Embed Size (px)
Citation preview

ENGLISH INSTRUCTIONS – page 1A Rapid Visual Test for the Qualitative Detection of InfectiousMononucleosis Heterophile Antibody.
DEUTSCHE ANLEITUNG – Siehe Seite 7Ein visueller Schnelltest für den qualitativen Nachweis von infektiösen,Mononukleose-typischen heterophilen Antikörpern.
INSTRUCCIONES EN ESPAÑOL – A partir de la página 13Una prueba visual rápida para la detección cualitativa del anticuerpoheterófilo de la mononucleosis infecciosa.
NOTICE EN FRANÇAIS – voir page 19 Test visuel rapide pour la détection qualitative des anticorps hétérophilesde la mononucléose infectieuse.
ISTRUZIONI IN ITALIANO – Vedere pagina 25Un test rapido a lettura visiva per la determinazione qualitativadell’anticorpo eterofilo della mononucleosi infettiva.
INSTRUÇÕES EM PORTUGUÊS – consulte a página 31Um teste visual rápido para a detecção qualitativa do anticorpo heterófiloda mononucleose infecciosa.
SVENSKA INSTRUKTIONER – se sid. 37Ett snabbt, visuellt test för kvalitativ detektion av infektiös mononukleosheterofil antikropp.
DANSKE INSTRUKTIONER – se side 43En hurtig, visuel test til kvalitativ påvisning af heterofilt antistof for infektiøsmononucleosis.
MONO TEST

1
For CLIA waived - Test with Fingerstick Blood or Whole Blood Specimens
For Moderately Complex - Test with Serum, Plasma or Whole Blood Specimens
INTENDED USE
The SureStep Mono Test is a rapid test for the visual, qualitative detection of Heterophile antibodies specific to InfectiousMononucleosis (IM) in human serum, plasma or whole blood. This test kit is intended as an aid in the diagnosis of IM inpatients with characteristic clinical symptoms, and is intended for professional laboratory use only.
SUMMARY
Infectious Mononucleosis is an acute, self-limiting disease caused by the Epstein-Barr virus (EBV) (1-3). Infection with EBVin early life usually is asymptomatic (4-5). However, up to 50% of infection occurring in young adulthood and adolescencewill develop clinical manifestations associated with IM.
Diagnosis of IM is based on the evaluation of characteristic clinical symptoms and serological changes. Serologicaldiagnosis of IM has been demonstrated by the detection of heterophile and EBV specific antibodies (3-5). The Heterophileantibody is detectable at some point during IM in most adults. It is a widely accepted practice among physicians to usethe detection of heterophile antibodies as an aid in the diagnosis of IM (3-7). The SureStep Mono Test utilizes bovineerythrocyte extract which has a higher sensitivity and specificity than extracts from other species. (4)
TEST PRINCIPLE
The SureStep Mono Test has been designed to detect IM through visual interpretation of color development in thetest device, which is a sandwich solid phase gold conjugate immunoassay. The test device contains a membranestrip which is precoated with heterophile antigens on the test line region and goat anti-mouse antibody on thecontrol line region. The anti-human IgM antibody-colloidal gold conjugate pad is placed at the end of themembrane. A mixture of colloidal gold conjugate together with the sample and developer buffer will move alongthe membrane chromatographically by capillary action. When the IM heterophile antibodies are present in thepatient sample, the mixture will migrate to the test line region and form a visible line as the antibody complexeswith the heterophile antigen. When IM heterophile antibodies are absent from the sample, no visible color linewill form on test line region. Therefore, the presence of a colored line on the test line region indicates a positiveresult. A colored line will always appear at the control region. This control line serves as a procedural indicatorfor the proper performance of the test and the device.
STORAGE AND STABILITY
The test kit is to be stored either refrigerated or at room temperature 2-30°C (36-86°F) in the sealed pouch for the durationof the shelf-life.
PRECAUTIONS
• For In Vitro Diagnostic Use Only.
• For professional and laboratory use.• All patient samples should be treated as if capable of transmitting disease.• Do not mix reagents from different lots.• Do not open the foil pouch until ready to perform the test.• Do not use whole blood stored for more than three days.• Grossly hemolyzed samples should not be used.• Developer Buffer and Controls contain sodium azide. Sodium azide may react with lead or copper
CLIA COMPLEXITY INSTRUCTIONS ARE LISTED BELOW IN THE QUALITY CONTROL SECTION.PLEASE FOLLOW THE APPROPRIATE INSTRUCTIONS REQUIRED BY YOUR LABORATORY GUIDELINES.
MONO TEST
ENGLISH
Do not reuse test.

2
plumbing to form potentially explosive metal azides. When disposing of these solutions, always flush with copious amounts of water to prevent azide buildup.
Extraction Reagents are slightly caustic. Avoid contact with eyes or mucous membranes. In the event of accidentalcontact, wash thoroughly with water.
Positive and Negative Controls contain sodium azide which may react with lead or copper plumbing to form potentiallyexplosive metal azides. When disposing of these solutions, always flush with copious amounts of water to prevent azidebuild-up.
Warning: Potential Biohazardous MaterialEach donor unit of human plasma or serum used in the preparation of the Positive and Negative Controlswere tested by FDA-approved methods for the presence of anti-HIV-1/HIV-2, HBsAg and anti-HCV, andfound to be negative. However, caution should be used when handling and disposing of these items at biosafetylevel 2, as recommended in the Center for Disease Control/National Institutes of Health Manual, Biosafety inMicobiological and BiomedicalLaboratories, 1984.
REAGENTS AND MATERIALS SUPPLIED
• 25 individually wrapped test devices. Each device contains one test strip with heterophile antigen coated membrane and colored antibody pad.
• Developer Buffer (8 ml) : 0.1 M Phosphate Buffered Saline, with additives and 0.1% sodium azide.
• Negative Control (0.2 ml) : Normal human plasma or serum diluted in saline solution with 0.2%sodium azide.
• Positive Control (0.2 ml) : IM heterophile antibody positive human plasma or serum diluted in saline solution with 0.2% sodium azide.
• 25 disposable Transfer Pipets.
• One Instruction Sheet.
SPECIMEN COLLECTION AND HANDLING
Fingerstick 1. Clean the area to be lanced with an alcohol swab.2. Squeeze the end of the fingertip and pierce with a sterile lancet.3. Wipe away the first drop of blood with sterile gauze or cotton.4. Allow the second drop to flow directly into the sample well of the test device or use the pipette provided
to obtain fresh blood. Add 2 drops (about 40 µl) into the sample well.
Whole Blood1. A certified phlebotomist should collect whole blood into a purple, blue or green top collection tube (containing EDTA,
citrate or heparin, respectively) by venipuncture.2. The whole blood may be used for testing immediately or stored at 2-8°C up to three days.
Plasma1. A certified phlebotomist should collect whole blood into a purple, blue or green top collection tube (containing EDTA,
citrate or heparin, respectively) by venipuncture.2. Separate the plasma by centrifugation.3. Carefully withdraw the plasma for testing or label and store at 2-8°C for up to two weeks. Plasma may be frozen at -20°C for
up to one year.
Serum1. A certified phlebotomist should collect whole blood into a red top collection tube (containing no anti
coagulants) by venipuncture.2. Allow the blood to clot and separate serum by centrifugation.3. Carefully withdraw the serum for testing, or label and store at 2-8°C for up to two weeks. Serum may be
frozen at -20°C for up to one year.
BUF DEV

3
TEST PROCEDURE
3. Immediately add 3 - 4 drops of Developer Buffer.
4. After the addition of the Developer Buffer wait for the colored lines to appear. Depending on the concentration ofheterophile antibodies present, positive results may be observed within 3 minutes. However, to confirm a negativeresult, the complete reaction time of 5 minutes is required. Do not read test results after 8 minutes.
Test Device
Control Region Test Region
Sample Well
Test device, Developer Buffer, patient’s samples, controls shouldbe brought to room temperature (20 to 30°C) prior to testing.Do not open pouches until ready to perform the assay.
1. Remove the test device from its protective pouch. (Bring thedevice to room temperature before opening to avoidcondensation of moisture on the membrane). Label the devicewith patient or control identification.
2. Add specimen to sample well:
Fingerstick blood: Allow the second drop to flow directly intothe sample well of the test device or use the pipet provided toobtain fresh blood. Add 2 drops (about 40 µl) into the samplewell.
Whole blood samples in collection tubes: add two drops(about 40 µl) into the sample well, holding the provided transferpipet in a vertical position.
Plasma or serum samples: add 1 drop (about 20 µl) of serum orplasma into the sample well,holding the provided transfer pipet ina vertical position
Transfer Pipet
INTERPRETATION OF RESULTS
Positive:Two colored lines appear. One in the control region (C) and one colored line inthe test region (T). When testing with strong positive samples, the intensity of thecontrol line may be lighter than expected. It is not recommended to compare theintensity of the lines.
Negative:Only one colored line appears in the control line region. No apparent faint colored lineon the test line region (T).
Invalid:A total absence of a line in the control region is an indication of procedural error orthat test reagent deterioration has occurred. The test should be voided since animproper test procedure may have been performed or deterioration of reagents mayhave occurred. Repeat the test with a new device. If the problem persists, call AppliedBiotech’s Technical Service at (888) 578-7956 for calls inside the United States and(858) 713 - 9668 for international calls. Calls are answered by English speakingpersonnel. Other languages email [email protected].
C T
C T
C T
C T
C TMONO

4
QUALITY CONTROL
Internal Procedural ControlA procedural control is included in the test. A colored line appearing in the control region (C) is considered an internalprocedural control, indicating proper performance and reactive reagents.
A clear background in the result window is considered an internal negative control. However, when whole blood samples are tested, the background may appear slightly reddish due to the low level hemolysis of some red blood cells. This is acceptable as long as it does not interfere with the reading of the test. The test is invalid if the back ground fails to clear and obscures the reading of the test result.
External Quality ControlGood laboratory practice recommends the use of external controls to assure functionality of reagents and properperformance. Positive and Negative Controls are supplied in the kit. These controls are human serum or plasma based andare tested like a patient sample. When testing with the controls, add one drop (about 20 µl) of Positive or Negative Controlin the sample well using the provided transfer pipette by holding it in a vertical position. Immediately add 3 or 4 drops ofdeveloper buffer. A positive signal is indicated by two lines, one in the test region (T) and one in the control region (C). Anegative signal is indicated by only one line in the control region (C). Observe results at 5 minutes and do not interpretafter 8 minutes. If the controls do not perform as expected, do not interpret the test results. Repeat the test or contactTechnical Service.
FOR CLIA WAIVED LABORATORIES:
Both Positive and Negative external control must be tested when opening a new test kit. Each operator must test a positiveand negative external control once with each 25 test kit.
FOR MODERATELY COMPLEX LABORATORIES OR PROFESSIONAL HEALTH CARE PROVIDERS:
It is recommended that both a Postive and Negative Control be tested with every new shipment or new kit lot numberand as otherwise required by your state and local regulations.
LIMITATIONS
This test kit is to be used for the qualitative detection of IgM antibodies to IM heterophile antigen. A positive result suggeststhe presence of IgM antibodies to heterophile antigen.
This test kit should be used for symptomatic individuals suspected of having IM. Diagnosis o f IM should be made byconfirmation with other clinical findings.
A negative result does not rule out the possibility of IM because the antibodies to heterophile antigen may be absentor may not be present in sufficient quantity to be detected.
EXPECTED RESULTS
During the acute phase of IM, heterophile antibodies are detectable in 80-85% of patients. Heterophile antibodies are detectable during first month of illness and decrease rapidly after week four (5).
PERFORMANCE CHARACTERISTICS
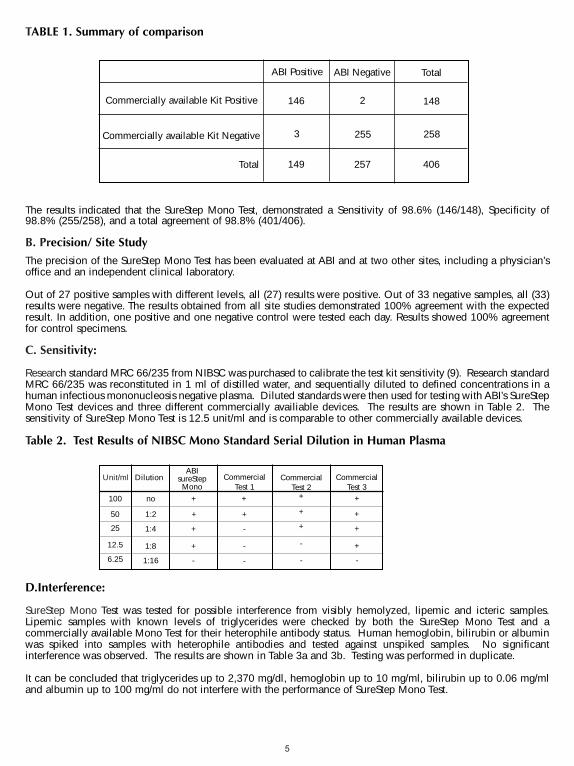
A. AccuracyThe accuracy of the SureStepTM Mono Test was evaluated in comparison to a commercially available qualitative colorimmuno-chromatographic assay for the detection of IM IgM heterophile antibodies in human serum, plasma or wholeblood. Four hundred and six human serum, plasma and whole blood samples (288 serum, 103 plasma and 15 whole blood)were used in the comparison study. The results are summarized in Table 1.
5
The results indicated that the SureStep Mono Test, demonstrated a Sensitivity of 98.6% (146/148), Specificity of98.8% (255/258), and a total agreement of 98.8% (401/406).
B. Precision/ Site Study
The precision of the SureStep Mono Test has been evaluated at ABI and at two other sites, including a physician’soffice and an independent clinical laboratory.
Out of 27 positive samples with different levels, all (27) results were positive. Out of 33 negative samples, all (33)results were negative. The results obtained from all site studies demonstrated 100% agreement with the expectedresult. In addition, one positive and one negative control were tested each day. Results showed 100% agreementfor control specimens.
C. Sensitivity:
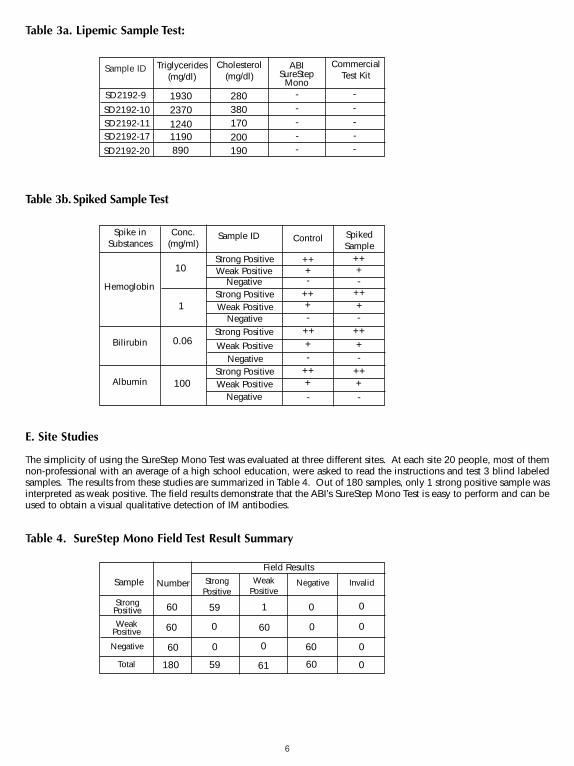
Research standard MRC 66/235 from NIBSC was purchased to calibrate the test kit sensitivity (9). Research standardMRC 66/235 was reconstituted in 1 ml of distilled water, and sequentially diluted to defined concentrations in ahuman infectious mononucleosis negative plasma. Diluted standards were then used for testing with ABI’s SureStepMono Test devices and three different commercially availiable devices. The results are shown in Table 2. Thesensitivity of SureStep Mono Test is 12.5 unit/ml and is comparable to other commercially available devices.
Table 2. Test Results of NIBSC Mono Standard Serial Dilution in Human Plasma
D.Interference:
SureStep Mono Test was tested for possible interference from visibly hemolyzed, lipemic and icteric samples.Lipemic samples with known levels of triglycerides were checked by both the SureStep Mono Test and acommercially available Mono Test for their heterophile antibody status. Human hemoglobin, bilirubin or albuminwas spiked into samples with heterophile antibodies and tested against unspiked samples. No significantinterference was observed. The results are shown in Table 3a and 3b. Testing was performed in duplicate.
It can be concluded that triglycerides up to 2,370 mg/dl, hemoglobin up to 10 mg/ml, bilirubin up to 0.06 mg/mland albumin up to 100 mg/ml do not interfere with the performance of SureStep Mono Test.
ABI Positive
Commercially available Kit Positive
Commercially available Kit Negative
ABI Negative Total
Total
146 2
255
148
258
406257
3
149
TABLE 1. Summary of comparison
Unit/ml DilutionABI
sureStepMono
CommercialTest 1
CommercialTest 2
CommercialTest 3
100
50
25
12.5
6.25
no
1:2
1:4
1:8
1:16
+
+
+
+
-
+
+
+
+
+
+
+
+
+
-
-
-
-
- -
6
Table 3a. Lipemic Sample Test:
Table 3b. Spiked Sample Test
E. Site Studies
The simplicity of using the SureStep Mono Test was evaluated at three different sites. At each site 20 people, most of themnon-professional with an average of a high school education, were asked to read the instructions and test 3 blind labeledsamples. The results from these studies are summarized in Table 4. Out of 180 samples, only 1 strong positive sample wasinterpreted as weak positive. The field results demonstrate that the ABI’s SureStep Mono Test is easy to perform and can beused to obtain a visual qualitative detection of IM antibodies.
Table 4. SureStep Mono Field Test Result Summary
Sample ID ABISureStep
Mono
CommercialTest Kit
1930
-
Triglycerides(mg/dl)
Cholesterol(mg/dl)
-
---280
380170200190890
119012402370
SD2192-9
SD2192-10
SD2192-20
SD2192-17SD2192-11
--
---
Spike inSubstances Control Spiked
Sample
+
-
Conc.(mg/ml)
Sample ID
-
-
-
-
-
-
-
++
100
0.06
1
10Strong Positive
Hemoglobin
Albumin
Bilirubin
Strong Positive
Strong Positive
Strong Positive
Weak Positive
Weak Positive
Weak Positive
Weak Positive
Negative
Negative
Negative
Negative
+
+
+
++
++
++
++++
++
++
++++
Sample WeakPositive
59
0
60
1
Number Negative Invalid
180
60
60
60
StrongPositive
Field Results
60
6059
0 0
0
0
0
00
0
61
StrongPositive
WeakPositive
Negative
Total

7
Für Tests mit Proben aus Kapillarblut oder Vollblut, die den CLIA-Bestimmungen nicht unterliegen
Für anspruchsvolle Tests mit Proben aus Serum, Plasma oder Vollblut
VERWENDUNGSZWECK
Der SureStep Mono Test ist ein Schnelltest für den visuellen, qualitativen Nachweis heterophiler Antikörper der infektiösenMononukleose (IM) in menschlichem Serum, Plasma oder Vollblut. Dieses Testkit dient als Unterstützung der Diagnose beiIM-Patienten mit typischen klinischen Symptomen und ist ausschließlich für die Anwendung durch medizinisch geschultesPersonal bestimmt.
ZUSAMMENFASSUNG
Die infektiöse Mononukleose ist eine akute, selbstlimitierende Krankheit, die durch das Epstein-Barr Virus verursacht wird(EBV) (1–3). In frühen Jahren verläuft eine Infektion mit EBV normalerweise asymptomatisch (4–5). Bei bis zu 50 % derInfektionen, die bei Jugendlichen oder jungen Erwachsenen auftreten, kommt es zu klinischen Symptomen im Zusammenhangmit der IM.
Die Diagnose der IM beruht auf der Auswertung typischer klinischer Symptome und serologischer Veränderungen. Dieserologische Diagnose der IM wurde durch den Nachweis heterophiler und EBV-spezifischer Antikörper bestätigt (3–5). Beiden meisten Erwachsenen ist der heterophile Antikörper zu einem gewissen Zeitpunkt während einer IM nachweisbar. DerNachweis von heterophilen Antikörpern als Unterstützung der Diagnose von IM ist eine allgemein anerkannte Methodeunter Ärzten (3–7). Der SureStep Mono Test verwendet Rindererythrozyten, die eine höhere Sensitivität und Spezifität alsExtrakte von anderen Spezies aufweisen. (4)
TESTPRINZIP
Der SureStep Mono Test wurde zum Nachweis von IM durch die visuelle Interpretation einer Farbentwicklung inder Testeinheit entwickelt, bekannt als Sandwich-Immunoassay mit einer soliden Phase und einem Goldkonjugat.Die Testeinheit besteht aus einem Membranstreifen, der im Bereich des Testfelds mit heterophilen Antigenen undim Bereich des Kontrollfelds mit Anti-Maus-Antikörpern (Ziege) beschichtet ist. Am Ende der Membran befindetsich ein Anti-Human-IgM-Antikörper-kolloidales Goldkonjugat-Polster. Eine Mischung aus dem kolloidalenGoldkonjugat und der Probe und dem Entwicklungspuffer wandert durch Kapillaraktivität chromatografisch ander Membran entlang. Wenn sich in der Patientenprobe IM-heterophile Antikörper befinden, wandert dasGemisch auf das Testfeld zu und formt eine sichtbare Linie, da die Antikörper mit den heterophilen AntigenenKomplexe bilden. Wenn sich keine IM-heterophilen Antikörper in der Probe befinden, bildet sich keine sichtbareFarblinie im Testfeld. Deshalb deutet das Vorhandensein einer Farblinie im Testfeld auf ein positives Ergebnis. ImKontrollfeld erscheint immer eine Farblinie. Diese Kontrolllinie zeigt an, dass der Test ordnungsgemäßdurchgeführt und die Testeinheit sachgemäß verwendet wurde.
AUFBEWAHRUNG UND STABILITÄT
Bis zum Verfallsdatum muss der Test in der verschlossenen Verpackung entweder gekühlt oder bei Raumtemperatur (2–30 °C) aufbewahrt werden.
VORSICHTSMASSNAHMEN
• Nur zur In-vitro-Diagnostik.
• Nur zur Anwendung durch medizinisches Fachpersonal und im Rahmen des Klinik- oder Ambulanzbetriebs• Alle Patientenproben sollten als potenziell krankheitsübertragend behandelt werden.• Keine Reagenzien aus verschiedenen Chargen mischen.
DIE ANWEISUNGEN ZUR CLIA-KOMPLEXITÄT SIND UNTEN IM ABSCHNITT QUALITÄTSKONTROLLEAUFGEFÜHRT. BITTE DIE ANWEISUNGEN DER VORGESCHRIEBENEN RICHTLINIEN DES LABORS BEFOLGEN.
MONO TEST
DEUTSCH
Testkit nicht wiederverwenden.

