Embed Size (px)
Citation preview

The roles of hyperoxia and mechanical deformation
in alveolar epithelial injury and repair
Stuart Robert McKechnie
Doctor of Philosophy
The University of Edinburgh
2007

ii
Declaration of Authorship
The work contained in this thesis is my own except where stated. This work has not been
submitted for any other degree or professional qualification.
Stuart Robert McKechnie
Edinburgh 2007

iii
Abstract
The alveolar epithelium is a key functional component of the air-blood barrier in the
lung. Comprised of two morphologically distinct cell types, alveolar epithelial type I
(ATI) and type II (ATII) cells, effective repair of the alveolar epithelial barrier following
injury appears to be an important determinant of clinical outcome. The prevailing view
suggests this repair is achieved by the proliferation of ATII cells and the
transdifferentiation of ATII cells into ATI cells.
Supplemental oxygen and mechanical ventilation are key therapeutic interventions in the
supportive treatment of respiratory failure following lung injury, but the effects of
hyperoxia and mechanical deformation in the injured lung, and on alveolar epithelial
repair in particular, are largely unknown. The clinical impression however, is that poor
outcome is associated with exposure of injured (repairing) epithelium to such iatrogenic
‘hits’. This thesis describes studies investigating the hypothesis that hyperoxia &
mechanical deformation inhibit normal epithelial repair.
The in vitro data presented demonstrate that hyperoxia reversibly inhibits the
transdifferentiation of ATII-like cells into ATI-like cells with time in culture. Whilst
confirming that hyperoxia is injurious to alveolar epithelial cells, these data further
suggest the ATII cell population harbours a subpopulation of cells resistant to
hyperoxia-induced injury. This subpopulation of cells appears to generate fewer reactive
oxygen species and express lower levels of the zonula adherens protein E-cadherin.
Using a panel of antibodies to ATI (RTI40) and ATII (MMC4 & RTII70) cell-selective
proteins, the effect of hyperoxia on the phenotype of the alveolar epithelium in a rat
model of resolving S. aureus-induced lung injury was investigated. These in vivo studies
support the view that, under normoxic conditions, alveolar epithelial repair occurs
through ATII cell proliferation & transdifferentiation of ATII cells into ATI cells, with
transdifferentiation occurring via a novel intermediate (MMC4/RTI40-coexpressing)
immunophenotype. However, in S. aureus-injured lungs exposed to hyperoxia, the
resolution of ATII cell hyperplasia was impaired, with an increase in ATII cell-staining

iv
membrane and a reduction in intermediate cell-staining membrane compared to injured
lungs exposed to normoxia alone. As hyperoxia is pro-apoptotic and known to inhibit
ATII cell proliferation, these data support the hypothesis that hyperoxia impairs normal
epithelial repair by inhibiting the transdifferentiation of ATII cells into ATI cells in vivo.
The effect of mechanical deformation on alveolar epithelial cells in culture was
investigated by examining changes in cell viability following exposure of epithelial cell
monolayers to quantified levels of cyclic equibiaxial mechanical strain. In the central
region of monolayers, deformation-induced injury was a non-linear function of
deformation magnitude, with significant injury occurring only following exposure to
strains greater than those associated with inflation of the intact lung to total lung
capacity. However, these studies demonstrate for the first time that different epithelial
cell phenotypes within the same culture system have different sensitivities to
deformation-induced injury, with spreading RTI40-expressing cells in the peripheral
region of epithelial cell monolayers and in the region of ‘repairing’ wounds being
injured even at physiological levels of mechanical strain. These findings are consistent
with the hypothesis that alveolar epithelial cells in regions of epithelial repair are highly
susceptible to deformation-induced injury.

v
Acknowledgements
I would like to express my sincere thanks to my supervisors Dr Mary McElroy and
Professor David Harrison without whose personal investment, advice and kindly support
the work in this thesis would not have been possible. Thanks also to Professors Tim
Walsh, Chris Haslett and Donald Salter who have all been very supportive and whose
help was instrumental in getting this project off the ground.
Bob Morris, Susan McIntyre and Kate Harris provided constant technical and logistic
support, while Kirsty Tyrell and Linda Franklin deserve special thanks for all their
patient help in the lab. Thanks to Spike Clay and Sharon Rossiter for their advice and
practical help in setting up the haemorrhagic shock model, Linda Wilson for assistance
with confocal microscopy and Susan Harvey for cutting the lung tissue sections. Chris
Baig kindly provided the foetal lung tissue sections used.
The work presented in this thesis describing the establishment of a haemorrhagic shock-
induced model of acute lung injury and the identification of MMC4/ RTI40–expressing
alveolar epithelial cells was performed in collaboration with Gareth Clegg. I would like
to thank Gareth for his help, good company, moral support and friendship throughout.
I am grateful to Professor Leland Dobbs for kindly providing anti-RTI40 and anti-RTII70
antibodies, Professor Michael Beers for anti-pro-SP-C antibody and Dr Tim Foster for S.
aureus strain 8325-4.
Finally, heartfelt thanks are due to my family and friends. To Jo, who gives so much to
everyone, for all her ‘wee ways’, and to Mum and Dad for just being there. Thank you.
This work was supported by a Wellcome Trust Clinical Research Training Fellowship.

vi
Table of contents
Declaration of Authorship .......................................................................................................ii
Abstract .................................................................................................................................... iii
Acknowledgements ...................................................................................................................v
Table of contents ......................................................................................................................vi
Figure List ................................................................................................................................xii
Abbreviations..........................................................................................................................xiv
1. Introduction ...........................................................................................................................1
1.1 Structure and function of the alveolus ......................................................................................1
1.1.1 Structure of the air-blood barrier...............................................................................................2
1.1.2 Pulmonary endothelium ..............................................................................................................2
1.1.3 Alveolar epithelium......................................................................................................................3
1.1.3.1 Alveolar epithelial type I (ATI) cells .......................................................................................3
1.1.3.2 Alveolar epithelial type II (ATII) cells ....................................................................................4
1.1.4 Alveolar epithelial cell junctions ................................................................................................4
1.1.4.1 Tight junctions ..........................................................................................................................5
1.1.4.2 Gap junctions ............................................................................................................................5
1.1.5 Extracellular matrix and structural features of the alveolar wall............................................6
1.1.5.1 Alveolar basement membranes ................................................................................................6
1.1.5.2 Alveolar interstitial connective tissue .....................................................................................6
1.1.5.3 Structural features of the alveolar wall...................................................................................7
1.1.6 Alveolar epithelial transport .......................................................................................................7
1.1.6.1 The role of sodium transport....................................................................................................8
1.1.6.2 The role of aquaporin water channels ....................................................................................8
1.1.7 Air-blood barrier function...........................................................................................................9
1.2 The role of the alveolar epithelium in disease pathogenesis ................................................11
1.2.1 The consequences of alveolar epithelial injury .......................................................................11
1.2.2 Acute respiratory distress syndrome (ARDS) & acute lung injury (ALI) ..............................12
1.2.2.1 Historical perspective and definitions...................................................................................12
1.2.2.2 Epidemiology of acute lung injury.........................................................................................13
1.2.2.3 Pathogenesis of acute lung injury .........................................................................................14
1.2.2.4 Treatment of acute lung injury...............................................................................................16
1.2.2.5 The significance of the alveolar epithelium in acute lung injury ........................................17
1.2.3 Bacterial pneumonia..................................................................................................................17

vii
1.2.3.1 Definition, aetiology and clinical presentation ....................................................................17
1.2.3.2 Epidemiology of bacterial pneumonia ..................................................................................19
1.2.3.3 Pathogenesis of bacterial pneumonia ...................................................................................19
1.2.3.4 The significance of the alveolar epithelium in bacterial pneumonia ..................................20
1.2.4 Idiopathic Pulmonary fibrosis ..................................................................................................21
1.2.4.1 Definition.................................................................................................................................21
1.2.4.2 Epidemiology of idiopathic pulmonary fibrosis ...................................................................22
1.2.4.3 Pathogenesis & the role of the alveolar epithelium in IPF .................................................23
1.3 Alveolar epithelial repair...........................................................................................................24
1.3.1 Historical perspective of cell cycle kinetics in the alveolar epithelium.................................24
1.3.2 Current model of alveolar epithelial repair.............................................................................25
1.3.3 Stem cells responsible for alveolar epithelial repair ..............................................................26
1.3.3.1 Alveolar stem cells..................................................................................................................26
1.3.3.2 Extra-alveolar stem cells........................................................................................................27
1.3.4 Alveolar epithelial repair and lung fibrosis.............................................................................28
1.4 The use of cell-selective markers in lung injury and repair.................................................30
1.4.1 Biomarkers and immunotargeting ............................................................................................30
1.4.2 Alveolar epithelial cell-selective markers ................................................................................31
1.4.2.1 ATI cell-specific and cell-selective proteins .........................................................................31
1.4.2.2 ATII cell-specific and cell-selective proteins........................................................................32
1.4.3 Alveolar epithelial cell-selective markers in the investigation of lung injury .......................33
1.4.3.1 Cell-selective markers in quantifying epithelial cell numbers and/or injury .....................33
1.4.3.2 Specific regulation of cell selective markers in lung injury.................................................35
1.4.3.3 Cell-selective markers and the phenotype of repairing epithelium.....................................35
1.5 Hyperoxia and the alveolar epithelium ...................................................................................37
1.5.1 Oxygen homeostasis ..................................................................................................................37
1.5.2 Hyperoxia ...................................................................................................................................38
1.5.3 Hyperoxia-induced lung injury .................................................................................................39
1.5.3.1 The effects of hyperoxia on the alveolar epithelium.............................................................40
1.6 Mechanical forces and the alveolar epithelium......................................................................43
1.6.1 Quantification of mechanical forces in situ .............................................................................43
1.6.2 The role of mechanical forces in lung development ................................................................44
1.6.2.1 The effect of mechanical forces on lung growth...................................................................44
1.6.2.2 Alveolar epithelial phenotype & differentiation...................................................................44
1.6.3 The effect of mechanical forces on the alveolar epithelium....................................................45
1.6.3.1 Surfactant secretion................................................................................................................45

viii
1.6.3.2 Phenotype................................................................................................................................46
1.6.3.3 Cytokine production ...............................................................................................................46
1.6.3.4 Apoptosis .................................................................................................................................47
1.6.3.5 Deformation-induced injury...................................................................................................47
1.6.4 Mechanical ventilation & lung injury: ventilator induced lung injury (VILI).......................48
1.6.4.1 Mechanisms of ventilator-induced lung injury .....................................................................49
1.6.4.2 The role of the alveolar epithelium in VILI...........................................................................52
1.7 Summary, thesis hypotheses and aims ....................................................................................53
1.7.1 Summary.....................................................................................................................................53
1.7.2 Hypotheses .................................................................................................................................54
1.7.3 Aims ............................................................................................................................................54
2. Materials and methods.......................................................................................................56
2.1 Primary alveolar epithelial type II cell isolation & culture.......................................................56
2.1.1 Alveolar epithelial type II cell isolation...................................................................................56
2.1.2 Modified Papanicolau staining & assessment of ATII purity.................................................57
2.1.3 Standard ATII cell culture.........................................................................................................58
2.1.4 ATII cell culture under hyperoxic conditions ..........................................................................59
2.1.5 ATII cell culture using the Bioflex! cell culture system..........................................................59
2.2 Protein quantification assay ........................................................................................................59
2.3 Measurement of cell viability in adherent cells ..........................................................................60
2.4 Lactate dehydrogenase (LDH)-cytotoxicity assay ......................................................................60
2.5 Feulgen staining & assessment of apoptosis ..............................................................................61
2.6 Estimation of reactive oxygen species (ROS) generation ..........................................................62
2.7 Estimation of ROS generation in response to oxidative stress ..................................................62
2.8 Quantification of antigens by ELISA-based dot blot assay........................................................63
2.9 Immunohistochemistry for fluorescence microscopy..................................................................64
2.9.1 General protocol for staining frozen sections .........................................................................65
2.9.2 General protocol for staining cultured ATII cells ...................................................................66
2.9.3 Bromo-deoxyuridine (BrdU) immunhistochemistry ................................................................66
2.9.4 Image acquisition.......................................................................................................................66
2.9.5 z-plane scanning and three-dimensional image reconstruction .............................................67
2.9.6 Image analysis ...........................................................................................................................68
2.9.6.1 Image density slicing ..............................................................................................................68
2.9.6.2 Quantification of epithelial surface staining ........................................................................68
2.9.6.3 Cell counts...............................................................................................................................68
2.9.6.4 Cell surface area.....................................................................................................................69

ix
2.10 Rat model of staphylococcus aureus pneumonia......................................................................69
2.10.1 Preparation of bacterial instillate ..........................................................................................69
2.10.2 Bacterial instillation................................................................................................................70
2.10.3 Initial recovery and randomisation ........................................................................................70
2.10.4 Housing under normoxic and hyperoxic conditions..............................................................70
2.10.5 Harvesting of blood, bronchoalveolar lavage (BAL) fluid and lung tissue.........................71
2.10.6 Bronchoalveolar lavage fluid processing ..............................................................................71
2.10.7 Lung tissue processing for biochemical analysis ..................................................................72
2.10.8 Lung tissue processing for morphological analysis ..............................................................72
2.11 Rat model of haemorrhagic shock-induced acute lung injury .................................................73
2.11.1 Animal preparation..................................................................................................................73
2.11.2 Experimental protocol.............................................................................................................74
2.12 Statistics.......................................................................................................................................74
3. The effect of hyperoxia on alveolar epithelial cells in culture .....................................76
3.1 Introduction & objectives ............................................................................................................76
3.2 Results ...........................................................................................................................................77
3.2.1 Characterisation of freshly isolated ATII cells........................................................................77
3.2.2 Change of morphology and phenotype of ATII cells in culture..............................................77
3.2.3 Identification of a novel intermediate cell phenotype .............................................................78
3.2.4 The immunoselectivity of the monoclonal antibody MMC6....................................................78
3.2.5 The effect of hyperoxia on ATII cells in culture.......................................................................80
3.2.6 The reversibility of hyperoxia-induced effects on ATII cells in culture .................................84
3.3 Discussion .....................................................................................................................................84
3.3.1 ATII cells transdifferentiate into ATI-like cells in culture ......................................................84
3.3.2 Identification of a novel intermediate cell phenotype .............................................................85
3.3.3 The MMC6 antigen appears to be ATI-selective .....................................................................85
3.3.4 Hyperoxia reversibly inhibits transdifferentiation in vitro .....................................................86
3.3.5 Hyperoxia is cytotoxic to alveolar epithelial cells ..................................................................87
3.3.6 A subpopulation of cells appears resistant to hyperoxia-induced injury...............................87
3.3.7 E-cadherin negative cells appear resistant to hyperoxia-induced injury ..............................88
3.3.8 Hyperoxia-resistant cells generate fewer ROS........................................................................89
3.3.9 Hyperoxia reversibly inhibits alveolar epithelial cell proliferation.......................................89
3.4 Summary .......................................................................................................................................90
4. The effect of hyperoxia on alveolar epithelial repair in vivo........................................91
4.1 Introduction & objectives ............................................................................................................91
4.2 Results ...........................................................................................................................................92

x
4.2.1 S. aureus-induced lung injury and the effect of hyperoxia......................................................92
4.2.2 The phenotype of the alveolar epithelium in control lung ......................................................93
4.2.3 The effect of hyperoxia on the proportion of ATI, ATII and intermediate cell-staining
membrane following S. aureus-induced lung injury .........................................................................94
4.2.4 The effect of hyperoxia on lung RTI40 concn following S.aureus-induced lung injury .........95
4.3 Discussion .....................................................................................................................................96
4.3.1 The effect of hyperoxic exposure on the resolution of lung injury .........................................96
4.3.2 The phenotype of the alveolar epithelium in control lungs.....................................................97
4.3.3 The phenotype of the alveolar epithelium in resolving S. aureus-induced lung injury.........97
4.3.4 The effect of hyperoxia on phenotype of the alveolar epithelium in resolving S. aureus-
induced lung injury .............................................................................................................................99
4.3.5 General discussion...................................................................................................................100
4.4 Summary .....................................................................................................................................102
5. Mechanical deformation-induced injury of alveolar epithelial cells ........................104
5.1 Introduction & objectives ..........................................................................................................104
5.2 Results .........................................................................................................................................105
5.2.1 Culture of alveolar epithelial cells in a mechanically active environment..........................105
5.2.2 Deformation-induced injury and magnitude of mechanical strain ......................................106
5.2.3 Deformation-induced injury and cell phenotype ...................................................................106
5.2.4 Deformation-induced injury in repairing wound...................................................................106
5.3 Discussion ...................................................................................................................................107
5.3.1 Deformation-induced injury is a non-linear function of deformation magnitude ...............107
5.3.2 Spreading RTI40-expressing cells in the peripheral region of epithelial cell monolayers are
susceptible to deformation-induced injury ......................................................................................108
5.3.3 Epithelial cells in the region of repairing epithelial monolayers appear susceptible to
deformation-induced injury ..............................................................................................................109
5.3.4 General discussion...................................................................................................................110
5.4 Summary .....................................................................................................................................112
6. Epithelial injury in a model of haemorrhagic shock-induced acute lung injury....113
6.1 Introduction ................................................................................................................................113
6.2 Results .........................................................................................................................................114
6.2.1 Characterisation of haemorrhagic shock-induced acute lung injury...................................114
6.2.2 Alveolar epithelial cell injury following haemorrhagic shock .............................................115
6.3 Discussion ...................................................................................................................................115
6.3.1 Haemorrhagic shock results in acute lung injury..................................................................115
6.3.2 Haemorrhagic shock is associated with ATI cell injury........................................................116

xi
6.4 Summary .....................................................................................................................................117
7. Summary ............................................................................................................................119
7.1 Hyperoxia inhibits the transdifferentiation of ATII cells into ATI cells..................................119
7.2 Spreading RTI40-expressing cells are susceptible to deformation-induced injury .................121
7.3 The multiple-hit hypothesis ........................................................................................................122
8. Future work .......................................................................................................................123
Figures ....................................................................................................................................125
Appendix I ..............................................................................................................................169
(A) Alveolar epithelial type I cell markers in adult lung ................................................................169
(B) Alveolar epithelial type II cell markers in adult lung...............................................................170
Appendix II Manufacturers and Suppliers.......................................................................171
Appendix III Buffers and Solutions ...................................................................................173
Bibliography .........................................................................................................................174

xii
Figure List
1. Schematic representation of alveolar epithelial repair
2. Primary ATII cell isolation
3. The immunophenotype of alveolar epithelial cells in culture
4. MMC4 & RTI40 coexpression in intermediate cells
5. Control lung stained with MMC6, RTI40 & TO-PRO-3
6. Control lung stained with MMC6, RTI40 & MMC4
7. 3D localisation of MMC6 in control lung
8. E16 lung stained with MMC6, RTI40 & TO-PRO-3
9. E17 lung stained with MMC6, RTI40 & TO-PRO-3
10. E20 lung stained with MMC6 & RTII70
11. E20 lung stained with MMC6, RTI40 & TO-PRO-3
12. MMC6 expression in alveolar epithelial cells in culture
13. 3D localisation of MMC6 in ATI-like cells
14. The effect of hyperoxia on adherent cell viability
15. The effect of hyperoxia on cell spreading
16. Hyperoxia & ATI cell-selective protein expression - immunofluoresence
17. Hyperoxia & ATI cell-selective protein expression - ELISA
18. Hyperoxia & ATII cell-selective protein expression - immunofluoresence
19. Hyperoxia & ATII cell-selective MMC4 expression - ELISA
20. E-cadherin expression in normoxia- & hyperoxia-exposed cells
21. The effect of hyperoxia on ROS generation
22. The effect of hyperoxia on alveolar epithelial cell proliferation
23. Hyperoxia-induced cell cytotoxicity
24. Hyperoxia-induced apoptosis in alveolar epithelial cells
25. Experimental protocol for S. aureus pneumonia model
26. BALF protein content in S. aureus pneumonia model
27. BALF cell counts in S. aureus pneumonia model
28. BALF RTI40 in S. aureus pneumonia model
29. Lung homogenate RTI40 in S. aureus pneumonia model
30. Quantification of alveolar epithelial surface staining
31. Expression of ATI & ATII cell-selective markers in control lung

xiii
32. Tiled image of normoxia-exposed S. aureus pneumonia
33. Epithelial repair in normoxia-exposed S. aureus pneumonia
34. Tiled image of hyperoxia-exposed S. aureus pneumonia
35. Epithelial repair in hyperoxia-exposed S. aureus pneumonia
36. ATI & ATII-staining membrane in S. aureus pneumonia model
37. Frequency distribution of ATII cell-staining membrane lengths
38. RTI40/MMC4 coexpressing membrane in S. aureus pneumonia model
39. The Bioflex! cell culture system & Flexercell FX-4000T strain unit"
40. Deformation-induced injury – Live/Dead! staining
41. Effect of deformation magnitude on deformation-induced injury
42. Relationship between lung volume and # epithelial BM surface area
43. Deformation-induced injury in central and peripheral monolayer regions
44. Cell viability data in central and peripheral monolayer regions
45. Phalloidin staining of central and peripheral monolayer regions
46. Immunophenotype of central and peripheral monolayer regions
47. Deformation-induced injury in repairing monolayer wounds
48. Cell viability data in repairing epithelial monolayer wounds
49. Protocol for haemorrhagic shock-induced acute lung injury model
50. Blood pressure profile of haemorrhagic shock and resuscitation
51. BALF protein content in haemorrhagic shock model
52. BALF cell counts in haemorrhagic shock model
53. BALF ATI cell-selective proteins in haemorrhagic shock model

xiv
Abbreviations
ALI acute lung injury
ANTU $-naphthylthiourea
APN aminopeptidase N
AQP aquaporin
ARDS acute respiratory distress syndrome
ARDSnet acute respiratory distress syndrome network
ATI alveolar epithelial type I
ATII alveolar epithelial type II
ATP adenosine triphosphate
BAB blood agar base
BALF bronchoalveolar lavage fluid
Bax Bcl-associated x protein
BB blocking buffer
BBT blocking buffer with triton
Bcl-2 B-cell lymphoma-2
BM basement membrane
BrdU 5-bromo-2’-deoxyuridine
BTS British Thoracic Society
Calcein AM calcein acetoxymethylester
CAP community acquired pneumonia
CC10 Clara cell-specific protein
CCSP Clara cell secretory protein
CMH2DCFDA 5-(6-)-chloromethyl-2’,7’-dichlorodihydrofluorescein diacetate
CO2 carbon dioxide
CRP C-reactive protein
CT computed tomographic
Cx connexin
d day/s

xv
DDW double distilled water
DIC differential interference contrast
DMEM Dulbecco’s modified eagle’s medium
DNA deoxyribonucleic acid
du densitometry units
EBMSA epithelial basement membrane surface area
ECM extracellular matrix
EDTA ethylenediaminetetraacetic acid
EGF epidermal growth factor
EGTA ethylene glycol tetraacetic acid
ELISA enzyme-linked immunosorbent assay
ENaC amiloride sensitive ion channel
EthD-1 ethidium homodimer-1
FBS foetal bovine serum
FGF fibroblast growth factor
FRC functional residual capacity
fu fluorescent units
GFP green fluorescent protein
GSF granulocyte colony-stimulating factor
GSH glutathione
h hour/s
HBSS Hank’s balanced salt solution
HEPES 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid
HGF hepatocyte growth factor
HIF-1$ hypoxia-inducible factor-1alpha
3HT tritiated thymidine
HTI56 human alveolar epithelial type I cell 56 kDa protein
ICAM-1 intracellular adhesion molecule-1
ICU intensive care unit
IFN-% interferon-gamma

xvi
IgG immunoglobulin G
IGF insulin-like growth factor
IL interleukin
IL-1& interleukin-1 beta
IL-ra interleukin-receptor antagonist
INT iodotetrazolium chloride
IPF idiopathic pulmonary fibrosis
KGF keratinocyte growth factor
LDH lactate dehydrogenase
LEL lectin lycoperiscon esculentum
LPS lipopolysaccharide
LSCM laser scanning confocal microscope
mRNA messenger ribonucleic acid
Mab monoclonal antibody
MIF macrophage migration inhibitory factor
MIP macrophage inflammatory protein
MMC Mary McElroy
MODS multiple organ dysfunction syndrome
MOF multiple organ failure
NAD &-nicotinamide adenine dinucleotide, oxidised form
NADH &-nicotinamide adenine dinucleotide, reduced form
Na+/K
+-ATPase sodium-potassium adenosine triphophatase
NF-'B nuclear factor-kappa B
NIH National Institute of Health
NO nitric oxide
NO2 nitrogen dioxide
O2 oxygen
OCT optimal cutting temperature compound
PAMP pathogen-associated molecular pattern
Pap Papanicolau

xvii
PBS phosphate buffered saline
PDGF platelet-derived growth factor
PEEP positive end-expiratory pressure
PKC protein kinase C
PMS phenazine methosulphate
pO2 partial pressure of oxygen
Pro-SP-C pro-surfactant protein-C
PRR pattern recognition receptor
PVDF polyvinylidene difluoride
RGB red green blue
RNS reactive nitrogen species
ROS reactive oxygen species
RTI40 rat alveolar epithelial cell type I 40 kDa protein
RTII70 rat alveolar epithelial cell type II 70 kDa protein
S. aureus Staphylococcus aureus
SA surface area
#SA change in surface area
SOD superoxide dismutase
SP-A/B/C/D surfactant protein-A/B/C/D
SSA sulfasalazine
T1$ (alveolar) type I cell alpha protein
TBS tris(hydroxymethyl)aminomethane-buffered saline
TGF-& transforming growth factor-beta
TIFF tagged image file format
TJ tight junction
TLC total lung capacity
TLR toll-like receptor
TNF-$ tumour necrosis factor-alpha
TRIS tris(hydroxymethyl)aminomethane
UK United Kingdom of Great Britain and Northern Ireland

xviii
US/USA United States/United States of America
VAP ventilator-associated pneumonia
VILI ventilator-induced lung injury
ZO zonna occludens

1
1. Introduction
Injury to the lung, resulting in impaired gas exchange and acute respiratory failure, is an
important cause of morbidity and mortality (Lewandowski et al. 1995; Behrendt 2000).
The alveolar epithelium plays a central role in the pathogenesis of this injury in a range
of common clinical conditions (Berthiaume et al. 1999; Newman et al. 2000; Ware and
Matthay 2000; Hippenstiel et al. 2006), with effective repair of the alveolar epithelial
barrier following injury an important determinant of clinical outcome (Adamson et al.
1988; Matthay and Wiener-Kronish 1990; Ware and Matthay 2001). Supplemental
oxygen and mechanical ventilation are key components of the supportive treatment of
respiratory failure but the effects of these therapeutic interventions in the injured lung
and on alveolar epithelial repair following injury are largely unknown. The clinical
impression however, is that poor outcome is associated with exposure of injured
(repairing) epithelium to such iatrogenic ‘hits’ (Ranieri et al. 1999; ARDSNet 2000;
Frank et al. 2002).
This thesis describes studies investigating the hypothesis that hyperoxia & mechanical
deformation inhibit normal epithelial repair following injury. In order to put these
studies into context, the introduction outlines the structure and function of the normal
alveolus and discusses the role of the alveolar epithelium in the pathogenesis of disease.
The process of alveolar epithelial repair is then discussed followed by a review of the
effects of hyperoxia and mechanical deformation in the lung.
1.1 Structure and function of the alveolus
The primary function of the lung is gas exchange. Inspired gases pass into the trachea
and through a series of branching conducting airways until they reach the respiratory
bronchioles, which have occasional alveoli budding from their walls, and finally the
alveolar ducts which are completely lined with alveoli. This alveoli-containing region of

2
the lung is known as the respiratory zone, and is where gas exchange occurs. In the
human lung, this region contains 300 million or so alveoli, with a total surface area in
the region of 50-100m2 (West, Weibel & Gomez 1962).
1.1.1 Structure of the air-blood barrier
Oxygen (O2) moves from air within alveoli into venous blood, and carbon dioxide (CO2)
from venous blood into alveoli. This process occurs by passive diffusion across the air-
blood barrier. This is a three-layered structure comprising pulmonary capillary
endothelium, extracellular matrix (ECM) and alveolar epithelium.
1.1.2 Pulmonary endothelium
The pulmonary vessels are lined by a squamous unistratified epithelium of mesodermal
origin, the endothelium. Alveolar capillary endothelial cells are large attenuated cells
(surface area (1000-1300µm2), with their nuclei usually oriented in the direction of
blood flow. Ultrastructurally, endothelial cells demonstrate a relative paucity of
intracellular organelles but have a large number of plasmalemmal transport vesicles,
thought to reflect adaptation of the vascular endothelium to its role in the transport of
macromolecules (Simionescu 1997). Joined by tight junctions, alveolar capillary
endothelial cells form a continuous (non-fenestratred) polarised monolayer with the
luminal surface of the cells in contact with blood, and the abluminal surface resting on
basement membrane.
In addition to its structural role as part of the air-blood barrier, the pulmonary
endothelium plays a pivotal role in regulating blood flow, coagulation and fibrinolysis
(Fishman 1982; Cines et al. 1998). Of central importance, is the role the endothelium
plays in controlling leucocyte migration from the blood, through the alveolar
interstitium, and into the alveolar airspace.
Leucocyte migration across the endothelial barrier occurs in many tissues as part of
normal immunosurveillance. However, the level of this cell migration can be increased

3
dramatically following endothelial cell activation - a term used to describe a change in
chemokine production and adhesion molecule expression on the luminal surface of the
endothelium in response to stimuli such as injury or infection (Cines et al. 1998).
Human lung microvascular endothelial cell activation has been demonstrated in vitro
following stimulation with tumour necrosis factor-$ (TNF-$) and interferon-% (IFN-%)
(Hillyer et al. 2003), and results in increased chemokine production and increased
expression of the adhesion molecules E-selectin and intercellular adhesion molecule-1
(ICAM-1). E-selectin, a member of the selectin family of transmembrane glycoproteins,
initiates the tethering and rolling of leucocytes on the endothelial cell surface (McEver et
al. 1995). Subsequent leucocyte activation enables &2 integrins on leucocytes to bind to
ICAM-1 (Springer 1995), and this is followed by migration of leucocytes between
endothelial cells into the tissues. These processes are thought to be of key importance in
the pathogenesis of acute lung injury (section 1.2.2.3) (Zimmerman et al. 1999).
1.1.3 Alveolar epithelium
The alveoli are lined by a continuous layer of epithelium comprised of two
morphologically distinct epithelial cell types: alveolar epithelial type I (ATI) and
alveolar epithelial type II (ATII) cells (Low 1952). Morphometric studies have
demonstrated that the distribution and morphology of these cell types is remarkably
similar across mammalian species including the rat and human (Crapo et al. 1983; Stone
et al. 1992).
1.1.3.1 Alveolar epithelial type I (ATI) cells
ATI cells comprise only (8-9% of the parenchymal cell population within the lung, but
cover (95% of the alveolar surface (Haies et al. 1981; Crapo et al. 1982).
Morphologically, ATI cells are large (luminal surface (5100µm2), flat (thickness
(0.2µm) and highly branched, with their thin attenuated cytoplasm facilitating passive
diffusion of gases across the cell (Weibel and Gomez 1962; Haies et al. 1981; Weibel

4
1984). Ultrastructurally, ATI cells appear relatively simple, with a small nucleus, a few
small mitochondria and inconspicuous endoplasmic reticulum and Golgi apparatus
(Schneeberger 1991). This paucity of organelles was thought to suggest little functional
role for ATI cells other than the passive facilitation of gas exchange, but more recently
ATI cells have been shown to have an important role in regulating alveolar fluid balance
(section 1.1.6) and, through mechanotransduction, the control of pulmonary surfactant
secretion (Dobbs et al. 1998; Johnson et al. 2002; Williams 2003).
1.1.3.2 Alveolar epithelial type II (ATII) cells
ATII cells comprise (15% of lung parenchymal cells, but cover only (3-5% of the
alveolar surface (Haies et al. 1981; Crapo et al. 1982). Unlike ATI cells, they are
cuboidal in shape (luminal surface (60µm2, thickness (3µm) with apical microvilli and
characteristic surfactant-containing organelles known as lamellar bodies (Crapo et al.
1983; Mason and Williams 1991). Typically located in the corners of alveoli, these cells
usually occur singly flanked by ATI cells. Known functions of ATII cells include the
synthesis, secretion and recycling of pulmonary surfactant, regulation of alveolar fluid
and electrolyte balance (section 1.1.6), host defence and immunomodulation (section
1.2.3.4) (Fehrenbach 2001). In addition to their synthetic and secretory capacities, ATII
cells function as the stem cell of the alveolar epithelium and have a central role in
alveolar epithelial repair (section 1.3.3.1).
1.1.4 Alveolar epithelial cell junctions
ATI and ATII cells are joined by both tight junctions (TJ) and gap junctions
(Schneeberger 1991).

5
1.1.4.1 Tight junctions
Tight junctions are gasket-like strands of protein connecting the apical portion of
alveolar epithelial cells and separating the apical and basolateral surfaces of the
epithelium (Schneeberger and Lynch 1992). They are thought to provide the primary
rate-limiting barrier to paracellular transport, with the majority of the barrier function
being provided by the integral membrane protein occludin (Saitou et al. 1997) and a
family of TJ proteins, the claudins (Wang et al. 2003). Tight junction-associated proteins
zona occludens (ZO)-1, -2 and -3, are intracellular proteins thought to act as a link
between the TJ and cytoskeletal proteins, although the functional significance of these
interactions is not known (Denker and Nigam 1998; Goodenough 1999).
Alveolar epithelial TJs are not static structures and appear to be highly regulated, with
the cytoskeleton playing a central role (Schneeberger and Lynch 1992). For example,
low levels of extracellular calcium and low levels of intracellular ATP (ATPi) have both
been shown to result in TJ dissociation, whilst activation of protein kinase C (PKC)
decreases TJ permeability (Gonzalez-Mariscal et al. 1985; Bacallao et al. 1994; Denker
and Nigam 1998). TJ dissociation, with reduced resistance of the paracellular pathway,
occurs in both hydrostatic and injury-induced pulmonary oedema and has been shown to
occur in response to inflammatory mediators in mice (Gon et al. 2005), hypoxia (Bouvry
et al. 2006) and pathological levels of mechanical strain in primary rat alveolar epithelial
cells (Cavanaugh et al. 2001).
1.1.4.2 Gap junctions
Gap junctions are transcellular channels that allow the passive diffusion of small
molecules and ions between adjacent cells. In vertebrates, gap junctions are composed of
hexamers of a family of gap junction proteins, the connexins (Cx) (Kumar and Gilula
1996; Alberts 2002). At least six different connexins are expressed in alveolar epithelial
cells, with ATI and ATII cells having different expression profiles (Abraham et al. 1999;
Isakson et al. 2003). The differential expression and post-translational modification of

6
these proteins is thought to influence gap junction permeability and function, and
appears to be highly regulated, although the functional significance of this remains
unclear (Isakson et al. 2003).
1.1.5 Extracellular matrix and structural features of the alveolar wall
Extracellular matrix (ECM) may be divided into two categories: basement membrane
(BM) and interstitial connective tissue (Kumar et al. 2004).
1.1.5.1 Alveolar basement membranes
Basement membranes are condensed sheet-like structures separating endothelial and
epithelial cells from interstitial connective tissue. In both the rat and human lung, the
alveolar epithelial BM and alveolar capillary BM has been shown to consist of highly
cross-linked type IV collagen, laminin and fibronectin (Gil and Martinez-Hernandez
1984; Kumar et al. 2004). The composition of the BM is thought to play a significant
role in preserving normal alveolar structure and function. In vitro, the ECM on which
alveolar epithelial cells are cultured has been shown to have a significant effect on cell
phenotype, with laminin-rich surfaces favouring retention of the differentiated ATII-like
phenotype and fibronectin-rich surfaces accelerating loss of the ATII phenotype and
acquisition of an ATI-like phenotype (Rannels and Rannels 1989; Olsen et al. 2005).
Fibronectin is believed to be involved in epithelial cell regeneration in many tissues
(Fujikawa et al. 1981; Clark et al. 1982), and comparison of the level and distribution of
fibronectin expression in normal and fibrotic lungs supports the hypothesis that
fibronectin plays an important role in alveolar epithelial cell repair (Torikata et al. 1985).
1.1.5.2 Alveolar interstitial connective tissue
The major components of alveolar interstitial connective tissue are types I collagen, type

7
III collagen and fibronectin (Gil and Martinez-Hernandez 1984; Kumar et al. 2004).
1.1.5.3 Structural features of the alveolar wall
In the rat lung, the alveolar wall has been shown to consist of thin areas, where capillary
endothelium and alveolar epithelium are separated by only a single fused alveolar and
capillary basement membrane, and thick areas, where endothelium and epithelium are
separated by their respective basement membranes and connective tissue of the
interstitial space (Vaccaro and Brody 1981). These differences suggest that the alveolar
basement membrane may contain functional and structural microdomains that regulate
alveolar epithelial cell phenotype: the single basement membrane separating capillary
blood from alveolar air in areas of thin alveolar wall provides the thinnest possible
barrier to gaseous diffusion, and during normal development and repair these areas
appear to favour the acquisition of an ATI phenotype. In contrast, areas of thick alveolar
wall, typically situated along alveolar septa and in the corners of alveoli, favour the
retention of an ATII phenotype (Sannes 1984; Lwebuga-Mukasa 1991). This is thought
to represent a plausible hypothesis to explain the relatively constant numerical ratio of
ATI and ATII cells and the non-random localisation of ATII cells within the lung
(Lwebuga-Mukasa 1991). Further evidence supporting the concept of basement
membrane-associated microdomains is the finding that the BM underlying ATI and ATII
cells differs in the distribution of cytochemically detected sulphate esters (Sannes 1984).
1.1.6 Alveolar epithelial transport
Current evidence suggests that the alveolar epithelium is at least an order of magnitude
less permeable to protein and small solutes than the pulmonary endothelium, and hence
is the main regulator of fluid and solute exchange across the air-blood barrier (Taylor
and Gaar 1970; Normand et al. 1971). The alveolar epithelium is more than a passive
barrier however, and plays an active role both in fluid homeostasis in the normal
alveolus and critically, in the reabsorption of fluid that enters the alveolus following

8
injury (Carden et al. 1998; Crandall and Matthay 2001).
1.1.6.1 The role of sodium transport
Active sodium transport appears to be the primary mechanism driving fluid reabsorption
within the alveolus. Sodium ions enter the apical membrane of alveolar epithelial cells,
at least in part, through amiloride-sensitive ion channels (ENaC) and are actively
transported across the basolateral membrane by Na+/K
+-ATPase. Chloride and water
follow the transport of sodium through both transcellular and paracellular pathways to
maintain electrochemical and osmotic neutrality across the alveolar epithelial membrane
(Crandall and Matthay 2001).
Initial in situ immunocytochemical studies demonstrated expression of Na+/K
+-ATPase
in ATII cells but not ATI cells, leading to the inference that alveolar fluid transport was
regulated largely by ATII cells, with ATI cells playing only a passive role (Schneeberger
and McCarthy 1986). However, subsequent in vitro studies have shown that ATI cells
not only have the highest known water permeability of any mammalian cell type, but
also express Na+/K
+-ATPase, suggesting that ATI cells may also play a significant role
in vectorial ion and water transport (Dobbs et al. 1998; Johnson et al. 2002).
1.1.6.2 The role of aquaporin water channels
Water transport down the osmotic gradient generated by active sodium transport is
thought to occur predominantly through aquaporin (AQP) water channels. The
aquaporins are a family of hydrophobic integral membrane proteins, four members of
which have been shown to be expressed in the lung (Verkman et al. 2000). AQP1 has
been immunolocalised to the microvascular endothelium and to a limited extent ATII
cells (Effros et al. 1997), AQP3 has been shown to be expressed on the basolateral
membrane of ATII cells (Kreda et al. 2001), whilst both AQP4 and AQP5 appear to be
expressed on the apical surface of ATI cells (Kreda et al. 2001; Williams 2003).

9
The precise role of aquaporins in the maintenance of alveolar fluid homeostasis remains
unclear. Deletions of AQP1 and AQP5 produce a dramatic decrease in osmotically
driven water transport, although not in isosmolar fluid transport (Bai et al. 1999; Ma et
al. 2000), and although AQP4 is thought to be the main aquaporin responsible for
removal of fluid from the neonatal alveolus, AQP1, AQP4 and AQP5 knock out mice
appear to have the same ability to clear lung fluid in the perinatal period as wild type
mice (Song et al. 2000). In addition, deletion of AQP1, AQP4 and AQP5 does not effect
the formation or clearance of lung oedema following acid- or hyperoxia-induced lung
injury (Song et al. 2000).
1.1.7 Air-blood barrier function
Because the epithelial barrier is much less permeable than the endothelial barrier,
epithelial injury removes a significant physical barrier to the passage of fluid into the
alveolus (Wiener-Kronish et al. 1991). In addition, loss of alveolar epithelial integrity
significantly disrupts normal epithelial fluid transport, impairing alveolar fluid clearance
capacity (Ware and Matthay 2001; Matthay et al. 2002; Matthay et al. 2005). It is
therefore unsuprising that preservation of alveolar epithelial barrier integrity appears to
have significant prognostic value in acute lung injury (ALI), with patients who maintain
a functional epithelial barrier having a better chance of survival than those with loss of
epithelial barrier integrity (Matthay and Wiener-Kronish 1990; Carden et al. 1998).
The majority of patients who die from acute lung injury do not do so from respiratory
failure, but from multiple organ failure (MOF)(Montgomery et al. 1985; Wyncoll and
Evans 1999). One mechanism postulated for this finding is that disruption of the air-
blood barrier allows the release of pro-inflammatory cytokines produced in the lung into
the pulmonary circulation with the subsequent development of systemic inflammation
and multiple organ dysfunction syndrome (MODS) (Douzinas et al. 1997; Slutsky and
Tremblay 1998). Loss of compartmentalisation of alveolar TNF-$ has been
demonstrated following $-naphthylthiourea (ANTU)-induced lung injury in rats (Tutor

10
et al. 1994), and also after Pseudomonas aeruginosa pneumonia in rabbits (Kurahashi et
al. 1999). In humans with acute lung injury, conventional mechanical ventilation is
associated with both a pulmonary and systemic cytokine response. Both of these
responses are attenuated by lung-protective ventilation designed to reduce further injury
to the air-blood barrier, supporting the concept that release of pro-inflammatory
cytokines from the lung into the systemic circulation may be related to integrity of the
air-blood barrier (section 1.6.4.1.1) (Ranieri et al. 1999).
Loss of alveolar compartmentalisation following disruption of the air-blood barrier has
also been shown to facilitate bacterial translocation from the alveoli into the systemic
circulation in both hamsters and dogs (Johanson et al. 1985; Nahum et al. 1997).
Changes in the expression of adhesion molecules following activation or injury of the
alveolar epithelium may play an important part in the further recruitment and migration
of inflammatory cells across the epithelial barrier. In support of this concept, acid
induced-injury, hyperoxia, and the inflammatory cytokines interleukin-1 (IL-1) and
TNF-$ have all been shown to upregulate alveolar epithelial cell expression of ICAM-1
(Tosi et al. 1992; Ohwada et al. 2003). The role of the endothelium in controlling
leucocyte migration from the blood, through the alveolar interstitium, and into the
alveolar air space is discussed above (section 1.1.2).

11
1.2 The role of the alveolar epithelium in disease pathogenesis
1.2.1 The consequences of alveolar epithelial injury
The alveolar epithelium has key structural and functional roles in maintaining normal
lung function. Alveolar epithelial injury, with disruption of the integrity of the air-blood
barrier, has profound consequences:
i. Loss of epithelial barrier function: Epithelial cell injury and tight junction disruption
removes the most significant physical barrier to the passage of fluid into the alveolus
and alveolar flooding results. Upregulation of adhesion molecules following injury
also facilitates inflammatory cell recruitment and trafficking across the air-blood
barrier (section 1.1.7).
ii. Impaired alveolar fluid clearance: Disruption of normal epithelial fluid transport
impairs the removal of oedema fluid from the alveolar air spaces (sections 1.1.6 &
1.1.7).
iii. Surfactant abnormalities: ATII cell injury impairs normal production, secretion and
recycling of pulmonary surfactant. This leads to loss of surface tension-reducing
phospholipids, alveolar instability and collapse (Lewis and Jobe 1993; Greene et al.
1999).
iv. Impaired innate immunity: The alveolar epithelium forms a significant mechanical
barrier to microbial invasion, and plays a central role in pathogen recognition and the
initiation of the innate immune response. Injury, together with surfactant depletion,
impairs this response (section 1.2.3.4).
v. Loss of alveolar compartmentalisation: Physical disruption of the epithelial barrier
may result in loss of compartmentalisation within the lung allowing the release of
bacteria and inflammatory mediators into the systemic circulation (section 1.1.7).
vi. Pulmonary fibrosis: In severe epithelial injury, disorganised or inadequate epithelial
repair may lead to fibrosis (section 1.3.4)(Adamson et al. 1988; Bitterman 1992).
It is unsurprising given the extent of these consequences that alveolar epithelial injury,

12
and effective repair of the alveolar epithelium following injury, play a central role in the
pathogenesis and resolution of a wide range of clinical conditions. Three such
conditions, with relevance to the work presented in this thesis are acute lung injury,
bacterial pneumonia and & idiopathic pulmonary fibrosis.
1.2.2 Acute respiratory distress syndrome (ARDS) & acute lung injury (ALI)
1.2.2.1 Historical perspective and definitions
The first description of respiratory distress syndrome was published in 1967, when
Ashbaugh and colleagues described patients in intensive care with acute respiratory
distress, hypoxaemia refractory to oxygen therapy, decreased lung compliance, and
diffuse alveolar infiltrates on chest radiograph (Ashbaugh et al. 1967). Initially called
the adult respiratory distress syndrome (Petty and Ashbaugh 1971), the term acute
respiratory distress syndrome (ARDS) was subsequently adopted, as the condition is
also known to occur in children. In 1994, the American-European Consensus
Conference Committee on ARDS recommended a new, and now widely accepted
definition, in which the severity of clinical lung injury was recognised. Based upon this
definition, patients with less severe hypoxaemia (defined by a ratio of the partial
pressure of arterial oxygen to the fraction of inspired oxygen) are considered to have
acute lung injury (ALI), whilst those with more severe hypoxaemia are considered to
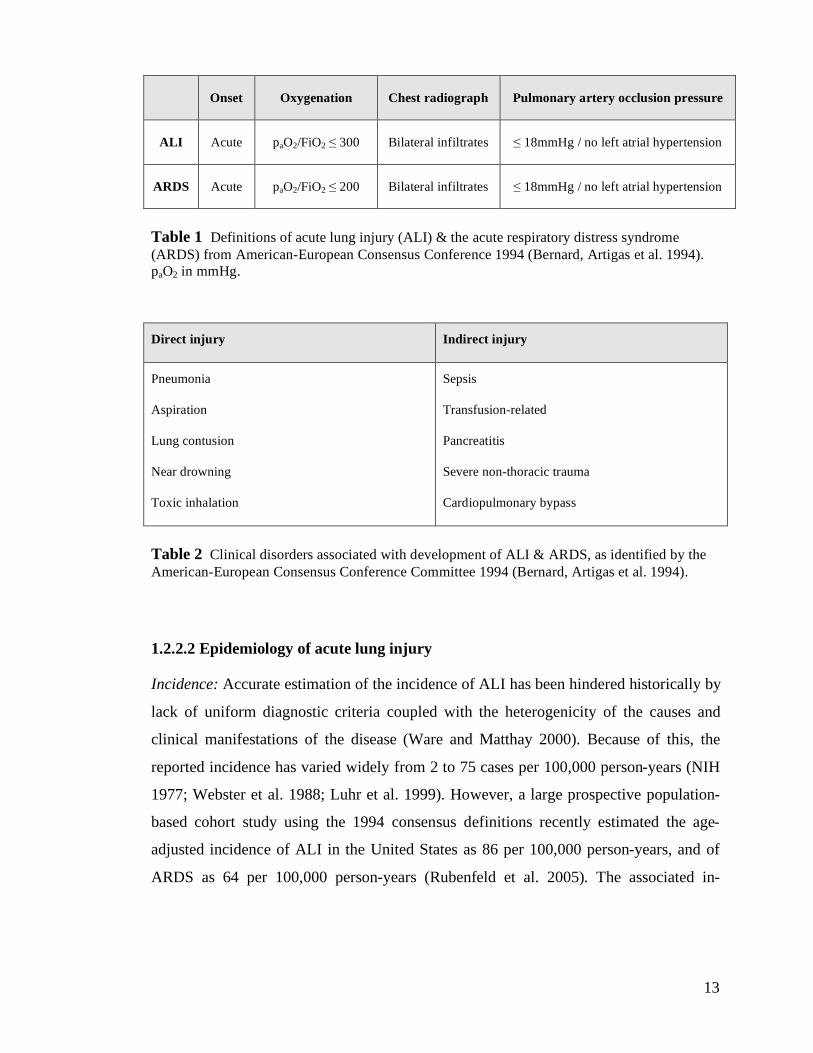
have the acute respiratory distress syndrome (table 1) (Bernard et al. 1994). In addition,
the American-European Consensus Conference definition recognised that ALI is
associated with a precipitating or predisposing event. These events can be separated into
those associated with direct injury to the lung, such as pneumonia, and those causing
indirect injury in the setting of a systemic process, such as sepsis or haemorrhage (table
2).
13
Onset Oxygenation Chest radiograph Pulmonary artery occlusion pressure
ALI Acute paO2/FiO2 ! 300 Bilateral infiltrates ! 18mmHg / no left atrial hypertension
ARDS Acute paO2/FiO2 ! 200 Bilateral infiltrates ! 18mmHg / no left atrial hypertension
Table 1 Definitions of acute lung injury (ALI) & the acute respiratory distress syndrome
(ARDS) from American-European Consensus Conference 1994 (Bernard, Artigas et al. 1994).
paO2 in mmHg.
Direct injury Indirect injury
Pneumonia
Aspiration
Lung contusion
Near drowning
Toxic inhalation
Sepsis
Transfusion-related
Pancreatitis
Severe non-thoracic trauma
Cardiopulmonary bypass
Table 2 Clinical disorders associated with development of ALI & ARDS, as identified by the
American-European Consensus Conference Committee 1994 (Bernard, Artigas et al. 1994).
1.2.2.2 Epidemiology of acute lung injury
Incidence: Accurate estimation of the incidence of ALI has been hindered historically by
lack of uniform diagnostic criteria coupled with the heterogenicity of the causes and
clinical manifestations of the disease (Ware and Matthay 2000). Because of this, the
reported incidence has varied widely from 2 to 75 cases per 100,000 person-years (NIH
1977; Webster et al. 1988; Luhr et al. 1999). However, a large prospective population-
based cohort study using the 1994 consensus definitions recently estimated the age-
adjusted incidence of ALI in the United States as 86 per 100,000 person-years, and of
ARDS as 64 per 100,000 person-years (Rubenfeld et al. 2005). The associated in-

14
hospital mortality in this study was 39%, in keeping with that reported in previous
studies of 40-60% (Ware and Matthay 2000). These data suggest that there are 200,000
cases of ALI each year in the United States, associated with 74,500 deaths.
Outcome: ARDS has a mortality of 30-40% in the UK (Abel et al. 1998). Of the patients
who die, the majority of deaths are attributed to sepsis or multiple organ failure rather
than primary respiratory failure (Montgomery et al. 1985; Wyncoll and Evans 1999). It
is likely, however, that pulmonary dysfunction contributes significantly to these
processes. The respiratory failure associated with ALI often necessitates mechanical
ventilatory support, and the recent therapeutic success of ventilation with low tidal
volumes appears to suggest that in some cases death may be directly related to a
combination of existing and iatrogenic lung injury (ARDSNet 2000).
Patients who survive ALI have been shown to have a reduced health-related quality of
life (Weinert et al. 1997) and persistent functional limitation, largely as a result of
muscle wasting and weakness (Herridge et al. 2003). Although mild restrictive patterns
on lung-function testing, and mild-moderate reductions in carbon monoxide diffusion
capacity have been demonstrated, these abnormalities are usually asymptomatic
(Herridge et al. 2003), and in most patients, pulmonary function returns to normal within
6-12 months (McHugh et al. 1994).
1.2.2.3 Pathogenesis of acute lung injury
The acute phase of ALI, corresponding to the first few days following the onset of
injury, is characterised by the influx of protein-rich oedema fluid and inflammatory cells
into the alveolar air spaces with the formation of characteristic hyaline membranes. This
has been termed the exudative phase of ALI, and arises as a consequence of both
endothelial and epithelial cell injury with increased permeability of the alveolar-
capillary barrier (Pugin et al. 1999; Ware and Matthay 2000; Tomashefski 2003). The
importance of endothelial injury and increased vascular permeability in the development
of pulmonary oedema has been well established, but the most conspicuous structure

15
damaged in the acute phase of ALI appears to be the alveolar epithelium, particularly
ATI cells (Bachofen and Weibel 1982; Berthiaume et al. 1999; Ware and Matthay
2000). The significance of epithelial injury is discussed below (section 1.2.2.5).
The second histopathological stage in the natural history of ALI, known as the
proliferative phase, is defined by the proliferation of ATII cells along alveolar septa and
the onset of epithelial regeneration (Tomashefski 2003). In uncomplicated cases,
denuded basement membrane is reepithelialised with restoration of normal alveolar
architecture and epithelial fluid transport capacity and rapid reabsorption of alveolar
oedema. However, in some patients alveolar oedema persists, mesenchymal cells fill the
alveolar spaces with subsequent organisation and progression to fibrotic lung injury
(Bachofen and Weibel 1982; Ware and Matthay 2000).
1.2.2.3.1 Inflammatory mechanisms in the pathogenesis of acute lung injury
Cytokines: A complex balance of pro- and anti-inflammatory cytokines appears to
initiate and regulate the inflammatory response in acute lung injury (Ware and Matthay
2000; Park et al. 2001). Macrophage migration inhibitory factor (MIF) is released from
the anterior pituitary and alveolar macrophages in response to a range of known
precipitants of lung injury such as trauma and sepsis (Joshi et al. 2000; Lehmann et al.
2001). MIF not only inhibits the anti-inflammatory effects of glucocorticoids (Calandra
et al. 1995), but also stimulates the release of the major neutrophil chemoattractant,
interleukin-8 (IL-8), and the pro-inflammatory mediators TNF-$, interleukin-1 (IL-1)
and interleukin-6 (IL-6) from alveolar macrophages (Donnelly et al. 1997; Abraham et
al. 2000).
Specific inhibitors of the proinflammatory cytokines, including IL-1-receptor antagonist
(IL-1ra), soluble TNF receptor (sTNF-R) and autoantibodies against IL-8 have been
described. In conjunction with the non-specific anti-inflammatory cytokines interleukin-
10 (IL-10) and interleukin-11 (IL-11), these mediators are thought to modulate
inflammatory lung injury, a hypothesis supported by the finding that low concentrations

16
of IL-10 and IL-1ra in the bronchoalveoalr fluid of patients with ARDS appears to be
associated with increased mortality (Donnelly et al. 1996; Pittet et al. 1997; Ware and
Matthay 2000; Park et al. 2001).
The role of the neutrophil: Histological studies have consistently shown a marked
accumulation of polymorphonuclear neutrophils in the acute phase of ALI, suggesting
that neutrophils play an important role in the pathogenesis of lung injury (Bachofen and
Weibel 1982; Abraham et al. 2000; Lee and Downey 2001; Martin 2002). Many animal
models of ALI appear to be neutrophil-dependent, and inhibiting neutrophil recruitment
by blocking IL-8 appears protective against acid-induced lung injury (Folkesson et al.
1995). However whether the neutrophil is the cause or result of lung injury remains
unclear. Some animal models appear neutrophil-independent, and patients with profound
neutropenia may still develop ALI (Laufe et al. 1986; Wiener-Kronish et al. 1991). In
addition, studies using granulocyte colony-stimulating factor (G-CSF) in humans with
severe pneumonia have demonstrated that increasing the number of circulating
neutrophils is not associated with an increase in either the incidence or severity of lung
injury (Nelson et al. 1998).
1.2.2.4 Treatment of acute lung injury
For many years, treatment of ALI was limited to the supportive treatment of organ
failure and treatment of the underlying cause of lung injury. Improvements in this
supportive treatment, rather than the success of a particular therapeutic intervention, are
thought to account for the recently observed decline in mortality from (40-60% to (30-
40% (Abel et al. 1998; Ware and Matthay 2000).
Despite significant advances in the understanding of the pathogenesis of ALI, specific
pharmacological interventions, including surfactant therapy, inhaled nitric oxide,
glucocorticoids and other anti-inflammatory agents, have shown little benefit (Adhikari
et al. 2004). However, a study by the ARDS Network (ARDSNet) comparing
conventional mechanical ventilation with a lung protective strategy of ventilation

17
reported a 22% reduction in mortality in patients ventilated using protective ventilation
(section 1.6.4.1.2) (ARDSNet 2000).
1.2.2.5 The significance of the alveolar epithelium in acute lung injury
The importance of epithelial injury in the development of ALI is now widely recognised
(Bachofen and Weibel 1982; Wiener-Kronish et al. 1991; Berthiaume et al. 1999; Ware
and Matthay 2000). In human studies, acute lung injury is associated with a significant
rise in both plasma and bronchoalveolar lavage fluid levels of the human ATI cell-
specific apical plasma membrane protein, HTI56, and the extent of alveolar epithelial
injury, as determined by the degree of impairment of alveolar fluid clearance, appears to
be an important predictor of outcome (Newman et al. 2000; Ware and Matthay 2001).
Alveolar epithelial recovery appears to be a key feature in surviving ALI, with
restoration of intact epithelial barrier function and normal alveolar fluid transport, being
associated with improved outcome (Matthay and Wiener-Kronish 1990; Ware and
Matthay 2001). However, abnormal alveolar epithelial repair appears to be a key
pathogenic mechanism in the development of fibrosis (Adamson et al. 1988).
Protective lung ventilation is the only therapeutic intervention of any proven benefit in
the treatment of ARDS (ARDSNet 2000), and the knowledge that the alveolar
epithelium plays a central role in the pathogenesis of ventilator-induced lung injury
(section 1.6.4.2), further highlights the central role of the alveolar epithelium in the
pathogenesis and recovery from acute lung injury.
1.2.3 Bacterial pneumonia
1.2.3.1 Definition, aetiology and clinical presentation
Pneumonia is defined as an inflammation of the lung parenchyma. It is typically caused

18
by microbial pathogens, including bacteria, viruses, fungi and parasites. A number of
modifying terms are used to classify pneumonia: acute or chronic dependent on the
duration of symptoms; lobar or bronchopneumonia dependent on chest radiograph
findings; community-acquired or hospital-acquired (also called nosocomial pneumonia)
dependent on the place of acquisition (Weatherall et al. 2003). Ventilator-associated
pneumonia (VAP), is a common form of hospital-acquired pneumonia, and describes
pneumonia occurring in patients who have required mechanical ventilation for at least
48 hours. The common aetiological agents causing pneumonia are listed in tables 3 & 4
(Bartlett et al. 1986; BTS 2001).
Aetiological organism %
Streptococcus pneumoniae
Haemophilus influenzae
Legionella spp
Staphylococcus aureus
Gram negative bacilli
Mycoplasma pneumoniae
Chlamydia psittaci
Influenza A & B
No pathogen isolated
36.0
10.2
0.4
0.8
1.3
1.3
1.3
8.1
45.3
Table 3 Causes of community-acquired pneumonia (CAP) (BTS 2001).
Aetiological organism %
Gram negative bacilli
Anaerobic bacteria
Staphylococcus aureus
Streptococcus pneumoniae
47
35
31
26
Table 4 Causes of hospital-acquired pneumonia (Bartlett, O’Keefe et al. 1986).
The clinical presentation of pneumonia varies according to the aetiological agent, but
typical features include cough and fever with the variable presence of sputum, dyspnoea
and pleuritic chest pain. Haematological investigations typically demonstrate a raised
white cell count and C reactive protein (CRP). Although radiological changes may lag

19
behind the clinical course, a chest radiograph confirming areas of consolidation remains
the most reliable clinical investigation in the diagnosis. Confirmation of the microbial
cause of pneumonia typically depends on sputum culture, blood culture and serology.
However, the aetiological agent remains unidentified in (45% of cases (BTS 1992; BTS
2001; Weatherall et al. 2003).
1.2.3.2 Epidemiology of bacterial pneumonia
Pneumonia is the third leading cause of death worldwide and the leading cause of death
due to infectious disease in industrialised countries (Hippenstiel et al. 2006). The annual
incidence of community acquired pneumonia (CAP) in the UK is thought to be 5-11 per
1000, with (20-40% of these cases requiring hospital admission (Woodhead et al. 1987;
Guest and Morris 1997). The mortality rate varies widely, but appears to be (5-12% in
those requiring hospitalisation, rising to more than 50% in patients requiring admission
to an intensive care unit (ICU) (Woodhead et al. 1985; BTS 1992; BTS and Service
1992; Hirani and Macfarlane 1997). Hospital-acquired pneumonia occurs in up to 10%
of patients admitted to hospital, with ventilator-associated pneumonia having an
incidence of (8-28% and an associated mortality of (24-50% (Chastre and Fagon 2002).
1.2.3.3 Pathogenesis of bacterial pneumonia
In animal models, microbial pathogens trigger an inflammatory response resulting in
both epithelial and endothelial cell injury (Seeger et al. 1990; McElroy et al. 1995;
Clegg et al. 2005). In bacterial pneumonia, the primary inflammatory trigger for this
appears to be the bacterial cell wall, with pepdtidoglycan binding to alveolar
macrophage cell surface receptors initiating a pro-inflammatory cytokine cascade
(McCullers and Tuomanen 2001). This leads to the release of the neutrophil
chemoattractants macrophage inflammatory protein-2 (MIP-2) and IL-8, activation of
the transcription factor nuclear factor kappa B (NF-'B) and release of the pro-

20
inflammatory cytokines TNF-$, IL-1 and IL-6 (Spellerberg et al. 1996; Delclaux and
Azoulay 2003). Direct activation of the alternative complement pathway by bacterial
cell wall components, such as teichoic acid, provides a further chemotactic signal for
leucocytes (Winkelstein and Tomasz 1978).
Disruption of the air-blood barrier results in flooding of the alveolar air space with a
protein-rich exudate containing bacteria, macrophages, neutrophils and erythrocytes. In
areas of confluent injury, this leads to lung consolidation known as red hepatisation
(Kumar et al. 2004). As erythrocytes disintegrate, the influx of leucocytes continues and
bacteria are cleared slowly by inefficient complement-mediated opsonisation and
phagocytosis. The pathological appearance changes from red to grey hepatisation during
this time.
Reduction of inflammation and resolution of pneumonia is dependent upon the
appearance of specific anti-microbial antibodies enabling efficient opsonisation and
phagocytosis by alveolar macrophages. Bacterial clearance is accompanied by
enzymatic and cellular degradation of consolidated alveolar exudate, with restoration of
normal alveolar structure and function (McCullers and Tuomanen 2001; Kumar et al.
2004).
1.2.3.4 The significance of the alveolar epithelium in bacterial pneumonia
Localised at the interface between the environment and the host, the pulmonary
epithelium not only forms a significant mechanical barrier to microbial invasion, but
also appears to play a central role in pathogen recognition and the initiation of the innate
immune response (Hippenstiel et al. 2006).
Transmembrane toll-like receptors (TLRs) (Akira and Takeda 2004; Armstrong et al.
2004), together with a family of cytosolic receptors known as the NLR family (Martinon
and Tschopp 2005), act as pattern recognition receptors (PRRs), detecting a variety of
highly conserved pathogen-associated molecular patterns (PAMPs) (Janeway and
Medzhitov 2002). The recognition of PAMPs by PRRs activates a network of signal

21
transduction pathways, a central aspect of which appears to be the upregulation of NF-
'B with resultant cytokine and chemokine production and neutrophilic inflammation
(Sadikot et al. 2003; Hippenstiel et al. 2006). This pathogen-related activation of airway
epithelial cells results in auto- and paracrine stimulation of further inflammatory
mediators (Cooper et al. 2001) and upregulation of adhesion molecules (Cooper et al.
2001; Neff et al. 2006), complement component C3 (Cortes et al. 2002) and
antimicrobial peptides such as the defensins, cathelicidins and lactoferrin (Bals and
Hiemstra 2004).
Many clinically relevant pathogens secrete cytotoxic virulence factors, such as
pneumolysin (Streptococcus pneumoniae), alpha toxin (Staphylococcus aureus) and the
type III secretion system-associated toxins (Pseudomonas aeruginosa). In combination
with the inflammatory mediators released as part of the host inflammatory response,
these virulence factors lead to significant alveolar epithelial cell injury (Duane et al.
1993; Rubins et al. 1993; McElroy et al. 1995; McElroy et al. 1999). In a model of
Pseudomonas aeruginosa pneumonia, McElroy et al. demonstrated that the alveolar
fluid content of the ATI cell-specific protein RTI40 correlated with both increased
permeability of the air-blood barrier and morphological assessment of epithelial injury,
suggesting that ATI cell injury is a central feature in the pathogenesis of Pseudomonas
aeruginosa pneumonia (McElroy et al. 1995). Significant epithelial injury has also been
demonstrated in models of Streptococcus pneumoniae and Staphylococcus aureus
pneumonia (McElroy et al. 1999; Clegg et al. 2005).
1.2.4 Idiopathic Pulmonary fibrosis
1.2.4.1 Definition
Idiopathic pulmonary fibrosis (IPF), also known as cryptogenic fibrosing alveolitis, is a
progressive and devastating interstitial lung disease. The disorder may be defined as a
clinicopathological syndrome characterised by cough, exertional dyspnoea, bilateral

22
inspiratory crepitations on auscultation, a restrictive defect on pulmonary function
testing, honeycombing on computed tomographic (CT) scan and a histological diagnosis
of usual interstitial pneumonia on lung biopsy (table 5) (ATS 2000; Selman et al. 2004).
Cardinal feature Ancillary features
Heterogeneous involvement
Predilection for subpleural & basilar regions
Bilateral involvement
Fibroblastic foci
Honeycomb cysts
Thickened, distorted alveolar walls
Excessive collagen & extracellular matrix
Scattered inflammatory cells
Bronchiolectasis ± bronchiectasis
Smooth muscle hypertrophy
Hyper- & metaplasia of ATII cells
Mucostasis
Secondary pulmonary hypertensive changes
Absence of granulomas or vasculitis
Absence of microorganisms or minerals
Table 5 Histopathological features of idiopathic pulmonary fibrosis (Selmann, Thannickal et
al. 2004).
1.2.4.2 Epidemiology of idiopathic pulmonary fibrosis
IPF is rare, with a prevalence of 3-20 cases per 100,000 people. The disease occurs
predominantly from middle age onwards, and is more prevalent in males. A familial
variant of the disease has been recognised but, other than the age of onset, is clinically
indistinguishable from non-familial cases.
Following presentation, clinical symptoms worsen over months to years with
progressive fibrosis and destruction of lung parenchyma. Most patients die from
respiratory failure within 3-8 years of the onset of symptoms. Current treatments,
typically using anti-inflammatory or immunosuppressive agents, remain of unproven
benefit and there is no evidence that any therapy alters the natural history of the disease
(Selman et al. 2004).

