Embed Size (px)
Citation preview

Progress in Neurobiology 89 (2009) 277–287
The influence of microglia on the pathogenesis of Parkinson’s disease
Caitrıona M. Long-Smith, Aideen M. Sullivan, Yvonne M. Nolan *
Department of Anatomy and Neuroscience, University College Cork, Cork, Ireland
Contents
1. Parkinson’s disease . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 278
1.1. Symptoms of Parkinson’s disease. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 278
1.2. Pathological features of Parkinson’s disease . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 278
2. Microglia . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 278
2.1. Microglia in the healthy brain . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 279
2.2. Microglial activation . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 279
2.2.1. Activating stimuli . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 279
2.2.2. Consequences of microglial activation . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 279
2.2.3. Reactive microgliosis . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 280
3. Role of inflammation in Parkinson’s disease . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 280
3.1. Inflammation in Parkinson’s disease patients . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 280
3.1.1. Activated microglia in Parkinson’s disease . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 281
3.1.2. Pro-inflammatory cytokines in Parkinson’s disease . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 281
3.2. Inflammation in animal models of Parkinson’s disease . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 281
3.2.1. MPTP model. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 281
3.2.2. 6-OHDA model. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 281
3.2.3. LPS model . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 282
4. Anti-inflammatory therapies . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 283
5. Conclusion . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 284
References . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 284
A R T I C L E I N F O
Article history:
Received 14 May 2009
Received in revised form 8 August 2009
Accepted 10 August 2009
Keywords:
Parkinson’s disease
Inflammation
Microglia
Anti-inflammatory therapies
Interleukin-1bCytokine
Animal models
A B S T R A C T
Parkinson’s disease (PD) is characterised by degeneration of dopaminergic neurons in the substantia
nigra pars compacta (SNpc). Inflammation may be associated with the neuropathology of PD due to the
following accumulating evidence: excessive microglial activation and increased levels of the pro-
inflammatory cytokines tumour necrosis factor-a and interleukin-1b in the SNpc of patients with PD;
the emergence of PD-like symptoms following influenza infection; the increased susceptibility to PD
associated with bacterial vaginosis; the presence of inflammatory mediators and activators in animal
models of PD; the ability of anti-inflammatory drugs to decrease susceptibility to PD; and the emerging
possibility of the use of microglial activation inhibitors as a therapy in PD. In this review, we will discuss
the role of inflammation in PD. We will focus on the influence of microglia in the pathogenesis of PD and
discuss potential therapeutic interventions for PD, that target microglia.
� 2009 Elsevier Ltd. All rights reserved.
Abbreviations: 6-OHDA, 6-hydroxydopamine; AD, Alzheimer’s disease; APC, antigen-presenting cells; ATP, adenosine triphosphate; BBB, blood–brain barrier; BDNF, brain-
derived neurotrophic factor; CNS, central nervous system; COX, cyclooxygenase; DAT, dopamine transporter; GDNF, glial cell-line-derived neurotrophic factor; IL,
interleukin; IL-1ra, IL-1R antagonist; LB, Lewy body; LPS, lipopolysaccharide; MHC, major histocompatibility complex; MMP-3, matrix metalloproteinase-3; MPP+, 1-methyl-
4-phenylpyridinium; MPTP, 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine; MS, multiple sclerosis; NADPH, nicotinamide adenine dinucleotide phosphate; NF-kB, nuclear
Contents lists available at ScienceDirect
Progress in Neurobiology
journa l homepage: www.e lsev ier .com/ locate /pneurobio
factor kappa B; NO, nitric oxide; NOS, nitric oxide synthase; NSAID, non-steroidal anti-inflammatory drug; PD, Parkinson’s disease; PEP, post-encephalytic parkinsonism;
PPARg, proliferator-activated receptor-g; ROS, reactive oxygen species; SAID, steroidal anti-inflammatory drug; SNpc, substantia nigra pars compacta; TLR, toll-like receptor;
TNF-a, tumour necrosis factor-g; TNFR, TNF receptor.
* Corresponding author. Tel.: +353 21 490 2787; fax: +353 21 427 3518.
E-mail address: [email protected] (Y.M. Nolan).
0301-0082/$ – see front matter � 2009 Elsevier Ltd. All rights reserved.
doi:10.1016/j.pneurobio.2009.08.001

C.M. Long-Smith et al. / Progress in Neurobiology 89 (2009) 277–287278
1. Parkinson’s disease
Parkinson’s disease (PD) was first described in 1817 by Dr.James Parkinson, a London physician, in ‘An Essay on the ShakingPalsy’ (Parkinson, 1817). It is the second most common neurode-generative disorder, after Alzheimer’s disease (AD). The incidenceof PD is age-related; in developed countries, it affects approxi-mately 1% of the population aged over 60 years (Nussbaum andEllis, 2003) and increases sharply with age after 60 years, rising toover 4% in the oldest populations (de Lau and Breteler, 2006). It hasbeen estimated that the number of people aged over 60 willincrease within the next 50 years, from 11% of the total populationin 2007 to 22% in 2050 (http://www.un.org). In 90–95% of cases, PDoccurs in an idiopathic manner, whilst in the remaining 5–10% ofcases, a genetic mutation is present (Toulouse and Sullivan, 2008).There is currently no cure for PD, thus it is imperative that researchon the causes and treatments of this disease is supportedappropriately within the coming years, both for humanitarianand economic purposes.
1.1. Symptoms of Parkinson’s disease
PD primarily affects areas of the brain which are involved inmotor control, and initially manifests clinically as a slight rhythmictremor, usually of a limb (Fahn, 2003; Wolters, 2008). As thedisease progresses, bradykinesia, tremor at rest, gait disturbances,postural instability and rigidity develop. Loss of facial expressionand micrographia are also common. The symptoms usually start onone side of the body and later become bilateral. During theprogression of the disease, non-motor areas of the brain becomeaffected, potentially leading to depression, sleep disorders andcognitive impairment. The Unified Parkinson’s Disease Rating Scale(UPDRS), which includes quantification of motor functions,complications of therapy, activities of daily life, behaviour andmood, is the most widely used standardised measure for stagingthe disease (Fahn and Elton, 1987).
1.2. Pathological features of Parkinson’s disease
PD is characterised by the degeneration of dopaminergicneurons of the substantia nigra pars compacta (SNpc) in themidbrain, and loss of their ascending projection to the striatum.This decrease in dopaminergic tone leads to the loss of control ofvoluntary movements. By the time a patient has been diagnosedwith PD, approximately 80% of striatal dopamine has been lost andthe disease is quite advanced. Although the loss of dopaminergicneurons within the SNpc is the primary pathological feature of PD,widespread neuronal loss also occurs in the locus coeruleus, thedorsal motor nucleus of the vagus and glossopharyngeal nerves,the nucleus basalis of Meynert, and in later stages, neuronal lossoccurs in the neocortex (Braak et al., 2003). However, the loss ofdopaminergic neurons in the SNpc is most acute and is responsiblefor the majority of the clinical manifestations of the disease. It is ofnote that the lateral SNpc shows more vulnerability than themedial part (Fearnley and Lees, 1991), possibly due to differentialmRNA profiles in cell death-related genes, mitochondrial complex Igenes, glutathione genes and pro-inflammatory cytokine genes,amongst others (Duke et al., 2007).
Braak et al. (2003) examined 110 brains from PD patients andnoted that the disease progression followed a ‘predeterminedsequence’ of events, and which were categorised into what is nowknown as Braaks Staging. Stages 1 and 2 involve pathology in theolfactory structures, medulla oblongata and part of the pontinetegmentum. Stages 3 and 4 see the involvement of SNpc pathologyand affect some of the proencephalic areas. Stages 5 and 6 involvedegeneration of higher-order sensory areas of the cortex and
premotor areas. Due to the involvement of non-motor areas, suchas the olfactory system, in the early stages of the disease, it hasbeen postulated that the use of olfactory testing may be a goodearly diagnostic tool for PD (Doty, 2007).
In the post-mortem PD brain, there are abundant roundeosinophilic insoluble cytoplasmic inclusions called Lewy bodies(LBs) in perikarya, and dystrophic thread-like insoluble neurites(Lewy neurites) in neuronal and glial processes within the SNpc(Braak et al., 1995). LBs were first described by Frederich Lewy in1912 (Gibb and Poewe, 1986) and are also found in other diseases,such as dementia with LBs and diffuse LB disease (Braak et al.,2003). LBs in PD patients have been shown to contain a-synuclein(Spillantini et al., 1997) and ubiquitin (Lowe et al., 1988) as well asseveral other proteins (Robinson, 2008). PD is also characterised bythe presence of an accumulation of activated microglia within theSNpc (McGeer et al., 1988). However, the exact reasons for theneurodegeneration and specific cellular manifestations of sporadicPD are unknown.
There is currently no cure for this disease—current treatmentsfor PD, such as L-DOPA, which is the mainstay of PD therapy, aremainly aimed at replacing dopamine in the striatum and do nothave any effect on the neurodegeneration. Furthermore, themajority of these drugs are effective for a limited number of years,and are associated with debilitating side effects. Thus, it isimperative that research on the causes and treatments of thisdisease is supported appropriately within the coming years, bothfor humanitarian and economic purposes.
2. Microglia
Microglia are the resident immune-competent cells of the CNSand have a role in monitoring the brain for immune insults andinvading pathogens. Ramon and Cajal considered microglia to bepart of the ‘third element’ of the CNS, being neither neuronal norastrocytic (Cajal, 1913). In the 1930s, Pio del Rio Hortega, a studentof Cajal, estimated that they make up approximately 12% of thecells in the brain (del Rio Hortega, 1932). Microglia have amesohaemopoietic origin and are likely to arise from myeloidtissue, however there is much controversy surrounding theirexact lineage (Cuadros and Navascues, 1998). Historically, theywere thought to invade the brain perinatally, from circulatingblood monocytes, yet microglial progenitors have been demon-strated in situ in the embryonic brain before the circulatorysystem has developed (Morris et al., 1991). It has been suggestedthat microglial progenitors are recruited from the periphery uponCNS damage (Ladeby et al., 2005b), while it has also been shownthat microglia resident in the CNS can self-renew and undergoesmitosis to increase the numbers of microglia in the affected areaduring insult (Ajami et al., 2007). Studies of microglial prolifera-tion in the hippocampus following a lesion of the perforantpathway in mice also showed a large increase in microglialnumber and that the vast majority of the resultant microglialpopulation was due to the proliferation of a subpopulation ofendogenous microglia and that bone marrow-derived macro-phages only contributed in small numbers (Ladeby et al., 2005a).Due to the ability of microglia to self-renew under certaincircumstances, microglia are thought of by some as an undiffer-entiated population of cells in the adult brain. For example, afterstimulation with growth factors in vitro, microglia from adult micewere shown to differentiate to either a dendritic cell-likephenotype or a peripheral macrophage phenotype (Santambrogioet al., 2001). It is postulated therefore, that there may be twopopulations of microglia within the brain: those which developpre-vascularisation and are generally amoeboid during develop-ment and those derived from blood monocytes post-natally,although the precise lineage has yet to be determined.

