Embed Size (px)
Citation preview

Mitokondrie-sygdomme Center for Små Handicapgrupper
Kilde: Annette, Sänger Center for små Handicap (CSH) Side 1
Juni 2001
Indholdsfortegnelse
Introduktion ……………..………………………………..…………………..…….2
Forekomst - Resume
Mitokondrie-sygdomme, der skyldes
mtDNA-defekter…………………………………..……………….………….……3
Hvad er mitokondrielle DNA-defekter?……………………………..……….……...3
CPEO (Chronic progressive external ophthalmoplegia) ……...……………….….3
Kearns-Sayre Syndrom (KSS) …………………….…………..……..…….……....4
Leighs syndrom (subacute necrotising encephalomyelopathy) ………………..…..4
LHON (Leber Hereditary Optic Neuropathy, LeberOptic Atrophy, Lebers
Hereditære synsnerveatrofi) …………………………………………..…………....5
MELAS (Myopathy, encephalopathy,lactic acidosis and stroke-like
episodes) ………..……….……………………..………………..…………..……6
MERRF (Myoclonic epilepsy with ragged red fibres) ……………..……………...6
Pearsons syndrom (Pearson marrow-pancreas syndrome) ………..………….…....7
Kompleks-defekterne ……………..…...……………..……………..……………..8
Biologien bag …………………………..…….……………………………………..9
Hvad er mitokondrier? ……………….…………………...…………………………....9
Hvad bestemmer hvordan syndromerne viser sig? ….……….….……………………10
Arvegang …………………………..…………………….…………………..……...10
Maternel arvegang …….……………………………………….………..…………..11
Recessiv arvegang ……………………………………………………..…..………..11
Et liv med en mitokondrie-sygdom ………………………..…………..…….11
Symptomerne ……………………………………..….…….………....…….……….11
Diagnosen ………………………………………..….……………….…………...…13
Behandling og forebyggelse ……………………….…………………..…….….…...15
Genetisk rådgivning …………………….…….…………..…………………...…….15
Muligheder for støtte ifølge den sociale lovgivning ……..……….……...16
Børn og unge ……………………..…………………………..……....………..……16
Voksne ………………………..……………………………………..………..……..17
Mere viden …………………………………..…………………..…….…....……18
Kontaktordning og patientforening ………………………………………..…....…..18
Adresser ……………………….…………..……………………..……..….…..……18
Internetadresser …………………………………..………………..….….…..….….19
Litteratur . ..………………………….…………………………………..……………..……....21

Mitokondrie-sygdomme Center for Små Handicapgrupper
Kilde: Annette, Sänger Center for små Handicap (CSH) Side 2
Introduktion Gruppen af mitokondrie-sygdomme omfatter en lang række syndromer, der kan komme meget forskel-
ligt til udtryk, men som har det til fælles, at mitokondrierne, som er kroppens energi-kraftværk, ikke
fungerer som de skal.
Nogle af syndromerne har et efterhånden kendt sygdomsbillede, og har fået et eget navn, mens andre
syndromer er mere uspecifikke og inddeles efter hvor fejlen er placeret - fx rækken af kompleks syn-
dromer (I-V).
Nogle af sygdommene skyldes fejl i de gener, der findes i cellekernen mens andre skyldes fejl i gener,
der findes i mitokondrierne selv. Denne beskrivelse tager udgangspunkt i sidstnævnte: dvs. de mitokon-
drie-sygdomme, som skyldes fejl i de gener, der findes i mitokondrierne selv. Mange af informationerne
i denne beskrivelse gælder dog for begge slags mitokondrie-sygdomme.
Nedenfor er der en liste over de fleste af de syndromer, der skyldes fejl i mitokondriernes egne gener.
De omtalte syndromer omtales også på vores hjemmeside www.csh.dk.
Forekomst
Man antager, at der i alt fødes et barn med en mitokondrie-sygdom for hver 10.000 fødsler. Hvor stor en
andel af dem, der skyldes mutationer i mitokondriet selv, er usikkert. Man må desuden gå ud fra, at der
findes en række personer, der ikke får diagnosen, bl.a. fordi deres symptomer er så ubetydelige, at de
ikke giver anledning til en diagnostisk udredning. Og endelig må man gå ud fra, at der er en række
spædbørn, der bliver så alvorligt ramt, at de dør umiddelbart efter fødslen, inden de når at få en diagno-
se.
Resumé Mitokondrier er cellernes - og dermed hele kroppens kraftværk, der producerer energi. Mitokondrierne
findes i flere hundrede eksemplarer i hver celle - og nogle celler har helt op til ca. 10.000 eksemplarer.
Nogle organer er mere afhængige af energien fra mitokondrierne end andre; det drejer sig om hjertet,
musklerne og hjernen (centralnervesystemet). Derfor vil man med en mitokondrie-sygdom også primært
have symptomer fra disse organer. Men også andre organer kan være involveret, som fx lever, øjne,
bugspytkirtel og tarme.
Mitokondrie-sygdomme er det, man kalder multi-system-sygdomme. Dvs. at flere forskellige organer
kan være påvirket samtidig. Mens nogle måske udelukkende har sukkersyge og migræne, er andre må-
ske mentalt retarderede eller har hjertefejl. Endelig er der også tilfælde, der er så alvorlige, at barnet dør
umiddelbart efter fødslen.
Der findes ingen helbredende behandling. Motion og forskellige former for kosttilskud kan være en
hjælp for nogle. Rygning og infektioner bør så vidt muligt undgås, da det kan forværre tilstanden.
Ægdonation kan være aktuelt for de familier, hvor syndromerne optræder i en arvelig form.

