Embed Size (px)
Citation preview

Medical genetics:
Identification of hidden structural variants
with long-read sequencing
Alexander HoischenAssistant Professor Immuno-Genomics
Scientific Director Radboud Genomics Technology Center
Departments of Human Genetics and Internal MedicineRadboud University Medical Center,
Nijmegen, The Netherlands
Contact: [email protected]/en/immunogenomics
@ahoischen
Engineer/PhD-student/Postdocjobs in bioinformatics available!

Full Disclosure
This project is a collaborationbetween RUMC and PacBio Inc. in which reagents costs were shared.

Finding the answer in the genome
6 billion nucleotides46 chromosomes
2 people differ at >4 million positions
1 variant (mutation) can result in disease*
Genome sequencing: All variants in one experiment!
* With all variant types known to cause disease: karyotype aberrations, SVs, indels, SNVs

De novo mutations in ID• Intellectual disability (ID = IQ <70) is a model for severe, sporadic disorders
• Similar to autism spectrum disorder (ASD), epilepsy and other (neuro-) developmental disorders (NDDs)
• >60% of severe ID is caused by de novo mutations
• De novo mutation rate for SNVs is ca. 1.8x10-8, i.e. 30-100 de novo SNVs per
genome per generation (i.e. 1-2 per exome); • De novo mutation rate for large CNVs ca. 0.2 per generation• De novo mutation rate for SVs/large indels – largely unknown!
Nat Rev Genet. 2012 Jul 18;13(8):565-75
De novo mutations reduce genome complexity/wealth of variation greatlySevere, sporadic diseases offer opportunity to identify novel paradigms

Gilissen et al. Nature 2014
De Ligt et al. NEJM 2012
De Vries et al. AJHG 2006& Vulto-van Silfhoutet al. Hum Mut 2013
New Genomic Technologies elucidate Intellectual Disability

Intellectual disability
42%3
11.6%1
27%2
Genomic microarray
Exome sequencing
2014Whole genome
sequencing
Single gene test~1-5%
% of ID patients with a
diagnosis
62%
±1,500 ID patients
Nodiagnosis
De novoSNVs
De novoSVsInherited
1Vulto-van Silfhout et al. Hum Mutat. 2013; 2De Ligt et al. NEJM. 20123Gilissen et al. Nature 2014
Majority is de novo!
2012
2008 38%

...hidden SVs – i.e. long reads?

Hidden de novo SVs in unsolved ID trios?
Patient cohort:
• A clinically well-characterized patient population with intellectual disability. These samples have been previously analyzed extensively:• CNV-microarrays (Vulto-van Silfhout et al. Hum Mut 2013)
• Whole exome sequencing (de Ligt et al. NEJM 2012)
• Short-read whole genome sequencing (Gilissen et al. Nature 2014)
• NovaSeq 30-40x whole genome sequencing
• All previous analyses failed to detect a causal variant
This study:
• Here we perform long-read SMRT sequencing on the Sequel platform in 5 such patient-parent trios
Hypothesis:
• Hidden, previously undetected, de novo SVs may explain disease

DNA• 5 trios
• Fresh gDNA from whole blood• Final libraries: fragment sizes of 40-70kb
Genomic DNAs Sheared DNAs
Michael Kwint

Our first data
5 first trios: • 1 trio with ~40x sequenced in Menlo Park (PacBio)• 4 trios with ~15x in Nijmegen
• Output:• On average 4.7Gb/SMRT cell*• 11.6kb average read length*
• All trios were also sequenced by short-read WGS (Complete Genomics, 80x & Illumina NovaSeq, 40x)
• BioNano mapping data has been generated for one trio
*Sequel 2.0 chemistry with mixture of 4.0 and 5.0 software.

New developments, latest WGS samples
Express library:
• Lower input, higher yield, less hands-on; amenable to automation
• 25 SMRT cells:• Average read length 19kb• Average output Gb per SMRT cell:
6.8Gb (Max. 9.2Gb)
Michael Kwint

Gigabase output – 234 SMRT cells
Sequel 2.0 chemistry with mixture of 4.0 and 5.0 software.
10X
20X
0
20
40
60
80
100
120
s1 s2 s3 s4 s5 s6 s7 s8 s9 s10 s11 s12 s13 s14 s15Samples
Base
s (
Gb) Trio
Trio1
Trio3
Trio4
Trio5
Trio7
Mapped bases per sample
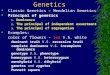
Coverage comparison with NovaSeq short reads
• 28Mb of the human reference genome is only covered reliably
by long reads;
• 12Mb overlaps with genic regions;
• 757kb coding sequence
0
5
10
15
20
25
30
Trio5 S1 Trio5 S2 Trio5 S3
Me
ga b
ase
s
Genomic
Genic
Exonic

