Embed Size (px)
Citation preview

00ournal ofNeurology, Neurosurgery, and Psychiaty 1993;56:900-905
Mitochondrial encephalomyopathy: variableclinical expression within a single kindred
D Crimmins, J G L Morris, G L Walker, C M Sue, E Byrne, S Stevens, B Jean-Francis,C Yiannikas, R Pamphlett
AbstractThe clincal manifestations of mitochon-drial encephalomyopathy are describedin four generations of a single kindred.The age of onset of major neurologicaldisturbance varied from 3-70 years. Insome patients, deafness was the onlymanifestation; in others, recurrent boutsof status epilepticus associated with focalneurological deficits and headache,caused severe disability or death.Examples of all three adult formsof mitochondrial encephalomyopathy:MELAS, MERFF and Kearns Sayresyndrome, were represented within thekindred. Associated features includeddeafness, short stature, non-insulin-dependent diabetes mellitus,migraine, peptic ulceration and severeconstipation. The nt 3243 A-G MELASmutation was detected in two membersof the kindred. This study highlights thediversity of clinical expression of a mito-chondrial mutation within a singlekindred.
(J Neurol Neurosurg Psychiatry 1993;56:900-905)
Mitochondrial encephalomyopathy takesmany clinical forms and can begin at any age.
Department of The onset is from birth or early childhood inNeurology, Westmead Leigh's disease (subacute necrotisingHospital, Sydney, encephalomyopathy),' Alper's disease (pro-
D Crimmins gressive cerebral poliodystrophy)2 andJ G L Morris Canavan's disease (spongy degeneration ofG L Walker the white matter).3 Patients presenting inC M Sue adolescence or adult life have previously beenC Yiannikas thought to conform to one of three types:Department of Kearns Sayre syndrome, with external oph-Neurology, St
41Vincent's Hospital, thalmoplegia45; mitochondrial encephalomy-Melbourne, Australia opathy with lactic acidosis and stroke likeE Byrne episodes (MELAS); and myoclonic epilepsyDepartment of with red ragged fibres (MERRF). While indi-Neuropathology,University of Sydney, vdual patients can usually be fitted ito oneSydney, Australia of these categories, it is uncertain whether theS Stevens same can be said for whole kindreds.f8 WeB Jean-Francis present a family with features of all three
Correspondence to: syndromes of adult onset mitochondrialDr Morris, Departnent of encephalomyopathy. The age of onset ofHeurolol, Westmead, major symptoms varied from childhood to oldNSW 2145, Australia age. Deafness, short stature, non-insulinReceived 8 October 1991 dependent diabetes mellitus, migraine, pepticand in final revised form ulceration and severe constipation were12 October 1992Accepted 16 October 1992 associated features.
MethodsFollowing identification of the proband, 14 of24 living members of the family were exam-ined by a neurologist (DC). CT scans wereperformed on 11 patients, audiometry onseven and EMG/nerve conduction studies onfive. Blood glucose levels were measured in10 patients, C-peptides, pyruvate and lactatelevels in six patients. CSF pyruvate and lac-tate levels were measured in two patients.Open muscle biopsies were performed in
eight patients. These were examined withstandard light microscopy, histochemistryand electron microscopy. The histochemicalstudies were performed on fresh frozen mus-cle obtained, under local anaesthesia, fromthe vastus lateralis or the deltoid muscle.Routine stains were performed including Hand E; myofibrillar ATPase at pH 9-4, 4-6and 4-3; NADH; succinate dehydrogenase;PAS; Sudan black; phosphorylase; Gomorritrichrome and acid phosphatase. Staining forcytochrome C oxidase was performed onsome of the later biopsies.The biochemical studies were performed
using standard methods.9 A total of 250muscle biopsies from a referral centre forneuromuscular disease, which were normalon routine histochemical stains, were used ascontrols. The mean and two standard devia-tions for each assay were calculated. Totalcell DNA was extracted from muscle biopsyspecimens in all cases. Polymerase ChainReaction (PCR) amplification and restrictionenzyme analysis was used to probe for mtDNA mutations. The nucleotide (nt) 3243A-G mutation (MELAS)I0 was sought using aprimer pair BLF corresponding to nt2826-2849 of the light strand and ND2H cor-responding to nt 5459 to 5482 of the heavystrand. The alternative MELAS mutation(A-G ntpos 11084) and the MERRF muta-tion (A-G ntpos 8344) were probed for aspreviously described.l 12 PCR amplified DNAfragments were examined by electrophoresison Agarose gels for the fragments in theregion of mt DNA associated with theMELAS and MERRF mutations.
