Embed Size (px)
Citation preview

Molecular Modeling and Drug Discovery
Judith Klein-Seetharaman
Assistant ProfessorDepartment of Pharmacology
University of Pittsburgh School of Medicine
andSchool of Computer ScienceCarnegie Mellon University
USA
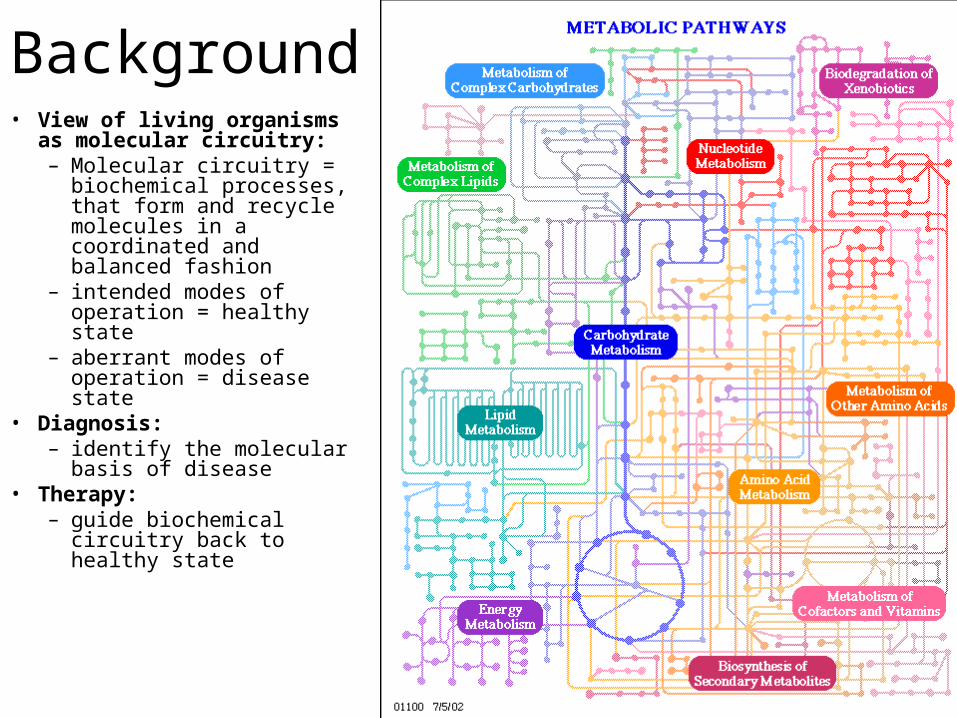
Background• View of living organisms as
molecular circuitry: – Molecular circuitry =
biochemical processes, that form and recycle molecules in a coordinated and balanced fashion
– intended modes of operation = healthy state
– aberrant modes of operation = disease state
• Diagnosis:– identify the molecular
basis of disease• Therapy:
– guide biochemical circuitry back to healthy state
Information Sources
• New technology generates massive amounts of data (often stored in publicly accessible databases): Genomics and Proteomics– Protein and DNA
sequences / Whole genome sequences
– Protein structure data– Protein pathways and
networks– Protein interaction data– Expression data
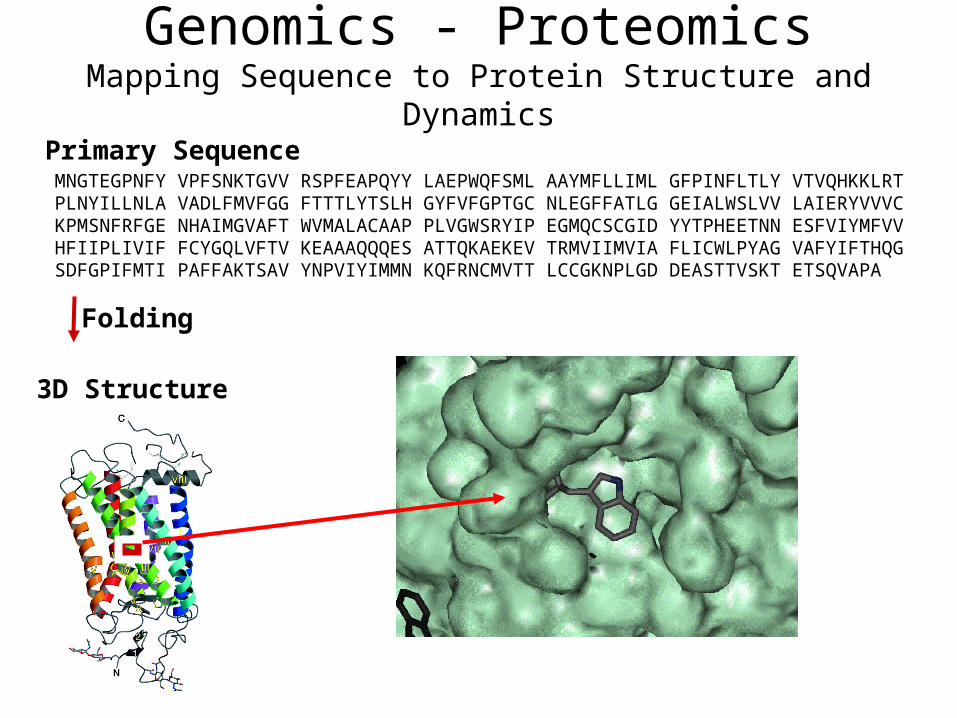
Genomics - ProteomicsMapping Sequence to Protein Structure and Dynamics
Primary SequenceMNGTEGPNFY VPFSNKTGVV RSPFEAPQYY LAEPWQFSML AAYMFLLIML GFPINFLTLY VTVQHKKLRT PLNYILLNLA VADLFMVFGG FTTTLYTSLH GYFVFGPTGC NLEGFFATLG GEIALWSLVV LAIERYVVVC KPMSNFRFGE NHAIMGVAFT WVMALACAAP PLVGWSRYIP EGMQCSCGID YYTPHEETNN ESFVIYMFVV HFIIPLIVIF FCYGQLVFTV KEAAAQQQES ATTQKAEKEV TRMVIIMVIA FLICWLPYAG VAFYIFTHQG SDFGPIFMTI PAFFAKTSAV YNPVIYIMMN KQFRNCMVTT LCCGKNPLGD DEASTTVSKT ETSQVAPA
3D Structure
Folding
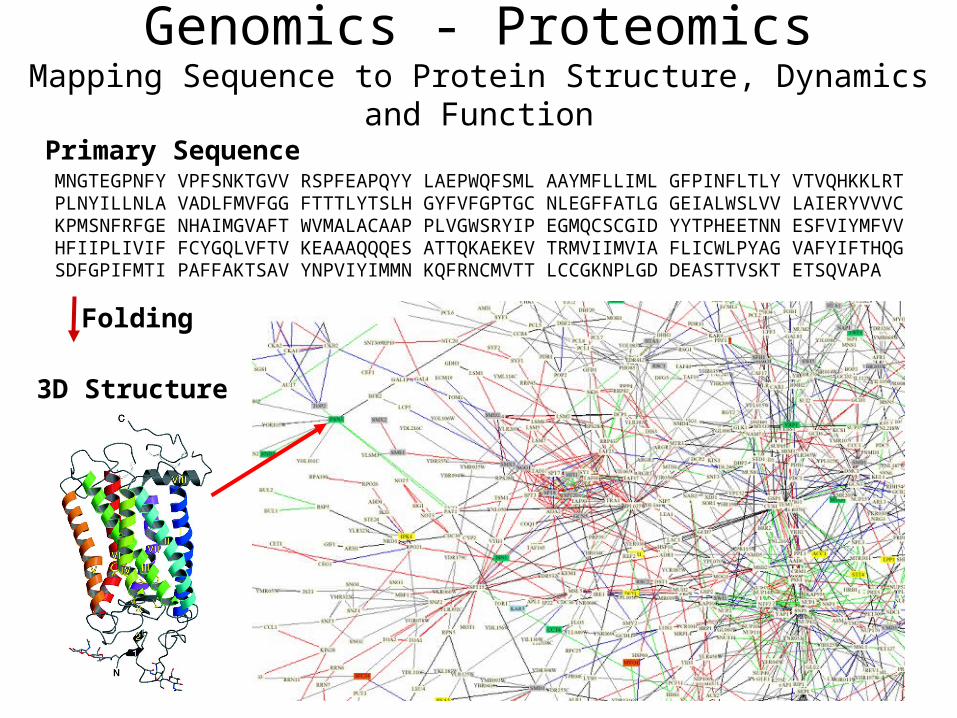
Genomics - ProteomicsMapping Sequence to Protein Structure, Dynamics and Function
Primary SequenceMNGTEGPNFY VPFSNKTGVV RSPFEAPQYY LAEPWQFSML AAYMFLLIML GFPINFLTLY VTVQHKKLRT PLNYILLNLA VADLFMVFGG FTTTLYTSLH GYFVFGPTGC NLEGFFATLG GEIALWSLVV LAIERYVVVC KPMSNFRFGE NHAIMGVAFT WVMALACAAP PLVGWSRYIP EGMQCSCGID YYTPHEETNN ESFVIYMFVV HFIIPLIVIF FCYGQLVFTV KEAAAQQQES ATTQKAEKEV TRMVIIMVIA FLICWLPYAG VAFYIFTHQG SDFGPIFMTI PAFFAKTSAV YNPVIYIMMN KQFRNCMVTT LCCGKNPLGD DEASTTVSKT ETSQVAPA
3D Structure
Folding
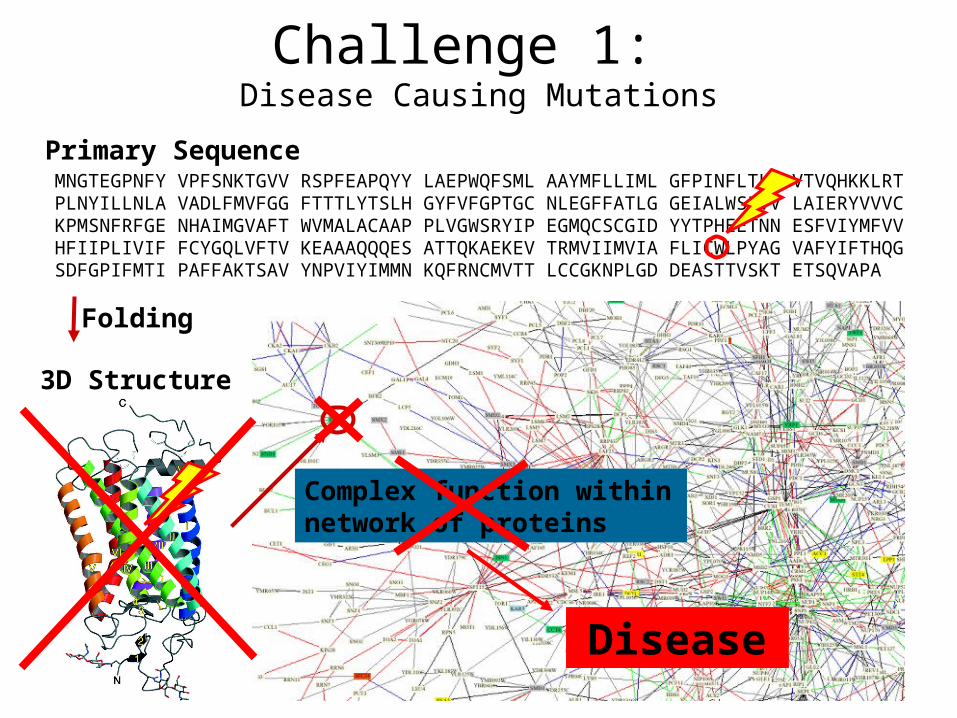
Primary SequenceMNGTEGPNFY VPFSNKTGVV RSPFEAPQYY LAEPWQFSML AAYMFLLIML GFPINFLTLY VTVQHKKLRT PLNYILLNLA VADLFMVFGG FTTTLYTSLH GYFVFGPTGC NLEGFFATLG GEIALWSLVV LAIERYVVVC KPMSNFRFGE NHAIMGVAFT WVMALACAAP PLVGWSRYIP EGMQCSCGID YYTPHEETNN ESFVIYMFVV HFIIPLIVIF FCYGQLVFTV KEAAAQQQES ATTQKAEKEV TRMVIIMVIA FLICWLPYAG VAFYIFTHQG SDFGPIFMTI PAFFAKTSAV YNPVIYIMMN KQFRNCMVTT LCCGKNPLGD DEASTTVSKT ETSQVAPA
3D Structure
Folding
Complex function within network of proteins
Disease
Challenge 1: Disease Causing Mutations
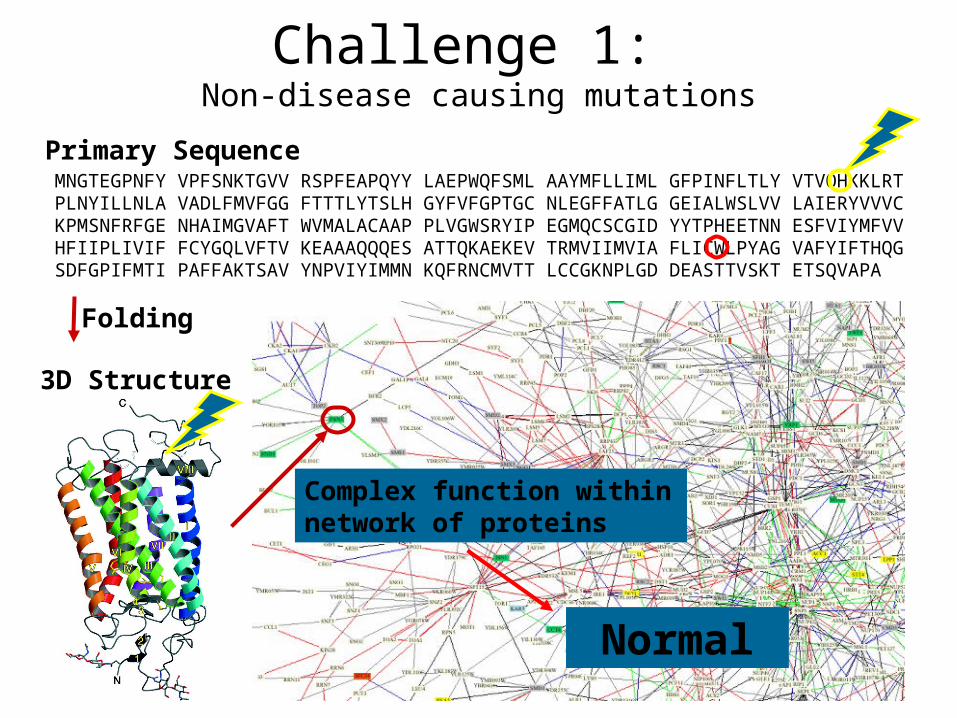
Primary SequenceMNGTEGPNFY VPFSNKTGVV RSPFEAPQYY LAEPWQFSML AAYMFLLIML GFPINFLTLY VTVQHKKLRT PLNYILLNLA VADLFMVFGG FTTTLYTSLH GYFVFGPTGC NLEGFFATLG GEIALWSLVV LAIERYVVVC KPMSNFRFGE NHAIMGVAFT WVMALACAAP PLVGWSRYIP EGMQCSCGID YYTPHEETNN ESFVIYMFVV HFIIPLIVIF FCYGQLVFTV KEAAAQQQES ATTQKAEKEV TRMVIIMVIA FLICWLPYAG VAFYIFTHQG SDFGPIFMTI PAFFAKTSAV YNPVIYIMMN KQFRNCMVTT LCCGKNPLGD DEASTTVSKT ETSQVAPA
3D Structure
Folding
Complex function within network of proteins
Normal
Challenge 1: Non-disease causing mutations

