2%(Po0.05) in H9c2 cells and 4.4%, 14.6% (Po0.001),
Figure 3. lncRNA-ROR aggravates hypoxia/reoxygenation (H/R)-induced cell injury by regulating cell viability and apoptosis. H9c2 cellsand human cardiomyocytes (HCM) were transfected with lncRNA-ROR overexpression vector (lncRNA-ROR) and inhibition vector(si-lncRNA-ROR). After H/R treatment for 24 h, (A) cell viability, (B) apoptosis, and (C) and (D) apoptosis-related factors Bax, Bcl-2,cytochrome C, Smac/Diablo, cleaved-caspase-3, and cleaved-caspase-9 were measured by MTT, flow cytometry, qRT-PCR, and westernblot, respectively. GAPDH served as the internal control. Data are reported as means±SE. *Po0.05, **Po0.01, ***Po0.001 (ANOVA).
Braz J Med Biol Res | doi: 10.1590/1414-431X20186555
Role of lncRNA-ROR in myocardial I/R injury 5/10
18.1% (Po0.01), and 9.1% (Po0.01) in HCM. Flow cytom-etry showed that overexpression of lncRNA-ROR couldfurther aggravate H/R-induced cell apoptosis (Figure 3B).
To further explore the potential molecular mechanismof action of lncRNA-ROR, expression of apoptosis-relatedproteins such as Bax, Bcl-2, cytochrome C, Smac/Diablo,cleaved-caspase-3, and cleaved-caspase-9 were exam-ined by qRT-PCR and western blot. Results revealed thatH/R markedly increased Bax, cytochrome C, Smac/Diablo,cleaved-caspase-3, and cleaved-caspase-9 expressions,but decreased Bcl-2 expression. Overexpression of lncRNA-ROR further increased the expression of these five factors(Po0.05) and decreased the level of expression of Bcl-2(Po0.01; Figure 3C and D). However, suppressionof lncRNA-ROR showed a contrary result. These dataindicated that lncRNA-ROR aggravated H/R-inducedcell injury by decreasing cell viability and increasingapoptosis.
