Embed Size (px)
Citation preview

36626 - Next Generation Sequencing Analysis
Preprocessing and SNP calling Natasja S. Ehlers, PhD student Center for Biological Sequence Analysis Functional Human Variation Group
Metagenomics & metagenomic assembly
Simon Rasmussen36626: Next Generation Sequencing analysis
DTU Bioinformatics

36626 - Next Generation Sequencing Analysis
What is metagenomics?
Metagenomics (Environmental Genomics, Ecogenomics or Community Genomics) is the study of genetic material
recovered directly from environmental samples.
Metagenomics is application of modern genomic techniques to the study of communities of microbial
organisms directly in their natural environments, bypassing the need for isolation and lab cultivation of
individual species
Chen &Pachter,2005

36626 - Next Generation Sequencing Analysis
Genomics to Metagenomics
Genomics
Metagenomics
E. coli, Science, 1997 Human, Nature/Science, 2001
Saragasso sea, Science, 2004 Human gut, Nature, 2010

36626 - Next Generation Sequencing Analysis
Environments

36626 - Next Generation Sequencing Analysis
Why metagenomics?• New antibiotics
• New molecules with new functions
• New enzymes and bioactive molecules
• Diversity of life
• Interplay between humans and microbes
• How do microbial communities work?
• …
36626 - Next Generation Sequencing Analysis
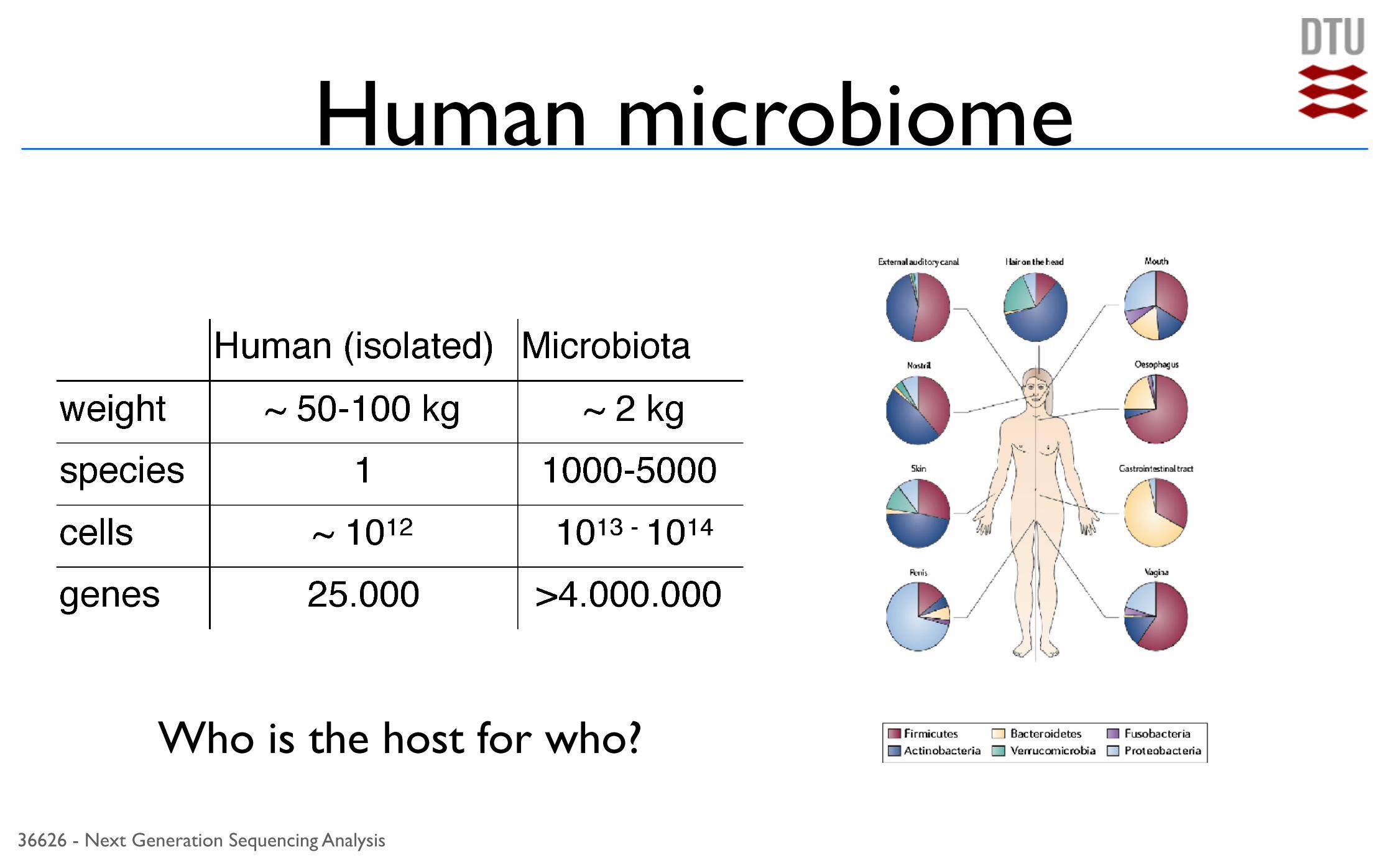
Human microbiome
Who is the host for who?

36626 - Next Generation Sequencing Analysis
Human microbiome• Associated with diseases
• Obesity• Diabetes
• Inflammatory bowel diseases
• Behavior
• Autism? Depression?• Schizophrenia?
36626 - Next Generation Sequencing Analysis
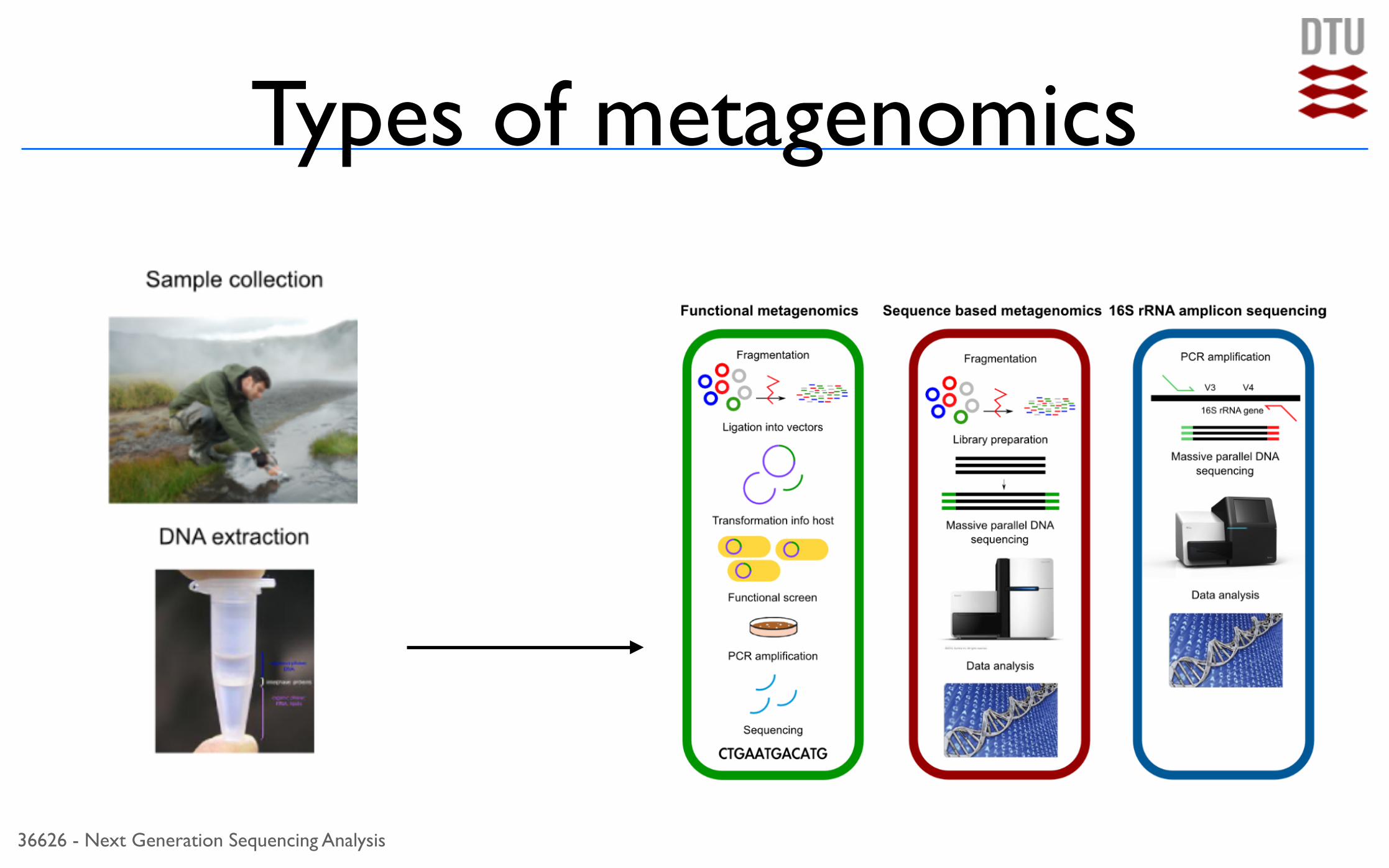
Types of metagenomicsHow to?How to?

