Embed Size (px)
Citation preview

Manipulating Unconventional CH-Based Hydrogen Bonding in aMethyltransferase via Noncanonical Amino Acid MutagenesisScott Horowitz,†,‡,§ Upendra Adhikari,∥ Lynnette M. A. Dirk,⊥ Paul A. Del Rizzo,¶ Ryan A. Mehl,#
Robert L. Houtz,⊥ Hashim M. Al-Hashimi,‡,□,△ Steve Scheiner,∥ and Raymond C. Trievel*,¶
†Howard Hughes Medical Institute, Ann Arbor, Michigan 48109, United States
Departments of ‡Biophysics, §Molecular, Cellular, and Developmental Biology, ¶Biological Chemistry, and □Chemistry, University ofMichigan, Ann Arbor, Michigan 48109, United States∥Department of Chemistry and Biochemistry, Utah State University, Logan, Utah 84322, United States⊥Department of Horticulture, University of Kentucky, Lexington, Kentucky 40546, United States#Department of Biochemistry and Biophysics, Oregon State University, Corvallis, Oregon 97331, United States
*S Supporting Information
ABSTRACT: Recent studies have demonstrated that the active sites of S-adenosylmethionine (AdoMet)-dependent methyltransferases form strong carbon−oxygen (CH···O) hydrogen bonds with the substrate’s sulfonium group that areimportant in AdoMet binding and catalysis. To probe these interactions, we substitutedthe noncanonical amino acid p-aminophenylalanine (pAF) for the active site tyrosine inthe lysine methyltransferase SET7/9, which forms multiple CH···O hydrogen bonds toAdoMet and is invariant in SET domain enzymes. Using quantum chemistrycalculations to predict the mutation’s effects, coupled with biochemical and structuralstudies, we observed that pAF forms a strong CH···N hydrogen bond to AdoMet that isoffset by an energetically unfavorable amine group rotamer within the SET7/9 activesite that hinders AdoMet binding and activity. Together, these results illustrate that theinvariant tyrosine in SET domain methyltransferases functions as an essential hydrogenbonding hub and cannot be readily substituted by residues bearing other hydrogen bondacceptors.
AdoMet-dependent methylation is essential to numerouscellular processes involving metabolism and signal trans-
duction. In this reaction, the methyl group is transferred fromthe AdoMet sulfonium cation to a nucleophilic acceptor atomvia an SN2 reaction mechanism. Recent work suggests thatunconventional CH···O hydrogen bonds1 are important inmultiple aspects of this reaction, as AdoMet is capable offorming CH···O hydrogen bonds that are stronger than thosetypically observed in biological systems due to its cationiccharacter.2,3 A crystallographic survey illustrated that the CH···O hydrogen bonds are conserved across different classes ofAdoMet-dependent methyltransferases through apparent con-vergent evolution, suggesting that these interactions may beideally suited to facilitating methyl transfer.3 To investigate thispossibility, we characterized the functions of CH···O hydrogenbonds between AdoMet and active site residues in the lysinemethyltransferase SET7/9 and found that these interactions areimportant to substrate binding and electrostatic transition statestabilization, as well as restricting the motion of the AdoMetmethyl group, presumably promoting catalysis.3 Among theseactive site residues, the hydroxyl group of Tyr335, an invariantresidue in the SET domain class, acts as an acceptor for manyCH···O hydrogen bonds, in addition to donating a hydrogenbond to the backbone carbonyl group of Ala295 in SET7/9
(Figure 1A). Mutation of this residue to a phenylalanine(Y335F) severely impaired AdoMet binding affinity, demon-strating the importance of the CH···O hydrogen bonds tosubstrate recognition. Interestingly, the methyl transfer rate wasnot appreciably altered by this mutation, indicating that theTyr335-mediated CH···O hydrogen bonding with AdoMetstabilizes the enzyme−substrate complex and transition statescomparably. Building from this initial characterization, wesought to evaluate the relative importance of the OH···O andfour CH···O hydrogen bonds formed by Tyr335 in the SET7/9active site (Figure 1A) to gain a more detailed understanding ofthe roles of CH···O hydrogen bonding in methylation.Toward this end, we undertook a combinatorial approach