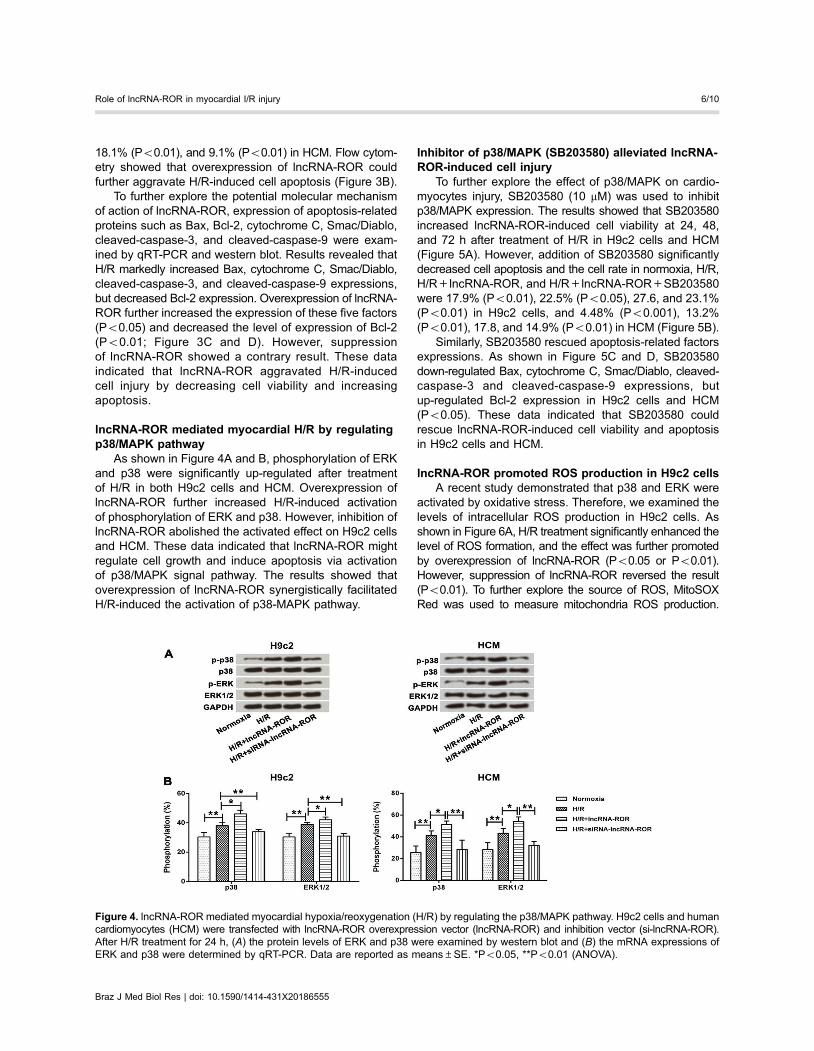
lncRNA-ROR mediated myocardial H/R by regulatingp38/MAPK pathway
As shown in Figure 4A and B, phosphorylation of ERKand p38 were significantly up-regulated after treatmentof H/R in both H9c2 cells and HCM. Overexpression oflncRNA-ROR further increased H/R-induced activationof phosphorylation of ERK and p38. However, inhibition oflncRNA-ROR abolished the activated effect on H9c2 cellsand HCM. These data indicated that lncRNA-ROR mightregulate cell growth and induce apoptosis via activationof p38/MAPK signal pathway. The results showed thatoverexpression of lncRNA-ROR synergistically facilitatedH/R-induced the activation of p38-MAPK pathway.
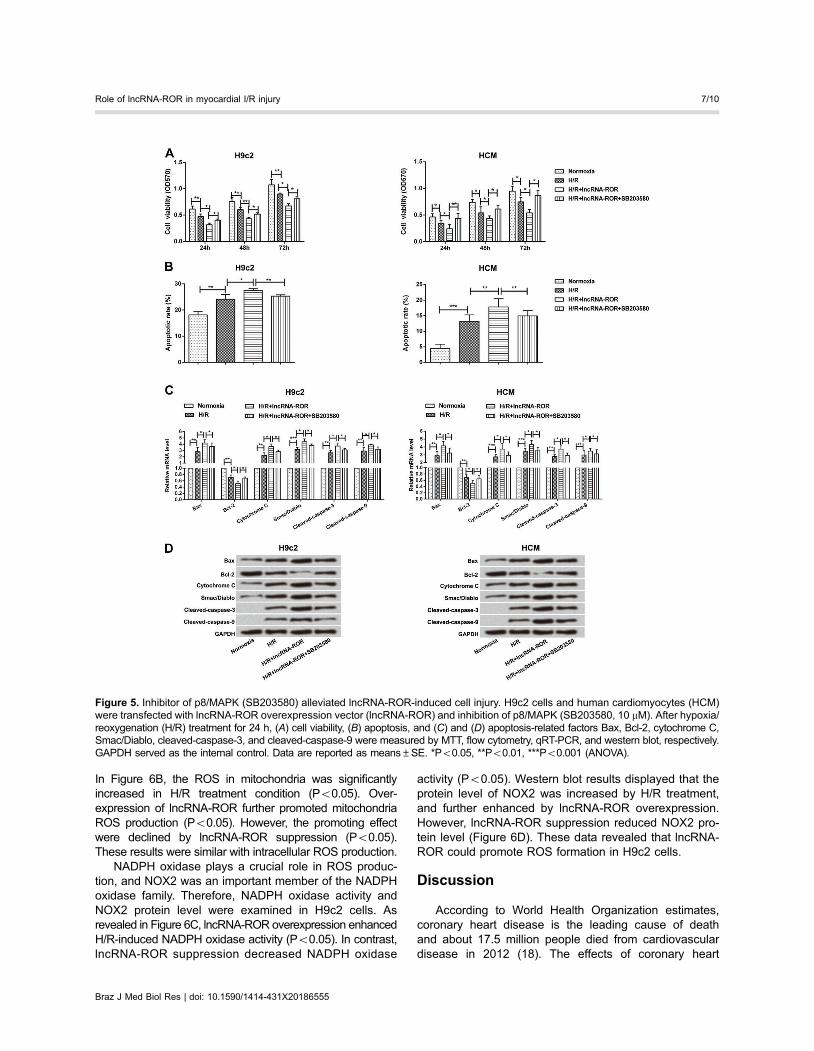
Inhibitor of p38/MAPK (SB203580) alleviated lncRNA-ROR-induced cell injury
To further explore the effect of p38/MAPK on cardio-myocytes injury, SB203580 (10 mM) was used to inhibitp38/MAPK expression. The results showed that SB203580increased lncRNA-ROR-induced cell viability at 24, 48,and 72 h after treatment of H/R in H9c2 cells and HCM(Figure 5A). However, addition of SB203580 significantlydecreased cell apoptosis and the cell rate in normoxia, H/R,H/R+lncRNA-ROR, and H/R+lncRNA-ROR+SB203580were 17.9% (Po0.01), 22.5% (Po0.05), 27.6, and 23.1%(Po0.01) in H9c2 cells, and 4.48% (Po0.001), 13.2%(Po0.01), 17.8, and 14.9% (Po0.01) in HCM (Figure 5B).