Challenge 1
How can we distinguish functional from non-functional protein sequences?
Needed: sequence to structure and function mapping
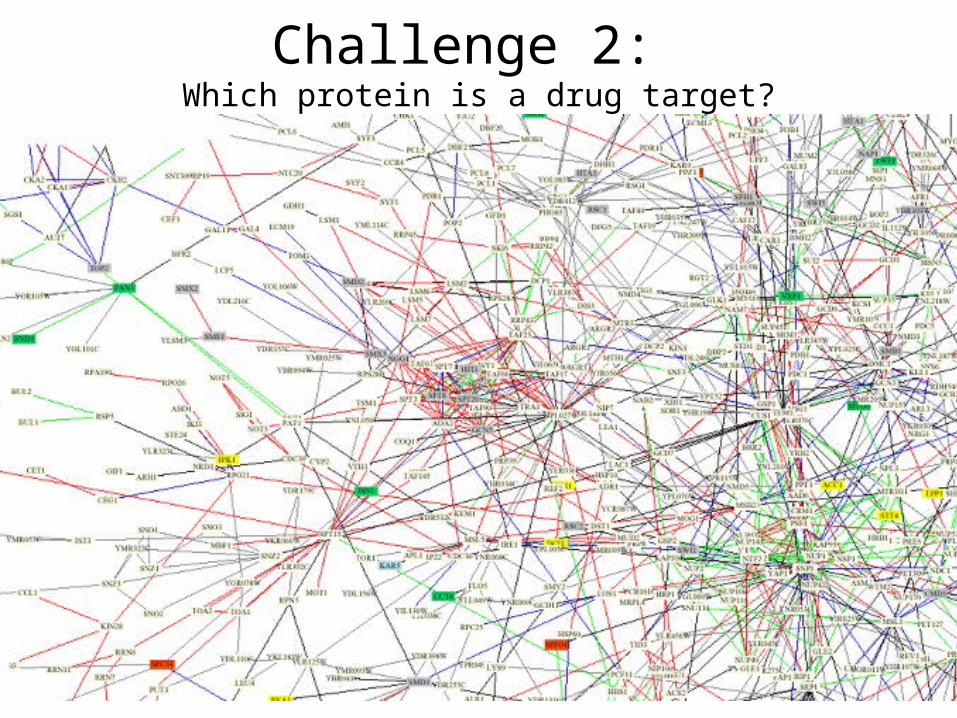
Challenge 2: Which protein is a drug target?
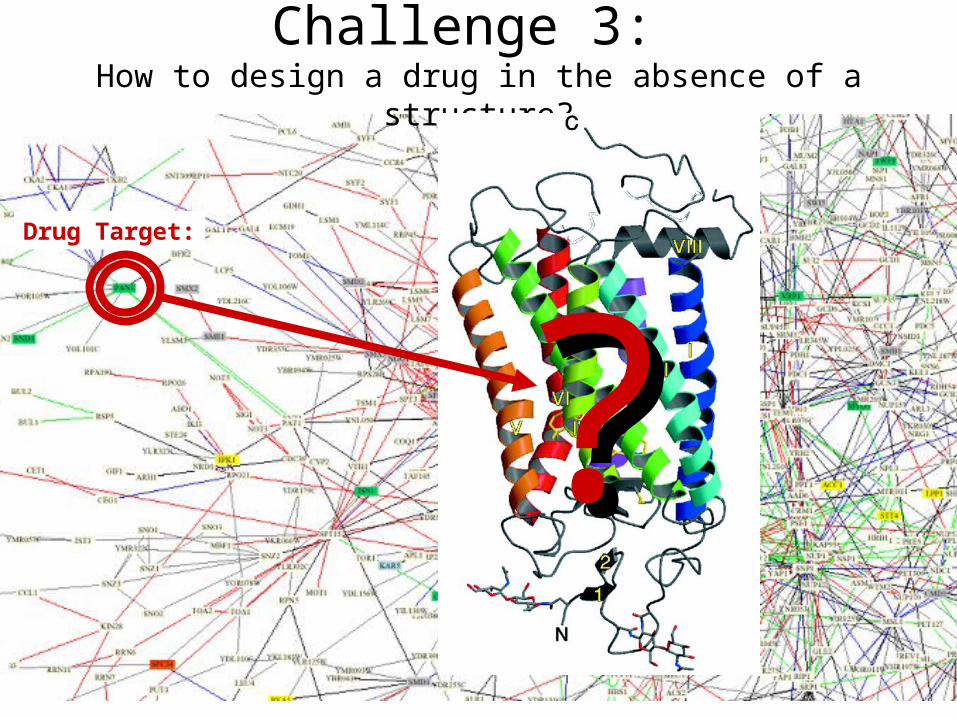
Challenge 3: How to design a drug in the absence of a structure?
Drug Target:
??
Challenge 4: Drug action, efficacy and side effects?
Drug Target:

Challenges
1.How can we distinguish functional from non-functional protein sequences?
2.Which protein is a drug target?3.How to design a drug in the absence of a
structure?4.Understanding drug action, efficacy and side
effects
Fundamental Scientific Challenge: Mapping the relationship between genome sequence and protein structures, dynamics and functions in complex cellular environments

Meaning for drug discovery
• If one could predict the structure of proteins from sequence, one could discover new drugs at a fast pace
• If one could predict the relationship between isozyme and tissue expression, one could design drugs specific to certain tissues
• If one could predict the interactions of proteins in different protein networks, one could interpret complex data such as animal models
• If one could…
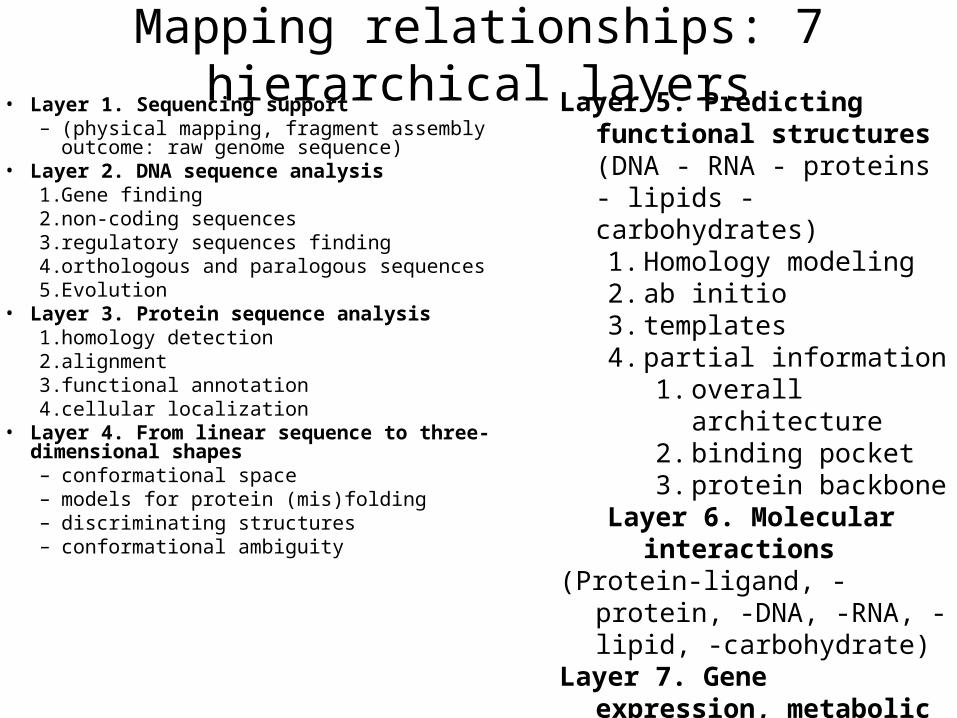
Mapping relationships: 7 hierarchical layers• Layer 1. Sequencing support
– (physical mapping, fragment assembly outcome: raw genome sequence)
• Layer 2. DNA sequence analysis1. Gene finding2. non-coding sequences3. regulatory sequences finding4. orthologous and paralogous sequences5. Evolution
• Layer 3. Protein sequence analysis 1. homology detection2. alignment3. functional annotation4. cellular localization
• Layer 4. From linear sequence to three-dimensional shapes– conformational space– models for protein (mis)folding– discriminating structures– conformational ambiguity
Layer 5. Predicting functional structures (DNA - RNA - proteins - lipids - carbohydrates) 1. Homology modeling2. ab initio3. templates4. partial information
1. overall architecture2. binding pocket3. protein backbone
Layer 6. Molecular interactions
(Protein-ligand, -protein, -DNA, -RNA, -lipid, -carbohydrate)
Layer 7. Gene expression, metabolic and regulatory networks

Specific Challenges for Bioinformatics in Drug Discovery
• Data needs to be organized, mined and visualized to allow scientific discovery
• Linking variety of databases
• Linking the different layers
• Interpretation of data
• Drug discovery

• use the information in the databases and infer information that is not provided directly by genomics and proteomics data: higher level information=> piece together all available information- to get detailed picture of a molecular process (or disease)- to identify new protein targets- to develop drugs
• based on chemical similarity of known drugs• rational (structure-based) drug design interactively on
computer screen• molecular docking (automatic, systematic computer-based
prediction of structure and binding affinity of complex)• high-throughput screening and combinatorial chemistry
Outline Drug Discovery Approach

Molecular modeling in drug discovery
I. Two case studies for sequence to structure mapping:
– Small changes in protein sequence cause dramatic difference in drug binding: COX inhibitors
– Large changes in protein sequence still maintain similar structure: G protein coupled receptors
II. Protein Structure Prediction
III. Ligand Docking to Protein Structures

Molecular modeling in drug discovery
I. Two case studies for sequence to structure mapping:
– Small changes in protein sequence cause dramatic difference in drug binding: COX inhibitors
– Large changes in protein sequence still maintain similar structure: G protein coupled receptors
II. Protein Structure Prediction
III. Ligand Docking to Protein Structures