C.M. Long-Smith et al. / Progress in Neurobiology 89 (2009) 277–287 279
2.1. Microglia in the healthy brain
In the adult brain, the majority of microglia are postulated to bein the ‘resting’ state, and have a characteristic ramified morphol-ogy. Kreutzberg (1996) proposed a classification system of thestaging of microglia in vivo, from a resting ramified morphologywith small cell bodies and many slim branchings, containing up to20 spines, to an active amoeboid morphology with truncatedbranches. Due to the difficulty in studying ‘resting’ microglia in anon-pathogenic environment, little is known about their function.This difficulty arises during dissection of microglia from tissue andculturing them in vitro, which inevitably results in some level ofactivation due to trauma. In an attempt to produce a population ofmicroglia that were in the resting state, one group preparedprimary cultures of rat microglia in a serine/glycine-free mediumand the resulting cells appeared to be in a functionally restingstate. Moreover, when the cells were co-cultured with neurons,they facilitated neuronal survival (Tanaka et al., 1998). Anothergroup added astrocyte-conditioned medium to murine microglialcultures to induce a ramified, resting phenotype and found adramatic change of morphology from ‘‘ramified’’ to ‘‘resting’’ afterjust a few hours of treatment (Eder et al., 1999). Immunohisto-chemical staining of brain slices offers a better chance of capturingmicroglial cells in their normal surveillance mode. In vivo imagingstudies using two-photon microscopy have shown resting micro-glia in as normal a state as possible, and illustrated the ability ofmicroglia to rapidly become activated and reorganise theirarchitectural structure (Davalos et al., 2005; Nimmerjahn et al.,2005). Some authors consider that microglia in the perinatal stateare not resting, but are in an intermediate phase of activation(Hanisch, 2002). Indeed the presence of partially activated ratmicroglia in untreated cultures has been observed (Eder et al.,1999).
Resting microglia, in contrary to what their name suggests, arenot static, dormant cells, they are proposed to constantly move andto monitor the area in which they reside for pathogens and changesin their microenvironment. Two studies that used time-lapseimaging in mice in vivo, showed that microglial cell bodies do notmove during normal surveillance, but that the microglial processesextend and retract rapidly and dynamically (Davalos et al., 2005;Nimmerjahn et al., 2005). Following activation to engulf cellulardebris, microglia release growth factors to support the surroundingneurons. Activation of microglia also occurs during developmentand remodelling of the healthy brain. Apoptosis occurs duringembryonic and early post-natal development of the brain; anyneurons that have not successfully made synaptic connections aredisposed of by microglia, and the remaining neurons are supportedby the growth factors produced by microglia (Batchelor et al., 1999).
2.2. Microglial activation
2.2.1. Activating stimuli
Microglia respond to activating stimuli in the extracellularenvironment which bind to a diverse selection of cell-surfacereceptors including receptors for endotoxin, cytokines, chemo-kines, mis-folded proteins, serum factors and ATP. One of the mostcommonly used methods of activating microglia both in vitro andin vivo is the application of the endotoxin lipopolysaccharide (LPS).LPS binds to toll-like receptor (TLR) 4, a member of the TLR familyof receptors involved in detecting microbial infection, of whichmicroglia are reported to express nine of the 12 members (Jacket al., 2005). LPS-induced activation of microglia results in theproduction of cytokines and chemokines such as interleukin (IL)-1b, IL-1 receptor antagonist (IL-1ra), IL-6, IL-8, IL-10, IL-12, IL-18,macrophage colony stimulating factor, macrophage inflammatoryprotein (MIP)-1a, MIP-1b, monocyte chemoattractant protein-1,
transforming growth factor-b, and tumour necrosis factor-a (TNF-a) by microglia. These cytokines in turn potentiate microglialactivation by binding to their receptors, which are expressed onmicroglia (Kim and de Vellis, 2005). Mis-folded or aberrantproteins such as amyloid-b are capable of activating microglia viascavenger receptors, which are up-regulated in the brains of ADpatients (El Khoury et al., 1998), or via initiation of microglialphagocytosis of the pathological protein (Rogers et al., 2002). It hasalso been shown that microglial phagocytosis occurs in response toaggregated a-synuclein, the major component of LBs in PD (Zhanget al., 2005). The serum factors thrombin and immunoglobulinsinitiate microglial activation through protease-activated receptor1 and Fc receptors, respectively (Stangel and Compston, 2001; Suoet al., 2002). ATP released from damaged neurons, has beendemonstrated to activate microglia (Davalos et al., 2005) bybinding to purinergic receptors, which are expressed on microglia(Brautigam et al., 2005). Matrix metalloproteinase-3 (MMP-3),which degrades extracellular macromolecules, and neuromelanin,the neuronal pigment released from dying dopaminergic neuronsin PD, have also been shown to induce microglial activation (Kimet al., 2005; Wilms et al., 2003), although the exact mechanisms ofhow these compounds regulate the activation process have yet tobe determined.
It has also been suggested that activation of microglia occursdue to a ‘‘switching-off’’ of an inhibitory effect that neurons exerton microglia under resting conditions. Neurons express CD200, acell-surface transmembrane glycoprotein, which binds to micro-glia expressing the receptor CD200R (Barclay et al., 2002), andhelps maintain microglia in a quiescent state in the healthy brain(Hoek et al., 2000). A down-regulation of CD200 expression hasbeen shown in neurons exposed to inflammatory conditions, whileinhibition of CD200 causes microglial activation, demonstrating aneuronal-directed mechanism for regulation of microglial activa-tion (Lyons et al., 2007). The presence of neurons can also decreasethe microglial response to low-dose LPS-stimulation, demon-strated by a decrease in nitric oxide (NO) and TNF-a production in
vitro (Chang et al., 2000). This finding concurs with the idea thathealthy neurons have an inhibitory effect on microglial activationwhile damaged neurons can promote their activation.
2.2.2. Consequences of microglial activation
Activated microglia have been shown to play key roles in boththe developing and adult CNS. During CNS development theyphagocytize neurons and cellular debris as a genetically deter-mined event to prevent excessive cell production (Ashwell, 1990).Upon activation in the adult CNS, microglia act primarily asscavengers and in brain tissue remodelling to restore and protectbrain structures and functions. Resting microglia are rapidlyactivated after insults such as changes in the extracellularenvironment of the injured or diseased brain and undergomorphological changes from a resting, ramified shape to an activeamoeboid shape to facilitate proliferation, migration and phago-cytosis (Nimmerjahn et al., 2005). Local microglia extend out theirprocesses to surround the area of insult (Davalos et al., 2005) andas a result damaged cells are engulfed by the microglia viaphagocytosis, removing any potentially damaging material fromthe area and protecting the neighbouring cells. Thus, microglia actas antigen-presenting cells (APCs) and present engulfed patho-genic material to invading T-cells to generate an adaptive immuneresponse; the presence of major histocompatibility complex(MHC) class II antigen expression on activated microglia corrobo-rates this theory (Hickey and Kimura, 1988; McGeer et al., 1988).The ability of endogenous microglia to act as APCs has recentlybeen questioned however, with some suggesting that perivascularmacrophages or invading dendritic cells are responsible for thistask (Perry, 1998).

C.M. Long-Smith et al. / Progress in Neurobiology 89 (2009) 277–287280
Microglia, in common with other cells of the myeloid lineage,have the ability to secrete a multitude of immunomodulatorymolecules such as cytokines, chemokines, neurotrophins andreactive oxygen and nitrogen species, which communicate signalsto surrounding cells (Garden and Moller, 2006). Cytokines are lowmolecular-weight proteins that are usually classified as either pro-or anti-inflammatory and are thought to signal both by paracrineand autocrine methods. While pro-inflammatory cytokines havethe ability to elicit a sustained immune response, anti-inflamma-tory cytokines act to down-regulate an immune response bybinding to appropriate receptors expressed on microglia andinitiating an autocrine signalling process. Cytokines have numer-ous effects on CNS function including growth promotion, inhibi-tion and proliferation of astrocytes and oligodendrocytes (Hanisch,2002), modulation of neurotransmitter release (Zalcman et al.,1994), long-term potentiation (Nolan et al., 2005), and behaviouralalterations such as memory impairment (Yirmiya et al., 2002),anhedonia (Konsman et al., 2002) and anxiety (Anisman andMerali, 1999). Chemokines act primarily as chemoattractants todraw additional microglia to the site of injury, while neurotrophicfactors such as nerve growth factor, brain-derived neurotrophicfactor and glial-derived neurotrophic factor released from micro-glia have been proposed to participate in the survival andregeneration of neurons (Batchelor et al., 2002; Nagata et al.,1993) as well as to prolong the existence of microglia and toregulate their function (Elkabes et al., 1996). Activated microgliacan also produce and release both reactive oxygen and nitrogenspecies due to catalysis by nicotinamide adenine dinucleotidephosphate (NADPH) oxidase (Babior, 1999) and these highlyreactive free radicals can kill surrounding pathogens. It has alsobeen reported however, that microglial-derived free radicals cancause neuronal cell death and so they have been implicated in thepathogenesis of neurodegenerative conditions (Chao et al., 1992).
2.2.3. Reactive microgliosis
Activation of microglia is vital to normal brain function in orderto control the neuronal microenvironment. Mild activation hasapparent beneficial effects on the surrounding cells but whenmicroglia are continuously activated or over-activated; damagingeffects ensue to inappropriately kill otherwise viable cells,particularly neurons. Due to the possible involvement of micro-glial-mediated inflammation in PD and other neurodegenerativediseases, it has been postulated that a vicious cycle of inflamma-tion may occur, regardless of the initial insult. When the microgliabecome activated, whether through direct activation via a toxin,pathogen or endogenous protein, or indirectly via signals fromdamaged neurons, these activated microglia may persist due topositive feedback from dying neurons, even if the initial insult hasceased. Thus, microglial activation, and hence neuroinflammation,may be propagated and prolonged inappropriately to amplify thedestruction of neurons; a process referred to as reactive micro-gliosis and which is a common characteristic of neurodegenerativediseases (Gao and Hong, 2008).
As of yet, it is unclear what mechanisms propel reactivemicrogliosis but there is some evidence to suggest that toxicfactors released from dying neurons into the microglial micro-environment are responsible. The active form of MMP-3 that isreleased from injured dopaminergic neurons was shown to inducethe production of superoxide in microglia and consequentlyaugment neuronal demise (Kim et al., 2007). The aggregatedproteins a-synuclein and amyloid-b, which are both products ofneuronal degeneration in PD and AD, respectively, activatemicroglia and in turn induce neuronal cell death (Floden et al.,2005; Zhang et al., 2005). Macrophage antigen complex-1, amicroglial surface receptor and member of the b2 intergrin familyhas also recently been identified as mediating reactive microgliosis
(Hu et al., 2008) and recent work has shown that necrotic neuronsactivate microglia in a manner dependent on the TLR-associatedadapter molecule myeloid differentiation primary response gene(MyD88) (Pais et al., 2008). However, a mediating stimulusbetween the neurons and microglia has not yet been determined inthis paradigm. Interruption to brain homeostasis due to microglial‘priming’, which occurs during the aging process itself, has alsobeen suggested as a factor that amplifies the microglial response.In this regard (Perry et al., 2007), have proposed that chronicexposure to pro-inflammatory signals from systemic infection thatoccur throughout an individual’s lifetime, promotes an exagger-ated microglial response that contributes to neuronal deteriorationinstead of facilitating a protective homeostatic response. Ongoingresearch thus highlights that degenerating neurons have thecapacity to reactivate microglia and that activated microglia arecapable of augmenting neuronal loss in neurodegenerativediseases, and as such, this cycle of perpetuating deterioration inCNS function is a potential target for therapeutic intervention.
