Embed Size (px)
Citation preview

PHYTOPHTHORA INFESTANS eEF3 FUNCTIONALLY COMPLEMENTS FOR LOSS OF FUNCTION OF SACCHAROMYCES CEREVISIAE eEF3
Tanique Bennett, Kaitlyn Boyle, Emma Carlson, Arjun Gupta, Enoch Jiang, Ryan Jin, Aishwarya Kalyanaraman, Jeferson Mendoza, Pooja Nahar, Danielle Pergola, Siri
Uppuluri, Sophia Velasquez
Advisor: Dr. Stephen DunawayAssistant: Jal Trivedi
ABSTRACT
The complex world of invasive mycoses has only recently emerged at the forefront of biomedical research as a serious threat to human health. Due to the increasing susceptibility of immunocompromised populations to invasive fungal infections (IFIs) and the ineffectiveness of current treatment methods, the field of medical mycology is in dire need of a novel therapeutic approach. In this paper, we focus on Eukaryotic Elongation Factor 3 (eEF3), one of three proteins in a family of translation factors known as eukaryotic elongation factors. eEF3 plays a critical role in polypeptide synthesis, aiding in the removal of deacetylated tRNAs from the ribosomal complex and recruiting aminoacetylated tRNAs from the cytosol to continue elongation. Curiously, it has been discovered that eEF3 is highly conserved in unicellular eukaryotes and is absent from, or at least functionally inactive in, more complex members of the domain. The functional exclusivity of eEF3 makes it a prime candidate for antifungal drug targeting, permitting the design of highly effective therapeutic compounds with minimal toxicity to host cells. However, the structural conservation of eEF3 varies among different species, and it is first necessary to determine the functional conservation of the protein as well as the conservation of integral functional domains. The current study presents an intuitive approach to answering these questions, employing the “plasmid shuffle” technique to transform P. infestans eEF3 into S. cerevisiae while forcing the latter species to forfeit its endogenous eEF3 gene. The results of the experiment indicated that, when placed under stringent environmental pressure, S. cerevisiae was able to survive on the P. infestans eEF3 homologue, suggesting that P. infestans eEF3 can functionally substitute for S. cerevisiae eEF3. This study provides a preliminary foundation for eEF3 targeting research and will hopefully have significant implications for antimicrobial drug development to combat fungal and other eukaryotic pathogens.
KEYWORDS
Eukaryotic elongation factor 3, eEF3, Phytophthora infestans, Saccharomyces cerevisiae, invasive fungal infections, antifungal drug targeting, Gibson Assembly Protocol, plasmid shuffle
[5-1]

INTRODUCTION
The contribution of fungal pathogens to the collective pool of human disease has been historically overlooked and has only recently captured the attention of the medical community, emerging in a variety of biomedical research avenues as an issue in need of addressing. Though the incidence of deaths related solely to invasive mycoses is comparably lower than that of other medical afflictions, the vast majority of these deaths occurs in patients with compromised immune systems due to pre-existing conditions, most notably Human Immunodeficiency Virus/Acquired Immunodeficiency Syndrome (HIV/AIDS) (1). This deadly combination of immunocompromisation and the resulting vulnerability to invasive fungal infections (IFIs) is responsible for over 50% of all AIDS-related deaths, earning a spotlight on the global medical stage (1, 2). The risk of contracting an IFI is also particularly heightened for transplant patients, preterm infants, and other individuals with underdeveloped/weakened immune systems or those undergoing immunosuppressive therapy (1,3). The net mortality rate due to both independently-acting invasive mycoses and IFIs is staggering; the seven most lethal fungal pathogens alone contribute to at least 1,600,000 recorded deaths every year (4).
The current state of affairs regarding the prophylaxis, diagnosis, and treatment of fungal infections is insufficient to combat the growing number of cases worldwide. The recognition and identification of a fungal infection is difficult and often results in a delayed diagnosis or no diagnosis at all (3). Our notable inability to distinguish among many existing fungal pathogens in microbiological cultures and the lack of simple serological tests further increases the threat that fungal pathogens pose to humans (3). Despite the availability of a vast array of antifungal therapeutics, an exponential increase in the administration of various antifungal drugs, and the expansion of the field of medical mycology, the susceptibility of human hosts to fungal pathogens continues to grow at an alarmingly rapid rate (1,5).
Fungal pathogens can also have devastating effects on non-human hosts such as crops, whose destruction often results in crippling economic implications and even widespread famine. P. infestans is an oomycete that causes potato late blight, a disease with the ability to wipe out countless potato crops, as well as other crops such as tomato (Solanum lycopersicum), pear melon (S. muricatum), and naranjilla (S. quitoense) (6). The horrific consequences of a P. infestans epidemic were epitomized by the Irish Potato Famine (also referred to as The Great Famine) in the mid-1840s, when the fungus destroyed over 75% of the potato harvest over the course of eight years. The potato crops were a major source of nutrition as well as a significant source of revenue for Irish farmers, and their loss resulted in nearly one million deaths, deep economic recession, and mass emigration (6). Even today, P. infestans remains an extraordinary threat to farmers around the world; in 1985, an outbreak in the state of Washington cost between $106.77 and $226.85 per acre affected, with a total management cost of nearly $30 million. The International Potato Center has estimated that in developing countries, P. infestans results in a minimum 15% harvest loss, or a total production loss of around $2.75 billion per year (6).
[5-2]