8
• Die Folienverpackung erst unmittelbar vor Durchführung des Tests öffnen.• Kein Vollblut verwenden, das länger als drei Tage gelagert wurde.• Keine übermäßig hämolysierten Blutproben verwenden.• Die Entwicklungspuffer und Kontrollen enthalten Natriumazid. Natriumazid kann mit Blei- oder Kupferrohren
reagieren und potenziell explosive Metallazide bilden. Beim Entsorgen dieser Lösungen stets mit viel Wassernachspülen, um eine Anhäufung dieser Azide zu vermeiden.
Die Extraktionsreagenzien sind leicht ätzend. Kontakt mit Augen und Schleimhäuten vermeiden. Im Fall eines zufälligenKontakts sofort mit viel Wasser auswaschen.
Die Positiv- und Negativkontrollen enthalten Natriumazid, das mit Blei- und oder Kupferrohren reagieren und potenziellexplosive Metallazide bilden kann. Beim Entsorgen dieser Lösungen stets mit viel Wasser nachspülen, um eine Anhäufungdieser Azide zu vermeiden.
Warnung: Potenziell biogefährliches MaterialJede Einheit von Spenderserum/-plasma, die zur Vorbereitung der Positiv- und Negativkontrollen verwendet wurde, wurdemittels einer von der FDA (US-amerikanische Nahrungsmittel- und Medikamentenbehörde) genehmigten Methode auf Anti-HIV-1/HIV2, HBsAg und Anti-HCV getestet und für negativ befunden. Bei der Handhabung und Entsorgung dieser Produktevom Biosicherheitsgrad 2 ist jedoch Vorsicht geboten, entsprechend den Anweisungen der Centers for DiseaseControl/National Institutes of Health Manual “Biosafety in Micobiological and Biomedical Laboratories”, 1984.
REAGENZIEN UND ENTHALTENE MATERIALIEN
• 25 einzeln verpackte Testeinheiten. Jede Testeinheit enthält einen Streifen mit einer mit heterophilem Antigen beschichtetenMembran und einem gefärbten Antikörper-Polster.
• Entwicklungspuffer (8 ml) : mit 0,1 M Phosphat gepufferter Kochsalzlösung mit Zusätzen und 0,1 % Natriumazid.
• Negativkontrolle (0,2 ml) : Normales menschliches Plasma oder in Kochsalzlösung mit 0,2 % Natriumazidverdünntes Serum.
• Positivkontrolle (0,2 ml) : IM-heterophiler Antikörper, positives menschliches Plasma oder in Kochsalzlösungmit 0,2 % Natriumazid verdünntes Serum.
• 25 Wegwerf-Transferpipetten
• Ein Anleitungsblatt.
PROBENNAHME UND VERARBEITUNG
Kapillarblut 1. Die vorgesehene Einstichstelle mit einem alkoholgetränkten Tupfer reinigen.2. Die Fingerbeere zusammendrücken und mit einer sterilen Stechhilfe punktieren.3. Den ersten Bluttropfen mit steriler Gaze oder steriler Watte wegwischen.4. Den zweiten Tropfen direkt in die Probenvertiefung der Testeinheit fließen lassen oder die gelieferte Pipette benutzen,
um frisches Blut zu erhalten. Zwei Tropfen (etwa 40 µl) in die Probenvertiefung geben.
Vollblut1. Ein zertifizierter Phlebotomist sollte durch Venenpunktion Vollblut abnehmen und das Blut in ein Sammelröhrchen (mit
EDTA, Zitrat oder Kochsalzlösung) mit violettem, blauem oder grünem Deckel geben.2. Das Vollblut kann für sofortige Tests benutzt werden oder bei 2–8 °C bis zu drei Tagen gelagert werden.
Plasma1. Ein zertifizierter Phlebotomist sollte durch Venenpunktion Vollblut abnehmen und das Blut in ein Sammelröhrchen (mit
EDTA, Zitrat oder Kochsalzlösung) mit violettem, blauem oder grünem Deckel geben.2. Durch Zentrifugieren das Plasma abtrennen.3. Das Plasma sorgfältig zum Testen entziehen oder mit einem Etikett versehen und bei 2–8 °C bis zu zwei Wochen
lagern. Gefroren bei –20 °C kann Plasma bis zu einem Jahr gelagert werden.
Serum1. Ein zertifizierter Phlebotomist sollte durch Venenpunktion Vollblut abnehmen und das Blut in ein Sammelröhrchen
geben, das keine antikoagulierenden Substanzen enthält.2. Dem Blut zum Gerinnen Zeit lassen und das Serum durch Zentrifugieren abscheiden.3. Das Serum sorgfältig zum Testen aspirieren oder mit einem Etikett versehen und bei 2–8 °C bis zu zwei Wochen
lagern. Gefroren kann Serum bei –20 °C bis zu einem Jahr gelagert werden.
BUF DEV

9
TESTVERFAHREN
3. Sofort 3–4 Tropfen Entwicklungspuffer zugeben.
4. Nach der Zugabe des Entwicklungspuffers warten, bis die Farblinien erscheinen. Je nach Konzentration dervorhandenen heterophilen Antikörper sind positive Ergebnisse bereits nach 3 Minuten ablesbar. Ein negativesTestergebnis gilt jedoch erst nach einer Reaktionszeit von 5 Minuten als gesichert. Nach 8 Minuten kann dasTestergebnis nicht mehr sicher ausgewertet werden.
Testeinheit
Kontrollfeld Testfeld
Probenvertiefung
Testeinheit, Entwicklungspuffer, Patientenproben und dieKontrollen sollten vor der Durchführung des Tests aufRaumtemperatur (20 bis 30 °C) gebracht werden. DieVerpackung erst unmittelbar vor Durchführung des Tests öffnen.
1. Die Testeinheit aus der Schutzverpackung nehmen. (Um einekondensationsbedingte Feuchtigkeitsbildung auf der Membran zuverhindern, die Verpackung erst öffnen, wenn die TesteinheitRaumtemperatur erreicht hat). Die Testeinheit mit einem Patienten-bzw. Kontrolletikett kennzeichnen.
2. Die Probe in die Probenvertiefung geben:
Kapillarblut: Den zweiten Tropfen direkt in dieProbenvertiefung der Testeinheit fließen lassen oder diePipette benutzen, um frisches Blut zu erhalten. Zwei Tropfen(etwa 40 µl) in die Probenvertiefung geben.
Vollblutproben in Sammelröhrchen: Zwei Tropfen (etwa 40 µl) indie Probenvertiefung geben und dabei die gelieferte Transfer-Pipette senkrecht halten.
Plasma- oder Serumproben: Einen Tropfen (etwa 20 µl) Serum oderPlasma in die Probenvertiefung geben und dabei die gelieferteTransfer-Pipette senkrecht halten.
Transfer-Pipette
AUSWERTUNG DER ERGEBNISSE
Positiv:Es erscheinen zwei farbige Linien. Eine im Kontrollfeld (C) und eine im Testfeld (T).Beim Testen stark positiver Proben kann die Farbintensität der Kontrolllinie heller alserwartet ausfallen. Die Farbintensität der beiden Linien sollte nicht verglichenwerden.
Negativ:Es erscheint nur eine farbige Linie im Kontrollfeld. Keine sichtbare schwach gefärbteLinie im Testfeld (T).
Ungültig:Das Fehlen einer Linie im Kontrollfeld ist als Hinweis auf einen Verfahrensfehler oderauf unbrauchbar gewordene Testreagenzien zu werten. Der Test sollte als ungültigbetrachtet werden, da es Hinweise auf einen Verfahrensfehler oder auf unbrauchbargewordene Reagenzien gibt. Den Test mit einer neuen Testeinheit wiederholen.Bestehen die Probleme fort, bitte den technischen Kundendienst von Applied Biotechanrufen. Innerhalb der USA (888) 578-7956, international +1 858 713-9668. Anrufewerden auf Englisch beantwortet. Für andere Sprachen bitte eine Nachricht [email protected] senden.
C T
C T
C T
C T
C TMONO

10
QUALITÄTSKONTROLLE
Interne VerfahrenskontrollenDer Test enthält eine testimmanente Verfahrenskontrolle. Eine Farblinie im Kontrollfeld (C) gilt als interneVerfahrenskontrolle und als Hinweis auf einen ordnungsgemäßen Testablauf und reaktive Reagenzien.
Ein unverfärbter Hintergrund im Ergebnisfenster gilt als negative interne Verfahrenskontrolle. Wenn Vollblutproben getestetwerden, kann der Hintergrund jedoch wegen der tiefen Hämolyse einiger roter Blutzellen leicht rötlich erscheinen. Das istakzeptierbar, so lange das Testergebnis gut abgelesen werden kann. Der Test ist ungültig, wenn sich der Hintergrund nichtklärt und das Ablesen des Testergebnisses behindert.
Externe QualitätskontrolleGute Laborpraktiken empfehlen externe Qualitätskontrollen, um die Funktionalität der Reagenzien und eine sachgemäßeDurchführung zu garantieren. Mit dem Kit werden Positiv- und Negativkontrollen mitgeliefert. Diese Kontrollen beruhenauf menschlichem Serum oder Plasma und werden wie Patientenproben getestet. Beim Testen mit den Kontrollen einenTropfen (etwa 20 µl) der Positiv- oder Negativkontrolle mit der mitgelieferten Transferpipette in die Probenvertiefung geben,indem die Pipette senkrecht gehalten wird. Sofort 3 oder 4 Tropfen Entwicklungspuffer zugeben. Ein positives Ergebnis wirddurch zwei Linien angezeigt: eine im Testfeld (T) und eine im Kontrollfeld (C). Ein negatives Ergebnis wird durch nur eineLinie im Kontrollfeld (C) angezeigt. Die Ergebnisse können nach 5 Minuten abgelesen werden und sollten nach 8 Minutennicht mehr ausgewertet werden. Wenn die Kontrollen anders als erwartet ausfallen, dürfen die Ergebnisse nicht ausgewertetwerden. Den Test wiederholen oder den technischen Kundendienst informieren.
FÜR LABORS, DIE DEN CLIA-BESTIMMUNGEN NICHT UNTERLIEGEN:
Bei jedem neuen Testkit muss sowohl eine externe Negativ- als auch Positivkontrolle getestet werden. Jeder Benutzer musspro Kit mit 25 Tests eine externe Positiv- und Negativkontrolle testen.
FÜR LABORS MIT ANSPRUCHSVOLLEN TESTS UND FÜR DEN KLINIK- ODER AMBULANZBETRIEB:
Es empfiehlt sich, mit jeder neuen Sendung oder jeder neuen Chargennummer eine externe Positiv- und eineNegativkontrolle zu testen und den jeweils geltenden örtlichen Bestimmungen Folge zu leisten.
EINSCHRÄNKUNGEN
Dieses Testkit ist zum qualitativen Nachweis von IgM-Antikörpern des IM-heterophilen Antigens bestimmt. Ein positivesErgebnis deutet auf das Vorhandensein von IgM-Antikörpern des heterophilen Antigens hin.
Das Testkit sollte für Patienten benutzt werden, bei denen aufgrund der Symptome eine IM vermutet wird. Die Diagnoseeiner IM sollte im Zusammenhang mit anderen klinischen Befunden gestellt werden.
Ein negatives Ergebnis schließt eine mögliche IM nicht aus, da Antikörper des heterophilen Antigens abwesend sein könnenoder nicht in genügender Anzahl zum Nachweisen vorhanden sein können.
ERWARTETE WERTE
Während der Akutphase der IM sind die heterophilen Antikörper in 80–85 % aller Patienten nachweisbar. Die heterophilenAntikörper sind im ersten Krankheitsmonat nachweisbar und verringern sich sehr schnell nach der vierten Woche (5).
LEISTUNGSMERKMALE
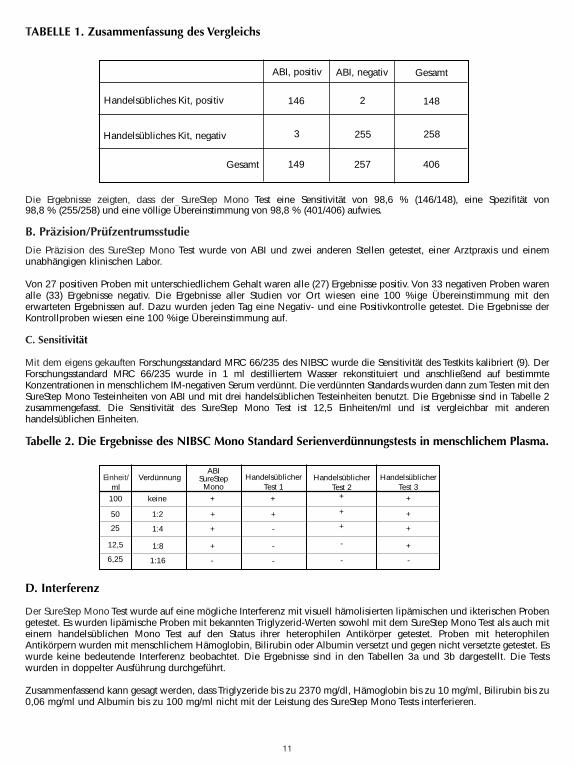
A. GenauigkeitDie Genauigkeit des SureStepTM Mono Test wurde im Vergleich mit einem handelsüblichen qualitativen,chromatographischen Farb-Immunoassay zum Nachweis von IM IgM-heterophilen Antikörpern in menschlichem Serum,Plasma und Vollblut, verglichen. In der Vergleichsstudie wurden vierhundertsechs Proben aus menschlichem Serum,Plasma und Vollblut (288 Serum, 103 Plasma und 15 Vollblut) getestet. Die Ergebnisse werden in Tabelle 1zusammengefasst.
11
Die Ergebnisse zeigten, dass der SureStep Mono Test eine Sensitivität von 98,6 % (146/148), eine Spezifität von98,8 % (255/258) und eine völlige Übereinstimmung von 98,8 % (401/406) aufwies.
B. Präzision/Prüfzentrumsstudie
Die Präzision des SureStep Mono Test wurde von ABI und zwei anderen Stellen getestet, einer Arztpraxis und einemunabhängigen klinischen Labor.
Von 27 positiven Proben mit unterschiedlichem Gehalt waren alle (27) Ergebnisse positiv. Von 33 negativen Proben warenalle (33) Ergebnisse negativ. Die Ergebnisse aller Studien vor Ort wiesen eine 100 %ige Übereinstimmung mit denerwarteten Ergebnissen auf. Dazu wurden jeden Tag eine Negativ- und eine Positivkontrolle getestet. Die Ergebnisse derKontrollproben wiesen eine 100 %ige Übereinstimmung auf.
C. Sensitivität
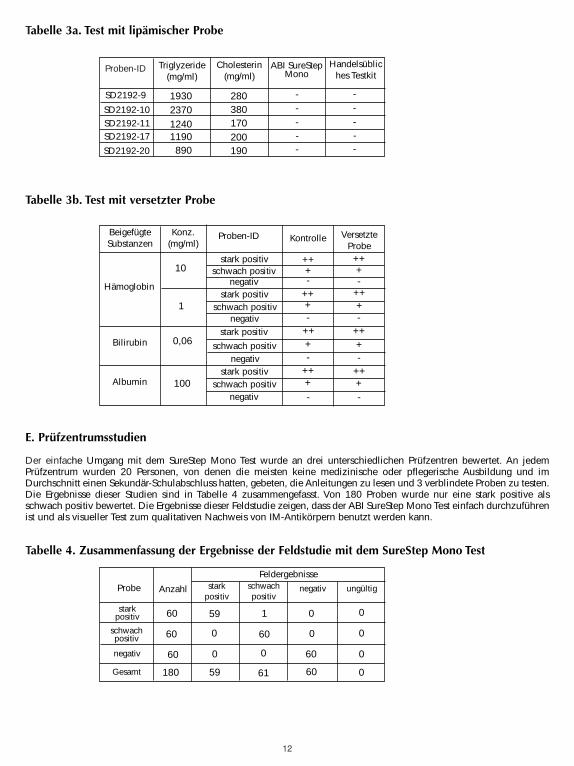
Mit dem eigens gekauften Forschungsstandard MRC 66/235 des NIBSC wurde die Sensitivität des Testkits kalibriert (9). DerForschungsstandard MRC 66/235 wurde in 1 ml destilliertem Wasser rekonstituiert und anschließend auf bestimmteKonzentrationen in menschlichem IM-negativen Serum verdünnt. Die verdünnten Standards wurden dann zum Testen mit denSureStep Mono Testeinheiten von ABI und mit drei handelsüblichen Testeinheiten benutzt. Die Ergebnisse sind in Tabelle 2zusammengefasst. Die Sensitivität des SureStep Mono Test ist 12,5 Einheiten/ml und ist vergleichbar mit anderenhandelsüblichen Einheiten.
Tabelle 2. Die Ergebnisse des NIBSC Mono Standard Serienverdünnungstests in menschlichem Plasma.
D. Interferenz
Der SureStep Mono Test wurde auf eine mögliche Interferenz mit visuell hämolisierten lipämischen und ikterischen Probengetestet. Es wurden lipämische Proben mit bekannten Triglyzerid-Werten sowohl mit dem SureStep Mono Test als auch miteinem handelsüblichen Mono Test auf den Status ihrer heterophilen Antikörper getestet. Proben mit heterophilenAntikörpern wurden mit menschlichem Hämoglobin, Bilirubin oder Albumin versetzt und gegen nicht versetzte getestet. Eswurde keine bedeutende Interferenz beobachtet. Die Ergebnisse sind in den Tabellen 3a und 3b dargestellt. Die Testswurden in doppelter Ausführung durchgeführt.
Zusammenfassend kann gesagt werden, dass Triglyzeride bis zu 2370 mg/dl, Hämoglobin bis zu 10 mg/ml, Bilirubin bis zu0,06 mg/ml und Albumin bis zu 100 mg/ml nicht mit der Leistung des SureStep Mono Tests interferieren.
ABI, positiv
Handelsübliches Kit, positiv
Handelsübliches Kit, negativ
ABI, negativ Gesamt
Gesamt
146 2
255
148
258
406257
3
149
TABELLE 1. Zusammenfassung des Vergleichs
Einheit/ml
VerdünnungABI
SureStep Mono
HandelsüblicherTest 1
Handelsüblicher Test 2
HandelsüblicherTest 3
100
50
25
12,5
6,25
keine
1:2
1:4
1:8
1:16
+
+
+
+
-
+
+
+
+
+
+
+
+
+
-
-
-
-
- -
12
Tabelle 3a. Test mit lipämischer Probe
Tabelle 3b. Test mit versetzter Probe
E. Prüfzentrumsstudien
Der einfache Umgang mit dem SureStep Mono Test wurde an drei unterschiedlichen Prüfzentren bewertet. An jedemPrüfzentrum wurden 20 Personen, von denen die meisten keine medizinische oder pflegerische Ausbildung und imDurchschnitt einen Sekundär-Schulabschluss hatten, gebeten, die Anleitungen zu lesen und 3 verblindete Proben zu testen.Die Ergebnisse dieser Studien sind in Tabelle 4 zusammengefasst. Von 180 Proben wurde nur eine stark positive alsschwach positiv bewertet. Die Ergebnisse dieser Feldstudie zeigen, dass der ABI SureStep Mono Test einfach durchzuführenist und als visueller Test zum qualitativen Nachweis von IM-Antikörpern benutzt werden kann.
Tabelle 4. Zusammenfassung der Ergebnisse der Feldstudie mit dem SureStep Mono Test
Proben-ID ABI SureStepMono
Handelsübliches Testkit
1930
-
Triglyzeride(mg/ml)
Cholesterin(mg/ml)
-
---280
380170200190890
119012402370
SD2192-9
SD2192-10
SD2192-20
SD2192-17SD2192-11
--
---
BeigefügteSubstanzen Kontrolle Versetzte
Probe
+
-
Konz.(mg/ml)
Proben-ID
-
-
-
-
-
-
-
++
100
0,06
1
10stark positiv
Hämoglobin
Albumin
Bilirubin
stark positiv
stark positiv
stark positiv
schwach positiv
schwach positiv
schwach positiv
schwach positiv
negativ
negativ
negativ
negativ
+
+
+
++
++
++
++++
++
++
++++
Probe schwachpositiv
59
0
60
1
Anzahl negativ ungültig
180
60
60
60
stark positiv
Feldergebnisse
60
6059
0 0
0
0
0
00
0
61
starkpositiv
schwachpositiv
negativ
Gesamt

13
Para exentos de complejidad CLIA : probar con muestras capilares o de sangre entera
Para moderadamente complejos: probar con muestras de suero, plasma o sangre entera
USO PREVISTO
La prueba SureStep Mono es una prueba rápida para la detección visual y cualitativa de anticuerpos heterófilos específicosde la mononucleosos infecciosa (MI) en suero, plasma o sangre entera humanos. Este equipo de prueba se ha diseñadosólo para su uso profesional en laboratorio como una ayuda en el diagnóstico de los pacientes de MI que presentansíntomas clínicos característicos.
RESUMEN
La mononucleosis infecciosa es una afección aguda y autolimitada causada por el virus de Epstein-Barr (VEB) (1–3). Lainfección por el VEB a una edad temprana suele ser asintomática (4–5). Sin embargo, hasta el 50 % de las infecciones que seproducen al principio de la edad adulta y durante la adolescencia desarrollan manifestaciones clínicas asociadas con la MI.
El diagnóstico de la MI se basa en la evaluación de síntomas clínicos característicos y cambios serológicos. El diagnósticoserológico de la MI se ha demostrado por la detección de anticuerpos heterófilos y específicos del VEB (3–5). En la mayoríade los adultos, el anticuerpo heterófilo puede detectarse en algún momento durante la MI. Una práctica ampliamenteaceptada entre los médicos es usar la detección de los anticuerpos heterófilos como ayuda en el diagnóstico de la MI (3–7).La prueba SureStep Mono utiliza extracto de eritrocito de origen bovino, que presenta una mayor sensibilidad yespecificidad que los extractos de otras especies. (4)
PRINCIPIO DE LA PRUEBA
La prueba SureStep Mono se ha diseñado para detectar la MI mediante la interpretación visual del desarrollo de un coloren el dispositivo de prueba, un inmunoensayo de sándwich de conjugado de oro de fase sólida. El dispositivo de pruebacuenta con una lámina de membrana que está recubierta previamente con antígenos heterófilos en la zona de la línea deprueba y de anticuerpo de cabra antirratón en la zona de la línea de control. Al final de la membrana se sitúa la almohadillade anticuerpo IgM antihumano conjugado con oro coloidal. Una mezcla de conjugado de oro coloidal, junto con lamuestra y el tampón de revelado, recorren la membrana cromatográficamente por capilaridad. Si los anticuerposheterófilos de la MI están presentes en la muestra del paciente, la muestra migra hasta la zona de la línea de prueba y formauna línea visible a medida que el anticuerpo forma complejo con el antígeno heterófilo. Si la muestra no contiene losanticuerpos heterófilos de la MI, no se forma ninguna línea coloreada en la zona de la línea de prueba. Por tanto, lapresencia de una línea coloreada en la zona de la línea de prueba indica un resultado positivo. La zona de controlpresentará siempre una línea coloreada. La línea de control sirve como indicador de procedimiento, para confirmar elfuncionamiento adecuado de la prueba y del dispositivo.
ALMACENAMIENTO Y ESTABILIDAD
El equipo de prueba debe almacenarse refrigerado o a temperatura ambiente 2–30 °C en su bolsa hermética durante elperiodo de validez.
PRECAUCIONES
• Sólo para uso diagnóstico in vitro.
• Para uso profesional y en laboratorio.• Todas las muestras de los pacientes deben tratarse teniendo en cuenta su posible capacidad infecciosa.• No mezclar reactivos de lotes diferentes.• No abrir la bolsa hasta el momento de realizar la prueba.
LAS INSTRUCCIONES REFERIDAS A LA COMPLEJIDAD CLIA SE ENUMERAN A CONTINUACIÓN, DENTRO DE LA SECCIÓN DE CONTROL DE CALIDAD.
SIGA LAS INSTRUCCIONES ADECUADAS ACORDE CON LAS DIRECTIVAS DE SU LABORATORIO.
PRUEBA MONO
ESPAÑOL
No reutilizar la prueba.