Case study COX
A Wonder Drug: What is the most commonly-taken drug today? It is an effective painkiller. It reduces fever and inflammation when the body gets overzealous in its defenses against infection and damage. It slows blood clotting, reducing the chance of stroke and heart attack in susceptible individuals. It may be an effective addition to the fight against cancer.
http://www.rcsb.org/pdb/molecules/pdb17_1.html
Aspirin has been used professionally for a century, and traditionally since ancient times. A similar compound found in willow bark, salicylic acid, has a long history of use in herbal treatment. But only in the last few decades have we understood how aspirin works, and how it might be improved

ProstaglandinsAs you might expect from a drug with such diverse actions, aspirin blocks a central process in the body: Aspirin blocks the production of prostaglandins, key hormones that are used to carry local messages. Unlike most hormones, which are produced in specialized glands and then delivered throughout the body by the blood, prostaglandins are created by cells and then act only in the surrounding area before they are broken down. Prostaglandins control many of these neighborhood processes, including the constriction of muscle cells around blood vessels, aggregation of platelets during blood clotting, and constriction of the uterus during labor. Prostaglandins also deliver and strengthen pain signals and induce inflammation. These many different processes are all controlled by different prostaglandins, but all created from a common precursor molecule.
http://www.rcsb.org/pdb/molecules/pdb17_1.html
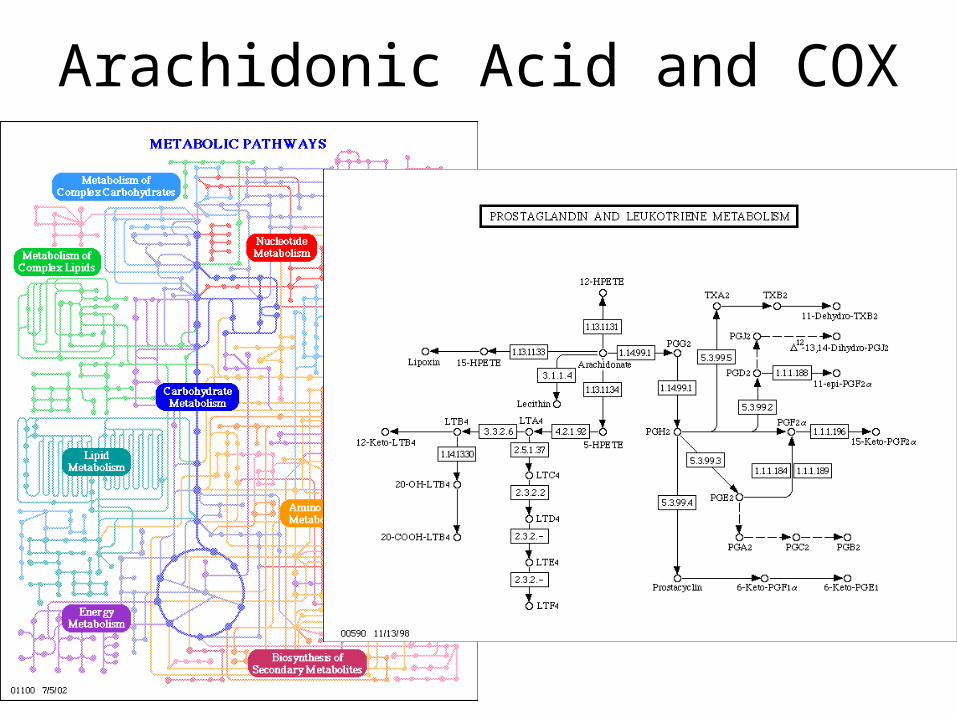
Arachidonic Acid and COX

What does COX do?
http://www.rcsb.org/pdb/molecules/pdb17_1.html
COX = Cyclooxygenase (PDB entry 1prh) performs the first step in the creation of prostaglandins from a common fatty acid.
It adds two oxygen molecules to arachidonic acid, beginning a set of reactions.
Aspirin blocks the binding of arachidonic acid in the cyclooxygenase active site. The normal messages are not delivered, so we don't feel the pain and don't launch an inflammation response.

Structural Organization of COX
Two different active sites, collectively prostaglandin synthase: 1, the cyclooxygenase active site discussed; 2, is has an entirely separate peroxidase site, which is needed to activate the heme groups that participate in the cyclooxygenase reaction.
Dimer of identical subunits (two cyclooxygenase active sites and two peroxidase active sites in close proximity)
Each subunit has a small carbon-rich knob, pointing downward anchoring the complex to the membrane of the endoplasmic reticulum, shown in light blue.
The cyclooxygenase active site is buried deep within the protein, and is reachable by a tunnel that opens out in the middle of the knob. This acts like a funnel, guiding arachidonic acid out of the membrane and into the enzyme for processing.
http://www.rcsb.org/pdb/molecules/pdb17_1.htmlPDB entry 4cox

Why is there a COX-1 and COX-2?
COX-1 and COX-2 are made for different purposes.
COX-1 is built in many different cells to create prostaglandins used for basic housekeeping messages throughout the body.
COX-2 is built only in special cells and is used for signaling pain and inflammation.
Aspirin attacks both. Since COX-1 is targeted, aspirin can lead to unpleasant complications, such as stomach bleeding.
Needed: specific compounds that block just COX-2, leaving COX-1 to perform its essential jobs. These drugs are selective pain-killers and fever reducers, without the unpleasant side-effects.

Active site Cox 1 (1pth)
1pth
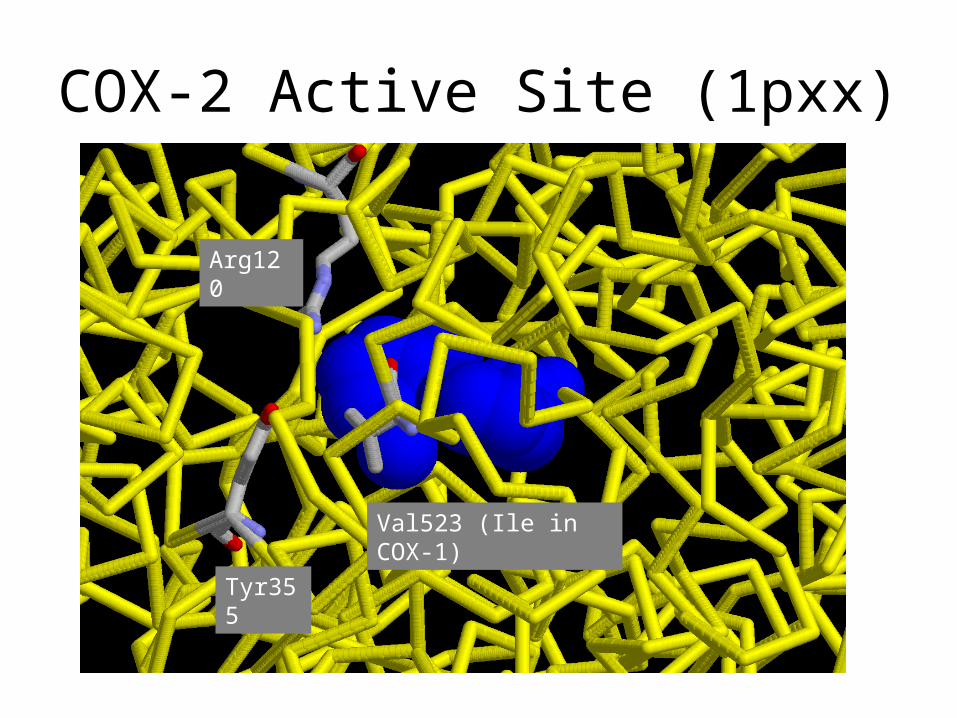
COX-2 Active Site (1pxx)
Tyr355
Arg120
Val523 (Ile in COX-1)
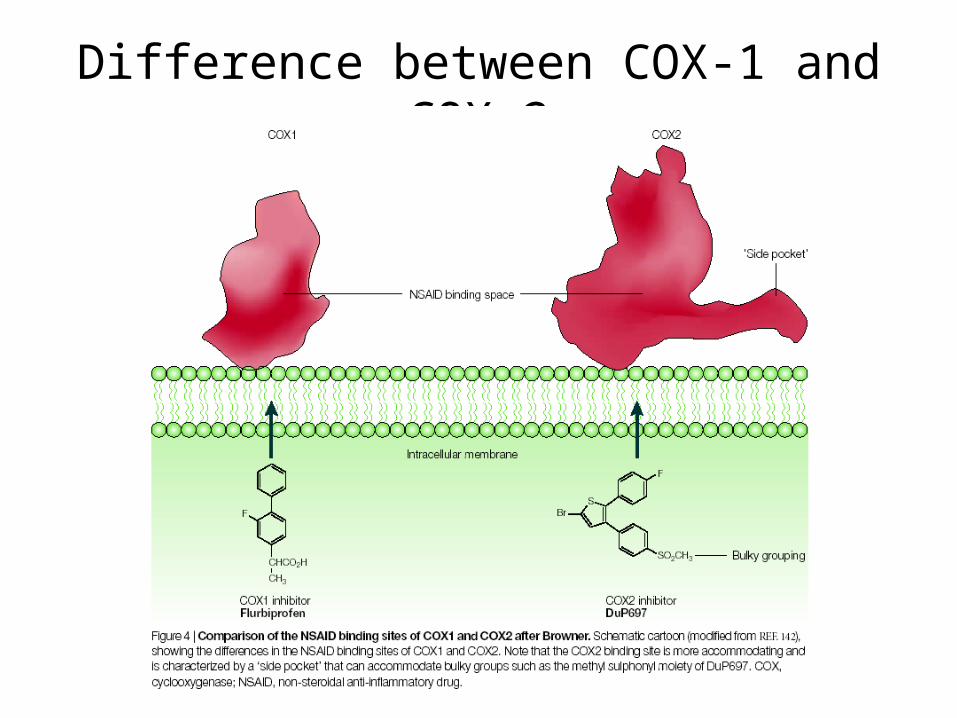
Difference between COX-1 and COX-2