Currently, antifungal drugs catered to treat human diseases are typically sorted into one of two categories: systemic or topical (7). Topical drugs are applied directly to the surface of the skin, most commonly in the form of a cream or spray, whereas systemic drugs target the organism as a whole. The most common systemic antifungal drug used to treat life-threatening infections is amphotericin B; however, amphotericin B is linked to side effects including nausea, vomiting and fever (7). Researchers have developed lipid formulations of this drug in an attempt to decrease nephrotoxicity and other adverse effects. These lipid formulations, however, have not demonstrated much of an advantage, and are much more expensive than the typical amphotericin B. Due to the resounding structural similarities between fungi and mammalian cells, very few myco-specific agents exist as options for highly selective drug treatment (7). The majority of antifungal therapeutics target minute differences in cell membrane composition that distinguish some fungal species from non-fungal cells (8). However, this approach is limited in its application as it is not validated for all known fungal pathogens and is highly susceptible to the emergence of antifungal resistance strains.
The devastating effects of fungal pathogens on both human and non-human hosts have necessitated the search for novel diagnostic and therapeutic approaches against invasive mycoses and drug-resistant phenotypes. To address this concern, the present study focuses on intracellular translational machinery in an attempt to identify a highly conserved pathway in lower order eukaryotic organisms that may serve as a potential drug target. One of the driving forces behind this research is the search for such a molecule that is active and necessary for life in lower order eukaryotes, including pathogenic fungi, but is absent or functionally inactive in higher order eukaryotes - specifically the hosts of fungal infection. The answer lies in the central dogma of biology.
The central dogma delineates a very straightforward, albeit complex process that permits living organisms to produce proteins necessary for life using a universal genetic code. More commonly known as protein synthesis, the conversion of DNA to mRNA to polypeptide is comprised of two major steps: transcription and translation (9). Transcription is the process by which mRNA is synthesized by RNA polymerase using genomic DNA as a template. Assisted by additional transcription factors, RNA polymerase binds to a promoter region on the template strand and reads the nucleotide sequence in the 3’ to 5’ direction, synthesizing the mRNA strand in the 5’ to 3’ direction (9). Posttranscriptional modifications to the mRNA transcript include the addition of a 5’ guanosine cap and the polymerization of numerous adenines onto the 3’ tail, allowing the transcript to escape degradation by RNases while it is translated in the cytosol (9).
The translation stage of eukaryotic protein synthesis involves three major steps: initiation, elongation, and termination. During initiation, a eukaryotic ribosome dissociates into two subunits denoted 40S and 60S (10). Initiation Factor 3 (IF3) then binds to an initiator tRNA carrying a methionine and the anticodon loop sequence UAC, creating a complex which binds to the 40S ribosome subunit. Initiation Factor 4 (IF4) then binds to the 5’ end of mRNA, guiding
[5-3]

the 40S subunit with the initiator RNA and associated IF3 to the mRNA. The complex travels along the mRNA until it reaches the AUG start codon, and upon annealing of the anticodon to the codon, the 60S subunit is signalled to localize to the transcript-subunit complex and start elongation (10). Elongation begins when an acetylated tRNA molecule, through recognition of the complementary mRNA codon, binds to the A site (aminoacyl site) of the ribosome complex (11). The tRNA in the A site carries an amino acid which it then attaches to the polypeptide chain carried in the P site (peptidyl site). Elongation factors assist the tRNA in the A site to move to the P site, where it will adopt the growing polypeptide chain (11). The tRNA molecule in the P site then releases its polypeptide chain and moves to the E site (exit site), where the deacetylated tRNA will be released from the ribosome complex into the cytosol, allowing it to restart the process with a new amino acid. Termination of translation occurs when the ribosome reaches a stop codon such as UAG, termination proteins bind to the ribosome, and the polypeptide chain is released (11).
A critical component of translation in eukaryotes is the involvement of various eukaryotic elongation factors, which assist the tRNA molecules in moving between the ribosomal binding sites (Fig. 1). The roles of different elongation factors, however, are not equally conserved among different eukaryotic species, making them a particularly interesting object of study. One such elongation factor, eukaryotic elongation factor 3 (eEF3), is present and active exclusively in lower order eukaryotic cells (12). eEF3 is responsible for transporting deacetylated tRNA out of the E site, allowing the subsequent tRNA molecule to move from the P site to the E site. This action is pivotal to the continuation of the elongation phase of translation as it continuously frees the E site, and thus the P site and the A site, once the tRNA is ready to exit the ribosome complex, allowing new, charged tRNAs to bind the complex and continue synthesis of the polypeptide (12).
Figure 1 | Various roles of Eukaryotic Elongation Factors in translation. The process of translation in eukaryotes is facilitated by a class of proteins known as eukaryotic elongation factors (eEFs). eEF3 hydrolyzes ATP to remove deacylated tRNA from the E site of the ribosome-mRNA complex, allowing subsequent tRNAs to move into their appropriate locations and continue the synthesis of the polypeptide product (12, 13). It has been further suggested that eEF3 plays a direct role in recruiting aminoacetylated tRNAs to the A site of the ribosome, allowing elongation to continue (12, 13).
[5-4]

