Embed Size (px)
Citation preview

March 14, 2017
Dockets Management Branch (HFA-305)
Food and Drug Administration
5630 Fishers Lane, Rm. 1061
Rockville, MD 20852
Re: Docket No. FDA-2016-D-4460: Multiple Endpoints in Clinical Trials
Dear Sir/Madam:
The Biotechnology Innovation Organization (BIO) thanks the Food and Drug Administration
(FDA) for the opportunity to submit comments on the Draft Guidance “Multiple Endpoints in
Clinical Trials” (Draft Guidance).
BIO is the world's largest trade association representing biotechnology companies, academic
institutions, state biotechnology centers and related organizations across the United States
and in more than 30 other nations. BIO members are involved in the research and
development of innovative healthcare, agricultural, industrial, and environmental
biotechnology products.
This important Draft Guidance was well written and provides instrumental guidance and
recommendations to handle major multiplicity problems in clinical trials. BIO believes that
the Draft Guidance is a very helpful document to Sponsors. However, we note that the Draft
Guidance fails to address a few points that are worthy of attention:
multiplicity adjustments at the interim analysis (either directly in this Draft Guidance or
via reference to ICH E9 or the Guidance for Industry Adaptive Design Clinical Trials for
Drugs and Biologics);
multiplicity adjustment on safety endpoints when assessing important safety signals as
part of the objectives of the trial (e.g., with p-values reported) to control the overall
false discovery rate of safety signals;
elucidation of the calculation methods to be used with the truncated Holm and truncated
Hochberg procedures (e.g., for the truncated Hochberg example, the α passed to the
next level); and
basic principles, such as closed test or partition principles, which many of the multiple
comparison methods in the guidance are based upon.
BIO further believes that it would be helpful if the Draft Guidance stated at the beginning,
perhaps more clearly and emphatically than it does now, the areas that are mentioned in
other sections as being out of scope. It would also be helpful to explain why these areas are
outside of the scope. For example, the Draft Guidance states that multiplicity in interim
analysis is not within the scope of the document (lines 180-184). This is something that
should also be stated in the introduction. Simulation based multiplicity adjustment is
another area that is not included in the guidance. If related topics are outside the scope but
they have been the subject of previous FDA guidance, such guidance should be referenced.

BIO Comments on Multiple Endpoints in Clinical Trials FDA Docket: FDA-2016-D-4460, March 14, 2017, Page 2 of 21
We note that in multiple places in the document, reference is made to the ‘endpoint’ when
we believe that reference is intended to be made to the hypothesis or the hypothesis test
(e.g., lines 252-253, 491-492, and 885). This language is likely sufficiently clear for many
readers of the guidance. However, FDA may consider correcting this language throughout
when revising the guidance. Alternatively, a statement early in the document can be
inserted which explains this use of terminology as interchangeable in the document. This
would help improve the clarity of the Draft Guidance.
Further, the Draft Guidance does not clearly indicate whether the Hochberg procedure is
recommended in the case of more than two correlated endpoints. Under the multivariate
central limit theorem, the joint density of the multiple test statistics for three non-negatively
correlated endpoints will follow a multivariate normal distribution in confirmatory trials
where large sample sizes are typically planned. Therefore, the Hochberg procedure
applicable to two correlated endpoints should apply to more than two non-negatively
correlated endpoints. It would be helpful if the Draft Guidance discusses and clarifies this
topic.
Additionally, we believe that “Section IV Statistical Methods” can be improved upon via
incorporation of alpha recycling (see below citations1). We suggest that FDA consider adding
a statement that incorporation of alpha recycling, as shown in the appendix on graphical
approaches, may provide extensions of the methods described in this section. We suggest
that FDA consider adding a paragraph referring to the appendix for graphical approaches
that can be used to improve the methods described in this section.
Regarding the graphical approach, BIO notes that many testing procedures described in the
Draft Guidance, including the Bonferroni method, the Holm procedure, the fixed-sequence
method, and the Fallback method, are special cases of multiple testing procedure. We
suggest FDA make the connection between the graphical approach and these special cases.
The Draft Guidance also considers the graphical approach as a visual presentation tool
instead of recognizing it as an extension of many commonly used statistical methods. It is
an important multiple testing procedure. Given the importance of the graphical approach
and its connection to other methods, including gate keeping testing strategies, it is worth
having a small section in the main text body (similar to re-sampling test method), yet still
retain the details in the appendix. Discussing it solely in the appendix will convey to some
readers that it is a less favored method. It provides uniformly more power across many
statistical situations. It also provides much greater flexibility in organizing the hypothesis
testing and the amount of alpha available to each hypothesis test. It also allows the clinical
importance of each endpoint to be taken into account. Finally, the graphical approach is also
extremely valuable as a communication tool. It communicates complex, hypothesis testing
plans that cannot be clearly communicated in text. However, because of its extensive
flexibility, it is not always clear which endpoints are the primary endpoints and which ones
are the secondary endpoints. The guidance should encourage sponsors that when they are
1 Burman CF, Sonesson C, Guilbaud O. A recycling framework for the construction of Bonferroni-based multiple tests. Statistics in Medicine. 2009; 28:739--761. Bretz F, Maurer W, Brannath W, Posch M. A graphical approach to sequentially rejective multiple test procedures. Statistics in Medicine} 2009; 28:586--604. Millen B, Dmitrienko A. Chain procedures: A class of flexible closed testing procedures with clinical trial applications. Statistics in Biopharmaceutical Research 2011; 3:14--30.

BIO Comments on Multiple Endpoints in Clinical Trials FDA Docket: FDA-2016-D-4460, March 14, 2017, Page 3 of 21
using the graphical approach, that they should clearly state the intended role of endpoints
(i.e., primary basis for approval vs. supportive). This will help to prevent misunderstandings
between the sponsor and the Agency about what is necessary for approval.
