Embed Size (px)
Citation preview

Metabolism At a Glance and in 1
Hour !
Simon Heales
Panda SA Workshop, Roodevallei,
Pretoria, May 2016
Mitochondrial
Enzymes – LSDs, GSDs....
Metabolic – UOA, AAs, Carnitines..
CSF Neurotransmitters
Vitamins & Antioxidants
Newborn Screening
“Routine”
Immunology
Haematology
Histopathology
Microbiology
Paediatric malignancy
Accredited Services
Chemical Pathology – Lysosomal, Special
Routine, Newborn Screening, Metabolic Profiling…
Haematology
Haematology – Cellular & Molecular
Diagnostic Service
Immunology
Histopathology
Microbiology & Virology
Genetics
Neurometabolic Unit (National) – Mitochondrial, CSF Neurotransmitters, Vitamins…
www.labs.gosh.nhs.uk

A B C D Enz
CoF
Enz
CoF
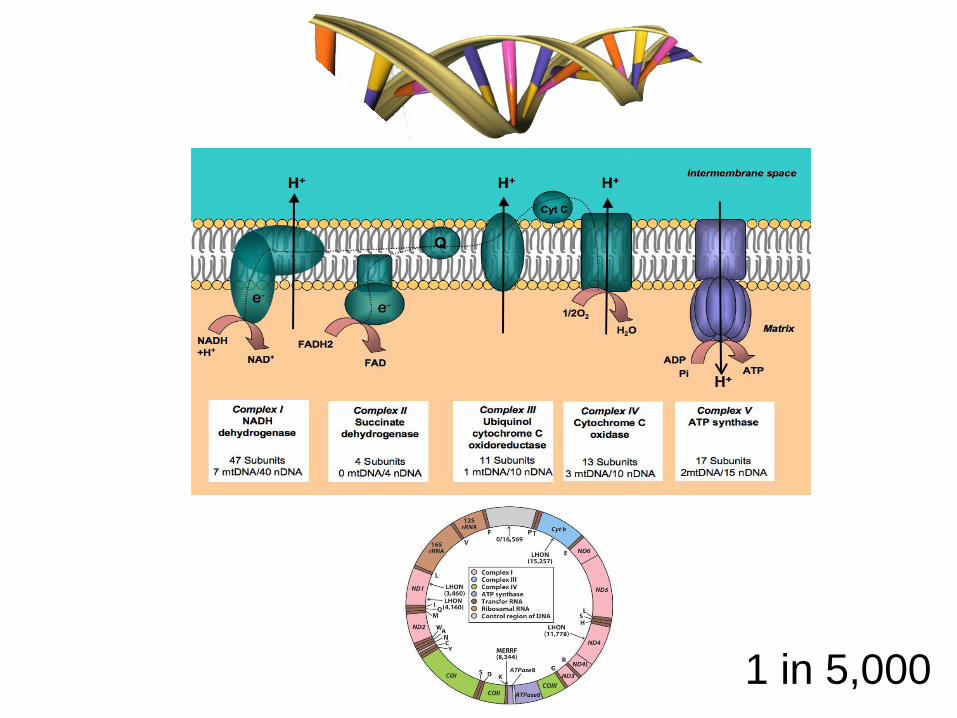
Inborn Errors of Metabolism
Enz
CoF
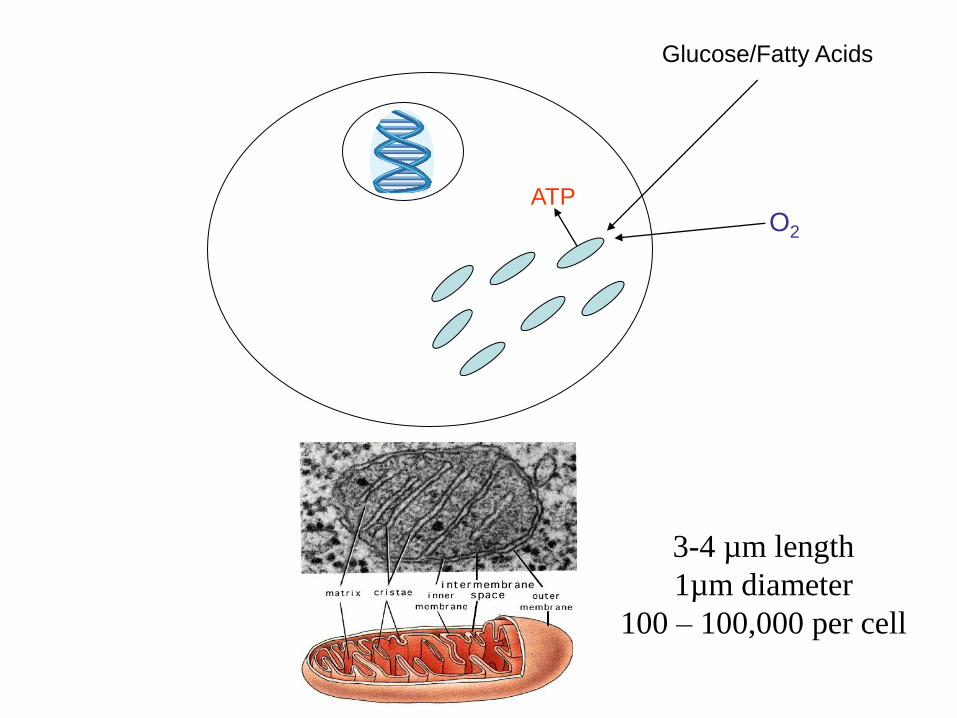
Glucose/Fatty Acids
ATP O2
3-4 µm length
1µm diameter
100 – 100,000 per cell
1 in 5,000
Clinical Picture
Biochemistry
Histochemistry