using quantum mechanical (QM) calculations, crystallography,and biochemistry with the goal of utilizing the QM calculationsto model the active site energetics of wild type (WT) SET7/9and Tyr335 mutants, which could then be directly comparedwith experimentally measured dissociation constants. Thisapproach enabled us to rationally perturb the active sitehydrogen bonding pattern and then analyze the resulting effects
Received: February 17, 2014Accepted: June 10, 2014Published: June 10, 2014
Letters
pubs.acs.org/acschemicalbiology
© 2014 American Chemical Society 1692 dx.doi.org/10.1021/cb5001185 | ACS Chem. Biol. 2014, 9, 1692−1697

upon methyltransferase function. To introduce a differenthydrogen bonding functionality into the active site, weemployed an amber stop-codon suppression strategy togenetically substitute Tyr335 in SET7/9 with the noncanonicalamino acid (ncAA) p-aminophenylalanine (pAF).4 This aminoacid is isosteric to tyrosine but replaces the hydroxyl group withan amine moiety. We reasoned that pAF would act as a CH···N
hydrogen bond acceptor to AdoMet, while simultaneouslyreplacing the OH···O hydrogen bond of the WT enzyme withan NH···O hydrogen bond to the Ala295 carbonyl group, thuslimiting alterations in the protein’s structure and stability.Although the Y335pAF mutant should be capable of acceptinghydrogen bonds, it possesses one less lone pair of electronscompared to a tyrosine hydroxyl group, thus limiting the abilityof pAF to act as a multifurcated hydrogen bond acceptor.Conversely, the extra hydrogen atom in the pAF amine groupwould potentially introduce additional geometric and stericconstraints within the SET7/9 active site.Prior to undertaking the QM calculations to predict the local
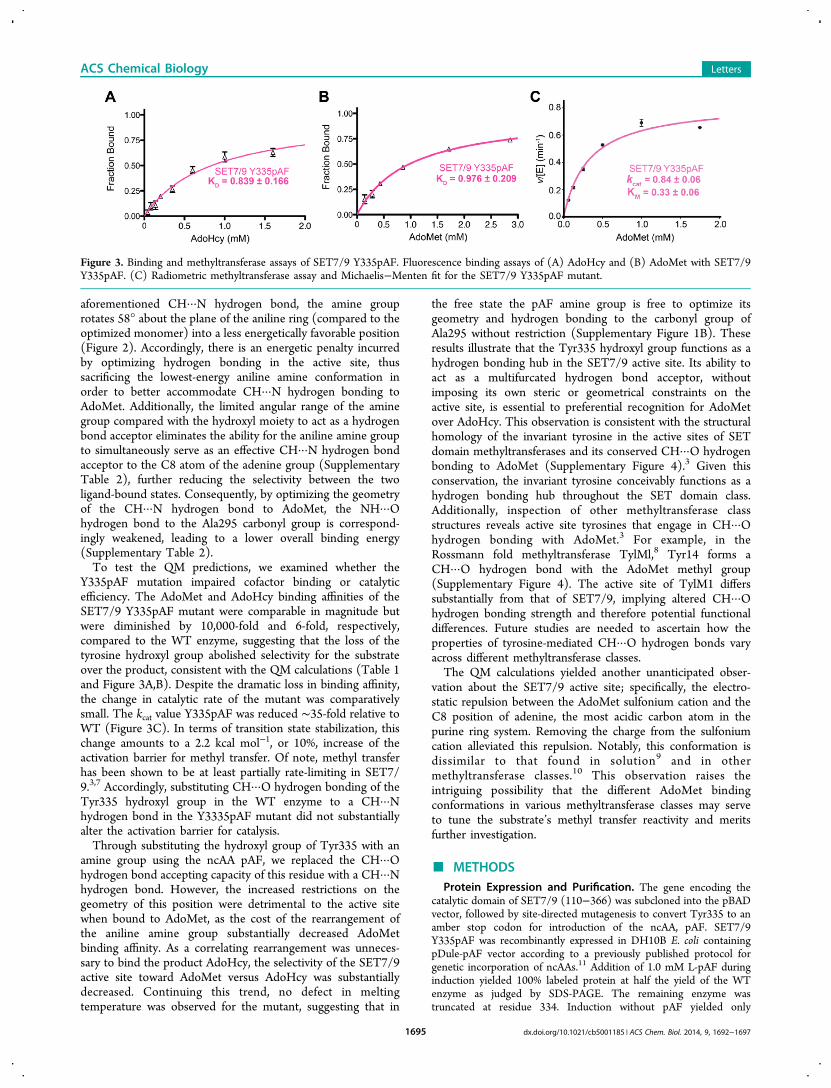
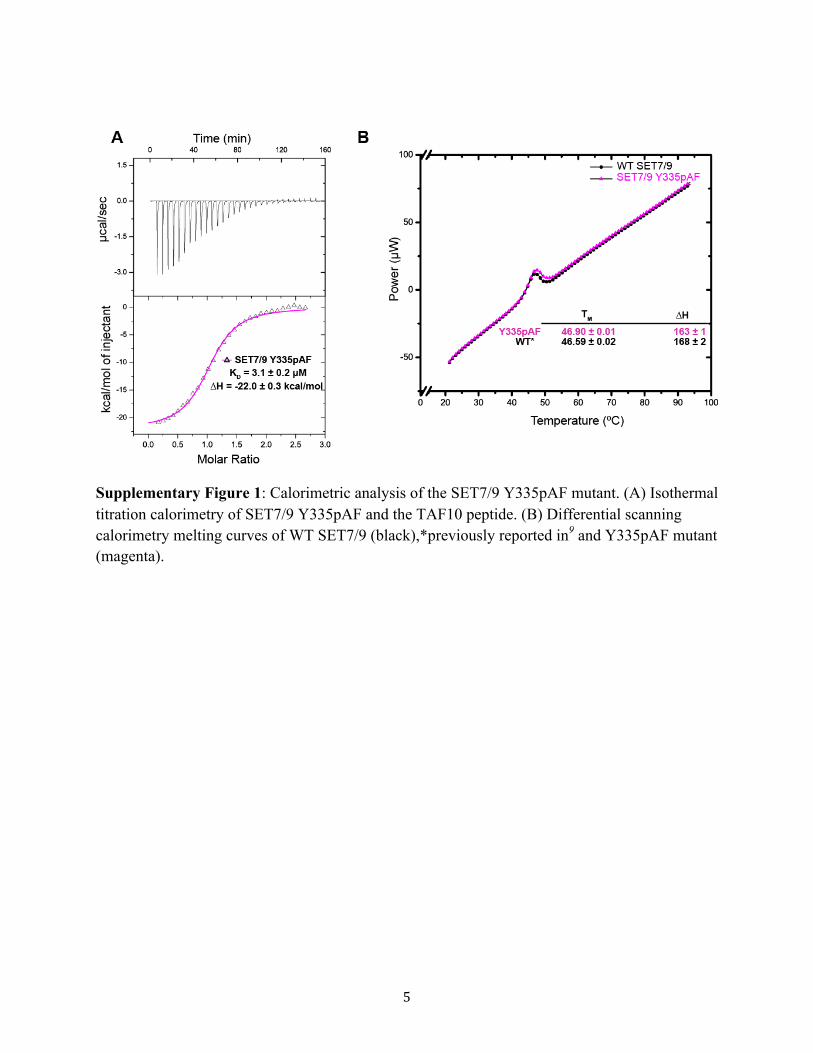
effects of this mutation, we performed a series of controlexperiments to determine whether the SET7/9 Y335pAFmutant would be suitable for probing SET7/9 active site CH···O hydrogen bonding. Isothermal titration calorimetry experi-ments demonstrated the binding affinity of the Y335pAFmutant to a substrate peptide representing the methylation sitein the general transcription factor TAF10 was essentiallyidentical to that of WT SET7/9 (Supplementary Figure 1A). Inaddition, differential scanning calorimetry illustrated that theWT enzyme and Y335pAF mutant exhibited essentiallyidentical stabilities, consistent with the formation of a hydrogenbond between the Ala295 carbonyl group and pAF aminegroup, as well as the maintenance of the neutral charge on theY335 side chain (Supplementary Figure 1B and SupplementaryText). To further examine the effects of this mutation on theenzyme’s overall and active site structures, we determined itscocrystal structure in complex with AdoHcy and a TAF10peptide at 1.6 Å resolution (Figure 1 and Supplementary Table1).5 The coordinates of this complex and the analogous WTcomplex superimpose with a Cα RMSD of 0.14 Å, illustratingthat the WT enzyme and mutant are structurally homologous.Further, the structural hydrogen bond between Tyr335 and thecarbonyl group of Ala295 is preserved by the pAF substitution(Figure 1B). The conventional hydrogen bonds between theenzyme and its ligands, AdoHcy and the TAF10 peptide, alsoappear to be preserved, as are the overall conformations theactive site residues compared with the WT enzyme. Together,these results demonstrate that the SET7/9 Y335pAF mutantretains the structure and protein substrate binding properties ofthe WT enzyme.To develop a molecular-level understanding of the effect of
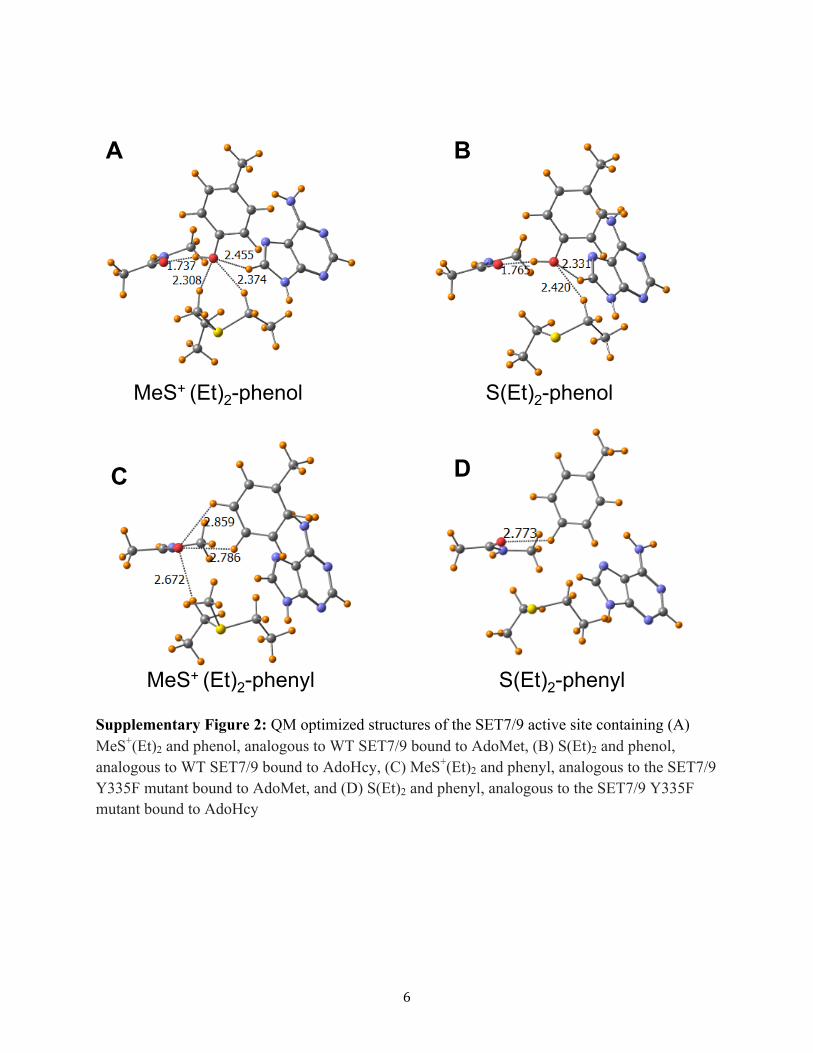