Mitokondrie-sygdomme Center for Små Handicapgrupper
Kilde: Annette, Sänger Center for små Handicap (CSH) Side 3
Mitokondrie-sygdomme,
der skyldes mtDNA-defekter Mitokondriernes funktion er ligesom resten af kroppens funktioner styret af generne. Disse gener befin-
der sig som udgangspunkt i celle-kernen (kaldet nDNA eller blot DNA), men hvert mitokondrie inde-
holder også selv nogle gener kaldet mtDNA. Denne beskrivelse handler som sagt hovedsagelig om de
syndromer, der skyldes mutationer (fejl) i mitokondriets DNA (mtDNA).
Afsnittet "Biologien bag" side 9 beskriver, hvad mitokondrier er.
Hvad er mitokondrielle DNA-defekter?
Mitokondrielle syndromer opstår, når mitokondrierne ikke danner al den energi (ATP), der er nødvendig
for, at kroppen kan fungere optimalt.
Årsagen kan enten ligge i mitokondriets eget DNA eller i cellekernens DNA - som begge koder for pro-
teiner til mitokondrierne. Når årsagen ligger i mitokondriets eget DNA (mtDNA'et) kaldes det for en
mitokondriel DNA defekt.
Der er forskel på de sygdomme, som skyldes en defekt i mitokondriets DNA og dem, der skyldes en
defekt i cellekernens DNA. Vi kommer nærmere ind på nogle af disse forskelle senere.
Nogle af de mitokondriesygdomme, der skyldes fejl i mtDNA'et, er listet herunder. Listen er ikke kom-
plet.
* CPEO (Chronic progressive external ophthalmoplegia)
* Kearns-Sayre Syndrom (KSS)
* Leighs syndrom (subacute necrotising encephalomyelopathy)
* LHON (Lebers hereditary optic neuropathy)
* MELAS (Myopathy, encephalopathy, lactic acidosis and strokelike episodes)
* MERRF (Myoclonic epilepsy with ragged red fibres)
* Pearsons syndrom
CPEO (Chronic Progressive External ophthalmoplegia)
CPEO er en kronisk fremadskridende lammelse af de ydre øjenmuskler, mens de indre øjenmuskler er
intakte. Hvis lammelsen forekommer dobbeltsidigt, kan det medføre et stift ansigtsudtryk med hængen-
de øjenlåg. Dertil kan man også have ændret pigmentering af nethinden (retinitis pigmentosa) svage
lemmer og generelt være plaget af forøget træthed.
CPEO viser sig efter 20-års-alderen. Med tiden kan man udvikle døvhed, miste sine reflekser, få forhøjet
mælkesyreindholdet i blodet, få koordinationsbesvær og noget der ligner slagtilfælde. CPEO beskrives
ofte som en mildere grad af Kearns-Sayre Syndrom, hvor der er flere mitokondrier med muteret DNA.
Og i modsætning til Kearns-Sayre syndrom, har CPEO ofte et godartet forløb.
Diagnosen stilles bl.a. ved hjælp af arbejdstests, hvor man bl.a. måler kroppens oxygen-optagelse, blod-
prøver, muskelbiopsier og genetiske analyser. Muskelbiopsier giver ofte entydige svar i den diagnosti-
ske udredning af CPEO. Der findes ingen helbredende behandling, kun symptombehandling.
CPEO skyldes sædvanligvis en spontan mutation, og man vil derfor sjældent have oplevet syndromet
tidligere i familiens historie.

Mitokondrie-sygdomme Center for Små Handicapgrupper
Kilde: Annette, Sänger Center for små Handicap (CSH) Side 4
Kearns-Sayre Syndrom (KSS)
Symptomerne på Kearns-Sayre syndrom viser sig typisk i teenageårene med lammelse af øjenmusklerne
og svækkelse af andre muskler, især hjertet, i form af rytmeforstyrrelser, samt synsnedsættelse og gang-
og koordinationsbesvær. Man kan desuden udvikle døvhed, diabetes og andre stofskifteproblemer. Di-
agnosen stilles bl.a. ved en muskelprøve.
Personer, der har symptomerne i en mild grad, eller som ikke opfylder alle symptomkriterier for Kearns-
Sayre syndrom, diagnosticeres ofte CPEO. Derfor kan det også være vanskeligt at angive præcis, hvor
mange der fødes i Danmark med Kearns-Sayre syndrom hvert år. Men man regner med ca. et tilfælde
om året.
Syndromet optræder i forskellige sværhedsgrader, er sædvanligvis ikke arvelig, men optræder sporadisk.
Der findes ingen helbredende behandling, men medicinsk behandling kan i nogle tilfælde bedre tilstan-
den. I de sværeste tilfælde er levetiden nedsat.
Leighs syndrom (subacute necrotising encephalomyelopathy)
Leighs syndrom er en fremadskridende tilstand med karakteristiske forandringer i hjernen, der kan
ramme børn. Det er primært de basale ganglier og områder i hjerne stammen, der rammes. Med tiden
udvikles forandringer flere steder i hjernen og der kan opstå hulrum pga. celledød (cellenekrose).
Synsnerven bliver ofte påvirket og med tiden også rygmarven. Mælkesyreindholdet i blodet er ofte for-
højet. Tilfældene er ofte så alvorlige, at de første symptomer allerede viser sig før etårs alderen, og det
fører ofte til, at barnet dør inden femårs alderen.
Der fødes ca. en med Leighs syndrom hvert andet år i Danmark. Dvs. gennemsnitligt mindre end en om
året. Cirka halvdelen af tilfældene af Leighs syndrom skyldes en mutation i det DNA, der findes i celle-
kernen, mens halvdelen skyldes en mutation i DNA' et fra itokondrierne. Det typiske forløb begynder oftest med spisebesvær og muskelsvaghed, derefter kommer der symptomer
som fx åndedrætsbesvær, og måske ligefrem anfald med åndedræts-stop (apnø). Øjenmuskulaturen
svækkes og der kan forekomme skelen, hængende øjenlåg (ptose), vanskeligheder med balancen, mang-
lende koordinering af bevægelserne (ataxi) og spasticitet.
Med tiden kan barnet miste almindelige færdigheder som fx at sidde og opnå kontakt. Barnets syn
svækkes, muligheden for at opnå kontakt med barnet bliver mindre, og barnet kan få kramper. Hvis
symptomerne starter senere i livet, udvikles syndromet langsommere. Symptomerne kan så være sen-
psykisk udvikling, balancevanskeligheder, motoriske problemer, psykiske symptomer og nedsat syn. En
almindelig infektion kan indlede eller forværre symptomerne.
Genetisk rådgivning er en stor hjælp i forbindelse med udredningen af risikoen for at få endnu et barn
med samme syndrom.
Diagnosen stilles bl.a. ved hjælp af scanning af hjernen, urin prøver, blodprøver, muskelbiopsier og ge-
netiske analyser. Der findes ingen helbredende behandling, kun symptombehandling. Det forhøjede
mælkesyreindhold i blodet kan forsøges behandlet medicinsk.

Mitokondrie-sygdomme Center for Små Handicapgrupper
Kilde: Annette, Sänger Center for små Handicap (CSH) Side 5
LHON
(Leber Hereditary Optic Neuropathy, Leber Optic Atrophy, Lebers hereditære
synsnerveatrofi) Syndromet viser sig i de fleste tilfælde hos unge mænd i form af pludselig nedsat syn (centralt skotom)
pga. nedbrydning af cellerne i synsnerven.
Afhængig af DNAfejlen (mutationen) kan det med forskellig hastighed føre til blindhed. Synsnedsættel-
sen kan ramme begge øjne samtidig, men oftest med et par måneders interval. Hos nogle enkelte kan
synsnedsættelsen / blindheden forsvinde igen.
Selvom synsnedsættelsen oftest er det eneste symptom, kan det forekomme, at personer med LHON og
deres slægtninge på mødrene side også oplever en række symptomer som fejl i hjertets ledningsevne,
knogleforandringer og neurologiske symptomer som fx ændrede reflekser, vanskeligheder med koordi-
nering af bevægelserne (ataxi) og føleforstyrrelser (neuropati).
LHON følger en maternel arvegang, hvilket vil sige, at syndromet udelukkende nedarves via kvinderne i
familien. Genetisk rådgivning er en stor hjælp i forbindelse eventuelt ønske om familieforøgelse.
Diagnosen stilles ved hjælp af øjenundersøgelser, blodprøver og genetiske analyser. Der findes ingen
helbredende behandling, kun symptombehandling.
Der er fundet 4 forskellige DNA mutationer i mitokondriet, der kan føre til LHON. Desuden er der be-
skrevet et antal mutationer, hvis rolle for udviklingen af LHON endnu er uvis.
Afhængig af hvilken mutation(er) der er årsagen, kan forløbet variere meget. Nogle mitokondrier vil
være normale mens andre vil indeholde det muterede mtDNA. Der skal en stor andel af muteret mtDNA
til, for at syndromet kommer til udtryk, og derfor kan nogle også være symptomfrie bærere.
På Statens øjenklinik (se adressen s. 18) findes der et komplet register over danske LHON slægter op til
200 år tilbage i tiden.
MELAS
(Myopathy, encephalopathy, lactic acidosis and stroke-like episodes)
Sygdommen viser sig sædvanligvis i 5-15 års alderen ved anfaldsvis hovedpine, opkastning og kramper
somme tider med efterfølgende lammelse og blindhed, der dog ikke altid er permanent. Efterhånden
optræder også muskelsvækkelse, væksthæmning, hørehæmning, balanceproblemer og en vis grad af
mental udviklingshæmning. Ca. 60 % har også epilepsi og mange har diabetes mellitis. Sygdommen er
fremadskridende.
Diagnosen stilles ved bl.a. påvisning af forhøjet mælkesyreindhold i blod og hjernevæske, hjernescan-
ning, undersøgelse af muskelprøver samt genetiske analyser. Der findes ingen helbredende behandling. I
Danmark fødes der i gennemsnit mindre end et barn om året, og der lever ca. 40 med syndromet.
Levetiden er nedsat.
Syndromet opstår hyppigst uden fortilfælde i familien. I de enkelte tilfælde, der er arvelige, vil det, som
ved de fleste mitokondrielle sygdomme, primært være en maternel arvegang. Det vil sige, at det er kvin-