Molecular Biology
Diabetes Thyroid
Disease Myopathy
Peripheral
Neuropathy
Deafness
Seizures /
Developmental delay
Respiratory Failure Optic Atrophy / Retinitis
Pigmentosa
Cardiomyopathy
Short Stature
Marrow Failure
Liver
Failure

ETC Defects
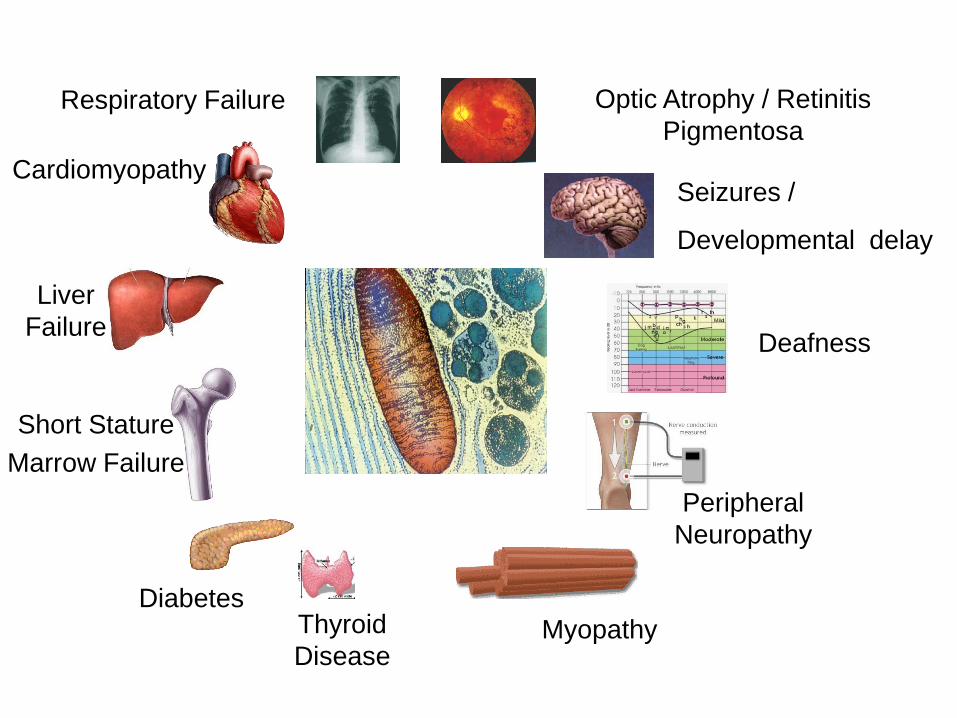
Complexity of genetics can lead to diverse clinical
features:
Any Symptom
Any Organ or Tissue
Any Age of Presentation
Any mode of inheritance
Munich et al., 1992

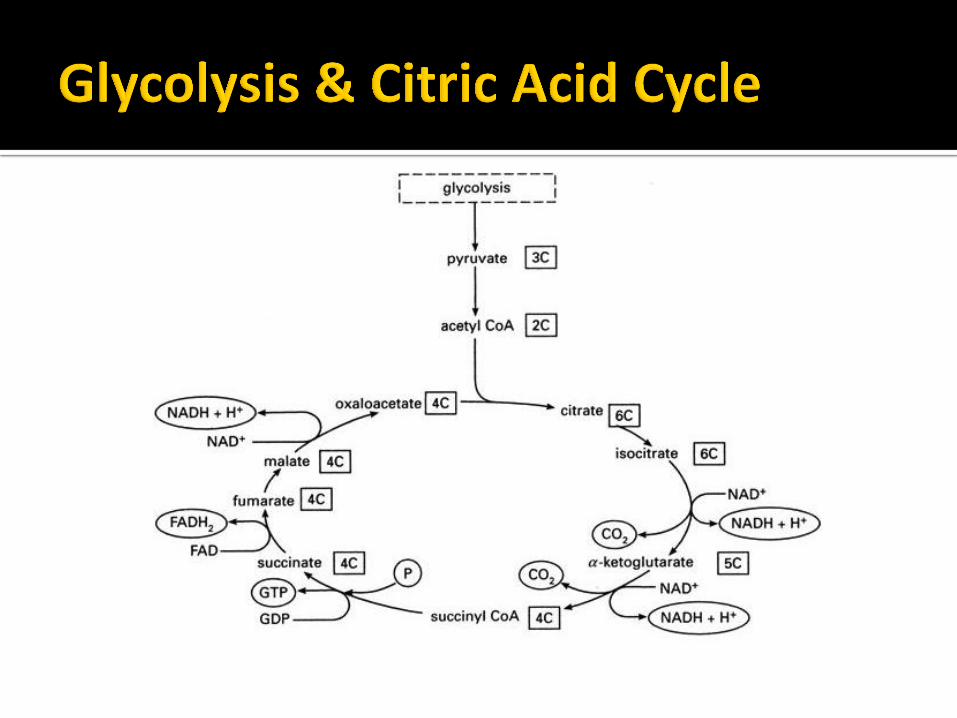
ETC DEFECTS
• ATP Depletion.
• Cytochrome c release.
• CNS Folate Deficiency
• Increased Oxidative Stress
• Cell death & tissue/organ failure.
• Dependent on Energetic Requirement of tissue.
• 1 in 5,000 live births
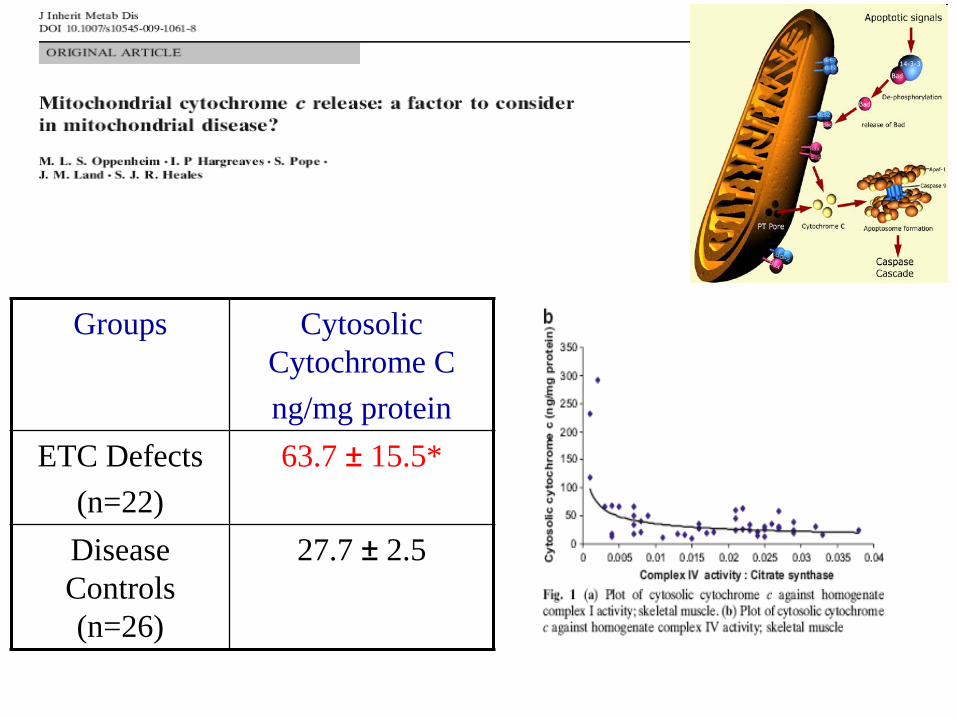
Groups Cytosolic
Cytochrome C
ng/mg protein
ETC Defects
(n=22)
63.7 ± 15.5*
Disease
Controls
(n=26)
27.7 ± 2.5
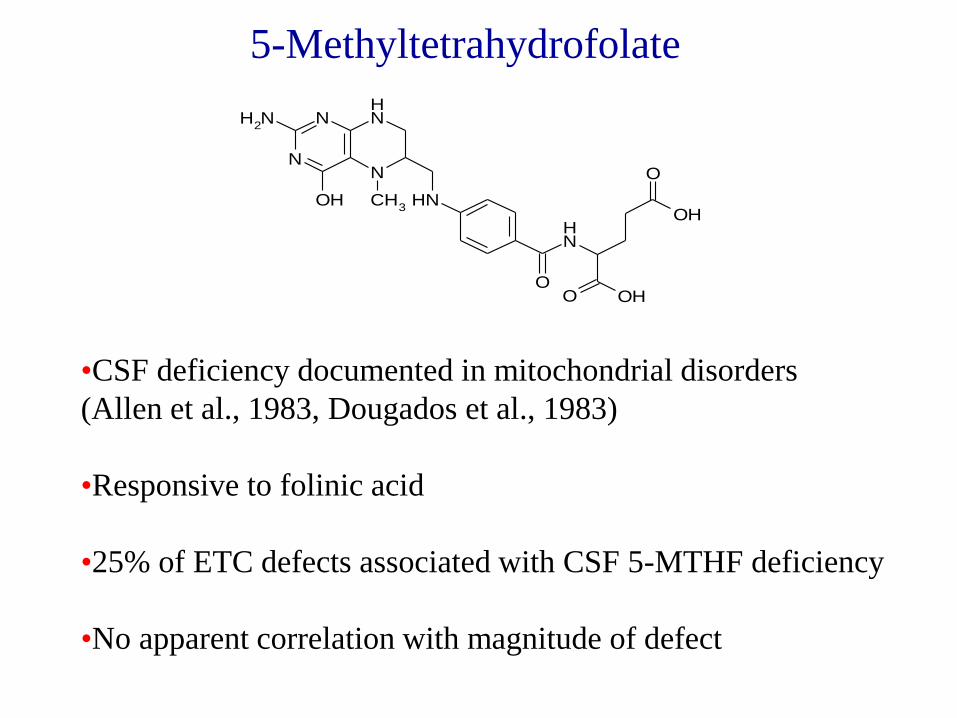
N
N
N
NH
NH2
NH
O
NH
O OH
OH
O
OH CH3
5-Methyltetrahydrofolate
