Upload
others
View
14
Download
0
Embed Size (px)
Citation preview
UNIT 4.4Microscopy and Image AnalysisGeorge McNamara,1 Michael Difilippantonio,2 Thomas Ried,3
and Frederick R. Bieber4
1Biomedical Consultant, Baltimore, Maryland2Division of Cancer Treatment and Diagnosis, National Cancer Institute, National Institutesof Health, Bethesda, Maryland
3Section of Cancer Genomics, Genetics Branch, Center for Cancer Research, NationalCancer Institute, National Institutes of Health, Bethesda, Maryland
4Brigham and Women’s Hospital, Boston, Massachusetts
This unit provides an overview of light microscopy, including objectives, lightsources, filters, film, and color photography for fluorescence microscopy andfluorescence in situ hybridization (FISH). We believe there are excellent oppor-tunities for cytogeneticists, pathologists, and other biomedical readers, to takeadvantage of specimen optical clearing techniques and expansion microscopy—we briefly point to these new opportunities. C© 2017 by John Wiley & Sons,Inc.
Keywords: light microscopy � digital imaging � fluorescence in situ hybridiza-tion � functional genomics
How to cite this article:McNamara, G., Difilippantonio, M., Ried, T., & Bieber, F. R. (2017).
Microscopy and image analysis. Current Protocols in Human Genetics,94, 4.4.1–4.4.89. doi: 10.1002/cphg.42
INTRODUCTIONThis unit provides an overview of light mi-
croscopy, including objectives, light sources,filters, and imaging for fluorescence mi-croscopy and fluorescence in situ hybridiza-tion (FISH). We encourage thinking outsidethe usual magnification range of 10× to100× objective lenses, by ranging from sin-gle molecules to whole mice and humans.Traditionally, clinical FISH was chromosomeand single or dual gene DNA FISH. Singlemolecule RNA FISH is now a research tool,and has opportunities in the clinic. Genome en-gineering with clustered regularly interspacedshort palindromic repeats (CRISPR)/Cas9,transcription activation-like effector nucleases(TALENs), and zinc finger nucleases requiresboth on-target validation and off-target safetychecks. Computerized image analysis systemscurrently used in clinical cytogenetics are alsodiscussed, and connected with the larger trendof digital slide scanning in pathology and cy-tology. We discuss how functional genomicscan contribute to cytogenetics and vice versa.Photophysics of fluorescence, and applicationsof specialized F-techniques, are well reviewed
by Ishikawa-Ankerhold, Ankerhold, & Drum-men (2012), and Liu, Ahmed, & Wohland(2008). Scanning and transmission electronmicroscopy as well as confocal microscopyand multi-photon excitation microscopy arenot covered in this unit despite their useful-ness as invaluable tools for contemporary stud-ies of biological systems; see Diaspro, 2001;Matsumoto, 2002; Minsky, 1988; Paddock,1999; Pawley, 2005; Shotton, 1993; van derVoort, Valkenburg, van Spronsen, Woldringh,and Brakenhoff, 1987, for further informationon confocal and multi-photon microscopies.Since the 2005 version of this unit (UNIT 4.4,McNamara, Difilippantonio, & Ried, 2005),Nobel Prizes have been awarded for fluores-cent proteins and super-resolution. We haveadded a table and select references for fluores-cent proteins. Super-resolution microscopiesare out of the scope of this chapter; we rec-ommend Schermelleh, Heintzmann, & Leon-hardt (2010) and Turkowyd, Virant, and En-desfelder (2016) for reviews. We summarizerecent advances in light sheet imaging andexpansion microscopy, and how these can beuseful. We believe there are excellent oppor-
Current Protocols in Human Genetics 4.4.1–4.4.89, July 2017Published online July 2017 in Wiley Online Library (wileyonlinelibrary.com).doi: 10.1002/cphg.42Copyright C© 2017 John Wiley & Sons, Inc.
Cytogenetics
4.4.1
Supplement 94
gmcnamaraTypewritten TextOpen Access
tunities for cytogeneticists, pathologists, andother biomedical readers, to take advantage ofspecimen optical clearing techniques and ex-pansion microscopy—we briefly point to thesenew opportunities.
NANO, MICRO, AND MACROSCALES
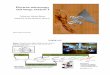
Figure 4.4.1 illustrates the nano, micro, andmacro scale. The left side is a mouse tis-sue section, with original dimensions of 40 ×14 mm, acquired on a Meyer Instruments Path-scan Enabler digital slide scanner at the equiv-alent of a 4× objective lens (Masson trichromestain, prepared slide from Carolina BiologicalSupply).
The right side top and second panels are sin-gle molecule RNA FISH of GAPDH mRNAof pancreatic cancer cells; top is single planeraw data, the second panel is a Microvo-lution 100 iteration quantitative deconvolu-tion (www.microvolution.com; Biosearch Stel-laris FISH oligonucleotides probe set, Quasar570 dye, detected with a Texas Red fil-ter set on a Leica DMI 6000 microscopewith Hamamatsu Flash 4.2 scientific com-plementary metal-oxide semiconductor [sC-MOS] camera, additional details and dataat https://works.bepress.com/gmcnamara/55).The four bright dots in the left side nucleus aremost likely multiple pre-messenger RNA andmRNA molecules from transcriptional burstsat the gene loci (aneuploidy or late S or G2phase of cell cycle). Fluorescence imaging ofsingle biological molecules on a research flu-orescence microscope should now be consid-ered routine. The quantitative deconvolutionimage processing (second panel) slightly im-proves spatial resolution (of the raw data), anddramatically increases contrast.
The right middle is a SKY spectral kary-otype of a metaphase from a MDA-MB-435subline (modal chromosome number �90,this metaphase is �400), acquired on aNikon Eclipse fluorescence microscope with40×/1.3 NA objective lens (width �250 μm).The image was acquired by Carrie Viars andSteve Goodison (University of California, SanDiego) and George McNamara, experiment re-lated to Urquidi et al. (2002).
Finally, the right side fourth and bottompanels are raw and quantitative deconvolutionimage data of human osteosarcoma cell lineSaOS2, double immunofluorescence (RUNX2red, osteocalcin green) plus Hoechst DNA nu-clear counterstain (blue), acquired by JaredMortus and George McNamara. Three Z-series acquisition time (MetaMorph software,
Leica microscope, Flash 4.2 sCMOS camera)and deconvolution time were each �17 sec(www.microvolution.com). Shown is a maxi-mum projection image. See Appendix 4.4.3(see Supporting Materials) for larger field ofview.
HISTORICAL FOUNDATIONS OFMICROSCOPY
Even in medieval times it was understoodthat curved mirrors and hollow glass spheresfilled with water had a magnifying effect. Inthe early 17th century, men began experiment-ing with lenses to increase magnification. Acompound telescope, with weak convex lensat one end and a concave lens as the eye-piece, was demonstrated by a Dutch spectaclemaker to the court at The Hague in September1608 (Ruestow, 1996). News quickly spreadthroughout Europe. Galileo made his owncompound telescope in 1609, turned it to theplanet Jupiter and discovered moons. Galileosoon turned his telescope around and observedflies with it. Credit for the now standard twoconvex lens microscope goes to the son andfather team of Janssen and Janssen. Natural-ists Jan Swammerdam (1637 to 1680) and Ne-hemiah Grew (1641 to 1712), anatomist Reg-nier Graaf (1641 to 1673), and physiologistMarcello Malpighi (1628 to 1694) made im-portant discoveries using magnifying lenses,especially tiny, strong single lenses (Ruestow,1996).
Robert Hooke’s book, Micrographia, pub-lished in 1665, contains beautiful drawingsbased on his microscopic observations. Hisexperimental demonstrations to the RoyalSociety were interrupted by the 1666 fire ofLondon, after which he and his friend andbusiness partner Christopher Wren had majorroles in the surveying and rebuilding ofthe city (Jardine, 2004). Also in 1666 SirIsaac Newton found that a prism separateswhite light into distinct colors, and crucially,brilliantly, discovered the rainbow could be re-combined into white light with a second prism(Newton, 1672, 1730). In 1683, the Dutchmerchant, Anton van Leeuwenhoek (1632 to1723), using his own meticulously preparedlenses, published his first of many papers tothe Philosophical Transactions of the RoyalSociety of London (Leewenhoeck, 1683).van Leeuwenhoek’s publications of “animal-cules,” blood cells, sperm and more, were thefirst multi-decade high throughput microscopyproject. In 1773 a Danish microbiologist, OttoMuller (1730 to 1784), used the microscope to
Microscopy andImage Analysis
4.4.2
Supplement 94 Current Protocols in Human Genetics
http://www.microvolution.com;https://works.bepress.com/gmcnamara/55http://www.microvolution.com
Figure 4.4.1 (legend appears on next page) Cytogenetics
4.4.3
Current Protocols in Human Genetics Supplement 94
describe the forms and shapes of variousbacteria. In 1833, Robert Brown (1773 to1858) discovered the consistent presence ofnuclei in plant cells. Brown also reported onthe microscopic behavior of tiny, non-livingclay particles, now called Brownian motion,which Einstein discussed in one of his classic1905 papers.
In 1856, William Perkin discovered mauve,the first useful synthetic dye. He used his firstbatch to stain silk. After trial and error he foundhe could use tannins to color fast stain mauveonto wool and cotton. His successful commer-cialization revolutionized the textile industry,fashion, and rapidly led to the modern chem-ical and pharmaceuticals industries (Garfield,2001; Perkin, 1906). The success of Perkin af-ter dropping out of scholarly chemistry stud-ies has parallels a century later with anothersuccessful William: Bill Gates of Microsoft.Mauve was a serendipitous discovery but in1865 F. August Kekule (1829 to 1896) dreamthis theory of the benzene ring, which togetherwith an understanding of the stoichiometry ofmolecules and chemical reactions led to chem-istry becoming a rational science.