3. Role of inflammation in Parkinson’s disease
3.1. Inflammation in Parkinson’s disease patients
The first evidence for a role for inflammation in PD came from apost-mortem study – in 1998, McGeer and colleagues foundactivated microglia and T-lymphocytes in the SNpc of a PD patient.Since then, there have been numerous studies which support a rolefor neuroinflammatory processes in PD (Hirsch and Hunot, 2009;McGeer and McGeer, 2004; Orr et al., 2002; Tansey et al., 2007). Inaddition to the presence of activated microglia and pro-inflam-matory cytokines (both of which are discussed below), enzymesassociated with inflammation, such as inducible nitric oxidesynthase (iNOS) and cyclooxygenase 2 (COX2), have been found inthe post-mortem PD brain (Hunot et al., 1996; Knott et al., 2000).Support for an involvement of neuroinflammation in PD comesfrom studies which show a link between infection and neurode-generation. Neurodegenerative diseases tend to be exacerbated bysystemic infections, and activated microglia may be involved inthis process. For example, system infection has been shown toinduce microglial activation in multiple sclerosis (MS) (Perry,2004). Also, periods of worsening dementia are known to emergein AD patients following infections such as pneumonia, andpatients suffering from MS experience a deterioration of symptomsfollowing similar infections (Cunningham et al., 2005; Perry et al.,2007). Therefore, it is possible that the added insult of infectionexacerbates the ongoing inflammatory and degenerative pro-cesses. It is not clear whether or not PD patients suffer from aworsening of symptoms after experiencing systemic infection.However, there are two lines of evidence which suggest thatinfection can increase the risk of developing PD. The first is theobservation of parkinsonism following encephalitis. Towards theend of World-War-1, a pandemic of influenza virus caused anunusually large increase in the number of cases of post-encephalytic parkinsonism (PEP), or von Economo encephalitis(Rail et al., 1981). Similarly, people who are infected with Japanese-encephalitis virus for a prolonged period are likely to develop PEP(Ogata et al., 2000). Japanese-encephalitis-induced parkinsonismshares many similarities with PD and has been used to create amodel of PD in rats (Ogata et al., 1997). The second line of evidencefor a link between systemic infection and the risk of developingparkinsonism comes from animal studies. Rat fetuses exposed toLPS were found to have fewer than normal dopaminergic neuronsat birth and were more susceptible to subsequent 6-hydroxydo-pamine (6-OHDA; a neurotoxin used to model PD in rats) exposurein adulthood (Ling et al., 2002; Ling et al., 2004). The authorshypothesised that children born to mothers who experienced

C.M. Long-Smith et al. / Progress in Neurobiology 89 (2009) 277–287 281
bacterial vaginosis during pregnancy were at a higher risk ofdeveloping PD in later life and that they may be more susceptibleto additional insults which may precipitate the development ofthis disease. Similarly, a recent study by (Gao et al., 2008) showedthat formylmethionyl-leucyl-phenylalanine, a bacterial-derivedchemoattractant involved in systemic infection, caused the deathof dopaminergic neurons in mouse midbrain cultures via theactivation of microglia.
It is thought that oxidative stress plays an important role indopaminergic neuronal death in PD (Jenner et al., 1992). Basallevels of lipid peroxidation are increased in the SN of PD patients(Dexter et al., 1989), which suggests that this area of brain isparticularly vulnerable to excess free radicals and ROS which maybe produced by activated microglia as part of an inflammatoryresponse.
3.1.1. Activated microglia in Parkinson’s disease
The presence of inflammation is generally indicated by theaccumulation of activated microglia in damaged areas of the brain.Particularly high numbers of activated microglia have been foundin the brains of PD patients’ post-mortem, predominantly in theSNpc in the vicinity of the degenerating dopaminergic neurons, butalso in the hippocampus, transentorhinal cortex, cingulate cortexand temporal cortex where neuronal loss is also prevalent (Banatiet al., 1998; Imamura et al., 2003; McGeer et al., 1988; Sawadaet al., 2006). Indeed in PD with dementia, hippocampal volume asdetected by MRI studies is diminished (Laakso et al., 1996). Whileincreased levels of activated microglia have been found in SNpc ofthe general elderly population (Beach et al., 2007), it is possiblethat this ‘normal’ aging pattern is exacerbated in PD. It is likely thatnigral neurons which are under stress or are damaged, in thepresence of already ‘‘primed’’ microglia, can exacerbate microglialactivation and induce them to release neurotoxic factors.Substances which are produced by dying dopaminergic neuronsand which can activate microglia include: a-synuclein aggregates(Zhang et al., 2005), ATP (Davalos et al., 2005), MMP-3 (Kim et al.,2007; Kim et al., 2005) and neuromelanin (Wilms et al., 2003). Asmentioned earlier, the dopaminergic neurons of the SNpc areparticularly susceptible to degeneration. Rodent nigral neuronshave been shown to be more vulnerable than either hippocampalor cortical neurons to LPS-induced degeneration in vivo and in vitro,and this was shown to be due to the higher number of activatedmicroglia per unit area in the SNpc compared with the other twobrain areas (Kim et al., 2000). Indeed, the SNpc contains the highestconcentration of microglia of any brain area (McGeer et al., 1988).It is also of note that activated microglia present in thehippocampus and cerebral cortex of PD patients may alsocontribute to the non-motor symptoms of the disease, the mostdebilitating of which is dementia (Emre, 2003). While substantialresearch has been carried out on the role of hippocampalinflammation in cognitive impairment (Lynch, in press), to datehowever, there is limited information on the contribution ofinflammatory processes to the intellectual demise specific to PD. Ithas been postulated that a vicious cycle of inflammation mayexacerbate the debilitating effects of dopaminergic neuron loss inPD, regardless of whether the inflammation is a cause orconsequence of the disease (Block and Hong, 2007). Evidence ofinflammation from post-mortem PD brains is primarily fromterminal stage cases and so it remains unknown if microglialactivation occurs only at this late stage of the disease as aconsequence of substantial neuronal loss, or at an earlier stage ofdisease progression to precipitate or possibly even preventneuronal loss. It has also been suggested that the observedmicroglial activation reflects their state in the final hours ofpatients’ lives rather than in the months or years before (McGeerand McGeer, 2008). More recent findings from animal and tissue
culture models however, and also from epidemiological studiessupport the notion that microglia contribute to the disease processat an early stage (Liu, 2006).
3.1.2. Pro-inflammatory cytokines in Parkinson’s disease
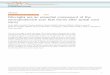
The pro-inflammatory cytokines, IL-1b, TNF-a, IL-2 and IL-6,are expressed at very low levels in healthy brain, but have beenfound at much higher levels in PD patients, in the post-mortembrain as well as in serum and cerebrospinal fluid in vivo (Boka et al.,1994; Dobbs et al., 1999; Mogi et al., 1994a; Mogi et al., 1994b;Stypula et al., 1996). The death-signalling receptor, TNFR-1, hasbeen found to be expressed on dopaminergic neurons in humanSNpc (Boka et al., 1994; Mogi et al., 2000). Animal studies supportan involvement of these pro-inflammatory cytokines in PD. Forexample, induction of chronic expression of IL-1b in adult rat SNpcusing a recombinant adenovirus resulted in dopaminergic celldeath after three weeks (Ferrari et al., 2006). A study usingneutralising antibodies to IL-1b and TNF-a showed that approxi-mately 50% of LPS-induced dopaminergic neuronal cell death inprimary cultures of rat midbrain was mediated by the productionof these two cytokines (Gayle et al., 2002). In support of a role forIL-1b in the demise of dopaminergic neurons, we have found thatLPS-stimulated primary cultures of rat microglial cells release IL-1b (Fig. 1b), that IL-1R1 is expressed on primary cultures ofmidbrain dopaminergic neurons (unpublished observation), andthat treatment of these dopaminergic neurons with IL-1b results insignificant cell death, comparable to that induced by 6-OHDA(Fig. 1c). Taken together, these findings support the hypothesisthat release of IL-1b from activated microglia are involved indopaminergic neuronal degeneration (Fig. 1a).
3.2. Inflammation in animal models of Parkinson’s disease
3.2.1. MPTP model
Methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) is a neu-rotoxin which rapidly induces severe parkinsonian-like symptomsin humans by selectively killing dopaminergic neurones of theSNpc (Langston et al., 1983; Langston and Ballard, 1983). MPTPitself is not toxic, but it crosses the blood–brain barrier (BBB) and isspontaneously oxidised to 1-methyl-4-phenylpyridinium (1-MPP+) by monoamine oxidase-B, mainly in glial cells. MPP+ isreleased into the extracellular space and actively taken up intodopaminergic neurons by the dopamine transporter (DAT),whereby it potently inhibits mitochondrial complex I of theelectron transport chain, causing an increase in production ofreactive oxygen species (ROS) and a drop in ATP levels, leading tocell death (Przedborski and Vila, 2003). Peripheral injection ofMPTP in monkeys or mice is commonly used to model PD.Activated microglia have been demonstrated in the brains of bothmonkeys (McGeer et al., 2003) and mice (Liberatore et al., 1999)after systemic injection of MPTP. Infiltration of T-lymphocytes hasalso been detected in the brains of MPTP-treated mice (Brochardet al., 2009; Kurkowska-Jastrzebska et al., 1999). It is thought thatROS produced by microglial NADPH play an important role inMPTP-induced neurotoxicity (Gao et al., 2003; Wu et al., 2003). Ithas recently been proposed that angiotensin II is responsible forthe inflammation induced by MPTP in mouse mesencephalicdopaminergic neurones in vitro and in vivo (Joglar et al., 2009).
3.2.2. 6-OHDA model
6-OHDA is a hydroxylated analogue of dopamine, which isactively taken up into dopaminergic neurons via DAT on the nerveterminals and selectively kills these cells via the generation of freeradicals and oxidative stress. Intracerebral application of 6-OHDAto nigrostriatal dopaminergic neurones, by injection at the site oftheir cell bodies, processes or terminals, induces degeneration of

Fig. 1. . (a) Schematic representation of the impact of microglial activation on dopaminergic neuronal deterioration through release of IL-1b. (b) IL-1b released from
microglial-enriched primary cultures of post-natal rat cortex which were treated with 0, 50, 100 or 500 ng/ml LPS for 24 h (n = 2). (c) Percentage of dopaminergic (tyrosine
hydroxylase-positive) neurons in embryonic rat midbrain neuronal-enriched cultures after treatment for 1 h in the absence or presence of IL-1b (10 ng/ml) or 6-OHDA
(40 mM) (n = 3). Data expressed as means with standard errors, *p < 0.05, **p < 0.01, ***p < 0.001, vs. control (ANOVA with post-hoc Dunnett’s test).
C.M. Long-Smith et al. / Progress in Neurobiology 89 (2009) 277–287282
nigral dopaminergic neurones and depletion of striatal dopaminelevels, and is the most widely used animal model of PD. We haverecently shown a significant increase in the number of activatedmicroglia, identified using immunocytochemistry for MHC class II,in the SNpc of 6-OHDA-lesioned rats at 10 and 28 days post-lesion(Crotty et al., 2008). The presence of activated microglia in thebrains of 6-OHDA-lesioned rats has previously been reported byother groups (Akiyama and McGeer, 1989; Depino et al., 2003; Heet al., 2001). The study by Depino et al. in 2003, which showedmicroglial cell activation in the 6-OHDA-lesioned rat brain, foundno increase in IL-1b at the protein level, although IL-1b mRNA wasincreased, and no increase in TNF-a at either mRNA or proteinlevel. In support of a role for pro-inflammatory cytokines in theneuronal death induced by 6-OHDA, blockade of the soluble formof the TNF-a receptor, but not the transmembrane form, was foundto attenuate the death of dopaminergic neurons in 6-OHDA-lesioned rats (McCoy et al., 2006).