23
1.2.4.3 Pathogenesis & the role of the alveolar epithelium in IPF
The aetiology and pathogenesis of IPF remain unknown. Early hypotheses focused on an
unidentified insult to the lung initiating a cycle of chronic alveolar inflammation leading
to fibrosis and destruction of normal lung architecture (Crystal et al. 1976). Based upon
this hypothesis, the alveolar epithelial alterations characteristic of IPF - namely, loss of
ATI cells and proliferation of ATII cells - are caused by an unresolved inflammatory
process in the surrounding microenvironment.
More recent hypotheses, however, suggest that IPF, rather than being a chronic
inflammatory disorder, results from an aberrant host response to wound healing
orchestrated by epithelial cell activation and abnormal epithelial-mesenchymal
interactions (Geiser 2003; Selman et al. 2004; Selman and Pardo 2006). Alveolar
epithelial hyperplasia and the presence of several altered cell phenotypes is a striking
feature of IPF, suggesting that alveolar reepithelialisation is severely disturbed (section
1.3.4). This is thought to disrupt normal epithelial-mesenchymal interactions generating
a profibrotic environment leading to the migration, proliferation and activation of
mesenchymal cells with the formation of active fibroblastic/myofibroblastic foci and
excessive accumulation of extracellular matrix (Selman and Pardo 2006).

24
1.3 Alveolar epithelial repair
1.3.1 Historical perspective of cell cycle kinetics in the alveolar epithelium
The concept of the ATII cell as a stem cell was first raised by Kapanci and colleagues
following study of the histological changes in the lungs of monkeys exposed to 100%
oxygen (Kapanci et al. 1969). These studies demonstrated that ATI epithelium,
destroyed after 4 days exposure to 100% oxygen, was initially replaced by cuboidal
ATII epithelium followed by the recovery of histologically normal lung upon re-
exposure of the animals to normal room air.
However, it is studies by Evans and colleagues (Evans et al. 1972; Evans et al. 1975),
combining the use of thymidine labelling and electron microscopy, which provide the
foundations for the current understanding of the response of the alveolar epithelium to
injury (Uhal 1997). They examined the ultrastructural changes occurring in the alveolar
epithelium following 48 h exposure to the oxidant gas nitrogen dioxide. The exposure
was followed immediately by an intraperitoneal injection of tritiated thymidine (3HT) to
achieve a pulse of label available for incorporation into newly synthesised DNA during
the S phase of the cell cycle. In lung sections obtained 1 h after labelling, virtually all
labelled cells were ATII cells. By 24 h after labelling, labelled ATI cells began to
appear, and, by 48 h the percentage of ATI cells had increased 10-fold compared to that
observed at 1 h. Over the same time period, the percentage of ATII cells had fallen to a
third of the original. In addition, a population of 3HT labelled ‘transitional’ cells with
morphological characteristics of both ATII and ATI cells could be identified. These data
suggested that alveolar epithelial repair following injury was achieved by ATII cell
division and the transition of ATII cells into ATI cells.
Similar studies by Adamson and colleagues (Adamson and Bowden 1974; Adamson and
Bowden 1975), in which mice were exposed to 90% oxygen for 6 days followed by
recovery in room air, examined the cell kinetics of alveolar epithelial repair using a
combination of 3HT labelling and colchicine-induced mitotic arrest. They found that

25
alveolar epithelial injury appeared to be limited to ATI cells, with relative sparing of
ATII cells. 3HT labelling of the alveolar epithelium became maximal 2 days following
termination of exposure to 90% oxygen, at which time-point mitoses were observed in
the ATII cell population. At this same time-point, neither 3HT labelling nor mitoses were
found in ATI cells, supporting the hypothesis that proliferation during recovery was
confined to the ATII cell population. By 3 days after exposure, labelling was observed in
the initially unlabelled ATI cell population with a concomitant decrease in the number of
labelled ATII cells. Analysis of bronchoalveolar lavage fluid (BALF) showed that ATII
cells were not being sloughed into the airspaces. Together with the observation of
‘intermediate’ cells exhibiting morphological characteristics of both ATII and ATI cells,
these data supported the notion of a direct transition of ATII cells to ATI cells during
alveolar epithelial repair.
Further evidence supporting the hypothesis that ATI cells must be derived from ATII
cells was the demonstration that ATI cells appear incapable of cell division, with
Adamson and colleagues unable to find evidence of ATI cell mitoses despite the
presence of abundant mitoses in neighbouring ATII cells following oxygen-induced
injury (Adamson and Bowden 1974; Adamson and Bowden 1975).
Support for this concept has been provided by in vitro studies of primary ATII cell
isolates, in which the ATII-like morphology of cells observed immediately after
isolation is replaced over a period of several days in culture by cells with an ATI-like
morphology (Kikkawa and Yoneda 1974). Immunohistochemistry has been used to
substantiate this observation, showing that primary ATII cells lose the expression of
ATII-selective proteins and gain the expression of ATI-selective proteins with time in
culture (Dobbs et al. 1985; Dobbs et al. 1988; Fehrenbach 2001).
1.3.2 Current model of alveolar epithelial repair
Based on these historical studies, it has been widely accepted for many years that an
important function of the ATII cell compartment is re-epithelialisation of denuded

26
basement membrane following alveolar injury, with damaged ATI cells being replaced
by the proliferation of ATII cells and the subsequent differentiation of a subpopulation
of daughter cells into ATI cells (figure 1A) (Uhal 1997).
However, more recent studies have refined the role of ATII cells in alveolar epithelial
repair (section 1.3.3.1), and also raised alternative mechanisms of ATI cell repair
(figure 1B) (McElroy and Kasper 2004).
It appears injured ATI cells may also be replaced by non-ciliated bronchiolar epithelial
progenitor cells and bone marrow cells (section 1.3.3.2). The concept that ATI cells are
terminally differentiated has also been challenged by studies examining the role of
mechanical load in the foetal lung, which suggest that ATI cells have the potential to
transdifferentiate into ATII cells (Flecknoe et al. 2002). The role this process plays in
epithelial repair remains unclear, and it is not known whether ATI cells are capable of
replacing ATII cells in the adult lung. Whilst the transformation of ATII cells into ATI
cells may be reversible, re-entry into the cell cycle appears to require reversion to the
ATII phenotype (Uhal 1997).
1.3.3 Stem cells responsible for alveolar epithelial repair
1.3.3.1 Alveolar stem cells
It is generally accepted that during homeostasis, and in response to injury, ATII cells
serve as progenitor cells within the alveolar compartment (Otto 2002). However, there
may be subpopulations of ATII cells with different susceptibilities to injury and different
proliferative capabilities following injury. Reddy and colleagues have suggested that
ATII cells isolated from hyperoxia-treated rat lungs can be segregated based on their
levels of E-cadherin expression (Reddy et al. 2004). E-cadherin negative cells are
damage resistant, proliferative and exhibit high levels of telomerase activity, suggesting
that they may represent a transiently amplifying progenitor subpopulation of the ATII

27
cell compartment (Reddy et al. 2004).
1.3.3.2 Extra-alveolar stem cells
Following severe injury to the alveolus, it is possible that repair mechanisms may
involve progenitor cells originating from outwith the alveolus (Otto 2002).
Tissue-specific stem cells: Examination of distal airway epithelium in mice following
bleomycin-induced lung injury revealed a subpopulation of cells containing Clara cell
specific protein (CC10) mRNA in alveolar-like structures (Daly et al. 1998). This was
thought to be the result of Clara cell migration or outpocketing, and the inference drawn
by Daly and colleagues was that alveolar epithelial progenitors could arise from
bronchiolar epithelium. This hypothesis is supported by the finding of pollutant-resistant
Clara cell secretory protein (CCSP)-expressing cells localised to the bronchoalveolar
duct junction that retain multipotent differentiation potential (Giangreco et al. 2002;
Reynolds et al. 2004).
Non-tissue-specific stem cells: Recent studies in experimental models and in humans
following lung transplant suggest that circulating bone marrow-derived cells may
participate in alveolar epithelial repair. Engraftment by circulating pluripotent cells
seems to occur in the lung parenchyma following injury (Ishizawa et al. 2004; Yamada
et al. 2004), and studies of bone marrow graft recipients have demonstrated numerous
donor-derived epithelial and endothelial cells in the lungs (Kleeberger et al. 2003; Suratt
et al. 2003).
However, the contribution of circulating bone marrow-derived stem cells to epithelial
repair remains controversial. Using a lineage-specific reporter system based on
transgenic mice that express green fluoresecent protein (GFP) only in ATII cells
(surfactant protein-C (SP-C)-GFP), Kotton and colleagues demonstrated that there was
no detectable reconstitution of alveolar epithelial cells by unfractionated bone marrow or
purified haematopoietic stem cells (Kotton et al. 2005).

28
1.3.4 Alveolar epithelial repair and lung fibrosis
Abnormal alveolar epithelial repair following injury appears to be a key pathogenic
mechanism in the development of fibrosis (Adamson et al. 1988). Normal alveolar
epithelial repair, on the other hand, appears to inhibit the development of pulmonary
fibrosis. Keratinocyte growth factor (KGF), a fibroblast-derived mitogen, has been
shown to stimulate the proliferation of ATII cells in normal rat lungs (Ulich et al. 1994),
and pre-treatment with KGF prior to bleomycin-induced injury has been shown to
facilitate epithelial repair and prevent fibrosis (Deterding et al. 1997; Guo et al. 1998).
Facilitation of epithelial repair by inhibiting ATII cell apoptosis has also been shown to
inhibit fibrosis in rats following bleomycin-induced injury (Wang et al. 2000).
In a comparison of models of normal repair (following hyperoxia-induced injury) and
abnormal repair with fibrosis (following bleomycin-induced injury) in the mouse,
Adamson and colleagues observed that, following ATI cell necrosis, normal repair was
associated with a significant, but short-lived, peak in ATII cell division. This was not
associated with fibrosis. However, abnormal repair was associated with an extended
period of ATII proliferation accompanied by fibroblast proliferation and collagen
deposition (Adamson et al. 1988). The number of epithelial-interstitial cell contacts in
these experiments appeared to be inversely related to the rate of ATII cell division,
suggesting that disruption of normal epithelial-fibroblast interactions may be a key event
in the progression to abnormal epithelial repair with fibrosis (Adamson et al. 1990).
The concept that abnormal repair is orchestrated by aberrant epithelial-mesenchymal
interactions is now widely accepted (Selman and Pardo 2002). Epithelial and
mesenchymal cells, functioning as a notional 'epithelial-mesenchymal trophic unit’,
signal each other dynamically and bidirectionally either by the release of soluble
mediators in a paracrine fashion or by direct cell-cell contact through spanning apertures
in the basement membrane (Evans et al. 1999; Fang 2000; Sirianni et al. 2003).
Epithelial cells are known to release transforming growth factor TGF-& and platelet-
derived growth factor (PDGF), both of which regulate fibroblast migration, proliferation,
matrix deposition and apoptosis. In turn, fibroblasts influence epithelial cell function by

29
releasing epidermal growth factor (EGF), keratinocyte growth factor (KGF) and
hepatocyte growth factor (HGF), all of which regulate ATII migration, proliferation,
differentiation and deposition of matrix proteins (Fang 2000).
It seems clear that normal repair following lung injury requires ordered and timely
reepithelialisation to restore alveolar epithelial integrity and normalise epithelial-
mesenchymal interactions. Failure of these processes appears to generate profibrotic
microenvironments within the lung leading to the migration, proliferation and activation
of mesenchymal cells and the formation of active fibroblastic foci (Bitterman 1992;
Fang 2000; Selman and Pardo 2006).

30
1.4 The use of cell-selective markers in lung injury and repair
Studies of changes in gas exchange, lung mechanics, pulmonary oedema formation,
alveolar protein leak and alveolar fluid clearance provide important information
regarding the functional consequences of lung injury. However, these studies provide
little information of disease process at the cellular level (Parker and Townsley 2004).
Light microscopy can provide qualitative evidence of lung injury, but accurate cellular
identification, particularly of attenuated ATI cells, is difficult at the level of resolution of
the light microscope. Because of this, most historical studies of alveolar epithelial injury
have relied on the use of electron microscopy (Evans et al. 1972; Evans et al. 1975;
McElroy and Kasper 2004).
Whilst use of the electron microscope to identify characteristic ultrastructural markers
does allow accurate identification of alveolar epithelial cells in both normal and
repairing lungs (Evans et al. 1975), it is only practicable to study very small areas of
lung using this approach, making representative sampling and accurate quantification
difficult. Given the heterogeneity of many forms of lung injury (Maunder et al. 1986;
Rouby et al. 2003), this presents obvious problems, and combining adequate numerical
analysis with precise cellular identification has presented significant difficulty in
studying the behaviour of the alveolar epithelium (Adamson and Bowden 1974).
1.4.1 Biomarkers and immunotargeting
The use of monoclonal antibodies as biomarkers to identify antigens specific to cells of a
particular lineage or stage of differentiation, has been applied to a variety of cell types
and organ systems including the alveolar epithelium (Funkhouser and Peterson 1989;
Williams and Dobbs 1990). Funkhouser and colleagues described this approach as
‘immunotargeting’, and when combined with immunofluoresence and laser scanning
confocal microscopy, this provides a powerful tool for characterising the alveolar

31
epithelium both in control lungs and in repairing lungs following injury, during lung
development and in primary cell culture.
1.4.2 Alveolar epithelial cell-selective markers
A number of proteins expressed by only one alveolar epithelial cell type have been
identified (Appendix I). These have been described as either cell-specific, when
expressed by the target cell only in the lung, or cell-selective, when expressed by the
target cell and other lung cells but not other alveolar epithelial cells (McElroy and
Kasper 2004). The cell-specific and cell-selective proteins used in the studies in this
thesis are described below.
1.4.2.1 ATI cell-specific and cell-selective proteins
Rat Type I 40-kDa protein (RTI40) or Type I cell alpha protein (T1"): RTI40 is an apical
membrane sialoglycoprotein expressed specifically by ATI cells in the lung. It is also
expressed in the choroid plexus and ciliary epithelium (Williams et al. 1996). The
function of RTI40 is unknown, but mice with targeted mutations of the T1$ locus die at
birth with respiratory failure, and histological examination of the lungs of these animals
demonstrate reduced numbers of ATI cells and defective alveolar formation (Ramirez et
al. 2003). Over expression of T1$ in endothelial cells and the homologous protein
PA2.26 in keratinocytes promotes the formation of long thin cell extensions and
increases migration rate in wounding experiments (Schacht et al. 2003). Based on these
data it has been speculated that RTI40 may regulate distal airway cell proliferation during
lung development and, in the adult lung, may be important in initiating and maintaining
the characteristic extended and flattened ATI phenotype (Williams 2003; McElroy and
Kasper 2004).
Aquaporin 5 (AQP5): AQP5 is a water channel protein located in the apical membrane
of ATI cells and bronchiolar epithelial cells in the lung (King and Agre 1996; Nielsen et

32
al. 1997). Studies using AQP5-deficient mice demonstrate that AQP5 provides the
principal pathway for the passive osmotically-driven movement of water across the
apical surface of the alveolar epithelium (Ma et al. 2000), although deletion appears to
have no significant effect on active alveolar fluid clearance out of the airspaces or the
accumulation of extravascular fluid arising from increases in hydrostatic pressure (Ma et
al. 2000).
MMC6 Antigen: MMC6 is a monoclonal antibody that was generated as a product of
work to develop novel antibodies recognising antigens expressed on the surface of rat
ATII cells (Boylan et al. 2001). The function of the MMC6 antigen is unknown, but
initial studies suggest that it is a lung-specific novel apical membrane protein expressed
by ATI cells (unpublished data McElroy & Franklin). Studies of the immunoselectivity
of MMC6 both in vitro and ex vivo form part of the work described in this thesis
(chapter 3). These studies confirm that the MMC6 antigen is ATI-specific.
1.4.2.2 ATII cell-specific and cell-selective proteins
Rat Type II 70-kDa protein (RTII70): Anti-RTII70 is a monoclonal antibody recognising
the rat ATII cell-specific apical membrane protein RTII70 (Dobbs et al. 1997). The
function of RTII70 is unknown.
MMC4 antigen or aminopeptidase N (APN) or p146: Aminopeptidase N (APN),
previously known as p146, is an apical membrane protein expressed in ATII and Clara
cells in the lung (Funkhouser et al. 1991). Boylan and colleagues described a novel
monoclonal antibody, MMC4, which selectively recognised an integral membrane
protein on the apical surface of rat ATII and Clara cells in the lung, proximal tubular
epithelial cells in the kidney and epithelial cells in the small intestine. Subsequent
characterisation has shown that the MMC4 antigen is APN (Franklin et al. 2004). The
function of MMC4 or APN in the lung remains unclear, although its main function in
other organs appears to be post-translational modification of proteins.
Pro-surfactant protein C (pro-SP-C): Surfactant protein-C (SP-C) is a lung specific

33
hydrophobic protein accounting for (4% of the protein content of pulmonary surfactant.
Beyond its role in reducing alveolar surface tension however, the precise role of
surfactant protein-C remains unknown, and difficulty purifying the protein together with
its tendency to denature and aggregate have made studying the protein difficult (Ikegami
and Jobe 1998). Alveolar SP-C (3.5 kD) is secreted by rat ATII cells, both in vivo and in
vitro, following synthesis and proteolytic cleavage of the precursor molecule pro-SP-C
(21 kD) (Beers and Fisher 1992; Beers and Lomax 1995). Anti-pro-SP-C is a cell-
specific polyclonal antibody recognising intracellular pro-SP-C within ATII cells (Beers
et al. 1992).
1.4.3 Alveolar epithelial cell-selective markers in the investigation of lung injury
1.4.3.1 Cell-selective markers in quantifying epithelial cell numbers and/or injury
ATI markers: Ultrastructural studies in injured lungs have confirmed that necrotic ATI
cells are sloughed from the basement membrane into the airspaces, and several studies
suggest that the quantity of ATI cell-specific protein recovered from bronchoalveolar
lavage fluid (BALF) can be used to quantify the extent of ATI cell necrosis following
injury (McElroy and Kasper 2004). McElroy and colleagues have shown that in a model
of severe lung injury caused by Pseudomonas aeruginosa, BALF RTI40 levels were
elevated 80-fold above controls. This correlated with widespread ATI cell necrosis on
morphological analysis by EM and complete abolition of alveolar fluid clearance, a
functional marker of epithelial integrity (McElroy et al. 1995). In contrast, in a model of
focal ATI cell injury using nitrogen dioxide, BALF RTI40 levels were elevated only 2-
fold above controls and alveolar fluid clearance was stimulated (McElroy et al. 1997).
Plasma levels of RTI40 have also been shown to correlate with the degree of alveolar
epithelial injury following acid-induced lung injury (Frank et al. 2002).
HTI56 is a human ATI-specific integral membrane protein that has been shown to be
significantly elevated in the alveolar oedema fluid of patients with pulmonary oedema

34
associated with epithelial cell injury (ARDS), but not in patients with pulmonary
oedema occurring in the absence of epithelial injury (hydrostatic pulmonary oedema).
This suggests that HTI56 could be used as a biochemical marker of ATI injury in humans
(Newman et al. 2000).
ATII markers: The use of ATII-selective surfactant proteins as markers of cell injury has
been particularly studied in the context of acute lung injury (Hermans and Bernard
1999). Both SP-A and SP-B BALF levels have been shown to decrease in patients with
ARDS (Gregory et al. 1991; Gunther et al. 1996), and in the case of SP-A, in those at
risk of developing ARDS (Gregory et al. 1991). By contrast no alteration was observed
in BALF levels of SP-A and SP-B in patients with cardiogenic pulmonary oedema
(Gunther et al. 1996). In traumatic lung injury, a transient decrease in BALF SP-A has
been demonstrated, with levels returning to normal over a time course dependent on the
severity of injury (Pison et al. 1992). In addition to the reduction of SP-A and SP-B in
BALF suggestive of ATII cell damage, both SP-A and SP-B levels have been shown to
increase in the serum of patients with ARDS (Doyle et al. 1995; Doyle et al. 1997). The
elevation of SP-A and SP-B in the serum appears to correlate negatively with both
oxygenation and clinical outcome, and is thought to reflect disruption of the air-blood
barrier (Doyle et al. 1997).
KL-6 is a human circulating high molecular weight glycoprotein. The gene for KL-6 has
been classified as belonging to the MUC-1 gene family which encodes membrane-
associated mucins (Hermans and Bernard 1999). In the lung, KL-6 is ATII cell-selective
(Kohno et al. 1988). KL-6 is elevated in both sera and BALF in patients with idiopathic
pulmonary fibrosis and pulmonary alveolar proteinosis and this elevation appears to
correlate with the clinical activity of disease (Kobayashi and Kitamura 1995). ATII cell
hyperplasia is one of the major histopathological findings in these conditions and
immunohistochemical studies suggest that the origin of the elevated levels KL-6 is
proliferating ATII cells (Takahashi et al. 1998).

35
1.4.3.2 Specific regulation of cell-selective markers in lung injury
Changes in levels of cell-selective proteins may not simply reflect cell damage or a
change in cell number, but rather specific regulation.
Hyperoxia has frequently been used as a model of lung injury, and is known to cause
ATI damage (Crapo 1986). However, Cao and colleagues investigating the levels of the
ATI-selective proteins in the lung following hyperoxic exposure found that both RTI40
and AQP5 levels were significantly increased despite ATI cell injury, suggesting
upregulation of these proteins by ATI cells in response to hyperoxic stress (Cao et al.
2003).
In contrast, in a viral model of pneumonia, Towne and colleagues demonstrated that
levels of AQP5 and its mRNA transcript were decreased in the absence of significant
ATI cell necrosis, suggesting downregulation of AQP5 gene expression following viral
pneumonia (Towne et al. 2000). AQP5 also appears to be downregulated following
lipopolysaccharide (LPS)-induced lung injury (Jiao et al. 2002).
ICAM-1 is ATI cell selective in the lung, but has been shown to be expressed by ATII
cells in culture as they become ATI-like (Christensen et al. 1993). ICAM-1 expression
also dramatically increases in ATII cells following both pneumonia and hyperoxia-
induced lung injury, and it has been suggested that this upregulation of ICAM-1 by ATII
cells in-vivo could represent an early differentiation event (McElroy and Kasper 2004).
p172 is a rat ATII- and Clara cell-selective apical membrane protein recognised by the
monoclonal antibody 3F9. Although the function of p172 is unknown, Girod and
colleagues have demonstrated that this protein is upregulated just prior to birth and
following hyperoxia-induced injury in the adult lung suggesting an adaptive function
(Girod et al. 1996).
1.4.3.3 Cell-selective markers and the phenotype of repairing epithelium
Following injury to the lung, the alveolar wall is populated by proliferating ATII cells,

36
ATI cells and a population of alveolar epithelial intermediate cells (McElroy and Kasper
2004). Although little is known about the proteins expressed by these cells, the use of
cell-selective proteins has been used in an attempt to quantify the relative numbers of the
different cell populations and characterise their interactions at sites of active injury
repair.
Using antibodies to the ATII selective SP-D and the ATI-selective lectin lycoperiscon
esculentum agglutinin (LEL), Fehrenbach and colleagues studied the epithelial
remodelling that occurs following (KGF)-induced ATII cell hyperplasia. They found
that the number of ATII cells was maximal at 2-3 days following KGF instillation, with
ATII cells covering (45% of the alveolar surface compared to (4% in control lungs. By
day 3 following KGF instillation, a population of intermediate cells expressing both SP-
D and LEL was apparent. These intermediate cells increased in number to cover 7% of
the alveolar surface by day 7 post-KGF instillation, compared to (0.1-0.8% in control
lungs, at which time alveolar architecture was almost normal with the majority of the
alveolar surface covered by ATI cells. Alveolar epithelial cell apoptosis was increased
from day 2, and appeared maximal at 7 days post-KGF instillation. These data suggest
that the epithelial remodelling following KGF-induced ATII cell hyperplasia was
achieved in vivo by both the differentiation of hyperplastic ATII cells into ATI cells and
ATII cell apoptosis (Fehrenbach et al. 1999).
Examination of repairing alveolar epithelium following Staphylococcus aureus-induced
lung injury by Clegg and colleagues, revealed that at day 3 post-inoculation the alveolar
wall was thickened secondary to ATII cell hyperplasia with a 5-fold increase in the
fractional surface area of the alveolar wall expressing the ATII-selective proteins RTII70
and MMC4 and a decrease in the area expressing the ATI-selective RTI40. S.aureus
treated lungs also contained RTI40/MMC4/RTII70 and RTI40/MMC4 coexpressing cells
not observed in control lungs. These cells were thought to be intermediates in the
transition of ATII cells into ATI cells (Clegg et al. 2005).

37
1.5 Hyperoxia and the alveolar epithelium
1.5.1 Oxygen homeostasis
The sequential reduction of molecular oxygen within the mitochondria during oxidative
phosphorylation generates a group of molecules and free radicals known collectively as
reactive oxygen species (ROS). ROS include the superoxide anion (O2-.), hydrogen
peroxide (H2O2) and the hydroxyl radical (OH.). ROS may react with other radicals
including nitric oxide (NO.) to generate further potent oxidants, such as peroxynitrite
(ONOO-). The products derived from NO
. are known collectively as reactive nitrogen
species (RNS). Small fluctuations in the steady-state concentrations of ROS appear to
play a role in intracellular signalling (Suzuki et al. 1997), but large increases lead to free
radical chain reactions that indiscriminately target cellular proteins, lipids,
polysaccharides and DNA, often leading to cell death (Thannickal and Fanburg 2000;
Turrens 2003).
All aerobic organisms face the threat of oxidative injury and have developed antioxidant
systems and gene-regulated adaptive responses to ensure oxygen homeostasis (Haddad
2002). The term ‘oxidative stress’ is frequently used to describe the deleterious
processes resulting from imbalances between ROS/RNS generation and these defence
mechanisms (Turrens 2003).
The principal antioxidant systems include the superoxide dismutases (SOD), a family of
metalloenzymes which enzymatically convert the superoxide anion to hydrogen
peroxide, and glutathione (GSH), a ubiquitous essential tripeptide (L-%-glutamyl-L-
cysteine-glycine) able to act as a substrate in the reduction of many free radicals and
ROS. Glutathione-associated metabolism in particular, provides a crucial equilibrium
interface between oxidative stress and the adaptive responses arising from changes in
gene expression (Haddad 2002; Turrens 2003).
Variations in the partial pressure of oxygen appear to regulate the pattern of gene

38
expression within cells primarily through the activation of the redox-sensitive
transcription factors hypoxia-inducible factor-1$ (HIF-1$) and nuclear factor-'B (NF-
'B). These are differentially regulated during oxidative stress, with HIF-1$ activity
increased under hypoxic conditions and NF-'B under hyperoxic conditions (D'Angio
and Finkelstein 2000; Haddad 2002; Michiels et al. 2002).
1.5.2 Hyperoxia
Hyperoxia can be defined as exposure of body tissues to higher than normal partial
pressures of oxygen. Although the brain and the preterm retina are susceptible to
hyperoxic injury, it is the lung which is normally exposed to the highest concentrations
of oxygen and therefore at greatest risk of oxidative injury.
The percentage of oxygen in air is a relatively constant 21%, and at normal atmospheric
pressure, the partial pressure of oxygen (pO2) in dry inspired air is therefore (21 kPa. As
air is transported into the lungs, through the conducting airways and into the alveoli, the
pO2 falls as a consequence of humidification of inspired gas, the uptake of O2 and the
admixture of carbon dioxide. By the time the inspired gas reaches the alveolus, the
normal alveolar partial pressure of oxygen (normoxia) is (13-14 kPa (West 2004).
Relative hyperoxia occurs in the neonate during the rapid transition from an intrauterine
pO2 of (3-4 kPa to an alveolar pO2 of (13-14 kPa following birth. Key developmental
changes in late gestation prepare for this ontogenic event by increasing antioxidant
defences in the lung, and the absence of these protective changes is thought to explain
the relative sensitivity of the preterm lung to oxidant injury and the development of
bronchopulmonary dysplasia (BPD) (O'Donovan and Fernandes 2004).
In adults, hyperoxia is invariably a consequence of the clinical use of therapeutic oxygen
to prevent hypoxaemia, and in severe respiratory failure, the prolonged use of high
fractions of inspired oxygen can result in an alveolar pO2 >90 kPa, with the alveolar
epithelium exposed to significant hyperoxia and oxidative stress as a result.

39
1.5.3 Hyperoxia-induced lung injury
Continuous exposure of normal adult rats to 100% oxygen causes characteristic
progressive lung injury, respiratory failure and eventually death (Crapo 1986; Clerch
and Massaro 1993). Atelectasis occurs within the first few hours of exposure due to
absence of nitrogen within the alveoli, and although there is little clinical evidence of
pulmonary oedema or inflammatory cell influx within the first 48 h, significant alveolar
protein leak has been demonstrated in the human lung within 24 h of exposure (Davis et
al. 1983). Between 48 and 60 h of exposure, pulmonary oedema and pleural effusions
develop, with animals beginning to die at 60 h. By 72 h, the majority of animals are dead
(Clerch and Massaro 1993). Examination of the lungs in these animals reveals a pattern
of injury similar to ARDS, with endothelial damage, extensive ATI cell necrosis,
alveolar-capillary oedema, lung protein leak, neutrophil infiltration and hyaline
membrane formation (Crapo 1986; Fracica et al. 1991).
Hyperoxia-induced lung injury appears to be mediated by a combination of the cytotoxic
effects of ROS and activation of redox-sensitive transcription factors, most notably NF-
'B, with subsequent upregulation of stress-response genes and changes in cell fate
signalling pathways. Upregulation of the pro-inflammatory cytokines TNF-$, IL-1&, IL-
6 and IL-8 occurs as part of this response, reinforcing the concept that there is a link
between oxidative stress and the evolution of an inflammatory response in the lung
(D'Angio and Finkelstein 2000; Haddad 2002; Michiels et al. 2002; Sue et al. 2004).
The rate of mitochondrial ROS generation increases linearly with oxygen concentration.
However, the rate of release of ROS from mitochondria appears to be biphasic,
increasing dramatically when cells are exposed to concentrations of oxygen exceeding
60% (Turrens 2003). The slower release of ROS at lower pO2 suggests that antioxidant
defences are able to compensate for moderate hyperoxia, but become overwhelmed at
higher pO2. This concept is supported by the finding that pre-exposure to 85% O2
confers resistance to hyperoxia-induced injury through an adaptive increase in lung

40
antioxidant content (Haddad 2002), and may explain the observation that prolonged
exposure of normal lungs to oxygen concentrations )60% appears to be injurious, whilst
concentrations *50% do not (Hayatdavoudi et al. 1981; Crapo et al. 1983).
1.5.3.1 The effects of hyperoxia on the alveolar epithelium
The alveolar surface is a key target for oxidant injury, and alveolar epithelial injury is a
characteristic feature of hyperoxia-induced lung injury (Crapo et al. 1980; Crapo 1986).
Continuous exposure to 100% O2 has been shown to cause complete destruction of the
alveolar epithelium in the lungs of normal monkeys (Kapanci et al. 1969; Crapo 1986),
and sublethal (<85%) exposure to cause significant ATI cell necrosis in rodents
(Adamson and Bowden 1974; Crapo et al. 1980).
1.5.3.1.1 Hyperoxia and ATII cell proliferation
Repair of damaged epithelium following hyperoxia-induced injury is dependent on the
initiation of ATII cell proliferation and the differentiation of ATII cells into ATI cells
(Adamson and Bowden 1974; Thet et al. 1986). ATII cell proliferation has been clearly
demonstrated in animals exposed to 100% O2 and then recovered in room air (Adamson
and Bowden 1974; Thet et al. 1986), and stimulation of ATII cell proliferation by the
ATII cell mitogen KGF has been shown to reduce both mortality and severity of lung
injury following hyperoxic exposure in rodents (Panos et al. 1995; Barazzone et al.
1999). However, exposure to 100% O2 itself appears to inhibit ATII proliferation (Crapo
1986).
The mechanisms regulating ATII cell proliferation during and following hyperoxia have
been difficult to study in vitro, because isolated primary ATII cells only proliferate to a
limited degree and rapidly lose their differentiated characteristics in culture (Dobbs
1990). However, in the transformed (Simian virus) SV40 ATII cell line, hyperoxia has
been shown to inhibit proliferation by post-transcriptional regulation of the late cell

41
cycle genes histone and thymidine kinase and to induce G1 cell cycle arrest through
transforming growth factor-& (TGF-&) signalling and increased expression of p21
(Clement et al. 1992; Corroyer et al. 1996). Further studies in mice have demonstrated
that hyperoxia causes nuclear DNA damage and increases the alveolar epithelial
expression of both p21 and p53 in vivo, supporting the hypothesis that hyperoxia inhibits
ATII cell proliferation through cell cycle checkpoint activation (O'Reilly et al. 1998;
O'Reilly et al. 1998).
Transcriptional regulation of the insulin growth factor (IGF) system also appears to play
a role in the arrest of ATII cell proliferation induced by hyperoxia. IGF-I, IGF-II and
IGF-IR have been shown to be upregulated by hyperoxia in vivo (Narasaraju et al.
2006), and in vitro the expression of IGF-II, IGF-IIR and IGF-binding protein-2
(IGFBP-2) increases in cells whose proliferation has been arrested by hyperoxia, falling
rapidly to control levels when cells are allowed to resume proliferation in normoxia
(Cazals et al. 1994).
1.5.3.1.2 Hyperoxia-induced alveolar epithelial apoptosis and necrosis
Alveolar epithelial cell injury is an important feature of hyperoxia-induced lung injury,
and although necrosis is known to occur following hyperoxic exposure, apoptosis
appears to be the principal mode of cell death in vivo (Kazzaz et al. 1996; Barazzone et
al. 1998).
Several studies suggest that the differential expression of signalling factors determining
cell fate in the alveolar epithelium is redox-sensitive (Haddad and Land 2000; Ward et
al. 2000; Buccellato et al. 2004), and that ROS act as (pro-apoptotic) upstream signalling
molecules in determining this fate (Lee and Choi 2003).
In support of this hypothesis, activation of the pro-apoptotic Bcl-associated x protein
(Bax) and expression of the pro-apoptotic cell cycle regulator p53 have both been shown
to increase in alveolar epithelial cells in parallel with increasing pO2 (Haddad and Land
2000), and hyperoxia-induced Bax activation has been shown to require the generation

42
of ROS (Buccellato et al. 2004). In a study using IL-6 over-expressing transgenic mice,
Ward and colleagues highlighted the significance of these findings by demonstrating that
IL-6 markedly diminished hyperoxia-induced lung injury and cell death and this
protective effect was associated with increased expression of the anti-apoptotic Bcl-2 but
not the pro-apoptotic Bax (Ward et al. 2000).
The pattern of hyperoxia-induced cell death in vitro is less clear. ATII cells isolated
from the lungs of rats exposed to sublethal hyperoxia prior to isolation demonstrate
DNA damage and increased levels of the pro-apoptotic proteins p21, p53 and Bax
(Buckley et al. 1998). However, this may simply reflect the fact that sublethal exposure
of ATII cells in vivo is a pro-apoptotic stimulus, damaging the cells sufficiently to result
in apoptosis in ex vivo culture. Other in vitro studies suggest that hyperoxia causes
necrosis or a cellular death pathway sharing features of both apoptosis and necrosis
(Kazzaz et al. 1996; Wang et al. 2003; De Paepe et al. 2005).
Interestingly, it appears that the alveolar epithelium may harbour a subpopulation of
cells resistant to hyperoxia-induced cell injury. In a study examining the phenotype of
ATII cells isolated from hyperoxia-treated rats, Reddy and colleagues found that the
isolated ATII cell population could be functionally segregated based on their expression
of E-cadherin (Reddy et al. 2004). The E-cadherin positive subpopulation contained
damaged cells and was non-proliferative with low levels of telomerase activity, whilst in
contrast, the E-cadherin negative subpopulation was undamaged, proliferative and
expressed high levels of telomerase activity. These data suggest that the ATII cell
population isolated from hyperoxia-treated rats is heterogeneous, with E-cadherin
expressing cells more sensitive to hyperoxia-induced injury than cells with low levels of
E-cadherin expression. The functional significance of these findings remains unclear,
but Reddy and colleagues have speculated that the E-cadherin negative subpopulation
may be the source of the transiently amplifying cells responsible for epithelial repair
following injury.

43
1.6 Mechanical forces and the alveolar epithelium
The lung is a dynamic organ that is subjected to mechanical forces throughout
development and adult life, and mechanical deformation appears to play an important
role in regulating both normal and pathological responses of the lung. The alveolar
epithelium appears to play a key role in regulating these responses (Riley et al. 1990;
Wirtz and Dobbs 2000; Edwards 2001).
1.6.1 Quantification of mechanical forces in situ
Several approaches have been used in an attempt to quantify the mechanical
deformations experienced by alveolar epithelial cells in situ. These include measurement
of changes in the surface area of the air-tissue interface using alveolar three-dimensional
reconstruction in serially sectioned rat lungs (Mercer et al. 1987) and measurement of
changes in epithelial basement membrane surface area (EBMSA) at different points on
the pressure-volume curve using morphometric analysis of electron micrographs
(Tschumperlin and Margulies 1999). These studies suggest that when the rat lung is
inflated from functional residual capacity (FRC) changes in epithelial cell basal surface
area are relatively small at low lung volumes increasing to between 16% (Bachofen et al.
1987) and 34% (Mercer and Crapo 1990; Tschumperlin and Margulies 1999) at total
lung capacity (TLC). Support for the magnitude of these changes in EBMSA with lung
volume comes from morphological studies of collagen fibril geometry in the alveolar
septa of lungs fixed at functional residual capacity (Mercer and Crapo 1990). These
studies found that collagen fibrils could theoretically be extended by (16% before the
wavelike structure of the fibrils straighten limiting further extension. This change in
fibril length translates to an increase in surface area of (32% (Mercer and Crapo 1990;
Tschumperlin and Margulies 1999).

44
1.6.2 The role of mechanical forces in lung development
1.6.2.1 The effect of mechanical forces on lung growth
During intrauterine life, the foetus makes foetal breathing movements generating
intrathoracic pressure changes similar to those generated in postnatal breathing
(Kitterman 1996). In addition, fluid is actively secreted into the lumen of the developing
lung generating a constant distending pressure in the potential air spaces (Kitterman
1996). It appears that both of these forces regulate normal growth and maturation of the
lung. Increasing foetal lung distension in sheep by chronic tracheal ligation has been
shown to both increase and accelerate lung growth (Alcorn et al. 1977; Hooper et al.
1993; Joe et al. 1997), whilst reducing foetal lung distension by chronic tracheal
drainage inhibits normal growth (Hooper et al. 1993). Abolition of foetal breathing
movements by cervical spinal cord transection also appears to inhibit lung growth
(Liggins et al. 1981; Harding et al. 1993).
Mechanical forces are also thought to play a role in the compensatory lung growth that
occurs following pneumonectomy in humans (Hsia 2004).
1.6.2.2 Alveolar epithelial phenotype & differentiation
Development of a normal alveolar epithelium is dependent on the regulated proliferation
and subsequent differentiation of alveolar epithelial cells into separate ATI and ATII cell
populations. Mechanical forces are thought to play a key role in the regulation of these
processes (Wirtz and Dobbs 2000). Increasing foetal lung distension by tracheal ligation
stimulates ATII cell proliferation (Hooper et al. 1993; De Paepe et al. 1999) but
qualitatively decreases the proportion of ATII cells in the foetal lung, whilst tracheal
drainage appears to increase the proportion of ATII cells (Alcorn et al. 1977; Joe et al.
1997). Electron microscopy reveals that the reduction in the number of ATII cells
following tracheal ligation in sheep is accompanied by a significant increase in the

45
number of cells of an intermediate cell phenotype, displaying characteristics of both
ATII and ATI cells, and this is followed, in a time-dependent fashion, by a fall in the
number of intermediate cells and a significant increase in the number of ATI cells. These
findings suggest that foetal lung distension stimulates the differentiation of ATII cells
into ATI cells via an intermediate cell type (Flecknoe et al. 2000).
Further evidence from animal models supporting the hypothesis that mechanical
distension stimulates the expression of the ATI cell phenotype and inhibits the ATII cell
phenotype is provided by the finding that mechanical distension of foetal lung decreases
the mRNA of the ATII cell-specific surfactant proteins SP-A, SP-B and SP-C whilst
increasing the mRNA of the ATI cell-specific protein RTI40 (Gutierrez et al. 1999; Lines
et al. 1999).
Intriguingly, Flecknoe and colleagues have also shown that reducing foetal lung
expansion in sheep stimulates the transdifferentiation of ATI cells into ATII cells,
challenging the long-accepted concept that ATI cells are terminally differentiated
(section 1.3.2) (Flecknoe et al. 2002).
1.6.3 The effect of mechanical forces on the alveolar epithelium
1.6.3.1 Surfactant secretion
The predominant physiological trigger for surfactant release appears to be the
mechanical forces occurring during breathing. Hyperpnoea during exercise, and the
application of a single deep inflation to isolated perfused rat lungs have both been shown
to cause a significant increase in surfactant release and a reduction in the volume density
of ATII cell lamellar bodies (Nicholas et al. 1982; Nicholas et al. 1982; Massaro and
Massaro 1983). Mechanical distension of primary rat ATII cells also stimulates
surfactant release, with stretch causing a transient increase in intracellular calcium and
sustained release of phosphatidylcholine (Wirtz and Dobbs 1990; Edwards et al. 1999).

46
1.6.3.2 Phenotype
Mechanical distension of foetal ovine alveolar epithelial cells in utero appears to favour
expression of the ATI cell phenotype and inhibit expression of the ATII cell phenotype
(section 1.6.2.2). The hypothesis that strain induces differentiation of ATII into ATI
cells is also supported by several in vitro studies. Sustained tonic distension of primary
rat ATII cells significantly increases the expression of the ATI cell-specific protein
RTI40 whilst reducing the expression of the ATII cell-specific proteins SP-B and SP-C
(Gutierrez et al. 1998). Sustained tonic contraction appears to have the reverse effect,
favouring expression of the ATII phenotype with reduced RTI40 expression and
increased expression of SP-A, SP-B and SP-C (Gutierrez et al. 2003). These effects
appear to be mediated at the transcriptional level (Gutierrez et al. 1999).
1.6.3.3 Cytokine production
Mechanical strain of the alveolar epithelium may contribute to the pathogenesis of
ventilator-induced lung injury (VILI) (section 1.6.4) by stimulating the release of pro-
inflammatory cytokines. Cyclical mechanical strain has been shown to upregulate the
expression of IL-8 in human lung carcinoma-derived A549 (ATII-like) cells (Vlahakis et
al. 1999; Li et al. 2003; Jafari et al. 2004), with this effect inhibited by the antioxidant
glutathione (Jafari et al. 2004).
However, in the majority of studies, stretch alone does not appear to significantly effect
cytokine expression, and DNA microarray analysis of changes in gene expression
following exposure of human alveolar epithelial cells to mechanical strain appears to
confirm this, with the expression of the commonly studied cytokines either not
significantly effected or decreased (Waters et al. 2002; dos Santos et al. 2004).

47
1.6.3.4 Apoptosis
ATII cells undergo apoptosis as part of normal cell turnover in the lung. ATII cell
apoptosis also appears to be a key mechanism in controlling the number of ATII cells in
the developing lung (Schittny et al. 1998), and in the restoration of normal alveolar
epithelium following ATII cell proliferation in repairing epithelium (Bardales et al.
1996; Fehrenbach et al. 1999; Fehrenbach et al. 2000; Fine et al. 2000). Mechanical
forces are thought to play a key role regulating this process (Edwards 2001).
Tracheal occlusion, with sustained foetal lung distension, has been shown to
significantly increase ATII cell apoptosis (De Paepe et al. 2004), and this, together with
the transdifferentiation of ATII cells into ATI cells (Flecknoe et al. 2000), is thought to
regulate the number of ATII and ATI cells in the developing alveolar epithelium. Cyclic
mechanical strain of primary rat ATII cells, and transformed human ATII-like (A549)
cells has also been shown to induce ATII cell apoptosis (Edwards et al. 1999;
Hammerschmidt et al. 2004). This effect appears to be attenuated by nitric oxide (NO)
released from co-cultured alveolar macrophages suggesting a physiological role of
alveolar macrophages in protecting ATII cells from stretch-induced apoptosis (Edwards
et al. 2000).
1.6.3.5 Deformation-induced injury
In addition to inducing apoptosis, mechanical deformation of the alveolar epithelial
basement membrane has been shown to induce necrotic cell death (Tschumperlin and
Margulies 1998; Hammerschmidt et al. 2004). This deformation-induced injury has been
implicated in the disruption of the air-blood barrier which occurs in animal models
during the development of ventilator-induced lung injury (Egan 1982; Tschumperlin and
Margulies 1998).
Tschumperlin and colleagues assessed the vulnerability of primary rat ATII cells in
culture to mechanical deformation-induced injury by subjecting cells to equibiaxial
deformations of 12, 24, 37 and 50%, representing lung inflation to ~60, 80, 100 and

48
>100% of total lung capacity respectively. Deformation-induced injury increased with
deformation magnitude and frequency in these studies, increasing rapidly with
deformations exceeding those associated with inflation to total lung capacity. This
appears to support the hypothesis that the alveolar epithelium undergoes irreversible cell
damage at high lung volumes (Tschumperlin and Margulies 1998; Tschumperlin et al.
2000). In addition, Tschumperlin and colleagues found that ATII-like cells (primary
ATII cells after 1 day in culture) appeared more sensitive to deformation-induced injury
than ATI-like cells (primary ATII cells after 5 days in culture), suggesting that the
morphological and phenotypic changes of ATII cells with time in culture also
significantly affect the vulnerability of alveolar epithelial cells to deformation
(Tschumperlin and Margulies 1998).
The mechanism underlying deformation-induced epithelial cell injury remains unclear,
although plasma membrane stress failure (Vlahakis and Hubmayr 2000), and oxidative
stress secondary to stretch-induced antioxidant depletion have both been implicated
(Jafari et al. 2004). A recent study by Chapman and colleagues, demonstrating increased
ROS generation in immortalised human alveolar epithelial (A549) cells in response to
mechanical deformation, supports the concept that oxidative stress may play a central
role in deformation-induced epithelial cell injury (Chapman et al. 2005).
1.6.4 Mechanical ventilation & lung injury: ventilator induced lung injury (VILI)
Mechanical ventilation is routinely used to provide respiratory support in critically ill
patients. However, both clinical and animal studies have shown that mechanical
ventilation can worsen existing lung injury and produce an iatrogenic injury known as
ventilator-induced lung injury (VILI) (Parker et al. 1993; Dreyfuss and Saumon 1998;
Slutsky 1999; Dos Santos and Slutsky 2000).
The clinical significance of VILI has been highlighted by several clinical trials
demonstrating that protective ventilatory strategies (section 1.6.4.1.2) are associated
with lower levels of both pulmonary and systemic inflammatory mediators, reduced

49
levels of organ dysfunction, and reduced mortality (Amato et al. 1998; Ranieri et al.
1999; ARDSNet 2000).
1.6.4.1 Mechanisms of ventilator-induced lung injury
For many years, ventilator-induced lung injury and barotrauma were thought to be
synonymous, with high airway pressures causing physical disruption of the alveolar
wall, air leaks and accumulation of extra-alveolar air (Dreyfuss and Saumon 1998;
Slutsky 1999). However, morphological examination of the lungs following ventilator-
induced injury reveals that more subtle changes occur following injurious ventilation,
with severe alveolar damage and changes in endothelial and epithelial permeability that
are indistinguishable from other forms of lung injury (Dreyfuss et al. 1985).
Both high pressure and high volume ventilation cause VILI in experimental models
(Dreyfuss and Saumon 1998; 1999), but alveolar overdistension (volutrauma) rather than
high airway pressures (barotrauma) appears to be the primary determinant of VILI
(Dreyfuss et al. 1988), with repeated alveolar collapse and reopening (atelectrauma)
contributing by the application of large shear forces (Mead et al. 1970; Webb and
Tierney 1974; Argiras et al. 1987).
These factors are of particular significance in acute lung injury, where patchy
parenchymal consolidation results in an uneven distribution of aerated, poorly aerated
and non-aerated lung (Maunder et al. 1986; Rouby et al. 2003). Positive pressure
ventilation in this context is thought to result in alveolar overdistension in uninjured
(aerated) alveoli with repeated alveolar opening and collapse secondary to cyclic
alveolar recruitment in poorly aerated regions. It is postulated that the resulting
ventilator-induced injury plays a key role in determining the poor outcome in patients
requiring mechanical ventilation through the development of a systemic inflammatory
response (Ranieri et al. 1999) and multiple organ failure (Slutsky and Tremblay 1998;
Tremblay and Slutsky 1998; Imai et al. 2003).

50
1.6.4.1.1 Inflammatory mediators in ventilator-induced injury: biotrauma
Barotrauma, volutrauma and atelectrauma are thought to be mechanical injuries caused
largely by stress failure of the plasma membrane and of the endothelial and epithelial
barriers (Slutsky 1999; Uhlig 2002). However, there is increasing evidence that
mechanical ventilation also leads to injury that is cell and inflammatory mediator based.
Slutsky and colleagues have coined the term biotrauma to describe this form of
ventilator-induced injury (Tremblay and Slutsky 1998).
Kawano and colleagues found that conventional ventilation of surfactant-depleted
rabbits resulted in lung injury characterised by the sequestration of large numbers of
neutrophils. This injury was attenuated by neutrophil depletion (Kawano et al. 1987).
Further evidence for the central role of the neutrophil in VILI has been provided by the
work of Zhang and colleagues, in which alveolar lavage fluid taken from ventilated
patients with ARDS was incubated with neutrophils from normal volunteers. Markers of
neutrophil activation were significantly higher in those ventilated conventionally
compared to those ventilated with a lung protective strategy (Zhang et al. 2002).
The alveolar macrophage may also play a role in the development of VILI. Mechanical
deformation of human alveolar macrophages in vitro, has been shown to increase the
expression of IL-8 and matrix-metalloproteinase-9 (Pugin et al. 1998), and in a rat model
of acute lung injury, Lentsch and colleagues demonstrated that alveolar macrophage
depletion suppressed NF-'B activation in the lung which appears to be an important step
in mediator release in response to overdistension (Lentsch et al. 1999; Held et al. 2001).
Work by Frank and colleagues also suggests macrophage activation is an early and
critical event in the initiation of VILI, with BALF from rats exposed to only 20 minutes
injurious ventilation capable of activating naïve primary alveolar macrophages in culture
and macrophage depletion decreasing ventilator-induced endothelial and epithelial
injury (Frank et al. 2006).
In animal studies, injurious ventilation of normal lungs has been shown to increase
concentrations of the pro-inflammatory cytokines TNF-$ and IL-6 in lavage fluid
(Tremblay et al. 1997; Veldhuizen et al. 2001; Tremblay et al. 2002) and in the perfusate

51
of isolated-perfused lungs (von Bethmann et al. 1998). In-situ hybridisation suggests that
the alveolar epithelium is a major source of these cytokines (Tremblay et al. 2002).
Clinical studies seem to support the concept that injurious ventilation promotes a
pulmonary inflammatory response and cytokine decompartmentalisation. Ranieri and
colleagues reported significant decreases in BALF and plasma concentrations of many
inflammatory mediators (TNF-$, IL-6 and IL-8) in patients ventilated with a lung
protective strategy (Ranieri et al. 1999), and in the ARDSnet trial, patients ventilated
with a lung protective strategy had a greater decrease in levels of plasma IL-6
(ARDSNet 2000).
1.6.4.1.2 The effect of protective ventilatory strategies
The avoidance of alveolar overdistension by limiting tidal volume and/or airway
pressures, with the addition of positive end inspiratory pressure (PEEP) to maintain
alveolar recruitment throughout the respiratory cycle, form the basis of lung protective
ventilatory strategies. Both clinical and animal studies have demonstrated the ability of
these manoeuvres to reduce VILI.
In a rat model of acid-induced lung injury, Frank and colleagues demonstrated that tidal
volume reduction, maintaining the same level of PEEP, significantly reduced both
endothelial and epithelial injury and this was associated with lower histological lung
injury severity scores (Frank et al. 2002).
Ranieri and colleagues reported significant decreases in the levels of both pulmonary
and systemic inflammatory mediators in patients ventilated using a lung protective
strategy (Ranieri et al. 1999), and in the ARDSnet trial, mortality was reduced by 22%
in patients ventilated with lower tidal volumes (6 ml/kg) compared to those ventilated
with traditional tidal volumes (12 ml/kg) (ARDSNet 2000). The protective effect of
PEEP has been highlighted in a study by Amato and colleagues, demonstrating that an
“open-lung” approach to mechanical ventilation, in which alveolar recruitment was
optimised with the use of high levels of PEEP, resulted in improved 28 day mortality

52
(Amato et al. 1998).
1.6.4.2 The role of the alveolar epithelium in VILI
The precise mechanisms of ventilator-induced lung injury remain unclear. It appears that
physical disruption with stress failure of plasma membranes and stress failure of the air-
blood barrier occurs in conjunction with cellular activation and the release of
inflammatory mediators (Slutsky 1999; Uhlig 2002). Neutrophils and alveolar
macrophages have both been implicated in the pathogenesis, but there is compelling
evidence that the alveolar epithelium plays a key regulatory role in the initiation and
regulation of the inflammatory response in VILI (Tremblay and Slutsky 1998; Slutsky
1999; Frank et al. 2006).
Localised at the air interface, the alveolar epithelial surface is clearly exposed to the
pressures generated during positive pressure ventilation, and morphometric analysis of
lungs exposed to injurious ventilation reveals significant epithelial injury, with
widespread ATI cell injury and ATII cell proliferation. The structural and functional
consequences of epithelial barrier disruption are discussed above (section 1.2.1).
Whether mechanical ventilation has a significant impact on epithelial repair following
injury remains unknown.

53
1.7 Summary, thesis hypotheses and aims
1.7.1 Summary
The alveolar surface of the lung is lined by the alveolar epithelium. This is comprised of
two morphologically distinct cell types: ATI and ATII cells. Joined by tight junctions,
these cells together constitute a key structural and functional component of the air-blood
barrier (section 1.1). Injury to the alveolar epithelium, with disruption of the integrity of
the air-blood barrier, is a central feature in the pathogenesis of a range of clinical
conditions (section 1.2).
ATI cells are more susceptible to injury than ATII cells but appear incapable of cell
division. The accepted paradigm of alveolar epithelial repair following injury posits that
damaged ATI cells are replaced by proliferation of ATII cells and the differentiation of a
subpopulation of daughter cells into ATI cells (section 1.3).
Timely and effective epithelial repair, with restoration of an intact epithelial barrier and
normal alveolar fluid transport, is a key determinant of clinical outcome following lung
injury. Abnormal epithelial repair on the other hand appears to be a key pathogenic
mechanism in progression to pulmonary fibrosis.
Oxygen and mechanical ventilation are key components in the supportive treatment of
respiratory failure and the prevention of hypoxaemia. However, both hyperoxia (section
1.5) and the lung deformations associated with mechanical ventilation (sections 1.6) are
potentially injurious to the lung. Localised at the air interface, the alveolar epithelial
surface is exposed to oxidative stress with the use of supplemental oxygen and
significant mechanical force during positive pressure ventilation. As such, it is a key
target for oxygen- and ventilation-induced iatrogenic injury.
Exposure of the normal lung to significant hyperoxia is known to cause ATI cell
necrosis and inhibit ATII cell proliferation. Its effect on the differentiation of ATII cells
into ATI cells has not been studied and is unknown. The effects of hyperoxic exposure

54
on the injured lung and on epithelial repair are also not known.
Similarly, although clinical experience suggests that mechanical ventilation is more
damaging to injured (repairing) lung than to normal lung, the effects of mechanical
deformation (± hyperoxia) on alveolar epithelial repair are not known.
The use of a panel of ATI and ATII cell-selective antibodies as biomarkers provides a
powerful tool for characterising the phenotype of alveolar epithelial cells in vitro and of
the alveolar epithelium ex vivo (section 1.4). It also enables the quantification of
individual cell phenotypes within the alveolar compartment. As normal epithelial repair
is associated with characteristic changes in epithelial cell number and cell phenotype
(ATII cell proliferation and differentiation of ATII cells into ATI cells), this
immunotargeting approach offers a novel means of extracting both qualitative and
quantitative data regarding the cell populations present in regions of alveolar epithelial
repair and of investigating the factors influencing alveolar epithelial repair in vivo.
1.7.2 Hypotheses
1. Hyperoxia inhibits normal alveolar epithelial repair by inhibiting the
transdifferentiation of ATII cells into ATI cells.
2. Alveolar epithelial cells in regions of epithelial repair are more susceptible to
mechanical deformation-induced injury than normal alveolar epithelial cells.
1.7.3 Aims
1. Establish the isolation and culture of primary alveolar epithelial cells (chapter 3).
2. Use a panel of cell-selective antibodies to characterise the immunophenotype of
alveolar epithelial cells in culture (chapter 3).
3. Investigate the immunoselectivity of a new monoclonal antibody (MMC6) in the
developing and adult lung and in alveolar epithelial cells in culture (chapter 3).

55
4. Determine the effect of hyperoxia on the transdifferentiation of alveolar epithelial
cells from an ATII- to an ATI-like phenotype in culture (chapter 3).
5. Further characterise an established model of resolving direct lung injury
(Staphylococcus aureus-induced pneumonia) to investigate exposure of the repairing
lung to hyperoxia (chapter 4).
6. Use a panel of cell-selective antibodies combined with immunofluoresence and laser
scanning confocal microscopy to quantify the proportion of the alveolar surface
associated with ATI, ATII and intermediate cell phenotypes in a model of resolving
lung injury and determine whether hyperoxia delays normal epithelial repair by
inhibiting the transdifferentiation of ATII cells into ATI cells in vivo (chapter 4).
7. Establish the primary cell culture of alveolar epithelial cells in a mechanically active
environment and determine the sensitivity of these cells to mechanical deformation-
induced injury (chapter 5).
8. Using an in vitro model, determine the susceptibility of cells in the ‘repairing’ region
of wounded primary alveolar epithelial cell monolayers to deformation-induced
injury (chapter 5).
9. Establish a comparative recovery model of indirect acute lung injury for investigation
of the effects of mechanical ventilation ± hyperoxia on alveolar epithelial injury and
repair (chapter 6).

56
2. Materials and methods
Reagents were obtained from Sigma-Aldrich Ltd unless otherwise indicated. Anaesthetic
agents and veterinary pharmaceuticals were supplied by Genusxpress. A list of
manufacturers and suppliers is given in Appendix II. Details of buffers, stock solutions
and culture media are given in Appendix III. Animal work was carried out in
accordance with the Animals Scientific Procedures Act 1986.
2.1 Primary alveolar epithelial type II cell isolation & culture
2.1.1 Alveolar epithelial type II cell isolation
ATII cells were isolated from the lungs of 150-250g male Sprague-Dawley specific
pathogen-free rats (Harlan) using a modified version of the method described by Dobbs
et al. (Dobbs et al. 1980; Dobbs et al. 1986; Dobbs 1990). Two rats were used per
isolation, with isolated cells being pooled prior to immunoglobulin G (IgG) panning.
Quantities stated are per rat. Isolation buffers and culture media were pre-warmed to
37+C, elastase (Roche) solution to 39+C.
Rats were anaesthetised by intraperitoneal injection of a mixture of midazolam (5
mg/kg) and ketamine (100 mg/kg). Heparin sulphate (500 iu) was administered by
separate intraperitoneal injection. Once anaesthetised, laparotomy was performed and
rats exsanguinated by abdominal aortotomy. The chest was opened, trachea cannulated
(18G Monoject blunt needle, Harvard Apparatus) and the lungs mechanically ventilated.
The left atrium was opened, and the lungs perfused until marble white by injection of
isolation buffer through the right ventricle. The heart and lungs were then explanted and
endobronchial lavage performed by inflation to total lung capacity (TLC) ten times with
Ca/Mg-free isolation buffer followed by four times with standard isolation buffer. The

57
lungs were then inflated to TLC with a solution of elastase (5mg in 20ml isolation
buffer) and incubated in saline at 39+C for 15 minutes. Remaining elastase solution was
then infused intratracheally over a further 15 minutes. Individual lobes of lung were
dissected from residual tissue, placed in DNAse solution (4mg in 2ml isolation buffer,
Roche) and elastase activity inhibited by addition of 4 ml foetal bovine serum (FBS).
The lungs were then chopped into (3 mm cubes, the resulting tissue suspension made up
to 40 ml with isolation buffer and hand shaken for three minutes, before sequential
filtration through 2 and 4-layer surgical gauze followed by 150, 20 and 10 µm nylon
mesh filters (Precision Textiles Ltd.). The resulting cell suspension was centrifuged
(257g, 6 min), resuspended in 20 ml serum-free Dulbecco’s modified eagle’s medium
(DMEM) and incubated at 37+C for 60 minutes on IgG coated bacteriological dishes
(Greiner Bio-One) to remove contaminant macrophages. The cells remaining unattached
at the end of this period were centrifuged (257g, 6 min) and resuspended in ATII culture
media.
Cell viability, assessed by exclusion of trypan blue, was determined by differential cell
count in a coverslipped Neubauer haemacytometer. Only isolations with a final cell
suspension cell viability > 90% were used in these studies.
Cell yield was assessed by cell count of crystal violet stained cells in a Neubauer
haemocytometer. The mean yield was 12.8 ± 3.1 x 106 cells per rat.
2.1.2 Modified Papanicolau staining & assessment of ATII purity
ATII cell purity was assessed by modified Papanicolau (Pap) staining of freshly isolated
cells (Dobbs 1990). (3x105 cells in 200µl media were cytospun (300 rpm, 3 min) onto
glass slides and air-dried. Slides were dipped in haematoxylin (Harris formulation) for
3mins, rinsed in DDW and dipped in saturated lithium carbonate (256mg Li2CO3 in 20
ml DDW) for 2min. After rinsing in DDW, slides were then sequentially dipped in 50%
ethanol for 90sec, 80% ethanol for 15sec, 95% ethanol for 15sec, 100% ethanol for
30sec, xylene:ethanol (1:1) for 30sec and finally 100% xylene for 1min. Cells were then

58
coverslipped using Pertex mounting medium.
Differential cell counts were performed at high magnification (100x oil immersion
objective), with ATII cells being identified as cells containing )3 purple-staining
inclusion (lamellar) bodies. Macrophages, neutrophils and lymphocytes were identified
by their characteristic cell morphology. A minimum of 500 cells was counted and
differential cell counts expressed as a percentage of totals.
2.1.3 Standard ATII cell culture
ATII cells were seeded at a density of 0.5 x 106 cells per cm
2 onto 13mm diameter
fibronectin-coated glass coverslips (Fisher Scientific) in ATII culture media and
incubated at 37°C in a humidified atmosphere of 95% air and 5% CO2 (normoxia) unless
otherwise stated. After 6 h, cells were washed 3x with serum free DMEM and the media
replaced with fresh ATII culture media. Media was subsequently changed every 48
hours throughout time in culture. The pO2, pCO2 and pH of culture media was measured
(Bayer RapidLab® 248 analyser) every 12 h during experiments under both normoxic
and hyperoxic conditions, with the results shown in table 6.
Normoxia Hyperoxia
pO2 (kPa) 16.3 ± 1.6 95.4 ± 4.3
pCO2 (kPa) 6.2 ± 0.7 6.4 ± 0.3
pH 7.50 ± 0.07 7.51 ± 0.02
Table 6 Culture media pH and partial pressures of oxygen and carbon dioxide
throughout time in culture under normoxic and hyperoxic conditions. Measurements
made using a Bayer RapidLab® 248 analyser (n=12).