SV variant calling
• Solo vs. joint calling using pbsv caller (early access by PacBio) • Joint calling: call the three samples of one trio together
Number of SVs/genome
0
5,000
10,000
15,000
20,000
25,000
30,000
Trio1 S1 Trio1 S2 Trio1 S3
Solo calling
Joint calling
Initial results at ±6x coverage:
[n]

Number of SVs (>50bp)
0
5,000
10,000
15,000
20,000
25,000
30,000
35,000
Trio1 Trio3 Trio4 Trio5 Trio7
Solo
Joint
Trio 1 Trio 2 Trio 3 Trio 4 Trio 5
[n]

Structural variants: 50bp – 1kb
0
5000
10000
15000
20000
25000
Insertions
Deletions
Alu insertions and deletions
[n]
[size bp]

SVs: 1kb – 10kb
0
200
400
600
800
1000
1200
1400
1600
INS
DEL
LINE elements
[n]
[size bp]

SV comparison with 40x NovaSeq
• ~70% novel SVs:Of >25,000 SVs – ca. 17,500 are novel; only ~30% of SVs also called in NovaSeq data using Manta*
• ~80% of insertions are novel, ~55% of deletions are novel (i.e. not called in NovaSeq data)
• There are ~1,000 SVs that are called by NovaSeq data only (~100 insertions; ~900 deletions)
*Overlaps done using Survivor: https://github.com/fritzsedlazeck/SURVIVOR

Variants missed by short read sequencing
PacBio 40x child
PacBio 40x father
PacBio 40x mother
NovaSeq 40x child
116bp heterozygous deletion
Paternally inherited
Not called.Ambigiuous reads?

Variants missed by short read sequencing
PacBio 40x child
PacBio 40x father
PacBio 40x mother
NovaSeq40x child
334bp insertion
Maternallyinherited
Not called.
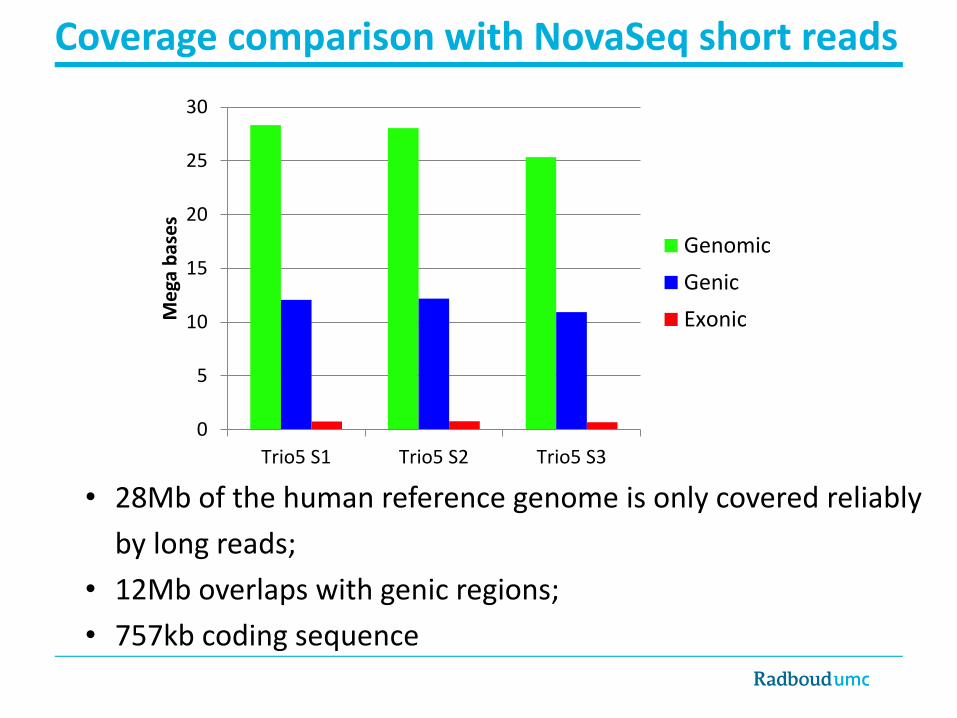
• Comparison with 40X WGS data (NovaSeq, called with Manta**): Using SURVIVOR*
How many SVs are novel?
*https://github.com/fritzsedlazeck/SURVIVOR**https://github.com/Illumina/manta/
15,430
(68.8%)
5,897
(26.3%)
1,092
(4.8%)60% in simple
repeats >100 bp81% in simplerepeats >100 bp
35% in simple
repeats >100 bp
Structural variants ≥50 bp PacBio vs Illumina NovaSeq

