Embed Size (px)
Citation preview

4 April 2017
MOBILE APPS & SOFTWARE
TO SUPPORT DRUG DEVICE
COMBINATION PRODUCTS
Current use and future outlook

2
Experience Expertise
Highlights
• 12+ years in the industry of Active
Implantable Medical Device (AIMD)
• 20+ in the field of MD development &
Regulatory Affairs
• High risk, class III medical devices
• Clinical development plan, Clinical Trial
Management & Regulatory processes
• Medical Device vigilance management
& reporting processes
• Neurologic diseases (i.e. pain
management, pelvic health, auditory
deficiency, Parkinsons, etc.)
• Senior Director – Voisin Consulting
Life Sciences
• Manager for clinical operations Europe
- Boston Scientific
• Field clinical specialist - Advanced
Bionics
Senior Director, Medical Devices, Combo Products & SW
• Conducting of global regulatory
strategic analysis for innovative
devices
• Management of international FIM
clinical trials with sophisticated
high risk devices
• Set-up and implementation of
global MD vigilance system for
Europe
• Member of Regulatory Affairs
Professionals Society (RAPS),
TOPRA Medical Devices
Regulatory Affairs, European
Confederation of Pharmaceutical
Entrepreneurs (EUCOPE),
European Association for
Bioindustries (EuropaBio),
Medical Devices Startups,
Neuromodulation Network, PSE
EPFL Medical device
+33 141 318 662
Christophe Amiel, M.Sc. – MD, Combo & Software
Christophe is leading design & implementation of regulatory strategies
for medical devices (including SW), combination (drug, biologic) &
borderline products. His areas of expertise encompass clinical
development, EU/US market approval & MD vigilance.
3
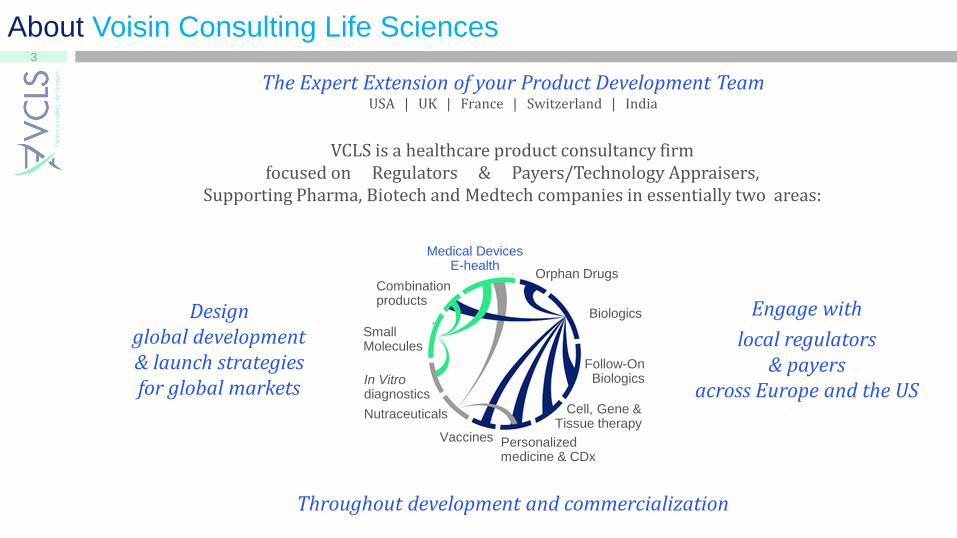
About Voisin Consulting Life Sciences
VCLS is a healthcare product consultancy firm focused on Regulators & Payers/Technology Appraisers,
Supporting Pharma, Biotech and Medtech companies in essentially two areas:
The Expert Extension of your Product Development Team USA | UK | France | Switzerland | India
Design global development & launch strategies for global markets
Engage with
local regulators & payers
across Europe and the US
Small Molecules
Combination products
Medical Devices E-health
In Vitro diagnostics
Nutraceuticals
Vaccines Personalized medicine & CDx
Cell, Gene & Tissue therapy
Follow-On Biologics
Biologics
Orphan Drugs
Throughout development and commercialization

