Embed Size (px)
Citation preview

Motion prediction:Molecular Dynamics (MD) and Normal Mode Analysis
(NMA)Lecture 10
Structural BioinformaticsDr. Avraham Samson
81-871
1

Normal mode analysis

3
Basic theory of normal modes• Normal modes analysis (NMA) is a technique for
calculating long time dynamics of biomolecules.• In contrast to molecular dynamics which solves
the exact equation of motion approximately, NMA solves the approximate equation of motion exactly.
• Even with its limitations, such as the neglect of the solvent effect and the use of harmonic approximation of the potential energy function, NMA have provided much useful insight into protein dynamics.
4
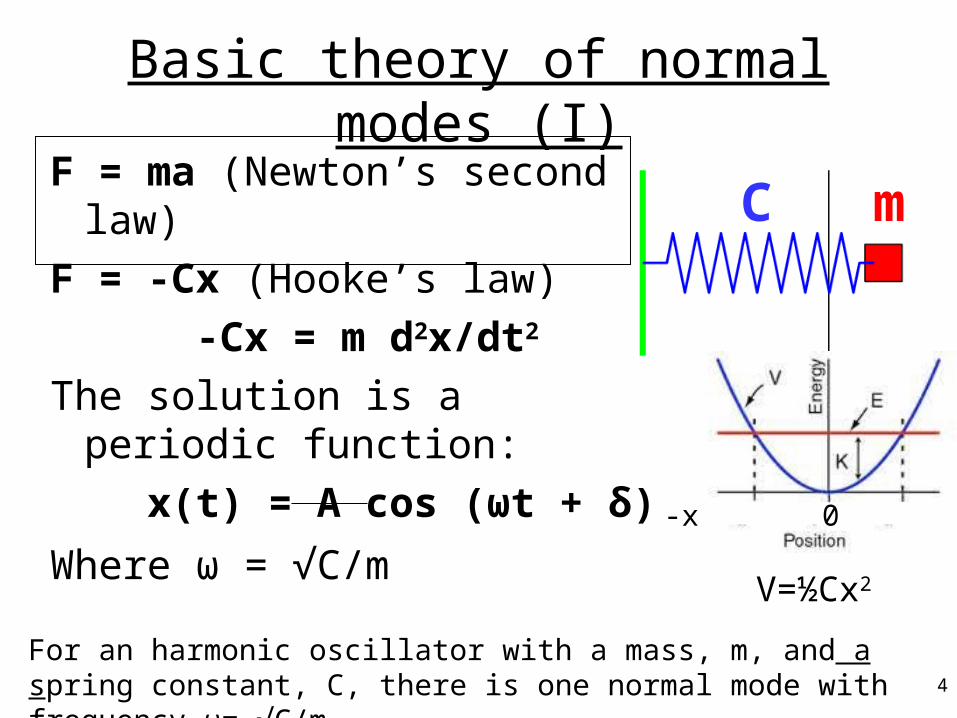
Basic theory of normal modes (I)
F = ma (Newton’s second law)
F = -Cx (Hooke’s law)
-Cx = m d2x/dt2
The solution is a periodic function:
x(t) = A cos (ωt + δ)
Where ω = √C/m
For an harmonic oscillator with a mass, m, and a spring constant, C, there is one normal mode with frequency ω= √C/m
mC
-x 0 x
V=½Cx2
5
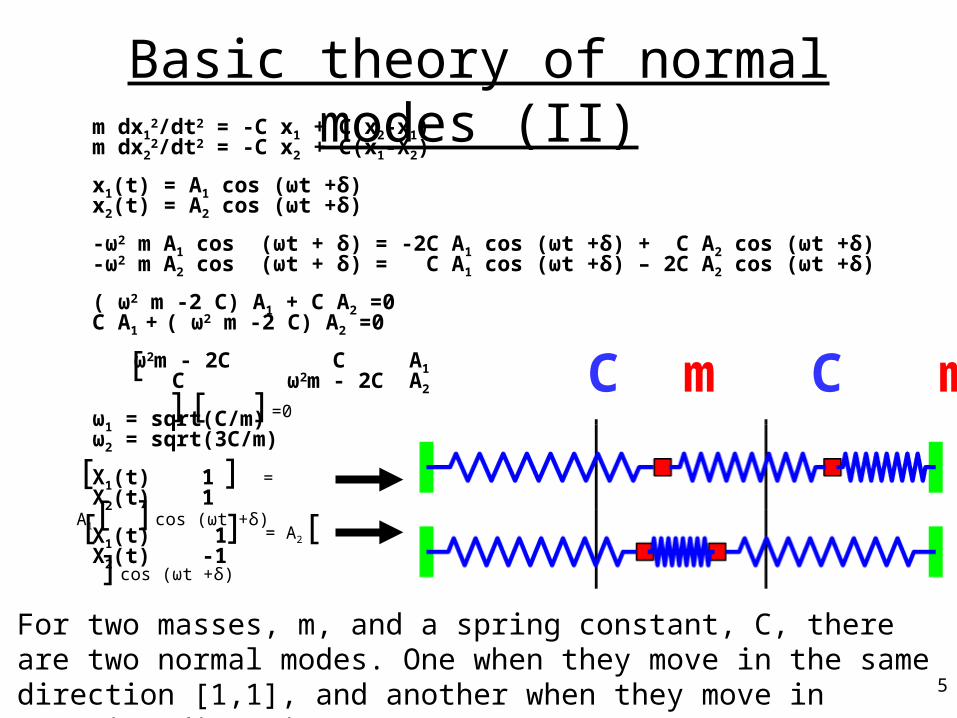
C m C m C
m dx12/dt2 = -C x1 + C(x2-x1)
m dx22/dt2 = -C x2 + C(x1-X2)
x1(t) = A1 cos (ωt +δ)x2(t) = A2 cos (ωt +δ) -ω2 m A1 cos (ωt + δ) = -2C A1 cos (ωt +δ) + C A2 cos (ωt +δ)-ω2 m A2 cos (ωt + δ) = C A1 cos (ωt +δ) – 2C A2 cos (ωt +δ)
( ω2 m -2 C) A1 + C A2 =0C A1 + ( ω2 m -2 C) A2 =0
ω2m - 2C C A1 C ω2m - 2C A2
ω1 = sqrt(C/m)ω2 = sqrt(3C/m)
X1(t) 1X2(t) 1
X1(t) 1X2(t) -1
[ ][ ]=0
[ ] = A1[ ]cos (ωt +δ)
[ ] = A2[ ]cos (ωt +δ)
Basic theory of normal modes (II)
For two masses, m, and a spring constant, C, there are two normal modes. One when they move in the same direction [1,1], and another when they move in opposite directions [1,-1].
6
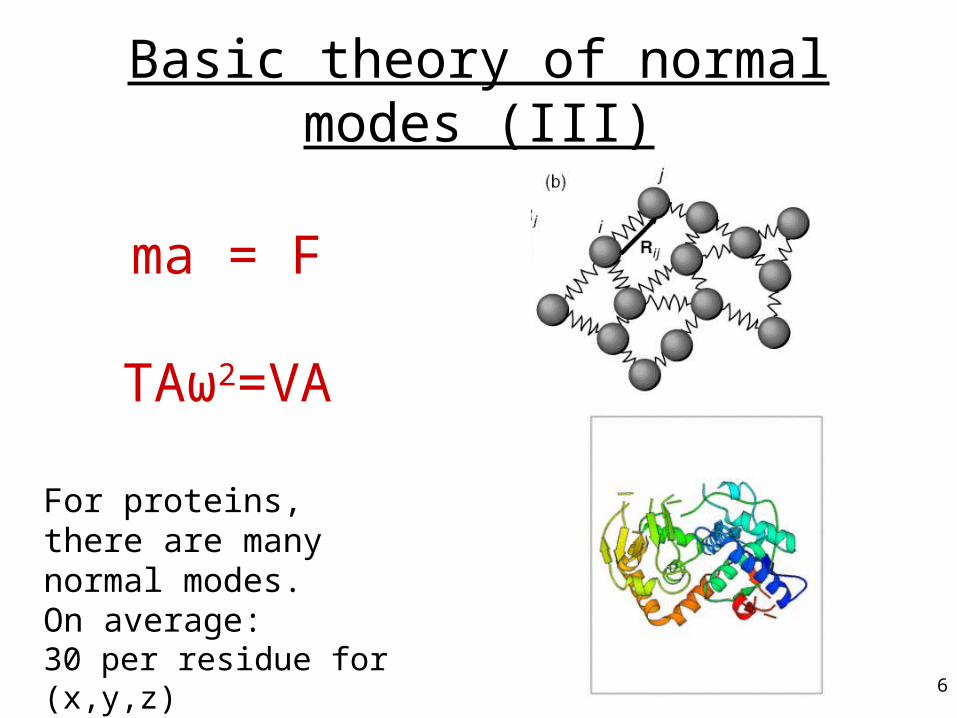
Basic theory of normal modes (III)
ma = F
TAω2=VA
For proteins, there are many normal modes. On average:30 per residue for (x,y,z)4 per residue for (φ,ψ,χ)

