Embed Size (px)
Citation preview

1p36 (1p36 microdeletion syndrome)
22q11.2 (Velocardiofacial / DiGeorge syndrome)
4p16 (Wolff-Hirschorn syndrome)
5p15.2 (Cri du Chat syndrome)
7q11.23 (Williams-Beuren syndrome) Diseño 1
15q11 (Prader Willi / Angelman syndrome [UBE3A])
15q11-q13 (Prader Willi / Angelman syndrome [SNRPN])
16p13.3 (ATR syndrome)
17p11.2-p12 (Charcot Marie Tooth / HNPP syndrome)
17p11.2 (Smith Magenis syndrome)
17p13.3 (Miller Dieker syndrome)
21q21.3 (APP locus duplication [Alzheimer])
Xp22.3 (X-linked ichthyosis syndrome)
17q11.2 (Neurofibromatosis Type1)
Xp22.31 (Kallman)Yp11.31 (SRY Microdeletion)
12p (Pallister Killian)
Microdeleciones-Microduplicaciones –SMD-
7q11.23 (Williams-Beuren syndrome) Diseño 2
21q22 (Down Syndrome Critical Region])

El Síndrome de Microdeleción 1p36 es el más común de los Síndromes de Deleción Terminal, afectando 1/5000 nacimientos. La región distal de 1p es muy rica en genes por lo que hay gran cantidad de posibles candidatos que podrían estar involucrados en esta anomalía. LIVe ofrece un kit
que abarca dos regiones críticas cuya deleción parece ser clave en la ocurrencia de este síndrome (Heilstedt
et al. 2003). El control positivo de reacción es 1qter.
Citas (References): Population data suggest that deletions of 1p36 are a relatively common chromosome abnormality. Heilstedt HA, Ballif BC, Howard LA, Kashork CD, Shaffer LG. Clin
Genet. 2003 Oct;64(4):310-6.
Physical map of 1p36, placement of breakpoints in monosomy
1p36, and clinical characterization of the síndrome. Heilstedt HA, Ballif BC, Howard LA, Lewis RA, Stal S, Kashork CD, Bacino CA, Shapira SK, Shaffer LG. Am J Hum Genet. 2003 May;72(5):1200-12. Epub
2003 Apr 8.
CDC2L2
1p 36
FLJ13052
CDC2L1
GNB1
SLC35E2
110K
B11
9KB
SHGC-79970
D1S1372
AL033717
1q44
Sonda Microdeleción 1p36
Lexel in Vitrowww.lexelmedical.com
Inicio Microdeleciones

4p 16.3 CTBP1
SPON2
MGC21675
LOC285498
FLJ35816
LETM1
WHSC1
W-H
regi
onII
La región crítica del Síndrome de microdeleción Wolf-Hirschhorn
se encuentra en 4p16. El genotipo-fenotipo de este síndrome se
correlaciona con el tamaño de la región delecionada (deleciones menores a 3.5Mb resultan en fenotipos sin malformaciones físicas (Zollino
et al.). En el 95% de los casos estas microdeleciones pueden ser detectadas por FISH con sondas específicas. LIVe ofrece en su kit
dos sondas que hibridan sobre regiones críticas de esta anomalía
, aumentando notablemente la definición del análisis. Además, un control positivo hibrida sobre 4q12.
-Am J Med Genet. 2000;94;254-61
W-H
regi
onI
600Kb
Lexel in Vitrowww.lexelmedical.com
Sonda Microdeleción Wolff-Hirschhorn
4q 12
Inicio Microdeleciones