4
1 Role of m-Health in Combination Products
2 EU/US Regulatory Framework for m-Health
3 Case Studies from the Market
L
4 Take Home Messages
Today’s agenda

5
1 Role of m-Health in Combination Products
2 EU/US Regulatory Framework for m-Health
3 Case Studies from the Market
L
4 Take Home Messages
Today’s agenda
6
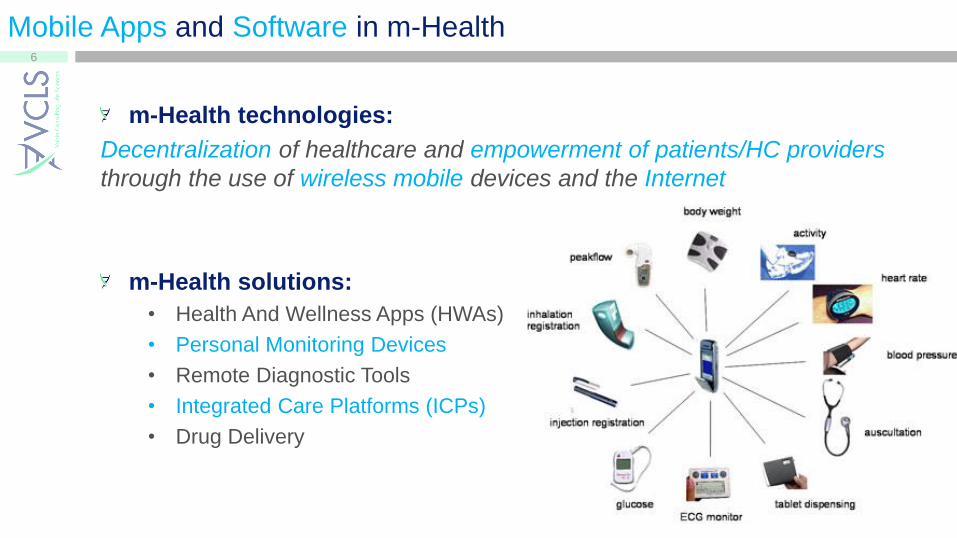
Mobile Apps and Software in m-Health
m-Health technologies:
Decentralization of healthcare and empowerment of patients/HC providers
through the use of wireless mobile devices and the Internet
m-Health solutions:
• Health And Wellness Apps (HWAs)
• Personal Monitoring Devices
• Remote Diagnostic Tools
• Integrated Care Platforms (ICPs)
• Drug Delivery
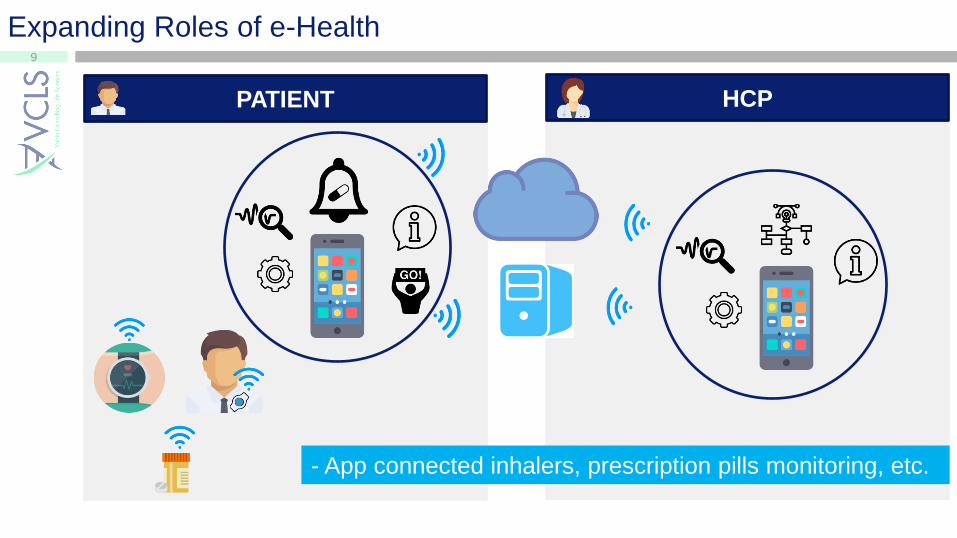
9
Expanding Roles of e-Health
PATIENT HCP
- App connected inhalers, prescription pills monitoring, etc.