Summary COX Case Study
• Being able to model the effect of small changes in sequence (isoforms) is essential for drug development

Molecular modeling in drug discovery
I. Two case studies for sequence to structure mapping:
– Small changes in protein sequence cause dramatic difference in drug binding: COX inhibitors
– Large changes in protein sequence still maintain similar structure: G protein coupled receptors
II. Protein Structure Prediction
III. Ligand Docking to Protein Structures
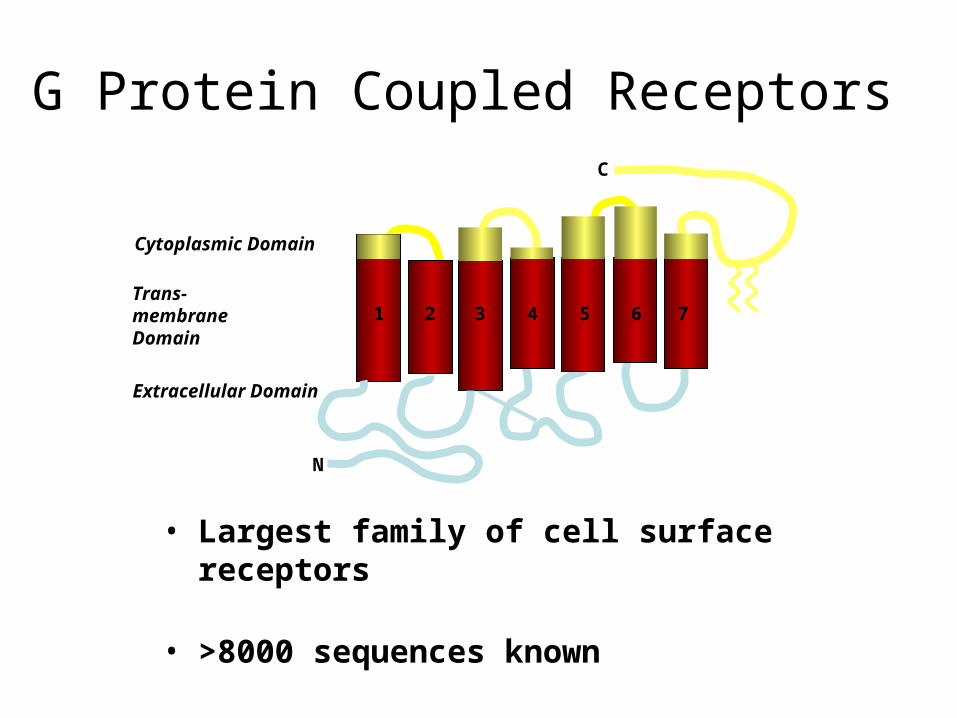
• Largest family of cell surface receptors
• >8000 sequences known
• 60% of all known drugs target GPCR
C
N
1 2 3 4 5 6 7
Cytoplasmic Domain
Trans-membraneDomain
Extracellular Domain
G Protein Coupled Receptors
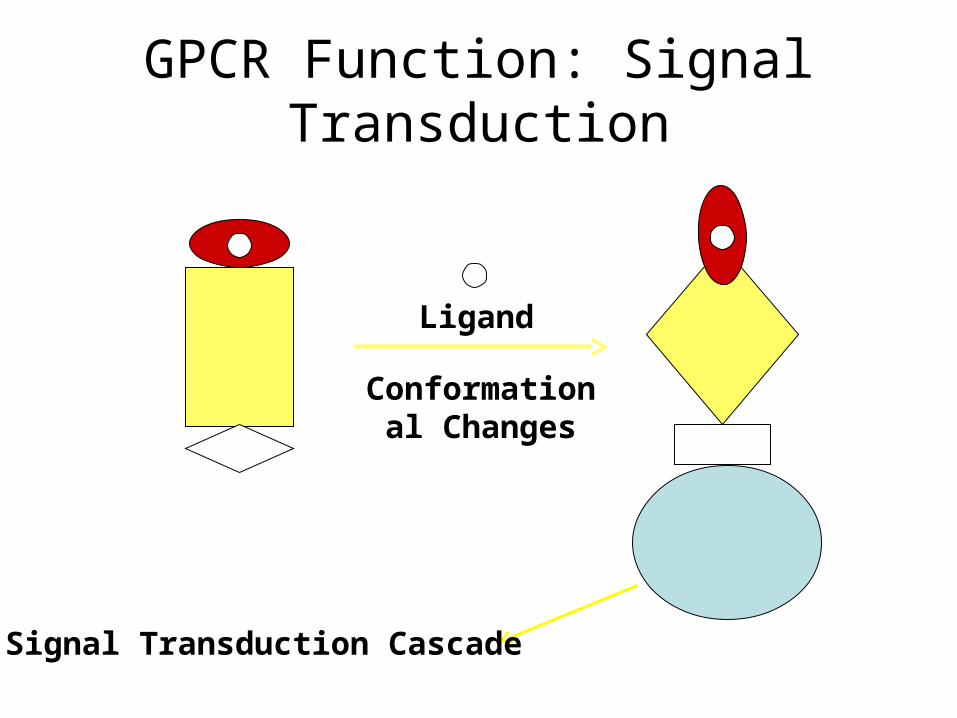
Ligand
Conformational Changes
Signal Transduction Cascade
GPCR Function: Signal Transduction