While we understand that the guidance cannot cover every statistical method that could be
relevant for multiple endpoints, we do believe it would be helpful if the guidance provided
some information about the criteria used to decide which methods to include. For instance,
a variety of innovative study designs are receiving greater attention, such as Umbrella
designs, Basket designs, and Platform designs. They are considered to have great potential
for making drug development more efficient. FDA should consider mentioning these in the
guidance. Additionally, it would be helpful if FDA provided some information about what
distinguishes the methods that were included in the main document versus the appendix.
Finally, we believe that an extension of this guidance or an additional guidance which more
broadly discusses the problem of “multiplicity” would be beneficial. In particular, we believe
that additional guidance regarding the interpretation of subgroup analyses would be helpful
in this guideline both for trials that did or did not meet their primary objective in the overall
population, respectively. Other topics of interest that could be addressed in an additional
guideline include analyses of multiple time-points (e.g., group-sequential trials), patient
selection (biomarker) subgroups, and multi-arm trials including trials with multiple doses
which may be modelled.
BIO appreciates this opportunity to submit comments on the Draft Guidance “Multiple
Endpoints in Clinical Trials”. We provide additional specific, detailed comments to improve
the clarity of the Draft Guidance in the following chart. We would be pleased to provide
further input or clarification of our comments, as needed.
Sincerely,
/S/
Cartier Esham, Ph.D.
Executive Vice President, Emerging Companies Section &
Vice President, Science & Regulatory Affairs
Biotechnology Innovation Organization

BIO Comments on Multiple Endpoints in Clinical Trials FDA Docket: FDA-2016-D-4460, March 14, 2017, Page 4 of 21
SPECIFIC COMMENTS
SECTION ISSUE PROPOSED CHANGE
I. INTRODUCTION
II. BACKGROUND AND SCOPE
A. Introduction to Study Endpoints
B. Demonstrating the Study Objective of Effectiveness
Line 130: The null hypotheses is presented as 2-sided, but the
alternative hypothesis is presented as 1-sided.
BIO asks FDA to make the null hypothesis 1-sided to be
consistent with the alternative hypothesis in this paragraph
and the other one refer to Line 142 to Line 145 in the last
paragraph of the section.
C. Type I Error
Lines 154-156,
158-162:
A definition of Type I error probability for the 2-sided
hypothesis tests is given at Lines 154-156 without
clearly stating this is a definition applied to the 2-
sided hypothesis tests. The same definition is
repeated at Lines 158-160 followed by a slightly
different definition of Type I error rate for 1-sided
hypothesis tests.
BIO suggest that FDA:
(1) re-write the sentence at Lines 154-156 to read as
“Under two-sided hypothesis tests, the probability of
concluding that there is a difference (beneficial or
harmful) between the drug and control when there is
no difference, is called the Type I error probability or
rate, denoted as alpha (α). and
(2) delete the first sentence in the 2nd paragraph of
Section II.C (Lines 158-160) as it repeats the same
definition that has been given at Lines 154-156.
Lines 168-170: The rationale as to why the use of 2-sided test would
provide strong assurance against the possibility of a
false-positive result (i.e., no more than 1 chance in
40), is a bit unclear (especially for non-statisticians).
BIO suggests re-writing this sentence to read:
“Use of a two-sided test with an alpha of 0.05 generally
allocates the alpha in a symmetric way, such also ensures
that the probability of falsely concluding the drug effect in
each of the two possible directions (benefit or harm) is no
more than 2.5 percent. benefit This ensures that the
BIO Comments on Multiple Endpoints in Clinical Trials FDA Docket: FDA-2016-D-4460, March 14, 2017, Page 5 of 21
SECTION ISSUE PROPOSED CHANGE
probability of falsely concluding drug benefit when there is
none is no more than approximately 2.5 percent (1 chance
in 40).”
Lines 170-172: The Draft Guidance reads, “…especially when
substantiated by another study or other confirmatory
evidence.”
BIO notes that this is not always the case, and it is
preferable to acknowledge that single studies may be
acceptable.
BIO suggests editing the text to read:
“....especially when substantiated by another study or other
confirmatory evidence. It should be noted that for orphan
diseases or diseases with high unmet medical needs, a single
study may be acceptable for regulatory approval.”
D. Relationship Between the Observed and True Treatment Effects
Lines 203-204: The Draft Guidance reads:
“An essential element of type I error rate control is
the prospective specification of:
all endpoints that will be tested and”
Since hypothesis testing is sometimes a part of
evaluating exploratory objectives which are out of
scope from a multiple testing perspective, we
recommend clarifying that the statement applies to
tests of ‘primary and secondary hypothesis tests’
rather than ‘all’ tests.
In order to clarify the Draft Guidance, BIO suggests editing
the text to read:
“An essential element of type I error rate control is the
prospective specification of:
all endpoints primary and secondary hypotheses that
will be tested and”
Lines 207-209: The Draft Guidance discusses the analysis plan for
multiple endpoints studies.
These lines in the draft guidance attempt to clarify
details needed in an analysis plan to pre-specify and
communicate a multiple testing plan. To more
BIO suggests replacing the text in this section with the
following:
The analysis plan should describe the multiple testing
procedure for the hypotheses being tested. This may include
specifying the ordering of testing the hypotheses, the initial
BIO Comments on Multiple Endpoints in Clinical Trials FDA Docket: FDA-2016-D-4460, March 14, 2017, Page 6 of 21
SECTION ISSUE PROPOSED CHANGE
completely accommodate the broad range of methods
to be conveyed, we suggest additions to this
statement which will be helpful for both reviewers
and practitioners in industry.
alpha allocation to the hypotheses, and the alpha
propagation rules (i.e., how alpha is passed from one
hypothesis test to another). These details may be
communicated in text and/or pictorials/graphics. (See
appendix for description of a graphical approach of
representing multiple testing procedures. Other conventions
are available as well. See, for example, Khordzhakia et al
(2012) and Dmitrienko et al (2013).) An alternative means
of describing the multiple testing procedure is to provide the
closed testing representation or partition testing
representation of the procedure. Any of these approaches is
sufficient to fully specify and communicate the details of the
procedure.