Similarly, SB203580 rescued apoptosis-related factorsexpressions. As shown in Figure 5C and D, SB203580down-regulated Bax, cytochrome C, Smac/Diablo, cleaved-caspase-3 and cleaved-caspase-9 expressions, butup-regulated Bcl-2 expression in H9c2 cells and HCM(Po0.05). These data indicated that SB203580 couldrescue lncRNA-ROR-induced cell viability and apoptosisin H9c2 cells and HCM.
lncRNA-ROR promoted ROS production in H9c2 cellsA recent study demonstrated that p38 and ERK were
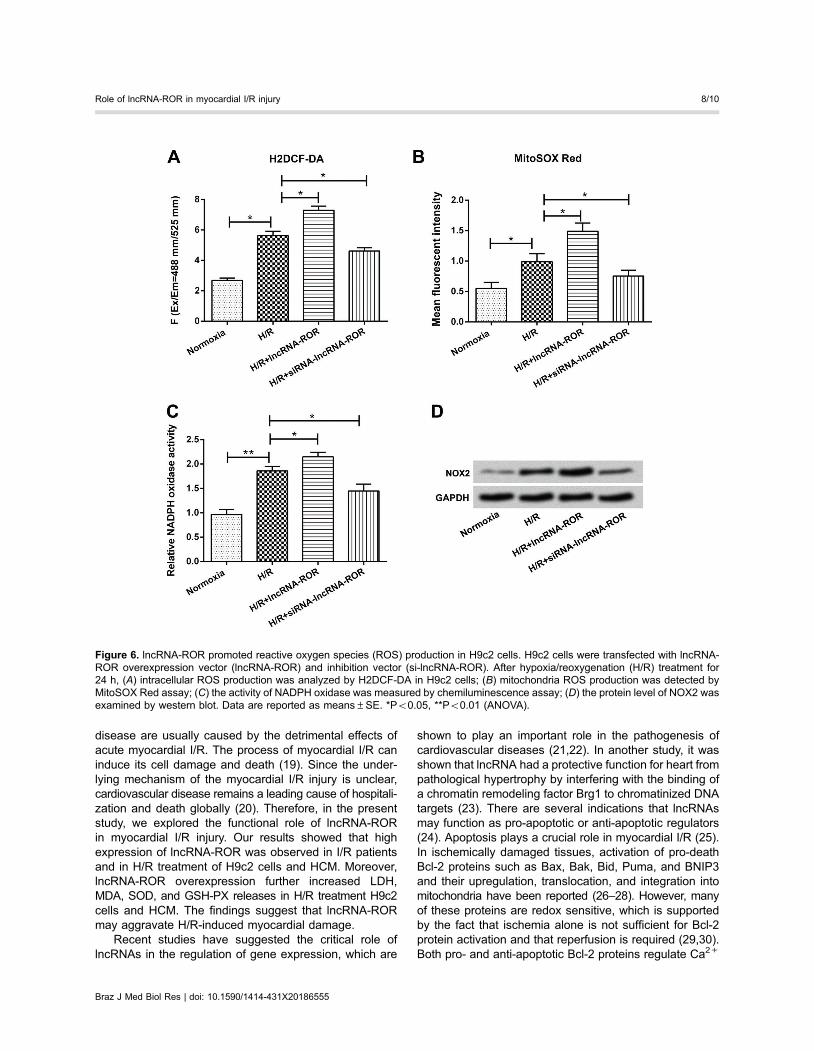
activated by oxidative stress. Therefore, we examined thelevels of intracellular ROS production in H9c2 cells. Asshown in Figure 6A, H/R treatment significantly enhanced thelevel of ROS formation, and the effect was further promotedby overexpression of lncRNA-ROR (Po0.05 or Po0.01).However, suppression of lncRNA-ROR reversed the result(Po0.01). To further explore the source of ROS, MitoSOXRed was used to measure mitochondria ROS production.
