Embed Size (px)
Citation preview

BRIEF REPORTOFFICIAL JOURNAL
www.hgvs.org
Mutations in NOTCH2 in Families with Hajdu-CheneySyndrome
Jacek Majewski,1 Jeremy A. Schwartzentruber,1 Aurore Caqueret,2 Lysanne Patry,2 Janet Marcadier,3 Jean-Pierre Fryns,7
Kym M. Boycott,3 Louis-Georges Ste-Marie,4 Fergus E. McKiernan,5 Ivo Marik,6 Hilde Van Esch,7
FORGE Canada Consortium,† Jacques L. Michaud,2 and Mark E. Samuels2,8∗1Department of Human Genetics, McGill University and Genome Quebec Innovation Centre, Canada; 2Centre de Recherche de l’HopitalSte-Justine, 3175, Cote Ste-Catherine, Montreal, Canada; 3Department of Genetics, Children’s Hospital of Eastern Ontario, Ottawa, Canada;4Universite de Montreal, Centre de Recherche du CHUM, Hopital Saint-Luc, 264 Rene Levesque boulevard east, Montreal, Canada; 5Center forBone Disease, Marshfield Clinic, Marshfield, Wisconsin; 6Ambulatory Centre for Defects of Locomotor Apparatus, Czech Republic; 7Center forHuman Genetics, University Hospitals Leuven, Leuven, Belgium; 8Department of Medicine, University of Montreal, Montreal, Canada
Communicated by Garry R. CuttingReceived 4 April 2011; accepted revised manuscript 1 June 2011.Published online 6 June 2011 in Wiley Online Library (www.wiley.com/humanmutation).DOI: 10.1002/humu.21546
ABSTRACT: Hajdu-Cheney syndrome (HCS) is a rare ge-netic disorder whose hallmark is acro-osteolysis, short-ening of terminal phalanges, and generalized osteoporosis.We assembled a cohort of seven families with the conditionand performed whole exome resequencing on a selected setof affected patients. One protein-coding gene, NOTCH2,carried heterozygous truncating variants in all patients andtheir affected family members. Our results replicate re-cently published studies of HCS and further support thisas the causal gene for the disorder. In total, we identifiedfive novel and one previously reported mutation, all clus-tered near the carboxyl terminus of the gene, suggesting anallele specific genotype-phenotype effect since other mu-tations in NOTCH2 have been reported to cause a form ofAlagille syndrome. Notch-mediated signaling is known toplay a role in bone metabolism. Our results support a po-tential therapeutic role for Notch pathways in treatmentof osteoporosis.Hum Mutat 32:1114–1117, 2011. C© 2011 Wiley-Liss, Inc.
KEY WORDS: Hajdu-Cheney Syndrome; NOTCH2; os-teoporosis; acro-osteolysis
Hajdu-Cheney syndrome (HCS), also known as acro-osteolysiswith osteoporosis and changes in skull and mandible [MIM#102500], is a rare genetic disorder whose prominent features, in-clude progressive digital shortening, especially of the fingers, dueto resorption of the waist of the terminal phalanges, together with
Additional Supporting Information may be found in the online version of this article.†FORGE Steering Committee listed in Acknowledgements∗Correspondence to: Mark E. Samuels, Centre de Recherche de l’Hopital Ste-Justine,
3175, Cote Ste-Catherine, Montreal, QC H3T 1C5, Canada. E-mail: mark.e.samuels@
umontreal.ca
Contract grant sponsor: Canadian Institutes of Health Research; Genome Canada;
Genome Quebec; Genome British Columbia; Ontario Genomics Institute; Centre de
Recherche du CHU Ste-Justine (to M.E.S.).
other skeletal bone abnormalities. There is a fairly broad range ofclinical presentations, with some patients also exhibiting renal, car-diac, or other organ involvement. HCS was originally describedby Hajdu [Hajdu and Kauntze, 1948], then by Cheney [Cheney,1965], and since then there have been many published case reportsof sporadic or familial cases. Its mode of transmission in families isconsistent with autosomal dominant transmission, with many ap-parently sporadic cases, and its overall incidence is rare, as expectedfor a monogenic disorder. Recently, two independent studies havereported mutations in the same gene, NOTCH2, in patients withHCS [Isidor et al., 2011; Simpson et al., 2011].