SV distribution over samples
0
2000
4000
6000
8000
10000
12000
14000
16000
18000
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15
Co
un
t o
f SV
s
Individuals with respective SV
SVs >50bp
common/reference issues
rare/private

Filter for de novo SVs – value of trios
Manual curation/high qual.: ~5-10 candidate de novo SV per patient
~40 SVs not in
parents
~1000 SVs not in
population
All ~25,000
SVs

De novo calling of SVs: DNM shortlist
Trio1 Trio3 Trio4 Trio5 Trio7
DEL INS INV DEL INS INV DEL INS INV DEL INS INV DEL INS INV0
5
10
15
# d
en
ovo
ca
nd
ida
tes
Lenient
Strict
De novo candidatesTrio 1 Trio 2 Trio 3 Trio 4 Trio 5

Chr16: 65bp deletion intragenic KLHDC4
PacBio 17x child
PacBio 20x father
PacBio 17x mother

De novo candidate SVs• 554bp deletion in intergenic region
PacBio 15x child
PacBio 15x father
PacBio 15x mother
544bp deletion

Validations for de novo candidates are ongoing
Last example: 544bp intergenic deletion
Conclusion:This is a true 544bp deletion
But it is inherited from the healthy father
wt allele (1210bp)
deletion allele 666bp)
patient father mother

Validations so far
• >30 validations; only one FP; all inherited so far
• No validated de novo SV in any patient yet
• In some regions PCR-primer design challenging