The structure and function of eukaryotic elongation factor 3 (eEF3) in S. cerevisiae has been studied in great depth, and prior research has established a sound foundation on which the present study is based. The interaction of eEF3 with deacetylated and aminoacylated tRNAs is critical to the proper progression of elongation (12). eEF3 not only removes deacylated tRNAs from the ribosomal E site, but also recruits charged tRNAs localized near the site of translation to the A site, where they are annealed to complementary mRNA codons. This multifaceted activity of eEF3 is mediated by various conformational changes made possible by ATP hydrolysis, exposing different functional domains and allowing eEF3 to interact with a host of targets (12). Recognizing the complexity of the protein, prior researchers have presented a crystal structure of eEF3 in S. cerevisiae and have mapped the various motifs to their respective amino acid regions in the original, pre-functional polypeptide (Fig. 2). This information gives significant insight into the activity of the translation factor in vivo and its various mechanisms of action (12).
Figure 2 | Structures of S. cerevisiae eEF3 amino acid sequence and tertiary protein (12). a, Schematic representation of functional domains mapped to regions on primary amino acid sequence (12). b, Crystal structure of eEF3 bound to ADP in S. cerevisiae (12). Colored regions from Fig. 2a corresponding colored domains in tertiary structure. It is important to note that ABC1 and ABC2 are ATP binding cassettes, which allow eEF3 to hydrolyze ATP and make conformational changes. The HEAT, 4HB, and Chromo motifs interact with rRNA and tRNAs to facilitate elongation.
eEF3 was first observed in S. cerevisiae and has since been discovered as an active component of various other species (14). Under cursory observation of eEF3’s role in lower order eukaryotes, it would seem logical to conclude that the function of the protein is so critical to eukaryotic translation and, by proxy, to the existence of eukaryotic life forms that it must be conserved ubiquitously throughout the domain. However, simple protein analysis has revealed the startling reality that eEF3 is only present in single-celled eukaryotes (15). The absence of eEF3 from higher order eukaryotes contributes immensely to its potential to serve as a drug
[5-5]

target for potent, broad-spectrum antifungal agents. This is primarily because administering an eEF3 inhibitor via a systemic drug delivery system would interfere with a fundamental life process in single-celled fungal pathogens with predictably lethal consequences without affecting translational activities in the cells of the host.
However, the eEF3 gene varies greatly between unicellular eukaryotic species, even those within the same genus (15). This raises an alarming question regarding the viability of eEF3 as a potential drug target for broad spectrum antifungal agents: are the specific roles of eEF3 in translation conserved throughout pathogenic fungi, and if so, is the structural conservation of eEF3 sufficient to allow researchers to design a drug targeting a functional motif that, if blocked, will render eEF3 nonfunctional in all pathogenic species?
The purpose of this study is to provide relevant, preliminary insights and to open the door to an avenue of research that we hope will one day provide the answer to that question. As representative models of the evolution of eEF3 in fungi and fungi-like species, the P. infestans eEF3 gene and the S. cerevisiae eEF3 gene are being compared. A quick alignment of their respective amino acid sequences using the NCBI Basic Local Alignment Search Tool (BLAST) revealed a 47% identity conservation and a 63% positive (physicochemical property) conservation. Furthermore, it identified the specific regions in the amino acid sequence which were most closely maintained between the two species. Applying this information in conjunction with the mapping of the amino acid sequence of S. cerevisiae eEF3 to its ultimate motifs in the tertiary structure (Fig. 2) permits us to make an educated guess regarding the conservation of functional domains between P. infestans eEF3 and S. cerevisiae eEF3.
Taking into account the high accuracy alignment, especially in regions encoding for critical functional domains like the ATP binding site, it can be hypothesized that the function of eEF3 is conserved between P. infestans and S. cerevisiae, despite significant structural differences. Results supporting our hypothesis would indicate more specifically the most necessary motifs within the eEF3 structure that contribute to maintenance of its activity in vitro. Our hope is that the results of this study provide significant implications for the appropriation of eEF3 as a viable drug target for antifungal agents with minimal toxicity to the host.
EXPERIMENTAL THEORY
The synthesis of recombinant DNA is essential to this study’s analysis of eEF3 functionality in both P. infestans and S. cerevisiae. In order to determine whether or not eEF3 holds the potential to become a target for antifungal infection drugs, a molecularly engineered plasmid must be introduced into S. cerevisiae. The basic procedure used to engineer this vector is to amplify an empty plasmid marked with the LEU2 gene by introducing it into Escherichia coli. This transformation is more formally known as the Gibson Assembly process. In this particular experiment, this process synthesizes DNA plasmids necessary to transform empty gel purified plasmids into a vector that incorporates P. infestans’ eEF3 gene, resulting in the desired vector.
[5-6]