Normal mode analysis tools
• ElNemo
http://igs-server.cnrs-mrs.fr/elnemo
• Nomad Ref
http://lorentz.immstr.pasteur.fr/nma/submission.php
http://lorentz.immstr.pasteur.fr/gromacs/nma_submission.php
• STAND (by appointment only)
7

Example 1.Example 1. Normal Mode Dynamics Normal Mode Dynamics of the Acetylcholine Receptorof the Acetylcholine Receptor
Samson A. & Levitt M. (2008) Biochemistry 47:4065-70

9
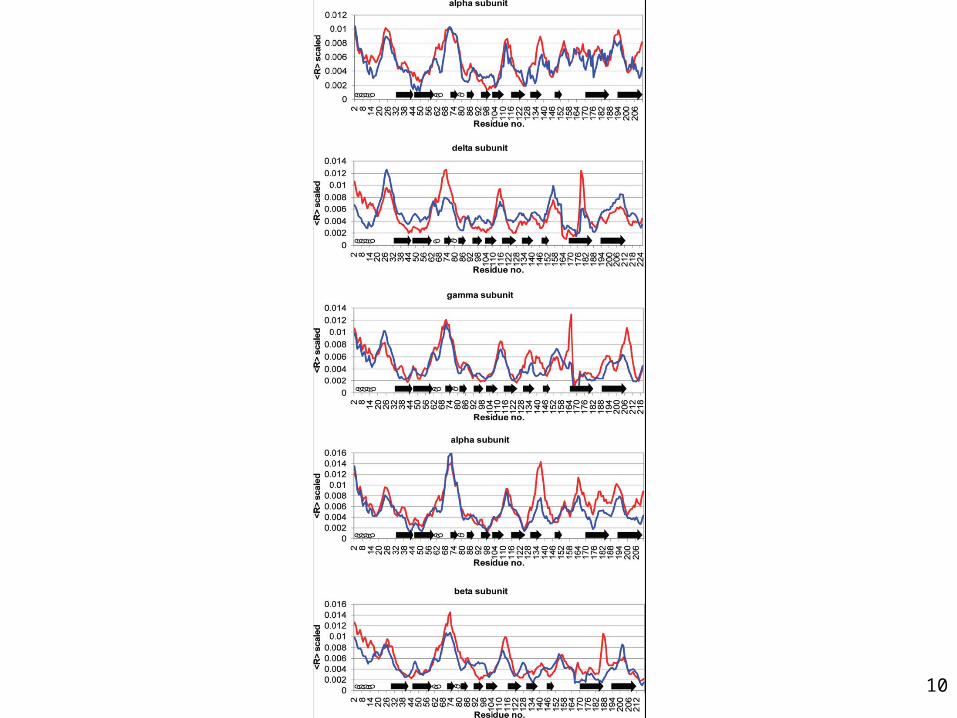
Normal mode of acetylcholine receptor
•The receptor displays an twist like motion, responsible for the axially symmetric opening and closing of the ion channel
10
11
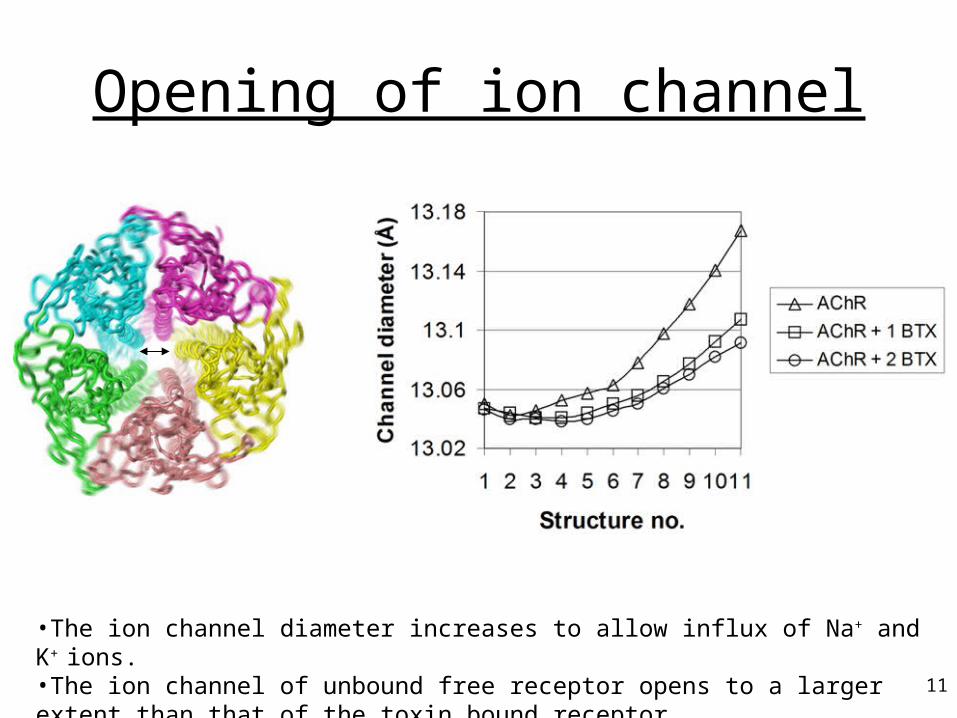
Opening of ion channel
•The ion channel diameter increases to allow influx of Na+ and K+ ions. •The ion channel of unbound free receptor opens to a larger extent than that of the toxin bound receptor.

12
Dynamics of AChR are inhibited by α-Bungarotoxin
•Twist like motion is not observed in the modes of the toxin bound receptor. •The toxins seem to lock together adjacent α and δ, and α and γ subunits •One toxin is sufficient to prevent the opening motion.

Example 2.Example 2. Normal Mode Dynamics Normal Mode Dynamics of Prion Proteinsof Prion Proteins
Samson A. & Levitt M. (2011) Biochemistry 50:2243-8
14
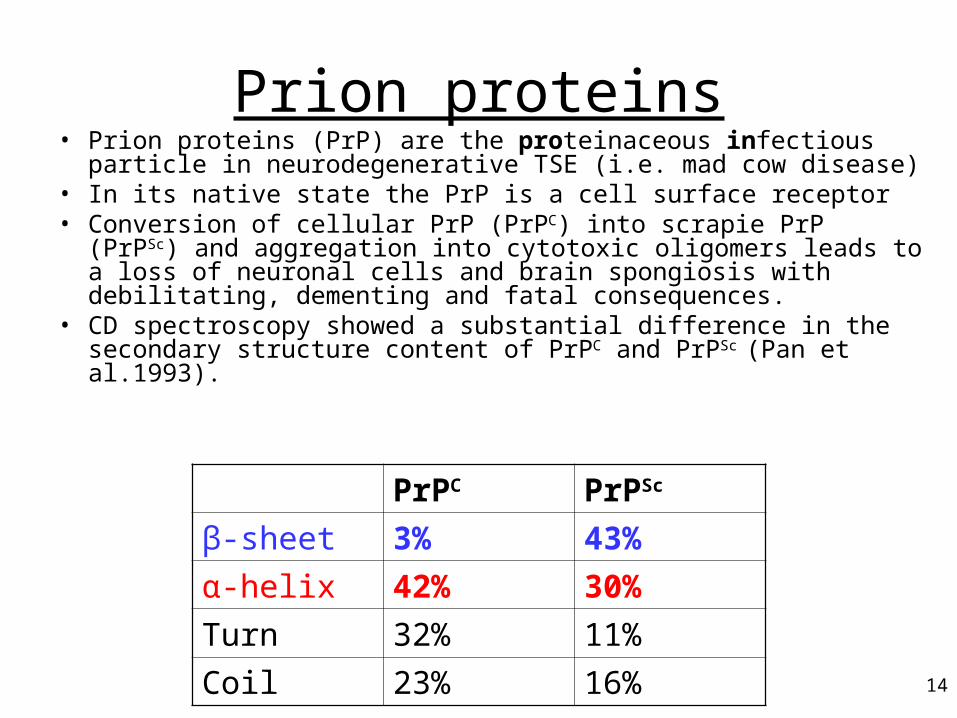
Prion proteins• Prion proteins (PrP) are the proteinaceous infectious particle in
neurodegenerative TSE (i.e. mad cow disease)• In its native state the PrP is a cell surface receptor • Conversion of cellular PrP (PrPC) into scrapie PrP (PrPSc) and
aggregation into cytotoxic oligomers leads to a loss of neuronal cells and brain spongiosis with debilitating, dementing and fatal consequences.
• CD spectroscopy showed a substantial difference in the secondary structure content of PrPC and PrPSc (Pan et al.1993).
PrPC PrPSc
β-sheet 3% 43%
α-helix 42% 30%
Turn 32% 11%
Coil 23% 16%