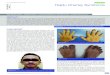
We assembled a cohort of mostly new families with typical HCS(Fig. 1; Supp. Table S1). Families 1 and 2 represent unrelated newsporadic cases ascertained in Quebec, of French-Canadian ethnicity.Family 3 represents a new multiplex family of Caucasian-Europeanethnicity. Family 4 represents a new sporadic case of CaucasianEuropean ethnicity. Family 5 represents a previously reported mul-tiplex family of Armenian ethnicity [Charvat et al., 2010; Mariket al., 2006]. Families 6 and 7 represent cases of Caucasian Europeanethnicity ascertained in the Unites States; family 7 has been reportedpreviously [McKiernan, 2007]. All patients showed at least a subsetof the typical facial features, including downslanted palpebral fis-sures, flat and broad nasal base, long philtrum, low set ears, widespaced eyes, and micrognathia (e.g., see Fig. 1). With the exceptionof the youngest patient who at 10 years of age showed broadeningof the finger tips but yet no changes on X-rays, all the other pa-tients showed acro-osteolysis (Fig. 1). The adult patients were alsoaffected by periodontal disease and osteoporosis, which resulted insome fragility fractures. One patient showed cystic kidneys, previ-ously reported in an approximately 10% of HCS subjects. Additionalclinical features, including coarctation of the aorta and early-onsetscoliosis, were found in single families (Supp. Table S1). Althoughpatent ductus arteriosus and septal defects can be found in HCS,coarctation of the aorta or other hemodynamic congenital heartdefects have not yet been reported in such patients.
The diverse origins of the families made a single founder effectunlikely, therefore we chose to sequence the entire protein-codingexomes of selected patients in hopes of identifying a unique candi-date causal gene. This approach has been successful for rare geneticdisorders where the affected patients are either sporadic cases orcome from small families [Choi et al., 2009; Ng et al.]. We per-formed whole exome sequencing on samples from six patients, one
C© 2011 WILEY-LISS, INC.

Figure 1. Familial and clinical characteristics of Hajdu-Cheney patients. A andB: Family trees showing the segregation of the syndrome andof the mutations in the two multiplex families 3 (A) and 5 (B). In family 5, mutation in exome-sequenced proband was not verified by Sanger assufficient sample was no longer available; mutation was verified in two other family members as shown. Patients were identified in the course ofclinical practice. All sampled family members provided informed consent to participate in the study. DNA was obtained from blood samples usingroutine extraction methods. All procedures were in accordance with ethical and methodological standards for human experimentation. Approvalfor this study was obtained from the Research Ethics Board of the CHU Ste-Justine. C,D, and E: Clinical characteristics of a sporadic case fromfamily 1. This patient showed downslanted palpebral fissures, flat and broad nasal base, wide spaced eyes, long philtrum, and low set ears (C).She also showed short and broad distal phalanges (D). X-ray studies confirmed the presence of acro-osteloysis in this patient (E). F: Locations ofmutations in NOTCH2.
from family 1, three from multiplex family 3, one from family 4,and one from multiplex family 5. An average of 14.5 gigabases (Gb)of raw sequence were acquired for each sample. After aligning readsto the human genome using an implementation of the Burroughs-Wheeler algorithm (BWA), mean and median read depths of bases
in consensus coding sequence exons were 134 and 114, respectively.Each sample had at least 10-fold coverage for 90% or more of codingexon bases. We quality filtered initial variant calls using Samtools,requiring at least 3 and >20% variant reads. As has been observedby other groups, each sample individually contained approximately
HUMAN MUTATION, Vol. 32, No. 10, 1114–1117, 2011 1115

20,000 variants in protein-coding elements (plus two bases of adja-cent intronic sequence). Of these, on average 9,851 were predictedto change protein structure (nonsynonymous, i.e., missense, non-sense, frameshift, or canonical splice site changes), and on average550 were novel, that is, private to each sample.
Because HCS is a rare highly penetrant disorder consistentwith dominant familial transmission, we hypothesized that disease-causing variants would be present in all samples, likely in heterozy-gous form. To identify candidate genes, we applied three stepwisefilters: the mutation should not be present in dbSNP (v.131), nor inthe 1,000 Genomes Project variant set (some of which is includedin dbSNP v.131), nor in 56 samples whose exomes we had previ-ously sequenced for other projects; the identical mutation shouldbe seen in all three sequenced samples from multiplex family 3; thesame gene should have a private potentially pathogenic, though notnecessarily the same, variant in all sequenced families.
