Embed Size (px)
Citation preview

MD
F
riwsssf
IHoisalplsihhic
TSmtoapdissd
FHcSDu
1
echanisms for the initiation of human atrial fibrillationavid E. Krummen, MD, FACC, Sanjiv M. Narayan, MB, MD, FHRS
rom the University of California and Veterans Affairs Medical Center, San Diego, California.
aid
Kr
(
Understanding of the mechanisms underlying atrial fibrillation (AF)emain limited. However, both persistent and paroxysmal AF likelynitiate via the interaction of triggers such as premature atrial beatsith ‘substrate’, that includes static components such as fibrosis andcar, as well as dynamic alterations that may include the rate-re-ponse of repolarization and conduction, autonomic modulation andtretch. This article attempts to synthesize concepts for AF initiation
CAAet
MIagtomesAP
LAsttsd
tsSi(dntsicsd.edu.
547-5271/$ -see front matter. Published by Elsevier Inc. on behalf of the Heart
pply them to the spectrum of disease processes that comprise clin-cal AF. Further investigation is urgently needed to translate theseiscoveries into effective therapy.
EYWORDS Atrial fibrillation; Action potential duration; Electricalestitution; Repolarization alternans; Human; Remodeling
Heart Rhythm 2009;6:S12–S16) Published by Elsevier Inc. on
rom in silico, in vitro and in vivo animal data and human studies and behalf of the Heart Rhythm Society.ntroductionuman atrial fibrillation (AF) likely encompasses a heter-geneous group of diseases, each resulting from a complexnteraction between triggers and functional and structuralubstrates.1 While great strides have been made in catheterblation for AF in various populations,2 the pathophysio-ogical targets for ablation are understood clearly only inatients with short-lived paroxysms of AF.3 The extensiveesions needed to treat longstanding AF4,5 and the limiteduccess of medications that target plausible and definedonic mechanisms6 reaffirm our limited understanding ofuman AF. Notable studies in the basic science literatureave revealed mechanisms to explain AF in animals,7,8 anddentifying those that contribute to human AF is an urgentlinical challenge.
rigger versus substrateince the seminal observation that pulmonary veins (PVs)ay act as sources of triggering ectopy for AF,3 PV isola-
ion has become a cornerstone of invasive therapy for par-xysmal as well as persistent AF.2 Nevertheless, the mech-nistic role of the PVs is not defined, particularly forersistent AF. The fact that the majority of potential triggerso not initiate AF2 suggests some role for an AF substraten most patients. However, it is unclear whether AF sub-trates are fixed and related to anatomic complexity9 orcar10 or functional and related to repolarization and con-uction dynamics and other dynamic factors.1,7,11
Supported by grants from the American College of Cardiology–Merckoundation (to Dr. Krummen) and the National Institutes of Health (nos.L70529, HL83359), Doris Duke Foundation, and American Heart Asso-
iation (to Dr. Narayan). Address reprint requests and correspondence:anjiv M. Narayan, M.D., Cardiology Section 111A, 3350 La Jolla Villagerive, San Diego, California 92161. E-mail address: snarayan@
urrent mechanistic hypotheses for human AFt the present time, two hypotheses predominate to explainF: the multiwavelet reentry and localized source hypoth-
ses. Evidence exists to support both, and they may poten-ially coexist in certain types of AF.
ultiwavelet reentryn this mechanism, leading circle reentry produces multipletrial circuits that may each meander, interact, and extin-uish. Evidence for multiwavelet reentry comes from mul-ielectrode mapping in dogs,11 from intraoperative mappingf AF in humans,12 and from the success of atrial compart-entalization via the Cox-Maze surgical procedure.13 Nev-
rtheless, clinical evidence remains indirect,8 since earlyurgical mapping was performed in patients without clinicalF and the Cox-Maze III-IV procedure also isolates theVs and denervates the atria.