Lines 225-230: The Draft Guidance states, “The narrower the
confidence interval, and the further away its lower
bound is from the null hypothesis of no treatment
effect (T-C = 0 or T/C = 1), the more confident we
are of both the magnitude and existence of the
treatment effect. Generally, the farther the lower
bound of the confidence interval is from zero (or 1),
the more persuasive (smaller) the p-value is and the
lower the likelihood that the effectiveness finding was
a chance occurrence.”
Only the lower bound of CI’s is mentioned but in many
situations, the upper bound is of interest (for example, for
hazard ratios of survival endpoints). BIO suggests editing
the text to read:
“The narrower the confidence interval, and the further away
its lower or upper bound, respectively, (depending on the
direction of the effect of interest) is from the null hypothesis
of no treatment effect (T-C = 0 or T/C = 1), the more
confident we are of both the magnitude and existence of the
treatment effect. Generally, the farther the lower or upper
bound of the confidence interval is from zero (or 1), the
more persuasive (smaller) the p-value is and the lower the
likelihood that the effectiveness finding was a chance
occurrence.”
Lines 243-246: The Draft Guidance discusses confidence intervals for
testing and reads,
The text referencing confidence intervals for testing here
may be confusing to readers as referring to multiplicity-
adjusted confidence sets, which is generally out of scope for

BIO Comments on Multiple Endpoints in Clinical Trials FDA Docket: FDA-2016-D-4460, March 14, 2017, Page 7 of 21
SECTION ISSUE PROPOSED CHANGE
“…it is critical to ensure that confidence intervals
appropriately reflect multiplicity of hypothesis tests.”
the document. Use of a confidence interval for testing does
not present a special case to be addressed separately. The
guidance on testing is wholly adequate. As such, BIO
suggests removing these lines to avoid confusion or have the
following revision:
“…it is critical to ensure that the confidence level of
confidence intervals appropriately reflect multiplicity of
hypothesis tests.”
E. Multiplicity
Lines 269-271: The stated type I error rate of 0.1 is approximate.
The exact error rate is given just prior to this
statement. The proposed text corrects this minor
error.
BIO suggests editing the text to read:
“Without correction, the chance of making a type I error for
the study as a whole would be approximately 0.1 and…”
Lines 309-310: The text states that “post hoc analyses by
themselves cannot establish effectiveness.” However,
there are cases where the agency has exercised
flexibility in this regard. For example, Pravagard PAC
was approved on the basis of a post hoc meta-
analysis of individual patient data from previous
clinical trials of the same compound. Hence, this
statement may have to be modified to accommodate
unusual situations.
BIO suggests editing the text to read:
“Consequently, post hoc analyses by themselves generally
cannot establish effectiveness.”
III. MULTIPLE ENDPOINTS: GENERAL PRINCIPLES
A. The Hierarchy of Family of Endpoints
Lines 391-393: The Draft Guidance states, “The collection of all
secondary endpoints is called the secondary endpoint
family. Secondary endpoints are those that may
provide supportive information about a drug’s effect
BIO also asks FDA to please provide additional detail and
examples here or in Lines 327-334 on the requirements for
endpoints, beyond the primary endpoint, that can be
included in physician labeling.

BIO Comments on Multiple Endpoints in Clinical Trials FDA Docket: FDA-2016-D-4460, March 14, 2017, Page 8 of 21
SECTION ISSUE PROPOSED CHANGE
on the primary endpoint or demonstrate additional
effects on the disease or condition.”
Endpoints “that may provide supportive information
about a drug’s effect on the primary endpoint” and
endpoints that “demonstrate additional effects on the
disease or condition” seem quite different, and it is
not clear why the former should be considered a
secondary endpoint (i.e., require multiplicity control).
In addition, this seems inconsistent with lines 327-
337 which imply that such endpoints (i.e., those that
describe other attributes of a drug’s effects) can be
included in physician labeling without multiplicity
control. Therefore, the distinction between endpoints
that do and do not require multiplicity control is
unclear.
Lines 409-413: The Draft Guidance states, “Moreover, the Type I
error rate should be controlled for any preplanned
analysis of pooled results across studies; pooled
analyses are rarely conducted for the planned
primary endpoint, but are sometimes used to assess
lower frequency events, such as cardiovascular
deaths, where the individual trials used a composite
endpoint, such as death plus hospitalization.
Statistical testing strategies to accomplish this are
discussed in section IV.”
While the Draft Guidance indicates that discussions
on preplanned pooled analysis to assess lower
frequency events as secondary endpoints and
methods to control Type I error, whether separately
These lines refer to type I error rate control for analyses
based on pooled results across studies. BIO asks FDA to
provide more detail, if possible, on what is expected in this
setting.

BIO Comments on Multiple Endpoints in Clinical Trials FDA Docket: FDA-2016-D-4460, March 14, 2017, Page 9 of 21
SECTION ISSUE PROPOSED CHANGE
or using a pre-planned alpha allocation, are discussed
in section IV, this does not appear to be the case.
Lines 431-433: The draft Guidance reads, “…becomes increasingly
small as the number of endpoints increases”
This statement is not accurate because the statement
is not necessary true for some multiplicity adjustment
methods. For example, in the fixed-sequence
procedure, the full alpha is passed to the next
endpoint if the previous endpoint is statistically
significant at the 0.05 level. Therefore, the chance of
demonstrating an effect on a series of sequential
endpoints depends on the method used for
multiplicity adjustment.
BIO suggests the text be edited to read:
“could becomes increasingly small as the number of
endpoints increases.”
Lines 443-445: The Draft Guidance reads, “The study’s likelihood of
avoiding Type II error (1-β), if the drug actually has
the specified effect, is called study power. The
desired power is an important factor in determining
the sample size”
The type II error is β, not 1-β.
The study power is 1-β.
Power for the secondary endpoints is not typically
considered as power loss after passing the primary
endpoints. Power should be associated with the
sample size for the primary endpoint.
BIO suggest the editing the text to read:
“The study’s likelihood of avoiding Type II error (1-β), if the
drug actually has the specified effect, is called study power
(1-β). The desired power is an important factor in
determining the sample size, especially for the primary
endpoints.”