changing the Tyr335 hydrogen bonding pattern, QMcalculations were performed to model the effects ofsubstitutions at the Tyr335 position on ligand binding. Webegan with a comparison of the energetics of binding asulfonium cation mimic of AdoMet, MeS+(Et)2, to that of athioether mimic of AdoHcy, S(Et)2. Notably, the sulfur atom ofAdoHcy is a neutral thioether moiety due to the loss of themethyl group, unlike the sulfonium cation of AdoMet. Due tothe highly homologous AdoMet and AdoHcy binding modes inthe WT enzyme and Tyr335 mutants, which is reflected in thecalculations (see below), trends in the difference in bindingaffinities of AdoHcy and AdoMet between the differentmutants should be well represented by calculations that focuson the active site. Completing the model active site using thecrystal structure of SET7/9, a phenol molecule was inserted inthe position of Tyr335, and an N-methylacetamide (NMA)molecule was placed in the position of Ala295 so as to simulatethe hydrogen bonding of its peptide group. The adenine groupof the cofactors was also included to account for the CH···Ohydrogen bond between its C8 atom and the Tyr335 hydroxyl
Figure 1. Structure of WT SET7/9 and Y335pAF mutant bound toAdoHcy and TAF10 peptide. (A) WT SET7/9 bound to AdoMet andTAF10 K189A (4J83.pdb). CH···O hydrogen bonds formed byAdoMet and the OH···O hydrogen bond between Ala295 are denotedby orange and cyan dashes, respectively.3 All four CH···O hydrogenbonds accepted by the hydroxyl group of Tyr335 are probed by thepAF mutation. (B) Active site of the SET7/9 Y335pAF mutant (PDBcode: 4J7F). The NH···O hydrogen bond between Ala295 andpAF335 and van der Waals contact between pAF335 and the AdoHcyadenine group are denoted by cyan and magenta dashes, respectively.(C) Simulated annealing omit map of the SET7/9 Y335pAF active sitecontoured at 2.0σ.
ACS Chemical Biology Letters
dx.doi.org/10.1021/cb5001185 | ACS Chem. Biol. 2014, 9, 1692−16971693

group (Supplementary Figure 2A,B). Since a full geometryoptimization of this four-unit model, without the restraints ofthe rest of the system, would likely take the geometry far afieldfrom the structure in the enzyme, several selected atoms werefixed in their X-ray positions during geometry optimizations(Supplementary Figure 3; please see the Supplementary Textfor a discussion of model parameters and dielectric).The total binding energy was calculated as the difference in
energy between the optimized complex and the sum of theenergies of the four subunits, each with its geometry optimizedseparately. This quantity reflects not only the direct interactionof the AdoMet, or other moiety, with the active site but alsoany changes induced by the AdoMet on the three other groupsin the site. This total binding energy can also be decomposedinto pairwise terms that reflect the strength of the interactionbetween any given pair of subunits to assist in the analysis. Thisquantity was computed first for the AdoHcy and AdoMetsystems and then for the Y335pAF and Y335F mutants inwhich the phenol group in the WT complex was substitutedwith an aniline and a benzene molecule, respectively (Figure 2and Supplementary Figure 2C,D).Examining the wild type substrate and product models, the
active site energy is more favorable for the MeS+(Et)2-containing complex compared to S(Et)2 complex by 11 kcalmol−1, consistent with past experiments (Table 1).3,6
Considering the pairwise energies, this preference arises dueto the strong attractive interaction of the NMA and Tyr335groups with the positively charged AdoMet (SupplementaryTable 2). In contrast, in the benzene and aniline mutations, theselectivity for MeS+(Et)2 over S(Et)2 is dramatically reduced(Table 1). This loss of selectivity is similar in magnitudebetween the two mutants. However, analyzing the pairwiseinteractions in the two mutants, the origin of the loss of bindingenergy differential is clearly different in each case (Supple-
mentary Table 2). In the case of the phenylalanine mutant, theprimary cause for the loss in selectivity is the abrogation of theTyr335-mediated hydrogen bonds, similar to our previousinterpretation.3 In contrast, the aniline group forms a strongCH···N hydrogen bond to the sulfonium group. The loss inbinding energy in the aniline case instead stems primarily froma change in conformation of the aniline amine group. Toaccommodate the methyl group and simultaneously form the
Figure 2. QM calculations of the aniline ring in the SET7/9 Y335pAF active site. Active sites of (A and C) MeS+(Et)2 and (B and D) S(Et)2,depicting the rotation of the amine group by 58° compared with the (E) optimized monomer aniline structure.