36626 - Next Generation Sequencing Analysis
Shotgun metagenomics
• Investigate all organisms (and genomic elements)
• Known/unknown
• Culture / Non-culturable (large fraction non-cult.)
Bacteria
plasmids
phages
genomicislands
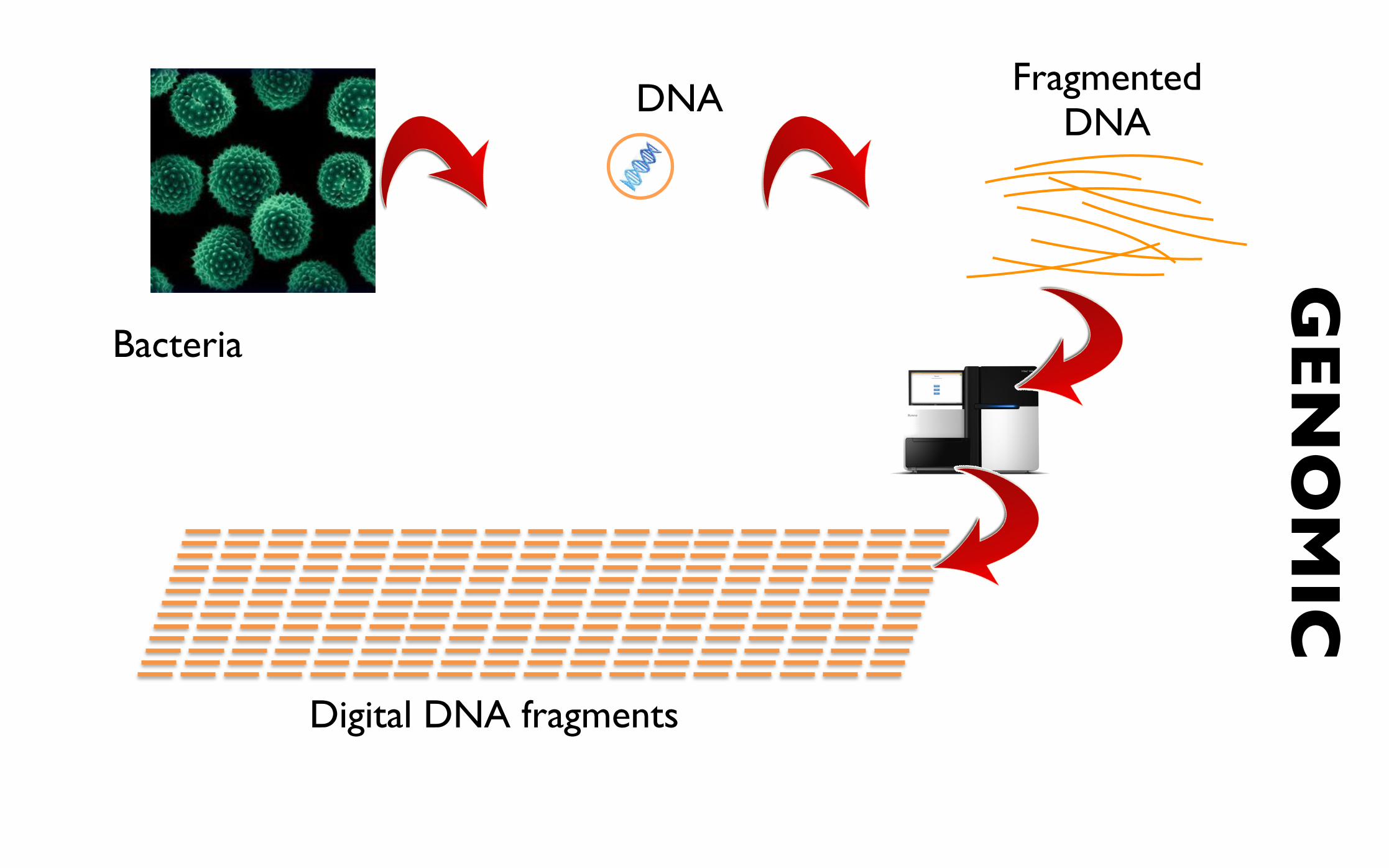
Bacteria
Fragmented DNA
Digital DNA fragments
GEN
OM
ICDNA
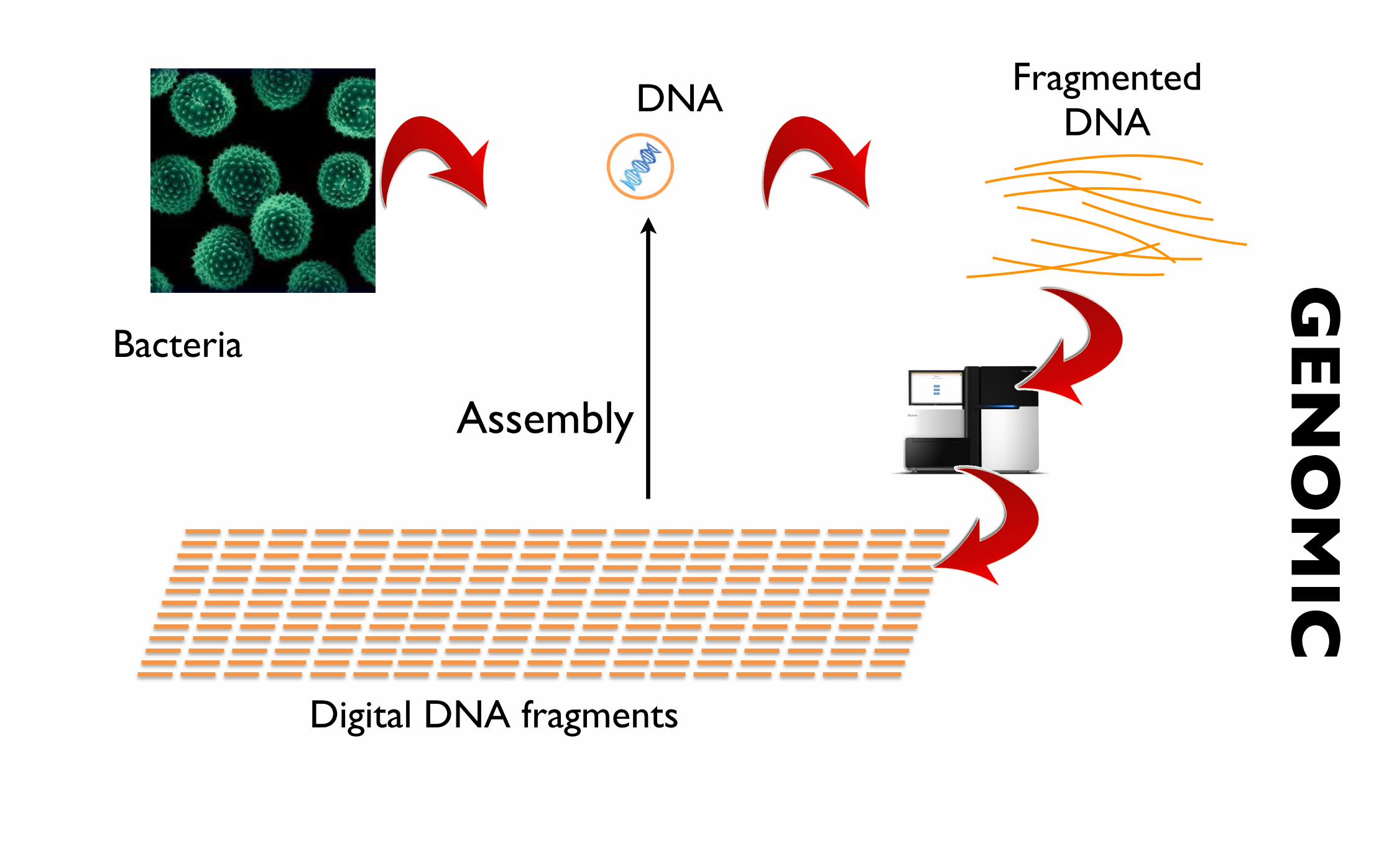
