Embed Size (px)
Citation preview

Mutual Interactions of the Methyl and Methylperoxy Radicals Studied by Flash
Photolysis and Kinetic Spectroscopy
HIROYUKI ADACHI, N. BASCO, and D. G. L. JAMES Department of Chemistry, University of British Columbia, Vancouver, B.C., Canada
V 6 T 1 Y6
Abstract
The decadic extinction coefficient of the methyl radical a t 216.4 nm and the rate constant for mutual combination were redetermined as:
~(216.4) = (9.5 f 0.4) X lo3 l./mol cm
k e = (3.2 f 0.4) X 1 O l o l./mol sec
The application of the Beer-Lambert law to these measurements was justified experimentally. The absorption spectrum of the methylperoxy radical was characterized as a weak, broad, structureless band, having a maximum at 240 nm with ~(240) = 1.55 X lo3 l./mol cm. The mutual interaction of methylperoxy radicals leads to the generation of methoxy and hydro- peroxy radicals as a consequence of the nonterminating interaction
2 CH30O + 2 CH30. + 0 2
CH30 + 0 2 -j HCHO + HOO
Each derivative radical may consume a significant fraction of the methylperoxy radicals, and either of these cross interactions may be made predominant by a suitable choice of oxygen pressure. The mutual interaction was studied under both conditions. The overall mechanism was analyzed by a precise computational method, and the rate constant of the total mutual interaction
2 CH3OO - all products
was estimated as
k q = (3.5 f 0.3) X lo8 l./mol sec
Introduction
The accurate estimation of the rate constants for mutual interaction is hindered, both for the methyl radical and for the methylperoxy radical, by intrinsic properties of the radicals themselves. A significant deviation from the Beer-Lambert law for the absorbance of the methyl radical may result from the use of a finite band pass for the monitoring light. A large fraction
International Journal of Chemical Kinetics, Vol. XII, 949-977 (1980) 0 1980 John Wiley & Sons, Inc. 0538-8066/80/0012-0949$02.90

950 ADACHI, BASCO, AND JAMES
of the mutual interaction of methylperoxy radicals is nonterminating and generates both methoxy and hydroperoxy radicals that interact with the methylperoxy radicals in a complex pattern of reactions. This article de- scribes an investigation of these difficulties and shows how they have been overcome to allow the evaluation of the rate constants.
The absorption of the spectrum of the CH; radical in the gas phase has been characterized by Herzberg [1,2]. The band at 216 nm comprises two diffuse maxima at 215.76 and 216.36 nm; the decadic extinction coefficient €(A) has a maximum value of (1.0 f 0.1) X lo4 l./mol cm, and estimates for the oscillator strength range from 0.010 to 0.0137 [3-51. The absorbance of this band has been used to measure the concentration of the methyl radical, and thereby to estimate the rate constant for its mutual combina- tion in several recent investigations [3-71, yielding a set of results which are in only moderate agreement [5].
The methyl radical is generated conveniently by the photolysis of azo- methane:
(1) CH3-N=N-CHB + hu -+ 2CH; + Nz
Conditions may be chosen so that the sole significant reaction of the methyl radical is mutual combination:
(2) 2CH3 + M + CHBCHB + M
The kinetic analysis of such a system is unambiguous as this reaction is terminating. The derivation of the concentration from the absorbance A(X) and the optical path length 1 has usually employed the relationship A(X) = E(X)[CH~]Z, involving the assumption that the Beer-Lambert law may be used. However, the absorption bands are comparatively narrow, and €(A) cannot be considered to maintain a unique value if the band pass of the monitoring light is increased beyond a certain limit. Accordingly, Si- mons and co-workers have used the empirical relationship A(X) = E~,,([CH~]Z)~, where both cap, and n depend upon the size of the band pass. They estimated the value of n to be 0.59 f 0.04 [7] and 0.72 f 0.07 [8] with a band pass of 0.20 nm in two sets of experiments in one apparatus, and derived a value of the rate constant k2 = (3.37 f 0.46) X 1O1O l./mol sec [7]. In the present apparatus the value of n has been estimated under a variety of conditions by both the variable band pass method and the variable path length method. Under our normal operating conditions the deviation of n from unity was found to be negligible, and this conclusion was fully supported by independent theoretical calculations. The values of €(A) and k2 reported in this study are consequently independent of the dispersion of the optical system, and a valid comparison with corresponding results of other workers is made possible.