The plasmid will be extracted and will undergo electrophoresis to confirm that the preferred vector has indeed been amplified. This process is then going to be repeated, with the added step of bacterial transformation. This transformation results in a plasmid engineered to contain P. infestans’ eEF3 gene with a LEU2 marker (16).
Gel electrophoresis is a useful method in analyzing the plasmid deoxyribonucleic acid essential to this experiment. The procedure allows scientists to detect and preliminarily characterize DNA. This process begins with the insertion of the dyed plasmids of interest into a porous 1% agarose gel. When an electric field is applied to this gel, the fragments of DNA migrate at a rate related inversely to their molecular weights (17). In this experiment, gel electrophoresis will be used to confirm that the plasmid has been isolated from E. coli, utilizing the method’s ability to separate DNA from other components. It will then be used again to determine if the plasmid does, as intended, take up the P. infestans eEF3 gene by comparing the results to an expected model.
Bacterial transformation is a process involved in the horizontal transfer of genetic material, and is integral to this experiment, for it allows the experimenters to amplify, alter, and mark genetic material (18). Transformation refers to a cell’s ability to uptake exogenous DNA and incorporate that DNA into its own genetic material (18). In this experiment, E. coli will be transformed with the recombinant plasmid containing a P. infestans eEF3 gene in order to promote amplification. After amplification, S. cerevisiae cells will be transformed with the recombinant plasmid.
After successfully cultivating S. cerevisiae with LEU2 and URA4 gene markers, it is necessary to determine whether or not transformation was successful by selecting for S. cerevisiae cells containing plasmids with the LEU2 marker rather than the URA4 marker. This selection was done with 5-fluoroorotic acid (5-FOA), a chemical that when in contact with URA4 becomes cytotoxic. When introduced to 5-FOA, cells containing URA4 can either expel the plasmid that contains this gene and remain with the plasmid with LEU2 or die. The functionality of 5-FOA at this point in the experiment is to filter out any S. cerevisiae cells that have not expelled the wild type vector containing URA4 (19). It was hypothesized that recombinant P. infestans eEF3 gene would be able to functionally serve as a substitute for the S. cerevisiae wild-type eEF3 gene, and that S. cerevisiae would subsequently survive with the recombinant gene.
The goal of this experiment is to determine whether the P. infestans eEF3 gene can functionally complement the S. cerevisiae gene. In a larger scope, the objective is to examine specific sequencing regions of fungal pathogenic eEF3 gene in the hopes of finding potential antifungal drug targets. Additionally, this study expands upon the previously existing breadth of knowledge regarding functions of the eEF3 gene, allowing further exploration and discussion of undiscovered functions of this gene.
[5-7]

MATERIALS AND METHODS
Prior to the experiment, Escherichia coli was transformed with a previously synthesized plasmid. The E. coli served to multiply the construct that would serve as the vector for the P. infestans eEF3 gene.
MiniPrep of Plasmid BackboneTo isolate the vector from the bacterial cells, the QIAprep Spin Miniprep Kit was used.
Initially, 1.5 milliliters of the bacterial cells were centrifuged at a speed of 8000 rotations per minute for 3 minutes at room temperature. The cells were then resuspended with 250 microliters of Buffer P1. After resuspension, 250 microliters of Buffer P2 was added to the cells and mixed by inverting the microcentrifuge tube six times. Next, 350 microliters of Buffer N3, a neutralization buffer, was added and the microcentrifuge tube was again inverted 6 times. The cells were then centrifuged for ten minutes at 13,000 rotations per minute, after which 800 microliters of the supernatant was taken from the microcentrifuge tube and then applied to the QIAprep 2.0 spin column. The cells were centrifuged for sixty seconds, and all flow through was discarded. The spin column was then washed again by adding .75 milliliters of Buffer PE and centrifuged for 60 seconds. The spin column was centrifuged for an additional minute to remove residual wash buffer. Then, the spin column was transferred to an empty 1.5 milliliter microcentrifuge tube and the DNA inside the spin column was eluted by adding fifty microliters of water. The DNA was again centrifuged for 1 minute.
Restriction Enzyme DigestionThe resulting DNA from the Miniprep was digested using restriction enzymes to prepare
the plasmid DNA for gel electrophoresis. The DNA was placed in a microcentrifuge tube, to which the restriction enzymes Xho1 and BamH1, from New England Biolabs, were added. The DNA was then incubated at fifty degrees Celsius for thirty minutes.
Gel Electrophoresis of Plasmid BackboneTo confirm that the plasmid of interest was isolated from the E. coli cells, the digested
DNA was run on a porous 1% agarose gel using gel electrophoresis. In addition to running the digested DNA, the undigested DNA samples were also run to confirm that the plasmid of interest was present prior to the digestion. Dye was added to the samples to ensure that they would be visible on the clear gel.
Gel PurificationThe agarose containing the DNA was excised from the gel and placed into a
microcentrifuge tube. ADB was then added to the agarose in a 3 to 1 ratio. The agarose containing the DNA was incubated for 10 minutes at 50 degrees Celsius. After incubation, the melted agarose was transferred to a Zymo-Spin column and centrifuged for 60 seconds. The flow
[5-8]