After applying these filters, only one gene remained, and ap-peared to harbor valid deleterious mutations in all the sequencedsamples. This was NOTCH2 [MIM# 600275], encoding a mem-ber of the Notch family of transmembrane receptor proteins. Allsix sequenced samples contained a heterozygous truncating muta-tion located in exon 35, the final protein-coding exon of the gene.The three samples from family 3 shared novel stop codon allelec.6622C>T, p.(Gln2208∗) (all mutations numbered with referenceto NCBI accession number NM_024408.3, encoding NOTCH2 iso-form 1; +1 as the A of ATG initiation codon 1), as did the patientsample from unrelated family 5. Multiple discordant homozygousSNPs near the gene were observed between these two families, in-dicating that the mutation probably arose independently on differ-ent haplotypes ancestrally to the two families. The samples fromfamilies 1 and 4 contained different truncating alleles, c.7078C>T,p.(Gln2360∗), and c.6457delT, p.(Ser2153Profs∗2), respectively (seeFig. 1F, Supp. Fig S1A–C, Supp. Table S2 for PCR primer details).The three mutations identified by exome sequencing were confirmedby PCR-based Sanger capillary sequencing, and segregated with thedisease state in multiplex families 3 and 5 (Fig. 1, Supp. Fig. S2 A, C,and D). Sequencing exon 35 in singleton patients from the three ad-ditional families identified truncating mutations in each of them: alarge deletion insertion leading to a frameshift, c.6655_6840delinsG,p.(Pro2219Glyfs∗10) in family 2; a small frameshifting deletionc.6403_6404delCT, p.(Leu2135Glufs∗2) in family 6; and stop codonc.6667C>T, p.(Gln2223∗) in family 7 (Table S1, Supp. Fig. S2 B, E,and F). Parental samples were not available for any of the singletoncases. In the course of our work, two publications appeared doc-umenting similar C-terminal truncating mutations in NOTCH2 indifferent HCS cohorts [Isidor et al., 2011; Simpson et al., 2011]. Themutation p.Gln2208∗, found in our families 3 and 5, was also iden-tified previously in two unrelated patients [Simpson et al., 2011].
Mutations in NOTCH2 have been reported as causal for a differ-ent genetic disorder, a variant form of Alagille Syndrome (ALGS2;MIM# 610205) [McDaniell et al., 2006]. There are substantial differ-ences between ALGS and HCS; the hallmark of ALGS is cholestasisdue to a developmental reduction in number of bile ducts lead-ing to accumulation of cholesterol and metabolites in the liver,a clinical presentation not associated with HCS. Both syndromescan include cardiac involvement, including atrial septal defects. Asmentioned above, the renal problems found in HCS are polycystickidneys. In ALGS, the renal problems are either structural (smallkidney, ureteropelvic obstruction, renal cysts) or functional (mostcommonly renal tubular acidosis) [Crosnier et al., 2000]. Both syn-dromes lead to subtle though different facial dysmorphologies. TheHCS face is different and becomes coarser with age. In contrast, theALGS face includes a prominent forehead, deep-set eyes with mod-
erate hypertelorism, pointed chin, and saddle or straight nose witha bulbous tip. These features give the face the appearance of an in-verted triangle. ALGS can include skeletal defects, typically butterflyvertebrae, and mineralization defects and while shortened phalangeshave been reported, these are not caused by acro-osteolysis as theyare in HCS. An increase in lower limb fractures in ALGS patientshas been noted, consistent with the hallmark bone weakening ob-served in HCS [Bales et al., 2010]. It seems likely that the verydifferent presentations of NOTCH2 mutations in HCS versus ALGSare allele-specific and reflect different effects on protein function.
Given the variable presentation of HCS in our patients, we ex-amined our exome sequencing data for novel potentially functionalcoding variants in 56 genes known or suggested to play importantroles in Notch pathway signaling, either extracellular ligands or in-tracellular modifiers or downstream targets [Fiuza and Arias, 2007].Such mutations could function as secondary phenotypic modifiers.Only two interesting private (i.e., not in dbSNP or the 1000 GenomesProject data sets) variants were identified, p.Gln608Arg in putativeligand coding gene DLL4 [MIM# 605185] and p.Val613Leu in in-teracting gene DTX1 [MIM# 602582], each in a single patient (seeSupp. Table S3 for complete list of genes examined). It is not yet clearwhether these variants are important in the clinical presentations ofthese particular patients.