3.2.3. LPS model
Intranigral injection of LPS in rats in vivo results in dopami-nergic neuronal loss and can be used as a model of PD (Castanoet al., 1998). Systemic administration of LPS has also been found toinduce progressive degeneration of nigral dopaminergic neuronesin rats (Qin et al., 2007). Furthermore, it has been reported thatprenatal exposure to LPS in rats results in the development offewer than normal nigral dopaminergic neurons (Ling et al., 2004).In vitro studies on rat mesencephalic cultures suggest thatdopaminergic neurons are twice as sensitive to LPS as non-
dopaminergic neurons and that the toxicity of LPS occurs viamicroglial activation (Bronstein et al., 1995; Gayle et al., 2002).Although many in vitro studies have supported an involvement ofNO in microglial-mediated dopaminergic neuronal death due toLPS-treatment (Chao et al., 1992; Gibbons and Dragunow, 2006),others have suggested that NO is not involved (Castano et al., 1998;Gayle et al., 2002). The pro-inflammatory cytokines IL-1b and TNF-a are thought to be involved in LPS-mediated toxicity (Gayle et al.,2002). In support of a role for pro-inflammatory cytokines in theneurotoxic action of LPS, we have found a dose-dependent releaseof IL-1b in rat microglial cultures following LPS-treatment(Fig. 1b). Furthermore, blockade of the soluble form of the TNF-a receptor has been reported to reduce microglial activation in thein vivo LPS model of PD (McCoy et al., 2006). Also, Ling and co-workers found that the decreased numbers of nigral dopaminergicneurons in rats after prenatal exposure to LPS, was accompanied byelevated levels of TNF-a in the striatum (Ling et al., 2004).
A recent study by (Koprich et al., 2008) showed that injection ofa non-toxic low-dose of LPS into adult rat SNpc resulted inmicroglial activation and increased levels of IL-1b, without causingdeath of dopaminergic neurons in vivo, but that subsequentinjection of 6-OHDA into the striatum resulted in a large loss ofdopaminergic neurons compared with that in animals treated with6-OHDA alone. This exacerbation of 6-OHDA-induced neuronalloss by LPS appeared to be partly mediated by IL-1, since treatmentwith LPS plus IL-1ra rescued some of the dopaminergic neuronsfrom 6-OHDA-induced death (Koprich et al., 2008). Another recentstudy showed that 6-OHDA injection into the adult rat striatum

C.M. Long-Smith et al. / Progress in Neurobiology 89 (2009) 277–287 283
plus a subsequent non-toxic LPS injection into the SNpc caused anincreased level of dopaminergic neuronal death and motor deficitscompared with those induced by either toxin alone (Godoy et al.,2008). In that study, increased levels of IL-1b, but not TNF-a, werefound in the SNpc of the 6-OHDA plus LPS-treated group. Both ofthese studies postulated that the initial insult caused priming ofmicroglia, while the second insult resulted in fully activatedmicroglia.
4. Anti-inflammatory therapies
As the evidence accumulates for a detrimental role of inflamma-tion in the pathogenesis of PD, a host of anti-inflammatory agents arenow under investigation (Table 1). Indeed data generated from useof non-steroidal anti-inflammatory drugs, microglial inhibitors andanti-inflammatory cytokines in animal and cellular studies, havesupported the notion that control of neuroinflammation is a strategyworth pursuing in the effort to retard or even prevent degenerationof dopaminergic neurons in PD. It should be noted however thatagents which exert their effect by absolute inhibition of microglialactivation may in the long-term be detrimental rather thantherapeutic to PD patients, because a reduction in the beneficialeffects of microglial activation such as immune surveillance andtissue repair, may render the patient defenceless if exposed to CNSinjuries or other harmful stimuli.
Table 1Neuroprotective effects of anti-inflammatory agents in animal models of Parkinson’s d
Agent Mode of action Species PD model Effects
Dexamethasone SAID Mouse MPTP 1. Prev
2. Prot
Rat LPS 1. Prev
2. Prot
Aspirin NSAID Mouse MPTP Preven
Rat 6-OHDA Preven
Salicylic acid NSAID Mouse MPTP 1. Atte
2. Prev
change
nucleu
Indomethacin NSAID Mouse MPTP 1. Prev
2. Prot
Celecoxib NSAID Rat 6-OHDA Revers
fibre a
Minocycline Microglial activation
inhibitor
Mouse MPTP 1. Prot
2. Prev
striatu
Rat 6-OHDA 1. Red
2. Prot
Rat LPS Protec
Interleukin-10 Anti-inflammatory
cytokine
Rat LPS Protec
Rat 6-OHDA 1. Prot
2. Prev
3. Red
Naloxone Opioid receptor
antagonist
Rat LPS Protec
Pioglitazone PPARg agonist Mouse MPTP Protec
Rat LPS 1. Prev
2. Prot
Rosiglitazone PPARg agonist Mouse MPTP 1. Prev
2. Prot
3. Part
VP025 Phosphotidylglycerol
phospholipid
Rat 6-OHDA 1. Prot
2. Prev
3. Red
Rat Proteasome
inhibition
1. Atte
2. Prev
While steroidal anti-inflammatory drugs (SAIDS) such asdexamethasone have been reported to have protective effects inrodent models of PD (Castano et al., 2002; Kurkowska-Jastrzebskaet al., 2004), the potential side-effects preclude their long-termclinical usage. Non-steroidal anti-inflammatory drugs (NSAIDS)such as aspirin, ibuprofen and indomethacin inhibit COX activity toblock the production of the pro-inflammatory lipid mediators,prostaglandins. It has also been shown that NSAIDS scavenge ROSin neuronal cells (Grilli et al., 1996), inactivate the pro-inflammatory transcription factor nuclear factor kappa B (NF-kB) (Kopp and Ghosh, 1994) and activate the peroxisomeproliferator-activated receptor-g (PPARg), a member of thenuclear receptor superfamily and mediator of anti-inflammatoryactivity, in microglia (Bernardo et al., 2005). Numerous experi-mental cell and animal models of PD have demonstrated thatNSAID pre-treatment protects against 6-OHDA- and MPTP-induced dopaminergic neuronal degeneration and associatedsymptoms (Esposito et al., 2007). While epidemiological studieshave determined that the use of NSAIDS is associated with adecreased risk of developing PD (Ton et al., 2006), there is noevidence of their efficacy in the treatment of the disease.
Other agents currently under investigation for their potentialneuroprotective effects in models of PD primarily act to suppressmicroglial activation and inhibit the production of neurotoxicfactors and pro-inflammatory cytokines. Minocycline is a tetra-
isease.
References
ented striatal dopamine depletion Kurkowska-Jastrzebska
et al. (2004)ected dopaminergic neurons in SN
ented striatal dopamine depletion Castano et al. (2002)
ected dopaminergic neurons in SN
ted striatal dopamine depletion Aubin et al. (1998)
ted striatal dopamine depletion Di Matteo et al. (2006)
nuated akinesia and catalepsy Mohanakumar et al. (2000)
ented dopamine depletion and
s in dopamine turnover in
s caudatus putamen
ented striatal dopamine depletion Kurkowska-Jastrzebska
et al. (2002)ected dopaminergic neurons in SN
ed striatal dopaminergic neuronal
nd nigral dopaminergic neuronal cell loss
Sanchez-Pernaute et al. (2004)
ected dopaminergic neurons in SN Du et al. (2001)
ented dopamine depletion in the
m and nucleus accumbens
uced apomorphine-induced rotations Quintero et al. (2006)
ected dopaminergic neurons in SN
ted dopaminergic neurons in SN Tomas-Camardiel et al. (2004)
ted dopaminergic neurons in SN Arimoto et al. (2006)
ected dopaminergic neurons in SN Johnston et al. (2008)
ented striatal dopamine depletion
uced apomorphine-induced rotations
ted dopaminergic neurons in SN Liu et al. (2000b)
ted dopaminergic neurons in SN Dehmer et al. (2004)
ented striatal dopamine depletion Hunter et al. (2007)
ected dopaminergic neurons in SN
ented errors in beam traversal test Schintu et al. (2009)
ected dopaminergic neurons in SN
ially prevented striatal dopamine depletion
ected dopaminergic neurons in SN Crotty et al. (2008)
ented striatal dopamine depletion
uced apomorphine-induced rotations
nuated impairment on accelerating rotarod Fitzgerald et al. (2008)
ented striatal dopamine depletion

C.M. Long-Smith et al. / Progress in Neurobiology 89 (2009) 277–287284
cycline analog, which due to its lipophilicity, can easily cross theBBB where it is reported to have anti-inflammatory andneuroprotective activity (Kim and Suh, 2009). As well as inhibitingmicroglial activation, proliferation and subsequent release of pro-inflammatory mediators (Griffin et al., 2006; Tikka et al., 2001;Tikka and Koistinaho, 2001), minocycline is reported to exert aneuroprotective action by inhibiting both the release of theapoptotic mediator cytochrome c from mitochondria (Zhu et al.,2002) and cytoplasmic caspase-1 and caspase-3 expression (Chenet al., 2000). In animal models of PD, minocycline has been shownto be effective in preventing MPTP-, 6-OHDA- and LPS-induceddopaminergic neurodegeneration (Du et al., 2001; He et al., 2001;Quintero et al., 2006; Tomas-Camardiel et al., 2004). However,there have also been contradictory reports regarding its efficacy inprotecting striatal dopaminergic fibres (Diguet et al., 2004; Yanget al., 2003). Recent results from a Phase II clinical trial did notshow any adverse events in early PD patients and suggest thatminocycline should be considered for Phase III trials (NINDS NET-PD Investigators, 2008).
The anti-inflammatory cytokine IL-10 has been shown to holdtherapeutic potential; pre-treatment of cultures of mesencephalicneuroglia with IL-10 inhibited LPS-stimulated microglial activationand degeneration of dopaminergic neurons (Qian et al., 2006).Similar neuroprotective effects were observed in vivo after chronicinfusion of IL-10 into the SNpc of rats that were challenged with LPS(Arimoto et al., 2006). More recently, gene therapy approaches havebeen developed to deliver IL-10 or a dominant negative TNF proteininto the rat SNpc, and both of these methods have proved effective inattenuating the neuronal loss and behavioural deficits in the 6-OHDA-rat model of PD (Johnston et al., 2008; McCoy et al., 2008).