through was then discarded and 200 microliters of DNA wash buffer was added to the spin column. The DNA was centrifuged for 30 seconds and the flow through was again discarded. The DNA was washed again and eluted by adding 6 microliters of DNA elution buffer to the column matrix. The mixture was then centrifuged for 60 seconds.
Gibson Assembly ReactionThe Gibson Assembly reaction was used to insert the P. infestans eEF3 gene into the
digested plasmid (Fig. 3). The P. infestans eEF3 gene was ordered from the NCBI genome library in two pieces. The two pieces are each 1600 base pairs long, with overlapping ends to ensure that the gene is complete and can hybridize with the cut ends of the plasmid. To perform the Gibson Assembly reaction, first 2 microliters of GB1 and and 2 microliters of GB2 were added to a microcentrifuge tube. Then, 2 microliters of the gel purified DNA plasmid were added to the tube. Lastly, 4 microliters of distilled deionized water and 10 microliters of the New England Biolabs Gibson Assembly Master Mix were added. The solution was then run at 55 degrees Celsius for 15 minutes and incubated.
Figure 3 | Gibson Assembly Reaction. The Gibson Assembly reaction allowed the insertion of the P. infestans eEF3 gene into the linearized vector. Crossed lines (X) indicate hybridization due to overlapping nucleotide sequences. Colored regions of the linearized DNA correspond to the colored regions they form in the recircularized plasmid. Blue and yellow segments represent the P. infestans eEF3 gene that has been split and extended with regions of hybridization. Purple, red, and orange segments represent the linearized vector.
[5-9]

Transformation of E. coli After the P. infestans eEF3 gene was inserted into the plasmid of interest, E. coli was
transformed with the recombinant plasmid to amplify the plasmid. This was done by first using 20 microliters of competent E. coli cells. Competent E. coli cells differ in that the cells have been processed to weaken the cell walls of individual cells, making the cells more likely to incorporate foreign DNA into their genomes. The competent cells were then mixed with 4 microliters of the assembled plasmid, and left on ice for 30 minutes. Then, the cells were heat shocked at 42 degrees Celsius for 30 seconds. The heat shock served to further weaken the cell walls of the bacterial cells, increasing the likelihood of the bacteria taking up the recombinant plasmid. After being heat shocked, the cells were placed on ice for 5 minutes and then mixed with 950 microliters of SOC media. The SOC media serves to increase the efficiency of the transformation of the competent cells. After the addition of the SOC media, the cells were shaken at 37 degrees Celsius for 60 minutes. After being shaken, the cells were then centrifuged at full speed, or about 13,300 rotations per minute, for 30 seconds. Then, 800 microliters of SOC media were removed, and the pellet was resuspended in the remaining SOC media. The transformed E. coli bacterial broth was then plated on an LB agar plate with the antibiotic ampicillin. This was done to select for transformed E. coli colonies with ampicillin resistance, indicating that these colonies had indeed taken up the plasmid of interest.
Miniprep of Recombinant PlasmidAfter allowing the bacteria to grow, certain colonies were used to obtain about 1.5
milliliters of E. coli bacterial culture. Then, the QIAprep Spin Miniprep Kit was again used to isolate the recombinant plasmid from the transformed E. coli cells. The same procedure was followed as was for initially isolating the empty vector from the E. coli cells.
Restriction Enzyme Digestion Of Recombinant PlasmidSimilar to the procedure after the first Miniprep, the recombinant plasmid was again
digested with restriction enzymes, in preparation for processing the plasmid fragments by gel electrophoresis.
Gel Electrophoresis of Recombinant PlasmidAfter the recombinant plasmid was digested using restriction enzymes, it was run on a
porous 1% agarose gel to determine if the Gibson Assembly reaction had worked and the plasmid had taken up the P. infestans eEF3 gene. The DNA was then excised from the gel and used to transform the S. cerevisiae cells.
Transformation of S. cerevisiae
To insert the recombinant plasmid into the S. cerevisiae cells, the recombinant cells were first centrifuged at 3000 rotations per minute for 5 minutes. The remaining media was poured off, and the pellet was resuspended in 25 milliliters of water. After resuspension, the cells were again centrifuged at 3000 rotations per minute for another 5 minutes. The cells were then
[5-10]

