Embed Size (px)
Citation preview

Maximum Likelihood Pedigree Reconstruction using
Integer Linear Programming
James Cussens1, Mark Bartlett1, Elinor M. Jones2, and Nuala A. Sheehan2
1Department of Computer Science, University of York, York, North
Yorkshire, YO10 5GH, United Kingdom
2Department of Health Sciences, University of Leicester, Leicester,
Leicestershire, LE1 6TP, United Kingdom
Running Title: Pedigree Reconstruction using ILP
Corresponding Author: Dr Nuala Sheehan,
Department of Health Sciences,
University of Leicester,
Adrian Building, University Road,
Leicester, LE1 7RH
Tel: 00 44 116 229 7271
Email: [email protected]
1

Abstract
Large population biobanks of unrelated individuals have been highly successful in
detecting common genetic variants affecting diseases of public health concern. However,
they lack the statistical power to detect more modest gene-gene and gene-environment
interaction effects or the effects of rare variants for which related individuals are ideally
required. In reality, most large population studies will undoubtedly contain sets of
undeclared relatives, or pedigrees. Although a crude measure of relatedness might
sometimes suffice, having a good estimate of the true pedigree would be much more
informative if this could be obtained efficiently. Relatives are more likely to share
longer haplotypes around disease susceptibility loci and are hence biologically more
informative for rare variants than unrelated cases and controls. Distant relatives are
arguably more useful for detecting variants with small effects since they are less likely to
share masking environmental effects. Moreover, the identification of relatives enables
appropriate adjustments of statistical analyses that typically assume unrelatedness.
We propose to exploit an integer linear programming (ILP) optimisation approach
to pedigree learning, which is adapted to find valid pedigrees by imposing appropriate
constraints. Our method is not restricted to small pedigrees and is guaranteed to return
a maximum likelihood pedigree. With additional constraints, we can also search for
multiple high probability pedigrees and thus account for the inherent uncertainty in
any particular pedigree reconstruction. The true pedigree is found very quickly by
comparison with other methods when all individuals are observed. Extensions to more
complex problems seem feasible.
Keywords: Constraint based optimisation, Genetic marker data, Bayesian networks,
Model uncertainty
2

1 Introduction
Identification of relatives amongst a group of individuals from genetic marker data is relevant
to areas as diverse as evolution and conservation research, epidemiological and genealogical
research, and forensic science identification problems [Blouin, 2003; Jones and Ardren, 2003;
Meagher and Thompson, 1987; Olaisen et al., 1997]. In medical research, large population
biobank studies of unrelated individuals have been set up worldwide to investigate the ge-
netic risk factors underlying the common complex diseases of major public health concern. It
is now well understood that they do not have sufficient statistical power to detect the effects
of rare genes and the more modest effects of gene-gene and gene-environment interactions.
Sets of relatives are more likely to share longer haplotypes around susceptibility loci and are
hence biologically more informative for rare variants than unrelated ‘cases’ and ‘controls’. In
fact, distant, rather than close, relatives are more likely to be useful for detecting genes with
small effects as the genetic effects are less likely to be masked by the shared environment
effects typically arising in near relatives. In the absence of exact information, general pair-
wise relatedness, rather than specific relationships, can be estimated from population sample
data using approaches based on identity by descent (IBD) methods [McPeek and Sun, 2000;
Sieberts et al., 2002; Leutenegger et al., 2003; Stankovich et al., 2005; Browning and Brown-
ing, 2010; Powell et al., 2010; Thompson, 2008b]. Such methods are particularly efficient at
detecting misspecified relationships: assessing the true relationship is more difficult. How-
ever, large population studies undoubtedly contain pedigrees (i.e. family trees) [Thompson,
2008a] so if it were possible to reconstruct (extended) pedigrees easily from population sam-
ple genetic data, we would have far richer information and sets of distant relatives could be
readily proposed.
Pedigree information is also relevant to standard statistical association testing where fail-
ure to take account of existing relationships can yield overly high false positive results [New-
man et al., 2001; Stankovich et al., 2005; Choi et al., 2009; Thornton and McPeek, 2010].
Likewise, some ‘true’ associations could be missed: a susceptibility gene for which different
variants are segregating in different groups of relatives would never be detected in an asso-
ciation analysis on unrelated individuals. Moreover, even in linkage analyses where pedigree
3

data are required, undeclared relatedness amongst the founders of a particular family and
between founders of different families can heavily bias the results if appropriate adjustments
are not made [Genin and Clerget-Darpoux, 1996; Sheehan and Egeland, 2008; Thompson,
2008a].
It is sometimes convenient to consider the pedigree identification problem as one of de-
termining the most likely pedigree from a set of possible alternatives. In theory, estimating
the most likely pedigree for a given set of individuals from genetic marker data simply
requires consideration of all possible relationships amongst them and computing the like-
lihood for each [Cannings and Thompson, 1981; Thompson, 1975]. However, due to the
potentially enormous number of possible pedigrees, simple enumeration rapidly becomes im-
practical [Egeland et al., 2000; Sheehan and Egeland, 2007]. When general features of the
pedigree are of more interest than precise estimation of the entire set of relationships, ‘se-
quential’ (i.e. greedy) algorithms can be used which produce a single high likelihood (but
not necessarily maximum likelihood) reconstruction by starting from a position where all
individuals are assumed to be unrelated and gradually accepting sibships on the basis of
the increase attained in log-likelihood [Thompson, 1976a]. In order to give good results,
the above approach requires information on age (or at least an age ordering), sex, average
sibship size, typical generation gap and age disparity among parents. Other approaches
to pedigree reconstruction which produce a high likelihood pedigree include simulated an-
nealing [Almudevar, 2003; Riester et al., 2009] and Markov chain Monte Carlo (MCMC)
methods [Almudevar, 2007].
In contrast, the method presented in this paper will yield a guaranteed maximum like-
lihood pedigree, rather than just a high-probability pedigree. This has also been achieved
through Bayesian network learning using dynamic programming by Cowell [2009]. The ap-
proach we propose in this paper uses constraint based methods which have been used for
Mendelian consistency checking [Sanchez et al., 2008] but not to our knowledge for pedigree
reconstruction. More specifically, we use integer linear programming (ILP) to search for
maximum likelihood pedigrees. For clarity and direct comparison with Cowell [2009], we
have chosen to cast the problem as one of Bayesian network learning noting that while this
is not necessary for the situation presented here, such a representation will undoubtedly be
4

useful for more complex applications.
There is no guarantee that the most probable pedigree is the true pedigree [Thompson,
1976b]. This is due to the probabilistic nature of genetic inheritance whereby marker data
on unrelated individuals may indicate that they are, in fact, related and where the observed
data could assign higher likelihood to an incorrect relationship than the true one. Failure
to acknowledge such inevitable uncertainty could lead to spuriously confident inferences and
so, in addition to finding the most likely pedigree, we will also show how our approach can
be used to search for many high likelihood pedigrees.
The structure of the paper is as follows. In Section 2, we outline how a pedigree can be
represented as a Bayesian network and present the likelihood function to be optimised. We
then explain how the pedigree reconstruction problem can be encoded as an ILP optimisation
following the argument presented in Cussens [2010]. Results for this encoding are given for
a straightforward situation in Section 3 and compared with competing methods. The paper
concludes in Section 4 with a discussion of the issues and challenges for future work, including
possible extensions to more complex problems.
2 Material and Methods
2.1 Pedigrees and Bayesian networks
A pedigree can easily be represented as a directed acyclic graph (DAG) [Lange and Elston,
1975], where each node corresponds to a pedigree member and a directed connection (i.e.
arrow, or directed edge) between two individuals represents a parent-child relationship. We
note that this defines a special class of DAG with well defined structural constraints: each
node in the graph is known to have exactly two parent nodes (although either may be latent)
and these parents must be of opposite sexes. A node with no parents is a pedigree founder.
Figure 1(a) shows a simple pedigree in conventional ‘family tree’ form, with males represented
as squares and females as circles. The corresponding DAG is shown in Figure 1(b) and the
marriage node graph [Thomas, 1985; Cannings et al., 1978] in Figure 1(c).
[Figure 1 about here.]
5