Aliquots of purified mt DNA fragmentswere digested with the appropriate restrictionenzyme at 37 degrees overnight (ApaI for nt3243[MELAS], Bgl 1 for nt 8344[MERRF]).Digested DNA was electrophoresed onagarose gels, stained with ethidium bromideand photographed under UV light. The elec-
900 on M
arch 11, 2022 by guest. Protected by copyright.
http://jnnp.bmj.com
/J N
eurol Neurosurg P
sychiatry: first published as 10.1136/jnnp.56.8.900 on 1 August 1993. D
ownloaded from
Mitochondrial encephalomyopathy: variable clinical expression within a single kindred
trophoresed DNA was then transferred ontoa nylon membrane, hybridised with theappropriate mt DNA fragment probe follow-ing random-prime labelling with (32P)dATPand visualised after exposure to Fuji Rx X-rayfilm at 70°C.
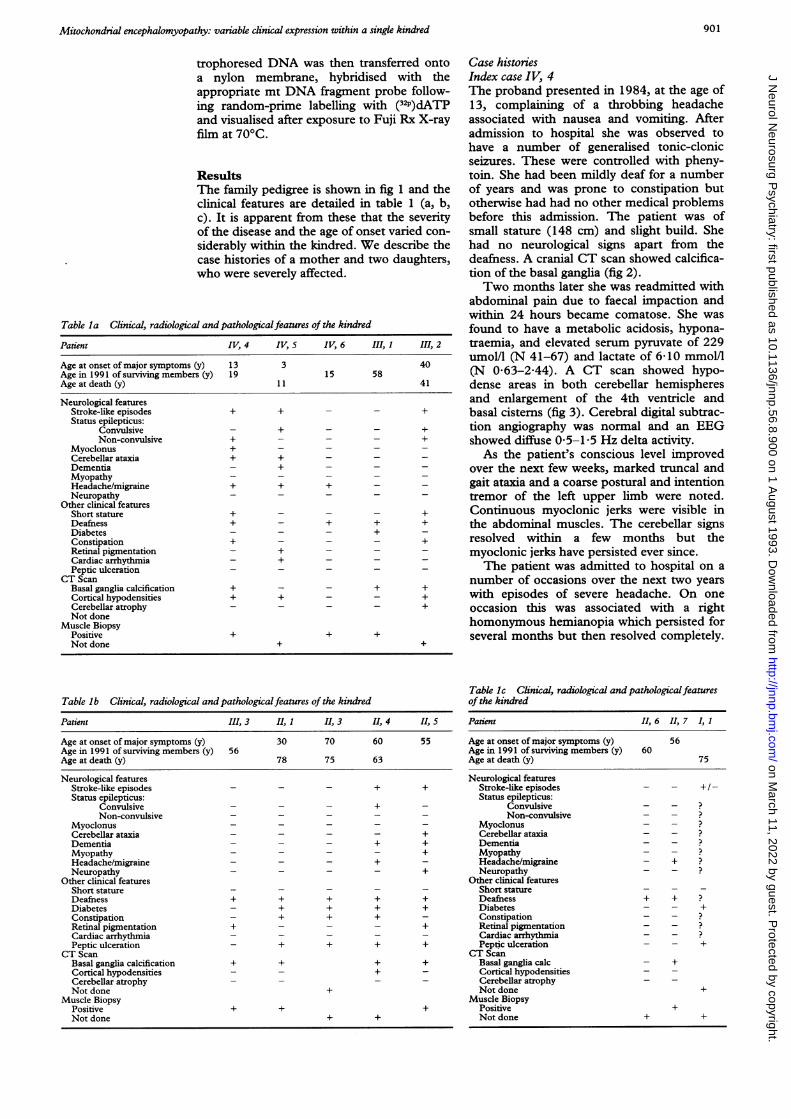
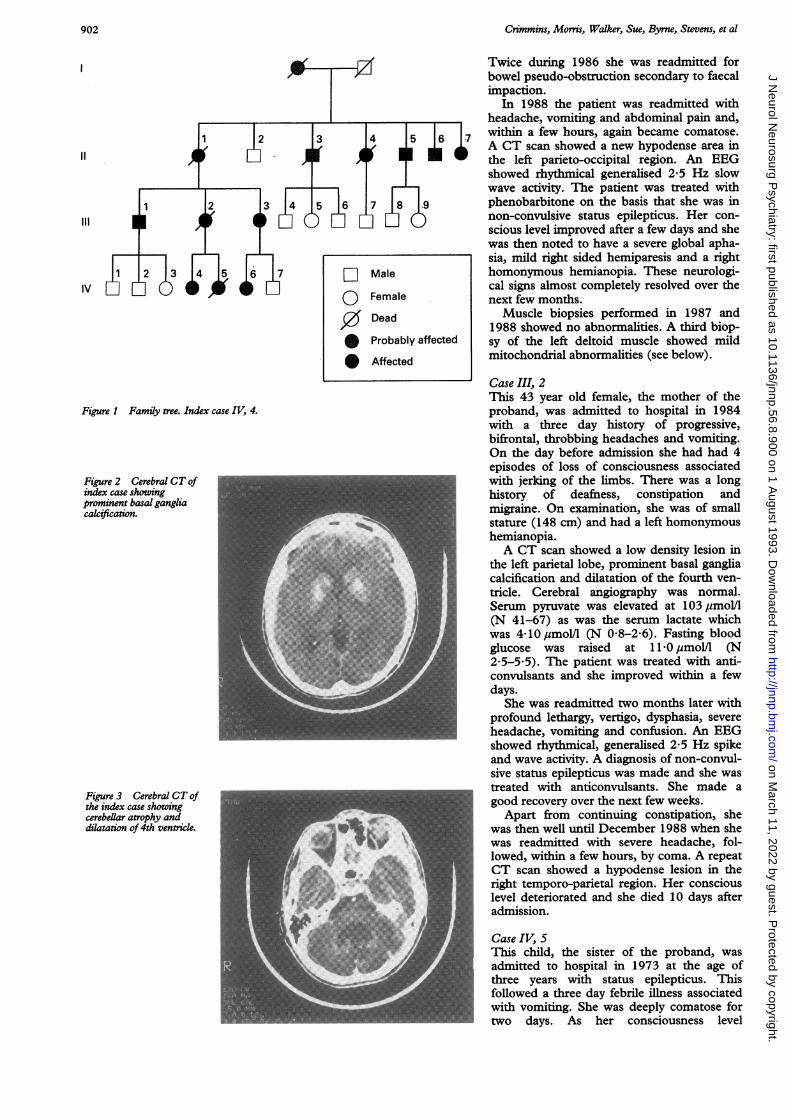
ResultsThe family pedigree is shown in fig 1 and theclinical features are detailed in table 1 (a, b,c). It is apparent from these that the severityof the disease and the age of onset varied con-siderably within the kindred. We describe thecase histories of a mother and two daughters,who were severely affected.
Table la Clinical, radiological and pathologicalfeatures of the kindred
Patient IV, 4 IV, 5 IV, 6 III, 1 III, 2
Age at onset of major symptoms (y) 13 3 40Age in 1991 of surviving members (y) 19 15 58Age at death (y) 1 1 41
Neurological featuresStroke-like episodes + + - - +Status epilepticus:
Convulsive - + - - +Non-convulsive + - - - +
Myoclonus +Cerebellar ataxia + +Dementia - +MyopathyHeadache/migraine + + +Neuropathy
Other clinical featuresShort stature + - - - +Deafness + - + + +Diabetes - - - +Constipation + - - - +Retinal pigmentation - +Cardiac arrhythmia - + - - -Peptic ulceration
CT ScanBasal ganglia calcification + - - + +Cortical hypodensities + + - - +Cerebellar atrophy - - - - +Not done
Muscle BiopsyPositive + + +Not done + +
Table lb Clinical, radiological and pathologicalfeatures of the kindred
Patient III, 3 II, 1 II, 3 11, 4 II, 5
Age at onset of major symptoms (y) 30 70 60 55Age in 1991 of surviving members (y) 56Age at death (y) 78 75 63
Neurological featuresStroke-like episodes - - - + +Status epilepticus:
Convulsive - - - +Non-convulsive - - -
Myoclonus - - -
Cerebellar ataxia - - - - +Dementia - - - + +Myopathy - - - - +Headache/migraine - - - +Neuropathy - - - - +
Other clinical featuresShort stature - - -
Deafness + + + + +Diabetes - + + + +Constipation - + + +Retinal pigmentation + - - - +Cardiac arrhythmiaPeptic ulceration - + + + +
CT ScanBasal ganglia calcification + + + +Cortical hypodensities - - +Cerebellar atrophyNot done +
Muscle BiopsyPositive + + +Not done + +
Case historiesIndex case IV, 4The proband presented in 1984, at the age of13, complaining of a throbbing headacheassociated with nausea and vomiting. Afteradmission to hospital she was observed tohave a number of generalised tonic-clonicseizures. These were controlled with pheny-toin. She had been mildly deaf for a numberof years and was prone to constipation butotherwise had had no other medical problemsbefore this admission. The patient was ofsmall stature (148 cm) and slight build. Shehad no neurological signs apart from thedeafness. A cranial CT scan showed calcifica-tion of the basal ganglia (fig 2).Two months later she was readmitted with
abdominal pain due to faecal impaction andwithin 24 hours became comatose. She wasfound to have a metabolic acidosis, hypona-traemia, and elevated serum pyruvate of 229umol/l (N 41-67) and lactate of 6 10 mmol/l(N 0 63-2A44). A CT scan showed hypo-dense areas in both cerebellar hemispheresand enlargement of the 4th ventricle andbasal cisterns (fig 3). Cerebral digital subtrac-tion angiography was normal and an EEGshowed diffuse 0-5-1 5 Hz delta activity.As the patient's conscious level improved
over the next few weeks, marked truncal andgait ataxia and a coarse postural and intentiontremor of the left upper limb were noted.Continuous myoclonic jerks were visible inthe abdominal muscles. The cerebellar signsresolved within a few months but themyoclonic jerks have persisted ever since.The patient was admitted to hospital on a
number of occasions over the next two yearswith episodes of severe headache. On oneoccasion this was associated with a righthomonymous hemianopia which persisted forseveral months but then resolved completely.