Table 1. Binding and Kinetic Data and QM EnergyCalculations of WT SET7/9 and Tyr335 Mutantsa
KD (μM)
WT Y335pAF Y335F
AdoMet 0.053 ± 0.003b 980 ± 210 52 ± 26c
AdoHcy 134 ± 36c 840 ± 170 714 ± 197c
TAF10 4.9 ± 0.1d 3.1 ± 0.2 8.8 ± 0.3c
Kinetic Parameters
WT Y335pAF Y335F
kcat (min−1) 29.6 ± 0.9c 0.84 ± 0.06 45.2 ± 3.6c
KM (μM) 2.5 ± 0.3c 330 ± 60 150 ± 30c
kcat/KM (μM min−1) 12.0 ± 1.5c 0.0026 ± 0.0005 0.31 ± 0.068c
QM energy calculations (kcal mol−1)
phenol aniline benzene
MeS+(Et)2 complex energy −27.9 −16.1 −10.8S(Et)2 complex energy −16.9 −10.1 −5.5
aThe KM and kcat/KM values are reported with respect to AdoMet. QMenergies shown are the total binding energies of the model complexes,minus the energy of the individual optimized monomer structuresusing M06-2X/6-31+G**. bThis research was originally published inref 23. Copyright the American Society for Biochemistry andMolecular Biology. cReproduced from ref 3. dThis research wasoriginally published in ref 5. Copyright the American Society forBiochemistry and Molecular Biology.
ACS Chemical Biology Letters
dx.doi.org/10.1021/cb5001185 | ACS Chem. Biol. 2014, 9, 1692−16971694
aforementioned CH···N hydrogen bond, the amine grouprotates 58° about the plane of the aniline ring (compared to theoptimized monomer) into a less energetically favorable position(Figure 2). Accordingly, there is an energetic penalty incurredby optimizing hydrogen bonding in the active site, thussacrificing the lowest-energy aniline amine conformation inorder to better accommodate CH···N hydrogen bonding toAdoMet. Additionally, the limited angular range of the aminegroup compared with the hydroxyl moiety to act as a hydrogenbond acceptor eliminates the ability for the aniline amine groupto simultaneously serve as an effective CH···N hydrogen bondacceptor to the C8 atom of the adenine group (SupplementaryTable 2), further reducing the selectivity between the twoligand-bound states. Consequently, by optimizing the geometryof the CH···N hydrogen bond to AdoMet, the NH···Ohydrogen bond to the Ala295 carbonyl group is correspond-ingly weakened, leading to a lower overall binding energy(Supplementary Table 2).To test the QM predictions, we examined whether the
Y335pAF mutation impaired cofactor binding or catalyticefficiency. The AdoMet and AdoHcy binding affinities of theSET7/9 Y335pAF mutant were comparable in magnitude butwere diminished by 10,000-fold and 6-fold, respectively,compared to the WT enzyme, suggesting that the loss of thetyrosine hydroxyl group abolished selectivity for the substrateover the product, consistent with the QM calculations (Table 1and Figure 3A,B). Despite the dramatic loss in binding affinity,the change in catalytic rate of the mutant was comparativelysmall. The kcat value Y335pAF was reduced ∼35-fold relative toWT (Figure 3C). In terms of transition state stabilization, thischange amounts to a 2.2 kcal mol−1, or 10%, increase of theactivation barrier for methyl transfer. Of note, methyl transferhas been shown to be at least partially rate-limiting in SET7/9.3,7 Accordingly, substituting CH···O hydrogen bonding of theTyr335 hydroxyl group in the WT enzyme to a CH···Nhydrogen bond in the Y3335pAF mutant did not substantiallyalter the activation barrier for catalysis.Through substituting the hydroxyl group of Tyr335 with an
amine group using the ncAA pAF, we replaced the CH···Ohydrogen bond accepting capacity of this residue with a CH···Nhydrogen bond. However, the increased restrictions on thegeometry of this position were detrimental to the active sitewhen bound to AdoMet, as the cost of the rearrangement ofthe aniline amine group substantially decreased AdoMetbinding affinity. As a correlating rearrangement was unneces-sary to bind the product AdoHcy, the selectivity of the SET7/9active site toward AdoMet versus AdoHcy was substantiallydecreased. Continuing this trend, no defect in meltingtemperature was observed for the mutant, suggesting that in
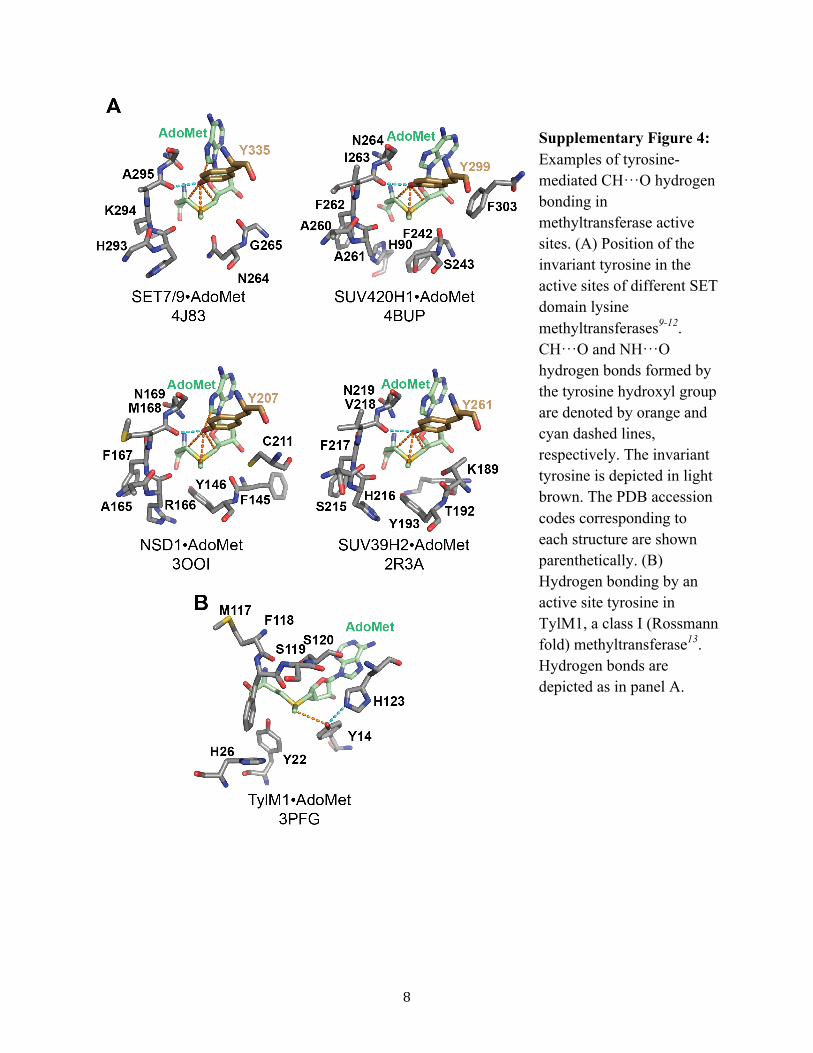
the free state the pAF amine group is free to optimize itsgeometry and hydrogen bonding to the carbonyl group ofAla295 without restriction (Supplementary Figure 1B). Theseresults illustrate that the Tyr335 hydroxyl group functions as ahydrogen bonding hub in the SET7/9 active site. Its ability toact as a multifurcated hydrogen bond acceptor, withoutimposing its own steric or geometrical constraints on theactive site, is essential to preferential recognition for AdoMetover AdoHcy. This observation is consistent with the structuralhomology of the invariant tyrosine in the active sites of SETdomain methyltransferases and its conserved CH···O hydrogenbonding to AdoMet (Supplementary Figure 4).3 Given thisconservation, the invariant tyrosine conceivably functions as ahydrogen bonding hub throughout the SET domain class.Additionally, inspection of other methyltransferase classstructures reveals active site tyrosines that engage in CH···Ohydrogen bonding with AdoMet.3 For example, in theRossmann fold methyltransferase TylMl,8 Tyr14 forms aCH···O hydrogen bond with the AdoMet methyl group(Supplementary Figure 4). The active site of TylM1 differssubstantially from that of SET7/9, implying altered CH···Ohydrogen bonding strength and therefore potential functionaldifferences. Future studies are needed to ascertain how theproperties of tyrosine-mediated CH···O hydrogen bonds varyacross different methyltransferase classes.The QM calculations yielded another unanticipated obser-
