Embed Size (px)
Citation preview

Marino et al., 2006 / ssAnPRT
1
Structural and Mutational Analysis of Substrate Complexation by Anthranilate
Phosphoribosyltransferase from Sulfolobus solfataricus
Marco Marino1&, Miriam Deuss2&, Dmitri I. Svergun3, Petr V. Konarev3, Reinhard Sterner2,
Olga Mayans1*
1Division of Structural Biology, Biozentrum, University of Basel, Klingelbergstrasse 70, CH-4056 Basel, Switzerland; 2Universität Regensburg, Institut für Biophysik und physikalischeBiochemie, Universitätsstrasse 31, D-93053 Regensburg & Universität zu Köln, Institut fürBiochemie, Otto-Fischer-Strasse 12-14, D-50674 Köln, Germany; 3European Molecular BiologyLaboratory, Hamburg Outstation, c/o DESY, Notkestrasse 85, D-22603 Hamburg, Germany &Institute of Crystallography, Russian Academy of Sciences, Leninsky pr. 59, 117333 Moscow,Russia
Running title: Substrate complexation by AnPRT& These authors contributed equally
*Author for correspondence: Olga Mayans, Division of Structural Biology, Biozentrum,University of Basel, Klingelbergstrasse 70, CH-4056 Basel, Switzerland, Tel: +41 61 2672083, Fax: +41 61 267 2109, Email: [email protected].
The metabolic synthesis and
degradation of essential nucleotide compounds
i s p r i m a r i l y c a r r i e d o u t b y
phosphoribosyltransferases (PRT) and
nucleoside phosphorylases (NP), respectively.
Despite the resemblance of their reactions, five
classes of PRTs and NPs exist, where
anthranilate PRT (AnPRT) constitutes the only
evolutionary link between synthesis and
degradation processes. We have characterized
the active site of dimeric AnPRT from
Sulfolobus solfataricus by elucidating crystal
structures of the wild-type enzyme complexed
to its two natural substrates: anthranilate and
PRPP/Mg2+
. These bind into two different
domains within each protomer and are brought
together during catalysis by rotational domain
motions as shown by small angle X-ray
scattering data. Steady-state kinetics of
mutated AnPRT variants address the role of
active site residues in binding and catalysis.
Results allow the comparative analysis of PRT
and pyrimidine NP families and expose related
s t r u c t u r a l m o t i f s i n v o l v e d i n
nucleotide/nucleoside recognition by these
enzyme families.
Nucleotide compounds are central to the
production of genetic material, the amino acids
histidine and tryptophan, and cofactors such as
NAD. Their metabolism is largely based on the
reversible transfer of a phosphoribosyl group to
aromatic bases. Although forward and reverse
ribosylation processes share a close resemblance
(Fig 1), they are carried out by different enzymes
and in the context of distinct metabolic pathways.
Synthesis reactions are performed by
phosphoribosyltransferases (PRT). These use 5-
phosphoribosyl-1-pyrophosphate (PRPP) as
universal phosphoribosyl donor. These reactions,
which are dependent on metal ions, involve the
displacement of the 1-pyrophosphate group of
PRPP and formation of a N-1’-glycosidic bond to
a nitrogenated base specific of each PRT.
Conversely, the release of aromatic bases from
nucleotides involves first the production of a
nucleoside intermediate by nucleotidases or
phosphatases, followed by the cleavage of the
glycosidic bond by either nucleoside
phosphorylases (NP) or nucleoside hydrolases
(NH). NP enzymes are key to nucleotide salvage
both in prokaryotes and in eukaryotes. They
catalyze the reversible phosphorolysis of the
glycosidic bond of nucleosides to yield free bases
and ribose 1-phosphate.
Despite the chemical resemblance of the
compounds intervening in synthesis and
degradation reactions by PRTs and NPs (Fig 1),
respectively, the enzymes involved in these
catalyses lack structural similarity. Three diverse
PRT classes that act on aromatic bases are known
to date: i) those with a canonical fold classified as
type PRT-I, which comprises most of the PRTs for
which an atomic structure is available (1); ii) type
PRT-II comprising quinolate PRT (2-3) and
http://www.jbc.org/cgi/doi/10.1074/jbc.M601403200The latest version is at JBC Papers in Press. Published on May 19, 2006 as Manuscript M601403200
Copyright 2006 by The American Society for Biochemistry and Molecular Biology, Inc.
by guest on March 19, 2020
http://ww
w.jbc.org/
Dow
nloaded from