ocalized sourcelternatively, AF may be maintained by rapid localized
ources that activate too rapidly for the surrounding atrium,hus leading to AF via fibrillatory conduction. Experimen-ally, stable and regular microreentrant sources have beenhown to sustain AF in isolated sheep heart7 and in vivo inogs.8
In humans, evidence for localized sources comes fromhe observation of intra-atrial gradients in AF rate. Theeminal intraoperative mapping studies of Wu et al14 andahadevan et al (summarized in reference 8) showed local-
zed sites of rapid, regular activation in AF surrounded bymore typical) irregular activation. Percutaneous endocar-ial mapping studies confirmed rate gradients,15 althoughone of these studies demonstrated a functional role forhese sites. Indeed, a recent study showed that ablation atuch sites (identified by high spectral dominant frequency as
n animal AF rotors7) did not terminate AF.16Rhythm Society. doi:10.1016/j.hrthm.2009.03.004
ootsrdi
DAAEpTswpsa
AsgvmrAics
AnTtos
broMpcplaharr
psih
cptosepdadbt
TSefadmfa
FhbsSAaA
S13Krummen and Narayan Mechanisms for Human AF
At this time, it is not clear whether the clinical limitationsf spectral dominant frequency reflect technical factors17,18
r confounding issues such as atrial movement and interac-ions between multiple mechanisms. It is also possible thatpatial gradients in AF rate reflect nonuniform multiwaveleteentry, from differences in atrial refractoriness and con-uction, rather than localized sources. Further human stud-es are clearly needed to address these issues.
o clinical scenarios that promote AF explainF substrates?F begets AFlectrical remodeling refers to alterations in atrial electro-hysiology as a result of AF that actually promote AF.11
he primary and consistent effect of remodeling is tohorten action potential duration (APD), which shortens theavelength for reentry and may exaggerate the spatial dis-ersion of repolarization and thus promote AF. Conductionlowing is part of electrical remodeling in some but not allnimal models, and its role in humans is uncertain.11
It is uncertain whether electrical remodeling is akin to anF substrate. If remodeling equated with AF substrate, AF
hould invariably progress over time. However, recent lon-itudinal studies show that AF not only progresses at aariable pace between individuals but may regress inany.19 Moreover, although effects of remodeling on APD
everse over time, the vulnerability to AF may not.20,21
ccordingly, the strategy of prompt termination of AF ep-sodes has had limited clinical success.22 Further studies arelearly needed to better define electrical remodeling and AFubstrates and to define their interaction.
F initiation may be dependent on the autonomicervous systemhe observation of AF episodes at times of high sympa-
hetic or parasympathetic balance, such as during exerciser after a meal, suggests a role of the autonomic nervousystem in clinical AF.
Vagal stimulation has long been shown to facilitate AF,y shortening APD and increasing the dispersion of atrialepolarization through mechanisms including abnormalitiesf muscarinic receptor-mediated activation of IK,Ach.23
oreover, initiation of AF in the canine model is facilitatedrogressively by the infusion of adrenergic, cholinergic, andombined adrenergic-cholinergic agents.24 Combined sym-athovagal stimulation in dogs leads to intracellular calciumoading that, in the PVs, causes APD shortening, earlyfter-depolarizations (EADs), triggered beats, and AF.25 Itas been shown that patients with AF also exhibit similarbnormalities in cellular calcium handling,26 which mayeflect functional abnormalities (phosphorylation of keyegulatory proteins27).