Bacteria
Fragmented DNA
Digital DNA fragments
GEN
OM
ICDNA
Assembly
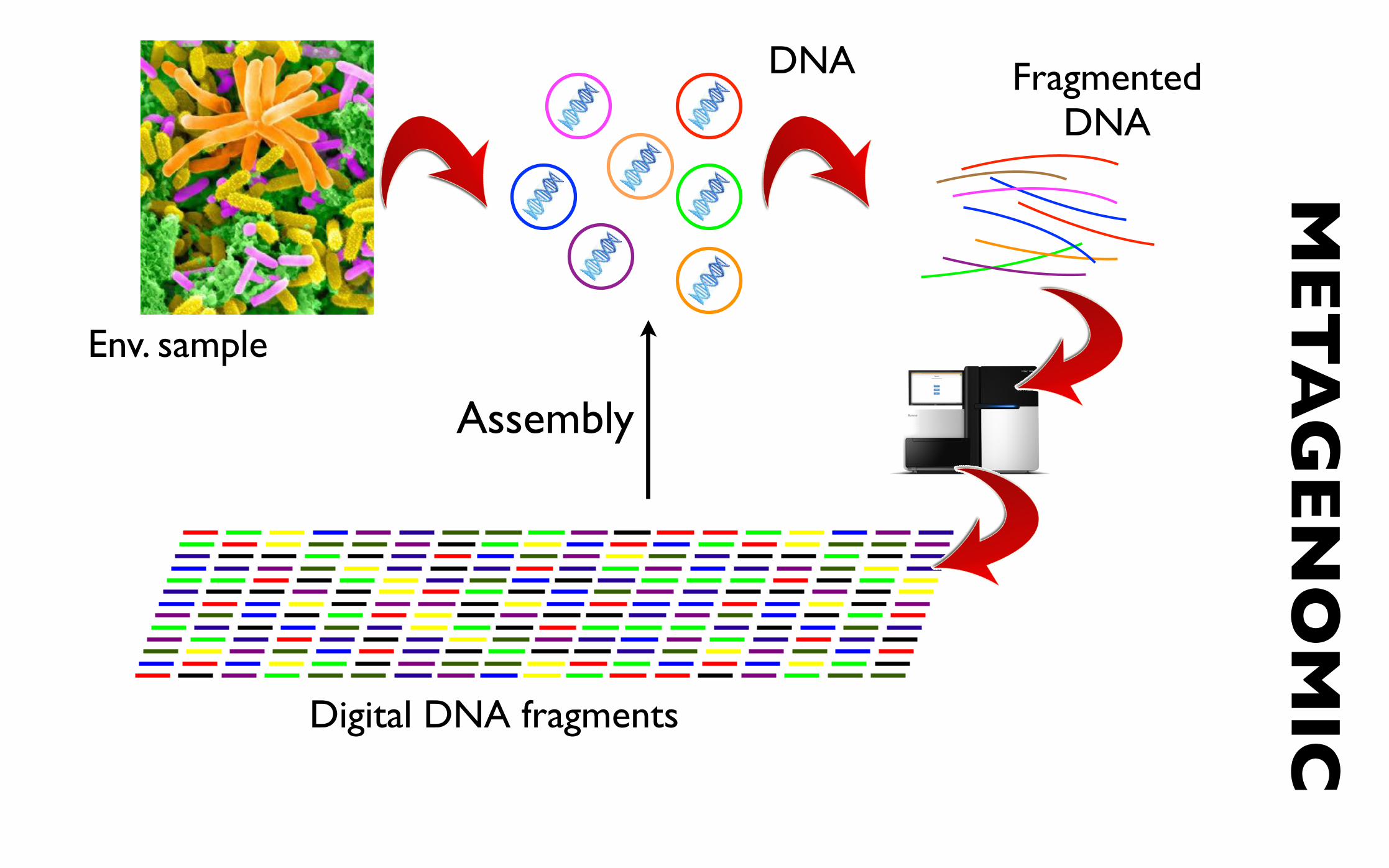
Env. sample
DNA Fragmented DNA
Digital DNA fragments Digital DNA fragments
MET
AG
ENO
MIC
Assembly
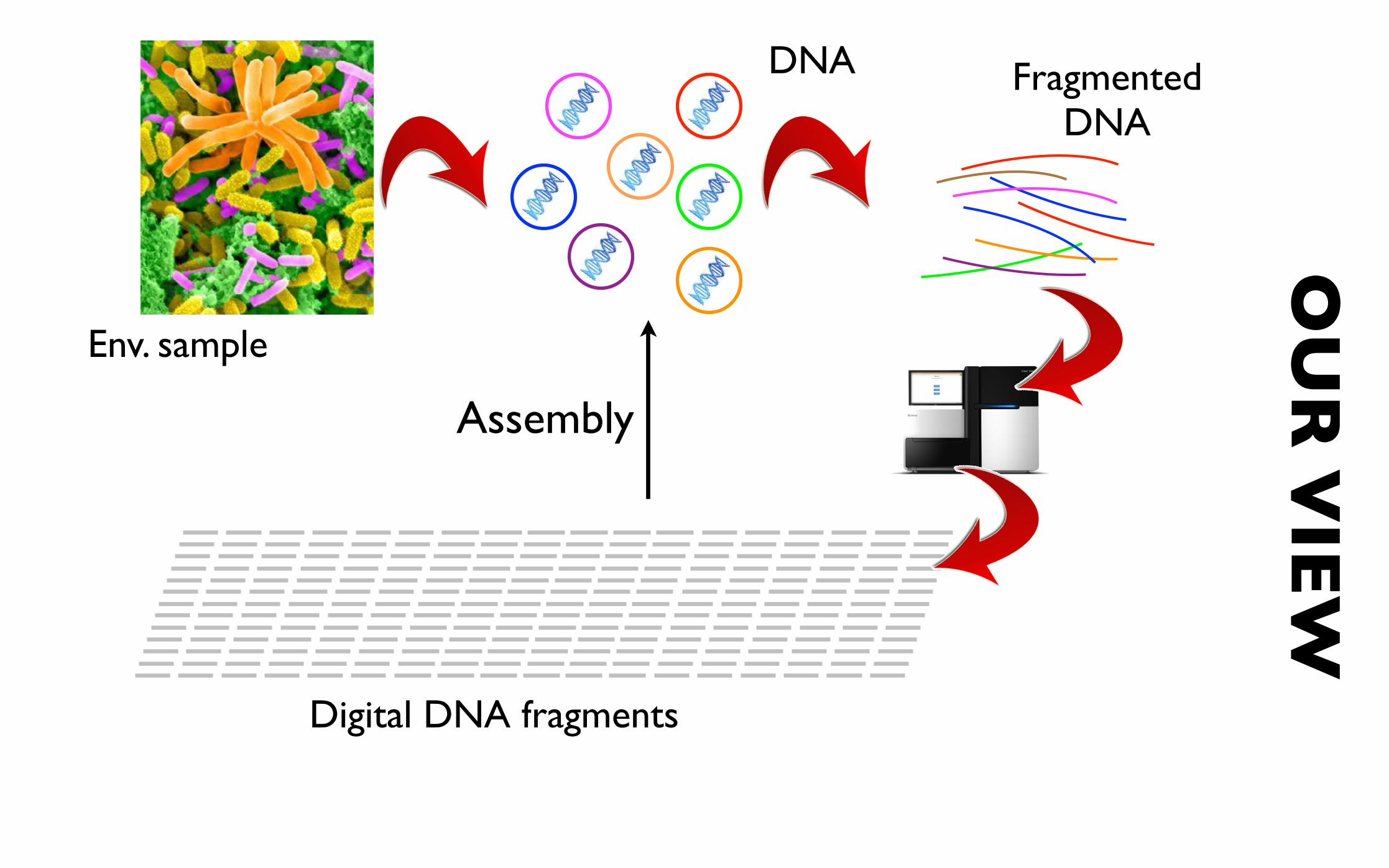
Env. sample
Fragmented DNA
OU
R V
IEW
Digital DNA fragments Digital DNA fragments
DNA
Assembly