The absorption spectrum of the methylperoxy radical in the gas phase has been described by Parkes and co-workers [9-121, by Hochanadel and

METHYL AND METHYLPEROXY RADICAL INTERACTIONS 951
co-workers [13], and by Calvert and co-workers [14,15], and is broad, structureless, and comparatively weak. However, these sources differ in their quantitative characterization of the absorption maximum, which may be represented by the respective values of the decadic molar extinction coefficient €(A): ~(238) = (1.44 f 0.26) X lo3, ~(235) = 0.87 X lo3 and r(239) = 0.82 X lo3 l./mol cm. The higher set of values of €(A) given by Parkes and co-workers is supported by a mean value at a single wavelength, 4250) = 1.13 X lo3 l./mol cm, reported recently by Cox and Tyndall [16]. There is no reason to doubt that the Beer-Lambert law will be valid for the methylperoxy radical. This radical was formed from the methyl radical by the reaction
(3) CH; + 0 2 + M -+ CHBOO. + M
with virtually complete efficiency under suitable conditions. The decline in the absorbance of the methylperoxy radical conformed to a second-order graph, and an apparent rate constant may be defined in relation to its gradient as k c = (~(A)-1/2)d[l/A(X)]/dt. Unfortunately, there is no simple relationship between 124' and k 4 , the rate constant for mutual interac- tion:
(4) 2 CH300. - all products
The problem arises because a fraction of this reaction is nonterminating; estimates of this fraction were reviewed by Parkes in 1977 [l l] , and the principal results are 0.33 [lo], 0.43 [17], and 0.48 [18]. The nonterminating interaction generates the methoxy radical
(44 2 C H 3 0 0 - 2 CH30 + 0 2
A proportion of this species reacts with oxygen to yield the hydroperoxy radical
(5) CH30. + O2 - HCHO + HOO.
and k5 = 4 X lo5 l./mol sec [19].
This proportion can be varied easily over a wide range by an appropriate choice of oxygen concentration. Finally, a fraction of each of these daughter radicals consumes methylperoxy radicals, principally by dis- proportionation:
(6)
(7)
CH;OO' + CH30. - CH;OOH + HCHO
CH;OO- + HOO. -+ CH3OOH + 0 2
Consequently, the ratio k 4 ' / k 4 must be computed by numerical integration of the rate equations associated with a comprehensive mechanism for this system, and an appropriate procedure has been developed in this investi- gation. Furthermore, either the methoxy radical or the hydroperoxy

952 ADACHI, BASCO, AND JAMES
radical can be made to act as the kinetically dominant daughter species by a suitable choice of oxygen concentration. Accordingly, two sets of mea- surements were made in complementary systems corresponding to these conditions; the comparison of the resultant values of k4 provided a further test of the procedure.
We have also investigated the influence of the bimolecular reaction
(14) CH; + 02 -+ HCHO + O H
upon the balance between the other reactions. This balance may be altered in two significant ways: (1) by the consumption of the methyl radical without equivalent generation of the methylperoxy radical, and (2) by the consumption of the methylperoxy radical by methathesis with the highly reactive hydroxyl radical:
(15) CH300 + O H -+ CH;O. + HOO
This reaction is not terminating, and further consumption of the methyl- peroxy radical by the daughter radicals in reactions (6) and (7) will result. Estimates of the magnitude of k14 vary widely. Our group has found evi- dence that k14 is not much greater than 2 X lo5 l./mol sec at 295 K [20a], and more recently Klais and co-workers showed that k14 Q 2 X lo5 l./mol sec at 368 K using the technique of flash photolysis and resonance fluo- rescence [20b]. This means that the influence of reaction (15) is negligible in the present investigation. A low value has been confirmed by very- low-pressure pyrolysis studies by Golden and co-workers [21]. In a recent review [22], Walker has attempted to correlate results obtained in eight studies of the shock-initiated oxidation of methane. The points from four of these studies lie close to an Arrhenius plot of the equation:
1214 = exp(-12.7 X 103/RT) l./mol sec
which extrapolates to 0.8 l./mol sec at 298 K. A very different Arrhenius expression was proposed by Washida and Bayes in 1976 [23]:
1214 = (108.24*0.35) exp[(-1.87 f 0.50) X 103/RT] l./mol sec
between 259 and 339 K, corresponding to 7.4 X lo6 l./mol sec at 298 K, and the latter value is supported by a later estimate of (1.0 f 0.7) X lo7 l./mol sec reported by Washida in 1979 [24]. Such high values were questioned by Baldwin and Golden in 1978 [25]. These authors concluded that k14 is certainly less than 3 X lo5 l./mol sec, with El4 < 25 kcal/mol, and such limits confirm our conclusion that reaction (15) is negligible in both the previous and the present investigations. However, Washida and Bayes have claimed that the consumption of the hydroxyl radical by reaction (15) was significant in our previous system [20], and that its neglect led to a systematic error in our estimate of k14. Consequently, an investigation of the influence of reaction (15) is required in relation to the estimation both

METHYL AND METHYLPEROXY RADICAL INTERACTIONS 953
of k4 and of k14, and this was performed in the present investigation with the aid of a comprehensive computer program.