These studies clearly make sympathovagal stimulation alausible mechanism for AF. Indeed, a recent canine studyuggested that peri-PV ganglionic plexi may explain themportance of the PVs to AF maintenance.28 Clinically,
owever, the evidence for an autonomic nervous system aontribution to AF remains controversial. It is clinicallylausible that the initiation of paroxysmal AF may be au-onomically driven. Ablation to denervate peri-PV gangli-nic plexi2 may eliminate AF, and a recent human studyhowed that vagomimetic intervention may create atriallectrogram fragmentation,29 considered by many to be aromising target for AF ablation.2 However, ablation toenervate the PVs also isolates them, and vagal responsesre often subsequently absent even when AF recurs.30 Ad-itional human studies are thus needed to assess the contri-ution of the autonomics to atrial repolarization, conduc-ion, and initiation of clinical AF.
he impact of atrial stretch on AF initiationtretch is an attractive mechanism for AF. Clinically, AFpisodes are more frequent at times of worsened heartailure,2 while elevated left atrial pressure may explain thessociation between lone AF and left ventricular diastolicysfunction31 and contribute to the link between AF anditral valve disease.2 Stretch likely reduces APD and re-
ractoriness (to facilitate AF) via stretch-activated channels,nd, in rabbit hearts, this may be inhibited by gadolinium
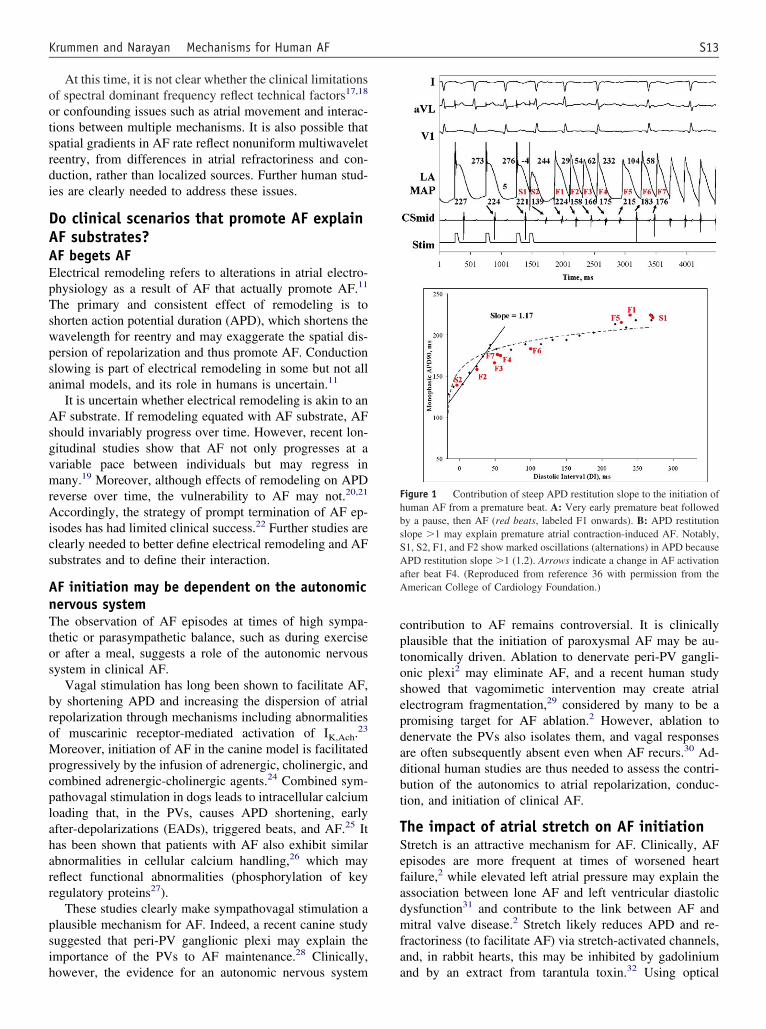
igure 1 Contribution of steep APD restitution slope to the initiation ofuman AF from a premature beat. A: Very early premature beat followedy a pause, then AF (red beats, labeled F1 onwards). B: APD restitutionlope �1 may explain premature atrial contraction-induced AF. Notably,1, S2, F1, and F2 show marked oscillations (alternations) in APD becausePD restitution slope �1 (1.2). Arrows indicate a change in AF activation
fter beat F4. (Reproduced from reference 36 with permission from themerican College of Cardiology Foundation.)