36626 - Next Generation Sequencing Analysis
Metagenomic assembly ...is even harder than single genome assembly

36626 - Next Generation Sequencing Analysis
Why?
Can you think of why?
Groups of 2-3 3 mins

36626 - Next Generation Sequencing Analysis
Why?
• Shared/similar regions between genomes works as repeats
• Different abundances
• High diversity, very large meta-genomes
• Which contigs belongs to which organism?
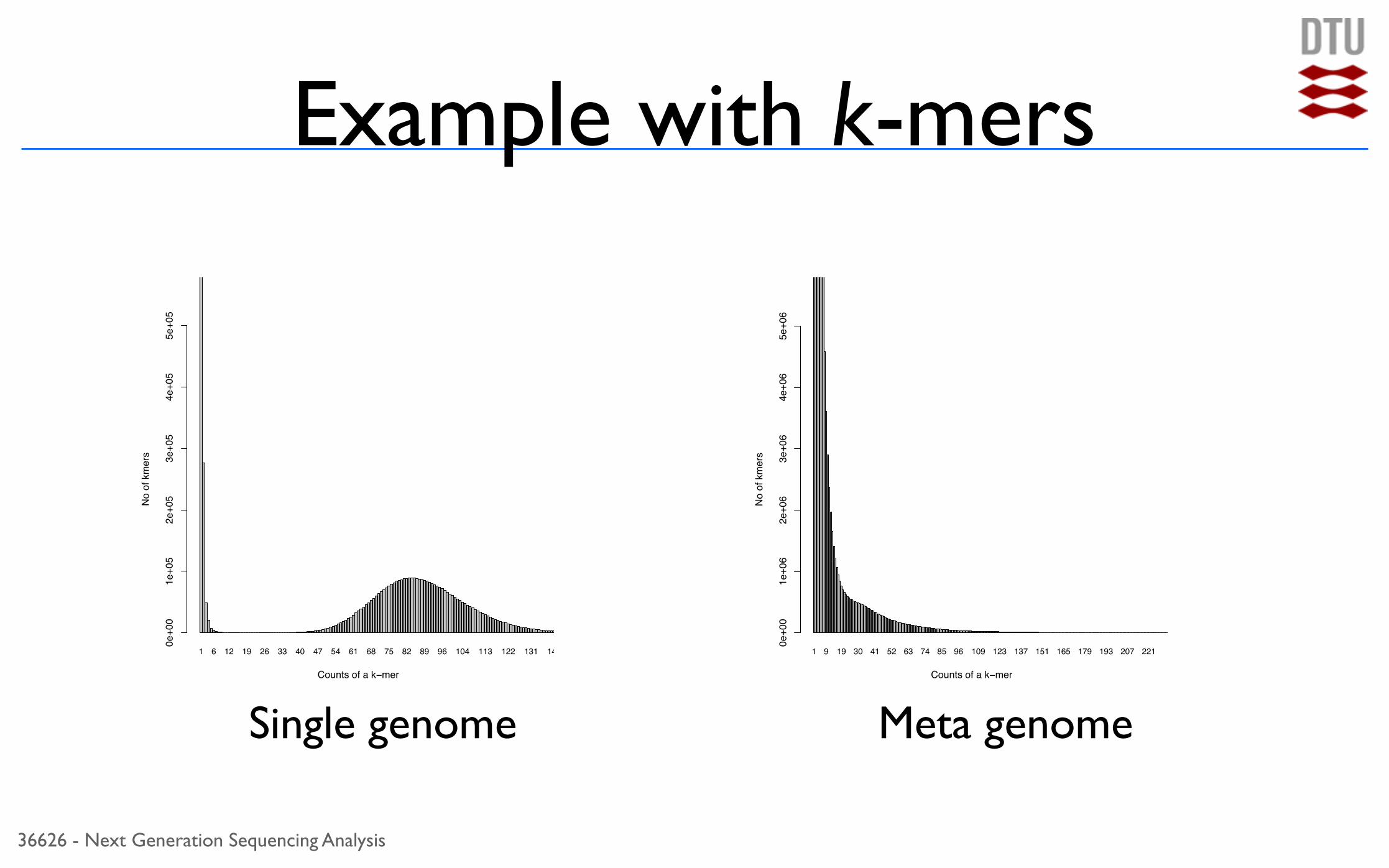
36626 - Next Generation Sequencing Analysis
Example with k-mers
1 9 19 30 41 52 63 74 85 96 109 123 137 151 165 179 193 207 221
Counts of a k−mer
No
of k
mer
s
0e+0
01e
+06
2e+0
63e
+06
4e+0
65e
+06
1 6 12 19 26 33 40 47 54 61 68 75 82 89 96 104 113 122 131 140
Counts of a k−mer
No
of k
mer
s
0e+0
01e
+05
2e+0
53e
+05
4e+0
55e
+05
Single genome Meta genome

36626 - Next Generation Sequencing Analysis
Which assembler then?
• A couple of metagenomic assemblers:
• SPAdes, Megahit, IDBA-ud (Illumina)
• MetaVelvet (Illumina, Solid, 454)