Mitokondrie-sygdomme Center for Små Handicapgrupper
Kilde: Annette, Sänger Center for små Handicap (CSH) Side 6
derne i familien, som kan bære sygdommen videre. Genetisk rådgivning er en stor hjælp i forbindelse
med eventuelt ønske om familieforøgelse.
MERRF
(Myoclonic epilepsy with ragged red fibres)
MERRF er et arvelig fremadskridende syndrom, der bl.a. rammer centralnervesystemet og musklerne.
Symptomer som ufrivillige rykvise muskelsammentrækninger (myoklonier), vanskeligheder med koor-
dinering af bevægelserne (ataksi), muskelsvaghed, epilepsi, nedsat hørelse og fedtknuder i nakken, kan
være tegn på syndromet.
Symptomerne viser sig hos nogle før et års alderen, mens andre først får symptomer som midaldrende.
Små børns første symptomer kan være uspecifikke i form af epileptiske anfald, udviklingshæmning og
derefter tab af indlærte færdigheder. Hos nogle kan syndromet udvikle sig til Leighs syndrom (se s. 4).
Hos ældre børn og voksne kan de første symptomer være ufrivillige rykvise muskelsammentrækninger
(myoklonier) eller epileptiske anfald. Andre tidlige symptomer kan være muskelsvaghed, nedsat kondi-
tion, vanskeligheder med koordinering af bevægelserne (ataksi) og nedsat hørelse. Med tiden kan intel-
lektet også rammes, nogle får føleforstyrrelser (neuropatier) og endelig kan musklerne i hjertet også
svækkes (kardiomyopati).
Forløbet kan skride fremad med forskellig hastighed. Mens det kan strække sig over årtier hos nogle,
kan det hos andre gå stærkere og betyde forkortet levetid. Ikke alle med MERRF udvikler alle de nævnte
symptomer.
Diagnosen stilles bl.a. ved hjælp af blodprøver, genetiske analyser og muskelbiopsier. Sidste del af syn-
dromnavnet - Ragged Red Fibres - hentyder til det at muskelfibrene ser "lasede" og "flossede" ud i mi-
kroskop ved muskelbiopsiundersøgelse.
Der findes ingen helbredende behandling, kun symptombehandling. MERRF kan gå i arv via kvinderne
i familien - syndromet følger det, man kalder en maternel arvegang. Genetisk rådgivning er en stor hjælp
i forbindelse med eventuelt ønske om familieforøgelse.
Pearson’s syndrom
(Pearson marrow-pancreas syndrome) Et af de primære symptomer ved Pearsons syndrom er en alvorlig blodmangel, der kræver gentagne
blodtransfusioner. Dette skyldes, at de røde blodlegemer dør inden de er færdigudviklede (sideroblast
anæmi), hvilket forårsager en ophobning af jern i blodlegemernes mitokondrier. Et andet primært symp-
tom er manglende funktion af bugspytkirtlen, der bl.a. kan ses ved at barnet udvikler en forstørret bug-
spytkirtel (fibrosis pancreatis) og sukkersyge (diabetes mellitus), der skal behandles med insulin. Andre
symptomer kan være lammelse af øjets motoriske nerver til både de indvendig og de udvendige øjen-
muskler (oftalmoplegi), nyresvigt (Faneonis syndrom), der bl.a. betyder at mange betydningsfulde næ-
ringsstoffer (der fra nyrerne gerne skulle gå tilbage til blodet) udskilles med urinen, og endelig kan man
ofte se et højt mælkesyreindhold i blodet.
Symptomerne viser sig lige efter fødslen. Udviklingen kan gå to veje: symptomerne kan være så alvorli-
ge at barnet ikke lever særlig længe, eller barnet overlever, men det vil med tiden udvikle Kearns-Sayre
syndrom (KSS) (se s. 4).

Mitokondrie-sygdomme Center for Små Handicapgrupper
Kilde: Annette, Sänger Center for små Handicap (CSH) Side 7
Diagnosen stilles bl.a. ved hjælp af blodprøver, muskelbiopsier og genetiske analyser. Ved undersøgel-
ser af muskelbiopsier i mikroskop vil man se, at muskelfibrene i forbindelse med specielle prøver ofte
farves røde (Ragged Red Fibres). Der findes ingen helbredende behandling, kun symptombehandling.
Syndromet skyldes at noget af mtDNA' et mangler (mtDNA-deletion). Syndromet opstår sporadisk hos
både drenge og piger. Der ses altså ikke nogle arvelig mønstre for syndromet.
Kompleks-defekterne
Ud over de ovennævnte sygdomme/syndromer, findes der en række defekter, hvor man udelukkende
kan konstatere, at et eller flere af de fem komplekser i mitokondriernes respirationskæde ikke fungerer,
som de skal (se afsnittet "Biologien bag" side 8).
Symptomerne for en kompleks-defekt er mange af de samme som for de specifikke syndromer (fx
CPEO, Kearns-Sayre syndrom, Leighs syndrom, LHON, MELAS, MERRF og Pearsons syndrom).
Diagnosen stilles primært på baggrund af biokemiske analyser. Men på grund af fremskridtene inden for
genteknologien, vil flere med tiden få en diagnose på baggrund af DNA-undersøgelser.
Man kan ikke give noget entydigt symptom-billede på hvert enkelt af kompleks-defekterne. Og nogle
personer har også defekter i flere end et kompleks. Men overordnet gælder det, at symptomerne for
kompleksdefekterne primært falder inden for nedenstående grupper:
Fatal infantile multisystem disorder, der viser sig ved udviklingshæmning,
muskelsvaghed, hjertesygdom, medfødt højt mælkesyreindhold
i blodet og vejrtrækningsproblemer
Fatal infantil encefalomyopati, der viser sig ved medfødt forhøjet mælkesyre i blodet, nedsat mu-
skelspænding (hypotoni), dårlig ernæringstilstand, slagtilfælde og koma.
Muskelsvækkelse og -smerter (myopati) startende i barndommen eller i voksenalderen. Det viser
sig især ved fysiske udfoldelser eller muskelsvaghed generelt og bliver efterhånden til kronisk mu-
skelsvaghed.
Eventuelt også forhøjet mælkesyreindhold i blodet.
Muskel- og hjernesvækkelse (encefalopati og myopati), der starter i barndommen eller senere i
voksenlivet og viser sig ved en kombination af generel muskelsvaghed, lille højde, manglende ko-
ordinering af bevægelserne (ataksi), demens, nedsat hørelse, føleforstyrrelser (neuropati), pig-
mentforandringer i nethinden og tegn på beskadigelse af nerveceller (pyramideceller) i rygmarven
i form af bl.a. spastisk lammelse og refleksforandringer.
Dårlig trivsel, forsinket udvikling, nedsat muskelspænding (hypotoni), "sovesyge" (letargi), vejr-
trækningsproblemer, manglende koordinering af bevægelser (ataksi) og kramper (myokloni). Des-
uden forhøjet mælkesyreindhold i blodet. Kan føre til Leighs syndrom (se s. 4).
Svækkelse af hjertemusklen begyndende i barndommen (kardiomyopati).
Kompleks I-defekt kaldes også for NADH-dehydrogenase (NADH CoQ red uctase ) -mangel.
Kompleks II-defekt kaldes også for Succinat-dehydrogenase-mangel. Kompleks III-defekt kaldes også for Ubiquinon-cytochrom c-oxireductase mangel.
Kompleks IV-defekt kaldes også for COX-mangel eller Cytochrom c-oxidasemangel.
Kompleks V-defekt kaldes også for ATP-syntase-mangel.