•CSF deficiency documented in mitochondrial disorders
(Allen et al., 1983, Dougados et al., 1983)
•Responsive to folinic acid
•25% of ETC defects associated with CSF 5-MTHF deficiency
•No apparent correlation with magnitude of defect

CSF 5-MTHF Deficiency and
Mitochondrial Disorders
Female, 15yrs 29 46-160 nmol/L
Male, 9yrs 5 72-172 nmol/L
Male, 8 yrs 44 72-172 nmol/L
Female, 2 yrs 17 52-178 nmol/L
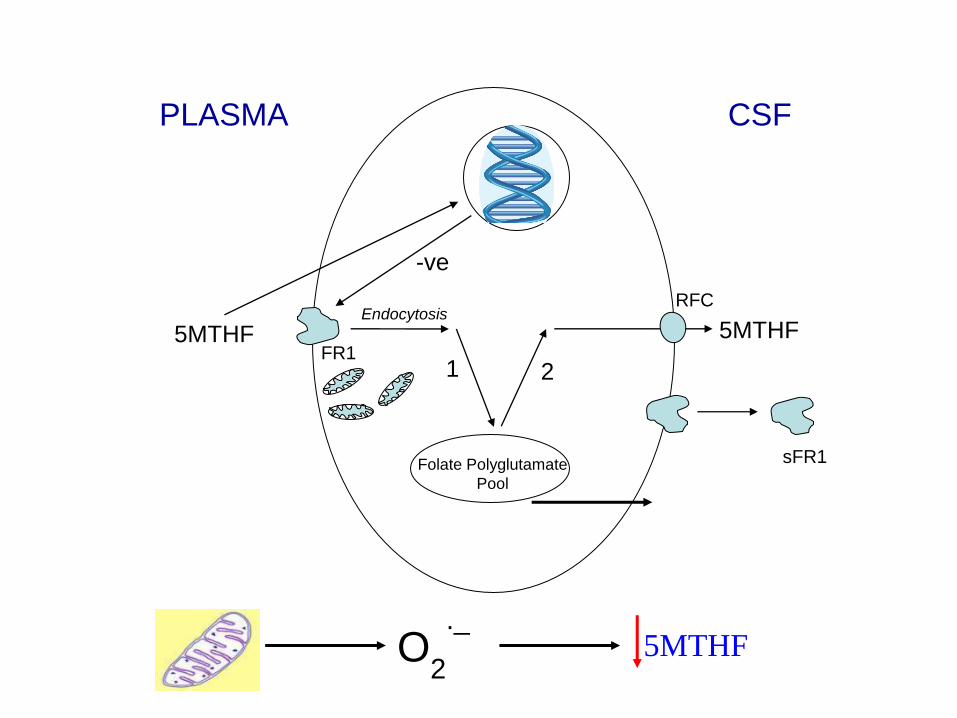
5MTHF Endocytosis
Folate Polyglutamate
Pool
5MTHF FR1
RFC
sFR1
-ve
PLASMA CSF
1 2
O2
._
5MTHF
0
2
4
6
8
10
12
14n
mo
l/m
g Control
ETC
Defect
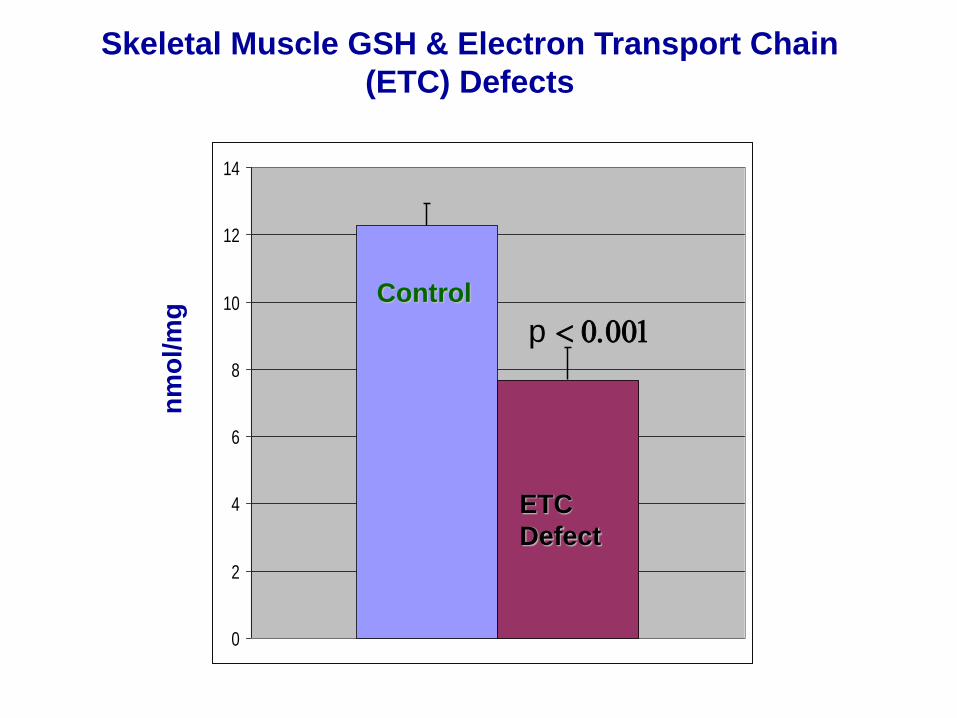
Skeletal Muscle GSH & Electron Transport Chain
(ETC) Defects
p < 0.001
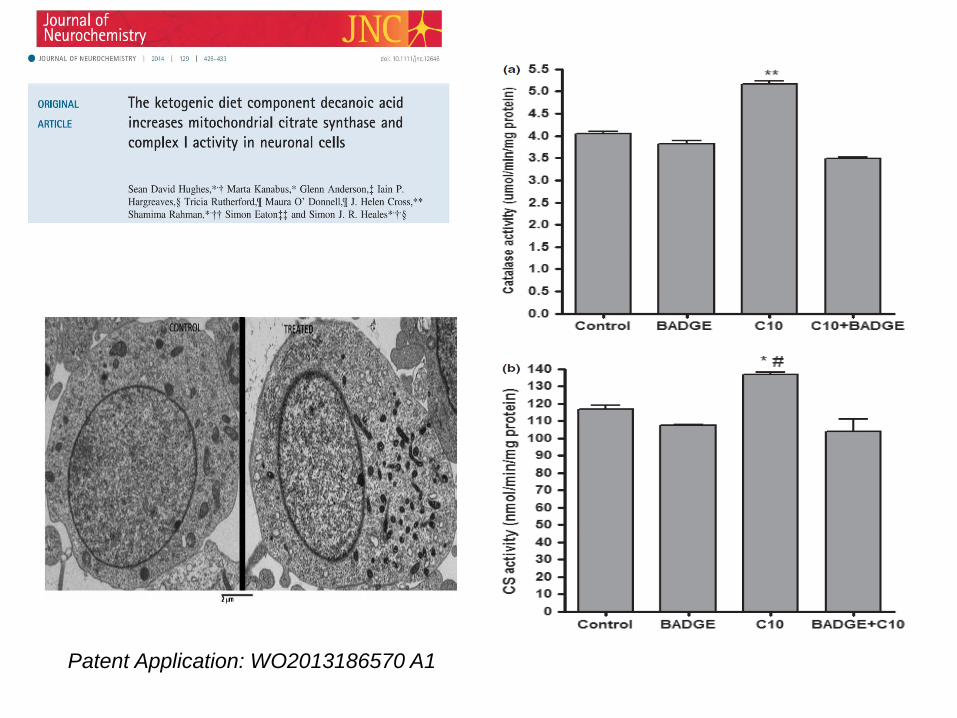
Patent Application: WO2013186570 A1

The presented research is supported by the NIHR Great Ormond Street Hospital Biomedical Research Centre
Disorders of Lysosomal Metabolism
•About 50 known disorders
•Individually rare.