Figure 4. lncRNA-ROR mediated myocardial hypoxia/reoxygenation (H/R) by regulating the p38/MAPK pathway. H9c2 cells and humancardiomyocytes (HCM) were transfected with lncRNA-ROR overexpression vector (lncRNA-ROR) and inhibition vector (si-lncRNA-ROR).After H/R treatment for 24 h, (A) the protein levels of ERK and p38 were examined by western blot and (B) the mRNA expressions ofERK and p38 were determined by qRT-PCR. Data are reported as means±SE. *Po0.05, **Po0.01 (ANOVA).
Braz J Med Biol Res | doi: 10.1590/1414-431X20186555
Role of lncRNA-ROR in myocardial I/R injury 6/10
In Figure 6B, the ROS in mitochondria was significantlyincreased in H/R treatment condition (Po0.05). Over-expression of lncRNA-ROR further promoted mitochondriaROS production (Po0.05). However, the promoting effectwere declined by lncRNA-ROR suppression (Po0.05).These results were similar with intracellular ROS production.
NADPH oxidase plays a crucial role in ROS produc-tion, and NOX2 was an important member of the NADPHoxidase family. Therefore, NADPH oxidase activity andNOX2 protein level were examined in H9c2 cells. Asrevealed in Figure 6C, lncRNA-ROR overexpression enhancedH/R-induced NADPH oxidase activity (Po0.05). In contrast,lncRNA-ROR suppression decreased NADPH oxidase
activity (Po0.05). Western blot results displayed that theprotein level of NOX2 was increased by H/R treatment,and further enhanced by lncRNA-ROR overexpression.However, lncRNA-ROR suppression reduced NOX2 pro-tein level (Figure 6D). These data revealed that lncRNA-ROR could promote ROS formation in H9c2 cells.
Discussion
According to World Health Organization estimates,coronary heart disease is the leading cause of deathand about 17.5 million people died from cardiovasculardisease in 2012 (18). The effects of coronary heart
Figure 5. Inhibitor of p8/MAPK (SB203580) alleviated lncRNA-ROR-induced cell injury. H9c2 cells and human cardiomyocytes (HCM)were transfected with lncRNA-ROR overexpression vector (lncRNA-ROR) and inhibition of p8/MAPK (SB203580, 10 mM). After hypoxia/reoxygenation (H/R) treatment for 24 h, (A) cell viability, (B) apoptosis, and (C) and (D) apoptosis-related factors Bax, Bcl-2, cytochrome C,Smac/Diablo, cleaved-caspase-3, and cleaved-caspase-9 were measured by MTT, flow cytometry, qRT-PCR, and western blot, respectively.GAPDH served as the internal control. Data are reported as means±SE. *Po0.05, **Po0.01, ***Po0.001 (ANOVA).