Literature values of the apparent rate constant k41 seem at first glance to be concordant: (3.3 f 0.6) X lo8 [ll], (2.65 f 0.6) X lo8 [12] (2.3 f 0.4) X lo8 [13], (2.5 f 0.3) X los [14,15], (3.1 f 0.4) X lo8 [16], and (2.2 f 0.2) X lo8 [26] l./mol sec. However, the apparent agreement of the second es- timate with the third, fourth and sixth ones is due to the chance compen- sation of disparities in the factors €(A) and k4</4X).
The diversity of current values for the extinction coefficient and the rate constants k4’ and k4 reveals a need for their reevaluation, and this is the chief aim of the present investigation.
Experimental
General Procedure
Absorbance measurements were made with the photoelectric apparatus described in detail elsewhere [27]. High sensitivity was achieved by using a pulsed xenon lamp as the analytical light source in conjunction with a dual-beam optical system including twin cells and balanced photomulti- pliers. The reaction and reference cells were made of Pyrex glass and fitted with Suprasil end windows to transmit monitoring light down to below 200 nm. Each cell was cylindrical in shape, with a length of 914 mm and an internal diameter of 20 mm. A double gas filter containing chlorine and bromine was placed before the slit to remove radiation between 280 and 520 nm. These precautions restricted the photolytic radiation to X > 280 nm and substantially reduced the intensity of scattered light within the spectrometer. Consequently, the absorbance of the methyl radical could be monitored during the period of the photoflash, and this capacity was used in the evaluation of its extinction coefficient. Kinetic xeasurements of the mutual interaction of methyl radicals began when the photoflash was virtually extinct, some 25 psec after the firing. Some confirmatory spectral measurements were made by plate photometry; the relevant photographic technique has been described earlier [3].
Methyl radicals were generated by the flash photolysis of azomethane, and n-pentane was used as a moderating gas in all experiments performed in the absence of oxygen. Azomethane was supplied by Merck, Sharp and Dohme (Canada) Ltd.; analysis by vapor phase chromatography showed that the sole impurities were traces of nitrogen, methane, and ethane, and these were removed by exhaustive degassing in freeze-thaw cycles under vacuum. The n-pentane supplied by Matheson, Coleman, and Bell Manufacturing Chemists was purified, analyzed and stored as described elsewhere [28]. The oxygen supplied by the Matheson Company was 99.95% pure, and was used without further purification.

954 ADACHI, BASCO, A N D JAMES
The only significant products of the flash photolysis of azomethane in the presence of n-pentane were nitrogen and ethane. The nitrogen was isolated by condensing the other constituents of the reaction mixture in a series of traps immersed in liquid nitrogen, which was further cooled by the passage of a stream of precooled helium gas. The number of moles of nitrogen was measured with a McLeod Gauge, and used to estimate C,,, from the equation Cma, = 2n(N2)/V7 where C,,, is the hypothetical con- centration corresponding to the simultaneous presence in the reaction cell of all the methyl radicals generated by the photoflash, and V is the volume of the cell.