In 1869, Friedrich Meischer isolated nu-cleic acids. Of the many dyes invented andused in the following decades, we mention thatfluorescein was discovered and synthesized in1871 (see Clark & Kasten, 1983 for a his-tory of staining). In 1879 to 1880 Paul Ehrlichperformed a series of studies on the natureof cell and tissue staining by acidic and ba-sic dyes and later went on to found immunol-ogy and the idea of magic bullet chemother-apies. Ernst Abbe described the mathematicsof diffraction and optimal lens constructionin 1876. The firm of Carl Zeiss quickly man-ufactured high quality oil immersion lensesof Abbe’s designs. In 1893, August Köhler,a German zoologist, described the principlesof what we now refer to as Köhler illumina-tion (APPENDIX 3N, Salmon & Canman, 2003).This was a critical step in generating a uniformfield of illumination and providing optimal im-age resolution. Rudolf Virchow (1821 to 1902)founded modern cell physiology in the midstof these twin revolutions in dye chemistry andmicroscope optics. Waldeyer coined the term
Figure 4.4.1 (image appears on previous page) Tiki_Goddess. Example of scanning an entiretissue section. This Masson trichrome stained mouse tissue section was scanned in “one click”using a Meyer Instruments Pathscan Enabler microscope slide scanner. The tissue section is 40× 14 mm (�10 µm thick), and the scan dimension length limit is 36 mm, so really a click, rotateslide, click, open in Adobe Photoshop, and a few more clicks and drags to merge, rotate vertically,white balance to taste, and save. The entire process took under five minutes. We emphasize herethe value of having “the whole story” in one image.
“chromosomes” in 1888 to refer to those col-ored bodies he saw in dividing cells.
Köhler and Moritz von Rohr made the firstultraviolet microscope, to try to take advan-tage of shorter wavelengths producing bet-ter resolution (summarized in Kohler, 2016).In the course of their studies they made thefirst fluorescence microscope. In the followingdecades, the firms of Zeiss and Reichert madethe first fluorescence microscopes using trans-mitted light illumination and simple filters.Among the earliest use of epi-illumination wasSinger’s (1932) observations on live tissues.While Singer preferred a therapeutic sunshinecarbon arc, his use of epi-illumination witha heat absorbing filter, excitation filter, mir-ror, and emission filter is organized along thelines of a modern fluorescence microscope.In the 1930s and 1940s Strugger and othersinvestigated biological fluorescence stainingwith acridine orange and other fluorophores.In 1934 Marrack conjugated pathogen-specificantibodies to a dye, however, the weak label-ing was hard to see on the bright backgroundof standard transmitted light microscopy. Al-bert Coons (1912 to 1978) and colleagues(see Coons, 1961; Coons, Creech, & Jones,1941; Coons, Creech, Jones, & Berliner, 1942)adapted Marrack’s dye coupling idea with afluorescent covalent labeling, introducing theconcept of immunofluorescence, which en-abled staining with unparalleled molecularspecificity. Coons and Kaplan (1950) intro-duced the use of a fluorescent secondary an-tibody to detect the antigen specific primaryantibody. Secondary antibodies both amplifiedthe staining intensity per antigen, and made thetechnique generic in that one batch labeled sec-ondary antibody (i.e., goat anti rabbit) couldbe used to detect many different primary anti-bodies on different specimens (i.e., rabbit anti-pneumococcus, rabbit anti-streptococcus). Akey advance when using formalin-fixed tis-sue sections for either immunohistochemistrywith chromogenic subtracts, or immunofluo-rescence, was the development of antigen re-trieval (Shi, Gu, & Taylor, 2000). A recentdevelopment may have further improved anti-gen retrieval (Vollert, Moree, Gregory, Bark,& Eriksen, 2015).
Microscopy andImage Analysis
4.4.4
Supplement 94 Current Protocols in Human Genetics
In 1924, Feulgen and Rossenbeck reportedon their use of pararosaniline, a close relativeof Perkin’s mauve, and their use of a quanti-tative depurination reaction to make a Schiff’sbase, to produce a reliable way to quantifyDNA in cell nuclei and bacteria. This ulti-mately led to the discovery of distinct stagesof eukaryotic cell cycle, constancy of DNA inmost mammalian cell lineages, proved that ga-metogenesis involves a reduction division, andstimulated many developments in the 1930s to1950s of quantitative microscopy by Caspers-son and others.
Waldeyer’s colored bodies were subdi-vided in the 1930s by Bridge’s observation ofchromosome banding in Drosophila chromo-somes. Hsu (1952), capitalizing on a serendip-itous buffer dilution error, invented a chromo-some spreading technique based on hypotonicswelling of metaphase cells. Tjio and Levan(1956) used the hypotonic swelling/cell drop-ping method to correctly enumerate the 46human chromosomes. Lejeune, Gautier, andTurpin (1959) quickly discovered trisomy 21in (most) Down syndrome patients. Caspers-son et al. (1968) published their methodof fluorescently banding chromosomes usingquinacrine mustard to uniquely identify eachof the 23 pairs of human chromosomes. Histor-ically, clinical chromosome banding was donewith the Perkin era nonfluorescent dye Giemsa(G-banding). Using an A:T specific fluores-cent DNA dye, with digital acquisition by sC-MOS, charge-coupled device (CCD), or con-focal microscopy, enables a similar bandingimage, after digital inverse contrast in the com-puter. Such images can be improved further byeither fast unsharp masking (Adobe Photoshopor FIJI ImageJ) or quantitative spatial decon-volution (e.g., www.microvolution.com) to ob-tain higher spatial resolution and contrast. Akey advantage of using DAPI over Giemsa isthat fluorescence further enables—using mul-tiple probe fluorescence hybridization, suchas specific DNA FISH probe sets, or wholegenome—spectral karyotyping.
MICROSCOPY IN MODERNHUMAN GENETICS
Microscopy currently plays a crucial rolein both research and diagnostic aspects ofmodern genetics. This typically involves theuse of light microscopes for the analysis ofmicrobiological, cytological, and pathologicalspecimens, as well as the cytogenetic analy-sis of metaphase and interphase chromosomes(APPENDIX 3N; Salmon & Canman, 2003). With
recent advances in fluorescence technology,there has been growth, even in clinical labora-tories, in the use of fluorescence microscopy.Spectral karyotyping and Multiplex-FISH in-struments have made their way into manyclinical labs, but in narrow niches. Confocalmicroscopy was invented by Minsky (1957,1988), and re-invented by Eggar and Petran(1967), but their achievements were not widelyappreciated. The first successful confocal mi-croscope was developed around the confo-cal laser scanning microscope introduced byWhite, Amos, and Fordham (1987). The his-tory of MRC confocal microscopes, and con-text of cell analysis and antibody develop-ments, has been reviewed by the inventors,Amos and White (2003). Confocal microscopyimproves lateral resolution by a factor of �2,that is, by 1.414, but more importantly, pro-vides optical sectioning by blocking out offocus light from reaching the detector. Thedownside is that some of the in-focus lightis also blocked. Most of the fluorescence mi-croarray readers, i.e., Affymetrix, now foundin clinical core labs, are based around con-focal scanner designs. Denk, Strickler, andWebb, (1990) introduced multi-photon excita-tion microscopy (MPEM). MPEM is an intrin-sically optical sectioning technique, where flu-orescence only occurs in a diffraction-limitedspot where a high intensity near infra-red lasercomes to a focal point. Although most MPEMmicroscopes are based on, and can be used as,confocal microscopes, the pinhole is typicallykept open to collect more light. A few con-focal microscopes are within the budget rangeof a well-endowed clinical lab, but commercialmultiphoton microscopes currently use expen-sive Ti:Sapphire pulsed lasers. The biomedicaloptics community is busy pushing the limits ofmicroscopy into nanoscopy, a field reviewedby Garini, Vermolen, and Young (2005), andawarded the 2014 Nobel Prize in Chemistry toEric Betzig, Stefan W. Hell, and William E.Moerner.
Analysis of cell types in blood and biopsyspecimens, apoptosis assays in diseased tis-sue, cytogenetic analysis, and even surgicalprocedures using fluorescence have all beenreported. Because this unit deals primarilywith the use of microscopy and contempo-rary image analysis in mammalian cytoge-netics, a brief explanation of some of theapplications of fluorescence microscopy inthis field is relevant. The ability to label nu-cleic acids with fluorescent molecules and de-tect them in situ was developed in the early1980s (Langer-Safer, Levine, & Ward, 1982;
Cytogenetics
4.4.5
Current Protocols in Human Genetics Supplement 94
http://www.microvolution.com
Manuelidis, Langer-Safer, & Ward, 1982).Fluorescence in situ hybridization (FISH;UNIT 4.3; Knoll & Lichter, 2005) technologyhas been applied to many different areas ofcytogenetic investigation (Lichter & Ward,1990). The first use involved determination ofchromosome copy number in interphase nucleiusing centromere-specific fluorescent probes(Cremer et al., 1986; Devilee et al., 1988;Manuelidis, 1985). This was later extendedto the identification of aberrant or markerchromosomes (Blennow et al., 1995; Tani-waki et al., 1993; Thangavelu, Pergament, Es-pinosa, & Bohlander, 1994), or microdeletions(Ried et al., 1990) using chromosome paint-ing probes or single-copy probes known tomap to specific chromosomes. Giemsa band-ing was not compatible with fluorescence hy-bridization techniques, thereby making it dif-ficult to obtain simultaneous identification ofthe chromosomes. Methods were soon de-veloped, however, whereby banding patternscould be obtained through the hybridizationof fluorescently labeled repetitive sequencesin both humans (Baldini & Ward, 1991) andmice (Arnold, Bhatt, Ried, Wienberg, & Ward,1992; Boyle, Ballard, & Ward, 1990). Con-current with these advances came the startof the Human Genome Project and the ap-plication of FISH to gene mapping (Lichteret al., 1990, 1992; Otsu et al., 1993; Ried,Baldini, Rand, & Ward, 1992; Trask et al.,1993; Ward et al., 1991). FISH mapping is auseful technique for the identification of othergenes in a gene family, including functionaland non-functional genes (Giordano et al.,1993), and for the mapping of genes acrossspecies barriers, a technique referred to asZoo-FISH (Chowdhary, Raudsepp, Fronicke,& Scherthan, 1998; Fronicke & Scherthan,1997; O’Brien et al., 1997; Raudsepp, Fron-icke, Scherthan, Gustavsson, & Chowdhary,1996; Wienberg, Jauch, Stanyon, & Cremer,1990).