59
2.1.4 ATII cell culture under hyperoxic conditions
ATII cells were seeded as for standard ATII cell culture. Cell culture plates were then
sealed in a humidified modular incubator chamber (MIC-101, Billups-Rothenberg inc.)
and the chamber flushed with filtered medical grade gas (95% O2, 5% CO2) from
cylinder supply (BOC Gases) at a flow rate of 20 l/min (Dual Flow Meter DFM 3002,
Billups-Rothenberg inc.) for 10 min. After 6 h, cells were washed 3x with serum free
DMEM and the media replaced with fresh ATII culture media. Media was subsequently
changed every 48 hours throughout time in culture. Media changes were carried out as
rapidly as possible under standard (normoxic) conditions. The pH and partial pressures
of O2 and CO2 measured in culture media during experiments under hyperoxic
conditions are outlined in table 6.
2.1.5 ATII cell culture using the Bioflex, cell culture system
Freshly isolated ATII cells were seeded onto the fibronectin-coated silastic membranes
of Bioflex, 6-well culture plates (Dunn Labortechnik GmbH) at a density of 0.5x106
cells/cm2. In all experiments, cell attachment was confined to the central portion of the
membrane by seeding within a removable silicon gasket (Grace Bio-Labs). Cells were
washed 6 h after seeding and silicon gaskets removed after 24 h in culture. In other
respects cell culture was identical to that for standard ATII cell culture.
Mechanical deformation of cells in culture was achieved using the Flexercell FX-
4000T" plates (Dunn Labortechnik GmbH) and is described in section 5.2.1.
2.2 Protein quantification assay
Protein concentrations in aqueous solution were determined using the Bradford (Bio-
Rad Laboratories) protein assay, a dye-binding assay based on the concentration-
dependent shift of the absorbance maximum of an acidic solution of Coomassie Brilliant
Blue G-250 from 465 nm to 595 nm upon binding protein (Bradford 1976).

60
Protein concentrations were obtained from the linear range of a standard curve
constructed by plotting absorbance against concentration (0-1.4 mg/ml) of bovine serum
albumin. Absorbance of samples was read at 630 nm using an ELISA plate reader (MRX
Microplater Reader, Revelation 3.04 software, Dynatech Laboratories). Each sample
was tested at two different dilutions with three repeats of each dilution.
2.3 Measurement of cell viability in adherent cells
The viability of adherent cells was assessed using fluorescent microscopy and a
LIVE/DEAD, viability/cytotoxicity kit (Molecular Probes, Invitrogen) comprising
ethidium homodimer-1 (EthD-1) and calcein acetoxymethyl ester (calcein AM). EthD-1
is excluded by the intact plasma membranes of live cells, but enters cells with damaged
membranes undergoing fluoresecent enhancement upon binding to nucleic acids and
producing red fluorescence (ex/em (495nm/(635nm). Cell permeant Calcein AM
undergoes enzymatic conversion by intracellular esterases present within live cells to the
polyanionic dye calcein. Calcein is well retained within live cells, producing a uniform
green fluorescence (ex/em (495nm/(515 nm). Adherent cells were washed 3x with
warmed serum-free DMEM containing 20mM 4-(2-hydroxyethyl)-1-
piperazineethanesulfonic acid (HEPES), and then incubated at 37 °C with 4µM EthD-1
and 2µM calcein AM in HEPES-containing DMEM for 60min. Cells were examined
with an inverted epifluorescnt microscope and images acquired from 3 random locations
using a digital-imaging system (Metamorph, Molecular Devices). The total number of
EthD-1 and calcein AM staining cells in each field was counted and data expressed as
percentage dead (EthD-1 positive) or viable (calcein AM positive) of total.
2.4 Lactate dehydrogenase (LDH)-cytotoxicity assay
Measurement of the increase in lactate dehydrogenase (LDH) activity following release
of cytoplasmic LDH from damaged cells is a widely accepted method of quantifying cell

61
cytotoxicity (Korzeniewski and Callewaert 1983). LDH activity may be determined
using a coupled enzymatic reaction in which the tetrazolium salt iodotetrazolium
chloride (INT) (yellow) is reduced to formazan (red) by NADH produced during the
LDH-catalysed conversion of lactate to pyruvate. Increases in LDH activity are directly
proportional to the amount of formazan produced in this coupled reaction, and this can
be determined colorimetrically.
After gentle agitation, cell supernatants were collected and remaining adherent cells
lysed by the addition of 100µl of 0.1% Triton X-100 solution to each culture well. After
15 min, culture wells were gently scraped with a cell scraper and the lysate removed.
Supernatant and lysate samples were stored at -70°C prior to analysis. LDH assays were
performed in triplicate on supernatant and paired lysate samples in 96 well culture
plates. 20µl Tris (hydroxymethyl)aminomethane buffer, 10µl sample and 50µl LDH
substrate were added to each well, mixed and incubated at 37°C for 5 min. 20µl of
freshly prepared chromagen solution was then added to each well, mixed and plates
incubated at 37°C for a further 5 min. 150µl 0.5M HCl was then added to each well,
mixed and formazan absorption at 540 nm read immediately (MRX Microplater Reader,
Revelation 3.04 software, Dynatech Laboratories). Samples were compared to control
wells in which sample was replaced with 10µl blank oxalate solution.
The percentage of cell death was calculated by determining the LDH activity in the
supernatant of each well as a percentage of total LDH activity (supernatant + lysate).
2.5 Feulgen staining & assessment of apoptosis
Apoptosis was assessed by nuclear morphology following Feulgen’s staining. Cells were
fixed in modified Bouin’s fixative at 4°C overnight then denatured by incubation with
5M HCl for 45 minutes at room temperature. After rinsing in tap water, cells were
incubated in Schiff’s reagent (BDH) for 1 hour and rinsed thoroughly ((15 min) in tap
water until red-purple DNA staining developed. Cytoplasm was counterstained with
0.1% light green, cells air-dried and mounted in cedarwood oil. Apoptotic cells were

62
distinguished by nuclear morphology using light microscopy (Bellamy et al. 1997). A
minimum of 500 cells was counted and results expressed as percentage of apoptosis.
2.6 Estimation of reactive oxygen species (ROS) generation
ROS generation was assessed by intracellular oxidation of 5-(and-6-)-chloromethyl-
2’,7’-dichlorodihydrofluorescein diacetate (CMH2DCFDA) (Molecular Probes,
Invitrogen). Cleavage of the lipophilic blocking groups of this cell-permeant probe by
intracellular esterases yields a charged form of the probe that is retained intracellularly.
Subsequent intracellular oxidation of nonfluorescent dichlorodihydrofluorescein to
(fluorescent) fluorescein can then be quantified spectrofluorometrically.
ATII cells on glass coverslips within 24 well plates (Greiner Bio-One) were washed 3x
with warmed phenol red-free Hank’s balanced salt solution (HBSS) (Invitrogen) and
500µl HBSS containing 10µM CMH2DCFDA added to each well. CMH2DCFDA was
omitted from otherwise identically treated control wells. Fluorescence was followed
continuously over 60min using a thermostated plate reading spectrofluorometer
(Fluoroskan Ascent, Thermo Scientific) at excitation 486 nm and emission 530 nm. The
protein content of each well was determined and the average rate of CMH2DCFDA
fluorescence corrected for variation in total protein between wells. Fluorescence
detected in control wells was subtracted from measurements obtained, and ROS
production reported as fluorescent units (fu)/mg protein/min.
2.7 Estimation of ROS generation in response to oxidative stress
ROS generation in response to oxidative stress was assessed by the oxidation of
CMH2DCFDA during exposure of ATII cells to hydrogen peroxide (H2O2). Controlled
exposure to H2O2 was produced by the addition of 50 milliunits of glucose oxidase
(catalysing the conversion of glucose within HBSS to D-glucono-1, 5-lactone and H2O2)
(Fluka and Riedel, Sigma-Aldrich) to each well containing 500µl HBSS and 10µM

63
CMH2DCFDA. ROS generation by ATII cells in response to H2O2 was then followed
continuously over 60min as detailed in section 2.6. Control wells contained HBSS and
CMH2DCFDA without glucose oxidase but were otherwise identical. Fluorescence
detected in control wells was subtracted from measurements obtained, and ROS
production reported as fu/mg protein/min.
2.8 Quantification of antigens by ELISA-based dot blot assay
The level of RTI40, MMC4 and MMC6 expression was determined semiquantitatively
using enzyme-linked immunoabsorbent assay (ELISA)-based dot blots. Samples were
immobilised by vacuum onto polyvinylidene difluoride (PVDF) Immobilon-P
membranes (Millipore) using a dot blot manifold (Schleicher & Schuell, Whatman
International). Immobilised samples were then blocked for 1.5 hours at room
temperature with 0.4% non-fat milk (casein) in tris(hydroxymethyl)aminomethane-
buffered saline (TBS, 154 mM, pH 8.2), incubated with primary antibody (1:500 RTI40
in DMEM; MMC4 hybridoma supernatant; MMC6 hybridoma supernatant) for 30
minutes, washed with TBS containing 0.05% Tween-20 (TBS-T) for 30 minutes and
then incubated with sheep anti-mouse secondary antibody conjugated to horseradish
peroxidase (1:3000, Amersham Pharmacia). Blots were subsequently washed again for
30 minutes with TBS-T before being developed with enhanced chemiluminescence
reagents (ECL, Amersham Biosciences) and developed to film (Kodak, X-omat AR
film, Sigma). Secondary only controls, in which incubation with primary antibody was
omitted, were performed as part of each assay. All samples for comparison were loaded
onto one membrane to allow for variation between blots. Semiquantitative data, reported
as densitometry or relative densitometry units per mg protein (du or rdu/mg protein),
were obtained from blots by determination of the absorbance of individual dots at 600
nm using a plate reader (MRX Microplate Reader, Revelation 3.04 software, Dynatech
Laboratories) normalised for sample protein concentration.
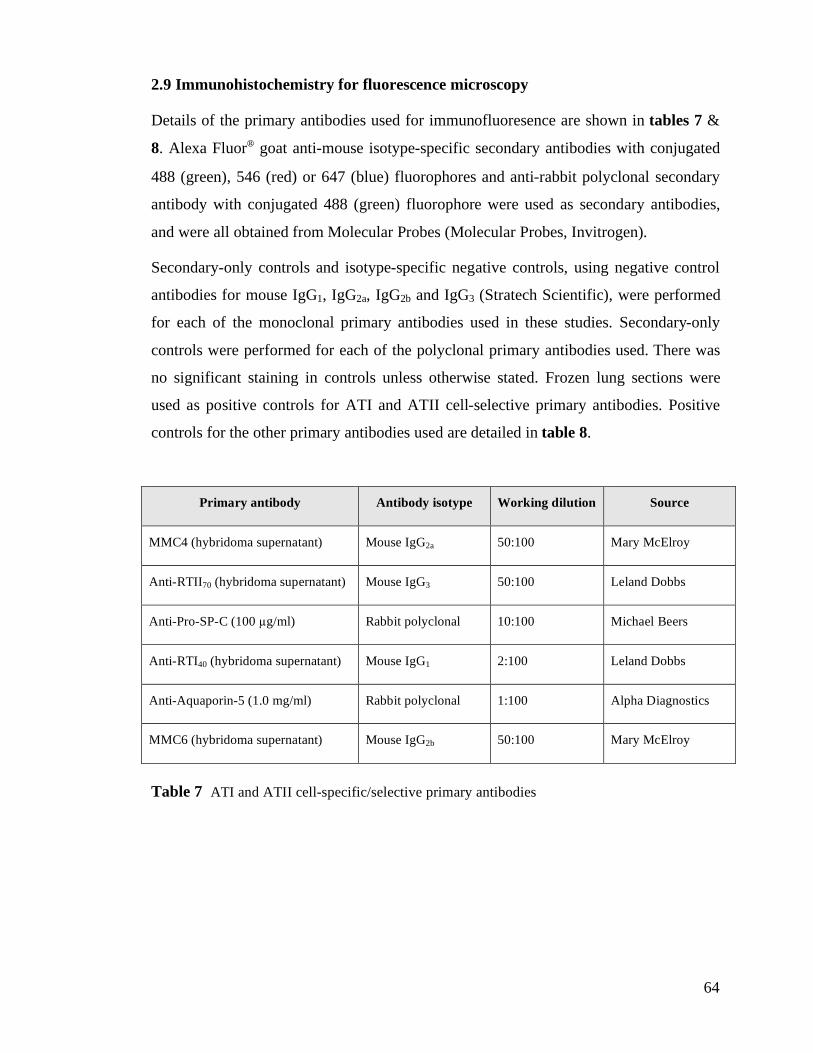
64
2.9 Immunohistochemistry for fluorescence microscopy
Details of the primary antibodies used for immunofluoresence are shown in tables 7 &
8. Alexa Fluor, goat anti-mouse isotype-specific secondary antibodies with conjugated
488 (green), 546 (red) or 647 (blue) fluorophores and anti-rabbit polyclonal secondary
antibody with conjugated 488 (green) fluorophore were used as secondary antibodies,
and were all obtained from Molecular Probes (Molecular Probes, Invitrogen).
Secondary-only controls and isotype-specific negative controls, using negative control
antibodies for mouse IgG1, IgG2a, IgG2b and IgG3 (Stratech Scientific), were performed
for each of the monoclonal primary antibodies used in these studies. Secondary-only
controls were performed for each of the polyclonal primary antibodies used. There was
no significant staining in controls unless otherwise stated. Frozen lung sections were
used as positive controls for ATI and ATII cell-selective primary antibodies. Positive
controls for the other primary antibodies used are detailed in table 8.
Primary antibody Antibody isotype Working dilution Source
MMC4 (hybridoma supernatant) Mouse IgG2a 50:100 Mary McElroy
Anti-RTII70 (hybridoma supernatant) Mouse IgG3 50:100 Leland Dobbs
Anti-Pro-SP-C (100 "g/ml) Rabbit polyclonal 10:100 Michael Beers
Anti-RTI40 (hybridoma supernatant) Mouse IgG1 2:100 Leland Dobbs
Anti-Aquaporin-5 (1.0 mg/ml) Rabbit polyclonal 1:100 Alpha Diagnostics
MMC6 (hybridoma supernatant) Mouse IgG2b 50:100 Mary McElroy
Table 7 ATI and ATII cell-specific/selective primary antibodies
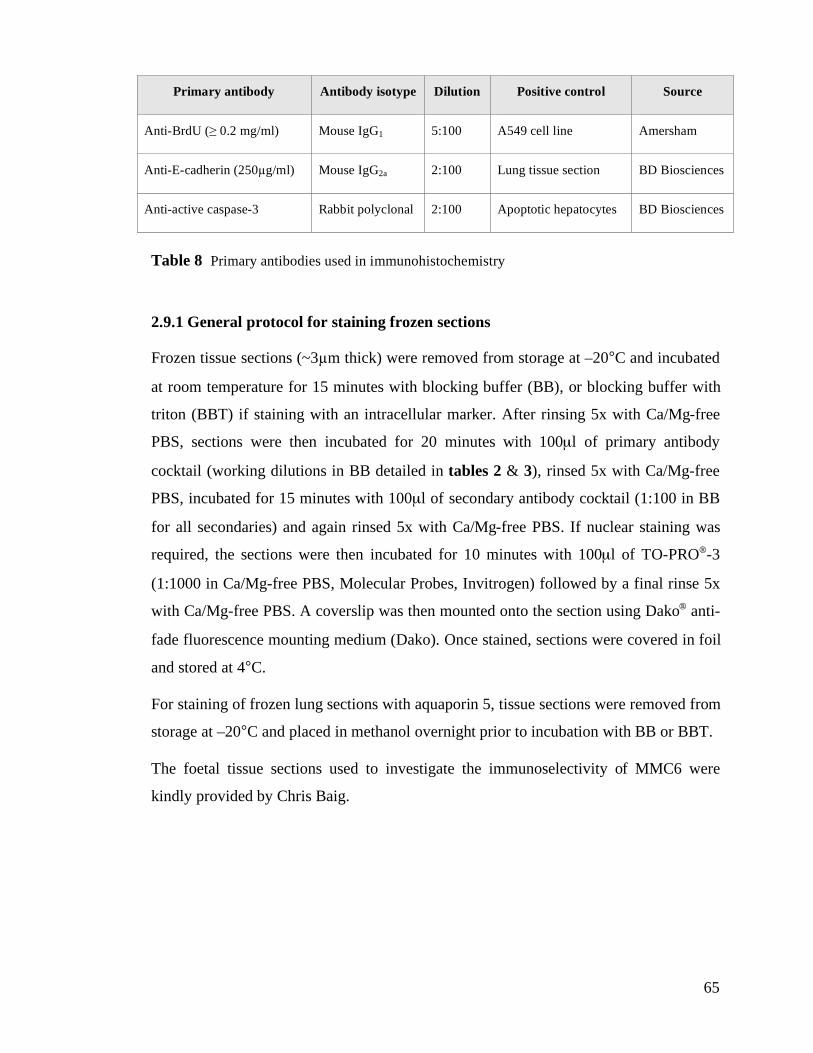
65
Primary antibody Antibody isotype Dilution Positive control Source
Anti-BrdU (# 0.2 mg/ml) Mouse IgG1 5:100 A549 cell line Amersham
Anti-E-cadherin (250"g/ml) Mouse IgG2a 2:100 Lung tissue section BD Biosciences
Anti-active caspase-3 Rabbit polyclonal 2:100 Apoptotic hepatocytes BD Biosciences
Table 8 Primary antibodies used in immunohistochemistry
2.9.1 General protocol for staining frozen sections
Frozen tissue sections (~3"m thick) were removed from storage at –20°C and incubated
at room temperature for 15 minutes with blocking buffer (BB), or blocking buffer with
triton (BBT) if staining with an intracellular marker. After rinsing 5x with Ca/Mg-free
PBS, sections were then incubated for 20 minutes with 100µl of primary antibody
cocktail (working dilutions in BB detailed in tables 2 & 3), rinsed 5x with Ca/Mg-free
PBS, incubated for 15 minutes with 100µl of secondary antibody cocktail (1:100 in BB
for all secondaries) and again rinsed 5x with Ca/Mg-free PBS. If nuclear staining was
required, the sections were then incubated for 10 minutes with 100µl of TO-PRO,-3
(1:1000 in Ca/Mg-free PBS, Molecular Probes, Invitrogen) followed by a final rinse 5x
with Ca/Mg-free PBS. A coverslip was then mounted onto the section using Dako, anti-
fade fluorescence mounting medium (Dako). Once stained, sections were covered in foil
and stored at 4°C.
For staining of frozen lung sections with aquaporin 5, tissue sections were removed from
storage at –20°C and placed in methanol overnight prior to incubation with BB or BBT.
The foetal tissue sections used to investigate the immunoselectivity of MMC6 were
kindly provided by Chris Baig.

66
2.9.2 General protocol for staining cultured ATII cells
ATII cells cultured on glass coverslips in 24-well plates were fixed in 4%
paraformaldehyde or methanol (aquaporin 5 staining only) and stored at 4°C. Once
fixed, coverslips were removed from 24-well plates using fine forceps, rinsed 5x with
Ca/Mg-free PBS and stained in an identical fashion to that for frozen sections by placing
the coverslip cell-side down onto a drop of the required solution (BB, primary antibody
cocktail etc.) pre-positioned on a piece of Parafilm M. Rinsing between steps was
performed by gentle irrigation holding the coverslip in fine forceps.
ATII cells cultured in BioFlex, 6-well culture plates were fixed in 4% paraformaldehyde
and stored at 4°C. Cells were then stained in-situ within the 6-well culture plates. The
sequence followed was similar to that for frozen sections with the exception that 500µl
aliquots of primary and secondary antibody cocktails and TO-PRO,-3 were required to
incubate the cells within the culture wells. Once stained, cells were covered with Ca/Mg-
free PBS and imaged with a Zeiss 510 laser scanning confocal microscope equipped
with an upright Zeiss Axioskop microscope and dipping lens.
2.9.3 Bromo-deoxyuridine (BrdU) immunhistochemistry
ATII cells were incubated with 10µM BrdU (Amersham Biosciences) in ATII culture
medium for 6 hours and fixed in 4% paraformaldehyde. After rinsing 5x with Ca/Mg-
free PBS, cells were incubated for 45 mins at room temperature in 5M HCl, rinsed 5x
5min in Ca/Mg-free PBS, incubated in BBT for 15min and then 60min with mouse anti-
BrdU Alexa Fluor, 488 conjugate (1:20 in BBT, Molecular Probes). After rinsing 5x
with Ca/Mg-free PBS, nuclei were counter-stained with TO-PRO,-3 and mounted as
detailed above.
2.9.4 Image acquisition
Images were captured using a confocal laser scanning microscope (LSM) with Zeiss

67
LSM 510 scanning head and an Axiovert 100M microscope unless stated otherwise.
Three lasers were used for specimen excitation: argon/krypton (488nm), helium/neon
(543nm) and helium/neon (633nm). To eliminate spectral spill, scanning with each laser
was performed sequentially (multi-track imaging). Comparative images were obtained
with identical laser and detector settings and images used for quantification were not
further digitally enhanced. Secondary antibodies had emission spectra with peak
wavelengths at 488nm (Alexa 488, green), 546nm (Alexa 546, red), and 647nm (Alexa
647, blue). Alexa 488 emissions were filtered through a Band Pass 500-530nm filter,
Alexa 546 emissions through a Band Pass 565-615nm filter and Alexa 647 emissions
through a Low Pass 650nm filter. Images were captured as three channel (red, green, and
blue) 8-bit grey scale LSM files at a resolution of 1024x1024 pixels, and subsequently
exported in tagged image file format (TIFF). A differential interference contrast (DIC)
image was also captured to record morphology at the transmitted light level.
For quantification purposes, a minimum of 3 large contiguous areas of specimen (up to
585 µm2
or 4096x4096 pixels with the 63x objective) were imaged as 4x4 contiguous
fields of view using the image tiling facility of the LSM.
2.9.5 z-plane scanning and three-dimensional image reconstruction
After determining the focal plane of the top and bottom (maximum and minimum z-
coordinates) of a specimen, a series of sequential images was taken by scanning at
incrementally changing z-positions between the maximum and minimum z-coordinates
(xy-coordinates constant). The resulting optical sections, arranged to form an image
stack, were viewed as a three-dimensional image using Volocity 3.0 (Improvision).
To improve image resolution, particularly in the z-plane, image stacks were acquired
using Nyquist settings. The raw data obtained were deconvoluted using Huygens 2 (SVI)
software and reconstructed using Imaris 4.0 software (Bitplane AG).

68
2.9.6 Image analysis
Images used for quantification were acquired using identical LSM settings and were not
digitally enhanced. Openlab 3.5.2 (Improvision) software was used for quantification.
2.9.6.1 Image density slicing
Positive staining with fluorochromes was determined by density slicing using the
Density Slice… function in Openlab. RGB TIFF files (max 4000x4000 pixels) were split
into constituent red, green and blue channels (Layers>Select All>Split RGB). The lower
threshold of positive staining in each channel was determined by polling the pixel value
of a positive area using the HSI (Hue, Saturation, Intensity) Colorspy palette, and a
binary layer of positive staining generated using the Image>Density slice… function.
Binary layers from different channels were combined using the Boolean Operations…
function to produce binary layers of fluorochrome colocalisation.
2.9.6.2 Quantification of epithelial surface staining
Binary layers, representing positive staining with ATII and/or ATI-selective markers,
were generated using image density slicing as described in section 2.9.6.1. Quantitative
assessment of alveolar epithelial composition was then made by measurement of the
length of epithelial surface positively staining in these binary layers using the
Measurement… functions in Openlab. The contribution of individual cell phenotypes to
the alveolar epithelial surface was reported as a percentage of total alveolar surface
measured.
2.9.6.3 Cell counts
In vitro, fixed cells staining positively with fluorescent markers were counted after
image density slicing using the Measurement… functions of Openlab. Total cell number

69
was determined using the nuclear stain To-Pro-3. Counts of cell viability using the
Live/Dead, assay are described in section 2.3.
2.9.6.4 Cell surface area
Cell surface area was determined by tracing around the cell membrane of E-cadherin or
phalloidin-stained cells using the Region of Interest… measurement tool of Openlab.
2.10 Rat model of Staphylococcus aureus pneumonia
The Staphylococcus aureus strain used in these study was wild-type strain 8325-4. This
was kindly provided by Dr. Tim Foster, Department of Microbiology, Moyne Institute of
Preventive Medicine, Trinity College, Dublin, Ireland. The experimental protocol used
is outlined in figure 15, and was based upon the model previously established and
characterised by McElroy et al. (McElroy et al. 1999).
2.10.1 Preparation of bacterial instillate
Bacterial stocks were stored in peptone broth containing 25% glycerol at –70°C. 36 h
before planned bacterial instillation, a scraping of glycerol stock was spread onto blood
agar base (BAB) plates (BD Biosciences) and incubated at 37°C for 18 h. 3ml of Todd
Hewitt broth (BD Biosciences) was then inoculated with a single bacterial colony
scraped from the BAB plate and the resulting broth incubated and shaken (220rpm) at
37°C for 18 h. On the day of instillation, 50µl/rat ((108cfu/rat) of the resulting
suspension was aliquoted into a sterile Eppendorf, washed twice with sterile endotoxin-
free PBS with Ca/Mg, centrifuging (11300g, 3min) between washes, and the bacterial
pellet resuspended in 1.5ml sterile endotoxin-free PBS with Ca/Mg and kept on ice. The
dose of viable bacteria instilled, expressed as colony forming units (cfu), was
determined by plating 50µl of 10-3
and 10-6
dilutions of instillate onto BAB plates and

70
incubating at 37°C overnight.
2.10.2 Bacterial instillation
Specific pathogen-free male Sprague-Dawley rats (250-300g; Harlan) were
anaesthetised by intraperitoneal injection of a mixture of fentanyl citrate (1mg/kg),
fluanisone (33mg/kg) and midazolam hydrochloride (17mg/kg). Once anaesthetised, rats
were suspended on a horizontal wire by the upper incisors and an intubation wedge
(modified 2ml Eppendorf; Fisher Scientific) placed in the mouth. A cannula (9cm
length, 0.96mm OD, Portex Fine Bore Polythene Tubing, Smiths Medical), with distal
end mounted on a 21G microlance needle, was inserted into the trachea and 0.5ml
bacterial suspension (~108 cfu) instilled into the lungs using a 1ml syringe. Control
animals were instilled with 0.5ml of sterile PBS. Instillation of bacterial suspension (or
PBS only in controls) was followed by injection of 0.5ml of air to facilitate dispersion of
bacteria to distal airways. Successful tracheal cannulation was distinguished by passage
of the tracheal cannula over palpable cartilagenous rings, and successful instillation was
typically accompanied by a brief episode of apnoea followed by tachypnoea.
2.10.3 Initial recovery and randomisation
Rats were given a subcutaneous injection of 1-2ml warmed sterile saline and recovered
on a heating pad inside a class II safety cabinet for (6 h before transfer to standard
housing. 72 h following instillation, animals were transferred to custom-built housing
units and randomised to either normoxic (air) or hyperoxic (93±2% O2) conditions for a
further 48 h.
2.10.4 Housing under normoxic and hyperoxic conditions
Units used to house rats under normoxic and hyperoxic conditions were comprised of

71
inner and outer clear acrylic chambers. Rats were housed within the inner chamber, and
this placed within the outer chamber. Vent holes in the sides of the inner chamber
allowed gas flow between chambers. Fresh gas was supplied to the outer chamber from
cylinder supply (medical-grade air normoxia, 100% O2 hyperoxia, BOC Gases) at 3
l/min and circulated throughout both inner and outer chambers by means of a coaxial fan
positioned on the external surface of the inner chamber. The outer box was vented to
room air ensuring normobaric conditions. CO2 accumulation was prevented through
absorption by soda lime placed between the outer and inner chambers. Rats had free
access to food and water. Temperature, lighting and humidity were maintained within
usual limits throughout. Once placed within the chambers, rats were left undisturbed for
48 h. O2 and CO2 tensions within the chambers were monitored continuously using a
Datex Normocap 200 sensor.
2.10.5 Harvesting of blood, bronchoalveolar lavage (BAL) fluid and lung tissue
At the end of the 48 h period in normoxic or hyperoxic conditions (5 days post-bacterial
instillation), rats were terminally anaesthetised by intraperitoneal injection of sodium
pentobarbitone (45mg/kg) mixed with heparin sulphate (500iu/rat). Laparotomy was
performed, and blood collected from the abdominal aorta. The chest was opened and
trachea cannulated (18G Monoject blunt needle, Harvard Apparatus). The lungs were
lavaged with 2x10ml sterile endotoxin- and Ca/Mg-free PBS, the volume of BALF
recovered recorded, and the lungs removed. Lungs for morphological analysis were
fixed (without lavage) by inflation to TLC with 4% paraformaldehyde in Ca/Mg-free
PBS, the trachea clamped to retain lung inflation, and the lungs removed and placed in
4% paraformaldehyde.
2.10.6 Bronchoalveolar lavage fluid processing
1ml of BALF was centrifuged (5000g, 5min) and the resulting cell pellet resuspended in
100-1000µl crystal violet. Total cell count was then determined using a

72
haemocytometer. Remaining BALF was centrifuged (212g, 6min) and the supernatant
aliquoted and stored at –70°C. The residual cell pellet was resuspended in Ca/Mg-free
PBS and (3x105 cells in 200µl cytospun (300rpm, 3mins) onto glass slides, air-dried and
stained with Diff-Quick, a modified Wright-Giemsa stain. Differential cell counts were
performed under oil immersion (100x) using characteristic cell morphology. A minimum
of 500 cells was counted and differential cell counts expressed as percentage of totals.
BALF protein concentration was measured using a Bradford protein assay (section 2.2).
2.10.7 Lung tissue processing for biochemical analysis
The left upper lobe was used in controls. When harvesting lungs from animals instilled
with bacteria, inflamed and/or consolidated lung was identified macroscopically and
separated. After weighing, lung tissue was stored at –70°C. Lung tissue was thawed on
ice, added to 5x lung weight of homogenisation buffer and homogenised (3x10sec,
PowerGen 125 Homogeniser, Fisher Scientific). The resulting homogenate was
centrifuged (300rpm, 3mins) to remove larger tissue fragments, aliquoted and stored at –
70°C. Homogenised lung protein concentration was measured using a Bradford protein
assay.
2.10.8 Lung tissue processing for morphological analysis
Lungs were left in 4% paraformaldehyde at 4°C overnight and then selected tissue cut
into 5mm cubes. Samples of left upper lobe were taken from control animals, and of
macroscopically inflamed and/or consolidated lung (typically left lung) from animals
instilled with bacteria. Tissue cubes were left in 30% sucrose solution at 4°C overnight
for cryoprotection. The following day blocks of tissue were submerged in optimal
cutting temperature compound (OCT), frozen in Freon 22 and then plunged into liquid
nitrogen. ~3"m sections were cut using a Bright cryostat (Huntington) by Susan Harvey.
Tissue sections were stored at -20°C until used.

73
2.11 Rat model of haemorrhagic shock-induced acute lung injury
The rat haemorrhagic shock model was developed jointly with Gareth Clegg.
Experiments were performed jointly, but subsequent characterisation of the
haemorrhagic shock-induced lung injury and biochemical analysis of bronchoalveolar
lavage fluid presented in this thesis was my own work. The experimental protocol used
is outlined in figure 39.
2.11.1 Animal preparation
Specific pathogen-free male Sprague-Dawley rats (250-300g, Harlan) were
anaesthetised by intraperitoneal injection of midazolam (5mg/kg) and ketamine (75-
100mg/kg) and placed supine on a temperature-regulated heating pad. A transverse
incision of the neck was made and the trachea, right common carotid artery and left
jugular veins exposed by blunt dissection. The common carotid was clamped proximally
and cannulated using PE50 tubing (BD Medical). This allowed continuous measurement
of arterial blood pressure (modified Propaq Encore monitor, WelchAllyn) and
intermittent arterial blood sampling. The left internal jugular vein was cannulated in a
similar manner, without proximal clamping, allowing central venous pressure
monitoring (modified Propaq Encore monitor, WelchAllyn) and the administration of
drugs and fluids. Anaesthesia was maintained with an intravenous infusion of propofol
(0.5mg/kg/min) and muscle relaxation with pancuronium bromide (2mg/kg/h). Normal
saline was administered intravenously such that intravenous fluids, including
medications, totalled 1.5ml/h. A tracheostomy was performed, the trachea cannulated
(18G Monoject blunt needle, Harvard Apparatus), and rats ventilated using a volume-
controlled ventilator (Small Animal Ventilator Model 683, Harvard Apparatus): tidal
volume 6ml/kg; positive end expiratory pressure (PEEP) 5cmH20; fractioned inspired
oxygen (FiO2) 1.0; respiratory rate 55-60 breaths/min adjusted to maintain pH between
7.30 and 7.45.

74
2.11.2 Experimental protocol
After a 30min stabilisation period, blood was removed from the carotid artery in 500µl
aliquots lowering the mean arterial blood pressure (MAP) to 30-40mmHg. This level of
haemorrhagic shock was maintained for 30min followed by resuscitation over 15-30min
with warmed intravenous normal saline restoring MAP to #80mmHg, whilst maintaining
CVP*10cmH2O. Rats were subsequently ventilated for an additional 4h and then
exsanguinated. Blood was collected from the abdominal aorta and the lungs lavaged
with 2x10ml sterile endotoxin- and Ca/Mg-free PBS to obtain BALF for analysis.
Control animals underwent surgery without subsequent haemorrhage.
BAL fluid was analysed for protein content (section 2.2), total and differential cell
counts (section 2.10.6) and, using ELISA-based dot blot assays, levels of the cell
selective proteins RTI40, MMC6 and MMC4 (section 2.8).
2.12 Statistics
Unless otherwise stated, data are expressed as the mean (taken from at least 3
independent experiments) ± standard deviation. Statistical analysis was carried out using
Graphpad Instat, 3.0 for Macintosh. Comparison of two means was performed using
unpaired t-tests assuming a two-tail p value. Comparison of multiple means was
performed using one-way ANOVA with post-test analysis and Bonferroni’s multiple
comparisons test where appropriate. p * 0.05 was considered statistically significant.
Data was tested for normality using the Kolmogorov-Smirnov test. There were too few
values to determine normality in the following data sets: adherent cell viability under
normoxic and hyperoxic conditions (figures 14); semiquantitative determination of
RTI40, MMC6 and MMC4 expression under normoxic and hyperoxic conditions (figures
17 & 19); LDH release and apoptosis of cells in culture under normoxic and hyperoxic
conditions (figures 23 & 24); ROS generation in response to standardised oxidative

75
stress by cells cultured under normoxic and hyperoxic conditions (figure 21B); BALF
protein, total cell count, differential cell counts and RTI40 content following S. aureus
instillation (figures 26, 27, 28 & 29); the proportion of the alveolar epithelial surface
with ATI-, ATII- and transitional-staining membrane following S. aureus instillation
(figures 36 & 38); deformation-induced injury in repairing monolayer wounds (figure
48); BALF protein, total cell count, differential cell counts, RTI40 and MMC6 content
following haemorrhagic shock (figures 51, 52 & 53). Data from these experiments was
also analysed using unpaired t-tests or one-way ANOVA, on the assumption of
normality.

76
3. The effect of hyperoxia on alveolar epithelial cells in culture
3.1 Introduction & objectives
Oxygen therapy is a potentially life-saving intervention in severe respiratory failure.
However, the therapeutic use of high fractions of inspired oxygen to prevent
hypoxaemia results in exposure of the alveolar epithelium to significant levels of
hyperoxia and oxidative stress. Hyperoxia is known to be injurious to the normal lung,
with prolonged exposure leading to a pattern of injury similar to ARDS (Crapo 1986;
Fracica et al. 1991). Damage to the alveolar epithelium is a central feature of this
hyperoxia-induced injury (Crapo 1986; Kazzaz et al. 1996; De Paepe et al. 2005).
The established paradigm postulates that ATI cells are more susceptible to injury than
ATII cells, and that following injury, damaged alveolar epithelium is replaced by the
proliferation of ATII cells and the differentiation of a subpopulation of daughter cells
into ATI cells (Uhal 1997). Hyperoxia has been shown to inhibit ATII cell proliferation
in vivo (Crapo 1986; O'Reilly et al. 1998) and to inhibit the differentiation of other cell
types in vitro (Zhang et al. 1999), but the effect of hyperoxia on the transdifferentiation
of ATII cells into ATI cells is not known.
The objective of the work outlined in this chapter was to use a series of in vitro
experiments to investigate the hypothesis that hyperoxia inhibits normal epithelial repair
by inhibiting the transdifferentiation of ATII cells into ATI cells.
Primary rat ATII cells are known to gradually lose their mature morphologic and
biochemical characteristics and adopt an ATI-like phenotype with time in culture
(Dobbs et al. 1985; Dobbs 1990). This transition from an ATII-like phenotype to an
ATI-like phenotype has obvious parallels to the processes thought to underlie normal
alveolar epithelial repair in vivo, and offers an in vitro model for the study of ATII cells,
ATI-like cells and the factors influencing the transition between the two cell types in
culture.
The first aim of the studies outlined in this chapter was therefore to establish the

77
isolation and culture of primary rat ATII cells, and using a panel of cell-selective
antibodies, characterise the phenotype of alveolar epithelial cells in culture. Having
characterised this in vitro system, the subsequent aim was to investigate the effect of
hyperoxia on the transition of ATII-like cells into ATI-like cells in culture.
The use of multiple cell-selective biomarkers working in the same assay facilitates the
correct assignment of cell type using this approach. MMC6 is a novel monoclonal
antibody (raised by McElroy) that appears to be recognised by ATI cells
(immunofluoresence screening during development). Preliminary data suggest that the
MMC6 antigen is lung-specific and distinct from RTI40 (Franklin and McElroy,
unpublished). To investigate the possibility that MMC6 may be a useful biomarker, a
further objective of the work outlined in this chapter was to characterise the expression
of the MMC6 antigen in both the adult and developing lung and by primary alveolar
epithelial cells in culture.
3.2 Results
3.2.1 Characterisation of freshly isolated ATII cells
The viability of freshly isolated cell suspensions at the time of seeding, determined by
exclusion of crystal violet, was >90% in all isolations used in these experiments. ATII
cell purity, determined by modified Papanicolau staining of cytospun freshly isolated
cell suspensions, was 68.8 ± 3.6% (figure 2A), rising to 82.3 ± 3.8% after rinsing 6 h
post-seeding (n=12). The major contaminants of ATII cell isolations were macrophages,
lymphocytes and neutrophils. The relative proportions of these are shown in (figure 2B).
Freshly isolated ATII cells co-expressed the ATII cell markers RTII70 and MMC4 but
did not express the ATI markers RTI40 or aquaporin 5 (figure 3A) (n=6).
3.2.2 Change of morphology and phenotype of ATII cells in culture
ATII cells cultured under normoxic conditions for 3 days, flattened and spread to form a

78
confluent monolayer. These cells expressed the ATI cell-selective markers RTI40 and
aquaporin 5. After 3 days in culture, cells did not express the ATII cell-selective markers
RTII70 and MMC4 (figure 3B) (n=6).
3.2.3 Identification of a novel intermediate cell phenotype
Loss of expression of the ATII cell marker RTII70 occurred by 24 h in culture, before
significant expression of the ATI marker aquaporin 5 was detected. However, a
subpopulation of cells (5.9 ± 1.4%) co-expressed the ATII marker MMC4 and the ATI
marker RTI40 after 24h in culture (n=3). Three dimensional imaging of RTI40/MMC4 co-
expressing cells revealed a more flattened shape than typical MMC4-expressing ATII
cells (figure 4A), with RTI40 expression localised to the spreading edge of the cells
(figure 4B).
3.2.4 The immunoselectivity of the monoclonal antibody MMC6
Localisation of MMC6 antigen expression in the adult lung: In control lungs, the
majority of the apical alveolar epithelial surface was comprised of thin attenuated cells
staining positively for the ATI-selective marker RTI40. These cells co-expressed MMC6
(figures 5, 6 & 7A) (n=3). RTII70 & MMC4 co-expressing cells were typically located
in the corners of the alveolar airspaces and were flanked by RTI40/MMC6 expressing
cells. RTII70/MMC4 co-expressing cells did not express RTI40 or MMC6 (figures 6 &
7B) (n=3). Endothelial cells, Clara cells and bronchiolar epithelial cells did not stain
positively for MMC6.
Developmental expression of MMC6 antigen: Foetal rat sections at different stages of
development were stained with combinations of MMC6, ATI-selective anti-RTI40 and
ATII-selective MMC4 and anti-RTII70 (n=3 for each developmental stage and

79
combination of primary antibodies used).
E16: At embryonic day 16 (E16), the size of the embryo enabled imaging of an entire
saggital section. No RTI40 or MMC6 staining was detected in the lung. The choroid
plexus and parts of the developing central nervous system stained positively for RTI40 as
previously described by Williams and colleagues (Williams et al. 1996), but did not stain
for MMC6. No MMC4 staining was detected at this time point (figure 8).
E17: At E17, RTI40 staining was detected on the apical surface of cuboidal cells lining
tubules within the embryonic lung. No significant MMC6 or MMC4 staining was
detected (figure 9).
E20: By E20 the embryonic lung had discernible airspaces with thick cellular alveolar
septae. The airspaces were lined with squamous cells staining positively for both RTI40
and MMC6 (figure 10). Separating these RTI40/MMC6 co-expressing cells were small
clusters of cells staining positively for RTII70 and MMC4. These cells did not stain for
either RTI40 or MMC6 (figure 11).
Expression of MMC6 antigen by alveolar epithelial cells in culture: Freshly isolated
ATII cells co-expressed the ATII cell markers RTII70 and MMC4 but did not express the
ATI cell marker RTI40 as described previously. Freshly isolated ATII cells did not
express MMC6 (figure 12A) (n=3). Loss of expression of the ATII cell marker RTII70
occurred by 24h in culture. At this time point a subpopulation of cells co-expressed
RTI40/MMC4 but MMC4/MMC6 coexpression was not detected (n=3).
After 3 days in culture, cells had flattened and spread to form a confluent monolayer of
ATI-like cells expressing RTI40 and MMC6 (figure 12B) (n=3). These cells did not
express the ATII cell markers RTII70 or MMC4. Deconvolution and 3D reconstruction
of the images obtained from MMC6/RTI40 coexpressing cells suggest that MMC6 is an
apical membrane protein (figure 13).

80
3.2.5 The effect of hyperoxia on ATII cells in culture
To determine the effect of hyperoxia on alveolar epithelial cells in culture, freshly
isolated ATII cells were cultured under either normoxic (media pO2 16.3 ± 1.6 kPa) or
hyperoxic (media pO2 95.4 ± 4.3 kPa) conditions for 3 days as described in materials
and methods (sections 2.1.3 & 2.1.4).
Adherent cell viability: There was no statistically significantly difference in the viability
of adherent cells cultured under normoxic and hyperoxic conditions after 3 d in culture
(98.0 ± 1.0% normoxia vs 93.4 ± 0.6% hyperoxia, p>0.05, n=3) (figure 14).
Cell spreading: Hyperoxia markedly impaired cell spreading. Cells cultured for 3 days
under normoxic conditions flattened and spread to form a confluent monolayer. These
cells had a mean surface area of 1801 ± 463µm2. In contrast, cells cultured under
hyperoxic conditions did not spread, but remained as small non-confluent clusters of
cells with a mean surface area of 583 ± 245µm2 (p<0.0001, n=3) (figure 15).
Expression of ATI markers: Hyperoxia inhibited the expression of the ATI-selective cell
markers, RTI40, aquaporin 5 and MMC6. Immunofluorescence analysis of normoxic-
exposed cells demonstrated that RTI40 (n=5), aquaporin 5 (n=3) and MMC6 (n=3) were
expressed on flattened alveolar epithelial cells after 3 days in culture. In contrast,
expression of each of these markers was greatly reduced in hyperoxic-exposed cells
(figure 16). The decrease in both RTI40 and MMC6 expression was (semi-) quantified
using ELISA-based dot blot assays. The concentration of RTI40 present in cells cultured
under normoxic conditions for three days was 13.0 ± 1.4 du/mg protein compared to
0.25 ± 0.2 du/mg protein in cells cultured under hyperoxic conditions, p<0.0001 (n=3)
(figure 17A). The concentration of MMC6 was 11.4 ± 1.7 du/mg protein for cells
cultured under normoxic conditions compared to 1.5 ± 1.1 du/mg protein for cells
cultured under hyperoxic conditions, p<0.05 (n=3) (figure 17B).

81
RTI40 expression in ATI-like cells: To determine the effect of hyperoxia on RTI40
expression in ATI-like cells, primary ATII cells were cultured under normoxic
conditions for 5 days and then exposed to hyperoxia for a further 2 days. There was no
significant change in morphology or difference in the level of RTI40 expression
determined by an ELISA-based dot blot assay between cells cultured under normoxic
conditions for 7 days and those switched to hyperoxic culture conditions on day 5 (10.8
± 1.9 du/mg protein normoxia vs 9.8 ± 0.4 du/mg protein d5 switch, p=0.3, n=3).
Expression of ATII markers: Hyperoxic exposure appeared to maintain the expression of
the ATII cell-selective markers MMC4 (n=3) and pro-SP-C (n=3) but not RTII70 (n=3)
in cultured ATII cells, with MMC4 and pro-SP-C detectable in epithelial cells cultured
under hyperoxic conditions for 3 days by immunofluorescence (figure 18). RTII70 was
not detectable by immunofluorescence in either normoxic- or hyperoxic- exposed cells
after 3 days in culture. Using an ELISA-based dot blot assay, the concentration of
MMC4 protein was determined to be more than 3-fold higher in cells cultured under
hyperoxic conditions compared with cells cultured under normoxic conditions (MMC4
55 ± 2 du/mg protein normoxia vs 187 ± 56 du/mg protein hyperoxia, p<0.01, n=3)
(figure 19).
Expression of E-cadherin: Reddy and colleagues have speculated that the ATII cell
population isolated from hyperoxia-treated rats is heterogeneous, with E-cadherin-
expressing cells more sensitive to hyperoxia-induced injury than cells with low levels of
E-cadherin expression (Reddy et al. 2004). To investigate the hypothesis that E-cadherin
negative cells may be more resistant to hyperoxia-induced injury, E-cadherin expression
in cells cultured under normoxic and hyperoxic conditions was examined by
immunofluoresence. Both cells cultured under normoxic and hyperoxic conditions
appeared to express E-cadherin heterogeneously. This pattern of staining was

82
reproducible, with strong expression at certain cell-cell contacts and weak expression at
others. Cells cultured under hyperoxic conditions appeared to have lower levels of E-
cadherin expression, with fewer strongly E-cadherin positive cells compared to cells
cultured under normoxic conditions (n=3) (figure 20).
Generation of ROS species: Hyperoxia-induced cytotoxicity is thought to be mediated
by the generation of reactive oxygen species. To investigate the hypothesis that cells
resistant to hyperoxia-induced injury generate fewer ROS, steady state ROS production
and the generation of ROS in response to oxidative stress was investigated in cells
cultured under normoxic and hyperoxic conditions. Day-3 cells cultured under hyperoxic
conditions demonstrated significantly lower steady state levels of ROS production than
those cultured under normoxic conditions (16 ± 5 vs 8 ± 3 fu/mg protein/min, p<0.05,
n=3). Hyperoxic-exposed cells also generated fewer ROS in response to standardised
exposure to oxidative stress (72 ± 8 vs 26 ± 4 fu/mg protein/min, p<0.001, n=3) (figure
21).
Proliferation: A small percentage (3.5 ± 1.0 %) of normoxia-exposed cells had BrdU-
incorporating nuclei. No BrdU-positive nuclei were detected in hyperoxic-exposed cells
(figure 22) (n=3).
Hyperoxia-induced cell cytotoxicity: Cell death, as determined by quantification of LDH
release, was observed in cells cultured under both normoxic and hyperoxic conditions at
all time points. There was no significant difference in the level of cell death determined
after 6 h (64.5 ± 2.1% normoxia vs 64.3 ± 10.7% hyperoxia, p>0.05, n=3) and 24 h in
culture (76.0 ± 4.4% normoxia vs 73.3 ± 17.2% hyperoxia, p>0.05, n=3), but after 3
days, there was significantly greater LDH release in cells cultured under hyperoxic
conditions (23.3 ± 8.3% normoxia vs 72.0 ± 2.6% hyperoxia, p<0.05, n=3). Significantly
greater LDH release was also observed under hyperoxic conditions after 5 days in

83
culture (16.0 ± 6.1% normoxia vs 70.5 ± 3.5% hyperoxia, p<0.05, n=3) (figure 23).
Hyperoxia-induced apoptosis: There was no statistically significant increase in the level
of apoptosis determined by Fuelgen staining with time in culture under normoxic
conditions, but the rate of apoptosis did increase with time in culture for cells cultured
under hyperoxic conditions. The difference in rate of apoptosis between cells cultured
under normoxic and hyperoxic conditions was not statistically significant after 6 h (1.7 ±
0.6% normoxia vs 2.7 ± 0.6% hyperoxia, p>0.05, n=3) and 24 h in culture (3.0 ± 1.7%
normoxia vs 5.7 ± 2.1% hyperoxia, p>0.05, n=3), but after 3 days, there was
significantly greater apoptosis in cells cultured under hyperoxic conditions (1.7 ± 0.6%
normoxia vs 6.3 ± 2.1% hyperoxia, p<0.05, n=3). Significantly greater rates of apoptosis
were also observed after 5 d in culture (3.0 ± 1.0% normoxia vs 7.7 ± 1.5% hyperoxia,
p<0.05, n=3) (figure 24).
The dose-response effect of the partial pressure of oxygen: The partial pressure of
oxygen in the culture media in these experiments was 95.4 ± 4.3 kPa under hyperoxic
conditions, and 16.3 ± 1.6 kPa under normoxic conditions, suggesting that these
experiments were conducted over a range of clinically relevant partial pressures of
oxygen. The effect of cell culture under hypoxic conditions (culture media pO2 4.3 ± 0.5
kPa) was investigated qualitatively in preliminary studies and appeared to have no effect
on adherent cell viability or the rate of transition from an ATII- to an ATI-like
phenotype with time in culture. Exposure of cells to 60% oxygen (culture media pO2
62.3 ± 1.9 kPa) also had no effect on adherent cell viability, but did qualitatively appear
to have an inhibitory effect on cell spreading and the expression of the ATI cell-selective
proteins RTI40 and MMC6. This effect was less dramatic than the effects described
following exposure to 95% oxygen and was not quantified.

84
3.2.6 The reversibility of hyperoxia-induced effects on ATII cells in culture
To determine the reversibility of the effects of hyperoxia on alveolar epithelial cells in
culture, freshly isolated ATII cells were cultured under hyperoxic conditions for 3 days
and then switched to normoxic conditions for a further 2 days.
The effects of hyperoxia on alveolar epithelial cell morphology and phenotype was
reversible. Cells cultured under hyperoxic conditions for 3 days and then transferred to
normoxic conditions spread and became confluent by day 5 in culture. These ‘d3
switched’ cells were viable (adherent cell viability 96.3 ± 3.1%, n=3) (figure 14) and
expressed the ATI cell-selective markers RTI40 (n=5), aquaporin 5 (n=3) and MMC6
(n=3) (figures 16 & 17) but not the ATII cell-selective markers MMC4 (n=3) and
RTII70 (n=3) (figure 19). There was a significant increase in cell proliferation following
the switch from hyperoxia to normoxia (13.1 ± 1.9% BrdU positive nuclei in switched
cells vs 0% in hyperoxia exposed cells, p<0.0001, n=3) (figure 22).
3.3 Discussion
The yield, viability and purity of the primary ATII cells used in these experiments was
in keeping with those published by other investigators (Dobbs et al. 1986; Dobbs 1990;
Chen et al. 2004).
3.3.1 ATII cells transdifferentiate into ATI-like cells in culture
Freshly isolated ATII cells co-expressed the ATII cell-selective markers RTII70 and
MMC4 (aminopeptidase N) but not the ATI cell-selective markers RTI40, aquaporin 5 or
MMC6, reflecting an ATII-like phenotype in freshly isolated cells. After 3 days in
culture under normoxic conditions this pattern of cell-selective protein expression
changed, with cells expressing the ATI cell-selective markers RTI40, aquaporin 5 and
MMC6 but not the ATII cell-selective markers RTII70 and MMC4. This change in
expression of cell-selective proteins was accompanied by a change in morphology as

85
cells flattened and spread. The loss of characteristics associated with the ATII cell
phenotype and acquisition of characteristics of the ATI cell phenotype under normoxic
conditions supports the widely held belief that primary ATII cells transdifferentiate from
an ATII-like phenotype to an ATI-like phenotype with time in culture (Dobbs et al.
1985; Dobbs et al. 1988; Borok et al. 1998; Borok et al. 1998; Campbell et al. 1999).
3.3.2 Identification of a novel intermediate cell phenotype
The finding of a subpopulation of cells coexpressing the ATII cell-selective protein
MMC4 and the ATI cell-selective protein RTI40 after 1 day in culture and before cells
could be defined as ATI-like is consistent with this population of cells representing an
intermediate or transition cell phenotype in the ATII to ATI cell transition. Although
others have described putative intermediate cell phenotypes (Fehrenbach et al. 1999;
McElroy and Kasper 2004), the coexpression of MMC4 and RTI40 is a novel finding.
The functions of MMC4 and RTI40 are unknown, and it is not known whether the
coexpression of MMC4 and RTI40 has any functional significance, or simply reflects the
fact that the MMC4 antigen has a longer half-life than RTII70. 3D visualisation of
MMC4/RTI40 coexpressing cells demonstrates that these cells have begun to spread and
flatten with RTI40 expression appearing localised to the spreading cell edge. Coupled
with the findings that RTI40 appears to be associated with increased migration rates in
wounding experiments (Schacht et al. 2003) and is associated with organisation of the
actin cytoskeleton (Scholl et al. 1999), these data support the speculation that RTI40 has
a role in cell spreading and migration.
3.3.3 The MMC6 antigen appears to be ATI-selective
In control lungs, the MMC6 antigen appeared to be coexpressed on the apical surface of
RTI40-expressing ATI cells. The MMC6 antigen did not appear to be expressed by ATII
cells, lung parenchymal cells or inflammatory cells. In vitro data, showing that the

86
MMC6 antigen was expressed by alveolar epithelial cells with an ATI-like phenotype
but not by freshly isolated primary ATII cells, provides further support for the
hypothesis that the MMC6 antigen is ATI-selective.
The MMC6 antigen appeared to be developmentally regulated, being co-expressed with
RTI40 in epithelial cells by E20, but absent in RTI40-expressing cells at E17. The fact
that RTI40 expressing cells did not bind MMC6 MAb in E17 lungs or E16 choroid
plexus suggests that RTI40 and MMC6 are different antigens.
Together, these data suggest that the MMC6 MAb is ATI-selective and recognises an
antigen distinct from RTI40. This suggests that MMC6 may be a useful additional
biomarker for the assignment of alveolar epithelial cell type both in vivo and in vitro.
Work to characterise the antigen is ongoing (Franklin & McElroy).
3.3.4 Hyperoxia reversibly inhibits transdifferentiation in vitro
Hyperoxia inhibited both the expression of ATI cell-selective proteins and the change in
morphology characteristic of the transition of alveolar epithelial cells from an ATII- into
an ATI-like phenotype with time in culture. The inhibition of transdifferentiation by
hyperoxia is a novel finding in relation to alveolar epithelial cells, although hyperoxia
has been shown to inhibit the differentiation (stellation) of astrocytes in the retina
(Zhang et al. 1999).
Hyperoxia also appeared to inhibit loss of expression of the ATII cell-selective marker
MMC4, although it did not appear to inhibit loss of RTII70 expression. Whether
inhibition of the loss of MMC4 expression has any functional significance, or simply
reflects the fact that the MMC4 antigen has a longer half-life than RTII70 under
hyperoxic conditions is not known, but after 3 days in culture under hyperoxic
conditions, alveolar epithelial cells appeared relatively undifferentiated. It has been
suggested that the dedifferentiation of ATII cells, with the gradual lose of differentiated
morphological and biochemical characteristics, is independently regulated from the
subsequent transdifferentiation into ATI cells (Gonzalez et al. 2005). Whether

87
dedifferentiation is an in vitro phenomenon remains unknown, but these data support the
hypothesis that hyperoxia inhibits the acquisition of an ATI-like phenotype
(differentiation), and may also inhibit the loss of an ATII-like phenotype
(dedifferentiation) in culture.
The finding that hyperoxia did not significantly affect the morphology or expression of
RTI40 in ATI-like cells suggests that hyperoxia was not simply a signal resulting in loss
of alveolar epithelial cell differentiation. It also suggests that the observed inhibition of
ATII transdifferentiation is not solely attributable to a global hyperoxia-induced
inhibition of protein synthesis. The fact that the inhibition of transdifferentiation was
reversible, with cells switched from hyperoxia to normoxia able to spread, flatten and
adopt an ATI-like phenotype, also suggests that the effect of hyperoxic exposure was not
caused by irreversible cell cytotoxicity.
3.3.5 Hyperoxia is cytotoxic to alveolar epithelial cells
The release of greater levels of LDH in hyperoxia-exposed cells compared to those
cultured under normoxic conditions confirms that prolonged exposure to hyperoxia is
cytotoxic (at least to some cells) and results in significant alveolar epithelial cell
necrosis. The increase in the rate of apoptosis following prolonged exposure to
hyperoxia also suggests that hyperoxic exposure is pro-apoptotic, and that the
mechanism of hyperoxia-induced cell death is, at least in part, apoptotic. These data lend
support to the concept suggested by other in vitro studies that hyperoxia may activate a
cellular death pathway sharing features of both apoptosis and necrosis (Kazzaz et al.
1996; Wang et al. 2003; De Paepe et al. 2005).
3.3.6 A subpopulation of cells appears resistant to hyperoxia-induced injury
Cell death in vitro is typically accompanied by cell detachment from the culture surface,
and the finding that there was no difference in adherent cell membrane integrity

88
(viability) between cells cultured under hyperoxic and normoxic conditions supports
this. However, these data also suggest that hyperoxia is not universally cytotoxic, and
that some cells may have a greater propensity to withstand hyperoxic insult.
3.3.7 E-cadherin negative cells appear resistant to hyperoxia-induced injury
In a study examining the phenotype of ATII cells isolated from hyperoxia-treated rats,
Reddy and colleagues found that the isolated ATII cell population could be functionally
segregated based on their expression of E-cadherin (Reddy et al. 2004). The E-cadherin
positive subpopulation contained damaged cells and was non-proliferative with low
levels of telomerase activity, whilst in contrast, the E-cadherin negative subpopulation
was undamaged, proliferative and expressed high levels of telomerase activity. These
data suggest that the ATII cell population isolated from hyperoxia-treated rats is
heterogeneous, with E-cadherin expressing cells more sensitive to hyperoxia-induced
injury than cells with low levels of E-cadherin expression. On the basis of these
findings, Reddy and colleagues speculated that the E-cadherin negative subpopulation
might be the source of the transiently amplifying cells responsible for epithelial repair
following injury.
To investigate the hypothesis that E-cadherin negative alveolar epithelial cells may be
more resistant to hyperoxia-induced injury, E-cadherin expression in cells cultured under
normoxic and hyperoxic conditions was investigated in the studies outlined in this
chapter. The pattern of E-cadherin expression observed by immunofluoresence was
heterogeneous in both normoxia and hyperoxia-exposed cells, with strong expression at
certain cell-cell contact points but not at others. However there appeared to be less E-
cadherin expression, with fewer E-cadherin positive cells, in cells cultured under
hyperoxic conditions compared to those cultured under normoxic conditions. This
finding may simply reflect downregulation of E-cadherin by hyperoxia, but would also
support the concept that hyperoxia-induced injury of E-cadherin positive cells resulted in
cell death and detachment of these cells leaving an adherent cell population with a

89
greater proportion of E-cadherin negative cells.
3.3.8 Hyperoxia-resistant cells generate fewer ROS
In addition to differing levels of E-cadherin expression, cells cultured under hyperoxic
conditions appeared to have a lower steady state production of ROS than cells cultured
under normoxic conditions, and to generate fewer ROS in response to oxidative stress.
ROS are thought to play a key role in mediating hyperoxia-induced cell injury; both
through a direct cytotoxic effect and through activation of the redox-sensitive
transcription factor NF-'B. Reducing ROS production in the face of hyperoxic exposure
is therefore likely to have a cytoprotective effect. The finding of reduced ROS
production in cells cultured under hyperoxic conditions may reflect such an adaptive
response, with fewer mitochondria or upregulation of antioxidant defences.
Alternatively, lower ROS production may further support the concept of a
heterogeneous ATII cell population, and simply reflect the fact that the cells surviving 3
days of hyperoxic exposure are phenotypically distinct and constitutively generate fewer
ROS in response to oxidative stress.
In an attempt to understand the role of ROS and NF-'B in the mechanism of hyperoxia-
induced inhibition of ATII cell transdifferentiation, preliminary studies were conducted
in which hyperoxia-exposed cells were incubated with the antioxidant and free radical
scavenger N-acetylcysteine and NF-'B inhibited by the addition of sulfasalazine (SSA)
and the inhibitory peptide SN-50. Neither the addition of antioxidant or inhibition of
NF-'B appeared to attenuate the effect of hyperoxia on ATII cell transdifferentiation
determined by change in cell morphology and RTI40 expression.
3.3.9 Hyperoxia reversibly inhibits alveolar epithelial cell proliferation
Primary ATII cells are thought to proliferate to a limited degree in culture (Dobbs 1990),
and this concept is supported by the finding of low proliferative rates in cells cultured

90
under normoxic condition in these studies. Whilst exposure of animals to 100% oxygen
has previously been shown to inhibit ATII cell proliferation in vivo (Crapo 1986), the
effect of hyperoxia on the proliferation of primary ATII cells in culture does not appear
to have been investigated previously. The studies presented in this chapter demonstrate
for the first time that hyperoxia is a potent inhibitor of ATII cell proliferation in vitro.
Switching cells from hyperoxic to normoxic conditions was followed by a proliferative
response in these studies, with BrdU incorporating cells going on to express the ATI
cell-selective protein RTI40. This is a novel finding, and supports the concept that short
term exposure of ATII cells to sublethal hyperoxia inhibits proliferation, with removal of
this inhibitory effect upon switching cells to normoxic conditions being followed by
ATII cell proliferation and transdifferentiation (Adamson and Bowden 1974; Thet et al.
1986; Yee et al. 2006).
3.4 Summary
The data presented in this chapter demonstrate for the first time that clinically relevant
levels of hyperoxia reversibly inhibit the normal transition of ATII-like cells into ATI-
like cells with time in culture. This suggests that hyperoxia may significantly inhibit
alveolar epithelial repair following injury in vivo. The findings of these in vitro studies
also support the hypothesis that the ATII cell population harbours a subpopulation of
cells, with lower levels of E-cadherin expression and ROS generation, resistant to
hyperoxia-induced injury. This subpopulation of hyperoxia-resistant cells may reflect
the presence of a transiently amplifying cell population cell within the isolated ATII cell
population.