14
• No usar sangre entera que haya estado almacenada durante más de tres días.• No se deben utilizar muestras muy hemolizadas.• El tampón de revelado y los controles contienen azida sódica. La azida sódica puede reaccionar con las
conducciones de plomo o cobre y formar azidas metálicas potencialmente explosivas. Al eliminar estas soluciones,lavarlas siempre con grandes cantidades de agua para evitar la acumulación de azida.
Los reactivos de extracción son ligeramente cáusticos. Evitar el contacto con los ojos o las mucosas. En caso de contactoaccidental, lavar meticulosamente con agua.
Los controles negativo y positivo contienen azida sódica que puede reaccionar con las conducciones de plomo o cobre yformar azidas metálicas potencialmente explosivas. Al eliminar estas soluciones, lavarlas siempre con grandes cantidadesde agua para evitar la acumulación de azida.
Advertencia: Material potencialmente biopeligrosoCada una de las bolsas de plasma humano o suero utilizadas para la preparación de los controles positivo o negativo fueronanalizadas con los métodos autorizados por la FDA de los EE.UU. para la detección de la presencia de anti-VIH-1/VIH-2,AgsHB y anti-VHC, obteniéndose resultados negativos. Sin embargo, se debe tener cuidado al manipular y eliminar estoselementos con el nivel 2 de bioseguridad recomendado en el documento Biosafety Microbiological and BiomedicalLaboratories (Bioseguridad en laboratorios microbiológicos y biomédicos), editado en 1984 por los organismos Centers forDisease Control y National Institutes of Health de los EE.UU.
REACTIVOS Y MATERIALES SUMINISTRADOS
• 25 dispositivos de prueba envasados individualmente. Cada dispositivo de prueba contiene una tira de prueba que cuentacon una membrana recubierta con antígeno heterófilo y una almohadilla de anticuerpo coloreada.
• Tampón de revelado (8 ml) : Solución salina tamponada con fosfato a una concentración de 0,1 M,con aditivos y azida sódica al 0,1 %.
• Control negativo (0,2 ml) : Plasma o suero humano normal diluido en una solución salina con azidasódica al 0,2 %.
• Control positivo (0,2 ml) : Plasma o suero humano positivo en anticuerpos heterófilos de MI,diluido en una solución salina con azida sódica al 0,2 %.
• 25 pipetas de transferencia desechables.
• Un prospecto de instrucciones.
OBTENCIÓN Y MANIPULACIÓN DE LA MUESTRAMuestra capilar 1. Limpiar con algodón humedecido en alcohol la zona de la punción.2. Apretar la yema del dedo y realizar la punción con una lanceta estéril.3. Con gasa estéril o algodón, eliminar la primera gota de sangre.4. Dejar que la segunda gota caiga directamente en el pocillo de muestra del dispositivo de prueba o usar la pipeta
incluida en el equipo para obtener sangre fresca. Verter dos gotas (aproximadamente 40 µl) en el pocillo de muestra.
Sangre entera1. Extraer la sangre entera mediante punción venosa y depositarla en un tubo de obtención con tapón morado, azul
o verde (que contienen EDTA, citrato o heparina, respectivamente).2. La sangre entera puede usarse inmediatamente para la prueba o conservarse hasta un máximo de tres días a una
temperatura de 2–8 °C.
Plasma1. Extraer la sangre entera mediante punción venosa y depositarla en un tubo de obtención con tapón morado, azul
o verde (que contienen EDTA, citrato o heparina, respectivamente).2. Separar el plasma por centrifugado.3. Tomar cuidadosamente el plasma necesario para la prueba o etiquetarlo y conservarlo a 2–8 °C hasta un máximo de dos
semanas. El plasma puede permanecer congelado a una temperatura de -20 °C hasta un año como máximo.
Suero1. Recoger la sangre entera mediante punción venosa y depositarla en un tubo de obtención con tapón rojo
(que no contiene anticoagulantes).2. Esperar a que la sangre se coagule y separar el suero por centrifugado.3. Tomar cuidadosamente el suero necesario para la prueba o etiquetarlo y conservarlo a 2–8 °C hasta un máximo de dos
BUF DEV

15
semanas. El suero puede permanecer congelado a una temperatura de -20 °C hasta un año como máximo.PROCEDIMIENTO DE LA PRUEBA
3. Verter inmediatamente 3 ó 4 gotas de tampón de revelado.
4. Tras verter el tampón de revelado, esperar a que aparezcan las líneas coloreadas. Dependiendo de la concentraciónpresente de anticuerpos heterófilos, los resultados positivos pueden observarse a los tres minutos. Sin embargo, paraconfirmar un negativo, se requiere el tiempo completo de reacción, de 5 minutos. No deben interpretarse los resultadosde la prueba una vez transcurridos más de 8 minutos.
Dispositivo dela prueba
Zona de control Zona de prueba
Pocillo demuestra
El dispositivo de prueba, el tampón de revelado, las muestras delos pacientes y los controles deben alcanzar la temperaturaambiente (20–30 °C) antes de la prueba. No abrir las bolsashasta el momento de realizar la prueba.
1. Extraer el dispositivo de prueba de su bolsa protectora (esperar a queel dispositivo alcance la temperatura ambiente antes de abrir labolsa para evitar la condensación de humedad sobre la membrana).Marcar el dispositivo con la identificación del paciente o del control.
2. Dispensar la muestra en el pocillo de muestra.
Sangre de muestra capilar: dejar que la segunda gota caigadirectamente en el pocillo de muestra del dispositivo de prueba ousar la pipeta incluida en el equipo para obtener sangre fresca.Verter dos gotas (aproximadamente 40 µl) en el pocillo de muestra.
Muestras de sangre entera en tubos de obtención: verter dos gotas(aproximadamente 40 µl) en el pocillo de muestra, sosteniendo enposición vertical la pipeta de transferencia incluida en el equipo.
Muestras de plasma o suero: verter una gota (aproximadamente20 µl) en el pocillo de muestra, sosteniendo en posición vertical lapipeta de transferencia incluida en el equipo.
Pipeta detransferencia
INTERPRETACIÓN DE LOS RESULTADOS
Positivo:Aparecen dos líneas coloreadas. Una en la zona de control (C) y una línea coloreadaen la zona de la prueba (T). Al evaluar muestras con positivos fuertes, la intensidadde la línea de control puede ser menor de la esperada. No se recomienda compararla intensidad de las líneas.
Negativo:Sólo aparece una línea coloreada en la zona de la línea de control. No apareceninguna línea coloreada débil en la zona de la línea de prueba (T).
No válido:La ausencia total de una línea en la zona de control indica un error en el procedimientoo que el reactivo de la prueba se ha deteriorado. La prueba debe considerarse nuladebido a la posibilidad de que el procedimiento seguido fuera incorrecto o de que sehayan deteriorado los reactivos. Repetir la prueba con otro dispositivo de prueba. Silos problemas persisten, llamar al Servicio Técnico de Applied Biotech, al número(888) 578-7956 para llamadas desde los EE.UU. o al número +1 858 713-9668 parallamadas desde otros países. Las llamadas serán contestadas por personal de hablainglesa. Para los demás idiomas, contactar por correo electrónico [email protected].
C T
C T
C T
C T
C TMONO

16
CONTROL DE CALIDAD
Control interno de procedimientoLa prueba incluye un procedimiento de control. La aparición de una línea coloreada en la zona de control (C) se consideraun control interno de procedimiento que indica un funcionamiento correcto y la reacción de los reactivos.
Un fondo transparente en la ventana de resultados se considera un control interno negativo. Sin embargo, si la prueba serealiza con muestras de sangre entera, el fondo puede presentar un tono ligeramente rojizo debido a la hemólisis de bajonivel de algunos glóbulos rojos. Se considera aceptable siempre y cuando no interfiera con la lectura del resultado de laprueba. La prueba se considera no válida si el color del fondo sigue siendo intenso y dificulta la lectura del resultado dela prueba.
Control de calidad externoLas prácticas de laboratorio adecuadas recomiendan el uso de controles externos para verificar la funcionalidad y laactuación correcta de los reactivos. El equipo de prueba se suministra con un control positivo y uno negativo. Estoscontroles se basan en el suero o el plasma humano y se prueban siguiendo el mismo método que con las muestras de lospacientes. Al probar los controles, verter una gota (aproximadamente 20 µl) de control positivo o negativo en el pocillode muestra usando la pipeta de transferencia incluida en el equipo, manteniéndola en una posición vertical. Verterinmediatamente 3 ó 4 gotas de tampón de revelado. Los positivos se indican con dos líneas, una en la zona de la prueba(T) y otra en la zona de control (C). Los negativos se indican con una sola línea en la zona de control (C). Observar elresultado a los 5 minutos y no interpretar una vez transcurridos más de 8 minutos. Si los controles no actúan de la formaesperada, no interpretar los resultados de las pruebas. Repetir la prueba o ponerse en contacto con el Servicio Técnico.
PARA LABORATORIOS EXENTOS DE COMPLEJIDAD CLIA:
Es necesario hacer tanto un control externo positivo como uno negativo al abrir un nuevo equipo de prueba. Cada operariodebe realizar un control externo positivo y uno negativo cada 25 equipos de prueba.
PARA LABORATORIOS MODERADADMENTE COMPLEJOS O SERVICIOS SANITARIOSPROFESIONALES:
Se recomienda realizar un control externo positivo y uno negativo con cada suministro o al cambiar de número de lote deequipos, además de en las situaciones exigidas por la normativa estatal y local.
LIMITACIONES
Este equipo de prueba debe usarse para la detección cualitativa de anticuerpos IgM del antígeno heterófilo de la MI. Unresultado positivo sugiere la presencia de anticuerpos IgM del antígeno heterófilo.
Este equipo de prueba debe usarse con personas que presenten síntomas que hagan sospechar que padecen la MI. Eldiagnóstico de la MI debe hacerse con una confirmación a través de otros resultados clínicos.
Un resultado negativo no excluye totalmente la posibilidad de la MI, dado que los anticuerpos del antígeno heterófilopueden no estar presentes o aparecer en una cantidad insuficiente para su detección.
EXPECTATIVA DE RESULTADOS
Durante la fase aguda de la MI, los anticuerpos heterófilos pueden detectarse en el 80–85 % de los pacientes. Losanticuerpos heterófilos pueden detectarse durante el primer mes de la afección y su número decrece rápidamente despuésde la cuarta semana (5).
CARACTERÍSTICAS DE FUNCIONAMIENTO
A. ExactitudLa exactitud de la prueba SureStepTM Mono fue evaluada en comparación con un ensayo inmunocromatográfico cualitativocomercial para la detección de anticuerpos heterófilos IgM de MI en suero, plasma o sangre entera humanos. El estudiocomparativo incluía cuatrocientas muestras de suero, plasma o sangre entera humanos (288 de suero, 103 de plasma y15 de sangre entera). Los resultados se resumen en la Tabla 1.
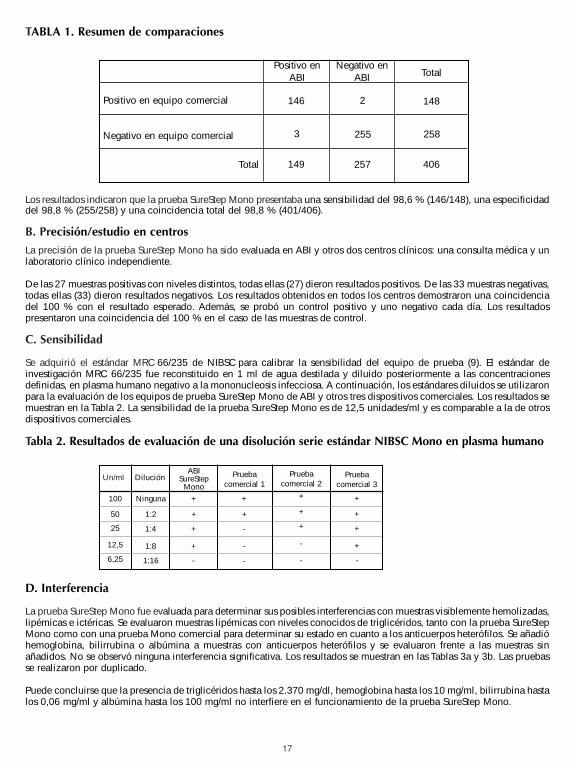
17
Los resultados indicaron que la prueba SureStep Mono presentaba una sensibilidad del 98,6 % (146/148), una especificidaddel 98,8 % (255/258) y una coincidencia total del 98,8 % (401/406).
B. Precisión/estudio en centros
La precisión de la prueba SureStep Mono ha sido evaluada en ABI y otros dos centros clínicos: una consulta médica y unlaboratorio clínico independiente.
De las 27 muestras positivas con niveles distintos, todas ellas (27) dieron resultados positivos. De las 33 muestras negativas,todas ellas (33) dieron resultados negativos. Los resultados obtenidos en todos los centros demostraron una coincidenciadel 100 % con el resultado esperado. Además, se probó un control positivo y uno negativo cada día. Los resultadospresentaron una coincidencia del 100 % en el caso de las muestras de control.
C. Sensibilidad
Se adquirió el estándar MRC 66/235 de NIBSC para calibrar la sensibilidad del equipo de prueba (9). El estándar deinvestigación MRC 66/235 fue reconstituido en 1 ml de agua destilada y diluido posteriormente a las concentracionesdefinidas, en plasma humano negativo a la mononucleosis infecciosa. A continuación, los estándares diluidos se utilizaronpara la evaluación de los equipos de prueba SureStep Mono de ABI y otros tres dispositivos comerciales. Los resultados semuestran en la Tabla 2. La sensibilidad de la prueba SureStep Mono es de 12,5 unidades/ml y es comparable a la de otrosdispositivos comerciales.
Tabla 2. Resultados de evaluación de una disolución serie estándar NIBSC Mono en plasma humano
D. Interferencia
La prueba SureStep Mono fue evaluada para determinar sus posibles interferencias con muestras visiblemente hemolizadas,lipémicas e ictéricas. Se evaluaron muestras lipémicas con niveles conocidos de triglicéridos, tanto con la prueba SureStepMono como con una prueba Mono comercial para determinar su estado en cuanto a los anticuerpos heterófilos. Se añadióhemoglobina, bilirrubina o albúmina a muestras con anticuerpos heterófilos y se evaluaron frente a las muestras sinañadidos. No se observó ninguna interferencia significativa. Los resultados se muestran en las Tablas 3a y 3b. Las pruebasse realizaron por duplicado.
Puede concluirse que la presencia de triglicéridos hasta los 2.370 mg/dl, hemoglobina hasta los 10 mg/ml, bilirrubina hastalos 0,06 mg/ml y albúmina hasta los 100 mg/ml no interfiere en el funcionamiento de la prueba SureStep Mono.
Positivo enABI
Positivo en equipo comercial
Negativo en equipo comercial
Negativo enABI Total
Total
146 2
255
148
258
406257
3
149
TABLA 1. Resumen de comparaciones
Un/ml DiluciónABI
SureStepMono
Pruebacomercial 1
Pruebacomercial 2
Pruebacomercial 3
100
50
25
12,5
6,25
Ninguna
1:2
1:4
1:8
1:16
+
+
+
+
-
+
+
+
+
+
+
+
+
+
-
-
-
-
- -
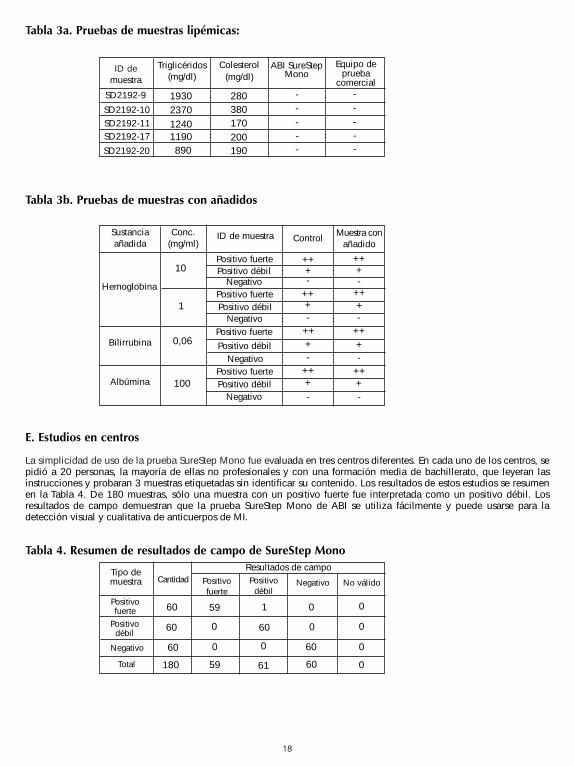
18
Tabla 3a. Pruebas de muestras lipémicas:
Tabla 3b. Pruebas de muestras con añadidos
E. Estudios en centros
La simplicidad de uso de la prueba SureStep Mono fue evaluada en tres centros diferentes. En cada uno de los centros, sepidió a 20 personas, la mayoría de ellas no profesionales y con una formación media de bachillerato, que leyeran lasinstrucciones y probaran 3 muestras etiquetadas sin identificar su contenido. Los resultados de estos estudios se resumenen la Tabla 4. De 180 muestras, sólo una muestra con un positivo fuerte fue interpretada como un positivo débil. Losresultados de campo demuestran que la prueba SureStep Mono de ABI se utiliza fácilmente y puede usarse para ladetección visual y cualitativa de anticuerpos de MI.
Tabla 4. Resumen de resultados de campo de SureStep Mono
ID demuestra
ABI SureStepMono
Equipo deprueba
comercial1930
-
Triglicéridos(mg/dl)
Colesterol(mg/dl)
-
---280
380170200190890
119012402370
SD2192-9
SD2192-10
SD2192-20
SD2192-17SD2192-11
--
---
Sustanciaañadida Control Muestra con
añadido
+
-
Conc.(mg/ml)
ID de muestra
-
-
-
-
-
-
-
++
100
0,06
1
10Positivo fuerte
Hemoglobina
Albúmina
Bilirrubina
Positivo fuerte
Positivo fuerte
Positivo fuerte
Positivo débil
Positivo débil
Positivo débil
Positivo débil
Negativo
Negativo
Negativo
Negativo
+
+
+
++
++
++
++++
++
++
++++
Tipo demuestra Positivo
débil
59
0
60
1
Cantidad Negativo No válido
180
60
60
60
Positivofuerte
Resultados de campo
60
6059
0 0
0
0
0
00
0
61
Positivofuerte
Positivodébil
Negativo
Total

19
Tests CLIA waived – Test à partir d’un prélèvement de sang capillaire au bout du doigt ou despécimens de sang entier
Tests modérément complexes – Test à partir de spécimens de sérum, de plasma ou de sang entier
UTILISATION PRÉVUE
Le test SureStep Mono est un test rapide pour la détection visuelle qualitative des anticorps hétérophiles spécifiques à lamononucléose infectieuse (MI) dans le sérum, le plasma ou le sang entier humain. Ce kit de test est conçu comme uninstrument pour le diagnostic de la MI chez les patients présentant des symptômes cliniques caractéristiques. Il est réservéà un usage laboratoire professionnel.
RÉSUMÉ
La mononucléose infectieuse est une maladie aiguë spontanément résolutive causée par le virus Epstein-Barr (VEB) (1–3).L’infection au VEB pendant l’enfance est généralement asymptomatique (4–5). Cependant, jusqu’à 50 % des infections semanifestant chez les jeunes adultes et les adolescents entraînent des manifestations cliniques associées à la MI.
Le diagnostic de la MI est basé sur l’évaluation de symptômes cliniques et de modifications sérologiques caractéristiques.Le diagnostic sérologique de la MI est fondé sur la détection d’anticorps hétérophiles et spécifiques au VEB (3–5). Lesanticorps hétérophiles sont détectables chez la plupart des adultes au bout d’un certain temps après l’apparition de la MI.La détection des anticorps dans le but de diagnostiquer la MI est une méthode largement adoptée par les médecins (3–7).Le test SureStep Mono utilise un extrait d’érythrocytes bovins qui présentent un seuil de sensibilité et de spécificitésupérieur à celui d’autres espèces. (4)
PRINCIPE DU TEST
Le test SureStep Mono est conçu pour détecter la MI via l’interprétation visuelle de la variation de couleur dans le dispositifde test, lequel consiste en un dosage immunologique à conjugué phase solide-or (méthode sandwich). Le test consiste enune bandelette membranaire recouverte d’anticorps hétérophiles dans la zone de test et d’anticorps de chèvre anti-sourisdans la zone de contrôle. Le conjugué anticorps IgM anti-humain-or colloïdal est placé à l’extrémité de la membrane. Unmélange combinant le conjugué d’or colloïdal, le spécimen et le tampon révélateur migrera chromatographiquement lelong de la membrane par action capillaire. Si le spécimen du patient présente des anticorps hétérophiles de la MI, lemélange migre vers la zone de test et forme une ligne visible où les anticorps se complexent aux antigènes hétérophiles.Si le spécimen ne présente aucun anticorps hétérophile de la MI, aucune ligne colorée visible ne se forme dans la zone detest. Par conséquent, la présence d’une ligne colorée dans la zone de test indique un résultat positif. Une ligne coloréeapparaîtra toujours dans la zone de contrôle. Cette ligne de contrôle sert d’indicateur lors de la procédure afin d’assurer labonne performance du dispositif et de bons résultats de test.
STOCKAGE ET STABILITÉ
Le kit de test doit être soit réfrigéré soit conservé à température ambiante (2–30 °C) dans une poche hermétiquement ferméependant toute sa durée de validité.
PRÉCAUTIONS
• Réservé au diagnostic in vitro.
• Réservé aux usages professionnels et de laboratoire.• Manipuler tous les spécimens de patient comme des agents pouvant transmettre des maladies.• Ne pas mélanger les réactifs de lots différents.• Ouvrir la poche en aluminium juste avant d’effectuer le test.
LES INSTRUCTIONS DU CLIA SONT LISTÉES CI-DESSOUS DANS LE PARAGRAPHE CONTRÔLE QUALITÉ.VEUILLEZ SUIVRE LES INSTRUCTIONS APPROPRIÉES REQUISES PAR VOS DIRECTIVES DE LABORATOIRE
TEST MONO
FRANÇAIS
Produit à usage unique.