Mitokondrie-sygdomme Center for Små Handicapgrupper
Kilde: Annette, Sänger Center for små Handicap (CSH) Side 8
Biologien bag Hvad er mitokondrier?
Mitokondrierne er cellernes små kraftværker, der producerer energi til cellerne og dermed til det meste
af kroppen. Der kan være op til flere tusind mitokondrier i hver celle. Når cellen skal bruge energi, ned-
brydes ATP, og der frigives energi. Vi har brug for energi til stort set alt, hvad vi foretager os: når vi
skal bevæge os, tænke, vokse osv.
Det har efterhånden længe været kendt, at vores krops funktioner styres af vores arveanlæg - også kaldet
vores DNA eller gener. DNA'et er pakket sammen i cellekernen i form af kromosomer. I 1963 opdagede
man dog, at der også findes DNA inde i mitokondrierne. Det DNA, der findes i mitokondrierne, kaldes
for mitokondrie DNA eller mtDNA.
A B C
Figur 1: Illustration af en celle med DNA i både cellekernen(A) og inde i mitokondrieme (B,C).
(Illustration: Pia Wognsen)
I mitokondriernes respirationskæde, som findes i mitochondriernes membran, omdannes nedbrydnings-
produkter til energi. Denne energi kaldes også for ATP. Respirationskæden er en form for 5-trins-proces
- trinene denne proces kaldes kompleks I til kompleks V. Og det er fejl i en eller flere af disse komplek-
ser, der lægger navn til kompleks-defekterne.
Kompleks I består af 42 enheder: syv enheder kodes af gener i mitokondriet.
Kompleks II består af 4 enheder: ingen enheder kodes af generne i mitokondriet.
Kompleks III består af 11 enheder: en enhed kodes af et gen i mitokondriet.
Kompleks IV består af 13 enheder: tre enheder kodes af gener i mitokondriet.
Kompleks V består af 14 enheder: to enheder kodes af gener i mitokondriet.
MtDNA koder kun for mitokondriernes egen funktion, heriblandt for strukturen af 13 af de mere end 80
enheder (proteiner), som skal til for at danne de fem komplekser i mitokondriernes respirationskæde. De
resterende ca. 70 enheder (proteiner)
cellekernen. i mitokondriernes respirationskæde kodes af DNA'et i cellekernen.
Mitokondrie-sygdomme Center for Små Handicapgrupper
Kilde: Annette, Sänger Center for små Handicap (CSH) Side 9
Hvad bestemmer, hvordan syndromerne viser sig? I modsætning til den DNA, som findes i celle-kernen i et eksemplar, er der flere mitokondrier og derfor
også flere kopier af mitokondrie-DNA' et i hver celle - se figur 1. Hos personer med et mitokondrielt
syndrom vil nogle af mitokondrierne være med muteret mtDNA og nogle af dem være med normalt
mtDNA.
Samme syndrom kan godt komme meget forskelligt til udtryk - selv når der er tale om personer inden
for samme familie. Sammenhængen mellem, hvordan fejlen i generne ser ud (genotypen), og hvordan
den kommer til udtryk hos personen (fænotypen), er altså ikke entydig. For eksempel er det af betyd-
ning, hvor meget muteret mtDNA, der er i cellerne i forhold til normalt mtDNA. Generelt gælder det, at
jo mere muteret mtDNA der er, jo tydeligere kommer syndromet til udtryk.
Bestemte mutationer i mtDNA'et er som udgangspunkt relateret til bestemte syndromer, men samtidig
kan samme mutationer godt give forskellige syndromer - og forskellige mutationer kan også give samme
syndrom. Det er der ikke nogen entydig forklaring på! Men det viser, at der er andre faktorer end gener-
ne, der har indflydelse på, hvordan synd romerne kommer til udtryk.
Arvegang
Langt de fleste personer med et mitokondrielt syndrom har fået det spontant/ sporadisk, og sandsynlig-
heden for at give det videre til næste generation er disse tilfælde næsten lig med nul. Men i nogle få til-
fælde kan man dog fastslå, at syndromet har vist sig i flere generationer i en familie, og der derfor er tale
om en arvelig form. I de tilfælde, hvor en mtDNA sygdom går i arv, er det ofte punktmutationer, der er
skyld i syndromet. De andre former for mutationer er sjældent arvelige.
I de få tilfælde hvor man kan tale om arvelighed, kan man ikke angive nogen entydig arvegang for kom-
pleks syndromerne. Det afhænger bl.a. af, hvor gen-fejlen befinder sig. Hvis fejlen ligger i en af de en-
heder, der kodes af mitokondriernes egne gener, kan syndromet føres videre til næste generation via en
maternel arvegang.
Er fejlen placeret i en af generne i cellekernen, er arvegangen oftest recessiv.
Matemel arvegang
Ved mutationer i det mitokondrielle DNA er det kun kvinderne i en familie, som kan føre mutationen
videre gennem generationerne. Dette kaldes maternel arvegang. DNA' et i mitokondrierne føres nemlig
videre til næste generation via moderens ægcelle. Faderens (sædcellens) mitokondrielle DNA føres slet
ikke med over i den befrugtede ægcelle. Derfor vil det også være moderen, der videregiver mutationen
til alle sine børn - drenge såvel som piger. Men det er kun hendes døtre, som via ægcellerne kan give
mutationen videre til næste generation igen.
Recessiv arvegang
Ved en recessivarvegang skal begge forældre have arveanlægget før barnet har risiko for at få sygdom-
men (begge forældre er raske anlægsbærere). Ved hver fødsel vil der være 50 % sandsynlighed for, at
barnet kun får anlægget fra en af forældrene og dermed også bliver rask bærer af arveanlægget. Der vil
være 25 % sandsynlighed for, at barnet får anlægget fra begge forældre og dermed får sygdommen. Med
25 % sandsynlighed vil barnet hverken få anlægget fra faderen eller moderen.
Mitokondrie-sygdomme Center for Små Handicapgrupper
Kilde: Annette, Sänger Center for små Handicap (CSH) Side 10
Et liv med en mitokondrie-sygdom Der er meget stor variation i, hvordan mitokondrie-sygdomme kommer til udtryk. Derfor er det også
svært at sige noget generelt om den indflydelse, syndromet vil have på personen og de pårørende. Da
spektret af symptomer. er så utrolig bredt og behandlingsforløbene meget individuelle, er det vigtigt at
involvere specialister fra det øjeblik, de første symptomer viser sig, diagnosen skal stilles og behandlin-
gen igangsættes. Det er ligeledes vigtigt med kontakt mellem og koordinering af de sociale, pædagogi-
ske og sundhedsfaglige instanser, for at den enkelte kan få bedst mulig støtte. Nogle personer vil opleve
meget lange udredningsforløb.
Symptomerne Nogle organer er mere afhængige af energien fra mitokondrierne end andre. Det drejer sig om hjertet, de
øvrige muskler og hjernen og dermed centralnervesystemet, øjnene og det indre øre. Derfor vil mito-
kondrie-sygdommene også primært ramme disse organer. Men også andre organer, som fx lever, bug-
spytkirtel og tarme, kan være involveret.
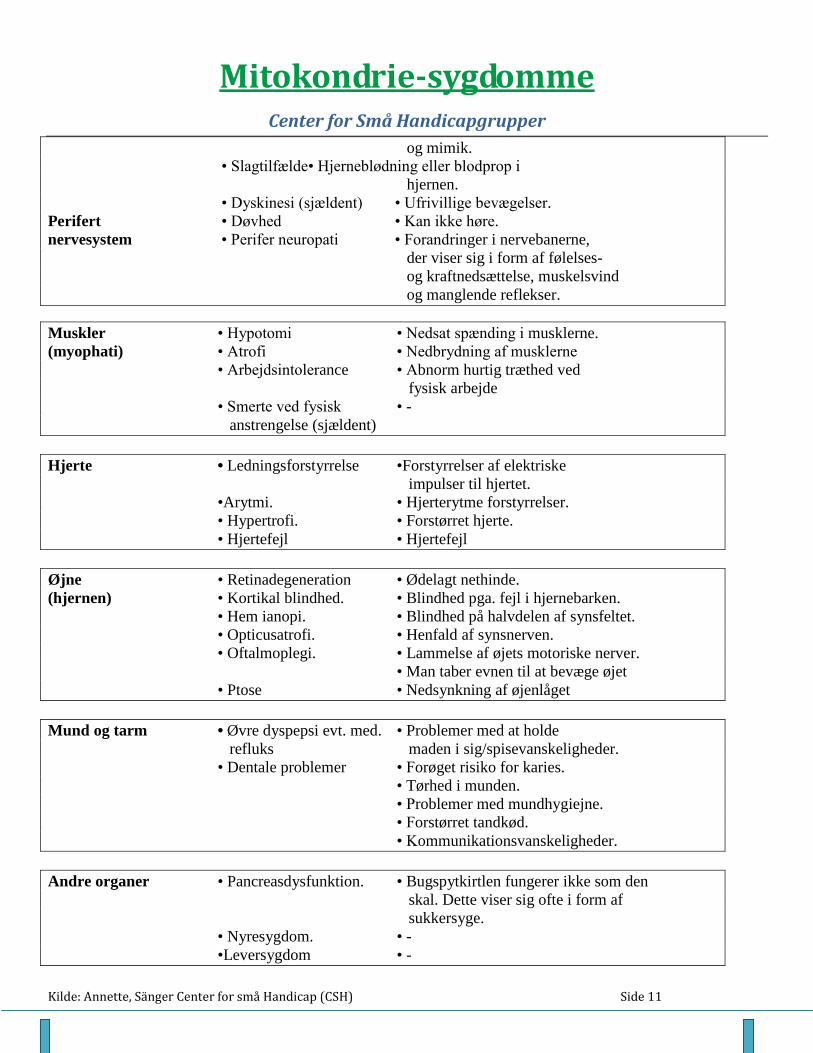
De symptomer, der listes i tabellen herefter, er de fleste af de kliniske symptomer, man er stødt på i hele
gruppen af mitokondrie-sygdomme. En person med en mitokondrie-sygdom, har oftest kun en del af
disse symptomer, men symptomerne kan skifte over tid hos den enkelte.
Tabel 1: Kliniske symptomer ved fejl i mitokondrierne. Nogle symptomer er synlige allerede fra føds-
len, andre kan opstå - afhængig af hvilket syndrom der er tale om.
ORGAN MEDICINSK FORKLARING
BETEGNELSE
Generelt •”Floppy infant” • Nyfødt barn med meget slappe
muskler og stor bevægelighed
i leddene.
• Migræne • Anfald af kraftig hovedpine.
• Vækstretardering • Barn er lille fra fødslen og vokser
ikke som det skal.
• Opkastning • Barnet kan ikke holde maden i sig.
• Hypertermi • Usædvanlig høj legemstemperatur.
• Polydipsi • Stor tørst.
• Dårlig trivsel • -
Centrale • Mental retarderet • Udviklingshæmmet.
nervesystem • Demens • Svækket intelligens.
(hjernen, • Kramper og myokloni • Forskellige former for kramper.
encefalopati) • Ataksi • Manglende koordination af sine
bevægelser – rammer fx ved
siden af tilsigtet mål.
• Spasticitet • Man har spastiske bevægelser.
• Stupor • Ekstrem sløv, uden bevægelse
Mitokondrie-sygdomme Center for Små Handicapgrupper
Kilde: Annette, Sänger Center for små Handicap (CSH) Side 11
og mimik.
• Slagtilfælde• Hjerneblødning eller blodprop i
hjernen.
• Dyskinesi (sjældent) • Ufrivillige bevægelser.
Perifert • Døvhed • Kan ikke høre.
nervesystem • Perifer neuropati • Forandringer i nervebanerne,
der viser sig i form af følelses-
og kraftnedsættelse, muskelsvind
og manglende reflekser.
Muskler • Hypotomi • Nedsat spænding i musklerne.
(myophati) • Atrofi • Nedbrydning af musklerne
• Arbejdsintolerance • Abnorm hurtig træthed ved
fysisk arbejde
• Smerte ved fysisk • -
anstrengelse (sjældent)
Hjerte • Ledningsforstyrrelse •Forstyrrelser af elektriske
impulser til hjertet.
•Arytmi. • Hjerterytme forstyrrelser.
• Hypertrofi. • Forstørret hjerte.
• Hjertefejl • Hjertefejl
Øjne • Retinadegeneration • Ødelagt nethinde.
(hjernen) • Kortikal blindhed. • Blindhed pga. fejl i hjernebarken.
• Hem ianopi. • Blindhed på halvdelen af synsfeltet.
• Opticusatrofi. • Henfald af synsnerven.
• Oftalmoplegi. • Lammelse af øjets motoriske nerver.
• Man taber evnen til at bevæge øjet
• Ptose • Nedsynkning af øjenlåget
Mund og tarm • Øvre dyspepsi evt. med. • Problemer med at holde
refluks maden i sig/spisevanskeligheder.
• Dentale problemer • Forøget risiko for karies.
• Tørhed i munden.
• Problemer med mundhygiejne.
• Forstørret tandkød.
• Kommunikationsvanskeligheder.
Andre organer • Pancreasdysfunktion. • Bugspytkirtlen fungerer ikke som den
skal. Dette viser sig ofte i form af
sukkersyge.
• Nyresygdom. • -
•Leversygdom • -