5p15.2
600K
B
SEMA5A
AF009306BC047112
TAS2R1
SNORD123
D5S721
D5S23
La mayoría de las deleciones del brazo corto del cromosoma 5 son asociadas con el síndrome de Cri
du
Chat. Se sabe que existe una severidad progresiva de manifestaciones clínicas y retardo físico-motor relacionada directamente con el incremento del tamaño de la deleción (Mainardi
et al. 2001). La sonda LIVe 5p15.2 hibrida sobre marcadores claves en esta microdeleción. Esta sonda está
acompañada por la sonda EN 5 (5 q11.2)
Citas
(Referentes):Clinical and molecular characterisation
of 80 patients with 5p deletion: genotype-phenotype correlationP Cerruti Mainardi, C Perfumo, A Calì, G Coucourde, G Pastore, S Cavani, F Zara, J Overhauser,M Pierluigi, F Dagna BricarelliJ Med Genet 2001;38:151–158
Lexel in Vitrowww.lexelmedical.com
Sonda Microdeleción Cri-Du-Chat
5q11.2
Inicio Microdeleciones
Lexel in Vitrowww.lexelmedical.com
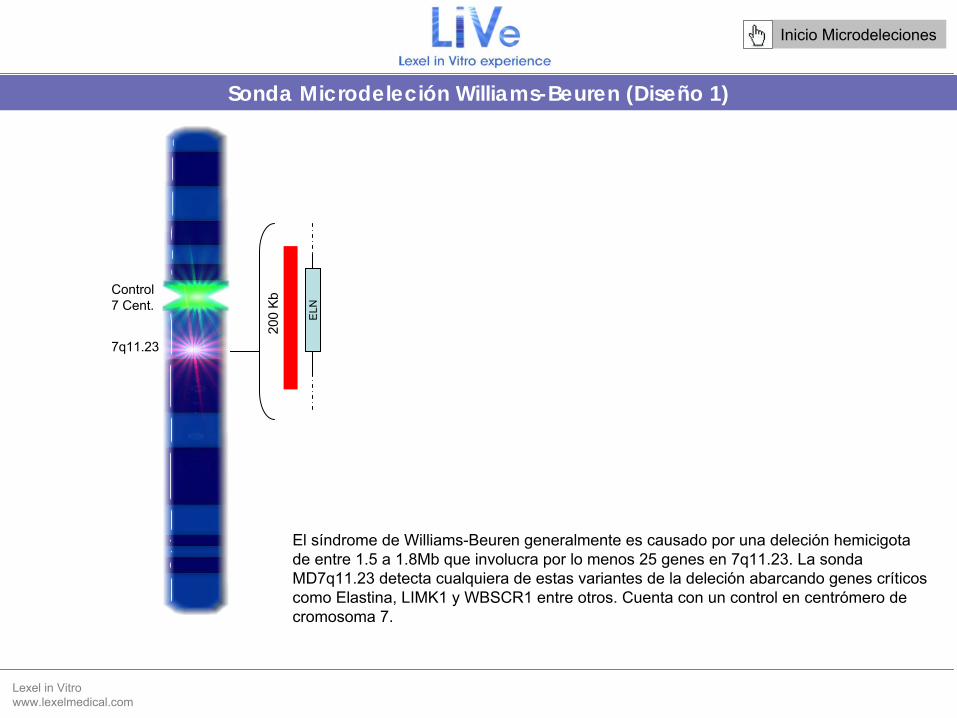
Sonda Microdeleción Williams-Beuren (Diseño 1)
Control 7 Cent.
200
Kb
ELN
El síndrome de Williams-Beuren
generalmente es causado por una deleción hemicigota
de entre 1.5 a 1.8Mb que involucra por lo menos 25 genes en 7q11.23. La sonda MD7q11.23 detecta cualquiera de estas variantes de la deleción abarcando genes críticos como Elastina, LIMK1 y WBSCR1 entre otros. Cuenta con un control
en centrómero de cromosoma 7.
7q11.23
Inicio Microdeleciones