A Bayesian network (BN) is a directed acyclic graph whose nodes, v ∈ V , represent
random variables. There are several ways of expressing a pedigree as a BN [Lauritzen
and Sheehan, 2003] but one of the simplest is to consider the graph in Figure 1(b) where
the nodes no longer represent the pedigree members themselves but random variables that
assign genotypes to these individuals. These random variables have a joint distribution that
depends on the pedigree structure and the mode of inheritance for the relevant genetic locus
which can be factorised into a product of conditional distributions for each node given its
parent nodes.
We will introduce our reconstruction approach with a focus, as other authors have done,
on the most straightforward case [Almudevar, 2003; Cowell, 2009; Thompson, 1976a]. Specif-
ically, we assume that the sample of individuals is complete by which we mean that each
observed individual has fully observed genetic data and each unobserved parent is assumed
to be unrelated to any other individual in the sample and is hence an unobserved pedigree
founder. We will also adopt the common assumption of random union of gametes indicating
that founder genes are independent of each other with the consequent implication of Hardy-
Weinberg proportions for founder genotypes. Finally, we will assume that genetic markers
at the different loci under consideration are segregating independently. While perfectly ad-
equate for the independent marker data that we consider here, we note that the above BN
representation of a pedigree will not suffice for more complex genetic models involving linked
loci, for example, where more general graphical model representations are more appropriate.
We note that it also implicitly assumes simple Mendelian inheritance of genes in offspring
from parents [Lauritzen and Sheehan, 2003].
For a given pedigree graph, G, let V be the set of individuals and identify v ∈ V with a
random variable Gv assigning a genotype at a single locus to v. Let fv and mv be the labels
of the nodes representing the father and mother of v, respectively. Then
τ(gv | gfv, gmv
) ≡ P (Gv = gv |Gfv= gfv
, Gmv= gmv
)
denotes the probability that v has genotype gv given the parental genotype combination
of gfvand gmv
. In the event where only one parent, e.g. mv, is observed, we assume that
6

the unobserved parent is a founder and sum over all possible genotypes for that parent.
Specifically,
τ(gv | gmv) =
∑
gfv
τ(gv | gfv, gmv
)p(gfv)
where p(gfv) — the marginal probability that a founder individual fv has genotype gfv
—
is derived from the population allele frequencies and the assumption of Hardy-Weinberg
proportions. This is equivalent to assuming that the gene inherited from the unobserved
parent is randomly drawn from the population allele frequency distribution. In general, we
can express these probabilities as
τ(v, Pa(v,G))
where Pa(v,G) denotes the parent set of v in G which will comprise 0, 1 or 2 nodes and
where τ(v, ∅) ≡ p(gv) represents the marginal probability that v has genotype gv. Note
that these probabilities are just the usual Mendelian transmission probabilities of 0, 1
4, 1
2
or 1 for non-founding pedigree members with two observed parents. Since a pedigree is
completely specified by parent-child relationships, the probability of any single configuration
of genotypes on a pedigree, and hence the likelihood under the assumption of a complete
sample and Mendelian inheritance, decomposes into a product of conditional probabilities
and can be written as
L(G) =∏
v∈V
τ(v, Pa(v,G)). (1)
Note that this is precisely the form of the standard decomposition of a pedigree likelihood
into triplet products under the same assumptions. (See Thompson [2000], for example.)
The aim of a maximum likelihood pedigree reconstruction is to find G such that L(G) is
maximised. Given that the factorisation of the joint distribution in (1) defines a Bayesian
network [Lauritzen, 1996], this is equivalent to searching for an optimal Bayesian network
where the search is constrained to find networks that are valid pedigrees. It is often more
convenient to work with the log-likelihood so our optimisation problem in that case is to find
7

G to maximise
l(G) = log L(G) =∑
v∈V
log τ(v, Pa(v,G)). (2)
Since our multilocus genotype is based on independently segregating markers, by as-
sumption, the joint likelihood of interest is simply a product of likelihoods (Equation (1))
calculated for each locus separately and the log-likelihood is a sum of the relevant expres-
sions (Equation (2)). For convenience, we will thus focus on a single locus to describe our
approach.
2.2 An ILP formulation for likelihood maximisation
An integer linear program (ILP) is defined by:
1. a set of variables X, representing unknown quantities, some of which will be restricted
to take only integer values,
2. a linear objective function of the form∑
x∈X cxx where the coefficients cx are fixed
constants, and
3. linear equations and inequalities putting joint constraints of the form∑
x∈X axx ≤ b
on the values the variables can take where the values ax and b are fixed constants.
The ILP optimisation problem is to find an assignment of values to the variables which
maximises the objective function while respecting all constraints. This is an NP-hard prob-
lem in the general case [Karp, 1972] which means there is no known polynomial time algo-
rithm to solve it in the general case. It is however still possible to solve the problem exactly,
thus guaranteeing that the optimal pedigree is found. One simple algorithm that does so
is to enumerate all potential solutions and examine them in turn. This would of course be
impractically slow for any reasonably sized problem.
Decades of research (both academic and commercial) have gone into creating highly opti-
mised ILP solvers based on the simplex algorithm which achieve good temporal performance
in practice [Wolsey, 1998]. Given increasingly large problems, these solvers will eventually
8

run out of computer memory or take too long to be of practical use. However, provided that
the solvers are given sufficient time and resources, it can be guaranteed that they will always
find the optimal solution. The typically good performance of these solvers means that they
present an attractive option for maximum likelihood pedigree reconstruction provided the
latter can be efficiently coded as an ILP problem.
Such an encoding is indeed possible but is far from obvious and so it will be described in
some detail here for the pedigree likelihood shown in Equation (1). For illustrative purposes
we will describe how our encoding applies to a very simple pedigree reconstruction problem
involving n = 3 individuals, v1, v2 and v3, under the assumption that marker data are
available for all three but there is no additional information concerning their sex, age or
relatedness.
The first step is to define variables which allow a numeric encoding of any possible
pedigree. From the simple observation that a pedigree specifies the parents of an individual,
indicator functions of the form I(W → v)(G) are created for each possible parentage for each
individual. Specifically,
I(W → v)(G) =
1 if v has parents W in G
0 otherwise(3)
where W ⊆ V \ {v} and |W | ≤ 2. In our tiny example there are 12 such variables:
I({} → v1) I({v2} → v1) I({v3} → v1) I({v2, v3} → v1)
I({} → v2) I({v1} → v2) I({v3} → v2) I({v1, v3} → v2)
I({} → v3) I({v1} → v3) I({v2} → v3) I({v1, v2} → v3)
where, for example, I({v2} → v1) = 1 indicates that v2 is the only parent of v1 in V .
[Figure 2 about here.]
Since each pedigree member has only one set of parents, any possible pedigree G deter-
mines a joint instantiation of the binary variables, I(W → v) in Equation (3) by setting
exactly |V | = n of them to 1 and all the others to 0. For example, if we consider the
pedigree for our toy example in Figure 2(a), we have I({} → v1) = 1 since v1 has no par-
9

ents (i.e. is a founder), I({v1} → v2) = 1 since v2 has one parent v1 in the pedigree and
I({v2} → v3) = 1 to represent that v3 has v2 as its only parent. Analogously, I({} → v1) = 1,
I({v1, v3} → v2) = 1 and I({} → v3) = 1 for Figure 2(b) and I({} → v1) = 1, I({} → v2) = 1
and I({v2} → v3) = 1 for Figure 2(c). This approach allows any pedigree to be mapped to
a set of variables of length n + n(n− 1) + 1
2n(n− 1)(n− 2) each with a value of zero or one.
However, the mapping is not one-to-one as most such sets of variables do not correspond to a
valid pedigree; there is no pedigree that corresponds to all variables being ones, for example.
Noting that I(W → v)(G) only takes the value 1 when W = Pa(v,G), the pedigree
log-likelihood shown in Equation (2) can now be rewritten in terms of these binary variables
as
l(G) = log L(G) =∑
v,W
log τ(v,W ) [I(W → v)(G)] . (4)
The maximum likelihood pedigree reconstruction problem is thus reformulated as a prob-
lem of finding an instantiation of I(W → v) to maximise the quantity shown in Equation (4)
subject to the constraint that this instantiation represents a valid pedigree, G. Since all
the variables are integer-valued and the objective function is linear in these variables, this
satisfies the definition of an integer linear programming problem as long as the constraints
on the I(W → v) can also be expressed as linear equations and inequalities. We show below
that such an expression is indeed possible.
Constraints are required to rule out sets of variable assignments that do not correspond
to a valid pedigree. There are three properties that must be met for a set of assignments
to encode a valid pedigree. First, each individual must have exactly one set of parents.
Second, no individual can ever be his own ancestor. Third, it must be possible to assign sex
consistently to each member of the pedigree. We will now show how each of these properties
can be expressed in the form of linear constraints.
Ensuring one set of parents The most basic constraint, as noted above, is that each in-
dividual, v, has exactly one parent set. This can be expressed concisely by n linear equations
of the form
10