20
• Ne pas utiliser de sang entier stocké depuis plus de trois jours.• Ne pas utiliser de spécimens grossièrement hémolysés.• Le tampon révélateur et les contrôles contiennent de l’azide de sodium. L'azide de sodium peut réagir avec la
tuyauterie en plomb ou en cuivre et former des azides de métal explosifs. Lors de l’élimination de ces solutions,toujours faire couler une grande quantité d’eau afin d’éviter toute accumulation d’azide.
Les réactifs d’extraction sont légèrement caustiques. Éviter tout contact avec les yeux ou les membranes muqueuses.Dans l’éventualité d’un contact accidentel, laver à grande eau.
Les contrôles positif et négatif contiennent de l’azide de sodium qui peut réagir avec la tuyauterie en plomb ou en cuivreet former des azides de métal explosifs. Lors de l’élimination de ces solutions, toujours faire couler une grande quantitéd’eau afin d’éviter toute accumulation d’azide.
Mise en garde : matière présentant un danger biologiqueChaque spécimen de plasma ou de sérum humain utilisé dans la préparation des contrôles positif et négatif a été testé selondes méthodes homologuées par la FDA (agence américaine de réglementation des produits alimentaires et pharmaceutiques)pour détecter toute présence éventuelle d’anticorps anti-VIH-1/VIH-2, d’antigènes d’enveloppe d’hépatite B (HBsAg) etd’anticorps du virus de l’hépatite C (anti-VHC), et ont donné des résultats négatifs. Cependant, faire attention lors de lamanipulation et de l’élimination de ces substances au niveau de biosécurité 2, tel que recommandé dans le Center forDisease Control/National Institutes of Health Manual, Biosafety in Micobiological and BiomedicalLaboratories, 1984.
RÉACTIFS ET MATÉRIELS FOURNIS
• 25 dispositifs de test sous emballage individuel. Chaque dispositif contient une bandelette de test comprenant unemembrane recouverte d’antigènes hétérophiles et un tampon d’anticorps coloré.
• Tampon révélateur (8 ml) : solution salée tamponnée par du phosphate (0,1 M) avec additifs et 0,1 %d’azide de sodium.
• Contrôle négatif (0,2 ml) : sérum ou plasma humain normal dilué dans une solution salée avec 0,2 %d’azide de sodium.
• Contrôle positif (0,2 ml) : sérum ou plasma humain testé positif aux anticorps hétérophiles dela MI dilué dans une solution salée avec 0,2 % d’azide de sodium.
• 25 pipettes de transfert jetables.
• Une notice.
PRÉLÈVEMENT DES ÉCHANTILLONS ET MANIPULATION
Prélèvement de sang capillaire au bout du doigt1. Nettoyer la zone à ponctionner avec un écouvillon imbibé d’alcool.2. Presser l’extrémité du doigt et le percer avec une lancette stérile.3. Essuyer la première goutte de sang avec de la gaze ou du coton.4. Attendre que la seconde goutte tombe directement dans le puits échantillon du dispositif du test ou utiliser la
pipette fournie pour obtenir du sang frais. Verser 2 gouttes (environ 40 µl) dans le puits échantillon.
Sang entier1. Un phlébotomiste agréé recueillera du sang entier dans un tube de prélèvement à bouchon violet, bleu ou vert
(contenant de l’EDTA, du citrate ou de l’héparine, respectivement) par ponction veineuse.2. Le sang entier peut être testé immédiatement ou conservé entre 2 et 8 °C pendant trois jours maximum.
Plasma1. Un phlébotomiste agréé recueillera du sang entier dans un tube de prélèvement à bouchon violet, bleu ou vert
(contenant de l’EDTA, du citrate ou de l’héparine, respectivement) par ponction veineuse.2. Séparer le plasma par centrifugation.3. Extraire délicatement le plasma en vue du test ou l’étiqueter et le conserver entre 2 et 8 °C pendant deux semaines
maximum. Le plasma peut être congelé à -20 °C jusqu’à un an.
Sérum1. Un phlébotomiste agréé recueillera du sang entier dans un tube à bouchon rouge (ne contenant aucun
anticoagulant) par ponction veineuse.2. Attendre que le sang se coagule et séparer le sérum par centrifugation.3. Extraire délicatement le plasma en vue du test ou l’étiqueter et le conserver entre 2 et 8 °C pendant deux
semaines maximum. Le sérum peut être congelé à -20 °C jusqu’à une période d’un an.
BUF DEV

21
PROCÉDURE DU TEST
3. Verser immédiatement 3 à 4 gouttes du tampon révélateur.
4. Ensuite, patienter jusqu’à l’apparition des lignes colorées. En fonction de la concentration des anticorps hétérophilesprésents, un résultat positif peut être observé dans les 3 minutes. Cependant, pour confirmer un résultat négatif, ilest nécessaire de respecter un délai de réaction de 5 minutes. Ne pas lire les résultats après 8 minutes.
Dispositif de test
Zone de contrôle Zone de test
Puitséchantillon
Attendre que le dispositif de test, le tampon révélateur, lesspécimens de patient et les contrôles atteignent la températureambiante (20–30 °C) avant de procéder au test. Ouvrir lespoches juste avant d’effectuerle test.
1. Retirer le dispositif de test de sa poche protectrice. (Ramener ledispositif à température ambiante avant de l’ouvrir afin d’évitertoute condensation d’humidité sur la membrane.) Apposer uneétiquette identifiant le patient ou le contrôle sur le dispositif.
2. Verser le spécimen dans le puits échantillon :
Prélèvement de sang capillaire au bout du doigt : Attendre que laseconde goutte tombe directement dans le puits échantillon dudispositif du test ou utiliser la pipette fournie pour obtenir du sangfrais. Verser 2 gouttes (environ 40 µl) dans le puits échantillon.
Spécimens de sang entier dans les tubes de prélèvement : verserdeux gouttes (environ 40 µl) dans le puits échantillon en tenant lapipette de transfert fournie en position verticale.
Spécimens de plasma ou de sérum : verser une goutte (environ20 µl) de sérum ou de plasma dans le puits échantillon en tenant lapipette de transfert fournie en position verticale.
Pipette detransfert
INTERPRÉTATION DES RÉSULTATS
Positif :Deux lignes colorées apparaissent, l’une dans la zone de contrôle (C) et l’autredans la zone de test (T). Lors des tests avec des spécimens fortement positifs,l’intensité de la couleur de la ligne de contrôle peut être plus claire que prévu. Il n’estpas recommandé de comparer l’intensité des lignes.
Négatif :Une seule ligne colorée apparaît dans la zone de contrôle. Aucune ligne de faibleintensité de couleur n’apparaît dans la zone de test (T).
Non valide :L’absence complète de ligne dans la zone de contrôle indique une erreur de procédureou la détérioration du réactif de test. Le test doit être annulé car une mauvaiseprocédure a été realisée ou il y a eu une détérioration des réactifs. Recommencer letest avec un dispositif de test. Si le problème persiste, appeler le support techniqued’Applied Biotech au (888) 578-7956 (pour les appels en provenance des États-Unis)ou au +1 858 713-9668 (pour les appels internationaux). Les appels sont reçus pardes agents parlant anglais uniquement. Pour toute autre langue, envoyer un courrierélectronique à [email protected].
C T
C T
C T
C T
C TMONO

22
CONTRÔLE DE QUALITÉ
Contrôle de procédure interneLe test comprend un contrôle de procédure. Une ligne colorée apparaissant dans la zone de contrôle (C) est considéréecomme un contrôle de procédure interne indiquant la bonne performance du test et la bonne qualité des réactifs.
Un fond clair dans la fenêtre du résultat est considéré comme un contrôle négatif interne. Cependant, lorsque desspécimens de sang entier sont testés, le fond peut apparaître légèrement rouge en raison de la faible hémolyse de certainsglobules rouges. Ceci est acceptable tant que ce phénomène n’affecte pas la lecture du résultat du test. Le test n’est pasvalide si le fond ne s’éclaircit pas et empêche la visibilité du résultat du test.
Contrôle de qualité externeDe bonnes pratiques de laboratoire prévoient l’usage de contrôles externes afin d’assurer la fonctionnalité des réactifs etune bonne performance. Des contrôles positif et négatif sont fournis avec le kit. Ces contrôles contiennent du sérum ou duplasma humain et sont testés comme des spécimens de patient. Pour effectuer le test avec ces contrôles, verser une goutte(environ 20 µl) du contrôle positif ou négatif dans le puits échantillon. Pour ce faire, utiliser la pipette de transfert fournieet la tenir en position verticale. Verser immédiatement 3 à 4 gouttes du tampon révélateur. Un signal positif est indiqué parla présence de deux lignes – l’une dans la zone de test (T), l’autre dans la zone de contrôle (C). Un signal négatif est indiquépar la présence d’une seule ligne dans la zone de contrôle (C). Observer les résultats au bout de 5 minutes et ne faireaucune interprétation après 8 minutes. Si les contrôles ne fonctionnent pas comme prévu, ne pas interpréter les résultatsdu test. Recommencer le test ou contacter le support technique.
RÉSERVÉ AUX LABORATOIRES CLIA WAIVED :
Les contrôles externes positif et négatif doivent être testés à l’ouverture de tout nouveau kit de test. Chaque opérateur doittester un contrôle externe positif et négatif une fois avec chacun des 25 kits de test.
RÉSERVÉ AUX LABORATOIRES EFFECTUANT DES TESTS MODÉRÉMENT COMPLEXES OU AUXPRESTATAIRES DE SOINS DE SANTÉ :
Il est recommandé de tester les contrôles positif et négatif pour chaque nouvel envoi ou nouveau numéro de kit et tel quela réglementation locale en vigueur le prévoit.
LIMITATIONS
Ce kit de test est indiqué pour la détection qualitative d’anticorps IgM aux antigènes hétérophiles de la MI. Un résultatpositif indique la présence d’anticorps IgM aux antigènes hétérophiles.
Ce kit de test doit être utilisé chez les individus symptomatiques susceptibles d’être atteints d’une MI. Le diagnostic de laMI doit être confirmé par d’autres résultats d’analyses cliniques.
Un résultat négatif n’écarte pas la possibilité d’une MI car les anticorps aux antigènes hétérophiles peuvent être soit absentssoit présents en trop faible quantité pour être détectés.
RÉSULTATS ATTENDUS
Pendant la phase aiguë de la MI, les anticorps hétérophiles sont détectables chez 80 à 85 % des patients. Les anticorpshétérophiles sont détectables au cours du premier mois de la maladie et diminuent rapidement en nombre après laquatrième semaine (5).
PERFORMANCES
A. ExactitudeL’exactitude du test SureStepTM Mono a été évaluée et comparée à celle d’un test immuno-chromatographique qualitatifcoloré disponible sur le marché indiqué pour la détection des anticorps hétérophiles IgM de la MI dans le sérum, le plasma oule sang entier humain. Quatre cent six spécimens de sérum, plasma et sang entier humains (288 de sérum, 103 de plasmaet 15 de sang entier) ont été utilisés pour l’étude de comparaison. Les résultats sont résumés dans le tableau 1.
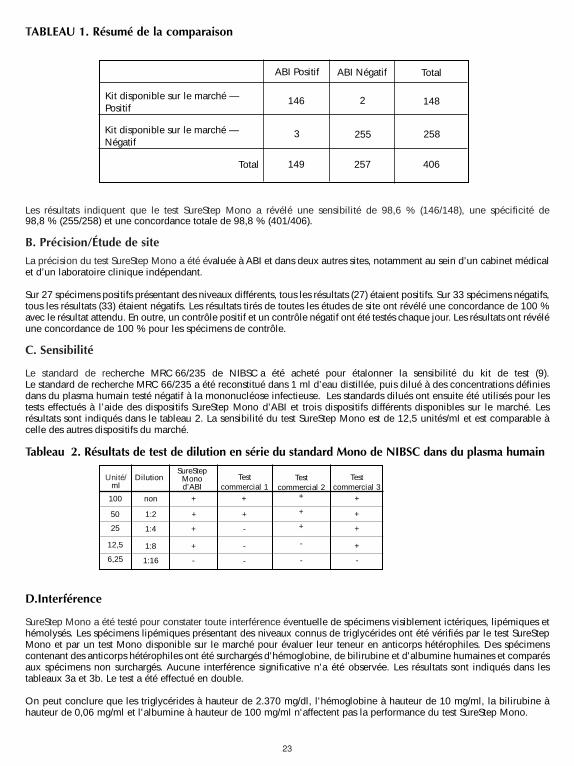
23
Les résultats indiquent que le test SureStep Mono a révélé une sensibilité de 98,6 % (146/148), une spécificité de98,8 % (255/258) et une concordance totale de 98,8 % (401/406).
B. Précision/Étude de site
La précision du test SureStep Mono a été évaluée à ABI et dans deux autres sites, notamment au sein d’un cabinet médicalet d’un laboratoire clinique indépendant.
Sur 27 spécimens positifs présentant des niveaux différents, tous les résultats (27) étaient positifs. Sur 33 spécimens négatifs,tous les résultats (33) étaient négatifs. Les résultats tirés de toutes les études de site ont révélé une concordance de 100 %avec le résultat attendu. En outre, un contrôle positif et un contrôle négatif ont été testés chaque jour. Les résultats ont révéléune concordance de 100 % pour les spécimens de contrôle.
C. Sensibilité
Le standard de recherche MRC 66/235 de NIBSC a été acheté pour étalonner la sensibilité du kit de test (9). Le standard de recherche MRC 66/235 a été reconstitué dans 1 ml d’eau distillée, puis dilué à des concentrations définiesdans du plasma humain testé négatif à la mononucléose infectieuse. Les standards dilués ont ensuite été utilisés pour lestests effectués à l’aide des dispositifs SureStep Mono d’ABI et trois dispositifs différents disponibles sur le marché. Lesrésultats sont indiqués dans le tableau 2. La sensibilité du test SureStep Mono est de 12,5 unités/ml et est comparable àcelle des autres dispositifs du marché.
Tableau 2. Résultats de test de dilution en série du standard Mono de NIBSC dans du plasma humain
D.Interférence
SureStep Mono a été testé pour constater toute interférence éventuelle de spécimens visiblement ictériques, lipémiques ethémolysés. Les spécimens lipémiques présentant des niveaux connus de triglycérides ont été vérifiés par le test SureStepMono et par un test Mono disponible sur le marché pour évaluer leur teneur en anticorps hétérophiles. Des spécimenscontenant des anticorps hétérophiles ont été surchargés d'hémoglobine, de bilirubine et d'albumine humaines et comparésaux spécimens non surchargés. Aucune interférence significative n'a été observée. Les résultats sont indiqués dans lestableaux 3a et 3b. Le test a été effectué en double.
On peut conclure que les triglycérides à hauteur de 2.370 mg/dl, l'hémoglobine à hauteur de 10 mg/ml, la bilirubine àhauteur de 0,06 mg/ml et l'albumine à hauteur de 100 mg/ml n'affectent pas la performance du test SureStep Mono.
ABI Positif
Kit disponible sur le marché —Positif
Kit disponible sur le marché —Négatif
ABI Négatif Total
Total
146 2
255
148
258
406257
3
149
TABLEAU 1. Résumé de la comparaison
Unité/ml
DilutionSureStep
Monod’ABI
Testcommercial 1
Test commercial 2
Testcommercial 3
100
50
25
12,5
6,25
non
1:2
1:4
1:8
1:16
+
+
+
+
-
+
+
+
+
+
+
+
+
+
-
-
-
-
- -
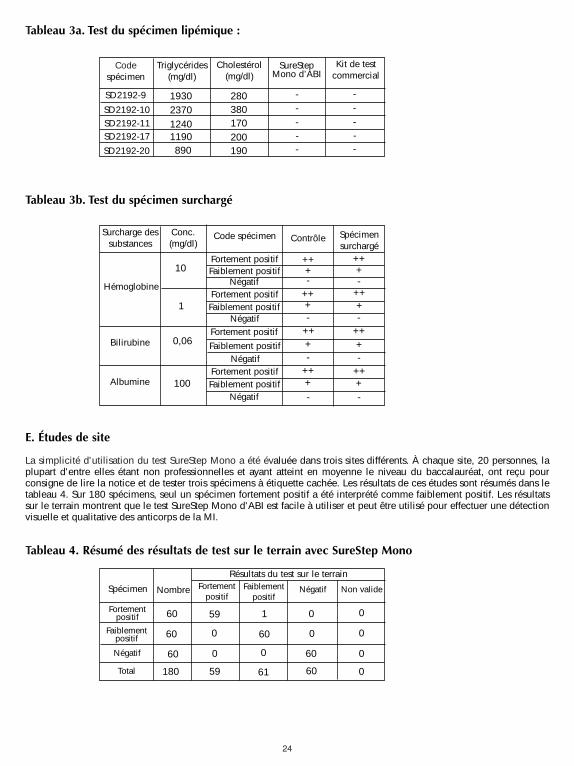
24
Tableau 3a. Test du spécimen lipémique :
Tableau 3b. Test du spécimen surchargé
E. Études de site
La simplicité d'utilisation du test SureStep Mono a été évaluée dans trois sites différents. À chaque site, 20 personnes, laplupart d'entre elles étant non professionnelles et ayant atteint en moyenne le niveau du baccalauréat, ont reçu pourconsigne de lire la notice et de tester trois spécimens à étiquette cachée. Les résultats de ces études sont résumés dans letableau 4. Sur 180 spécimens, seul un spécimen fortement positif a été interprété comme faiblement positif. Les résultatssur le terrain montrent que le test SureStep Mono d'ABI est facile à utiliser et peut être utilisé pour effectuer une détectionvisuelle et qualitative des anticorps de la MI.
Tableau 4. Résumé des résultats de test sur le terrain avec SureStep Mono
Codespécimen
SureStepMono d’ABI
Kit de testcommercial
1930
-
Triglycérides(mg/dl)
Cholestérol(mg/dl)
-
---280
380170200190890
119012402370
SD2192-9
SD2192-10
SD2192-20
SD2192-17SD2192-11
--
---
Surcharge dessubstances Contrôle Spécimen
surchargé
+
-
Conc.(mg/dl)
Code spécimen
-
-
-
-
-
-
-
++
100
0,06
1
10Fortement positif
Hémoglobine
Albumine
Bilirubine
Fortement positif
Fortement positif
Fortement positif
Faiblement positif
Faiblement positif
Faiblement positif
Faiblement positif
Négatif
Négatif
Négatif
Négatif
+
+
+
++
++
++
++++
++
++
++++
Spécimen Faiblementpositif
59
0
60
1
Nombre Négatif Non valide
180
60
60
60
Fortementpositif
Résultats du test sur le terrain
60
6059
0 0
0
0
0
00
0
61
Fortementpositif
Faiblementpositif
Négatif
Total

25
Per i laboratori “CLIA waived” (esenti) – Test con campioni di sangue ottenuto tramitedigitopuntura o sangue intero
Per i laboratori “Moderately Complex” (a complessità moderata) – Test con campioni di siero,plasma o sangue intero
USO PREVISTO
Il Test SureStep Mono è un esame rapido a lettura visiva per la determinazione qualitativa degli anticorpi eterofili specificidella mononucleosi infettiva (in sigla, MI) nel siero, plasma o sangue intero di origine umana. Il presente kit per analisi vautilizzato come strumento ausiliario nella diagnosi della MI in pazienti con sintomi clinici caratteristici, ed è destinatoesclusivamente all’uso professionale e di laboratorio.
SPIEGAZIONE
La mononucleosi infettiva è una patologia a decorso acuto, causata dal virus di Epstein-Barr (EBV) (1–3). Nei primi anni divita, l’infezione da EBV è in genere asintomatica (4–5). Nel 50% dei casi, tuttavia, le infezioni contratte nel corsodell’adolescenza e della prima giovinezza sono destinate a sviluppare manifestazioni cliniche associate alla MI.
La diagnosi della MI si basa sulla valutazione dei sintomi clinici caratteristici e sulle alterazioni sierologiche. La diagnosisierologica della MI è stata dimostrata mediante determinazione degli anticorpi eterofili e specifici del virus EBV (3–5).L’anticorpo eterofilo può essere individuato in un momento specifico del decorso della MI nella maggior parte degli adulti.È pratica comune tra i medici l’impiego delle tecniche di rivelazione degli anticorpi eterofili a supporto della diagnosi dellaMI (3–7). Il Test SureStep Mono utilizza un estratto di eritrociti bovini, che è caratterizzato da una maggiore sensibilità especificità rispetto ad estratti di altre specie. (4)
PRINCIPIO DELL’ANALISI
Il Test SureStep Mono è utilizzato per la determinazione della MI mediante interpretazione visiva dell’evoluzione del colorenel dispositivo per analisi: in pratica, è un immunodosaggio a sandwich con fase solida e coniugato di oro. Il dispositivocontiene una membrana preliminarmente rivestita con antigeni eterofili nella zona della linea del test e con anticorpicaprini anti-topo nella zona della linea di controllo. Il tampone assorbente con anticorpo IgM anti-umano e oro colloidaleviene collocato al termine della membrana. Insieme al campione e al tampone di sviluppo, una miscela di coniugato dioro colloidale si sposta lungo la membrana per azione capillare, secondo il processo cromatografico. Quando gli anticorpieterofili della MI sono presenti nel campione del paziente, la miscela si trasferisce nella zona della linea del test,disegnando una linea visibile via via che l’anticorpo forma complessi con l’antigene eterofilo. Quando gli anticorpi eterofilidella MI non sono presenti nel campione del paziente, non si formano linee colorate visibili nella zona della linea del test.Di conseguenza, la presenza di una linea colorata nella zona della linea del test indica un risultato positivo. Nella zona dicontrollo comparirà sempre una linea colorata, ad indicare che la procedura di analisi seguita è corretta e che il dispositivofunziona.
CONSERVAZIONE E STABILITÀ
Il kit per analisi può essere conservato in frigorifero o a temperatura ambiente (2-30°C) nella confezione originale sigillata,fino alla data di scadenza indicata.
PRECAUZIONI
• Solo per uso diagnostico in vitro.
• Solo per uso professionale e di laboratorio.• Tutti i campioni dei pazienti devono essere manipolati come materiale potenzialmente infetto.• Non mischiare reagenti di lotti diversi.
LE ISTRUZIONI “CLIA COMPLEXITY” SONO RIPORTATE PIÙ AVANTI, NELLA SEZIONE “CONTROLLO DI QUALITÀ”. ATTENERSI ALLE ISTRUZIONI CORRISPONDENTI ALLE LINEE
GUIDA ADOTTATE DAL PROPRIO LABORATORIO.
TEST MONO
ITALIANO
Non riutilizzare il test