nd by an extract from tarantula toxin.32 Using optical

iiabnfn
EsEfibfimptaah
tpnd
EcfIpmw
em
SepAbotsira
ePmtasaAAs
DpCtaA
Fh4
S14 Heart Rhythm, Vol 6, No 8S, August Supplement 2009
maging in sheep atria, Vaquero et al7 showed that stretchncreased both the rate of AF rotors emanating from the PVsnd the relative contribution of the PVs over the left atrialody to the perpetuation of AF. Thus, human studies areeeded to confirm this mechanism, particularly in heartailure patients with AF who are likely to experience sig-ificant volume changes.
vidence for structural factors in the AFubstratelegant mathematical and experimental33 studies show thatxed spatial dispersion of refractoriness may facilitate wavereak and initiate reentry. In humans, atrial scarring andbrosis in older patients34 and those with heart failure35
ay explain their propensity for AF, and they also occur inatients with AF.10 At a cellular level, APD and the effec-ive refractory period are shorter in canine left than righttrium owing in part to larger IKr, thus allowing the lefttrium to activate faster than the right and thus potentiallyarbor localized sources of AF.36
However, while fixed factors may clearly facilitate AF,hey cannot explain why AF is episodic or why manyatients with these factors (particularly older individuals) doot experience AF. This makes a logical case for the role ofynamic factors in the initiation of human AF.
vidence for dynamic repolarization andonduction properties as mechanismsor human AFn the ventricle, rate-dependent electrophysiology may ex-lain the transition from rapid rhythms to fibrillation,33 whyalignant arrhythmias occur in some but not all patients
igure 2 Contribution of APD alternans to human AF. The patient shoigh right atrium, HRA) that became exaggerated in amplitude before cond1 with permission from the American Heart Association).
ith structural disease, and why they are sporadic. Recent p
vidence suggests that dynamic atrial electrophysiologyay contribute to human AF.
teep repolarization (APD) restitution mayxplain initiation of paroxysmal AF fromremature beats
central clinical question is why some premature atrialeats initiate AF, while others simply conduct to the atriumr block. APD restitution describes the relationship of APDo the diastolic interval separating a beat from its predeces-or and may provide an explanation. Marked APD shorten-ng at rapid rates and short diastolic intervals (i.e., steepestitution, slope �1) may lead to self-amplifying APDlternans and ultimately to conduction block and reentry.33
We recently reported that patients with paroxysmal AFxhibit APD restitution slope �1 (steep restitution) near theVs.37 Figure 1 depicts a patient with paroxysmal AF andaximum APD restitution slope �1.2. Notably, the prema-
ure beat (paced) had a very short APD and was followed bypause then a beat with a long APD, then a beat with a
horter APD, then AF. Notably, the premature beat inter-cted with APD restitution slope �1 to cause exaggeratedPD oscillations that led to AF, potentially via wave break.ll but one of our patients with paroxysmal AF demon-
trated maximum APD restitution slope �1.37
ynamic conduction slowing in patients withersistent AFonversely, the maximum APD restitution slope was rela-
ively shallow in patients with persistent AF (0.7 � 0.2),nd no patient exhibited slope �1.37 We found that shallowPD restitution may have been caused by the fact that
ical atrial flutter with APD alternans at the cavotricupid isthmus (but notlock (asterisked), period multiplying, and AF (reproduced from reference
wed typuction b
atients with persistent AF exhibited marked conduction

slt
rHtc
RCahet
wmacv�psfaclw
apcwa
SHeorctdTadw
R
1
1
1
1
1
1
1
1
1
1
2
2
2
2
2
2
2
2
2
2
S15Krummen and Narayan Mechanisms for Human AF
lowing for early premature atrial contractions, thus pro-onging their diastolic intervals and truncating the left por-ion of the curve (i.e., short diastolic intervals).37
Both experimental11 and human38,39 studies suggest thategional conduction slowing contributes to AF substrate.owever, the precise mechanism by which this contributes
o AF remains unclear. One possibility, which will be dis-ussed below, is via the initiation of APD alternans.