Braz J Med Biol Res | doi: 10.1590/1414-431X20186555
Role of lncRNA-ROR in myocardial I/R injury 7/10
disease are usually caused by the detrimental effects ofacute myocardial I/R. The process of myocardial I/R caninduce its cell damage and death (19). Since the under-lying mechanism of the myocardial I/R injury is unclear,cardiovascular disease remains a leading cause of hospitali-zation and death globally (20). Therefore, in the presentstudy, we explored the functional role of lncRNA-RORin myocardial I/R injury. Our results showed that highexpression of lncRNA-ROR was observed in I/R patientsand in H/R treatment of H9c2 cells and HCM. Moreover,lncRNA-ROR overexpression further increased LDH,MDA, SOD, and GSH-PX releases in H/R treatment H9c2cells and HCM. The findings suggest that lncRNA-RORmay aggravate H/R-induced myocardial damage.
Recent studies have suggested the critical role oflncRNAs in the regulation of gene expression, which are
shown to play an important role in the pathogenesis ofcardiovascular diseases (21,22). In another study, it wasshown that lncRNA had a protective function for heart frompathological hypertrophy by interfering with the binding ofa chromatin remodeling factor Brg1 to chromatinized DNAtargets (23). There are several indications that lncRNAsmay function as pro-apoptotic or anti-apoptotic regulators(24). Apoptosis plays a crucial role in myocardial I/R (25).In ischemically damaged tissues, activation of pro-deathBcl-2 proteins such as Bax, Bak, Bid, Puma, and BNIP3and their upregulation, translocation, and integration intomitochondria have been reported (26–28). However, manyof these proteins are redox sensitive, which is supportedby the fact that ischemia alone is not sufficient for Bcl-2protein activation and that reperfusion is required (29,30).Both pro- and anti-apoptotic Bcl-2 proteins regulate Ca2+
Figure 6. lncRNA-ROR promoted reactive oxygen species (ROS) production in H9c2 cells. H9c2 cells were transfected with lncRNA-ROR overexpression vector (lncRNA-ROR) and inhibition vector (si-lncRNA-ROR). After hypoxia/reoxygenation (H/R) treatment for24 h, (A) intracellular ROS production was analyzed by H2DCF-DA in H9c2 cells; (B) mitochondria ROS production was detected byMitoSOX Red assay; (C) the activity of NADPH oxidase was measured by chemiluminescence assay; (D) the protein level of NOX2 wasexamined by western blot. Data are reported as means±SE. *Po0.05, **Po0.01 (ANOVA).
Braz J Med Biol Res | doi: 10.1590/1414-431X20186555
Role of lncRNA-ROR in myocardial I/R injury 8/10

homeostasis, which influences I/R injury (31). Our resultswere in line with these findings, which showed that due tooverexpression of lncRNA-ROR the level of expressionof Bcl-2 was decreased, which in turn led to a higherapoptosis rate. Furthermore, overexpression of lncRNA-ROR further increased the level of expression of Bax proteins.These findings indicated that lncRNA-ROR increased cardi-omyocyte apoptosis.
To further illustrate the underlying molecular mech-anism for apoptosis, which is mediated by lncRNA-ROR,MAPKs such as p38 and ERK were measured. Severalstudies have indicated that activation of p38 occurs duringI/R (32,33), whereas inhibition of p38 has shown reduc-tion in I/R-induced cell death (34,35). We observed thatlncRNA-ROR mediated myocardial H/R by regulatingthe p38/MAPK pathway. This was further proved by theimpact of addition of p38 inhibitor (SB203580) to theH9c2 cells. It was observed that SB203580 could rescuelncRNA-ROR-induced cell viability, expression of Bax andBcl-2, and reduce apoptotic cells rate. These findings aresimilar to the results obtained in other conditions such asrenal I/R injury cells (34), brain cells (35), and chronicmyelogenous leukemia K562 cells (36).
Recent studies have demonstrated that ROS is closelyrelated to diverse signal pathways including p38/MAPK (37).
Moreover, the production of ROS has been proven tobe involved in regulation of myocardial I/R injury (38). In arecent study, Kim et al. (39) demonstrated that PICOTalleviated myocardial I/R injury via decreasing intracellularlevels of ROS. Furthermore, mitochondrial and NADPHoxidase are important sources of ROS, and the NADPHoxidase family member of NOX2 exerted an important rolein ROS production (40). Based on these studies, we fur-ther explored the effect of lncRNA-ROR on ROS formationin myocardial I/R injury. We found that lncRNA-RORoverexpression significantly increased the production ofintracellular ROS and mitochondrial ROS. Moreover, theNADPH oxidase activity and NOX2 protein level were alsopromoted by lncRNA-ROR overexpression in H9c2 cells.These data indicated that lncRNA-ROR-promoted myo-cardial I/R injury might be associated with the inductionof ROS generation. Further studies still need to clarify thehypothesis.