The fluorescent labeling and hybridizationof entire genomes is useful for a number of dif-ferent areas of investigation. Somatic cell hy-brid lines (see Chapter 3; Haines et al., 2017)were historically very useful for isolating andmapping disease genes in both mice and hu-mans (Harris, 1995). Comparative genome hy-bridization (CGH) is another technique involv-ing the fluorescent labeling of entire genomes.In this instance, the genomes may be fromkaryotypically normal reference and mutant(i.e., tumor) populations. The genomes, la-beled with different fluorophores, are pooled
prior to hybridization. The hybridization ra-tio of the two fluorophores along the lengthof each chromosome is calculated to deter-mine the gain or loss of chromosomal regionsin the mutant cells (UNIT 4.6; DeVries, Gray,Pinkel, Waldman, & Sudar, 1995; Forozan,Karhu, Kononen, Kallioniemi, & Kallioniemi1997; Kallioniemi et al., 1992; Ried et al.,1997). This technique is extremely useful incases involving multiple chromosomal rear-rangements for which specific bands cannotbe identified by Giemsa staining, or for de-tecting small insertions or deletions (largerthan 1 Mb). Comparative cytogenetics is thestudy of changes in chromosome number andcomposition in different species as a functionof their evolutionary divergence from one an-other. Chromosome painting has proved veryuseful in identifying homologous chromo-some regions between species and has led to abetter understanding of the evolutionary rear-rangement of genomes (Wienberg et al., 1990).Many fluorescent dyes have now been created,each with different excitation and emissioncharacteristics. This has allowed for the si-multaneous hybridization and discernment ofmultiple probes on a single slide (Johnson, Mc-Neil, Carter, & Lawrence, 1991; Ried et al.,1992). A natural extension of this procedureinvolves the labeling of different probes withvarious combinations of fluorophores, therebyenabling the hybridization of more probesthan there are distinguishable dyes. With Nfluorophores, the number of possible label-ing combinations is given by 2N – 1. Thiscombinatorial labeling of individual chromo-somes using five different fluorophores is usedfor spectral karyotyping (SKY; UNIT 4.9; Lee,Rens, & Yang, 2000; Garini et al., 1996; Liyan-age et al., 1996; Schröck et al., 1996) and M-FISH (Azofeifa et al., 2000; Jentsch, Geigl,Klein, & Speicher, 2003; Karhu et al., 2001;Speicher, Ballard, & Ward, 1996) analysis ofmouse and human metaphase chromosomes.25 − 1 = 31 combinations are more than the24 human or 21 types of mouse chromosomes.This technique has proved very useful for theidentification of chromosome aberrations inhuman tumors and mouse models of tumori-genesis (Barlow et al., 1996; Coleman et al.,1997; Ghadimi et al., 1999; Veldman, Vignon,Schröck, Rowley, & Ried, 1997). SKY analy-sis has also been applied to evolutionary stud-ies and will improve the analysis of genomicrelationships (Schröck et al., 1996). Anotherapproach has been to label probes not onlyby using combinations of dyes, but also byMicroscopy and
Image Analysis
4.4.6
Supplement 94 Current Protocols in Human Genetics
varying the ratios in which they are used (Ned-erlof, van der Flier, Vrolijk, Tanke, & Raap,1992). If K different concentrations are usedfor each fluorophore, and if the highest concen-tration used is 1 and the other concentrationsare defined as (1/2)1, (1/2)2 . . . .(1/2)K−2 and0, the total number of possible valid combi-nations as described by (Garini, Gil, Bar-Am,Cabib, & Katzir, 1999) is given by:
K N − (K − 1)N
where “valid” refers to combinations that havedifferent fluorophore-concentration ratios. Inother words, ratios of (1,1) and (0.5,0.5) fora two dye scheme have identical concentra-tion ratios of 1. Extending Nederlof’s methodto enough dyes and ratios to paint each hu-man chromosome uniquely is called combinedbinary ratio labeling fluorescence in situ hy-bridization (Tanke et al., 1999). The methodhas been used for adding a unique gene (HPV)FISH probe (Szuhai et al., 2000), painting pversus q arms (Wiegant et al., 2000), and a to-tal of 49 colors for all chromosome arms (48combinations involving 6 fluorophores) plusan HPV genome-specific FISH probe with along Stokes shift dye that did not cross-talkwith the other fluorophores (Brink et al., 2002).Using subtelomeric COBRA-labeled BAC andPAC FISH probes, all 41 unique subtelomereshave been imaged on single human metaphasespreads (Engels et al., 2003).
Fluorescence technology is also contribut-ing to advances in the areas of cell biology,and in studies to determine nuclear topogra-phy (Carter et al., 1993; Lawrence, Carter, &Xing, 1993; Lawrence, Singer, & Marselle,1989; Xing, Johnson, Dobner, & Lawrence,1993) and chromatin organization (Sachs, vanden Engh, Trask, Yokota, & Hearst, 1995;Yokota, Singer, van den Engh, & Trask, 1997;Yokota, van den Engh, Hearst, Sachs, & Trask,1995). Studies designed to analyze gene func-tion also incorporate advances in fluorescencemicroscopy, but the fluorescence in this caseis not from a fluorophore conjugated to a nu-cleic acid, but from a green fluorescent pro-tein (GFP) isolated from jellyfish. By mak-ing constructs encoding the gene of interestfused to the GFP gene, researchers are ableto determine the cellular sub-localization oftheir “glowing” gene product (Chalfie, Tu, Eu-skirchen, Ward, & Prasher, 1994). GFP rev-olutionized biology and was justly awardedthe Nobel Prize in Chemistry in 2008 to Os-amu Shimomura, Roger Y. Tsien, and MartinChalfie. GFP has also been used as a reporter
gene in transgenic mice to determine the devel-opmental stage and tissue specific transcrip-tional activation of promoters (Fleischmannet al., 1998). The fusion of GFP to the CENPBgene, the product of which is known to local-ize to all human centromeres, has been usedin conjunction with time-lapse fluorescencemicroscopy to follow the movement of cen-tromeres throughout the cell cycle (Sullivan &Shelby, 1999).
Subsequent in vitro modifications of theAequorea GFP gene protein sequence haveresulted in the development of other fluores-cent proteins, including blue, cyan, and yel-low whereby enabling the simultaneous useof multiple fluorescently tagged proteins inthe same living cell (reviewed by Tsien, 1998,2005). A key discovery was made by a Russiangroup that cloned, from Discosoma coral, theisolate of a red fluorescent protein (DsRed;Matz et al., 1999). This was followed bycharacterization of the genetic diversity ofthe colorful fluorescent protein family (Labaset al., 2002), isolation of more useful mu-tants of DsRed, such as “Timer” (Terskikhet al., 2000), a monomeric version, mRFP1(Campbell et al., 2002), and mRFP1’s fruityspectrum of mutants from orange through far-red mPlum derivatives (Shaner et al., 2004;Wang, Jackson, Steinbach, & Tsien, 2004).Niu et al. (2016) used spectral karyotypingand FUCCI cell cycle specific fluorescent pro-tein reporters to produce better insight intotumor DNA ploidy changes (“giant cell cycle”hypothesis).
The ability to fuse cDNAs of fluorescentproteins with proteins of interest and/or withother fluorescent proteins, has been the en-abling technology for many localization andcolocalization studies at the cell and organ-ismal level. The majority of fluorescent pro-tein fluorescence resonance energy transfer(FRET) papers now use 37°C stable versionsof cyan (Heim, Prasher, & Tsien, 1994) andyellow (Ormö et al., 1996) fluorescent pro-teins, with some biosensors switching to state-of-the-art green (mNeonGreen or mClover3)and red (mScarlet or mRuby3; Bajar et al.,2016; Bindels et al., 2017; McNamara &Boswell, 2007; Newman, Fosbrink, & Zhang,2011; Okumoto, Jones, & Frommer, 2012;Tewson, Martinka, Shaner, Hughes, & Quinn,2016; see Table 4.4.1 at end of article). Atthe cell level, the interaction of cyan fluo-rescent protein (CFP)-sensor-yellow fluores-cent protein (YFP) fusion with a molecule ofinterest, or CFP-protein1 plus YFP-protein2interactions, can answer questions with
Cytogenetics
4.4.7
Current Protocols in Human Genetics Supplement 94
nanometer precision. These studies take ad-vantage of FRET in which the energy releasedby the excitation of one fluorescent moleculein the cell can directly cause the excitation ofa different fluorescent molecule if they are inclose enough proximity to one another. Theeffective range of Förster-type energy transferis less than 10 nm, making FRET an excel-lent spectroscopic ruler for molecular inter-actions in cells (Stryer & Haugland, 1967).For example, Miyawaki and colleagues usedCFP-calmodulin-M13-YFP as a calcium sen-sor, called CaMeleon. In the absence of cal-cium, the CFP and YFP were far enough apartand/or oriented such that little FRET occurredfrom CFP to YFP. In the presence of calcium,the binding of four calcium ions to the calmod-ulin component resulted in a specific interac-tion between calmodulin and the M13 pep-tide, which resulted in CFP and YFP becomingclose enough and/or orienting their dipole mo-ments such that FRET increased (Miyawakiet al., 1997).
FRET is an excellent imaging tool whenlarge changes occur as in the binding orunbinding of calcium and CaMeleon, orthe loss of FRET on cleavage of a proteasesubstrate in a CFP-substrate-YFP proteinfusion. However, subtle features of the sensormodule and/or the fluorescent protein(s)can have an enormous impact on successor failure. Making the effort to optimizethe FRET system can pay off with largedynamic range sensor responses, as in theoutstanding twenty times dynamic range fora FRET CFP-caspase substrate-YFP sensorfound by mutagenesis and flow sort screeningfor color derivatives of the two fluorescentproteins (Nguyen & Daugherty, 2005). Thismethodology has been utilized to identify invivo molecular interactions between differentproteins in transgenic organisms, such ascell-type and temporal-specific expression ofcalcium ion sensing CaMeleons (Hasan et al.,2004) or calpain protease activation (Stock-holm et al., 2005) in mice. A comprehensivereview of FRET methods and calculations hasbeen published by Jares-Erjiman and Jovin(2003). Fluorescent protein biosensors arereviewed by Newman et al. (2011); Frommer(http://biosensor.dpb.carnegiescience.edu);McNamara (http://www.geomcnamara.com/fluorescent-biosensors); and Table 4.4.1.