Other agents with a variety of mechanisms of actions have alsobeen shown to have both anti-inflammatory and neuroprotectiveactivities in models of PD. LPS-stimulated microglial activation anddegeneration of nigral dopaminergic neurons in vitro (Liu et al.,2000a) and in vivo (Liu et al., 2000b; Lu et al., 2000) is reduced aftertreatment with the non-selective opioid receptor antagonistnaloxone. The anticonvulsant and mood stabiliser valproate, andother histone deacetylase inhibitors, have recently been demon-strated to inhibit microglial activation and to increase theexpression of glial cell-line-derived neurotrophic factor (GDNF)and brain-derived neurotrophic factor (BDNF) in astrocytes (Wuet al., 2008), which acts to protect dopaminergic neurons from LPS-induced death in vitro (Chen et al., 2006; Chen et al., 2007; Penget al., 2005). Pioglitazone and rosiglitazone are agonists of thenuclear hormone receptor PPARg, and are currently approved forthe treatment of type II diabetes. In the CNS they exhibitneuroprotective effects in models of neurodegenerative disorders,including PD, by preventing inflammation, oxidative damage andapoptosis (Chaturvedi and Beal, 2008). Specifically, pioglitazoneprevents MPTP-induced activation of microglia and dopaminergicneuronal cell loss in murine SNpc in vivo (Dehmer et al., 2004), anaction which has been shown to occur through inhibition ofmonoamine oxidase B (Quinn et al., 2008), the enzyme responsiblefor conversion of MPTP to its toxic metabolite MPP+. Whenpioglitazone was administered to rats that were also injectedintrastriatally with LPS, the resultant LPS-induced microglialactivation and dopaminergic degeneration was attenuated (Hunteret al., 2007). Recently, the neuroprotective effects of rosiglitazonehave been shown in the MPTP mouse model of PD; chronicadministration of the drug prevented behavioural deficits,dopaminergic neuronal loss and microglial activation in the SNpcin vivo (Schintu et al., 2009). Motor impairments, nigraldopaminergic neurodegeneration and striatal dopamine lossobserved in the 6-OHDA rat model were reversed by pre-treatmentwith VP025, a drug formulation based on phospholipid nanopar-ticles incorporating phosphatidylglycerol (Crotty et al., 2008).
VP025 has also been shown to attenuate motor impairments andloss of striatal dopamine in the proteasome inhibitor model of PD(Fitzgerald et al., 2008). This compound, which is designed tointeract with APCs, was shown to prevent activation of microglia(Crotty et al., 2008) suggesting a combined anti-inflammatory andneuroprotective mechanism, a phenomenon that is apparent in thenumerous potential therapeutically beneficial compounds underinvestigation for the treatment of PD.
5. Conclusion
The death of dopaminergic neurons in the SNpc is the keypathology of PD. Therefore, it is imperative that research isundertaken, not only in areas which could provide protectivestrategies for the remaining neurons, or which involve dopami-nergic neuronal cell replacement therapies, but also into under-standing the fundamental mechanisms by which these cells die.Although the precise role of inflammation in the pathogenesis ofPD remains unclear, an array of evidence from the clinic and fromanimal models now points to its substantial involvement in thisdebilitating disease. In addition, rapid advances in diagnostic toolsare being made to detect dopaminergic neuronal degeneration, thevast majority of which has already occurred before patients arecurrently diagnosed with PD. Thus, it is likely that a deeperunderstanding of microglial activation and the consequentinflammatory response, will contribute to the advancement oftherapeutics aimed at halting the demise of dopaminergic neuronsbefore substantial clinical manifestations appear.
References
Ajami, B., Bennett, J.L., Krieger, C., Tetzlaff, W., Rossi, F.M., 2007. Local self-renewalcan sustain CNS microglia maintenance and function throughout adult life. Nat.Neurosci. 10, 1538–1543.
Akiyama, H., McGeer, P.L., 1989. Microglial response to 6-hydroxydopamine-induced substantia nigra lesions. Brain Res. 489, 247–253.
Anisman, H., Merali, Z., 1999. Anhedonic and anxiogenic effects of cytokine expo-sure. Adv. Exp. Med. Biol. 461, 199–233.
Arimoto, T., Choi, D.Y., Lu, X., Liu, M., Nguyen, X.V., Zheng, N., Stewart, C.A., Kim, H.C.,Bing, G., 2006. Interleukin-10 protects against inflammation-mediated degenera-tion of dopaminergic neurons in substantia nigra. Neurobiol. Aging 28, 894–906.
Ashwell, K., 1990. Microglia and cell death in the developing mouse cerebellum.Brain Res. Dev. Brain Res. 55, 219–230.
Aubin, N., Curet, O., Deffois, A., Carter, C., 1998. Aspirin and salicylate protect againstMPTP-induced dopamine depletion in mice. J. Neurochem. 71, 1635–1642.
Babior, B.M., 1999. NADPH oxidase: an update. Blood 93, 1464–1476.Banati, R.B., Daniel, S.E., Blunt, S.B., 1998. Glial pathology but absence of apoptotic
nigral neurons in long-standing Parkinson’s disease. Mov. Disord. 13, 221–227.Barclay, A.N., Wright, G.J., Brooke, G., Brown, M.H., 2002. CD200 and membrane
protein interactions in the control of myeloid cells. Trends Immunol. 23, 285–290.Batchelor, P.E., Liberatore, G.T., Wong, J.Y., Porritt, M.J., Frerichs, F., Donnan, G.A.,
Howells, D.W., 1999. Activated macrophages and microglia induce dopaminer-gic sprouting in the injured striatum and express brain-derived neurotrophicfactor and glial cell line-derived neurotrophic factor. J. Neurosci. 19, 1708–1716.
Batchelor, P.E., Porritt, M.J., Martinello, P., Parish, C.L., Liberatore, G.T., Donnan, G.A.,Howells, D.W., 2002. Macrophages and microglia produce local trophic gradi-ents that stimulate axonal sprouting toward but not beyond the wound edge.Mol. Cell. Neurosci. 21, 436–453.
Beach, T.G., Sue, L.I., Walker, D.G., Lue, L.F., Connor, D.J., Caviness, J.N., Sabbagh, M.N.,Adler, C.H., 2007. Marked microglial reaction in normal aging human substantianigra: correlation with extraneuronal neuromelanin pigment deposits. ActaNeuropathol. 114, 419–424.
Bernardo, A., Ajmone-Cat, M.A., Gasparini, L., Ongini, E., Minghetti, L., 2005. Nuclearreceptor peroxisome proliferator-activated receptor-gamma is activated in ratmicroglial cells by the anti-inflammatory drug HCT1026, a derivative of flurbi-profen. J. Neurochem. 92, 895–903.
Block, M.L., Hong, J.S., 2007. Chronic microglial activation and progressive dopa-minergic neurotoxicity. Biochem. Soc. Trans. 35, 1127–1132.
Boka, G., Anglade, P., Wallach, D., Javoy-Agid, F., Agid, Y., Hirsch, E.C., 1994.Immunocytochemical analysis of tumor necrosis factor and its receptors inParkinson’s disease. Neurosci. Lett. 172, 151–154.
Braak, H., Braak, E., Yilmazer, D., Schultz, C., de Vos, R.A., Jansen, E.N., 1995. Nigraland extranigral pathology in Parkinson’s disease. J. Neural Transm. 46, 15–31.
Braak, H., Del Tredici, K., Rub, U., de Vos, R.A., Jansen Steur, E.N., Braak, E., 2003.Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol.Aging 24, 197–211.

C.M. Long-Smith et al. / Progress in Neurobiology 89 (2009) 277–287 285
Brautigam, V.M., Frasier, C., Nikodemova, M., Watters, J.J., 2005. Purinergic receptormodulation of BV-2 microglial cell activity: potential involvement of p38 MAPkinase and CREB. J. Neuroimmunol. 166, 113–125.
Brochard, V., Combadiere, B., Prigent, A., Laouar, Y., Perrin, A., Beray-Berthat, V.,Bonduelle, O., Alvarez-Fischer, D., Callebert, J., Launay, J.M., Duyckaerts, C.,Flavell, R.A., Hirsch, E.C., Hunot, S., 2009. Infiltration of CD4+ lymphocytes intothe brain contributes to neurodegeneration in a mouse model of Parkinsondisease. J. Clin. Invest. 119, 182–192.
Bronstein, D.M., Perez-Otano, I., Sun, V., Mullis Sawin, S.B., Chan, J., Wu, G.C., Hudson,P.M., Kong, L.Y., Hong, J.S., McMillian, M.K., 1995. Glia-dependent neurotoxicityand neuroprotection in mesencephalic cultures. Brain Res. 704, 112–116.
Cajal, S., 1913. Contribucion al conocimiento de la neuroglia del cerebro humano.Trab. Lab. Investig. Biol. 11, 255–315.
Castano, A., Herrera, A.J., Cano, J., Machado, A., 1998. Lipopolysaccharide intranigralinjection induces inflammatory reaction and damage in nigrostriatal dopami-nergic system. J. Neurochem. 70, 1584–1592.
Castano, A., Herrera, A.J., Cano, J., Machado, A., 2002. The degenerative effect of asingle intranigral injection of LPS on the dopaminergic system is prevented bydexamethasone, and not mimicked by rh-TNF-alpha, IL-1beta and IFN-gamma.J. Neurochem. 81, 150–157.
Chang, R.C., Hudson, P., Wilson, B., Haddon, L., Hong, J.S., 2000. Influence of neuronson lipopolysaccharide-stimulated production of nitric oxide and tumor necrosisfactor-alpha by cultured glia. Brain Res. 853, 236–244.
Chao, C.C., Hu, S., Molitor, T.W., Shaskan, E.G., Peterson, P.K., 1992. Activatedmicroglia mediate neuronal cell injury via a nitric oxide mechanism. J. Immu-nol. 149, 2736–2741.
Chaturvedi, R.K., Beal, M.F., 2008. PPAR: a therapeutic target in Parkinson’s disease.J. Neurochem. 106, 506–518.
Chen, M., Ona, V.O., Li, M., Ferrante, R.J., Fink, K.B., Zhu, S., Bian, J., Guo, L., Farrell, L.A.,Hersch, S.M., Hobbs, W., Vonsattel, J.P., Cha, J.H., Friedlander, R.M., 2000.Minocycline inhibits caspase-1 and caspase-3 expression and delays mortalityin a transgenic mouse model of Huntington disease. Nat. Med. 6, 797–801.
Chen, P.S., Peng, G.S., Li, G., Yang, S., Wu, X., Wang, C.C., Wilson, B., Lu, R.B., Gean,P.W., Chuang, D.M., Hong, J.S., 2006. Valproate protects dopaminergic neuronsin midbrain neuron/glia cultures by stimulating the release of neurotrophicfactors from astrocytes. Mol. Psychiatry 11, 1116–1125.
Chen, P.S., Wang, C.C., Bortner, C.D., Peng, G.S., Wu, X., Pang, H., Lu, R.B., Gean, P.W.,Chuang, D.M., Hong, J.S., 2007. Valproic acid and other histone deacetylaseinhibitors induce microglial apoptosis and attenuate lipopolysaccharide-induced dopaminergic neurotoxicity. Neuroscience 149, 203–212.
Crotty, S., Fitzgerald, P., Tuohy, E., Harris, D.M., Fisher, A., Mandel, A., Bolton, A.E.,Sullivan, A.M., Nolan, Y., 2008. Neuroprotective effects of novel phosphatidyl-glycerol-based phospholipids in the 6-hydroxydopamine model of Parkinson’sdisease. Eur. J. Neurosci. 27, 294–300.
Cuadros, M.A., Navascues, J., 1998. The origin and differentiation of microglial cellsduring development. Prog. Neurobiol. 56, 173–189.