∀v :∑
W
I(W → v) = 1 (5)
For our simple example, these three constraints are as follows:
I({} → v1) + I({v2} → v1) + I({v3} → v1) + I({v2, v3} → v1) = 1
I({} → v2) + I({v1} → v2) + I({v3} → v2) + I({v1, v3} → v2) = 1
I({} → v3) + I({v1} → v3) + I({v2} → v3) + I({v1, v2} → v3) = 1.
To understand why these constraints work, note that for a valid pedigree, exactly one of
the variables in each equation will be equal to 1 (the variable encoding the correct parent
set) and the remaining variables will each be equal to 0. If a variable were to have no parent
set, then the equation for that variable would have its left hand side equal to 0 and hence the
constraint would be violated. Similarly, if a variable were assigned two or more parent sets,
the left hand side would sum to more than 1 and again the constraint would be violated.
Ruling out cycles While an instantiation of the I(W → v) satisfying Equation (5) will
represent a graph where no node can have more than one parent set, these constraints are
not enough to rule out graphs with (directed) cycles i.e. where an individual node can be its
own ancestor or descendant. In our example, setting I({v3} → v1) = 1, I({v1} → v2) = 1
and I({v2} → v3) = 1 would clearly satisfy the constraints in Equation (5) but does not
result in a valid pedigree.
One way to rule out directed cycles, and the approach we use in this paper, is to assign
generation numbers to the individuals. A founder member has generation number zero and
every other individual must have a generation number that is greater than that of each of
its parents. In the absence of any prior knowledge on the maximum generation number, m,
it is possible to use m = n − 1.
For each individual v in the problem, let us introduce a new variable, gen(v), to represent
the generation number of that individual. If gen(v) denotes the generation number assigned
to individual v and u is a parent of v, then we must find assignments to these variables such
11

that gen(v) ≥ gen(u) + 1. Consider the following n(n − 1) constraints:
∀u, v : gen(v) − gen(u) ≥ −m + (m + 1)∑
W :u∈W
I(W → v) (6)
for two distinct individuals labelled u and v. Note that if u is not a parent of v, the
summation∑
W :u∈W I(W → v) on the RHS is 0 and the constraint is vacuously satisfied.
If u is a parent of v, the summation — and hence the entire RHS — is 1 effecting the
desired constraint, gen(v)−gen(u) ≥ 1, on a parent-child relationship. Furthermore, for any
ancestor w of v, it follows that gen(w) < gen(v). A directed cycle necessarily implies that
at least two individuals are their own ancestors leading to an obvious inconsistency. The
constraints in Equation (6) are hence sufficient to rule out such cycles.
The form given in Equation (6) does not correspond to that required for an ILP problem,
but after rearrangement we can write:
∀u, v : (m + 1)∑
W :u∈W
I(W → v) − gen(v) + gen(u) ≤ m (7)
which is in the desired form.
Returning to the example with n = 3 individuals, the 6 new constraints obtained are:
3 [I({v2} → v1) + I({v2, v3} → v1)] − gen(v1) + gen(v2) ≤ 2
3 [I({v1} → v2) + I({v1, v3} → v2)] − gen(v2) + gen(v1) ≤ 2
3 [I({v3} → v1) + I({v2, v3} → v1)] − gen(v1) + gen(v3) ≤ 2
3 [I({v1} → v3) + I({v1, v2} → v3)] − gen(v3) + gen(v1) ≤ 2
3 [I({v3} → v2) + I({v1, v3} → v2)] − gen(v2) + gen(v3) ≤ 2
3 [I({v2} → v3) + I({v1, v2} → v3)] − gen(v3) + gen(v2) ≤ 2
Ensuring sex consistency
[Figure 3 about here.]
The constraints given by (5) together with those in (7) are sufficient to ensure that only
variable assignments encoding directed acyclic graphs are allowed. However, not all such
12

graphs are valid pedigrees. The simple example in Figure 3 (taken from Cowell [2009]) shows
a DAG that is not a valid pedigree. The problem arises because of the fact that each of the
parent pairs (1, 2), (1, 3) and (2, 3) must be of opposing sexes and these clearly cannot be
consistently assigned.
In order to ensure that a sex can be consistently assigned to all individuals in the pedigree,
a further binary variable If (u) is created for each individual u such that If (u) = 1 if and
only if u is a female. Constraint (8) states that if an individual v has two parents, then
at most one is female and constraint (9) states that in this situation at least one parent is
female. Note that in both cases, if I({u,w} → v) = 0 (i.e. v does not have {u,w} as parents)
then the constraints are vacuously satisfied.
∀u, v, w : I({u,w} → v) + If (u) + If (w) ≤ 2 (8)
∀u, v, w : I({u,w} → v) − If (u) − If (w) ≤ 0. (9)
With these constraints in place, the maximum (log) likelihood pedigree reconstruction
problem can now be reformulated as follows from Equation (4):
Maximise∑
v,W log τ(v,W )I(W → v)
subject to
∀v :∑
W I(W → v) = 1
∀u, v : (m + 1)∑
W :u∈W I(W → v) − gen(v) + gen(u) ≤ m
∀u, v, w : I({u,w} → v) + If (u) + If (w) ≤ 2
∀u, v, w : I({u,w} → v) − If (u) − If (w) ≤ 0
where
|W | ≤ 2
∀v,W : I(W → v) ∈ {0, 1} ∀v : If (v) ∈ {0, 1} ∀v : gen(v) ∈ [0,m].
(10)
Any assignment to the variables that meets these constraints corresponds to a valid
13

pedigree. A particular assignment to the variables that also maximises the objective function
is guaranteed to be a maximum likelihood pedigree.
Obviously, for the tiny running example of n = 3 individuals just considered, one could
easily construct and score each of the 25 possible pedigrees and avoid the procedure de-
scribed above. However, that would not scale to larger numbers of individuals. For the ILP
formulation with n individuals, there are only 1
2n3 − 1
2n2 + 3n variables and n3 − 2n2 + 2n
constraints so, in principle, the method should extend to reasonably large problems.
Note that, for any given situation, the number of variables and constraints to be consid-
ered may in fact be lower. We consider a genetic model in which there is no mutation; this
means that τ(v,W ) = 0 for many combinations of parent set W and child u. A valid pedigree
will hence not contain such relationships and the value of the corresponding I(W → v) has
to be 0. Once these variables are fixed at 0, some of the constraints featuring these variables
may be provably satisfied by any valid assignment of values to the remaining variables. Such
redundant constraints can then be removed.
2.3 Additions and improvements to the ILP formulation
Having formulated the problem of finding the most likely pedigree as an ILP problem, we
introduce some slight modifications that improve the solving performance and allow for the
k most likely pedigrees to be found.
At least one founder Preliminary experiments revealed that explicitly adding the implied
constraint that there has to be at least one pedigree founder greatly increases the performance
of the algorithm. This is given as
∑
v
I({} → v) ≥ 1. (11)
This is always trivially true since pedigrees are acyclic and so a maximum likelihood
pedigree will still be returned.
Finding the kth most likely pedigree As already noted, finding a single maximum
likelihood reconstruction does not take account of model uncertainty, and for this reason,
14

we wish to find multiple high likelihood pedigrees. Specifically, we want to find the k most
likely pedigrees, for some arbitrary value k. This can be done by repeatedly solving the ILP
problem given in (10), adding one additional constraint each time to prevent a previously
found solution from being returned again. In this way, repeated solutions of the problem
yield valid pedigrees in order of descending likelihood.
The additional constraint added each time is as follows:
∀ W, v s.t. I(W → v)(G) = 1 :∑
W,v
I(W → v) < n (12)
where G is the previously found pedigree. The constraint simply states that at least one
parent set in the new pedigree has to differ from the previous one. For the kth most likely
pedigree, there will be k−1 such constraints, one for each of the previously found pedigrees.
2.4 Marker data and pedigree structures
In order to assess how well the method performs, the results reported in Section 3 are based
on simulated datasets about which there is no uncertainty. To generate each dataset, genetic
profiles were created for each individual of a given pedigree based on a given distribution of
population allele frequencies under the assumptions stated in Section 2.1. We simulated 100
datasets for each combination of pedigree structure and set of allele frequencies considered.
Allele Frequencies We simulated data from two sets of allele frequencies, both based on a
typical forensic set of microsatellite markers. We used the original (not rounded) Caucasian
allele frequencies for the 15 autosomal short tandem repeat loci (the 13 CODIS core loci plus
D19S433 and D2S1338) corresponding to Table 1 of Butler et al. [2003] as an example of
a realistic distribution. These allele frequencies were previously used for the same purpose
in Cowell [2009]. For our second set of frequencies, we considered the same 15 marker set
but assigned equal probabilities to the listed alleles for each marker. This is what we will
refer to as our ‘uniform frequency distribution’. In both cases, we thus have 15 markers with
allele numbers ranging between 7 and 15.
15

Pedigrees
[Figure 4 about here.]
[Figure 5 about here.]
[Figure 6 about here.]
Several pedigree structures are considered. For direct comparison with the results re-
ported in Cowell [2009], we used the two small pedigrees of 20 individuals considered in
that paper and reproduced in Figure 4. As an example of a larger structure, we took the
59-member pedigree of Almudevar [2003], reproduced here in Figure 5, as a reasonable repre-
sentation of the complexity to be expected in a typical human pedigree. In order to consider
a pedigree complexity effect, we also considered the cyclic half-sib, cyclic first cousin and
quadruple second cousin regular mating structures of Wright [1921], each with 64 individuals
i.e. 7 generations descended from 8 founders. The half-sib structure is the most inbred of
the three. However, although there are no matings between close relatives in the quadru-
ple second cousin pedigree shown in Figure 6, it requires only four generations before all
individuals are related to all eight founders.
3 Results
Initial reported results using integer linear programming for maximum likelihood pedigree
reconstruction are encouraging [Cussens, 2010]. Here, we will perform a more detailed ex-
ploration where we aim to assess how well our algorithm performs on a range of pedigrees.
We will consider the time needed by this approach to find the most likely pedigree and also
the time needed to find the kth most likely pedigree.
Further to this, we examine the suitability of any approach which searches for maximum
likelihood pedigrees by examining the similarity between the real pedigree and the maximum
likelihood one. This is developed into considering the distribution of likelihood over the
k most likely pedigrees. Finally, we compare our approach to those presented by Cowell
[2009], Riester et al. [2009] and Almudevar [2003].
16