15
_________________________________________________________________ . . . . . SEQ 125 LGGYMLGSAMSRPIIHFGSDYEDRYYRENMHRYPNQVYYRPMDEYSNQNN 174 Native EE HHHHHHHHHHGGG EE GGGGG HHH Distorted EE HHHHHHHHHHGGG EE GGGGG HHH . . . . . SEQ 175 FVHDCVNITIKQHTVTTTTKGENFTETDVKMMERVVEQMCITQYERESQA 224 Native HHHHHHHHHHHHHHHHHHH HHHHHHHHHHHHHHHHHHHHHHHHH Distorted HHHHHHHHHHHHHHHHHHH HHHHHHHHHHHHHHHHHHHHHHHH _________________________________________________________________
(PDB ID 3HES)
Normal mode of M129 prion (WT)
16
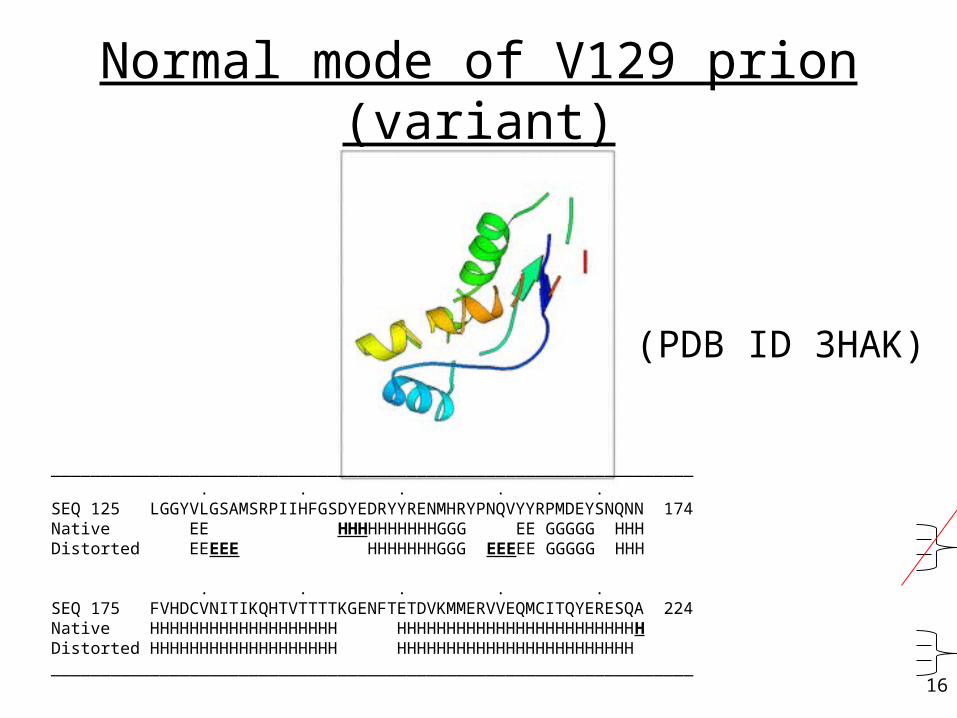
Normal mode of V129 prion (variant)
_________________________________________________________________ . . . . . SEQ 125 LGGYVLGSAMSRPIIHFGSDYEDRYYRENMHRYPNQVYYRPMDEYSNQNN 174 Native EE HHHHHHHHHHGGG EE GGGGG HHH Distorted EEEEE HHHHHHHGGG EEEEE GGGGG HHH . . . . . SEQ 175 FVHDCVNITIKQHTVTTTTKGENFTETDVKMMERVVEQMCITQYERESQA 224 Native HHHHHHHHHHHHHHHHHHH HHHHHHHHHHHHHHHHHHHHHHHHH Distorted HHHHHHHHHHHHHHHHHHH HHHHHHHHHHHHHHHHHHHHHHHH _________________________________________________________________
(PDB ID 3HAK)

Trajectory path using NMA
http://lorentz.immstr.pasteur.fr/joel/index.php
Coiled coils of hemagglutinin!
17

Molecular dynamics

Molecular dynamics
• Why simulate motion of molecules?
• Energy model of conformation
• Two main approaches:– Monte Carlo - stochastic– Molecular dynamics - deterministic
• Applications of MD
• Speeding up MD
19

Why simulate motion?
• Predict structure• Understand interactions• Understand properties• Learn about normal modes of vibration• Design of bio-nano materials• Experiment on what cannot be studied
experimentally• Obtain a movie of the interacting molecules
20
Aquaporin simulation
2004 Winner of Visualization Challenge in Science and Engineering, Organized by the National Science Foundation and Science Magazine.
21