26
• Non aprire le confezioni con troppo anticipo prima dell’analisi.• Non utilizzare sangue intero conservato da più di tre giorni.• Non utilizzare campioni emolizzati.• Il tampone di sviluppo e i controlli contengono sodio azide, una sostanza che può reagire con il piombo e il rame
delle tubature per formare azidi metalliche potenzialmente esplosive. Durante lo smaltimento di queste soluzioni,utilizzare sempre acqua in abbondanza per prevenire l’accumulo di azide.
I reagenti per l’estrazione sono leggermente corrosivi. Evitare il contatto con gli occhi e le membrane mucose. In caso dicontatto accidentale, sciacquare accuratamente con acqua.
I controlli positivi e negativi contengono sodio azide, una sostanza che può reagire con il piombo e il rame delle tubatureper formare azidi metalliche potenzialmente esplosive. Durante lo smaltimento di queste soluzioni, utilizzare sempreacqua in abbondanza per prevenire l’accumulo di azide.
Avvertenza: materiale a potenziale rischio biologicoLe unità di plasma e siero di tutti i donatori che sono state utilizzate per la preparazione dei controlli positivi e negativisono state sottoposte ai test opportuni approvati dalla Food and Drug Administration (FDA), così da escludere la presenzadi anticorpi anti-HIV-1/HIV-2, HBsAg e anti-HCV. Tutte le unità sono risultate negative. Si raccomanda tuttavia di prestarela massima attenzione durante la manipolazione e lo smaltimento del materiale con sicurezza biologica di livello 2,secondo le direttive del Center for Disease Control/National Institutes of Health, nella pubblicazione Biosafety inMicrobiological and Biomedical Laboratories, 1984.
REAGENTI E MATERIALI FORNITI
• 25 dispositivi per analisi in confezioni singole. Ogni dispositivo contiene una striscia per analisi con una membranarivestita di antigene eterofilo e un tampone assorbente con anticorpo colorato.
• Tampone di sviluppo (8 ml) : soluzione salina tamponata con fosfato 0,1 M, contenente additivi e sodioazide 0,1%.
• Controllo negativo (0,2 ml) : plasma o siero umano non patologico diluito in soluzione salina con sodioazide 0,2%.
• Controllo positivo (0,2 ml) : plasma o siero umano positivo all’anticorpo eterofilo MI, diluito insoluzione salina con sodio azide 0,2%.
• 25 pipette monouso.
• Un foglietto illustrativo.
RACCOLTA E TRATTAMENTO DEI CAMPIONIDigitopuntura1. Con un batuffolo imbevuto di alcol, pulire la zona su cui verrà praticata la puntura.2. Spremere delicatamente la punta del dito e pungere la cute con una lancetta.3. Asciugare la prima goccia di sangue con garza o cotone sterile.4. Fare in modo che la seconda goccia confluisca direttamente nel pozzetto per campioni del dispositivo per analisi,
oppure utilizzare la pipetta acclusa per ottenere sangue fresco. Aggiungere 2 gocce (circa 40 µl) nel pozzetto per campioni.
Sangue intero1. Il prelievo del sangue intero deve essere effettuato tramite venopuntura da un flebotomista professionista, che utilizzerà
provette per la raccolta dotate di tappo viola, blu o verde (contenenti rispettivamente EDTA, citrato o eparina).2. Il sangue intero può essere analizzato immediatamente oppure conservato a 2-8°C per un massimo di tre giorni.
Plasma1. Il prelievo del sangue intero deve essere effettuato tramite venopuntura da un flebotomista professionista, che utilizzerà
una provetta per la raccolta dotata di tappo viola, blu o verde (contenenti rispettivamente EDTA, citrato o eparina).2. Il sangue intero deve essere centrifugato per separare il plasma.3. Dopo avere raccolto delicatamente il plasma, è possibile analizzarlo subito oppure etichettarlo e conservarlo a 2-8°C
fino a due settimane. Il plasma può essere congelato a -20°C e conservato per un anno al massimo.
Siero1. Il prelievo del sangue intero deve essere effettuato tramite venopuntura da un flebotomista professionista, che utilizzerà
provette per la raccolta dotate di tappo rosso (prive di anticoagulanti).2. Attendere che il sangue si coaguli, quindi centrifugarlo per separare il siero.3. Dopo avere raccolto delicatamente il siero, è possibile analizzarlo subito oppure etichettarlo e conservarlo a 2-8°C per
un massimo di due settimane. Il siero può essere congelato a -20°C e conservato per un anno al massimo.
BUF DEV

27
PROCEDURA DI ANALISI
3. Aggiungere immediatamente 3 o 4 gocce di tampone di sviluppo.
4. Dopo l’aggiunta del tampone di sviluppo, attendere la comparsa delle linee colorate. A seconda della concentrazionedi anticorpi eterofili presenti, si possono osservare risultati positivi già dopo 3 minuti. Per avere la conferma di unrisultato negativo, occorre invece attendere un tempo di reazione completo di 5 minuti. Non leggere i risultati se sonotrascorsi più di 8 minuti.
Dispositivo peranalisi
Zona controllo Zona test
Pozzettocampioni
Il dispositivo per analisi, il tampone di sviluppo, i campioni deipazienti e i controlli devono essere portati a temperaturaambiente (20°-30°C) prima di iniziare l’analisi. Aprire leconfezioni solo un attimo prima di eseguire il test.
1. Estrarre il dispositivo per analisi dall’involucro protettivo. Prima diaprire la confezione, portare il dispositivo a temperatura ambientein modo da impedire la formazione di umidità e condensa sullamembrana. Applicare al dispositivo un’etichetta con i datiidentificativi del paziente o del controllo.
2. Aggiungere i campioni nel pozzetto per campioni.
Sangue ottenuto tramite digitopuntura: fare in modo che laseconda goccia confluisca direttamente nel pozzetto percampioni del dispositivo per analisi, oppure utilizzare lapipetta acclusa per ottenere sangue fresco. Aggiungere 2gocce (circa 40 µl) nel pozzetto per campioni.
Campioni di sangue intero in provette per la raccolta:aggiungere 2 gocce (circa 40 µl) nel pozzetto per campioni,mantenendo la pipetta acclusa in posizione verticale.
Campioni di plasma o siero: aggiungere 1 goccia (circa 20 µl)di siero o plasma nel pozzetto per campioni, mantenendo lapipetta acclusa in posizione verticale.
Pipetta per ladispensazione
INTERPRETAZIONE DEI RISULTATI
Positivo:Compaiono due linee colorate, una nella zona di controllo (C) e una nella zona deltest (T). Durante l’analisi con campioni positivi-alti, la linea di controllo potrebbe appariremeno intensa del previsto. Si consiglia di non confrontare l’intensità delle linee.
Negativo:Compare una sola linea colorata nella zona della linea di controllo. Non è visibilenessuna linea dalla colorazione debole nella zona della linea del test (T).
Non valido:L’assenza di una linea nella zona di controllo indica che è stato commesso un erroredi procedura o che il reagente utilizzato potrebbe essersi deteriorato. Il test èinvalidato, in quanto la procedura seguita per l’analisi non è corretta oppure ireagenti utilizzati potrebbero essersi deteriorati. Ripetere il test con un nuovodispositivo. Se il problema persiste, contattare l’assistenza tecnica Applied Biotech alnumero (888) 578-7956 per le chiamate dagli Stati Uniti o al numero +1 858 713-9668per le chiamate internazionali. Risponderanno alle chiamate operatori di lingua inglese.Per comunicazioni in altre lingue, inviare un messaggio e-mail all’[email protected].
C T
C T
C T
C T
C TMONO

28
CONTROLLO DI QUALITÀ
Controllo procedurale internoIl test include un controllo della procedura eseguita. La linea colorata che compare nella zona di controllo (C) è considerataun controllo procedurale interno, in quanto indica la correttezza della procedura e dei reattivi impiegati.
Uno sfondo chiaro nella finestra di lettura del risultato è considerato un controllo interno negativo. Quando si analizzanocampioni di sangue intero, è tuttavia possibile che lo sfondo assuma una colorazione rosso pallido, dovuta all’emolisi dibasso livello di alcuni globuli rossi. Tale fenomeno è accettabile, purché non interferisca con la lettura del risultato. Il testè da ritenersi non valido se lo sfondo non si schiarisce, impedendo la lettura del risultato.
Controllo di qualità esternoÈ buona pratica di laboratorio verificare il corretto funzionamento dei reagenti e del test mediante una serie di controlliesterni. Nel kit sono presenti controlli positivi e negativi. Tali controlli sono costituiti da siero o plasma di origine umana esono trattati e analizzati come normali campioni dei pazienti. Nelle sedute analitiche con i controlli, aggiungere unagoccia (circa 20 µl) di controllo positivo o negativo nel pozzetto per campioni utilizzando la pipetta acclusa nel kit.Mantenere la pipetta in posizione verticale. Aggiungere immediatamente 3 o 4 gocce di tampone di sviluppo. Un risultatopositivo è segnalato dalla comparsa di due linee, una nella zona del test (T) e una nella zona di controllo (C). Un risultatonegativo è segnalato dalla comparsa di una sola linea, nella zona di controllo (C). Leggere i risultati dopo 5 minuti e noninterpretarli se sono trascorsi più di 8 minuti. Se i controlli non si comportano nel modo previsto, non interpretare i risultatidell’analisi. Ripetere il test o contattare l’assistenza tecnica.
PER I LABORATORI CLASSIFICATI COME “CLIA WAIVED” (ESENTI):
All’apertura di ogni nuovo kit è necessario analizzare un controllo esterno positivo e un controllo esterno negativo. Tutti glioperatori sono tenuti ad analizzare un controllo esterno positivo e un controllo esterno negativo dopo ogni kit da 25 test.
PER I LABORATORI CLASSIFICATI COME “MODERATELY COMPLEX” OPPURE GLI OPERATORISANITARI PROFESSIONISTI:
Si consiglia di analizzare un controllo esterno positivo e un controllo esterno negativo ad ogni nuova consegna o ad ogninuovo numero di lotto, oltre che nelle modalità e nei tempi previsti dalle normative locali in materia.
LIMITI
Questo kit per analisi è impiegato per la determinazione qualitativa degli anticorpi IgM dell’antigene eterofilo MI. Unrisultato positivo rivela la presenza di anticorpi IgM dell’antigene eterofilo.
Questo kit per analisi deve essere utilizzato in caso di sospetta infezione da MI. La diagnosi della MI deve essereconfermata da altri riscontri clinici.
Un risultato negativo non esclude la possibilità che l’infezione da MI sia in atto, in quanto gli anticorpi dell’antigeneeterofilo potrebbero essere assenti oppure potrebbero essere presenti in quantità non rilevabili.
VALORI ATTESI
Nella fase acuta della MI, gli anticorpi eterofili sono rinvenibili nell’80-85% dei pazienti. Gli anticorpi eterofili possonoessere individuati nel corso del primo mese della malattia, ma diminuiscono rapidamente dopo la quarta settimana (5).
CARATTERISTICHE PRESTAZIONALI
A. AccuratezzaL’accuratezza del Test SureStepTM Mono è stata valutata in rapporto ai saggi qualitativi immunocromatografici disponibili incommercio per la determinazione degli anticorpi eterofili IgM da MI nel siero, plasma o sangue intero di origine umana.Ai fini dello studio di comparazione sono stati utilizzati quattrocentosei (406) campioni di siero, plasma e sangue intero diorigine umana, così suddivisi: 288 campioni di siero, 103 campioni di plasma e 15 campioni di sangue intero. I risultatisono riassunti nella Tabella 1.
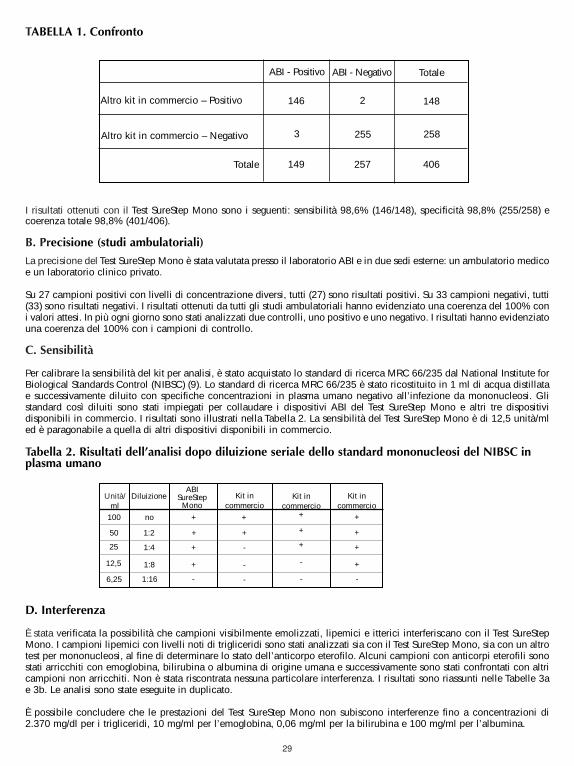
29
I risultati ottenuti con il Test SureStep Mono sono i seguenti: sensibilità 98,6% (146/148), specificità 98,8% (255/258) ecoerenza totale 98,8% (401/406).
B. Precisione (studi ambulatoriali)
La precisione del Test SureStep Mono è stata valutata presso il laboratorio ABI e in due sedi esterne: un ambulatorio medicoe un laboratorio clinico privato.
Su 27 campioni positivi con livelli di concentrazione diversi, tutti (27) sono risultati positivi. Su 33 campioni negativi, tutti(33) sono risultati negativi. I risultati ottenuti da tutti gli studi ambulatoriali hanno evidenziato una coerenza del 100% coni valori attesi. In più ogni giorno sono stati analizzati due controlli, uno positivo e uno negativo. I risultati hanno evidenziatouna coerenza del 100% con i campioni di controllo.
C. Sensibilità
Per calibrare la sensibilità del kit per analisi, è stato acquistato lo standard di ricerca MRC 66/235 dal National Institute forBiological Standards Control (NIBSC) (9). Lo standard di ricerca MRC 66/235 è stato ricostituito in 1 ml di acqua distillatae successivamente diluito con specifiche concentrazioni in plasma umano negativo all’infezione da mononucleosi. Glistandard così diluiti sono stati impiegati per collaudare i dispositivi ABI del Test SureStep Mono e altri tre dispositividisponibili in commercio. I risultati sono illustrati nella Tabella 2. La sensibilità del Test SureStep Mono è di 12,5 unità/mled è paragonabile a quella di altri dispositivi disponibili in commercio.
Tabella 2. Risultati dell’analisi dopo diluizione seriale dello standard mononucleosi del NIBSC inplasma umano
D. Interferenza
È stata verificata la possibilità che campioni visibilmente emolizzati, lipemici e itterici interferiscano con il Test SureStepMono. I campioni lipemici con livelli noti di trigliceridi sono stati analizzati sia con il Test SureStep Mono, sia con un altrotest per mononucleosi, al fine di determinare lo stato dell’anticorpo eterofilo. Alcuni campioni con anticorpi eterofili sonostati arricchiti con emoglobina, bilirubina o albumina di origine umana e successivamente sono stati confrontati con altricampioni non arricchiti. Non è stata riscontrata nessuna particolare interferenza. I risultati sono riassunti nelle Tabelle 3ae 3b. Le analisi sono state eseguite in duplicato.
È possibile concludere che le prestazioni del Test SureStep Mono non subiscono interferenze fino a concentrazioni di2.370 mg/dl per i trigliceridi, 10 mg/ml per l’emoglobina, 0,06 mg/ml per la bilirubina e 100 mg/ml per l’albumina.
ABI - Positivo
Altro kit in commercio – Positivo
Altro kit in commercio – Negativo
ABI - Negativo Totale
Totale
146 2
255
148
258
406257
3
149
TABELLA 1. Confronto
Unità/ml
DiluizioneABI
SureStepMono
Kit incommercio
Kit incommercio
Kit incommercio
100
50
25
12,5
6,25
no
1:2
1:4
1:8
1:16
+
+
+
+
-
+
+
+
+
+
+
+
+
+
-
-
-
-
- -
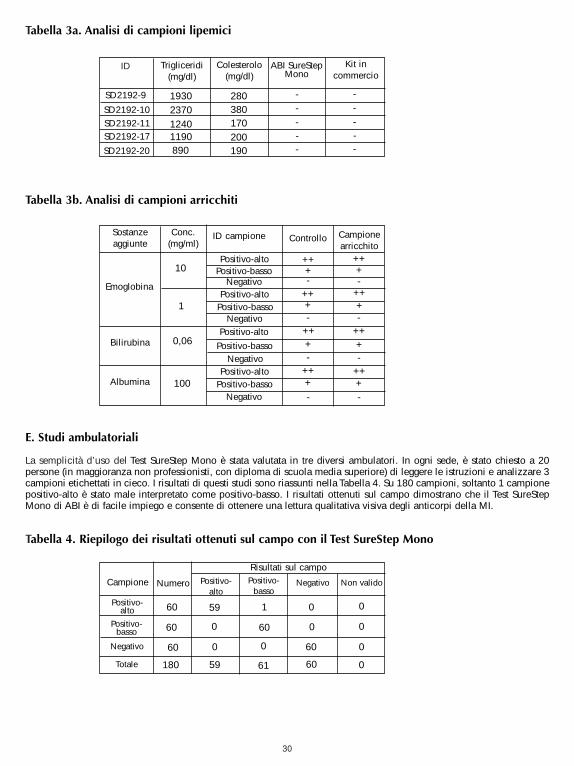
30
Tabella 3a. Analisi di campioni lipemici
Tabella 3b. Analisi di campioni arricchiti
E. Studi ambulatoriali
La semplicità d’uso del Test SureStep Mono è stata valutata in tre diversi ambulatori. In ogni sede, è stato chiesto a 20persone (in maggioranza non professionisti, con diploma di scuola media superiore) di leggere le istruzioni e analizzare 3campioni etichettati in cieco. I risultati di questi studi sono riassunti nella Tabella 4. Su 180 campioni, soltanto 1 campionepositivo-alto è stato male interpretato come positivo-basso. I risultati ottenuti sul campo dimostrano che il Test SureStepMono di ABI è di facile impiego e consente di ottenere una lettura qualitativa visiva degli anticorpi della MI.
Tabella 4. Riepilogo dei risultati ottenuti sul campo con il Test SureStep Mono
ID ABI SureStepMono
Kit incommercio
1930
-
Trigliceridi(mg/dl)
Colesterolo(mg/dl)
-
---280
380170200190890
119012402370
SD2192-9
SD2192-10
SD2192-20
SD2192-17SD2192-11
--
---
Sostanzeaggiunte Controllo Campione
arricchito
+
-
Conc.(mg/ml)
ID campione
-
-
-
-
-
-
-
++
100
0,06
1
10Positivo-alto
Emoglobina
Albumina
Bilirubina
Positivo-alto
Positivo-alto
Positivo-alto
Positivo-basso
Positivo-basso
Positivo-basso
Positivo-basso
Negativo
Negativo
Negativo
Negativo
+
+
+
++
++
++
++++
++
++
++++
Campione Positivo-basso
59
0
60
1
Numero Negativo Non valido
180
60
60
60
Positivo-alto
Risultati sul campo
60
6059
0 0
0
0
0
00
0
61
Positivo-alto
Positivo-basso
Negativo
Totale

31
Para laboratórios com derrogação CLIA – Teste com amostras de sangue por picada do dedo ou desangue total
Para laboratórios de complexidade moderada – Teste com amostras de sangue total, soro e plasma
UTILIZAÇÃO PRETENDIDA
O teste SureStep Mono é um teste rápido para a detecção visual qualitativa de anticorpos heterófilos específicos damononucleose infecciosa (MI) em sangue total, soro ou plasma humano. Este kit de teste destina-se a ser utilizadoexclusivamente em laboratórios, como auxiliar no diagnóstico da MI em pacientes com sintomas clínicos característicos.
RESUMO
A mononucleose infecciosa é uma doença auto-limitadora aguda causada pelo vírus Epstein-Barr (EBV) (1–3). A infecçãocom o EBV em idades precoces é assintomática (4–5). No entanto, até 50 % da infecção na fase da adolescência e inícioda vida adulta desenvolverá manifestações clínicas associadas à MI.
O diagnóstico da MI baseia-se na avaliação dos sintomas clínicos característicos e nas alterações serológicas. Odiagnóstico serológico da MI foi demonstrado através da detecção de anticorpos heterófilos e anticorpos específicos doEBV (3–5). O anticorpo heterófilo é detectável em dada altura durante a MI na maioria dos adultos. É uma prática correnteentre médicos utilizarem a detecção de anticorpos heterófilos como auxiliar no diagnóstico da MI (3–7). O teste SureStepMono utiliza extractos de eritrócitos de bovinos, os quais apresentam maior sensibilidade e especificidade do que osextractos de outras espécies. (4)
PRINCÍPIO DO TESTE
O teste SureStep Mono destina-se a detectar a MI através da interpretação visual do desenvolvimento de cores nodispositivo de teste, que é um imunoensaio sandwich de conjugado de ouro de fase sólida. O dispositivo de teste écomposto por uma tira de membrana pré-revestida com antigénios heterófilos na região da linha de teste e com anticorpoanti-rato de cabra na região da linha de controlo. Na extremidade da membrana encontra-se uma almofada de conjugadode ouro coloidal/anticorpo IgM anti-humano. Uma mistura de conjugado de ouro coloidal, juntamente com a amostra etampão revelador, deslocar-se-á cromatograficamente ao longo da membrana por acção capilar. Sempre que houveranticorpos heterófilos da MI presentes na amostra do paciente, a mistura migrará para a região da linha de teste e formaráuma linha visível à medida que o anticorpo complexa com o antigénio heterófilo. Sempre que os anticorpos heterófilos daMI estiverem ausentes da amostra, não se forma qualquer linha de cor visível na região da linha de teste. Por conseguinte,a presença de uma linha colorida na região da linha de teste indica um resultado positivo. Aparecerá sempre uma linhacolorida na região de controlo. Esta linha de controlo funciona como indicador processual para o adequado desempenhodo teste e do dispositivo.
ARMAZENAMENTO E ESTABILIDADE
O kit de teste deve ser armazenado refrigerado ou à temperatura ambiente (2°–30 °C) no saco selado durante todo o prazode validade do kit.
PRECAUÇÕES
• Exclusivamente para uso em diagnóstico in-vitro.
• Para utilização por profissionais e em laboratórios.• Todas as amostras dos pacientes devem ser tratadas como se fossem passíveis de transmitir doenças. • Não misture reagentes de diferentes lotes.• Não abra o saco de alumínio até estar preparado para efectuar o teste.• Não use sangue total que tenha sido armazenado durante mais de três dias.
AS INSTRUÇÕES DE COMPLEXIDADE DA CLIA ENCONTRAM-SE NA SECÇÃO REFERENTE AO CONTROLO DAQUALIDADE ABAIXO. SIGA AS INSTRUÇÕES ADEQUADAS, EXIGIDAS PELAS NORMAS DO SEU LABORATÓRIO.
TESTE MONO
PORTUGUÊS
Não reutilize o teste.