Lexel in Vitrowww.lexelmedical.com
Sonda Microdeleción Williams-Beuren (Diseño 2)
El síndrome de Williams-Beuren generalmente es causado por una deleción hemicigota de entre 1.5 a 1.8 Mb que involucra por lo menos 25 genes en 7q 11.23. LIVe ofrece un kit con dos sondas para secuencias críticas de esta región, abarcando, tanto el gen de la elastina (ELN) como otros genes involucrados en esta anomalía. Un control sobre 7p22.3 completa el kit.
Citas (References):Osborne LR, Li M, Pober B, Chitayat D, Bodurtha J, Mandel A, Costa T, Grebe T, Cox S, Tsui LC, Scherer SW . A 1.5 million-base pair inversion polymorphism in families with Williams-Beuren syndrome. Nat Genet. 2001;29;321-5.
Schubert C, Laccone F. Williams-Beuren syndrome: determination of deletion size using quantitative real-time PCR. Int J Mol Med. 2006 Nov;18(5):799-806.
Gilbert-Dussardier B. Williams-Beuren syndrome. Rev Prat. 2006 Dec 15;56(19):2102-6.
El síndrome de Williams-Beuren generalmente es causado por una deleción hemicigota de entre 1.5 a 1.8 Mb que involucra por lo menos 25 genes en 7q 11.23. LIVe ofrece un kit con dos sondas para secuencias críticas de esta región, abarcando, tanto el gen de la elastina (ELN) como otros genes involucrados en esta anomalía. Un control sobre 7p22.3 completa el kit.
Citas (References):Osborne LR, Li M, Pober B, Chitayat D, Bodurtha J, Mandel A, Costa T, Grebe T, Cox S, Tsui LC, Scherer SW . A 1.5 million-base pair inversion polymorphism in families with Williams-Beuren syndrome. Nat Genet. 2001;29;321-5.
Schubert C, Laccone F. Williams-Beuren syndrome: determination of deletion size using quantitative real-time PCR. Int J Mol Med. 2006 Nov;18(5):799-806.
Gilbert-Dussardier B. Williams-Beuren syndrome. Rev Prat. 2006 Dec 15;56(19):2102-6.
70 K
b
WB
SC
R23
GTF
2IR
D1
ELN
LIM
K1
NTA
L
200
Kb
7q11.23
7p22.3
Inicio Microdeleciones

12p13.33
156
Kb
Control12qter
Sonda Duplicación Pallister-Killan
Lexel in Vitrowww.lexelmedical.com
El síndrome Pallister-Killan
es causado por un desorden cromosómico caracterizado por tetrasomía
12p como consecuencia de un isocromosoma. Este material extra es generalmente encontrado en mosaico y su presencia está
casi exclusivamente restringida a fibroblastos, lo cual permite detectar tempranamente esta anomalía en muestras de líquido amniótico.LIVe
diseñó
el kit
MD12p
compuesto por una sonda específica para 12p33 y un control sobre 12qter. Este kit
está
especialmente optimizado para lograr resultados óptimos en muestras de isopado
bucal o cualquier tipo de muestra celular sin procesar (células con citoplasma).
Inicio Microdeleciones

15q11-q13
2.4Mb
200K
b20
0Kb
UBE3A
D15S839
D15S164E
CYFIP1
D15S1035
D15S18
BP1
BP2
BP3
Los desórdenes genómicos de imprinting
conocidos como Prader-Willi
/ Angelman
están asociados a alteraciones genómicas de la región 15q11-q13. Aproximadamente el 70% de los pacientes presentan una microdeleción intersticial de 5-7Mb en esta región. LIVe ofrece dos kits con sondas para la región 15q11-q13 que hibridan sobre genes claves cercanos a los 3 puntos de rupturas (BP1, 2, y 3) eventualmente involucrados en los síndromes de Prader
Willi
/ Angelman. Estas sondas están diseñadas para examinar más de una región por vez, maximizando la definición del análisis. 15q26.3 se utiliza como control positivo en ambos kits.
Control15q26.3
Lexel in Vitrowww.lexelmedical.com
Sonda Microdeleción Prader Willi / Angelman (UBE3A)
Inicio Microdeleciones