Aquaporin simulation
Structure, Dynamics, and Function of Aquaporins
http://www.ks.uiuc.edu/Research/aquaporins/
22

Method of Molecular Dynamics
1. Use physics to find the potential energy between all pairs of atoms.
2. Move atoms to the next state.
3. Repeat.
HOW???23
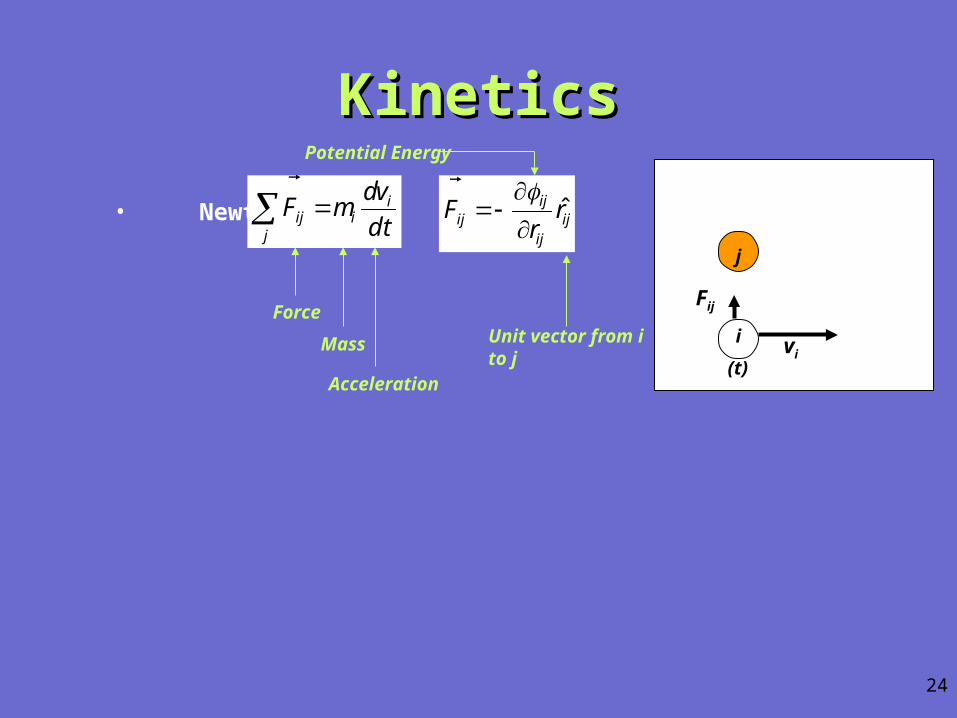
• Newton:dt
vdmF i
ij
ij
ij
ij
ijij r
rF ˆ
i
j
Fij
vi(t)
Force
Mass
Acceleration
Potential Energy
Unit vector from i to j
KineticsKinetics
24

• Newton:dt
vdmF i
ij
ij
ij
ij
ijij r
rF ˆ
• Updated Position:
i
iiii m
tFttvttrttr
)(
2
1)()()( 2
new position old
position old velocity
old force
KineticsKinetics
i
Fij
vi
i
(t)
(t + t)
j
25

• Newton:dt
vdmF i
ij
ij
ij
ij
ijij r
rF ˆ
• Updated Position:
i
iiii m
tFttvttrttr
)(
2
1)()()( 2
• Updated Velocity:
i
iii m
tFttv
ttv
)(
2
1)(
2
i
iii m
ttFt
ttvttv
)(
2
1
2)(
first half of time step second half of time step
KineticsKinetics
i
Fij
vi
i
(t)
Fij
vi
(t + t)
j
26

• Newton:dt
vdmF i
ij
ij
ij
ij
ijij r
rF ˆ
• Updated Position:
i
iiii m
tFttvttrttr
)(
2
1)()()( 2
• Updated Velocity:
i
iii m
tFttv
ttv
)(
2
1)(
2
i
iii m
ttFt
ttvttv
)(
2
1
2)(
• System Temperature:
TNkmv B
N
ii 2
3
2
1
1
2
N
iii
B
vmNk
T1
2
3
1
system kinetic energy
equipartition theorem
KineticsKinetics
i
Fij
vi
i
(t)
Fij
vi
(t + t)
j
27

Carey, V.P., Statistical Thermodynamics and Microscale Thermophysics, New York: Cambridge University Press, 1999.
• No long range interactions
• Interact via elastic collisions
“Hard Sphere” Potential
Gases
• Electric dipole ~ r -3
• 2 dipoles ~ r -6
• Repulsive nuclear forces ~ r -12
• Total Potential = (Attractive) + (Repulsive)
“Lennard-Jones 6-12 Potential”
Dense Gases, Liquids and Solids
Intermolecular PotentialsIntermolecular Potentials
28