Table Ic Clinical, radiological and pathologicalfeaturesof the kindred
Patient 1, 6 II, 7 I, 1
Age at onset of major symptoms (y) 56Age in 1991 of surviving members (y) 60Age at death (y) 75
Neurological featuresStroke-like episodes - - + /-Status epilepticus:
Convulsive - -Non-convulsive - -
Myoclonus - - ?Cerebellar ataxia - -Dementia - -Myopathy - -Headache/migraine - +Neuropathy - -
Other clinical featuresShort statureDeafness + + ?Diabetes - - +Constipation - - ?Retinal pigmentation - -Cardiac arrhythmia - -
_Peptic ulceration - - +CT Scan
Basal ganglia calc - +Cortical hypodensities - -Cerebellar atrophyNot done +
Muscle BiopsyPositive +Not done + +
901 on M
arch 11, 2022 by guest. Protected by copyright.
http://jnnp.bmj.com
/J N
eurol Neurosurg P
sychiatry: first published as 10.1136/jnnp.56.8.900 on 1 August 1993. D
ownloaded from
Crimmins, Morris, Walker, Sue, Byrne, Stevens, et al
1 ~~2
1 2 3 4 5 6
Figure 1 Family tree. Index case IV, 4.
Figure 2 Cerebral CT ofindex case showingprominent basal gangliacalcification.
Figure 3 Cerebral CT ofthe index case showingcerebellar atrophy and Udilatation of 4th ventricle.
Twice during 1986 she was readmitted forbowel pseudo-obstruction secondary to faecalimpaction.
In 1988 the patient was readmitted withheadache, vomiting and abdominal pain and,within a few hours, again became comatose.A CT scan showed a new hypodense area inthe left parieto-occipital region. An EEGshowed rhythmical generalised 2-5 Hz slowwave activity. The patient was treated withphenobarbitone on the basis that she was innon-convulsive status epilepticus. Her con-scious level improved after a few days and shewas then noted to have a severe global apha-sia, mild right sided hemiparesis and a righthomonymous hemianopia. These neurologi-cal signs almost completely resolved over thenext few months.
Muscle biopsies performed in 1987 and1988 showed no abnormalities. A third biop-sy of the left deltoid muscle showed mildmitochondrial abnormalities (see below).
Case III, 2This 43 year old female, the mother of theproband, was admitted to hospital in 1984with a three day history of progressive,bifrontal, throbbing headaches and vomiting.On the day before admission she had had 4episodes of loss of consciousness associatedwith jerking of the limbs. There was a longhistory of deafness, constipation andmigraine. On examination, she was of smallstature (148 cm) and had a left homonymoushemianopia.A CT scan showed a low density lesion in
the left parietal lobe, prominent basal gangliacalcification and dilatation of the fourth ven-tricle. Cerebral angiography was normal.Serum pyruvate was elevated at 103,mol/1(N 41-67) as was the serum lactate whichwas 4 10 pmol/1 (N 0 8-2 6). Fasting bloodglucose was raised at 11 0 jsmol/l (N2 5-5 5). The patient was treated with anti-convulsants and she improved within a fewdays.
She was readmitted two months later withprofound lethargy, vertigo, dysphasia, severeheadache, vomiting and confusion. An EEGshowed rhythmical, generalised 2 5 Hz spikeand wave activity. A diagnosis of non-convul-sive status epilepticus was made and she wastreated with anticonvulsants. She made agood recovery over the next few weeks.
Apart from continuing constipation, shewas then well until December 1988 when shewas readmitted with severe headache, fol-lowed, within a few hours, by coma. A repeatCT scan showed a hypodense lesion in theright temporo-parietal region. Her consciouslevel deteriorated and she died 10 days afteradmission.