Marino et al., 2006 / ssAnPRT
2
nicotinate PRT (4); and iii) anthranilate PRT
(AnPRT) (5-7) (Fig 1a). NPs are also classified in
two groups with distinct folds: i) NP-I, which act
primarily on purine nucleosides but also accept the
pyrimidine uridine and ii) NP-II which degrade
thymidine and/or uridine depending on the
organism of origin (8) (Fig 1b) (throughout this
text NP-II enzymes will be termed pyrimidine NP
- PyNP - irrespective of their substrate specificity).
One single evolutionary link has been encountered
among these five protein families, exemplified by
AnPRT and PyNP, enzymes that share little or no
sequence homology, but which display significant
architectural similarities. AnPRT synthesizes an
intermediate in the biosynthesis pathway of the
essential amino acid tryptophan. It ribosylates the
aromatic base anthranilate (AA) using PRPP to
convert it into a nucleotide-like product, 5’-
phosphoribosyl-anthranilate, which constitutes the
basic skeleton of tryptophan (Fig 1a). Atomic
structures of AnPRTs from Sulfolobus solfataricus
( 5 ) , Pectobacterium c a r o t o v o r u m (6),
Mycobacterium tuberculosis (7), T h e r m u s
thermophilus (PDB entry 1V8G) and Nostoceae
(1VQU) have become available recently, with the
structure from M. tuberculosis (mtAnPRT) being
complexed to its PRPP substrate and magnesium
ions (7). Although the sequence similarity between
AnPRT and PyNP is restricted to their N-termini
and a few confined motifs (5, 9), their
architectural correspondence is significant. Both
enzymes share a common fold comprising a small
N-terminal, ! -helical domain made up of six
helices and a large !/" domain formed by a central
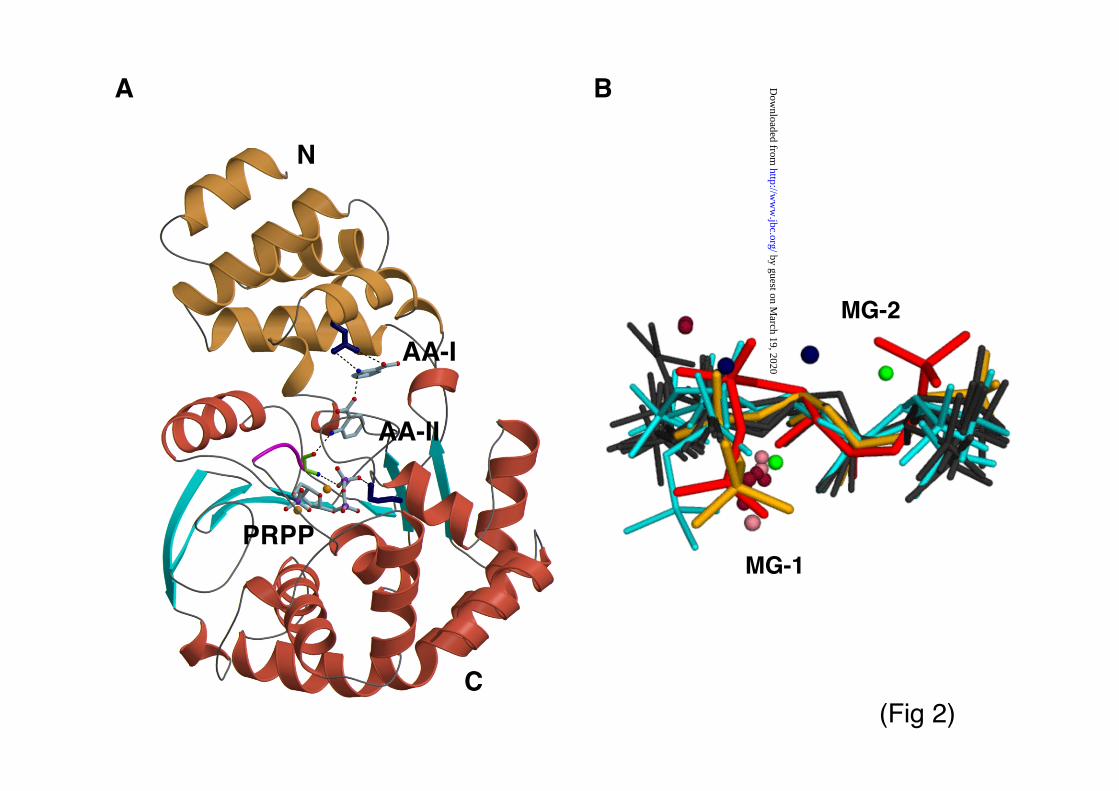
"-sheet and a cluster of eight !-helices (Fig 2a).
They also exhibit a similar quaternary structure
consisting of head-to-head dimers with equivalent
interfaces. A topological comparison of their
architectures shows that a particular feature of
AnPRT is the presence of a short loop within the
substrate binding cleft, which carries a DE
sequence motif absolutely conserved within this
family (5). This motif, which is not present in
PyNPs, has been proposed to confer AnPRT the
ability of binding and activating the PRPP
substrate central to PRT catalysis (5).
PRT and NP enzymes are of marked
biomedical relevance, either for being connected
to major human disorders (such as rheumatoid
arthritis, metastatic processes in several cancers
and the Lesch-Nyhan syndrome, among many
others) or for their potential as antiparasitic targets
(for example against malaria, Chagas disease and
trichosomiasis). Recently, AnPRT has also
emerged as a candidate for biomedical
app l i ca t ions . Studies on M y c o b a c t e r i u m
tuberculosis showed that strains with disrupted
AnPRT presented attenuated virulence but lead to
protection in mouse models equal to vaccination
with virulent strains (10-11). In order to betterunderstand the mode of action of AnPRT and to
investigate further the general catalytic strategies
across PRT and NP nucleotide/nucleoside
processing enzymes, we have now characterized
the active site of S. solfataricus AnPRT
(ssAnPRT) by studying its complexation of
natural substrates using X-ray crystallography,
small angle X-ray scattering and steady-state
enzyme kinetics of mutated variants. Our results
indicate that, despite architectural and functional
differences in these protein families, their tactics
for substrate recognition are related.
Experimental procedures
Crystal structure elucidation - ssAnPRT was
overexpressed, purified and crystallized as
reported (12). Substrate complexation was in the
crystalline state using soaking at 4 oC, where
crystals of the apo enzyme were immersed in
cryo-solutions containing mother liquor
supplemented with 22% (v/v) glycerol (as cryo-
protectant) and ligands. AA and PRPP substrates
were purchased from Sigma-Aldrich. Specific
soaking conditions and X-ray data statistics are
given in Table I. For X-ray data collection,
crystals were flash-frozen at 100 K. The APRV
interface (13) to XDS (14) and CNS (15) was used
for data processing and interpretation, with initial
phases derived from apo-ssAnPRT (PDB entry
1O17) (5). A model for anthranilate was obtained
from the Cambridge Structural Database, while
PRPP initially derived from PDB entry 1FSG.
Manual rebuilding of protein and substrate models
was in O (16). For cross-validation, diffraction
data were divided into a working and a test set in
XSCALE (14). NCS restraints across domains
were applied to all models. Water molecules were
built using ARP/wARP (17) and validated visually
in electron density maps.
Coordinates for ssAnPRT in complex with
AA, PRPP/Mg2+
and AA/PRPP/Mg2+
have been
deposited at the PDB with accession codes 2GVQ,
1ZXY and 1ZYK, respectively.
Small angle X-ray scattering - SAXS data werecollected on the EMBL camera X33 (DORIS III,
by guest on March 19, 2020
http://ww
w.jbc.org/
Dow
nloaded from

Marino et al., 2006 / ssAnPRT
3
DESY) with a linear gas detector (18). Sampleswere in 10 mM HEPES pH 8.0 at substrate
binding conditions as for crystal soaking (Table I).
Data were recorded at multiple proteinconcentrations (1.8 to 15.0 mg/ml) at atemperature of 4.0± 0 .2°C in the range ofmomentum transfer 0.15 < s < 3.2 nm-1 (s = 4#
sin($)/%, where 2$ is the scattering angle and % =0.15 nm is the X-ray wavelength). The data,collected in 15 successive 1-minute frames,showed no significant variation as a function oftime, indicating the absence of both radiationdamage and catalysis on the substrates. Data wereprocessed using the program PRIMUS (19). Theforward scattering I(0), radius of gyration Rg,maximum dimension Dmax and the distancedistribution function p(r) were evaluated with theprogram GNOM (20). The molecular masses(MM) of the solutes were derived by comparisonof I(0) with that from reference solutions ofbovine serum albumin (MM = 66 kDa).
Multiple models of ssAnPRT in open andclosed conformations were constructed bysystematic rigid-body rotation of the C-terminal! /" domain relative to its fixed N-terminalcounterpart. The interdomain hinge region inssAnPRT was taken as loops !4-"1, !8’-!8 and!9-"4 (definition as in (5)). The rotation axis wascalculated from non-crystallographic differencesin apo crystal models (1O17) using DYNDOM(21). With hinge and rotation axis so defined, the!/"-domain was swung in 5O increments fromextremely open to maximally closedconformations. The scattering patterns of 225generated symmetric and asymmetric dimers, allkeeping the dimerization interface as in apo-ssAnPRT, were compared to the experimental datausing CRYSOL (22).
Site-directed mutagenesis - Mutants K106Q,R164A, R170A, and D223N were produced byoverlap extension PCR using the plasmidpQE40_sstrpD (12) as template. Mutants H107Aand E224Q were produced with the QuikChangemethod (Stratagene), again using pQE40 sstrpD asthe template. Double mutants R164A+H154A andP178A+H107A were generated by overlapextension PCR, using the templatespQE40_ss trpD-R164A and pQE40_sstrpD -H107A. The details of the PCR reactions,including the DNA sequences of theoligonucleotides used, are provided assupplementary material. Using BamHI and HindIIIrestriction sites, the mutated genes were cloned
into pQE40. Gene expression from pQE40 resultsin the addition of an N-terminal His6-tag to theproduced protein. To confirm the introduced basesubstitutions and to exclude further inadvertentpoint mutations, all mutated genes were entirelysequenced.
Heterologous expression and purification of
ssAnPRT mutants - ssAnPRT variants were
heterologously expressed at 37°C in E. coli strain
W3110 trpEA2, containing the repressor plasmid
pDM (23). The resulting protein products were
purified from the soluble fraction of the crude
extract by heat precipitation of the host proteins
and metal chelate affinity chromatography. N-
terminal His6-tags were removed by proteolysis.
Details of gene expression and protein purification
are provided as supplementary material. As judged
by SDS-PAGE, all samples were at least 95%
pure. Yields were 0.2-0.8 mg protein per g wet cell
mass. The proteins were dripped into liquid
nitrogen and stored at –80°C.
Steady State Kinetics - The ssAnPRT reaction was
followed at 60°C by a fluorimetric assay (CARY
Eclipse fluorescence spectrophotometer, Varian)
(12) performed in 50 mM Tris-HCl pH 7.5 at
various concentrations of MgCl2. The Michaelis
constants KMAA
and KMPRPP
were determined by
analyzing saturation curves that were deduced
from initial velocity measurements recorded in the
presence of an excess (10xKM) of the second
substrate. The turnover number kcat was obtained
by dividing the maximum catalytic rate by the
total concentration of active sites.
RESULTS
Crystal structures of wild-type ssAnPRT in
complex with its natural substrates
Atomic models of ssAnPRT in complexwith i ) AA, i i ) PRPP/Mg2+ and iii)AA/PRPP/Mg2+, the latter showing the active siteat full occupancy, have been obtained by crystalsoaking at 4oC where catalysis by thisthermophilic enzyme is not detectable (12). Thecrystal form employed in this study contained twoAnPRT dimers in its asymmetric unit. X-ray dataand model refinement statistics are given in TableI.
Recognition of Anthranilic acid: ssAnPRT bindstwo molecules of AA per protomer (Fig 2a, c & e).
by guest on March 19, 2020
http://ww
w.jbc.org/
Dow
nloaded from