32
• Não deverão ser utilizadas amostras muito hemolizadas. • Os controlos e tampão revelador contêm azida de sódio. A azida de sódio poderá reagir com a tubagem de chumbo
ou cobre e formar azidas de metal, potencialmente explosivas. Para a eliminação destas soluções, faça sempre correruma grande quantidade de água para evitar a acumulação de azidas.
Os reagentes para extracção são ligeiramente cáusticos. Evite o contacto com os olhos ou mucosas. Em caso de contactoacidental, lave minuciosamente com água.
Os controlos positivo e negativo contêm azida de sódio que poderá reagir com a tubagem de chumbo ou cobre e formarazidas de metal, potencialmente explosivas. Para a eliminação destas soluções, faça sempre correr uma grande quantidadede água para evitar a acumulação de azidas.
Aviso: material com potencial risco biológicoTodas as unidades doadoras de plasma e soro humano usadas na preparação dos controlos positivo e negativo foramtestadas por métodos aprovados pela FDA dos EUA relativamente à presença de anti-VIH-1/VIH-2, HBsAg e anti-HCV,tendo-se revelado negativas. Todavia, dever-se-á tomar todas devidas as precauções durante a manipulação e eliminaçãodestes artigos com nível de segurança biológica 2, conforme recomendado no manual do Center for DiseaseControl/National Institutes of Health, Biosafety in Micobiological and BiomedicalLaboratories, 1984.
REAGENTES E MATERIAIS FORNECIDOS
• 25 dispositivos de teste embalados individualmente. Cada dispositivo contém uma tira de teste com membranarevestida com antigénio heterófilo e uma almofada com anticorpo colorido.
• Tampão revelador (8 ml) :solução-tampão salina de fosfato 0,1 M, com aditivos e 0,1% de azida de sódio.
• Controlo negativo (0,2 ml) : soro ou plasma humano normal diluído em solução salina com 0,2% deazida de sódio.
• Controlo positivo (0,2 ml) : soro ou plasma humano positivopara anticorpos heterófilos de MI ou sorodiluído em solução salina com 0,2% de azida de sódio.
• 25 pipetas de transferência descartáveis.
• Um folheto com instruções.
COLHEITA E MANUSEAMENTO DA AMOSTRA
Picada do dedo 1. Limpe a área a lancetar com um cotonete com álcool.2. Aperte a ponta do dedo e perfure com uma lanceta estéril.3. Limpe a primeira gota de sangue com uma compressa ou algodão esterilizados.4. Deixe a segunda gota de sangue cair directamente no poço da amostra do dispositivo de teste ou use a pipeta de
transferência fornecida para obter sangue fresco. Coloque 2 gotas (aproximadamente de 40 µl) no poço da amostra.
Sangue total1. Um flebotomista qualificado deve colher o sangue total num tubo de colheita com tampa púrpura, azul ou verde
(contendo EDTA, citrato ou heparina, respectivamente) por punção venosa.2. O sangue total pode ser imediatamente usado no teste ou armazenado a 2–8°C durante um período máximo de três dias.
Plasma1. Um flebotomista qualificado deve colher o sangue total num tubo de colheita com tampa púrpura, azul ou verde
(contendo EDTA, citrato ou heparina, respectivamente) por punção venosa.2. Separe o plasma por centrifugação.3. Retire cuidadosamente o plasma para o teste, ou rotule e armazene a 2–8°C durante um período máximo de duas
semanas. O plasma pode ser congelado a -20°C durante um período máximo de um ano.
Soro1. Um flebotomista qualificado deve colher o sangue total num tubo de colheita com tampa vermelha (não contendo
qualquer anti coagulante) por punção venosa.2. Deixe o sangue coagular e separe o soro por centrifugação. 3. Retire cuidadosamente o soro para o teste, ou rotule e armazene a 2–8°C durante um período máximo de duas semanas.
O soro pode ser congelado a -20°C durante um período máximo de um ano.
BUF DEV

33
PROCEDIMENTO DE TESTE
3. Adicione imediatamente 3–4 gotas de tampão revelador.
4. Depois do tampão revelador ter sido adicionado, irão começar a aparecer linhas coloridas. Dependendo daconcentração de anticorpos heterófilos presentes, os resultados positivos poderão ser observados ao fim de 3 minutos.No entanto, para confirmar um resultado negativo, é necessário um tempo de reacção de 5 minutos. Não leia resultadosdecorridos 8 minutos.
Dispositivo de Teste
Região deControlo
Região de Teste
Poço daAmostra
O dispositivo de teste, o tampão revelador, as amostras dospacientes e controlos deverão ser colocados à temperaturaambiente (20 a 30°C) antes do teste. Não abra os sacos até estarpreparado para efectuar o teste.
1. Retire o dispositivo de teste do respectivo saco protector. (Permitaque o dispositivo atinja a temperatura ambiente antes de abrir o sacopara evitar a condensação de humidade na membrana). Rotule odispositivo com a identificação do paciente ou do controlo.
2. Coloque amostra no respectivo poço:
Sangue por picada do dedo: deixe a segunda gota de sangue cairdirectamente no poço da amostra do dispositivo de teste ou use apipeta de transferência fornecida para obter sangue fresco.Coloque 2 gotas (aproximadamente de 40 µl) no poço da amostra.
Amostras de sangue total em tubos de colheita: coloque duas gotas(aproximadamente 40 µl) no poço da amostra, mantendo a pipetade transferência fornecida numa posição vertical.
Amostras de soro ou plasma: coloque uma gota (aproximadamente20 µl) de soro ou plasma no poço da amostra, mantendo a pipetade transferência fornecida numa posição vertical.
Pipeta detransferência
INTERPRETAÇÃO DOS RESULTADOS
Positivo:Aparecem duas linhas coloridas. Uma na região de controlo (C) e outra na região deteste (T). Quando se testam amostras fortemente positivas, a intensidade da linha decontrolo pode ser mais clara do que o esperado. Não se recomenda a comparaçãoda intensidade das linhas.
Negativo:Só aparece uma linha colorida na região da linha de controlo. Não existe qualquerlinha colorida esbatida aparente na região da linha de teste (T).
Inválido:A ausência de uma linha na região de controlo é uma indicação de que poderá terocorrido um erro no procedimento ou deterioração do reagente de teste. O testedeverá ser considerado nulo, uma vez que poderá ter sido levado a cabo umprocedimento de teste incorrecto ou os reagentes poderão ter-se deteriorado. Repitao teste com um novo dispositivo de teste. Se o problema persistir, contacte aassistência técnica da Applied Biotech pelo telefone (888) 578-7956 para chamadasnos E.U.A. ou pelo telefone +1 858 713-9668 para chamadas internacionais. Aschamadas são atendidas por funcionários que falam inglês. Para outros idiomasdeverá enviar um e-mail para o endereço [email protected].
C T
C T
C T
C T
C TMONO

34
CONTROLO DE QUALIDADE
Controlo interno do procedimentoO teste inclui um controlo do procedimento. O aparecimento de uma linha colorida na região de controlo (C) éconsiderado um controlo interno do procedimento, indicando um desempenho e reagentes reactivos correctos.
Um fundo transparente na janela de resultado é considerado como um controlo negativo interno. Todavia, quando setestam amostras de sangue total, o fundo pode ficar ligeiramente avermelhado devido ao baixo nível de hemólise de algunsglóbulos vermelhos. Tal é aceitável desde que não interfira com a interpretação do resultado do teste. O teste é inválido seo fundo não se limpar e obscurecer a leitura do resultado.
Controlo de qualidade externoA boa prática laboratorial recomenda a utilização de controlos externos para garantir a funcionalidade dos reagentes e umdesempenho adequado. No kit são fornecidos controlos positivos e negativos. Estes controlos são à base de soro ou plasmahumano e são testados como uma amostra de um paciente. Durante o teste com os controlos, coloque uma gota(aproximadamente 20 µl) de controlo positivo ou negativo no poço da amostra utilizando a pipeta de transferênciafornecida e segurando-a na vertical. Adicione imediatamente 3 ou 4 gotas de tampão revelador. Duas linhas indicam umsinal positivo: uma na região de teste (T) e uma na região de controlo (C). Apenas uma linha na região de controlo indicaum sinal negativo (C). Leia os resultados passado 5 minutos e não os interprete se tiverem decorrido 8 minutos. Se oscontrolos não apresentarem o desempenho esperado, não faça a interpretação dos resultados. Repita o teste ou contactea assistência técnica.
PARA LABORATÓRIOS COM DERROGAÇÃO CLIA:
Recomenda-se que se teste um controlo externo Positivo e Negativo com cada novo kit de teste. Cada utilizador deverátestar um controlo externo positivo e negativo de 25 em 25 kits de teste.
PARA LABORATÓRIOS DE COMPLEXIDADE MODERADA OU PROFISSIONAIS DE SAÚDE:
Recomenda-se o teste de um controlo externo positivo e negativo em cada nova remessa ou kit com novo número de lote,bem como sempre que os regulamentos locais e nacionais assim o exigirem.
LIMITAÇÕES
Este kit de teste destina-se a ser usado para a detecção qualitativa de anticorpos IgM para o antigénio heterófilo da MI. Umresultado positivo sugere a presença de anticorpos IgM para o antigénio heterófilo.
Este kit de teste deve ser usado para indivíduos sintomáticos que se suspeite estarem infectado com MI. O diagnóstico daMI deverá ser feito através da confirmação com outros achados clínicos.
Um resultado negativo não exclui a possibilidade da MI, porque os anticorpos para o antigénio heterófilo podem estarausentes ou podem não estar presentes em quantidade suficiente para serem detectados.
RESULTADOS ESPERADOS
Durante a fase aguda da MI, os anticorpos heterófilos são detectáveis em 80 a 85% dos pacientes. Os anticorpos heterófilossão detectáveis durante o primeiro mês da doença e diminuem rapidamente após a quarta (4) semana.
CARACTERÍSTICAS DE DESEMPENHO
A. PrecisãoA precisão do teste SureStepTM Mono foi avaliada em comparação com um teste qualitativo imunocromatográfico a coresà venda no mercado, para a detecção de anticorpos heterófilos IgM da MI em sangue total, soro ou plasma humano. Noestudo comparativo foram utilizadas quatrocentas e seis amostras de soro, plasma e sangue total (288 soro, 103 plasma e15 sangue total). Os resultados estão sintetizados na tabela 1.
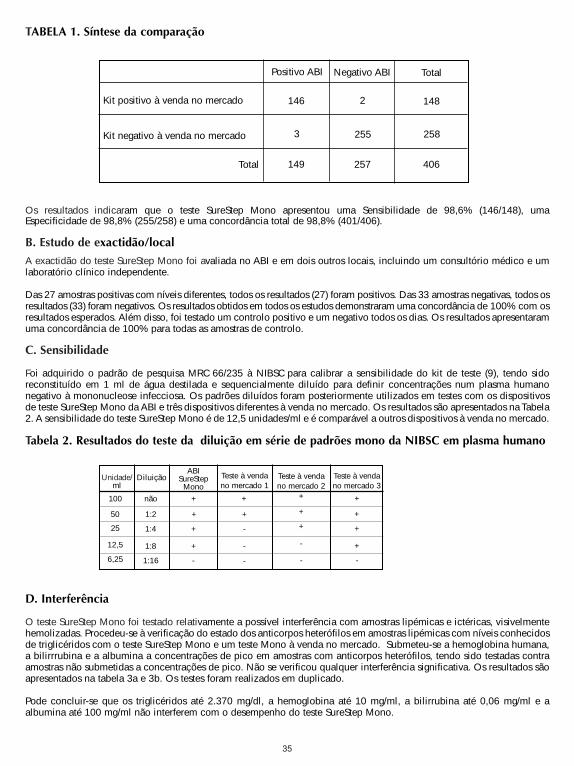
35
Os resultados indicaram que o teste SureStep Mono apresentou uma Sensibilidade de 98,6% (146/148), umaEspecificidade de 98,8% (255/258) e uma concordância total de 98,8% (401/406).
B. Estudo de exactidão/local
A exactidão do teste SureStep Mono foi avaliada no ABI e em dois outros locais, incluindo um consultório médico e umlaboratório clínico independente.
Das 27 amostras positivas com níveis diferentes, todos os resultados (27) foram positivos. Das 33 amostras negativas, todos osresultados (33) foram negativos. Os resultados obtidos em todos os estudos demonstraram uma concordância de 100% com osresultados esperados. Além disso, foi testado um controlo positivo e um negativo todos os dias. Os resultados apresentaramuma concordância de 100% para todas as amostras de controlo.
C. Sensibilidade
Foi adquirido o padrão de pesquisa MRC 66/235 à NIBSC para calibrar a sensibilidade do kit de teste (9), tendo sidoreconstituído em 1 ml de água destilada e sequencialmente diluído para definir concentrações num plasma humanonegativo à mononucleose infecciosa. Os padrões diluídos foram posteriormente utilizados em testes com os dispositivosde teste SureStep Mono da ABI e três dispositivos diferentes à venda no mercado. Os resultados são apresentados na Tabela2. A sensibilidade do teste SureStep Mono é de 12,5 unidades/ml e é comparável a outros dispositivos à venda no mercado.
Tabela 2. Resultados do teste da diluição em série de padrões mono da NIBSC em plasma humano
D. Interferência
O teste SureStep Mono foi testado relativamente a possível interferência com amostras lipémicas e ictéricas, visivelmentehemolizadas. Procedeu-se à verificação do estado dos anticorpos heterófilos em amostras lipémicas com níveis conhecidosde triglicéridos com o teste SureStep Mono e um teste Mono à venda no mercado. Submeteu-se a hemoglobina humana,a bilirrrubina e a albumina a concentrações de pico em amostras com anticorpos heterófilos, tendo sido testadas contraamostras não submetidas a concentrações de pico. Não se verificou qualquer interferência significativa. Os resultados sãoapresentados na tabela 3a e 3b. Os testes foram realizados em duplicado.
Pode concluir-se que os triglicéridos até 2.370 mg/dl, a hemoglobina até 10 mg/ml, a bilirrubina até 0,06 mg/ml e aalbumina até 100 mg/ml não interferem com o desempenho do teste SureStep Mono.
Positivo ABI
Kit positivo à venda no mercado
Kit negativo à venda no mercado
Negativo ABI Total
Total
146 2
255
148
258
406257
3
149
TABELA 1. Síntese da comparação
Unidade/ml
DiluiçãoABI
SureStepMono
Teste à vendano mercado 1
Teste à vendano mercado 2
Teste à vendano mercado 3
100
50
25
12,5
6,25
não
1:2
1:4
1:8
1:16
+
+
+
+
-
+
+
+
+
+
+
+
+
+
-
-
-
-
- -
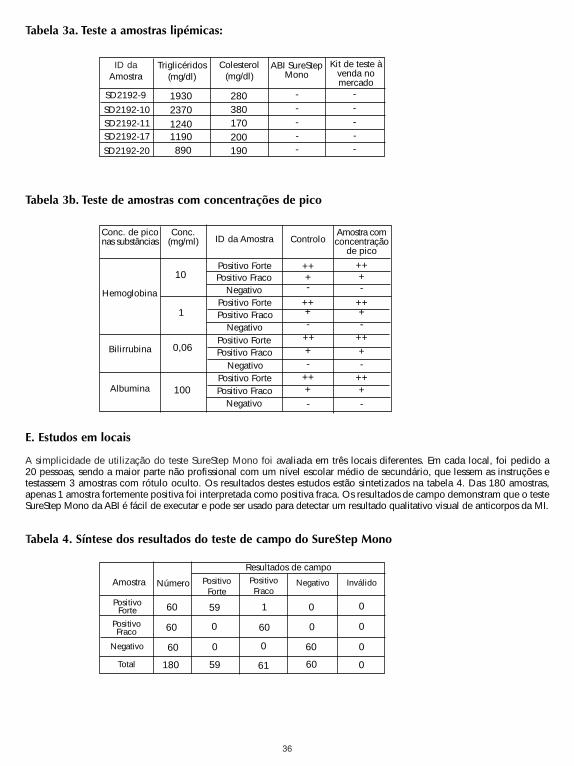
36
Tabela 3a. Teste a amostras lipémicas:
Tabela 3b. Teste de amostras com concentrações de pico
E. Estudos em locais
A simplicidade de utilização do teste SureStep Mono foi avaliada em três locais diferentes. Em cada local, foi pedido a 20 pessoas, sendo a maior parte não profissional com um nível escolar médio de secundário, que lessem as instruções etestassem 3 amostras com rótulo oculto. Os resultados destes estudos estão sintetizados na tabela 4. Das 180 amostras,apenas 1 amostra fortemente positiva foi interpretada como positiva fraca. Os resultados de campo demonstram que o testeSureStep Mono da ABI é fácil de executar e pode ser usado para detectar um resultado qualitativo visual de anticorpos da MI.
Tabela 4. Síntese dos resultados do teste de campo do SureStep Mono
ID daAmostra
ABI SureStepMono
Kit de teste àvenda nomercado
1930
-
Triglicéridos(mg/dl)
Colesterol(mg/dl)
-
---280
380170200190890
119012402370
SD2192-9
SD2192-10
SD2192-20
SD2192-17SD2192-11
--
---
Conc. de piconas substâncias Controlo
Amostra comconcentração
de pico
+
-
Conc.(mg/ml) ID da Amostra
-
-
-
-
-
-
-
++
100
0,06
1
10Positivo Forte
Hemoglobina
Albumina
Bilirrubina
Positivo Forte
Positivo Forte
Positivo Forte
Positivo Fraco
Positivo Fraco
Positivo Fraco
Positivo Fraco
Negativo
Negativo
Negativo
Negativo
+
+
+
++
++
++
++++
++
++
++++
Amostra PositivoFraco
59
0
60
1
Número Negativo Inválido
180
60
60
60
PositivoForte
Resultados de campo
60
6059
0 0
0
0
0
00
0
61
PositivoForte
PositivoFraco
Negativo
Total

37
För laboratorier som undantagits av CLIA – Test med blodprov från finger eller helblodsprov
För måttligt komplexa laboratorier – Test med serum, plasma eller helblodsprov
AVSEDD ANVÄNDNING
SureStep Mono-test är ett snabbtest för visuell, kvalitativ detektion av heterofila antikroppar, som är specifika för infektiösmononukleos (IM) i humanserum, plasma eller helblod. Denna testutrustning är avsedd som ett hjälpmedel vid diagnos avpatienter med infektiös mononukleos, med karakteristiska, kliniska symptom och är endast avsedd för professionellanvändning i laboratorium.
SAMMANFATTNING
Infektiös mononukleos är en akut, självbegränsande sjukdom, som orsakas av Epstein-Barr-viruset (EBV) (1–3). Infektionmed EBV tidigt i livet är vanligen asymptomatiskt (4–5). Upp till 50 % av infektioner, som förekommer hos tonåringar ochunga personer, utvecklar manifestationer som associeras med infektiös mononukleos.
Diagnos av infektiös mononukleos baseras på utvärderingen av karakteristiska kliniska symptom och serologiskaförändringar. Serologisk diagnos av infektiös mononukleos har påvisats genom detektion av heterofila och EBV-specifikaantikroppar (3–5). Den heterofila antikroppen är detekterbar vid någon tidpunkt under infektiös mononukleos hos de flestavuxna. Det är allmänt accepterad praxis bland läkare att använda detektion av heterofila antikroppar som ett hjälpmedelvid diagnos av infektiös mononukleos (3–7). SureStep Mono Test använder sig av bovint erytrocytextrakt, som har en högrekänslighet och specificitet än extrakt från andra arter. (4)
TESTPRINCIP
SureStep Mono-testet har utformats för att detektera infektiös mononukleos genom visuell tolkning av färgutveckling itestutrustningen, som är en immunanalys av sandwichtyp i fast fas på guldkonjugat. Testet består av en membranremsa, somför-belagts med heterofila antigener på testlinjefältet och get-anti-musantikropp på kontrollinjefältet. Den anti-humana IgM antikroppen på kolloidal guldkonjugatdyna är placerad på membranets ände. En blandning av kolloidalt guldkonjugat,tillsammans med provet och framkallningsbuffert, rör sig sedan kromatografiskt längs membranet genom kapillär aktivitet.När IM-heterofila antikroppar är närvarande i patientprovet, kommer blandningen att vandra till testlinjefältet och bilda ensynlig linje som antikroppskomplexen med den heterofila antigenen. När IM-heterofila antikroppar är frånvarande i provetbildas ingen synligt färgad linje på testlinjefältet. Närvaro av en färgad linje i testlinjefältet tyder därför på ett positivtresultat. En färgad linje kommer alltid att framträda i kontrollfältet. Denna kontrollinje tjänar som en indikator på att testetoch testutrustningen fungerade på rätt sätt.
FÖRVARING OCH HANTERING
Testutrustningen kan förvaras antingen kyld eller i rumstemperatur 2–30 °C i den förslutna förpackningen under denangivna hållbarhetstiden.
FÖRSIKTIGHETSÅTGÄRDER
• Endast för in vitro-diagnostik.
• För professionell användning och användning i laboratorium.• Alla patientprov bör hanteras som potentiellt smittsamma.• Blanda inte reagenser från olika partier.• Öppna inte folieförpackningen förrän allt är klart för analys.• Använd inte helblod som lagrats i mer än tre dagar.• Kraftigt hemolytiska prover bör ej användas.
CLIA KOMPLEXITETINSTRUKTIONER ANGES NEDAN I AVSNITTET KVALITETSKONTROLL. VAR VÄNLIG FÖLJDE LÄMPLIGA INSTRUKTIONER SOM ERFORDRAS ENLIGT RIKTLINJERNA FÖR DITT LABORATORIUM.
MONOTEST
SVENSKA
Återanvänd inte detta test.