Fluorescent proteins have also been fusedwith, or otherwise made to interact with, re-combinant luciferases. These recombinantsrecapitulate the native system of Aequorea,in which the calcium-activated blue emitting
photoprotein aequorin (a one shot luciferase),excites non-covalently bound GFP by biolumi-nescence resonance energy transfer (BRET),to produce predominantly green light. Re-searchers have made use of BRET specifically,and luciferases in general, to monitor protein-protein interactions in homogeneous biochem-ical assays, live cells, and from cells insidemice (e.g., Contag et al., 1995; Ray, De, Min,Tsien, & Gambhir, 2004; Takai et al., 2015;Waud et al., 2001; Xu, Piston, & Johnson,1999). While luciferase and GFP labeled cellsare not going to become general clinical tools,the ability to look at scales from moleculesto mice, from nanoseconds to weeks, is calledmolecular imaging. For example, the abilityof study transplanted stem cells, and theirprogeny, to differentiate in mouse models, isgreatly facilitated by luciferase and/or fluores-cent protein tagging. Stem cell progeny canbe tracked non-invasively by bioluminescenceimaging (Wang et al., 2003). At the end of theexperiment, the proof of identify, and pres-ence or lack of fusion with host cells, ide-ally needs to be proven by combined GFPfluorescence, cell type marker-specific fluo-rescence immunocytochemistry and sex chro-mosome and species-specific fluorescence insitu hybridization.
TYPES OF MICROSCOPYOne source for an excellent and detailed
discussion of microscopy and live cell analysis(in addition to other topics) is Cells: A Labora-tory Manual (Spector, Goldman, & Leinwand,1998), or, in its condensed form, Live CellImaging (Goldman & Spector, 2004). In gen-eral, the ability to generate a clear image is de-pendent on magnification, contrasts betweeninternal and external milieu, and the abilityto resolve discrete objects. There are manydifferent microscope arrangements that can beused to enhance the contrast of specimens. Themajor imaging modes are epi-fluorescence,transmitted brightfield (APPENDIX 3N; Salmon& Canman, 2003), phase contrast (APPENDIX3N; Salmon & Canman, 2003), polarized light,Nomarski, or differential interference con-trast (DIC; APPENDIX 3N; Salmon & Canman,2003), darkfield illumination, and reflectedlight illumination. Most of these configura-tions are used in conjunction with fluorescencemicroscopy.
With respect to fluorescence, the olderliterature referred to “diascopic” fluores-cence in the case of transmitted lightfluorescence, or trans-fluorescence, and
Microscopy andImage Analysis
4.4.8
Supplement 94 Current Protocols in Human Genetics
http://biosensor.dpb.carnegiescience.eduhttp://www.geomcnamara.com/fluorescent-biosensorshttp://www.geomcnamara.com/fluorescent-biosensors
Figure 4.4.2 The concepts of (A) epi-illumination (incident), and (B) transmitted light as appliedto fluorescence microscopy. Many of the major components are similar to those used with eithertype of brightfield microscopy (with the elimination of the filters which are labeled c and i here).Indicated in the diagrams are the (a) light source, now should be light emitting diode(s) (LEDs) orfor specialty research microscopes, lasers, (b) stage condenser, (c) excitation filter, (d) dichroicmirror, (e) selected excitation wavelength, (f) objective, (g) microscope slide with specimen, (h) epi-illumination (in A) or transmitted (in B) illumination (short wave; darker) and emitted fluorescence(long wave; lighter), (i) barrier or emission filter, (j) eyepiece condenser, and (k) eyepiece or camera.In 2016, the standard monochrome digital camera should be a 4.2 or 5.5 megapixel scientificCMOS camera (sCMOS). These are available from Andor, Hamamatsu, PCO, and Photometrics.USB3 interface enables full chip imaging at 30 frames per second (fps); more expensive interfacesenable 100 fps. 30 fps is fine for “real-time” focusing by eye. Exposure time for acquisition of a singlefluorophore might be a few milliseconds for DAPI interphase nuclei or metaphase chromosomes;10s of milliseconds for bright immunofluorescence or overexpressed state of the art fluorescentproteins (e.g., mNeonGreen-Histone H2B), 100 milliseconds for “typical” immunofluorescence orDNA or single molecule RNA FISH on a current generation research or cytogenetics microscope,1000 milliseconds (1 sec) for dim fluorescence such as single molecule RNA FISH with a non-optimized microscope.
episcopic, incident light fluorescence, orepi-fluorescence (APPENDIX 3N, Salmon &Canman, 2003), in the case of reflected lightexcitation. In incident light microscopy, the in-coming light is first reflected down through thespecimen and then back up through it into theobjective (Fig. 4.4.2). Note that reflected lightis useful for imaging thick objects that can-not be trans-illuminated (such as skin tumors,cells in live mice, or circulating blood cells us-ing the handheld CytoScan, CapiScope HVCSHandheld Video Capillaroscopy System), oron microscope slides with monochromatic po-
larized illumination and a crossed polarizer forimaging highly scattering silver grains or im-munogold. This latter configuration has highcontrast and is commercially available frommicroscope and filter companies as an IGS fil-ter cube (immunogold/silver staining). Sincethe excitation side polarizer only reduces ex-citation by 50%, a custom IGS cube has beenmade for the authors by Chroma Technologythat has UV-green excitation + polarizer, a10% reflection/90% transmission 45 degreebeamsplitter, and green-near-infrared (NIR)+ cross-polarizer emission filter. This custom Cytogenetics
4.4.9
Current Protocols in Human Genetics Supplement 94
IGS can be used in standard IGS mode withgreen (546 nm) exciter, or with UV exciterfor quantum dot fluorescence, or with No-marski differential interference contrast (DIC)transmitted light where the emission polar-izer serves as the DIC analyzer. This has theadditional advantage of automating the DICsystem on the authors’ Leica RMRXA/RF8(8-filter cube turret) microscope while elim-inating the many-micrometer pixel shift in-troduced by the standard Leica DIC analyzerslider. The single-pass exciter filters were cho-sen to also enable single channel imaging witha triple DAPI/fluorescein/Cy3 filter set in thesame microscope. The custom IGS cube wasinspired by the non-reflective reflector fluores-cence filter cube of Sawano, Hama, Saito, andMiyawaki (2002).
Oblique illumination, common on stere-omicroscopes with a ring light or fiber op-tic gooseneck, is also possible for microscopeslides, using the DarkLite illuminator (MicroVideo Instruments) to illuminate through theedges of the slide. The DarkLite illuminatorprovides barely enough light for imaging sil-ver particles and tissue by scattered light, butis not suitable for fluorescence when used withits standard light bulb. A special type of flu-orescence excitation, total internal reflectionfluorescence (TIRF), uses the glass surface thecells or molecules are on as a light guide. Es-sentially, the light is trapped in the high refrac-tive index glass: The same principle is used infiber optics in the communication industry. InTIRF, the light is trapped, propagating parallelto the glass surface, but fluorophores within�100 nm in the low refractive index mountingmedia (i.e., cell culture fluid) are excited by theevanescent wave in the latter media. The ad-vantage of TIRF is that only fluorophores veryclose to the glass-mounting medium interfaceare excited. This can include the basal plasmamembrane of cells in close apposition to theinterface. The evanescent wave energy falls offexponentially with distance into the mountingmedium, and the depth can be controlled byadjusting the angle of light entering the glassslide. TIRF can be set up using a prism to cou-ple the excitation light into the glass, or a highnumerical aperture objective lens can be used,with the illumination restricted to the high NAportion. Special TIRF lenses are commerciallyavailable from Zeiss (NA = 1.45) and Olym-pus (NA = 1.45 or, with special expensivecoverglass and toxic immersion media, NA =1.65). See reviews by Oheim (2001); Axelrod,Burghardt, and Thompson (1984); Axelrod,
(2003, 2013); or Jaiswal and Simon (2003)for additional details on TIRF.
Köhler illumination (APPENDIX 3N; Salmon& Canman, 2003) results in an evenly lit fieldof view and is used for all light microscopy.Epi-fluorescence microscopes use the objec-tive lens for both excitation and emission. Theexcitation optical path of current fluorescencemicroscopes are equipped with field and nu-merical apertures, which are placed at appro-priate conjugate optical planes for imaging. Aslong as they are reasonably well centered, andthe numerical aperture fully opened to max-imize brightness, epi-Köhler illumination isassured. For a few specific applications, suchas high magnification, high numerical aper-ture imaging of single metaphase spreads orinterphase nuclei for FISH, the field aper-ture can and should be reduced to just out-side the diameter of the object of interest.This will reduce glare arising from excita-tion of auto-fluorescent mounting media in thearea around the object, dramatically improvingcontrast. Using fresh mounting medium andoptimizing FISH wash steps, will also reduceautofluorescence.
With respect to trans-Köhler illumination,there are many terms associated with mi-croscopy that indicate the direction of the in-coming light with respect to the angle at whichit intercepts the sample and the side of the sam-ple through which it first passes (Fig. 4.4.2).Brightfield microscopy (APPENDIX 3N; Salmon& Canman, 2003) is the most commonly usedlight-microscopic technique, and is the basisfor phase contrast, polarization, and NomarskiDIC contrast methods. As the name indicates,in brightfield, the surrounding background isbright and the object is dark. The objects maybe dark because of their scattering proper-ties, endogenous pigments, or exogenous dyes.Light from the illumination source is transmit-ted along a pathway parallel to the optical axisdirectly through the sample into the objective.As light encounters the specimen, the intensity(or amplitude) is reduced compared to its sur-roundings, resulting in a darker appearance.The location of the illumination source de-fines the two types of brightfield microscopy.In transmitted light brightfield microscopy, theillumination source is directly below the sam-ple for an upright microscope. As such, thelight passes through the sample only once onits way from the source to the objective. Köhlerillumination is the standard configuration fortransmitted illumination of nearly all biomed-ical microscopes. However, because theMicroscopy and
Image Analysis
4.4.10
Supplement 94 Current Protocols in Human Genetics
condenser can be lowered or raised, and movedlaterally, it is up to the user to adjust thecondenser field aperture centration and fo-cus and numerical aperture setting. Becausemany samples do not contain sufficient ab-sorption properties to be discerned with nor-mal brightfield microscopy, one can generatecontrast and reveal structures with low reso-lution by slightly rotating the condenser tur-ret. This technique is referred to as oblique,anaxial, or asymmetric illumination contrast(Kachar, 1985).