36626 - Next Generation Sequencing Analysis
Meta-approach• Differences in coverage between
genomes
• Use of mate-pair / paired end reads to dis-entagle the graph
• After assembly: Sequence composition binning
• Abundance estimation
[2] Detection of multiple peaks on k-mer frequencies:2. Calculate the empirical distribution of
‘length-weighted frequencies’ of node coverages,where a node coverage is assigned to each nodeby Velvet on the construction of the de Bruijngraph (Figure 5).
3. Approximate the empirical distribution by amixture of Poisson distributions and detectmultiple peaks in the Poisson mixture. Then, thehighest peak of expected coverage is chosen asthe ‘primary expected coverage’, and the nexthighest is chosen as the ‘secondary expectedcoverage’.
4. Classify every node into one distribution of thePoisson mixture by calculating its posterior prob-ability for the node coverage value.
[3] Decomposition of the de Bruijn graph:5. (Decomposition by connectivity) Decompose the
initial de Bruijn graph into connected subgraphs.6. (Decomposition by coverage value) If the coverage
of a node belongs to the primary expectedcoverage, the node is classified as a ‘primarynode’. Subsequently, the primary nodes arelabeled as ‘ON’ and the other nodes are labeledas ‘OFF’. Then, a chimeric node is detected as
having two incoming edges whose origin nodesare labeled ON and OFF, and two outgoingedges whose destination nodes are labeled ONand OFF, and having a coverage value mostlyequal (within 5% difference by default) to theaverage between the sum of the coverage valuesof the two origin nodes and the sum of the twodestination nodes. Second, check the consistencyof the ON and OFF labeling for the two originnodes and two destination nodes using paired-endinformation. If the consistency is satisfied, resolveevery chimeric node by separating the nodeinto two nodes with only one incoming edgeand one outgoing edge, whose origin and destin-ation nodes have the same label, as shown inFigure 4. After separating the chimeric nodes,further decompose the resulting graph into con-nected subgraphs.
7. If a connected subgraph consists of more thanx% (a predefined parameter, the default is set to100%) of nodes labeled ‘ON’, the subgraph isunmasked. All other subgraphs are masked.
[4] Assembly of contigs and scaffolds:8. Apply the Velvet functions to the unmasked
subgraphs to build contigs and then applyPebble and Rock Band functions to buildscaffolds.
9. Remove the unmasked subgraphs and recursivelyapply Step 2–8 to the remaining de Bruijn graphuntil no node remains.
It might be thought that in Substep 3 above, a chimericnode could have the highest expected coverage. However,the contigs of chimeric nodes are very short comparedwith the unique nodes; therefore, the length-weightedfrequencies of coverage values for the chimeric nodes donot form any significant peaks.
EXPERIMENTAL RESULTS
The performance of the MetaVelvet assembler was testedon simulated datasets and on real metagenome datasetsobtained from human gut microbiome. The method wascompared with the naive use of two single-genome as-semblers, Velvet (15) and SOAPdenovo (22), and therecently proposed metagenome assembler Meta-IDBA(6). Furthermore, for the simulated datasets, wecompared our results with those of a single-genomeassembly from pure sequence reads of each single-isolategenome. We compared the following standard statisticalmeasures to evaluate the performance of the assemblersfor short read assembly and metagenome assembly: thenumber of scaffolds, the total length of scaffolds andN50, where N50 indicates the scaffold length such that50% of the de novo assembled sequences lie in scaffoldsof this size or larger. The precise definition of N50 is asfollows. Let jAj denote the length of a sequence (contig,scaffold or genome) A. Let S1, S2, . . . , Sn denote the list ofscaffolds in descending order of length as output by anassembler. Let L denote the total length of all scaffolds,
90.8
28.5
59.6
31.3
62.7
expected coverageprimary : 60
secondary : 30
OFF
ON
OFF
ON
28.5
59.6
31.3
62.7
OFF
ON
OFF
ON
separating
Chimericnode
Figure 4. An example of a chimeric node and its resolution byseparating the node. The node is chimeric because [(28.5+59.6)+(31.3+62.7)]/2. 90.8.
Node coverages
Leng
th-w
eigh
ted
Freq
uenc
y
10 30 60
PrimarySecondary
Figure 5. Detection of multiple peaks in the histogram of coveragevalues of the nodes.
PAGE 5 OF 12 Nucleic Acids Research, 2012, Vol. 40, No. 20 e155
by guest on May 31, 2013
http://nar.oxfordjournals.org/D
ownloaded from

36626 - Next Generation Sequencing Analysis
Abundance estimation
• Even with metagenome assemblers it is very hard!
• Map reads back to estimate abundance on genes or contigs
Sample A Sample B
Gene 1 10 9
Gene 2 20 20
Gene 3 10 20
Gene 4 5 0
Species abundanceFunctional abundance
Re-assemblyAnalogous to RNAseq

36626 - Next Generation Sequencing Analysis
Binning
• Which contigs goes together?
• Binning:
• Similarity (alignment)
• Sequence composition & co-abundance of contigs (clustering)

36626 - Next Generation Sequencing Analysis
Similarity (alignment)
• Align (BLAST/BWA) all contigs to reference database
• Quite accurate, but
• Databases are huge! and growing
• Majority of sequences are unknown (30%-99%)
• We can not find what is not there!
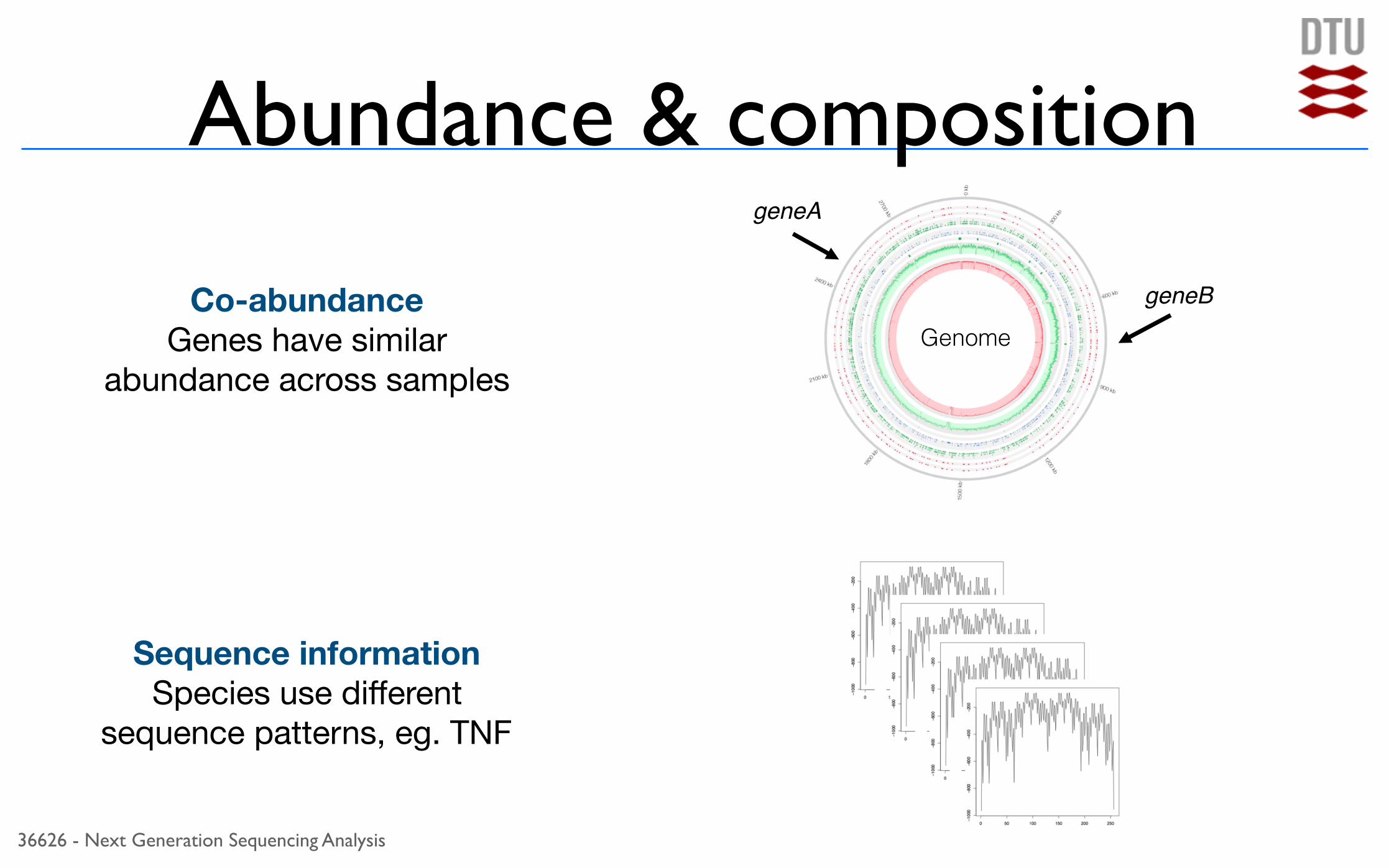
36626 - Next Generation Sequencing Analysis
Abundance & compositiongeneA
geneB
GenomeCo-abundance
Genes have similar abundance across samples
Sequence information Species use different
sequence patterns, eg. TNF