Marino et al., 2006 / ssAnPRT
4
The two AA molecules are housed within the N-terminal, !-helical domain in consecutive bindingpockets (sites AA-I and AA-II). Site I is definedby enzyme residues N109, A150, H154, M157 andR164, located at the N-terminal end of the bindingcleft. Site II, positioned at the middle point of thebinding groove, is close to a hinge region at thecenter of the enzyme architecture. It is formed byprotein residues A78, G79, H107, G108, N109,G177 and P178. Protein-AA interactions both atsites I and II are mostly of hydrophobic character.The charged, lateral groups of AA are involvedonly in few specific interactions, so that theircontribution to the productive orientation of thiscompound in either pocket appears to bemoderate. AA-I is oriented primarily via itscarboxyl group by protein residues R164 andN109. AA-II interacts with N109 via its carboxylgroup, whereas its amino group is orientedtowards the PRPP binding cavity via a hydrogenbond to the backbone carbonyl group of G79 (Fig2e). As a further contribution to substrateorientation, the imidazole rings of H154 and H107at sites I and II, respectively, act as #-acceptors ofthe aromatic CH groups of anthranilate. Thesecontacts are better classified as weak hydrogenbonds than as unspecific hydrophobic interactions(24-25). Both histidine residues are similarlyarranged with respect to their corresponding AAligands. Besides protein-ligand contacts, the twoAA molecules interact directly with each other viaan electrostatic interaction between the aminogroup of molecule AA-I and the carboxyl group ofAA-II. This is a prominent contribution to theorientation of these molecules in the bindinggroove of ssAnPRT.
In order to clarify whether ssAnPRT bindsAA preferentially at a given pocket, a titrationseries was carried out in the crystal at AA soakingconcentrations of 0.2, 0.5, 1 and 2 mM over 30minutes to ensure equilibration. Binding becameinterpretable only at the highest concentration of 2mM and in two out of the four ssAnPRT copies inthe asymmetric unit. The analysis of electrondensity maps indicated that the enzyme within thecrystal lattice favors binding of AA at pocket IIand that the AA binding mode is the same at highand low substrate concentrations. A visualinspection of the lattice suggested that this result isunlikely to be due to impaired accessibility ofpocket I.
Protein groups involved in AAcoordination at sites I and II are highly conserved(>85%) within the AnPRT family (Fig 3). Glycine
residues at positions 79 and 177 are required toprevent steric blockage of substrate binding topocket II. Of all residues involved, only R164 atsite I is absolutely conserved across the AnPRTand PyNP families.
Recognition of PRPP and magnesium counterions:
ssAnPRT coordinates one PRPP molecule per
subunit (Fig 2a, d & f). The binding site for this
substrate is located in a deep depression within the
large !/" domain. The most prominent change in
the enzyme upon PRPP binding is the ordering of
the glycine-rich loop 79GTGGD83 (Fig 2a & d),
which is unstructured in the apo form of the
enzyme (5). This loop constitutes one of the
signature motifs of the AnPRT family (Fig 3).
Ordering appears to be caused by interactions of
residues G79 and D83 with PRPP. Contacts
established between enzyme and substrate include
in addition residues T87, N89, S91, T92 and K106
(Fig 2f). The pyrophosphate moiety of PRPP in
these structures is similarly coordinated as the PPi
leaving group previously reported (5-6).Given that divalent ions mediate PRPP
binding in all other PRT families (1) and thatssAnPRT requires magnesium for catalysis (12),we expected Mg2+ ions to also coordinate PRPP inthe current complex. Since atoms of this lightmetal could not be resolved in electron densitymaps at the working resolution, we used insteadmanganese in soaking studies to experimentallylocate metal sites using anomalous differenceFourier maps calculated with phases from the apo-model. Eight ions were identified with peakheights of 7.8& , 7.6&, 7.5&, 7.0 & 6.7&, 6.3& ,
5.5&, 4.8&, corresponding to two metal ions perPRPP molecule. A first metal atom (MG-1) iscoordinated by the hydroxyl groups of the riboseand pyrophosphate oxygens, so that it is internal toPRPP with no protein groups being involved (Fig2f). This is likely to correspond to the high-affinitymetal binding site present in the PRPP compound,which forms in solution a predominant mono-magnesium complex thought to be the truesubstrate of PRTs (26-27). Accordingly, crystalstructures of the unrelated PRT-I and -II incomplex with PRPP reveal an equivalent metal ionat this site, also lacking direct protein contacts (Fig2b) (1, 3). In ssAnPRT, the coordinated action ofMG-1 and a hydrogen bond between the aminogroup of G79 and a phosphate oxygen induce an
eclipsed conformation in the pyrophosphate group
(Fig 2d & f). Eclipsed arrangements are also
common in PRPP coordination by the canonical
by guest on March 19, 2020
http://ww
w.jbc.org/
Dow
nloaded from

Marino et al., 2006 / ssAnPRT
5
PRT-I and it probably provides electrostatic
assistance to catalysis (1). In ssAnPRT, a secondmetal site (MG-2) reconciles binding of the 5-phosphate group of PRPP to a cluster ofnegatively charged protein residues comprisingD223 and E224 (Fig 2d, f), which constitute anabsolutely conserved motif in the AnPRT family.PRPP in solution has a low-affinity metal bindingsite at its 5-phosphate group and can form di-magnesium complexes in metal rich media,although with very low propensity (26). A metalcounterion at this position, however, has not beenobserved previously in PRPP coordination byother PRTs (Fig 2b).
Interestingly, even that most residuesinvolved in PRPP/Mg2+ coordination are highlyconserved across members of the AnPRT family,the binding site for PRPP in the enzymes from S.
solfataricus and M. tuberculosis differs somewhat(Fig 3). PRPP bound to ssAnPRT bends uponitself into an “S”-shaped arrangement, while it iscoordinated in an “extended” form in other PRTsas well as in mtAnPRT (Fig 2b). Curiously,neither of the magnesium sites proposed formtAnPRT in complex with its PRPP substrateagree with those proposed here for ssAnPRT orwith those encountered in any other PRT (Fig 2b).
Active site at full occupancy: Crystal structures offully complexed ssAnPRT protomers (Fig 2a)reveal that no significant structural changes otherthan the ordering of the glycine-rich loop, which isinduced by the binding of PRPP/Mg2+, take placein the enzyme upon full substrate binding. Domaindisplacements are not detectable when comparingmodels of the apo enzyme, single liganded formsand the complete complex. Despite, bindingpockets for the AA and PRPP:2Mg2+ substrates inthese structures are too far apart to allow forcatalysis to occur. The nitrogen groups of AA-Iand -II are separated by 14.6 Å and 8.6 Å,respectively, from the C1 ribose atom of PRPP.For the synthesis reaction to proceed, substratesmust be brought together, probably by means of ahinge motion where the N- and C-terminaldomains close upon each other. Such domainrearrangements are likely to be hindered within thecrystal lattice.
Domain motion upon substrate complexation
Hinge motion events caused by substrate
complexation were monitored in solution by small
angle X-ray scattering (SAXS). Samples were kept
at 4oC throughout the irradiation experiments to
prevent catalysis (12). The overall parameters of
free and liganded ssAnPRT solutions are given in
Table II and scattering patterns are shown in Fig 4.
The molecular mass of the solute was 72±5 kDa
for all cases, in agreement with the theoretical
value for dimeric ssAnPRT (73.6 kDa). Based on
the radii of gyration (Rg), maximum molecular
dimensions (Dmax) and distance distribution
functions p(r), the apo enzyme and its complex
with PRPP have a similar structure in solution. In
both cases, the separation between the centers of
the two protomers (Dinter, given by the position of
the shoulder in p(r)) is ~7 nm (Fig 4b). In complex
with AA, the enzyme exhibits a similar Dmax but
slightly reduced Dinter and Rg values, hinting at a
minor structural rearrangement upon AA binding.
In contrast, the fully complexed enzyme shows
Dmax and Dinter values reduced by ~1 nm and a
significantly smaller Rg, indicating that the
enzyme adopts here a much more compact
conformation. Consequently, the comparison of
experimental data with the scattering computed
from the crystal structure of apo ssA n P R T
(defined by the discrepancy 'C in Table II) shows
that the latter is a close representative of the apo
enzyme in solution and its complex with a single
substrate, but differs from the overall
conformation of the fully liganded enzyme. It can
be concluded that while coordination of one single
substrate type does not induce significant domain
motions in ssAnPRT, the enzyme undergoes a
noticeable compaction upon full occupancy of its
active site. This suggests that the inherent
flexibility of the hinge region is central to the
catalytic function of ssAnPRT.
In order to reconstruct a model ofssAnPRT in its closed, catalytically activeconformation, 225 models were generated wherethe larger !/" domains had been systematicallyrotated in 5o intervals relative to the small ! -helical domains (see Methods). The dimerizationinterface between subunits, which engages the !-helical, N-terminal domains of both protomers in ahead-to-head fashion, was kept constant. Giventhat this interface is remote from the active sites ofthe individual protomers, it is unlikely that it willundergo alterations during catalysis. Resultingmodels comprised both symmetric arrangements,where concerted rotations had been applied to both!/" domains in the dimer, as well as asymmetricstructures, where different rotations had beenintroduced in the protomers. Scattering curveswere computed from all models and compared
by guest on March 19, 2020
http://ww
w.jbc.org/
Dow
nloaded from