Other SVs: Indels? Inversions?
• How many smaller events can we find?• Indels 20-50bp
• How many inversions per genome?
trio
1_f
ath
er
trio
1_m
oth
er
trio
1_p
atie
nt
trio
2_f
ath
er
trio
2_m
oth
er
trio
2_p
atie
nt
trio
3_m
oth
er
trio
3_p
atie
nt
trio
3_f
ath
er
trio
4_p
atie
nt
trio
4_f
ath
er
trio
4_m
oth
er
trio
5_f
ath
er
trio
5_m
oth
er
trio
5_p
atie
nt
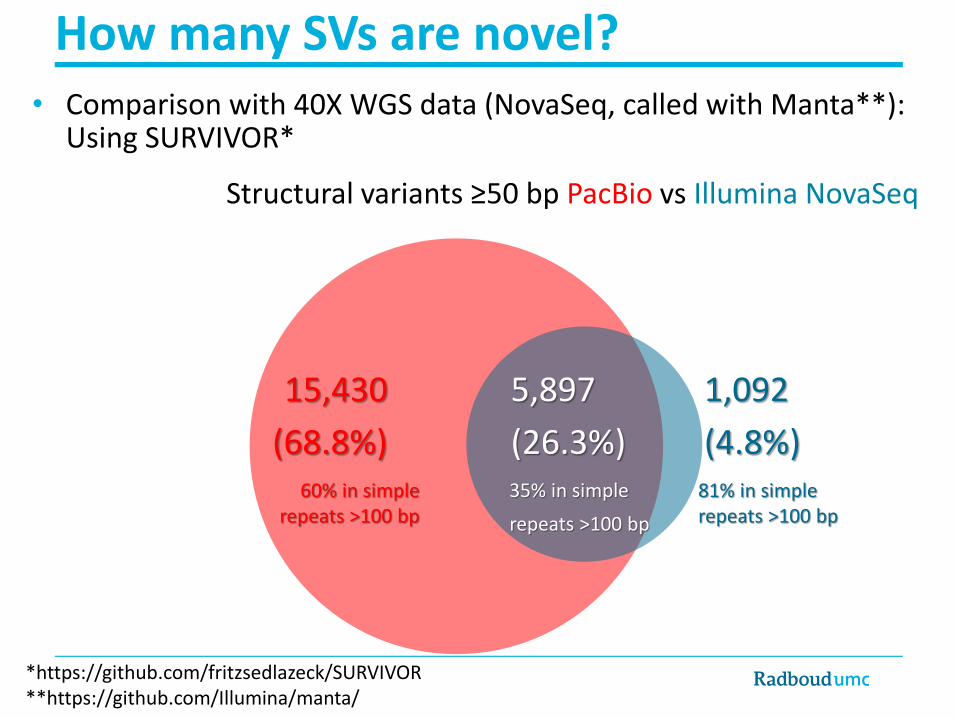
SVs/indels >20bp
>33-40,000 SVs/genome (20-50bp)
PBSV caller with lower cut-off of 20bp
0
10,000
20,000
30,000
40,000
50,000
60,000
70,000
80,000
Solo
Joint
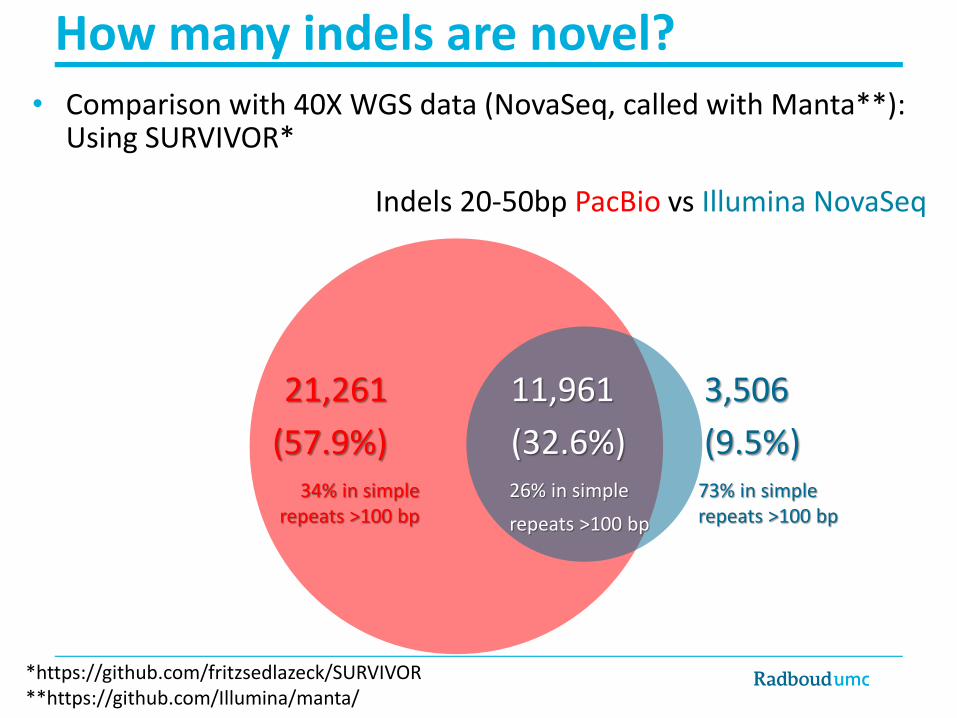
How many indels are novel?
*https://github.com/fritzsedlazeck/SURVIVOR**https://github.com/Illumina/manta/
21,261
(57.9%)
11,961
(32.6%)
3,506
(9.5%)34% in simple
repeats >100 bp73% in simplerepeats >100 bp
26% in simple
repeats >100 bp
Indels 20-50bp PacBio vs Illumina NovaSeq
• Comparison with 40X WGS data (NovaSeq, called with Manta**): Using SURVIVOR*

SV distribution over samples
rare/private
0
5000
10000
15000
20000
25000
30000
35000
40000
45000
50000
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15
Co
un
t o
f SV
s
Individuals with respective SV
SVs > 20bp
How many are inherited?
Up to 74% in patient& parent (18% random)
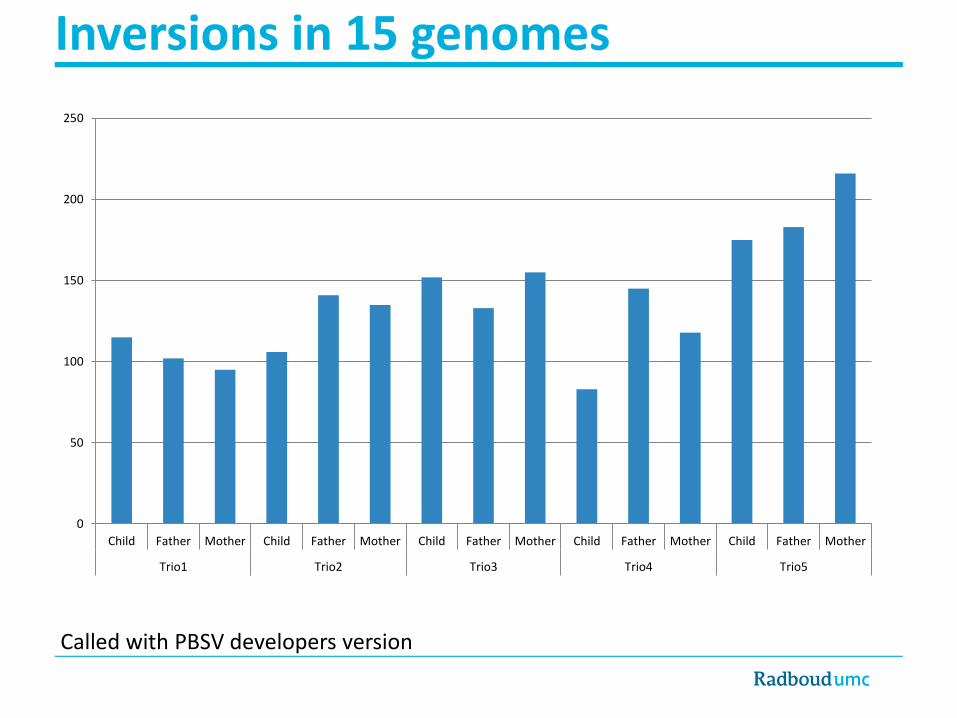
Inversions in 15 genomes
0
50
100
150
200
250
Child Father Mother Child Father Mother Child Father Mother Child Father Mother Child Father Mother
Trio1 Trio2 Trio3 Trio4 Trio5
Called with PBSV developers version