1Mb
130K
b18
0Kb
BP2
BP3
SNRPN
SNURF
MKRN3
Lexel in Vitrowww.lexelmedical.com
Los desórdenes genómicos de imprinting
conocidos como Prader-Willi
/ Angelman
están asociados a alteraciones genómicas de la región 15q11-q13. Aproximadamente el 70% de los pacientes presentan una microdeleción intersticial de 5-7Mb en esta región. LIVe ofrece dos kits con sondas para la región 15q11-q13 que hibridan sobre genes claves cercanos a los 3 puntos de rupturas (BP1, 2, y 3) eventualmente involucrados en los síndromes de Prader
Willi
/ Angelman. Estas sondas están diseñadas para examinar más de una región por vez, maximizando la definición del análisis. 15q26.3 se utiliza como control positivo en ambos kits.
Sonda Microdeleción Prader Willi / Angelman (SNRPN)
15q11-q13
Control15q26.3
Inicio Microdeleciones
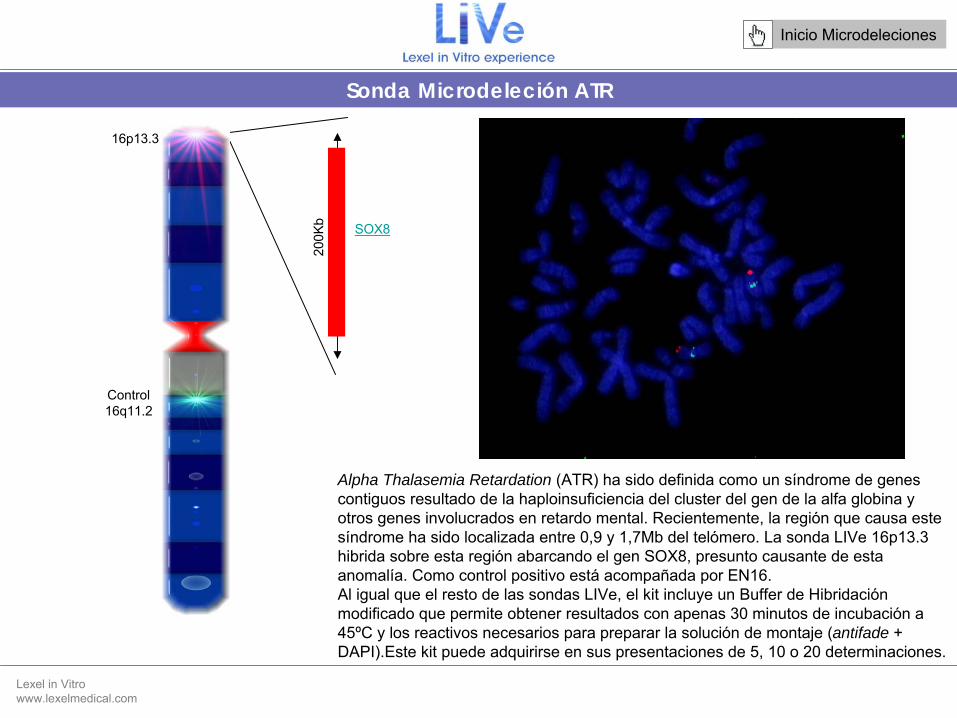
Alpha Thalasemia Retardation (ATR) ha sido definida como un síndrome de genes contiguos resultado de la haploinsuficiencia
del cluster del gen de la alfa globina y otros genes involucrados en retardo mental. Recientemente, la región que causa este síndrome ha sido localizada entre 0,9 y 1,7Mb del telómero. La sonda LIVe 16p13.3 hibrida sobre esta región abarcando el gen SOX8, presunto causante de esta anomalía. Como control positivo está
acompañada por EN16.Al igual que el resto de las sondas LIVe, el kit
incluye un Buffer de Hibridación modificado que permite obtener resultados con apenas 30 minutos de incubación a 45ºC
y los reactivos necesarios para preparar la solución de montaje (antifade + DAPI).Este kit
puede adquirirse en sus presentaciones de 5, 10 o 20 determinaciones.
16p13.3
200K
b SOX8
Control16q11.2
Lexel in Vitrowww.lexelmedical.com
Sonda Microdeleción ATR
Inicio Microdeleciones

LIS1
17p13.3
MET10
ControlCent
17
El Sindrome
Miller-Dieker
es una afección de malformaciones múltiples, caracterizado por licencefalia
y asociado con deleciones visibles o submicroscópicas en 17p13.3. Estas deleciones varían en tamaño entre 0,1 y 2,9Mb y siempre involucran al gen LIS1. La sonda LIVe 17p13.3 hibrida sobre el gen LIS1 y viene acompañada por la sonda de enumeración EN17 en 17p11.2 como control de reacción.Al igual que el resto de las sondas LIVe, el kit
incluye un Buffer de Hibridación modificado que permite obtener resultados con apenas 30 minutos de incubación a 45ºC
y los reactivos necesarios para preparar la solución de montaje (antifade + DAPI).Este kit
puede adquirirse en sus presentaciones de 5, 10 o 20 determinaciones.
Lexel in Vitrowww.lexelmedical.com
200K
b
Sonda Microdeleción Miller-Dieker
Inicio Microdeleciones