91
4. The effect of hyperoxia on alveolar epithelial repair in vivo
4.1 Introduction & objectives
The finding that hyperoxia inhibits the change in cell morphology and expression of
cell-selective proteins associated with the transition of alveolar epithelial cells from an
ATII-like phenotype to an ATI-like phenotype with time in culture suggests that
hyperoxia may significantly inhibit the transdifferentiation of ATII cells during alveolar
epithelial repair (chapter 3). However, a recent study demonstrating that freshly isolated
ATI cells, ATII cells and cultured ATII cells have distinct molecular phenotypes
(Gonzalez et al. 2005), highlights the potential limitations inherent in extrapolating
biological relevance from these in vitro studies.
To investigate the hypothesis that hyperoxia inhibits alveolar epithelial repair in vivo, the
studies outlined in this chapter examined the effect of hyperoxia on the resolution of
lung injury in a characterised rat model of Staphylococcus aureus (S. aureus)-induced
pneumonia.
The model used was based upon a model of S. aureus-induced lung injury established
and characterised by McElroy and colleagues, in which rats are inoculated by
intratracheal instillation of S. aureus and then allowed to recover in standard (normoxic)
conditions (Bachofen et al. 1987; McElroy et al. 1999; Clegg et al. 2005). S. aureus is a
common cause of both pneumonia and (direct) acute lung injury (sections 1.2.2 and
1.2.3), and in the McElroy model, animals appear to develop a reproducible
inflammatory injury with damage to the air-blood barrier and ATI cell necrosis a central
feature. Previous characterisation of this S. aureus-induced injury has shown significant
ATII cell hyperplasia at day 3 post-instillation which resolves by day 7 post-instillation
as a result of the transdifferentiation of ATII cells into ATI cells and (probably) ATII
cell apoptosis (Clegg et al. 2005).
To examine the effect of hyperoxia on the resolution of the ATII cell hyperplasia
observed at day 3 in this model, rats were inoculated with S. aureus and recovered for 3
days in standard (normoxic) conditions in an identical manner, but then randomised to

92
either normoxic (air) or hyperoxic (95% O2) custom-built animal chambers for a further
2 days as detailed in materials and methods (section 2.10) (figure 25). The hypothesis
was that hyperoxic exposure would inhibit the normal resolution of ATII hyperplasia.
Having established this model, the first objective of the studies described in this chapter
was to characterise the lung injury at day 5 following instillation, comparing the extent
of injury in animals exposed to hyperoxia with that in animals exposed to normoxia
alone. The second objective was to use a panel of ATI- (RTI40) and ATII-selective
(RTII70 and MMC4) antibodies, coupled with laser scanning confocal microscopy, to
determine the proportion of the alveolar surface associated with ATI, ATII and
intermediate (MMC4/RTI40-coexpressing) cells to quantify the effect of hyperoxic
exposure on the changes in these cell populations during the resolution of lung injury.
4.2 Results
A total of 24 animals were randomised to either PBS-instilled control (n=8, 8 analysed),
S. aureus installation followed by recovery in normoxia for 120h (n=8, 7 analysed) or S.
aureus installation with recovery in normoxia for 72h followed by 48h in hyperoxia
(n=8, 8 analysed). In the S. aureus-instilled groups, rats received a dose of (3.2 ± 1.1) x
108 cfu. One animal in the S. aureus normoxia group appeared distressed with marked
tachypnoea 24h following S. aureus installation and was withdrawn from the
experimental protocol for the purposes of animal welfare. This animal was excluded
from subsequent analysis.
4.2.1 S. aureus-induced lung injury and the effect of hyperoxia
To characterise the effect of hyperoxia on the resolution of S. aureus-induced injury
total BALF protein, BALF cell counts and BALF RTI40 were determined.
BALF protein: The total protein recovered from BALF of control (PBS-instilled) lungs

93
was 1.1 ± 0.2mg (n=5). In S. aureus-instilled lungs exposed to normoxia alone this rose
to 2.8 ± 1.0mg (n=4), whilst in lungs instilled with S. aureus recovered in normoxia for
3 days and then exposed to hyperoxia for 2 days the total amount of protein recovered
was 5.9 ± 2.4mg (n=5) (figure 26).
BALF cell counts: The total number of leucocytes recovered from BALF was 14 ± 7 x
104 in control lungs (n=5), 314 ± 169 x 10
4 in S. aureus-instilled lungs exposed to
normoxia alone (n=4) and 530 ± 303 x104 in S. aureus-instilled lungs exposed to
hyperoxia (n=5) (figure 27A). Differential cell counts revealed that the majority of
leucocytes recovered were alveolar macrophages, but the presence of significant
numbers of neutrophils in the S. aureus-instilled lungs (0.3 ± 0.6% control lungs, 8.0 ±
2.8% instilled lungs exposed to normoxia alone and 9.3 ± 7.9% instilled lungs exposed
to hyperoxia) confirmed the presence of an acute inflammatory injury in these lungs
(figure 27B).
BALF RTI40: To determine whether there was significant ATI cell injury, BALF RTI40
content was measured. The total amount of RTI40 in BALF recovered from control lungs
was 9.8 ± 3.0 du (n=5). In S. aureus-instilled lungs exposed to normoxia alone this fell
to 7.8 ± 3.6 du (n=4), whilst in lungs instilled with S. aureus and exposed to hyperoxia
the total amount of RTI40 recovered was 5.6 ± 4.5 du (n=5). The observed differences in
BALF RTI40 content were not statistically significant (p=0.4) (figure 28).
4.2.2 The phenotype of the alveolar epithelium in control lung
The method used for quantification of alveolar epithelial surface staining with individual
cell-selective markers was based upon that previously described by Clegg and
colleagues (Clegg et al. 2005), as detailed in materials and methods (section 2.9.6) and
illustrated in figure 30.

94
ATI (RTI40) staining membrane: In control lungs (n=3), 93.6 ± 1.6% of the apical
alveolar epithelial surface stained positively for the ATI cell-selective antibody RTI40
(figures 31 & 36A).
ATII (RTII70 and MMC4) staining membrane: 2.4 ± 0.4% of the alveolar epithelial
surface in control lungs stained positively for both of the ATII-selective proteins RTII70
& MMC4. RTII70 & MMC4 co-expressing cells were typically located in the corners of
the alveolar airspaces and were flanked by RTI40-expressing cells (figure 31 & 36B).
The lengths of alveolar epithelial surface co-expressing RTII70 & MMC4 in control lung
was normally distributed with a mean of 7.6 ± 3.6µm, representing the mean apical
surface length of ATII cells (figure 31 & 37). Small airways in control lung sections
were lined with cuboidal, non-ciliated epithelial cells, morphologically typical of Clara
cells. These cells stained positively for MMC4 but not for RTII70 or RTI40 as described
previously (Boylan et al. 2001) (figure 31).
Intermediate cell (RTI40 and MMC4) staining membrane: In control lungs, 0.6 ± 0.1% of
the alveolar epithelial surface stained positively for both RTI40 and MMC4 (figure 38).
The majority of this positively staining membrane was <1µm in length (mean 0.8 ±
1.0µm), and occurred at the junction of ATI and ATII cells.
4.2.3 The effect of hyperoxia on the proportion of ATI, ATII and intermediate cell-
staining membrane following S. aureus-induced lung injury
ATI (RTI40) staining membrane: In S. aureus-instilled lungs exposed to normoxia alone
(n=3), 84.4 ± 2.6% of the alveolar epithelial surface stained positively for the ATI-
selective protein RTI40. This fell to 73.4 ± 5.3% in S. aureus-instilled lungs exposed to
hyperoxia (n=3, p<0.05) (figures 32, 33, 34, 35 & 36).

95
ATII (RTII70 and MMC4) staining membrane: In S. aureus-instilled lungs exposed to
normoxia alone (n=3), 7.7 ± 1.1% of the alveolar epithelial surface stained positively for
both RTII70 and MMC4 (figures 32, 33 & 36). The mean length of RTII70/MMC4 co-
expressing membrane was 7.3 ± 6.8µm. However, the distribution of the lengths of
RTII70 and MMC4 staining membrane was markedly different from that observed in
control lungs, with a greater range and a negative skew (figure 37). In S. aureus-instilled
lungs exposed to hyperoxia (n=3), 15.5 ± 1.2% of the alveolar epithelial surface stained
positively for both RTII70 and MMC4 (figures 34, 35 & 36), with a mean length of
positively staining membrane of 9.6 ± 6.6µm. The distribution of lengths of positively
staining membrane was binomial, with peaks around (7, 14µm (figure 37).
Intermediate cell (RTI40 and MMC4) staining membrane: In S. aureus-instilled lungs
exposed to normoxia alone (n=3), 11.9 ± 4.2% of the alveolar epithelial surface co-
expressed RTI40 and MMC4 (figures 32, 33 & 38), with a mean length of positively
staining membrane of 6.5 ± 11.2µm. In S. aureus-instilled lungs exposed to hyperoxia
(n=3), 3.0 ± 0.8% of the alveolar epithelial surface stained positively for RTI40 and
MMC4 (figures 34, 35 & 38), with a mean length of co-expressing membrane of 3.3 ±
4.8µm.
4.2.4 The effect of hyperoxia on lung RTI40 concentration following S.aureus-
induced lung injury
Lung samples were obtained from the left upper lobe (controls) and macroscopically
inflamed regions (S. aureus-instilled lungs) and homogenised as detailed in materials
and methods (section 2.10.7). The concentration of RTI40 showed a two-fold decrease in
S. aureus-instilled lungs exposed to hyperoxia compared to those exposed to normoxia
alone (41.4 ± 19.8 vs 18.8 ± 10.4 du/mg protein, n=3, p<0.05). Both lungs exposed to
hyperoxia, and those exposed to normoxia alone, had significantly lower concentrations
of RTI40 than control lung (330.3 ± 88.0 du/mg protein, n=3, p<0.001) (figure 29). In
contrast, PBS-instilled (control) lungs exposed to hyperoxia had a significantly higher

96
concentration of RTI40 compared to control lungs exposed to normoxia alone (566.0 ±
32.5 vs 330.3 880 du/mg protein, n=3, p<0.05).
4.3 Discussion
4.3.1 The effect of hyperoxic exposure on the resolution of lung injury
S. aureus instillation resulted in an inflammatory injury to the lungs in animals
recovered in air (normoxia) alone and in those recovered in air for three days then
switched to hyperoxia (90-95% O2). This was evident by an increase in the total protein
content and number of leucoytes, both neutrophils and macrophages, in BALF recovered
from S. aureus-instilled lungs. Injury was most significant in the lungs of animals
exposed to hyperoxia. Previous characterisation of the model used in these studies has
demonstrated that, under normoxic conditions, the inflammatory response to S. aureus
(ie. the total number of BALF leucocytes) and alveolar wall damage (as assessed by the
total amount of protein in BALF) is maximal at day 1, resolving by day 3 and almost
completely resolved by day 7 following instillation (McElroy et al. 1999; Clegg et al.
2005). At day 3 following S. aureus instillation, the total protein recovered in BALF was
elevated 1.6-fold above control values in these studies compared to a 6.8-fold elevation
at day 1 post-instillation and the total number of BALF leucocytes was elevated 2.5-fold
at day 3 compared to a 57-fold elevation at day 1. This is consistent with the finding
that, although still elevated, the alveolar protein content and number of leucocytes
recovered from S. aureus-instilled lungs exposed to normoxia alone in these studies was
not significantly different from controls at day 5 following instillation. The fact that
hyperoxic exposure of S. aureus-instilled lungs resulted in a significant increase in these
markers of lung injury however, suggests that hyperoxia was either causing an
inflammatory lung injury itself and/or inhibiting the normal resolution of the pre-
existing lung injury.
Continuous exposure of rats to 100% oxygen has been shown in several studies to cause
a progressive inflammatory lung injury, with alveolar epithelial injury and ATI cell

97
necrosis a characteristic feature (Crapo et al. 1982; Crapo 1986; Fracica et al. 1991).
However, these studies refer to the effect of hyperoxia on the normal lung and not it’s
effect on injured lung, and the finding that the amount of RTI40 recovered from BALF, a
well-characterised marker of ATI cell necrosis (McElroy et al. 1995; McElroy et al.
1997), was not significantly elevated in hyperoxia-exposed animals suggests ongoing
ATI cell necrosis was not a central feature of the injury observed.
4.3.2 The phenotype of the alveolar epithelium in control lungs
In control lungs, the alveoli were lined with RTI40-positive (ATI) and RTII70/MMC4-
positive (ATII) cells as reported previously (Boylan et al. 2001; Clegg et al. 2005). The
distribution and extent of the alveolar epithelial surface covered by the two distinct
epithelial cell types determined using the immunotargeting methodology adopted in
these studies was in keeping with conventional (EM) morphological analysis, with Haies
and colleagues reporting an ATI:ATII surface area ratio of 39:1 in normal lungs, almost
identical to that observed in these studies (93.6% ATI: 2.4% ATII) (Haies et al. 1981;
Crapo et al. 1982).
The individual lengths of RTII70/MMC4 coexpressing membrane in control lungs were
normally distributed, with a mean (and mode) of (7µm, reflecting the mean apical
length of an ATII cell. This is also in keeping with the findings of the morphological
studies conducted by Haies and colleagues, which (excluding microvilli and assuming a
circular surface) suggest an apical ATII surface diameter of 8-9µm (Haies et al. 1981).
4.3.3 The phenotype of the alveolar epithelium in resolving S. aureus-induced lung
injury
Examination of the phenotype of S. aureus-instilled lungs exposed to normoxia alone
revealed a significant decrease in ATI cell-staining (RTI40-positive) membrane
consistent with a significant decrease in the proportion of the alveolar epithelial surface

98
covered by ATI cells in these lungs. The observed decrease in lung homogenate RTI40
concentration is consistent with this finding.
There was a 3-fold increase in ATII cell-staining (RTII70/MMC4-positive) alveolar
membrane in normoxia-exposed lungs in keeping with an increase in the population of
ATII cells following injury. However, there was also a dramatic increase in the
proportion of the epithelial surface coexpressing the ATI cell-selective protein RTI40 and
the ATII cell-selective protein MMC4 (11.9% vs 0.6% controls). Deconvolution of high
magnification images has previously confirmed that these proteins colocalise within the
limits of resolution of confocal microscopy ((0.5µm), confirming coexpression on the
same cell membrane, rather than apparent coexpression as an artefact of ATI cells
overlaying ATII cells (Clegg et al. 2005). The finding of RTI40/MMC4-coexpressing
cells both ex vivo in repairing lungs and in vitro as cultured ATII cells lose ATII cell
phenotypic characteristics and gain ATI-like characteristics (see chapter 3), supports the
concept that these cells are kinetically competent to be intermediate cells. The
observation that a significant number of the RTI40/MMC4-positive cells had a luminal
surface greater than that of ATII cells in control lungs, also suggests that these cells are
in the process of becoming attenuated ATI cells.
Together, these data are consistent with the accepted paradigm of alveolar epithelial
repair: damaged ATI cells being replaced by the proliferation of ATII cells and the
subsequent resolution of ATII cell hyperplasia by the transdifferentiation of a
subpopulation of these cells into ATI cells (Uhal 1997). Comparison of the data obtained
in these studies with that obtained by Clegg and colleagues from characterisation of
alveolar epithelium at day 3 following S. aureus instillation supports this interpretation,
suggesting that the alveolar epithelial repair occurring under normoxic conditions
between days 3 to 5 following instillation is associated with an increase in ATI-staining
membrane (from ~80% at day 3 in the studies of Clegg and colleagues to 84% in these
studies) and decrease in ATII-staining membrane (from ~13% at day 3 in the studies of
Clegg and colleagues to 8% in these studies) (Clegg et al. 2005).

99
4.3.4 The effect of hyperoxia on phenotype of the alveolar epithelium in resolving S.
aureus-induced lung injury
In S. aureus-instilled lungs exposed to hyperoxia, both the proportion of ATI cell-
staining membrane and lung RTI40 content were reduced significantly in comparison to
controls and to S. aureus-instilled animals exposed to normoxia alone. This is unlikely to
reflect downregulation of RTI40 expression by hyperoxia because PBS-instilled (control)
lungs exposed to hyperoxia and non-inflamed lung from hyperoxia exposed S. aureus-
instilled lungs showed a significant increase in the concentration of RTI40. Published
work by Cao and colleagues also suggests that RTI40 (T1$) in mice is upregulated by
hyperoxic exposure (Cao et al. 2003). It therefore seems likely that the observed
decrease in ATI cell-staining membrane reflects a significant decrease in the proportion
of the alveolar epithelial surface covered by ATI cells in hyperoxia-exposed S. aureus-
instilled lungs. Consistent with this, there was a 6-fold increase in the proportion of ATII
cell-staining alveolar membrane in these lungs.
In S. aureus-instilled lungs exposed to hyperoxia, not only was there a significant
increase in the proportion of ATII cell-staining membrane, but there was also a
difference in the distribution of RTII70/MMC4 coexpressing membrane lengths, with a
significant number of longer lengths of coexpressing membrane and a trinomial
distribution with peaks around 7, 14 and 22µm (figure 27). Assuming that the surface
area of ATII cells is comparable in both control and injured lungs, this finding is
consistent with the concept of significant ATII cell hyperplasia in the injured lung, with
2 and (less frequently) 3 ATII cells lying adjacent to one another.
Hyperoxia has been shown to inhibit ATII cell proliferation both in vivo (Crapo 1986;
O'Reilly et al. 1998; O'Reilly et al. 1998) and in vitro (see chapter 3), and this would
suggest that the ATII cell proliferation observed in hyperoxia-exposed lungs occurred
during the 3-day recovery of the S. aureus-instilled animals in normoxia and prior to
transfer into hyperoxic conditions. This, in combination with the fact that the proportion
of ATII cell-staining membrane was significantly greater in hyperoxia-exposed animals
compared to those exposed to normoxia alone suggests that hyperoxia significantly

100
inhibits the resolution of ATII hyperplasia and the restoration of normal cell numbers
which occurs during normal repair.
The ATII cell hyperplasia associated with normal alveolar epithelial repair is thought to
be resolved by a combination of apoptosis and differentiation of ATII cells into ATI
cells (Fehrenbach et al. 1999; Fine et al. 2000). Failure of this resolution has been
speculated to be a key event in the disruption of epithelial-fibroblast interactions and
progression to abnormal epithelial repair with fibrosis (see section 1.3.4) (Adamson et
al. 1988).
As hyperoxia is pro-apoptotic to ATII cells (see chapter 3) (Haddad and Land 2000;
Lee and Choi 2003; Buccellato et al. 2004), this suggests that the lack of resolution of
the ATII cell hyperplasia observed in hyperoxia-exposed S. aureus-instilled lungs is
largely due to the inhibition of the differentiation of ATII cells into ATI cells. This
concept is supported by the in vitro data presented in this thesis (see chapter 3), and also
by the finding of significantly less intermediate (RTI40/MCC4 coexpressing) membrane
in the lungs of animals exposed to hyperoxia compared to those exposed to normoxia
alone (11.9% vs 3.0%).
4.3.5 General discussion
The mechanism by which hyperoxia inhibits the differentiation of ATII cells into ATI
cells in the repairing lung was not investigated. However several studies have shown
that upregulation of insulin-like growth factor (IGF) components occurs during
hyperoxia-induced lung injury (Besnard et al. 2001; Chetty and Nielsen 2002;
Narasaraju et al. 2006), and a recent study has demonstrated that addition of antibodies
to the IGF-1 receptor to ATII cells in culture inhibits the transition of cells from an
ATII-like to an ATI-like phenotype with time in culture (Narasaraju et al. 2006). The
change in expression profile of the IGF system might therefore be speculated to have a
central role in the mechanism. It has also been suggested that transdifferentiation is
modulated by signalling through the Smad-dependent TGF-&1 pathway (Bhaskaran et al.

101
2007), raising the possibility that hyperoxia may inhibit transdifferentiation through
inhibition of TGF-&1 signalling.
Intratracheal instillation of S. aureus produced a reproducible, heterogeneous pattern of
injury most frequently affecting the left lung. This is in keeping with other published
models of pulmonary infection in the rat (Bakker-Woudenberg 2003). The left upper
lobe was taken consistently for biochemical and morphological analysis from control
animals, but in S. aureus-instilled animals macroscopically inflamed lung was readily
identifiable and was separated from non-inflamed lung prior to tissue processing as
described in materials and methods (section 2.10.7 & 8). A limitation of this approach,
inherent in the morphological analysis of any model producing a heterogeneous pattern
of injury, was that initial tissue sampling was based upon identification of
macroscopically inflamed areas and was not strictly random. This increases the
likelihood of sampling error. However, the aim of these studies was to compare discrete
areas of repairing lung with relatively homogeneous uninjured (control) lung. As such,
the data presented are not reflective of the whole lung, but rather a representative region
of injured repairing lung and its reponse to hyperoxia.
Hyperoxic exposure in these experiments was achieved by means of animal housing in
custom-built chambers as described in materials and methods (section 2.10.4). The
oxygen tension within the chambers was recorded continuously throughout hyperoxic
exposure and remained >90% at all times (mean 93 ± 2%). In spontaneously breathing
rats with normal (unconsolidated) lungs, this fraction of inspired oxygen would result in
an alveolar partial pressure of oxygen in the region of ( 90kPa. In the heterogeneously
injured lung however, in which there are consolidated and underventilated regions, it is
likely that there are alveoli in which the alveolar partial pressure of oxygen is
significantly less than this. As the partial pressure of oxygen within individual alveoli
cannot be determined, it is not possible to correlate the observed changes in alveolar
epithelial phenotype with a precise knowledge of the partial pressure of oxygen to which
the epithelium was exposed. However, despite this drawback, the model of an animal
with a heterogeneous lung injury breathing high concentrations of oxygen remains

102
clinically relevant, and it does not seem unreasonable to compare the effects of
‘hyperoxic’ and ‘normoxic’ exposure.
4.4 Summary
The studies outlined in this chapter demonstrate the novel use of immunotargetting to
investigate the effect of hyperoxia on the phenotype of the alveolar epithelium during
resolution of S. aureus-induced lung injury. The use of cell-selective markers, combined
with laser scanning confocal microscopy and image analysis of large contiguous areas of
lung tissue, enabled the extraction of quantitative data demonstrating the proportion of
the alveolar epithelial surface associated with ATI cells, ATII cells and cells with an
intermediate immunophenotype and the effect hyperoxic exposure has on the changes in
these cell populations during the resolution of lung injury.
Characterisation of the phenotype of the repairing epithelium during resolution of S.
aureus-induced injury in lungs exposed to normoxia alone revealed a reduction in the
proportion of ATI(RTI40)-staining epithelial surface with an increase in
ATII(RTII70/MMC4)-staining surface. Inflamed regions in these lungs were
characterised by a significant proportion of the epithelial surface having an intermediate
or transitional phenotype expressing both RTI40 and MMC4. This intermediate cell-
staining membrane was not present to any significant degree in control lungs, but was
observed in vitro as ATII-like cells became ATI-like cells with time in culture (chapter
3). Together, these data are consistent with the accepted paradigm of alveolar epithelial
repair: damaged ATI cells being replaced by the proliferation of ATII cells and the
subsequent resolution of ATII cell hyperplasia by the transdifferentiation of a
subpopulation of these cells into ATI cells (Uhal 1997).
In S. aureus-instilled animals exposed to hyperoxia, the repairing epithelium had a
different phenotype from both control and S. aureus-instilled lungs exposed to normoxia
alone. Inflamed regions in these lungs were still characterised by a reduction in the
proportion of ATI-staining surface and an increase in the proportion of ATII-staining
membrane, but the increase in ATII-staining membrane was significantly greater than

103
that observed in S. aureus-instilled lungs exposed to normoxia alone. As hyperoxia is
pro-apoptotic and known to inhibit ATII cell proliferation, and coupled with the finding
that hyperoxia-exposed lungs had significantly less intermediate cell-staining membrane,
these data suggest a reduction in resolution of ATII cell hyperplasia in hyperoxia-
exposed lungs due to inhibition of the transdifferentiation of ATII cells into ATI cells.
These data support the in vitro findings presented in this thesis (chapter 3), and are
consistent with the hypothesis that clinically relevant levels of hyperoxia inhibit normal
alveolar epithelial repair by inhibiting the differentiation of ATII cells into ATI cells.

104
5. Mechanical deformation-induced injury of alveolar epithelial cells
5.1 Introduction & objectives
The basement membrane underlying the alveolar epithelium undergoes marked
expansion as the lung is inflated toward total lung capacity (Tschumperlin and
Margulies 1999). This suggests that alveolar epithelial cells may be exposed to
significant mechanical deformation during ventilation at high lung volumes. Whilst
mechanical deformation of alveolar epithelial cells is known to have important
functional and developmental roles (Riley et al. 1990; Wirtz and Dobbs 2000), it is also
potentially injurious (Tschumperlin and Margulies 1998; Tschumperlin et al. 2000), and
deformation-induced epithelial injury is thought to play a central role in the disruption of
the air-blood barrier that occurs during the development of ventilator-induced lung
injury (VILI) (Tremblay and Slutsky 1998; Slutsky 1999). This epithelial injury is
thought to result from the alveolar overdistension in aerated alveoli during positive
pressure ventilation of the heterogeneously injured lung.
The clinical signficance of this is highlighted by the fact that following acute lung
injury, lung protective strategies of ventilation, designed to limit alveolar overdistension,
are associated with lower levels of epithelial and endothelial injury (Frank et al. 2002),
lower organ dysfunction scores and reduced mortality (Amato et al. 1998; Ranieri et al.
1999; ARDSNet 2000).
Whilst both clinical and animal studies have shown that mechanical ventilation can
worsen existing lung injury and produce an iatrogenic injury, the effect that mechanical
ventilation has on the mechanisms underlying alveolar epithelial repair is not known.
The Flexercell FX-4000T" is a commercially available apparatus that allows the
application of uniform radial and circumferential strain to cells cultured on flexible
silastic membranes in the BioFlex! cell culture system. Both the magnitude and
frequency of the strain applied to cells is computer regulated, allowing the exposure of
cells to uniform and quantifiable deformations in vitro and enabling a simple correlation
between deformation magnitude and cell response.

105
The first aim of the studies outlined in this chapter was to establish the culture of
primary alveolar epithelial cells in the BioFlex! cell culture system and to characterise
the use of the Flexercell FX-4000T by determining the vulnerability of primary alveolar
epithelial cell monolayers to deformation-induced injury. Having established the
relationship between cell injury and deformation magnitude, the objective was to use
this in vitro system to investigate the hypothesis that the migrating and flattening cells in
regions of ‘repairing’ epithelial cell monolayers are more susceptible to deformation-
induced injury than ‘normal’ epithelium.
5.2 Results
5.2.1 Culture of alveolar epithelial cells in a mechanically active environment
Primary alveolar epithelial cells were seeded at constant density on the fibronectin-
coated silastic membranes of BioFlex! cell culture plates. In all experiments, cell
attachment was successfully confined to the central portion of the membrane by seeding
within a removable silicon gasket placed in the centre of the culture well. After 6 h in
culture, cells appeared to be fully adherent to culture substratum. Gaskets were removed
24 h post-seeding. After 5 days in culture, cells formed a confluent monolayer
comprising two broadly distinct cell types: a central area of small densely grouped cells
(SA 490 ± 116µm2), surrounded by a peripheral region of larger cells (SA 1963 ±
527µm2), which had flattened and spread (figure 45).
Day-5 alveolar epithelial cell monolayers were subject to 1 h of cyclic equibiaxial
mechanical strain at a frequency of 15 cycles per minute using a Flexercell FX-4000T
strain unitTM
(figure 39). Deformation-induced injury was assessed 1 h after completion
of this strain protocol using a Live/DeadTM
cell viability assay.
Cells remained attached to the culture surface during all the deformation protocols used,
and by restricting the position of cell attachment to the central portion of the membrane
all cells were subjected to identical strains in both radial and circumferential directions
(figure 39).

106
5.2.2 Deformation-induced injury and magnitude of mechanical strain
To determine the extent of cell injury following exposure to a range of clinically
relevant mechanical deformations, day-5 alveolar epithelial cell monolayers were
subjected to 1 h of cyclic equibiaxial mechanical strain at physiological magnitudes of
12 and 22% #SA and at the pathological magnitude of 44% #SA. In the central region
of cell monolayers, there was no difference in cell viability between unstrained controls
and cells strained at 12% change in basement membrane surface area (#SA) (1.4 ± 0.7%
control vs 1.4 ± 0.6% 12% #SA, p>0.05, n=5). Cell death increased slightly at 22% #SA
but this was not statistically significant compared to unstrained controls (4.3 ± 0.8%,
p>0.05, n=5). At 44% #SA there was a significant increase in deformation-induced
injury (48.9 ± 12.5%, p<0.001, n=5) (figures 40 & 41).
5.2.3 Deformation-induced injury and cell phenotype
In the central region of day-5 epithelial cell monolayers, there was no significant
deformation-induced injury in cells strained at 12% #SA, however, even at this
physiological level there was significant cell death in cells located in the peripheral
region of the same monolayers (1.4 ± 0.6% central vs 49.5 ± 12.2% peripheral, p<0.001,
n=5) (figures 43 & 44).
Both centrally located cells and those towards the periphery of the monolayer lacked
lamellar bodies. Cells towards the periphery of the monolayer expressed the ATI-
selective protein RTI40 but not the ATII-selective proteins MMC4 or RTII70. Centrally
located cells did not express RTI40, but stained weakly for MMC4 (n=3) (figure 46).
5.2.4 Deformation-induced injury in repairing wound
Alveolar epithelial cell monolayers were ‘wounded’ by scoring across the central section
of day-3 monolayers with a 200µl pipette tip. Wounded monolayers were then cultured

107
under standard conditions for a further 48 h prior to mechanical deformation of
‘repairing’ wounds at 12% #SA.
Cells in the repairing region of epithelial cell monolayers appeared to express RTI40
(n=1) and spread to occupy a significantly greater surface area in comparison to cells in
the unwounded central portion of the same monolayers (1440 ± 319µm2 vs 487 ±
191µm2, p<0.05, n=3). Deformation caused significant injury of cells in repairing
wounds in comparison to the same region of repairing alveolar epithelium in unstrained
controls (38.0 ± 8% cell death vs 8.3 ± 1.6% unstrained controls, p < 0.005) (figures 47
& 48).
5.3 Discussion
5.3.1 Deformation-induced injury is a non-linear function of deformation
magnitude
Mechanical deformation-induced injury was found to be a non-linear function of
deformation magnitude in these studies, with no statistically significant cell death
occurring with cyclic (physiological) deformations up to and including 22% #SA, but
significant cell death occurring at (pathological) deformations of 44% #SA.
Reassuringly, these data are in broad agreement with the only other known report of
changes in day-5 alveolar epithelial cell viability with quantified deformation
(Tschumperlin and Margulies 1998). These findings also make intuitive sense when
interpreted in light of the changes in lung volumes that these deformations are thought to
represent (figure 42).
In the isolated rat lung, inflation from functional residual capacity (FRC) to 80% of total
lung capacity (TLC) has been shown to result in a change in the epithelial basement
membrane surface area of (25%, whilst inflation to 100% TLC results in a change of
(30-40% (Mercer and Crapo 1990; Tschumperlin and Margulies 1999). Mechanical
deformation producing a 22% #SA might therefore be reasonably interpreted as
physiological in magnitude, whilst a 44% #SA is greater than that experienced in

108
inflation of the normal lung to TLC and is likely to be pathological. A 44% #SA was
chosen for these experiments because it was thought to reflect the deformations likely to
occur during alveolar overdistension resulting from positive pressure ventilation of the
heterogeneously injured lung.
Whilst there are limitations in the use of alveolar epithelial cells in culture as a surrogate
for the alveolar epithelial barrier, it is reassuring that there appears to be a close
correlation between the finding of ATI cell injury in electron micrographs of rat lungs
inflated beyond TLC (Dreyfuss et al. 1988) and the findings in these studies of a
dramatic increase in deformation-induced injury when deformations exceed those
associated with inflation to TLC.
5.3.2 Spreading RTI40-expressing cells in the peripheral region of epithelial cell
monolayers are susceptible to deformation-induced injury
The finding that a physiological level of strain (12% #SA) produces significant cell
death in the peripheral region of an alveolar epithelial cell monolayer, but not in the
central region of the same monolayer, is a novel finding. Primary ATII cells typically
flatten and spread in culture, losing their lamellar bodies and expression of ATII-
selective proteins whilst simultaneously acquiring expression of ATI-selective proteins
(chapter 3) (Dobbs et al. 1985; Danto et al. 1992; Paine and Simon 1996). This
transition from an ATII- into an ATI-like phenotype typically occurs within 3 d of
seeding, suggesting that a monolayer of day-5 alveolar epithelial cells may uniformly
represent this ATI-like phenotype. However, in these studies, cells were seeded at
relatively high density within removable gaskets in order to constrain cell attachment to
the central portion of the culture membrane. Whilst this ensured that all cells within the
monolayer were exposed to uniform patterns of strain, an artefact of this method of
seeding was the development of different cell phenotypes within the monolayer, as upon
removal of the silicon gasket, cells in the peripheral region were free to flatten and
spread onto unoccupied culture surface, whilst cells in the central region were prevented
from doing so by neighbouring cells. After 5 d in culture, cells in the peripheral region

109
of the monolayer resembled ATI cells both morphologically and phenotypically, being
large, attenuated and RTI40-expressing. These findings are consistent with the
morphologic and phenotypic changes known to occur in ATII cells with time in culture.
However, cells in the central region of the monolayer appeared relatively
undifferentiated. They were less spread and demonstrated only weak MMC4 expression.
Cells in the central region of the monolayer did not express either RTII70 or RTI40. The
finding of different levels of cell injury in the two regions suggests that the different
alveolar epithelial cell phenotypes observed (relatively undifferentiated and ATI-like)
have different susceptibilities to deformation-induced injury, with the larger RTI40-
expressing (ATI-like) cells being more susceptible to injury.
Although there are limitations inherent in the extrapolation of any in vitro model, there
are clear parallels between the system of ATII cell culture used in these studies and the
mechanisms thought to operate in the alveolar epithelium during epithelial repair in vivo
– namely re-epithelialisation of denuded basement membrane by the transition of a
population of ATII cells into ATI cells, possibly via some kind of undifferentiated cell
phenotype (figure 1). Viewed in this light, it is clear that spreading RTI40-expressing
cells in the peripheral region of monolayers share many similarities with alveolar
epithelial cells in regions of epithelial repair. These data would then support the concept
that the spreading and flattening cells in regions of active epithelial repair are susceptible
to deformation-induced injury even at physiological levels of mechanical strain.
5.3.3 Epithelial cells in the region of repairing epithelial monolayers appear
susceptible to deformation-induced injury
The ‘repair’ of wounded d 3 epithelial cell monolayers appeared to occur by migration
and spreading of cells from the wound edge. By 48 h following wounding, cells in the
region of the repairing wound displayed similar characteristics to those of cells in the
peripheral region of the monolayers, having adopted an ATI-like (RTI40-expressing)
phenotype and spread to occupy a greater surface area. The finding that spreading RTI40-
expressing cells in regions of repairing wounds appear susceptible to deformation-

110
induced injury at physiological levels of deformation supports the concept that cells in
regions of active alveolar epithelial repair may also be highly susceptible to
deformation-induced injury in vivo. These data may explain the clinical observation that
patterns of mechanical ventilation which are not injurious in the normal lung appear to
cause significant injury in the already injured repairing lung (ARDSNet 2000; Frank et
al. 2002).
5.3.4 General discussion
Freshly isolated ATII cells did not attach well to uncoated silastic membranes, and the
BioFlex, culture plates were therefore pre-coated with extracellular matrix protein prior
to seeding. In preliminary experiments, pre-coating the culture plates with fibronectin,
collagen or laminin, the major components of the alveolar epithelial basement
membrane, did not appear to significantly alter either cell attachment or the pattern of
deformation-induced cell injury observed. Fibronectin was therefore chosen because it
has been shown to stimulate alveolar epithelial cell spreading in vitro and is found in
elevated concentrations in the basement membrane of the repairing lung (Sugahara et al.
1993).
Because cells in these studies were exposed to uniform patterns of strain, it is reasonable
to assume that the differences in the cellular response observed are a reflection of
differences in cellular properties. Cell morphology, phenotype, cytoskeletal structure,
cell-cell and cell-matrix attachments may all play a role in this heterogeneity of
response. Cells in the peripheral region of alveolar epithelial cell monolayers had a
significantly greater surface area than those in the central region, and Tschumperlin and
colleagues have previously reported a dependence of deformation-induced injury on
mean cell size, with injury of ATI-like cells being greatest in cells with largest surface
area (Tschumperlin et al. 2000). However, these investigators also found that day-1
alveolar epithelial (ATII-like) cells, with a significantly smaller size, were more
sensitive to deformation-induced injury than day-5 (ATI-like) cells suggesting that it is
not only cell surface area which determines the sensitivity of individual cells to injury.

111
Whilst previous studies suggest that ATII-like cells may be more sensitive to mechanical
deformation-induced injury than ATI-like cells in vitro, these studies have assumed the
phenotype of the cells based on time in culture alone rather than the expression of cell-
selective proteins. The correlation between the expression of RTI40 and increased
sensitivity to injury within the same cell culture is a novel finding.
The function of RTI40 is unknown, but it has been speculated to have a functional role in
cell spreading and migration (chapter 3) (Williams 2003). It increases migration rates in
wounding experiments (Schacht et al. 2003), and time-lapse studies of the transition of
ATII cells into ATI-like cells in culture, reveal that RTI40 is initially expressed in the
leading or spreading cell edge (McKechnie and Clegg unpublished data and figure 4).
The finding that RTI40 expressing cells are more susceptible to deformation-induced
injury may therefore lend support to the hypothesis that cells in the process of spreading
and migrating are more vulnerable to injury.
The outcome measure used to assess deformation-induced injury in these studies was
loss of cell membrane integrity, which leads to cell death. To determine whether cells
were able to recover from the membrane disruption allowing ethidium homodimer-1 to
enter cells, a number of wells were followed for 24 h following completion of
deformation with the addition of fresh calcein AM. Cells labelled with ethidium
homodimer-1 did not appear to recover their ability to concentrate calcein, suggesting
that the deformation-induced injury in these cells was irreversible.
Alveolar epithelial damage due to deformation is likely to occur as a continuum of cell
responses, from loss of normal cell function through to gross structural failure. Cell
death represents the extreme end of this continuum, and more subtle forms of injury,
such as disruption of tight junction function and alveolar epithelial transport, may well
contribute to deformation-induced damage of the air-blood barrier. These more subtle
forms of cell injury were not investigated in these studies.
In addition, the mechanism of cell injury was not formally examined. Although
mechanical strain has previously been shown to induce apoptosis in ATII cells (Edwards
et al. 1999), nuclear staining did not reveal significant numbers of apoptotic nuclei in
these experiments, and preliminary studies using a polyclonal anti-active caspase-3

112
antibody did not show significant caspase-3 activation. These findings suggest that the
deformation-induced cell injury in these studies was necrotic rather than apoptotic, most
likely resulting from plasma membrane stress failure (Vlahakis and Hubmayr 2000).
Oxidative stress secondary to stretch-induced antioxidant depletion and increased ROS
generation in response to mechanical deformation has been implicated as a potential
mechanism underlying deformation-induced epithelial cell injury (Jafari et al. 2004;
Chapman et al. 2005). These studies suggest that cells generating fewer ROS in response
to deformation may be more resistant to deformation-induced injury. Intriguingly, such
cells also appear resistant to hyperoxia-induced injury (section 3.3.8).
5.4 Summary
The findings of these in vitro studies support the hypothesis that at high lung volumes
alveolar epithelial cells undergo irreversible cell damage. The finding that different
alveolar epithelial phenotypes within the same culture show different sensitivities to
deformation-induced injury, with spreading RTI40-expressing cells more susceptible to
injury, suggests that epithelial cells in the process of spreading and adopting an ATI
phenotype in regions of epithelial repair may be more susceptible to deformation-
induced injury, and may be injured even at physiological levels of mechanical strain.
The increased susceptibility of cells in the region of ‘repairing’ epithelial monolayer
wounds to deformation-induced injury further supports this hypothesis. These data
suggest that alveolar epithelial deformation-induced injury may play a central role in the
development of ventilator-induced lung injury during mechanical ventilation of the
already injured lung.

113
6. Alveolar epithelial injury in a rat haemorrhagic shock-induced
model of acute lung injury
6.1 Introduction
Haemorrhagic shock is a common cause of acute lung injury. Unlike pneumonia
however, haemorrhage results in an indirect injury to the lung (Bernard et al. 1994;
Ware and Matthay 2000).
The distinction between direct and indirect lung injury has been recognised for some
time. Disorders resulting in ‘direct’ injury to the alveolar air-blood barrier (most
commonly gastric aspiration and pulmonary infection) appear to inflict damage to the
alveolar-capillary membrane through a local inflammatory response. Conversely
‘indirect’ injury to the lung (caused most often by sepsis, trauma or massive blood
transfusion) is thought to result from circulating mediators generated by extra-
pulmonary sources of inflammation. Although the distinctions in pathogenesis are
largely speculative, there are differences in the clinical behaviour of direct and indirect
injuries, and clinical observation supports the existence of two distinct pathogenic
pathways with different morphological aspects, respiratory mechanics and responses to
ventilatory strategies (Gattinoni et al. 1998; Pelosi et al. 2003).
There is experimental evidence that haemorrhagic shock and fluid resuscitation are
associated with lung endothelial injury (Todd et al. 1978; Michel et al. 1981), and that
severe haemorrhage alters alveolar epithelial barrier function (Pittet et al. 1996; Pittet et
al. 1996; Modelska et al. 1997), but few studies have examined whether haemorrhage is
injurious to the alveolar epithelium.
The use of cell-selective markers in investigating epithelial injury is not without
precedent (see section 1.4), and offers the advantage of indicating the degree to which
individual epithelial cell populations are affected during injury (McElroy and Kasper
2004). This approach has not been used to study haemorrhagic shock-induced lung
injury.

114
The initial objective of the work outlined in this chapter was to establish a (non-
recovery) rat model of haemorrhagic shock-induced lung injury, characterise the injury
and, using epithelial cell-selective proteins, assess the severity of epithelial injury.
Having achieved this, the objective was to develop the model into a recovery model that
could be used in combination with cell-selective biomarkers to compare the effects of
hyperoxia (± mechanical ventilation) on the phenotype of the repairing alveolar
epithelium following direct (pneumonia-induced, chapter 4) and indirect (haemorrhagic
shock-induced) injuries.
The haemorrhagic shock protocol chosen in this study (figure 49) was based upon that
established by Modelska et al. (Modelska et al. 1997). The ventilatory strategy used was
clinically applicable and has previously been shown not to cause lung injury in isolation
(Frank et al. 2002).
6.2 Results
A total of 6 animals, 3 undergoing haemorrhage and 3 controls (surgery without
haemorrhage), completed the experimental protocol (figure 49). Blood loss during
surgery was minimal. Only animals with normal acid-base status at the end of the
stabilisation period were included in the study. The blood pressure profile of a typical
animal undergoing surgery, haemorrhage and fluid resuscitation is shown in (figure 50).
6.2.1 Characterisation of haemorrhagic shock-induced acute lung injury
BALF Protein: BALF recovered from animals undergoing haemorrhage had
significantly (5-fold) higher levels of total protein (13.2 ± 2.1mg haemorrhage vs 2.7 ±
1.1 control, p<0.01) (figure 51), indicative of a loss of alveolar epithelial integrity.

115
BALF cell counts: There was no statistically significant difference in the total cell counts
in BALF between the two groups (86.2 ± 13.9x104
cells haemorrhage vs 97.3 ± 19.9
control, p=0.24) (figure 52A). However, differential cell counts revealed a significantly
higher proportion of neutrophils in the BALF recovered from animals undergoing
haemorrhage (12.7 ± 2.1% haemorrhage vs 0.1 ± 0.1% control, p<0.001), with a
decrease in the proportion of alveolar macrophages (82.3 ± 2.9% haemorrhage vs 98.3 ±
0.6% control, p<0.001) (figure 52B). There was no statistically significant difference
between the number of other cell types identified in the BALF in the two groups.
6.2.2 Alveolar epithelial cell injury following haemorrhagic shock
ATI-selective proteins: The amount of the ATI-selective integral membrane protein
RTI40 showed a 7-fold increase in BALF recovered from animals undergoing
haemorrhage (108 ± 54 du haemorrhage vs 15 ± 9 du control, p<0.05) (figure 53A).
There was also a 2-fold increase in the ATI-selective protein MMC6 (6.9 ± 0.8 du
haemorrhage vs 2.6 ± 0.8 control, p<0.05) (figure 53B). Neither RTI40 nor MMC6 were
detected in plasma samples from either control or haemorrhaged rats.
ATII-selective proteins: Although there was an increase in the level of the ATII-selective
protein MMC4 in BALF recovered from rats undergoing haemorrhage (125 ± 76 du
haemorrhage vs 63 ± 39 control, p=0.16), this did not reach statistical significance.
6.3 Discussion
6.3.1 Haemorrhagic shock results in acute lung injury
Loss of alveolar air-blood barrier integrity, with accumulation of protein-rich oedema
fluid and neutrophils within the airspaces, is a pathognomonic feature of the acute phase

116
of acute lung injury (Ware and Matthay 2000). The data presented in this chapter,
demonstrating a significant increase in the level of protein and number of neutrophils in
BALF following haemorrhage, confirm that a clinically relevant model of haemorrhagic
shock produces significant lung injury, and that it does so within six hours of the onset
of haemorrhage.
6.3.2 Haemorrhagic shock is associated with ATI cell injury
The use of alveolar epithelial cell-selective proteins as markers of acute lung injury
offers the advantage of determining which of the specific alveolar epithelial cell
populations is affected (McElroy and Kasper 2004). For example, elevation of the ATI-
selective integral membrane protein RTI40 in BALF has been shown to provide a
biochemical marker of ATI cell injury in a Pseudomonas aeruginosa-induced model of
lung injury. In this model, the degree of RTI40 elevation correlated with both the degree
of injury, assessed morphologically, and the severity of functional disruption of the
alveolar air-blood barrier (McElroy et al. 1995).
The 7-fold elevation of the ATI-selective protein RTI40 within 6 hours of the onset of
haemorrhage is a novel finding and suggests that haemorrhagic shock resulted in
significant alveolar type I cell injury in this model. Further evidence for this is provided
by the finding of a statistically significant increase in the level of MMC6.
The ATII-selective marker MMC4 was elevated 2-fold, which although suggestive of
alveolar type II cell injury, was not statistically significant. However,
immunophenotyping of injured lungs following S. aureus instillation (see chapter 4)
revealed that MMC4 is expressed by activated alveolar macrophages in addition to ATII
cells in the inflamed lung, suggesting that BALF MMC4 content is unlikely to prove a
useful marker of ATII cell injury.
The mechanism of haemorrhagic shock-induced alveolar epithelial injury was not
investigated in detail. The finding of significant alveolar neutrophil infiltration suggests
that the mechanism is, at least in part, inflammatory in nature. This would be consistent

117
with the prevailing view that neutrophils have an important early immunomodulatory
role in haemorrhage-induced acute lung injury, being responsible for the initial
activation of nuclear factor-'B (NF-'B) and subsequent increase in proinflammatory
cytokines such as interleukin-1& (IL-1&) and tumour necrosis factor-$ (TNF-$)
(Shenkar and Abraham 1993; Shenkar et al. 1996; Abraham et al. 2000).
Bacterial translocation, in which viable bacteria or bacterial endotoxins pass from the
gastrointestinal tract to extraintestinal sites as a consequence of gut ischaemia, has been
implicated in the pathogenesis of sepsis (Rush et al. 1988; Sori et al. 1988), and may
play a role in the pathogenesis of acute lung injury following haemorrhage. In a rat
model of haemorrhagic shock, Tani and colleagues demonstrated bacterial translocation
and upregulation of the proinflammatory cytokine tumour necrosis factor-$ (TNF-$)
within 1h of haemorrhage (Tani et al. 2000). However, these effects were prevented by
hyperoxia (inhaled 100% O2) early in the course of haemorrhagic shock, suggesting that
bacterial translocation is unlikely to have played a significant role in this model. This
hypothesis is supported by the fact that blood cultures taken from animals completing
the study were negative in all cases.
6.4 Summary
The objective of this study was to establish a reproducible model of indirect acute lung
injury and investigate the affect of this injury on the alveolar epithelium. It was hoped
the model could then be modified to establish a recovery model enabling the study of
alveolar epithelial repair following indirect lung injury. Unfortunately, the model proved
difficult to establish with a significant animal mortality rate, and only the biochemical
characterisation of the lung injury was possible.
Whilst the work presented in this chapter reflects only the early stages in the
development of a model of haemorrhagic shock-induced acute lung injury, it does
suggest that haemorrhage is injurious to the alveolar epithelium, leading to significant

118
ATI cell necrosis. As such, it establishes an important proof of principle and provides a
positive platform for further work.

119
7. Summary
Oxygen and mechanical ventilation are key therapeutic interventions in the supportive
treatment of respiratory failure following lung injury. Although both hyperoxia and the
mechanical deformations associated with mechanical ventilation are known to be
potentially injurious to the normal lung, their effects on alveolar epithelial repair
following injury are not known. The clinical impression however, is that poor outcome
is associated with exposure of injured (repairing) epithelium to these iatrogenic ‘hits’.
This thesis describes in vitro (chapter 3) and in vivo (chapter 4) studies investigating
the hypothesis that hyperoxia inhibits normal alveolar epithelial repair by inhibiting the
transdifferentiation of ATII cells into ATI cells, and in vitro studies addressing the
hypothesis that (spreading and flattening) alveolar epithelial cells in regions of epithelial
repair are susceptible to mechanical deformation-induced injury (chapter 5).
7.1 Hyperoxia inhibits the transdifferentiation of ATII cells into ATI cells
The in vitro data presented demonstrate for the first time that hyperoxia reversibly
inhibits the changes in morphology and cell-selective protein expression characteristic of
the transition of ATII-like cells into ATI-like cells with time in culture. These studies
also confirm that hyperoxia inhibits proliferation and is cytotoxic to alveolar epithelial
cells, with cell death occuring by both apoptosis and necrosis following prolonged
exposure. However, it appears that hyperoxia is not universally cytotoxic, and these
studies suggest that the (isolated) ATII cell population harbours a subpopulation of cells
that have proliferative potential and are less susceptible to hyperoxia-induced injury.
These cells appear to generate fewer ROS and have lower levels of E-cadherin
expression.
The effect of hyperoxia on alveolar epithelial repair in vivo was investigated using a
novel immunotargeting approach and quantitative comparison of the phenotype of
repairing alveolar epithelium following S. aureus-induced lung injury.

120
In S. aureus-instilled lungs exposed to normoxia alone, these studies revealed a
reduction in the proportion of ATI(RTI40)-staining epithelial surface and an increase in
ATII(RTII70/MMC4)-staining surface, consistent with ATI cell injury and ATII cell
proliferation. Inflamed regions in these lungs were also characterised by a significant
proportion of the alveolar epithelial surface having an intermediate immunophenotype
expressing both the ATI-selective RTI40 and the ATII-selective MMC4. This
RTI40/MMC4-coexpressing membrane was not present to any significant degree in
control lungs, but was observed in vitro as ATII-like cells became ATI-like cells with
time in culture. These data are consistent with the accepted paradigm of normal alveolar
epithelial repair: damaged ATI cells being replaced by the proliferation of ATII cells and
the subsequent differentiation of a subpopulation of daughter cells into ATI cells, with
this differentiation of ATII into ATI cells occurring via cells with an intermediate
(RTI40/MMC4-coexpressing) cell phenotype.
Markers of lung injury were elevated to a greater degree in S. aureus-instilled lungs
exposed to hyperoxia than in those exposed to normoxia alone, suggesting that
hyperoxia was either causing an inflammatory lung injury in the repairing lung itself
and/or inhibiting normal resolution of the pre-existing lung injury. Examination of the
alveolar epithelium in S. aureus-instilled lungs exposed to hyperoxia revealed significant
differences in the immunophenotype, with a greater proportion of ATII-staining alveolar
epithelial surface. As hyperoxia is known to be pro-apoptotic and to inhibit ATII cell
proliferation, both factors likely to reduce the ATII cell population, the observed
increase in ATII-staining alveolar epithelial surface following hyperoxic-exposure
suggests that hyperoxia inhibits normal epithelial repair by inhibiting the differentiation
of ATII cells into ATI cells, preventing effective resolution of ATII cell hyperplasia.
The finding that hyperoxia-exposed lungs had significantly less intermediate cell-
staining membrane is consistent with this interpretation.
The in vitro and in vivo studies described support the hypothesis that prolonged exposure
to clinically relevant levels of hyperoxia is injurious to the alveolar epithelium and
inhibits normal alveolar epithelial repair by inhibiting the differentiation of ATII cells
into ATI cells. This is a novel finding.

121
7.2 Spreading RTI40-expressing cells are susceptible to deformation-induced injury
The effect of mechanical deformation on alveolar epithelial cells was investigated by
examining changes in cell viability following exposure of cells to uniform, quantified
equibiaxial strain. The studies performed to characterise the in vitro system used for
these experiments demonstrated that deformation-induced injury was a non-linear
function of deformation magnitude, with significant injury occurring following exposure
of cells to strains greater than those associated with inflation of the lung to total lung
capacity (>40% #SA), but not over a range of more physiological levels of strain (<30%
#SA). These findings are in broad agreement with previously published data linking
deformation-induced injury with deformation magnitude and animal studies
demonstrating significant ATI cell injury following inflation of the lungs beyond total
lung capacity.
Subsequent studies demonstrated that different alveolar epithelial cell phenotypes within
the same culture system had different sensitivities to deformation-induced injury, with
spreading RTI40-expressing cells in regions of ‘repairing’ epithelial cell monolayers
susceptible to injury even at physiological levels of mechanical strain. This is a novel
observation.
Although there are limitations inherent in the extrapolation of any in vitro system, there
are parallels between the cell phenotypes observed in the cell culture system used for
these experiments and those present in the repairing epithelium, with a population of
ATII cells losing their mature ATII cell phenotype whilst spreading and flattening to
cover denuded basement membrane and acquiring an ATI (RTI40-expressing) cell
phenotype. Viewed in this light, these data are consistent with the hypothesis that
repairing epithelium is susceptible to deformation-induced injury even at physiological
levels of strain.

122
7.3 The multiple-hit hypothesis
Based upon clinical observation, it has been speculated that initial injury to the lung
‘primes’ the epithelium for an excessive or abnormal response following exposure to a
second insult (hit). This has been termed the ‘two-’ or ‘multiple-hit’ hypothesis (Moore
and Moore 1995; Vuichard et al. 2005). The data presented in this thesis support this
hypothesis by demonstrating that the clinically relevant ‘hits’ of mechanical deformation
and hyperoxia are potentially injurious to the alveolar epithelium and inhibit the
processes underlying normal alveolar epithelial repair.

123
8. Future work
The mechanism underlying the hyperoxia-induced inhibition of ATII cell
transdifferentiation observed in these studies remains unknown and warrants further
investigation. A recent study has suggested that transdifferentiation of ATII cells into
ATI cells is modulated by signalling through the Smad-dependent TGF-&1 pathway
(Bhaskaran et al. 2007). This raises the possibility that hyperoxia inhibits
transdifferentiation through inhibition of TGF-&1 signalling and suggests one avenue for
further work.
Several studies have demonstrated that upregulation of the insulin growth factor (IGF)
system occurs during hyperoxia-induced lung injury, and transcriptional regulation of
this system appears to play a role in the arrest of ATII cell proliferation induced by
hyperoxia. Interestingly, a recent study has demonstrated that addition of IGF-1 receptor
antibodies to ATII cells in culture also inhibits the ATII to ATI transition (Narasaraju et
al. 2006), and it seems reasonable to speculate that changes in the expression profile of
the IGF system may have a central role in the mechanism of hyperoxia-induced
inhibition of ATII cell transdifferentiation.
Hyperoxia-induced injury is thought to be mediated by a combination of ROS
cytotoxicity and activation of the redox-sensitive transcription factor NF-'B (D'Angio
and Finkelstein 2000; Haddad 2002; Haddad 2002). Studies in which hyperoxia-exposed
cells were incubated with the free radical scavenger N-acetylcysteine did not appear to
attenuate the effect of hyperoxia on ATII cell differentiation. Similarly, NF-'B
inhibition by sulfasalazine and the inhibitory peptide SN-50 appeared to have little effect
on the inhibition of ATII cell differentiation. However, these experiments were
preliminary and warrant further investigation.
Oxidative stress secondary to stretch-induced ROS generation has been implicated as a
potential mechanism underlying deformation-induced epithelial cell injury (Chapman et
al. 2005). The role of ROS generation in the deformation-induced injury observed in
these studies was not investigated but warrants further study. Intriguingly, it appears that

124
ROS generation by alveolar epithelial cells may play a central role in determining
sensitivity to both hyperoxia- and mechanical deformation-induced injury.
Integrin and extracellular matrix interactions have been shown to play an important role
in epithelial cell migration and spreading and in the repair of distal airway epithelium
(White et al. 1999). In vitro, laminin-rich culture surfaces favour retention of an ATII-
like cell phenotype whilst fibronectin-rich surfaces accelerate loss of the ATII phenotype
(Rannels and Rannels 1989). Although the precise role of integrin/matrix interactions in
the transdifferentiation of ATII cells is not known, these data suggest that
integrin/matrix interactions play an important role in the process of alveolar epithelial
cell repair. The effect of hyperoxia on these interactions is not known.
The data presented in this thesis suggest that functional segregation of the ATII cell
population into E-cadherin-positive and-negative populations may be possible prior to
hyperoxic-exposure. A similar approach has been used to segregate the population of
ATII cells isolated from hyperoxia-exposed rats (Reddy et al. 2004). This approach
would allow further investigation of the hyperoxia-resistant epithelial cell phenotype.
The ‘multiple-hit’ hypothesis postulates that initial injury to the lung primes the
epithelium for an excessive or abnormal response following exposure to a second insult
(hit) (Moore and Moore 1995; Vuichard et al. 2005). Both hyperoxia and mechanical
deformation are clinically relevant second ‘hits’ but the clinical reality is that the
epithelium is exposed to both with the use of supplemental oxygen and positive pressure
ventilation. Whether these hits act synergistically in the context of lung injury remains
unknown, and an obvious extension to the work presented in this thesis is investigation
of the effects of mechanical deformation in combination with hyperoxia (± hypoxia) in
the injured and repairing lung.