All reported results were produced on a normal desktop computer (2.8GHz quad-core
running Windows 7) with the Gurobi ILP solver, for which a free academic licence is available.
3.1 Solution Times
For the four larger pedigrees (the 59 member one shown in Figure 5 and the three regular
mating structures studied), we first considered the time taken to find a most likely pedigree
for the 100 data sets simulated from both sets of allele frequencies. Side by side boxplots of
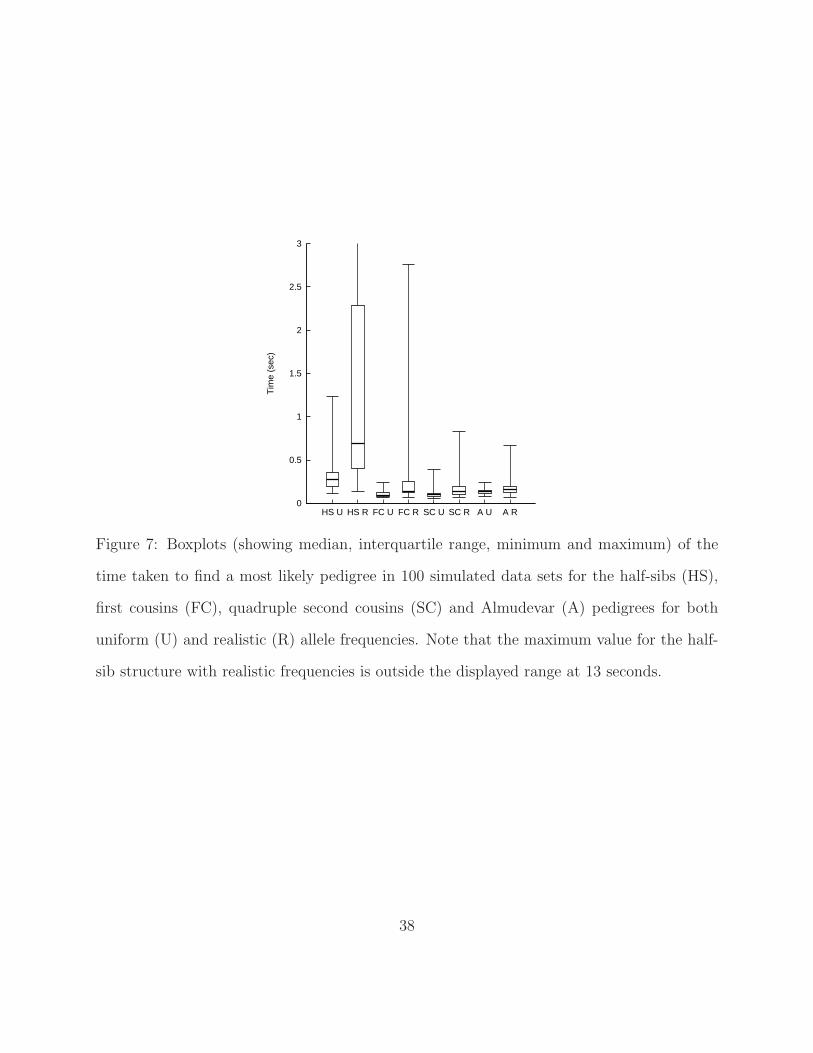
these time distributions are shown in Figure 7.
[Figure 7 about here.]
The results show that a maximum likelihood solution was usually found very quickly with
the most extreme case taking about 1.3 seconds when uniform allele frequencies were used.
Note that, despite the fact that we have differing numbers of alleles at each locus, uniform
allele frequencies clearly make the reconstruction problem easier. Typical solving times were
always slower and much more variable with realistic frequencies on all four pedigrees. The
cyclic half-sib pedigree proved to be the most challenging structure for the algorithm, in
terms of solving time, with an extreme value of 13 seconds when marker data were simulated
from the realistic allele frequency distribution. The pedigree in Figure 5 and the quadruple
second cousin structure in Figure 6 were the easiest with the maximum likelihood solution
always found within a second.
[Figure 8 about here.]
As noted earlier, a single maximum likelihood reconstruction is not always particularly
useful so we now consider the 100 most likely pedigrees. Figure 8 shows the median times
taken between finding the (k − 1)th and the kth most likely pedigree for all four structures
and both sets of allele frequencies. The overall pattern is that the maximum likelihood
pedigree (i.e. the first of the 100 pedigrees) is found most quickly and solving times tend to
increase for each additional pedigree sought. Solving times are slower for the realistic allele
frequencies than for the uniform ones and are more variable for the pedigree of Figure 5 and
the half-sib structure than for the other two regular structures. The cyclic half-sib structure
is markedly more difficult to solve with increasing number of pedigrees found.
17

3.2 Accuracy of the reconstructed pedigree
Assessing the value of a particular reconstruction depends to some extent on what it is
required for. Often, as would be the case for demographic research or ecological studies that
aim to design a breeding program, it is more important that the estimated pedigree reflect
general characteristics of the true structure, such as correct distribution of sibship sizes,
correct levels of multiple marriages and overall connectedness of the sampled individuals,
rather than be accurate in every detail [Thompson, 1986]. There are many such criteria
that may be important and these will differ between applications. We therefore focus on
a general property for assessing the quality of the reconstruction, while noting that this
may not be the best criterion in any given situation. Comparing the number of erroneous
parent-offspring links (or incorrect graph edges) is an obvious statistic and is commonly
used in BN learning. However, it should be noted that although a maximum likelihood
pedigree is often a reasonable approximation to the true pedigree, whether it is or not has
no bearing on the speed and exactness of a method for finding it. Our goal is to find high
likelihood pedigrees quickly so it could be argued that error rates such as the mean number
of parental misspecifications are not the appropriate statistics for evaluating the success of
a likelihood approach. Nevertheless, they can be useful — particularly when several high
likelihood pedigrees are considered.
[Table 1 about here.]
Table 1 shows the sample distribution of incorrect parent-offspring links among the 100
maximum likelihood pedigrees for each of the 8 combinations of allele frequency and pedigree
structure. Specifically, we define a link to be incorrect if it is either missing or incorrectly
attributed. The maximum number of such errors is twice the number of individuals. As can
be seen, the maximum likelihood pedigrees had considerably fewer incorrect links when the
data were simulated under uniform allele frequencies than for the case with realistic frequen-
cies. The reconstruction of the pedigree in Figure 5, which is a more realistic representation
of a human pedigree structure, was most often correct or close to correct. Quadruple second
cousins were the most accurate of the regular mating systems we considered by this mea-
sure. The cyclic half-sib maximum likelihood reconstruction was the least likely to be the
18