Cunningham, C., Wilcockson, D.C., Campion, S., Lunnon, K., Perry, V.H., 2005. Centraland systemic endotoxin challenges exacerbate the local inflammatory responseand increase neuronal death during chronic neurodegeneration. J. Neurosci. 25,9275–9284.
Davalos, D., Grutzendler, J., Yang, G., Kim, J.V., Zuo, Y., Jung, S., Littman, D.R., Dustin,M.L., Gan, W.B., 2005. ATP mediates rapid microglial response to local braininjury in vivo. Nat. Neurosci. 8, 752–758.
de Lau, L.M., Breteler, M.M., 2006. Epidemiology of Parkinson’s disease. LancetNeurol. 5, 525–535.
Dehmer, T., Heneka, M.T., Sastre, M., Dichgans, J., Schulz, J.B., 2004. Protection bypioglitazone in the MPTP model of Parkinson’s disease correlates with I kappa Balpha induction and block of NF kappa B and iNOS activation. J. Neurochem. 88,494–501.
del Rio Hortega, P., 1932. Microglia. In: Penfield, W. (Ed.), Cytology and CellularPathology of the Nervous System. Hoeber, New York, pp. 481–534.
Depino, A.M., Earl, C., Kaczmarczyk, E., Ferrari, C., Besedovsky, H., del Rey, A., Pitossi,F.J., Oertel, W.H., 2003. Microglial activation with atypical proinflammatorycytokine expression in a rat model of Parkinson’s disease. Eur. J. Neurosci. 18,2731–2742.
Dexter, D.T., Carter, C.J., Wells, F.R., Javoy-Agid, F., Agid, Y., Lees, A., Jenner, P.,Marsden, C.D., 1989. Basal lipid peroxidation in substantia nigra is increased inParkinson’s disease. J. Neurochem. 52, 381–389.
Di Matteo, V., Pierucci, M., Di Giovanni, G., Di Santo, A., Poggi, A., Benigno, A., Esposito,E., 2006. Aspirin protects striatal dopaminergic neurons from neurotoxin-induced degeneration: an in vivo microdialysis study. Brain Res. 1095, 167–177.
Diguet, E., Fernagut, P.O., Wei, X., Du, Y., Rouland, R., Gross, C., Bezard, E., Tison, F.,2004. Deleterious effects of minocycline in animal models of Parkinson’sdisease and Huntington’s disease. Eur. J. Neurosci. 19, 3266–3276.
Dobbs, R.J., Charlett, A., Purkiss, A.G., Dobbs, S.M., Weller, C., Peterson, D.W., 1999.Association of circulating TNF-alpha and IL-6 with ageing and parkinsonism.Acta Neurol. Scand. 100, 34–41.
Doty, R.L., 2007. Olfaction in Parkinson’s disease. Parkinsonism Relat. Disord. 13(Suppl 3), S225–S228.
Du, Y., Ma, Z., Lin, S., Dodel, R.C., Gao, F., Bales, K.R., Triarhou, L.C., Chernet, E., Perry,K.W., Nelson, D.L., Luecke, S., Phebus, L.A., Bymaster, F.P., Paul, S.M., 2001.Minocycline prevents nigrostriatal dopaminergic neurodegeneration in theMPTP model of Parkinson’s disease. Proc. Natl. Acad. Sci. USA 98, 14669–14674.
Duke, D.C., Moran, L.B., Pearce, R.K., Graeber, M.B., 2007. The medial and lateralsubstantia nigra in Parkinson’s disease: mRNA profiles associated with higherbrain tissue vulnerability. Neurogenetics 8, 83–94.
Eder, C., Schilling, T., Heinemann, U., Haas, D., Hailer, N., Nitsch, R., 1999. Morpho-logical, immunophenotypical and electrophysiological properties of restingmicroglia in vitro. Eur. J. Neurosci. 11, 4251–4261.
El Khoury, J., Hickman, S.E., Thomas, C.A., Loike, J.D., Silverstein, S.C., 1998. Micro-glia, scavenger receptors, and the pathogenesis of Alzheimer’s disease. Neuro-biol. Aging 19, S81–84.
Elkabes, S., DiCicco-Bloom, E.M., Black, I.B., 1996. Brain microglia/macrophagesexpress neurotrophins that selectively regulate microglial proliferation andfunction. J. Neurosci. 16, 2508–2521.
Emre, M., 2003. Dementia associated with Parkinson’s disease. Lancet Neurol. 2,229–237.
Esposito, E., Di Matteo, V., Benigno, A., Pierucci, M., Crescimanno, G., Di Giovanni, G.,2007. Non-steroidal anti-inflammatory drugs in Parkinson’s disease. Exp. Neu-rol. 205, 295–312.
Fahn, S., 2003. Description of Parkinson’s disease as a clinical syndrome. Ann. N. Y.Acad. Sci. 991, 1–14.
Fahn, S., Elton, R., 1987. Members of UPDRS develoment committee. In: Fahn, S.,Marsden, C.D., Calne, D.B., Goldstein, M. (Eds.), Recent Developments in Par-kinson’s Disease. Macmillan Health Care Information, Florham Park, NJ.
Fearnley, J.M., Lees, A.J., 1991. Ageing and Parkinson’s disease: substantia nigraregional selectivity. Brain 114, 2283–2301.
Ferrari, C.C., Pott Godoy, M.C., Tarelli, R., Chertoff, M., Depino, A.M., Pitossi, F.J.,2006. Progressive neurodegeneration and motor disabilities induced bychronic expression of IL-1beta in the substantia nigra. Neurobiol. Dis. 24,183–193.
Fitzgerald, P., Mandel, A., Bolton, A.E., Sullivan, A.M., Nolan, Y., 2008. Treatment withphosphotidylglycerol-based nanoparticles prevents motor deficits induced byproteasome inhibition: implications for Parkinson’s disease. Behav. Brain Res.195, 271–274.
Floden, A.M., Li, S., Combs, C.K., 2005. Beta-amyloid-stimulated microglia induceneuron death via synergistic stimulation of tumor necrosis factor alpha andNMDA receptors. J. Neurosci. 25, 2566–2575.
Gao, H.M., Hong, J.S., 2008. Why neurodegenerative diseases are progressive:uncontrolled inflammation drives disease progression. Trends Immunol. 29,357–365.
Gao, H.M., Liu, B., Zhang, W., Hong, J.S., 2003. Critical role of microglial NADPHoxidase-derived free radicals in the in vitro MPTP model of Parkinson’s disease.FASEB J. 17, 1954–1956.
Gao, X., Hu, X., Qian, L., Yang, S., Zhang, W., Zhang, D., Wu, X., Fraser, A., Wilson, B.,Flood, P.M., Block, M., Hong, J.S., 2008. Formyl-methionyl-leucyl-phenylala-nine-induced dopaminergic neurotoxicity via microglial activation: a mediatorbetween peripheral infection and neurodegeneration? Environ. Health Per-spect. 116, 593–598.
Garden, G.A., Moller, T., 2006. Microglia biology in health and disease. J. Neuroim-mune Pharmacol. 1, 127–137.
Gayle, D.A., Ling, Z., Tong, C., Landers, T., Lipton, J.W., Carvey, P.M., 2002. Lipopo-lysaccharide (LPS)-induced dopamine cell loss in culture: roles of tumornecrosis factor-alpha, interleukin-1beta, and nitric oxide. Brain Res. Dev. BrainRes. 133, 27–35.
Gibb, W.R., Poewe, W.H., 1986. The centenary of Friederich H. Lewy 1885–1950.Neuropathol. Appl. Neurobiol. 12, 217–222.
Gibbons, H.M., Dragunow, M., 2006. Microglia induce neural cell death via aproximity-dependent mechanism involving nitric oxide. Brain Res. 1084, 1–15.
Godoy, M.C., Tarelli, R., Ferrari, C.C., Sarchi, M.I., Pitossi, F.J., 2008. Central andsystemic IL-1 exacerbates neurodegeneration and motor symptoms in a modelof Parkinson’s disease. Brain 131, 1880–1894.
Griffin, R., Nally, R., Nolan, Y., McCartney, Y., Linden, J., Lynch, M.A., 2006. The age-related attenuation in long-term potentiation is associated with microglialactivation. J. Neurochem. 99, 1263–1272.
Grilli, M., Pizzi, M., Memo, M., Spano, P., 1996. Neuroprotection by aspirin andsodium salicylate through blockade of NF-kappaB activation. Science 274,1383–1385.
Hanisch, U.K., 2002. Microglia as a source and target of cytokines. Glia 40, 140–155.He, Y., Appel, S., Le, W., 2001. Minocycline inhibits microglial activation and protects
nigral cells after 6-hydroxydopamine injection into mouse striatum. Brain Res.909, 187–193.
Hickey, W.F., Kimura, H., 1988. Perivascular microglial cells of the CNS are bonemarrow-derived and present antigen in vivo. Science 239, 290–292.
Hirsch, E.C., Hunot, S., 2009. Neuroinflammation in Parkinson’s disease: a target forneuroprotection? Lancet Neurol. 8, 382–397.
Hoek, R.M., Ruuls, S.R., Murphy, C.A., Wright, G.J., Goddard, R., Zurawski, S.M., Blom,B., Homola, M.E., Streit, W.J., Brown, M.H., Barclay, A.N., Sedgwick, J.D., 2000.Down-regulation of the macrophage lineage through interaction with OX2(CD200). Science 290, 1768–1771.
Hu, X., Zhang, D., Pang, H., Caudle, W.M., Li, Y., Gao, H., Liu, Y., Qian, L., Wilson, B., DiMonte, D.A., Ali, S.F., Zhang, J., Block, M.L., Hong, J.S., 2008. Macrophage antigencomplex-1 mediates reactive microgliosis and progressive dopaminergic neu-rodegeneration in the MPTP model of Parkinson’s disease. J. Immunol. 181,7194–7204.
Hunot, S., Boissiere, F., Faucheux, B., Brugg, B., Mouatt-Prigent, A., Agid, Y., Hirsch,E.C., 1996. Nitric oxide synthase and neuronal vulnerability in Parkinson’sdisease. Neuroscience 72, 355–363.
Hunter, R.L., Dragicevic, N., Seifert, K., Choi, D.Y., Liu, M., Kim, H.C., Cass, W.A.,Sullivan, P.G., Bing, G., 2007. Inflammation induces mitochondrial dysfunctionand dopaminergic neurodegeneration in the nigrostriatal system. J. Neurochem.100, 1375–1386.

C.M. Long-Smith et al. / Progress in Neurobiology 89 (2009) 277–287286
Imamura, K., Hishikawa, N., Sawada, M., Nagatsu, T., Yoshida, M., Hashizume, Y.,2003. Distribution of major histocompatibility complex class II-positive micro-glia and cytokine profile of Parkinson’s disease brains. Acta Neuropathol. 106,518–526.
Jack, C.S., Arbour, N., Manusow, J., Montgrain, V., Blain, M., McCrea, E., Shapiro, A.,Antel, J.P., 2005. TLR signaling tailors innate immune responses in humanmicroglia and astrocytes. J. Immunol. 175, 4320–4330.
Jenner, P., Dexter, D.T., Sian, J., Schapira, A.H., Marsden, C.D., 1992. Oxidative stressas a cause of nigral cell death in Parkinson’s disease and incidental Lewy bodydisease. Ann. Neurol. 32 (Suppl), S82–S87.