125
Figures
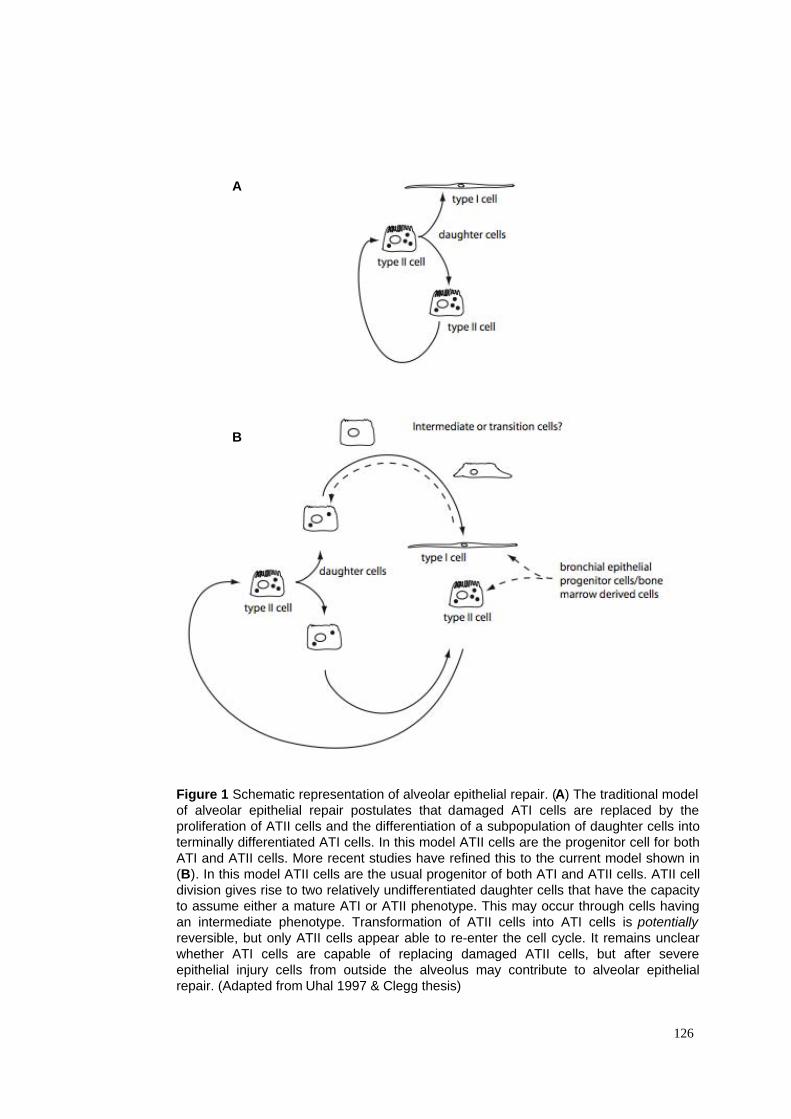
126
Figure 1 Schematic representation of alveolar epithelial repair. (A) The traditional model
of alveolar epithelial repair postulates that damaged ATI cells are replaced by the
proliferation of ATII cells and the differentiation of a subpopulation of daughter cells into
terminally differentiated ATI cells. In this model ATII cells are the progenitor cell for both
ATI and ATII cells. More recent studies have refined this to the current model shown in
(B). In this model ATII cells are the usual progenitor of both ATI and ATII cells. ATII cell
division gives rise to two relatively undifferentiated daughter cells that have the capacity
to assume either a mature ATI or ATII phenotype. This may occur through cells having
an intermediate phenotype. Transformation of ATII cells into ATI cells is potentially
reversible, but only ATII cells appear able to re-enter the cell cycle. It remains unclear
whether ATI cells are capable of replacing damaged ATII cells, but after severe
epithelial injury cells from outside the alveolus may contribute to alveolar epithelial
repair. (Adapted from Uhal 1997 & Clegg thesis)
A
B
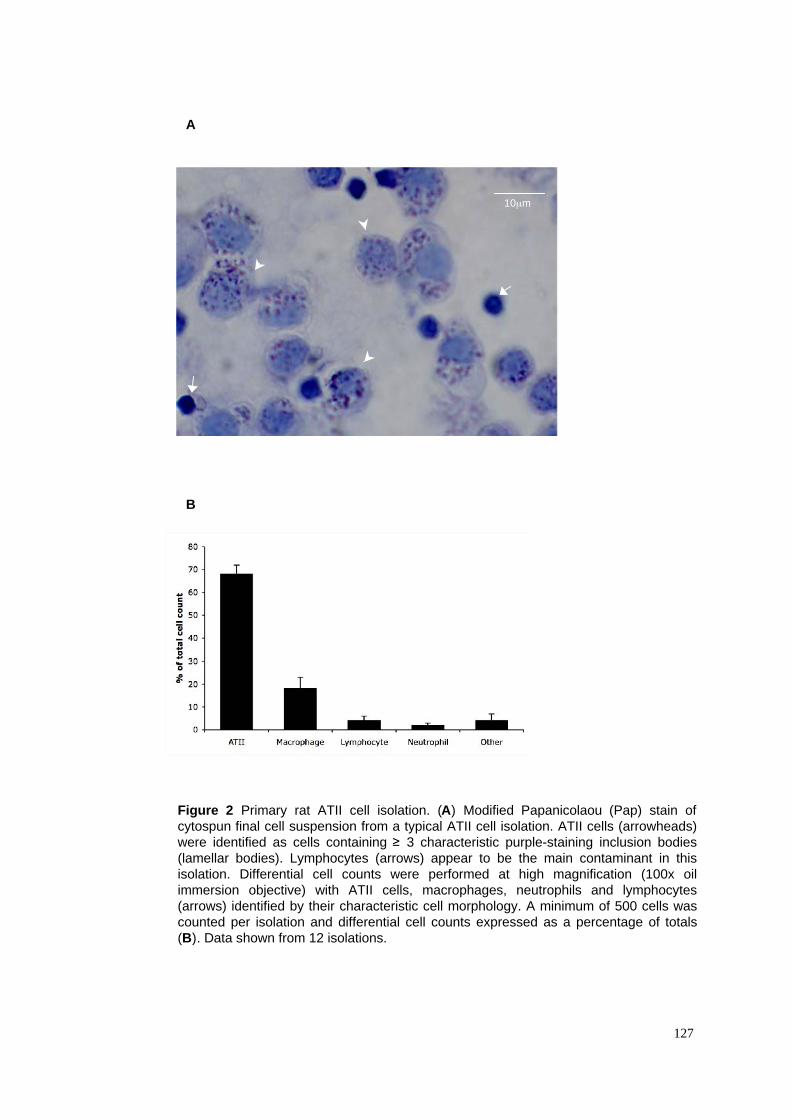
A
B
Figure 2 Primary rat ATII cell isolation. (A) Modified Papanicolaou (Pap) stain of
cytospun final cell suspension from a typical ATII cell isolation. ATII cells (arrowheads)
were identified as cells containing ! 3 characteristic purple-staining inclusion bodies
(lamellar bodies). Lymphocytes (arrows) appear to be the main contaminant in this
isolation. Differential cell counts were performed at high magnification (100x oil
immersion objective) with ATII cells, macrophages, neutrophils and lymphocytes
(arrows) identified by their characteristic cell morphology. A minimum of 500 cells was
counted per isolation and differential cell counts expressed as a percentage of totals
(B). Data shown from 12 isolations.
127
10µm
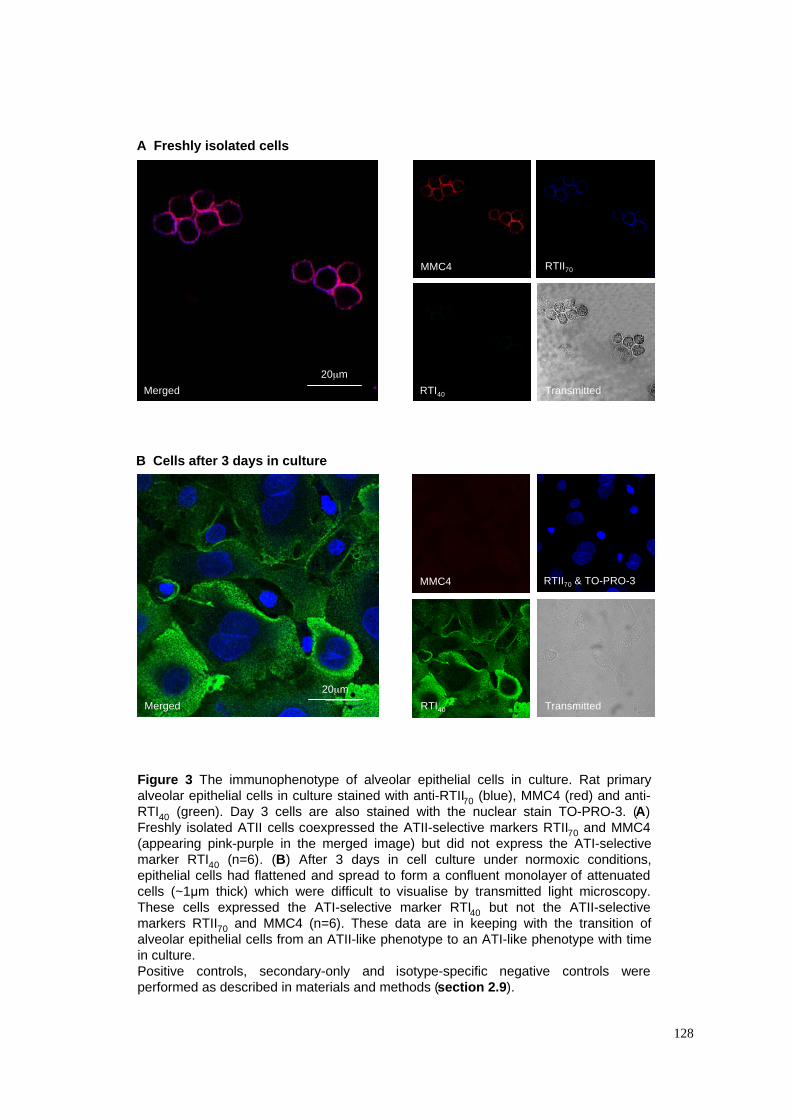
A Freshly isolated cells
B Cells after 3 days in culture
Figure 3 The immunophenotype of alveolar epithelial cells in culture. Rat primary
alveolar epithelial cells in culture stained with anti-RTII70 (blue), MMC4 (red) and anti-
RTI40 (green). Day 3 cells are also stained with the nuclear stain TO-PRO-3. (A)
Freshly isolated ATII cells coexpressed the ATII-selective markers RTII70 and MMC4
(appearing pink-purple in the merged image) but did not express the ATI-selective
marker RTI40 (n=6). (B) After 3 days in cell culture under normoxic conditions,
epithelial cells had flattened and spread to form a confluent monolayer of attenuated
cells (~1!m thick) which were difficult to visualise by transmitted light microscopy.
These cells expressed the ATI-selective marker RTI40 but not the ATII-selective
markers RTII70 and MMC4 (n=6). These data are in keeping with the transition of
alveolar epithelial cells from an ATII-like phenotype to an ATI-like phenotype with time
in culture.
Positive controls, secondary-only and isotype-specific negative controls were
performed as described in materials and methods (section 2.9).
128
20µm
20µm
MMC4
RTI40
RTII70
Merged
RTI40 Transmitted
MMC4 RTII70 & TO-PRO-3
Merged
Transmitted
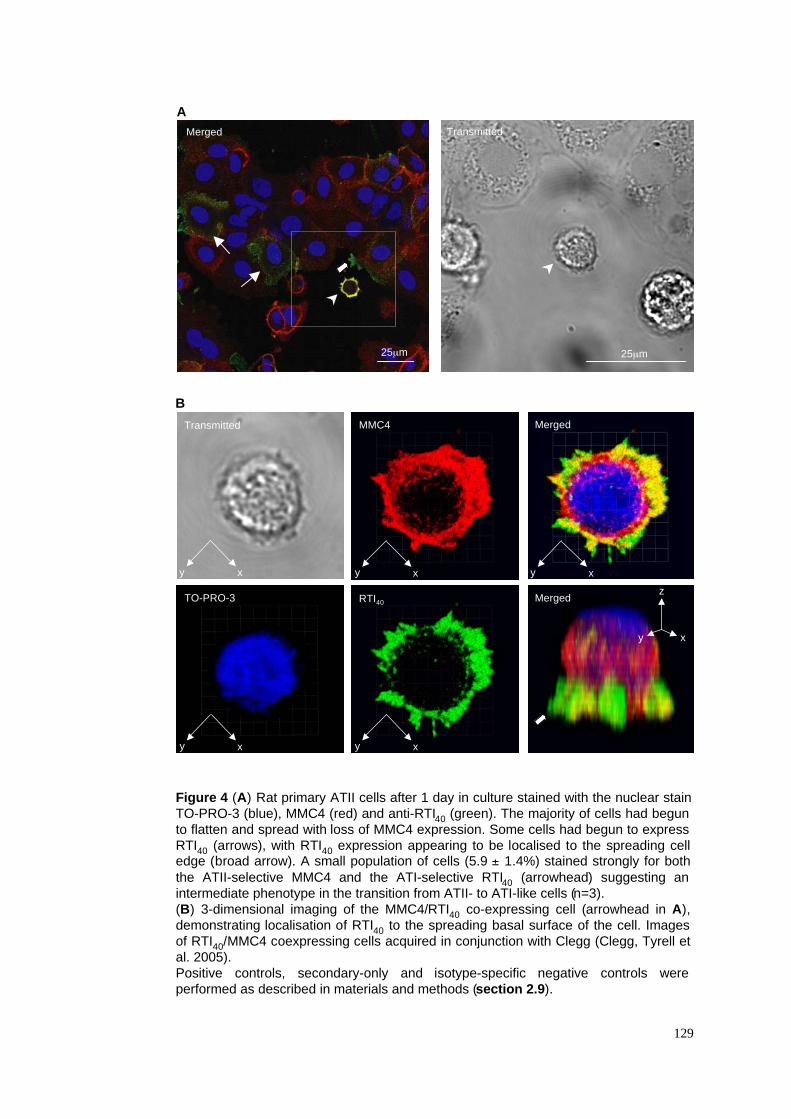
A
B
Figure 4 (A) Rat primary ATII cells after 1 day in culture stained with the nuclear stain
TO-PRO-3 (blue), MMC4 (red) and anti-RTI40 (green). The majority of cells had begun
to flatten and spread with loss of MMC4 expression. Some cells had begun to express
RTI40 (arrows), with RTI40 expression appearing to be localised to the spreading celledge (broad arrow). A small population of cells (5.9 ± 1.4%) stained strongly for both
the ATII-selective MMC4 and the ATI-selective RTI40 (arrowhead) suggesting an
intermediate phenotype in the transition from ATII- to ATI-like cells (n=3).
(B) 3-dimensional imaging of the MMC4/RTI40 co-expressing cell (arrowhead in A),
demonstrating localisation of RTI40 to the spreading basal surface of the cell. Images
of RTI40/MMC4 coexpressing cells acquired in conjunction with Clegg (Clegg, Tyrell et
al. 2005).
Positive controls, secondary-only and isotype-specific negative controls were
performed as described in materials and methods (section 2.9).
129
MMC4
RTI40
Transmitted
TO-PRO-3
Merged
xy
Merged
xy
z
TransmittedMerged
25µm 25µm
xy xy
xy xy
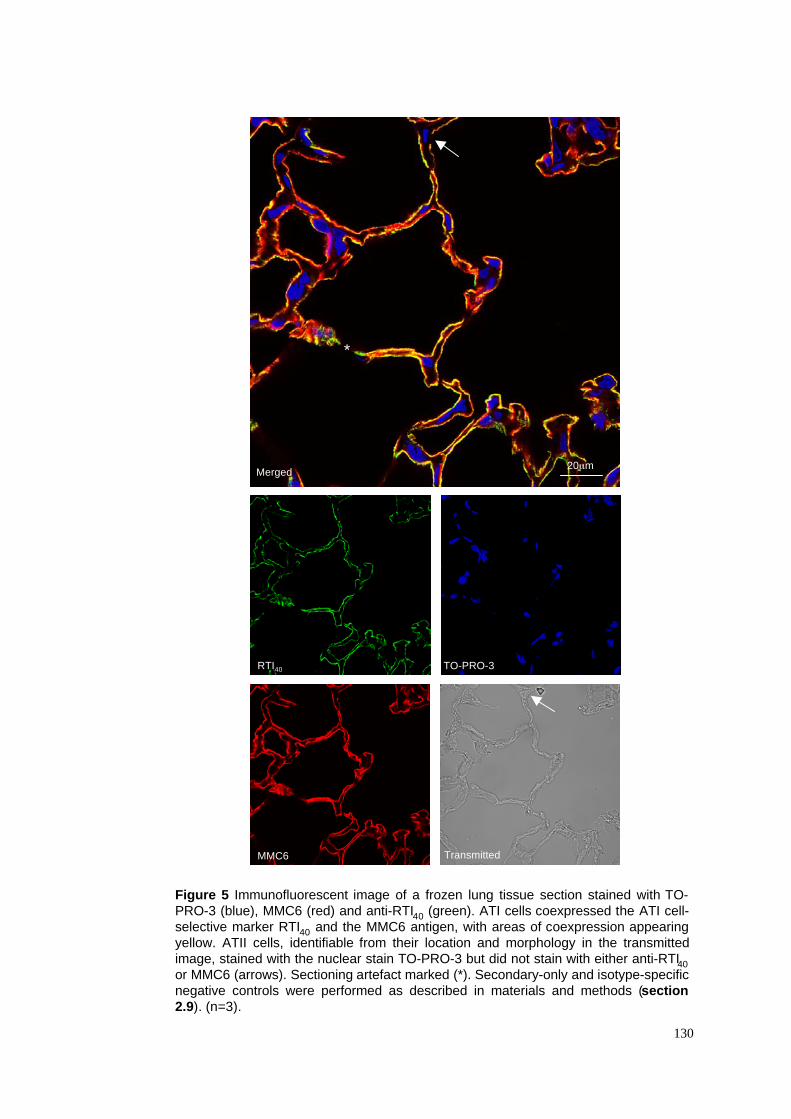
Figure 5 Immunofluorescent image of a frozen lung tissue section stained with TO-
PRO-3 (blue), MMC6 (red) and anti-RTI40 (green). ATI cells coexpressed the ATI cell-
selective marker RTI40 and the MMC6 antigen, with areas of coexpression appearing
yellow. ATII cells, identifiable from their location and morphology in the transmitted
image, stained with the nuclear stain TO-PRO-3 but did not stain with either anti-RTI40
or MMC6 (arrows). Sectioning artefact marked (*). Secondary-only and isotype-specific
negative controls were performed as described in materials and methods (section
2.9). (n=3).
130
Merged
MMC6
RTI40 TO-PRO-3
Transmitted
20µm
*
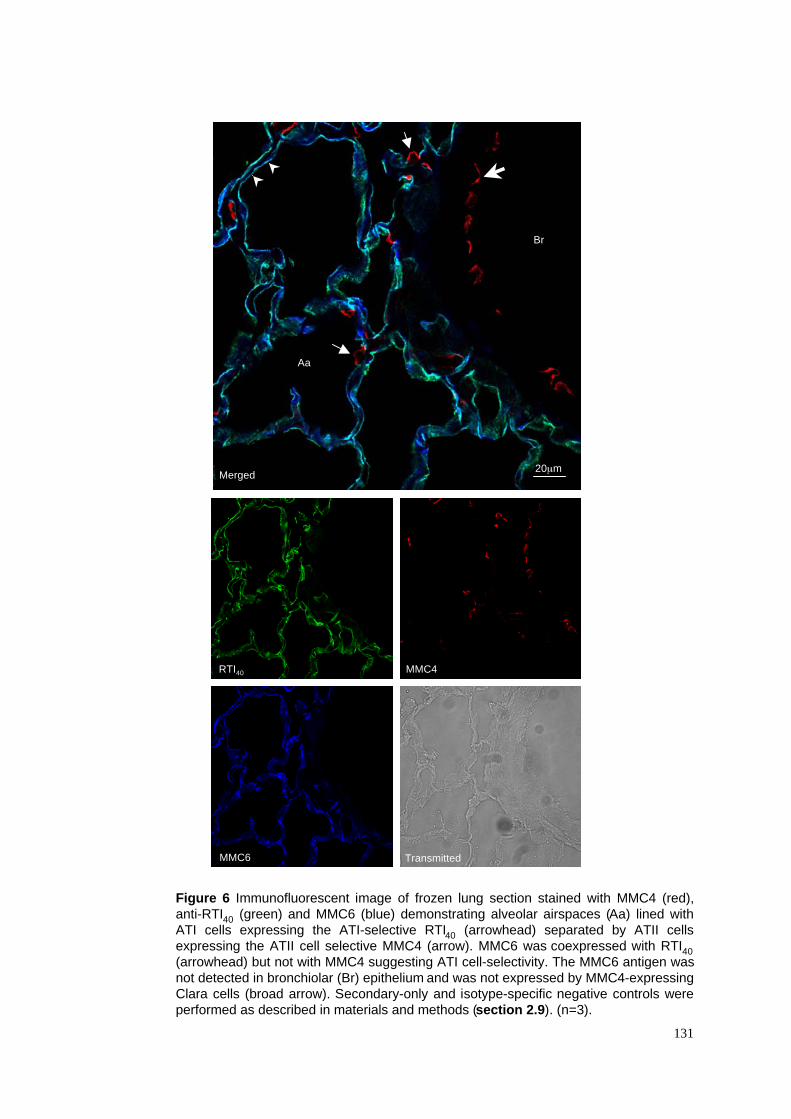
Figure 6 Immunofluorescent image of frozen lung section stained with MMC4 (red),
anti-RTI40 (green) and MMC6 (blue) demonstrating alveolar airspaces (Aa) lined with
ATI cells expressing the ATI-selective RTI40 (arrowhead) separated by ATII cells
expressing the ATII cell selective MMC4 (arrow). MMC6 was coexpressed with RTI40
(arrowhead) but not with MMC4 suggesting ATI cell-selectivity. The MMC6 antigen was
not detected in bronchiolar (Br) epithelium and was not expressed by MMC4-expressing
Clara cells (broad arrow). Secondary-only and isotype-specific negative controls were
performed as described in materials and methods (section 2.9). (n=3).
131
Merged
RTI40
TransmittedMMC6
20µm
MMC4
Br
Aa
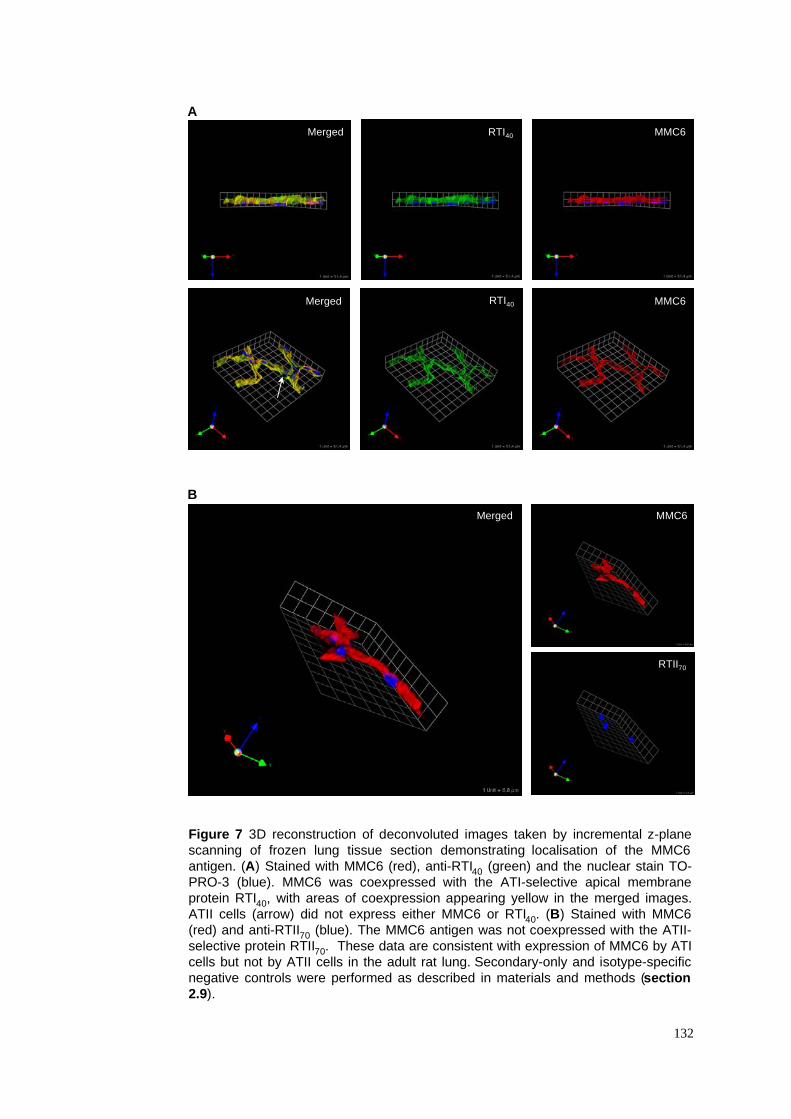
Figure 7 3D reconstruction of deconvoluted images taken by incremental z-plane
scanning of frozen lung tissue section demonstrating localisation of the MMC6
antigen. (A) Stained with MMC6 (red), anti-RTI40 (green) and the nuclear stain TO-
PRO-3 (blue). MMC6 was coexpressed with the ATI-selective apical membrane
protein RTI40, with areas of coexpression appearing yellow in the merged images.
ATII cells (arrow) did not express either MMC6 or RTI40. (B) Stained with MMC6
(red) and anti-RTII70 (blue). The MMC6 antigen was not coexpressed with the ATII-
selective protein RTII70. These data are consistent with expression of MMC6 by ATI
cells but not by ATII cells in the adult rat lung. Secondary-only and isotype-specific
negative controls were performed as described in materials and methods (section
2.9).
132
Merged
MMC6
RTI40 MMC6
RTI40 MMC6
Merged
A
B
Merged
RTII70
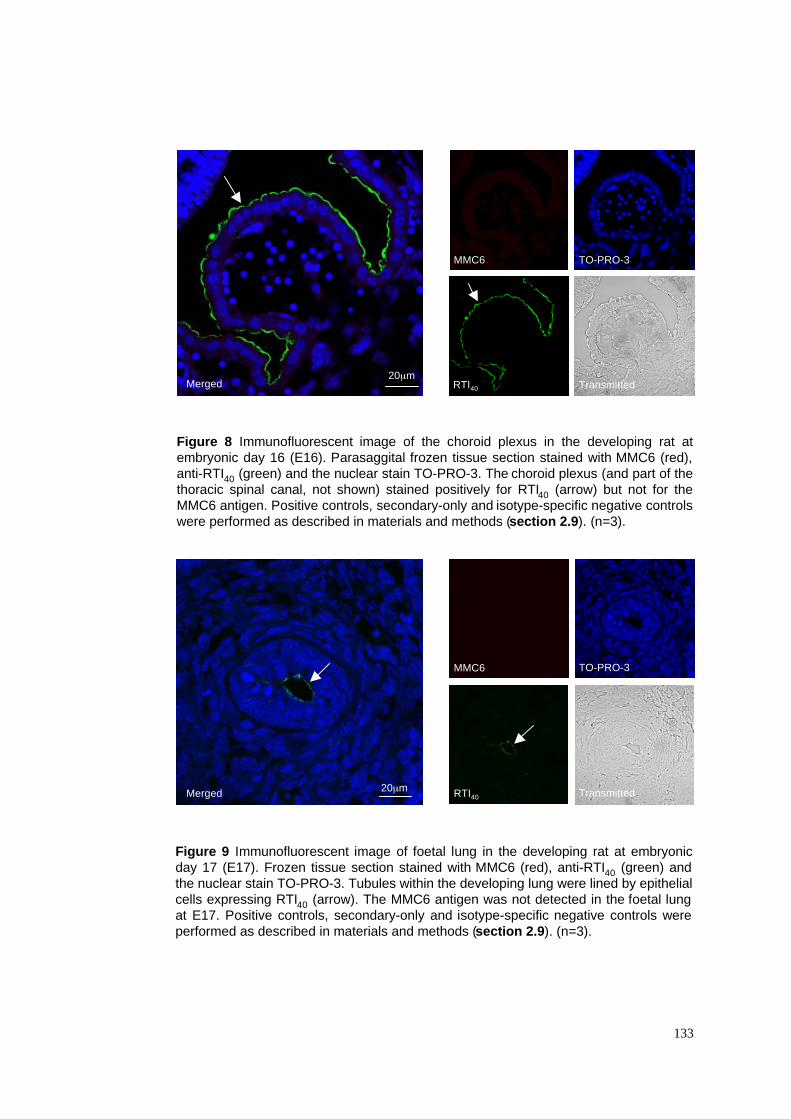
Figure 8 Immunofluorescent image of the choroid plexus in the developing rat at
embryonic day 16 (E16). Parasaggital frozen tissue section stained with MMC6 (red),
anti-RTI40 (green) and the nuclear stain TO-PRO-3. The choroid plexus (and part of the
thoracic spinal canal, not shown) stained positively for RTI40 (arrow) but not for the
MMC6 antigen. Positive controls, secondary-only and isotype-specific negative controls
were performed as described in materials and methods (section 2.9). (n=3).
133
Merged RTI40
TO-PRO-3
Transmitted
MMC6
20µm
Merged RTI40
TO-PRO-3
Transmitted
MMC6
20µm
Figure 9 Immunofluorescent image of foetal lung in the developing rat at embryonic
day 17 (E17). Frozen tissue section stained with MMC6 (red), anti-RTI40 (green) and
the nuclear stain TO-PRO-3. Tubules within the developing lung were lined by epithelial
cells expressing RTI40 (arrow). The MMC6 antigen was not detected in the foetal lung
at E17. Positive controls, secondary-only and isotype-specific negative controls were
performed as described in materials and methods (section 2.9). (n=3).
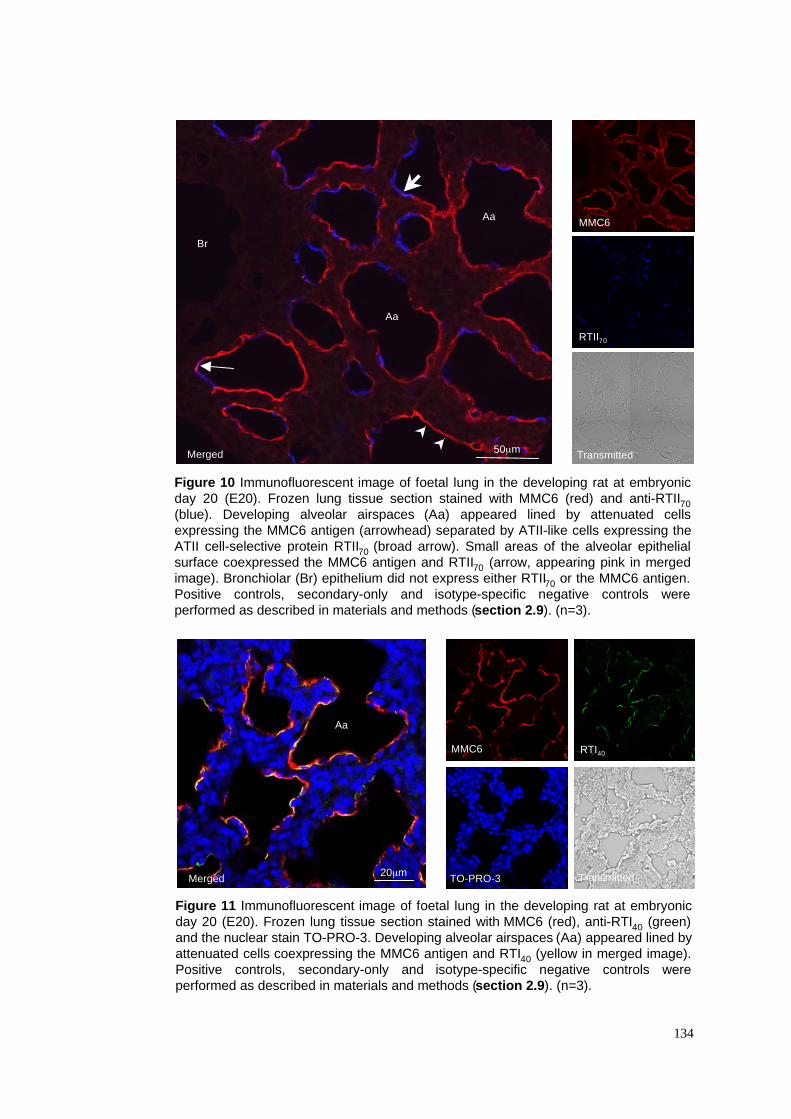
134
Figure 10 Immunofluorescent image of foetal lung in the developing rat at embryonic
day 20 (E20). Frozen lung tissue section stained with MMC6 (red) and anti-RTII70
(blue). Developing alveolar airspaces (Aa) appeared lined by attenuated cells
expressing the MMC6 antigen (arrowhead) separated by ATII-like cells expressing the
ATII cell-selective protein RTII70 (broad arrow). Small areas of the alveolar epithelial
surface coexpressed the MMC6 antigen and RTII70 (arrow, appearing pink in merged
image). Bronchiolar (Br) epithelium did not express either RTII70 or the MMC6 antigen.
Positive controls, secondary-only and isotype-specific negative controls were
performed as described in materials and methods (section 2.9). (n=3).
Figure 11 Immunofluorescent image of foetal lung in the developing rat at embryonic
day 20 (E20). Frozen lung tissue section stained with MMC6 (red), anti-RTI40 (green)
and the nuclear stain TO-PRO-3. Developing alveolar airspaces (Aa) appeared lined by
attenuated cells coexpressing the MMC6 antigen and RTI40 (yellow in merged image).
Positive controls, secondary-only and isotype-specific negative controls were
performed as described in materials and methods (section 2.9). (n=3).
Merged
RTI40
TO-PRO-3 Transmitted
MMC6
20µm
Merged
RTII70
Transmitted
MMC6
50µm
Aa
Aa
Br
Aa
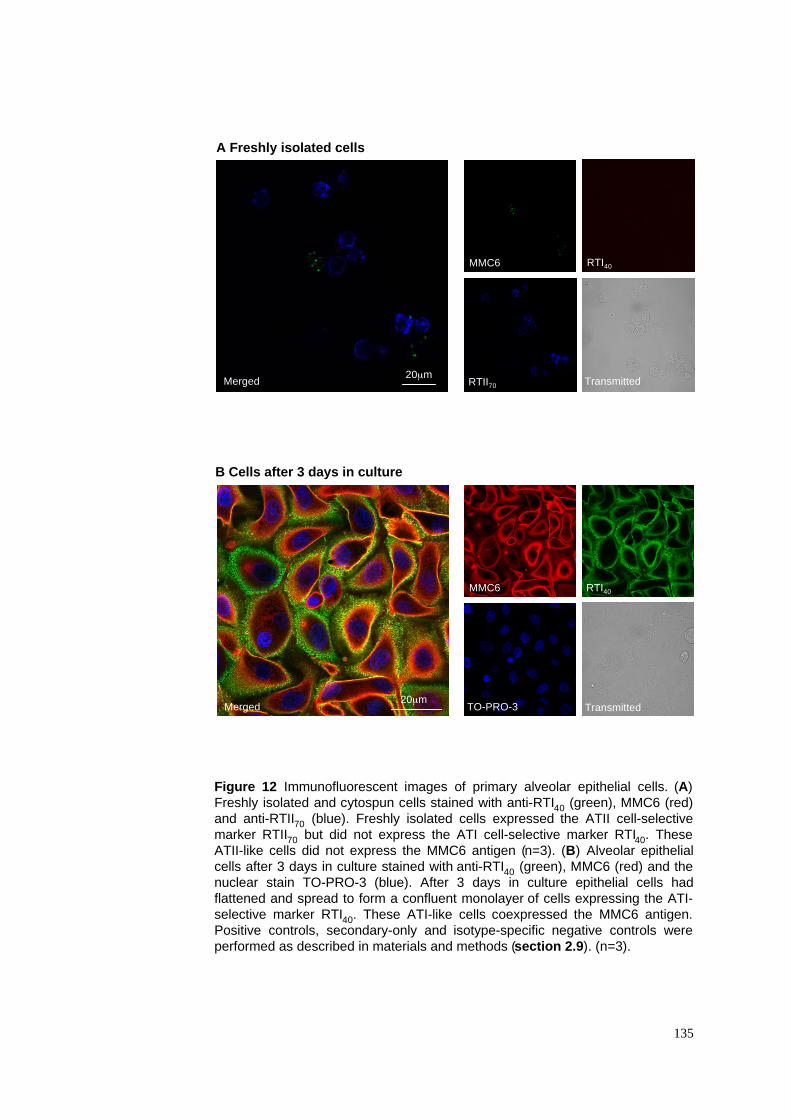
Figure 12 Immunofluorescent images of primary alveolar epithelial cells. (A)
Freshly isolated and cytospun cells stained with anti-RTI40 (green), MMC6 (red)
and anti-RTII70 (blue). Freshly isolated cells expressed the ATII cell-selective
marker RTII70 but did not express the ATI cell-selective marker RTI40. These
ATII-like cells did not express the MMC6 antigen (n=3). (B) Alveolar epithelial
cells after 3 days in culture stained with anti-RTI40 (green), MMC6 (red) and the
nuclear stain TO-PRO-3 (blue). After 3 days in culture epithelial cells had
flattened and spread to form a confluent monolayer of cells expressing the ATI-
selective marker RTI40. These ATI-like cells coexpressed the MMC6 antigen.
Positive controls, secondary-only and isotype-specific negative controls were
performed as described in materials and methods (section 2.9). (n=3).
135
20µmRTII70
Merged Transmitted
MMC6
Merged
RTI40
TO-PRO-3 Transmitted
MMC6
20µm
A Freshly isolated cells
B Cells after 3 days in culture
RTI40
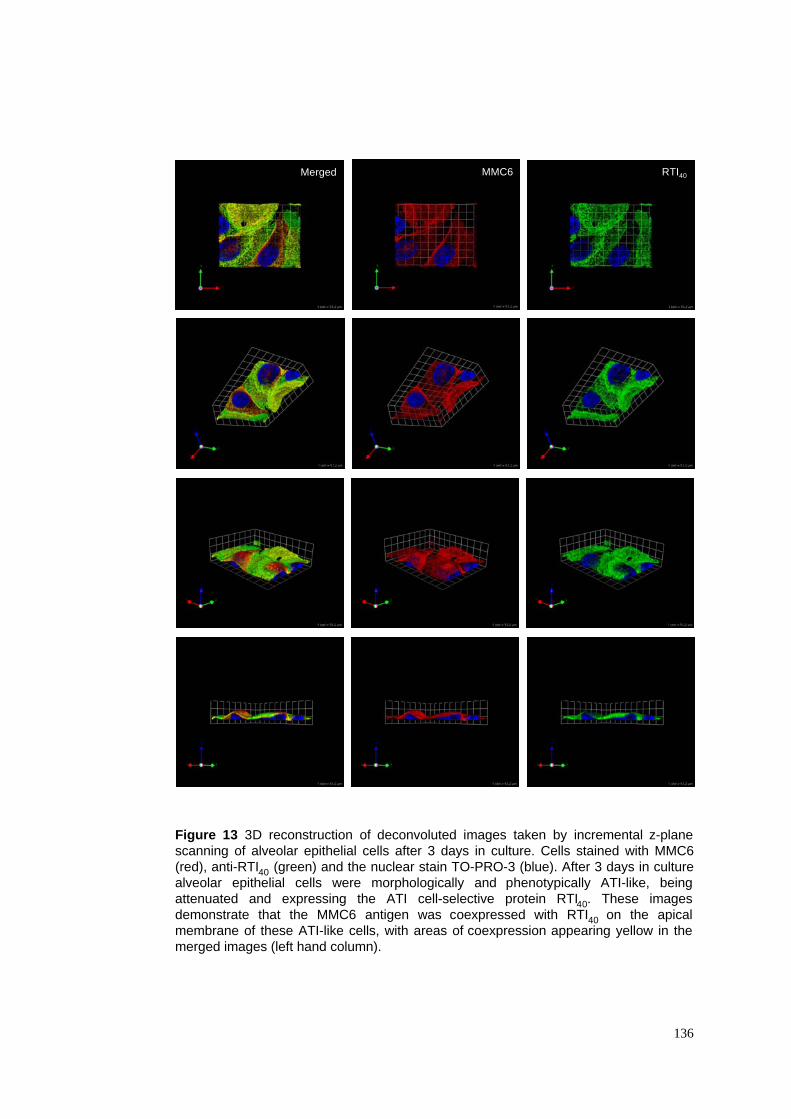
Figure 13 3D reconstruction of deconvoluted images taken by incremental z-plane
scanning of alveolar epithelial cells after 3 days in culture. Cells stained with MMC6
(red), anti-RTI40 (green) and the nuclear stain TO-PRO-3 (blue). After 3 days in culture
alveolar epithelial cells were morphologically and phenotypically ATI-like, being
attenuated and expressing the ATI cell-selective protein RTI40. These images
demonstrate that the MMC6 antigen was coexpressed with RTI40 on the apical
membrane of these ATI-like cells, with areas of coexpression appearing yellow in the
merged images (left hand column).
136
Merged RTI40MMC6
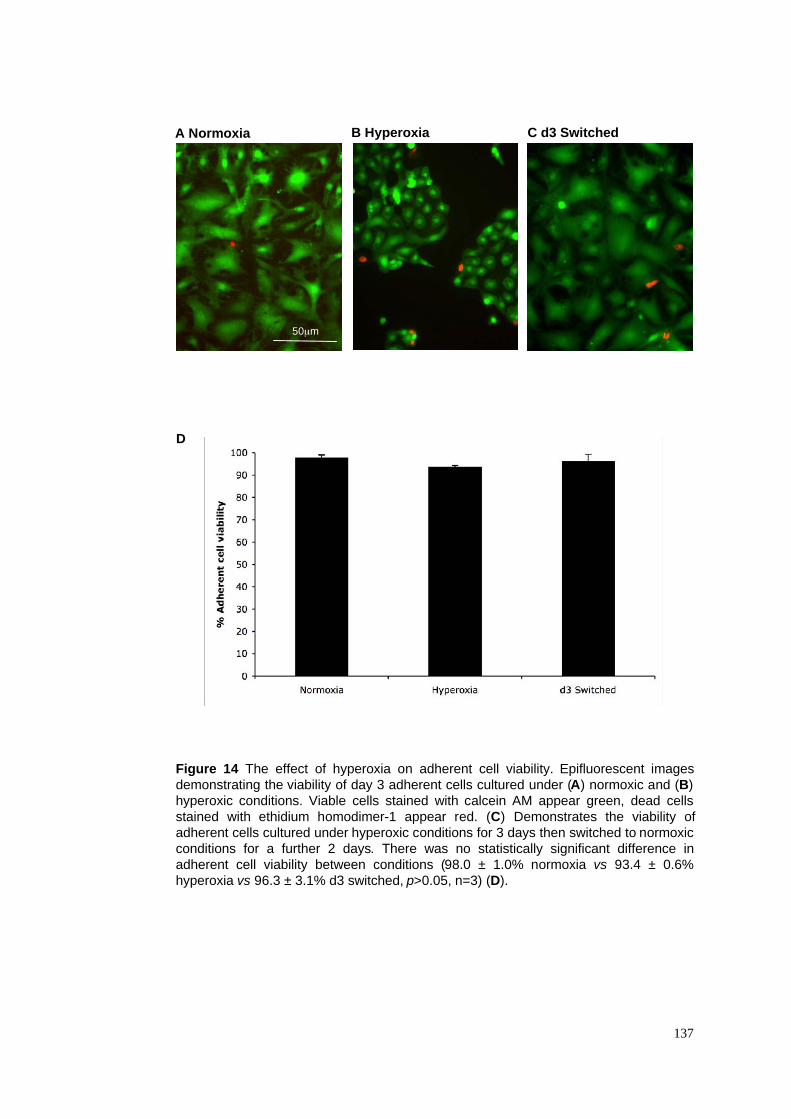
A Normoxia B Hyperoxia
Figure 14 The effect of hyperoxia on adherent cell viability. Epifluorescent images
demonstrating the viability of day 3 adherent cells cultured under (A) normoxic and (B)
hyperoxic conditions. Viable cells stained with calcein AM appear green, dead cells
stained with ethidium homodimer-1 appear red. (C) Demonstrates the viability of
adherent cells cultured under hyperoxic conditions for 3 days then switched to normoxic
conditions for a further 2 days. There was no statistically significant difference in
adherent cell viability between conditions (98.0 ± 1.0% normoxia vs 93.4 ± 0.6%
hyperoxia vs 96.3 ± 3.1% d3 switched, p>0.05, n=3) (D).
137
C d3 Switched
D
50µm
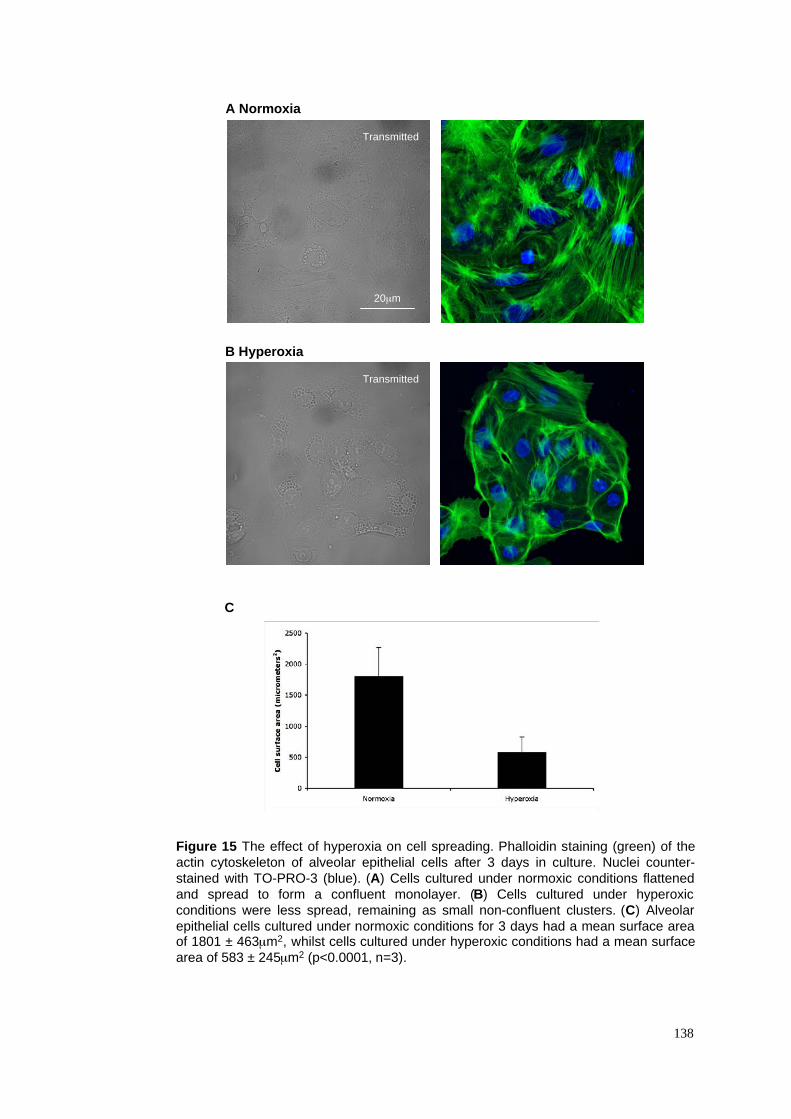
A Normoxia
B Hyperoxia
Figure 15 The effect of hyperoxia on cell spreading. Phalloidin staining (green) of the
actin cytoskeleton of alveolar epithelial cells after 3 days in culture. Nuclei counter-
stained with TO-PRO-3 (blue). (A) Cells cultured under normoxic conditions flattened
and spread to form a confluent monolayer. (B) Cells cultured under hyperoxic
conditions were less spread, remaining as small non-confluent clusters. (C) Alveolar
epithelial cells cultured under normoxic conditions for 3 days had a mean surface areaof 1801 ± 463µm2, whilst cells cultured under hyperoxic conditions had a mean surface
area of 583 ± 245µm2 (p<0.0001, n=3).
138
C
Transmitted
Transmitted
20µm
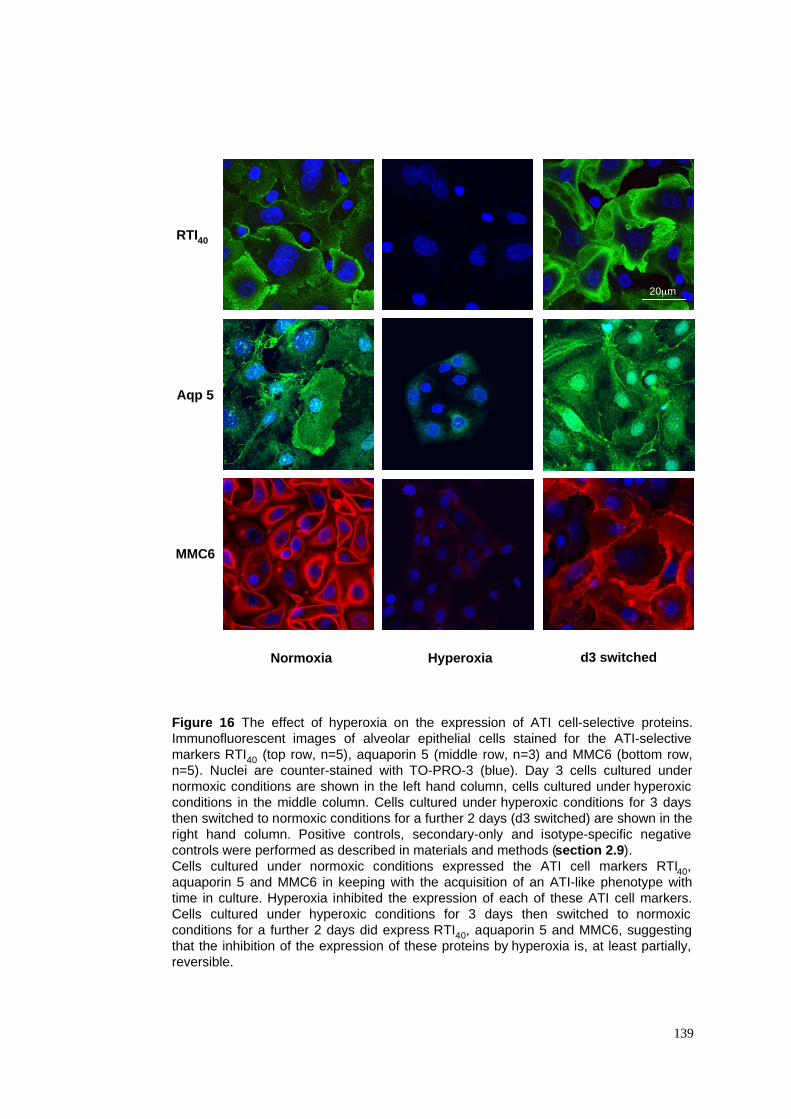
Figure 16 The effect of hyperoxia on the expression of ATI cell-selective proteins.
Immunofluorescent images of alveolar epithelial cells stained for the ATI-selective
markers RTI40 (top row, n=5), aquaporin 5 (middle row, n=3) and MMC6 (bottom row,
n=5). Nuclei are counter-stained with TO-PRO-3 (blue). Day 3 cells cultured under
normoxic conditions are shown in the left hand column, cells cultured under hyperoxic
conditions in the middle column. Cells cultured under hyperoxic conditions for 3 days
then switched to normoxic conditions for a further 2 days (d3 switched) are shown in the
right hand column. Positive controls, secondary-only and isotype-specific negative
controls were performed as described in materials and methods (section 2.9).
Cells cultured under normoxic conditions expressed the ATI cell markers RTI40,
aquaporin 5 and MMC6 in keeping with the acquisition of an ATI-like phenotype with
time in culture. Hyperoxia inhibited the expression of each of these ATI cell markers.
Cells cultured under hyperoxic conditions for 3 days then switched to normoxic
conditions for a further 2 days did express RTI40, aquaporin 5 and MMC6, suggesting
that the inhibition of the expression of these proteins by hyperoxia is, at least partially,
reversible.
139
Normoxia Hyperoxia d3 switched
RTI40
Aqp 5
MMC6
20µm
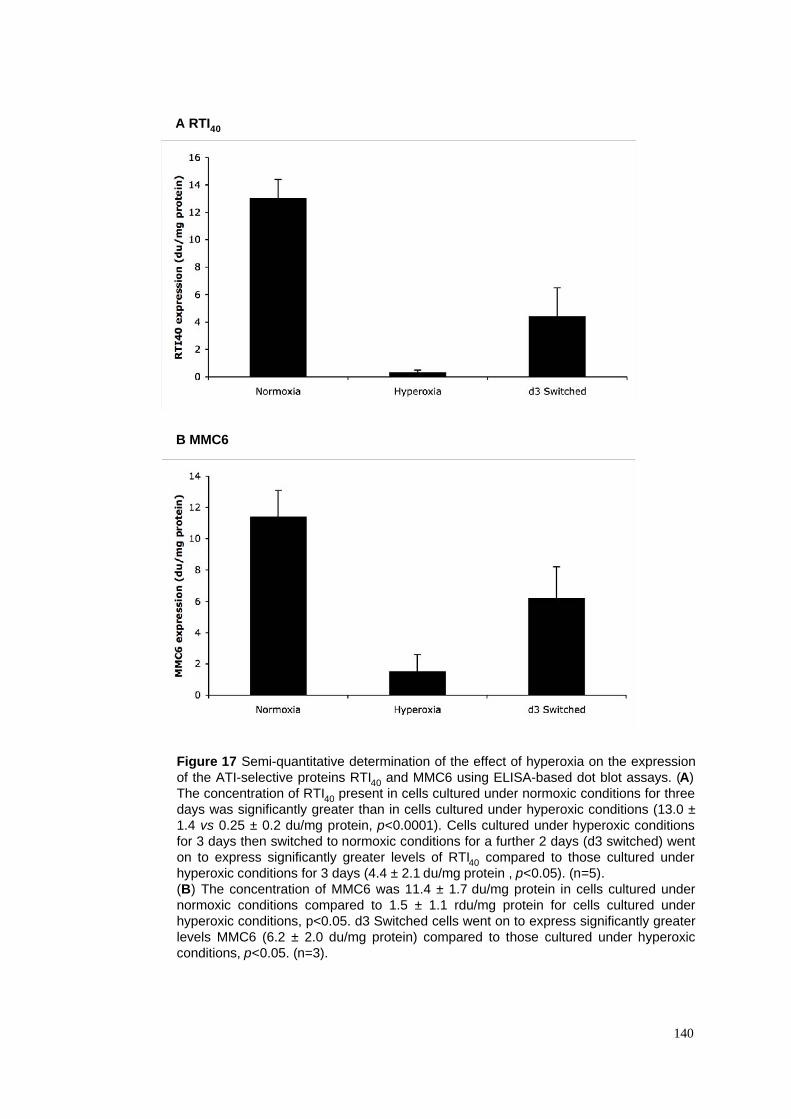
A RTI40
Figure 17 Semi-quantitative determination of the effect of hyperoxia on the expression
of the ATI-selective proteins RTI40 and MMC6 using ELISA-based dot blot assays. (A)
The concentration of RTI40 present in cells cultured under normoxic conditions for three
days was significantly greater than in cells cultured under hyperoxic conditions (13.0 ±
1.4 vs 0.25 ± 0.2 du/mg protein, p<0.0001). Cells cultured under hyperoxic conditions
for 3 days then switched to normoxic conditions for a further 2 days (d3 switched) went
on to express significantly greater levels of RTI40 compared to those cultured under
hyperoxic conditions for 3 days (4.4 ± 2.1 du/mg protein , p<0.05). (n=5).
(B) The concentration of MMC6 was 11.4 ± 1.7 du/mg protein in cells cultured under
normoxic conditions compared to 1.5 ± 1.1 rdu/mg protein for cells cultured under
hyperoxic conditions, p<0.05. d3 Switched cells went on to express significantly greater
levels MMC6 (6.2 ± 2.0 du/mg protein) compared to those cultured under hyperoxic
conditions, p<0.05. (n=3).
140
B MMC6
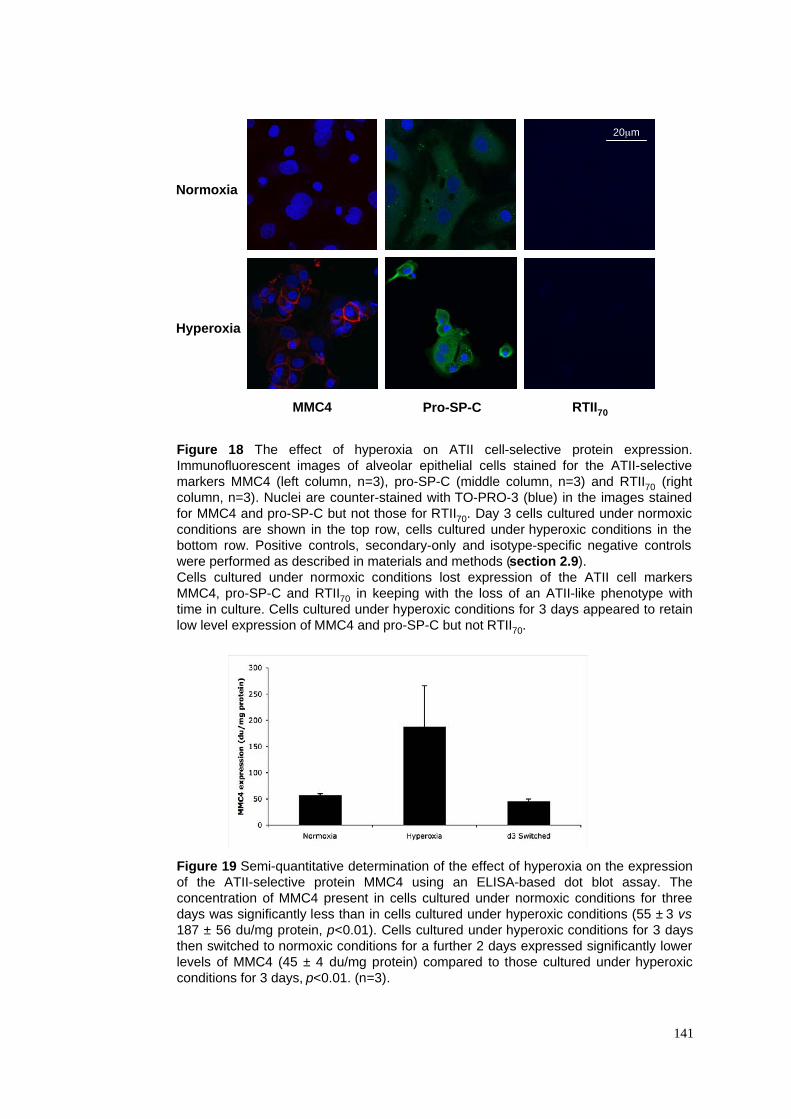
Figure 19 Semi-quantitative determination of the effect of hyperoxia on the expression
of the ATII-selective protein MMC4 using an ELISA-based dot blot assay. The
concentration of MMC4 present in cells cultured under normoxic conditions for three
days was significantly less than in cells cultured under hyperoxic conditions (55 ± 3 vs
187 ± 56 du/mg protein, p<0.01). Cells cultured under hyperoxic conditions for 3 days
then switched to normoxic conditions for a further 2 days expressed significantly lower
levels of MMC4 (45 ± 4 du/mg protein) compared to those cultured under hyperoxic
conditions for 3 days, p<0.01. (n=3).
141
Normoxia
Hyperoxia
MMC4 Pro-SP-C RTII70
Figure 18 The effect of hyperoxia on ATII cell-selective protein expression.
Immunofluorescent images of alveolar epithelial cells stained for the ATII-selective
markers MMC4 (left column, n=3), pro-SP-C (middle column, n=3) and RTII70 (right
column, n=3). Nuclei are counter-stained with TO-PRO-3 (blue) in the images stained
for MMC4 and pro-SP-C but not those for RTII70. Day 3 cells cultured under normoxic
conditions are shown in the top row, cells cultured under hyperoxic conditions in the
bottom row. Positive controls, secondary-only and isotype-specific negative controls
were performed as described in materials and methods (section 2.9).
Cells cultured under normoxic conditions lost expression of the ATII cell markers
MMC4, pro-SP-C and RTII70 in keeping with the loss of an ATII-like phenotype with
time in culture. Cells cultured under hyperoxic conditions for 3 days appeared to retain
low level expression of MMC4 and pro-SP-C but not RTII70.
20µm
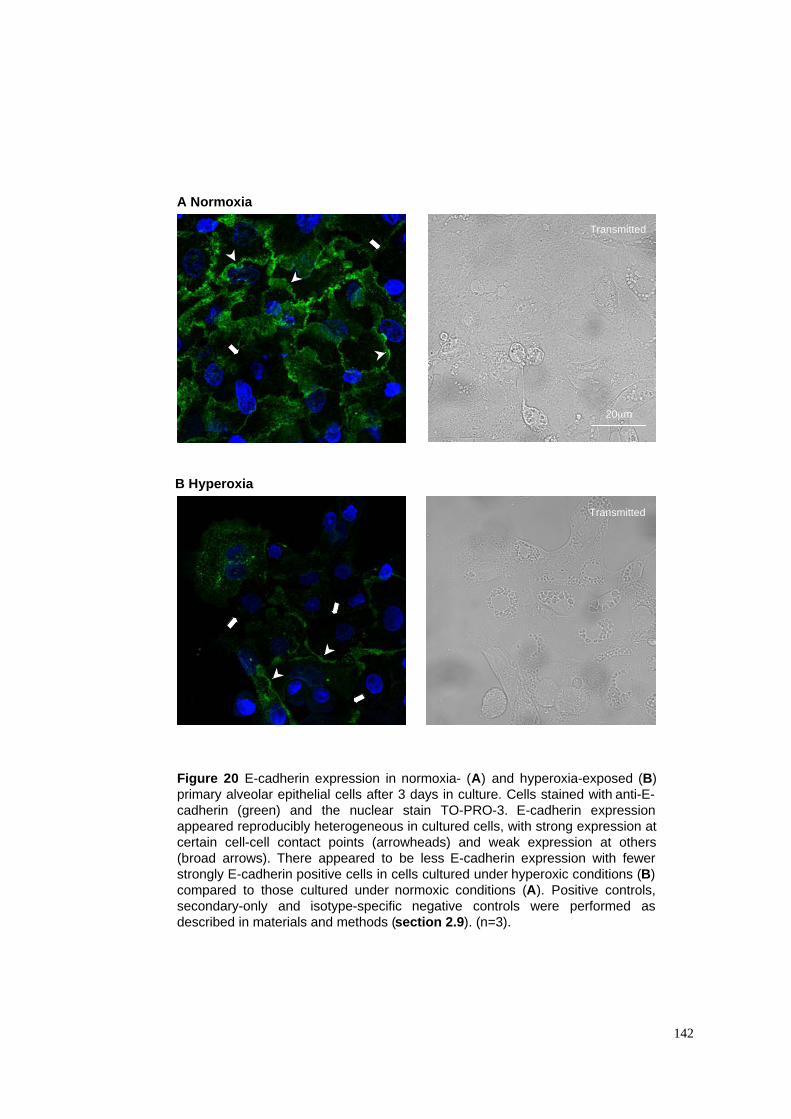
A Normoxia
Figure 20 E-cadherin expression in normoxia- (A) and hyperoxia-exposed (B)
primary alveolar epithelial cells after 3 days in culture. Cells stained with anti-E-
cadherin (green) and the nuclear stain TO-PRO-3. E-cadherin expression
appeared reproducibly heterogeneous in cultured cells, with strong expression at
certain cell-cell contact points (arrowheads) and weak expression at others
(broad arrows). There appeared to be less E-cadherin expression with fewer
strongly E-cadherin positive cells in cells cultured under hyperoxic conditions (B)
compared to those cultured under normoxic conditions (A). Positive controls,
secondary-only and isotype-specific negative controls were performed as
described in materials and methods (section 2.9). (n=3).
142
B Hyperoxia
Transmitted
Transmitted
20µm
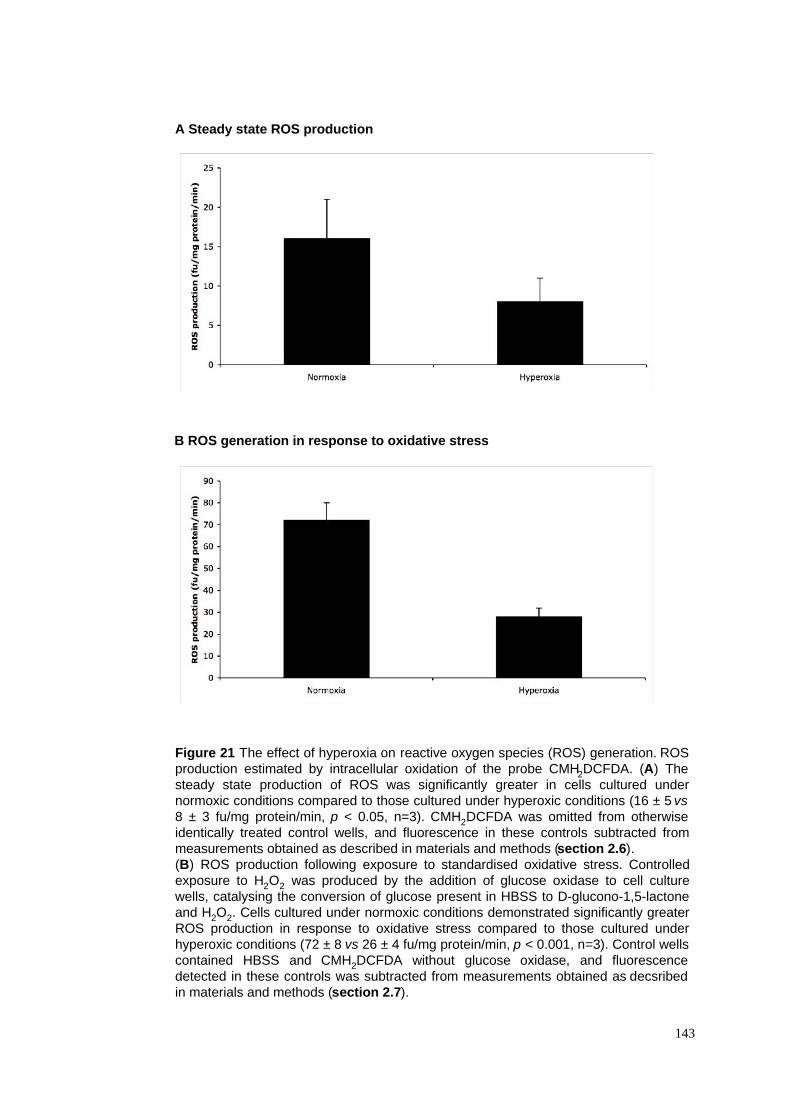
143
A Steady state ROS production
B ROS generation in response to oxidative stress
Figure 21 The effect of hyperoxia on reactive oxygen species (ROS) generation. ROS
production estimated by intracellular oxidation of the probe CMH2DCFDA. (A) The
steady state production of ROS was significantly greater in cells cultured under
normoxic conditions compared to those cultured under hyperoxic conditions (16 ± 5 vs
8 ± 3 fu/mg protein/min, p < 0.05, n=3). CMH2DCFDA was omitted from otherwise
identically treated control wells, and fluorescence in these controls subtracted from
measurements obtained as described in materials and methods (section 2.6).
(B) ROS production following exposure to standardised oxidative stress. Controlled
exposure to H2O2 was produced by the addition of glucose oxidase to cell culture
wells, catalysing the conversion of glucose present in HBSS to D-glucono-1,5-lactone
and H2O2. Cells cultured under normoxic conditions demonstrated significantly greater
ROS production in response to oxidative stress compared to those cultured under
hyperoxic conditions (72 ± 8 vs 26 ± 4 fu/mg protein/min, p < 0.001, n=3). Control wells
contained HBSS and CMH2DCFDA without glucose oxidase, and fluorescence
detected in these controls was subtracted from measurements obtained as decsribed
in materials and methods (section 2.7).
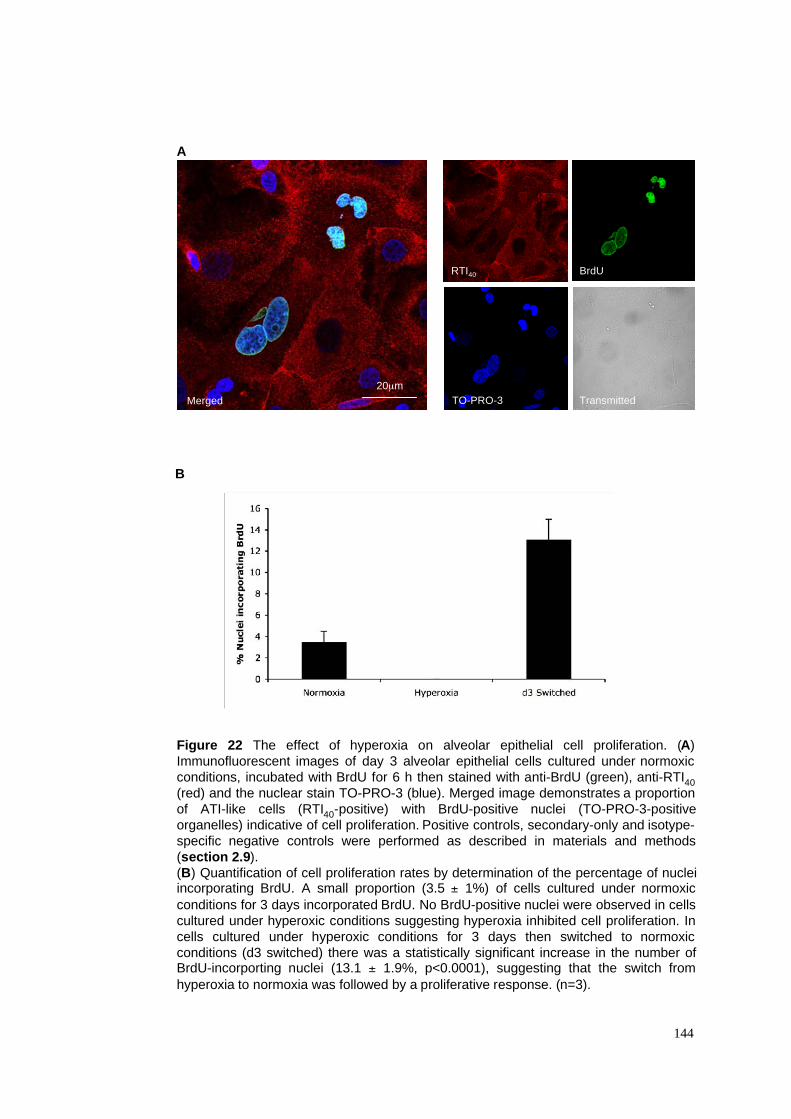
A
B
Figure 22 The effect of hyperoxia on alveolar epithelial cell proliferation. (A)
Immunofluorescent images of day 3 alveolar epithelial cells cultured under normoxic
conditions, incubated with BrdU for 6 h then stained with anti-BrdU (green), anti-RTI40
(red) and the nuclear stain TO-PRO-3 (blue). Merged image demonstrates a proportion
of ATI-like cells (RTI40-positive) with BrdU-positive nuclei (TO-PRO-3-positive
organelles) indicative of cell proliferation. Positive controls, secondary-only and isotype-
specific negative controls were performed as described in materials and methods
(section 2.9).
(B) Quantification of cell proliferation rates by determination of the percentage of nucleiincorporating BrdU. A small proportion (3.5 ± 1%) of cells cultured under normoxic
conditions for 3 days incorporated BrdU. No BrdU-positive nuclei were observed in cells
cultured under hyperoxic conditions suggesting hyperoxia inhibited cell proliferation. In
cells cultured under hyperoxic conditions for 3 days then switched to normoxic
conditions (d3 switched) there was a statistically significant increase in the number ofBrdU-incorporting nuclei (13.1 ± 1.9%, p<0.0001), suggesting that the switch from
hyperoxia to normoxia was followed by a proliferative response. (n=3).
144
Merged
20µm
RTI40
TO-PRO-3
BrdU
Transmitted
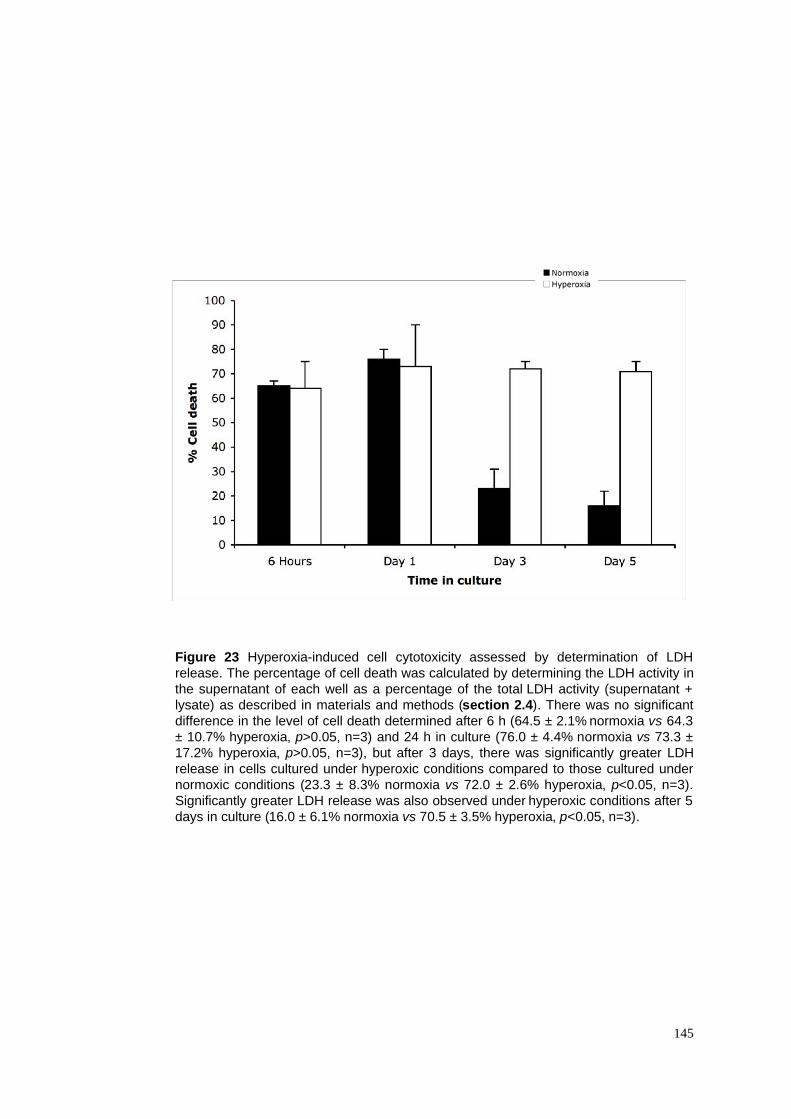
Figure 23 Hyperoxia-induced cell cytotoxicity assessed by determination of LDH
release. The percentage of cell death was calculated by determining the LDH activity in
the supernatant of each well as a percentage of the total LDH activity (supernatant +
lysate) as described in materials and methods (section 2.4). There was no significant
difference in the level of cell death determined after 6 h (64.5 ± 2.1% normoxia vs 64.3
± 10.7% hyperoxia, p>0.05, n=3) and 24 h in culture (76.0 ± 4.4% normoxia vs 73.3 ±
17.2% hyperoxia, p>0.05, n=3), but after 3 days, there was significantly greater LDH
release in cells cultured under hyperoxic conditions compared to those cultured under
normoxic conditions (23.3 ± 8.3% normoxia vs 72.0 ± 2.6% hyperoxia, p<0.05, n=3).
Significantly greater LDH release was also observed under hyperoxic conditions after 5
days in culture (16.0 ± 6.1% normoxia vs 70.5 ± 3.5% hyperoxia, p<0.05, n=3).
145
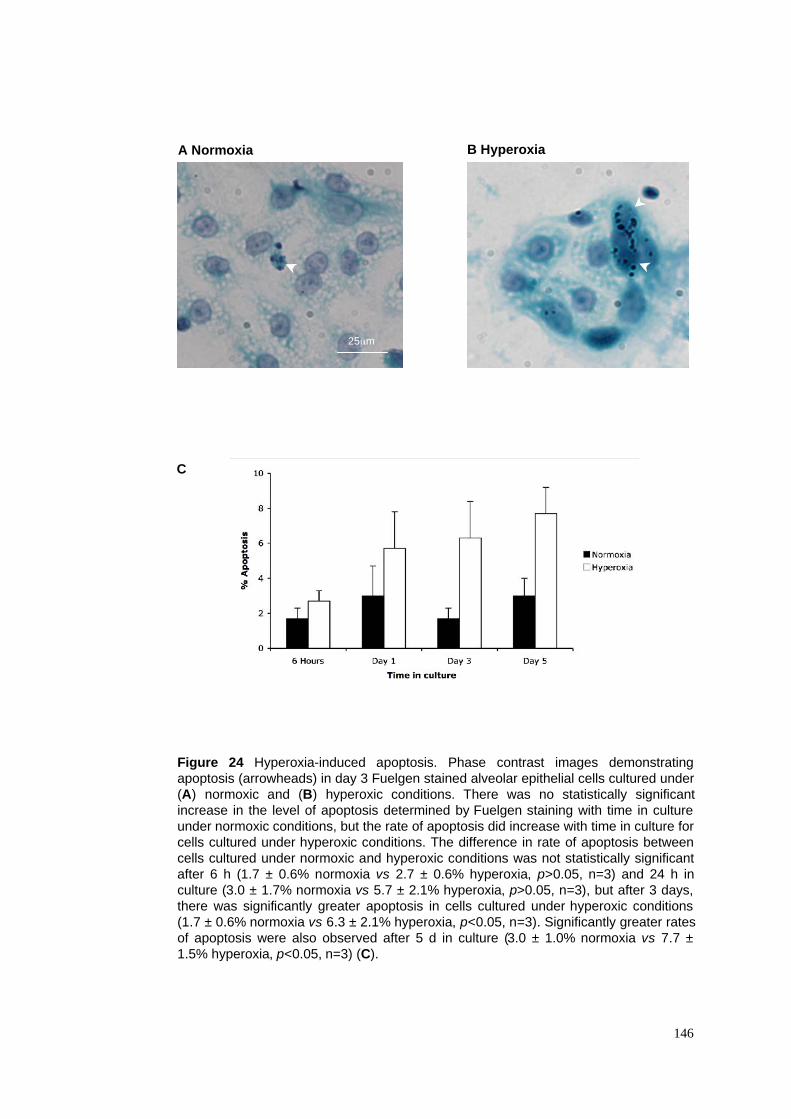
146
A Normoxia B Hyperoxia
Figure 24 Hyperoxia-induced apoptosis. Phase contrast images demonstrating
apoptosis (arrowheads) in day 3 Fuelgen stained alveolar epithelial cells cultured under
(A) normoxic and (B) hyperoxic conditions. There was no statistically significant
increase in the level of apoptosis determined by Fuelgen staining with time in culture
under normoxic conditions, but the rate of apoptosis did increase with time in culture for
cells cultured under hyperoxic conditions. The difference in rate of apoptosis between
cells cultured under normoxic and hyperoxic conditions was not statistically significant
after 6 h (1.7 ± 0.6% normoxia vs 2.7 ± 0.6% hyperoxia, p>0.05, n=3) and 24 h in
culture (3.0 ± 1.7% normoxia vs 5.7 ± 2.1% hyperoxia, p>0.05, n=3), but after 3 days,
there was significantly greater apoptosis in cells cultured under hyperoxic conditions
(1.7 ± 0.6% normoxia vs 6.3 ± 2.1% hyperoxia, p<0.05, n=3). Significantly greater rates
of apoptosis were also observed after 5 d in culture (3.0 ± 1.0% normoxia vs 7.7 ±
1.5% hyperoxia, p<0.05, n=3) (C).
C
25µm
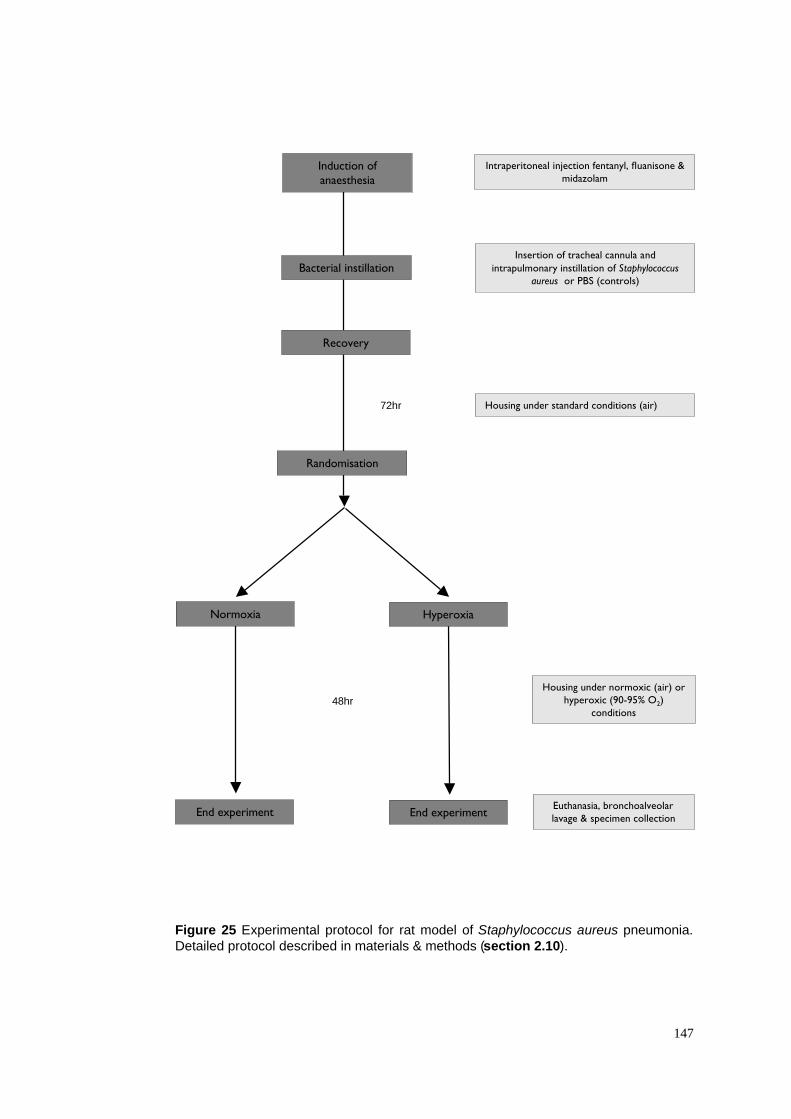
Figure 25 Experimental protocol for rat model of Staphylococcus aureus pneumonia.
Detailed protocol described in materials & methods (section 2.10).
147
Insertion of tracheal cannula and
intrapulmonary instillation of Staphylococcusaureus or PBS (controls)
Housing under standard conditions (air)
Induction ofanaesthesia
Bacterial instillation
72hr
48hr
Recovery
Randomisation
Intraperitoneal injection fentanyl, fluanisone &midazolam
Housing under normoxic (air) orhyperoxic (90-95% O2)
conditions
Euthanasia, bronchoalveolarlavage & specimen collection
Normoxia Hyperoxia
End experiment End experiment
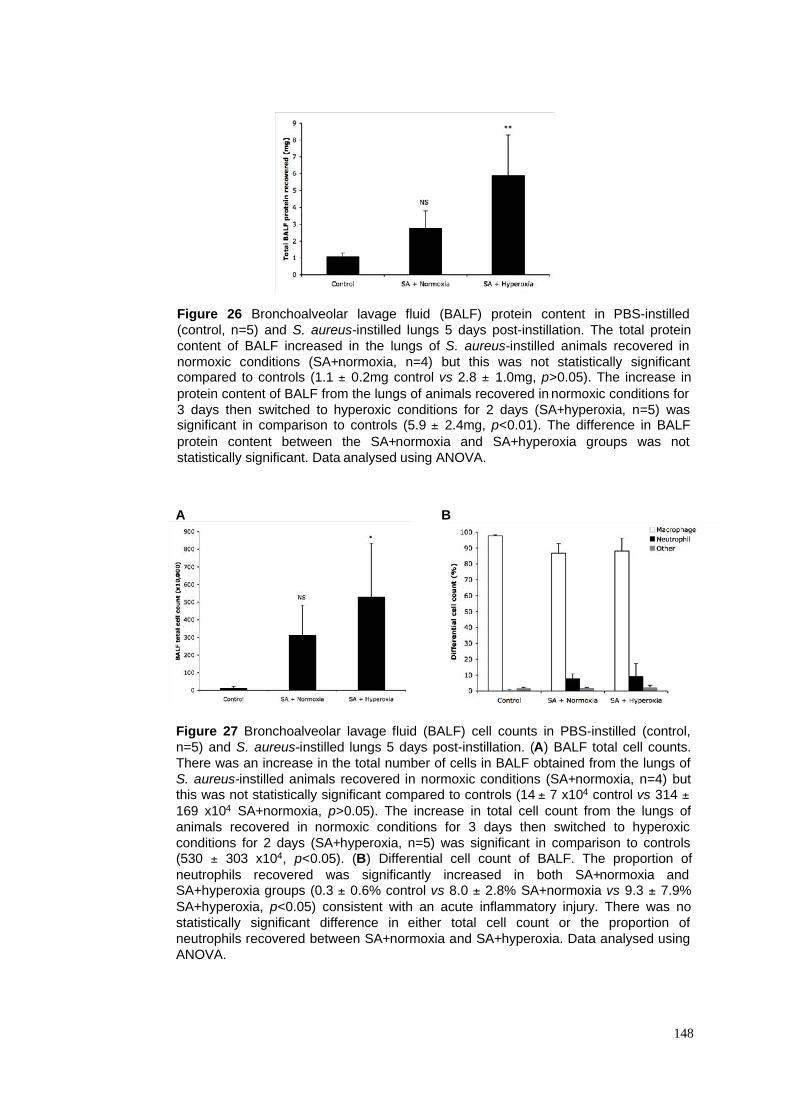
A B
Figure 27 Bronchoalveolar lavage fluid (BALF) cell counts in PBS-instilled (control,
n=5) and S. aureus-instilled lungs 5 days post-instillation. (A) BALF total cell counts.
There was an increase in the total number of cells in BALF obtained from the lungs of
S. aureus-instilled animals recovered in normoxic conditions (SA+normoxia, n=4) butthis was not statistically significant compared to controls (14 ± 7 x104 control vs 314 ±
169 x104 SA+normoxia, p>0.05). The increase in total cell count from the lungs of
animals recovered in normoxic conditions for 3 days then switched to hyperoxic
conditions for 2 days (SA+hyperoxia, n=5) was significant in comparison to controls(530 ± 303 x104, p<0.05). (B) Differential cell count of BALF. The proportion of
neutrophils recovered was significantly increased in both SA+normoxia andSA+hyperoxia groups (0.3 ± 0.6% control vs 8.0 ± 2.8% SA+normoxia vs 9.3 ± 7.9%
SA+hyperoxia, p<0.05) consistent with an acute inflammatory injury. There was no
statistically significant difference in either total cell count or the proportion of
neutrophils recovered between SA+normoxia and SA+hyperoxia. Data analysed using
ANOVA.
148
Figure 26 Bronchoalveolar lavage fluid (BALF) protein content in PBS-instilled
(control, n=5) and S. aureus-instilled lungs 5 days post-instillation. The total protein
content of BALF increased in the lungs of S. aureus-instilled animals recovered in
normoxic conditions (SA+normoxia, n=4) but this was not statistically significantcompared to controls (1.1 ± 0.2mg control vs 2.8 ± 1.0mg, p>0.05). The increase in
protein content of BALF from the lungs of animals recovered in normoxic conditions for
3 days then switched to hyperoxic conditions for 2 days (SA+hyperoxia, n=5) wassignificant in comparison to controls (5.9 ± 2.4mg, p<0.01). The difference in BALF
protein content between the SA+normoxia and SA+hyperoxia groups was not
statistically significant. Data analysed using ANOVA.
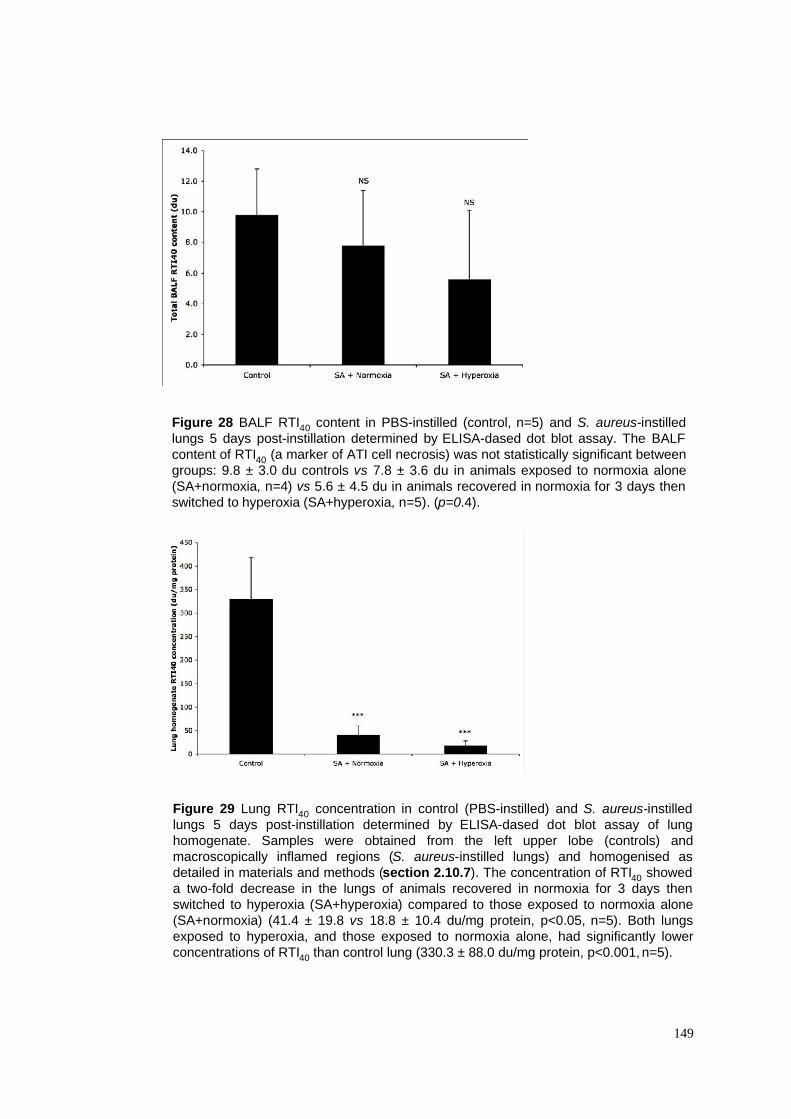
Figure 29 Lung RTI40 concentration in control (PBS-instilled) and S. aureus-instilled
lungs 5 days post-instillation determined by ELISA-dased dot blot assay of lung
homogenate. Samples were obtained from the left upper lobe (controls) and
macroscopically inflamed regions (S. aureus-instilled lungs) and homogenised as
detailed in materials and methods (section 2.10.7). The concentration of RTI40 showed
a two-fold decrease in the lungs of animals recovered in normoxia for 3 days then
switched to hyperoxia (SA+hyperoxia) compared to those exposed to normoxia alone
(SA+normoxia) (41.4 ± 19.8 vs 18.8 ± 10.4 du/mg protein, p<0.05, n=5). Both lungs
exposed to hyperoxia, and those exposed to normoxia alone, had significantly lower
concentrations of RTI40 than control lung (330.3 ± 88.0 du/mg protein, p<0.001, n=5).
149
Figure 28 BALF RTI40 content in PBS-instilled (control, n=5) and S. aureus-instilled
lungs 5 days post-instillation determined by ELISA-dased dot blot assay. The BALF
content of RTI40 (a marker of ATI cell necrosis) was not statistically significant between
groups: 9.8 ± 3.0 du controls vs 7.8 ± 3.6 du in animals exposed to normoxia alone
(SA+normoxia, n=4) vs 5.6 ± 4.5 du in animals recovered in normoxia for 3 days then
switched to hyperoxia (SA+hyperoxia, n=5). (p=0.4).
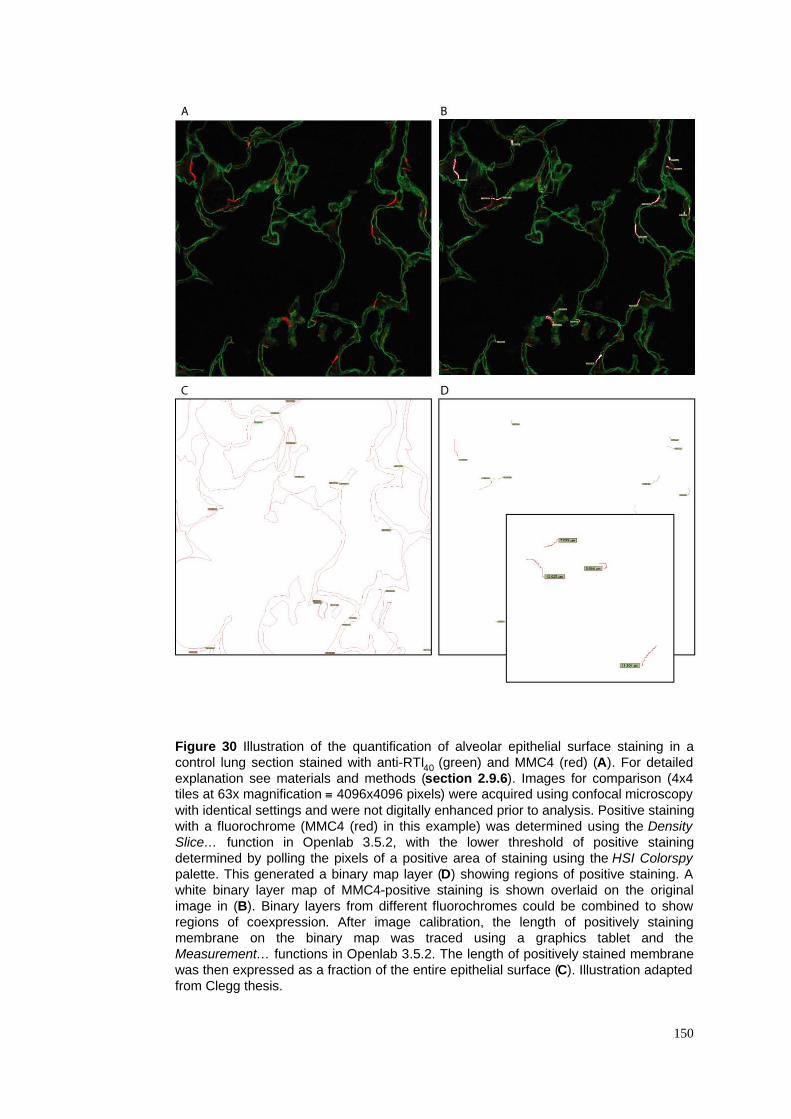
Figure 30 Illustration of the quantification of alveolar epithelial surface staining in a
control lung section stained with anti-RTI40 (green) and MMC4 (red) (A). For detailed
explanation see materials and methods (section 2.9.6). Images for comparison (4x4tiles at 63x magnification ! 4096x4096 pixels) were acquired using confocal microscopy
with identical settings and were not digitally enhanced prior to analysis. Positive staining
with a fluorochrome (MMC4 (red) in this example) was determined using the Density
Slice… function in Openlab 3.5.2, with the lower threshold of positive staining
determined by polling the pixels of a positive area of staining using the HSI Colorspy
palette. This generated a binary map layer (D) showing regions of positive staining. A
white binary layer map of MMC4-positive staining is shown overlaid on the original
image in (B). Binary layers from different fluorochromes could be combined to show
regions of coexpression. After image calibration, the length of positively staining
membrane on the binary map was traced using a graphics tablet and the
Measurement… functions in Openlab 3.5.2. The length of positively stained membrane
was then expressed as a fraction of the entire epithelial surface (C). Illustration adapted
from Clegg thesis.
150
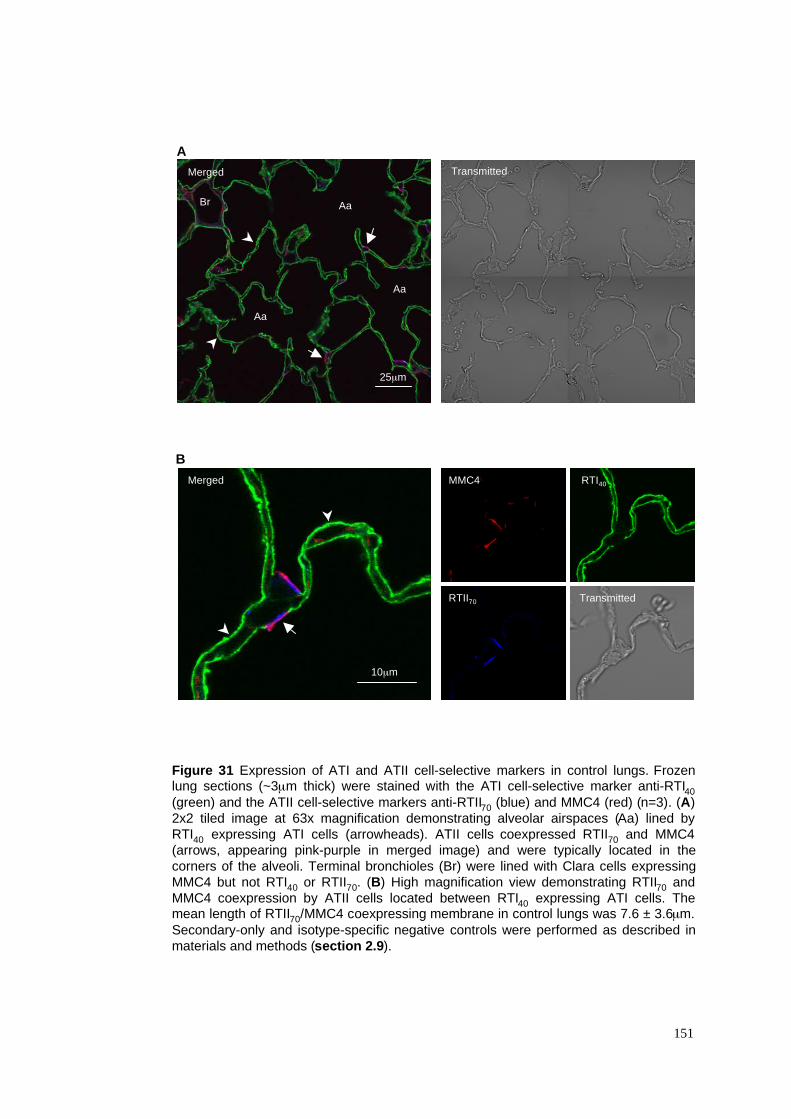
A
B
Figure 31 Expression of ATI and ATII cell-selective markers in control lungs. Frozenlung sections (~3µm thick) were stained with the ATI cell-selective marker anti-RTI40
(green) and the ATII cell-selective markers anti-RTII70 (blue) and MMC4 (red) (n=3). (A)
2x2 tiled image at 63x magnification demonstrating alveolar airspaces (Aa) lined by
RTI40 expressing ATI cells (arrowheads). ATII cells coexpressed RTII70 and MMC4
(arrows, appearing pink-purple in merged image) and were typically located in the
corners of the alveoli. Terminal bronchioles (Br) were lined with Clara cells expressing
MMC4 but not RTI40 or RTII70. (B) High magnification view demonstrating RTII70 and
MMC4 coexpression by ATII cells located between RTI40 expressing ATI cells. Themean length of RTII70/MMC4 coexpressing membrane in control lungs was 7.6 ± 3.6µm.
Secondary-only and isotype-specific negative controls were performed as described in
materials and methods (section 2.9).
151
MMC4 RTI40
25µm
Merged
Merged
Transmitted
TransmittedRTII70
10µm
Br Aa
Aa
Aa
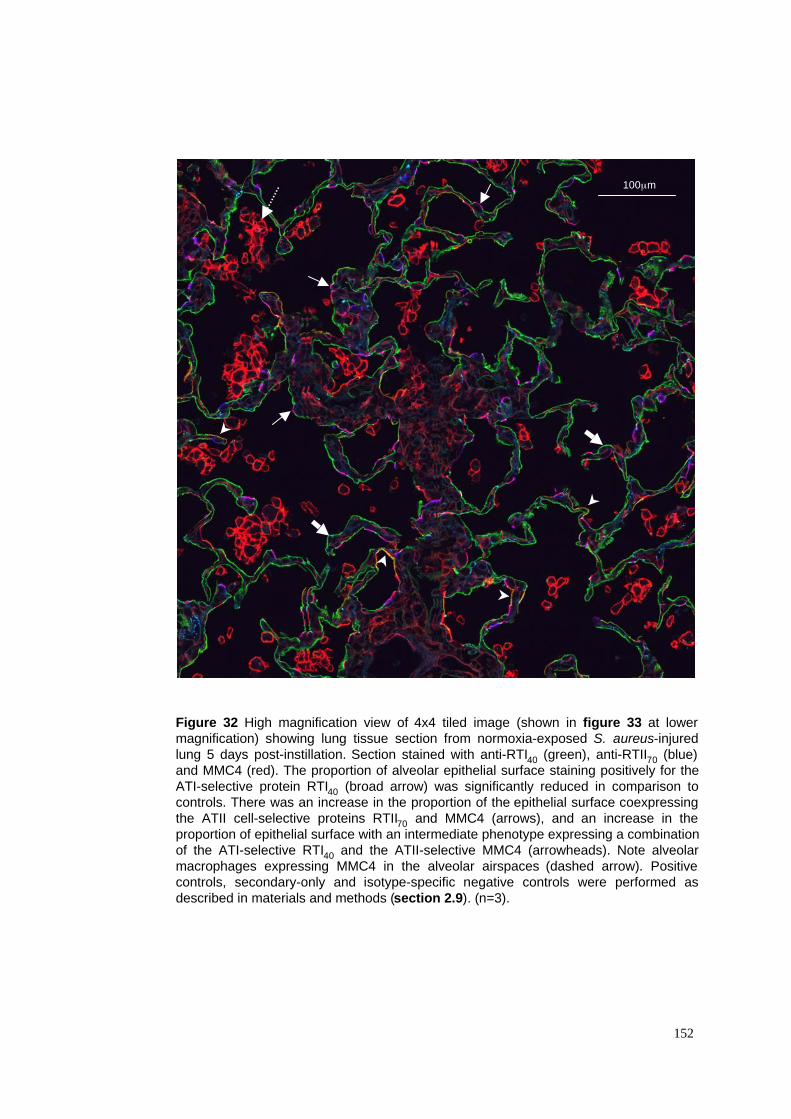
Figure 32 High magnification view of 4x4 tiled image (shown in figure 33 at lower
magnification) showing lung tissue section from normoxia-exposed S. aureus-injured
lung 5 days post-instillation. Section stained with anti-RTI40 (green), anti-RTII70 (blue)
and MMC4 (red). The proportion of alveolar epithelial surface staining positively for the
ATI-selective protein RTI40 (broad arrow) was significantly reduced in comparison to
controls. There was an increase in the proportion of the epithelial surface coexpressing
the ATII cell-selective proteins RTII70 and MMC4 (arrows), and an increase in the
proportion of epithelial surface with an intermediate phenotype expressing a combination
of the ATI-selective RTI40 and the ATII-selective MMC4 (arrowheads). Note alveolar
macrophages expressing MMC4 in the alveolar airspaces (dashed arrow). Positive
controls, secondary-only and isotype-specific negative controls were performed as
described in materials and methods (section 2.9). (n=3).
152
100µm
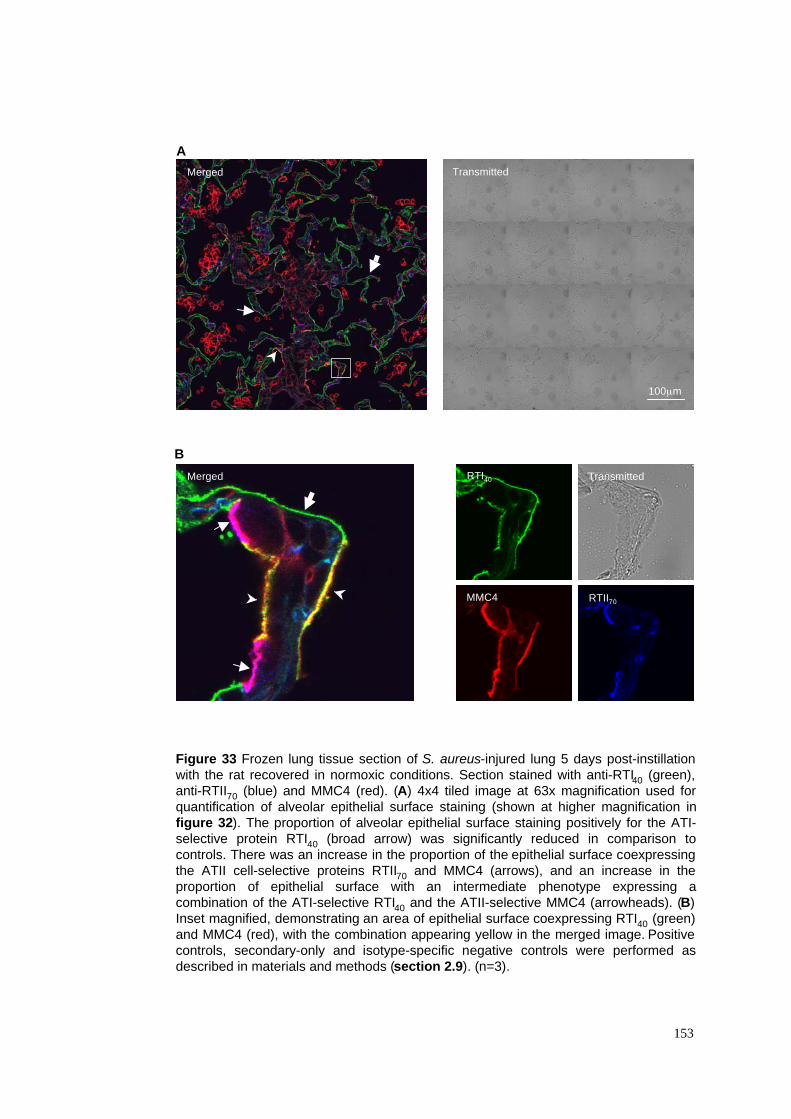
A
B
Figure 33 Frozen lung tissue section of S. aureus-injured lung 5 days post-instillation
with the rat recovered in normoxic conditions. Section stained with anti-RTI40 (green),
anti-RTII70 (blue) and MMC4 (red). (A) 4x4 tiled image at 63x magnification used for
quantification of alveolar epithelial surface staining (shown at higher magnification in
figure 32). The proportion of alveolar epithelial surface staining positively for the ATI-
selective protein RTI40 (broad arrow) was significantly reduced in comparison to
controls. There was an increase in the proportion of the epithelial surface coexpressing
the ATII cell-selective proteins RTII70 and MMC4 (arrows), and an increase in the
proportion of epithelial surface with an intermediate phenotype expressing a
combination of the ATI-selective RTI40 and the ATII-selective MMC4 (arrowheads). (B)
Inset magnified, demonstrating an area of epithelial surface coexpressing RTI40 (green)
and MMC4 (red), with the combination appearing yellow in the merged image. Positive
controls, secondary-only and isotype-specific negative controls were performed as
described in materials and methods (section 2.9). (n=3).
153
100µm
RTII70
Transmitted
Transmitted
MMC4
RTI40
Merged
Merged
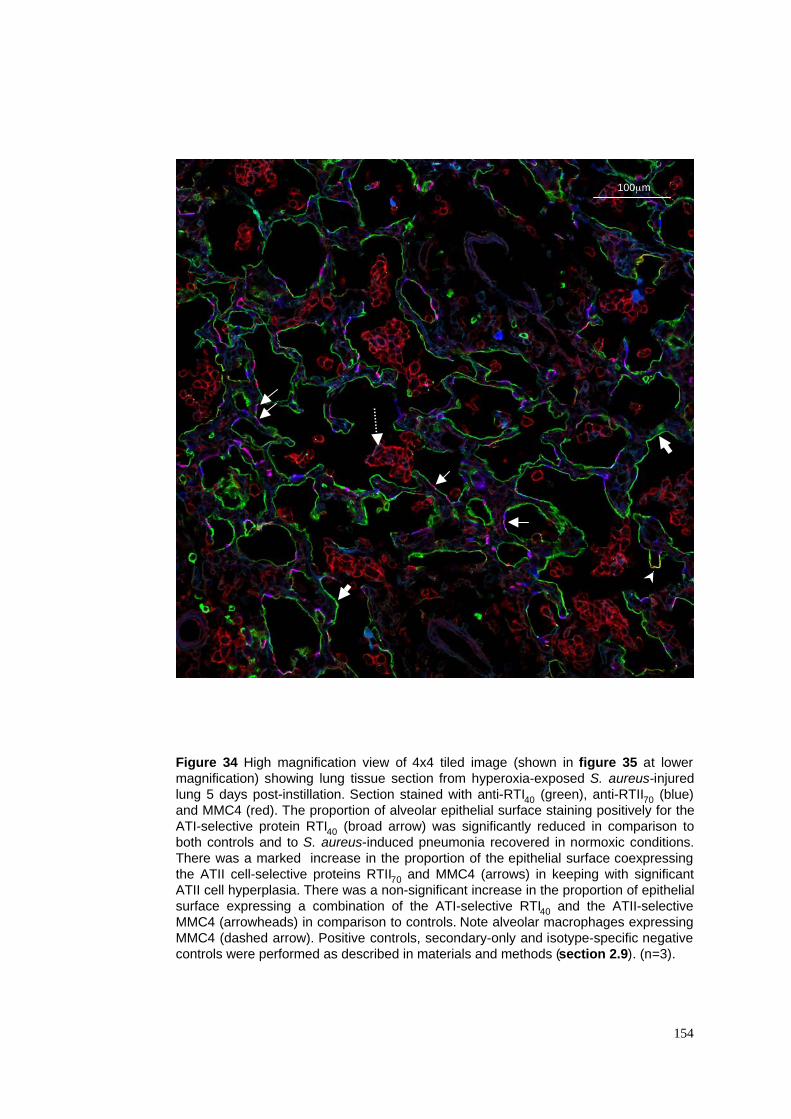
Figure 34 High magnification view of 4x4 tiled image (shown in figure 35 at lower
magnification) showing lung tissue section from hyperoxia-exposed S. aureus-injured
lung 5 days post-instillation. Section stained with anti-RTI40 (green), anti-RTII70 (blue)
and MMC4 (red). The proportion of alveolar epithelial surface staining positively for the
ATI-selective protein RTI40 (broad arrow) was significantly reduced in comparison to
both controls and to S. aureus-induced pneumonia recovered in normoxic conditions.
There was a marked increase in the proportion of the epithelial surface coexpressing
the ATII cell-selective proteins RTII70 and MMC4 (arrows) in keeping with significant
ATII cell hyperplasia. There was a non-significant increase in the proportion of epithelial
surface expressing a combination of the ATI-selective RTI40 and the ATII-selective
MMC4 (arrowheads) in comparison to controls. Note alveolar macrophages expressing
MMC4 (dashed arrow). Positive controls, secondary-only and isotype-specific negative
controls were performed as described in materials and methods (section 2.9). (n=3).
154
100µm
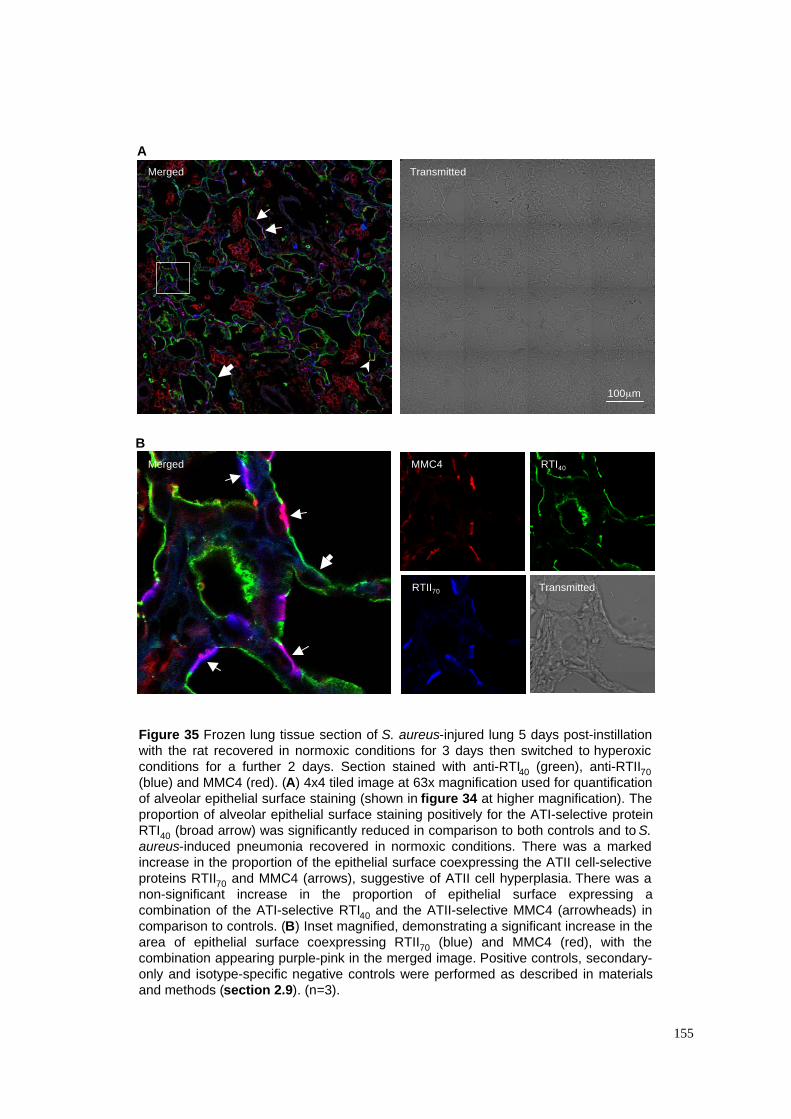
A
B
Figure 35 Frozen lung tissue section of S. aureus-injured lung 5 days post-instillation
with the rat recovered in normoxic conditions for 3 days then switched to hyperoxic
conditions for a further 2 days. Section stained with anti-RTI40 (green), anti-RTII70
(blue) and MMC4 (red). (A) 4x4 tiled image at 63x magnification used for quantification
of alveolar epithelial surface staining (shown in figure 34 at higher magnification). The
proportion of alveolar epithelial surface staining positively for the ATI-selective protein
RTI40 (broad arrow) was significantly reduced in comparison to both controls and to S.
aureus-induced pneumonia recovered in normoxic conditions. There was a marked
increase in the proportion of the epithelial surface coexpressing the ATII cell-selective
proteins RTII70 and MMC4 (arrows), suggestive of ATII cell hyperplasia. There was a
non-significant increase in the proportion of epithelial surface expressing a
combination of the ATI-selective RTI40 and the ATII-selective MMC4 (arrowheads) in
comparison to controls. (B) Inset magnified, demonstrating a significant increase in the
area of epithelial surface coexpressing RTII70 (blue) and MMC4 (red), with the
combination appearing purple-pink in the merged image. Positive controls, secondary-
only and isotype-specific negative controls were performed as described in materials
and methods (section 2.9). (n=3).
155
RTII70 Transmitted
MMC4 RTI40
Transmitted
100µm
Merged
Merged
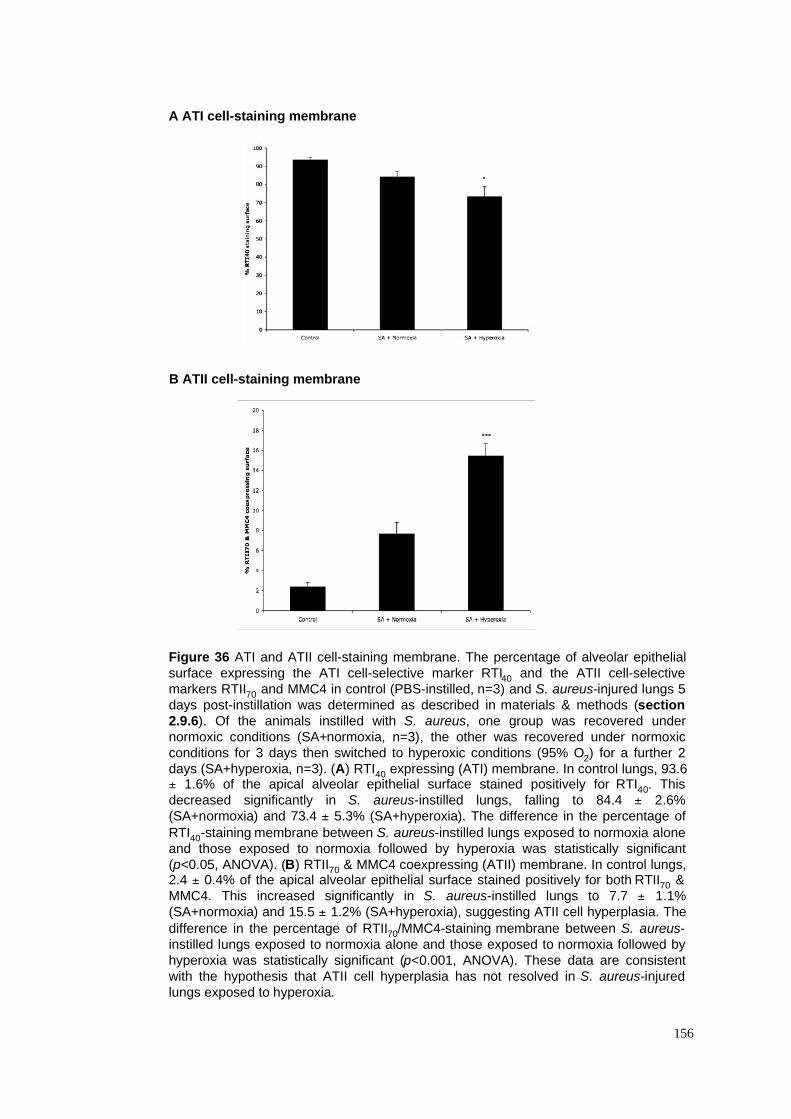
A ATI cell-staining membrane
B ATII cell-staining membrane
Figure 36 ATI and ATII cell-staining membrane. The percentage of alveolar epithelial
surface expressing the ATI cell-selective marker RTI40 and the ATII cell-selective
markers RTII70 and MMC4 in control (PBS-instilled, n=3) and S. aureus-injured lungs 5
days post-instillation was determined as described in materials & methods (section
2.9.6). Of the animals instilled with S. aureus, one group was recovered under
normoxic conditions (SA+normoxia, n=3), the other was recovered under normoxic
conditions for 3 days then switched to hyperoxic conditions (95% O2) for a further 2
days (SA+hyperoxia, n=3). (A) RTI40 expressing (ATI) membrane. In control lungs, 93.6± 1.6% of the apical alveolar epithelial surface stained positively for RTI40. This
decreased significantly in S. aureus-instilled lungs, falling to 84.4 ± 2.6%
(SA+normoxia) and 73.4 ± 5.3% (SA+hyperoxia). The difference in the percentage of
RTI40-staining membrane between S. aureus-instilled lungs exposed to normoxia alone
and those exposed to normoxia followed by hyperoxia was statistically significant
(p<0.05, ANOVA). (B) RTII70 & MMC4 coexpressing (ATII) membrane. In control lungs,2.4 ± 0.4% of the apical alveolar epithelial surface stained positively for both RTII70 &
MMC4. This increased significantly in S. aureus-instilled lungs to 7.7 ± 1.1%
(SA+normoxia) and 15.5 ± 1.2% (SA+hyperoxia), suggesting ATII cell hyperplasia. The
difference in the percentage of RTII70/MMC4-staining membrane between S. aureus-
instilled lungs exposed to normoxia alone and those exposed to normoxia followed by
hyperoxia was statistically significant (p<0.001, ANOVA). These data are consistent
with the hypothesis that ATII cell hyperplasia has not resolved in S. aureus-injured
lungs exposed to hyperoxia.
156
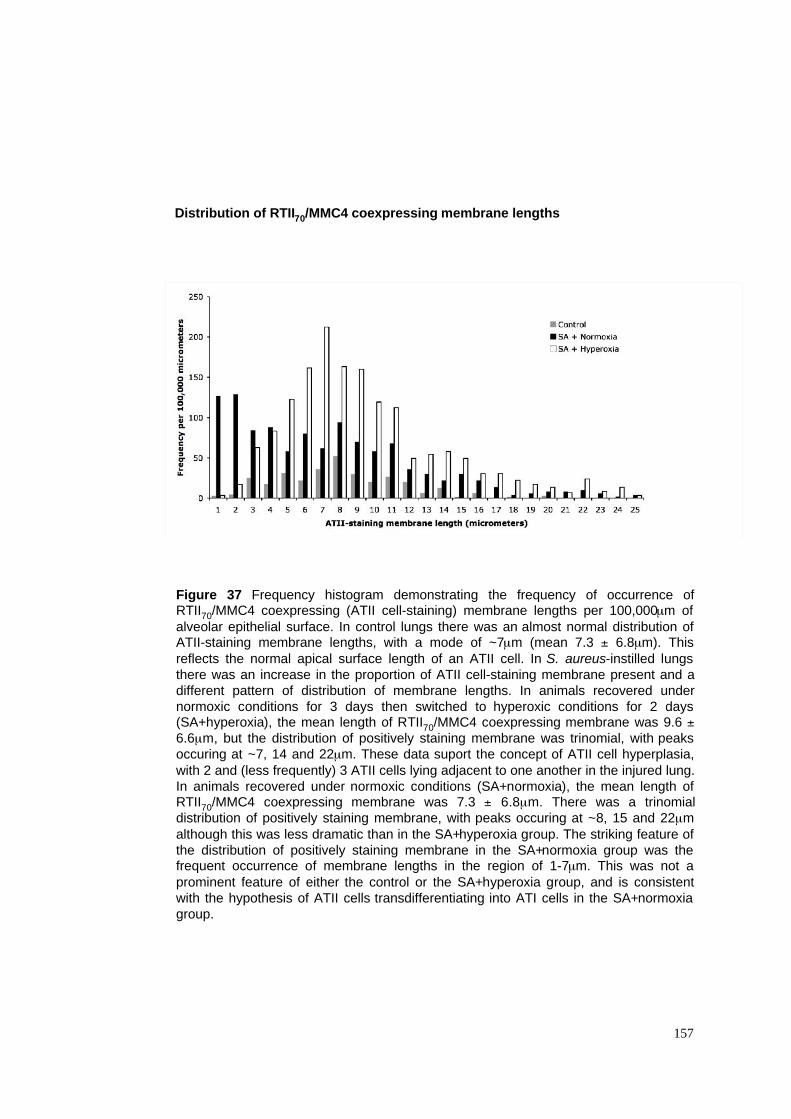
Figure 37 Frequency histogram demonstrating the frequency of occurrence ofRTII70/MMC4 coexpressing (ATII cell-staining) membrane lengths per 100,000µm of
alveolar epithelial surface. In control lungs there was an almost normal distribution ofATII-staining membrane lengths, with a mode of ~7µm (mean 7.3 ± 6.8µm). This
reflects the normal apical surface length of an ATII cell. In S. aureus-instilled lungs
there was an increase in the proportion of ATII cell-staining membrane present and a
different pattern of distribution of membrane lengths. In animals recovered under
normoxic conditions for 3 days then switched to hyperoxic conditions for 2 days(SA+hyperoxia), the mean length of RTII70/MMC4 coexpressing membrane was 9.6 ±
6.6µm, but the distribution of positively staining membrane was trinomial, with peaks
occuring at ~7, 14 and 22µm. These data suport the concept of ATII cell hyperplasia,
with 2 and (less frequently) 3 ATII cells lying adjacent to one another in the injured lung.
In animals recovered under normoxic conditions (SA+normoxia), the mean length ofRTII70/MMC4 coexpressing membrane was 7.3 ± 6.8µm. There was a trinomial
distribution of positively staining membrane, with peaks occuring at ~8, 15 and 22µm
although this was less dramatic than in the SA+hyperoxia group. The striking feature of
the distribution of positively staining membrane in the SA+normoxia group was thefrequent occurrence of membrane lengths in the region of 1-7µm. This was not a
prominent feature of either the control or the SA+hyperoxia group, and is consistent
with the hypothesis of ATII cells transdifferentiating into ATI cells in the SA+normoxia
group.
157
Distribution of RTII70/MMC4 coexpressing membrane lengths
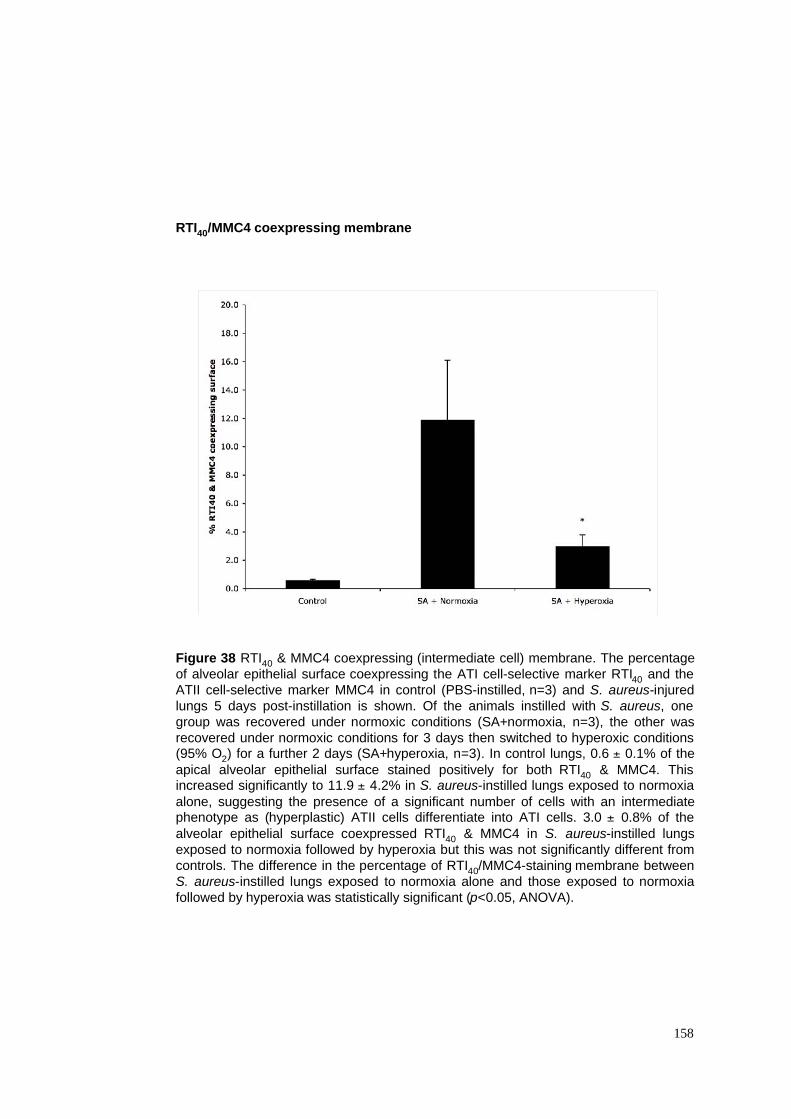
158
RTI40/MMC4 coexpressing membrane
Figure 38 RTI40 & MMC4 coexpressing (intermediate cell) membrane. The percentage
of alveolar epithelial surface coexpressing the ATI cell-selective marker RTI40 and the
ATII cell-selective marker MMC4 in control (PBS-instilled, n=3) and S. aureus-injured
lungs 5 days post-instillation is shown. Of the animals instilled with S. aureus, one
group was recovered under normoxic conditions (SA+normoxia, n=3), the other was
recovered under normoxic conditions for 3 days then switched to hyperoxic conditions(95% O2) for a further 2 days (SA+hyperoxia, n=3). In control lungs, 0.6 ± 0.1% of the
apical alveolar epithelial surface stained positively for both RTI40 & MMC4. Thisincreased significantly to 11.9 ± 4.2% in S. aureus-instilled lungs exposed to normoxia
alone, suggesting the presence of a significant number of cells with an intermediatephenotype as (hyperplastic) ATII cells differentiate into ATI cells. 3.0 ± 0.8% of the
alveolar epithelial surface coexpressed RTI40 & MMC4 in S. aureus-instilled lungs
exposed to normoxia followed by hyperoxia but this was not significantly different from
controls. The difference in the percentage of RTI40/MMC4-staining membrane between
S. aureus-instilled lungs exposed to normoxia alone and those exposed to normoxia
followed by hyperoxia was statistically significant (p<0.05, ANOVA).
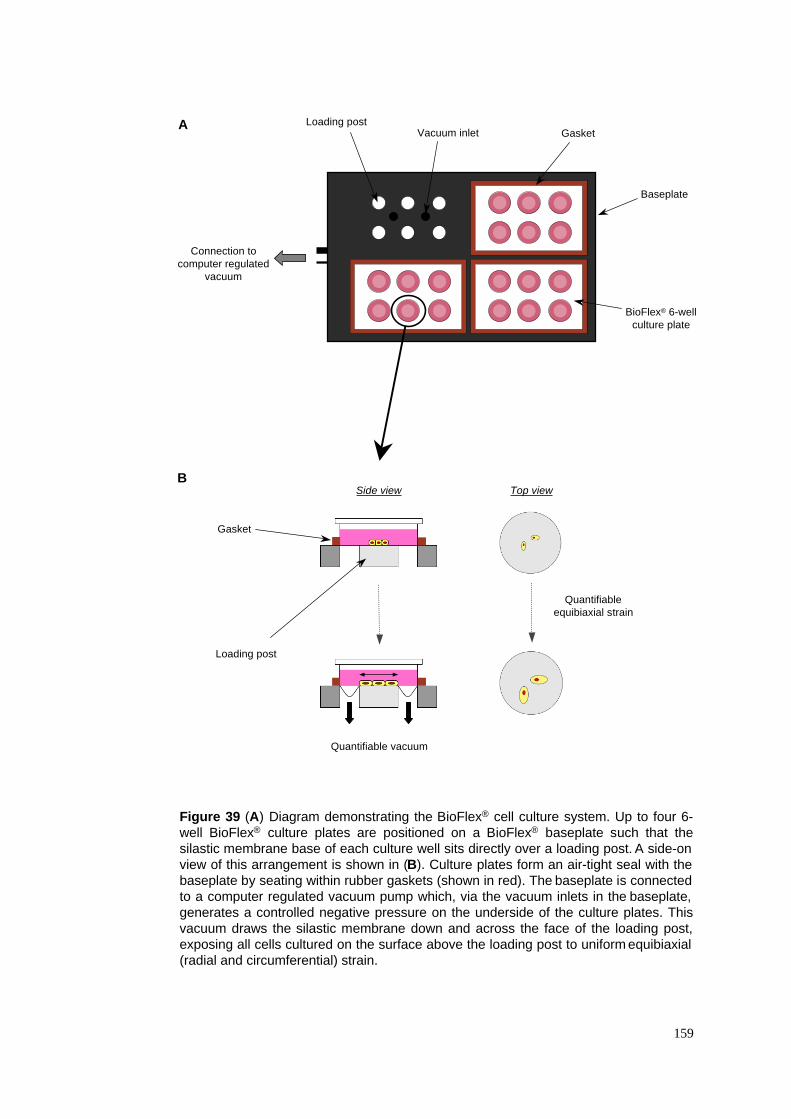
159
Figure 39 (A) Diagram demonstrating the BioFlex® cell culture system. Up to four 6-
well BioFlex® culture plates are positioned on a BioFlex® baseplate such that the
silastic membrane base of each culture well sits directly over a loading post. A side-on
view of this arrangement is shown in (B). Culture plates form an air-tight seal with the
baseplate by seating within rubber gaskets (shown in red). The baseplate is connected
to a computer regulated vacuum pump which, via the vacuum inlets in the baseplate,
generates a controlled negative pressure on the underside of the culture plates. This
vacuum draws the silastic membrane down and across the face of the loading post,
exposing all cells cultured on the surface above the loading post to uniform equibiaxial
(radial and circumferential) strain.
Connection to
computer regulated
vacuum
Baseplate
Loading postVacuum inlet Gasket
BioFlex® 6-well
culture plate
Gasket
Loading post
A
B
Quantifiable vacuum
Quantifiable
equibiaxial strain
Top viewSide view
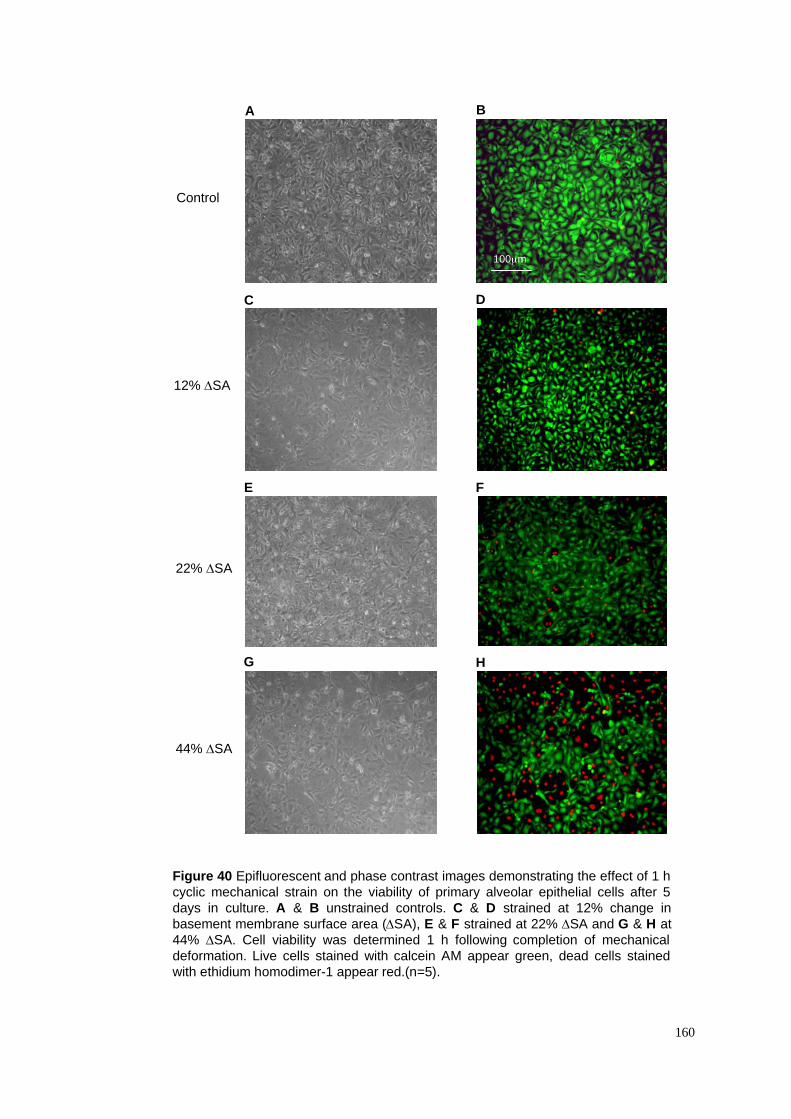
160
Figure 40 Epifluorescent and phase contrast images demonstrating the effect of 1 h
cyclic mechanical strain on the viability of primary alveolar epithelial cells after 5
days in culture. A & B unstrained controls. C & D strained at 12% change in
basement membrane surface area (!SA), E & F strained at 22% !SA and G & H at
44% !SA. Cell viability was determined 1 h following completion of mechanical
deformation. Live cells stained with calcein AM appear green, dead cells stained
with ethidium homodimer-1 appear red.(n=5).
D
A
E F
C
B
100µm
G H
Control
12% !SA
22% !SA
44% !SA
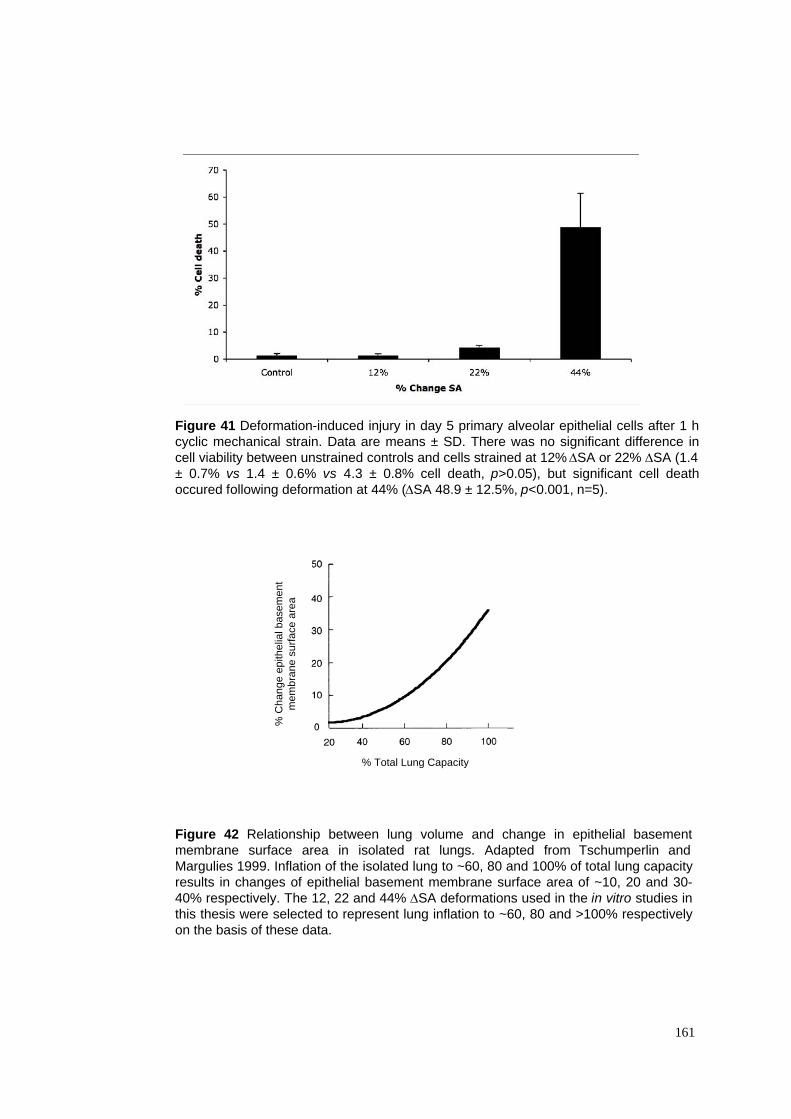
Figure 41 Deformation-induced injury in day 5 primary alveolar epithelial cells after 1 h
cyclic mechanical strain. Data are means ± SD. There was no significant difference in
cell viability between unstrained controls and cells strained at 12% !SA or 22% !SA (1.4
± 0.7% vs 1.4 ± 0.6% vs 4.3 ± 0.8% cell death, p>0.05), but significant cell death
occured following deformation at 44% (!SA 48.9 ± 12.5%, p<0.001, n=5).
161
% Total Lung Capacity
% C
ha
ng
e e
pith
elia
l b
ase
me
nt
me
mb
ran
e s
urf
ace
are
a
Figure 42 Relationship between lung volume and change in epithelial basement
membrane surface area in isolated rat lungs. Adapted from Tschumperlin and
Margulies 1999. Inflation of the isolated lung to ~60, 80 and 100% of total lung capacity
results in changes of epithelial basement membrane surface area of ~10, 20 and 30-
40% respectively. The 12, 22 and 44% !SA deformations used in the in vitro studies in
this thesis were selected to represent lung inflation to ~60, 80 and >100% respectively
on the basis of these data.
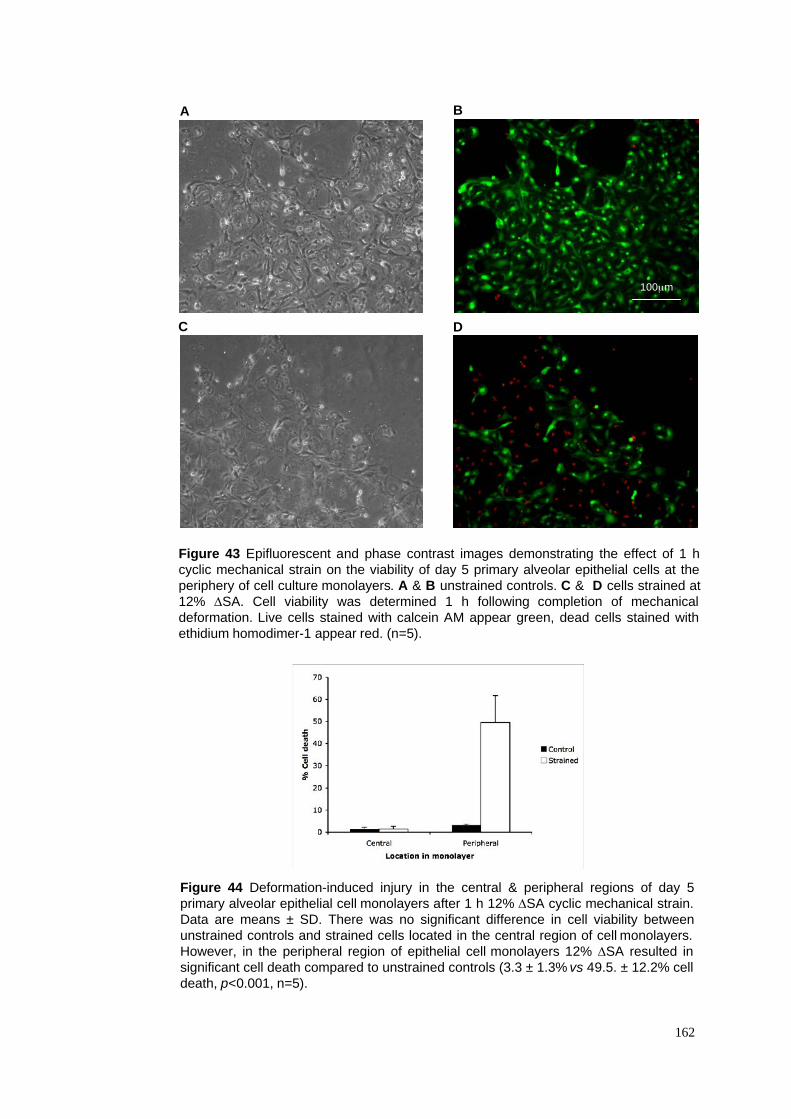
Figure 43 Epifluorescent and phase contrast images demonstrating the effect of 1 h
cyclic mechanical strain on the viability of day 5 primary alveolar epithelial cells at the
periphery of cell culture monolayers. A & B unstrained controls. C & D cells strained at
12% !SA. Cell viability was determined 1 h following completion of mechanical
deformation. Live cells stained with calcein AM appear green, dead cells stained with
ethidium homodimer-1 appear red. (n=5).
Figure 44 Deformation-induced injury in the central & peripheral regions of day 5
primary alveolar epithelial cell monolayers after 1 h 12% !SA cyclic mechanical strain.
Data are means ± SD. There was no significant difference in cell viability between
unstrained controls and strained cells located in the central region of cell monolayers.
However, in the peripheral region of epithelial cell monolayers 12% !SA resulted in
significant cell death compared to unstrained controls (3.3 ± 1.3% vs 49.5. ± 12.2% cell
death, p<0.001, n=5).
162
D
A
C
B
100µm
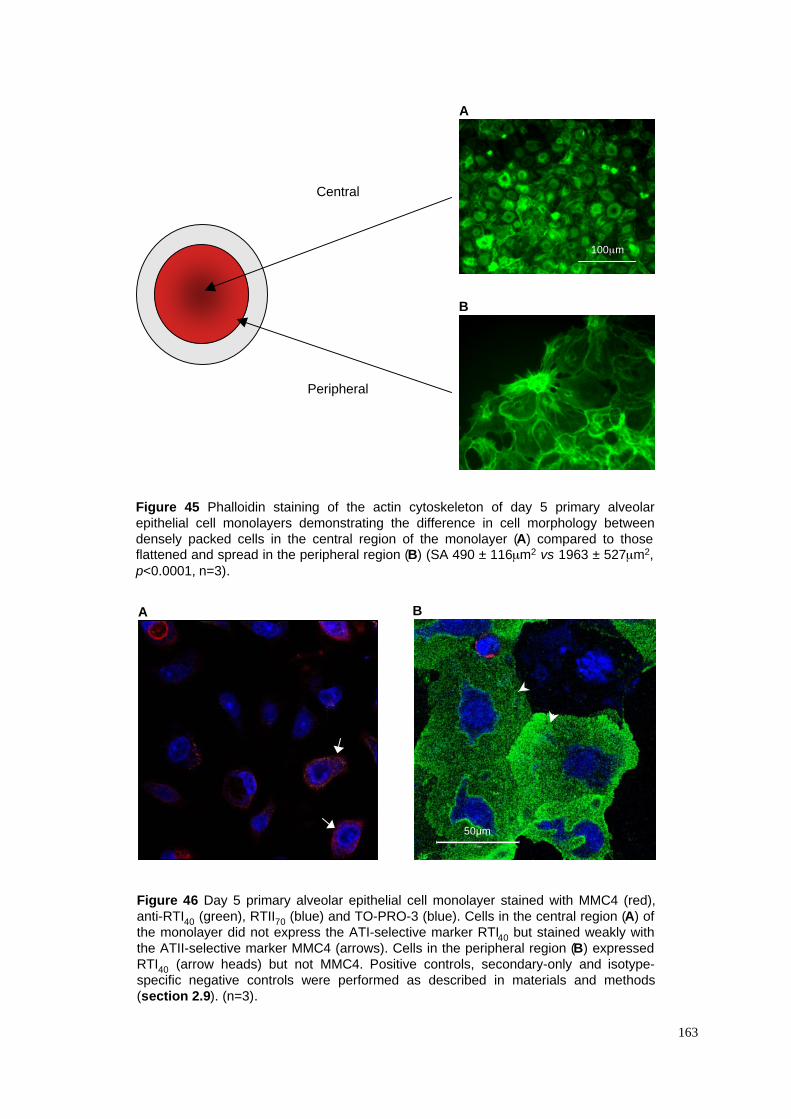
Central
Peripheral
B
A
Figure 45 Phalloidin staining of the actin cytoskeleton of day 5 primary alveolar
epithelial cell monolayers demonstrating the difference in cell morphology between
densely packed cells in the central region of the monolayer (A) compared to thoseflattened and spread in the peripheral region (B) (SA 490 ± 116µm2 vs 1963 ± 527µm2,
p<0.0001, n=3).
50!m
BA
Figure 46 Day 5 primary alveolar epithelial cell monolayer stained with MMC4 (red),
anti-RTI40 (green), RTII70 (blue) and TO-PRO-3 (blue). Cells in the central region (A) of
the monolayer did not express the ATI-selective marker RTI40 but stained weakly with
the ATII-selective marker MMC4 (arrows). Cells in the peripheral region (B) expressed
RTI40 (arrow heads) but not MMC4. Positive controls, secondary-only and isotype-
specific negative controls were performed as described in materials and methods
(section 2.9). (n=3).
163
100µm
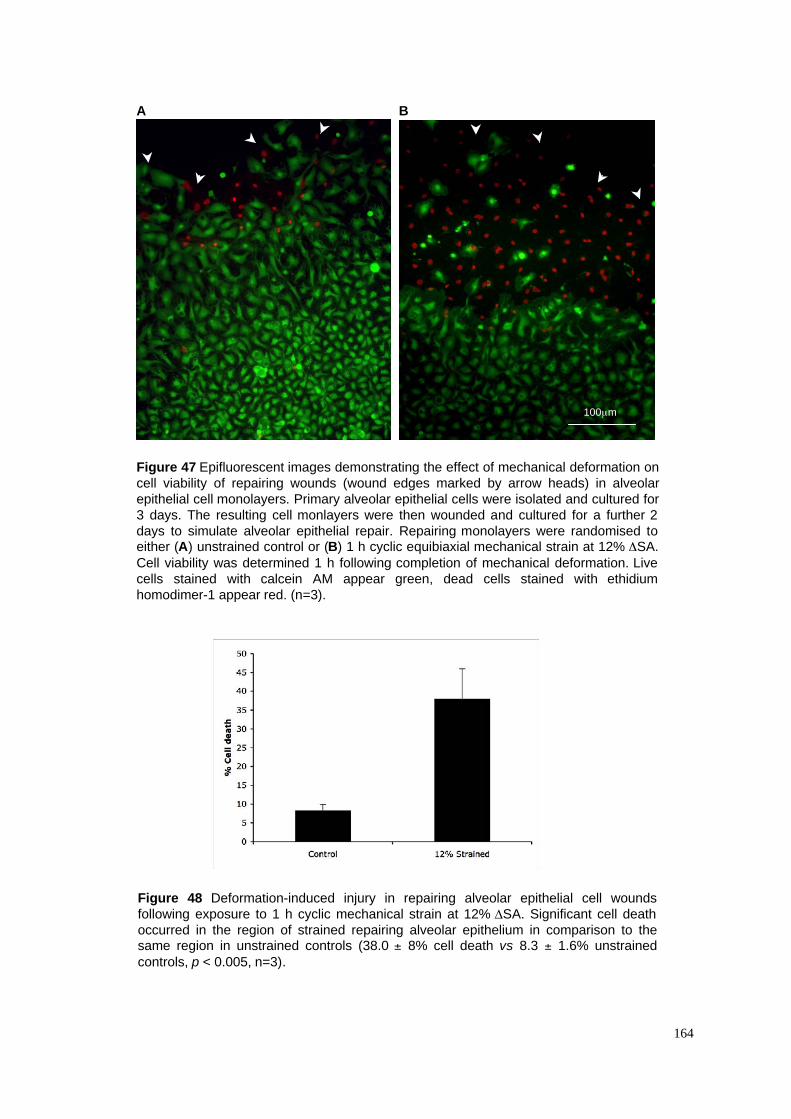
Figure 47 Epifluorescent images demonstrating the effect of mechanical deformation on
cell viability of repairing wounds (wound edges marked by arrow heads) in alveolar
epithelial cell monolayers. Primary alveolar epithelial cells were isolated and cultured for
3 days. The resulting cell monlayers were then wounded and cultured for a further 2
days to simulate alveolar epithelial repair. Repairing monolayers were randomised toeither (A) unstrained control or (B) 1 h cyclic equibiaxial mechanical strain at 12% !SA.
Cell viability was determined 1 h following completion of mechanical deformation. Live
cells stained with calcein AM appear green, dead cells stained with ethidium
homodimer-1 appear red. (n=3).
A B
Figure 48 Deformation-induced injury in repairing alveolar epithelial cell wounds
following exposure to 1 h cyclic mechanical strain at 12% !SA. Significant cell death
occurred in the region of strained repairing alveolar epithelium in comparison to thesame region in unstrained controls (38.0 ± 8% cell death vs 8.3 ± 1.6% unstrained
controls, p < 0.005, n=3).
164
100µm
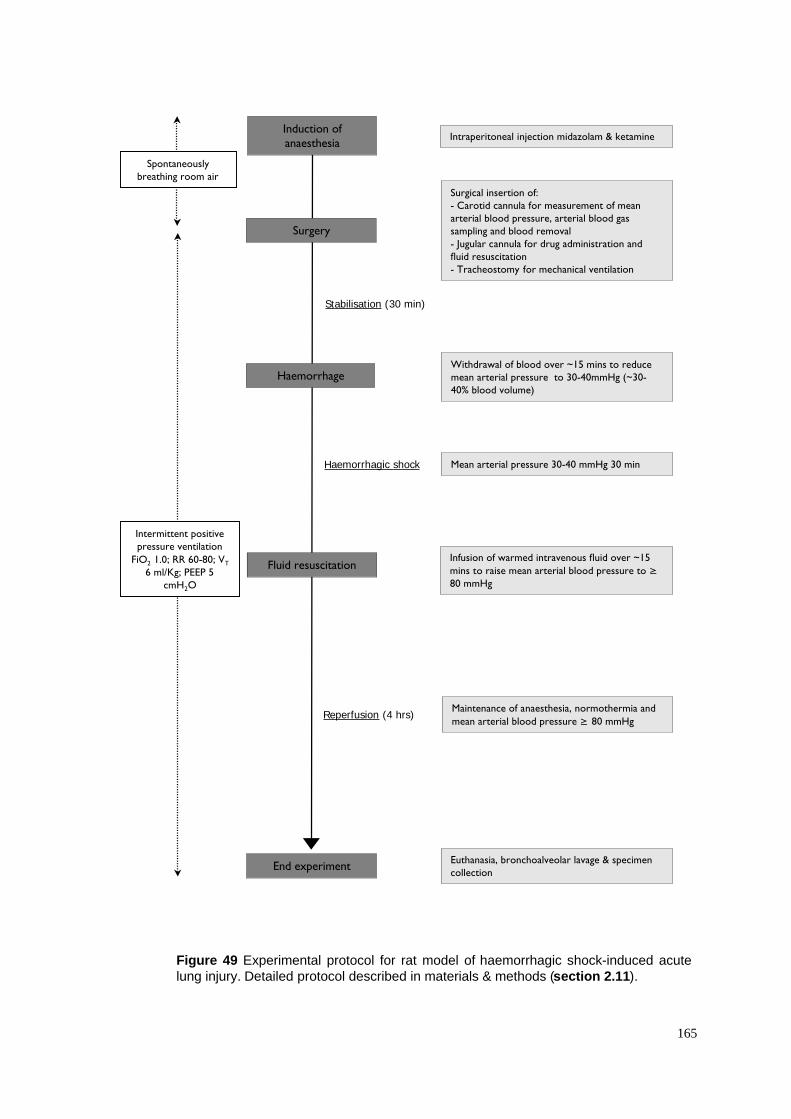
Figure 49 Experimental protocol for rat model of haemorrhagic shock-induced acute
lung injury. Detailed protocol described in materials & methods (section 2.11).
165
Surgical insertion of:- Carotid cannula for measurement of meanarterial blood pressure, arterial blood gassampling and blood removal- Jugular cannula for drug administration andfluid resuscitation- Tracheostomy for mechanical ventilation
Withdrawal of blood over ~15 mins to reducemean arterial pressure to 30-40mmHg (~30-40% blood volume)
Infusion of warmed intravenous fluid over ~15
mins to raise mean arterial blood pressure to !80 mmHg
Induction ofanaesthesia
Stabilisation (30 min)
Surgery
Haemorrhagic shock
Reperfusion (4 hrs)
End experiment
Haemorrhage
Fluid resuscitation
Intraperitoneal injection midazolam & ketamine
Maintenance of anaesthesia, normothermia and
mean arterial blood pressure ! 80 mmHg
Euthanasia, bronchoalveolar lavage & specimencollection
Spontaneouslybreathing room air
Intermittent positivepressure ventilation
FiO2 1.0; RR 60-80; VT
6 ml/Kg; PEEP 5cmH2O
Mean arterial pressure 30-40 mmHg 30 min
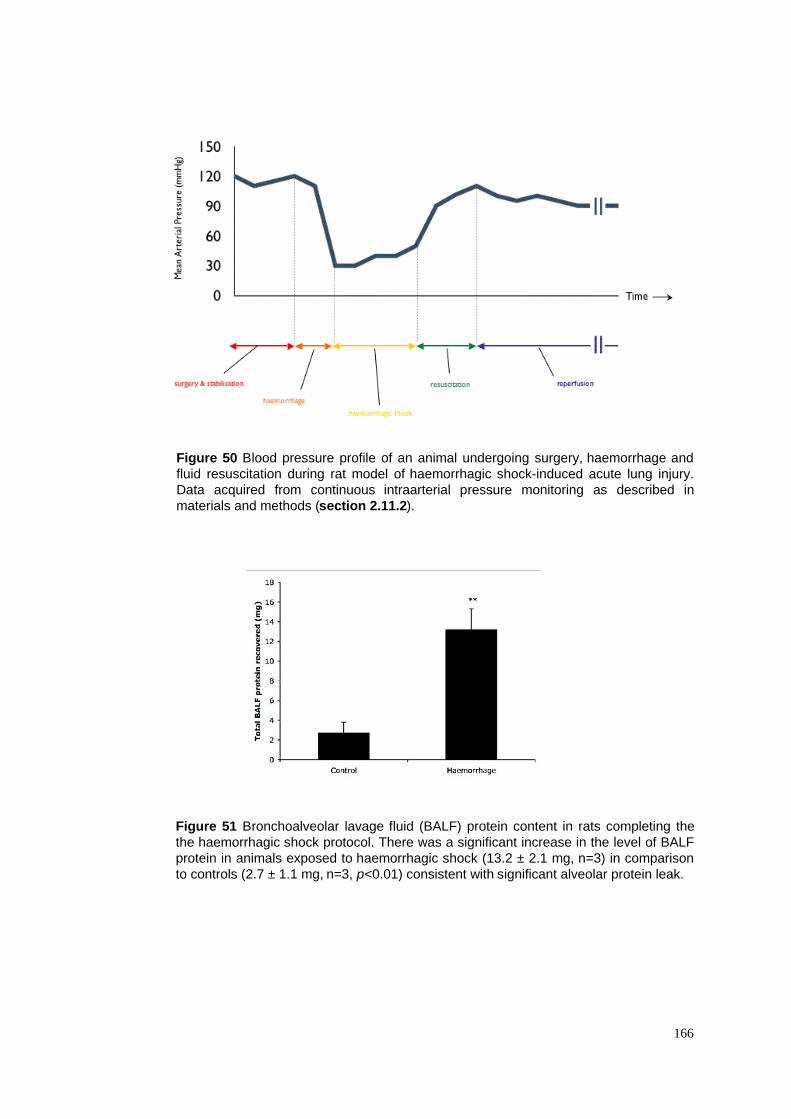
Figure 51 Bronchoalveolar lavage fluid (BALF) protein content in rats completing the
the haemorrhagic shock protocol. There was a significant increase in the level of BALF
protein in animals exposed to haemorrhagic shock (13.2 ± 2.1 mg, n=3) in comparison
to controls (2.7 ± 1.1 mg, n=3, p<0.01) consistent with significant alveolar protein leak.
166
Figure 50 Blood pressure profile of an animal undergoing surgery, haemorrhage and
fluid resuscitation during rat model of haemorrhagic shock-induced acute lung injury.
Data acquired from continuous intraarterial pressure monitoring as described in
materials and methods (section 2.11.2).
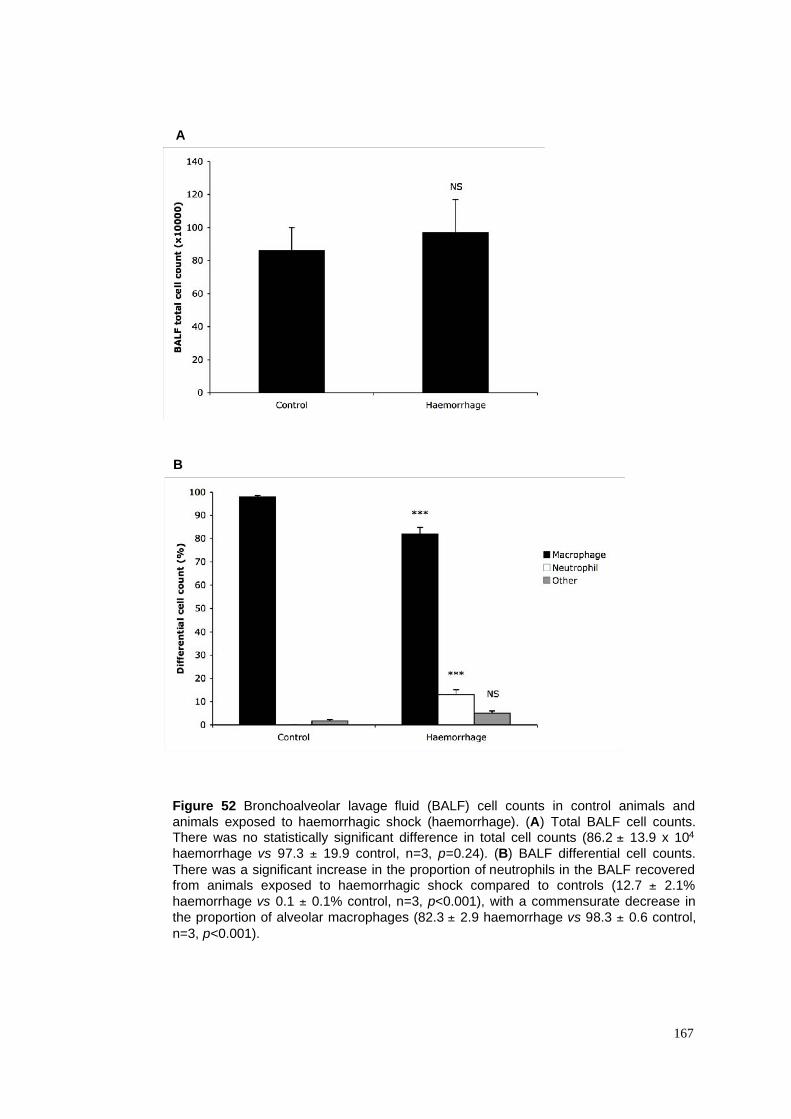
Figure 52 Bronchoalveolar lavage fluid (BALF) cell counts in control animals and
animals exposed to haemorrhagic shock (haemorrhage). (A) Total BALF cell counts.There was no statistically significant difference in total cell counts (86.2 ± 13.9 x 104
haemorrhage vs 97.3 ± 19.9 control, n=3, p=0.24). (B) BALF differential cell counts.
There was a significant increase in the proportion of neutrophils in the BALF recoveredfrom animals exposed to haemorrhagic shock compared to controls (12.7 ± 2.1%
haemorrhage vs 0.1 ± 0.1% control, n=3, p<0.001), with a commensurate decrease in
the proportion of alveolar macrophages (82.3 ± 2.9 haemorrhage vs 98.3 ± 0.6 control,
n=3, p<0.001).
167
A
B
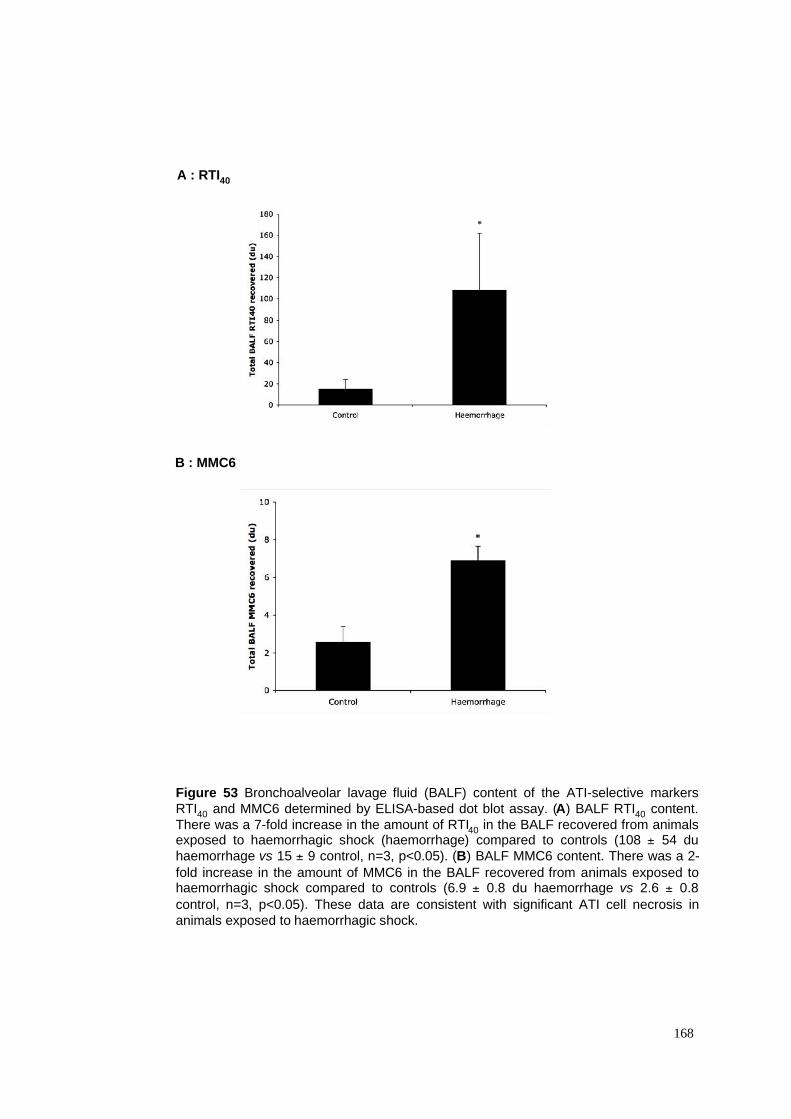
Figure 53 Bronchoalveolar lavage fluid (BALF) content of the ATI-selective markers
RTI40 and MMC6 determined by ELISA-based dot blot assay. (A) BALF RTI40 content.
There was a 7-fold increase in the amount of RTI40 in the BALF recovered from animalsexposed to haemorrhagic shock (haemorrhage) compared to controls (108 ± 54 du
haemorrhage vs 15 ± 9 control, n=3, p<0.05). (B) BALF MMC6 content. There was a 2-
fold increase in the amount of MMC6 in the BALF recovered from animals exposed tohaemorrhagic shock compared to controls (6.9 ± 0.8 du haemorrhage vs 2.6 ± 0.8
control, n=3, p<0.05). These data are consistent with significant ATI cell necrosis in
animals exposed to haemorrhagic shock.
168
A : RTI40
B : MMC6
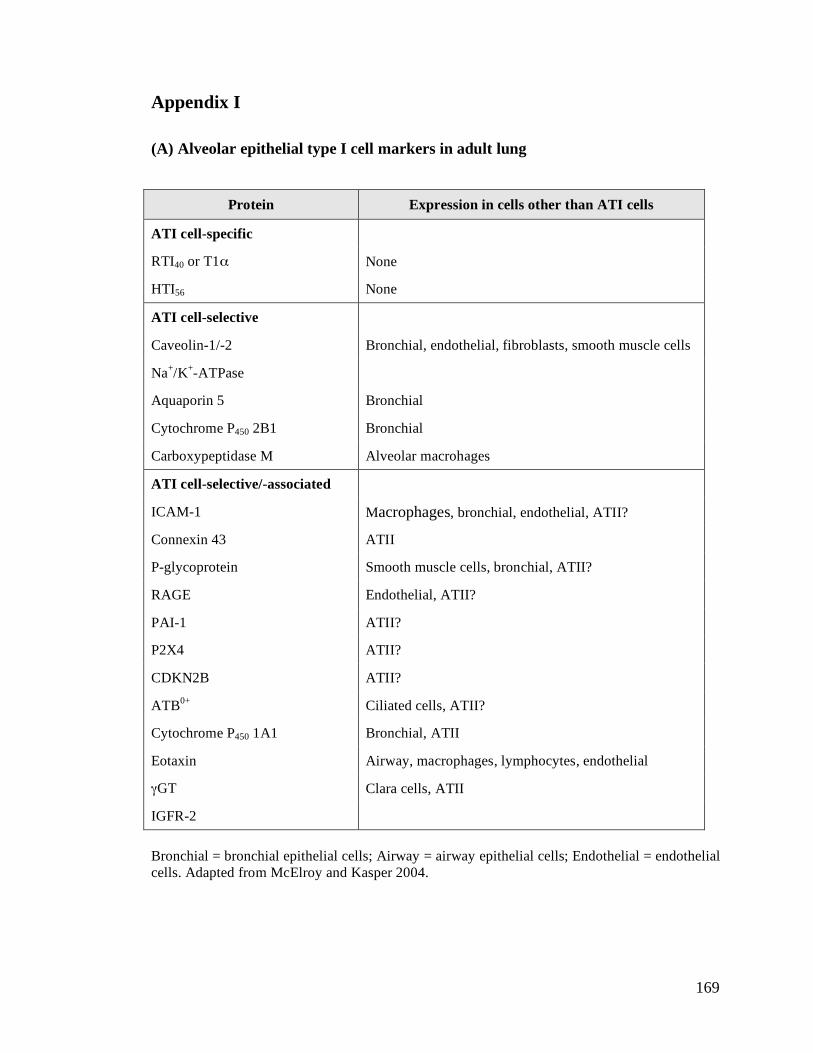
169
Appendix I
(A) Alveolar epithelial type I cell markers in adult lung
Protein Expression in cells other than ATI cells
ATI cell-specific
RTI40 or T1$ None
HTI56 None
ATI cell-selective
Caveolin-1/-2 Bronchial, endothelial, fibroblasts, smooth muscle cells
Na+/K
+-ATPase
Aquaporin 5 Bronchial
Cytochrome P450 2B1 Bronchial
Carboxypeptidase M Alveolar macrohages
ATI cell-selective/-associated
ICAM-1 Macrophages, bronchial, endothelial, ATII?
Connexin 43 ATII
P-glycoprotein Smooth muscle cells, bronchial, ATII?
RAGE Endothelial, ATII?
PAI-1 ATII?
P2X4 ATII?
CDKN2B ATII?
ATB0+
Ciliated cells, ATII?
Cytochrome P450 1A1 Bronchial, ATII
Eotaxin Airway, macrophages, lymphocytes, endothelial
%GT Clara cells, ATII
IGFR-2
Bronchial = bronchial epithelial cells; Airway = airway epithelial cells; Endothelial = endothelial
cells. Adapted from McElroy and Kasper 2004.
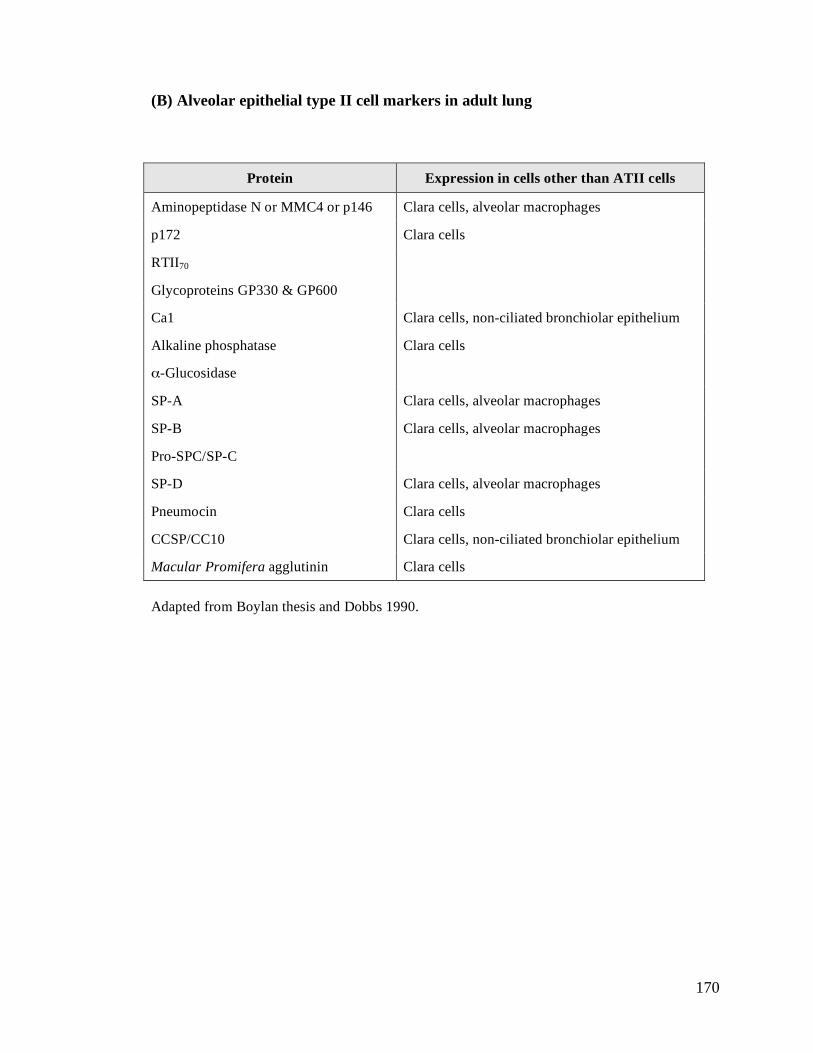
170
(B) Alveolar epithelial type II cell markers in adult lung
Protein Expression in cells other than ATII cells
Aminopeptidase N or MMC4 or p146 Clara cells, alveolar macrophages
p172 Clara cells
RTII70
Glycoproteins GP330 & GP600
Ca1 Clara cells, non-ciliated bronchiolar epithelium
Alkaline phosphatase Clara cells
$-Glucosidase
SP-A Clara cells, alveolar macrophages
SP-B Clara cells, alveolar macrophages
Pro-SPC/SP-C
SP-D Clara cells, alveolar macrophages
Pneumocin Clara cells
CCSP/CC10 Clara cells, non-ciliated bronchiolar epithelium
Macular Promifera agglutinin Clara cells
Adapted from Boylan thesis and Dobbs 1990.