B. Type II Error Rate and Multiple Endpoints

BIO Comments on Multiple Endpoints in Clinical Trials FDA Docket: FDA-2016-D-4460, March 14, 2017, Page 10 of 21
SECTION ISSUE PROPOSED CHANGE
Lines 483-484: The draft Guidance states, “The loss of power may
not be so severe when the endpoints are correlated
(i.e., not fully independent)”.
This statement is not accurate because the loss of
power may be severe for weak correlated endpoints
or negatively correlated endpoints if one endpoint is
successful. The statement is true for endpoints with
strong positive correlation. Therefore, we revised the
statement to make it accurate.
In addition, the phrase “(i.e., not fully independent)”
is problematic for this statement. If the correlation of
endpoints is not high, then the power loss may be
severe for the endpoint with small treatment effect
size. Thus, we suggest deleting the parenthetical
statement.
BIO suggests editing the text to read:
“The loss of power may not be so severe when the endpoints
are strongly positively correlated (i.e., not fully
independent).”
C. Types of Multiple Endpoints
Line 507 BIO notes that the term “co-primary endpoints” is
used specifically for the case of all-of-N, must be
statistically significant. In some therapeutic areas
that term is also used for 1 of N.
BIO thinks it would be useful for FDA to mention the use of
the term “co-primary endpoints” for 1 of N in this discussion.
BIO also suggests the FDA clarifies that in this Guidance, the
term “co-primary endpoints” is exclusively used for the case
of all-of-N.
Lines 554-558: It is possible to test for co-primary endpoints in a
manner which controls the overall type I error at the
required alpha level (e.g., 0.05) yet does not require
each endpoint to be tested at the 0.05 level. See
Kordzhakia et al, 2010, for example.
BIO asks FDA to consider broadening the language on co-
primary endpoints to allow for the possibility mentioned:
When using co-primary endpoints, however there is
generally only one result that is considered a study success,
namely, that all of the separate endpoints are statistically
significant. With this approach, there is no opportunity for

BIO Comments on Multiple Endpoints in Clinical Trials FDA Docket: FDA-2016-D-4460, March 14, 2017, Page 11 of 21
SECTION ISSUE PROPOSED CHANGE
It should be permissible to select significance levels
for testing the individual endpoints in any way that
controls the overall Type I error at the required level
α, even if one or more of these individual significance
levels are greater than α. The set of admissible
individual significance levels will depend on the
number of co-primary endpoints as well as the
specific formulation of the null hypothesis, and the
final choice should be justified by an objective
demonstration of overall Type I error control.
inflation of the type I error rate; rather the impact of co-
primary endpoint testing is to increase the type II error rate.
While statistical significance for each co-primary endpoint is
sufficient to satisfy the requirement of overall type I error
control, it is also possible to maintain overall type I error
control using other significance levels for the individual
hypotheses. (See Kordzhakia et al, 2010, for example.)
Such an approach may be considered in some cases where
power is greatly reduced when simply testing each co-
primary endpoint at level alpha.
Lines 564-566: The Draft Guidance states, “Relaxation of alpha is
generally not acceptable because doing so would
undermine the assurance of an effect on each disease
aspect considered essential to showing that the drug
is effective in support of approval.”
When co-primary endpoints are required yet not
highly correlated (e.g., histological evidence and
symptom improvement), not relaxing alpha for each
individual co-primary component may result in much
reduced study-wise Type I error rate and
unnecessary large sample size, increased drug
development timeline and cost.
BIO suggests that FDA consider clarifying the ability of
sponsors to engage in application specific discussions.
Lines 596-631: This section on composite endpoints may touch on
recent developments, especially analysis that takes
clinical importance into account, such as win-ratio
analysis or weighted analysis.
BIO suggests FDA consider elaborating on some of these
new developments and analyses.
Lines 608-614: “An important reason for using a composite endpoint
is that the incidence rate of each of the events may
BIO suggests editing the text to read:

BIO Comments on Multiple Endpoints in Clinical Trials FDA Docket: FDA-2016-D-4460, March 14, 2017, Page 12 of 21
SECTION ISSUE PROPOSED CHANGE
be too low to allow a study of reasonable size to have
adequate power; the composite endpoint can provide
a substantially higher overall event rate that allows a
study with a reasonable sample size and study
duration to have adequate power. Composite
endpoints are often used when the goal of treatment
is to prevent or delay morbid, clinically important but
uncommon events (e.g., use of an anti-platelet drug
in patients with coronary artery disease to prevent
myocardial infarction, stroke, and death).”
BIO notes that another reason for composite
endpoints is when a more serious (but less common)
event occurs in the absence of a documented more
common (but less serious) event, for example deaths
in the absence of disease progression or relapse. In
such cases, the more severe event should not be
ignored.
“An important One possible reason for using a composite
endpoint is that the incidence rate of each of the events may
be too low to allow a study of reasonable size to have
adequate power; the composite endpoint can provide a
substantially higher overall event rate that allows a study
with a reasonable sample size and study duration to have
adequate power. Composite endpoints are often used when
the goal of treatment is to prevent or delay morbid, clinically
important but uncommon events (e.g., use of an anti-
platelet drug in patients with coronary artery disease to
prevent myocardial infarction, stroke, and death).”
Lines 678-679: The Draft Guidance states, “Study power can be
adversely affected, however, if there is limited
correlation among the endpoints.”
It is not clear to BIO whether the document is
referring to the issue of reverse multiplicity or
differences in underlying effect sizes.
BIO suggests the FDA provides clarification on this point.
Lines 689-691: BIO notes that it is frequently meaningful to add
“worse outcomes” (i.e., rare but clinically critical
endpoints) as components to the primary endpoints
to avoid interpretational issues due to competing
risks. We suggest comment or acknowledgement of
this principle.