vation about the SET7/9 active site; specifically, the electro-static repulsion between the AdoMet sulfonium cation and theC8 position of adenine, the most acidic carbon atom in thepurine ring system. Removing the charge from the sulfoniumcation alleviated this repulsion. Notably, this conformation isdissimilar to that found in solution9 and in othermethyltransferase classes.10 This observation raises theintriguing possibility that the different AdoMet bindingconformations in various methyltransferase classes may serveto tune the substrate’s methyl transfer reactivity and meritsfurther investigation.
■ METHODSProtein Expression and Purification. The gene encoding the
catalytic domain of SET7/9 (110−366) was subcloned into the pBADvector, followed by site-directed mutagenesis to convert Tyr335 to anamber stop codon for introduction of the ncAA, pAF. SET7/9Y335pAF was recombinantly expressed in DH10B E. coli containingpDule-pAF vector according to a previously published protocol forgenetic incorporation of ncAAs.11 Addition of 1.0 mM L-pAF duringinduction yielded 100% labeled protein at half the yield of the WTenzyme as judged by SDS-PAGE. The remaining enzyme wastruncated at residue 334. Induction without pAF yielded only
Figure 3. Binding and methyltransferase assays of SET7/9 Y335pAF. Fluorescence binding assays of (A) AdoHcy and (B) AdoMet with SET7/9Y335pAF. (C) Radiometric methyltransferase assay and Michaelis−Menten fit for the SET7/9 Y335pAF mutant.
ACS Chemical Biology Letters
dx.doi.org/10.1021/cb5001185 | ACS Chem. Biol. 2014, 9, 1692−16971695

SET7/9 truncated at residue 334. SET7/9 Y335pAF was purified aspreviously described,12 with the exception that gel filtrationchromatography was performed using a HiLoad Superdex 75 column(GE Healthcare) for the pAF mutant. This chromatography effectivelyseparated the SET7/9 Y335pAF from the truncated enzyme based onthe difference in their elution profiles, permitting the latter to beeliminated as a contaminant.X-ray Crystallography and Biochemical Assays. Y335F
SET7/9 crystals were grown by hanging drop vapor diffusion aspreviously described3,5 (Supplementary Table 1), in which thecrystallization solution was mixed in a 1:1 ratio with 10−12 mgmL−1 SET7/9, 3.0−4.5 mM AdoHcy, and 2.0 mM of a 10-residuepeptide of TAF10 peptide in 20 mM TRIS pH 8.0, 100 mM NaCl, and2.0 mM Tris(2-carboxyethyl)phosphine. Crystallization solutionscontained 0.86−1.07 M sodium citrate with 100 mM imidazole, pH7.6−8.4 and 8.0−18 mM NiCl2. Crystals were flash frozen in 1.5 Msodium citrate, with 1.0 mM AdoHcy, 10−13 mM NiCl2, and 100 mMimidazole pH 8.2−8.7. Diffraction data were collected at the AdvancedPhoton Source Synchrotron beamline 21-IDG (LS-CAT), at 100 Kand wavelength 0.9786 nm and were processed using HKL2000.13 Thestructure was solved by molecular replacement using MOLREP,14 withthe previously reported protein coordinates of the SET7/9 ternarycomplex (PDB accession code 3M53)5 as the search model. Modelbuilding and refinement were carried out using Coot,15 MiFit (version2010.10, http://code.google.com/p/mifit), and REFMAC.16 Thequality of the final model was verified by Molprobity.17 CNS wasused to generate simulated annealing omit maps,18 with K189 of theTAF10 peptide, pAF, and AdoHcy omitted. RMSD values werecalculated and structural figure rendered in PyMol (Schrodinger,LLC).Radiometric methyltransferase kinetics assays (using 10 μM
Y335pAF mutant), tryptophan fluorescence binding experiments,ITC, and DSC were performed as described previously.3
QM Calculations. All quantum mechanical calculations werecarried out via the Gaussian-09 package.19 Density functional theorywith M06-2X variant of functional20 was used with the 6-31+G** basisset. This level of theory has been found to be in good agreement withexperimental values and MP2/aug-cc-pVDZ level of theory.2,21,22
Binding energies were calculated as the difference between the energyof the complex and sum of the optimized monomers and werecorrected for basis set superposition error using the counterpoiseprocedure. Structural figures were rendered in Chemcraft (Chem-craft).
■ ASSOCIATED CONTENT
*S Supporting InformationAdditional discussion of the protonation status of residue 335in SET7/9, QM modeling of the SET7/9 active site, effect ofthe dielectric constant on selectivity, and supplementary datatables and figures. This material is available free of charge viathe Internet at http://pubs.acs.org.
Accession CodesThe coordinates and structure factors for the SET7/9Y335pAF·AdoHcy·TAF10 peptide complex (accession code4J7F) have been deposited in the RCSB Protein Data Bank.