•1 in 5,000 collectively
•Treatments available, e.g. enzyme replacement therapy.
•Earlier diagnosis better outcome
•Commissioned service

Coarse facial features
Corneal clouding or related ocular abnormalities
Angiokeratoma
Umbilical/inguinal hernias
Short stature
Developmental delays
Joint or skeletal deformities
Organomegaly (especially liver and spleen)
Muscle weakness or lack of control (ataxia, seizures, etc.)
Neurologic failure/decline or loss of gained development
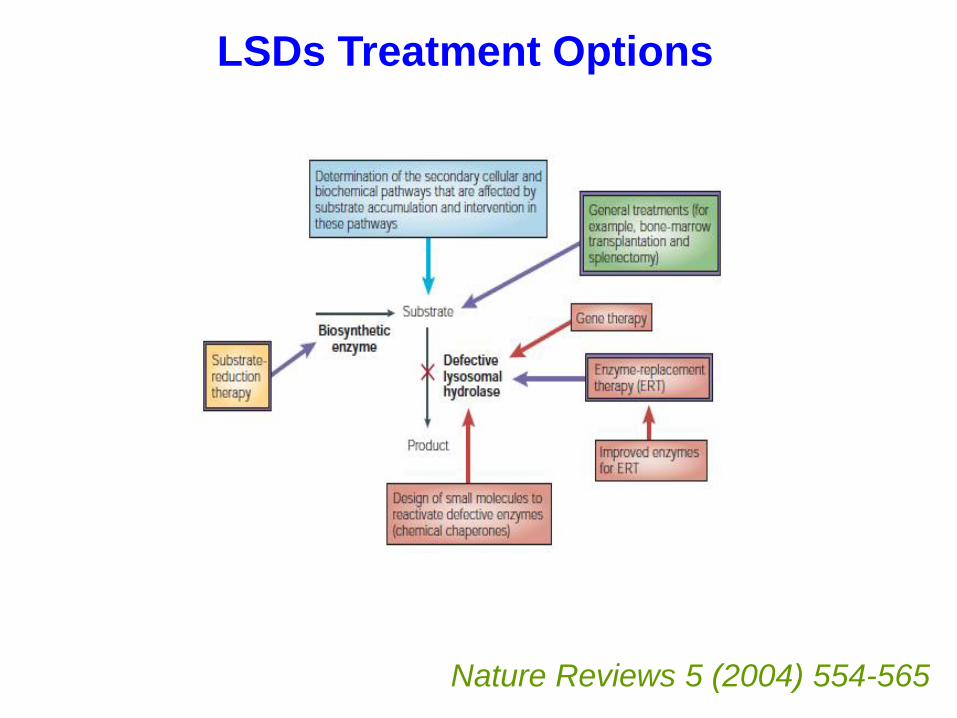
LSDs Treatment Options
Nature Reviews 5 (2004) 554-565
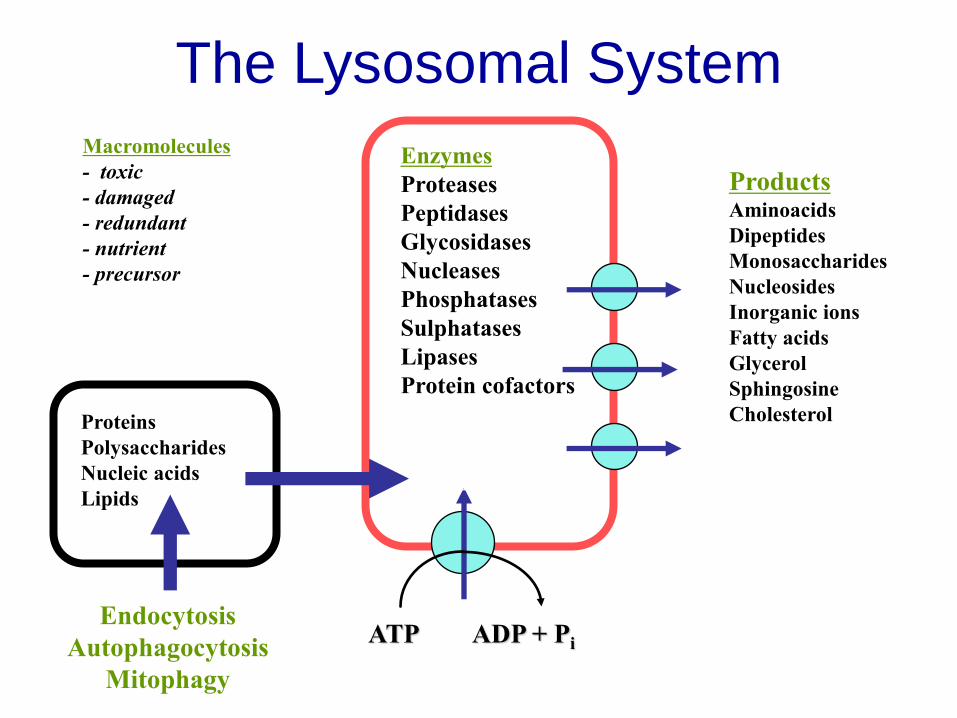
The Lysosomal System
Products Aminoacids
Dipeptides
Monosaccharides
Nucleosides
Inorganic ions
Fatty acids
Glycerol
Sphingosine
Cholesterol
Macromolecules
- toxic
- damaged
- redundant
- nutrient
- precursor
H+
ATP ADP + Pi
Enzymes
Proteases
Peptidases
Glycosidases
Nucleases
Phosphatases