Ama,(X) is the absorbance at the wavelength X corresponding to C,,,. The extrapolation procedure used to evaluate A,,,( A) has been described previously [3]. In the present investigation each absorbance curve between 0 and 36 psec was divided into 4-psec intervals, and the mean absorbance (A; ) was estimated for each interval from the smoothed curve. Summation over the nine intervals yields the equation
9
i= l Amax = A36 + (4 X 10-6)G C (A;)'
where A36 is the absorbance at 36 p e c and G = d(l/A)/dt is the gradient of the corresponding second-order plot observed after the extinction of the photoflash [28].
The extinction coefficient €(A) and the rate constant k2 may be derived from the equations ((A) = Amax(A)/lCmax and kz = */~Glc(X) , where 1 is the length of the reaction cell, only if the concentration of the methyl radical is related to its absorbance by the equation [CH;] = A(X)/lc(X). The va- lidity of this relationship at a band pass of 0.06 nm was therefore examined in detail.
Beer-Lambert Law for the Methyl Radical
Two independent methods were used for this investigation, each with the same photoflash energy of 1080 J:
Variable band pass method. Experiments of series 1 and 2 employed respective values of the band pass of 0.06 and 0.16 nm, each centered on 216.36 nm, and measurements were made at similar values of the signal- to-noise ratio. To obtain these conditions, the widths of the entrance and exit slits of the spectrometer and the voltage applied to the pulsed xenon arc were, respectively, 75 pm and 140 V and 200 pm and 60 V. The effective value of the extinction coefficient for a band pass of AA nm was defined as e,ff[AA] = A/[CH3]l. Corresponding values of k2/ceff[AX] were derived from the gradients of the appropriate second-order plots, and values of e,ff[AX] were calculated from approprisrte values of A,,, and the yield of nitrogen, each as described above. The values of 104[CH3N=NCH3] and 103[n-

METHYL AND METHYLPEROXY RADICAL INTERACTIONS 955
CsH12] fell within the respective ranges of 1.64-1.91 and 1.91-2.73 mole/ 1.
The effect of the signal-to-noise ratio upon the precision of measurement was examined with the aid of the experiments of series 3; these employed the same conditions of series 2, except that the voltage applied to the spectroflash was increased from 60 to 140 V.
Variable path length method. A band pass of 0.06 nm centered on 216.36 nm was used for this study. Values of the absorbance at 216.36 nm were measured when the whole length and when one-half the length of the reaction cell were exposed to illumination by the photoflash under otherwise identical conditions. One-half of the illuminated length of 914 nm was masked by sets of cylindrical masks of total length 457 mm according to three patterns: (i) a single mask, (ii) three equal masks, and (iii) 15 equal masks. The subdivided masks were arranged symmetrically along the cell to give alternating regions of light and darkness in patterns (ii) and (iii). In pattern (i) any asymmetry in the illumination of the cell by the photo- flash was compensated for by conducting experiments in pairs, in which the illuminated and masked regions of the reaction cell were interchanged. Moreover, the whole series of experiments was repeated, using the original reaction cell as the reference cell, and vice versa. These cells are of the same length, but differ in volume by 9.5%; an appropriate correction was made for this difference. The effect of asymmetry was found to be negligible, and no corresponding additional experiments were necessary for patterns (ii) and (iii). The values of 104[CH3N=NCH3] and 103[n-CsH12] fell within the respective range of 1.64-1.91 and 1.80-1.86 mol/l.
Definitive Measurements of €(A) and k 2 / ~ ( A ) for the Methyl Radical
The main series 4 and 5 comprise 48 sets of measurements designed to achieve greater precision by employing the most favorable conditions used in this study. In particular, the band pass was 0.06 nm, the spectroscopic lamp voltage was 140 V, and the photoflash energy was 360 J. The values of 104[CH3N=NCH3], [~z-C~H~~]/[CH~-N=N-CH~] and 107C,,, fell into three groups and were, respectively, 1.47,1.64, and 1.64 moleh.; 44,11, and 14; and 2.5,3.0, and 3.1 mole/l. The simpleequationsgiveninthesec- tion on general procedures were used to evaluate €(A) and k z / c ( A ) . For comparison, the relative absorption spectrum was recorded on a photo- graphic plate and evaluated by plate densitometry, following a procedure described previously [3].
Reaction Mechanism in the Presence of Oxygen
A reaction mechanism for the initial period of the flash photolysis of azomethane in the presence of excess oxygen at room temperature is given in Table I, with values for the rate constants from the literature, where these

956 ADACHI, BASCO, AND JAMES
TABLE I. Mechanism for the photolysis of azomethane in the presence of oxygen during the initial 4 msec.