epolarization alternans and human AFomputer models of the human atria have shown that APDlternans may cause repolarization dispersion and lead touman AF,40 analogous to experimental and computationalvidence linking APD alternans with fibrillation in the ven-ricle.33
APD alternans provides one potential mechanism byhich dynamic slowing may cause AF. Because rapid beatsay encounter varying conduction delay, those beats may
rrive at a particular region of the heart with variable (os-illating) timing, and those beats will then demonstratearying (oscillating) APD even if the restitution slope1.33,41 Indeed, we showed that the right atrial isthmus in
atients with typical atrial flutter, a region of documentedlow conduction, developed APD alternans in precisely thisashion.42 As shown in Figure 2, such APD alternans en-bled spontaneous and pace-induced transitions from typi-al atrial flutter to AF. However, there are currently no datainking APD alternans with AF in the left atrium of patientsith clinical AF.Alternans may reflect abnormal calcium handling,33,43,44
nd this mechanism, rather than simply repolarization dis-ersion, may link it with arrhythmias.43 However, at theurrent time, further studies are needed to determinehether strategies to improve calcium handling45,46 may
ttenuate APD alternans and reduce arrhythmic risk.
ummaryuman AF likely represents a constellation of related dis-
ases with varied mechanisms. Although each may dependn triggers and functional and structural substrates, theelative contribution of each may vary with the clinicalontext. Therefore, additional mechanistic studies in pa-ients with AF are urgently needed to complement elegantescriptions of AF mechanisms revealed experimentally.his translational approach is likely the best strategy toddress the heterogeneous milieu of human AF and toesign novel therapies for the increasing number of patientsith this condition.
eferences1. Nattel S. New ideas about atrial fibrillation 50 years on. Nature 2002;415:219–
226.2. Calkins H, Brugada J, Packer D, et al. HRS/EHRA/ECAS expert consensus
statement on catheter and surgical ablation of atrial fibrillation: recommenda-tions for personnel, policy, procedures and follow-up. A report of the HeartRhythm Society (HRS) Task Force on catheter and surgical ablation of atrialfibrillation.European Heart Rhythm Association (EHRA); European CardiacArrhythmia Scoiety (ECAS); American College of Cardiology (ACC); Ameri-can Heart Association (AHA); Society of Thoracic Surgeons (STS). Heart
Rhythm 2007;4:816–61.3. Haissaguerre M, Jais P, Shah DC, et al. Spontaneous initiation of atrial fibril-lation by ectopic beats originating in the pulmonary veins. N Engl J Med1998;339:659–666.
4. Haissaguerre M, Hocini M, Sanders P, et al. Catheter ablation of long-lastingPersistent atrial fibrillation: clinical outcome and mechanisms of subsequentarrhythmias. J Cardiovasc Electrophysiol 2005;16:1138–1147.
5. Oral H, Chugh A, Good E, et al. Radiofrequency catheter ablation of chronicatrial fibrillation guided by complex electrograms. Circulation 2007;115:2606–2612.
6. Singh BN, Singh SN, Reda DJ, et al. Amiodarone versus sotalol for atrialfibrillation. N Engl J Med 2005;352:1861–1872.
7. Vaquero M, Calvo D, Jalife J. Cardiac fibrillation: from ion channels to rotorsin the human heart. Heart Rhythm. Jun 2008;5(6):872–879.
8. Waldo AL, Feld GK. Inter-relationships of atrial fibrillation and atrial fluttermechanisms and clinical implications. J Am Coll Cardiol 2008;51:779–786.
9. Ho SY, Sanchez-Quintana D. The importance of atrial structure and fibers. ClinAnat. Jan 2009;22(1):52–63.
0. Chang SL, Tai CT, Lin YJ, et al. Biatrial substrate properties in patients withatrial fibrillation. J Cardiovasc Electrophysiol 2007;18:1134–1139.