10
1 Role of m-Health in Combination Products
2 EU/US Regulatory Framework for m-Health
3 Case Studies for the Market
L
4 Take Home Messages
Today’s agenda

12
m-Health Regulatory Framework
Highly regulated products if medical purpose claimed
Regulation not harmonized internationally
• EUROPE: National legislation discrepancies (Greatly
improved with dedicated MDR provisions)
• USA: Unique regulatory statuses (Medical Device Data
System MDDS, FDA enforcement discretion)

13
m-Health Regulatory Framework - EUROPE
To qualify as Medical Device, m-Health product shall:
• Have a medical purpose
• Perform an action on data (processing, Etc.) other than just storage, for the medical benefit of individual patients
• Generate/manage personalized alerts based on monitored patients vital parameters to drive clinical management (risk scoring, Etc.)
• Use of an algorithm to support/facilitate medical decisions by HCP (hospitalization, Etc.)
CE marking scope based on
claimed functionalities & medical purpose

14
Coming Regulatory changes with MDR (1) - EUROPE
MDR upgraded classification for standalone software
(Annex VII/ Rule 10a):
“Software intended to provide information which is used to take decisions with diagnosis or
therapeutic purposes, is in class IIa, except if such decisions have an impact that may
directly or indirectly cause:
• the death or an irreversible deterioration of the state of health, in which case it is in
class III;
• a serious deterioration of the state of health or a surgical intervention, in which case it is
in class IIb.
Software intended to monitor physiological processes is in class IIa, except if it is intended
for monitoring of vital physiological parameters, where the nature of variations is such that
it could result in immediate danger to the patient, in which case it is in class IIb.
All other software is in class I.”

15
Coming Regulatory changes with MDR (2) - EUROPE
Upclassification Notified body intervention: for CE certification of
class Im, IIa, IIb and III impacting costs and delays (Audits, MDR
redesignation…)
Traceability SW modifications management: notifications to NB
triggering potential additional interventions (change in design approval,
update of Tech File…)
Clinical evaluation Clinical investigation conducting: correlated
with levels of impact on the patient or public health (reinforced clinical
pre/post CE mark data,…)

17
m-Health Regulatory Framework - EUROPE
Various conformity assessment routes:
Risk
Classification
NB
intervention
Conformity
assessment
Clinical Evidence
required
Class I none • Self certification Yes, for safety and
performance claims
Class IIa yes • Certification audits Yes, for safety and
performance claims
Class IIb yes • Certification audits
• Technical file
reviews
Yes, for safety and
performance claims