Sulphatases
Lipases
Protein cofactors
Proteins
Polysaccharides
Nucleic acids
Lipids
Endocytosis
Autophagocytosis
Mitophagy
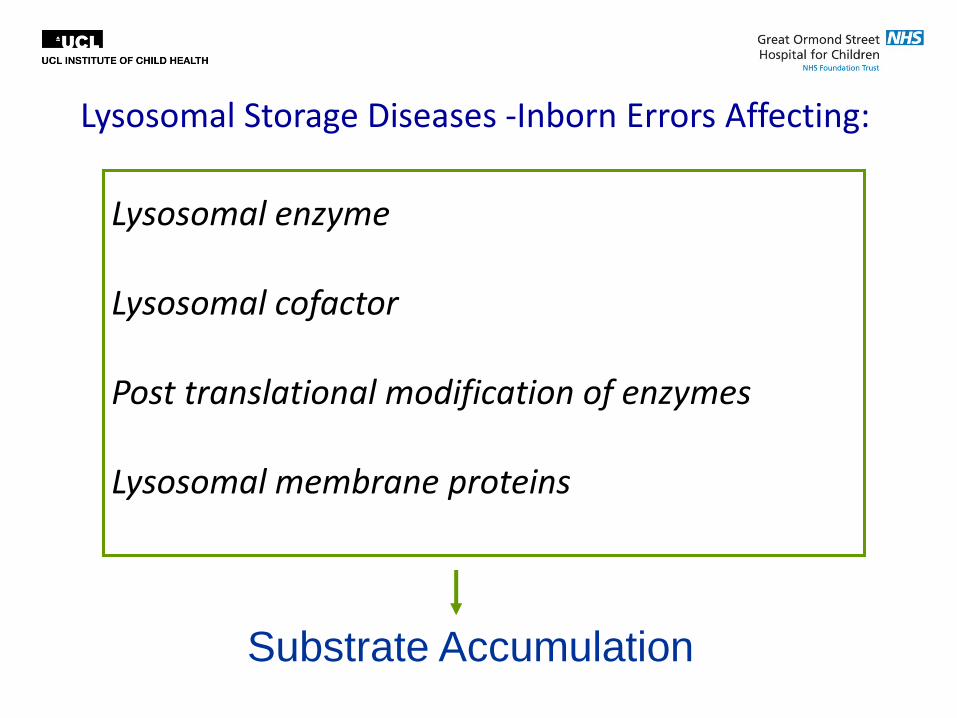
Lysosomal Storage Diseases -Inborn Errors Affecting:
Lysosomal enzyme Lysosomal cofactor Post translational modification of enzymes Lysosomal membrane proteins
Substrate Accumulation
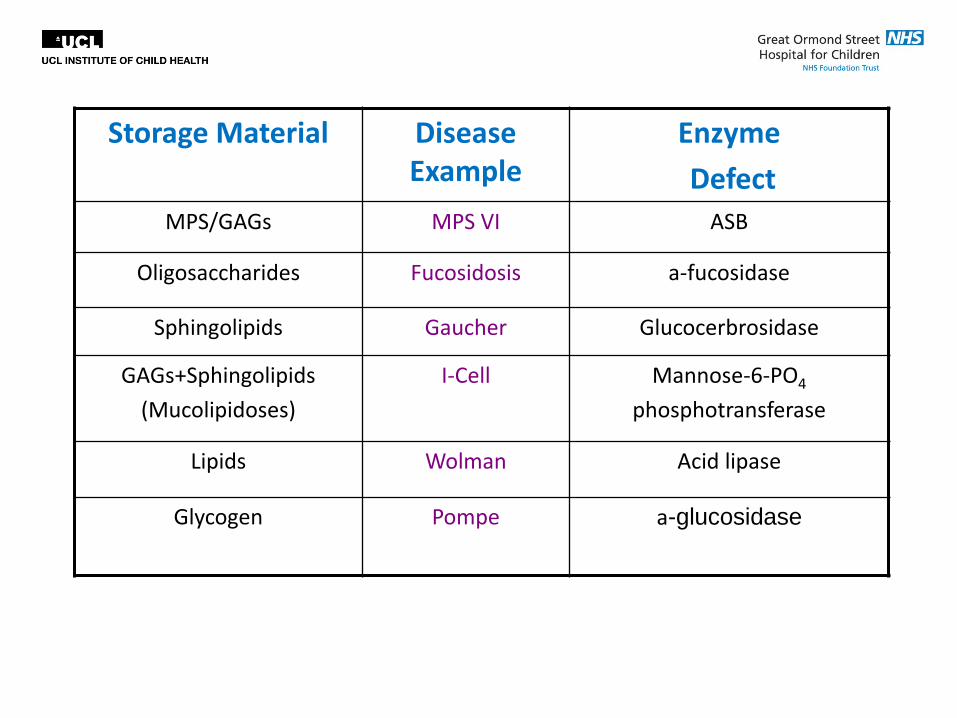
Storage Material Disease Example
Enzyme
Defect
MPS/GAGs MPS VI ASB
Oligosaccharides Fucosidosis a-fucosidase
Sphingolipids Gaucher Glucocerbrosidase
GAGs+Sphingolipids
(Mucolipidoses)
I-Cell Mannose-6-PO4
phosphotransferase
Lipids Wolman Acid lipase
Glycogen Pompe a-glucosidase
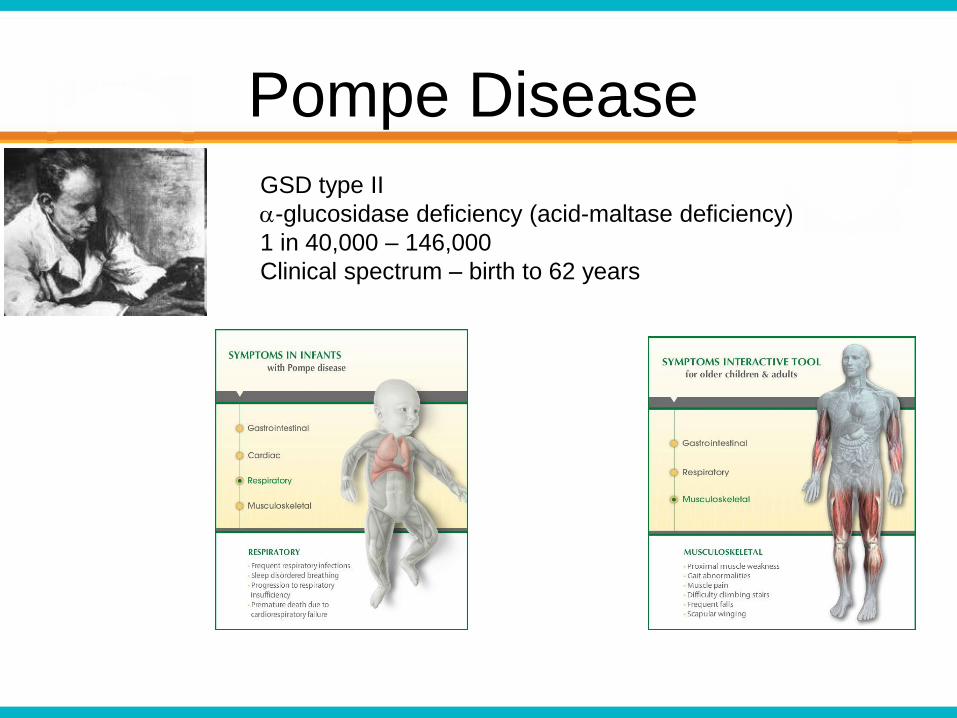
Pompe Disease
GSD type II
a-glucosidase deficiency (acid-maltase deficiency)
1 in 40,000 – 146,000
Clinical spectrum – birth to 62 years
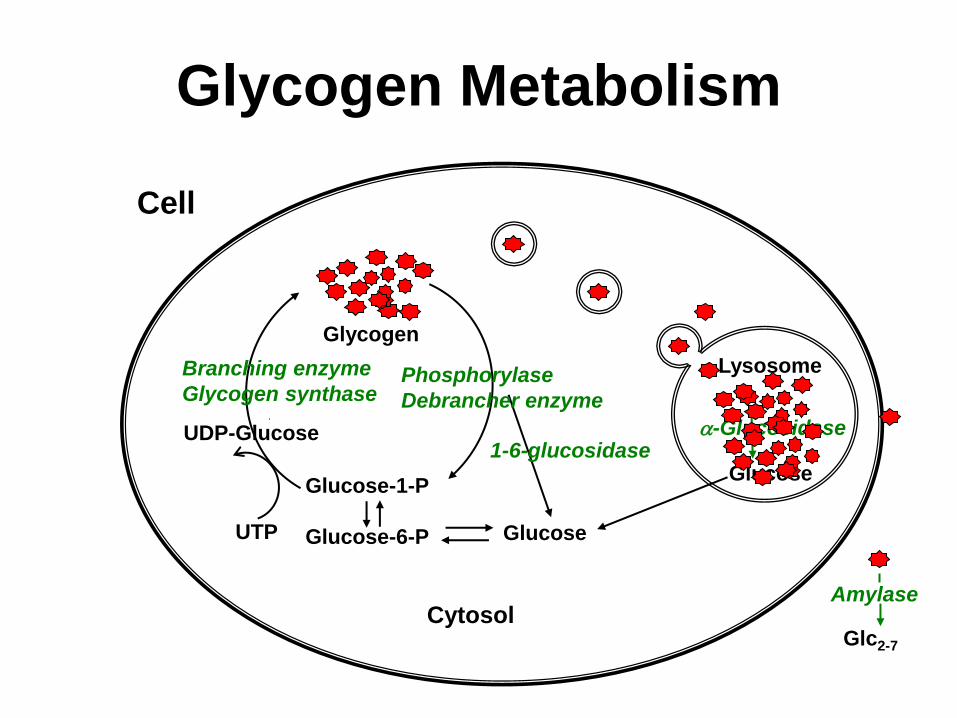