Both transmitted illumination and incidentillumination (epi-illumination) can be usedfor fluorescence. Older microscopes may usetransmitted fluorescence illumination (trans-fluorescence), as in its pioneering use a cen-tury ago by Köhler and von Rohr. Trans-fluorescence with a bright arc lamp could behazardous to the user if full intensity, un-filtered light were transmitted through theeyepieces. Trans-fluorescence went out offashion in the decades after Bas Ploem in-troduced the Ploem-pak filter cube (Ploem,1967), using a wavelength selective excita-tion filter, 45 degree angle dichroic beamsplit-ter, and emission filter. However, with mod-ern interference filters acting as exciter andemission—eliminating the need for a beam-splitter or cube assembly—trans-fluorescencehas uses, especially with a high numericalaperture condenser (Tran & Chang, 2001).Trans-fluorescence may be particularly usefulon microscopes that have an arc lamp with awavelength controller, i.e., filter wheel, shut-ter, and the Ellis light scrambler (fiber opticor liquid light guide to homogenize illumi-nation) for DIC or other high resolution mi-croscopies (Inoué & Spring, 1997; Reitz &Pagliaro, 1994). With a high sensitive CCD orsCMOS camera, and interference filters, evena standard tungsten-halogen lamp could serveas a light source for visible and near infra-red excitation (these lamps produce little UVhowever, making them practically useless forDAPI or blue fluorescent protein [BFP] ex-citation). Trans-fluorescence may be particu-larly useful for thick specimens, such as mousebrain slices, combined with large Stokes shiftdyes, the idea being that the shorter wave-length excitation light may penetrate throughonly part of the specimen, whereas the longerwavelength NIR fluorescence emission mayget through the specimen to the objective lensand detector. This could complement the in-frared DIC-videomicroscopy method of Dodtand Zieglgansberger (1994). Fluorescence mi-
croscopy is dominated by fluorophores thatemit in the visible, since “seeing is believ-ing.” With the increasing use of “no eyepiece”microscopes and high content imaging sys-tems, and especially to increase multiplexing,expanding the wavelength range is becomingmore popular. We discuss later both UV andNIR fluorescence applications.
Darkfield microscopy can be used to ob-tain resolution of objects or features that arenormally below the resolution of the light mi-croscope. This is only possible with trans-mitted illumination because no direct light isallowed to enter the objective. Only incom-ing light diffracted, refracted, or reflected bythe specimen enters the objective, resulting inbright objects on a dark background. One noteof importance is that the numerical apertureof the condenser must be higher than that ofthe objective. This technique is incompatiblewith the use of phase contrast microscopy inassociation with fluorescence. Darkfield mi-croscopy also requires scrupulously clean op-tics and slides, because any dirt will cause lightto be scattered into the objective and mar theimage quality. This is because darkfield hasan effectively large depth of field and manymicroscope optics accumulate dust and dan-der because the microscopes are not sealedto allow heat to escape. Conversely, the largedepth of field could be useful for tracking ob-jects in relatively large volume, though mostof the objects will appear out of focus (mak-ing them larger, which could in turn improvethe centroid XY location precision by imagemeasurement of the larger object).
Differential interference contrast (DIC;APPENDIX 3N; Salmon & Canman, 2003; Allen,David, & Nomarski, 1969) can be performedat high numerical apertures, gives better res-olution than darkfield microscopy, and canbe used in conjunction with fluorescence mi-croscopy and live-cell imaging. DIC is referredto as an optical sectioning technique, becausefor transparent objects, soon after it leavesthe focal plane it disappears. The DIC tech-nique involves polarized light plus a pair ofmatched Wollaston prisms (Nomarski DIC) orsimilar light path splitters (Smith DIC). Theincoming light is first passed through a polar-izing filter that only allows waves oriented inthe same direction to pass through the filter.Each plane-polarized light beam is then splitinto two separate beams containing perpen-dicularly oscillating components with a Wol-laston prism (composed of two quartz prismscemented together with their optical angles
Cytogenetics
4.4.11
Current Protocols in Human Genetics Supplement 94
oriented at 90 degrees with respect to eachother). Thickness and refractive index differ-ences within the specimen generate oppos-ing phase shifts in the two halves of the splitbeam. A second Wollaston prism placed afterthe objective recombines the halves of eachsplit beam. Constructive or destructive inter-ference occurs as a result of the phase shift be-tween the two separate beams. The light thenpasses through another polarizing filter (ana-lyzer) and is visualized as differences in gray-scale levels across the specimen. The bas-reliefof DIC is due to differences in refractive in-dex, and not the three-dimensional topographyof the specimen. Allen and coworkers (Allen,Travis, Allen, & Yilmaz 1981a; Allen, Allen,& Travis, 1981b), and Inoué (1981) inde-pendently introduced the advantages of videoenhanced polarization (VEC-Pol) and dif-ferential interference contrast (VEC-DIC)for imaging unstained cells. Holzwarth andcoworkers (Holzwarth, Webb, Kubinski, &Allen 1997; Holzwarth, Hill, & McLaughlin,2000) have described polarization-modulationDIC hardware that switches the bas-relief witheach image, followed by an image difference(plus offset) mathematical operation to doublethe edges. As anyone who has worn polarizedsunglasses knows, polarizers can reduce glare.In light microscopy, polarized light is used forimaging birefringent structures such as chro-mosomes and/or mitotic spindles (Inoué &Dan, 1951; see also Inoué & Spring, 1997).
Phase contrast microscopy (APPENDIX 3N;Salmon & Canman, 2003) is an alternativeto DIC that converts normally invisible phasechanges as light propagates into and out of in-terfaces of different refractive indices (culturemedia-plasma membrane, plasma membrane-cytoplasm, cytoplasm-organelle membrane,organelle membrane-contents, and back out)or through thick slabs of such contents (thinversus thick cytoplasm). Phase contrast isan excellent mode for reviewing metaphasespreads on microscope slides before decidingwhat areas on what slides to carry out FISHor Giemsa staining. The invention was justlyhonored with a Nobel Prize (Zernike, 1955).Zernike pointed out the irony that when bright-field imaging ultra-thin, transparent objects,the best focus makes the object invisible. Herecognized that the light propagating throughthe object would delay (or advance) the wave,and worked out a way to convert phase to in-tensity. The firm of Carl Zeiss famously ex-hibited a lack of interest, initially, but even-tually reversed itself. A phase annulus (ring)
in the condenser only allows a ring of light toreach the condenser. Focusing of this ring bythe condenser lens generates a hollow cone oflight that is projected onto the back focal planeof the objective. Some of the light waves areretarded as they pass through the sample. Thisis due to absorptive differences among cel-lular structures and differences in refractiveindex or thickness. As a result, their phase isshifted relative to those waves from the orig-inal light source, which have not encounteredphase dense objects. These phase shifts areusually not sufficient, however, to generate fullconstructive or destructive interference visiblewith normal brightfield microscopy. The stan-dard dark-light (DL) phase ring in the back fo-cal plane of the objective absorbs 70% to 80%of the non-diffracted rays. In the absence ofan object, this only has the effect of reducingbrightness slightly. The phase ring cruciallyshifts the phase by one quarter of the wave-length for the diffracted rays. This arrange-ment alters the amplitude and phase relation-ships of the diffracted versus non-diffractedlight, thereby enhancing the contrast. Regionswith a higher refractive index usually appeardarker, with the standard DL design. Objectswith too high a refractive index or thicknesscan result in a rather large phase shift and causea contrast reversal (i.e., a positive phase shiftof 1.5 λ would appear identical to a negativephase shift of 0.5 λ). The phase ring is op-timized for monochromatic light of 546 nm(a mercury arc lamp line), so best results areachieved with a monochromatic green filter,though satisfactory results are obtained with afairly broad pass green interference filter (GIF,or fluorescein emission filter, 510 to 540 nm).Either green filter will also enhance contrast ofGiemsa stained chromosomes or nuclei, andcan be useful for imaging hematoxylin andother absorption dyes.
Microscope manufacturers also offer lim-ited numbers of special phase contrast lenses,such as the dark-medium (DM) design. A keyadvantage for phase contrast when imagingsingle cells by eye is that, with the DL de-sign, a bright phase halo surrounds the darkcell, making the invisible angelic. However,the same halo results in difficulties for preciseimage analysis of the cells, especially in ultra-thin regions such as lamellipodia or filopodia,where the contrast of edge of the cell and halodisappear due to the weakness of the phase dif-ference. The phase ring in the objective lensabsorbs some of light. This is not an issue forphase contrast brightfield illumination, whereMicroscopy and
Image Analysis
4.4.12
Supplement 94 Current Protocols in Human Genetics
even a tungsten-halogen lamp provides plentyof light, but the ring does reduce somewhat theintensity of fluorescence.
In 2002, quantitative phase amplitude mi-croscopy (QPm), a mathematical method us-ing three (or more) brightfield images ofdifferent focus positions, was described byBarone-Nugent, Barty, and Nugent (2002).QPm can be performed in software with ei-ther conventional brightfield microscope im-ages or with the “free” (but not confocal op-tical sectioning) transmitted light image of aconfocal microscope (Cody et al., 2005). QPmcan be done as an off-line post processing, andproduces quantitative phase maps that are in-dependent of any absorbing features presentin the specimen (provided the entire specimenis not black). Source code for a version ofQPm was posted in the supplemental files ofPhillips, Baker-Groberg, and McCarty (2014).The phase maps can be used directly (refrac-tive index measurement) or can be recombinedin a user-adjustable manner with the absorb-ing features. The phase maps can also be dig-itally converted into Nomarski DIC or phasecontrast like images, with or without absorb-ing features. We have not seen QPm used onmetaphase chromosomes or cell nuclei, but themethod may produce interesting chromosomemaps or a novel form of nuclear texture analy-sis. Hopefully, in the future, the refractive in-dex measurements can be combined with dig-ital deconvolution algorithms to improve 3Ddeconvolution fluorescence microscopy.
There are many occasions when it is use-ful to follow the movement of cells or theirorganelles as a function of time. Studies ofcell division, movement of chromosomes andcentrosomes, and the polymerization of mi-totic tubules are a few examples. Such analysisinvolves the successive microscopic imagingof live cells, rather than a single image of afixed specimen. There are two different meth-ods for accomplishing such an analysis. Thefirst, time-lapse microscopy, involves the ac-quisition of individual images at distinct timepoints or intervals (e.g., every 10 min over an8 hr period). These images can then be inte-grated into a single composite image and dis-played simultaneously for an easy comparisonof changes as a function of time. Video mi-croscopy, however, involves near-continuousimaging over a prolonged time period, as onewould do with a standard video camera that hasa rate of 30 frames/sec. If continuous obser-vation is not required, a computer controlledshutter can be added to the system, and a fewvideo images may be captured and averaged,
or a single exposure with a digital CCD cam-era, can be captured on demand. A thoroughtreatment of this technique, which is beyondthe scope of this chapter, can be found in Spec-tor et al. (1998), Goldman and Spector (2004)or Yuste and Konnerth (2005).