In conclusion, we have shown that lncRNA-ROR playsa crucial role in myocardial I/R injury by regulation of thep38/MAPK signal pathway. Our results suggested thatlncRNA-ROR might be an important therapeutic targetfor myocardial I/R injury and this finding may help inthe development of a new strategy for the treatment ofmyocardial I/R injury.
References
1. Whelan RS, Kaplinskiy V, Kitsis RN. Cell death in the patho-genesis of heart disease: mechanisms and significance.Ann Rev Physiol 2010; 72: 19, doi: 10.1146/annurev.physiol.010908.163111.
2. Dabek J, Owczarek A, Gasior Z, Ulczok R, Skowerski M,Ku"ach A, et al. Oligonucleotide microarray analysis of genesregulating apoptosis in chronically ischemic and postinfarctionmyocardium. Biochem Genet 2008; 46: 241–247, doi: 10.1007/s10528-007-9137-3.
3. Thind GS, Agrawal PR, Hirsh B, Saravolatz L, Chen-ScarabelliC, Narula J, et al. Mechanisms of myocardial ischemia-reperfusion injury and the cytoprotective role of minocycline:scope and limitations. Future Cardiol 2015; 11: 61–76, doi:10.2217/fca.14.76.
4. Turer AT, Hill JA. Pathogenesis of Myocardial ischemia-reperfusion injury and rationale for therapy. Am J Cardiol2010; 106: 360–368, doi: 10.1016/j.amjcard.2010.03.032.
5. Zeng XC, Li XS, Wen H. Telmisartan protects against micro-vascular dysfunction during myocardial ischemia/reperfusioninjury by activation of peroxisome proliferator-activated receptorgamma. BMC Cardiovasc Disord 2013; 13: 39, doi: 10.1186/1471-2261-13-39.
6. Arslan F, Smeets MB, O’Neill LA, Keogh B, Mcguirk P,Timmers L, et al. Myocardial ischemia/reperfusion injury ismediated by leukocytic toll-like receptor-2 and reduced bysystemic administration of a novel anti-toll-like receptor-2antibody. Circulation 2010; 121: 80–90, doi: 10.1161/CIRCULATIONAHA.109.880187.
7. Cannon CP, Gibson CM, Lambrew CT, Shoultz DA, Levy D,French WJ, et al. Relationship of symptom-onset-to-balloon
time and door-to-balloon time with mortality in patients under-going angioplasty for acute myocardial infarction. JAMA2000; 283: 2941–2947, doi: 10.1001/jama.283.22.2941.
8. Arslan F, de Kleijn DP, Timmers L, Doevendans PA,Pasterkamp G. Bridging innate immunity and myocardialischemia/reperfusion injury: the search for therapeutic targets.Curr Pharmac Design 2008; 14: 1205–1216, doi: 10.2174/138161208784246090.
9. Sugden PH, Clerk A. ‘‘Stress-responsive’’ mitogen-acti-vated protein kinases (c-Jun N-terminal kinases and p38mitogen-activated protein kinases) in the myocardium. CircRes 1998; 83: 345–352, doi: 10.1161/01.RES.83.4.345.
10. Anversa P, Kajstura J. Myocyte cell death in the diseasedheart. Circ Res 1998; 82: 1231–1233, doi: 10.1161/01.RES.82.11.1231.
11. Mccully JD, Wakiyama H, Hsieh YJ, Jones M, Levitsky S.Differential contribution of necrosis and apoptosis in myo-cardial ischemia-reperfusion injury. Am J Physiol Heart CircPhysiol 2004; 286: H1923–H1935, doi: 10.1152/ajpheart.00935.2003.
12. Ponting CP, Oliver PL, Reik W. Evolution and functions oflong noncoding RNAs. Cell 2009; 136: 629, doi: 10.1016/j.cell.2009.02.006.