Glycogen Metabolism
Glucose-1-P
Glucose-6-P
Glycogen
Glucose
1-6-glucosidase UDP-Glucose
Phosphorylase
Debrancher enzyme
UTP
Branching enzyme
Glycogen synthase
Cytosol
Lysosome
Glucose
a-Glucosidase
Cell
Amylase
Glc2-7
Glc4 – A biomarker – found in Urine
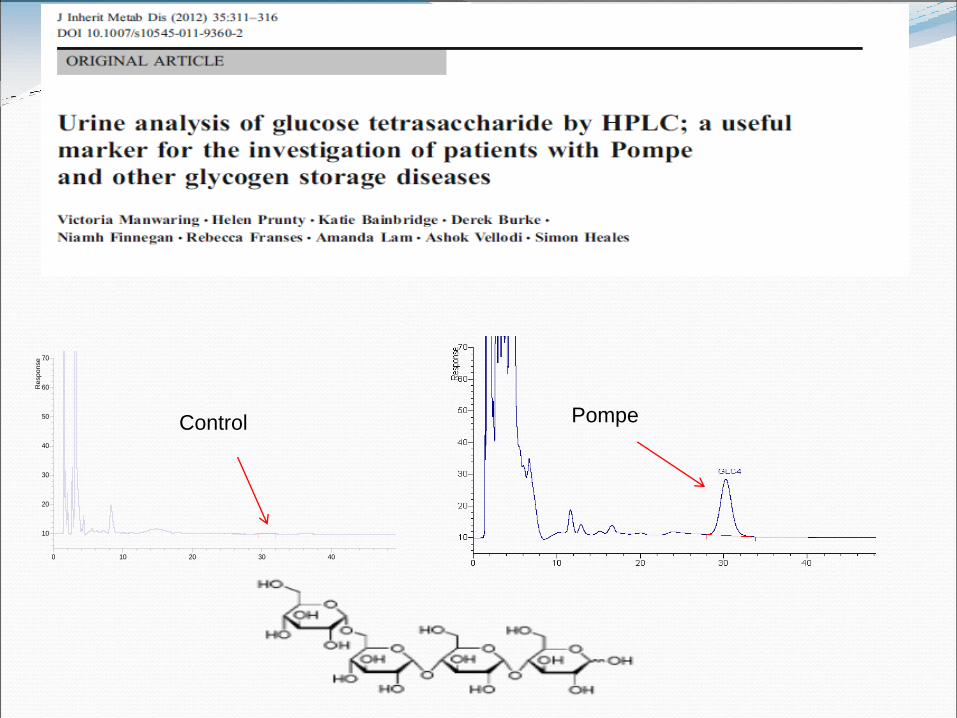
10C060779 (3,1) HPLC,Instrument2.GLC4_14JUN10,3,1,1
Acquired Monday, June 14, 2010 3:21:25 PM
10
20
30
40
50
60
70
Response
0 10 20 30 40 50
Retention time
Control Pompe
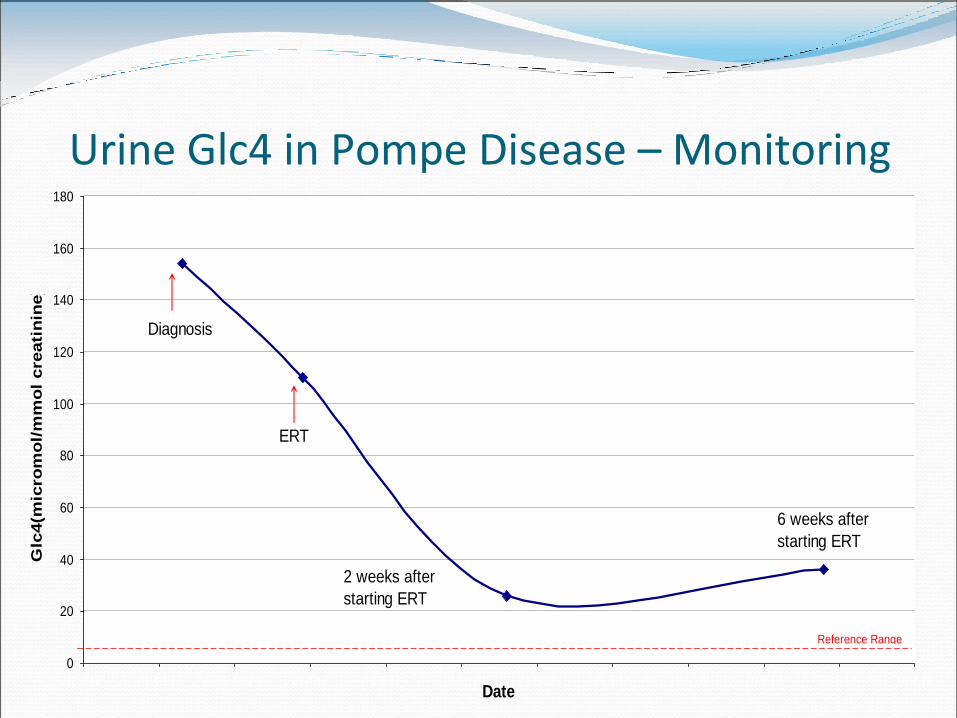
Urine Glc4 in Pompe Disease – Monitoring
0
20
40
60
80
100
120
140
160
180
22.3.12 1.4.12 11.4.12 21.4.12 1.5.12 11.5.12 21.5.12 31.5.12 10.6.12 20.6.12 30.6.12 10.7.12Date
Glc
4(m
icro
mo
l/m
mo
l cre
ati
nin
e)
Reference Range
Diagnosis
ERT
2 weeks after
starting ERT
6 weeks after
starting ERT

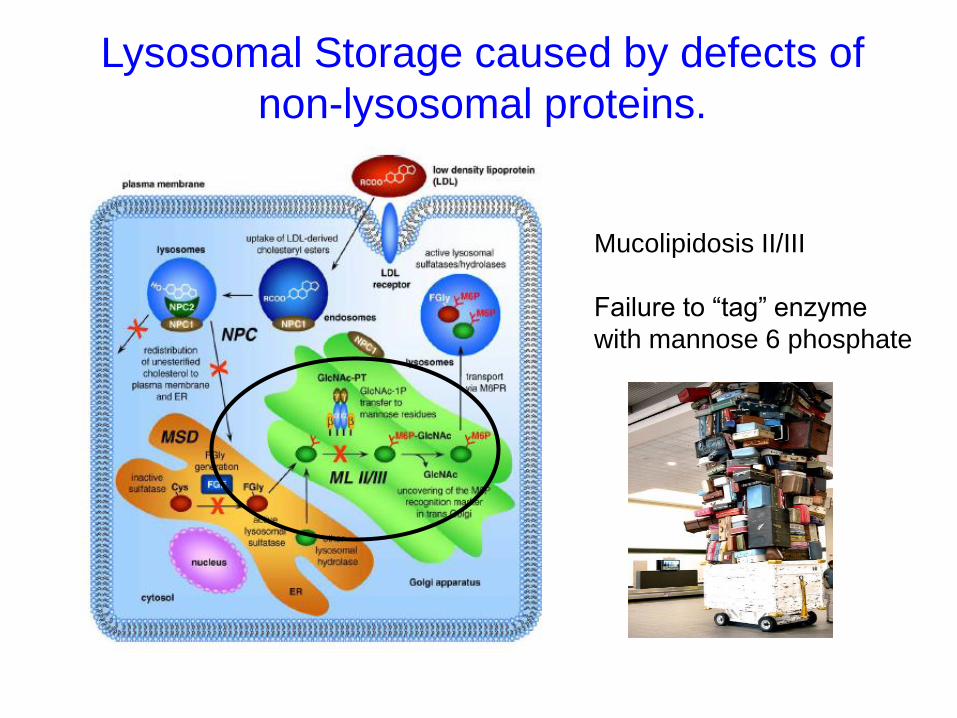
Lysosomal Storage caused by defects of
non-lysosomal proteins.