BIO suggests editing the text to read:
“For many serious diseases, there is an endpoint of such
great clinical importance that it is unreasonable not to collect
and analyze the endpoint data; the usual example is
mortality or major morbidity events (e.g., stroke, fracture,
BIO Comments on Multiple Endpoints in Clinical Trials FDA Docket: FDA-2016-D-4460, March 14, 2017, Page 13 of 21
SECTION ISSUE PROPOSED CHANGE
pulmonary exacerbation). Even if relatively few of these
events are expected to occur in the trial, they may be
included it is frequently beneficial from an interpretational
perspective to include them in a composite endpoint (see
section III.C.3) and they may also be designated as a
planned secondary endpoint to potentially support a
conclusion regarding effect on that separate clinical
endpoint, if the effect of the drug on the composite primary
endpoint is demonstrated.”
D. The Individual Components of Composite Endpoints
Lines 730-733: The Draft Guidance states, “One approach considers
only the initial event in each patient. This method
displays the incidence of each type of component
event only when it was the first event for a patient.
The sum of the first events across all categories will
equal the total events for the composite endpoint.”
BIO asks the Agency to consider clarifying that sponsors
have additional options regarding analysis based on only the
initial event in each patient, due to a serious methodological
issue. For example, consider the analysis of ESRD in RENAAL
as described in the document. The numbers for ESRD under
“Decomposition of the primary endpoint” represents the
numbers of patients who had ESRD that was not preceded
by Doubling of Serum Creatinine. A reduction in this number
is necessarily accompanied by an equal increase in the sum
of these two categories of patients: 1) Those without ESRD
at all (a favorable outcome); and 2) Those with Doubling of
Serum Creatinine followed by ESRD (an unfavorable
outcome). In other words, the approach discussed by the
FDA essentially creates a composite endpoint that comprises
both favorable and unfavorable outcomes. This violates what
should be an absolute criterion for creation of composite
endpoints, because it is unclear whether an increase or a
decrease in this endpoint would be of benefit to the patient.
Lines 741-767: The example is very long without conclusions and
may not provide any helpful information.

BIO Comments on Multiple Endpoints in Clinical Trials FDA Docket: FDA-2016-D-4460, March 14, 2017, Page 14 of 21
SECTION ISSUE PROPOSED CHANGE
Line 757 (Table
1):
The footnote says “Hazard ratio from Cox
proportional hazards time-to-event analysis”.
However, as competing risks are present for these
analyses, it should be clarified that this refers to
models for the cause-specific hazards functions.
Moreover, in the presence of competing risks,
alternative regression models such as the Fine and
Gray would also be applicable.
BIO believes that this issue merits further discussion
and possibly a reference to a paper on competing
risks and multi-stage models. E.g. The “Tutorial in
biostatistics: competing risks and multi-state
models.” published in Statistics in Medicine by
Geskus et al (2007).
More generally, we think that further guidance on the
topic of competing risks, which is only briefly
mentioned in Lines 770-776, would be beneficial.
BIO suggests editing the text to read:
“Hazard ratio from cause-specific Cox proportional hazards
time-to-event analysis accounting for competing events. Of
note, component endpoints could alternatively be analyzed
on the cumulative incidence scale using the Fine and Gray
regression model.”
Lines 779-784: The Draft Guidance refers to “decomposition
analyses” but not define the term.
BIO finds the meaning of “decomposition” in this context is
not clear. BIO asks FDA to please clarify.
IV. STATISTICAL METHODS
A. Type I Error Rate for a Family of Endpoints and Conclusions on Individual Endpoints
B. When the Type I Error Rate is Not Inflated or When the Multiplicity Problem is Addressed Without Statistical Adjustment or by Other
Methods
Lines 933-937: The Draft Guidance reads, “but it is difficult to
estimate the increase in error rate because the
results of these different analyses are likely to be
similar and it is unclear how many choices could have
BIO believes that the guidance should clearly state that one
analysis method must be the primary method and pre-
specified so that a biased choice cannot be made. The
BIO Comments on Multiple Endpoints in Clinical Trials FDA Docket: FDA-2016-D-4460, March 14, 2017, Page 15 of 21
SECTION ISSUE PROPOSED CHANGE
been made. As with other multiplicity problems,
prospective specification of the analysis method will
generally eliminate the concern about a biased
(result-driven) choice.”
difficulty in estimating the increase in Type 1 error rate is
irrelevant. As such, BIO suggests editing the text to read:
“but it is difficult to estimate the increase in error rate
because the results of these different analyses are likely to
be similar and it is unclear how many choices could have
been made. As with other multiplicity problems, prospective
specification of the analysis method will generally eliminate
the concern about a biased (result-driven) choice. One
method should be pre-specified as the primary analysis
method to eliminate the possibility of a result based on a
biased choice.”
C. Common Statistical Methods for Addressing Multiple Endpoint-Related Multiplicity Problems
Line 978: It will be helpful for both reviewers and practitioners
in industry to provide reference to general
construction of multiple testing procedures in addition
to the ones presented in the document.
BIO suggests adding the following text to this part of the
Statistical Methods section:
It is, of course, impossible to include discussion or examples
of all potential multiple testing procedures applicable to
clinical trials. However, two important principles which
allows for the construction of multiple testing procedures
which provide strong type I error are: the closed testing
principle (or closure principle) and the partitioning principle.
These principles may be used to develop procedures beyond
those outlined below.
Line 982-983: The Draft Guidance states, “Single-step procedures
tend to cause loss of study power, so that sample
sizes need to be increased in comparison to sample
sizes needed for a single-endpoint study.”
BIO asks FDA to clarify the intended meaning of this section.

BIO Comments on Multiple Endpoints in Clinical Trials FDA Docket: FDA-2016-D-4460, March 14, 2017, Page 16 of 21
SECTION ISSUE PROPOSED CHANGE
It is not clear that this is necessarily the case. For
example, if there are two independent endpoints, A
and B, each with 90% power at the 5% significance
level, the power for a single-step Bonferroni
procedure (i.e., the probability that either one of the
two would achieve a p-value < 0.025) would be
approximately 97%, which is greater than the power
for either as a single endpoint. The FDA might have
intended to say that a single-step test of hypotheses
A and B has less power for showing an effect on A
than a test of A as a single endpoint.