1. Allessie MA, Ausma J, Schotten U. Electrical, contractile and structural remod-eling during atrial fibrillation. Cardiovasc Res 2002;54:230–246.
2. Konings K, Kirchhof C, Smeets J, Wellens H, Penn O, Allessie M. High-densitymapping of electrically induced atrial fibrillation in humans. Circulation 1994;89:1665–1680.
3. Cox JL. The central controversy surrounding the interventional-surgical treat-ment of atrial fibrillation. J Thorac Cardiovasc Surg 2005;129:1–4.
4. Wu T-J, Doshi RN, Huang H-LA, et al. Simultaneous biatrial computerizedmapping during permanent atrial fibrillation in patients with organic heartdisease. J Cardiovasc Electrophysiol 2002;13:571–577.
5. Lazar S, Dixit S, Marchlinski FE, et al. Presence of left-to-right atrial frequencygradient in paroxysmal but not persistent atrial fibrillation in humans. Circula-tion 2004;110:3181–3186.
6. Sanders P, Berenfeld O, Hocini M, et al. Spectral analysis identifies sites ofhigh-frequency activity maintaining atrial fibrillation in humans. Circulation2005;112:789–797.
7. Narayan SM, Krummen DE, Kahn AM, et al. Evaluating fluctuations in humanatrial fibrillatory cycle length using monophasic action potentials. Pacing ClinElectrophysiol 2006;29:1209–1218.
8. Ng J, Kadish AH, Goldberger JJ. Technical considerations for dominant fre-quency analysis. J Cardiovasc Electrophysiol 2007;18:757–764.
9. Jahangir A, Lee V, Friedman PA, et al. Long-term progression and outcomeswith aging in patients with lone atrial fibrillation: a 30-year follow-up study.Circulation 2007;115:3050–3056.
0. Everett TH, IV, Li H, Mangrum JM, et al. Electrical, morphological, andultrastructural remodeling and reverse remodeling in a canine model of chronicatrial fibrillation. Circulation 2000:1454–1460.
1. Akar JG, Everett THI, Kok LC, et al. Effect of electrical and structural remod-eling on spatiotemporal organization in acute and persistent atrial fibrillation. J.Cardiovasc Electrophysiol 2002;13:1027–1034.
2. Fynn S, Todd D, Hobbs W, et al. Clinical evaluation of a policy of early repeatedinternal cardioversion for recurrence of atrial fibrillation. J Cardiovasc Electro-physiol 2002;13:135–141.
3. Dobrev D, Graf E, Wettwer E, et al. Molecular basis of downregulation ofG-protein-coupled inward rectifying K(�) current (I(K,ACh) in chronic humanatrial fibrillation: decrease in GIRK4 mRNA correlates with reduced I(K,ACh)and muscarinic receptor-mediated shortening of action potentials. Circulation2001;104:2551–2557.
4. Sharifov OF, Fedorov VV, Beloshapko GG, et al. Roles of adrenergic andcholinergic stimulation in spontaneous atrial fibrillation in dogs. J Am CollCardiol 2004;43:483–490.
5. Patterson E, Lazzara R, Szabo B, et al. Sodium-calcium exchange initiated bythe Ca2� transient: an arrhythmia trigger within pulmonary veins. J Am CollCardiol 2006;47:1196–1206.
6. Workman AJ, Kane KA, Rankin AC. The contribution of ionic currents tochanges in refractoriness of human atrial myocytes associated with chronic atrialfibrillation. Cardiovasc Res 2001;52:226–235.
7. El-Armouche A, Boknik P, Eschenhagen T, et al. Molecular determinants ofaltered Ca2� handling in human chronic atrial fibrillation. Circulation 2006;114:670–680.
8. Lemola K, Chartier D, Yeh YH, et al. Pulmonary vein region ablation inexperimental vagal atrial fibrillation: role of pulmonary veins versus autonomicganglia. Circulation 2008;117:470–477.