MICROSCOPE OBJECTIVES ANDEYEPIECE LENSES
The compound light microscope must beequipped with the highest-quality optics (ob-jectives and eyepieces), must be preciselyaligned, and must have the proper filters in-stalled to observe and record all relevant in-formation from the objects under study. Adetailed manual outlining the steps requiredto align the microscope is available from themanufacturer of the respective microscopeand is discussed in APPENDIX 3N (Salmon &Canman, 2003). The importance of properalignment cannot be overstated. An excellentdiscussion of microscopy and photography ispresented in The ACT Cytogenetics Labora-tory Manual (Barch, Knutsen, & Spurbeck,1997) and in Human Cytogenetics: A PracticalApproach (Rooney & Czepulkowski, 1992).Both references are invaluable resources forthe cytogenetics laboratory. High-quality ob-jective lenses are critical for obtaining max-imum information in the study of biologicalspecimens. Lenses condense light and mag-nify the image. The objective lens system musthave high resolving power and correction forlens aberrations. The resolving power, R, ofa lens is defined as the minimum distance bywhich two luminous points can be separatedand still be discerned as distinct objects usingthat objective. R is described by the theory ofoptical diffraction as:
R = 1.22λ/(2 × NA)
where λ is equal to the wavelength of the in-cident light and NA is the numerical aperture,a measure of the light cone entering the ob-jective at the fixed objective distance (James& Tanke, 1991; Rawlins, 1992). The value ofNA is given by:
NA = n sin α
with n equal to the refractive index of themedium between the objective and the sampleand α equal to half the vertical angle of the lightcone (Fig. 4.4.3). The NA is restricted for tech-nical reasons to a maximum of 1.35 to 1.44 forglass objectives and oil immersion media (re-fractive index 1.515) and is usually indicated
Cytogenetics
4.4.13
Current Protocols in Human Genetics Supplement 94
Figure 4.4.3 Diagrammatic explanation of numerical aperture. As α approaches 90 degrees, sinα approaches 1.0. The refractive index of the medium between the sample and the objective isdesignated as n. Standard fluorescence microscopes use epi-illumination and detection through acommon path (see Fig. 4.4.2A). Epi-illumination is superior for fluorescence than transmitted light(Fig. 4.4.1). Some applications benefit from total internal reflection fluorescence (TIRF) excitation,through the coverglass, or “light sheet” illumination from the side(s) of the specimen. The light is stillcollected the same way—numerical aperture applies to emission with TIRF or light sheet enablingnew applications. TIRF is briefly described in Appendix 4.4.2 (see Supporting Materials): Total In-ternal Reflection Fluorescence (TIRF) Microscopy. Light sheet applications are briefly described inthe text along with a major opportunity to synergize with expansion microscopy for single cells, opti-cally cleared tumor masses, and developing whole organisms (worms, zebrafish, and whole mice).
on the side of the objective. Special objectivelenses in the NA 1.42 to 1.47 NA range areavailable for the conditions and applications oftotal internal reflection fluorescence (TIRF),for fluorescence at a coverglass-aqueous so-lution interface, discussed in Appendix 4.4.2(see Supporting Materials). High NA resultsin the smallest lateral resolution, smallest ax-ial resolution, and maximum capture of lightphotons. This is particularly important in flu-orescence microscopy, where the amount ofemitted light is often very small. The bright-ness of the captured light is affected by manydifferent parameters, including the concen-tration of the fluorophore, the transmissionof light through the optics, the total magni-fication, and the numerical aperture of theobjective (and the condenser, in the case oftransmitted fluorescence). The relative imagebrightness in epi-fluorescence microscopy isgiven by the equation:
B = (N.A.obj)4
Mag2
when the so-called “object space” between theobjective and coverglass contains air (as witha “dry” objective), the numerical aperture can-not exceed 0.95. However, when immersionoil with a refractive index of 1.515 is used be-tween the two surfaces, an NA of 1.35 to 1.40can be obtained. This is because the refractiveindex of the oil is identical to that of the glassslide, coverglass and objective. This preventsthe light from being refracted as it passes fromthe specimen through these other materials.Immersion media include various natural and
synthetic oils (with varying n values), water (n= 1.333), and glycerol (n = 1.466 for 100%glycerol, 1.391 for 40% glycerol:60% water, at23°C). Immersion objectives are usually pro-duced for use with a specific type of immersionmedium and are so indicated on the side of theobjective; “dry” objectives will not function asimmersion objectives. Immersion oil with lowfluorescence is required for fluorescence mi-croscopy, and the objective manufacturer canhelp obtain the proper type.
In addition to resolving power, which isa function of both magnification and nu-merical aperture, modern light microscopeobjectives must correct for problems of spher-ical and chromatic aberration. Spherical aber-ration is produced by failure of the curvedsurface(s) of a lens to direct all light rayspassing through the lens to the same focalpoint. A coverglass of the incorrect thicknessor a refractive-index mismatch (i.e., wrong im-mersion oil) can also cause spherical aberra-tion and an inability to focus. Early micro-scopes (single or compound lenses) sufferedfrom loss of fine detail due to chromatic aber-ration that resulted in rings of color aroundsmall objects. White light passing through thelens is broken up into its constituent colors.Different wavelengths are diffracted to dif-ferent extents, and hence have different fo-cal points. After 1820, achromatic lenses weredeveloped, allowing great advances in biologyand medicine (Inoué & Spring, 1997; Kapitza,1996). Achromatic objectives are rather sim-ple in that spherical aberration is correctedfor the middle range of the light spectrum,
Microscopy andImage Analysis
4.4.14
Supplement 94 Current Protocols in Human Genetics
thereby directing all broken-up wavelengths tothe same focal point. Plan-achromatic objec-tives are more complex and have the advantageof less curvature-of-field aberration than ordi-nary achromatic objectives. Curvature of fieldis caused when light passing through the pe-riphery of the objective is focused closer to theback focal plane of the lens than light passingthrough the center. The result is a discrepancyin focal plane between the center and periph-ery of the field of view. Plan-apochromaticobjectives are costly, complex, flat-field ob-jectives that offer the greatest correction forchromatic and spherical aberration. The typeof correction supplied by the objective is alsoindicated on its side. In the past, fluorite (syn-thetic quartz) and plan-fluorite lenses wererequired for deep UV excitation, for exam-ple, for 340 nm excitation of Fura-2, becauseapochromat lenses had many UV-absorbingglass components. Modern (post 1997) plan-apochromat lenses often, but not always, trans-mit light down to 300 nm. If this may beimportant, as in Fura-2 or to truly maximizequantum dot excitation, transmission curvesshould be obtained from the manufacturer, orseveral lenses should be tested in the lab. Asbiologists expand our color palette, wantingto use our microscopes for imaging specimensfrom UV (
dyes bind well to DNA, resulting in high lo-cal concentrations, and the DNA shields thesetwo fluorophores from water, enhancing flu-orescence. As a consequence, while not hav-ing exceptional photophysics, DAPI and theHoechst dyes are brilliant DNA counterstains.DAPI only fluoresces in AT-rich DNA, whichresults in the characteristic R-banding pattern.
A new class of products—fluorescentnanocrystals, or quantum dots—are highlyphotostable, high absorbing, good quantumyield molecules (typically CdSe, CdS, CdTe,and/or ZnS) encased in a bio-compatible coat-ing that enable coupling to streptavidin, an-tibodies, other proteins, and haptens such asbiotin (Bruchez, Moronne, Gin, Weiss, &Alivisatos, 1998; Chan & Nie, 1998). In 2016,Qdots have not taken over any fluorescencemicroscopy market niche (i.e., cell biology,cytogenetics, flow cytometry, or pathology). Acombination of “old school is good enough,”aggregation issues with some (maybe many)production lots, atypical excitation → emis-sion requirements (405 or 488 nm excitation,Qdot### specific emission wavelengths), lackof synergy between Qdot reagent vendors, mi-croscope vendors, and spectral imager ven-dors, and lack of enthusiasm of those whoowned spectral imagers, may have each con-tributed. The new “great bright hope” are theBrilliant polymers (BD Biosciences, BioLe-gend), based on the Nobel Prize in Chem-istry (2000) work on conductive polymers byAlan J. Heeger, Alan MacDiarmid, and HidekiShirakawa (and colleagues and competitors).Both Qdots and Brilliants are tabulated withkey photophysics parameters, in Table 4.4.2,along with an extensive, though not exhaus-tive, list of conventional fluorescent dyes.
In order to grasp the principles of fluores-cence, it is necessary to further understand thelaws describing light. Energy behaves in ac-cordance with Planck’s law, which states:
E = hν = hc/λ
where E is energy, h equals Plank’s constant,v equals light frequency, c equals light ve-locity, and λ equals light wavelength. Thus,energy is linearly proportional to the light fre-quency and inversely proportional to its wave-length. The quantum of energy (E) is greaterfor radiations of shorter wavelengths, such asUV, than for radiations of longer wavelength,such as infrared. Wavelengths in the UV spec-trum (300 to 380 nm), visible light spectrum(380 to 700 nm), and near infrared (700 nmto 1000 nm) are nowadays used in fluores-
cence. The spectral characteristics of individ-ual fluorophores and fluorescent proteins aredependent upon the regions of the light spec-trum where absorption (excitation) and emis-sion of light energy occur. Stokes’ law statesthat the average wavelength of emitted flu-orescence is longer than the average excita-tion wavelength for any given fluorophore. Thelonger wavelength of the emitted light is dueto the rapid loss of some vibration energy inthe first picoseconds post-absorption (with aspectral confocal microscope and bright fluo-rescent objects, the anti-Stokes emission canbe detected—however this is a small fractionof the Stokes emission). Multi-photon exci-tation fluorescence works by having two lowenergy photons, for example 800-nm wave-length each, reach the molecule at the sametime (femtoseconds). This results in an excita-tion event equivalent to absorbing an �400-nmphoton, and fluorescence practically identicalto that of 1-photon excitation. At the quantummechanical level, the selection rules for 1-, 2-and 3-photon excitation do differ, which leadsto some dyes working poorly, and others—such as quantum dots—exceptionally well, formulti-photon excitation.