Marino et al., 2006 / ssAnPRT
6
with the experimental data. The best models andtheir scattering patterns are displayed in Fig 4,with Rg and ' values given in Table II. Data fromsolutions of apo ssAnPRT as well as fromssAnPRT in single complex with its PRPPsubstrate were best fitted by models with domainarrangements similar to those of the apo crystalstructure. Best-fitting models for the enzymecomplexed to AA displayed a relatively smallasymmetric domain closure of the protomerswithin the homodimer. In contrast, models fittingthe fully complexed ssAnPRT were markedlyasymmetric (Fig 4d). To check whether enzymepopulations in solution could consist of a mixtureof symmetrically open and closed dimers, anattempt was also made to fit the scattering data bylinear combinations of symmetric models withvarious degrees of hinge opening. Such mixtures,however, yielded poor fits (e.g. '=2.4 for the fullycomplexed enzyme). It was thus concluded thatbinding of AA together with PRPP induces anasymmetric hinge closure in the dimer, where oneof the subunits retains a hinge opening similar tothat of the apo form.
The reconstructed model of a ssAnPRTprotomer in its closed conformation shows howthe lower !/"-lobe swings to bring the PRPPsubstrate close to the AA molecules, in particularto AA at site II, implying that this site iscatalytically relevant (Fig 4d). The magnitude ofthe hinge rotation can be estimated as 25-33o. Thisis in close agreement with crystallographic data onPyNP from Bacillus stearothermophilus, wheredomain rotations of ~21o are observed uponbinding of a nucleoside analog (28). Based oncurrent data, it is difficult to envisage how AA atposition I could be brought in contact with PRPPby means of a simple rigid-body rotation of theenzyme domains.
Kinetic characterization of wild-type ssAnPRT
and mutated variants
According to crystallographic data, theactive site of a ssAnPRT protomer can host twoAA molecules, one PRPP compound and twometal sites in its full occupancy state. Thesubstrates can bind independently to the enzyme,
suggesting a random, sequential bi-bi mechanism.
This is in accordance with kinetic data on S .
cerevisiae AnPRT (29), which show no evidence
for ordered sequential or ping-pong mechanisms.To further examine the role of active site residuesin catalysis and substrate binding in ssAnPRT,
variants with single or double amino acidexchanges were analyzed by steady-state enzymekinetics. Residues were selected according to theirproximity to either one of the two AA moleculesor the PRPP/Mg2+ substrate. The measuredturnover numbers kcat and the Michaelis-constantsKM
AA and KMPRPP at 60°C are given in Table III. In
the course of these studies, it was observed thatthe wild-type enzyme and the variants involvingresidues unrelated to PRPP binding (R164A,R164A+H154A, H107A and H107A+P178A)showed unexpected inhibition by highconcentrations of Mg2+. Their kcat values aredecreased 3-10 fold in the presence of 2 mM Mg2+
compared to 25-500 µM of added Mg2+.Moreover, the PRPP saturation curves of thesevariants were sigmoidal, with drastically increasedKM
PRPP values (shown in Table III for wtssAnPRT). To account for this effect, steady-statemeasurements on these variants were performedwith added 50 µM Mg2+, whereas the remainingvariants were assayed in 2 mM Mg2+.
All ssAnPRT variants designed to test the
catalytic relevance of AA binding sites I and II
( R 1 6 4 A , R 1 6 4 A + H 1 5 4 A , H 1 0 7 A ,
H107A+P178A) (Fig 2c & e) showed increasedKM
AA values, while KM
PRPP values remained
similar to those of the wild-type, confirming that
the effect of these exchanges is local to the AA
binding pockets (Table III). The exchange R164Aled to more than 7000-fold increase in K M
AA,
although only a moderate 7-fold decrease in kcat,indicating that this residue is crucial for substratebinding but not for catalysis. Similarly, thesubstitution H107A+P178A, local to site AA-II,resulted in a 300-fold increase of KM
AA, which was
mainly caused by the P178A exchange, while kcat
remained nearly unchanged. These results showthat R164 and P178, which are absolutelyconserved across the AnPRT family (Fig 3), are
the main determinants of AA recognition by
ssAnPRT. Since these residues are local to a
different AA binding pocket, ssAnPRT probably
ligates AA at both sites I and II.
The exchanges H154A and H107A hadonly a limited effect on either KM
AA or kcat, provingthat histidine residues at these positions aredispensable. This is remarkable, given their high
conservation within the family (Fig 3) and that
coordination of nitrogenated bases via # ( #
interactions involving imidazole groups of
histidines is a common structural theme among
nucleotide/nucleoside processing enzymes
including PRTs, NPs and nucleoside hydrolases.
by guest on March 19, 2020
http://ww
w.jbc.org/
Dow
nloaded from