17p11.2-12
Entre el 70-80% de todos los casos de CMT tipo 1 son de la variante A (CMT1A) e involucra anomalías en el gen PMP22. Los casos de CMT1A son causados por la
duplicación de una región de 1.5Mb en la región 17p12 que contiene al gen PMP22. El producto recíproco de esta duplicación es una deleción del mismo tamaño que causa una neuropatía hereditaria denominada HNPP. La sonda LIVe 17p12 hibrida sobre
el gen PMP22 y está
diseñada para analizar metafases e interfases permitiendo detectar tanto la duplicación como la deleción de este gen. Como control está
acompañada por una sonda ubicada a 2.3Mb (que incluye el locus RAI).
2.3M
b
Lexel in Vitrowww.lexelmedical.com
Sonda Microduplicación 17p12 Charcot Marie Tooth (1A) / Microdeleción HNPP
PMP2
2R
AI1
Inicio Microdeleciones

17p11.2 95Kb
Todas las deleciones en 17p11.2 asociadas con el síndrome de Smith Magenis
(SMS) incluyen la deleción del gen RAI1. Por otro lado, estudios recientes detectaron que otros genes dentro de la misma región contribuyen a las características variables y la severidad general de este síndrome. La sonda LIVe
17p11.2 está
diseñada para detectar esta deleción con alta eficacia ya que no solamente abarca genes críticos de la región sino que incluye específicamente el gen RAI1. Como control de reacción se incluye la sonda Centromérica 17.
RA
I1
Lexel in Vitrowww.lexelmedical.com
Sonda Microdeleción Smith Magenis
ControlCent 17
Inicio Microdeleciones

459
Kb
17q11.2
La Neurofibromatosis Tipo 1 (NF-1) es un desorden autosómico
dominante con una incidencia de 1 en 2500 nacidos vivos. Aproximadamente 5% de los
pacientes presentan deleciones del gen NF-1 y otros genes contiguos, encontrándose una relación directa entre el tamaño de la deleción y la severidad fenotípica. Se han caracterizado 2 tipos de eventos que resultan en deleción del gen NF1. La deleción tipo I ocurre por recombinación intercromosómica
durante la meiosis, la denominada tipo II está
mediada por recombinación intracromosómica
en la mitosis. Esta última puede generar mosaicos somáticos.Lexel In vitro
ofrece un kit
de sondas específicas apuntadas a detectar la pérdida del gen NF1 como consecuencia de deleción cromosómica. Esta sonda está
acompañada de un control en la región centromérica 17.
Lexel in Vitrowww.lexelmedical.com
Sonda Microdeleción Neurofibromatosis Tipo 1
NF1
Inicio Microdeleciones

Lexel in Vitrowww.lexelmedical.com
Sonda Microduplicación 21q21.3 Early Onset Alzheimer
21q21.3AP
P
170
Kb
Estudios recientes han detectado que la duplicación del gen APP (Amyloid Precursor Protein) en el cromosoma 21 está
relacionado con la aparición temprana de la enfermedad de Alzheimer. Una duplicación de entre 0.58 y 6.37Mb se encontró
en pacientes con abundantes depósitos de peptidos
β-amiloides. Se ha comprobado que sondas de FISH específicas para el locus APP detectan correctamente esta duplicación. LIVe
ofrece la sonda 21q21.3 que hibrida completamente sobre el locus APP. Como
control de reacción está
acompañada por 21q22.3. Al igual que el resto de las sondas LIVe, el kit
incluye un Buffer de Hibridación modificado que permite obtener resultados con apenas 30 minutos de incubación a 45ºC
y los reactivos necesarios para preparar la solución de montaje (antifade + DAPI).Este kit
puede adquirirse en sus presentaciones de 5, 10 o 20 determinaciones.
Control21q22.3
Inicio Microdeleciones

Lexel in Vitrowww.lexelmedical.com
Duplicación 21q22 Región Crítica Sindrome Down
316
Kb
DS
CR
4D
SC
R8
ERG
21q22.2La sonda MD21q22
ha sido específicamente diseñada para la detección de dos regiones críticas del cromosoma 21 cuya ganancia está
directamente asociada con el Síndrome de Down. Esta sonda hibrida sobre 316kb abarcando completamente las regiones Down Syndrome Critical Region 2 y 4. Al igual que el resto de las sondas LIVe, el kit
incluye un Buffer de Hibridación modificado que permite obtener resultados con apenas 30 minutos de incubación a 45ºC
y los reactivos necesarios para preparar la solución de montaje (antifade + DAPI).Este kit
puede adquirirse en sus presentaciones de 5, 10 o 20 determinaciones
Células de Material de Aborto hibridadas con MD21q22. Izq. Célula normal, Der. Célula con ganancia para la región 21q22
Inicio Microdeleciones