We use as an example, DAPI (4,6- diamino-2-phenylindole), a fluorescent dye with affin-ity for A-T residues in DNA. This is oftenused to give a chromosome banding pattern(R-banding) complementary to that obtainedwith Giemsa staining (UNIT 4.2; Schreck &Distèche). A simple digital image-processingstep, “inverse contrast,” is thus able to trans-form a DAPI-stained metaphase spread intoa Giemsa-like staining pattern. An additionalsharpening step then improves the bandingto near G-band quality. It is also useful asa DNA counterstain for interphase nuclearstudies, where, for example, flat tissue cul-ture cell nuclei can be identified as human ormouse on the basis of dark nucleoli versusbright centromeres, respectively (the compari-son only works for human versus Mus muscu-lus nuclei; e.g., bovine, other mice have otherstructures). The energy absorbed at 350 nmraises an electron to a higher excitation state.Some of the energy is then lost to vibration.When the electron falls back to its starting, orground, state the energy is released as fluo-rescent light. Because the amount of energyis less than the input, the emitted light has alonger wavelength (the mode being 470 nm)and is said to be red-shifted. With fluorescencelifetime spectral imaging it is possible to mea-sure that the molecules that emit rapidly (1nsec) tend to emit shorter wavelengths (higher
Microscopy andImage Analysis
4.4.16
Supplement 94 Current Protocols in Human Genetics
energies) than those that emit slowly (5 nsec).The latter are red-shifted because they havehad more time to lose more vibration energy tothe surrounding molecules. Fluorophores canalso give up some or all of their energy toother molecules. Many excited fluorophorescan have the excited electron transition fromthe first excited singlet state (S1) to the slightlylower energy first triplet state (T1). This is re-ferred to as intersystem crossing. The tripletstate can be very long-lived, microseconds(10−6 sec) to milliseconds (10−3 sec), afterwhich delayed fluorescence (if it flips back tothe S1 state) or phosphorescence (emission di-rectly from the T1 to the ground state) can oc-cur. However, when in the T1 state, the energymay be transferred to a nearby normally tripletstate oxygen molecule (O23), which flips tothe highly reactive singlet state (O21). Thesinglet oxygen molecule can convert to oxy-gen radicals, all of which are highly reactive.Since the fluorophore and the reactive oxygenmolecule are in close proximity, they often re-act, resulting in destruction of the fluorophore.Sometimes the reaction can be taken advan-tage of, as in the in the reaction of superoxidewith non-fluorescent hydroethidine to makea fluorescent ethidium dye (Haugland, 2004and http://www.probes.com/handbook/). Moregenerally, an excited fluorophore may donateits energy to another molecule, the acceptor,which if it is an efficient fluorophore, may thenemit at its characteristic wavelength. This pro-cess, is non-radiative (the light is not emittedand then re-absorbed), and when the transferoccurs with an efficiency that falls off as dis-tance to the sixth power, is termed Förster-type fluorescence resonance energy transfer(FRET; Förster, 1965; Jares-Erijman & Jovin,2003).
Some fluorophores, however, may requirean excitation energy that overlaps with theemission energy of another fluorophore. Thisfact must be considered when setting up anexperiment involving the simultaneous use ofmore than one fluorophore. It is crucial to notethat spectral properties of some fluorophoresare subject to significant environmental effectsand vary depending on measuring conditions,e.g., medium, pH, and substrate (Haugland,1996; Mason, 1993).
Understanding the spectral properties ofvarious fluorophores is important when choos-ing components of the microscope used in flu-orescence microscopy. This includes the lightsource (Xe or Hg) as well as the filter(s). Togenerate and observe fluorescence requires afluorophore bound to a molecule. This can be a
fluorophore bound either directly to a nucleicacid probe or indirectly, through the use ofhaptens (such as biotin and digoxigenin) andfluorescently conjugated moieties with whichthey interact (avidin and anti-digoxigenin an-tibodies, respectively). Another source of flu-orescence tagging is fusion of the GFP or anyof its many derivatives directly to a protein ofinterest. A light source and optical filters arerequired to produce the correct wavelength(s)of light energy required for excitation of thefluorescent moiety. The light passing throughor being emitted by the sample must then passthrough another set of optical filters, such thatemitted light energy of only the desired wave-length reaches the detection system (i.e., eye oranother detector; Fig. 4.4.2). Details of theserequirements are elaborated in the followingsections.
LIGHT SOURCES
Old SchoolThe standard light source for brightfield mi-
croscopy has been a tungsten-filament bulb.Bulbs are now losing favor thanks to theimprovements in white light light-emittingdiodes (LEDs). The intensity of a bulb iscontrolled by changing the amount of cur-rent flowing to the lamp via a rheostat dial;this has the undesirable effect of changing thecolor temperature. The use of white light LEDsensures consistent color balance at any lightlevel.
The light sources used in fluorescence mi-croscopy used to be mercury or xenon arclamps, or metal-halide lamp. These are nowbeing superceded by high intensity LEDsor lasers (discussed below). The choice ofwhether mercury or xenon arc lamp was de-termined by the wavelengths of excitation en-ergy needed to excite the fluorescent moleculebeing used as a probe. In turn, this limiteduseful fluorophores to those that worked wellwith the mercury or xenon lamp and filtersets on hand. Mercury lamps have three mainpeaks of excitation light around 440, 550, and580 nm, whereas xenon lamps are more uni-form in their intensity across this range. Anadditional mercury peak around 365 nm inthe UV range is important for the imaging ofDAPI-stained objects (i.e., DNA). Both typesof bulbs have been available in a number of dif-ferent wattages. Brighter lamps result in moreintense fluorescence and therefore a shorterexposure time is required. A 100-W bulb alsohas a longer operating life than a 50-W bulb(200 hr versus 100 hr). LEDs have 10,000+ hr! Cytogenetics
4.4.17
Current Protocols in Human Genetics Supplement 94
http://www.probes.com/handbook/
Solid state lasers (diodes) have similarly longlifespans to LEDs, and laser light is monochro-matic (single nm wavelength, e.g., 488 nm).LEDs and lasers are discussed further below.Another important parameter is the gap be-tween the anode and cathode in the bulb itself.This is known as the arc gap. Small gaps pro-vide a small arc that can emulate a point sourcefor epi-Köhler illumination.
It is important to monitor the bulb use be-cause older bulbs result in weaker fluores-cence signals. This will affect exposure set-tings, which is more important when usingphotographic film, because it is easier to retakethe image with a digital-imaging device. Also,mercury bulbs should not be used >200 hrbecause there is a risk of explosion that candamage the microscope. Changing and align-ing the bulb requires patience and skill and isoften best performed by the microscope ser-vice representative.
New School and Why You ShouldSwitch
When purchasing a new light microscope,the transmitted light should now (i.e., 2017)be LED based. Fluorescence cost-benefit andperformance, are now favoring LED or laserover mercury, xenon, or Hg-Xe arc lamps,or metal halide lamps. Besides being energyinefficient (heat more than light), requiringreplacement every 100 to 1000 hr (typicalrating is 300 hr), high pressure Hg arc lampssometimes explode. This is most likely tohappen when someone is trying to adjust thelamp. While most lamps have some safety pro-tections so glass shards do not blast into yourface, why take the risk anymore. Even withoutan explosion and medical costs, the totalcost of ownership now favors LEDs or lasersfor fluorescence illumination. High intensityepi-fluorescence LED lamps are now as brightas arc lamps or metal halide. Combined withLED optimized filter set(s), these greatlyoutperform every lab’s “old” illuminationsetup. A basic fluorescence four-wavelengthLED with liquid light guide is available forunder $3,000 (https://www.thorlabs.com/newgrouppage9.cfm?objectgroup_id=3836;the microscope-specific adapter cost dependson the microscope, usually under $200), withmore and brighter LEDs and computer controladding to the price.
We are especially excited by recent devel-opments in laser light sources for fluorescencemicroscopy. Each modern laser is monochro-matic (1 to 3 nm full width at half maximumand the same as full width at 5% maximum),
providing a big “effective power per nm” ad-vantage over LEDs (10 to 30 nm band fullwidth at half maximum, but much wider fullwidth at 5% maximum, requiring a bandpassfilter to cut off these tails). Modern hard coatedfilter sets can in turn be optimized for thelaser lines, and then the emission light pathcan be optimized to take advantage of es-sentially all emission photons. For example,with a six laser line system, say 405, 440,488, 514, 550, 633 nm, with 5-nm blockingfor each wavelength, the emission range from410 to 810 nm, would have 25 nm blocked bythe laser safety emission bandpass (so for a400 nm range—25 nm = 375 nm collectiblephotons—the dichroic would also help makethe light path safer; a typical dichroic reflects99%, so it transmits �1% [1e-2], whereas anoptimized emission filter blocks 1e-6. A keyissue is the price point of this “laser stack”:Imagine if it is under $20,000). Conceivably,one filter cube could serve all six laser lines,then an emission filter (or six or more cameraswith appropriate image splitters), would en-able multiple wavelengths acquired per laserline. Such a setup would decrease the numberof moving parts: The upshot is that we expectwith LEDs, and especially lasers, and com-puter processing, to be able to acquire multi-ple channels in a shorter time than is done now.This will enable light microscopy to competewith both flow cytometry (which needs sin-gle cells, so no more spatial information) andother approaches, such as “imaging mass cy-tometry.” Multiplexing with speed and highspatial resolution could enable efficient multi-plex single gene DNA FISH, single moleculeRNA FISH, and high sensitivity immunofluo-rescence, to be imaged and quantified simulta-neously, on isolated cells, “touch preps,” fineneedle aspirates, tissue sections, and modestlythick tissues. In 1996 many cytogenetics andpathology labs acquired on film, printed onpaper, and cut out chromosomes and cells ofinterest with scissors; in 2006 CCD camerasand any color you wanted as long as theywere DAPI, fluorescein, Cy3, and Cy5 were invogue. In 2017, sCMOS and LEDs are becom-ing common in the research setting (althoughcytogenetics is lagging behind). In 2027, wehope everyone is able to acquire and processall their images in “one click.”