Marino et al., 2006 / ssAnPRT
7
For example, in PyNPs an absolutely conserved
histidine residue (H116 in human thymidine NP)
occupies a position structurally equivalent to that
of H107 in ssAnPRT (Fig 5b). In that family, this
residue has been suggested to be involved in
catalysis by mediating the transfer of the proton
from the lytic phosphate to the O2 position of
thymine (30). Similarly, in quinolinate PRT type IIfrom Salmonella typhimurium (2) and M .
tuberculosis (3) residues H174 and H161,respectively, coordinate quinolate (Fig 5b). Arelated substrate interaction is established by H82in the nucleoside hydrolase from Crithidia
fasciculate (31) . As in AnPRT, these histidineresidues are highly conserved within theirrespective protein families. Given that current data
indicate that the contribution of these interactions
to substrate coordination can be only minor in
AnPRTs, it should be investigated to which extent
they are decisive in other enzyme families.
The role in binding and catalysis of
residues mediating PRPP/Mg2+
recognition in
ssAnPRT was checked by characterizing the
variants K106Q, D223N and E224Q, which
involve absolutely conserved residues in this
family (Fig 3). When compared at optimal Mg2+
concentrations, all variants tested displayed a
KMPRPP
similar to that of the wild-type enzyme
(Table III). Furthermore, kcat values were
unchanged in the D223N and E224Q variants and
decreased only ~10-fold in K106Q. This suggests
that, remarkably, these residues are expendable for
PRPP binding and that none acts as key catalyst.
However, contrary to wt ssAnPRT, these variants
were not inhibited by high concentrations of Mg2+
.
Instead, their catalytic rates asymptotically
increased up to the highest tested Mg2+
concentration of 3 mM (data not shown). An
inhibition by magnesium has also been reported
for hypoxanthine PRT (class I), where free Mg2+
ions in the medium have been suggested to act as
competitive inhibitors of PRPP/Mg2+
species (32).In this enzyme, two metal ions interact with thepyrophosphate moiety of PRPP (33). An inhibition
of ssAnPRT by free Mg2+
is supported by
crystallographic models of AnPRT in complex
with PPi and Mg2+
or Mn2+
ions (5-6), which show
that two metal sites related to those in complexed
PRPP are present in the enzyme in the absence of
the 5’-phosphate-ribose fraction. To sum up, it is
unexpected that the substitution in ssAnPRT of the
K106 residue and the DE motif, both absolutely
conserved in this family, does not result in
significant alterations of steady-state enzyme
kinetic values. Although these residues do not
appear to play key functional roles, they, however,
render this enzyme sensitive to the levels of
magnesium in the medium. It is currently
unknown how this could be of biological
advantage to S. solfataricus.
To conclude, no active catalysts could be
identified in ssAnPRT during this study, which
suggests that the active site of ssAnPRT leads tocatalysis primarily by aiding the productiveorientation of substrates.
DISCUSSION
Substrate coordination by ssAnPRT
The active site of s s A n P R Taccommodates two substrates, AA andPRPP/Mg2+, which can bind independently to theenzyme. AA coordination exploits primarily thehydrophobic character of this compound, whilespecific interactions involving its charged, lateralgroups are scarce. Site-directed mutagenesis datareveal that a stacking interaction involving P178and, in particular, electrostatic contact to R164 aredecisive for productive AA binding (Fig 2c; TableIII). R164 establishes a sequential chain ofhydrogen bonds along the binding cleft that alignsboth AA molecules within the active site (Fig 2a).It coordinates both the carboxylate and aminogroups of AA-I; in turn, the latter forms a saltbridge with the carboxylate group of AA-II; andfinally the amino group of AA-II gets orientedtowards the PRPP substrate pocket via aninteraction of its amino group with the main chaincarbonyl group of G79 (Fig 2e). This residue,absolutely conserved and part of the 79GTGGD83
signature motif of the AnPRT family, alsointeracts with the pyrophosphate moiety of PRPPvia its main chain amino group (Fig 2f), so that itappears essential for the relative orientation ofPRPP and AA-II substrates and, thus, for AnPRTactivity.
The finding of two AA binding sites inssAnPRT agrees well with recent predictionsbased on the structure of mtAnPRT (7). However,it opens questions about the relative catalyticsignificance of the sites. Titration of apo ssAnPRTcrystals with AA indicated that this compoundpreferentially occupies pocket II close to the PRPP
by guest on March 19, 2020
http://ww
w.jbc.org/
Dow
nloaded from

Marino et al., 2006 / ssAnPRT
8
binding site, suggesting that AA-II might act assubstrate. The catalytic significance of site I iscurrently unclear. It could be speculated that AA-Iplays a structural role in the binding of the “true”substrate, judging by the fact that it constitutes themost prominent orientational anchor for AA-II.Since AA is a small metabolite that can diffuseeasily out of the cell, a redundant bindingmechanism would maximize AA intake from thecellular medium putatively acting as a “rescue”mechanism.
The coordination of PRPP by ssAnPRTdiffers from that by other PRTs in its metalbinding pattern as well as in the compactconformation imposed on the substrate by theenzyme. Coordination of PRPP via two metal ionsis rare in PRTases (Fig 2b). Commonly, a singlemetal site is present that interacts with thepyrophosphate and ribose moieties of PRPP,corresponding to the high-affinity metal site in thePRPP compound. In those cases where a secondmetal site is observed, this also involves thepyrophosphate group. In ssAnPRT, however, asecond metal site bridges the 5-phosphate group tothe negatively charged enzyme residues D223 andE224. This results in a compact arrangement of thebound PRPP, which could either lead to anincreased reactivity or simply reflect the flexibilityof this compound. This conformation of PRPP is,however, not observed in the complexed enzymefrom M. tuberculosis (7). In that case, a differentmetal binding pattern is also present, where twoion sites are encountered but none corresponds tothose previously observed in PRTs, including theconserved, high affinity site MG-I thought to beintrinsic to the PRPP substrate (Fig 2b). The basisfor the differences between these AnPRTs arecurrently unknown.
Loop structuring and domain motions
In its apo form, ssAnPRT adopts an“open” hinge conformation, where the AA andPRPP substrates bind into distantly locatedcavities of the N- and C-terminal domains,respectively. According to SAXS data, a structuralcompaction takes place in ssAnPRT upon bindingof both substrates (Fig 4; Table II), suggesting thatthe flexibility of the hinge region is essential forcatalysis. The binding pockets for PRPP and AA-II relate to each other via the conserved79GTGGD83 loop, where G79 binds both substratesand D83 binds PRPP (Fig 2). This loop is
disordered in the apo crystal structure of ssAnPRT(5) and it becomes structured upon binding ofPRPP. Although the apo models of AnPRT fromP. carotovorum (6) and T. thermophilus do notexhibit such disorder, the structures from M .
tuberculosis (7), which shows rearrangements inthis area upon PRPP binding, and Nostoceae,
which is heavily unstructured at this central hingeregion, suggest that PRPP-induced loopstructuring in ssAnPRT is not fortuitous.
The similarity in domain motions acrossAnPRT and PyNP families is manifest. Thedistance between the amino group of AA-II andthe C1-ribose atom of PRPP in open forms ofssAnPRT is )8.6 Å, equivalent to the distance of8–9 Å between the lytic phosphate group and thenucleoside cleavage site in open forms of PyNPenzymes (34). Upon hinge closure, comparabledomain rotations take place in both enzymes: )25-33o in ssAnPRT and )21o in B. stearothermophilus
PyNP (28), indicating that similar architecturalmechanisms underlie hinge motion.
Absence of indispensable catalytic residues in
AnPRT
Members of the AnPRT family comprisefour primary, highly conserved sequence features:i ) an arginine residue (R164) involved in AAcoordination; ii) a glycine rich-loop hosting a79GTGGD83 motif; iii) a 106KHGN109 "-strandinvolved in coordination of pyrophosphate andAA groups; and iv) a 223DE224 motif involved inmetal-phosphate binding (numbering according tossAnPRT; cf. Fig 3). Of these, we have tested thefunctional relevance of R164, K106, H107, D223and E224 using site-directed mutagenesis.Notably, none of these residues is essential forcatalysis (only R164A and K106A exchanges ledto a moderate decrease in kcat values) and onlyR164 is crucial for substrate binding. Given thatno active catalysts could be identified, it is
reasonable to conclude that the active site ofAnPRT might primarily serve as a template for theproductive orientation of substrates.
An arginine residue, equivalent to R164 inssAnPRT, is absolutely conserved also in thePyNP family (Fig 3 & 5b). There it has beenproposed to mediate catalysis by stabilizing thenegative charge arising in the base ring uponcleavage of the glycosidic bond (35). Accordingly,its replacement by glutamate resulted in enzyme
inactivation (36). However, this did not clarifywhether the residue is involved in catalysis or
by guest on March 19, 2020
http://ww
w.jbc.org/
Dow
nloaded from