Mitokondrie-sygdomme Center for Små Handicapgrupper
Kilde: Annette, Sänger Center for små Handicap (CSH) Side 12
Tabellen er udarbejdet med hjælp fra LF. Kennedy instituttet (OK) og Ågrenska (SE) (se adresse- og litteraturliste).
Diagnosen Nogle mitokondrie-sygdomme er så karakteristiske, at det ikke skulle være et problem at stille en diag-
nose (se afsnittene med beskrivelse af de enkelte syndromer). Men selvom diagnosen kan stilles ud fra
det kliniske billede, er en genetisk udredning alligevel vigtig. Dermed kan man fastslå, om det skulle
dreje sig om en arvelig form eller en spontant opstået form, og om genetisk rådgivning er relevant.
Når man mistænker, at en person har en mitokondriel DNA defekt, påbegyndes en række undersøgelser,
der skal hjælpe med at stille den endelige diagnose. Det er vigtigt, at man ikke stopper rækken af under-
søgelser, selvom de første undersøgelser måske ikke viser nogle tegn på et mitokondrielt syndrom. Der
er så mange undtagelser og individuelle udtryk inden for
gruppen, at diagnosen måske udelukkende skal sættes på baggrund af en enkelt undersøgelse.
Det er vigtigt at være opmærksom på, hvorfra på kroppen man tager sit undersøgelsesmateriale, da mu-
tationerne ikke med sikkerhed rammer alt væv. Muskel-væv vil næsten altid være ramt i forbindelse
med mitokondriesygdomme, og det er også tilgængeligt. I sjældne tilfælde kan det komme på tale at
tage væv fra leveren, men det er ikke normen.
Udredningen kan med fordel foretages efter følgende model:
1. Kliniske undersøgelser:
Undersøgelse af hjerte og lunger med stetoskop.
Undersøgelse af blodet for forskellige stoffer: kreatinkinase, laktat og glucose (mælkesyre og suk-
ker).
Evt. undersøgelse af urinen for forskellige stoffer som fx organiske syrer og aminosyrer
Undersøgelse af rygmarvsvæsken (spinalvæsken) for protein, mælkesyre mv.
Ultralydsundersøgelse af hjertet (Ekkokardiografi)
Undersøgelse af elektriske afladninger i hjernen (EEG), hjerte (EKG) og evt. nerver (EMG)
Hjernescanning (CT /MRI)
Ud fra disse kliniske undersøgelser, kan man muligvis fastlægge om der er en velkendt mitokon-
drie-sygdom.
2. Hvis den første kliniske undersøgelse fører til mistanke om et specifikt syndrom, foretager man en
DNA undersøgelse ved hjælp af en blodprøve for at konstatere, om der nu også er den mutation i
DNA' et, som man normalt forbinder med det mistænkte syndrom. Hvis det ikke er muligt at finde
frem til en velkendt mutation, går man videre med yderligere undersøgelser.
3. Man tager en muskelbiopsi til forskellige undersøgelser i mikroskop. Man laver desuden biokemi-
ske analyser, for at se om man kan lokalisere fejlen til en af respirationskædens 5 komplekser.
Samtidig foretager man arbejdstests på de personer, der er gamle nok til at udføre testene.
4. Hvis ovennævnte undersøgelser tyder på en mitokondrie-sygdom, uden man har kunnet stille en
egentlig diagnose, kan man undersøge for mere sjældne mutationer, som ikke er undersøgt under
punkt 2.