Energy Function
• Target function that MD tries to optimize
• Describes the interaction energies of all atoms and molecules in the system
• Always an approximation– Closer to real physics --> more realistic, more
computation time (I.e. smaller time steps and more interactions increase accuracy)
29
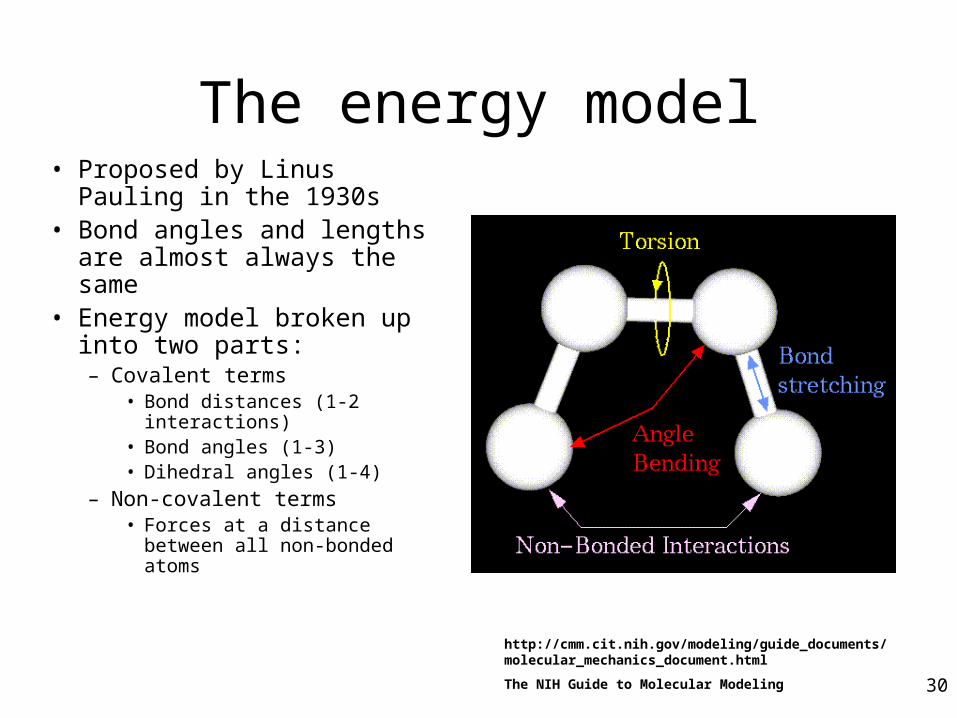
The energy model
http://cmm.cit.nih.gov/modeling/guide_documents/molecular_mechanics_document.html
The NIH Guide to Molecular Modeling
• Proposed by Linus Pauling in the 1930s
• Bond angles and lengths are almost always the same
• Energy model broken up into two parts:– Covalent terms
• Bond distances (1-2 interactions)
• Bond angles (1-3)• Dihedral angles (1-4)
– Non-covalent terms• Forces at a distance between
all non-bonded atoms
30