Case IV, SThis child, the sister of the proband, wasadmitted to hospital in 1973 at the age ofthree years with status epilepticus. Thisfollowed a three day febrile illness associatedwith vomiting. She was deeply comatose fortwo days. As her consciousness level
D Male
0 Female
0 Dead
* Probably affected
* Affected
902 on M
arch 11, 2022 by guest. Protected by copyright.
http://jnnp.bmj.com
/J N
eurol Neurosurg P
sychiatry: first published as 10.1136/jnnp.56.8.900 on 1 August 1993. D
ownloaded from

Mitochondrial encephalomyopathy: variable clinical expression within a single kindred
improved over the next few days it was notedthat she was blind. Pupillary responses werepreserved suggesting that this was corticalblindness. Her sight returned to normal overthe next four weeks. An EEG showed gener-alised slow wave activity. She had had a nor-mal birth and development had been normal.
Over the next three years the patient hadnumerous admissions to hospital for statusepilepticus, and episodes of dysphasia, rightsided weakness, vomiting and throbbingfrontal headache. In February 1978 she wasnoted to have an ataxic gait, intention tremorof both hands, mild right sided weakness andbrisk reflexes with bilateral extensor plantarresponses. An echocardiogram revealed mildmitral valve prolapse and an ECG showed thefeatures of Wolff-Parkinson-White syndrome.Marked macular pigmentation was evident onfumdoscopy. The serum lactate was 3-4 timesthe upper limit of the normal range.
In 1979 the patient stopped attendingschool because of progressive cognitiveimpairment and increasing lethargy. By theend of the year, she was unable to feed her-self. A CT scan showed multiple low densitylesions in both cerebral hemispheres, withdilatation of the ventricular system.The patient's condition continued to dete-
riorate with dementia, lethargy, incontinenceof urine and faeces, weakness, vomiting,headaches, and fits which became increasing-ly resistant to medication. The patient died inAugust 1982.
MUSCLE BIOPSY FINDINGSMusce morphology, histochemistry and biochemistryOpen muscle biopsies were performed ineight patients of which seven were abnormal.Abnormalities varied in severity from minorchanges (with only occasional fibres showingmitochondrial disorganisation or sub-sar-colemmal aggregation on succinate dehydro-genase and Gomorri trichrome stains) tomarked changes (including generalised fibreatrophy with some fibres undergoing degener-ation and up to 20% of the fibres showing
Table 2 Muscle biopsy biochemistry
Mitochondrial enzyme activity
Pyruvate 2-Oxoglutarate Glutamate Succinate Cytochromeand Malate Cyt Cyt "C" Cyt "C" Cyt "C" "C" Oxidase"C" Reductase Reductase Reductase Reductase
Normal Rangej mol/min/gm 0-20-1 0-15-1 0-15-1 3-10 4-30of muscle
Patient:IV, 4 0 70 0-18 0-63 2-70 8-0III, 1 0-72 0-72 0-28 0-72 3-9III, 3 0-36 0 50 0-19 0 90 1-9IV, 6 0-57 0-50 0-30 0-20 2-2II 1 0-45 0-46 0-18 0 75 2-3
Table 3 Senum pyruvate and lactate levels
Patient II, I III, 3 IV, 6 IV, 4 III, 2 IV, 5
Serum Lactate 1-30 1-20 1-10 6-10 4-10 7-32(N 0-63-2-44 mmol/l)
Serum Pyruvate 82 94 71 229 103 -
(N 41-67 umoLVl)
mitochondrial abnormalities with glycogenand lipid accumulation on appropriate histo-chemical stains). Cytochrome C oxidasestaining was performed on several of themore recent biopsies but did not yield addi-tional information. The fibre type was of nor-mal distribution.
Muscle biopsy abnormalities tended to bemost marked in older patients (for example,case II, 5), rather than in those with severeclinical manifestations (such as, case IV, 4,the index case).Mitochondrial mutation studiesThe nt 3243 A-G MELAS mutation wasdetected in two cases examined (patients: II,1and IV, 4). There was no evidence of thealternative A-G MELAS mutation (nt 11084)or the A-G 8344 (MERRF) mutation in anyof the cases. A full investigation of the mito-chondrial genome in symptomatic andasymptomatic members of this family is nowin progress and will be reported separately.The results of the muscle biochemistry on
crude extract are seen in table 2. Four of thepatients had cytochrome "C" oxidasedeficiency.
Serum lactate and pyruvate levelsTable 3 shows the serum pyruvate and lactatelevels for the group. Five patients had ele-vated pyruvate levels and three had elevatedlactate levels.
Muscle biopsy biochemistryIn three patients a cytochrome C oxidasedeficiency was demonstrated. One otherpatient had borderline cytochrome C oxidasedeficiency. Normal mitochondrial enzymeactivity was demonstrated in the otherpatients studied.