38
• Framkallningsbuffert och kontroller innehåller natriumazid. Natriumazid kan reagera med bly- eller kopparrör ochbilda högexplosiva metallazider. Vid kassering av dessa lösningar, spola alltid med rikliga mängder vatten för attundvika azidansamling.
Extraktionsreagenserna är lätt frätande. Undvik kontakt med ögon eller slemhinnor. Vid oavsiktlig kontakt, tvätta noggrantmed vatten.
Positiva och negativa kontroller innehåller natriumazid som kan reagera med bly- eller kopparrör och bilda högexplosivametallazider. Vid kassering av dessa lösningar, spola all t id med rikliga mängder vatten för att undvikaazidansamling.
Varning: Potentiellt bioriskmaterialVarje donatorenhet av humanplasma eller serum, som används vid framställningen av de positiva och negativakontrollerna, testades med metoder som godkänts av FDA (Food and Drug Administration) för närvaro av anti-HIV-1/HIV-2, HBsAg och anti-HCV och befanns vara negativa. Försiktighetsåtgärder bör emellertid vidtagas vidhantering och kassering av dessa artiklar vid biosäkerhetsnivå 2, som rekommenderas i Center for DiseaseControl/National Institutes of Health Manual, Biosafety in Micobiological and BiomedicalLaboratories, 1984.
MEDFÖLJANDE REAGENSER OCH MATERIAL
• 25 individuellt förpackade testutrustningar. Varje testutrustning innehåller en testremsa, vars membran ochfärgade antikroppsdyna belagts med heterofilt antigen.
• Framkallningsbuffert (8 ml) : 0,1 M fosfatbuffrad saltlösning, med tillsatsmedel och 0,1 %natriumazid.
• Negativ kontroll (0,2 ml) : Normal humanplasma eller serum utspätt i saltlösning med 0,2 %natriumazid.
• Positiv kontroll (0,2 ml) : IM-heterofil antikropp-positiv humanplasma eller serum utspätt isaltlösning med med 0,2 % natriumazid.
• 25 överföringspipetter för engångsbruk.
• Ett produktblad.
UPPSAMLING OCH HANTERING AV PROV
Blodprov från finger1. Rengör området som skall stickas med en spritkompress.2. Kläm åt runt fingertoppen och stick hål med en steril lancett.3. Torka bort den första bloddroppen med steril gasväv eller bomull.4. Låt den andra droppen rinna direkt ner i testutrustningens provbrunn eller använd den tillhandahållna pipetten
för att erhålla färskt blod. Tillsätt 2 droppar (ca 40 µl) till provbrunnen.
Helblod1. En utbildad laboratorieassistent bör uppsamla helblodet i ett uppsamlingsrör med lila, blått eller grönt lock
(innehållande EDTA, citrat respektive heparin) genom venpunktur.2. Helblodet kan användas för testning omedelbart eller förvaras vid 2–8 °C upp till tre dagar.
Plasma1. En utbildad laboratorieassistent bör uppsamla helblodet i ett uppsamlingsrör med lila, blått eller grönt lock
(innehållande EDTA, citrat respektive heparin) genom venpunktur.2. Separera plasman genom centrifugering.3. Ta försiktigt bort plasman som skall testas eller märk och förvara vid 2–8 °C upp till två veckor. Plasma kan
frysas vid -20 °C upp till ett år.
Serum1. En utbildad laboratorieassistent bör uppsamla helblodet i ett uppsamlingsrör med rött lock (som inte innehåller
anti koagulant) genom venpunktur.2. Låt blodet koagulera och separera serum genom centrifugering.3. Ta försiktigt bort serumet som skall testas eller märk och förvara vid 2–8 °C upp till två veckor. Plasma kan
frysas vid -20 °C upp till ett år.
BUF DEV

39
TESTPROCEDUR
3. Tillsätt omedelbart 3–4 droppar framkallningsbuffert.
4. Vänta tills de färgade linjerna syns efter tillsatsen av framkallningsbufferten. Beroende på koncentrationen av heterofilaantikroppar som är närvarande, kan positiva resultat vara synliga efter 3 minuter. Den fullständiga reaktionstiden på5 minuter är emellertid nödvändig för att bekräfta negativa resultat. Vänta högst 8 minuter med att avläsa resultaten.
Testutrustning
Kontrollfält Testfält
Provbrunn
Testanordning, framkallningsbuffert, patientprover och kontrollerbör återfå rumstemperatur (20–30 °C) innan testning sker.Öppna inte förpackningar förrän allt är klart för analys.
1. Ta ut kassetten ur sin skyddsförpackning. (Låt utrustningen återfårumstemperatur före öppnande för att undvika kondensation påmembranet). Märk utrustningen med patient- ellerkontrollidentifikation. i
2. Tillsätt prov till provbrunnen:
Blodprov från finger: Låt den andra droppen rinna direkt ner itestutrustningens provbrunn eller använd den tillhandahållnapipetten för att erhålla färskt blod. Tillsätt 2 droppar (ca 40 µl) tillprovbrunnen.
Helblodsprov i uppsamlingsrör: tillsätt två droppar (ca 40 µl) tillprovbrunnen, medan den tillhandahållna överföringspipetten hållesi vertikalt läge.
Plasma- eller serumprov: tillsätt en droppe (ca 20 µl) serum ellerplasma till provbrunnen, medan den tillhandahållnaöverföringspipetten hålles i vertikalt läge.
Överföringspipett
TOLKNING AV RESULTAT
Positivt:Två färgade linjer syns. En i kontrollfältet (C) och en annan färgad linje i testfältet (T).Vid testning med starkt positiva prov, kan kontrollinjens färgintensitet vara ljusare änväntat. Det rekommenderas inte att jämföra linjernas färgintensitet.
Negativt:Endast en färgad linje syns i kontrollinjefältet. Ingen synbar svagt färgad linje itestlinjefältet (T).
Ogiltigt:Total frånvaro av en linje i kontrollfältet är ett tecken på procedurfel eller atttestreagensen försämrats. Testet skall annulleras eftersom testproceduren kan hautförts på fel sätt eller reagenserna kan ha försämrats. Gör om testet med en nytestenhet. Om problemen kvarstår, ring Applied Biotechs Tekniska Service på telefon(888) 578-7956 för samtal inom USA och +1 858 713-9668 för internationella samtal.Samtalen besvaras av engelsktalande personal. För andra språk, sänd e-post [email protected].
C T
C T
C T
C T
C TMONO

40
KVALITETSKONTROLL
Intern procedurkontrollEn procedurkontroll ingår i testet. När en färgad linje framträder i kontrollfältet (C) betraktas det som en intern positivprocedurkontroll som tyder på korrekt funktion och reaktiva reagenser.
En klar bakgrund i resultatfönstret betraktas som en intern negativ procedurkontroll. När helblodsprov testas kan emellertidbakgrunden verka lätt rödaktig, på grund av den låga hemolysnivån hos vissa röda blodkroppar. Detta är acceptabelt sålänge det inte interfererar med avläsningen av testet. Testet är ogiltigt om bakgrunden inte klarnar upp och fördunklaravläsningen av testresultatet.
Extern kvalitetskontrollGod laboratoriepraxis rekommenderar användning av externa kontroller för att tillförsäkra reagensernas funktion ochprestanda. Positiva och negativa kontroller tillhandahålls i förpackningen. Dessa kontroller är baserade på humanserumeller plasma och testas som ett patientprov. Vid testning med kontrollerna, tillsätt en droppe (ca 20 µl) positiv eller negativkontroll i provbrunnen, med användning av den tillhandahållna överföringspipetten som hålles i vertikalt läge. Tillsättomedelbart 3–4 droppar framkallningsbuffert. Ett positivt resultat anges av två linjer, en i testfältet (T) och en ikontrollfältet (C). Ett negativt resultat anges av endast en linje i kontrollfältet (C). Läs av resultaten efter 5 minuter men väntahögst 8 minuter med avläsningen. Tolka inte testresultaten om kontrollerna inte fungerar som förväntat. Gör om testet ellerkontakta teknisk service.
FÖR LABORATORIER SOM UNDANTAGITS AV CLIA:
Både en positiv och en negativ extern kontroll måste testas när en ny testsats öppnas. Varje användare måste testa en positivoch en negativ kontroll en gång per 25 testsatser.
FÖR MÅTTLIGT KOMPLEXA LABORATORIER ELLER PROFESSIONELLA VÅRDGIVARE:
Det rekommenderas att både en positiv och en negativ kontroll testas med varje ny sändning eller nytt partinummer påförpackningen och i annat fall enligt lokala föreskrifter.
BEGRÄNSNINGAR
Denna testsats skall användas för kvalitativ detektion av IgM-antikroppar mot IM-heterofil antigen. Ett positivt resultat tyderpå närvaro av IgM-antikroppar mot heterofil antigen.
Denna testsats bör användas för symptomatiska patienter, som misstänks ha infektiös mononukleos. Diagnos av infektiösmononukleos bör göras genom bekräftelse med andra kliniska fynd.
Ett negativt resultat utesluter inte möjligheten av infektiös mononukleos eftersom antikroppar mot heterofil antigen kan varafrånvarande eller inte vara närvarande i tillräcklig mängd för att upptäckas.
FÖRVÄNTADE RESULTAT
Under den akuta fasen av infektiös mononukleos är heterofila antikroppar detekterbara hos 80–85 % av patienterna.Heterofila antikroppar är detekterbara under sjukdomens första månad och minskar snabbt efter vecka fyra (5).
PRESTANDAEGENSKAPER
A. PrecisionPrecisionen hos SureStep™ Mono-test utvärderades i jämförelse med en kommersiellt tillgänglig, kvalitativ färg-immunkromatografisk analys för detektionen av IM-IgM-heterofila antikroppar i humanserum, plasma eller helblod.Fyrahundrasex humanserum-, plasma- eller helblodsprover (288 serum, 103 plasma och 15 helblod) användes ijämförelsestudien. Resultaten finns sammanfattade i Tabell 1.
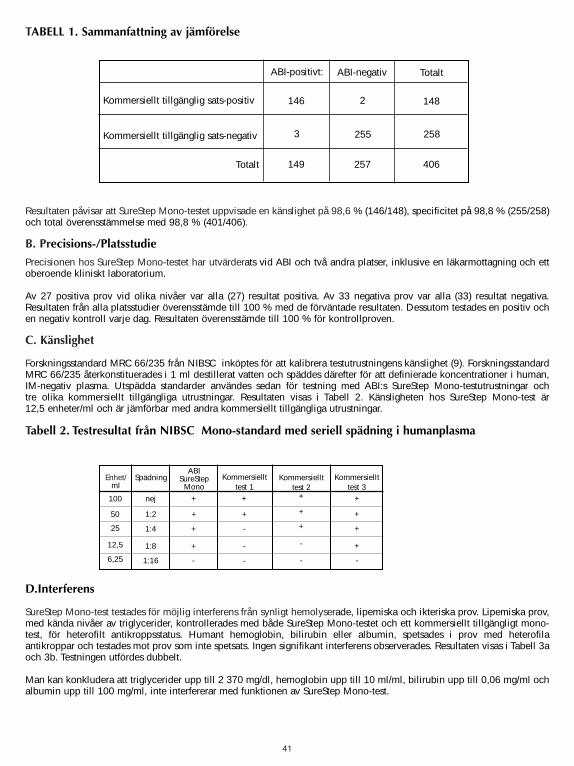
41
Resultaten påvisar att SureStep Mono-testet uppvisade en känslighet på 98,6 % (146/148), specificitet på 98,8 % (255/258)och total överensstämmelse med 98,8 % (401/406).
B. Precisions-/Platsstudie
Precisionen hos SureStep Mono-testet har utvärderats vid ABI och två andra platser, inklusive en läkarmottagning och ettoberoende kliniskt laboratorium.
Av 27 positiva prov vid olika nivåer var alla (27) resultat positiva. Av 33 negativa prov var alla (33) resultat negativa.Resultaten från alla platsstudier överensstämde till 100 % med de förväntade resultaten. Dessutom testades en positiv ochen negativ kontroll varje dag. Resultaten överensstämde till 100 % för kontrollproven.
C. Känslighet
Forskningsstandard MRC 66/235 från NIBSC inköptes för att kalibrera testutrustningens känslighet (9). ForskningsstandardMRC 66/235 återkonstituerades i 1 ml destillerat vatten och späddes därefter för att definierade koncentrationer i human,IM-negativ plasma. Utspädda standarder användes sedan för testning med ABI:s SureStep Mono-testutrustningar och tre olika kommersiellt tillgängliga utrustningar. Resultaten visas i Tabell 2. Känsligheten hos SureStep Mono-test är 12,5 enheter/ml och är jämförbar med andra kommersiellt tillgängliga utrustningar.
Tabell 2. Testresultat från NIBSC Mono-standard med seriell spädning i humanplasma
D.Interferens
SureStep Mono-test testades för möjlig interferens från synligt hemolyserade, lipemiska och ikteriska prov. Lipemiska prov,med kända nivåer av triglycerider, kontrollerades med både SureStep Mono-testet och ett kommersiellt tillgängligt mono-test, för heterofilt antikroppsstatus. Humant hemoglobin, bilirubin eller albumin, spetsades i prov med heterofilaantikroppar och testades mot prov som inte spetsats. Ingen signifikant interferens observerades. Resultaten visas i Tabell 3aoch 3b. Testningen utfördes dubbelt.
Man kan konkludera att triglycerider upp till 2 370 mg/dl, hemoglobin upp till 10 ml/ml, bilirubin upp till 0,06 mg/ml ochalbumin upp till 100 mg/ml, inte interfererar med funktionen av SureStep Mono-test.
ABI-positivt:
Kommersiellt tillgänglig sats-positiv
Kommersiellt tillgänglig sats-negativ
ABI-negativ Totalt
Totalt
146 2
255
148
258
406257
3
149
TABELL 1. Sammanfattning av jämförelse
Enhet/ml
SpädningABI
SureStepMono
Kommersiellttest 1
Kommersiellt test 2
Kommersiellttest 3
100
50
25
12,5
6,25
nej
1:2
1:4
1:8
1:16
+
+
+
+
-
+
+
+
+
+
+
+
+
+
-
-
-
-
- -
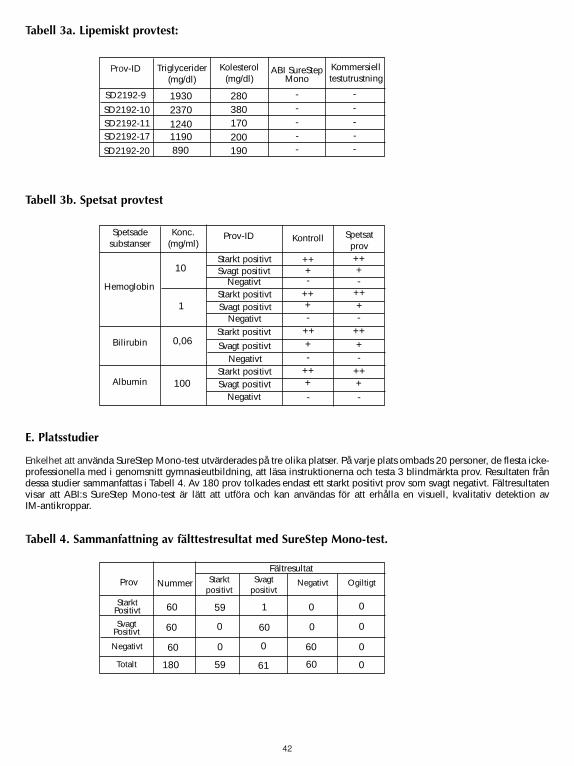
42
Tabell 3a. Lipemiskt provtest:
Tabell 3b. Spetsat provtest
E. Platsstudier
Enkelhet att använda SureStep Mono-test utvärderades på tre olika platser. På varje plats ombads 20 personer, de flesta icke-professionella med i genomsnitt gymnasieutbildning, att läsa instruktionerna och testa 3 blindmärkta prov. Resultaten fråndessa studier sammanfattas i Tabell 4. Av 180 prov tolkades endast ett starkt positivt prov som svagt negativt. Fältresultatenvisar att ABI:s SureStep Mono-test är lätt att utföra och kan användas för att erhålla en visuell, kvalitativ detektion av IM-antikroppar.
Tabell 4. Sammanfattning av fälttestresultat med SureStep Mono-test.
Prov-ID ABI SureStepMono
Kommersielltestutrustning
1930
-
Triglycerider(mg/dl)
Kolesterol(mg/dl)
-
---280
380170200190890
119012402370
SD2192-9
SD2192-10
SD2192-20
SD2192-17SD2192-11
--
---
Spetsadesubstanser Kontroll Spetsat
prov
+
-
Konc.(mg/ml)
Prov-ID
-
-
-
-
-
-
-
++
100
0,06
1
10Starkt positivt
Hemoglobin
Albumin
Bilirubin
Starkt positivt
Starkt positivt
Starkt positivt
Svagt positivt
Svagt positivt
Svagt positivt
Svagt positivt
Negativt
Negativt
Negativt
Negativt
+
+
+
++
++
++
++++
++
++
++++
Prov Svagtpositivt
59
0
60
1
Nummer Negativt Ogiltigt
180
60
60
60
Starktpositivt
Fältresultat
60
6059
0 0
0
0
0
00
0
61
StarktPositivt
SvagtPositivt
Negativt
Totalt

43
Til “CLIA waived” – test med fingerprikblod eller fuldblodsprøver
Til “Moderately Complex” – test med serum-, plasma- eller fuldblodsprøver
TILSIGTET BRUG
SureStep monotest er en hurtig, visuel test til kvalitativ påvisning af heterofilt antistof specifikt for infektiøs mononucleosis(IM) i humant serum, plasma eller fuldblod. Dette test-kit er beregnet som en hjælp ved diagnosticeringen af IM i patientermed karakteristiske, kliniske symptomer og bør kun bruges af professionelle laboratorier.
OPSUMMERING
Infektiøs mononucleosis er en akut, selvbegrænsende sygdom forårsaget af Epstein-Barr-virus (EBV) (1–3). Infektion medEBV tidligt i livet er sædvanligvis asymptomatisk (4–5). Dog vil op til 50 % af de infektioner, der opstår hos unge, udviklesig til kliniske manifestationer associeret med IM.
Diagnosticeringen af IM er baseret på evaluering af karakteristiske, kliniske symptomer og serologiske forandringer.Serologisk diagnosticering af IM er blevet demonstreret ved påvisning af heterofile og EBV-specifikke antistoffer (3–5). Deheterofile antistoffer er påviselige på et givent tidspunkt under IM hos de fleste voksne. Det er en almindeligt accepteretpraksis blandt læger at bruge påvisningen af heterofile antistoffer som et hjælpemiddel ved diagnosticeringen af IM (3–7).SureStep monotest bruger bovint erythrocyt-udtræk, der har en højere sensitivitet og specificitet end udtræk fra andre arter. (4)
TESTPRINCIP
SureStep monotest er designet til at påvise IM gennem visuel fortolkning af farveudviklingen i testanordningen, der er ensandwich af kompakt fase/guldkonjugat-immunoanalyse. Testanordningen indeholder en membranstrimmel, der er coatetmed heterofile antigener i testliniezonen og anti-mus-antistof fra geder i kontrolliniezonen. Den anti-human IgM antistof-kolloide guldkonjugatpude er anbragt for enden af membranen. En blanding bestående af kolloid guldkonjugat og prøven,bevæger sig sammen med en fremkalderbuffer kromatografisk langs med membranen ved kapillærvirkning. Når de IM-heterofile antistoffer er tilstede i patientprøven, migrerer blandingen til testliniezonen og danner en synlig linie, idetbindingen mellem antistoffet og det heterofile antigen dannes. Når IM-heterofile antistoffer ikke er tilstede i prøven, dannesder ingen synlig, farvet linie i testliniezonen. Dermed indikerer en farvet linie i testliniezonen et positivt resultat. Der vilaltid fremkomme en farvet linie i kontrolzonen. Denne kontrollinie er en proceduremæssig indikator, der viser, at både testog anordning fungerede korrekt.
OPBEVARING OG STABILITET
Test-kittet skal opbevares enten i køleskab eller ved stuetemperatur (2°–30°C) i den forseglede pose, så længe holdbarhedsdatoenikke er overskredet.
FORHOLDSREGLER
• Kun til in vitro-diagnostisk brug.
• Kun til professionel og laboratoriemæssig brug.• Alle patientprøver bør behandles som potentielle smittebærere. • Reagenser fra forskellige partier må ikke blandes.• Folieposen må ikke åbnes, før man er klar til at foretage testen.• Der må ikke anvendes fuldblod, der er blevet opbevaret i mere end tre dage.• Kraftigt hæmolyserede prøver bør ikke anvendes.
INSTRUKTIONER VEDR. CLIA-KOMPLEKSITET ER ANFØRT NEDENFOR I AFSNITTET OM KVALITETSKONTROL.FØLG DE NØDVENDIGE INSTRUKTIONER I OVERENSSTEMMELSE MED LABORATORIETS RETNINGSLINJER.
MONOTEST
DANSK
Må ikke genbruges.