171
Appendix II Manufacturers and Suppliers
Amersham Biosciences http://www.amershambiosciences.com
BD Biosciences http://www.bdbeurope.com
BD Medical http://www.bdeurope.com
Bio-Rad Laboratories http://www.bio-rad.com
Billups-Rothenberg http://www.brincubator.com
Bitplane AG http://www.bitplane.com
BOC Gases http://www.boc-gases.com
Dako http://www.dako.com
Dunn Labortechnik GmbH http://www.dunnlab.de
Dynatech Laboratories http://dynatechlaboratories.com
Fisher Scientific http://www.fisher.co.uk
Fluka and Riedel http://www.sigmaaldrich.com
Genusxpress http://www.touchaberdeen.com
Grace Bio Labs http://www.gracebio.com
Greiner Bio-One http://www.greinerbioone.com
Harlan (UK) http://www.arcned.nl
Harvard Apparatus http://www.harvardapparatus.co.uk
Improvision http://www.improvision.com
Invitrogen http://www.invitrogen.com
Portex http://www.smiths-medical.com
Roche http://www.roche-applied-science.com
Millipore http://www.millipore.com
Molecular Devices http://www.moleculardevices.com
Molecular Probes http://www.invitrogen.com
Schleicher & Schuell http://www.schleicher-schuell.com
Sigma-Aldrich http://www.sigmaaldrich.com
Stratech Scientific http://www.stratech.co.uk
172
Thermo Scientific http://www.thermo.com
WelchAllyn http://www.welchallyn.com
Zeiss http://www.zeiss.com/micro
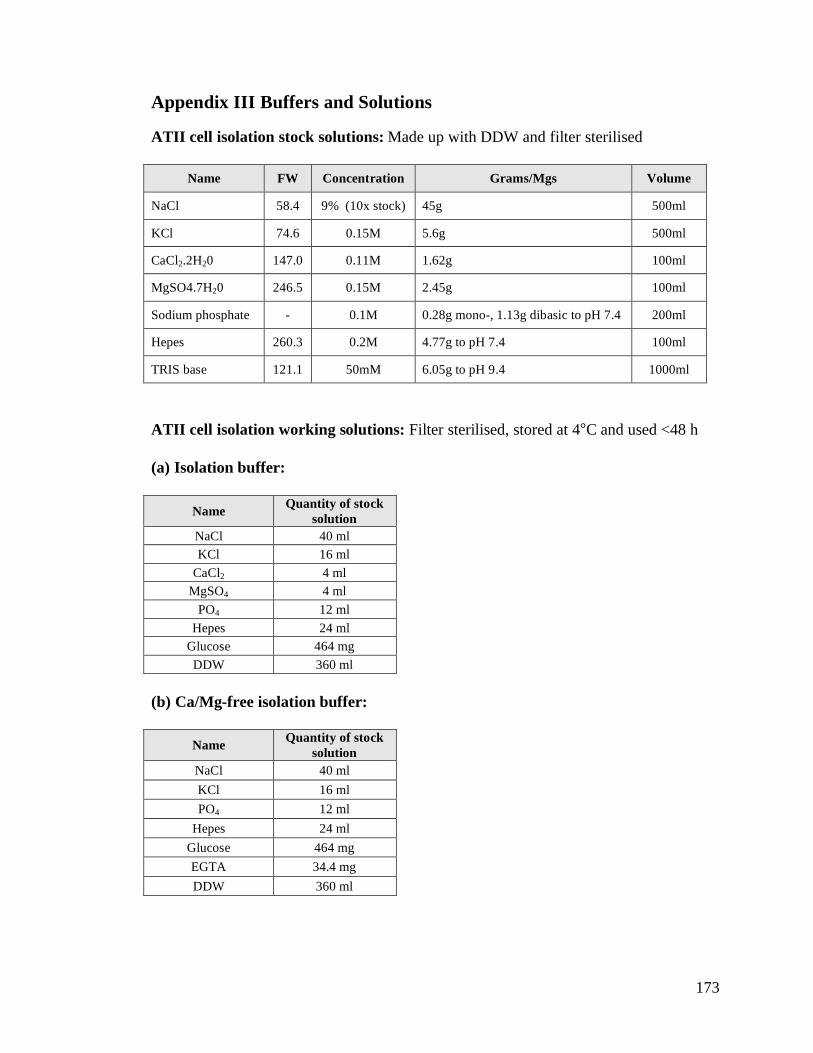
173
Appendix III Buffers and Solutions
ATII cell isolation stock solutions: Made up with DDW and filter sterilised
Name FW Concentration Grams/Mgs Volume
NaCl 58.4 9% (10x stock) 45g 500ml
KCl 74.6 0.15M 5.6g 500ml
CaCl2.2H20 147.0 0.11M 1.62g 100ml
MgSO4.7H20 246.5 0.15M 2.45g 100ml
Sodium phosphate - 0.1M 0.28g mono-, 1.13g dibasic to pH 7.4 200ml
Hepes 260.3 0.2M 4.77g to pH 7.4 100ml
TRIS base 121.1 50mM 6.05g to pH 9.4 1000ml
ATII cell isolation working solutions: Filter sterilised, stored at 4°C and used <48 h
(a) Isolation buffer:
(b) Ca/Mg-free isolation buffer:
Name Quantity of stock
solution
NaCl 40 ml
KCl 16 ml
CaCl2 4 ml
MgSO4 4 ml
PO4 12 ml
Hepes 24 ml
Glucose 464 mg
DDW 360 ml
Name Quantity of stock
solution
NaCl 40 ml
KCl 16 ml
PO4 12 ml
Hepes 24 ml
Glucose 464 mg
EGTA 34.4 mg
DDW 360 ml