Mitokondrie-sygdomme Center for Små Handicapgrupper
Kilde: Annette, Sänger Center for små Handicap (CSH) Side 13
Behandling og forebyggelse
Det er endnu ingen effektiv og helbredende behandling af mitokondriesygdomme.
Kosttilskud af forskellige vitaminer og mineraler kan muligvis afhjælpe nogle symptomer. Men der er
endnu ingen undersøgelser, der entydigt beviser, at disse kosttilskud har nogen effekt. Pointen med de
forskellige kosttilskud er at forsøge at øge ATP produktionen, at sænke mælkesyreindholdet og forbedre
stofskiftet. Kennedy Instituttet og neurologisk afdeling på Rigshospitalet (se adresserne bagest i beskri-
velsen) kan rådgive herom.
Eventuelle bivirkninger behandles individuelt. Fx kan nedsynkning af øjenlåget (ptose) afhjælpes ved
operation og ligeledes skal eventuel epilepsi eller sukkersyge (diabetes) selvfølgelig behandles.
Motion i moderate mængder har vist sig at være godt for personer med en mitokondrie-sygdom. Energi-
omsætningen i musklerne forbedres og musklernes styrke øges. Af samme årsager kan fysioterapeutisk
behandling som fx bassintræning/ svømning og ridning, være af stor gavn.
Infektioner tærer på energi-ressourcerne. Personer med en mitokondrie-sygdom kan opleve, at fx mu-
skelstyrke, sprog og udviklingen generelt bliver sat tilbage efter en infektion. I nogle tilfælde i en sådan
grad, at tabet kan være svært at indhente igen. Derfor er det vigtigt så vidt muligt at udgå infektioner.
Det kan være nyttigt bl.a. at inddrage talepædagog og fysioterapeut i
behandlingen.
Hvis der i spædbarnsalderen er problemer for barnet med at holde maden i sig, pga. opkastninger / dårlig
tarmfunktion, er det vigtigt at sørge for tilstrækkeligt med næringsrig kost til barnet. Dermed øges sand-
synligheden for at barnet trods alt får noget næring med den sparsomme kost, der indtages. En diætist
kan være en god hjælp. Der er ikke noget almindelig
mad, som er forbudt at spise, hvis man har en mitokondrie-sygdom.
Genetisk rådgivning
Ved syndromer med mutationer i mtDNA er det meget svært at foretage fosterdiagnostik. Der er heller
ikke umiddelbart udsigt til behandlingsmuligheder ved hjælp af den traditionelle genterapi.
Syndromer, der skyldes mutationer i det mitokondrielle DNA, kan som tidligere nævnt i denne beskri-
velse gå i arv via kvinderne i familien. Men det er ikke alle kvinder, der automatisk vil give sygdommen
videre til deres børn. Det afhænger af forskellige faktorer, som fx om der er tidligere tilfælde i familien
osv. Derfor kan kvinder med en mitokondrie-sygdom med fordel få genetisk rådgivning før planlægning
af eventuelle børn.
I skrivende stund omtales der en ny metode, hvor det er muligt at sortere de af kvindens ægcelle r fra,
der indeholder muteret mtDNA. Derefter kan de "raske" ægceller befrugtes og sættes tilbage i livmode-
ren. Alternativt kan ægdonation blive aktuelt for de kvinder, der ønsker at få børn, og som kommer fra
familier, hvor der er konstateret en arvelig form for mitokondrie-sygdom.