Genes/contigs = starsOrganisms = galaxies
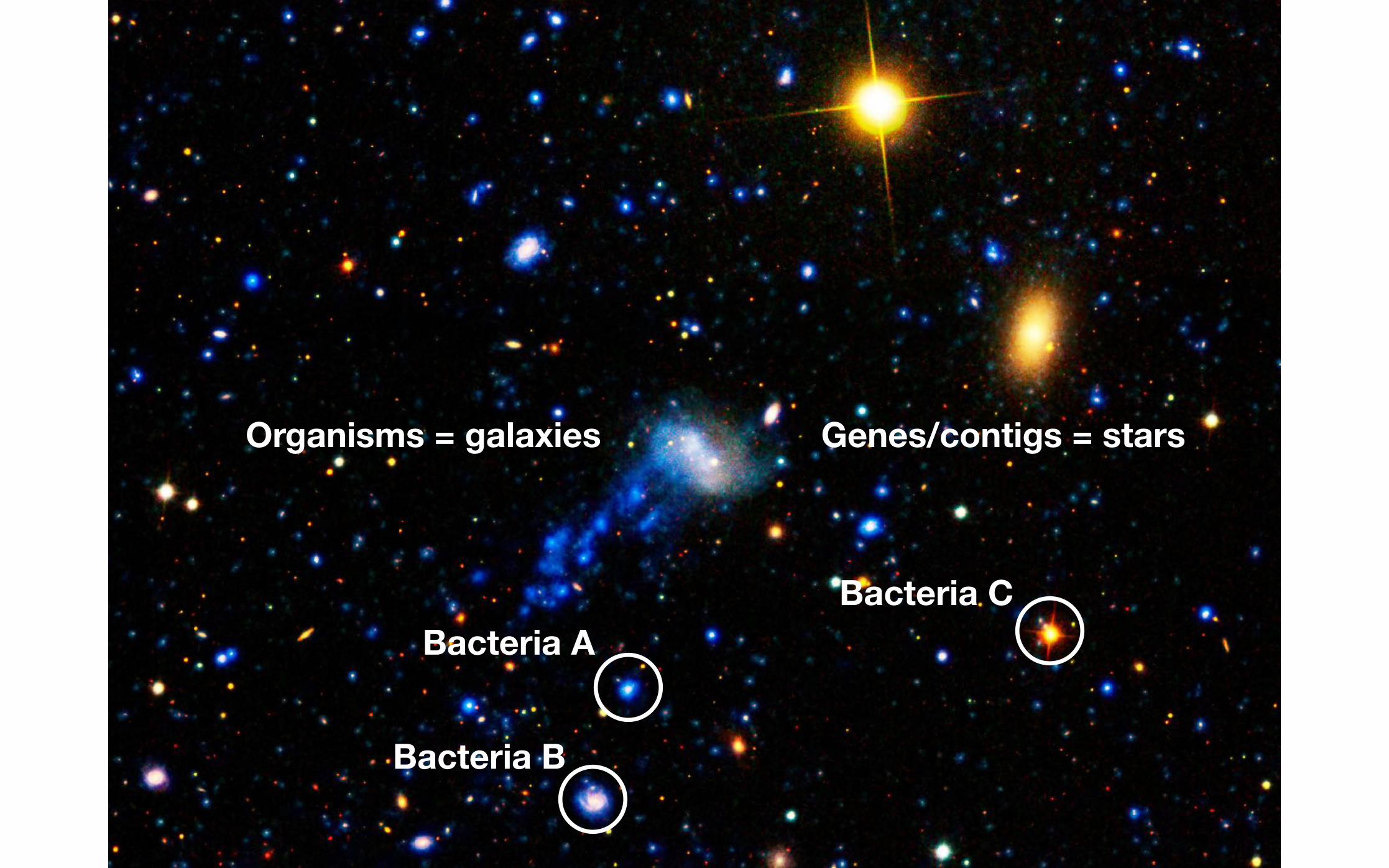
Genes/contigs = starsOrganisms = galaxies
Bacteria A
Bacteria B
Bacteria C
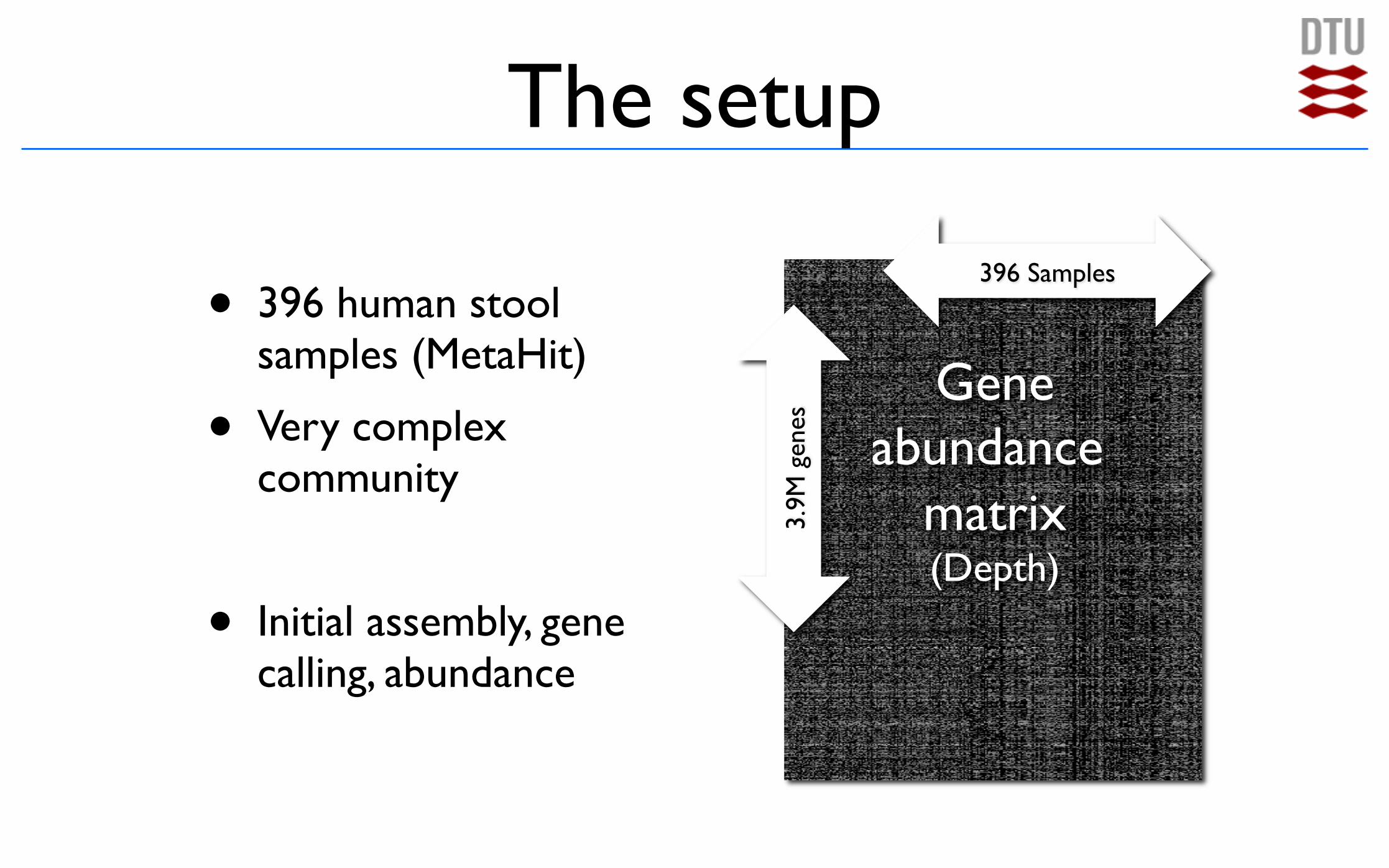
The setup
Gene!abundance
matrix!(Depth)
396 Samples
3.9M
gen
es
• 396 human stool samples (MetaHit)
• Very complex community
• Initial assembly, gene calling, abundance