Electron microscopyThe muscle biopsies examined by electronmicroscopy showed abnormalities varyingfrom severe to mild. Severe cases showedmyofibrillar degeneration with patchy lossand thinning of the myofibrils and associatedincrease in glycogen; in these areas themitochrondria tended to be pleomorphic. Insome cases, there were increased numbers oflipid vacuoles and occasionally some dilata-tion of the sarcoplasmic reticulum. Somefibres had aggregates of mitochondria, pleo-morphic and with inclusions, beneath the sar-colemmal membrane.The inclusions were constant in appear-
ance within this family and consisted ofround, uniformly stained, moderately densebodies without a limiting membrane or obvi-ous internal structure. Paracrystalline or"parking lot" inclusions were not seen. Theinclusions varied in number from case to caseand correlated with the severity of the otherchanges. In the more severely affected cases,numerous mitochondria within an affectedfibre would contain an inclusion, occasionallymore than one; in the mildly affected casesonly an occasional mitochondrion with aninclusion could be seen and then only after acareful search.
903 on M
arch 11, 2022 by guest. Protected by copyright.
http://jnnp.bmj.com
/J N
eurol Neurosurg P
sychiatry: first published as 10.1136/jnnp.56.8.900 on 1 August 1993. D
ownloaded from

Crimmins, Morris, Walker, Sue, Byrne, Stevens, et al
DiscussionThere seems little doubt of the diagnosis ofmitochondrial encephalomyopathy in thisfamily. Ten members had clinical featuresknown to be associated with this disorder; amaternal pattern of inheritance could beshown over four generations; seven membershad evidence of mitochondrial abnormalitieson muscle biopsy; a mitochondrial pointmutation was detected in the two cases whichwere examined. Of interest in this kindred isthe diversity of clinical expression in mem-bers, all of whom presumably had the samemitochondrial mutation. The index case hadepisodes of status epilepticus associated withmajor neurological deficits beginning in ado-lescence. By contrast, the only clinical evi-dence of the disease in case III,1, a man ofmiddle age, was deafness and diabetes. Incase II,5 myopathy, peripheral neuropathy,dementia and an extrapyramidal disorder hadtheir onset in late middle age.
Severely affected patients had a distinctivepresentation. Typically, an attack wouldbegin with a severe, unilateral, throbbingheadache associated with vomiting and insome cases constipation. Within 24-48 hours,the patients developed generalised tonic-clonic seizures usually leading to statusepilepticus. Two patients became comatosewithout preceding convulsions and werefound, on electroencephalography, to be innon-convulsive status epilepticus (IV,4 andIII,2). On recovery of consciousness, a neuro-logical deficit, such as homonymous hemi-anopia, dysphasia, hemiparesis or cerebellarataxia, was apparent. The cerebral CT scanat this time revealed low density lesions whichfitted no single vascular territory. These werenot associated with cerebral or extracranialangiographic abnormalities, a finding consis-tent with other reports."3 The recovery fromneurological deficits which, on both clinicaland radiological grounds were severe, wasoften surprisingly good. In this kindred, acuteneurological deficits were usually accompa-nied by fits. The term "stroke-like" episode,often used to describe such attacks78 does notconvey an accurate picture of their presenta-tion. Similarly, the epilepsy which occurs inthese patients is unusual in that it is charac-terised by episodes of status epilepticus sepa-rated by relatively fit-free intervals."3A number of patients within this kindred
had features of mitochondrial encephalomy-opathy which were not readily apparent.Deafness was present in 11, basal gangliacalcification (on CT scanning) in eight, shortstature in two, severe constipation in five,peptic ulceration in five, non-insulin depen-dent diabetes in six and migraine in five.Some of these features are common andtherefore could be coincidental; all, however,appeared in a pattern consistent with mito-chondrial inheritance and an association withME, with the exception of peptic ulceration,has been described in previous studies.78 "-"It seems unlikely that the deafness was coinci-dental for it always began in childhood orearly adulthood. Basal ganglia calcification
occurs in only 0-23-1-6% of patients16-18 andnon-insulin dependent diabetes in 1-2%.'9The constipation which occurred in this kin-dred was severe enough in itself to warranthospital admission in three patients (IV,4,II,3 and II,1). Patient II,1 had recurrentbouts of severe constipation beginning in her30s and culminating in death from pseudo-obstruction of the bowel at the age of 78.Severe constipation, associated with mito-chondrial myopathy has been reported byothers.20 Rather than listing these as associat-ed features of mitochondrial encephalomy-opathy it is probably more correct to viewthem as an integral part of a multi-system dis-ease, a mitochondrial cytopathy.