Number Reaction Rate constant Reference or
( t.male-lsec-‘) conrmenr
~~
(1) CH3-N-N-CH3 + hv + 2 CH3’ + N2 tm = 4 w e c
(2) 2 CH3’ + M + CH3CH3 + M 3 . 2 x l o l o [this word
(3) CH3’ + O2 + M + CH300’ + M 2 . 4 x 10’ [203; 0.80 atm
1.0 x 108 [20]; 0.038 atm.
(4) 2 CH300’ + a l l products 3 . 5 x 10’ [this vorkJb
( 4 4 2 C H 3 0 0 ’ + 2 CH300’ + O 2 f x k4 f = 0.50 or 0 . 3 3
( 5 ) CH3’ + CH300‘ .- + M + CH300CH3 + M 1. x l o l o K5 4 2 ( k 2 k 4 ) 7 ,a
(6) C H 3 0 0 ’ + CH30‘ + CH3WH + HCHO 9.2 x l o8 [ 301
(7) 2 CH30’ + CH30H + HCHO 1 . 0 x l o l o [19, 301
(8) CH3’ + CH30’ + CH4 + HCHO 2.0 x l o l o kg = 2 k:
(9) C H ~ O ‘ + o2 + HCHO + HOO‘ 4.0 1 0 ~ [19. 311
k10 - k ’ 4 ;lo) CH300’ + HOO’ + CH300H + O2 3 .5 x lo8 (11) CH30’ + HOO’ * CH30H + 0 1 . 0 x 1010 k l l = 1’12”
(12) 2 HOO’ + HWH + o2 2.0 x lo9 [32, 331
‘13 = “11“ (13) CH3’ + HOO‘ + CH4 + O2 2.0 x 1010
(14) C H ~ ‘ + o2 + HCHO + OH‘ 2.0 x lo5 POI 7 . 4 x 106 ~ 2 3 1
(15) C H ~ O O * + OH’ t C H ~ O ‘ + HOO’ 2.0 x lo9 k15 = k16/10a
(16) C H i O * + OH’ + HCHO + HOH 2.0 x 10”
2.0 x 1010 (17) 800’ + OH’ + O2 + HOH
(18) CH3’ + OH’ + M -t CH30H + H
‘16 - k17 - klF
2.0 x 1010 k18 - (19) 2 OH’ + M t HWH + M 2.9 l o 9 [34. 351
a Estimated value. Final value of an iterative process.
exist. The pseudo-second-order rate coefficient k; varies with pressure, and the higher and lower values quoted are valid for the systems with oxygen concentration of 3.23 X and 1.0 X mol/l., respectively. Such values are the lowest in literature and were adopted so that the sys- tems could be analyzed under the least favorable conditions for the con- version of the methyl radical to the methylperoxy radical. Two repre- sentative values of k 14 have been quoted, and duplicate sets of calculations have been performed to assess the influence of this parameter upon the course of the reaction. The values of k2 and k 4 are new ones which were measured in the present investigation. Other values are estimates and reflect a general order of reactivity: OH. - CH; Z CH30 > HOO- > CH300..

METHYL AND METHYLPEROXY RADICAL INTERACTIONS 957
The mechanism is similar to that proposed by Parkes in 1977 [ll], but is supplemented by the bimolecular reaction (14):
(14) CH; + 0 2 ---* HCHO + OH.
and five reactions of mutual or cross interaction of the hydroxyl radical. Metathetical reactions between each of the five radical species and either azomethane or a molecular product have been excluded as occurring to an insignificant extent during the reaction time of 4 msec because of the small size of either the rate constant or the product of concentrations, or both. For example, consider the rate of reaction (20):
(20) CH; + C H ~ N Z N C H ~ -+ CH4 + CH3NzNCHi
with k2o = 2.2 X lo2 l./mol sec [29] in relation to the rate of reaction (3); R20/R3 = 4 X mol/l. and [ 0 2 ] = 1.0 X mol/l., the lower value used in this study. Metathesis between the methylperoxy radical and azomethane has been treated as negligible for analogous reasons.
when [CH3N=NCH3] = 4.3 X
The Computer Program
The stoichiometric and kinetic calculations involved the solution of as many as 30 simultaneous differential equations derived from the mecha- nism of Table I, and we chose the GEARB program [36] because it is de- signed to solve the initial value problem for stiff systems of ordinary dif- ferential equations to a specified error tolerance [37].