Inversions – size distribution
0
50
100
150
200
250
300
0-100 100-200 200-300 300-400 400-500 >500
Inversions <1Kb, bin=100 bp
0
2
4
6
8
10
12
14
Inversions 1-10 Kb, bin=1 Kb

Next steps
• Are any candidate de novo SVs truly de novo?
• If they are, could they explain disease?• Genotype/phenotype recurrence in other cases?
• Do rare hidden SVs unmask recessive disease?

Extra ‘goodies’ of long-reads?
• Can we detect de novo SNVs?
• Phasing de novo mutations
• Phasing of candidate comp. het. variants

We can detect de novo SNVs
PacBio 40x child
PacBio 40x father
PacBio 40x mother
de novo c. 1685A>C; p.(His562Pro); TBKBP1

We can phase de novo SNVsde novo c. 1685A>C;
p.(His562Pro); TBKBP1
PacBio 40x child
PacBio 40x father
PacBio 40x mother
On same allele: Maternalheterozygous SNP
Phasing de novo mutations important to understand DNM biology
e.g.: Goldmann et al. Nat Genet 2016 & 2018

Compound heterozygous variants?
Allele 1Heterozygous variant
PacBio 10x childonly
Allele 2Heterozygous variant

Work in progress..
• Calling SV with other tools, e.g. sniffles• Calling single nucleotide variants• Assembly using the GRCh38 reference and full de novo assembly• Comparison with other technologies (10x genomics, bionano, etc.)

Summary
• Per genome: High quality coverage for 28Mb of previously uncovered sequence
• SMRT sequencing allows detection of ~25,000 SVs per genome
• Also: >33,000 indels (20-50bp) are called per genome
• Majority of those SVs/indels were not detected by short-read WGS
• Long reads to comprehend de novo mutation rates of indels/SVs – start to understand clinical relevance

PacBio in diagnostics
• Kornelia Neveling/Marcel Nelen:
Use long-range PCR for complex human regions:• HLA (5 amplicons), collab. with medical immunology• Pseudogenes• mtDNA• Repeat expansions
Launch of diagnostic PacBio assay for HLA: June 15th! ...others will follow in 2018
Full list of diagnostic tests: www.genomediagnosticsnijmegen.nl

EU: 15Mio€ H2020 grant – start in 2018
Ambition to solve a significant number of rare diseases (RD) that remained unsolved after exome/genome sequencing
Total RD cases ~20,000
Aim for >500 long-read genomes & >100 full-isoform seqs
Solving the unsolved Rare Diseases
www.SOLVE-RD.eu*Co-coordinated by: Tübingen, Leicester, Nijmegen

Goal:Latest genomics tools &
integrating other omics to understand biology and disease
https://www.x-omics.nl
https://www.solve-rd.eu

Acknowledgements
Radboud UMC
• Michael Kwint• Marc Pauper• Maartje van der Vorst
• Jordi Corominas Galbany• Marcel Nelen• Kornelia Neveling
• Christian Gilissen• Lisenka Vissers• Han Brunner
All patients and families
PacBio• Aaron M Wenger
• Primo Baybayan • Luke Hickey• Jonas Korlach
• Kevin Corcoran
PacBio EU
John Kuijpers, Gerard van de Burgt, Ralph
Vogelsang, David Stucki, Philip Lobb, John
Baeten, Gerrit Kuhn, Deepak Singh