GPCR Family and Their Ligands
Class A: Rhodopsin-like FamilyOpsins, Odorants, Monoamines, Lipid messengers, Purines, Neuropeptides, Peptide hormones (e.g. platelet activating factor, gonadotropin -releasing hormone, th yrotropin releasing hormone & melatonin), Glycoprotein hormones, Chemokines, Proteases, Cannabis, Viral
Class B: Secretin-like Family Glucagon, Calcitonin, parathyroid hormone, secretin
Class C: Metabotropic glutamate and Chemosensor FamilymGluR 1-7, Calcium sensors, GABA-B
Class D: Fungal pheromone Family
Class E: c-AMP receptor (Dictyostelium) Family
Class F: Frizzled/Smoothened family
Putative families:Ocular albinism proteins, Drosophila odorant receptors, Plant Mlo receptors,Nematode chemoreceptors, Vomeronasal receptors
Putative/ unclassified orphans
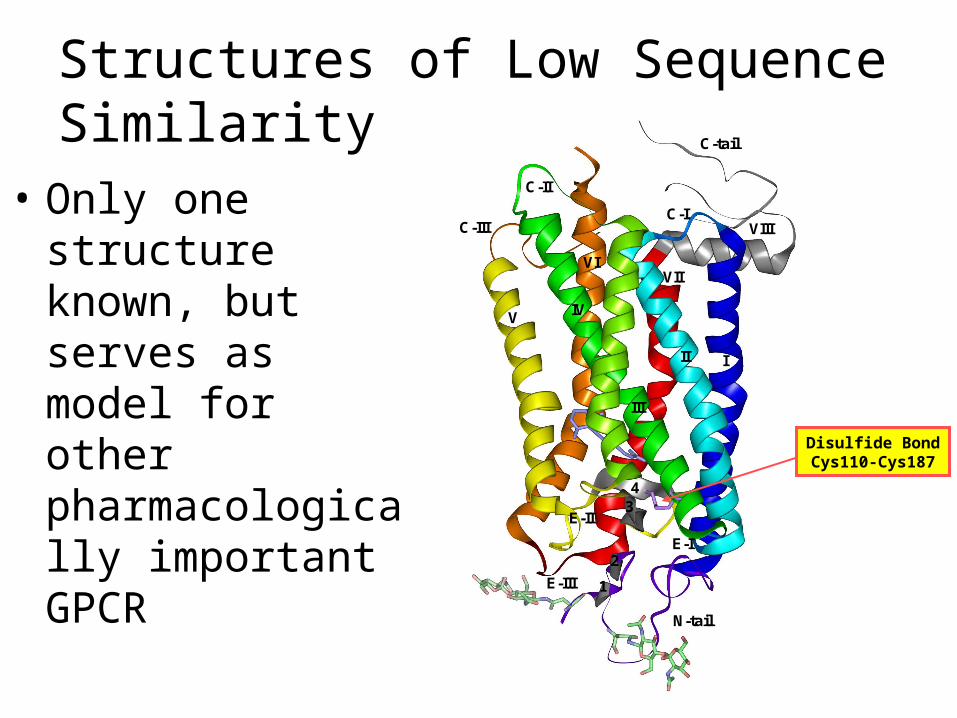
Structures of Low Sequence Similarity
• Only one structure known, but serves as model for other pharmacologically important GPCR
N-tail
E-I
C-I
C-II
E-II
E-III
C-III VIII
III
III
IVV
VIVII
C-tail
1
2
34
Disulfide BondCys110-Cys187
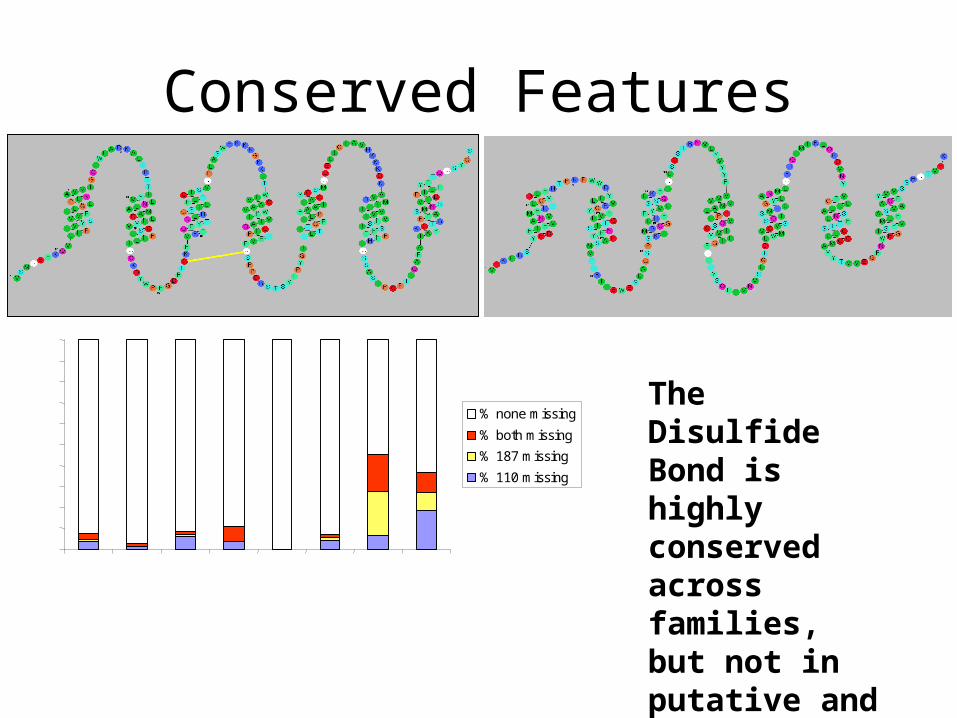
Conserved Features
0%
10%
20%
30%
40%
50%
60%
70%
80%
90%
100%
Class A
Class B
Class C
Class D
Class E
Frizze
d/Sm
ooth
ened
Fam
ily
Putat
ive F
amilie
s
Orpha
ns
% none missing
% both missing
% 187 missing
% 110 missing
The Disulfide Bond is highly conserved across families, but not in putative and orphan receptors

Summary GPCR Case Study
• Being able to model proteins with low sequence homology is essential to exploit structural information that is hard to get (membrane proteins) but where the impact is very high (>40% of R&D portfolios in companies)

Molecular modeling in drug discovery
I. Two case studies for sequence to structure mapping:
– Small changes in protein sequence cause dramatic difference in drug binding: COX inhibitors
– Large changes in protein sequence still maintain similar structure: G protein coupled receptors
II. Protein Structure Prediction
III. Ligand Docking to Protein Structures

Modeling Methods and Relation to Sequence Similarity
A. When no information but sequence and physical principles are used= ab initio structure prediction (Blue Gene IBM )B. When other information is used ("ab initio" methods that use pdb
information) Common features: "fold recognition“, requires a method for evaluating the compatibility of a given sequence with a given folding pattern1. 3D profiles2. Rosetta: conformations from short segments in pdb3. Including experimental structural constraints4. Threading (=sequence-structure alignment), 5. Inverse threading and folding experiments
a. using short-range informationb. using short- and long-range information
6. Predicting structural class only 7. Predicting active site only8. Predicting protein-protein interaction sites9. Predicting surface shape?

Modeling Methods ContinuedC. When a template with known structure must be available: homology
modeling D. Modeling structures based on experimental data
Both NMR and X-ray underdetermine the protein structure. To solve a structure one must minimize a combination of the deviation from the experimental data and the conformational energy:a. NMR (set of constraints on distances and angles)b. X-ray crystallography (Fourier transform of the electron density)
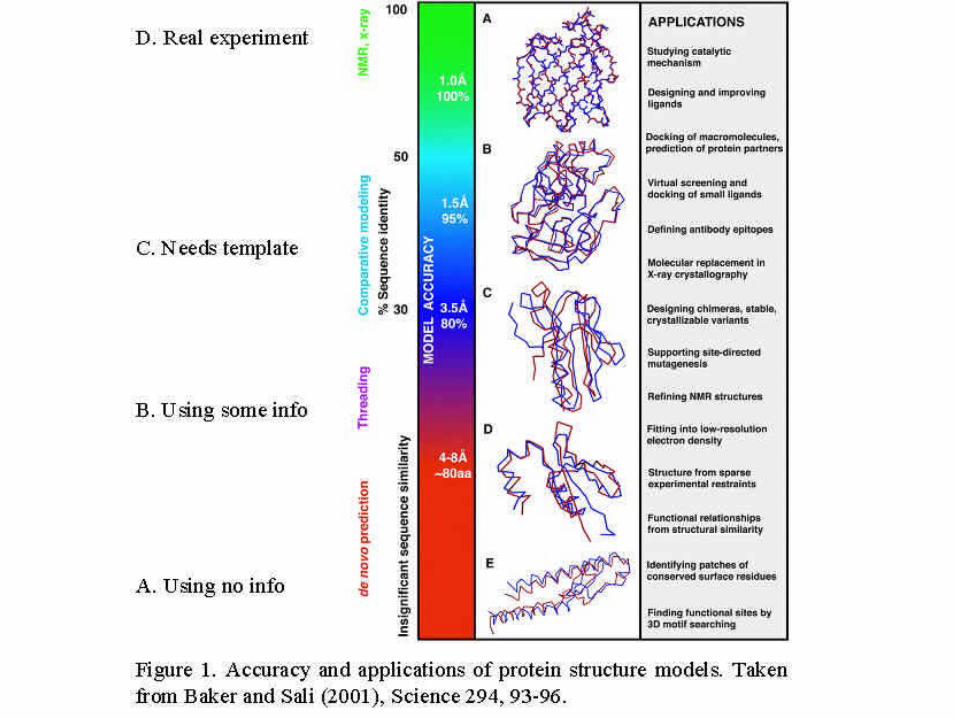

Evaluating structure prediction
• Use rmsd to known structures - defines structural similarity
• Critical Assessment of Structure Predictions (CASP) competitions
• EVA, EVA submits sequences automatically to different prediction servers shortly before structures are published in pdb

Homology Modeling• Database searching for homologous proteins
( Blast the query sequence towards the pdb database )
• Alignment (Pairwise/ Multiple Alignments)– needs minimum 30% sequence identity, but to be useful usually need
40-50%– note that ~30% of genomes have sequence identity of 20%
• Model Building– Modeller , Composer etc
• Model Refinement and Evaluation– Joy– Procheck etc

BLAST (Basic Local Alignment Search Tools)
BLAST is a heuristic search method that seeks words of length W (default = 3 in blastp) that score at least T when aligned with the query and scored with a substitution matrix. Words in the database that score T or greater are extended in both directions in an attempt to fina a locally optimal ungapped alignment or HSP (high scoring pair) with a score of at least S or an E value lower than the specified threshold. HSPs that meet these criteria will be reported by BLAST, provided they do not exceed the cutoff value specified for number of descriptions and/or alignments to report.