18
m-Health Regulatory Framework (1) - USA
To qualify as “regulated” Medical Device, the m-Health product shall:
• Meet the definition of a “device” meaning it is intended for use in the
diagnosis of disease or other conditions, or in the cure, mitigation, treatment,
or prevention of disease, in man or other animals
AND
• Be intended to be used as an accessory to a regulated medical device, or
transform a mobile platform into a regulated medical device.
Unique classification regulations applies to certain software products
• Medical Device Data Systems (MDDS) providing electronic transfer, storage,
exchange, retrieval, display and conversion of medical device data Class I MD
(510k exempt, Enforcement Discretion)
19
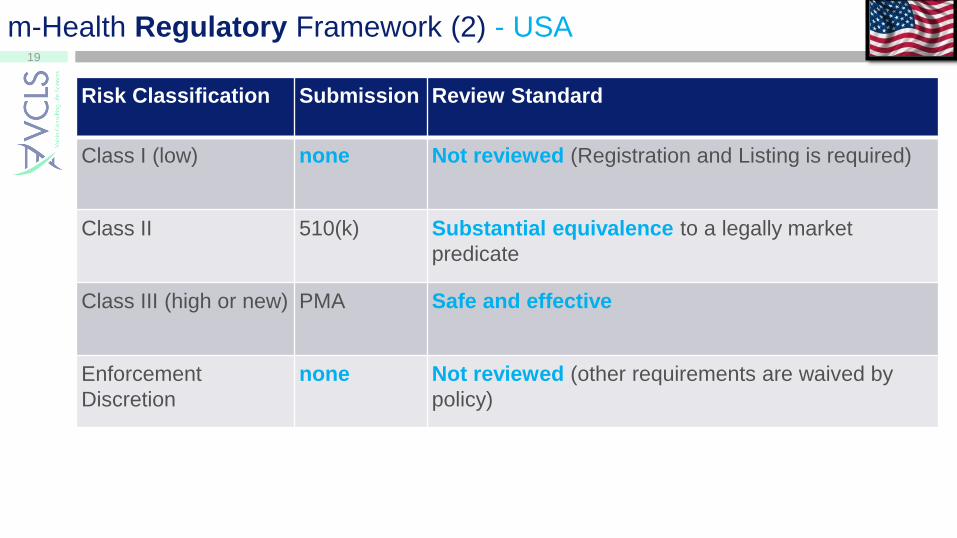
m-Health Regulatory Framework (2) - USA
Risk Classification Submission Review Standard
Class I (low) none Not reviewed (Registration and Listing is required)
Class II 510(k) Substantial equivalence to a legally market
predicate
Class III (high or new) PMA Safe and effective
Enforcement
Discretion
none Not reviewed (other requirements are waived by
policy)
20
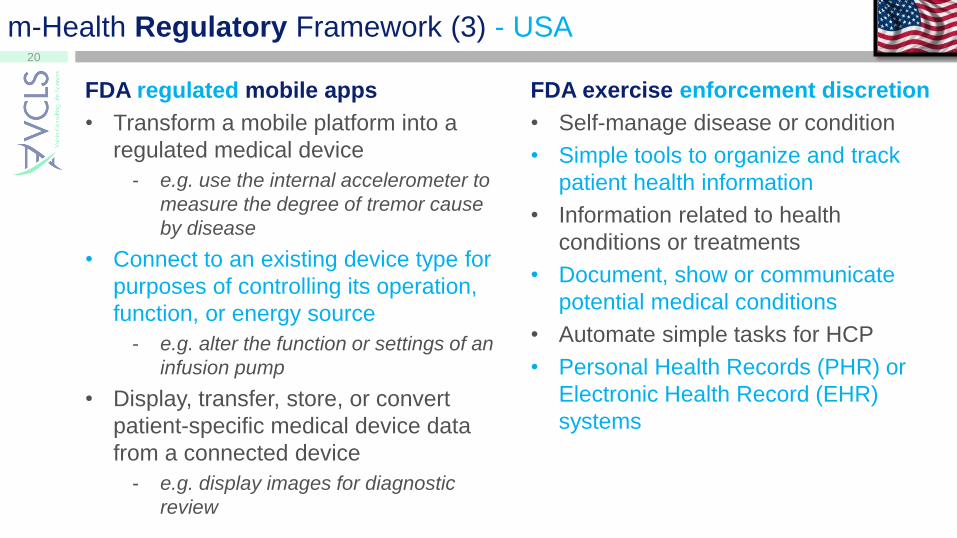
m-Health Regulatory Framework (3) - USA
FDA regulated mobile apps
• Transform a mobile platform into a
regulated medical device
- e.g. use the internal accelerometer to
measure the degree of tremor cause
by disease
• Connect to an existing device type for
purposes of controlling its operation,
function, or energy source
- e.g. alter the function or settings of an
infusion pump
• Display, transfer, store, or convert
patient-specific medical device data
from a connected device
- e.g. display images for diagnostic
review
FDA exercise enforcement discretion
• Self-manage disease or condition
• Simple tools to organize and track
patient health information
• Information related to health
conditions or treatments
• Document, show or communicate
potential medical conditions
• Automate simple tasks for HCP
• Personal Health Records (PHR) or
Electronic Health Record (EHR)
systems