Mucolipidosis II/III
Failure to “tag” enzyme
with mannose 6 phosphate
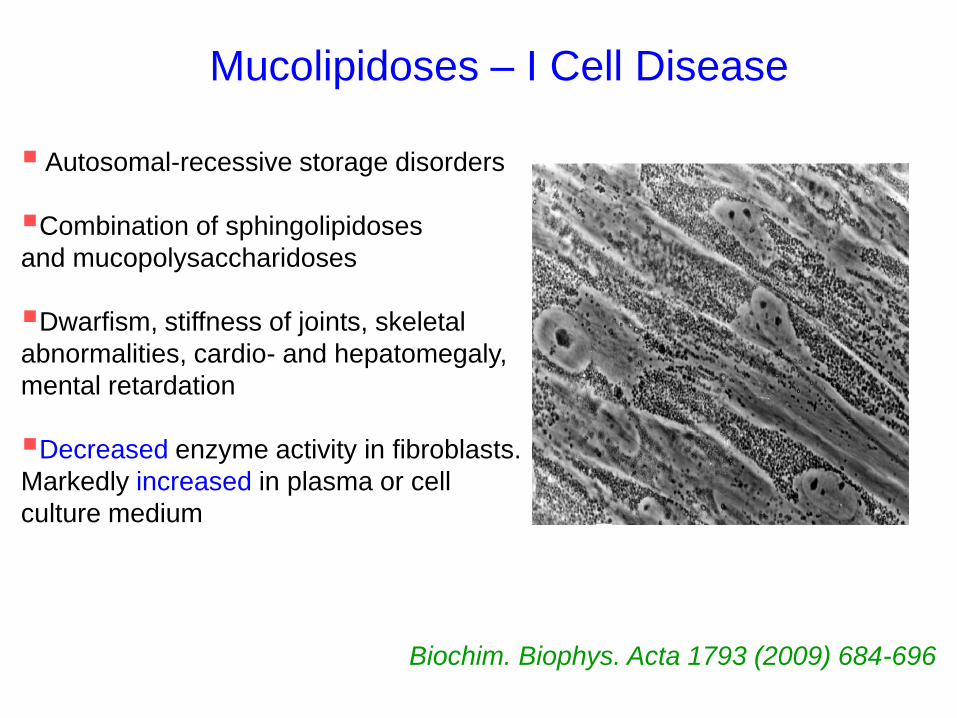
Mucolipidoses – I Cell Disease
Autosomal-recessive storage disorders
Combination of sphingolipidoses
and mucopolysaccharidoses
Dwarfism, stiffness of joints, skeletal
abnormalities, cardio- and hepatomegaly,
mental retardation
Decreased enzyme activity in fibroblasts.
Markedly increased in plasma or cell
culture medium
Biochim. Biophys. Acta 1793 (2009) 684-696
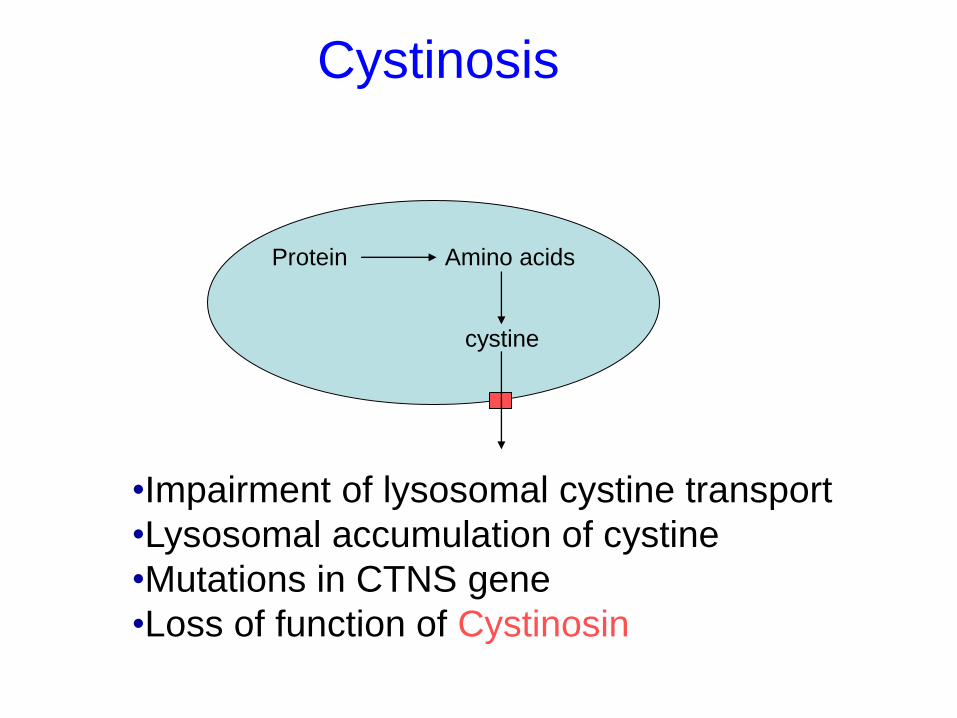
Cystinosis
Protein Amino acids
cystine
•Impairment of lysosomal cystine transport
•Lysosomal accumulation of cystine
•Mutations in CTNS gene
•Loss of function of Cystinosin

Cystinosis
Infantile - Nephropathy kidney wasting of water,
electrolytes, glucose, amino acids, carnitine
Cravings for the four P’s:-
Endocrine disturbances, myopathy, corneal crystals
(photophobia). Central nervous system involvement
Juvenile – neuropathy
Adult – corneal crystals
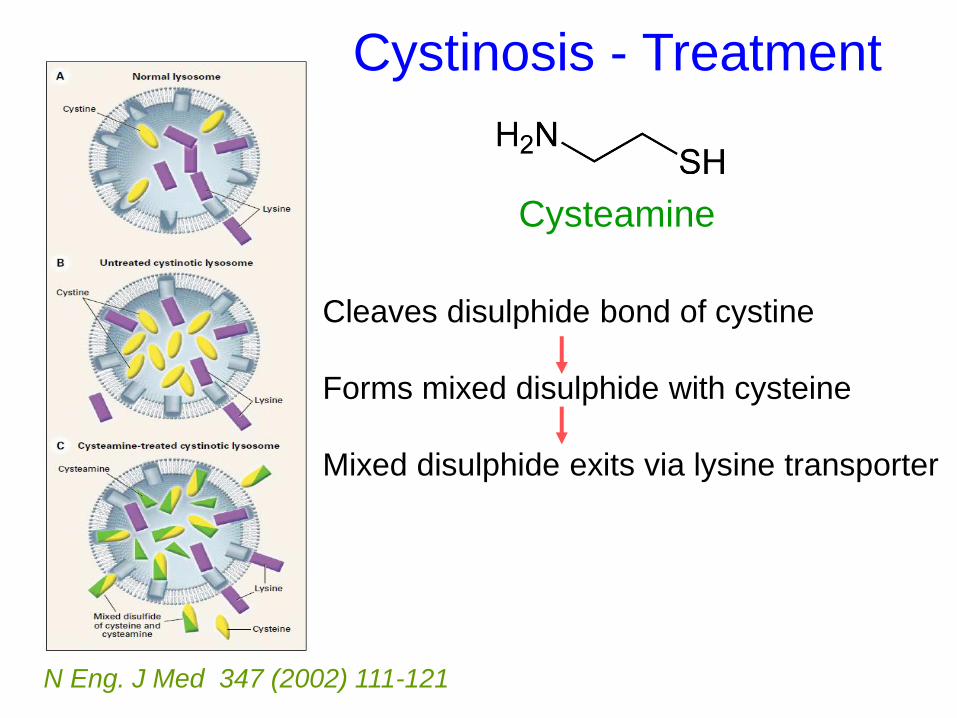
Cystinosis - Treatment
N Eng. J Med 347 (2002) 111-121
Cysteamine
Cleaves disulphide bond of cystine