Scoring matricesBLOSUM62 Substitution Scoring Matrix. The BLOSUM 62 matrix shown here is a 20 x 20 matrix of which a section is shown here in which every possible identity and substitution is assigned a score based on the observed frequencies of such occurences in alignments of related proteins. Identities are assigned the most positive scores. Frequently observed substitutions also receive positive scores and seldom observed substitutions are given negative scores.
The PAM family PAM matrices are based on global alignments of closely related proteins. The PAM1 is the matrix calculated from comparisons of sequences with no more than 1% divergence. Other PAM matrices are extrapolated from PAM1.
The BLOSUM family BLOSUM matrices are based on local alignments. BLOSUM 62 is a matrix calculated from comparisons of sequences with no less than 62% divergence. All BLOSUM matrices are based on observed alignments; they are not extrapolated from comparisons of closely related proteins. BLOSUM 62 is the default matrix in BLAST 2.0. Though it is tailored for comparisons of moderately distant proteins, it performs well in detecting closer relationships. A search for distant relatives may be more sensitive with a different matrix.
The relationship between BLOSUM and PAM substitution matrices. BLOSUM matrices with higher numbers and PAM matrices with low numbers are both designed for comparisons of closely related sequences. BLOSUM matrices with low numbers and PAM matrices with high numbers are designed for comparisons of distantly related proteins. If distant relatives of the query sequence are specifically being sought, the matrix can be tailored to that type of search.
http://www.ncbi.nlm.nih.gov/Education/

Sequence Alignment when homology is low
• Hidden Markov Models of Protein Families
• Secondary structure prediction methods
• Novel alignment methods
• Sequence conservation based on property conservation

Model Building
• Modeller (freeware, http://www.salilab.org/modeller/modeller.html)
• Spdbviewer Swissmodel–module (freeware, http://us.expasy.org/spdbv/)
• Composer (module of InsightII, commercial version of Modeller)

Model Building Principles
• Sequentially go from amino acid position to next position– if same amino acid, copy the coordinates– If different amino acid, if the new amino acid has
atoms in common with the template, those atoms will be copied, and the rest are computed
• At every step, check for steric clashes with previous amino acids– Minimization allowing the position of new amino acid
to change– Only at the final stage, bond energy is minimized

Model Refinement and Evaluationhttp://cgat.ukm.my/spores/Predictory/evaluation.html
• Verify3D (based on surface accessibility)
• Procheck (based on phi/psi angle, rmsd deviations)
• Joy (based on secondary structure assignments)
• WHAT IF (bond length, bond angles, chi values, etc.)

WHAT IF ChecklistA WHAT IF check report: what does it mean?
General points Administrative checks
Nomenclature Chain name Weights (occupancy) Missing atoms and C-terminal oxygens
Symmetry Consistency Cell conventions Matthews' Coefficient Higher symmetry Non crystallographic symmetry
Geometry Chirality Bond lengths Bond angles Torsion Angles: "Evaluation"; "Ramachandran"; "omega"; "Chi1/2" Rings and planarity: "Planarity"; "Proline Puckering"
Structure Inside/outside profile Bumps Packing quality Backbone: "number of hits"; "backbone normality"; "peptide flips" Sidechain rotamers Water molecules: "floating clusters"; "symmetry relations" B-factors: "average"; "low B-factors"; "B-factor distribution" Hydrogen bonds: "Flip check"; "HIS assignments"; "Unsatisfied"

Collection of homology models
• MODBASE– uses PSI-BLAST plus MODELLER to model
and stores coordinates in this database
• SWISS-MODEL– automatic structure prediction

Play with homology models• www.cs.cmu.edu/~blmt/Seminar/SeminarMaterials/COX
• Rasmol is also in this directory, just click on the raswin icon to start program
COX 2 Modelling :Template structure : 1PTH.pdb (cox1 in ovis aries)query seq:sequence of 1PXX.pdb (cox2 in mus musculus)model generated using modeller: 2cox.pdb
COX 1 Modelling:Template structure : 1PXX.pdb (cox2 in mus musculus)query seq:sequence of 1PTH.pdb (cox1 in ovis aries)model generated using modeller: 1cox.pdb
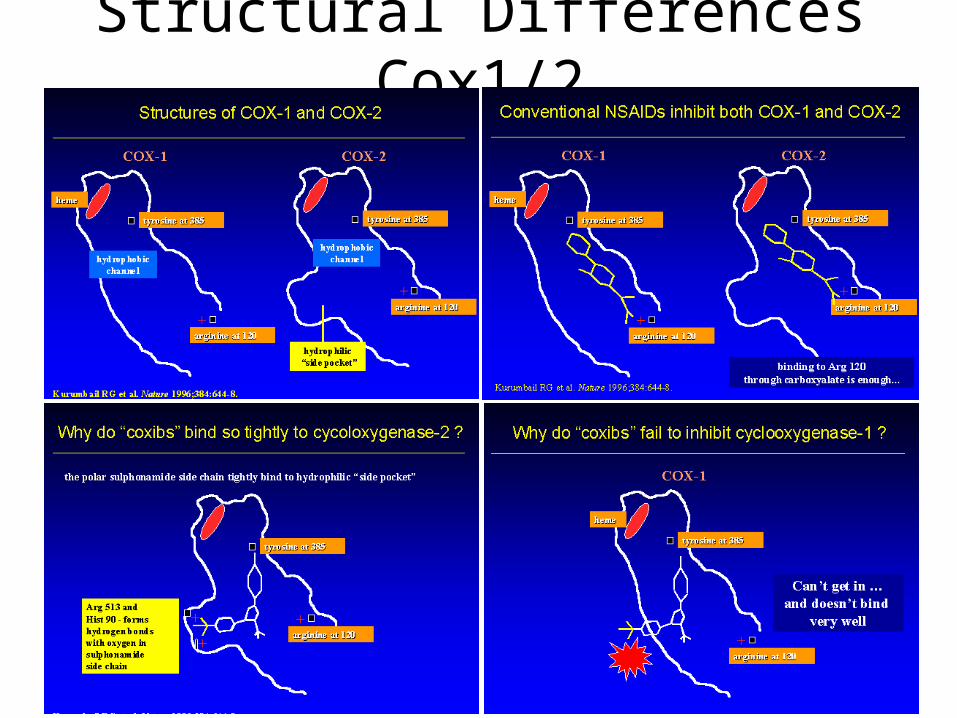
Structural Differences Cox1/2
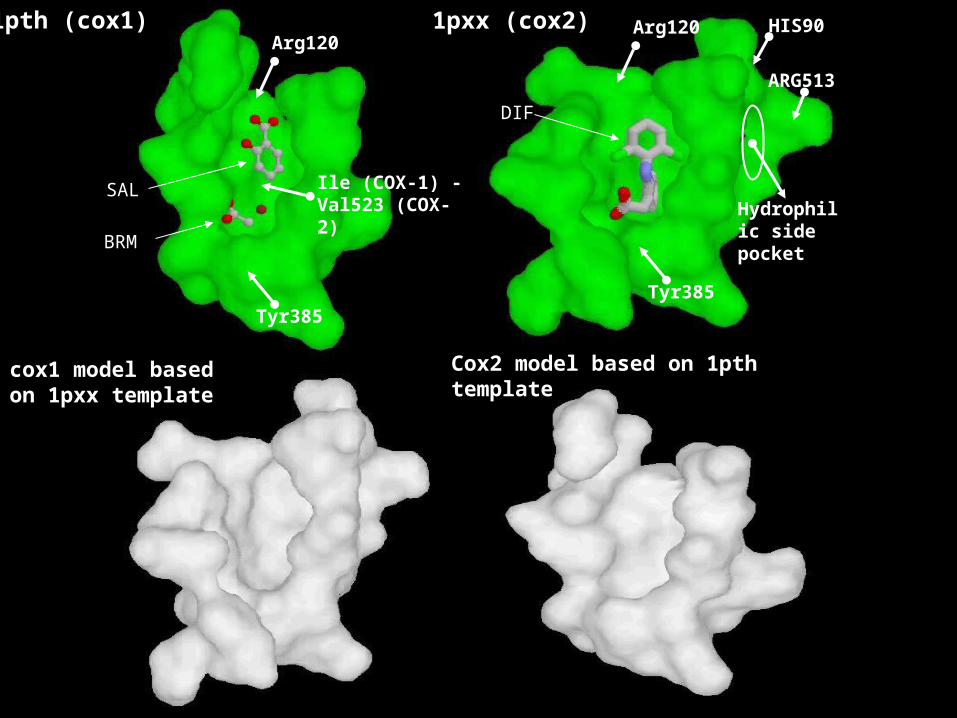
1pth (cox1)
Tyr385
Arg120
Hydrophilic side pocket
Ile (COX-1) - Val523 (COX-2)
BRM
SAL
DIF
Arg120
Tyr385
HIS90
ARG513
cox1 model based on 1pxx template
1pxx (cox2)
Cox2 model based on 1pth template
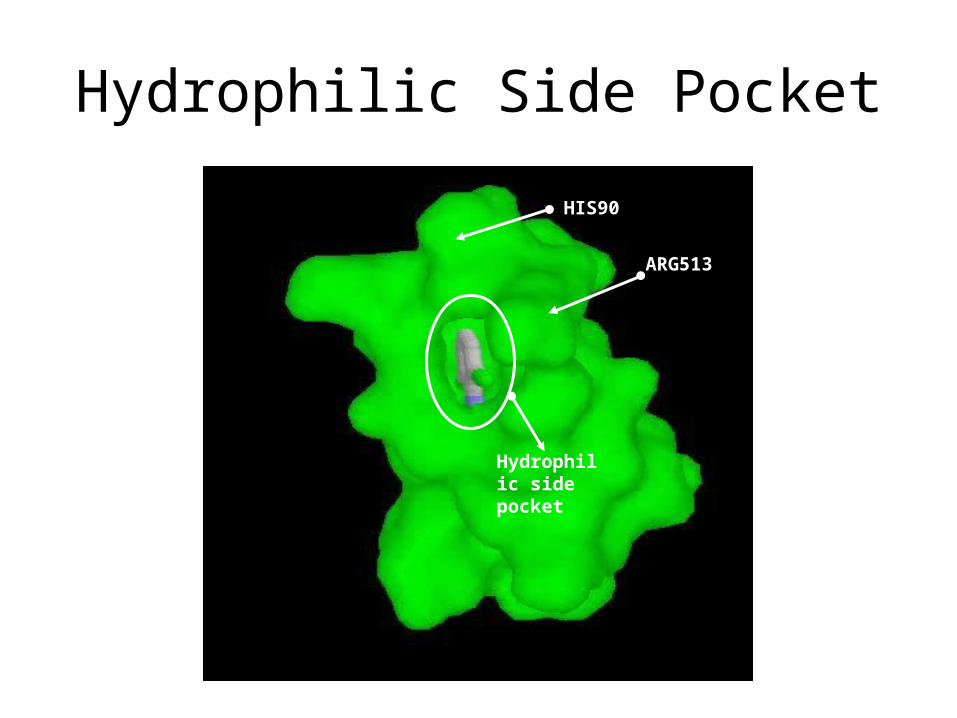
Hydrophilic Side Pocket
HIS90
ARG513
Hydrophilic side pocket