21
m-Health Regulatory Referential (1)
EU environment
• 93/42/EEC Directive of 14 June 1993 as amended concerning Medical
Devices
• MEDDEV 2.1/6 on Qualification & Classification of standalone software
• IMDRF/SaMD/ WG/N10 Dec 2013 Software as a Medical Device: Key Definitions
• IMDRF/SaMD/WG/N12FINAL:2014 Software as a Medical Device": Possible
Framework for Risk Categorization and Corresponding Considerations
• IMDRF/WG/N23 Proposed Document (PD1)R3 - Application of QMS
• NB-MED/2.2/Rec4 recommendation relative to medical software and computers
• Manual on borderline and classification in the Community regulatory
framework

22
m-Health Regulatory Referential (2)
Standards - IEC 62304:2006 - Medical Devices software – Life cycle processes
- IEC 62366:2007 - Medical Devices - Application of engineering
- IEC 14764:2006 - Software Engineering - Software Life Cycle Processes – Maintenance
- IEC 80001-1 - Application of risk management for IT-networks incorporating medical devices
- IEC TR 80002-1 Medical device software – Guidance on the application of ISO 14971 to
medical device software
- IEC 62443 - Industrial communication networks and system security
- IEC 62083, Medical electrical equipment – Requirements for the safety of radiotherapy
treatment planning systems
- ISO/IEC 27000:2009 - Information technology —Information security management systems
- IEC/ISO 10918-1:1994 Digital compression and coding of continuous-tone still images
- NEMA PS 3.1 - 3.20 (2011) Digital Imaging and Communications in Medicine (DICOM) Set
- NEMA XR 22-2006 "Quality Control Manual" Template for Manufacturers of Displays and
Workstations Labeled for Final Interpretation in Full-field Digital Mammography

23
m-Health Regulatory Referential (3)
US FDA
• Guidance on Mobile Medical Applications (2015)
• Medical Device Data Systems, Medical Image Storage Devices, and Medical
Image Communications Devices (2015)
• Content of Premarket Submissions for Management of Cybersecurity in
Medical Devices (2014)
• Content of Pre-market Submission for Software Contained in Medical Devices
(2005) & General principles of Software validation (2002)
• DRAFT: Deciding When to Submit a 510(k) for a Software Change to an Existing
Device - Draft Guidance for Industry and Food and Drug Administration Staff
(2016)
• DRAFT: Software as a Medical Device (SaMD): Clinical Evaluation [IMDRF]
(2016)
• DRAFT: Use of Electronic Health Record Data in Clinical Investigations (2016)

24
m-Health Regulatory Referential (4)
EU Guidelines
• UK - Guideline on Stand alone SW & m-Health products (MHRA, 2016)
• SWEDEN – Guidance for qualification and classification of stand alone software with medical purpose (2013)
• France – Study on Safety of medical devices software (ANSM, 2016)
Health Canada
• Notice - Software Regulated as a Class I or Class II Medical Device
Australian TGA
• Regulation of medical software and mobile medical applications (2013)

25
1 Role of m-Health in Combination Products
2 EU/US Regulatory Framework for m-Health
3 Case Studies from the Market
L
4 Take Home Messages
Today’s agenda

26
Current challenges with m-health development
From the regulator’s perspective: Rapid
development of m-health apps without
concomitant establishment of adapted/
harmonized regulations
From a company’s perspective: Develop easy-
to-use and reliable m-health systems to avoid
serious health consequences for patients