44
• Fremkalderbuffer og kontroller indeholder natriumazid. Natriumazid kan reagere med bly- eller kobberrør ogdannepotentielt eksplosive metalazider. Ved bortskaffelse af disse opløsninger skal der altid skylles med rigeligemængder vand for at forhindre azidophobning.
Ekstraktionsreagenser er lettere ætsende. Undgå kontakt med øjne eller slimhinder. I tilfælde af utilsigtet kontakt skyllesgrundigt med rigeligt vand.
Positive og negative kontroller indeholder natriumazid, som kan reagere med bly- eller kobberrør, og danne potentielteksplosive metalazider. Ved bortskaffelse af disse opløsninger skal der altid skylles med vand i rigelige mængder for atforhindre azidophobning.
Advarsel: Potentielt biologisk risikomaterialeAlle donorenheder af humant plasma eller der blev anvendt ved forberedelsen af de positive og negative kontrolemner, blevtestet efter FDA-godkendte metoder for tilstedeværelsen af anti-HIV-1/HIV-2, HBsAg og anti-HCV, og blev fundet negative.Der bør dog udvises forsigtighed ved håndtering og bortskaffelse af disse genstande i overensstemmelse med biologisksikkerhedsniveau 2 som anbefalet af Center for Disease Control/National Institutes of Health Manual, Biosafety inMicobiological and BiomedicalLaboratories, 1984.
LEVEREDE REAGENSER OG MATERIALER
• 25 separat indpakkede testanordninger. Hver anordning indeholder en teststrip med en heterofil antigen-coatetmembran og farvet antistof-polstring.
• Fremkalderbuffer (8 ml) : 0,1 M saltvand med phosphatbuffer, med tilsætningsstoffer og 0,1 %natriumazid.
• Negativ kontrol (0,2 ml) : Normalt humant serum eller plasma fortyndet i saltvandsopløsning med 0,2% natriumazid.
• Positiv kontrol (0,2 ml) : IM-heterofil antistof positivt humant plasma eller serum fortyndet isaltvandsopløsning med med 0,2 % natriumazid.
• 25 transferpipetter til engangsbrug.
• Et instruktionsark.
PRØVEUDTAGNING OG HÅNDTERING
Fingerprik 1. Det område, der skal prikkes, rengøres med en alkoholtampon.2. Pres enden af fingerspidsen og prik med en steril lancet.3. Tør den første dråbe blod af med sterilt gaze eller vat.4. Lad den anden dråbe blod falde direkte ned i testanordningens prøvebrønd eller brug den medfølgende pipette til
at opsamle friskt blod. Tilsæt 2 dråber (ca. 40 µl) til prøvebrønden.
Fuldblod1. Fuldblod opsamles ved venepunktur i et opsamlingsrør med lilla, blå eller grøn top (indeholder henholdsvis EDTA, citrat
eller heparin).2. Fuldblodet kan bruges til testen øjeblikkeligt, eller gemmes ved 2–8°C i op til 3 dage.
Plasma1. Fuldblod opsamles ved venepunktur i et opsamlingsrør med lilla, blå eller grøn top (indeholder henholdsvis EDTA, citrat
eller heparin).2. Plasmaet adskilles ved centrifugering.3. Det plasma, der skal testes, opsuges forsigtigt, eller det påsættes etiket og opbevares ved 2–8°C i op til to uger. Plasma kan
fryses ved -20°C i op til et år.
Serum1. Fuldblod opsamles ved venepunktur i et rødt opsamlingsrør (der ikke indeholder anti-koagulanter).2. Lad blodet koagulere, og adskil serumet ved centrifugering. 3. Det serum, der skal testes, opsuges forsigtigt, eller det påsættes etiket og opbevares ved 2–8°C i op til to uger. Serum kan
fryses ved -20°C i op til et år.
BUF DEV

45
TESTPROCEDURE
3. 3 eller 4 dråber fremkalderbuffer tilføres straks.
4. Efter tilsætning af fremkalderbufferen ventes der, til, de farvede linier viser sig. Afhængigt af koncentrationen af tilstedeværende,heterofile antistoffer kan positive resultater observeres inden for 3 minutter. For at bekræfte et negativt resultat krævesdog en samlet reaktionstid på 5 minutter. Foretag ikke resultataflæsning efter 8 minutter.
Testanordning
Kontrolzone Testzone
Prøvebrønd
Test-enheden, fremkalderbuffer, patientens prøver og kontrollerskal bringes op til stuetemperatur (20 til 30°C) inden testen udføres.Poserne må ikke åbnes, før man er klar til at udføre prøven.
1. Tag testudstyret ud af beskyttelsesposen. (Opvarm udstyret tilstuetemperatur, inden posen åbnes, for at forhindre fugtog kondensation på membranen). Mærk udstyret med patient-eller kontrolidentifikation.
2. Tilfør prøvepræparatet til prøvebrønden:
Fingerprikblod: Lad den anden dråbe blod falde direkte ned itestudstyrets prøvebrønd eller brug den medfølgende pipette tilat opsamle frisk blod. Tilsæt 2 dråber (ca. 40 µl) til prøvebrønden.
Fuldblodsprøver i opsamlingsrør: Tilsæt to dråber (ca. 40 µl) tilprøvebrønden, mens den medfølgende transfer pipette holdes i envertikal position.
Plasma- eller serumprøver: Tilsæt en dråbe (ca. 20 µl) serumeller plasma til prøvebrønden, mens den medfølgende transfer-pipette holdes i en vertikal position.
Transferpipette
FORTOLKNING AF RESULTATER
Positivt:To farvede streger vises. Én i kontrolzonen (C) og en anden farvet linie i testzonen (T).Når der testes med stærkt positive prøver, kan intensiteten af kontrollinien værelysere end forventet. Det anbefales ikke at sammenligne intensiteten af linierne.
Negativt:Der fremkommer kun én farvet linje i kontrolzonen. Der vises ingen synlig linie itestliniezonen (T).
Ugyldig:Fravær af en linie i kontrolzonen indikerer forkert fremgangsmåde eller muligforringelse af prøvereagensen. Testen bør erklæres ugyldig, da der kan være tale om,at der er anvendt en ukorrekt testprocedure eller sket en reagensforringelse. Testengentages med en ny testanordning. Hvis problemet vedbliver, ringes til Applied Biotech’s Technical Service på (888) 578-7956 for opringninger fra USA, og+1 858 713-9668 for internationale opringninger. Opringninger besvares afengelsktalende personale. Andre sprog bedes sende e-mail til:[email protected].
C T
C T
C T
C T
C TMONO

46
KVALITETSKONTROL
Intern procedurekontrolEn procedurekontrol er inkluderet i prøven. Fremkomst af en farvet linie i kontrolzonen (C) anses for en intern positivprocedurekontrol, som angiver korrekt udførelse og reaktive reagenser.
En klar baggrund i resultatvinduet anses for en intern negativ kontrol. Når fuldblodsprøver testes, kan baggrunden dogforekomme let rødlig på grund af det lave niveau af hæmolyse i visse røde blodceller. Dette er acceptabelt, så længe det ikkeforstyrrer fortolkningen af testresultaterne. Testen er ugyldig, hvis baggrunden ikke bliver klar, så resultatet kan aflæses.
Ekstern kvalitetskontrolI overensstemmelse med god laboratoriepraksis anbefales det at anvende eksterne kontroller for at sikre reagensernestilstrækkelige funktionsdygtighed og ydelse. Positive og negative kontroller leveres med kittet. Disse kontroller er baseret påhumant serum eller plasma og testes som patientprøver. Når der testes med kontroller, tilføres én dråbe (ca. 20 µl) positiveller negativ kontrol til prøvebrønden ved hjælp af den leverede transferpipette ved at holde den i lodret position. 3 eller4 dråber fremkalderbuffer tilføres straks. Et positivt signal angives ved to linier, én i testzonen (T) og én i kontrolzonen (C).Et negativt signal angives ved kun én linie i kontrolzonen (C). Resultaterne observeres efter 5 minutter og må ikke fortolkesefter 8 minutter. Hvis kontrollerne ikke reagerer som forventet, må testresultaterne ikke fortolkes. Gentag testen eller kontaktteknisk service.
FOR “CLIA WAIVED” LABORATORIER:
Både positive og negative eksterne kontroller skal testes, når der åbnes et nyt test-kit. Hver operatør skal teste en positiv ognegativ ekstern kontrol én gang for hvert 25 test-kit.
FOR "MODERATELY COMPLEX" LABORATORIER ELLER PROFESSIONELLE FRASUNDHEDSSEKTOREN:
Det anbefales, at både en positiv og en negativ kontrol testes for hver ny forsendelse eller hvert nyt kit-lotnummer og efterde øvrige krav fra statslige og lokale bestemmelser.
BEGRÆNSNINGER
Dette test-kit skal benyttes til kvalitativ påvisning af IgM-antistoffer til IM-heterofilt antigen. Et positivt resultat antydertilstedeværelsen af IgM antistof til heterofilt antigen.
Dette test-kit bør bruges til symptomatiske personer, der mistænkes for at have IM. Diagnosticering af IM bør kun foretagesved bekræftelse fra andre kliniske fund.
Et negativt resultat udelukker ikke muligheden for IM, da antistofferne til heterofilt antigen enten kan være fraværendeeller ikke tilstedeværende i tilstrækkelig mængde til at kunne påvises.
FORVENTEDE RESULTATER
Under den akutte fase af IM kan heterofile antistoffer påvises hos 80–85 % af patienterne. Heterofile antistoffer kan påvisesunder den første måneds sygdom, og niveauet falder hurtigt efter uge fire (5).
YDELSESKARAKTERISTIK
A. NøjagtighedNøjagtigheden af SureStepTM monotest blev evalueret ved sammenligning med en kommercielt tilgængelig kvalitativ, farve-immuno-kromatografisk analyse til påvisning af IgM-heterofile antistoffer for IM i humant serum, plasma eller fuldblod. Derblev anvendt 406 humane serum-, plasma- og fuldblodsprøver (288 serum, 103 plasma and 15 fuldblod) i densammenlignende undersøgelse. En oversigt over resultaterne findes i Tabel 1.
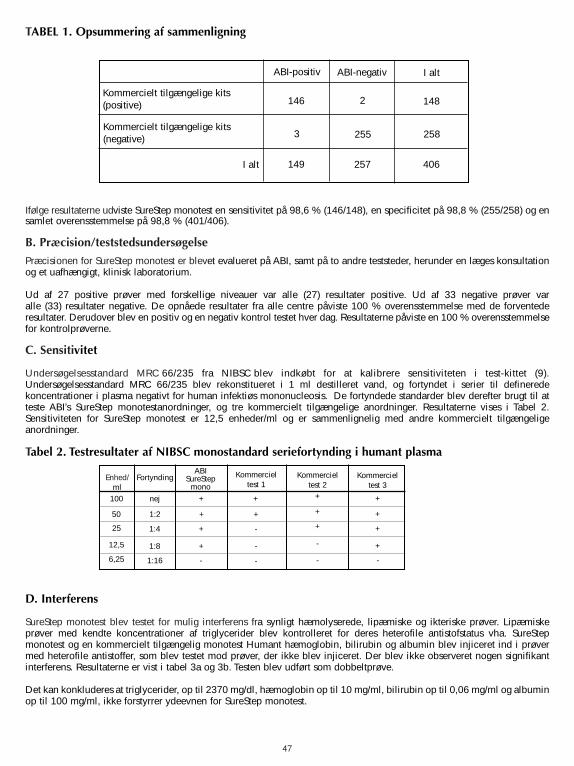
47
Ifølge resultaterne udviste SureStep monotest en sensitivitet på 98,6 % (146/148), en specificitet på 98,8 % (255/258) og ensamlet overensstemmelse på 98,8 % (401/406).
B. Præcision/teststedsundersøgelse
Præcisionen for SureStep monotest er blevet evalueret på ABI, samt på to andre teststeder, herunder en læges konsultationog et uafhængigt, klinisk laboratorium.
Ud af 27 positive prøver med forskellige niveauer var alle (27) resultater positive. Ud af 33 negative prøver var alle (33) resultater negative. De opnåede resultater fra alle centre påviste 100 % overensstemmelse med de forventederesultater. Derudover blev en positiv og en negativ kontrol testet hver dag. Resultaterne påviste en 100 % overensstemmelsefor kontrolprøverne.
C. Sensitivitet
Undersøgelsesstandard MRC 66/235 fra NIBSC blev indkøbt for at kalibrere sensitiviteten i test-kittet (9).Undersøgelsesstandard MRC 66/235 blev rekonstitueret i 1 ml destilleret vand, og fortyndet i serier til defineredekoncentrationer i plasma negativt for human infektiøs mononucleosis. De fortyndede standarder blev derefter brugt til atteste ABI’s SureStep monotestanordninger, og tre kommercielt tilgængelige anordninger. Resultaterne vises i Tabel 2.Sensitiviteten for SureStep monotest er 12,5 enheder/ml og er sammenlignelig med andre kommercielt tilgængeligeanordninger.
Tabel 2. Testresultater af NIBSC monostandard seriefortynding i humant plasma
D. Interferens
SureStep monotest blev testet for mulig interferens fra synligt hæmolyserede, lipæmiske og ikteriske prøver. Lipæmiskeprøver med kendte koncentrationer af triglycerider blev kontrolleret for deres heterofile antistofstatus vha. SureStepmonotest og en kommercielt tilgængelig monotest Humant hæmoglobin, bilirubin og albumin blev injiceret ind i prøvermed heterofile antistoffer, som blev testet mod prøver, der ikke blev injiceret. Der blev ikke observeret nogen signifikantinterferens. Resultaterne er vist i tabel 3a og 3b. Testen blev udført som dobbeltprøve.
Det kan konkluderes at triglycerider, op til 2370 mg/dl, hæmoglobin op til 10 mg/ml, bilirubin op til 0,06 mg/ml og albuminop til 100 mg/ml, ikke forstyrrer ydeevnen for SureStep monotest.
ABI-positiv
Kommercielt tilgængelige kits(positive)
Kommercielt tilgængelige kits(negative)
ABI-negativ I alt
I alt
146 2
255
148
258
406257
3
149
TABEL 1. Opsummering af sammenligning
Enhed/ml
FortyndingABI
SureStepmono
Kommercieltest 1
Kommerciel test 2
Kommerciel test 3
100
50
25
12,5
6,25
nej
1:2
1:4
1:8
1:16
+
+
+
+
-
+
+
+
+
+
+
+
+
+
-
-
-
-
- -
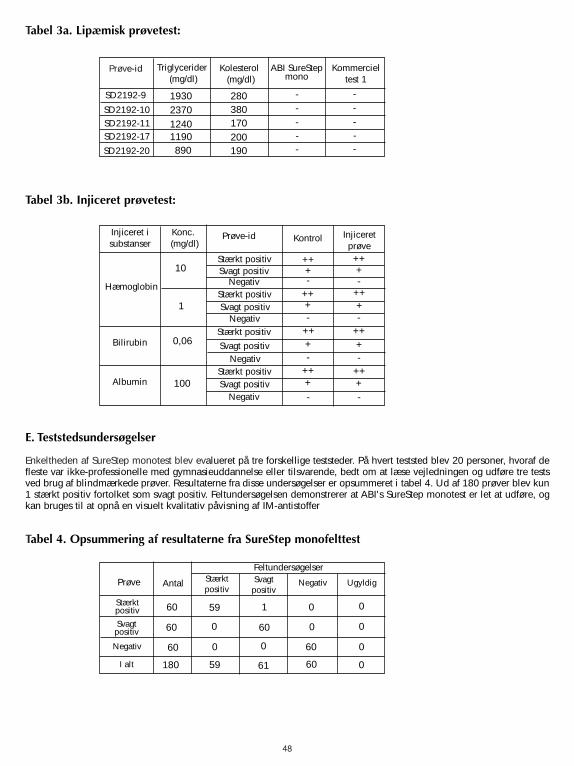
48
Tabel 3a. Lipæmisk prøvetest:
Tabel 3b. Injiceret prøvetest:
E. Teststedsundersøgelser
Enkeltheden af SureStep monotest blev evalueret på tre forskellige teststeder. På hvert teststed blev 20 personer, hvoraf defleste var ikke-professionelle med gymnasieuddannelse eller tilsvarende, bedt om at læse vejledningen og udføre tre testsved brug af blindmærkede prøver. Resultaterne fra disse undersøgelser er opsummeret i tabel 4. Ud af 180 prøver blev kun1 stærkt positiv fortolket som svagt positiv. Feltundersøgelsen demonstrerer at ABI's SureStep monotest er let at udføre, ogkan bruges til at opnå en visuelt kvalitativ påvisning af IM-antistoffer
Tabel 4. Opsummering af resultaterne fra SureStep monofelttest
Prøve-id ABI SureStepmono
Kommercieltest 1
1930
-
Triglycerider(mg/dl)
Kolesterol(mg/dl)
-
---280
380170200190890
119012402370
SD2192-9
SD2192-10
SD2192-20
SD2192-17SD2192-11
--
---
Injiceret isubstanser Kontrol Injiceret
prøve
+
-
Konc.(mg/dl)
Prøve-id
-
-
-
-
-
-
-
++
100
0,06
1
10Stærkt positiv
Hæmoglobin
Albumin
Bilirubin
Stærkt positiv
Stærkt positiv
Stærkt positiv
Svagt positiv
Svagt positiv
Svagt positiv
Svagt positiv
Negativ
Negativ
Negativ
Negativ
+
+
+
++
++
++
++++
++
++
++++
Prøve Svagtpositiv
59
0
60
1
Antal Negativ Ugyldig
180
60
60
60
Stærktpositiv
Feltundersøgelser
60
6059
0 0
0
0
0
00
0
61
Stærktpositiv
Svagtpositiv
Negativ
I alt

REFERENCES • LITERATUR • REFERENCIAS • BIBLIOGRAPHIE • RIFERIMENTI BIBLIOGRAFICI • REFERÊNCIAS • REFERENSER • HENVISNINGER
1. Evans, A. S. (1974) The History of Infectious Mononucleosis. The American Journal of the Medical Sciences, 267(3): 189-195.
2. Roberts, G.H. (1989) The Many Faces of Epstein-Barr Virus. Diagnostics & Clinical Testing, 27: 16-21.
3. Khanna, R., Burrows, S.R., and Moss, D.J. (1995) Immune Regulation in EpsteinBarr Virus-Association Diseases. Microbiological Review, 59:387-405.
4. Baehner, R.L. and Shulter, S.E. (1967) Infectious Mononucleosis in Childhood. Clinical Pediatrics, 6(7): 393-399.
5. Linde, A. (1996) Diagnosis of Epstein-Barr Virus Related Diseases. Scandinavian Journal of Infectious Disease Supplement, 100:83-88.
6. Evans, A.S., Niederman, J.C., Cenabre, L.C., West, B. and Richards, V.A. (1975) A Prospective Evaluation of Heterophile and Epstein-Barr Virus-Specific IgM Antibody Test in Clinical and Subclinical Infectious Mononucleosis: Specificity and Sensitivity of the Tests and Persistence of Antibody. The Journal of Infectious Diseases, 132: 546-554.
7. Patarca, R. and Fletcher, M.A. (1995) Structure and Pathophysiology of the Erythrocyte Membrane Associated Paul-Bunnell Heterophile Virus-Associated Disease. Critical Review in Oncogenesis, 6(3-6):305-326.
8. Fletcher, M.A. and Woolfolk, B.J. (1971) Immunochemical Studies of Infectious Mononucleosis. Isolation and Characterization of Heterophile Antigens from Hemoglobin-Free Stroma. The Journal of Immunology, 107(3): 842-853.
9. MRC research stand A, 66/235, for infectious mononucleosis serum: National Institute for Biological Standard and Control, Blanche Lane, South Mimms, Potters Bar, Hertfordshire, EN6 3QG, United Kingdom.
49


32-6906-A Oct 20036908 REF
CLIAwaived,Inc.4332 Corte de la Fonda San Diego, CA 92130 USATel: (858) 481-5031Fax (801) 720-7568
IVD
2
CE Mark/ CE-Kennzeichen/ Marca CE/ Marque CE/ Marchio CE/ Marca de Conformidade CE/ CE-märkning/ CE-mærke
Do not Reuse/ Nicht wiederverwenden/ No reutilizar/ Ne pas réutiliser/ Non riutilizzareNão Reutilizar/ Återanvänd ej/ Må ikke genbruges
Authorized Representative/ Autorisierter Vertreter/ Representante autorizado/ Représentant agréé/ Rivenditore autorizzato/ Representante Autorizado/ Auktoriserad representant/ Repræsentant
Lot number/Batch code/ Chargenbez./Loscode/ Número de lote/Código de remesa/ Numéro de lot/Code du lot/ Numero lotto/codice batch/ Número de Lote/Código do Lote/ Partinummer/satskod/ Lotnummer/batchkode
In vitro diagnostic device/ In-vitro Diagnostikum/ Dispositivo para diagnóstico/ Dispositif de diagnostic in vitro/ Dispositivo ad uso diagnostico in vitro/ Dispositivo para diagnóstico in vitro/Enhet för in vitro-diagnostik/ Udstyr til in vitro-diagnostik
Manufactured by/ Hersteller/ Fabricado por/ Fabriqué par/ Produttore/ Fabricado Por/ Tillverkad av/ Fremstillet af
Temperature Limitation/ Temperaturbeschränkung/ Límite de temperatura/ Limites de température/ Limitti di temperatura/ Limitação de Temperatura/ Temperaturbegränsning/ Temperaturebegrænsing
Use By/Expiration Date/ Verwendbar bis/Verfalldatum/ Utilizar antes de/Fecha de caducidad/ A utiliser jusque/Date d'expiration/ Utilizzare entro/Data di scadenza/ Utilizar Até/Prazo de Validade/ Används senast/utgångsdatum/ Holdbar til/udløbsdato
Consult Insert/ Packungsbeilage beachten/ Consulta el prospecto/ Consulter le texte inséré/ Consultare il foglietto illustrativo/ Consultar o Folheto Informativo/ Informationsbilaga/ Se indlægsseddel
Catalog Number/ Best.-Nr./ N˚ de referencia/ Référence Catalogue/ N. di catalogo/ Número de Catálogo/ KatalognummerREF
∑ Quantity/ Quantitãt/ Cantidad/ Quantité/ Quantitá/ Quantidade/ Antal
Attention/ Achtung/ Atención/ Attention/ Attenzione/ Atencão/ Observera/ Bemæk





