true structure with as many as 8 misspecified parent-offspring links even for the uniform
allele frequency scenario. Accuracy of reconstruction appears to deteriorate with increasing
levels of inter-relatedness. From the form of the optimisation function in (4), the algorithm
is choosing the ‘best’ parentage for each individual. The more inter-related the individuals,
the more genetically consistent choices there are for parent pairs or triplets. This is consis-
tent with the observation in Thompson [1986] that high exclusion probabilities for incorrect
parents are required in order to obtain an accurate reconstruction.
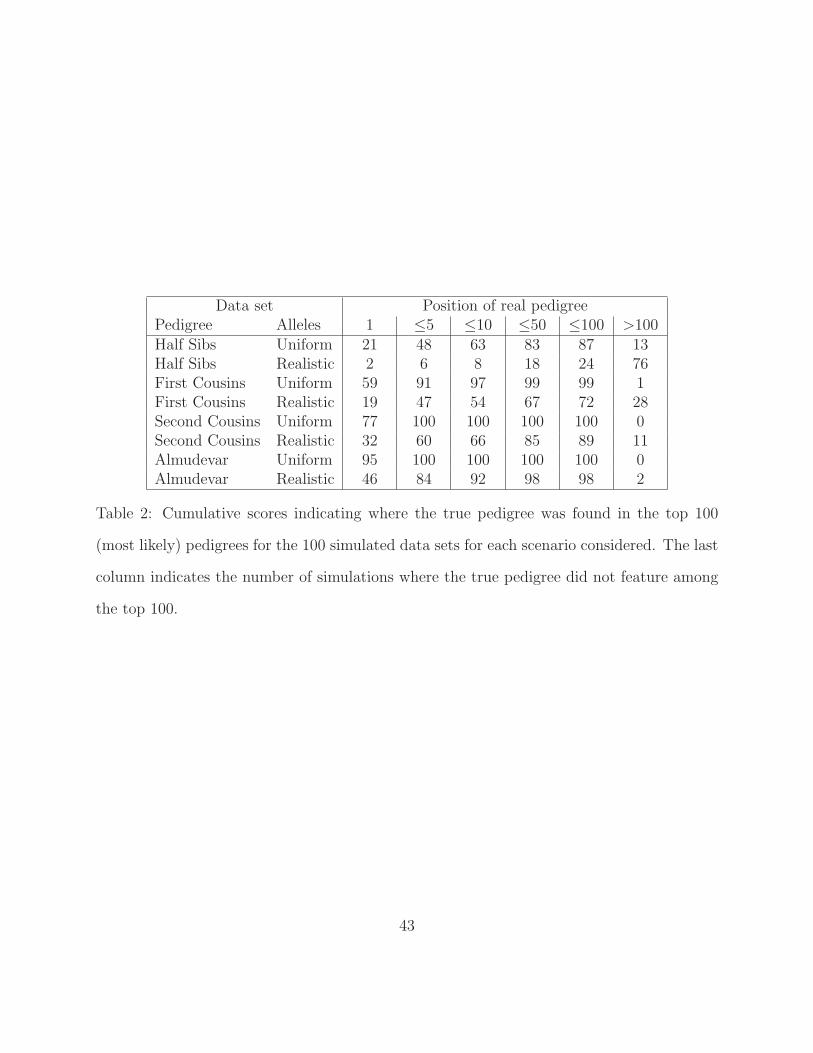
[Table 2 about here.]
It is reasonable to expect that while the true pedigree may not always have maximum
likelihood, the inferred relationships should include many of the true parent-child links and
so the true pedigree should often feature among the most likely ones. Table 2 shows where
the true pedigree was found among the 100 most likely pedigrees for all 100 simulated data
sets for each scenario considered. For the uniform allele frequency distribution, the maximum
likelihood pedigree was actually the true pedigree 95% of the time for the pedigree of Figure 5.
The story was quite different for the realistic frequencies where the true pedigree did not
appear in the top 100 reconstructions for 2 of the runs. There was a similar trend for the
regular mating structures with the cyclic half-sib pedigree being the hardest to get right: the
true structure was the first one found in 2 runs but 76 had failed to find it among the first
100 for the realistic allele frequency distribution. These results highlight the importance
of being able to generate multiple high probability pedigrees rather than rely on a single
reconstruction.
3.3 Distribution of likelihoods
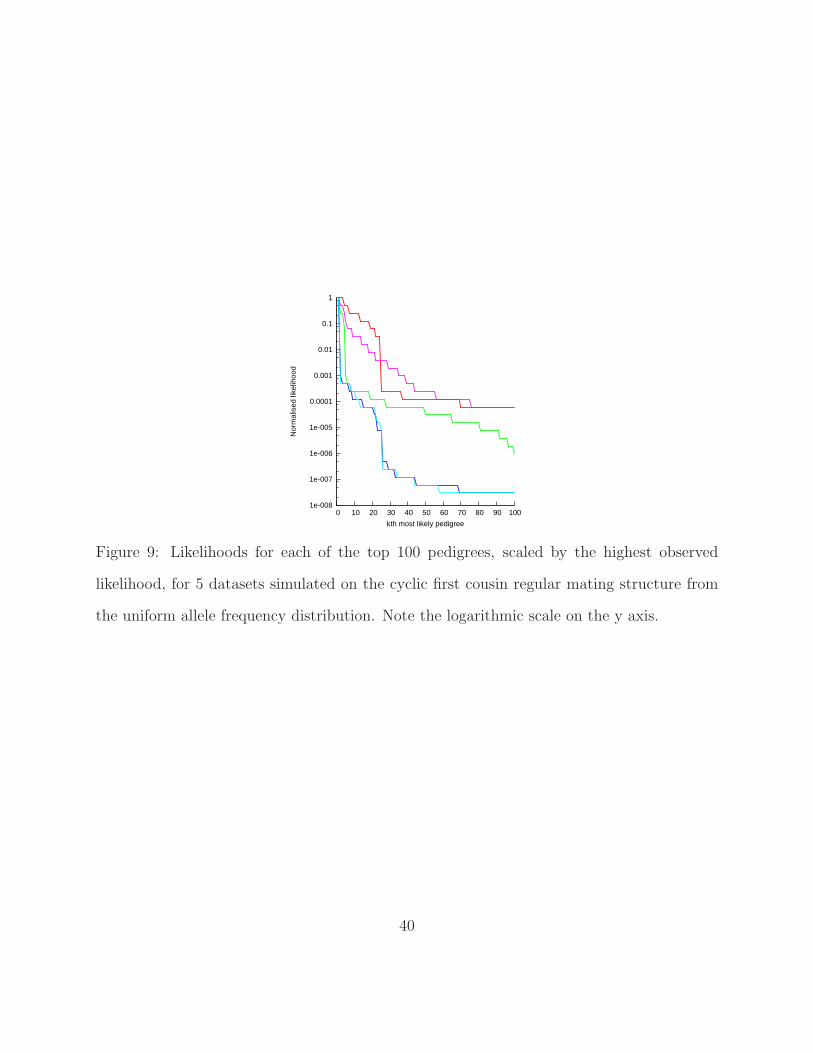
[Figure 9 about here.]
In order to get an idea of what the search space of pedigree likelihoods looks like, we
investigated the relative distribution of the likelihoods of the top 100 pedigrees. Because
likelihoods vary considerably with different combinations of simulated data and pedigree
structure, we divided the likelihood of the kth most likely pedigree in each of the 100 runs
19

by the value of the likelihood for the first pedigree found in that run. Examples of how
variable these are for some particular data sets are given in Figure 9 for the first cousin
64-member pedigree. As can be seen, the likelihoods tend to drop sharply soon after the top
pedigree is found, plateau out and then drop again several times to a tail of low likelihood
pedigrees. Median values across the 100 simulated datasets with interquartile range error
bars at selected intervals are displayed in Figure 10 for all four pedigree structures using the
uniform (a) and realistic (b) allele frequencies. The y-axis has been plotted on a log scale to
stretch out the lower values because of the sharp drop in likelihood at the outset.
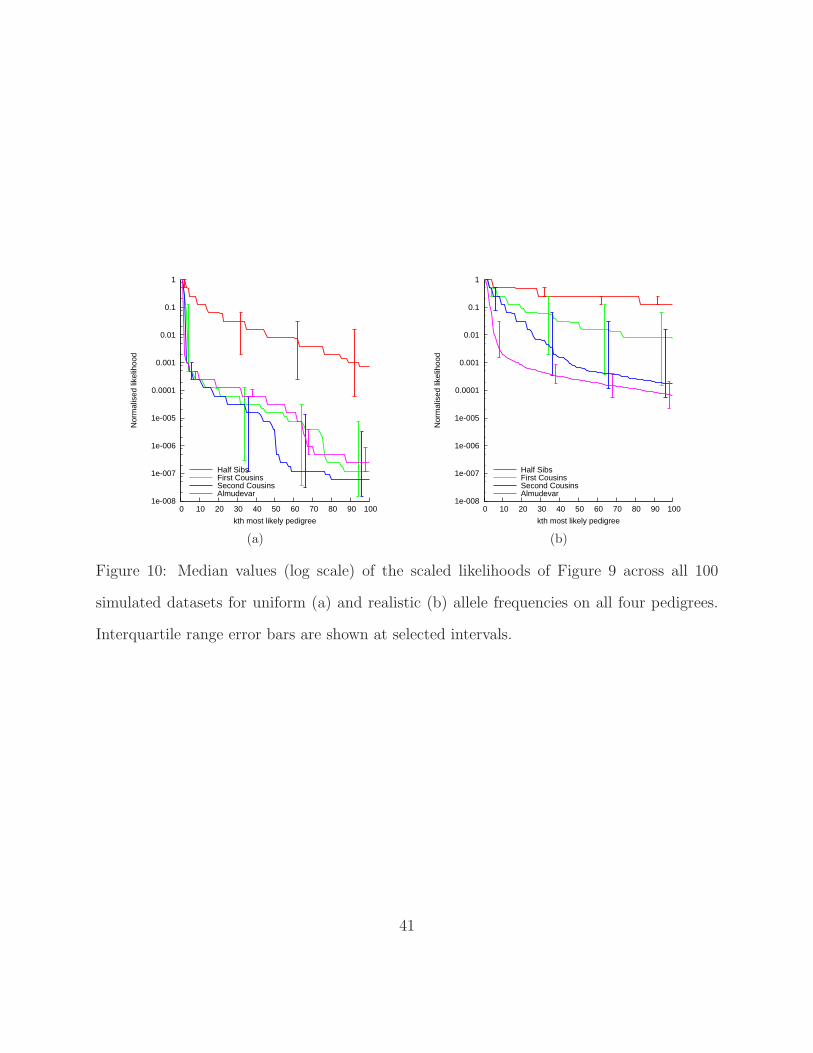
[Figure 10 about here.]
Likelihoods drop more quickly for the uniform allele frequencies, again reflecting the fact
that maximum likelihood pedigree reconstruction is easier with uniform frequencies than
with more realistic ones. For our set of realistic allele frequencies, the likelihoods of the
reconstructed cyclic half-sib pedigrees are not surprisingly the slowest to drop and are by far
the least variable indicating the difficulty of the optimisation problem.
3.4 Comparison with other approaches
There are three other notable approaches to maximum likelihood pedigree reconstruction [Al-
mudevar, 2003; Cowell, 2009; Riester et al., 2009] against which we will now compare our
approach.
The method described by Almudevar [2003] relies on simulated annealing to search for a
maximum likelihood pedigree. However, any method such as this that uses a heuristic search
cannot be guaranteed to find the actual maximum likelihood pedigree. The results shown in
Figure 10 illustrate the particular problem with this in the context of pedigree reconstruction;
the most likely pedigree can be orders of magnitude more likely than the next few most likely.
Methods that do not necessarily find the most likely pedigree may instead find pedigrees
significantly less likely than the best. Calculation times reported in Almudevar [2003] are
of the order of minutes, compared to sub-second times for reconstructing the same pedigree
using our method. It should also be noted that the evaluation presented in Almudevar [2003]
20