Marino et al., 2006 / ssAnPRT
9
merely substrate binding in that family. Our datashow that the conserved arginine in ssAnPRT is ofno catalytic relevance, accentuating the need forfurther comparative studies on the role ofconserved residues across AnPRT and PyNPfamilies.
Substrate binding motifs across PRTs and
PyNPs
Structural data suggest that AnPRTs andPyNPs have evolved from a common ancestor (5),but independently from the unrelated PRT familiesI and II (Fig 5a). Nonetheless, AnPRTs, PyNPs,PRTs-I and -II host related structural motifs attheir active sites for nucleotide/nucleosiderecognition (Fig 5b). Conserved arginine, lysine and histidinegroups as well as glycine-rich loops are frequentlyfound mediating binding of nitrogenated bases andphosphate moieties (Fig 5b). This is manifest inAnPRT and PyNP enzymes, which achievesubstrate coordination via: i) an absolutelyconserved arginine residue, which forms ahydrogen bond with the nitrogenated base; ii) a“KH” sequence that coordinates a phosphategroup and the nitrogenated base, respectively andiii) a glycine-rich loop. Glycine-rich loops, termedP-loops because of their binding of phosphategroups, are encountered across the broad spectrumof nucleotide binding proteins including kinases,phosphatases, tri- di- and mono- nucleotideprocessing enzymes (37 and references therein). Adistinct P-loop-like motif involved in phosphate orPRPP binding is found across PyNPs, PRTs-I andAnPRT (Fig 5b). It contains a consensus “TGG”motif flanked by residues whose conservation isfamily dependent (Fig 5b). Interestingly, therelative location of the “KH” and “TGG” motifs inAnPRTs and PyNPs is not conserved (Fig 3).While in AnPRT the KH motif is located in strand"2, ~20 residues away from the glycine-rich loop,in PyNPs the KH and TGG sequences arecontiguous at the C-terminus of "1. This indicatesthat the KH motif must have developed
independently during the divergent evolution of
these enzymes – highlighting that even in
homologous families recurrent motifs can result
from functional needs and no from residual
conservation. A related pattern of basic residues isfound in quinolinate PRT (type II), whosearchitecture is unrelated to that of AnPRTs andPyNPs (Fig 5a). In that family, conserved R and Hresidues (161HR162 in the enzyme from M.
tuberculosis) bind the nitrogenated base, while alysine residue (K140) interacts with the 5-phosphate group of PRPP (2, 4) (Fig 5b). Takentogether, these findings indicate a genericsignificance of these amino acid groups innucleoside/nucleotide recognition.
Canonical PRT-I do not show a markedconservation of R, H and K groups involved in thecoordination of their substrates. Instead, thestructural hallmark of these enzymes is thepresence of two acidic residues that coordinatemagnesium and the hydroxyl groups of the ribosefraction of PRPP (1). Similar protein-PRPPinteractions are established in the unrelatedquinolinate PRT-II by two non-consecutive acidicresidues: E201 and D222 in the enzyme from M.
tuberculosis (3). In ssAnPRT, on the contrary, theconserved doublet 223DE224, primarily coordinatesa second metal ion (MG-2), unique to this family,at the 5-phosphate of PRPP (Fig 2d,f; 5). Insummary, acidic doublets frequently recur inenzyme types involved in coordination ofphosphoribose moieties, but they do not mediateconserved interactions to the substrate.
This comparative analysis reveals that
PyNP and PRT enzyme classes share relatedstrategies for substrate recognition as a result ofindependent, convergent evolution. Within theseenzymes, AnPRT is a unique evolutionary link,which combines the overall architecture of PyNPswith the classical features of a PRT active site.Despite, it has adopted a different mode ofsubstrate binding with the use of a second metalion, compacting of the PRPP substrate andredundant aromatic base coordination.
Acknowledgements
We thank Rainer Bucher (Biozentrum,Basel) for making avai lable recentcrystallographic data on co-crystallized forms ofssAnPRT and its substrates, which did not revealdifferences to those reported here and, thus, servedfor validation of this work.
Our deepest gratitude goes to the staff atbeamlines X06SA (SLS), X11 (EMBL/DESY) andID14-2 (ESRF) for support during data collection.Special thanks go to Prof. Kasper Kirschner forcritical reading of this manuscript.
This work was supported by a grant of theDeutsche Forschungsgemeinschaft (STE 891/5-1)in the framework of the priority program 1170.
by guest on March 19, 2020
http://ww
w.jbc.org/
Dow
nloaded from

Marino et al., 2006 / ssAnPRT
10
REFERENCES
1. Sinha, S. C., and Smith, J. L. (2001). Curr Opin Struct Biol 11, 733-739.2. Eads, J. C., Ozturk, D., Wexler, T. B., Grubmeyer, C., and Sacchettini, J. C. (1997). Structure 5,
47-58.3. Sharma, V., Grubmeyer, C., and Sacchettini, J. C. (1998). Structure 6, 1587-1599.4. Shin, D. H., Oganesyan, N., Jancarik, J., Yokota, H., Kim, R., and Kim, S. H. (2005). J Biol
Chem 280, 18326-18335.5. Mayans, O., Ivens, A., Nissen, L. J., Kirschner, K., and Wilmanns, M. (2002). EMBO J 21, 3245-
3254.6. Kim, C., Xuong, N. H., Edwards, S., Madhusudan, Yee, M. C., Spraggon, G., and Mills, S. E.
(2002). FEBS Lett 523, 239-246.7. Lee, C. E., Goodfellow, C., Javid-Majd, F., Baker, E. N., Shaun Lott, J. (2006) J Mol Biol. 355,
784-97.
8. Pugmire, M. J., and Ealick, S. E. (2002). Biochem J 361, 1-25.9. Mushegian, A. R., and Koonin, E. V. (1994). Protein Sci 3, 1081-1088.10. Smith, D. A., Parish, T., Stoker, N. G., and Bancroft, G. J. (2001). Infect Immun 69, 1142-1150.11. Parish, T. (2003). J Bacteriol 185, 6702-6706.12. Ivens, A., Mayans, O., Szadkowski, H., Wilmanns, M., and Kirschner, K. (2001). Eur J Biochem
268, 2246-2252.13. Kroemer, M., Dreyer, M. K., and Wendt, K. U. (2004) Acta Crystallogr D60, 1679-82.
14. Kabsch, W. (1993). J Appl Cryst 26, 795-800.15. Brunger, A. T., Adams, P. D., Clore, G. M., DeLano, W. L., Gros, P., Grosse-Kunstleve, R. W.,
Jiang, J. S., Kuszewski, J., Nilges, M., Pannu, N. S., et al. (1998). Acta Crystallogr D54, 905-921.16. Jones, T. A., Zou, J. Y., Cowan, S. W., and Kjeldgaard M. (1991) Acta Cryst. A47, 110-119.17. Perrakis, A., Morris, R., and Lamzin, V. S. (1999). Nat Struct Biol 6, 458-463.18. Koch, M.H.J., and J. Bordas. (1983). Nucl. Instrum. Methods 208, 461-469.19. Konarev, P. V., Volkov, V. V., Sokolova, A. V., Koch, M. H. J., and Svergun, D. I. (2003). J
Appl Crystallogr 36, 1277-1282.20. Svergun, D. I. (1992). J Appl Crystallogr 25, 495 - 503.21. Hayward, S., and Berendsen, H. J. (1998). Proteins 30, 144-154.22. Svergun, D. I., Barberato, C., and Koch, M. H. J. (1995). J Appl Crystallogr 28, 768-773.23. Jurgens, C., Strom, A., Wegener, D., Hettwer, S., Wilmanns, M., and Sterner, R. (2000). Proc
Natl Acad Sci U S A 97, 9925-9930.24. Brandl, M., Weiss, M. S., Jabs, A., Suhnel, J., and Hilgenfeld, R. (2001). J Mol Biol 307, 357-
377.25. Steiner, T., and Koellner, G. (2001). J Mol Biol 305, 535-557.26. Thompson, R. E., Li, E. L., Spivey, H. O., Chandler, J. P., Katz, A. J., and Appleman, J. R.
(1978). Bioinorg Chem 9, 35-45.27. Meola, M., Yamen, B., Weaver, K., and Sandwick, R. K. (2003). J Inorg Biochem 93, 235-242.28. Pugmire, M. J., and Ealick, S. E. (1998). Structure 6, 1467-1479.29. Hommel, U., Lustig, A., and Kirschner, K. (1989). Eur J Biochem 180, 33-40.30. Mendieta, J., Martin-Santamaria, S., Priego, E. M., Balzarini, J., Camarasa, M. J., Perez-Perez, M.
J., and Gago, F. (2004). Biochemistry 43, 405-414.31. Degano, M., Gopaul, D. N., Scapin, G., Schramm, V. L., and Sacchettini, J. C. (1996).
Biochemistry 35, 5971-5981.32. Salerno, C., and Giacomello, A. (1981). J Biol Chem 256, 3671-3673.33. Heroux, A., White, E. L., Ross, L. J., Kuzin, A. P., and Borhani, D. W. (2000). Structure Fold Des
8, 1309-1318.
by guest on March 19, 2020
http://ww
w.jbc.org/
Dow
nloaded from