The energy equation
Energy =
Stretching Energy +
Bending Energy +
Torsion Energy +
Non-Bonded Interaction Energy
These equations together with the data (parameters) required to describe the behavior of different kinds of atoms and bonds, is called a force-field.
31
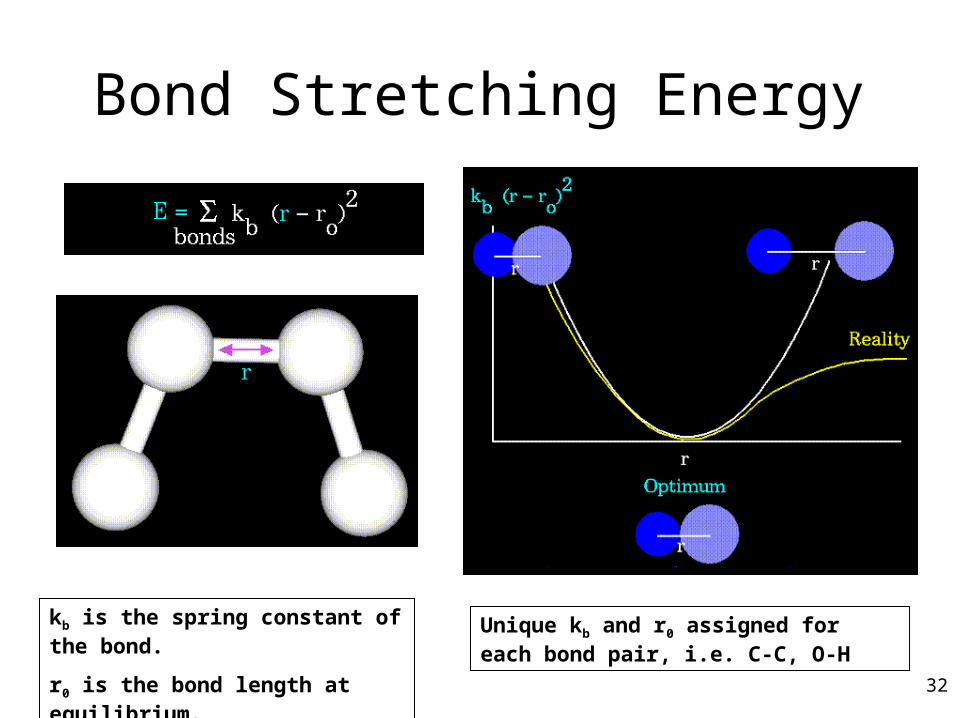
Bond Stretching Energy
kb is the spring constant of the bond.
r0 is the bond length at equilibrium.
Unique kb and r0 assigned for each bond pair, i.e. C-C, O-H
32
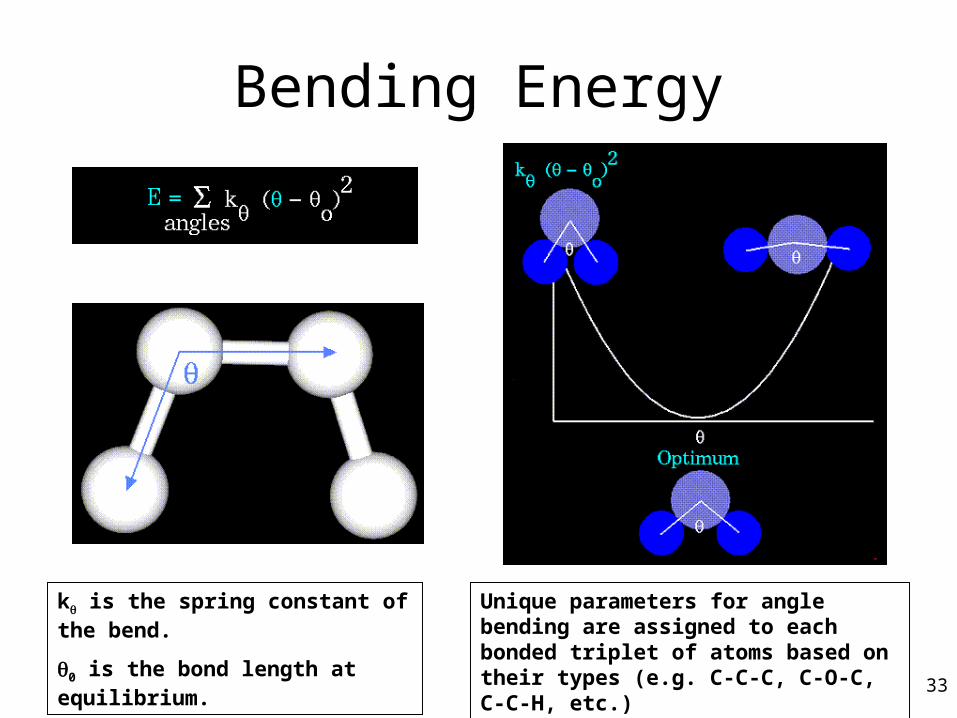
Bending Energy
k is the spring constant of the bend.
0 is the bond length at equilibrium.
Unique parameters for angle bending are assigned to each bonded triplet of atoms based on their types (e.g. C-C-C, C-O-C, C-C-H, etc.) 33