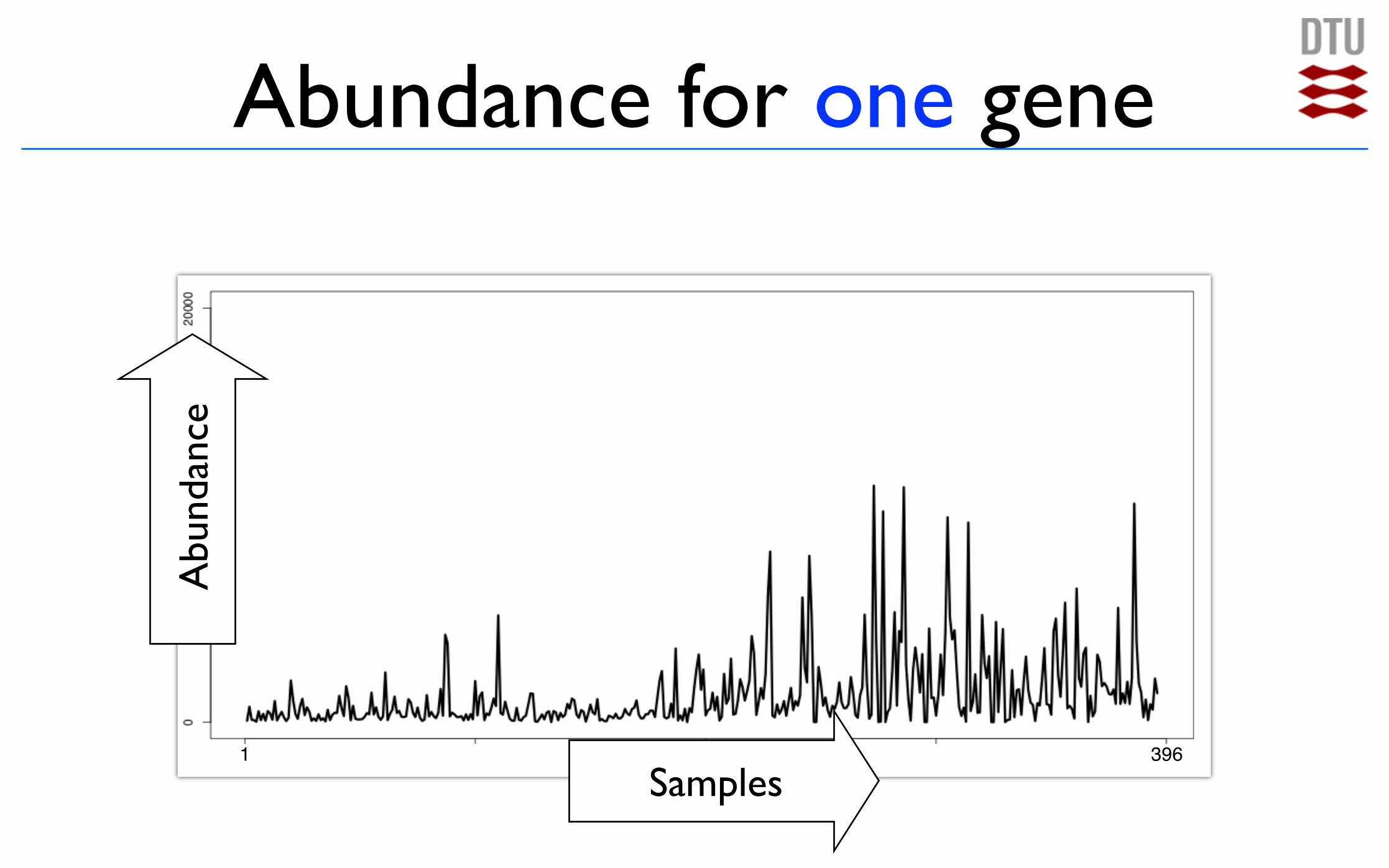
Abundance for one gene
1 396
Abu
ndan
ce
Samples
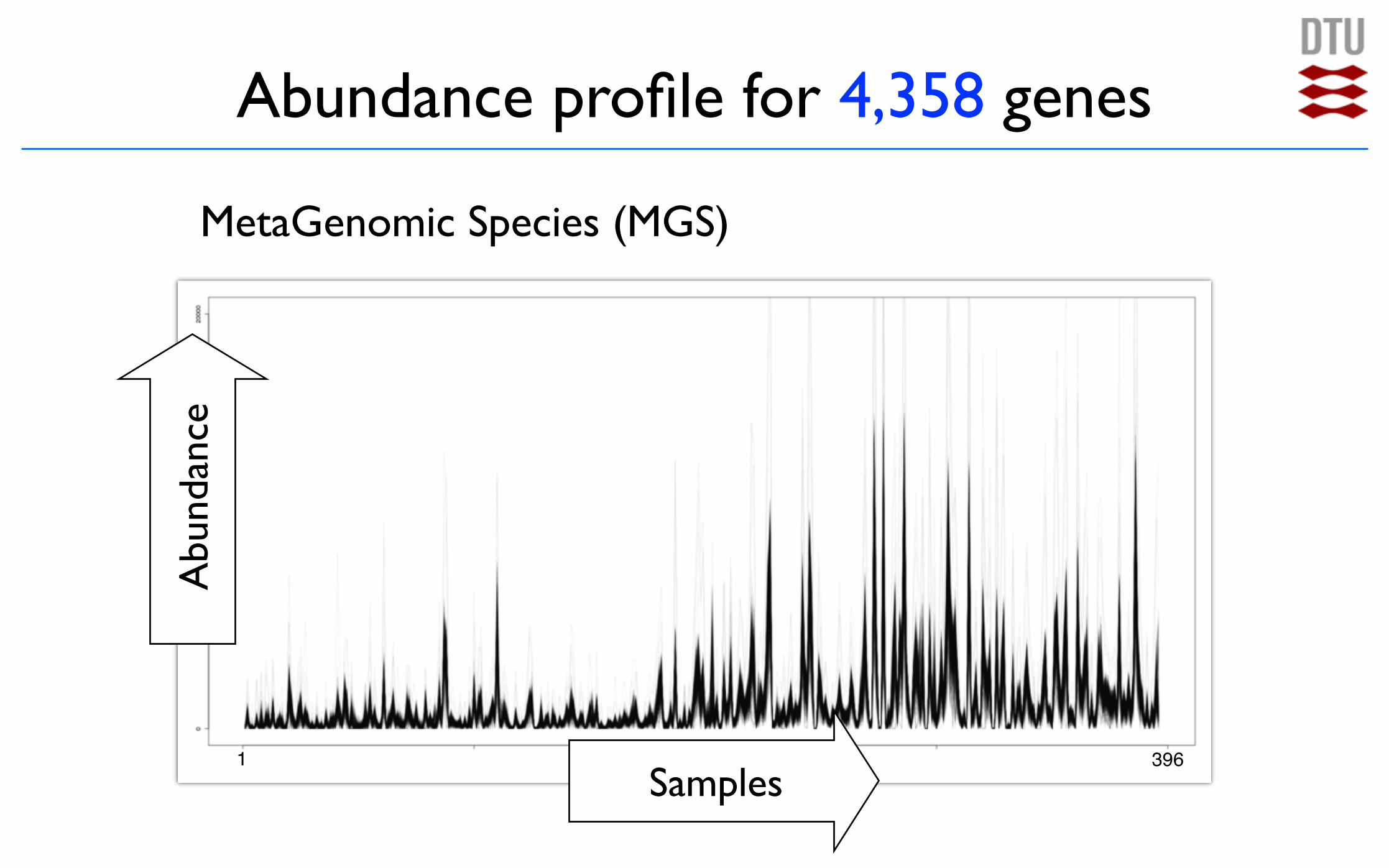
Abundance profile for 4,358 genes
1 396
Abu
ndan
ce
Samples
MetaGenomic Species (MGS)
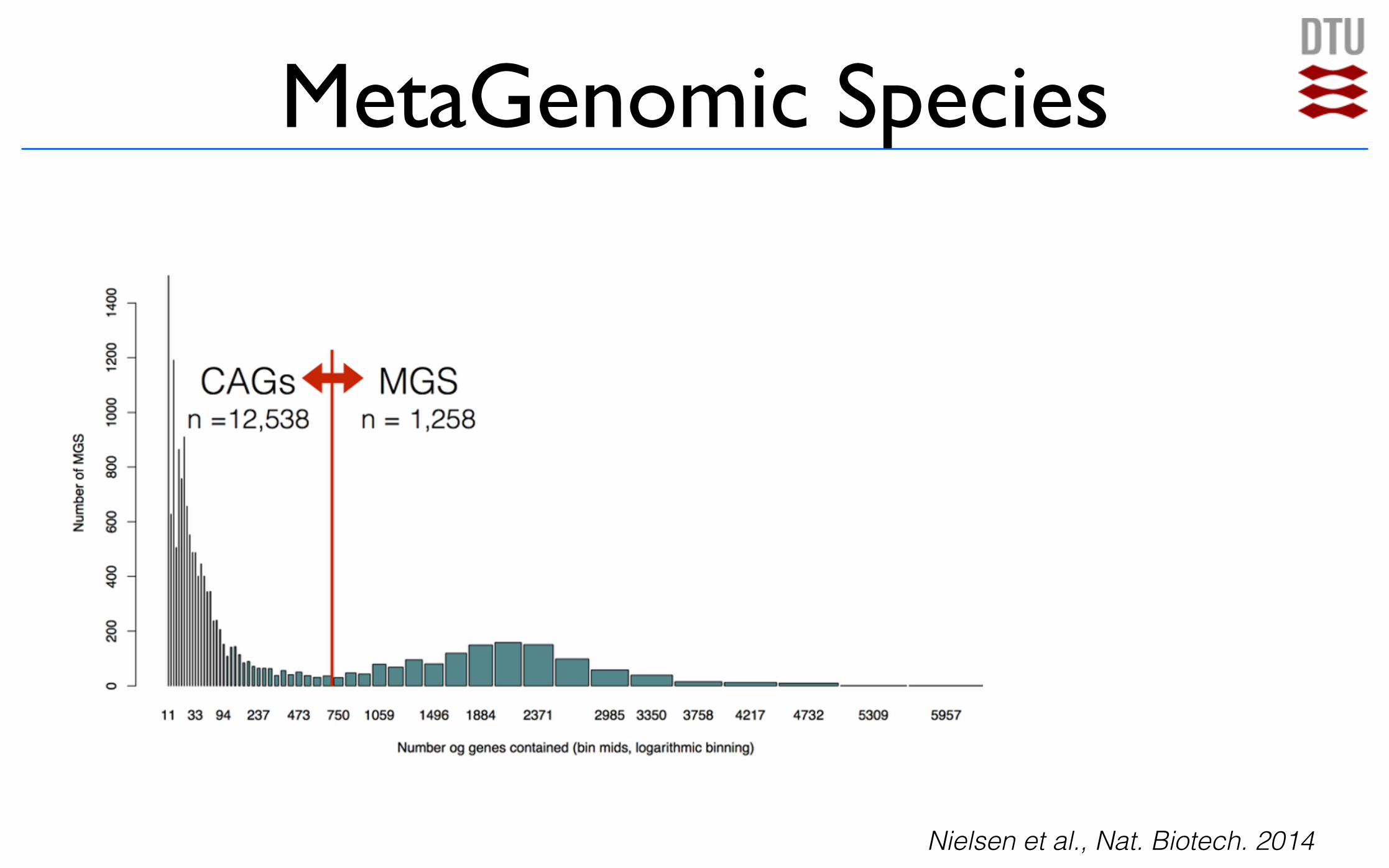
MetaGenomic Species
Nielsen et al., Nat. Biotech. 2014
Not just species gets binnedAlso plasmids and viruses... and lots of artifacts!