is conducted under the assumption of uniform allele frequency. Our results show that this
makes the pedigree reconstruction problem easier than it would be using realistic frequencies.
As for our method, the dynamic programming approach of Cowell [2009] guarantees
a maximum likelihood pedigree. However, that approach is restricted to small pedigrees
due to storage requirements (i.e. up to about 30 individuals). For direct comparison, the
dynamic programming approach to maximum likelihood pedigree reconstruction of Cowell
[2009] and the ILP algorithm of Section 2.2 were both run on 100 simulated data sets for
both sets of allele frequencies on the two pedigrees in Figure 4. Both algorithms were quick
(less that 1 second) to find a single maximum likelihood pedigree. Solving times for the
dynamic programming method were all consistently between 0.45 to 0.5 seconds while the
ILP approach was noticeably faster with 90% of the runs succeeding in less than 0.25 seconds.
The dynamic programming approach occasionally found a sex-inconsistent pedigree but this
never happened with the ILP algorithm as the search was constrained to rule out such
structures. Solving time was a little longer for the more inbred pedigree of Figure 4(b) and
this became more apparent when many high probability pedigrees were sought with the ILP
algorithm. In terms of the number of correctly inferred parent-offspring relationships, the
maximum likelihood pedigree was always very close to the true pedigree structure with ≤ 3
different links 95% of the time. The true pedigree almost always featured among the 10 most
likely pedigrees when these could be found and, not unexpectedly, the likelihood distribution
dropped off dramatically after the top 10 pedigrees, or so. For cyclic half-sib structures of
100 individuals, we found that the algorithm took considerably longer to find a maximum
likelihood pedigree. For realistic allele frequencies, 75% of the runs found a solution in less
than 100 seconds but some took as long as 4 hours. This is not too surprising, given that this
was by far the hardest structure we considered, and we would expect much better results for
less complex structures.
Riester et al. [2009] present a software tool for pedigree reconstruction. At the core of the
tool, a solution is found using either the method of Almudevar [2003] or that of Cowell [2009].
Hence, the tool suffers from the disadvantages discussed above: either the pedigree cannot
be guaranteed to be the most likely or the number of individuals that can be considered is
seriously limited. However, it does incorporate several features that we have not addressed.
21

The likelihoods of parentage triples are modified in a principled way to admit the possibility
of genotyping errors and to compensate for a finite number of mating individuals in the
population. These likelihoods are also modified during the search process based on the
current beliefs about population allele frequencies, rather than assuming such frequencies
are known accurately a priori. The tool also allows for potential missing data for some
individuals by repeatedly estimating values for such data during the search process using
Gibbs sampling.
To the best of our knowledge, our ILP algorithm is the only approach to maximum
likelihood pedigree reconstruction that can deliver the k most likely pedigrees and can thus
address the issue of model uncertainty.
4 Discussion
4.1 Extensions to more complex problems
The method we have presented here performs well under the assumptions of a complete sam-
ple, independently segregating genetic markers and Hardy-Weinberg proportions for founder
genotypes. In particular, when complete marker data are observed for all individuals, an
offspring genotype is independent of all non-descendant genotypes conditional on the geno-
types of its parents. This is generally known as the (directed local) Markov property for
Bayesian networks and results in the decomposition of the (log) likelihood function in Equa-
tions (1) and (4) which is the core of our ILP formulation. As noted in Lauritzen and Sheehan
[2003], Mendelian inheritance of offspring genotypes from parental types is also implicit in
this decomposition. Contrary to the statement in Cowell [2009], the desired decomposition
breaks down when markers are linked and siblings are no longer conditionally independent
given their parents. This can be easily seen for the case of two linked marker loci with a
recombination fraction r and alleles A1, A2, A3 and B1, B2, B3, respectively. Consider two
offspring from a doubly heterozygous (A1A2, B1B2) and doubly homozygous (A3A3, B3B3)
mating where parental phase is unknown. We are interested in the probability of the second
offspring, I2, having genotype (A1A3, B1B3). If the first offspring, I1 is also (A1A3, B1B3),
22

P (I2 = (A1A3, B1B3)|I1 = (A1A3, B1B3)) =1
4(1 − r)2 +
1
4r2
i.e. both offspring inherited both alleles from the same chromosome (no recombination) or
they both inherited them from different chromosomes. If I1 is (A1A3, B2B3), we have that
P (I2 = (A1A3, B1B3)|I1 = (A1A3, B2B3)) =1
2r(1 − r)
i.e. there was a recombination for one offspring and not for the other. In order for the Markov
property to hold, both these probabilities should be equal. This is clearly true only when
r = 1
2i.e. when the loci are unlinked.
Complete data will generally not be available when constructing pedigrees for linkage
analysis or for identifying distant relatives for homozygosity mapping from human population
studies. The same is true for wildlife management and forensic science applications. In these
cases, unobserved individuals that form ‘missing links’ between the individuals of interest
have to be accounted for and the likelihood in Equation (1) has to be modified by summing
over all possible combinations of genotypes on the unobserved individuals [Thompson, 2000].
The more unobserved individuals, the larger the sum and the more difficult the likelihood
computation. In order to extend our approach to include individuals with wholly or partially
missing marker data, the missing information could be represented using additional variables
so that the log-likelihood remains linear. The number of additional variables required should
be modest if there is no linkage and the approach should be feasible given the observed
efficiency of the algorithm. An alternative method is suggested by Riester et al. [2009]
whereby missing data are estimated during the reconstruction using Gibbs sampling.
Existing likelihood approaches to pedigree construction are all based on independently
segregating genetic markers. Likelihood calculations are hence simplified, as was the case
in this paper, since only one marker has to be considered at any one time. There are some
relationships, however, that have the same likelihood and are hence indistinguishable for
any number of unlinked markers, but are distinguishable with linked markers [Thompson,
1975; Egeland and Sheehan, 2008]. Given the ever increasing availability of dense sets of
SNPs from genetic association studies, reconstruction algorithms should aim to make use of
23

linked markers. Correlated markers naturally lead to a reduction in discriminatory power
when compared with (the same number of) unlinked markers so it is to be expected that
many more markers will be required. Preliminary work for distinguishing between particular
alternatives for pairwise relationships has shown that huge numbers of markers, as are now
readily available for genetic association studies, can be used to distinguish between quite
distant relationships [Skare et al., 2009]. In order to adapt our method for handling linked
markers, we could adopt a similar strategy to that described above for missing data by repre-
senting the unknown phase information with additional variables. This restores the desired
decomposition of the likelihood function and the optimisation procedure now returns a most
probable combination of pedigree structure and haplotypes. Note that the observed (un-
ordered) marker data put tight constraints on the possible haplotypes and these constraints
are easy to express in the ILP framework. It is no surprise that ILP has been successfully
used for inferring haplotypes in the case where the true pedigree is known [Brown and Har-
rower, 2006]. Likelihood calculations will be much more intensive for linked markers and we
may realistically need to consider approximations.
Finally, in addition to genetic marker data, prior population demographic information on
breeding patterns, average numbers of offspring and cultural prejudices, for example, as well
as specific knowledge about particular relationships may often be available in practice. It is
important to include such information when reconstructing pedigrees since structures that
fit the marker data the best may be unreasonable in other respects, such as by representing
improbably high levels of interrelatedness, for the population in question [Egeland et al.,
2000]. More importantly, a formal Bayesian approach permits a principled way of quantifying
model uncertainty using Bayesian model averaging [Kass and Raftery, 1995; Hoeting et al.,
1999].
Many existing approaches [Hadfield et al., 2006; Neff et al., 2001; Riester et al., 2009;
Thompson, 1976a; Thompson and Meagher, 1987] do incorporate some prior information,
but often in an informal way and at an interim stage of the reconstruction, for example
when the likelihood approach is favouring what is clearly an undesirable structure [Sheehan
and Egeland, 2007]. If sensible priors could be assigned to pedigrees [Egeland et al., 2000;
Sheehan and Egeland, 2007], the likelihood of any candidate pedigree L(G), based on the
24

observed marker data, can be combined with a prior probability π(G) to derive the posterior
probability p(G | data) ∝ π(G)L(G) using Bayes’ theorem. The pedigree reconstruction prob-
lem then reduces to a search for a pedigree with maximal posterior probability, often called
MAP estimation. This is equivalent to likelihood estimation in the case of a uniform prior
distribution over all possible pedigrees.
Extending our approach to deal with priors should be quite simple. If the prior encodes
‘hard’ information (e.g. an individual is female, or a particular individual is the parent of
another), this can be added to the ILP formulation as additional constraints. If the prior
encodes ‘soft’ information (e.g. a probability distribution over sibship sizes), it can be shown
that this can be accurately incorporated into the problem through the addition of extra log
prior terms to the objective function, provided that the prior can be encoded as a linear
function.
4.2 Conclusions
Family relationships, and hence pedigrees, are central to all gene mapping studies [Day-
Williams et al., 2011]. Establishing pedigree information through interviews and historical
records is time consuming and so it is appealing to be able to reconstruct pedigrees from
genetic marker data using available large population studies, especially since such studies
have a reasonable chance of representing extended pedigree information which is otherwise
difficult to obtain [Browning and Browning, 2010]. The ILP approach to pedigree reconstruc-
tion that we have presented in this paper has been shown to out-perform other approaches
in the standard setting where all individuals are observed, segregation is Mendelian, markers
are independent and founder genotypes are in Hardy-Weinberg equilibrium. It also extends
to quite large pedigrees, guarantees a maximum likelihood solution and can deal with un-
certainty in the reconstruction by providing the k most likely pedigrees. Most importantly,
we have a framework that seems more amenable to adaptation to harder, and hence more
interesting, reconstruction problems. Our algorithm usually finds a maximum likelihood
pedigree efficiently but is sensitive to the allele frequency distribution and the complexity of
the pedigree structure. We would warn that the use of uniform allele frequencies, as com-
monly arises in simulation studies, can be misleading as they make the problem deceptively
25