Marino et al., 2006 / ssAnPRT
11
34. Walter, M. R., Cook, W. J., Cole, L. B., Short, S. A., Koszalka, G. W., Krenitsky, T. A., andEalick, S. E. (1990). J Biol Chem 265, 14016-14022.
35. Rick, S. W., Abashkin, Y. G., Hilderbrandt, R. L., and Burt, S. K. (1999). Proteins 37, 242-252.36. Moghaddam, A., Zhang, H. T., Fan, T. P., Hu, D. E., Lees, V. C., Turley, H., Fox, S. B., Gatter,
K. C., Harris, A. L., and Bicknell, R. (1995). Proc Natl Acad Sci U S A 92, 998-1002.37. Prasad, G. S. (2001). Curr Protein Pept Sci 2,301-11.38. Norman, R. A, Barry, S. T., Bate, M., Breed, J., Colls, J. G., Ernill, R. J., Luke, R. W., Minshull,
C. A., McAlister, M. S., McCall, E. J., McMiken, H. H., Paterson, D. S., Timms, D., Tucker, J.A., Pauptit, R. A. (2004) Structure 12, 75-84.
FIGURES
Figure 1: Reactions catalyzed by anthranilate phosphoribosyltransferase (AnPRT) (a) and thymidinenucleoside phosphorylase (TNP) (b)
Figure 2: Substrate coordination by ssAnPRT
a. Overview (only one protomer of the homodimer is shown). Protein domains are color-coded wherethe N-terminal, !-helical domain is golden and the larger C-terminal !/" domain is orange and cyan.The active site is shown at full occupancy and key directional interactions are indicated by stippledlines. R164 (!-helical domain) and K106 (!/" domain), mediating interactions conserved acrossAnPRTs and PyNPs, are displayed in dark blue. G79 (in green) orients AA-II with respect to PRPP/Mg2+. The 79GTGGD83 loop, which becomes structured upon binding of PRPP/Mg2+, is shown inpurple. b. Superimposition of PRPP molecules in their bound state. Coordinates were obtained fromPDB entries 1ZVW, 1A95, 1D6N, 1ECC, 1FSG, 1I13, 1L1R, 1LH0, 1QPR, 1TC2 and 1OPR. PRPPcompounds are shown in dark grey, analogs in green; PRPP bound to ssAnPRT and mtAnPRT iscoloured red and orange, respectively. Magnesium and manganese ions are coloured purple and pink,respectively. The magnesium ions of ssAnPRT and mtAnPRT are labelled green and dark blue,respectively; c. Coordination of AA and d. PRPP/Mg 2+. (2Fo-Fc)!c electron density maps aredisplayed at 1& contour level. Domains and residues R164 and K106 are color-coded as in a; e.Protein-AA interactions; f. Protein-PRPP/Mg 2+ interactions. Dashed lines represent hydrogen bonds,red lines indicate the coordination of Mg2+.
Figure 3: Sequence conservation in the AnPRT and PyNP families
Structure-based sequence alignment of ssAnPRT and human thymidine NP (hsTNP; 1UOU) (38).Secondary structure elements are displayed and numbered, and their integrating residues are
underlined in the individual sequences. Absolutely conserved residues in each family are shown in
black, while green indicates >85% identity conservation. Conservation estimates are based on a
sequence alignment of 97 available sequences of AnPRTs and 57 sequences of PyNPs. Residues
coordinating anthranilate are indicated by “A”. “P” stands for residues binding phosphate groups of
PRPP in AnPRT or the lytic phosphate in PyNP. “M”, “R” and “N” represent residues that coordinate
a magnesium ion, the ribose moiety of PRPP or of a nucleoside substrate, and the nitrogenated base,
respectively. Residues reported to coordinate PRPP in mtAnPRT (7) are colored in orange.
Figure 4: SAXS data from apo and complexed ssAnPRT
a. Scattering patterns from apo ssAnPRT (1) and its complexes with PRPP (2), AA (3) and PRPP/AA(4); (5) scattering computed from the best-fitting models. b. Distance distribution function p(r) ofssAnPRT and its complexes calculated using the full experimental range 0.15 < s < 3.2 nm-1
(identification of curves as in a). c. Superposition of the five best-fitting models in open and d. closed
by guest on March 19, 2020
http://ww
w.jbc.org/
Dow
nloaded from

Marino et al., 2006 / ssAnPRT
12
conformation as modeled using SAXS data. For optimal view of the closure, dimers in c and d areshown slightly rotated with respect to each other. The upper protomers in c and d are equivalent. Tobetter visualize the motion of the large !/" domain in the lower protomer, relative moleculardistances are indicated by arrows. To illustrate the possible repositioning of substrates upon domainmotion, these are displayed in the lower protomer. Substrates in the open conformation (c) arearranged as observed in crystal structures. In the closed conformation (d), PRPP/Mg2+ is positioned inthe equivalent binding pocket of the rotated !/" domain.
Figure 5: Comparative features of substrate complexation by PyNPs, AnPRT, PRTs I and II
a. The structures from left to right are human thymidine NP (PyNP) with bound inhibitor TPI (5-chloro-6-[1-(2-iminopyrrolidinyl) methyl] uracil hydrochloride) (1UOU) and the lytic phosphategroup modeled as in the homologous PyNP from B. stearothermophilus (1BRW); ssAnPRT withbound AA and PRPP/Mg2+; M. tuberculosis quinolinate PRT with bound quinolinate and PRPP(1QPR); and human hypoxanthine PRT with bound inhibitor HPP (7-hydroxy[4,3-d] pyrazolopyrimidine) and PRPP (1D6N). Biological dimers are shown for all cases. Bound substrates aredisplayed to indicate location of active sites within the folds. Nitrogenated bases and nucleosides arein green, phosphate groups and PRPP in red, metals in cyan. For better visualization of substrates, therelated architectures of PyNP and AnPRT are shown here rotated 90o with respect to each other. b.Detail of the active sites of enzymes shown in a. Color code for substrates as in a. The TGG loopmotif is in yellow, the acidic doublets in magenta and R, H and K residues in dark blue. The presenceof conserved glycine-rich motifs as well as acidic doublets in the given families is explicitly displayedas sequence fragments, where “+” indicates that residues are not contiguous in sequence. Color codefor residues in these sequences are as in the models. To reflect the degeneracy in the diverse PRT-Ienzymes, three representative sequences occurring in the family are given, where the middle onecorresponds to the PRT displayed.
by guest on March 19, 2020
http://ww
w.jbc.org/
Dow
nloaded from

Table I: X-ray data collection and structure refinement statistics
Substrate AA PRPP /Mg2+ AA + PRPP/Mg2+
Soaking conditions
Substrate concentration 10 mM 10 mM PRPP / 5mM MgCl2 10 mM AA / 10 mM PRPP / 5mM MgCl2
Time 1 min 2 min 10 minTemperature 20 oC 4 oC 4 oC
X-ray Source XS06A (SLS) X11 (EMBL/DESY) ID14-2 (ESRF)
Detector MarCCD-165 MarCCD-165 ADSC Quantum CCD
Wavelength 0.951 Å 0.815 Å 0.933 Å
Symmetry P2 P2 P2 Unit cell (Å) a=91.81, b=65.62, c=115.61 a=92.69, b=65.81, c=116.72 a=92.56, b=66.10, c=117.07
!= 107.39° != 107.8° != 107.76°Resolution (Outer-shell) 20 – 2.43 (2.5 – 2.43) Å 20 –2.56 (2.67 – 2.56) Å 20 – 2.40 (2.50 – 2.40) ÅUnique reflections 48850 (3874) 41256 (3550) 51457 (5266)Rsym(I) (%) 6.5 (43.0) 7.0 (32.0) 6.2 (24.6)Multiplicity 3.84 (3.41) 3.6 (2.2) 3.76 (3.03)Completeness (%) 97.9 (96.2) 94.6 (69.3) 96.9 (87.1)<I/" (I)> 10.99 (3.1) 10.43 (3.22) 15.33 (5.45)
Content a.u. 4AnPRT:7AA 4AnPRT:4PRPP:8Mg2+ 4AnPRT:7AA:4PRPP:8Mg2+
No. of non-hydrogen atoms 10826 10752 11239Protein residues1 1366 1376 1375Water molecules 253 92 505Rwork / Rfree (%) 19.9 / 24.2 20.3 / 26.0 20.6 / 26.9No. R-free reflections 584 503 624RMSDbond (Å) 0.007 0.007 0.007RMSDangle
(o) 1.33 1.39 1.33Protein residues favoured (%)2 93.2 90.6 90.2Protein residues disallowed 2 0 0 0Disordered protein residues3 A81-A83,A344-A345 A345, B345 A345, B345
B81-B83, B344-B345 C345, D345 C344-C345C80-C83 D345
1One ssAnPRT protomer comprises 345 amino acids; 2Calculated using PROCHECK (Laskowski et al., 1993); 3Individual polypeptide chains in the asymmetricunit of this crystal form are indicated as A, B, C and D.
by guest on March 19, 2020
http://ww
w.jbc.org/
Dow
nloaded from