doi:10.1038/nbt.2939
Co-abundance groups vs metagenomic species
36626 - Next Generation Sequencing Analysis
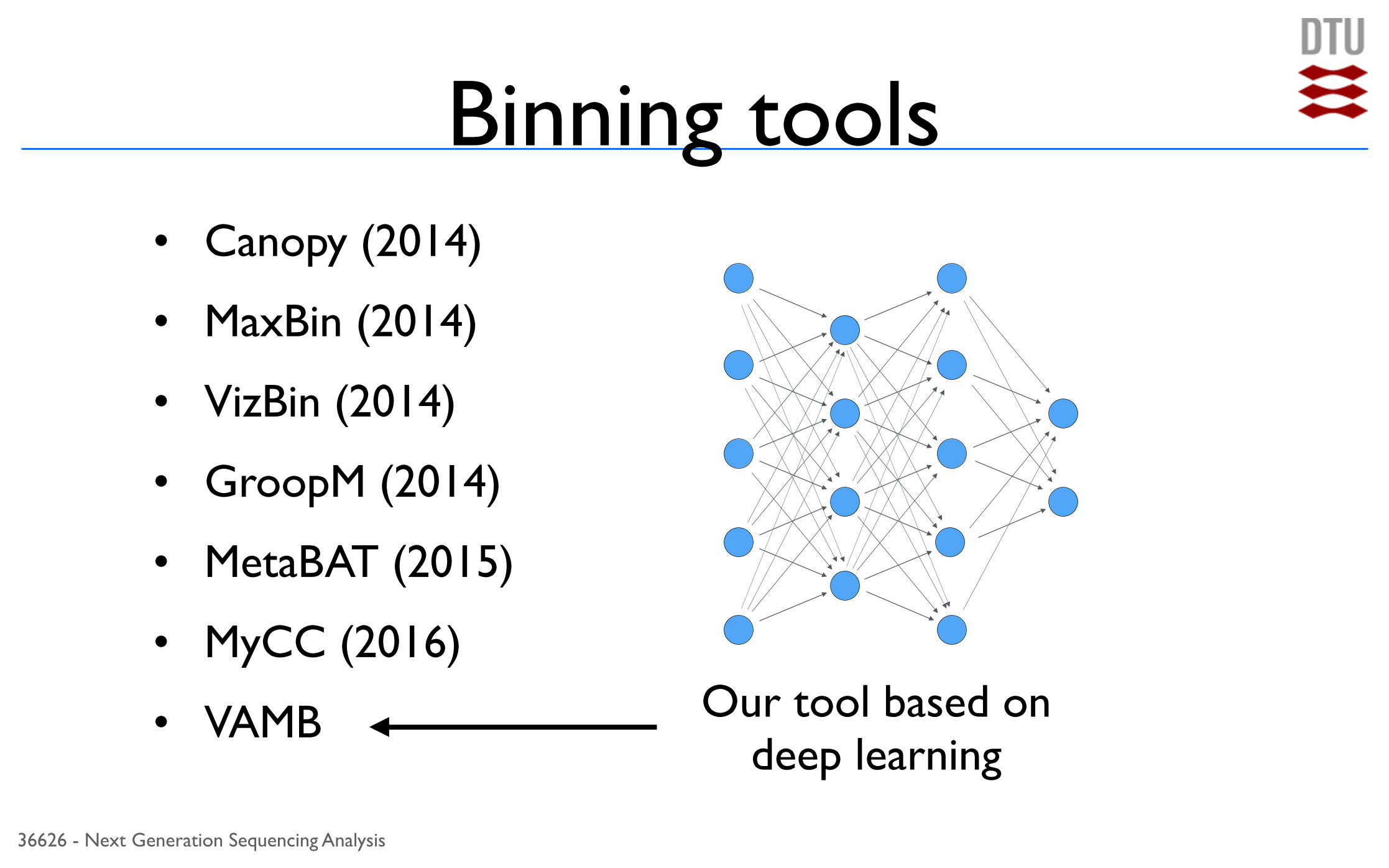
Binning tools• Canopy (2014)
• MaxBin (2014)
• VizBin (2014)
• GroopM (2014)
• MetaBAT (2015)
• MyCC (2016)
• VAMB Our tool based on deep learning





