27
Key Development Considerations
• The type and level of evidence is determined by:
o the intended use identify the medical purpose and claims
o the risks posed by the product rigorous proactive risk analysis
• As with any medical product, the key considerations:
o Quality Management System set-up
o Risks mitigation definiton
o Performance establishing (possibly clinical)
Top perceived barriers to mhealth technology adoption from HCP* - Absence of evidence/Trust in reliability of data collected by the devices = 27%
- Training for patients to use new systems/technologies = 24%
*Future Health Index Report 2016 – 2500+ HCP over 13 countries (i.e. Australia, Brazil, China, France, Germany, Japan, the Netherlands,
Singapore, South Africa, Sweden, the United Arab Emirates, the United Kingdom and the United States)

28
Quality Aspects
• Quality Management System
o Ensure rigor in generating evidence towards adequate validation, reliability &
usability of the mhealth product
o Europe ISO 13485 (taking into account SW specifics)
o US Quality System Regulation (21 CFR 820)
• Critical elements at the development and evaluation stages
o design controls (continuous improvement, ability to maintain quality)
o risk management (display media, servers/cloud)
o document/record control (iteration history)
o supplier management (SW coding & ISO62304)
• Post-market policies for managing complaints and safety incidents
o Continuous evaluation paradigm (real world performance)

29
Safety Aspects
• Software verification and validation plan
o Comprehensive & well-structured (as per ISO62304 requirements)
o Verification objective evidence that the design outputs meet specified
requirements as set by developer
o Validation objective evidence that software specifications consistently conform
to user needs and intended uses
• Cybersecurity
o Risks patient AND connected device & networks
o Robust protection mechanisms expected (strong regulatory FDA focus)
• Protection of sensitive patient information
o US HIPPA Privacy Rule

30
Performance Aspects
• Clinical effectiveness and Benefit
• Clinical evaluation method required for ALL medical app
• Level of evidence depends on SW risks when used as defined by the developer
• Intended purpose: Treat/Diagnose, Drive clinical management, Inform clinical
management
• Disease type/Patient condition: Life threatening, often curable, slow in progression, etc.
• Type of clinical evaluation as a function of impact on public health & SW specifics
• Key challenges:
• no gold standards for clinical evaluation of SW
• misalignment with SW development cycle times
• none static product (as with conventional hardware MDs)
• Workarounds:
• Premarket clinical evidence to be complemented by post market data
• Use of real world clinical evidence

31
Performance Aspects
• Case studies
• Online app version calculating a clinical score commonly used by HCP
• Ability of a SW to accurately and reliably generate the intended output, from the input data
(i.e. reproducibility, repeatability, etc.)
• Technical functional performance (ISO62304 requirements) Analytical validity
• Medical app assessing the risk a life-threating complication using novel algorithm
• Establish how well the output of the SW accurately correlates to the intended clinical
health care situation or condition of the intended use of the app
• Show evidence of the ability of the app to yield a clinically meaningful output (i.e.
diagnostic purpose)
• Controlled clinical trial (ISO14155 requirements) Scientific validity & Clinical
performance