13. Archer K, Broskova Z, Bayoumi AS, Teoh J, Davila A, TangY, et al. Long non-coding RNAs as master regulators incardiovascular diseases. Int J Mol Sci 2015; 16: 23651–23667, doi: 10.3390/ijms161023651.
14. Gao W, Wang ZM, Zhu M, Lian XQ, Zhao H, Zhao D,et al. Altered long noncoding RNA expression profiles in themyocardium of rats with ischemic heart failure. J Cardiovasc
Braz J Med Biol Res | doi: 10.1590/1414-431X20186555
Role of lncRNA-ROR in myocardial I/R injury 9/10

Med 2015; 16: 473–479, doi: 10.2459/JCM.0b013e32836499cd.
15. Derrien T, Johnson R, Bussotti G, Tanzer A, Djebali S,Tilgner H, et al. The GENCODE v7 catalog of human longnoncoding RNAs: Analysis of their gene structure, evolution,and expression. Genome Res 2012; 22: 1775, doi: 10.1101/gr.132159.111.
16. Wu Z, Qi Y, Guo Z, Li P, Zhou D. miR-613 suppressesischemia-reperfusion-induced cardiomyocyte apoptosis bytargeting the programmed cell death 10 gene. Biosci Trends2016; 10: 251–257, doi: 10.5582/bst.2016.01122.
17. Cariello M, Simone S, Loverre A, Gigante M, Incampo F,Pietanza S, et al. Coagulation activation is associatedwith nicotinamide adenine dinucleotide phosphate oxidase-dependent reactive oxygen species generation in hemodia-lysis patients. Antioxid Redox Signal 2012; 16: 428–439,doi: 10.1089/ars.2011.4062.
18. Cardiovascular diseases (CVDs). Available at: http://www.who.int/mediacentre/factsheets/fs317/en/.
19. Yellon DM, Hausenloy DJ. Myocardial reperfusion injury.N Engl J Med 2007; 357: 1121–1135, doi: 10.1056/NEJMra071667.
20. Barnett P, Hoff MJBVD. Cardiac regeneration: different cellssame goal. Med Biol Eng Comput 2011; 49: 723–732, doi:10.1007/s11517-011-0776-5.
21. Thum T, Condorelli G. Long noncoding RNAs and micro-RNAs in cardiovascular pathophysiology. Circ Res 2015;116: 751, doi: 10.1161/CIRCRESAHA.116.303549.
22. Condorelli G, Latronico MV, Cavarretta E. microRNAs incardiovascular diseases: current knowledge and the roadahead. J Am Coll Cardiol 2014; 63: 2177–2187, doi: 10.1016/j.jacc.2014.01.050.
23. Liu ZG, Tang B, Zeng Y, He N, Wang ZY, Han H, et al.Mitochondrial genome of a spontaneous multiple mye-loma bone cancer model mouse C57BL/KaLwRij strain.Mitochondrial DNA A DNA Mapp Seq Anal 2015; 27:4071–4072.
24. Gazzaniga FS, Blackburn EH. An anti-apoptotic role fortelomerase RNA in human immune cells independent oftelomere integrity or telomerase enzymatic activity. Blood2014; 124: 3675–3684, doi: 10.1182/blood-2014-06-582254.
25. Puyal J, Vaslin A, Mottier V, Clarke PG. Postischemictreatment of neonatal cerebral ischemia should target auto-phagy. Ann Neurol 2009; 66: 378–389, doi: 10.1002/ana.21714.
26. Wei Q, Yin XM, Wang MH, Dong Z. Bid deficiencyameliorates ischemic renal failure and delays animal deathin C57BL/6 mice. Am J Physiol Renal Physiol 2006; 290:F35–F42, doi: 10.1152/ajprenal.00184.2005.
27. Wu B, Qiu W, Wang P, Yu H, Cheng T, Zambetti GP, et al.p53 independent induction of PUMA mediates intestinalapoptosis in response to ischaemia-reperfusion. Gut 2007;56: 645, doi: 10.1136/gut.2006.101683.