easier.
It can be argued that likelihood approaches are not necessarily the most useful for pedi-
gree estimation, especially when general features or specific sections of the pedigree are of
more interest that the overall structure. Comparison of estimates is much less straightfor-
ward in these situations, although realising an appropriate one is generally simpler. For small
populations, multidimensional scaling of pairwise kinship coefficients has been suggested as
one way of representing general population structure [Thompson, 1986]. Where the overall
structure is of interest, however, we have verified that maximising the likelihood is a good
approach. Unless the structure is extremly complex, the maximum likelihood pedigree is
typically quite similar to the true structure with regard to the number of parent-offspring
links that are correctly assigned and usually features quite early in the list of most likely
pedigrees. We agree with the comment in Thompson [1986] that a single maximum likeli-
hood estimate may not be particularly helpful but useful inferences should be possible from
several high probability pedigrees. For example, reasonable probability estimates of particu-
lar pedigree features can be acquired from the proportion of high probability structures that
contain them.
Pedigree reconstruction has also been proposed using a two-step approach that involves
estimation of pairwise kinship coefficients followed by clustering of individuals into pedigrees
using graph learning methods [Cowell and Mostad, 2003; Day-Williams et al., 2011]. This ap-
proach is also sensitive to allele frequencies and the choice of cut-off value used to determine
the graph edges. For quantitative trait locus (QTL) linkage analysis, pedigree information is
only incorporated via local and global pairwise kinship coefficients so estimating these quan-
tities from marker data using dynamic programming and a method of moments approach,
respectively, can obviate the need for standard pedigrees in such analyses [Day-Williams
et al., 2011]. In fact, it has also been noted by Choi et al. [2009] that estimated kinship
coefficients can often be more accurate than pedigree-based quantities because they exploit
the observed sharing of genes between two individuals rather than the expected sharing im-
plied by the pedigree structure. The ILP approach presented here also uses the observed
sharing but has the advantage of considering all related individuals jointly in the likelihood
calculation. We would thus expect it to have an advantage over approaches based on pair-
26

wise estimates and, indeed, in the simple standard scenario we considered here, we do not
think that one could do much better. However, we need to extend the method to deal with
unobserved individuals and linked markers before a sensible comparison can be made. Also,
when extended pedigrees are of interest, genotyping error and/or mutation rates should also
be included. These are much harder reconstruction problems but ILP would seem to provide
the right framework in which to address them.
Acknowledgements
The authors would like to thank Robert Cowell for giving access to his code.
The authors acknowledge support from the Medical Research Council (Project Grant G1002312),
the Leverhulme Trust (Research Fellowship RF/9/RFG/2009/0062) and the BioSHaRE-EU
project (HEALTH-F4-2010-261433) funded by the European Commission under the Seventh
Framework Program (FP7/2007-2013).
References
Almudevar, A. (2003). A simulated annealing algorithm for maximum likelihood pedigree
reconstruction. Theoretical Population Biology, 63:63–75.
Almudevar, A. (2007). A graphical approach to relatedness inference. Theoretical Population
Biology, 71:213–229.
Blouin, M. S. (2003). DNA-based methods for pedigree reconstruction and kinship analysis
in natural populations. Trends in Ecology and Evolution, 18:503–511.
Brown, D. and Harrower, I. (2006). Integer programming approaches to haplotype inference
by pure parsimony. IEEE/ACM Transactions in Computational Biology and Bioinformat-
ics, 3(2):141–154.
Browning, S. R. and Browning, B. L. (2010). High-resolution detection of identity by descent
in unrelated individuals. American Journal of Human Genetics, 86:526–539.
27

Butler, J. M., Schoske, R., Vallone, P. M., Redman, J. W., and Kline, M. C. (2003). Allele
frequencies for 15 autosomal STR loci on U.S. Caucasian, African American, and Hispanic
populations. Journal of Forensic Sciences, 48(4).
Cannings, C. and Thompson, E. A. (1981). Genealogical and Genetic Structure. Cambridge
University Press.
Cannings, C., Thompson, E. A., and Skolnick, M. H. (1978). Probability functions on
complex pedigrees. Advances in Applied Probability, 10:26–61.
Choi, Y., Wijsman, E. M., and Weir, B. S. (2009). Case-control testing in the presence of
unknown relaionships. Genetic Epidemiology, 33:668–678.
Cowell, R. G. (2009). Efficient maximum likelihood pedigree reconstruction. Theoretical
Population Biology, 76(4):285–291.
Cowell, R. G. and Mostad, P. (2003). A clustering algorithm using DNA marker information
for sub-pedigee reconstruction. Journal of Forensic Sciences, 48:1239–1248.
Cussens, J. (2010). Maximum likelihood pedigree reconstruction using integer programming.
In Workshop on Constraint Based Methods for Bioinformatics (WCB-10), Edinburgh.
Day-Williams, A. G., Blangero, J., Dyer, T. D., Lange, K., and Sobel, E. M. (2011). Linkage
analysis without defined pedigrees. Genetic Epidemiology, 35(5):360–370.
Egeland, T., Mostad, P. F., Mevag, B., and Stenersen, M. (2000). Beyond traditional
paternity and identification cases: selecting the most probable pedigree. Forensic Science
International, 110:47–59.
Egeland, T. and Sheehan, N. (2008). On identification problems requiring linked markers.
Forensic Science International: Genetics, 2:219–225.
Genin, E. and Clerget-Darpoux, F. (1996). Consanguinity and the sib-pair method: an
approach using identity by descent between and within individuals. American Journal of
Human Genetics, 59:1149–1162.
28

Hadfield, J. D., Richardson, D. S., and Burke, T. (2006). Towards unbiased parentage
assignment: combining, genetic, behavioural and spatial data in a Bayesian framework.
Molecular Ecology, 15:3715–3730.
Hoeting, J. A., Madigan, D., Raftery, A. E., and Volinsky, C. T. (1999). Bayesian model
averaging: a tutorial. Statistical Science, 14(4):382–401.
Jones, A. G. and Ardren, W. R. (2003). Methods of parentage analysis in natural populations.
Molecular Ecology, 12:2511–2522.
Karp, R. (1972). Reducibility among combinatorial problems. In Miller, R. and Thatcher,
J., editors, Complexity of Computer Computations, pages 85–103. Plenum Press.
Kass, R. E. and Raftery, A. E. (1995). Bayes factors. Journal of the American Statistical
Association, 90(430):773–795.
Lange, K. and Elston, R. C. (1975). Extensions to pedigree analysis. I. Likelihood calculations
for simple and complex pedigrees. Human Heredity, 25:95–105.
Lauritzen, S. L. (1996). Graphical Models. Clarendon Press, Oxford, United Kingdom.
Lauritzen, S. L. and Sheehan, N. (2003). Graphical models for genetic analyses. Statistical
Science, 18:489–514.
Leutenegger, A. L., Prum, B., Genin, E., Verny, C., Lemainque, A., Clerget-Darpoux, F.,
and Thompson, E. A. (2003). Estimation of the inbreeding coefficient through use of
genomic data. American Journal of Human Genetics, 73:516–523.
McPeek, M. S. and Sun, L. (2000). Statistical test for detection of misspecified relationships
by use of genome-screen data. American Journal of Human Genetics, 66:1076–1094.
Meagher, T. R. and Thompson, E. A. (1987). Analysis of parentage for naturally established
seedlings of Chamaelirium luteum (liliaceae). Ecology, 68:803–812.
Neff, B. D., Repka, J., and Gross, M. R. (2001). A Bayesian framework for parentage
analysis: the value of genetic and other biological data. Theoretical Population Biology,
59:315–331.
29