Table II: Experimental and calculated scattering parameters of ssAnPRT and its
substrate complexes
Sample Rg(nm)a Rgt(nm)b Dmax(nm)c !Cd !d !M
d
(Apo) 1O17 3.44 11.4e
Apo-AnPRT 3.44±0.04 3.42 11.0±0.5 1.19 1.17 1.58
AnPRT/PRPP 3.42±0.05 3.40 11.0±0.5 1.22 1.21 1.59
AnPRT/AA 3.30±0.10 3.24 10.5±1.0 1.24 1.09 1.21
AnPRT/PRPP/AA 3.13±0.06 3.10 10.0±0.5 1.67 1.15 2.33
aRg, experimental radius of gyration; bRgt, radius of gyration of the model with lowest fitted
discrepancy to the experimental data; cDmax, maximum molecular dimensions; dBest fitted
discrepancies between experimental data and scattering curves computed from structural
models, where !C refers to experimental data versus the crystallographic model of apo-
ssAnPRT (1O17), ! accounts for experimental data versus best models obtained by rigid
body rotation, which in this case showed an asymmetric hinge closure, and !M corresponds
to experimental data versus mixtures of models in symmetrically opened and closed
conformations. Calculated values for 1O17 are given as reference, where e includes a plus 6
Å correction to account for the hydration shell.
by guest on March 19, 2020
http://ww
w.jbc.org/
Dow
nloaded from

Table III: Steady-state enzyme kinetic data for purified ssAnPRT variants.
Enzyme variant liganda Mg2+ [mM] KMAA [µM] KM
PRPP[mM] kcat [s--1]
Wild type 0.05 0.04 0.16 4.2
2 0.07 4.3 1.1
R164A AA-I 0.05 297 0.17 0.62
R164A+H154A AA-I 0.05 149 bn.d. 0.73
H107A AA-II 0.05 0.53 0.30 5.1
H107A+P178A AA-II 0.05 12.5 bn.d. 2.0
K106Q PRPP 2 0.75 0.26 0.48
D223N MG-2 2 0.16 0.07 6.0
E224Q MG-2 2 0.91 0.26 5.6
Conditions: 50mM Tris-HCl, pH 7.5 at 60°C, in the presence of saturating concentrations
(10 x KM) of the second substrate; aligand complexed by wild type residue; bn.d.: value
was not determined.
by guest on March 19, 2020
http://ww
w.jbc.org/
Dow
nloaded from

5-phosphoribosyl-1-pyrophosphate
A
B
(Fig 1)
by guest on March 19, 2020
http://ww
w.jbc.org/
Dow
nloaded from
A B
PRPP
AA-II
AA-I
N
C
(Fig 2)
MG-1
MG-2
by guest on March 19, 2020
http://ww
w.jbc.org/
Dow
nloaded from

R164
M157
G79
A78
P178
G177
H154
H107
G108
N109
A150
AA-I
AA-IIG79
K106
T92
N89
T87
D83
D223
E224
S91
MG-1
MG-2
C D
(Fig 2)
by guest on March 19, 2020
http://ww
w.jbc.org/
Dow
nloaded from

E F
MG-1
MG-2
(Fig 2)
by guest on March 19, 2020
http://ww
w.jbc.org/
Dow
nloaded from

P
AA
ssAnPRT ----MNINEILKKLINKSDLEINEAEELAKAIIRGEVPEILVSAILVALRMKGESKNEIVGFARAMRELAIKI-DV---P-NAIDTAGTGGDGLGT:87
hsTNP PKQL---PELIRMKRDGGRLSEADIRGFVAAVVNGSAQGAQIGAMLMAIRLRGMDLEETSVLTQALAQSGQQLEWPEAWRQQLVDKHSTGGVG---:122
PNR
P RP PAAA A A A A
ssAnPRT VNVSTASAILLSLV-NPVAKHGNRAVSGKSG-------SADVLEAL-GYNIIVPPERAKELVNKTNFVFLFAQ-YYHPAMKNV-ANVRKTLGI-RT-:171
hsTNP DKVSLVLAPALAACGCKVPMISGR-------GLGHTGGTLDKLESIPGFNVIQSPEQMQVLLDQAGCCIVGQSEQLVPADGILYA-ARDVTATVDSL:211
N N N
AA MM
ssAnPRT IFNILGPLT-N-PANAKYQLMGV------FS------KDHLDLLSKSAYELDFNKIILVYGEPGIDEVSPIGNTFMKIVSKRGIEEVKLNVTDFGIS:254
hsTNP PLITASILSKKLVEGLSALVVDVKFGGAAVFPNQEQARELAKTLVGVGASLG L-RVAAALTA--M-------------------------------:283
N N N N
ssAnPRT PIPIEKLI-VNSAEDSAIKIVRAFLGKD-EHVAEFIKINTAVALFALD-RVGDFREGYEYADHLI-E-KSLDKLNEIISMNGDVTKLKTIVVKSSG:345
hsTNP ---DKPLGRCVGHALEVEEALLCMDGAGPPDLRDLVTTLGGALLWLSGHA-GTQAQGAARVAAALDDGSALGRFERMLAAQG..............:480
!8’ !8!5 !6 !7"2 "3
!1 !2 !3 "1!4
!12 !13 !14 !15!11’’ !11
!10"4 "5 "6 "7 !11’!9
by guest on March 19, 2020
http://ww
w.jbc.org/
Dow
nloaded from

A B
C D
(Fig 4)
by guest on March 19, 2020
http://ww
w.jbc.org/
Dow
nloaded from

PyNP AnPRT PRT type II PRT type I
A
(Fig 5)
by guest on March 19, 2020
http://ww
w.jbc.org/
Dow
nloaded from

R202
K115
H116
118TGG
120
R164
H154
H107
K106
D223
E224
D222
E201
R162
H161
K140
R105!
80TGG
82
E133
D134
138TG
139
DKHSTGGXGD DXXGTGGD + DE E + D DDxxTGGT
EDxxDTGxx
DDxxDTGGT
B (Fig 5)
by guest on March 19, 2020
http://ww
w.jbc.org/
Dow
nloaded from

and Olga MayansMarco Marino, Miriam Deuss, Dmitri I. Svergun, Petr V. Konarev, Reinhard Sterner
phosphoribosyltransferase from sulfolobus solfataricusStructural and mutational analysis of substrate complexation by anthranilate
published online May 19, 2006J. Biol. Chem.
10.1074/jbc.M601403200Access the most updated version of this article at doi:
Alerts:
When a correction for this article is posted•
When this article is cited•
to choose from all of JBC's e-mail alertsClick here
Supplemental material:
http://www.jbc.org/content/suppl/2006/05/23/M601403200.DC1
by guest on March 19, 2020
http://ww
w.jbc.org/
Dow
nloaded from

![Nicotinamide phosphoribosyltransferase postpones rat bone ... · entering a senescent state during aging [1]. Accordingly, the aging of SCs is crucially implicated in individual aging](https://img.pdfslide.net/doc/110x75/5f1daac5b2c2c1053d52d773/nicotinamide-phosphoribosyltransferase-postpones-rat-bone-entering-a-senescent.jpg)



