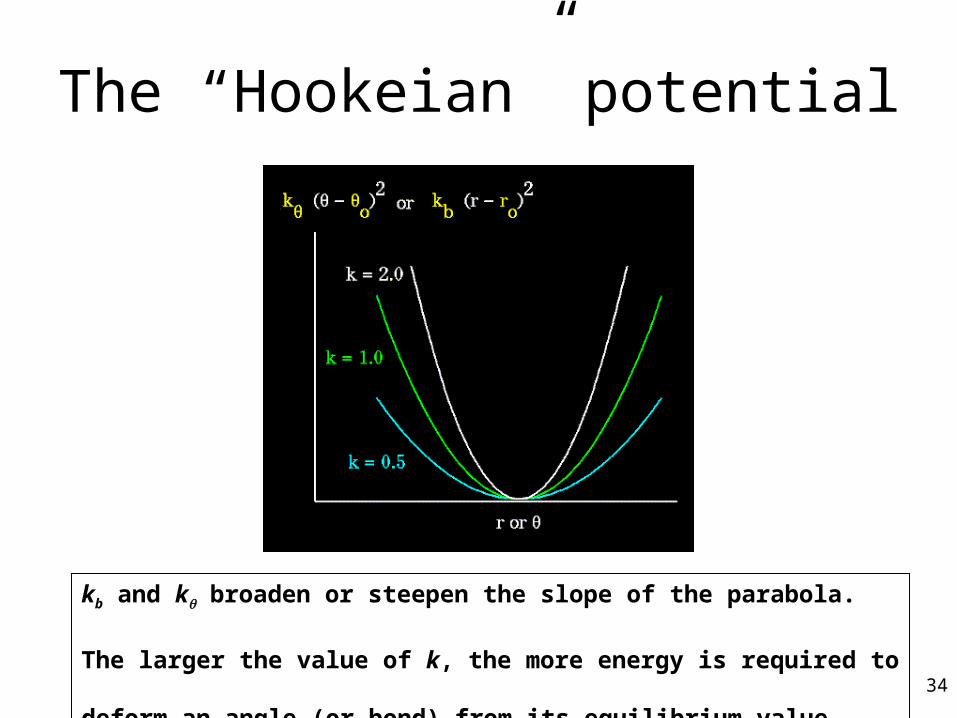
The “Hookeian” potential
kb and k broaden or steepen the slope of the parabola.
The larger the value of k, the more energy is required to deform an
angle (or bond) from its equilibrium value. 34
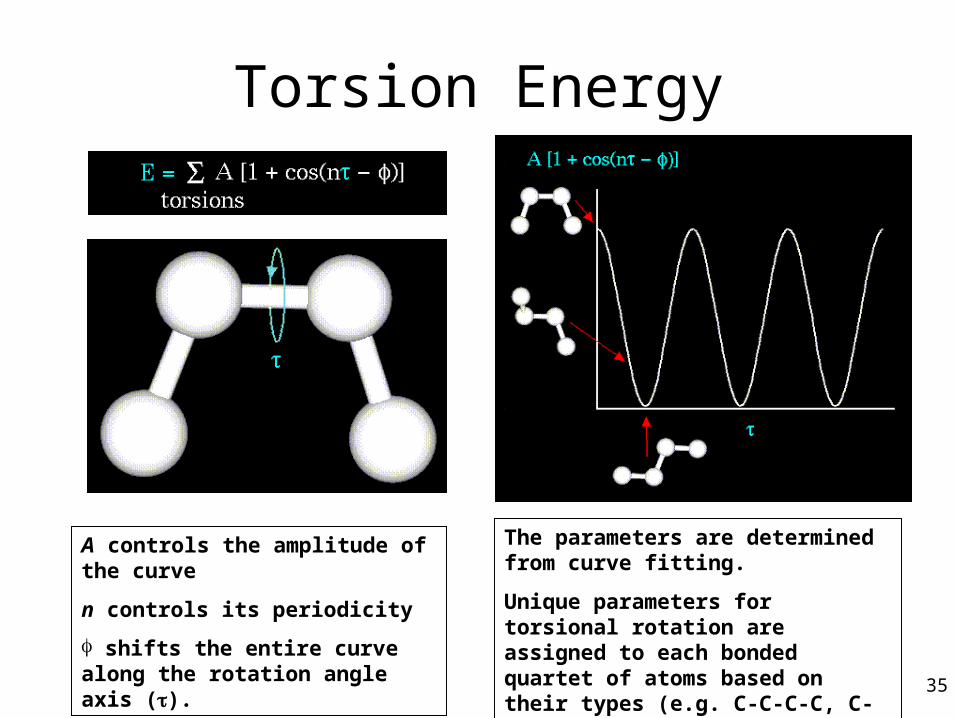
Torsion Energy
A controls the amplitude of the curve
n controls its periodicity
shifts the entire curve along the rotation angle axis ().
The parameters are determined from curve fitting.
Unique parameters for torsional rotation are assigned to each bonded quartet of atoms based on their types (e.g. C-C-C-C, C-O-C-N, H-C-C-H, etc.) 35