There are difficulties in fitting this kindredinto the current classification of mitochondri-al encephalomyopathy.'782' The index caseand patient IV,5 had features of MELAS:acute neurological deficits, epilepsy, lacticacidaemia and mitochondrial changes onmuscle biopsy. 'he index case, however, alsohad myoclonus and cerebellar ataxia, featuresmore consistent with MERRF.6 Patient IV,5had retinal changes and a heart conductiondefect, features associated with Kearns Sayresyndrome.4 The major manifestation of thedisease in patient II,5 was of a mitochondrialmyopathy with proximal weakness, ptosis andexternal ophthalmoplegia. In addition, he haddeafness, peripheral neuropathy, extrapyra-midal signs and ataxia. In this kindred therewas also marked variability in the age of onsetof major symptoms; the youngest presentedat three years, the oldest at 70 and fourpresented after the age of 40.
Others have also been struck by the vari-ability of clinical features and age of onsetwithin a single kindred. In reviewing 90patients with proven mitochondrial myopa-thy, Petty et aP4 found marked overlapbetween clinical categories. The kindred ofShimogi et at2 had features of MELAS andmitochondrial myopathy. The overlapbetween MERFF and MELAS has beenrecognised by a number of authors5 1' includ-ing Fukuhara, who first described MERFF asa clinical entity.2' The variability of clinicalexpression of mitochondrial encephalomy-opathy cannot be related to specific defects inthe respiratory chain2426 and remains thesubject of speculation.27-30The nt 3243 A-G mutation identified in
this family is identical to that reported in themajority of MELAS families in Japan.'0 It hasalso been encountered in at least one otherAustralian family. It is not present in allAustralian cases; an alternative A-G transi-tion mutation at nt 11084 has been identifiedin one MELAS family who lack the nt 3243mutation."
Mitochondrial encephalomyopathy is prob-ably under-recognised, particularly in itsminor manifestations. Many of its features arecommon and readily attributable to othercauses. It should be considered in the follow-ing circumstances:
1) Major manifestations: patients present-ing with an acute "stroke"-like episode with
904 on M
arch 11, 2022 by guest. Protected by copyright.
http://jnnp.bmj.com
/J N
eurol Neurosurg P
sychiatry: first published as 10.1136/jnnp.56.8.900 on 1 August 1993. D
ownloaded from

Mitochondrial encephalomyopathy: variable clinical expression within a single kindred
these additional features: a) status epilepticus,convulsive or non-convulsive; b) onset inchildhood or early adulthood; c) familialdeafness and/or diabetes mellitus d) shortstature; e) severe headache, vomiting andconstipation; f) calcification of the basal gan-glia on CT scan.
2) Minor manifestations: patients pre-senting with one or more of the followingconstellation: a) familial deafness; b) diabetesmellitus; c) intractable constipation; d)myopathy (a major manifestation in somepatients); e) possibly migraine and familialpeptic ulceration.
Having considered the diagnosis, confirma-tion depends upon the demonstration ofabnormal mitochondria on muscle biopsy, oridentification of a mitochondrial DNA muta-tion. Unfortunately, as exemplified by theproband, the patchy distribution of theabnormality may lead to a negative result onmuscle biopsy. Paradoxically, muscle abnor-malities may be most striking in olderpatients some ofwhom have only minor man-ifestations of the disease.
1 Leigh D. Subacute necrotizing encephalomyelopathy in aninfant. J Neurol Neurosurg Psychiat 1951;14:216-21.
2 Alpers B. Diffuse progressive degeneration of the greymatter of the cerebrum. Arch Neurol Psychiatry1931;25:469-505.
3 Gabetti P, Mellman W, Gonatas N. Familial spongydegeneration of the central nervous system (VonBogaert-Bertrondt disease). Acta Neuropathol 1969;12:103-8.
4 Kearns TP, Sayre GP. Retinitis pigmentosa, external oph-thalmoplegia and complete heart block: unusual syn-drome with histologic study in one of two cases. ArchOpthalmol 1958;60:280-9.
5 Rowland LP, Hays AP, Dimauro S, De Vivo DC, BehrensM. Diverse clinical disorders associated with morpho-logical abnormalities of mitochondria. In: Scarlato G,Cervi C, eds. Mitochondrial pathology in muscle diseases.Padua, Italy: Piccin Medical Books, 1983:212-8.
6 Fukuhara N, Tokiguchi S, Shirakawa K, Tsubaki T.Myoclonus epilepsy associated with ragged red fibres(mitochondrial abnormalities): disease entity or syn-drome? Light and electron microscopic studies of twocases and a review of the literature. Journal of NeurolSciences 1980;47:117-33.
7 Pavlakis SG, Phillips PC, Dimauro S, De Vivo DC,Rowland LP. Mitochondrial myopathy, encephalopathy,lactic acidosis and stroke like episodes: a distinctive clin-ical syndrome. Ann Neurol 1984;16:481-8.