22q11.22
22q13.32
Los síndromes de DiGeorge
y Velo-cardio-facial (DGS/VCFS) son causados por una microdeleción hemicigota
de entre 1.5 y 3Mb en el cromosoma 22 (q11.2). Se presume que dos de los principales genes responsables de la mayoría de las malformaciones físicas serían el gen TBX1 y el HIRA (también llamado TUPLE1). LIVe
ofrece la sonda MD22q11.2 que hibrida sobre los loci
TBX1 y Tuple1 (o HIRa) y contiene además un
control de reacción
sobre
22qter. Al igual que el resto de las sondas LIVe, el kit
incluye un Buffer de Hibridación modificado que permite obtener resultados con apenas 30 minutos de incubación a 45ºC
y los reactivos necesarios para preparar la solución de montaje (antifade + DAPI).Este kit
puede adquirirse en sus presentaciones de 5, 10 o 20 determinaciones.
Sonda Microdeleción Velocardiofacial o DiGeorge 22q11.2
Lexel in Vitrowww.lexelmedical.com
200
Kb
TBX
1H
IRA
Inicio Microdeleciones

170K
b
DXS6767
DXS7434E11
5Kb
DXS1354
DXS7956
La Ictiosis ligada al X (XLI), con una prevalencia estimada de 1/6000, es un desorden genético causado por un déficit en la enzima sulfatasa
esteroide (STS). Cerca del 90% de las pacientes con Ictiosis pierden el gen STS entero. La mayoría de estos pacientes con STS delecionada sólo tienen las características clínicas de XLI, pero hay casos con desórdenes mas complejos, incluyendo retardo mental, que son el resultado de deleciones intersticiales o terminales que también incluyen el gen VCX. La sonda LIVe Xp22.3 X-linked ichthyosis region probe hibrida no solo sobre el gen STS, sino también sobre VCX aumentando la definición del análisis. Como control de reacción está
acompañada por la sonda EN X (Xp11.2).
Xp22.3
Xp11.2
Lexel in Vitrowww.lexelmedical.com
STS
VCX
Sonda Microdeleción X-linked ichthyosis
Inicio Microdeleciones

160K
b
Xp22.31
Xp11.2
El síndrome de genes contiguos (CGS) presenta una serie de características clínicas como resultado de deleciones intersticiales o terminales de varios genes adyacentes. Varios genes importantes han sido identificados en la región Xp22.3 como responsables de enfermedades genéticamente heterogéneas, uno de ellos es KAL1. Se ha demostrado que las mutaciones en este gen ubicado en Xp22.3 resulta en la forma del síndrome ligada al cromosoma X. Varios tipos de anomalías genéticas en KAL1 han sido reportados en pacientes con síndrome de Kallman, incluyendo mutaciones missense y nonsense, mutaciones del sitio de empalme, deleciones intragénicas
y deleciones cromosómicas submicroscópicas
involucrando todo el gen KAL1.
Lexel in vitro
ofrece un kit
específico cuya sonda principal hibrida sobre la región del gen Kal1 y una sonda control en Xp11.2 (ENX).
Lexel in Vitrowww.lexelmedical.com
Sonda Microdeleción Kallman
Kal
1
DXS7470
DXS1090
Inicio Microdeleciones

Yp11.31
130K
bControlYq11.21
El gen SRY (Yp11.3) está
involucrado en diversos eventos relacionados al sexo. A escala cromosómica presenta principalmente dos anomalías, deleción y translocación. La deleción de la región cromosómica conteniendo este gen es un importante factor de riesgo en pacientes con Disgenecia
Gonadal completa ya que genera Carcinoma in situ y Gondadoblastoma
en el 10-15% de los casos. Por otro lado la translocación de este gen ya sea al cromosoma X o, menos frecuentemente a cualquier autosoma, provoca cariotipos masculinos 46,XX. Las consecuencias fenotípicas de estas anomalías pueden ser diversas pero frecuentemente conllevan infertilidad.Esta sonda está
acompañada por un control sobre Yq11.21
SRY
Lexel in Vitrowww.lexelmedical.com
Sonda Microdeleción / Translocación Locus SRY
Inicio Microdeleciones





