resuspended in 1 milliliter of 100 milli-Molar Lithium Acetate. The cells were then transferred to a microcentrifuge tube, and centrifuged at 13,300 rotations per minute for 30 seconds. The supernatant was then poured off, and the pellet was again resuspended in 400 microliters of 100 milli-Molar Lithium Acetate. To then set up the transformation reaction, 50 microliters of the S. cerevisiae cells were added to a microcentrifuge tube. Then, 240 microliters of Polyethylene Glycol, 36 microliters of 1 Molar Lithium Acetate, and 15 microliters of the recombinant DNA plasmid were added to the S. cerevisiae cells. The mixture of cells was vortexed and then incubated at 30 degrees Celsius for 30 minutes. After incubation, 40 microliters of dimethyl sulfoxide was added to the DNA solution and mixed. The cells were then heat shocked at 42 degrees Celsius for 30 minutes. The supernatant was removed and the pellet of cells was resuspended in 200 microliters of media.
After the transformation reaction was complete, the S. cerevisiae cells were plated on a surface containing the antibiotic ampicillin while in absence of leucine. The lack of leucine ensures that only those cells that have taken up the recombinant plasmid, which contains the LEU2 marker, would survive on the plate. From the resulting bacterial colonies, one colony was taken and streaked on a plate containing 5-Fluoroorotic Acid (5-FOA). The 5-FOA forces the S. cerevisiae cells to expel the wild type plasmid, while killing those S. cerevisiae cells still containing the wild-type plasmid with the URA4 marker, ensuring that the cells remaining on the plate contain only the recombinant plasmid.
RESULTS
Figure 4 | Gel electrophoresis of digested recombinant plasmid. The digested recombinant plasmid was run through gel electrophoresis to determine the success of the Gibson Assembly reaction. The size of the linearized vector was 7 kb, and the size of the inserted eEF3 gene was ~4 kb. Matching bands in three of the digested samples indicated successful Gibson Assembly. *Due to improper loading of the sample, fragment travel distance did not accurately represent fragment size . However, proportional travel distance of two distinct and expected fragments was consistent with that observed in other successful dig. samples.
[5-11]

The processing of the recombinant plasmid via gel electrophoresis resulted in two distinctly visible bands in lanes 2, 6, and 7. These lanes contained digested samples 1, 5 and 6. The fragments formed by digested samples 1 and 5 were consistent with the fragment sizes indicated by the marker. Both digested samples produced two fragments, one with a fragment size of 7 kilobases and one with a fragment size of 4 kilobases. Although the distance between the two fragments in sample 6 was consistent with that observed in samples 1 and 5, the overall distance traveled by the fragments did not correspond (Fig. 4). Digested samples 2, 3 and 4 did not produce any distinct bands.
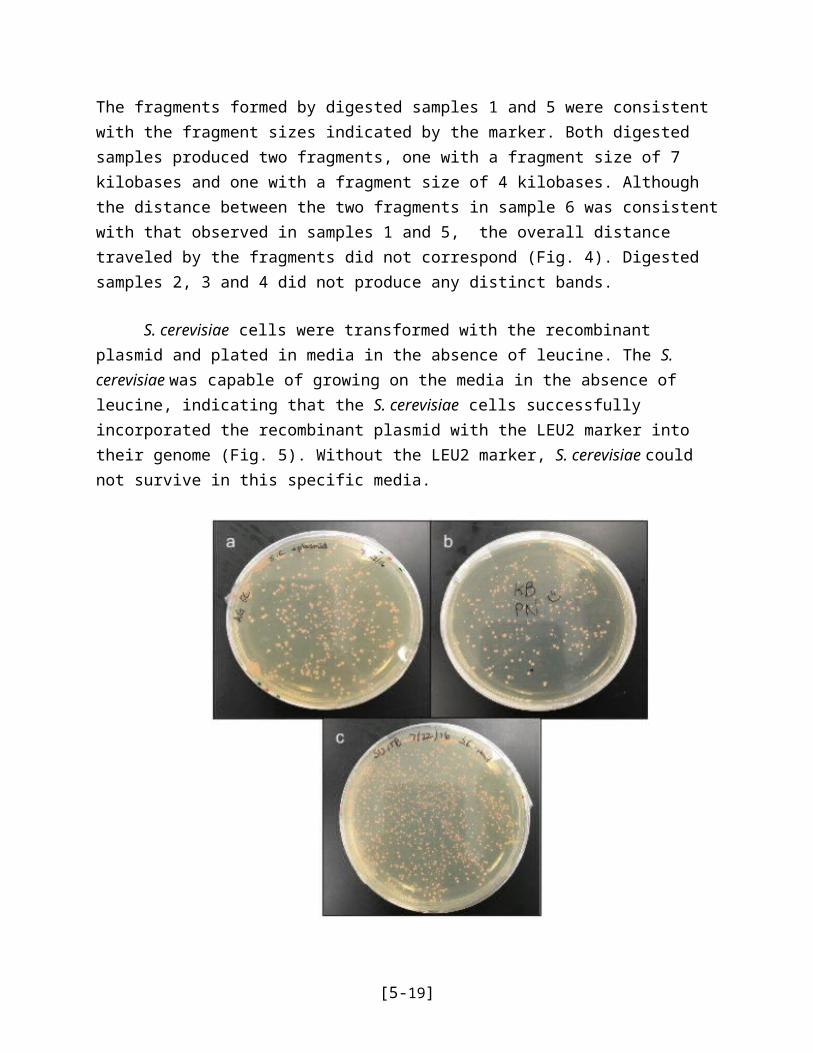
S. cerevisiae cells were transformed with the recombinant plasmid and plated in media in the absence of leucine. The S. cerevisiae was capable of growing on the media in the absence of leucine, indicating that the S. cerevisiae cells successfully incorporated the recombinant plasmid with the LEU2 marker into their genome (Fig. 5). Without the LEU2 marker, S. cerevisiae could not survive in this specific media.
Figure 5 | S. cerevisiae cultures in absence of leucine. a, b, c, Yeast transformed with the recombinant plasmid containing the P. infestans eEF3 gene was grown on LEU-/- media. The presence of colonies confirmed that the yeast successfully uptook the plasmid during transformation and expressed the exogenous LEU2 gene on the recombinant plasmid.
[5-12]