The stoichiometric calculations yield instantaneous values of actual or equivalent concentrations at specified intervals over a reaction period of 4 msec, and divide into two classes:
Class (i). The actual concentration [R] of each of the five radicals CH;, CH300*, CH30-, HOO-, and O H was evaluated by integrating the corre- sponding differential equation over the specified interval. As an example, a set of values of [CH3-] was derived from the equation
d[CH;]/dt = (C,,,t/t;) exp(-tlt,) - 2k2[CH;I2 - (k3 + k14)[CH3][02] - k~j[CHi][CH300-] - k*[CH;][CH30*] - k13[CH;][HOO] - k1a[CH;][OH*]
The first term is the generating function proposed by Basco, Callear, and Norrish [38] with the current values C,,, = 6.55 X mol/l. and t , = 4 psec. The ratio f = k4Jk4 was set at 0.50. Duplicate sets of calculations corresponding to the two estimates for k 1 4 in Table I were performed for comparative purposes; where two values are given for a parameter, the second corresponds to k14 = 2.0 X lo5 l./mol sec. However, the results quoted in Figures 1-3 were calculated with k14 = 2.0 X lo5 l./mol sec.
The rise and fall of the methyl radical concentration follow the profile
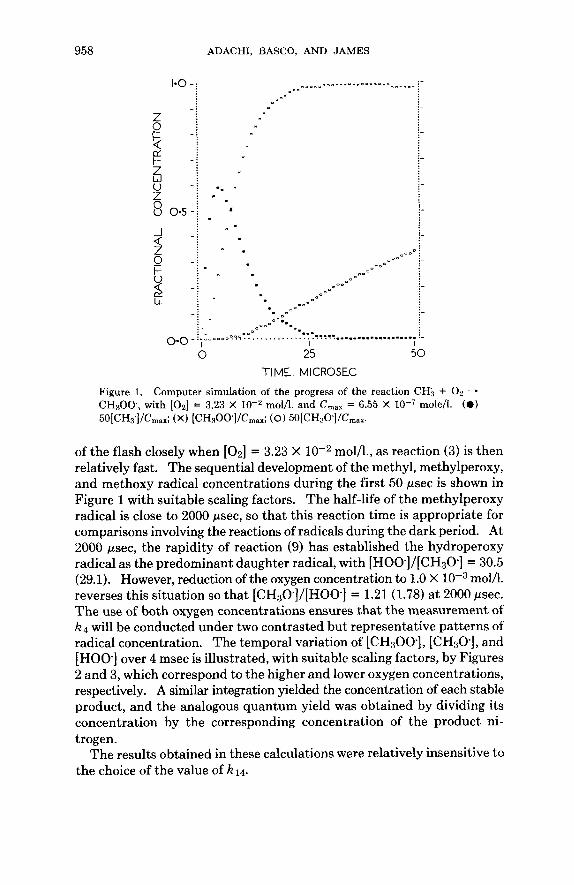
958 ADACHI, BASCO, AND JAMES
0 25 50 TIME. MICROSEC
Figure 1. Computer simulation of the progress of the reaction CH; + 0 2 - CH300., with [Oz] = 3.23 X moI/l. and C,,, = 6.55 X mole/l. (a) 50[CH3']/Cmax; (X) [CH,OO]/Cmax; (0) 5O[CH3O]/Cmax.
of the flash closely when [ 0 2 ] = 3.23 X mol/l., as reaction (3) is then relatively fast. The sequential development of the methyl, methylperoxy, and methoxy radical concentrations during the first 50 psec is shown in Figure 1 with suitable scaling factors. The half-life of the methylperoxy radical is close to 2000 psec, so that this reaction time is appropriate for comparisons involving the reactions of radicals during the dark period. At 2000 psec, the rapidity of reaction (9) has established the hydroperoxy radical as the predominant daughter radical, with [HOO.]/[CH30] = 30.5 (29.1). However, reduction of the oxygen concentration to 1.0 X mol/l. reverses this situation so that [CH30-]/[HOOe] = 1.21 (1.78) at 2000 psec. The use of both oxygen concentrations ensures that the measurement of k4 will be conducted under two contrasted but representative patterns of radical concentration. The temporal variation of [CH300-], [CHsO], and [HOO] over 4 msec is illustrated, with suitable scaling factors, by Figures 2 and 3, which correspond to the higher and lower oxygen concentrations, respectively. A similar integration yielded the concentration of each stable product, and the analogous quantum yield was obtained by dividing its concentration by the corresponding concentration of the product ni- trogen.