32
Example 1: Voluntis – Diabetes management
Source: http://www.voluntis.com/
Mobile Medical
App
33
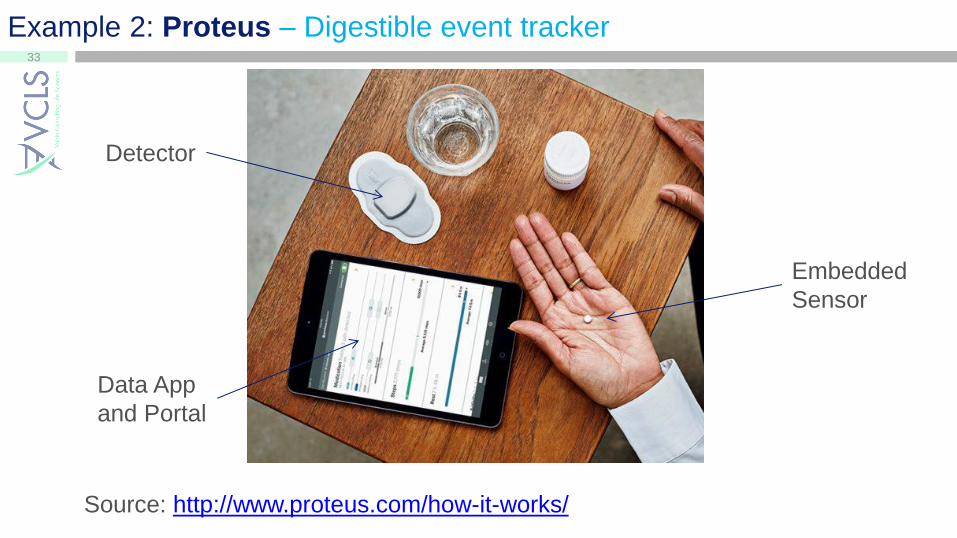
Example 2: Proteus – Digestible event tracker
Embedded
Sensor
Detector
Data App
and Portal
Source: http://www.proteus.com/how-it-works/
34
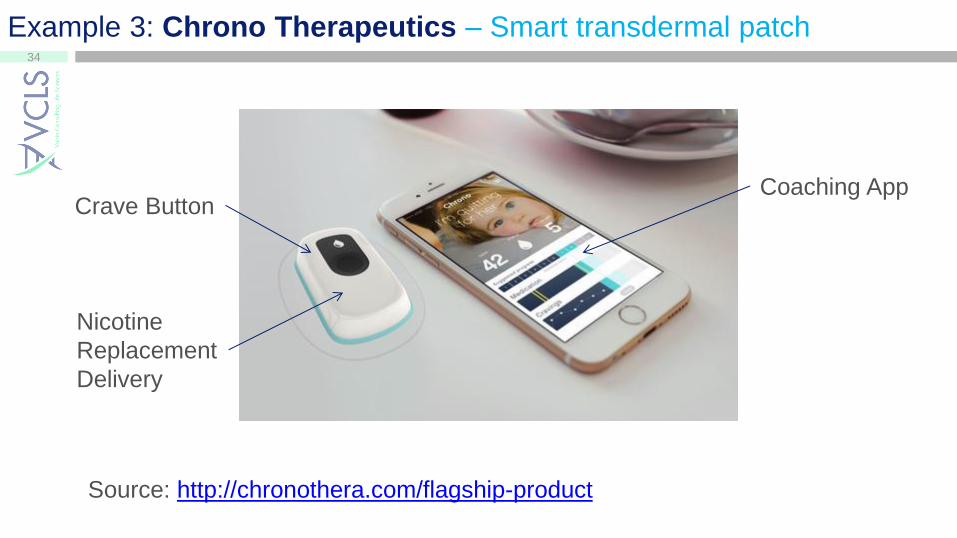
Example 3: Chrono Therapeutics – Smart transdermal patch
Nicotine
Replacement
Delivery
Crave Button Coaching App
Source: http://chronothera.com/flagship-product

37
1 Role of m-Health in Combination Products
2 EU/US Regulatory Framework for m-Health
3 Case Studies from the Market
L
4 Take Home Messages
Today’s agenda

38
m-Health product is NOT just a standard IT technology used for a
medical purpose... but to be regarded as any other MD
- Medical qualification Be clear on your m-Health product functionalities,
targeted indications for use and safety/performance claims
- Product development & Evaluation Follow-up appropriate framework
during the entire product lifecycle (Design, V&V, etc.)
More stringent regulatory landscape ahead for medical mobile apps...
- EUROPE: Reinforced clinical evidence (MDR provisions, IMDRF guideline)
- USA: Revised FDA enforcement discretion rules (More apps in the radar)
Take Home Messages

Voisin Consulting Life Sciences
voisinconsulting.com
linkedin.com/company/voisin-consulting





