Single colonies of S. cerevisiae were replated on a media containing 5-Fluoroorotic acid (5-FOA). These colonies were able to grow on this media, indicating the elimination of the wild-type eEF3 gene because they contain URA4 (Fig. 6). This also indicates successful substitution with the P. infestans eEF3 gene (Fig. 6).
Figure 6 | S. cerevisiae cultures in 5-FOA media. a, b, c, d, Successfully transformed yeast were streaked onto 5-FOA+/+ media. 5-FOA combines with URA4 to form a highly cytotoxic compound. All samples exhibited propagation of colonies, therefore indicating that the S. cerevisiae forfeited its own plasmid containing native eEF3 and URA4 in order to survive. Further, this indicates that the only source of eEF3 for these yeast, which is critical to survival, is most likely the P. infestans eEF3 gene on the recombinant plasmid.
[5-13]

DISCUSSION
The results of this experiment provide insight into the behavior of the eEF3 gene, as well as offer a further understanding into its potential as a therapeutic drug target for fungal infections. The transfection of the P. infestans eEF3 gene into S. cerevisiae first required verification that the vector contained the gene of interest. To do this, gel electrophoresis was performed on the digested vector. The results from the gel electrophoresis confirmed that the Gibson Assembly reaction successfully inserted the P. infestans eEF3 gene into the vector. Of the six digested samples that were processed, three samples showed two distinct bands that matched the predicted values. While sample 6 does show the expected distance between the two fragments, its distance does not accurately reflect fragment size (Fig. 4). This error could be due to improper loading of the digested sample.
While three of the digested samples showed DNA fragments confirming the success of the Gibson Assembly reaction, the remaining digested samples did not result in the formation of distinct DNA bands (Fig. 4). This can be attributed to either the shearing of the plasmid DNA during digestion, or improper loading of the digested samples.
After the success of the Gibson Assembly reaction was confirmed, the S. cerevisiae cells were transformed with the recombinant plasmid and plated in media lacking leucine. The resulting plates showed growth of yeast colonies (Fig. 5). The presence of colonies confirmed the presence of the recombinant plasmid within the bacterial genome of the S. cerevisiae cells, for the the plasmid contained a LEU2 marker that would allow cells to continue producing leucine despite nutrient deprivation. Therefore, the cells that survived must have taken up the recombinant plasmid.
[5-14]

Figure 7 | Schematic representation of plasmid shuffle technique. After being introduced to the 5-FOA+/+ media, the S. cerevisiae cells were forced expel the endogenous plasmid containing the URA4 marker in order to survive, as the URA4/5-FOA combination is cytotoxic.
In order to select for bacteria containing only the recombinant plasmid, the cells were exposed to a media containing 5-FOA (Fig. 6). Prior to this point, the petri dishes contained S. cerevisiae cells with both wild-type plasmid containing the URA4 and the S. cerevisiae eEF3 genes and the recombinant plasmid with the LEU2 and the P. infestans eEF3 gene. 5-FOA, when combined with URA4, becomes cytotoxic. The subsequent growth of colonies in this media demonstrated a successful plasmid shuffle, and indicated that the P. infestans eEF3 gene can functionally substitute for the wild-type S. cerevisiae eEF3 gene in yeast cells (Fig. 6, 7).
This experiment could be improved by using a larger time frame to study the long term effects of the yeast with the recombinant plasmid in the 5-FOA. This could potentially reveal more information not initially seen in the cells. Additionally, with a larger time frame to conduct the experiment, minor errors in measurements are less likely.
[5-15]

CONCLUSION
Pathogenic fungal infections are omnipresent within the scope of healthcare and medical research. The current treatment methods are largely insufficient in combating the spread and implications of harmful mycoses; thus, heightened research both at the molecular and clinical level has become necessary. The genetic makeup of some fungal species, including that of the potato late blight P. infestans, has come into examination as potential targets for antifungal gene treatments. With proper assessment of key genomic sequences that could result in fungal virulence, the ability to treat invasive fungal infections can be improved dramatically.
The crucial role of eukaryotic elongation factor 3, or eEF3, is underscored by its wide-ranging prevalence in single-celled eukaryotic organisms despite periods of evolutionary change. As demonstrated by the results of the study in which eEF3-containing plasmids were successfully transformed from the ancient species P. infestans to a more recent evolutionary eukaryotic counterpart, S. cerevisiae, the necessity of eEF3 extends far beyond its function in translation. Unexplored regions of P. infestans’ genome in relation to protein synthesis may serve as potential antifungal drug targets. Future experimental studies include further genome sequence examinations. In particular, assessments of genomic mutations may be useful in elucidating the functions of specific sequencing regions of eEF3. Due to the evolutionary retainment and translational applications of this particular gene in lower level eukaryotes, a novel realm of potential fungal infection combatants has been brought to light.
[5-16]