Mitokondrie-sygdomme Center for Små Handicapgrupper
Kilde: Annette, Sänger Center for små Handicap (CSH) Side 14
Muligheder for støtte ifølge den sociale lovgivning
Når der bliver behov for støtte rettes henvendelse til kommunens socialforvaltning. Det er vigtigt at der
fra begyndelsen træffes bestemmelse om, hvem der har ansvaret for koordinationen mellem de forskelli-
ge instanser. Kommune og Amtskommune har pligt til at yde gratis rådgivning til personer med nedsat
funktionsevne.
For mitokondrielle syndromer er der meget store forskelle i, hvordan sygdommen kommer til udtryk.
Deraf følger, at der vil være meget forskelligartede behov for støtte efter den sociale lovgivning.
Børn og unge
Bestemmelserne i serviceloven gælder for børn og unge med varigt nedsat fysisk/psykisk funktionsevne,
som forsøges i hjemmet. Der kan f.eks. være tale om:
1. Tabt arbejdsfortjeneste efter servicelovens § 29: Hvis det skønnes nødvendigt, at en af forældrene
passer barnet i hjemmet, kan der ydes kompensation for tabt arbejdsfortjeneste for kortere eller
længere tid, på deltid eller fuld tid. Det er vigtigt at være opmærksom på de problemstillinger, der
er i forholdet mellem A-kassesystemet og servicelovens § 29. Der kan ligeledes bevilges tabt ar-
bejdsfortjeneste, hvis barnet skal indlægges på hospital på grund af en eller flere af de alvorlige
symptomer, som syndromet kan medføre, hvis en eller begge forældre skal være til stede under
indlæggelsen.
2. Merudgifter efter servicelovens § 28: Merudgifter i forbindelse med barnets handicap, f.eks. til
kost og diætpræparater, medicin, befordring, særlig beklædning, handicap rettede kurser til foræl-
dre og andre pårørende.
Det er vigtigt, at barnets eller den unges udvikling følges nøje, og at der sættes tidligt ind med den nød-
vendige støtte, afhængig af, hvordan syndromet kommer til udtryk. Der kan f.eks. være tale om støtte i
skolen i forbindelse med nedsat syn eller hørelse. Men også i form af afløsning i hjemmet eller aflast-
ningsophold uden for hjemmet.
I nogle tilfælde vil barnet eller den unge få brug for hjælpemidler. Spørgsmålet kan rejses under syge-
husindlæggelse, fra kommunen eller fra forældrene, når behovet opstår. Hjælpemidler bevilges af kom-
mune eller amtskommune. Når der er tale om børn er det vigtigt at der lægges vægt på at barnet i videst
muligt omfang skal kunne fungere som andre børn.
Her er kun nævnt nogle eksempler på støttemuligheder. Er der yderligere spørgsmål, kan man kontakte
CSHs rådgivningsafdeling og til artiklen Støttemuligheder ifølge lovgivningen til børn og unge med
handicap. Se evt. også artiklen om Pligter og rettigheder for borgere og myndigheder i "Information om
Sjældne Handicap" eller på vores hjemmeside www.csh.dk.

Mitokondrie-sygdomme Center for Små Handicapgrupper
Kilde: Annette, Sänger Center for små Handicap (CSH) Side 15
Voksne
Bestemmelserne i serviceloven gælder for voksne med betydeligt og varigt nedsat fysisk/psykisk funkti-
onsevne, og formålet er at forebygge, at problemerne for den enkelte forværres og at forbedre den enkel-
tes sociale og personlige funktion og udviklingsmuligheder.
Bestemmelserne for voksne dækker støtte til f.eks. dækning af nødvendige merudgifter, personlig hjælp
og pleje, revalidering, botilbud og hjælpemidler.
Der kan blive behov for vejledning ved valg af uddannelse og erhverv under hensyn til de funktionsned-
sættelser, der kan følge med syndromet. Der kan yderligere blive tale om revalidering, som består i støt-
te til en person, hvis erhvervsevne er begrænset af fysiske, psykiske eller sociale årsager. Der kan være
tale om omskoling, uddannelse eller iværksættelse af selvstændig virksomhed. Målet er, at personen
opnår beskæftigelse på arbejdsmarkedet og dermed bliver i stand til at forsørge sig selv. Se artiklen om
Revalidering i "Information om Sjældne Handicap" eller på vores hjemmeside www.csh.dk.
Hvis erhvervsevnen er væsentlig nedsat kan der igangsættes ansøgning om førtidspension, men sagen
kan først påbegyndes når alle aktiverings-, revaliderings-, og behandlingsmæssige foranstaltninger er
forsøgt. Ansøgningen om førtidspension behandles og afgøres af kommunen.
Er der yderligere spørgsmål henvises til CSHs rådgivningsafdeling og til artiklen Støttemuligheder iføl-
ge lovgivningen for voksne med handicap. Se evt. også artiklen Pligter og rettigheder for borger og
myndigheder. Begge artikler findes Revalidering i "Information om Sjældne Handicap" eller på vores
hjemmeside www.csh.dk.
Mere viden
Kontaktordning og patientforening
Center for Små Handicapgruppers kontaktordning er et tilbud til mennesker med sjældne sygdomme og
handicap, som ikke har en forening eller andet netværk i Danmark at henvende sig til. Via kontaktord-
ningen tilbyder vi at formidle kontakt mellem personer eller familier, der lever med samme sjældne
handicap. Der findes kontaktperson(er) for nogle mitokondrie-sygdomme.
Du kan læse mere om kontaktordningen på vores hjemmeside www.csh.dk eller du kan kontakte os på
tlf. 86 76 30 22.
Der er også taget initiativ til at starte en patientforening. Og hvis man vil høre nærmere om hvordan det
arbejde går, kan man kontakte:
Mitokondrie-foreningen i Danmark
Ulla Isbye
Ved Bellahøj Syd 28 B, 1. th.
2700 Brønshøj
Tlf.: 47 36 19 12 e-mail: [email protected] eller: [email protected]

Mitokondrie-sygdomme Center for Små Handicapgrupper
Kilde: Annette, Sänger Center for små Handicap (CSH) Side 16
Adresser: Rigshospitalet
Blegdamsvej 9, 2100 København ø
Tlf.: 35 45 35 45
John Vissing (professor)
Flemming Skovby (professor, dr. med)
Statens Øjenklinik
Rymarksvej 1
2900 Hellerup
Tlf.: 39 45 24 00
Thomas Rosenberg (overlæge)
John F. Kennedy Instituttet
Gammel Landevej 7
2600 Glostrup
Tlf.: 43 26 01 00
Flemming Wibrand (biokemiker)
Elsebet Østergaard (læge)
Nina Horn (biokemiker / afd. leder)
(Tak til ovennævnte, der har alle bidraget til denne beskrivelse).
Internetadresser: Vær opmærksom på, at der kan bruges mange forskellige stavemåder, når man søger informationer på
nettet. En forkert stavning kan betyde, at man ikke finder noget som helst, eller udelukkende finder ma-
teriale på det sprog, som svarer til den stavemåde man har benyttet. Prøv derfor med flere forskellige
stavemåder (sprog) for at være sikker på at have fundet det du
søger.
http://www.sos.se/smkh
Den svenske socialstyrelses hjemmeside med beskrivelser af små og mindre kendte handicap.
www.umdf.org
United Mitokondrial Disease Foundation. Stedet indeholder flere links til engelsk/ amerikansk informa-
tion.
CPEO
www.orpha.net
For en generel information om CPEO, se den franske database Orphanet. Klik til venstre på 'Services to
health professionals', dernæst på 'Disease' øverst til venstre og skriv så " Progressive external ophthal-
moplegia" i søgefeltet.