174
Immunoglobulin G (IgG) solution & IgG coating of bacteriological plates
5mg reagent grade rat IgG (Sigma) dissolved in 10mls of 50mM tris base. Solution filter
sterilised. 5mls of IgG solution placed in bacteriological plates, agitated for 30 min at
room temperature then plates stored at 4°C prior to use within 24 hours. IgG solution
removed prior to use (solution filter sterilised and reused up to 4x), and plates rinsed 5x
with Ca/Mg free PBS.
ATII culture medium media
Dulbecco’s modified eagle’s medium (DMEM) (Sigma-Aldrich) 450ml
Foetal bovine serum (Gibco) 40ml filter sterilised
Penicillin G (10,000 units/ml) and streptomycin sulphate (10,000 g/ml) (Gibco) 5ml
1% L-glutamine (Gibco) 5ml filter sterilised
4% Paraformaldehyde
4g paraformaldehyde dissolved in 100mls Ca/Mg-free PBS in water bath at 70°C (30
min). Filter sterilised, stored at 4°C & used < 1 month or aliquoted and stored at -20°C.
Tris(hydroxymethyl)aminomethane-buffered saline (TBS)
(a) 20x TBS stock solution: 90g NaCl and 24.2g Tris base dissolved in 450ml DDW.
pH adjusted to 8.2 with HCl and final volume brought up to 500ml with DDW.
Stored at room temperature.
(b) TBS: 25ml 20x TBS to final volume 500ml with DDW
(c) TBS-T: 0.25ml Tween-20 added to 500ml TBS
Homogenisation buffer
1x Complete" protease inhibitor cocktail tablet (Roche) freshly dissolved in 50ml TBS

175
Blocking buffer (BB)
(a) Blocking buffer (BB): 44ml fish gelatin mixed in 100ml Ca/Mg-free PBS. 20ml
goat serum added to 80ml of resulting fish gelatine solution. Aliquoted & stored -20°C.
(b) Blocking buffer with triton (BBT): 10µl 10% tritonX (in Ca/Mg-free PBS) added
to 1ml BB. Used fresh.
30% Sucrose
15g sucrose dissolved in 50mls Ca/Mg-free PBS. Filter sterilised and used fresh.
Bouin’s Fixative (Modified)
25 ml glacial acetic acid added to 425 ml methanol followed by 50 ml 40%
formaldehyde. Stored at room temperature.
LDH stock solutions
(a) Tris buffer: 2.1g Tris base dissolved in 50ml DDW, adjusted to pH 8.5 with (6.3ml
5M HCl and made up to 100ml with DDW.
(b) LDH substrate: 0.01mol l-lactic acid made up to 50ml with DDW, adjusted to pH
5.5 with 1M NaOH and made up to 100ml with DDW.
(c) Chromagen solution: 12.5mg NAD added to 5mg iodotetrazolium chloride (INT)
dissolved in 2ml DDW. Immediately prior to use, 2.5mg of phenazine methosulphate
(PMS) weighed out and dissolved in 0.5ml DDW. 0.5ml of this PMS solution added to
the INT/NAD solution to make completed chromagen solution.
(d) Blank oxalate solution: 0.2g K2C2O4.H2O and 0.2g EDTA Na2H2.2H2O dissolved
in DDW and made up to 100ml.

176
Bibliography
(1999). "International consensus conferences in intensive care medicine: Ventilator-associated
Lung Injury in ARDS. This official conference report was cosponsored by the American
Thoracic Society, The European Society of Intensive Care Medicine, and The Societe de
Reanimation de Langue Francaise, and was approved by the ATS Board of Directors, July
1999." Am J Respir Crit Care Med 160(6): 2118-24.
Abel, S. J., S. J. Finney, S. J. Brett, B. F. Keogh, C. J. Morgan and T. W. Evans (1998).
"Reduced mortality in association with the acute respiratory distress syndrome (ARDS)." Thorax
53(4): 292-4.
Abraham, E., A. Carmody, R. Shenkar and J. Arcaroli (2000). "Neutrophils as early
immunologic effectors in hemorrhage- or endotoxemia-induced acute lung injury." Am J Physiol
Lung Cell Mol Physiol 279(6): L1137-45.
Abraham, V., M. L. Chou, K. M. DeBolt and M. Koval (1999). "Phenotypic control of gap
junctional communication by cultured alveolar epithelial cells." Am J Physiol 276(5 Pt 1): L825-
34.
Adamson, I. Y. and D. H. Bowden (1974). "The type 2 cell as progenitor of alveolar epithelial
regeneration. A cytodynamic study in mice after exposure to oxygen." Lab Invest 30(1): 35-42.
Adamson, I. Y. and D. H. Bowden (1975). "Derivation of type 1 epithelium from type 2 cells in
the developing rat lung." Lab Invest 32(6): 736-45.
Adamson, I. Y., C. Hedgecock and D. H. Bowden (1990). "Epithelial cell-fibroblast interactions
in lung injury and repair." Am J Pathol 137(2): 385-92.
Adamson, I. Y., L. Young and D. H. Bowden (1988). "Relationship of alveolar epithelial injury
and repair to the induction of pulmonary fibrosis." Am J Pathol 130(2): 377-83.
Adhikari, N., K. Burns and M. Meade (2004). "Pharmacologic therapies for adults with acute
lung injury and acute respiratory distress syndrome." Cochrane Database Syst Rev(4):
CD004477.
Akira, S. and K. Takeda (2004). "Toll-like receptor signalling." Nat Rev Immunol 4(7): 499-
511.
Alberts, B. (2002). "Molecular biology of the cell, 4th Edition."
Alcorn, D., T. M. Adamson, T. F. Lambert, J. E. Maloney, B. C. Ritchie and P. M. Robinson
(1977). "Morphological effects of chronic tracheal ligation and drainage in the fetal lamb lung."
J Anat 123(Pt 3): 649-60.
Amato, M. B., C. S. Barbas, D. M. Medeiros, R. B. Magaldi, G. P. Schettino, G. Lorenzi-Filho,
R. A. Kairalla, D. Deheinzelin, C. Munoz, R. Oliveira, T. Y. Takagaki and C. R. Carvalho

177
(1998). "Effect of a protective-ventilation strategy on mortality in the acute respiratory distress
syndrome." N Engl J Med 338(6): 347-54.
ARDSNet (2000). "Ventilation with lower tidal volumes as compared with traditional tidal
volumes for acute lung injury and the acute respiratory distress syndrome. The Acute
Respiratory Distress Syndrome Network." N Engl J Med 342(18): 1301-8.
Argiras, E. P., C. R. Blakeley, M. S. Dunnill, S. Otremski and M. K. Sykes (1987). "High PEEP
decreases hyaline membrane formation in surfactant deficient lungs." Br J Anaesth 59(10):
1278-85.
Armstrong, L., A. R. Medford, K. M. Uppington, J. Robertson, I. R. Witherden, T. D. Tetley and
A. B. Millar (2004). "Expression of functional toll-like receptor-2 and -4 on alveolar epithelial
cells." Am J Respir Cell Mol Biol 31(2): 241-5.
Ashbaugh, D. G., D. B. Bigelow, T. L. Petty and B. E. Levine (1967). "Acute respiratory distress
in adults." Lancet 2(7511): 319-23.
ATS (2000). "American Thoracic Society. Idiopathic pulmonary fibrosis: diagnosis and
treatment. International consensus statement. American Thoracic Society (ATS), and the
European Respiratory Society (ERS)." Am J Respir Crit Care Med 161(2 Pt 1): 646-64.
Bacallao, R., A. Garfinkel, S. Monke, G. Zampighi and L. J. Mandel (1994). "ATP depletion: a
novel method to study junctional properties in epithelial tissues. I. Rearrangement of the actin
cytoskeleton." J Cell Sci 107 ( Pt 12): 3301-13.
Bachofen, H., S. Schurch, M. Urbinelli and E. R. Weibel (1987). "Relations among alveolar
surface tension, surface area, volume, and recoil pressure." J Appl Physiol 62(5): 1878-87.
Bachofen, M. and E. R. Weibel (1982). "Structural alterations of lung parenchyma in the adult
respiratory distress syndrome." Clin Chest Med 3(1): 35-56.
Bai, C., N. Fukuda, Y. Song, T. Ma, M. A. Matthay and A. S. Verkman (1999). "Lung fluid
transport in aquaporin-1 and aquaporin-4 knockout mice." J Clin Invest 103(4): 555-61.
Bakker-Woudenberg, I. A. (2003). "Experimental models of pulmonary infection." J Microbiol
Methods 54(3): 295-313.
Bals, R. and P. S. Hiemstra (2004). "Innate immunity in the lung: how epithelial cells fight
against respiratory pathogens." Eur Respir J 23(2): 327-33.
Barazzone, C., Y. R. Donati, A. F. Rochat, C. Vesin, C. D. Kan, J. C. Pache and P. F. Piguet
(1999). "Keratinocyte growth factor protects alveolar epithelium and endothelium from oxygen-
induced injury in mice." Am J Pathol 154(5): 1479-87.
Barazzone, C., S. Horowitz, Y. R. Donati, I. Rodriguez and P. F. Piguet (1998). "Oxygen
toxicity in mouse lung: pathways to cell death." Am J Respir Cell Mol Biol 19(4): 573-81.

178
Bardales, R. H., S. S. Xie, R. F. Schaefer and S. M. Hsu (1996). "Apoptosis is a major pathway
responsible for the resolution of type II pneumocytes in acute lung injury." Am J Pathol 149(3):
845-52.
Bartlett, J. G., P. O'Keefe, F. P. Tally, T. J. Louie and S. L. Gorbach (1986). "Bacteriology of
hospital-acquired pneumonia." Arch Intern Med 146(5): 868-71.
Beers, M. F. and A. B. Fisher (1992). "Surfactant protein C: a review of its unique properties and
metabolism." Am J Physiol 263(2 Pt 1): L151-60.
Beers, M. F. and C. Lomax (1995). "Synthesis and processing of hydrophobic surfactant protein
C by isolated rat type II cells." Am J Physiol 269(6 Pt 1): L744-53.
Beers, M. F., A. Wali, M. F. Eckenhoff, S. I. Feinstein, J. H. Fisher and A. B. Fisher (1992). "An
antibody with specificity for surfactant protein C precursors: identification of pro-SP-C in rat
lung." Am J Respir Cell Mol Biol 7(4): 368-78.
Behrendt, C. E. (2000). "Acute respiratory failure in the United States: incidence and 31-day
survival." Chest 118(4): 1100-5.
Bellamy, C. O., A. R. Clarke, A. H. Wyllie and D. J. Harrison (1997). "p53 Deficiency in liver
reduces local control of survival and proliferation, but does not affect apoptosis after DNA
damage." Faseb J 11(7): 591-9.
Bernard, G. R., A. Artigas, K. L. Brigham, J. Carlet, K. Falke, L. Hudson, M. Lamy, J. R.
Legall, A. Morris and R. Spragg (1994). "The American-European Consensus Conference on
ARDS. Definitions, mechanisms, relevant outcomes, and clinical trial coordination." Am J
Respir Crit Care Med 149(3 Pt 1): 818-24.
Berthiaume, Y., O. Lesur and A. Dagenais (1999). "Treatment of adult respiratory distress
syndrome: plea for rescue therapy of the alveolar epithelium." Thorax 54(2): 150-60.
Besnard, V., S. Corroyer, G. Trugnan, K. Chadelat, E. Nabeyrat, V. Cazals and A. Clement
(2001). "Distinct patterns of insulin-like growth factor binding protein (IGFBP)-2 and IGFBP-3
expression in oxidant exposed lung epithelial cells." Biochim Biophys Acta 1538(1): 47-58.
Bhaskaran, M., N. Kolliputi, Y. Wang, D. Gou, N. R. Chintagari and L. Liu (2007). "Trans-
differentiation of alveolar epithelial type II cells to type I cells involves autocrine signaling by
transforming growth factor beta 1 through the Smad pathway." J Biol Chem 282(6): 3968-76.
Bitterman, P. B. (1992). "Pathogenesis of fibrosis in acute lung injury." Am J Med 92(6A): 39S-
43S.
Borok, Z., S. I. Danto, R. L. Lubman, Y. Cao, M. C. Williams and E. D. Crandall (1998).
"Modulation of t1alpha expression with alveolar epithelial cell phenotype in vitro." Am J
Physiol 275(1 Pt 1): L155-64.

179
Borok, Z., R. L. Lubman, S. I. Danto, X. L. Zhang, S. M. Zabski, L. S. King, D. M. Lee, P. Agre
and E. D. Crandall (1998). "Keratinocyte growth factor modulates alveolar epithelial cell
phenotype in vitro: expression of aquaporin 5." Am J Respir Cell Mol Biol 18(4): 554-61.
Bouvry, D., C. Planes, L. Malbert-Colas, V. Escabasse and C. Clerici (2006). "Hypoxia-induced
cytoskeleton disruption in alveolar epithelial cells." Am J Respir Cell Mol Biol 35(5): 519-27.
Boylan, G. M., J. G. Pryde, L. G. Dobbs and M. C. McElroy (2001). "Identification of a novel
antigen on the apical surface of rat alveolar epithelial type II and Clara cells." Am J Physiol
Lung Cell Mol Physiol 280(6): L1318-26.
Bradford, M. M. (1976). "A rapid and sensitive method for the quantitation of microgram
quantities of protein utilizing the principle of protein-dye binding." Anal Biochem 72: 248-54.
BTS (1992). "The aetiology, management and outcome of severe community-acquired
pneumonia on the intensive care unit. The British Thoracic Society Research Committee and
The Public Health Laboratory Service." Respir Med 86(1): 7-13.
BTS (2001). "BTS Guidelines for the Management of Community Acquired Pneumonia in
Adults." Thorax 56 Suppl 4: IV1-64.
BTS and T. B. T. S. R. C. a. T. P. H. L. Service (1992). "The aetiology, management and
outcome of severe community-acquired pneumonia on the intensive care unit." Respir Med
86(1): 7-13.
Buccellato, L. J., M. Tso, O. I. Akinci, N. S. Chandel and G. R. Budinger (2004). "Reactive
oxygen species are required for hyperoxia-induced Bax activation and cell death in alveolar
epithelial cells." J Biol Chem 279(8): 6753-60.
Buckley, S., L. Barsky, B. Driscoll, K. Weinberg, K. D. Anderson and D. Warburton (1998).
"Apoptosis and DNA damage in type 2 alveolar epithelial cells cultured from hyperoxic rats."
Am J Physiol 274(5 Pt 1): L714-20.
Calandra, T., J. Bernhagen, C. N. Metz, L. A. Spiegel, M. Bacher, T. Donnelly, A. Cerami and
R. Bucala (1995). "MIF as a glucocorticoid-induced modulator of cytokine production." Nature
377(6544): 68-71.
Campbell, L., A. J. Hollins, A. Al-Eid, G. R. Newman, C. von Ruhland and M. Gumbleton
(1999). "Caveolin-1 expression and caveolae biogenesis during cell transdifferentiation in lung
alveolar epithelial primary cultures." Biochem Biophys Res Commun 262(3): 744-51.
Cao, Y. X., M. I. Ramirez and M. C. Williams (2003). "Enhanced binding of Sp1/Sp3
transcription factors mediates the hyperoxia-induced increased expression of the lung type I cell
gene T1alpha." J Cell Biochem 89(5): 887-901.
Carden, D. L., J. S. Alexander and R. B. George (1998). "The pathophysiology of the acute
respiratory distress syndrome." Pathophysiology 5: 1-13.

180
Cavanaugh, K. J., Jr., J. Oswari and S. S. Margulies (2001). "Role of stretch on tight junction
structure in alveolar epithelial cells." Am J Respir Cell Mol Biol 25(5): 584-91.
Cazals, V., B. Mouhieddine, B. Maitre, Y. Le Bouc, K. Chadelat, J. S. Brody and A. Clement
(1994). "Insulin-like growth factors, their binding proteins, and transforming growth factor-beta
1 in oxidant-arrested lung alveolar epithelial cells." J Biol Chem 269(19): 14111-7.
Chapman, K. E., S. E. Sinclair, D. Zhuang, A. Hassid, L. P. Desai and C. M. Waters (2005).
"Cyclic mechanical strain increases reactive oxygen species production in pulmonary epithelial
cells." Am J Physiol Lung Cell Mol Physiol 289(5): L834-41.
Chastre, J. and J. Y. Fagon (2002). "Ventilator-associated pneumonia." Am J Respir Crit Care
Med 165(7): 867-903.
Chen, J., Z. Chen, T. Narasaraju, N. Jin and L. Liu (2004). "Isolation of highly pure alveolar
epithelial type I and type II cells from rat lungs." Lab Invest 84(6): 727-35.
Chetty, A. and H. C. Nielsen (2002). "Regulation of cell proliferation by insulin-like growth
factor 1 in hyperoxia-exposed neonatal rat lung." Mol Genet Metab 75(3): 265-75.
Christensen, P. J., S. Kim, R. H. Simon, G. B. Toews and R. Paine, 3rd (1993). "Differentiation-
related expression of ICAM-1 by rat alveolar epithelial cells." Am J Respir Cell Mol Biol 8(1):
9-15.
Cines, D. B., E. S. Pollak, C. A. Buck, J. Loscalzo, G. A. Zimmerman, R. P. McEver, J. S.
Pober, T. M. Wick, B. A. Konkle, B. S. Schwartz, E. S. Barnathan, K. R. McCrae, B. A. Hug, A.
M. Schmidt and D. M. Stern (1998). "Endothelial cells in physiology and in the pathophysiology
of vascular disorders." Blood 91(10): 3527-61.
Clark, R. A., J. M. Lanigan, P. DellaPelle, E. Manseau, H. F. Dvorak and R. B. Colvin (1982).
"Fibronectin and fibrin provide a provisional matrix for epidermal cell migration during wound
reepithelialization." J Invest Dermatol 79(5): 264-9.
Clegg, G. R., C. Tyrrell, S. R. McKechnie, M. F. Beers, D. Harrison and M. C. McElroy (2005).
"Coexpression of RTI40 with alveolar epithelial type II cell proteins in lungs following injury:
identification of alveolar intermediate cell types." Am J Physiol Lung Cell Mol Physiol 289(3):
L382-90.
Clement, A., M. Edeas, K. Chadelat and J. S. Brody (1992). "Inhibition of lung epithelial cell
proliferation by hyperoxia. Posttranscriptional regulation of proliferation-related genes." J Clin
Invest 90(5): 1812-8.
Clerch, L. B. and D. Massaro (1993). "Tolerance of rats to hyperoxia. Lung antioxidant enzyme
gene expression." J Clin Invest 91(2): 499-508.
Cooper, P., S. Potter, B. Mueck, S. Yousefi and G. Jarai (2001). "Identification of genes induced
by inflammatory cytokines in airway epithelium." Am J Physiol Lung Cell Mol Physiol 280(5):
L841-52.