Joglar, B., Rodriguez-Pallares, J., Rodriguez-Perez, A.I., Rey, P., Guerra, M.J., Laban-deira-Garcia, J.L., 2009. The inflammatory response in the MPTP model ofParkinson’s disease is mediated by brain angiotensin: relevance to progressionof the disease. J. Neurochem. 109, 656–669.
Johnston, L.C., Su, X., Maguire-Zeiss, K., Horovitz, K., Ankoudinova, I., Guschin, D.,Hadaczek, P., Federoff, H.J., Bankiewicz, K., Forsayeth, J., 2008. Human inter-leukin-10 gene transfer is protective in a rat model of Parkinson’s disease. Mol.Ther. 16, 1392–1399.
Kim, H.S., Suh, Y.H., 2009. Minocycline and neurodegenerative diseases. Behav.Brain Res. 196, 168–179.
Kim, S.U., de Vellis, J., 2005. Microglia in health and disease. J. Neurosci. Res. 81,302–313.
Kim, W.G., Mohney, R.P., Wilson, B., Jeohn, G.H., Liu, B., Hong, J.S., 2000. Regionaldifference in susceptibility to lipopolysaccharide-induced neurotoxicity in therat brain: role of microglia. J. Neurosci. 20, 6309–6316.
Kim, Y.S., Choi, D.H., Block, M.L., Lorenzl, S., Yang, L., Kim, Y.J., Sugama, S., Cho, B.P.,Hwang, O., Browne, S.E., Kim, S.Y., Hong, J.S., Beal, M.F., Joh, T.H., 2007. A pivotalrole of matrix metalloproteinase-3 activity in dopaminergic neuronal degen-eration via microglial activation. FASEB J. 21, 179–187.
Kim, Y.S., Kim, S.S., Cho, J.J., Choi, D.H., Hwang, O., Shin, D.H., Chun, H.S., Beal, M.F.,Joh, T.H., 2005. Matrix metalloproteinase-3: a novel signaling proteinase fromapoptotic neuronal cells that activates microglia. J. Neurosci. 25, 3701–3711.
Knott, C., Stern, G., Wilkin, G.P., 2000. Inflammatory regulators in Parkinson’sdisease: iNOS, lipocortin-1, and cyclooxygenases-1 and -2. Mol. Cell. Neurosci.16, 724–739.
Konsman, J.P., Parnet, P., Dantzer, R., 2002. Cytokine-induced sickness behaviour:mechanisms and implications. Trends Neurosci. 25, 154–159.
Kopp, E., Ghosh, S., 1994. Inhibition of NF-kappa B by sodium salicylate and aspirin.Science 265, 956–959.
Koprich, J.B., Reske-Nielsen, C., Mithal, P., Isacson, O., 2008. Neuroinflammationmediated by IL-1beta increases susceptibility of dopamine neurons to degen-eration in an animal model of Parkinson’s disease. J. Neuroinflammation 5, 8.
Kreutzberg, G.W., 1996. Microglia: a sensor for pathological events in the CNS.Trends Neurosci. 19, 312–318.
Kurkowska-Jastrzebska, I., Babiuch, M., Joniec, I., Przybylkowski, A., Czlonkowski, A.,Czlonkowska, A., 2002. Indomethacin protects against neurodegenerationcaused by MPTP intoxication in mice. Int. Immunopharmacol. 2, 1213–1218.
Kurkowska-Jastrzebska, I., Litwin, T., Joniec, I., Ciesielska, A., Przybylkowski, A.,Czlonkowski, A., Czlonkowska, A., 2004. Dexamethasone protects against dopa-minergic neurons damage in a mouse model of Parkinson’s disease. Int. Immu-nopharmacol. 4, 1307–1318.
Kurkowska-Jastrzebska, I., Wronska, A., Kohutnicka, M., Czlonkowski, A., Czlon-kowska, A., 1999. The inflammatory reaction following 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine intoxication in mouse. Exp. Neurol. 156, 50–61.
Laakso, M.P., Partanen, K., Riekkinen, P., Lehtovirta, M., Helkala, E.L., Hallikainen, M.,Hanninen, T., Vainio, P., Soininen, H., 1996. Hippocampal volumes in Alzhei-mer’s disease, Parkinson’s disease with and without dementia, and in vasculardementia: an MRI study. Neurology 46, 678–681.
Ladeby, R., Wirenfeldt, M., Garcia-Ovejero, D., Fenger, C., Dissing-Olesen, L., Dalmau,I., Finsen, B., 2005a. Microglial cell population dynamics in the injured adultcentral nervous system. Brain Res. Rev. 48, 196–206.
Ladeby, R., Wirenfeldt, M., Dalmau, I., Gregersen, R., Garcia-Ovejero, D., Babcock, A.,Owens, T., Finsen, B., 2005b. Proliferating resident microglia express the stemcell antigen CD34 in response to acute neural injury. Glia 50, 121–131.
Langston, J.W., Ballard, P., Tetrud, J.W., Irwin, I., 1983. Chronic parkinsonism inhumans due to a product of meperidine-analog synthesis. Science 219, 979–980.
Langston, J.W., Ballard Jr., P.A., 1983. Parkinson’s disease in a chemist working with1-methyl-4-phenyl-1,2,5,6-tetrahydropyridine. N. Engl. J. Med. 309, 310.
Liberatore, G.T., Jackson-Lewis, V., Vukosavic, S., Mandir, A.S., Vila, M., McAuliffe,W.G., Dawson, V.L., Dawson, T.M., Przedborski, S., 1999. Inducible nitric oxidesynthase stimulates dopaminergic neurodegeneration in the MPTP model ofParkinson disease. Nat. Med. 5, 1403–1409.
Ling, Z., Gayle, D.A., Ma, S.Y., Lipton, J.W., Tong, C.W., Hong, J.S., Carvey, P.M., 2002. Inutero bacterial endotoxin exposure causes loss of tyrosine hydroxylase neuronsin the postnatal rat midbrain. Mov. Disord. 17, 116–124.
Ling, Z.D., Chang, Q., Lipton, J.W., Tong, C.W., Landers, T.M., Carvey, P.M., 2004.Combined toxicity of prenatal bacterial endotoxin exposure and postnatal 6-hydroxydopamine in the adult rat midbrain. Neuroscience 124, 619–628.
Liu, B., 2006. Modulation of microglial pro-inflammatory and neurotoxic activity forthe treatment of Parkinson’s disease. AAPS J. 8, E606–621.
Liu, B., Du, L., Kong, L.Y., Hudson, P.M., Wilson, B.C., Chang, R.C., Abel, H.H., Hong, J.S.,2000a. Reduction by naloxone of lipopolysaccharide-induced neurotoxicity inmouse cortical neuron-glia co-cultures. Neuroscience 97, 749–756.
Liu, B., Jiang, J.W., Wilson, B.C., Du, L., Yang, S.N., Wang, J.Y., Wu, G.C., Cao, X.D., Hong,J.S., 2000b. Systemic infusion of naloxone reduces degeneration of rat sub-
stantia nigral dopaminergic neurons induced by intranigral injection of lipo-polysaccharide. J. Pharmacol. Exp. Ther. 295, 125–132.
Lowe, J., Blanchard, A., Morrell, K., Lennox, G., Reynolds, L., Billett, M., Landon, M.,Mayer, R.J., 1988. Ubiquitin is a common factor in intermediate filamentinclusion bodies of diverse type in man, including those of Parkinson’s disease,Pick’s disease, and Alzheimer’s disease, as well as Rosenthal fibres in cerebellarastrocytomas, cytoplasmic bodies in muscle, and mallory bodies in alcoholicliver disease. J. Pathol. 155, 9–15.
Lu, X., Bing, G., Hagg, T., 2000. Naloxone prevents microglia-induced degeneration ofdopaminergic substantia nigra neurons in adult rats. Neuroscience 97, 285–291.
Lynch, M.A. The multifaceted profile of activated microglia. Mol. Neurobiol., inpress.
Lyons, A., Downer, E.J., Crotty, S., Nolan, Y.M., Mills, K.H., Lynch, M.A., 2007. CD200ligand receptor interaction modulates microglial activation in vivo and in vitro:a role for IL-4. J. Neurosci. 27, 8309–8313.
McCoy, M.K., Martinez, T.N., Ruhn, K.A., Szymkowski, D.E., Smith, C.G., Botterman,B.R., Tansey, K.E., Tansey, M.G., 2006. Blocking soluble tumor necrosis factorsignaling with dominant-negative tumor necrosis factor inhibitor attenuatesloss of dopaminergic neurons in models of Parkinson’s disease. J. Neurosci. 26,9365–9375.
McCoy, M.K., Ruhn, K.A., Martinez, T.N., McAlpine, F.E., Blesch, A., Tansey, M.G.,2008. Intranigral lentiviral delivery of dominant-negative TNF attenuates neu-rodegeneration and behavioral deficits in hemiparkinsonian rats. Mol. Ther. 16,1572–1579.
McGeer, P.L., Itagaki, S., Boyes, B.E., McGeer, E.G., 1988. Reactive microglia arepositive for HLA-DR in the substantia nigra of Parkinson’s and Alzheimer’sdisease brains. Neurology 38, 1285–1291.
McGeer, P.L., McGeer, E.G., 2004. Inflammation and the degenerative diseases ofaging. Ann. N. Y. Acad. Sci. 1035, 104–116.
McGeer, P.L., McGeer, E.G., 2008. Glial reactions in Parkinson’s disease. Mov. Disord.23, 474–483.
McGeer, P.L., Schwab, C., Parent, A., Doudet, D., 2003. Presence of reactive microgliain monkey substantia nigra years after 1-methyl-4-phenyl-1,2,3,6-tetrahydro-pyridine administration. Ann. Neurol. 54, 599–604.
Mogi, M., Harada, M., Kondo, T., Riederer, P., Inagaki, H., Minami, M., Nagatsu, T.,1994a. Interleukin-1 beta, interleukin-6, epidermal growth factor and trans-forming growth factor-alpha are elevated in the brain from parkinsonianpatients. Neurosci. Lett. 180, 147–150.
Mogi, M., Harada, M., Riederer, P., Narabayashi, H., Fujita, K., Nagatsu, T., 1994b.Tumor necrosis factor-alpha (TNF-alpha) increases both in the brain and in thecerebrospinal fluid from parkinsonian patients. Neurosci. Lett. 165, 208–210.
Mogi, M., Togari, A., Kondo, T., Mizuno, Y., Komure, O., Kuno, S., Ichinose, H., Nagatsu,T., 2000. Caspase activities and tumor necrosis factor receptor R1 (p55) level areelevated in the substantia nigra from parkinsonian brain. J. Neural Transm. 107,335–341.
Mohanakumar, K.P., Muralikrishnan, D., Thomas, B., 2000. Neuroprotection bysodium salicylate against 1-methyl-4-phenyl-1,2,3, 6-tetrahydropyridine-induced neurotoxicity. Brain Res. 864, 281–290.
Morris, L., Graham, C.F., Gordon, S., 1991. Macrophages in haemopoietic and othertissues of the developing mouse detected by the monoclonal antibody F4/80.Development 112, 517–526.
Nagata, K., Takei, N., Nakajima, K., Saito, H., Kohsaka, S., 1993. Microglial condi-tioned medium promotes survival and development of cultured mesencephalicneurons from embryonic rat brain. J. Neurosci. Res. 34, 357–363.