ACKNOWLEDGEMENTS
We would like to thank Dr. Stephen Dunaway, our primary research mentor and professor, for welcoming us into his laboratory and teaching us the importance of molecular research. Additionally, we would like to thank Jal Trivedi for advising us throughout the course of our work, from performing the experiments to finalizing our paper and presentation. A special thanks goes out to the undergraduate students Justyna Pupek, McClellan Knapp, and Alexandria Garino for assisting with our experiment.
Finally, we would like to recognize the following sponsors of the New Jersey Governor’s School in the Sciences for their generous contributions that make research like this possible:
John and Laura Overdeck, Independent College Fund of America, Novartis Pharmaceuticals Corp., Bayer Healthcare, Johnson & Johnson, Celgene, AT&T, Allergan, Mango Concept, and the Parents and Alumni of the NJGSS
With their help, the NJGSS scholars can continue conducting incredible research and making great strides in the field of science.
[5-17]

REFERENCES
1. Brown GD, Meintjes G, Kolls JK, Gray C, Horsnell W. AIDS-related mycoses: the way forward. Trends in Microbiology. 2014;22(3):107–109.
2. Armstrong-James D, Meintjes G, Brown GD. A neglected epidemic: fungal infections in HIV/AIDS. Trends in Microbiology. 2014;22(3):120–127.
3. Hawkins C, Armstrong D. Fungal infections in the immunocompromised host. Clinical Haematology. 1984 Oct:599–630.
4. Fungal Disease Frequency. Gaffi.com. [accessed 2016 Jul 23]. http://www.gaffi.org/why/fungal-disease-frequency/
5. Martín-Peña A, Aguilar-Guisado M, Cisneros JM. Does the current treatment of invasive fungal infection need to be reviewed? Enfermedades Infecciosas y Microbiología Clínica. 2014;32(8):523–528.
6. Social Impact and Economic Importance of Late Blight. CGIAR.org. [accessed 2016 Jul 23]. https://research.cip.cgiar.org/confluence/display/gilbweb/social impact and economic importance of late blight
7. Dismukes WE. Introduction to Antifungal Drugs. Clinical Infectious Diseases. 2000;30(4):653–657.
8. Lewis RE. Current Concepts in Antifungal Pharmacology. Mayo Clinic Proceedings. 2011;86(8):805–817.
9. Crick F. Central Dogma of Molecular Biology. Nature. 1970;227(5258):561–563.
10. DNA-RNA-Protein. Nobelprize.org. 2016 [accessed 2016 Jul 23]. https://www.nobelprize.org/educational/medicine/dna/a/translation/initiation.html
11. Clancy S. DNA Transcription. Nature Education. 2008;1(1):41.
12. Andersen CBF, Becker T, Blau M, Anand M, Halic M, Balar B, Mielke T, Boesen T, Pedersen JS, Spahn CMT, et al. Structure of eEF3 and the mechanism of transfer RNA release from the E-site. Nature. 2006;443(7112):663–668.
13. Chakraburtty K. Elongation Factor 3 in Fungal Translation. Encyclopedia of Life Sciences. 2001.
[5-18]
14. Gontarek RR, Li H, Nurse K, Prescott CD. The N Terminus of Eukaryotic Translation Elongation Factor 3 Interacts with 18 S rRNA and 80 S Ribosomes. Journal of Biological Chemistry. 1998;273(17):10249–10252.
15. Kovalchuke O, Kambampati R, Pladies E, Chakraburtty* K. Competition and cooperation amongst yeast elongation factors. Eur J Biochem European Journal of Biochemistry. 1998;258(3):986–993.
16. Griffiths AJF. An introduction to genetic analysis. New York: W.H. Freeman; 2000. http://www.ncbi.nlm.nih.gov/books/nbk21881/
17. Meyers JA, Sanchez D, Elwell LP, Falkow S. Simple Agarose Gel Electrophoretic Method for the Identification and Characterization of Plasmid Deoxyribonucleic Acid. Journal of Bacteriology. 1976 [accessed 2016 Jul 22];127(3):1529–1537.
18. Lorenz MG, Wackernagel W. Bacterial gene transfer by natural genetic transformation in the environment. Microbiological Reviews. 1994;38(3):563–602.
19. Boeke JD, Trueheart J, Natsoulis G, Fink GR. 5-Fluoroorotic acid as a selective agent in yeast molecular genetics. Methods in Enzymology. 1987 [accessed 2016 Jul 24];154:164–175.
[5-19]















![AGR,IPLf]S fnaialab Og5+ O72g - faperta.uho.ac.idfaperta.uho.ac.id/agriplus/Fulltext/2006/AGP1602004.pdf · Nluhammod Tauftk dan Syalr : ISOLASI DNA PLASMID DENGAN METODE MINIPREP](https://img.pdfslide.net/doc/110x75/5cc4a2d988c993e82a8be274/agriplfs-fnaialab-og5-o72g-nluhammod-tauftk-dan-syalr-isolasi-dna-plasmid.jpg)