■ AUTHOR INFORMATION
Corresponding Author*E-mail: [email protected].
Present Address△Department of Biochemistry, Duke University, Durham, NC27710, USA.
NotesThe authors declare no competing financial interest.
■ ACKNOWLEDGMENTS
This work was supported by NSF-MCB-0448297 to R.A.M., bygrants from the University of Michigan’s Biomedical ResearchCouncil and the Office for the Vice President for Research andNSF (CHE-1213484) to R.C.T., the Kentucky AgricultureExperiment station Hatch Project #KY011031 to R.L.H. andL.M.A.D., and NSF-CHE-1026826 to S.S. Use of the AdvancedPhoton Source, an Office of Science User Facility operated forthe U.S. Department of Energy (DOE) Office of Science byArgonne National Laboratory, was supported by the U.S. DOEunder Contract No. DE-AC02-06CH11357. Use of the LS-CAT Sector 21 was supported by the Michigan EconomicDevelopment Corporation and the Michigan Technology Tri-Corridor (Grant 085P1000817).
■ REFERENCES(1) Horowitz, S., and Trievel, R. C. (2012) Carbon-oxygen hydrogenbonding in biological structure and function. J. Biol. Chem. 287,41576−41582.(2) Adhikari, U., and Scheiner, S. (2013) Magnitude and mechanismof charge enhancement of CH..O hydrogen bonds. J. Phys. Chem. A117, 10551−10562.(3) Horowitz, S., Dirk, L. M., Yesselman, J. D., Nimtz, J. S., Adhikari,U., Mehl, R. A., Scheiner, S., Houtz, R. L., Al-Hashimi, H. M., andTrievel, R. C. (2013) Conservation and functional importance ofcarbon-oxygen hydrogen bonding in AdoMet-dependent methyltrans-ferases. J. Am. Chem. Soc. 135, 15536−15548.(4) Mehl, R. A., Anderson, J. C., Santoro, S. W., Wang, L., Martin, A.B., King, D. S., Horn, D. M., and Schultz, P. G. (2003) Generation of abacterium with a 21 amino acid genetic code. J. Am. Chem. Soc. 125,935−939.(5) Del Rizzo, P. A., Couture, J. F., Dirk, L. M. A., Strunk, B. S.,Roiko, M. S., Brunzelle, J. S., Houtz, R. L., and Trievel, R. C. (2010)SET7/9 catalytic mutants reveal the role of active site water moleculesin lysine multiple methylation. J. Biol. Chem. 285, 31849−31858.(6) Couture, J. F., Hauk, G., Thompson, M. J., Blackburn, G. M., andTrievel, R. C. (2006) Catalytic roles for carbon-oxygen hydrogenbonding in SET domain lysine methyltransferases. J. Biol. Chem. 281,19280−19287.(7) Ibanez, G., McBean, J. L., Astudillo, Y. M., and Luo, M. K. (2010)An enzyme-coupled ultrasensitive luminescence assay for proteinmethyltransferases. Anal. Biochem. 401, 203−210.(8) Carney, A. E., and Holden, H. M. (2011) Molecular architectureof TylM1 from Streptomyces fradiae: an N,N-dimethyltransferaseinvolved in the production of dTDP-D-mycaminose. Biochemistry 50,780−787.(9) Markham, G. D., and Reczkowski, R. S. (2004) Structural studiesof inhibition of S-adenosylmethionine synthetase by slow, tight-binding intermediate and product analogues. Biochemistry 43, 3415−3425.(10) Schubert, H. L., Blumenthal, R. M., and Cheng, X. D. (2003)Many paths to methyltransfer: a chronicle of convergence. TrendsBiochem. Sci. 28, 329−335.(11) Hammill, J. T., Miyake-Stoner, S., Hazen, J. L., Jackson, J. C.,and Mehl, R. A. (2007) Preparation of site-specifically labeledfluorinated proteins for 19F-NMR structural characterization. Nat.Protoc. 2, 2601−2607.(12) Trievel, R. C., Beach, B. M., Dirk, L. M. A., Houtz, R. L., andHurley, J. H. (2002) Structure and catalytic mechanism of a SETdomain protein methyltransferase. Cell 111, 91−103.(13) Otwinowski, Z., and Minor, W. (1997) Processing of X-raydiffraction data collected in oscillation mode. Methods Enzymol. 276,307−326.(14) Vagin, A., and Teplyakov, A. (2000) An approach to multi-copysearch in molecular replacement. Acta Crystallogr. D 56, 1622−1624.(15) Emsley, P., Lohkamp, B., Scott, W. G., and Cowtan, K. (2010)Features and development of Coot. Acta Crystallogr. D 66, 486−501.
ACS Chemical Biology Letters
dx.doi.org/10.1021/cb5001185 | ACS Chem. Biol. 2014, 9, 1692−16971696

(16) Vagin, A. A., Steiner, R. A., Lebedev, A. A., Potterton, L.,McNicholas, S., Long, F., and Murshudov, G. N. (2004) REFMAC5dictionary: organization of prior chemical knowledge and guidelinesfor its use. Acta Crystallogr. D 60, 2184−2195.(17) Chen, V. B., Arendall, W. B., Headd, J. J., Keedy, D. A.,Immormino, R. M., Kapral, G. J., Murray, L. W., Richardson, J. S., andRichardson, D. C. (2010) MolProbity: all-atom structure validation formacromolecular crystallography. Acta Crystallogr. D 66, 12−21.(18) Brunger, A. T., Adams, P. D., Clore, G. M., DeLano, W. L.,Gros, P., Grosse-Kunstleve, R. W., Jiang, J. S., Kuszewski, J., Nilges, M.,Pannu, N. S., Read, R. J., Rice, L. M., Simonson, T., and Warren, G. L.(1998) Crystallography & NMR system: A new software suite formacromolecular structure determination. Acta Crystallogr. D 54, 905−921.(19) Frisch, M. J., Trucks, G. W., Schlegel, H. B., Scuseria, G. E.,Robb, M. A., Cheeseman, J. R., Scalmani, G., Barone, V., Mennucci, B.,Petersson, G. A., Nakatsuji, H., Caricato, M., Li, X., Hratchian, H. P.,Izmaylov, A. F., Bloino, J., Zheng, G., Sonnenberg, J. L., Hada, M.,Ehara, M., Toyota, K., Fukuda, R., Hasegawa, J., Ishida, M., Nakajima,T., Honda, Y., Kitao, O., Nakai, H., Vreven, T., Montgomery, J. A., Jr.,Peralta, J. E., Ogliaro, F., Bearpark, M., Heyd, J. J., Brothers, E., Kudin,K. N., Staroverov, V. N., Kobayashi, R., Normand, J., Raghavachari, K.,Rendell, A., Burant, J. C., Iyengar, S. S., Tomasi, J., Cossi, M., Rega, N.,Millam, J. M., Klene, M., Knox, J. E., Cross, J. B., Bakken, V., Adamo,C., Jaramillo, J., Gomperts, R., Stratmann, R. E., Yazyev, O., Austin, A.J., Cammi, R., Pomelli, C., Ochterski, J. W., Martin, R. L., Morokuma,K., Zakrzewski, V. G., Voth, G. A., Salvador, P., Dannenberg, J. J.,Dapprich, S., Daniels, A. D., Farkas, O., Foresman, J. B., Ortiz, J. V.,Cioslowski, J., and Fox, D. J. (2009) Gaussian 09, Revision B.01,Gaussian, Inc., Wallingford, CT.(20) Zhao, Y., and Truhlar, D. G. (2006) Comparative DFT study ofvan der waals complexes: Rare-gas dimers, alkaline-earth dimers, zincdimer, and zinc-rare-gas dimers. J. Phys. Chem. A 110, 5121−5129.(21) Vincent, M. A., and Hillier, I. H. (2011) The structure andinteraction energies of the weak complexes of CHClF2 and CHF3with HCCH: a test of density functional theory methods. Phys. Chem.Chem. Phys. 13, 4388−4392.(22) Majumder, M., Mishra, B. K., and Sathyamurthy, N. (2013) CHcenter dot center dot center dot pi and pi center dot center dot centerdot pi interaction in benzene-acetylene clusters. Chem. Phys. Lett. 557,59−65.(23) Horowitz, S., Yesselman, J. D., Al-Hashimi, H. M., and Trievel,R. C. (2011) Direct evidence for methyl group coordination bycarbon-oxygen hydrogen bonds in the lysine methyltransferase SET7/9. J. Biol. Chem. 286, 18658−18663.