Lines 1008-1009: The Draft Guidance states, “The most common form
of the Bonferroni method divides the available total
alpha (typically 0.05) equally among the chosen
endpoints.”
BIO asks FDA to clarify that the typical alpha=0.05 is two-
sided:
“The most common form of the Bonferroni method divides
the available total alpha (typically 0.05 two-sided) equally
among the chosen endpoints.”
Lines 1031-1033: The Draft Guidance reads, “When a multiple-arm
study design is used (e.g., with several dose-level
groups), there are methods that take into account
the correlation arising from comparing each
treatment group to a common control group.”
BIO notes that the language in this section is the Dunnett
method. However, FDA does not seem to refer to is as such.
The method’s name is mentioned later (line 1394), so it
should also be mentioned here. Making clear to the reader
that this is the Dunnett method will provide more clarity to
reader. A such, BIO suggests editing the text to read:
“When a multiple-arm study design is used (e.g., with
several dose-level groups), there are methods that take into
account the correlation arising from comparing each
treatment group to a common control group (i.e., Dunnett
method).”

BIO Comments on Multiple Endpoints in Clinical Trials FDA Docket: FDA-2016-D-4460, March 14, 2017, Page 17 of 21
SECTION ISSUE PROPOSED CHANGE
Lines 1053-1111
(Section IV C2
The Holm
Procedure)
BIO finds it is confusing when use of “endpoints 1, 2,
3, 4”, don’t correspond to the order in which they are
tested. Specifically, on line 1093, “endpoint 3” is the
second endpoint tested.
BIO suggests naming the endpoints as A, B, C, and D for
clarity.
Lines 1078-1080: The Draft Guidance reads, “The procedure stops,
however, whenever a step yields a non-significant
result. Once an ordered p-value is not significant, the
remaining larger p-values are not evaluated and it
cannot be concluded that a treatment effect is shown
for those remaining endpoints.”
BIO notes that the adjusted p-values for all
comparisons can be calculated, it is just all
comparisons after the first non-significant result are
non-significant. This also applies to the text in lines
1225-1226.
BIO asks FDA to please clarify whether testing stops or
whether further tests are not considered statistically
significant.
Lines 1238-1239: The Draft Guidance states, “The Holm test would not
find significant effects for additional endpoints either,
unless the p-value for endpoint A was p < 0.025.”
BIO notes that both "unless pA < 0.025" is equally as
true as "unless pC < 0.025"? While we believe that
neither statement is necessary, just giving one
causes confusion.
To reduce confusion BIO recommends either changing the
text to read:
“The Holm test would not find significant effects for
additional endpoints either, unless the p-value for either
endpoint A or C was p < 0.025.”
Or removing the latter section of the sentence altogether:
“The Holm test would not find significant effects for
additional endpoints either, unless the p-value for endpoint A
was p < 0.025.”
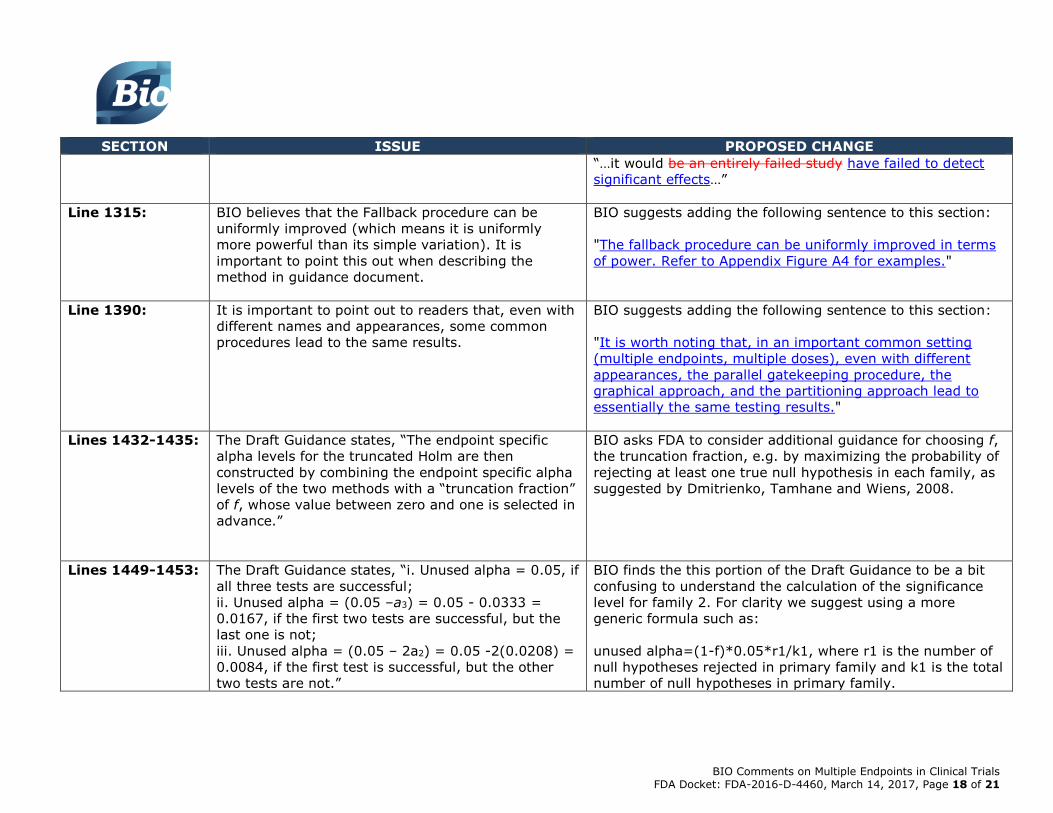
Lines 1240-1241: The Draft Guidance discusses failed studies.
However, BIO notes that a lack of statistical
significance does not make a study a failed study.
To ensure accuracy in the Draft Guidance, BIO recommends
editing the text to read:
BIO Comments on Multiple Endpoints in Clinical Trials FDA Docket: FDA-2016-D-4460, March 14, 2017, Page 18 of 21
SECTION ISSUE PROPOSED CHANGE
“…it would be an entirely failed study have failed to detect
significant effects…”
Line 1315: BIO believes that the Fallback procedure can be
uniformly improved (which means it is uniformly
more powerful than its simple variation). It is
important to point this out when describing the
method in guidance document.