9. Lellouche N, Buch E, Celigoj A, et al. Functional characterization of atrial
electrograms in sinus rhythm delineates sites of parasympathetic innervation in
3
3
3
3
3
3
3
3
3
3
4
4
4
4
4
4
4
S16 Heart Rhythm, Vol 6, No 8S, August Supplement 2009
patients with paroxysmal atrial fibrillation. J Am Coll Cardiol 2007;50:1324–1331.
0. Verma A, Saliba W, Lakkireddy D, et al. Vagal responses induced byendocardial left atrial autonomic ganglion stimulation before and after pul-monary vein antrum isolation for atrial fibrillation. Heart Rhythm 2007;4:1177–1182.
1. Jais P, Peng JT, Shah DC, et al. Left ventricular diastolic dysfunction in patients withso-called lone atrial fibrillation. J Cardiovasc Electrophysiol 2000;11:623–625.
2. Bode F, Sachs F, Franz MR. Tarantula peptide inhibits atrial fibrillation. Nature2001;409:35–36.
3. Weiss JN, Karma A, Shiferaw Y, et al. From pulsus to pulseless: the saga ofcardiac alternans (review). Circ Res 2006;98:1244–1253.
4. Sanders P, Morton JB, Kistler PM, et al. Electrophysiological and electroana-tomic characterization of the atria in sinus node disease: evidence of diffuseatrial remodeling. Circulation 2004;109:1514–1522.
5. Li D, Fareh S, Leung TK, et al. Promotion of atrial fibrillation by heartfailure in dogs: atrial remodelling of a different sort. Circulation 1999;100:87–95.
6. Li D, Zhang L, Kneller J, et al. Potential ionic mechanism for repolarizationdifferences between canine right and left atrium. Circ Res 2001;88:1168–1175.
7. Narayan SM, Kazi D, Krummen DE, et al. Repolarization and activation resti-tution near human pulmonary veins and atrial fibrillation initiation: a mechanism
for the initiation of atrial fibrillation by premature beats. J Am Coll Cardiol2008;52:1222–1230.8. Markides V, Schilling RJ, Yen Ho S, et al. Characterization of left atrialactivation in the intact human heart. Circulation 2003;107:733–739.
9. Roberts-Thomson KC, Stevenson IH, Kistler PM, et al. Anatomically deter-mined functional conduction delay in the posterior left atrium relationship tostructural heart disease. J Am Coll Cardiol 2008;51:856–862.
0. Gong Y, Xie F, Stein K, et al. Mechanism underlying initiation of paroxysmalatrial flutter/atrial fibrillation by ectopic foci: a simulation study. Circulation2007;115:2094–2102.
1. Franz MR. The electrical restitution curve revisited: steep or flat slope—whichis better? J Cardiovasc Electrophysiol 2003;14:S140–147.
2. Narayan SM, Bode F, Karasik PL, et al. Alternans of atrial action potentials asa precursor of atrial fibrillation. Circulation 2002;106:1968–1973.
3. Laurita KR, Rosenbaum DS. Mechanisms and potential therapeutic targets forventricular arrhythmias associated with impaired cardiac calcium cycling. J MolCell Cardiol 2008;44:31–43.
4. Narayan SM, Bayer J, Lalani G, et al. Action potential dynamics explain arrhythmicvulnerability in human heart failure: a clinical and modeling study implicatingabnormal calcium handling. J Am Coll Cardiol 2008;52:1782–1792.
5. Ohashi N, Mitamura H, Tanimoto K, et al. A comparison between calcium channelblocking drugs with different potencies for T- and L-type channels in preventingatrial electrical remodeling. J Cardiovasc Pharmacol 2004;44:386–392.
6. Mahajan A, Sato D, Shiferaw Y, et al. Modifying L-type calcium current
kinetics: consequences for cardiac excitation and arrhythmia dynamics. BiophysJ 2008;94:411–423.




