FiltersFluorescence microscopes use filters in sev-
eral positions in the excitation and emis-sion light paths to select specific wave-lengths. Modern microscope filters use an
Microscopy andImage Analysis
4.4.18
Supplement 94 Current Protocols in Human Genetics
https://www.thorlabs.com/newgrouppage9.cfmobjectgroup_id-3836https://www.thorlabs.com/newgrouppage9.cfmobjectgroup_id-3836
interference coating, with the glass substratecarrying vacuum-deposited thin layers ofmetallic salt compounds (see http://micro.magnet.fsu.edu/primer/java/filters/interference fortutorial).
These filters maximize the advantage of flu-orescent reagents: The excitation wavelengthsand emission wavelengths are different. Underalmost all conditions the emission is longerthan the excitation. For example, fluoresceinis typically excited �488 nm, close to its ex-citation spectrum peak, and emission wave-lengths �500 nm directed to the detector(eye, camera, photomultiplier tube). Tradi-tional mercury arc lamps, or a similar LED,might use a 480 to 500 nm excitation fil-ter (Fig. 4.4.2A, component c) and 510 to540 nm bandpass emission filter (Fig. 4.4.2A,component i). A key component in an epi-illumination microscope is the dichroic beam-splitter (Fig. 4.4.2A, component d): Dichroic istwo colors; in this configuration, short wave-lengths ( new sCMOS camera > newobjective lens(s). Any list of available fil-ters and/or filter sets would be incomplete
and therefore misleading to anyone gettingstarted with fluorescence microscopy. A listof vendors and their Web sites has thereforebeen included at the end of this unit (Ta-ble 4.4.3 at end of article) and a continu-ally updated version will be maintained onour Web site (http://www.geomcnamara.com/light-microscopy-websites). A discussion witheach manufacturer stating the nature of the ex-periment, illumination source available, andfluorophores to be used (particularly for sam-ples labeled with multiple dyes) is highly rec-ommended prior to the purchase of any fil-ter or filter set. Fluorophores commonly usedin fluorescence microscopy and for which fil-ters are typically needed are DAPI, Hoechst,quinacrine, or propidium iodide as DNA coun-terstains, as well as fluorescein isothiocyanate(FITC), Texas red, rhodamine, and other flu-orophores emitting in the far-red portion ofthe spectrum as probe-specific labels (seeTable 4.4.2 for an extensive list of fluo-rophores). Filters are usually supplied in sets,and consist of an excitation filter, a dichroicmirror, and an emission filter. These are of-ten contained within a device known as a filtercube, which are pre-aligned, easy to handle,and go easily into the filter holder or turret ofthe microscope.
Filter sets also differ in the amount of lightthey permit to pass. There is usually a trade-offbetween brightness and selectivity. Short-passfilters allow all light shorter than a particu-lar wavelength to pass through to the spec-imen, while long-pass filters only allow thepassage of longer wavelengths. These typesof filters are therefore not very restrictive intheir transmission range. Band-pass filters aremore selective in that they transmit one (ormore) particular region(s) or band(s) of thelight spectrum. This means that wavelengthsboth shorter and longer than the excitationwavelength are blocked. Narrowband-pass fil-ters have a much more restricted range oftransmitted wavelengths compared to wide-pass (or broad-pass) filters. Broad-pass filters,because they allow more light through, resultin a brighter image but include a broader spec-trum of wavelengths. Band-pass filters nowhave high light transmission values (>90%)and very narrow band characteristics thatallow selective excitation of one or morefluorophores. The use of lasers (confocal mi-croscopes, flow cytometers, some widefield re-search microscopes), simplify the excitationside: Each laser line is an �1 nm wide wave-length. The newest solid-state lasers (often
Cytogenetics
4.4.19
Current Protocols in Human Genetics Supplement 94
http://micro.magnet.fsu.edu/primer/java/filters/interferencehttp://micro.magnet.fsu.edu/primer/java/filters/interferencehttp://www.geomcnamara.com/light-microscopy-websiteshttp://www.geomcnamara.com/light-microscopy-websites
laser diodes) are electronically controlled and“instant” on and off, enabling use of a singlelaser line at a time. An all solid-state laser exci-tation microscope would eliminate the need forexcitation filters (Fig. 4.4.2A, c), simplify andmake more efficient the dichroic filter positionto be a notch or multi-notch filter (Fig. 4.4.2A,d) to simply reflect the laser lines (on excita-tion, to the specimen; on the return, unwantedreflections would be directed back along theexcitation path, that is, away from the emis-sion path). Such a “laser dichroic” or “lasermultichroic notch filter” would transmit moreemission wavelengths, for example, for fluo-rescein 488 nm excitation, “all but 488 nm”through the dichroic; 490 to 540 nm throughthe emission filter (Fig. 4.4.2A, i). Optionally,the emission filter could be fixed in place (witha multi-line notch filter, “one filter cube touse them all”) and an emission filter wheelcould handle the emission wavelength selec-tion (the wheel might be inside the microscope,or mounted between the microscope cameraport and camera).
Dual- and triple-band-pass filters, whichpermit concurrent visualization of two orthree fluorophore combinations, are available.These are mostly for use by eye, since the bestcameras are monochrome and are best used toimage one fluorophore at a time. These are use-ful for the simultaneous excitation and detec-tion of multiple fluorophores immunostainedor DNA or RNA probes hybridized to the samesample and abrogate the need to change filtersbetween imaging each fluorophore. The sim-plest application is the imaging of two gene-specific probes for mapping. To take advantageof these filter sets, one needs a means of imag-ing that distinguishes the different colors. Anadvanced example of a triple-pass filter cube isthe one used for spectral karyotyping (SKY).This contains filters that allow alternating re-gions of excitation and emission wavelengths.This is necessary due to the overlapping ex-citation and emission spectra of the five fluo-rophores used in the hybridization. Quadruple-pass filters do have some compromises, how-ever, in that the brightness may be affected (P.Millman, Chroma Technology, pers. comm.).Filters and filter sets can be purchased frommicroscope companies or directly (and usu-ally at a reduced cost) from filter manufac-turers (e.g., Chroma Technology and Semrockare the two leading fluorescence microscopefilters companies—the microscope manufac-turers now mostly sell filters manufactured bythese or similar companies). Other filters areimportant for altering the intensity of light en-
tering the system. Neutral density filters de-crease the overall amount of transmitted lightwithout altering the intensity ratios of dif-ferent wavelengths—mostly of use for colorbalancing tungsten-halogen transmitted lightimages (Fig. 4.4.2B), with such lamps nowbeing superceded by white light LEDs. Thismay be desired if the signal intensity is strongand the fluorophore is particularly sensitive tophotobleaching. Heat filters can be extremelyimportant in removing excessive heat radi-ating from lamps (especially arc and metalhalide lamps for fluorescence, also useful fortungsten-halogen lamps for transmitted light;LEDs and lasers usually handle heat dissipa-tion at the source). KG-1, BG-38, and Hot-Mirrors are some examples of heat filters. Eachhas a different wavelength at which it reducesthe transmitted heat. Choosing the correct heatfilter depends on the wavelengths one needsfor excitation of the sample. Requiring shorterexcitation wavelengths allows one to choosea heat filter that prevents a larger portion ofthe spectrum from reaching the sample. Heatfilters protect not only the specimen from dam-age, but are also useful for protecting sensitiveelements such as polarizers and other filters.
FROM FLUORESCENT DYES,QDOTS, AND POLYMERS TOFLUORESCENT PROTEINS
Fluorophores are available as small or-ganic dyes, various types of nanoparticles, in-cluding quantum dots (Qdots; Bruchez et al.,1998, Chan & Nie, 1998), organic poly-mers (based on the 2000 Nobel Prize inChemistry on electron conduction of organicplastic wires), and even single walled car-bon nanotubes (SWCNT, related to the fa-mous Buckyballs; O’Connell et al., 2002). Ta-ble 4.4.2 lists many fluorescent dyes and thesenewer compositions of bright matter.
Chalfie et al. (1994) ushered in a revolutionin cell imaging by the cloning and expressionof green fluorescent protein (GFP), enablingeasy DNA construction of gene cassettes ofGFP tagged protein, optionally with specificexpression promoters (now $65 per plasmid,http://www.addgene.org/fluorescent-proteins).An explosion of colors and biosensors hasfollowed. Table 4.4.1 lists many fluorescentproteins (FP) and their basic photophysicalproperties. Table 4.4.3 lists many of the pub-lished fluorescent protein biosensors and whatthey measure (see Frommer, 2016; Newmanet al., 2011, for references and background).We envision that 96-well plate(s), 384-well
Microscopy andImage Analysis
4.4.20
Supplement 94 Current Protocols in Human Genetics
http://www.addgene.org/fluorescent-proteins
plate, or single “well” reverse transfection cellmicroarrays (www.persomics.com) could beused to transfect or transduce FP localizationreporters, transcriptional activity reporters(e.g., promoter response elements → FP orFPB), or constitutive or linearizer-induciblepromoter → FP or FPB, to comprehensivelycharacterize control and patient-derived cells.We further suggest that localizing FPs or FPBswould enable better signal-to-noise ratio (e.g.,nucleolus is smaller volume than nucleus,which is smaller volume than nucleoplasm+ cytosol), multimerizing (mNeonGreen-mNeonGreen-mNeonGreen is three timesbrighter than single mNeonGreen, which inturn is over three times brighter than the 1996enhanced GFP), and enable multiplexing. FPscan also add value to FPBs. For example, ifa panel of dark-to-bright green FPBs wereexpressed from different plasmids, other 5color × 5 locations, would enable marking25 populations. If using combinatorial 5 × 5pairs, or triplets, this would greatly expandthe number of cell populations that could bemarked (i.e., green FPB, with 252 = 625, or253 = 16,525, recognizing that quantitativefluorescence microscopy could be used toseparate one times, two times, and threetimes brightness at the same location—muchas done in the past for COBRA-FISH onmetaphase chromosome spreads).
IMAGE ACQUISITIONAfter identifying an object that is wor-
thy of documenting, an image must beacquired, particular portions of the object re-solved and defined, and a careful analysis per-formed to generate useful information. In thepast, images were photographed or studied di-rectly at the microscope. Photographic filmwas high resolution, but low quantum effi-ciency (at bes