The results obtained in these calculations were relatively insensitive to the choice of the value of kid.
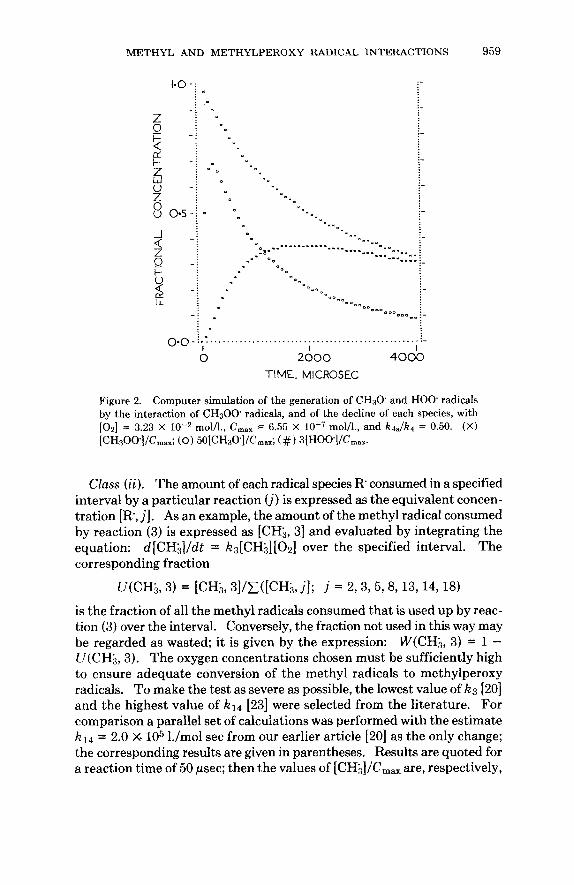
METHYL AND METHYLPEROXY RADICAL INTERACTIONS 959
............... .... .... ." . . . . . .-.. I.."
-*-.. ." ... --. - 8 - .-- ea
................................................. I I I 1 2 0 0 0 4000
TIME. MICROSEC
Figure 2. Computer simulation of the generation of CH30 and HOO radicals by the interaction of CH300 radicals, and of the decline of each species, with [ 0 2 ] = 3.23 X mol/l., C,,, = 6.55 X mol/l., and k4Jk4 = 0.50. (X) (CH300]/Cm,,; (0) 50[CH3O]/CmaX; (#) 3[HOO]/CmaX.
Class (ii). The amount of each radical species R consumed in a specified interval by a particular reaction G) is expressed as the equivalent concen- tration [R, j ] . As an example, the amount of the methyl radical consumed by reaction (3) is expressed as [CH;, 31 and evaluated by integrating the equation: cl[CH;]/dt = kS[CH;] [O,] over the specified interval. The corresponding fraction
U(CH,, 3) = [CH;, 3]/C([CH3,j]; j = 2,3,5,8, 13,14, 18)
is the fraction of all the methyl radicals consumed that is used up by reac- tion (3) over the interval. Conversely, the fraction not used in this way may be regarded as wasted; it is given by the expression: W(CH;, 3) = 1 - U(CH3,3). The oxygen concentrations chosen must be sufficiently high to ensure adequate conversion of the methyl radicals to methylperoxy radicals. To make the test as severe as possible, the lowest value of h~ [20] and the highest value of 1214 [23] were selected from the literature. For comparison a parallel set of calculations was performed with the estimate h 14 = 2.0 X. lo5 l./mol sec from our earlier article [20] as the only change; the corresponding results are given in parentheses. Results are quoted for a reaction time of 50 p e c ; then the values of [CH;]/C,, are, respectively,
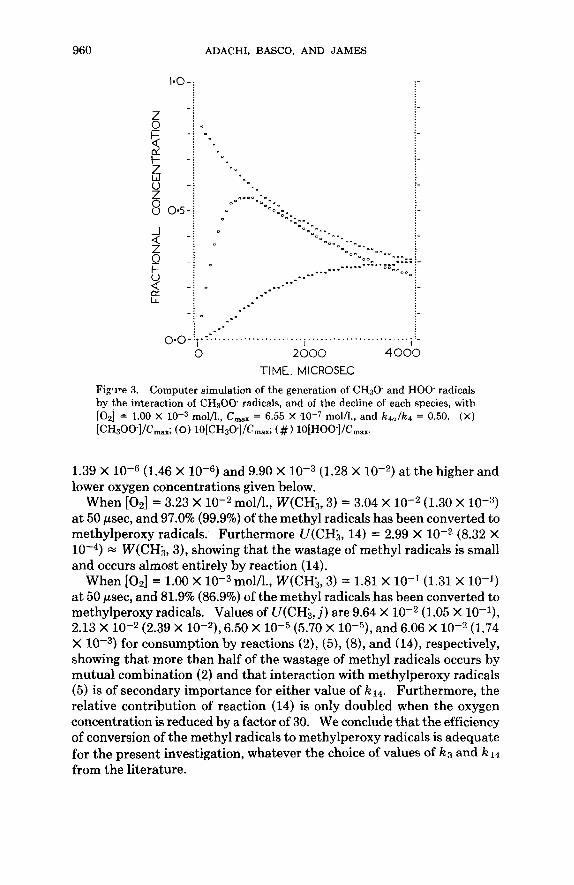
960 ADACHI, BASCO, AND JAMES
_ . . . .-
. - . - . - O.O-'.-' ................................................ :-
0 2 000 4000 TIME, MICROSEC
I I