ACS Chemical Biology Letters
dx.doi.org/10.1021/cb5001185 | ACS Chem. Biol. 2014, 9, 1692−16971697

1
Manipulating Unconventional CH-based Hydrogen Bonding in a
Methyltransferase via Non-Canonical Amino Acid Mutagenesis
Supporting Information
Scott Horowitz, Upendra Adhikari, Lynnette M.A. Dirk, Paul A. Del Rizzo, Ryan A. Mehl,
Robert L. Houtz, Hashim M. Al-Hashimi, Steve Scheiner, and Raymond C. Trievel

2
Supplementary Text: Protonation Status of Residue 335 in SET7/9
One consideration in substituting the active Tyr335 in SET7/9 with the non-canonical amino acid (ncAA) pAF is the possibility of introducing a positive charge into the enzyme’s active site by the protonation of the aniline side chain of pAF. It is worth noting that the pKA value of the aniline ammonium cation has a value of 4.61, implying that the aniline side chain of pAF would exist in the deprotonated, uncharged state at the neutral to basic pH values at which the SET7/9 Y335pAF mutant was characterized. Nonetheless, it is possible that the side chains of tyrosine and pAF differ in charge in the active site environment despite their isostericity and the relatively low pKA value of aniline2, 3. To examine this possibility, we analyzed the thermal stability, structure, and substrate binding properties of the SET7/9 Y335pAF mutant. Measurement of the dissociation constant of the TAF10 peptide substrate for WT SET7/9 and the Y335pAF mutant by isothermal titration calorimetry demonstrated that the pAF substitution did not disrupt peptide substrate binding (Supplementary Figure 1A and Table 1). This observation is important because the ε-ammonium cation of the Lys189 substrate in the TAF10 peptide is positioned within ~4 Å of the Tyr335 hydroxyl group and Y335pAF amine group (Figure 1B). Consequently, if the pAF side chain was protonated, it would presumably result in strong electrostatic repulsion with the Lys189 ε-ammonium cation, impairing the binding of the TAF10 peptide to the Y335pAF mutant. In addition, the differential scanning calorimetry experiments demonstrated that pAF substitution did not alter the thermal stability of the Y335pAF mutant compared to WT SET7/9 (Supplementary Figure 1B), consistent with a conservation of the charge at residue 335 in the active site. Finally, the 1.6 Å resolution crystal structure of the SET7/9 Y335pAF mutant illustrates that its overall active site structure is essentially unchanged compared to the structure of WT SET7/9 (Figure 1A-B). Based on these findings, we conclude that the Y335pAF mutation does not alter the charge at this position in the active site of SET7/9. QM Modeling of the SET7/9 Active Site and the Effect of the Dielectric Constant on Selectivity
Using a combination of QM modeling and experiment, we re-engineered the hydrogen bonding network within the active site of methyltransferase SET7/9. Of course, the reduction of the entire enzymatic system to a four-unit complex cannot be expected to recapitulate the subtleties of this entire system, nor the dynamic motions of groups within the enzyme. The calculations discussed in the main text also do not include the polarizing effects of surrounding enzymatic groups, nor of the solvent. Nonetheless, the trends in the QM calculations mirrored the experimental data, facilitating an understanding of the underlying causes of the selectivity of this system.
It is well established that the properties of a sulfonium group, and particularly its hydrogen bond donating capabilities, may in part depend on its solvation4, 5, and thus we performed tests to ensure that the predicted trends are robust through different dielectric environments. As may be seen in Table 1, the selectivity for MeS+(Et)2 over S(Et)2 is 11.0 kcal mol-1 for phenol, 6.0 for aniline, and 5.3 for benzene. In other words, phenol has a higher selectivity than the other two respective species by 5.0 and 5.7 kcal mol-1. These calculations were repeated within the context of a polarizable medium, so as to simulate the effects of the surrounding protein. When the environment is modeled by a dielectric constant of 4, a value that is commonly taken for a protein interior6-8, these values drop slightly to 4.5 and 1.9 kcal mol-1, retaining the higher selectivity of phenol. Even in the extreme situation of a very high dielectric

3
constant, viz. 80 as in water, the selectivity advantage of phenol over the other two species remains as high as 5.5 and 2.7 kcal mol-1. It is concluded that the effects observed in vacuo are qualitatively unaffected by immersion of the system in an environment more akin to a protein interior.