NINDS NET-PD Investigators, N.N.-P., 2008. A pilot clinical trial of creatine andminocycline in early Parkinson disease: 18-month results. Clin. Neuropharma-col. 31, 141–150.
Nimmerjahn, A., Kirchhoff, F., Helmchen, F., 2005. Resting microglial cells are highlydynamic surveillants of brain parenchyma in vivo. Science 308, 1314–1318.
Nolan, Y., Maher, F.O., Martin, D.S., Clarke, R.M., Brady, M.T., Bolton, A.E., Mills, K.H.,Lynch, M.A., 2005. Role of interleukin-4 in regulation of age-related inflamma-tory changes in the hippocampus. J. Biol. Chem. 280, 9354–9362.
Nussbaum, R.L., Ellis, C.E., 2003. Alzheimer’s disease and Parkinson’s disease. N.Engl. J. Med. 348, 1356–1364.
Ogata, A., Tashiro, K., Nukuzuma, S., Nagashima, K., Hall, W.W., 1997. A rat model ofParkinson’s disease induced by Japanese encephalitis virus. J. Neurovirol. 3,141–147.
Ogata, A., Tashiro, K., Pradhan, S., 2000. Parkinsonism due to predominant involve-ment of substantia nigra in Japanese encephalitis. Neurology 55, 602.
Orr, C.F., Rowe, D.B., Halliday, G.M., 2002. An inflammatory review of Parkinson’sdisease. Prog. Neurobiol. 68, 325–340.
Pais, T.F., Figueiredo, C., Peixoto, R., Braz, M.H., Chatterjee, S., 2008. Necrotic neuronsenhance microglial neurotoxicity through induction of glutaminase by aMyD88-dependent pathway. J. Neuroinflammation 5, 43.
Parkinson, J., 1817. An Essay on the Shaking Palsy. Whittingham & Rowland,London.
Peng, G.S., Li, G., Tzeng, N.S., Chen, P.S., Chuang, D.M., Hsu, Y.D., Yang, S., Hong, J.S.,2005. Valproate pretreatment protects dopaminergic neurons from LPS-induced neurotoxicity in rat primary midbrain cultures: role of microglia. BrainRes. 134, 162–169.
Perry, V.H., 1998. A revised view of the central nervous system microenvironmentand major histocompatibility complex class II antigen presentation. J. Neu-roimmunol. 90, 113–121.
Perry, V.H., 2004. The influence of systemic inflammation on inflammation in thebrain: implications for chronic neurodegenerative disease. Brain Behav. Immun.18, 407–413.

C.M. Long-Smith et al. / Progress in Neurobiology 89 (2009) 277–287 287
Perry, V.H., Cunningham, C., Holmes, C., 2007. Systemic infections and inflammationaffect chronic neurodegeneration. Nat. Rev. Immunol. 7, 161–167.
Przedborski, S., Vila, M., 2003. The 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridinemouse model: a tool to explore the pathogenesis of Parkinson’s disease. Ann. N.Y. Acad. Sci 991, 189–198.
Qian, L., Hong, J.S., Flood, P.M., 2006. Role of microglia in inflammation-mediateddegeneration of dopaminergic neurons: neuroprotective effect of interleukin10. J. Neural Transm. 70, 367–371.
Qin, L., Wu, X., Block, M.L., Liu, Y., Breese, G.R., Hong, J.S., Knapp, D.J., Crews, F.T.,2007. Systemic LPS causes chronic neuroinflammation and progressive neuro-degeneration. Glia 55, 453–462.
Quinn, L.P., Crook, B., Hows, M.E., Vidgeon-Hart, M., Chapman, H., Upton, N.,Medhurst, A.D., Virley, D.J., 2008. The PPARgamma agonist pioglitazone iseffective in the MPTP mouse model of Parkinson’s disease through inhibitionof monoamine oxidase B. Br. J. Pharmacol. 154, 226–233.
Quintero, E.M., Willis, L., Singleton, R., Harris, N., Huang, P., Bhat, N., Granholm, A.C.,2006. Behavioral and morphological effects of minocycline in the 6-hydroxy-dopamine rat model of Parkinson’s disease. Brain Res. 1093, 198–207.
Rail, D., Scholtz, C., Swash, M., 1981. Post-encephalitic parkinsonism: currentexperience. J. Neurol. Neurosurg. Psychiatry 44, 670–676.
Robinson, P.A., 2008. Protein stability and aggregation in Parkinson’s disease.Biochem. J. 413, 1–13.
Rogers, J., Strohmeyer, R., Kovelowski, C.J., Li, R., 2002. Microglia and inflammatorymechanisms in the clearance of amyloid beta peptide. Glia 40, 260–269.
Sanchez-Pernaute, R., Ferree, A., Cooper, O., Yu, M., Brownell, A.L., Isacson, O., 2004.Selective COX-2 inhibition prevents progressive dopamine neuron degenera-tion in a rat model of Parkinson’s disease. J. Neuroinflammation 1, 6.
Santambrogio, L., Belyanskaya, S.L., Fischer, F.R., Cipriani, B., Brosnan, C.F., Ricciardi-Castagnoli, P., Stern, L.J., Strominger, J.L., Riese, R., 2001. Developmental plas-ticity of CNS microglia. Proc. Natl. Acad. Sci. U. S. A. 98, 6295–6300.
Sawada, M., Imamura, K., Nagatsu, T., 2006. Role of cytokines in inflammatoryprocess in Parkinson’s disease. J. Neural Transm. 70, 373–381.
Schintu, N., Frau, L., Ibba, M., Caboni, P., Garau, A., Carboni, E., Carta, A.R., 2009.PPAR-gamma-mediated neuroprotection in a chronic mouse model of Parkin-son’s disease. Eur. J. Neurosci. 29, 954–963.
Spillantini, M.G., Schmidt, M.L., Lee, V.M., Trojanowski, J.Q., Jakes, R., Goedert, M.,1997. Alpha-synuclein in Lewy bodies. Nature 388, 839–840.
Stangel, M., Compston, A., 2001. Polyclonal immunoglobulins (IVIg) modulate nitricoxide production and microglial functions in vitro via Fc receptors. J. Neuroim-munol. 112, 63–71.
Stypula, G., Kunert-Radek, J., Stepien, H., Zylinska, K., Pawlikowski, M., 1996.Evaluation of interleukins, ACTH, cortisol and prolactin concentrations in theblood of patients with parkinson’s disease. Neuroimmunomodulation 3,131–134.
Suo, Z., Wu, M., Ameenuddin, S., Anderson, H.E., Zoloty, J.E., Citron, B.A.,Andrade-Gordon, P., Festoff, B.W., 2002. Participation of protease-activatedreceptor-1 in thrombin-induced microglial activation. J. Neurochem. 80,655–666.
Tanaka, J., Toku, K., Matsuda, S., Sudo, S., Fujita, H., Sakanaka, M., Maeda, N., 1998.Induction of resting microglia in culture medium devoid of glycine and serine.Glia 24, 198–215.
Tansey, M.G., McCoy, M.K., Frank-Cannon, T.C., 2007. Neuroinflammatory mechan-isms in Parkinson’s disease: potential environmental triggers, pathways, andtargets for early therapeutic intervention. Exp. Neurol. 208, 1–25.
Tikka, T., Fiebich, B.L., Goldsteins, G., Keinanen, R., Koistinaho, J., 2001. Minocycline,a tetracycline derivative, is neuroprotective against excitotoxicity by inhibitingactivation and proliferation of microglia. J. Neurosci. 21, 2580–2588.
Tikka, T.M., Koistinaho, J.E., 2001. Minocycline provides neuroprotection against N-methyl-D-aspartate neurotoxicity by inhibiting microglia. J. Immunol. 166,7527–7533.
Tomas-Camardiel, M., Rite, I., Herrera, A.J., de Pablos, R.M., Cano, J., Machado, A.,Venero, J.L., 2004. Minocycline reduces the lipopolysaccharide-induced inflam-matory reaction, peroxynitrite-mediated nitration of proteins, disruption of theblood-brain barrier, and damage in the nigral dopaminergic system. Neurobiol.Dis. 16, 190–201.
Ton, T.G., Heckbert, S.R., Longstreth Jr., W.T., Rossing, M.A., Kukull, W.A., Franklin,G.M., Swanson, P.D., Smith-Weller, T., Checkoway, H., 2006. Nonsteroidal anti-inflammatory drugs and risk of Parkinson’s disease. Mov. Disord. 21, 964–969.
Toulouse, A., Sullivan, A.M., 2008. Progress in Parkinson’s disease-where do westand? Prog. Neurobiol. 85, 376–392.
Wilms, H., Rosenstiel, P., Sievers, J., Deuschl, G., Zecca, L., Lucius, R., 2003. Activationof microglia by human neuromelanin is NF-kappaB dependent and involves p38mitogen-activated protein kinase: implications for Parkinson’s disease. FASEB J.17, 500–502.
Wolters, E., 2008. Variability in the clinical expression of Parkinson’s disease. J.Neurol. Sci. 266, 197–203.
Wu, D.C., Teismann, P., Tieu, K., Vila, M., Jackson-Lewis, V., Ischiropoulos, H.,Przedborski, S., 2003. NADPH oxidase mediates oxidative stress in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine model of Parkinson’s disease.Proc. Natl. Acad. Sci. U. S. A. 100, 6145–6150.
Wu, X., Chen, P.S., Dallas, S., Wilson, B., Block, M.L., Wang, C.C., Kinyamu, H., Lu, N.,Gao, X., Leng, Y., Chuang, D.M., Zhang, W., Lu, R.B., Hong, J.S., 2008. Histonedeacetylase inhibitors up-regulate astrocyte GDNF and BDNF gene transcriptionand protect dopaminergic neurons. Int. J. Neuropsychopharmacol. 11,1123–1134.
Yang, L., Sugama, S., Chirichigno, J.W., Gregorio, J., Lorenzl, S., Shin, D.H., Browne,S.E., Shimizu, Y., Joh, T.H., Beal, M.F., Albers, D.S., 2003. Minocycline enhancesMPTP toxicity to dopaminergic neurons. J. Neurosci. Res. 74, 278–285.
Yirmiya, R., Winocur, G., Goshen, I., 2002. Brain interleukin-1 is involved in spatialmemory and passive avoidance conditioning. Neurobiol. Learn. Mem. 78,379–389.
Zalcman, S., Green-Johnson, J.M., Murray, L., Nance, D.M., Dyck, D., Anisman, H.,Greenberg, A.H., 1994. Cytokine-specific central monoamine alterationsinduced by interleukin-1, -2 and -6. Brain Res. 643, 40–49.
Zhang, W., Wang, T., Pei, Z., Miller, D.S., Wu, X., Block, M.L., Wilson, B., Zhang, W.,Zhou, Y., Hong, J.S., Zhang, J., 2005. Aggregated alpha-synuclein activatesmicroglia: a process leading to disease progression in Parkinson’s disease.FASEB J. 19, 533–542.
Zhu, S., Stavrovskaya, I.G., Drozda, M., Kim, B.Y., Ona, V., Li, M., Sarang, S., Liu, A.S.,Hartley, D.M., Wu, D.C., Gullans, S., Ferrante, R.J., Przedborski, S., Kristal, B.S.,Friedlander, R.M., 2002. Minocycline inhibits cytochrome c release and delaysprogression of amyotrophic lateral sclerosis in mice. Nature 417, 74–78.