The clustering of mutations within the coding region of NOTCH2in our HCS patients and in other published reports suggest a stronggenotype–phenotype correlation. The extreme carboxyl terminusof NOTCH proteins includes a potential PEST domain, thoughtto be involved in protein turnover [Kopan and Ilagan, 2009]. Ab-sence of this domain could lead to dominant gain-of-function effectsthrough increased protein stability and excessive signaling activity.In the paralog NOTCH1, deletions of the C-terminal region contain-ing the PEST domain increased the half-life of the NOTCH1 intracel-lular domain, and were associated with T-cell acute lymphoblasticleukemia [Weng et al., 2004]. Alternatively, since the C-terminusalso functions in nuclear transcriptional regulation, these allelesmight represent mild loss-of-function effects. Mutations in mouseNotch2 have been engineered, and although heterozygous mice seemnormal, homozygotes for complete or partial loss of gene functionalleles die either as embryos or neonates with defects in kidney,heart, and eye development (McCright, 2002 #116; McCright, 2006#114). Mouse mutants akin to the C-terminal truncations observedin HCS patients have not yet been reported. A direct role of NOTCHgenes in bone metabolism and maintenance has been demonstrated(Engin et al., 2008; Engin and Lee, 2010). Our results support thesuggestion that modulation of the Notch pathway, and specificallyNOTCH2, may be a productive therapeutic mode for treatment oftypical age-related osteoporosis, although the pleiotropic nature ofthe gene’s activity is an issue of concern.
Acknowledgments
Foremost, we thank the families who generously contributed their time andmaterials to this research study. The FORGE Canada Consortium: Findingof Rare Disease Genes in Canada; Steering Committee consists of KymBoycott (leader; Ottawa, Ontario), Jan Friedman (co-leader; Vancouver,British Columbia), Jacques Michaud (co-leader; Montreal, Quebec), Fran-cois Bernier (Calgary, Alberta), Michael Brudno (Toronto, Ontario), BridgetFernandez (St Johns, Newfoundland), Bartha Knoppers (Montreal, Quebec),Mark Samuels (Montreal, Quebec), Steve Scherer (Toronto, Ontario). Wethank Dr. Cedric Le Caignec for sharing general project status in advance ofpublication. We would like to thank Janet Marcadier (Clinical Coordinator)and Chandree Beaulieu (Project Manager) for their contribution to the in-frastructure of the FORGE Canada Consortium. This work was funded by
1116 HUMAN MUTATION, Vol. 32, No. 10, 1114–1117, 2011

the Government of Canada through the Canadian Institutes of Health Re-search, Genome Canada, Genome Quebec, Genome British Columbia andthe Ontario Genomics Institute (OGI-049). M.E.S. was supported by theCentre de Recherche du CHU Ste-Justine.JMaj supervised analysis of next generation sequencing data. J.S. performedanalysis of next generation sequencing data. A.C. performed PCR-basedSanger sequencing to validate and identify new mutations. LP assisted withsample preparation and management. J.Mar. and J.P.D.F. assisted with clin-ical ascertainment. K.M.B., L.G.S.M., F.E.M., I.M., H.V.E., and J.L.M. per-formed clinical ascertainment and manuscript preparation. M.E.S. managedall aspects of the project and primary manuscript preparation.
References
Bales CB, Kamath BM, Munoz PS, Nguyen A, Piccoli DA, Spinner NB, Horn D, Shults J,Leonard MB, Grimberg A, Loomes KM. 2010. Pathologic lower extremity fracturesin children with Alagille syndrome. J Pediatr Gastroenterol Nutr 51:66–70.
Charvat J, Seydlova M, Dostalova T, Marik I. 2010. Characteristics of Hajdu-CheneySyndrome and specifics of the full denture: case report. Locomotor System 17:97–109.
Cheney WD. 1965. Acro-osteolysis. Am J Roentgenol Radium Ther Nucl Med 94:595–607.
Choi M, Scholl UI, Ji W, Liu T, Tikhonova IR, Zumbo P, Nayir A, Bakkaloglu A, Ozen S,Sanjad S, Nelson-Williams C, Farhi A, Mane S, Lifton RP. 2009. Genetic diagnosisby whole exome capture and massively parallel DNA sequencing. Proc Natl AcadSci U S A 106:19096–19101.
Coffey AJ, Kokocinski F, Calafato MS, Scott CE, Palta P, Drury E, Joyce CJ, LeproustEM, Harrow J, Hunt S, and others. 2011. The GENCODE exome: sequencing thecomplete human exome. Eur J Hum Genet.
Crosnier C, Lykavieris P, Meunier-Rotival M, Hadchouel M. 2000. Alagille syndrome.The widening spectrum of arteriohepatic dysplasia. Clin Liver Dis 4:765–778.
Engin F, Lee B. 2010. NOTCHing the bone: insights into multi-functionality. Bone46:274–280.