Supplementary Table 1: Crystallographic and refinement statistics for the structure of SET7/9 Y335pAF•AdoHcy•TAF10 peptide complex SET7/9 Y335pAF• AdoHcy•TAF10 peptide PDB Code 4J7F Data collection Space group P3221 Cell dimensions a, b, c (Å) 83.0, 83.0, 95.5 α, β, γ (°) 90, 90, 120 Resolution (Å) 50.0-1.60 (1.64-1.60) Rmerge (%) 7.7 (44.2) I/sI 26.7 (3.1) Completeness (%) 99.4 (94.9) Redundancy 10.7 (4.8) Refinement Resolution (Å) 1.60 No. of Reflections 48,193 Rwork/ Rfree 0.20/0.22 No. of Atoms 1889 Protein 1941 Ligand/ion 75 Water 171 B-factors 23.9 Protein 23.4 Ligand/ion 34.9 Water 32 R.M.S Deviations Bond lengths (Å) 0.014 Bond angles (º) 1.51 MolProbity Score Percentile 84% Resolution Range (Å) 1.59 ± 0.25 Ramachandran Favored (%) 95.4 Allowed (%) 4.6 Outliers (%) 0

4
Supplementary Table 2: Pairwise interaction energies (Y=phenol, aniline or benzene, X=MeS+(Et)2 or S(Et)2) in kcal mol-1
MeS+ (Et)2 complexes S(Et)2 complexes Phenol Aniline Benzene Phenol Aniline Benzene Adenine---X 4.57 4.59 4.51 -0.50 -0.53 -0.54 Adenine---NMA -1.44 -1.46 -1.45 -1.50 -1.50 -1.53 Adenine---Y -2.11 -0.27 0.12 -2.11 -2.60 0.09 Y---NMA -9.96 -2.08 -2.07 -10.47 -4.87 -2.43 Y---X -9.36 -11.70 -2.76 -1.44 -2.02 -0.38 NMA---X -10.15 -10.02 -9.94 -0.84 -0.78 -0.83 total -28.45 -20.94 -11.60 -16.86 -12.32 -5.62 Minus opt monomers -27.93 -16.08 -10.83 -16.92 -10.05 -5.53
5
Supplementary Figure 1: Calorimetric analysis of the SET7/9 Y335pAF mutant. (A) Isothermal titration calorimetry of SET7/9 Y335pAF and the TAF10 peptide. (B) Differential scanning calorimetry melting curves of WT SET7/9 (black),*previously reported in9 and Y335pAF mutant (magenta).
6
Supplementary Figure 2: QM optimized structures of the SET7/9 active site containing (A) MeS+(Et)2 and phenol, analogous to WT SET7/9 bound to AdoMet, (B) S(Et)2 and phenol, analogous to WT SET7/9 bound to AdoHcy, (C) MeS+(Et)2 and phenyl, analogous to the SET7/9 Y335F mutant bound to AdoMet, and (D) S(Et)2 and phenyl, analogous to the SET7/9 Y335F mutant bound to AdoHcy
MeS+ (Et)2-phenol S(Et)2-phenol
MeS+ (Et)2-phenyl S(Et)2-phenyl
A B
C D

7
Supplementary Figure 3: AdoMet complex where asterisks indicate atoms frozen into X-ray coordinates during optimization.
8
Supplementary Figure 4: Examples of tyrosine-mediated CH···O hydrogen bonding in methyltransferase active sites. (A) Position of the invariant tyrosine in the active sites of different SET domain lysine methyltransferases9-12. CH···O and NH···O hydrogen bonds formed by the tyrosine hydroxyl group are denoted by orange and cyan dashed lines, respectively. The invariant tyrosine is depicted in light brown. The PDB accession codes corresponding to each structure are shown parenthetically. (B) Hydrogen bonding by an active site tyrosine in TylM1, a class I (Rossmann fold) methyltransferase13. Hydrogen bonds are depicted as in panel A.

9
References
[1] Gross, K. C., and Seybold, P. G. (2000) Substituent effects on the physical properties and pKa of aniline, Int J Quantum Chem 80, 1107-1115.
[2] Gutbezahl, B., and Grunwald, E. (1953) The Effect of Solvent on Equilibrium and Rate Constants .2. The Measurement and Correlation of Acid Dissociation Constants of Anilinium and Ammonium Salts in the System Ethanol Water, J Am Chem Soc 75, 559-565.
[3] Bagno, A., and Terrier, F. (2001) Carbon and nitrogen basicity of aminothiophenes and anilines, J Phys Chem A 105, 6537-6542.
[4] Markham, G. D., and Bock, C. W. (1996) The interaction of water with sulfonium ions and the effects of hydration on the energetics of methyl group transfer: An ab initio molecular orbital study of the hydration of (CH3)(3) S+ and (CH3)(2) S+CH2CO2-, Struct Chem 7, 281-300.
[5] Adhikari, U., and Scheiner, S. (2013) Magnitude and mechanism of charge enhancement of CH..O hydrogen bonds, J Phys Chem A 117, 10551-10562.
[6] Wang, Z. X., and Duan, Y. (2004) Solvation effects on alanine dipeptide: A MP2/cc-pVTZ//MP2/6-31G** study of (Phi, Psi) energy maps and conformers in the gas phase, ether, and water, J Comput Chem 25, 1699-1716.
[7] Simonson, T., and Perahia, D. (1995) Internal and Interfacial Dielectric-Properties of Cytochrome-C from Molecular-Dynamics in Aqueous-Solution, Proc Natl Acad Sci USA 92, 1082-1086.
[8] Dwyer, J. J., Gittis, A. G., Karp, D. A., Lattman, E. E., Spencer, D. S., Stites, W. E., and Garcia-Moreno, B. (2000) High apparent dielectric constants in the interior of a protein reflect water penetration, Biophys J 79, 1610-1620.
[9] Horowitz, S., Dirk, L. M., Yesselman, J. D., Nimtz, J. S., Adhikari, U., Mehl, R. A., Scheiner, S., Houtz, R. L., Al-Hashimi, H. M., and Trievel, R. C. (2013) Conservation and functional importance of carbon-oxygen hydrogen bonding in AdoMet-dependent methyltransferases, J Am Chem Soc 135, 15536-15548.
[10] Southall, S. M., Cronin, N. B., and Wilson, J. R. (2014) A novel route to product specificity in the Suv4-20 family of histone H4K20 methyltransferases, Nucleic Acids Res 42, 661-671.
[11] Qiao, Q., Li, Y., Chen, Z., Wang, M., Reinberg, D., and Xu, R. M. (2011) The structure of NSD1 reveals an autoregulatory mechanism underlying histone H3K36 methylation, J Biol Chem 286, 8361-8368.
[12] Wu, H., Min, J., Lunin, V. V., Antoshenko, T., Dombrovski, L., Zeng, H., Allali-Hassani, A., Campagna-Slater, V., Vedadi, M., Arrowsmith, C. H., Plotnikov, A. N., and Schapira, M. (2010) Structural biology of human H3K9 methyltransferases, Plos One 5, e8570.
[13] Carney, A. E., and Holden, H. M. (2011) Molecular architecture of TylM1 from Streptomyces fradiae: an N,N-dimethyltransferase involved in the production of dTDP-D-mycaminose, Biochemistry 50, 780-787.





