181
Corroyer, S., B. Maitre, V. Cazals and A. Clement (1996). "Altered regulation of G1 cyclins in
oxidant-induced growth arrest of lung alveolar epithelial cells. Accumulation of inactive cyclin
E-DCK2 complexes." J Biol Chem 271(41): 25117-25.
Cortes, G., D. Alvarez, C. Saus and S. Alberti (2002). "Role of lung epithelial cells in defense
against Klebsiella pneumoniae pneumonia." Infect Immun 70(3): 1075-80.
Crandall, E. D. and M. A. Matthay (2001). "Alveolar epithelial transport. Basic science to
clinical medicine." Am J Respir Crit Care Med 163(4): 1021-9.
Crapo, J. D. (1986). "Morphologic changes in pulmonary oxygen toxicity." Annu Rev Physiol
48: 721-31.
Crapo, J. D., B. E. Barry, H. A. Foscue and J. Shelburne (1980). "Structural and biochemical
changes in rat lungs occurring during exposures to lethal and adaptive doses of oxygen." Am
Rev Respir Dis 122(1): 123-43.
Crapo, J. D., B. E. Barry, P. Gehr, M. Bachofen and E. R. Weibel (1982). "Cell number and cell
characteristics of the normal human lung." Am Rev Respir Dis 126(2): 332-7.
Crapo, J. D., B. A. Freeman, B. E. Barry, J. F. Turrens and S. L. Young (1983). "Mechanisms of
hyperoxic injury to the pulmonary microcirculation." Physiologist 26(3): 170-6.
Crapo, J. D., S. L. Young, E. K. Fram, K. E. Pinkerton, B. E. Barry and R. O. Crapo (1983).
"Morphometric characteristics of cells in the alveolar region of mammalian lungs." Am Rev
Respir Dis 128(2 Pt 2): S42-6.
Crystal, R. G., J. D. Fulmer, W. C. Roberts, M. L. Moss, B. R. Line and H. Y. Reynolds (1976).
"Idiopathic pulmonary fibrosis. Clinical, histologic, radiographic, physiologic, scintigraphic,
cytologic, and biochemical aspects." Ann Intern Med 85(6): 769-88.
D'Angio, C. T. and J. N. Finkelstein (2000). "Oxygen regulation of gene expression: a study in
opposites." Mol Genet Metab 71(1-2): 371-80.
Daly, H. E., C. M. Baecher-Allan, A. T. Paxhia, R. M. Ryan, R. K. Barth and J. N. Finkelstein
(1998). "Cell-specific gene expression reveals changes in epithelial cell populations after
bleomycin treatment." Lab Invest 78(4): 393-400.
Danto, S. I., S. M. Zabski and E. D. Crandall (1992). "Reactivity of alveolar epithelial cells in
primary culture with type I cell monoclonal antibodies." Am J Respir Cell Mol Biol 6(3): 296-
306.
Davis, W. B., S. I. Rennard, P. B. Bitterman and R. G. Crystal (1983). "Pulmonary oxygen
toxicity. Early reversible changes in human alveolar structures induced by hyperoxia." N Engl J
Med 309(15): 878-83.
De Paepe, M. E., B. D. Johnson, K. Papadakis and F. I. Luks (1999). "Lung growth response
after tracheal occlusion in fetal rabbits is gestational age-dependent." Am J Respir Cell Mol Biol
21(1): 65-76.

182
De Paepe, M. E., Q. Mao, Y. Chao, J. L. Powell, L. P. Rubin and S. Sharma (2005). "Hyperoxia-
induced apoptosis and Fas/FasL expression in lung epithelial cells." Am J Physiol Lung Cell
Mol Physiol 289(4): L647-59.
De Paepe, M. E., Q. Mao and F. I. Luks (2004). "Expression of apoptosis-related genes after
fetal tracheal occlusion in rabbits." J Pediatr Surg 39(11): 1616-25.
Delclaux, C. and E. Azoulay (2003). "Inflammatory response to infectious pulmonary injury."
Eur Respir J Suppl 42: 10s-14s.
Denker, B. M. and S. K. Nigam (1998). "Molecular structure and assembly of the tight junction."
Am J Physiol 274(1 Pt 2): F1-9.
Deterding, R. R., A. M. Havill, T. Yano, S. C. Middleton, C. R. Jacoby, J. M. Shannon, W. S.
Simonet and R. J. Mason (1997). "Prevention of bleomycin-induced lung injury in rats by
keratinocyte growth factor." Proc Assoc Am Physicians 109(3): 254-68.
Dobbs, L. G. (1990). "Isolation and culture of alveolar type II cells." Am J Physiol 258(4 Pt 1):
L134-47.
Dobbs, L. G., E. F. Geppert, M. C. Williams, R. D. Greenleaf and R. J. Mason (1980).
"Metabolic properties and ultrastructure of alveolar type II cells isolated with elastase." Biochim
Biophys Acta 618(3): 510-23.
Dobbs, L. G., R. Gonzalez, M. A. Matthay, E. P. Carter, L. Allen and A. S. Verkman (1998).
"Highly water-permeable type I alveolar epithelial cells confer high water permeability between
the airspace and vasculature in rat lung." Proc Natl Acad Sci U S A 95(6): 2991-6.
Dobbs, L. G., R. Gonzalez and M. C. Williams (1986). "An improved method for isolating type
II cells in high yield and purity." Am Rev Respir Dis 134(1): 141-5.
Dobbs, L. G., M. S. Pian, M. Maglio, S. Dumars and L. Allen (1997). "Maintenance of the
differentiated type II cell phenotype by culture with an apical air surface." Am J Physiol 273(2
Pt 1): L347-54.
Dobbs, L. G., M. C. Williams and A. E. Brandt (1985). "Changes in biochemical characteristics
and pattern of lectin binding of alveolar type II cells with time in culture." Biochim Biophys
Acta 846(1): 155-66.
Dobbs, L. G., M. C. Williams and R. Gonzalez (1988). "Monoclonal antibodies specific to apical
surfaces of rat alveolar type I cells bind to surfaces of cultured, but not freshly isolated, type II
cells." Biochim Biophys Acta 970(2): 146-56.
Donnelly, S. C., C. Haslett, P. T. Reid, I. S. Grant, W. A. Wallace, C. N. Metz, L. J. Bruce and
R. Bucala (1997). "Regulatory role for macrophage migration inhibitory factor in acute
respiratory distress syndrome." Nat Med 3(3): 320-3.
Donnelly, S. C., R. M. Strieter, P. T. Reid, S. L. Kunkel, M. D. Burdick, I. Armstrong, A.
Mackenzie and C. Haslett (1996). "The association between mortality rates and decreased

183
concentrations of interleukin-10 and interleukin-1 receptor antagonist in the lung fluids of
patients with the adult respiratory distress syndrome." Ann Intern Med 125(3): 191-6.
dos Santos, C. C., B. Han, C. F. Andrade, X. Bai, S. Uhlig, R. Hubmayr, M. Tsang, M. Lodyga,
S. Keshavjee, A. S. Slutsky and M. Liu (2004). "DNA microarray analysis of gene expression in
alveolar epithelial cells in response to TNFalpha, LPS, and cyclic stretch." Physiol Genomics
19(3): 331-42.
Dos Santos, C. C. and A. S. Slutsky (2000). "Invited review: mechanisms of ventilator-induced
lung injury: a perspective." J Appl Physiol 89(4): 1645-55.
Douzinas, E. E., P. D. Tsidemiadou, M. T. Pitaridis, I. Andrianakis, A. Bobota-Chloraki, K.
Katsouyanni, D. Sfyras, K. Malagari and C. Roussos (1997). "The regional production of
cytokines and lactate in sepsis-related multiple organ failure." Am J Respir Crit Care Med
155(1): 53-9.
Doyle, I. R., A. D. Bersten and T. E. Nicholas (1997). "Surfactant proteins-A and -B are elevated
in plasma of patients with acute respiratory failure." Am J Respir Crit Care Med 156(4 Pt 1):
1217-29.
Doyle, I. R., T. E. Nicholas and A. D. Bersten (1995). "Serum surfactant protein-A levels in
patients with acute cardiogenic pulmonary edema and adult respiratory distress syndrome." Am J
Respir Crit Care Med 152(1): 307-17.
Dreyfuss, D., G. Basset, P. Soler and G. Saumon (1985). "Intermittent positive-pressure
hyperventilation with high inflation pressures produces pulmonary microvascular injury in rats."
Am Rev Respir Dis 132(4): 880-4.
Dreyfuss, D. and G. Saumon (1998). "Ventilator-induced lung injury: lessons from experimental
studies." Am J Respir Crit Care Med 157(1): 294-323.
Dreyfuss, D., P. Soler, G. Basset and G. Saumon (1988). "High inflation pressure pulmonary
edema. Respective effects of high airway pressure, high tidal volume, and positive end-
expiratory pressure." Am Rev Respir Dis 137(5): 1159-64.
Duane, P. G., J. B. Rubins, H. R. Weisel and E. N. Janoff (1993). "Identification of hydrogen
peroxide as a Streptococcus pneumoniae toxin for rat alveolar epithelial cells." Infect Immun
61(10): 4392-7.
Edwards, Y. S. (2001). "Stretch stimulation: its effects on alveolar type II cell function in the
lung." Comp Biochem Physiol A Mol Integr Physiol 129(1): 245-60.
Edwards, Y. S., L. M. Sutherland and A. W. Murray (2000). "NO protects alveolar type II cells
from stretch-induced apoptosis. A novel role for macrophages in the lung." Am J Physiol Lung
Cell Mol Physiol 279(6): L1236-42.
Edwards, Y. S., L. M. Sutherland, J. H. Power, T. E. Nicholas and A. W. Murray (1999). "Cyclic
stretch induces both apoptosis and secretion in rat alveolar type II cells." FEBS Lett 448(1): 127-
30.

184
Effros, R. M., C. Darin, E. R. Jacobs, R. A. Rogers, G. Krenz and E. E. Schneeberger (1997).
"Water transport and the distribution of aquaporin-1 in pulmonary air spaces." J Appl Physiol
83(3): 1002-16.
Egan, E. A. (1982). "Lung inflation, lung solute permeability, and alveolar edema." J Appl
Physiol 53(1): 121-5.
Evans, M. J., L. J. Cabral, R. J. Stephens and G. Freeman (1975). "Transformation of alveolar
type 2 cells to type 1 cells following exposure to NO2." Exp Mol Pathol 22(1): 142-50.
Evans, M. J., R. J. Stephens, L. J. Cabral and G. Freeman (1972). "Cell renewal in the lungs of
rats exposed to low levels of NO2." Arch Environ Health 24(3): 180-8.
Evans, M. J., L. S. Van Winkle, M. V. Fanucchi and C. G. Plopper (1999). "The attenuated
fibroblast sheath of the respiratory tract epithelial-mesenchymal trophic unit." Am J Respir Cell
Mol Biol 21(6): 655-7.
Fang, K. C. (2000). "Mesenchymal regulation of alveolar repair in pulmonary fibrosis." Am J
Respir Cell Mol Biol 23(2): 142-5.
Fehrenbach, H. (2001). "Alveolar epithelial type II cell: defender of the alveolus revisited."
Respir Res 2(1): 33-46.
Fehrenbach, H., M. Kasper, R. Koslowski, T. Pan, D. Schuh, M. Muller and R. J. Mason (2000).
"Alveolar epithelial type II cell apoptosis in vivo during resolution of keratinocyte growth
factor-induced hyperplasia in the rat." Histochem Cell Biol 114(1): 49-61.
Fehrenbach, H., M. Kasper, T. Tschernig, T. Pan, D. Schuh, J. M. Shannon, M. Muller and R. J.
Mason (1999). "Keratinocyte growth factor-induced hyperplasia of rat alveolar type II cells in
vivo is resolved by differentiation into type I cells and by apoptosis." Eur Respir J 14(3): 534-44.
Fine, A., Y. Janssen-Heininger, R. P. Soultanakis, S. G. Swisher and B. D. Uhal (2000).
"Apoptosis in lung pathophysiology." Am J Physiol Lung Cell Mol Physiol 279(3): L423-7.
Fishman, A. P. (1982). "Endothelium: a distributed organ of diverse capabilities." Ann N Y
Acad Sci 401: 1-8.
Flecknoe, S., R. Harding, G. Maritz and S. B. Hooper (2000). "Increased lung expansion alters
the proportions of type I and type II alveolar epithelial cells in fetal sheep." Am J Physiol Lung
Cell Mol Physiol 278(6): L1180-5.
Flecknoe, S. J., M. J. Wallace, R. Harding and S. B. Hooper (2002). "Determination of alveolar
epithelial cell phenotypes in fetal sheep: evidence for the involvement of basal lung expansion."
J Physiol 542(Pt 1): 245-53.
Folkesson, H. G., M. A. Matthay, C. A. Hebert and V. C. Broaddus (1995). "Acid aspiration-
induced lung injury in rabbits is mediated by interleukin-8-dependent mechanisms." J Clin
Invest 96(1): 107-16.

185
Fracica, P. J., M. J. Knapp, C. A. Piantadosi, K. Takeda, W. J. Fulkerson, R. E. Coleman, W. G.
Wolfe and J. D. Crapo (1991). "Responses of baboons to prolonged hyperoxia: physiology and
qualitative pathology." J Appl Physiol 71(6): 2352-62.
Frank, J. A., J. A. Gutierrez, K. D. Jones, L. Allen, L. Dobbs and M. A. Matthay (2002). "Low
tidal volume reduces epithelial and endothelial injury in acid-injured rat lungs." Am J Respir Crit
Care Med 165(2): 242-9.
Frank, J. A., C. M. Wray, D. F. McAuley, R. Schwendener and M. A. Matthay (2006). "Alveolar
macrophages contribute to alveolar barrier dysfunction in ventilator-induced lung injury." Am J
Physiol Lung Cell Mol Physiol 291(6): L1191-8.
Franklin, L., A. D. Cronshaw, D. J. Harrison and M. C. McElroy (2004). "The type II and clara
cell specific monoclonal antibody (MMC4) recognises aminopeptidase N." Eur Respir J 24:
Suppl. 48: 719s.
Fujikawa, L. S., C. S. Foster, T. J. Harrist, J. M. Lanigan and R. B. Colvin (1981). "Fibronectin
in healing rabbit corneal wounds." Lab Invest 45(2): 120-9.
Funkhouser, J. D. and R. D. Peterson (1989). "Immunotargeting: a contemporary approach to the
study of lung development." Am J Physiol 257(6 Pt 1): L311-7.
Funkhouser, J. D., S. D. Tangada, M. Jones, S. J. O and R. D. Peterson (1991). "p146 type II
alveolar epithelial cell antigen is identical to aminopeptidase N." Am J Physiol 260(4 Pt 1):
L274-9.
Gattinoni, L., P. Pelosi, P. M. Suter, A. Pedoto, P. Vercesi and A. Lissoni (1998). "Acute
respiratory distress syndrome caused by pulmonary and extrapulmonary disease. Different
syndromes?" Am J Respir Crit Care Med 158(1): 3-11.
Geiser, T. (2003). "Idiopathic pulmonary fibrosis--a disorder of alveolar wound repair?" Swiss
Med Wkly 133(29-30): 405-11.
Giangreco, A., S. D. Reynolds and B. R. Stripp (2002). "Terminal bronchioles harbor a unique
airway stem cell population that localizes to the bronchoalveolar duct junction." Am J Pathol
161(1): 173-82.
Gil, J. and A. Martinez-Hernandez (1984). "The connective tissue of the rat lung: electron
immunohistochemical studies." J Histochem Cytochem 32(2): 230-8.
Girod, C. E., D. H. Shin, M. B. Hershenson, J. Solway, R. Dahl and Y. E. Miller (1996). "p172:
An alveolar type II and Clara cell specific protein with late developmental expression and
upregulation by hyperoxic lung injury." Am J Respir Cell Mol Biol 14(6): 538-47.
Gon, Y., M. R. Wood, W. B. Kiosses, E. Jo, M. G. Sanna, J. Chun and H. Rosen (2005). "S1P3
receptor-induced reorganization of epithelial tight junctions compromises lung barrier integrity
and is potentiated by TNF." Proc Natl Acad Sci U S A 102(26): 9270-5.

186
Gonzalez, R., Y. H. Yang, C. Griffin, L. Allen, Z. Tigue and L. Dobbs (2005). "Freshly isolated
rat alveolar type I cells, type II cells, and cultured type II cells have distinct molecular
phenotypes." Am J Physiol Lung Cell Mol Physiol 288(1): L179-89.
Gonzalez-Mariscal, L., B. Chavez de Ramirez and M. Cereijido (1985). "Tight junction
formation in cultured epithelial cells (MDCK)." J Membr Biol 86(2): 113-25.
Goodenough, D. A. (1999). "Plugging the leaks." Proc Natl Acad Sci U S A 96(2): 319-21.
Greene, K. E., J. R. Wright, K. P. Steinberg, J. T. Ruzinski, E. Caldwell, W. B. Wong, W. Hull,
J. A. Whitsett, T. Akino, Y. Kuroki, H. Nagae, L. D. Hudson and T. R. Martin (1999). "Serial
changes in surfactant-associated proteins in lung and serum before and after onset of ARDS."
Am J Respir Crit Care Med 160(6): 1843-50.
Gregory, T. J., W. J. Longmore, M. A. Moxley, J. A. Whitsett, C. R. Reed, A. A. Fowler, 3rd, L.
D. Hudson, R. J. Maunder, C. Crim and T. M. Hyers (1991). "Surfactant chemical composition
and biophysical activity in acute respiratory distress syndrome." J Clin Invest 88(6): 1976-81.
Guest, J. F. and A. Morris (1997). "Community-acquired pneumonia: the annual cost to the
National Health Service in the UK." Eur Respir J 10(7): 1530-4.
Gunther, A., C. Siebert, R. Schmidt, S. Ziegler, F. Grimminger, M. Yabut, B. Temmesfeld, D.
Walmrath, H. Morr and W. Seeger (1996). "Surfactant alterations in severe pneumonia, acute
respiratory distress syndrome, and cardiogenic lung edema." Am J Respir Crit Care Med 153(1):
176-84.
Guo, J., E. S. Yi, A. M. Havill, I. Sarosi, L. Whitcomb, S. Yin, S. C. Middleton, P. Piguet and T.
R. Ulich (1998). "Intravenous keratinocyte growth factor protects against experimental
pulmonary injury." Am J Physiol 275(4 Pt 1): L800-5.
Gutierrez, J. A., R. Ertsey, L. M. Scavo, E. Collins and L. G. Dobbs (1999). "Mechanical
distention modulates alveolar epithelial cell phenotypic expression by transcriptional
regulation." Am J Respir Cell Mol Biol 21(2): 223-9.
Gutierrez, J. A., R. F. Gonzalez and L. G. Dobbs (1998). "Mechanical distension modulates
pulmonary alveolar epithelial phenotypic expression in vitro." Am J Physiol 274(2 Pt 1): L196-
202.
Gutierrez, J. A., V. V. Suzara and L. G. Dobbs (2003). "Continuous mechanical contraction
modulates expression of alveolar epithelial cell phenotype." Am J Respir Cell Mol Biol 29(1):
81-7.
Haddad, J. J. (2002). "Oxygen homeostasis, thiol equilibrium and redox regulation of signalling
transcription factors in the alveolar epithelium." Cell Signal 14(10): 799-810.
Haddad, J. J. (2002). "Science review: Redox and oxygen-sensitive transcription factors in the
regulation of oxidant-mediated lung injury: role for nuclear factor-kappaB." Crit Care 6(6): 481-
90.

187
Haddad, J. J. and S. C. Land (2000). "The differential expression of apoptosis factors in the
alveolar epithelium is redox sensitive and requires NF-kappaB (RelA)-selective targeting."
Biochem Biophys Res Commun 271(1): 257-67.
Haies, D. M., J. Gil and E. R. Weibel (1981). "Morphometric study of rat lung cells. I.
Numerical and dimensional characteristics of parenchymal cell population." Am Rev Respir Dis
123(5): 533-41.
Hammerschmidt, S., H. Kuhn, T. Grasenack, C. Gessner and H. Wirtz (2004). "Apoptosis and
necrosis induced by cyclic mechanical stretching in alveolar type II cells." Am J Respir Cell Mol
Biol 30(3): 396-402.
Harding, R., S. B. Hooper and V. K. Han (1993). "Abolition of fetal breathing movements by
spinal cord transection leads to reductions in fetal lung liquid volume, lung growth, and IGF-II
gene expression." Pediatr Res 34(2): 148-53.
Hayatdavoudi, G., J. J. O'Neil, B. E. Barry, B. A. Freeman and J. D. Crapo (1981). "Pulmonary
injury in rats following continuous exposure to 60% O2 for 7 days." J Appl Physiol 51(5): 1220-
31.
Held, H. D., S. Boettcher, L. Hamann and S. Uhlig (2001). "Ventilation-induced chemokine and
cytokine release is associated with activation of nuclear factor-kappaB and is blocked by
steroids." Am J Respir Crit Care Med 163(3 Pt 1): 711-6.
Hermans, C. and A. Bernard (1999). "Lung epithelium-specific proteins: characteristics and
potential applications as markers." Am J Respir Crit Care Med 159(2): 646-78.
Herridge, M. S., A. M. Cheung, C. M. Tansey, A. Matte-Martyn, N. Diaz-Granados, F. Al-Saidi,
A. B. Cooper, C. B. Guest, C. D. Mazer, S. Mehta, T. E. Stewart, A. Barr, D. Cook and A. S.
Slutsky (2003). "One-year outcomes in survivors of the acute respiratory distress syndrome." N
Engl J Med 348(8): 683-93.
Hillyer, P., E. Mordelet, G. Flynn and D. Male (2003). "Chemokines, chemokine receptors and
adhesion molecules on different human endothelia: discriminating the tissue-specific functions
that affect leucocyte migration." Clin Exp Immunol 134(3): 431-41.
Hippenstiel, S., B. Opitz, B. Schmeck and N. Suttorp (2006). "Lung epithelium as a sentinel and
effector system in pneumonia--molecular mechanisms of pathogen recognition and signal
transduction." Respir Res 7: 97.
Hirani, N. A. and J. T. Macfarlane (1997). "Impact of management guidelines on the outcome of
severe community acquired pneumonia." Thorax 52(1): 17-21.
Hooper, S. B., V. K. Han and R. Harding (1993). "Changes in lung expansion alter pulmonary
DNA synthesis and IGF-II gene expression in fetal sheep." Am J Physiol 265(4 Pt 1): L403-9.
Hsia, C. C. (2004). "Signals and mechanisms of compensatory lung growth." J Appl Physiol
97(5): 1992-8.

188
Ikegami, M. and A. H. Jobe (1998). "Surfactant protein metabolism in vivo." Biochim Biophys
Acta 1408(2-3): 218-25.
Imai, Y., J. Parodo, O. Kajikawa, M. de Perrot, S. Fischer, V. Edwards, E. Cutz, M. Liu, S.
Keshavjee, T. R. Martin, J. C. Marshall, V. M. Ranieri and A. S. Slutsky (2003). "Injurious
mechanical ventilation and end-organ epithelial cell apoptosis and organ dysfunction in an
experimental model of acute respiratory distress syndrome." Jama 289(16): 2104-12.
Isakson, B. E., G. J. Seedorf, R. L. Lubman, W. H. Evans and S. Boitano (2003). "Cell-cell
communication in heterocellular cultures of alveolar epithelial cells." Am J Respir Cell Mol Biol
29(5): 552-61.
Ishizawa, K., H. Kubo, M. Yamada, S. Kobayashi, M. Numasaki, S. Ueda, T. Suzuki and H.
Sasaki (2004). "Bone marrow-derived cells contribute to lung regeneration after elastase-induced
pulmonary emphysema." FEBS Lett 556(1-3): 249-52.
Jafari, B., B. Ouyang, L. F. Li, C. A. Hales and D. A. Quinn (2004). "Intracellular glutathione in
stretch-induced cytokine release from alveolar type-2 like cells." Respirology 9(1): 43-53.
Janeway, C. A., Jr. and R. Medzhitov (2002). "Innate immune recognition." Annu Rev Immunol
20: 197-216.
Jiao, G., E. Li and R. Yu (2002). "Decreased expression of AQP1 and AQP5 in acute injured
lungs in rats." Chin Med J (Engl) 115(7): 963-7.
Joe, P., L. D. Wallen, C. J. Chapin, C. H. Lee, L. Allen, V. K. Han, L. G. Dobbs, S. Hawgood
and J. A. Kitterman (1997). "Effects of mechanical factors on growth and maturation of the lung
in fetal sheep." Am J Physiol 272(1 Pt 1): L95-105.
Johanson, W. G., Jr., J. H. Higuchi, D. E. Woods, P. Gomez and J. J. Coalson (1985).
"Dissemination of Pseudomonas aeruginosa during lung infection in hamsters. Role of oxygen-
induced lung injury." Am Rev Respir Dis 132(2): 358-61.
Johnson, M. D., J. H. Widdicombe, L. Allen, P. Barbry and L. G. Dobbs (2002). "Alveolar
epithelial type I cells contain transport proteins and transport sodium, supporting an active role
for type I cells in regulation of lung liquid homeostasis." Proc Natl Acad Sci U S A 99(4): 1966-
71.
Joshi, P. C., G. V. Poole, V. Sachdev, X. Zhou and Q. Jones (2000). "Trauma patients with
positive cultures have higher levels of circulating macrophage migration inhibitory factor
(MIF)." Res Commun Mol Pathol Pharmacol 107(1-2): 13-20.
Kapanci, Y., E. R. Weibel, H. P. Kaplan and F. R. Robinson (1969). "Pathogenesis and
reversibility of the pulmonary lesions of oxygen toxicity in monkeys. II. Ultrastructural and
morphometric studies." Lab Invest 20(1): 101-18.
Kawano, T., S. Mori, M. Cybulsky, R. Burger, A. Ballin, E. Cutz and A. C. Bryan (1987).
"Effect of granulocyte depletion in a ventilated surfactant-depleted lung." J Appl Physiol 62(1):
27-33.

189
Kazzaz, J. A., J. Xu, T. A. Palaia, L. Mantell, A. M. Fein and S. Horowitz (1996). "Cellular
oxygen toxicity. Oxidant injury without apoptosis." J Biol Chem 271(25): 15182-6.
Kikkawa, Y. and K. Yoneda (1974). "The type II epithelial cell of the lung. I. Method of
isolation." Lab Invest 30(1): 76-84.
King, L. S. and P. Agre (1996). "Pathophysiology of the aquaporin water channels." Annu Rev
Physiol 58: 619-48.
Kitterman, J. A. (1996). "The effects of mechanical forces on fetal lung growth." Clin Perinatol
23(4): 727-40.
Kleeberger, W., A. Versmold, T. Rothamel, S. Glockner, M. Bredt, A. Haverich, U. Lehmann
and H. Kreipe (2003). "Increased chimerism of bronchial and alveolar epithelium in human lung
allografts undergoing chronic injury." Am J Pathol 162(5): 1487-94.
Kobayashi, J. and S. Kitamura (1995). "KL-6: a serum marker for interstitial pneumonia." Chest
108(2): 311-5.
Kohno, N., M. Akiyama, S. Kyoizumi, M. Hakoda, K. Kobuke and M. Yamakido (1988).
"Detection of soluble tumor-associated antigens in sera and effusions using novel monoclonal
antibodies, KL-3 and KL-6, against lung adenocarcinoma." Jpn J Clin Oncol 18(3): 203-16.
Korzeniewski, C. and D. M. Callewaert (1983). "An enzyme-release assay for natural
cytotoxicity." J Immunol Methods 64(3): 313-20.
Kotton, D. N., A. J. Fabian and R. C. Mulligan (2005). "Failure of bone marrow to reconstitute
lung epithelium." Am J Respir Cell Mol Biol 33(4): 328-34.
Kreda, S. M., M. C. Gynn, D. A. Fenstermacher, R. C. Boucher and S. E. Gabriel (2001).
"Expression and localization of epithelial aquaporins in the adult human lung." Am J Respir Cell
Mol Biol 24(3): 224-34.
Kumar, N. M. and N. B. Gilula (1996). "The gap junction communication channel." Cell 84(3):
381-8.
Kumar, V., A. Abbas and N. Fausto (2004). "Robbins & Cotran's Pathologic Basis of Disease."
Kurahashi, K., O. Kajikawa, T. Sawa, M. Ohara, M. A. Gropper, D. W. Frank, T. R. Martin and
J. P. Wiener-Kronish (1999). "Pathogenesis of septic shock in Pseudomonas aeruginosa
pneumonia." J Clin Invest 104(6): 743-50.
Laufe, M. D., R. H. Simon, A. Flint and J. B. Keller (1986). "Adult respiratory distress
syndrome in neutropenic patients." Am J Med 80(6): 1022-6.
Lee, P. J. and A. M. Choi (2003). "Pathways of cell signaling in hyperoxia." Free Radic Biol
Med 35(4): 341-50.

190
Lee, W. L. and G. P. Downey (2001). "Neutrophil activation and acute lung injury." Curr Opin
Crit Care 7(1): 1-7.
Lehmann, L. E., U. Novender, S. Schroeder, T. Pietsch, T. von Spiegel, C. Putensen, A. Hoeft
and F. Stuber (2001). "Plasma levels of macrophage migration inhibitory factor are elevated in
patients with severe sepsis." Intensive Care Med 27(8): 1412-5.
Lentsch, A. B., B. J. Czermak, N. M. Bless, N. Van Rooijen and P. A. Ward (1999). "Essential
role of alveolar macrophages in intrapulmonary activation of NF-kappaB." Am J Respir Cell
Mol Biol 20(4): 692-8.
Lewandowski, K., J. Metz, C. Deutschmann, H. Preiss, R. Kuhlen, A. Artigas and K. J. Falke
(1995). "Incidence, severity, and mortality of acute respiratory failure in Berlin, Germany." Am
J Respir Crit Care Med 151(4): 1121-5.
Lewis, J. F. and A. H. Jobe (1993). "Surfactant and the adult respiratory distress syndrome." Am
Rev Respir Dis 147(1): 218-33.
Li, L. F., B. Ouyang, G. Choukroun, R. Matyal, M. Mascarenhas, B. Jafari, J. V. Bonventre, T.
Force and D. A. Quinn (2003). "Stretch-induced IL-8 depends on c-Jun NH2-terminal and
nuclear factor-kappaB-inducing kinases." Am J Physiol Lung Cell Mol Physiol 285(2): L464-75.
Liggins, G. C., G. A. Vilos, G. A. Campos, J. A. Kitterman and C. H. Lee (1981). "The effect of
spinal cord transection on lung development in fetal sheep." J Dev Physiol 3(4): 267-74.
Lines, A., L. Nardo, I. D. Phillips, F. Possmayer and S. B. Hooper (1999). "Alterations in lung
expansion affect surfactant protein A, B, and C mRNA levels in fetal sheep." Am J Physiol
276(2 Pt 1): L239-45.
Low, F. N. (1952). "Electronmicroscopy of the rat lung." Anat Rec 113: 437-4443.
Luhr, O. R., K. Antonsen, M. Karlsson, S. Aardal, A. Thorsteinsson, C. G. Frostell and J. Bonde
(1999). "Incidence and mortality after acute respiratory failure and acute respiratory distress
syndrome in Sweden, Denmark, and Iceland. The ARF Study Group." Am J Respir Crit Care
Med 159(6): 1849-61.
Lwebuga-Mukasa, J. S. (1991). "Matrix-driven pneumocyte differentiation." Am Rev Respir Dis
144(2): 452-7.
Ma, T., N. Fukuda, Y. Song, M. A. Matthay and A. S. Verkman (2000). "Lung fluid transport in
aquaporin-5 knockout mice." J Clin Invest 105(1): 93-100.
Martin, T. R. (2002). "Neutrophils and lung injury: getting it right." J Clin Invest 110(11): 1603-
5.
Martinon, F. and J. Tschopp (2005). "NLRs join TLRs as innate sensors of pathogens." Trends
Immunol 26(8): 447-54.

191
Mason, R. J. and M. C. Williams (1991). "The Lung: Scientific Foundations edited by R. G.
Crystal, J. B. West et al." 235-246.
Massaro, G. D. and D. Massaro (1983). "Morphologic evidence that large inflations of the lung
stimulate secretion of surfactant." Am Rev Respir Dis 127(2): 235-6.
Matthay, M. A., H. G. Folkesson and C. Clerici (2002). "Lung epithelial fluid transport and the
resolution of pulmonary edema." Physiol Rev 82(3): 569-600.
Matthay, M. A., L. Robriquet and X. Fang (2005). "Alveolar epithelium: role in lung fluid
balance and acute lung injury." Proc Am Thorac Soc 2(3): 206-13.
Matthay, M. A. and J. P. Wiener-Kronish (1990). "Intact epithelial barrier function is critical for
the resolution of alveolar edema in humans." Am Rev Respir Dis 142(6 Pt 1): 1250-7.
Maunder, R. J., W. P. Shuman, J. W. McHugh, S. I. Marglin and J. Butler (1986). "Preservation
of normal lung regions in the adult respiratory distress syndrome. Analysis by computed
tomography." Jama 255(18): 2463-5.
McCullers, J. A. and E. I. Tuomanen (2001). "Molecular pathogenesis of pneumococcal
pneumonia." Front Biosci 6: D877-89.
McElroy, M. C., H. R. Harty, G. E. Hosford, G. M. Boylan, J. F. Pittet and T. J. Foster (1999).
"Alpha-toxin damages the air-blood barrier of the lung in a rat model of Staphylococcus aureus-
induced pneumonia." Infect Immun 67(10): 5541-4.
McElroy, M. C. and M. Kasper (2004). "The use of alveolar epithelial type I cell-selective
markers to investigate lung injury and repair." Eur Respir J 24(4): 664-73.
McElroy, M. C., J. F. Pittet, L. Allen, J. P. Wiener-Kronish and L. G. Dobbs (1997).
"Biochemical detection of type I cell damage after nitrogen dioxide-induced lung injury in rats."
Am J Physiol 273(6 Pt 1): L1228-34.
McElroy, M. C., J. F. Pittet, S. Hashimoto, L. Allen, J. P. Wiener-Kronish and L. G. Dobbs
(1995). "A type I cell-specific protein is a biochemical marker of epithelial injury in a rat model
of pneumonia." Am J Physiol 268(2 Pt 1): L181-6.
McEver, R. P., K. L. Moore and R. D. Cummings (1995). "Leukocyte trafficking mediated by
selectin-carbohydrate interactions." J Biol Chem 270(19): 11025-8.
McHugh, L. G., J. A. Milberg, M. E. Whitcomb, R. B. Schoene, R. J. Maunder and L. D.
Hudson (1994). "Recovery of function in survivors of the acute respiratory distress syndrome."
Am J Respir Crit Care Med 150(1): 90-4.
Mead, J., T. Takishima and D. Leith (1970). "Stress distribution in lungs: a model of pulmonary
elasticity." J Appl Physiol 28(5): 596-608.
Mercer, R. R. and J. D. Crapo (1990). "Spatial distribution of collagen and elastin fibers in the
lungs." J Appl Physiol 69(2): 756-65.

192
Mercer, R. R., J. M. Laco and J. D. Crapo (1987). "Three-dimensional reconstruction of alveoli
in the rat lung for pressure-volume relationships." J Appl Physiol 62(4): 1480-7.
Michel, R. P., M. Laforte and J. C. Hogg (1981). "Physiology and morphology of pulmonary
microvascular injury with shock and reinfusion." J Appl Physiol 50(6): 1227-35.
Michiels, C., E. Minet, D. Mottet and M. Raes (2002). "Regulation of gene expression by
oxygen: NF-kappaB and HIF-1, two extremes." Free Radic Biol Med 33(9): 1231-42.
Modelska, K., M. A. Matthay, M. C. McElroy and J. F. Pittet (1997). "Upregulation of alveolar
liquid clearance after fluid resuscitation for hemorrhagic shock in rats." Am J Physiol 273(2 Pt
1): L305-14.
Montgomery, A. B., M. A. Stager, C. J. Carrico and L. D. Hudson (1985). "Causes of mortality
in patients with the adult respiratory distress syndrome." Am Rev Respir Dis 132(3): 485-9.
Moore, F. A. and E. E. Moore (1995). "Evolving concepts in the pathogenesis of postinjury
multiple organ failure." Surg Clin North Am 75(2): 257-77.
Nahum, A., J. Hoyt, L. Schmitz, J. Moody, R. Shapiro and J. J. Marini (1997). "Effect of
mechanical ventilation strategy on dissemination of intratracheally instilled Escherichia coli in
dogs." Crit Care Med 25(10): 1733-43.
Narasaraju, T. A., H. Chen, T. Weng, M. Bhaskaran, N. Jin, J. Chen, Z. Chen, M. R. Chinoy and
L. Liu (2006). "Expression profile of IGF system during lung injury and recovery in rats exposed
to hyperoxia: a possible role of IGF-1 in alveolar epithelial cell proliferation and differentiation."
J Cell Biochem 97(5): 984-98.
Neff, S. B., R. Z'Graggen B, T. A. Neff, M. Jamnicki-Abegg, D. Suter, R. C. Schimmer, C.
Booy, H. Joch, T. Pasch, P. A. Ward and B. Beck-Schimmer (2006). "Inflammatory response of
tracheobronchial epithelial cells to endotoxin." Am J Physiol Lung Cell Mol Physiol 290(1):
L86-96.
Nelson, S., S. M. Belknap, R. W. Carlson, D. Dale, B. DeBoisblanc, S. Farkas, N.
Fotheringham, H. Ho, T. Marrie, H. Movahhed, R. Root and J. Wilson (1998). "A randomized
controlled trial of filgrastim as an adjunct to antibiotics for treatment of hospitalized patients
with community-acquired pneumonia. CAP Study Group." J Infect Dis 178(4): 1075-80.
Newman, V., R. F. Gonzalez, M. A. Matthay and L. G. Dobbs (2000). "A novel alveolar type I
cell-specific biochemical marker of human acute lung injury." Am J Respir Crit Care Med 161(3
Pt 1): 990-5.
Nicholas, T. E., J. H. Power and H. A. Barr (1982). "Surfactant homeostasis in the rat lung
during swimming exercise." J Appl Physiol 53(6): 1521-8.
Nicholas, T. E., J. H. Power and H. A. Barr (1982). "The pulmonary consequences of a deep
breath." Respir Physiol 49(3): 315-24.

193
Nielsen, S., L. S. King, B. M. Christensen and P. Agre (1997). "Aquaporins in complex tissues.
II. Subcellular distribution in respiratory and glandular tissues of rat." Am J Physiol 273(5 Pt 1):
C1549-61.
NIH (1977). "Conference report: Mechanisms of acute respiratory failure." Am Rev Respir Dis
115(6): 1071-8.
Normand, I. C., R. E. Olver, E. O. Reynolds and L. B. Strang (1971). "Permeability of lung
capillaries and alveoli to non-electrolytes in the foetal lamb." J Physiol 219(2): 303-30.
O'Donovan, D. J. and C. J. Fernandes (2004). "Free radicals and diseases in premature infants."
Antioxid Redox Signal 6(1): 169-76.
O'Reilly, M. A., R. J. Staversky, B. R. Stripp and J. N. Finkelstein (1998). "Exposure to
hyperoxia induces p53 expression in mouse lung epithelium." Am J Respir Cell Mol Biol 18(1):
43-50.
O'Reilly, M. A., R. J. Staversky, R. H. Watkins and W. M. Maniscalco (1998). "Accumulation
of p21(Cip1/WAF1) during hyperoxic lung injury in mice." Am J Respir Cell Mol Biol 19(5):
777-85.
Ohwada, A., Y. Tsutsumi-Ishii, Y. Yoshioka, K. Iwabuchi, I. Nagaoka and Y. Fukuchi (2003).
"Acid exposure potentiates intercellular adhesion molecule-1 and e-cadherin expression on A549
alveolar lining epithelial cells." Exp Lung Res 29(6): 389-400.
Olsen, C. O., B. E. Isakson, G. J. Seedorf, R. L. Lubman and S. Boitano (2005). "Extracellular
matrix-driven alveolar epithelial cell differentiation in vitro." Exp Lung Res 31(5): 461-82.
Otto, W. R. (2002). "Lung epithelial stem cells." J Pathol 197(4): 527-35.
Paine, R., 3rd and R. H. Simon (1996). "Expanding the frontiers of lung biology through the
creative use of alveolar epithelial cells in culture." Am J Physiol 270(4 Pt 1): L484-6.
Panos, R. J., P. M. Bak, W. S. Simonet, J. S. Rubin and L. J. Smith (1995). "Intratracheal
instillation of keratinocyte growth factor decreases hyperoxia-induced mortality in rats." J Clin
Invest 96(4): 2026-33.
Park, W. Y., R. B. Goodman, K. P. Steinberg, J. T. Ruzinski, F. Radella, 2nd, D. R. Park, J.
Pugin, S. J. Skerrett, L. D. Hudson and T. R. Martin (2001). "Cytokine balance in the lungs of
patients with acute respiratory distress syndrome." Am J Respir Crit Care Med 164(10 Pt 1):
1896-903.
Parker, J. C., L. A. Hernandez and K. J. Peevy (1993). "Mechanisms of ventilator-induced lung
injury." Crit Care Med 21(1): 131-43.
Parker, J. C. and M. I. Townsley (2004). "Evaluation of lung injury in rats and mice." Am J
Physiol Lung Cell Mol Physiol 286(2): L231-46.

194
Pelosi, P., D. D'Onofrio, D. Chiumello, S. Paolo, G. Chiara, V. L. Capelozzi, C. S. Barbas, M.
Chiaranda and L. Gattinoni (2003). "Pulmonary and extrapulmonary acute respiratory distress
syndrome are different." Eur Respir J Suppl 42: 48s-56s.
Petty, T. L. and D. G. Ashbaugh (1971). "The adult respiratory distress syndrome. Clinical
features, factors influencing prognosis and principles of management." Chest 60(3): 233-9.
Pison, U., U. Obertacke, W. Seeger and S. Hawgood (1992). "Surfactant protein A (SP-A) is
decreased in acute parenchymal lung injury associated with polytrauma." Eur J Clin Invest
22(11): 712-8.
Pittet, J. F., T. J. Brenner, K. Modelska and M. A. Matthay (1996). "Alveolar liquid clearance is
increased by endogenous catecholamines in hemorrhagic shock in rats." J Appl Physiol 81(2):
830-7.
Pittet, J. F., S. Hashimoto, M. Pian, M. C. McElroy, G. Nitenberg and J. P. Wiener-Kronish
(1996). "Exotoxin A stimulates fluid reabsorption from distal airspaces of lung in anesthetized
rats." Am J Physiol 270(2 Pt 1): L232-41.
Pittet, J. F., R. C. Mackersie, T. R. Martin and M. A. Matthay (1997). "Biological markers of
acute lung injury: prognostic and pathogenetic significance." Am J Respir Crit Care Med 155(4):
1187-205.
Pugin, J., I. Dunn, P. Jolliet, D. Tassaux, J. L. Magnenat, L. P. Nicod and J. C. Chevrolet (1998).
"Activation of human macrophages by mechanical ventilation in vitro." Am J Physiol 275(6 Pt
1): L1040-50.
Pugin, J., G. Verghese, M. C. Widmer and M. A. Matthay (1999). "The alveolar space is the site
of intense inflammatory and profibrotic reactions in the early phase of acute respiratory distress
syndrome." Crit Care Med 27(2): 304-12.
Ramirez, M. I., G. Millien, A. Hinds, Y. Cao, D. C. Seldin and M. C. Williams (2003).
"T1alpha, a lung type I cell differentiation gene, is required for normal lung cell proliferation
and alveolus formation at birth." Dev Biol 256(1): 61-72.
Ranieri, V. M., P. M. Suter, C. Tortorella, R. De Tullio, J. M. Dayer, A. Brienza, F. Bruno and
A. S. Slutsky (1999). "Effect of mechanical ventilation on inflammatory mediators in patients
with acute respiratory distress syndrome: a randomized controlled trial." Jama 282(1): 54-61.
Rannels, D. E. and S. R. Rannels (1989). "Influence of the extracellular matrix on type 2 cell
differentiation." Chest 96(1): 165-73.
Reddy, R., S. Buckley, M. Doerken, L. Barsky, K. Weinberg, K. D. Anderson, D. Warburton
and B. Driscoll (2004). "Isolation of a putative progenitor subpopulation of alveolar epithelial
type 2 cells." Am J Physiol Lung Cell Mol Physiol 286(4): L658-67.
Reynolds, S. D., A. Giangreco, K. U. Hong, K. E. McGrath, L. A. Ortiz and B. R. Stripp (2004).
"Airway injury in lung disease pathophysiology: selective depletion of airway stem and

195
progenitor cell pools potentiates lung inflammation and alveolar dysfunction." Am J Physiol
Lung Cell Mol Physiol 287(6): L1256-65.
Riley, D. J., D. E. Rannels, R. B. Low, L. Jensen and T. P. Jacobs (1990). "NHLBI Workshop
Summary. Effect of physical forces on lung structure, function, and metabolism." Am Rev
Respir Dis 142(4): 910-4.
Rouby, J. J., L. Puybasset, A. Nieszkowska and Q. Lu (2003). "Acute respiratory distress
syndrome: lessons from computed tomography of the whole lung." Crit Care Med 31(4 Suppl):
S285-95.
Rubenfeld, G. D., E. Caldwell, E. Peabody, J. Weaver, D. P. Martin, M. Neff, E. J. Stern and L.
D. Hudson (2005). "Incidence and outcomes of acute lung injury." N Engl J Med 353(16): 1685-
93.
Rubins, J. B., P. G. Duane, D. Clawson, D. Charboneau, J. Young and D. E. Niewoehner (1993).
"Toxicity of pneumolysin to pulmonary alveolar epithelial cells." Infect Immun 61(4): 1352-8.
Rush, B. F., Jr., A. J. Sori, T. F. Murphy, S. Smith, J. J. Flanagan, Jr. and G. W. Machiedo
(1988). "Endotoxemia and bacteremia during hemorrhagic shock. The link between trauma and
sepsis?" Ann Surg 207(5): 549-54.
Sadikot, R. T., W. Han, M. B. Everhart, O. Zoia, R. S. Peebles, E. D. Jansen, F. E. Yull, J. W.
Christman and T. S. Blackwell (2003). "Selective I kappa B kinase expression in airway
epithelium generates neutrophilic lung inflammation." J Immunol 170(2): 1091-8.
Saitou, M., Y. Ando-Akatsuka, M. Itoh, M. Furuse, J. Inazawa, K. Fujimoto and S. Tsukita
(1997). "Mammalian occludin in epithelial cells: its expression and subcellular distribution." Eur
J Cell Biol 73(3): 222-31.
Sannes, P. L. (1984). "Differences in basement membrane-associated microdomains of type I
and type II pneumocytes in the rat and rabbit lung." J Histochem Cytochem 32(8): 827-33.
Schacht, V., M. I. Ramirez, Y. K. Hong, S. Hirakawa, D. Feng, N. Harvey, M. Williams, A. M.
Dvorak, H. F. Dvorak, G. Oliver and M. Detmar (2003). "T1alpha/podoplanin deficiency
disrupts normal lymphatic vasculature formation and causes lymphedema." Embo J 22(14):
3546-56.
Schittny, J. C., V. Djonov, A. Fine and P. H. Burri (1998). "Programmed cell death contributes
to postnatal lung development." Am J Respir Cell Mol Biol 18(6): 786-93.
Schneeberger, E. E. (1991). "The Lung: Scientific Foundations edited by R. G. Crystal, J. B.
West et al." 229-234.
Schneeberger, E. E. (1991). "The Lung: Scientific Foundations edited by R. G. Crystal, J. B.
West et al." 205-214.
Schneeberger, E. E. and R. D. Lynch (1992). "Structure, function, and regulation of cellular tight
junctions." Am J Physiol 262(6 Pt 1): L647-61.

196
Schneeberger, E. E. and K. M. McCarthy (1986). "Cytochemical localization of Na+-K+-
ATPase in rat type II pneumocytes." J Appl Physiol 60(5): 1584-9.
Scholl, F. G., C. Gamallo, S. Vilar inverted question marko and M. Quintanilla (1999).
"Identification of PA2.26 antigen as a novel cell-surface mucin-type glycoprotein that induces
plasma membrane extensions and increased motility in keratinocytes." J Cell Sci 112 ( Pt 24):
4601-13.
Seeger, W., R. G. Birkemeyer, L. Ermert, N. Suttorp, S. Bhakdi and H. R. Duncker (1990).
"Staphylococcal alpha-toxin-induced vascular leakage in isolated perfused rabbit lungs." Lab
Invest 63(3): 341-9.
Selman, M. and A. Pardo (2002). "Idiopathic pulmonary fibrosis: an epithelial/fibroblastic cross-
talk disorder." Respir Res 3: 3.
Selman, M. and A. Pardo (2006). "Role of epithelial cells in idiopathic pulmonary fibrosis: from
innocent targets to serial killers." Proc Am Thorac Soc 3(4): 364-72.
Selman, M., V. J. Thannickal, A. Pardo, D. A. Zisman, F. J. Martinez and J. P. Lynch, 3rd
(2004). "Idiopathic pulmonary fibrosis: pathogenesis and therapeutic approaches." Drugs 64(4):
405-30.
Shenkar, R. and E. Abraham (1993). "Effects of hemorrhage on cytokine gene transcription."
Lymphokine Cytokine Res 12(4): 237-47.
Shenkar, R., M. D. Schwartz, L. S. Terada, J. E. Repine, J. McCord and E. Abraham (1996).
"Hemorrhage activates NF-kappa B in murine lung mononuclear cells in vivo." Am J Physiol
270(5 Pt 1): L729-35.
Simionescu, M. (1997). "The Lung: Scientific Foundations Edited by R.G Crystal, J.B West et
al." (Second Edition): 615-628.
Sirianni, F. E., F. S. Chu and D. C. Walker (2003). "Human alveolar wall fibroblasts directly
link epithelial type 2 cells to capillary endothelium." Am J Respir Crit Care Med 168(12): 1532-
7.
Slutsky, A. S. (1999). "Lung injury caused by mechanical ventilation." Chest 116(1 Suppl): 9S-
15S.
Slutsky, A. S. and L. N. Tremblay (1998). "Multiple system organ failure. Is mechanical
ventilation a contributing factor?" Am J Respir Crit Care Med 157(6 Pt 1): 1721-5.
Song, Y., N. Fukuda, C. Bai, T. Ma, M. A. Matthay and A. S. Verkman (2000). "Role of
aquaporins in alveolar fluid clearance in neonatal and adult lung, and in oedema formation
following acute lung injury: studies in transgenic aquaporin null mice." J Physiol 525 Pt 3: 771-
9.
Sori, A. J., B. F. Rush, Jr., T. W. Lysz, S. Smith and G. W. Machiedo (1988). "The gut as source
of sepsis after hemorrhagic shock." Am J Surg 155(2): 187-92.

197
Spellerberg, B., C. Rosenow, W. Sha and E. I. Tuomanen (1996). "Pneumococcal cell wall
activates NF-kappa B in human monocytes: aspects distinct from endotoxin." Microb Pathog
20(5): 309-17.
Springer, T. A. (1995). "Traffic signals on endothelium for lymphocyte recirculation and
leukocyte emigration." Annu Rev Physiol 57: 827-72.
Stone, K. C., R. R. Mercer, P. Gehr, B. Stockstill and J. D. Crapo (1992). "Allometric
relationships of cell numbers and size in the mammalian lung." Am J Respir Cell Mol Biol 6(2):
235-43.
Sue, R. D., J. A. Belperio, M. D. Burdick, L. A. Murray, Y. Y. Xue, M. C. Dy, J. J. Kwon, M. P.
Keane and R. M. Strieter (2004). "CXCR2 is critical to hyperoxia-induced lung injury." J
Immunol 172(6): 3860-8.
Sugahara, K., T. Kiyota, R. A. Clark and R. J. Mason (1993). "The effect of fibronectin on
cytoskeleton structure and transepithelial resistance of alveolar type II cells in primary culture."
Virchows Arch B Cell Pathol Incl Mol Pathol 64(2): 115-22.
Suratt, B. T., C. D. Cool, A. E. Serls, L. Chen, M. Varella-Garcia, E. J. Shpall, K. K. Brown and
G. S. Worthen (2003). "Human pulmonary chimerism after hematopoietic stem cell
transplantation." Am J Respir Crit Care Med 168(3): 318-22.
Suzuki, Y. J., H. J. Forman and A. Sevanian (1997). "Oxidants as stimulators of signal
transduction." Free Radic Biol Med 22(1-2): 269-85.
Takahashi, T., M. Munakata, I. Suzuki and Y. Kawakami (1998). "Serum and bronchoalveolar
fluid KL-6 levels in patients with pulmonary alveolar proteinosis." Am J Respir Crit Care Med
158(4): 1294-8.
Tani, T., M. Fujino, K. Hanasawa, T. Shimizu, Y. Endo and M. Kodama (2000). "Bacterial
translocation and tumor necrosis factor-alpha gene expression in experimental hemorrhagic
shock." Crit Care Med 28(11): 3705-9.
Taylor, A. E. and K. A. Gaar, Jr. (1970). "Estimation of equivalent pore radii of pulmonary
capillary and alveolar membranes." Am J Physiol 218(4): 1133-40.
Thannickal, V. J. and B. L. Fanburg (2000). "Reactive oxygen species in cell signaling." Am J
Physiol Lung Cell Mol Physiol 279(6): L1005-28.
Thet, L. A., S. C. Parra and J. D. Shelburne (1986). "Sequential changes in lung morphology
during the repair of acute oxygen-induced lung injury in adult rats." Exp Lung Res 11(3): 209-
28.
Todd, T. R., E. Baile and J. C. Hogg (1978). "Pulmonary capillary and permeability during
hemorrhagic shock." J Appl Physiol 45(2): 298-306.

198
Tomashefski, J. F. J. (2003). Pulmonary Pathology of the Acute Respiratory Distress Syndrome
- Diffuse Alveolar Damage. Acute Respiratory Distress Syndrome. M. A. Matthay. New York,
Marcel Dekker. 179: 75-114.
Torikata, C., B. Villiger, C. Kuhn, 3rd and J. A. McDonald (1985). "Ultrastructural distribution
of fibronectin in normal and fibrotic human lung." Lab Invest 52(4): 399-408.
Tosi, M. F., J. M. Stark, C. W. Smith, A. Hamedani, D. C. Gruenert and M. D. Infeld (1992).
"Induction of ICAM-1 expression on human airway epithelial cells by inflammatory cytokines:
effects on neutrophil-epithelial cell adhesion." Am J Respir Cell Mol Biol 7(2): 214-21.
Towne, J. E., K. S. Harrod, C. M. Krane and A. G. Menon (2000). "Decreased expression of
aquaporin (AQP)1 and AQP5 in mouse lung after acute viral infection." Am J Respir Cell Mol
Biol 22(1): 34-44.
Tremblay, L., F. Valenza, S. P. Ribeiro, J. Li and A. S. Slutsky (1997). "Injurious ventilatory
strategies increase cytokines and c-fos m-RNA expression in an isolated rat lung model." J Clin
Invest 99(5): 944-52.
Tremblay, L. N., D. Miatto, Q. Hamid, A. Govindarajan and A. S. Slutsky (2002). "Injurious
ventilation induces widespread pulmonary epithelial expression of tumor necrosis factor-alpha
and interleukin-6 messenger RNA." Crit Care Med 30(8): 1693-700.
Tremblay, L. N. and A. S. Slutsky (1998). "Ventilator-induced injury: from barotrauma to
biotrauma." Proc Assoc Am Physicians 110(6): 482-8.
Tschumperlin, D. J. and S. S. Margulies (1998). "Equibiaxial deformation-induced injury of
alveolar epithelial cells in vitro." Am J Physiol 275(6 Pt 1): L1173-83.
Tschumperlin, D. J. and S. S. Margulies (1999). "Alveolar epithelial surface area-volume
relationship in isolated rat lungs." J Appl Physiol 86(6): 2026-33.
Tschumperlin, D. J., J. Oswari and A. S. Margulies (2000). "Deformation-induced injury of
alveolar epithelial cells. Effect of frequency, duration, and amplitude." Am J Respir Crit Care
Med 162(2 Pt 1): 357-62.
Turrens, J. F. (2003). "Mitochondrial formation of reactive oxygen species." J Physiol 552(Pt 2):
335-44.
Tutor, J. D., C. M. Mason, E. Dobard, R. C. Beckerman, W. R. Summer and S. Nelson (1994).
"Loss of compartmentalization of alveolar tumor necrosis factor after lung injury." Am J Respir
Crit Care Med 149(5): 1107-11.
Uhal, B. D. (1997). "Cell cycle kinetics in the alveolar epithelium." Am J Physiol 272(6 Pt 1):
L1031-45.
Uhlig, S. (2002). "Ventilation-induced lung injury and mechanotransduction: stretching it too
far?" Am J Physiol Lung Cell Mol Physiol 282(5): L892-6.

199
Ulich, T. R., E. S. Yi, K. Longmuir, S. Yin, R. Biltz, C. F. Morris, R. M. Housley and G. F.
Pierce (1994). "Keratinocyte growth factor is a growth factor for type II pneumocytes in vivo." J
Clin Invest 93(3): 1298-306.
Vaccaro, C. A. and J. S. Brody (1981). "Structural features of alveolar wall basement membrane
in the adult rat lung." J Cell Biol 91(2 Pt 1): 427-37.
Veldhuizen, R. A., A. S. Slutsky, M. Joseph and L. McCaig (2001). "Effects of mechanical
ventilation of isolated mouse lungs on surfactant and inflammatory cytokines." Eur Respir J
17(3): 488-94.
Verkman, A. S., M. A. Matthay and Y. Song (2000). "Aquaporin water channels and lung
physiology." Am J Physiol Lung Cell Mol Physiol 278(5): L867-79.
Vlahakis, N. E. and R. D. Hubmayr (2000). "Invited review: plasma membrane stress failure in
alveolar epithelial cells." J Appl Physiol 89(6): 2490-6;discussion 2497.
Vlahakis, N. E., M. A. Schroeder, A. H. Limper and R. D. Hubmayr (1999). "Stretch induces
cytokine release by alveolar epithelial cells in vitro." Am J Physiol 277(1 Pt 1): L167-73.
von Bethmann, A. N., F. Brasch, R. Nusing, K. Vogt, H. D. Volk, K. M. Muller, A. Wendel and
S. Uhlig (1998). "Hyperventilation induces release of cytokines from perfused mouse lung." Am
J Respir Crit Care Med 157(1): 263-72.
Vuichard, D., M. T. Ganter, R. C. Schimmer, D. Suter, C. Booy, L. Reyes, T. Pasch and B.
Beck-Schimmer (2005). "Hypoxia aggravates lipopolysaccharide-induced lung injury." Clin Exp
Immunol 141(2): 248-60.
Wang, F., B. Daugherty, L. L. Keise, Z. Wei, J. P. Foley, R. C. Savani and M. Koval (2003).
"Heterogeneity of claudin expression by alveolar epithelial cells." Am J Respir Cell Mol Biol
29(1): 62-70.
Wang, R., O. Ibarra-Sunga, L. Verlinski, R. Pick and B. D. Uhal (2000). "Abrogation of
bleomycin-induced epithelial apoptosis and lung fibrosis by captopril or by a caspase inhibitor."
Am J Physiol Lung Cell Mol Physiol 279(1): L143-51.
Wang, X., S. W. Ryter, C. Dai, Z. L. Tang, S. C. Watkins, X. M. Yin, R. Song and A. M. Choi
(2003). "Necrotic cell death in response to oxidant stress involves the activation of the
apoptogenic caspase-8/bid pathway." J Biol Chem 278(31): 29184-91.
Ward, N. S., A. B. Waxman, R. J. Homer, L. L. Mantell, O. Einarsson, Y. Du and J. A. Elias
(2000). "Interleukin-6-induced protection in hyperoxic acute lung injury." Am J Respir Cell Mol
Biol 22(5): 535-42.
Ware, L. B. and M. A. Matthay (2000). "The acute respiratory distress syndrome." N Engl J Med
342(18): 1334-49.

200
Ware, L. B. and M. A. Matthay (2001). "Alveolar fluid clearance is impaired in the majority of
patients with acute lung injury and the acute respiratory distress syndrome." Am J Respir Crit
Care Med 163(6): 1376-83.
Waters, C. M., P. H. Sporn, M. Liu and J. J. Fredberg (2002). "Cellular biomechanics in the
lung." Am J Physiol Lung Cell Mol Physiol 283(3): L503-9.
Weatherall, D., E. J. Benz, D. A. Warrell, T. M. Cox and F. J. D (2003). "Oxford Textbook of
Medicine."
Webb, H. H. and D. F. Tierney (1974). "Experimental pulmonary edema due to intermittent
positive pressure ventilation with high inflation pressures. Protection by positive end-expiratory
pressure." Am Rev Respir Dis 110(5): 556-65.
Webster, N. R., A. T. Cohen and J. F. Nunn (1988). "Adult respiratory distress syndrome--how
many cases in the UK?" Anaesthesia 43(11): 923-6.
Weibel, E. R. (1984). The pathway for oxygen. Cambridge, Massachusetts and London, Harvard
University Press.
Weibel, E. R. and D. M. Gomez (1962). "Architecture of the human lung. Use of quantitative
methods establishes fundamental relations between size and number of lung structures." Science
137: 577-85.
Weinert, C. R., C. R. Gross, J. R. Kangas, C. L. Bury and W. A. Marinelli (1997). "Health-
related quality of life after acute lung injury." Am J Respir Crit Care Med 156(4 Pt 1): 1120-8.
West, J. B. (2004). "Repiratory Physiology - the essentials."
White, S. R., D. R. Dorscheid, K. F. Rabe, K. R. Wojcik and K. J. Hamann (1999). "Role of very
late adhesion integrins in mediating repair of human airway epithelial cell monolayers after
mechanical injury." Am J Respir Cell Mol Biol 20(4): 787-96.
Wiener-Kronish, J. P., K. H. Albertine and M. A. Matthay (1991). "Differential responses of the
endothelial and epithelial barriers of the lung in sheep to Escherichia coli endotoxin." J Clin
Invest 88(3): 864-75.
Williams, M. C. (2003). "Alveolar type I cells: molecular phenotype and development." Annu
Rev Physiol 65: 669-95.
Williams, M. C., Y. Cao, A. Hinds, A. K. Rishi and A. Wetterwald (1996). "T1 alpha protein is
developmentally regulated and expressed by alveolar type I cells, choroid plexus, and ciliary
epithelia of adult rats." Am J Respir Cell Mol Biol 14(6): 577-85.
Williams, M. C. and L. G. Dobbs (1990). "Expression of cell-specific markers for alveolar
epithelium in fetal rat lung." Am J Respir Cell Mol Biol 2(6): 533-42.
Winkelstein, J. A. and A. Tomasz (1978). "Activation of the alternative complement pathway by
pneumococcal cell wall teichoic acid." J Immunol 120(1): 174-8.

201
Wirtz, H. R. and L. G. Dobbs (1990). "Calcium mobilization and exocytosis after one
mechanical stretch of lung epithelial cells." Science 250(4985): 1266-9.
Wirtz, H. R. and L. G. Dobbs (2000). "The effects of mechanical forces on lung functions."
Respir Physiol 119(1): 1-17.
Woodhead, M. A., J. T. Macfarlane, J. S. McCracken, D. H. Rose and R. G. Finch (1987).
"Prospective study of the aetiology and outcome of pneumonia in the community." Lancet
1(8534): 671-4.
Woodhead, M. A., J. T. Macfarlane, F. G. Rodgers, A. Laverick, R. Pilkington and A. D. Macrae
(1985). "Aetiology and outcome of severe community-acquired pneumonia." J Infect 10(3): 204-
10.
Wyncoll, D. L. and T. W. Evans (1999). "Acute respiratory distress syndrome." Lancet
354(9177): 497-501.
Yamada, M., H. Kubo, S. Kobayashi, K. Ishizawa, M. Numasaki, S. Ueda, T. Suzuki and H.
Sasaki (2004). "Bone marrow-derived progenitor cells are important for lung repair after
lipopolysaccharide-induced lung injury." J Immunol 172(2): 1266-72.
Yee, M., P. F. Vitiello, J. M. Roper, R. J. Staversky, T. W. Wright, S. A. McGrath-Morrow, W.
M. Maniscalco, J. N. Finkelstein and M. A. O'Reilly (2006). "Type II epithelial cells are critical
target for hyperoxia-mediated impairment of postnatal lung development." Am J Physiol Lung
Cell Mol Physiol 291(5): L1101-11.
Zhang, H., G. P. Downey, P. M. Suter, A. S. Slutsky and V. M. Ranieri (2002). "Conventional
mechanical ventilation is associated with bronchoalveolar lavage-induced activation of
polymorphonuclear leukocytes: a possible mechanism to explain the systemic consequences of
ventilator-induced lung injury in patients with ARDS." Anesthesiology 97(6): 1426-33.
Zhang, Y., R. M. Porat, T. Alon, E. Keshet and J. Stone (1999). "Tissue oxygen levels control
astrocyte movement and differentiation in developing retina." Brain Res Dev Brain Res 118(1-
2): 135-45.
Zimmerman, G. A., K. H. Albertine, H. J. Carveth, E. A. Gill, C. K. Grissom, J. R. Hoidal, T.
Imaizumi, C. G. Maloney, T. M. McIntyre, J. R. Michael, J. F. Orme, S. M. Prescott and M. S.
Topham (1999). "Endothelial activation in ARDS." Chest 116(1 Suppl): 18S-24S.





