BIO suggests adding the following sentence to this section:
"The fallback procedure can be uniformly improved in terms
of power. Refer to Appendix Figure A4 for examples."
Line 1390: It is important to point out to readers that, even with
different names and appearances, some common
procedures lead to the same results.
BIO suggests adding the following sentence to this section:
"It is worth noting that, in an important common setting
(multiple endpoints, multiple doses), even with different
appearances, the parallel gatekeeping procedure, the
graphical approach, and the partitioning approach lead to
essentially the same testing results."
Lines 1432-1435: The Draft Guidance states, “The endpoint specific
alpha levels for the truncated Holm are then
constructed by combining the endpoint specific alpha
levels of the two methods with a “truncation fraction”
of f, whose value between zero and one is selected in
advance.”
BIO asks FDA to consider additional guidance for choosing f,
the truncation fraction, e.g. by maximizing the probability of
rejecting at least one true null hypothesis in each family, as
suggested by Dmitrienko, Tamhane and Wiens, 2008.
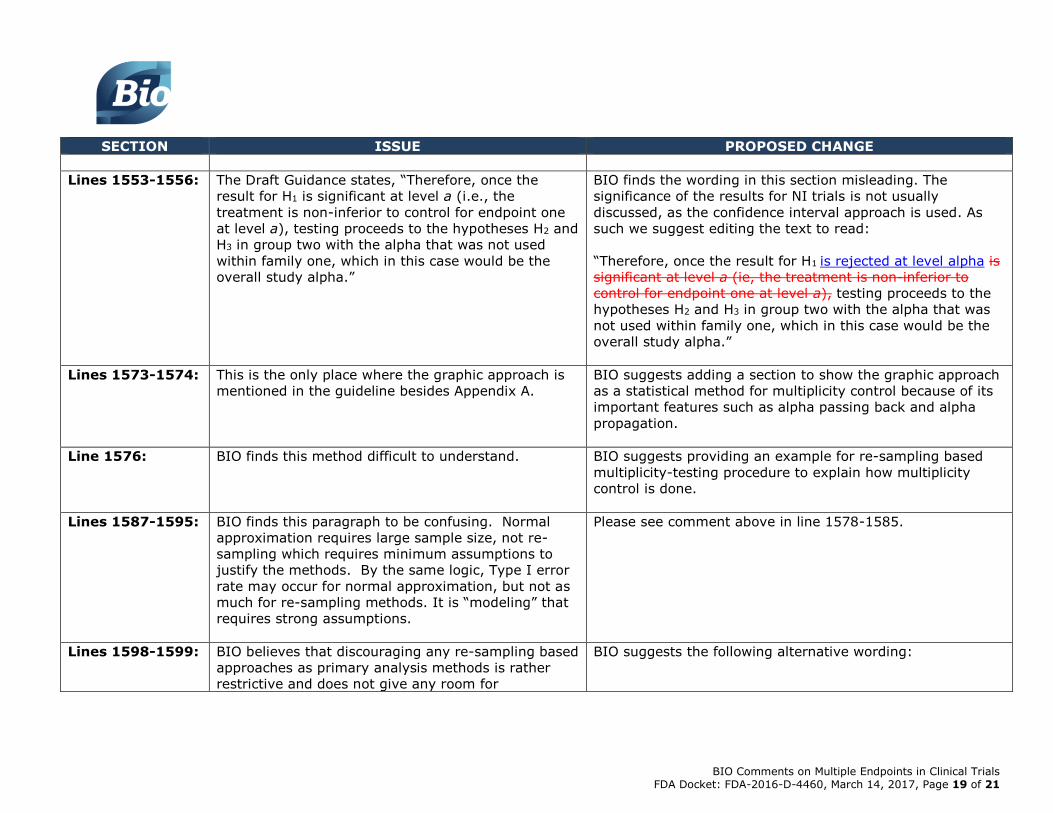
Lines 1449-1453: The Draft Guidance states, “i. Unused alpha = 0.05, if
all three tests are successful;
ii. Unused alpha = (0.05 –a3) = 0.05 - 0.0333 =
0.0167, if the first two tests are successful, but the
last one is not;
iii. Unused alpha = (0.05 – 2a2) = 0.05 -2(0.0208) =
0.0084, if the first test is successful, but the other
two tests are not.”
BIO finds the this portion of the Draft Guidance to be a bit
confusing to understand the calculation of the significance
level for family 2. For clarity we suggest using a more
generic formula such as:
unused alpha=(1-f)*0.05*r1/k1, where r1 is the number of
null hypotheses rejected in primary family and k1 is the total
number of null hypotheses in primary family.
BIO Comments on Multiple Endpoints in Clinical Trials FDA Docket: FDA-2016-D-4460, March 14, 2017, Page 19 of 21
SECTION ISSUE PROPOSED CHANGE
Lines 1553-1556: The Draft Guidance states, “Therefore, once the
result for H1 is significant at level a (i.e., the
treatment is non-inferior to control for endpoint one
at level a), testing proceeds to the hypotheses H2 and
H3 in group two with the alpha that was not used
within family one, which in this case would be the
overall study alpha.”
BIO finds the wording in this section misleading. The
significance of the results for NI trials is not usually
discussed, as the confidence interval approach is used. As
such we suggest editing the text to read:
“Therefore, once the result for H1 is rejected at level alpha is
significant at level a (ie, the treatment is non-inferior to
control for endpoint one at level a), testing proceeds to the
hypotheses H2 and H3 in group two with the alpha that was
not used within family one, which in this case would be the
overall study alpha.”
Lines 1573-1574: This is the only place where the graphic approach is
mentioned in the guideline besides Appendix A.
BIO suggests adding a section to show the graphic approach
as a statistical method for multiplicity control because of its
important features such as alpha passing back and alpha
propagation.
Line 1576: BIO finds this method difficult to understand.