28. Metukuri MR, Beerstolz D, Namas RA, Dhupar R, Torres A,Loughran PA, et al. Expression and subcellular localization
of BNIP3 in hypoxic hepatocytes and liver stress. Am JPhysiol Gastrointest Liver Physiol 2009; 296: G499, doi:10.1152/ajpgi.90526.2008.
29. Diwan A, Krenz M, Syed FM, Wansapura J, Ren X, KoestersAG, et al. Inhibition of ischemic cardiomyocyte apoptosisthrough targeted ablation of Bnip3 restrains postinfarc-tion remodeling in mice. J Clin Invest 2007; 117: 2825–2833, doi: 10.1172/JCI32490.
30. Ben-Ari Z, Pappo O, Cheporko Y, Yasovich N, Offen D,Shainberg A, et al. Bax ablation protects against hepaticischemia/reperfusion injury in transgenic mice. Liver Transpl2007; 13: 1181–1188, doi: 10.1002/lt.21221.
31. Scorrano L, Oakes SA, Opferman JT, Cheng EH, SorcinelliMD, Pozzan T, et al. BAX and BAK regulation of endo-plasmic reticulum Ca2+: A control point for apoptosis. Science2003; 300: 135–139, doi: 10.1126/science.1081208.
32. Harding SJ, Browne GJ, Miller BW, Prigent SA, Dickens M.Activation of ASK1, downstream MAPKK and MAPK isoformsduring cardiac ischaemia. Biochim Biophys Acta 2010; 1802:733–740, doi: 10.1016/j.bbadis.2010.06.005.
33. Takagi Y, Nozaki K, Sugino T, Hattori I, Hashimoto N.Phosphorylation of c-Jun NH(2)-terminal kinase and p38mitogen-activated protein kinase after transient forebrainischemia in mice. Neurosci Letters 2000; 294: 117, doi: 10.1016/S0304-3940(00)01552-4.
34. Rongshan LI, Ding T, Liu X, Caixia LI. Influence ofSB203580 on Cell apoptosis and P38MAPK in renalischemia/reperfusion injury. J Huazhong Univ Sci TechnologMed Sci 2006; 26: 50–52, doi: 10.1007/BF02828037.
35. Piao CS, Kim JB, Han PL, Lee JK. Administration of thep38 MAPK inhibitor SB203580 affords brain protection witha wide therapeutic window against focal ischemic insult.J Neurosci Res 2003; 73: 537–544, doi: 10.1002/jnr.10671.
36. Ha SH. Jellyfish extract induces apoptotic cell death throughthe p38 pathway and cell cycle arrest in chronic myelogen-ous leukemia K562 cells. PeerJ 2017; 5: e2895, doi:10.7717/peerj.2895.
37. Duan F, Yu Y, Guan R, Xu Z, Liang H, Hong L. Vitamin K2induces mitochondria-related apoptosis in human bladdercancer cells via ROS and JNK/p38 MAPK signal pathways.PlosOne 2016; 11: e0161886, doi: 10.1371/journal.pone.0161886.
38. Pisarenko O, Shulzhenko V, Studneva I, Pelogeykina Y,Timoshin A, Anesia R, et al. Structural apelin analogues:mitochondrial ROS inhibition and cardiometabolic protectionin myocardial ischaemia reperfusion injury. Br J Pharmacol2015; 172: 2933–2945, doi: 10.1111/bph.13038.
39. Kim J, Kim J, Kook H, Park WJ. PICOTalleviates myocardialischemia-reperfusion injury by reducing intracellular levels ofreactive oxygen species. Biochem Biophys Res Commun2017; 485: 807–813, doi: 10.1016/j.bbrc.2017.02.136.
40. Lin N, Zhang H, Su Q. Advanced glycation end-productsinduce injury to pancreatic beta cells through oxidative stress.Diabetes Metab 2012; 38: 250, doi: 10.1016/j.diabet.2012.01.003.
Braz J Med Biol Res | doi: 10.1590/1414-431X20186555
Role of lncRNA-ROR in myocardial I/R injury 10/10





