Molecular modeling in drug discovery
I. Two case studies for sequence to structure mapping:
– Small changes in protein sequence cause dramatic difference in drug binding: COX inhibitors
– Large changes in protein sequence still maintain similar structure: G protein coupled receptors
II. Protein Structure Prediction
III. Ligand Docking to Protein Structures

Protein-ligand docking
• First (if structure is known) or second (after structure prediction) step in a drug design project: find a lead structure (=small molecule which binds to a given target)
• docking problem - predicting the energetically most favorable complex between a protein and a putative drug molecule
• For a given protein structure, one can apply docking algorithms to virtually search through the space
• 2 questions:1. what does the protein-ligand complex look like2. what is the affinity with respect to other candidates?

What makes the docking problem hard to solve?
1. Scoring problem– = calculating binding affinity given a protein-ligand
complex– no general scoring function is available
2. Large number of degrees of freedom– most important degrees of freedom:
1. relative orientation of the two molecules2. conformation of the ligand 3. protein conformation4. water molecules can be between ligand and protein5. protonation state

Types of Docking Problems1. Macromolecular docking• = two macromolecules are docked, such as protein and DNA, or protein
and protein• large contact area• molecules have fixed overall shape• => methods based on geometric properties like shape
complementarities alone can be efficiently used to create energetically favorable complexes
2. Small molecule docking• = a small molecule is docked to a macromolecule• ligand is typically not fixed in shape (as opposed to macromolecular
docking)• typical ligand size has 5-12 rotatable bonds• often fragments of ligand are used for modeling, eg. combinatorial
libraries are docked by combining placement for individual building blocks of the library

Steps in Molecular Docking1. Find a set of compounds to start with - e.g. from inspecting known ligands for a protein (e.g.
substrate in an enzyme)2. compounds from a screening experiment of a combinatorial
library (in which there is usually a molecular fragment that is common between all molecules of the library, the core, and the fragments attached to the core are R-groups)
3. compounds from a filtering experiment using other software4. from varying other lead structures or known ligands5. virtual screening using a fast docking algorithm (typically
from a million molecules)6. de novo design using fragments of compounds
=> get several hundred to thousands of ligands to start with

Docking Methods
• Rigid-body docking algorithms– Historically the first approaches. – Protein and ligand are held fixed in conformational
space which reduces the problem to the search for the relative orientation fo the two molecules with lowest energy.
– All rigid-body docking methods have in common that superposition of point sets is a fundamental sub-problem that has to be solved efficiently:
– Superposition of point sets: minimize the RMSD• Flexible ligand docking algorithms
– most ligands have large conformational spaces with several low energy states
http://www-2.cs.cmu.edu/~blmt/Seminar/SeminarMaterials/interactions.html

Clique-search based approaches
• = matching characteristic features of the two molecules • use a graph to search for compatible matches: the
vertices of the graph are all possible matches and edges connect pairs of vertices representing compatible matches
• compatibility = distance compatibility with in a fixed tolerance epsilon
• The matches (p1,l1), (p2,l2) are distance-compatible if |d(p1,p2)-d(l1,l2)| < epsilon
• Example: DOCK program
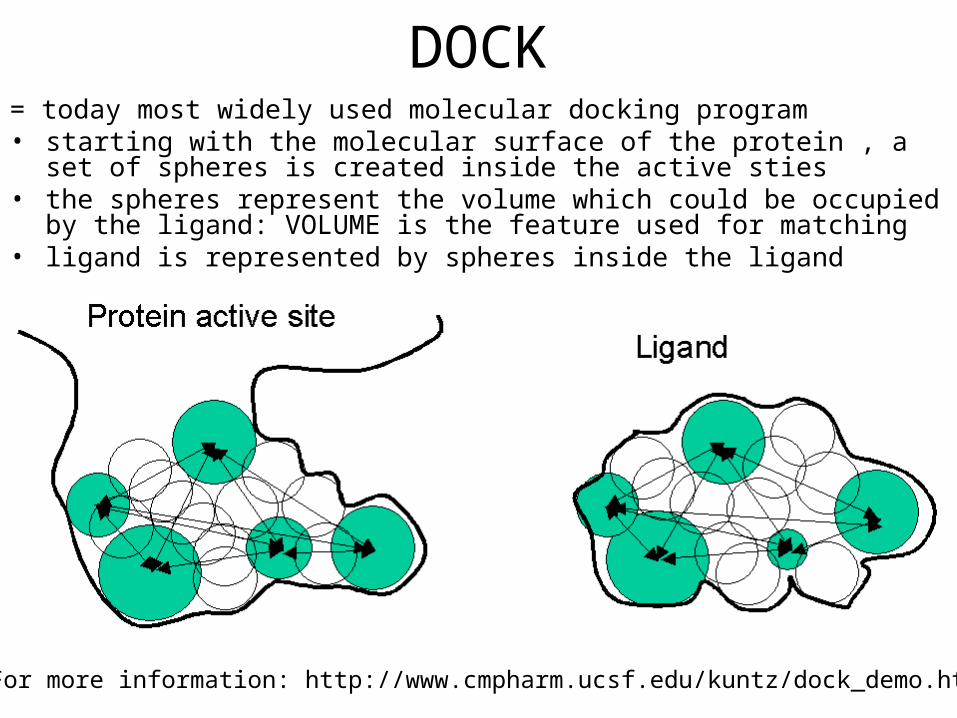
DOCK= today most widely used molecular docking program• starting with the molecular surface of the protein , a set of spheres is created
inside the active sties• the spheres represent the volume which could be occupied by the ligand:
VOLUME is the feature used for matching• ligand is represented by spheres inside the ligand
For more information: http://www.cmpharm.ucsf.edu/kuntz/dock_demo.html

Take home messages
• Structural and functional effects of small changes in sequences
• Conservation of structure despite large differences in sequences
• Prediction of structural and functional effects using computational pharmacology to understand disease mechanisms and drug action with the goal of identifying targets and designing drugs against them– Example: Specific Structure of COX and of GPCR– Current hot topics: Complex interactions of proteins within their
environment, differences between individuals• Even with lots of structural information available,
prediction of ligand binding affinities is challenging





