BIO suggests providing an example for re-sampling based
multiplicity-testing procedure to explain how multiplicity
control is done.
Lines 1587-1595: BIO finds this paragraph to be confusing. Normal
approximation requires large sample size, not re-
sampling which requires minimum assumptions to
justify the methods. By the same logic, Type I error
rate may occur for normal approximation, but not as
much for re-sampling methods. It is “modeling” that
requires strong assumptions.
Please see comment above in line 1578-1585.
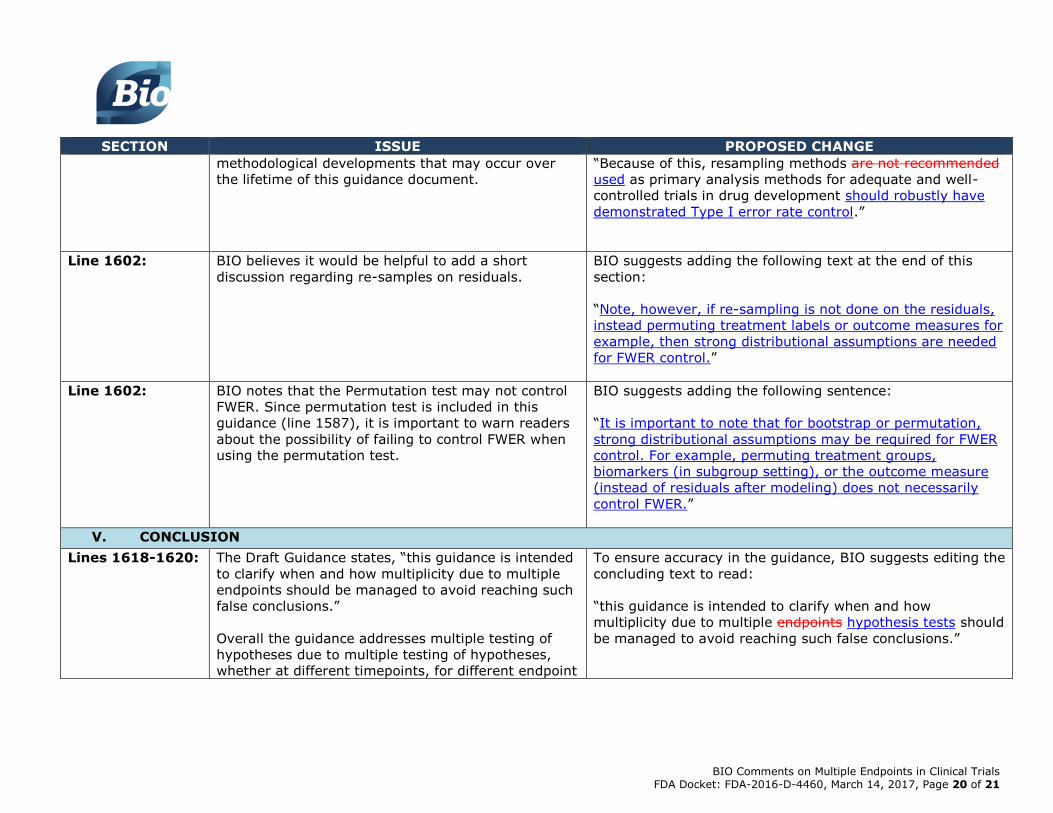
Lines 1598-1599: BIO believes that discouraging any re-sampling based
approaches as primary analysis methods is rather
restrictive and does not give any room for
BIO suggests the following alternative wording:
BIO Comments on Multiple Endpoints in Clinical Trials FDA Docket: FDA-2016-D-4460, March 14, 2017, Page 20 of 21
SECTION ISSUE PROPOSED CHANGE
methodological developments that may occur over
the lifetime of this guidance document.
“Because of this, resampling methods are not recommended
used as primary analysis methods for adequate and well-
controlled trials in drug development should robustly have
demonstrated Type I error rate control.”
Line 1602: BIO believes it would be helpful to add a short
discussion regarding re-samples on residuals.
BIO suggests adding the following text at the end of this
section:
“Note, however, if re-sampling is not done on the residuals,
instead permuting treatment labels or outcome measures for
example, then strong distributional assumptions are needed
for FWER control.”
Line 1602: BIO notes that the Permutation test may not control
FWER. Since permutation test is included in this
guidance (line 1587), it is important to warn readers
about the possibility of failing to control FWER when
using the permutation test.
BIO suggests adding the following sentence:
“It is important to note that for bootstrap or permutation,
strong distributional assumptions may be required for FWER
control. For example, permuting treatment groups,
biomarkers (in subgroup setting), or the outcome measure
(instead of residuals after modeling) does not necessarily
control FWER.”
V. CONCLUSION
Lines 1618-1620: The Draft Guidance states, “this guidance is intended
to clarify when and how multiplicity due to multiple
endpoints should be managed to avoid reaching such
false conclusions.”
Overall the guidance addresses multiple testing of
hypotheses due to multiple testing of hypotheses,
whether at different timepoints, for different endpoint
To ensure accuracy in the guidance, BIO suggests editing the
concluding text to read:
“this guidance is intended to clarify when and how
multiplicity due to multiple endpoints hypothesis tests should
be managed to avoid reaching such false conclusions.”

BIO Comments on Multiple Endpoints in Clinical Trials FDA Docket: FDA-2016-D-4460, March 14, 2017, Page 21 of 21
SECTION ISSUE PROPOSED CHANGE
measures, at different looks (interim or final), for
different populations, etc. BIO believes that the
concluding language regarding multiple endpoints
does not adequately reflect this.
APPENDIX: THE GRAPHICAL APPROACH
Line 1779: Correct typo of “alphas” to “alpha”.
Line 1882: The text reads “ash shown in Figure A5(a),” BIO suggests editing the text to read:
“as shown in Figures A5(a) and A5(b),”
Line 1922: BIO notes that the alpha for H3 in Figure A5 (c) should be
epsilon*alpha2.





