Newman, D. L., Abney, M., McPeek, M. S., Ober, C., and Cox, N. J. (2001). The importance
of genealogy in determining genetic associations with complex traits. American Journal
of Human Genetics, 69:1146–1148.
Olaisen, B., Stenersen, M., and Mevag, B. (1997). Identification by DNA analysis of the
victims of the August 1996 Spitsbergen civil aircraft disaster. Nature Genetics, 15:402–405.
Powell, J. E., Visscher, P. M., and Goddard, M. E. (2010). Reconciling the analysis of IBD
and IBS in complex trait studies. Nature Reviews:Genetics, 11:800–805.
Riester, M., Stadler, P. F., and Klemm, K. (2009). FRANz: reconstruction of wild multi-
generation pedigrees. Bioinformatics, 25:2134–2139.
Sanchez, M., Givry, S. d., and Schiex, T. (2008). Mendelian error detection in complex
pedigrees using weighted constraint satisfaction techniques. Constraints, 13:130–154.
Sheehan, N. A. and Egeland, T. (2007). Structured incorporation of prior information in
relationship identification problems. Annals of Human Genetics, 71:501–518.
Sheehan, N. A. and Egeland, T. (2008). Adjusting for founder relatedness in a linkage
analysis using prior information. Human Heredity, 65(4):221–231.
Sieberts, S. K., Wijsman, E. M., and Thompson, E. A. (2002). Relationship inference from
trios of individuals in the presence of typing error. American Journal of Human Genetics,
70:170–180.
Skare, O., Sheehan, N., and Egeland, T. (2009). Identification of distant family relationships.
Bioinformatics, 25:2376–2382.
Stankovich, J., Bahlo, M., Rubio, J. P., Wilkinson, C. R., Thomson, R., Banks, A., Ring, M.,
Foote, S. J., and Speed, T. P. (2005). Identifying nineteenth century links from genotypes.
Human Genetics, 117:188–199.
Thomas, A. (1985). Data structures, methods of approximation and optimal computation for
pedigree analysis. PhD thesis, Cambridge University.
30

Thompson, E. A. (1975). The estimation of pairwise relationships. Annals of Human Ge-
netics, 39:173–188.
Thompson, E. A. (1976a). Inference of genealogical structure. Social Science Information,
15:477–526.
Thompson, E. A. (1976b). A paradox of genealogical inference. Advances in Applied Proba-
bility, 8:648–650.
Thompson, E. A. (1986). Pedigree Analysis in Human Genetics. The Johns Hopkins Uni-
versity Press, Baltimore.
Thompson, E. A. (2000). Statistical Inference from Genetic Data on Pedigrees, volume 6 of
NSF-CBMS Regional Conference Series in Probability and Statistics. Institute of Mathe-
matical Statistics and the American Statistical Association, Beachwood, Ohio, USA.
Thompson, E. A. (2008a). Analysis of data on related individuals through inference of iden-
tity by descent. Technical Report 539, Department of Statistics, University of Washington.
Thompson, E. A. (2008b). The IBD process along four chromosomes. Theoretical Population
Biology, 73:369–373.
Thompson, E. A. and Meagher, T. R. (1987). Parent and sib likelihoods in genealogy
reconstruction. Biometrics, 43:585–600.
Thornton, T. and McPeek, M. S. (2010). ROADTRIPS: Case-control association testing with
partially or completely unknown population and pedigree structure. American Journal of
Human Genetics, 86:172–184.
Wolsey, L. A. (1998). Integer Programming. Wiley-Interscience Series in Discrete Mathe-
matics and Optimization. Wiley.
Wright, S. (1921). Systems of mating. Genetics, 6:111–178.
31

(a) (b) (c)
Figure 1: A simple pedigree in standard form (a); with corresponding representations as a
directed acyclic graph (b); and marriage node graph (c).
32

(a) (b) (c)
v1v1v1
v2
v2 v2
v3
v3
v3
Figure 2: Three of the twenty-five possible pedigrees for 3 individuals, where individuals
other than v1, v2 and v3 are not represented
33

1 2 3
4 5 6
Figure 3: A simple DAG where no node has more than two parent nodes but which is not
a pedigree as sex cannot be consistently assigned. Since 2 and 3 have a child together, they
must be of opposite sex. However as they each have a child with 1, they must have the same
sex. It is clearly impossible to assign sexes that meet these contradictory constraints.
34

1 2
2 4 5 6
16
7 8 9 10 1211
201918171513 14
(a)
1 2 3 4 5
6 7 8 9 19
11 12 13 14 15
16 17 18 19 20
(b)
Figure 4: The two pedigrees of 20 individuals corresponding to Figures 2 and 3 in Cowell
[2009] relabelled and drawn as marriage node graphs. Note that the pedigree in (a) has one
first cousin marriage and two sets of half-siblings (offspring of 7 and 12). The pedigree in
(b) is more inbred.
35

1 6 8 9
12 13 14 15 16 17 18 21 22 23 24
25 26 30
35 36 37 38 40 42 43 44 47
48 50 51 53 54 55 56 57 59
2 73 4 5
10 11 19 20
27 28 29 31 32 33 34
39 41 45 46
49 52 58
Figure 5: A 59-member pedigree featuring some first cousin marriages and half-siblings taken
from Almudevar [2003].
36

1 2 3 4 5 6 7 8
71 72 73 74 75 76 77 78
61 62 63 64 65 66 67 68
51 52 53 54 55 56 57 58
4847464544434241
31 32 33 34 35 36 37 38
2827262524232221
11 12 13 14 15 16 17 18
Figure 6: A 64-member pedigree constructed as a quadruple second cousin regular mating
structure as described in Wright [1921].
37
0
0.5
1
1.5
2
2.5
3
HS U HS R FC U FC R SC U SC R A U A R
Tim
e (s
ec)
Figure 7: Boxplots (showing median, interquartile range, minimum and maximum) of the
time taken to find a most likely pedigree in 100 simulated data sets for the half-sibs (HS),
first cousins (FC), quadruple second cousins (SC) and Almudevar (A) pedigrees for both
uniform (U) and realistic (R) allele frequencies. Note that the maximum value for the half-
sib structure with realistic frequencies is outside the displayed range at 13 seconds.
38

0
0.5
1
1.5
2
2.5
3
3.5
0 20 40 60 80 100
Tim
e (s
ec)
kth most likely pedigree
Half SibsFirst Cousins
Second CousinsAlmudevar
(a)
0
0.5
1
1.5
2
2.5
3
3.5
0 20 40 60 80 100
Tim
e (s
ec)
kth most likely pedigree
Half SibsFirst Cousins
Second CousinsAlmudevar
(b)
Figure 8: Time taken to find the kth most likely pedigree after finding the (k − 1)th. The
lines show median solving times (out of 100 simulated data sets) for each of the four larger
pedigree structures for both uniform, (a), and realistic, (b), allele frequencies. Interquartile
range error bars are shown at 4 selected intervals to give an indication of the variability of
these solving times.
39
1e-008
1e-007
1e-006
1e-005
0.0001
0.001
0.01
0.1
1
0 10 20 30 40 50 60 70 80 90 100
Nor
mal
ised
like
lihoo
d
kth most likely pedigree
Figure 9: Likelihoods for each of the top 100 pedigrees, scaled by the highest observed
likelihood, for 5 datasets simulated on the cyclic first cousin regular mating structure from
the uniform allele frequency distribution. Note the logarithmic scale on the y axis.
40
1e-008
1e-007
1e-006
1e-005
0.0001
0.001
0.01
0.1
1
0 10 20 30 40 50 60 70 80 90 100
Nor
mal
ised
like
lihoo
d
kth most likely pedigree
Half SibsFirst CousinsSecond CousinsAlmudevar
(a)
1e-008
1e-007
1e-006
1e-005
0.0001
0.001
0.01
0.1
1
0 10 20 30 40 50 60 70 80 90 100
Nor
mal
ised
like
lihoo
d
kth most likely pedigree
Half SibsFirst CousinsSecond CousinsAlmudevar
(b)
Figure 10: Median values (log scale) of the scaled likelihoods of Figure 9 across all 100
simulated datasets for uniform (a) and realistic (b) allele frequencies on all four pedigrees.
Interquartile range error bars are shown at selected intervals.
41

Data set Number of edges incorrectPedigree Alleles 0 1 2 3 4 5 6 7 8 >8Half Sibs Uniform 21 27 19 13 10 4 2 3 1 0Half Sibs Realistic 2 3 13 8 17 12 11 10 4 20First Cousins Uniform 59 32 4 3 2 0 0 0 0 0First Cousins Realistic 19 26 18 12 10 4 8 0 2 1Second Cousins Uniform 77 17 5 0 1 0 0 0 0 0Second Cousins Realistic 32 22 26 9 3 4 1 0 1 2Almudevar Uniform 95 4 1 0 0 0 0 0 0 0Almudevar Realistic 46 31 18 3 1 0 1 0 0 0
Table 1: Number of edges that differ between the true pedigree and the most likely pedi-
gree found for each of the 100 simulated data sets in each scenario. Each incorrect edge
corresponds to a mistake in the assignment of a single parent-child relationship.
42
Data set Position of real pedigreePedigree Alleles 1 ≤5 ≤10 ≤50 ≤100 >100Half Sibs Uniform 21 48 63 83 87 13Half Sibs Realistic 2 6 8 18 24 76First Cousins Uniform 59 91 97 99 99 1First Cousins Realistic 19 47 54 67 72 28Second Cousins Uniform 77 100 100 100 100 0Second Cousins Realistic 32 60 66 85 89 11Almudevar Uniform 95 100 100 100 100 0Almudevar Realistic 46 84 92 98 98 2
Table 2: Cumulative scores indicating where the true pedigree was found in the top 100
(most likely) pedigrees for the 100 simulated data sets for each scenario considered. The last
column indicates the number of simulations where the true pedigree did not feature among
the top 100.
43





