Forms mixed disulphide with cysteine
Mixed disulphide exits via lysine transporter
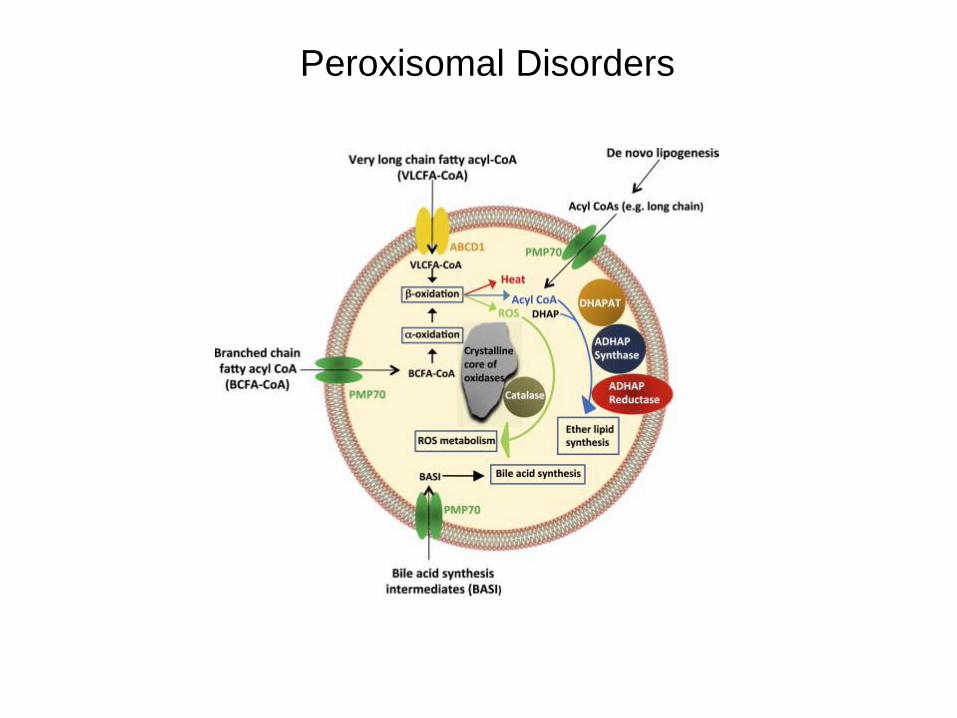
Peroxisomal Disorders
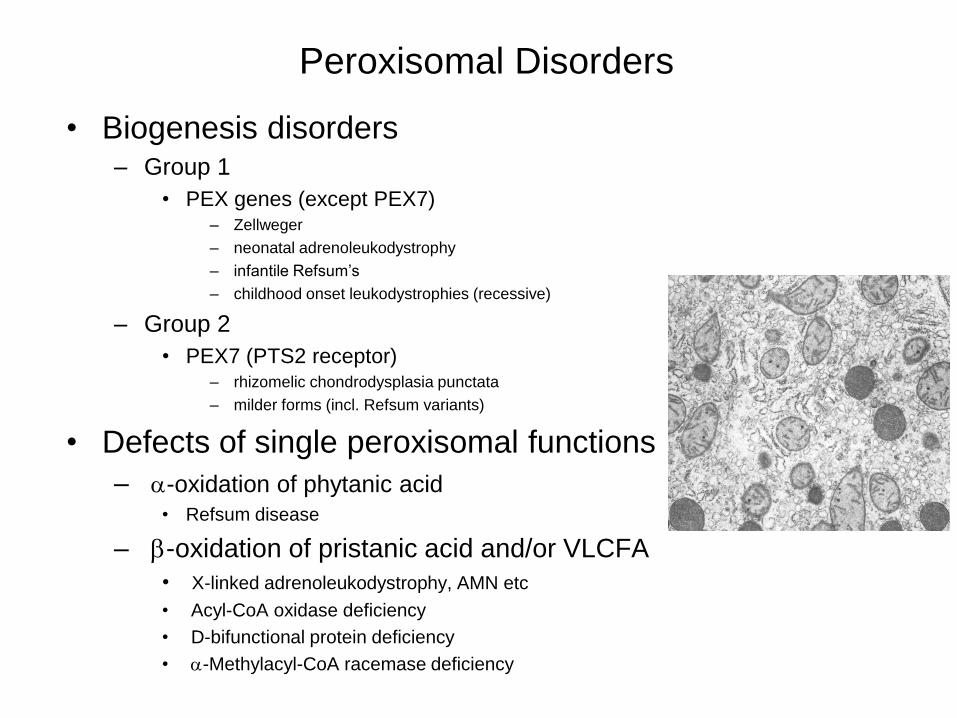
Peroxisomal Disorders
• Biogenesis disorders – Group 1
• PEX genes (except PEX7) – Zellweger
– neonatal adrenoleukodystrophy
– infantile Refsum’s
– childhood onset leukodystrophies (recessive)
– Group 2
• PEX7 (PTS2 receptor) – rhizomelic chondrodysplasia punctata
– milder forms (incl. Refsum variants)
• Defects of single peroxisomal functions
– a-oxidation of phytanic acid
• Refsum disease
– b-oxidation of pristanic acid and/or VLCFA
• X-linked adrenoleukodystrophy, AMN etc
• Acyl-CoA oxidase deficiency
• D-bifunctional protein deficiency
• a-Methylacyl-CoA racemase deficiency
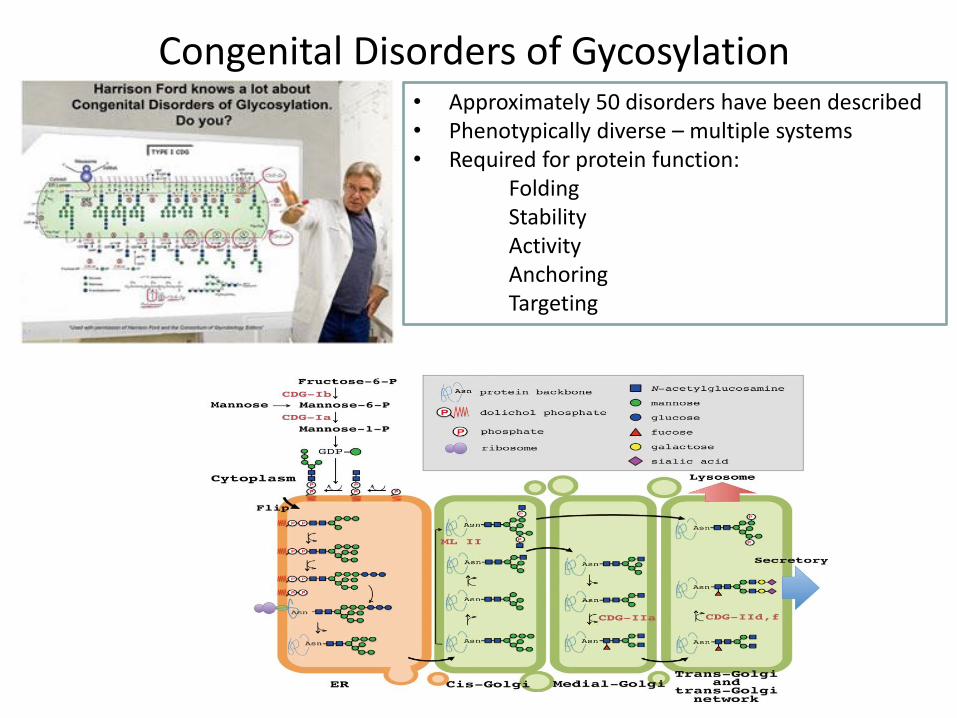
• Approximately 50 disorders have been described • Phenotypically diverse – multiple systems • Required for protein function: Folding Stability Activity Anchoring Targeting
Congenital Disorders of Gycosylation
Neurotransmitters – Coming Soon !





