Mitokondrie-sygdomme Center for Små Handicapgrupper
Kilde: Annette, Sänger Center for små Handicap (CSH) Side 17
Leighs syndrom
www.sos.se/smkh/nr981270.htm
Den svenske socialstyrelses database har en mere uddybende beskrivelse af Leigh’s syndrom.
www.stepstn.com/cgi-win
nord.exe ?proc=GetDocument&rectype=O&recnum=392
Den amerikanske database NORD
www.orpha.net
Den franske database Orphanet. Klik til venstre på 'Services to health professionals', dernæst på 'Disea-
se' øverst til venstre og skriv så "Leigh disease" i søgefeltet
www.ninds.nih.ovhealthandmedicaldisordersleihsdiseasedoc.htm
Den amerikanske hjemmeside National Institute of Neurological Disorders and Stroke
http://biochemgen.ucsd.edu/mmdc/lcc01.htm
Endvidere findes The official Newsletter of the Leigh' s Center for Children at University of California
San Diego
LHON
www.stepstn.com/cgi-win
nord.exe ?proc=GetDocument&rectype=0&recnum=534
Den amerikanske database NORD
www.orpha.net
Den franske database Orphanet. Klik til venstre på 'Services to health professionals', dernæst på 'Disea-
se' øverst til venstre og skriv så " Leber hereditary optic neuropathy" i søgefeltet.
www.leeder.demon.co.uk/pages/lhonhome.htm
Privat hjemmeside.
MELAS
www.sos.se/smkh/nr981271.HTM
Den svenske socialstyrelses database om små og mindre kendte handicapgrupper.
www.orpha.net
Den franske database Orphanet. Klik til venstre på 'Services to health professionals', dernæst på 'Disea-
se' øverst til venstre og skriv så "MELAS syndrome" i søgefeltet.
www.melas.org /mitoindex.htm
På The Jackson Family's Website Mitokondrial-Related Links findes der mange links om Melas syn-
drom og mitokondriesygdomme. Der er bl.a. postlister, chat rooms og kontaktadresser.
MERRF
www.sos.se/smkh/9912105/9912105.HTM
Den svenske Socialstyrelses database om små og mindre kendte handicapgrupper.

Mitokondrie-sygdomme Center for Små Handicapgrupper
Kilde: Annette, Sänger Center for små Handicap (CSH) Side 18
www.diseasedir.org.uk/genetic/mt02.htm
Engelsk Disease Directory med beskrivelse af MERRF syndrom.
www.icondata.com/health/pedbase/files/MYOCLONU.HTM
Den canadiske Pediatric database med generel information om syndromet:
www.stpstne com/cgi-win/nord.exe ?proc=GetDocument&rectype=O&recnum==965
Den amerikanske database NORD
www.orpha.net
Den franske database Orphanet. Sproget på hjemmesiden er engelsk og fransk. Klik til venstre på 'Ser-
vices to health professionals', dernæst på 'Disease' øverst til venstre og skriv så " MERRF syndrome" i
søgefeltet.
Pearson syndrome
www.orpha.net
For en generel information om syndromet, se den franske database Orphanet. Klik til venstre på skærm-
billedet: 'Services to health professionals', dernæst på 'Disease' øverst til venstre og skriv så " Pearson
syndrome" i søgefeltet.
Kompleks-defekterne
www.orpha.net
Den franske database Orphanet, for en generel information om kompleks syndromerne på fransk og en-
gelsk. Klik til venstre på skærmbilledet: 'Services to health professionals', dernæst på 'Disease' øverst til
venstre og skriv så:
Nadh coq reductase deficiencyi søgefeltet for kompleks I syndrom
Succinate coenzyme q reductase deficiency i søgefeltet for kompleks II syndrom
Coenzyme q cytochrome i søgefeltet for kompleks III syndrom
Cytochrome c oxidase deficiency i søgefeltet for kompleks IV syndrom
ATP synthetase deficiency i søgefeltet for kompleks V syndrom
Litteratur
"Mitokondrial Respiratory Chain Disorders 1: Mitokondrial DNA Defects", af J V Leonard, A H V
Schapira. The Lancet 2000; 355:299-304.
"Mitokondrial Respiratory Chain Disorders 2: Neurodegenerative Disorders and Nuclear Gene De-
fects", af J V Leonard, A H V Schapira. The Lancet 2000; 355:299-304.
"Clinicai Mitokondrial Genetics", af Chinnery et al. J. Med Genet 1999; 36:425-436.
"Does the Patient Have a Mitokondrial Encephalomyopathy", af DiMauro et. Al. Journal of Child Neu-
rology, supplement 1999; 14:s23-s35.
"Mitokondriella sjukdomar" Nr. 163. Udgivet 1999 af Ågrenska, Box 2058, 436 02 Hovås, Sverige.
www.agrenska.org

Mitokondrie-sygdomme Center for Små Handicapgrupper
Kilde: Annette, Sänger Center for små Handicap (CSH) Side 19
"Mitokondrial Myopathies and Encephalomyopathies" af A H VSchapira, H R Cock. European Journ-
mal af Clinical Investigation (1999) 29, 886-898.
Vissing J. "Mitochondrial Myopathies". In "Skeletal Muscle: Pathology, Diagnosis and management in
Disease", Preedy VR, Peters TJ (eds). London: Greenwich Medical Media Ltd, in press.
"Mendelianlnheritancein Man -A Catalog af Human Genes and Genetic Disorders" af McKusick VA.
Et al.The John Hopkins University Press (1998), 12th ed.
"Medicinsk Kompendium" red.: Lorenzen I, Bendixen G, Hansen Nordisk Forlag Arnold Busck A/S
(1999) 15. udg.
"Harrison's Principles of lnternal Medicine" af Isselbacher et al. Congress Cataloging-in-Publication
Data, (1994) 13th ed.

Mitokondrie-sygdomme Center for Små Handicapgrupper
Kilde: Annette, Sänger Center for små Handicap (CSH) Side 20
CenterforSmå Handicapgrupper
Dokumentation og formidling: Bredgade 25, opg. F, 5. sal 1260 København K Tlf. 33 91 40 20
Projekter og rådgivning: Vesterport 3, 3.sal 8000 Århus C Tlf. 86 76 30 22
e-mail: [email protected]. Internetadresse:www.csh.dk





