Engin F, Yao Z, Yang T, Zhou G, Bertin T, Jiang MM, Chen Y, Wang L, Zheng H,Sutton RE, Boyce BF, Lee B. 2008. Dimorphic effects of Notch signaling in bonehomeostasis. Nat Med 14:299–305.
Fiuza UM, Arias AM. 2007. Cell and molecular biology of Notch. J Endocrinol 194:459–474.
Griffiths-Jones S, Saini HK, van Dongen S, Enright AJ. 2008. miRBase: tools for mi-croRNA genomics. Nucleic Acids Res 36:D154–D158.
Hajdu N, Kauntze R. 1948. Cranio-skeletal dysplasia. Br J Radiol 21:42–48.
Isidor B, Lindenbaum P, Pichon O, Bezieau S, Dina C, Jacquemont S, Martin-CoignardD, Thauvin-Robinet C, Le Merrer M, Mandel JL, and others. 2011. Truncating mu-tations in the last exon of NOTCH2 cause a rare skeletal disorder with osteoporosis.Nat Genet 43:306–308.
Kopan R, Ilagan MX. 2009. The canonical Notch signaling pathway: unfolding theactivation mechanism. Cell 137:216–233.
Li H, Durbin R. 2009. Fast and accurate short read alignment with Burrows-Wheelertransform. Bioinformatics 25:1754–1760.
Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G,Durbin R. 2009. The Sequence Alignment/Map format and SAMtools. Bioinfor-matics 25:2078–2079.
Marik I, Kuklik M, Zemkowa D, Kozlowski K. 2006. Hajdu-Cheney syndrome: reportof a family and a short literature review. Australas Radiol 50:534–538.
McDaniell R, Warthen DM, Sanchez-Lara PA, Pai A, Krantz ID, Piccoli DA, SpinnerNB. 2006. NOTCH2 mutations cause Alagille syndrome, a heterogeneous disorderof the notch signaling pathway. Am J Hum Genet 79:169–173.
McKiernan FE. 2007. Integrated anti-remodeling and anabolic therapy for the osteo-porosis of Hajdu-Cheney syndrome. Osteoporos Int 18:245–249.
Ng PC, Henikoff S. 2003. SIFT: predicting amino acid changes that affect proteinfunction. Nucleic Acids Res 31:3812–3814.
Ng SB, Buckingham KJ, Lee C, Bigham AW, Tabor HK, Dent KM, Huff CD, ShannonPT, Jabs EW, Nickerson DA, Shendure J, Bamshad MJ. 2010. Exome sequencingidentifies the cause of a mendelian disorder. Nat Genet 42:30–35.
Pruitt KD, Harrow J, Harte RA, Wallin C, Diekhans M, Maglott DR, Searle S, FarrellCM, Loveland JE, Ruef BJ, Hart E, Suner MM, Landrum MJ, Aken B, Ayling S,Baertsch R, Fernandez-Banet J, Cherry JL, Curwen V, Dicuccio M, Kellis M, Lee J,Lin MF, Schuster M, Shkeda A, Amid C, Brown G, Dukhanina O, Frankish A, HartJ, Maidak BL, Mudge J, Murphy MR, Murphy T, Rajan J, Rajput B, Riddick LD,Snow C, Steward C, Webb D, Weber JA, Wilming L, Wu W, Birney E, Haussler D,Hubbard T, Ostell J, Durbin R, Lipman D. 2009. The consensus coding sequence(CCDS) project: identifying a common protein-coding gene set for the humanand mouse genomes. Genome Res 19:1316–1323.
Simpson MA, Irving MD, Asilmaz E, Gray MJ, Dafou D, Elmslie FV, Mansour S,Holder SE, Brain CE, Burton BK, Kim KH, Pauli RM, Aftimos S, Stewart H,Kim CA, Holder-Espinasse M, Robertson SP, Drake WM, Trembath RC. 2011.Mutations in NOTCH2 cause Hajdu-Cheney syndrome, a disorder of severe andprogressive bone loss. Nat Genet 43:303–305.
Wang K, Li M, Hakonarson H. 2010. ANNOVAR: functional annotation of ge-netic variants from high-throughput sequencing data. Nucleic Acids Res 38:e164.
Weng AP, Ferrando AA, Lee W, Morris JPt, Silverman LB, Sanchez-Irizarry C, BlacklowSC, Look AT, Aster JC. 2004. Activating mutations of NOTCH1 in human T cellacute lymphoblastic leukemia. Science 306:269–271.
HUMAN MUTATION, Vol. 32, No. 10, 1114–1117, 2011 1117