8 Di Mauro S, Bonilla E, Zeviani M, Nakagawa M, De VivoDC. Mitochondrial myopathies. Ann Neurol 1985;17:521-38.
9 Edwards RHT, Round JM, Jones DA. Needle biopsy ofskeletal muscle: a review of 10 years experience. MuscleNerve 1983;6:676-83.
10 Goto Y, Nonaku I, Horai S. A mutant in the tRNAleucine gene associated with the MELAS subgroup ofmitochondrial encephalomyopathies. Nature 1990;348:651-3.
11 Lertrit P, Noer AS, Jean-Francis MJB, et al. A new diseaserelated mutation for mitochondrial encephalomyopathy,lactic acidosis and stroke-like episodes syndrome affect-ing the ND4 subunit of complex I. Am J7 HumanGenetics 1992;51(3):457-68.
12 Noer AF, Sudoyo H, Lertrit P, Thyarajun D,Utthariaphol RP, Kapsa R, Byrne E, Marzuki S. tRNAmutation in mitochondrial DNA is the causative geneticlesion underlying Myoclonic Epilepsy and Ragged RedFibre syndrome. Am J Human Genetics 1991;49:715-22.
13 Montagna P, Gallassi R, Medori R, et al. MELAS syn-drome: Characteristic migrainous and epileptic featuresand maternal transmission. Neurology 1988;38:751-4.
14 Petty RKH, Harding AE, Morgan-Hughes JA. Theclinical features of mitochondrial myopathy. Brain1986;109:915-38.
15 Peterson PL, Martens ME, Lee CP. Mitochondrialencephalomyopathies. Neurol Clinics 1988;6:529-44.
16 Cegido A, Zimmerman R, Packer R, Balinuk L, Seigal K,Dangio G. Significance of basal ganglia calcification inchildren. Pediatr Neurosci 1988;14:64-70.
17 Murphy M. Clinical correlations of CT scan detected cal-cifications in the basal ganglia. Ann Neurol 1979;6:507-11.
18 Harrington MG, Macpherson P, Mcintosh WB, AllamBF, Bone I. The presence of the incidental finding ofbasal ganglia calcification on computerised tomography.J Neurol Neurosurg Psychiatry 1981;44:1168-70.
19 Unger RH, Foster DW. Diabetes Mellitus. In: Wilson JD,Foster DW, eds. William's textbook of endocrinology;8th ed. Philadelphia: Saunders, 1990;1739-59.
20 Cervera R, Bruix J, Bayes A, Blesa R, Illa I, Coll J, Carcia-Puges AM. Chronic intestinal pseudo-obstruction andophthalmoplegia in a patient with mitochondrialmyopathy. Gut 1988;29:544-7.
21 Driscoll P, Larsen PD, Gruber AB. MELAS syndromeinvolving a mother and two children. Arch Neurol1987;44:971-3.
22 Shimoji A, Katsuragi S, Miyakawa T, Hira R, WatanabeK, Miyakawa K, Ishitsu T, Miike T. Familial mitochon-drial encephalomyopathy with stroke like episodes andepisodic disturbances of consciousness. A study of apedigree including three generations with multisystemicabnormalities. Japanese J of Psychiat and Neurol1987;41: 1:47-55.
23 Fukuhara N. Stroke-like episodes in MERRF. Ann Neurol1985;18:368.
24 Riggs JE, Schochet SS, Fakades AV, et al. Mitochondrialencephalomyopathy with decreased succinate-cytochrome C reductase activity. Neurology 1984;34:48-53.
25 Di Mauro S, Hays AP, Eastwood AB. Different clinicalexpressions of cytochrome "C" oxidase deficiency. In:Scarlato G, Cerri C, eds. Mitochondrial pathology inmuscle diseases. Padua, Italy: Piccin Medical Books,1983,112-29.
26 Di Mauro S, Zeviani M, Servidei S, et al. Cytochrome oxi-dase deficiency: Clinical and biochemical heterogeneity.Ann NYAcad Sci 1986;488: 19-32.
27 Kroon A, Van den Bogert. Biogenesis of mitochondriaand genetics of mitochondrial defects. J Inherit MetabDis 1987;(Supp 1):54.
28 Darley-Usmar V. The molecular aetiology of human mito-chondrial myopathies. Biochem Soc Trans 1987;15:102.
29 Schon EA, Rizzuto R, Moraes CT, Nakase H, Leuiani M,Dimauro S. A direct repeat is a hotspot for large scaledeletion of human mitochondrial DNA. Science1989;244:346-9.
30 Grivel LA. Small, beautiful and essential. Nature1989;341:569-71.
905 on M
arch 11, 2022 by guest. Protected by copyright.
http://jnnp.bmj.com
/J N
eurol Neurosurg P
sychiatry: first published as 10.1136/jnnp.56.8.900 on 1 August 1993. D
ownloaded from