Torsion Energy parameters
A is the amplitude.
n reflects the type symmetry in the dihedral angle.
used to synchronize the torsional potential to the initial rotameric state of the molecule
36

Non-bonded Energy
A determines the degree the attractiveness
B determines the degree of repulsion
q is the charge
A determines the degree the attractiveness
B determines the degree of repulsion
q is the charge37
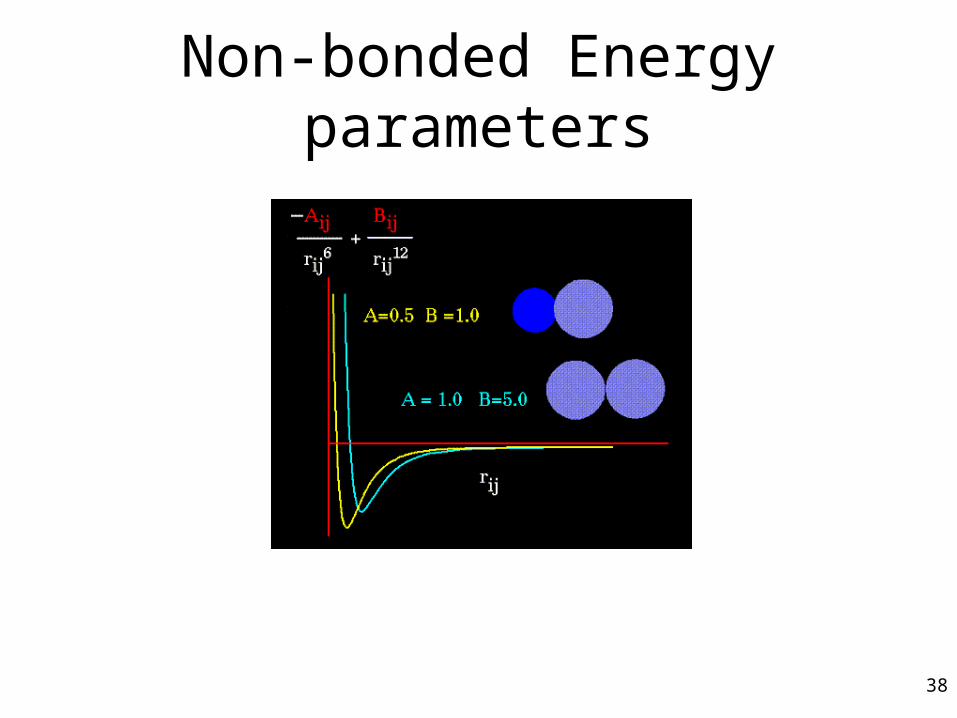
Non-bonded Energy parameters
38

Simulating In A Solvent• The smaller the system, the more particles on
the surface– 1000 atom cubic crystal, 49% on surface
– 10^6 atom cubic crystal, 6% on surface
• Would like to simulate infinite bulk surrounding N-particle system
• Two approaches:– Implicitly– Explicitly
• Ex. Periodic boundary conditions
Schematic representation of periodic boundary conditions.
http://www.ccl.net/cca/documents/molecular-modeling/node9.html
40

Monte CarloExplore the energy surface by randomly probing
the configuration spaceMetropolis method (avoids local minima):
1. Specify the initial atom coordinates.2. Select atom i randomly and move it by random
displacement.3. Calculate the change of potential energy, E corresponding
to this displacement.4. If E < 0, accept the new coordinates and go to step 2.5. Otherwise, if E 0, select a random R in the range [0,1]
and:1. If e-E/kT < R accept and go to step 2 2. If e-E/kT R reject and go to step 2
41

Properties of MC simulation
• System behaves as a Markov process– Current state depends only on previous.– Converges quickly.
• System is ergodic (any state can be reached from any other state).
• Accepted more by physicists than by chemists.– Not deterministic and does not offer time evolution of
the system.
42

Deterministic Approach
• Provides us with a trajectory of the system.
• Typical simulations of small proteins including surrounding solvent in the pico-seconds.
Fi E
x i
F m
a
43

Deterministic / MD methodology
• From atom positions, velocities, and accelerations, calculate atom positions and velocities at the next time step.
• Integrating these infinitesimal steps yields the trajectory of the system for any desired time range.
• There are efficient methods for integrating these elementary steps with Verlet and leapfrog algorithms being the most commonly used.
44

MD algorithm
• Initialize system– Ensure particles do not overlap in initial
positions (can use lattice)– Randomly assign velocities.
• Move and integrate.
{r(t), v(t)}
{r(t+t), v(t+t)}
Leapfrog algorithm
45





