Fi,pve 3. Computer simulation of the generation of CH30 and HOO. radicals by the interaction of C H 3 0 0 radicals, and of the decline of each species, with [Oz] = 1.00 X molh., C,,, = 6.55 X molh., and k4a/k4 = 0.50. (X) [CH3OO]/Cmax; (0) 10[CH30]/Cmax; (#) 10[H001/Cmax.
1.39 X (1.46 X 10-6) and 9.90 X (1.28 X at the higher and lower oxygen concentrations given below.
a t 50 psec, and 97.0% (99.9%) of the methyl radicals has been converted to methylperoxy radicals. Furthermore U(CH,, 14) = 2.99 X (8.32 X
= W(CH;, 3), showing that the wastage of methyl radicals is small and occurs almost entirely by reaction (14).
When [02 ] = 1.00 X molh., W(CH3, 3) = 1.81 X 16-1 (1.31 X 10-l) at 50 psec, and 81.9% (86.9%) of the methyl radicals has been converted to methylperoxy radicals. Values of U(CH;, j ) are 9.64 X loe2 (1.05 X lO- l ) , 2.13 X (2.39 X 6.50 X (5.70 X and 6.06 X (1.74 X for consumption by reactions (2), (5), (8), and (14), respectively, showing that more than half of the wastage of methyl radicals occurs by mutual combination (2) and that interaction with methylperoxy radicals (5) is of secondary importance for either value of k14. Furthermore, the relative contribution of reaction (14) is only doubled when the oxygen concentration is reduced by a factor of 30. We conclude that the efficiency of conversion of the methyl radicals to methylperoxy radicals is adequate for the present investigation, whatever the choice of values of k3 and k14 from the literature.
When [O,] = 3.23 X mol/l., W(CH3, 3) = 3.04 X lov2 (1.30 X

METHYL AND METHYLPEROXY RADICAL INTERACTIONS 961
The material balance ratio M(R) may be expressed in terms of the actual and equivalent radical concentrations defined above. As an example,
([HOO-] + C ([HOO., j]; j’ = 10,11,12,13, 17)) [CH30., 91 + [CHsOO-, 151
M(HO0.) =
as the hydroperoxy radical is generated in reactions (9) and (15). The precision of the GEARB program can be monitored continually through the values of the five material balance ratios. In fact, a value of 1.0000000 is maintained for each ratio throughout the program.
The kinetic calculations are designed to simulate the course of the decline of the absorbance of the methylperoxy radical during the dark period fol- lowing the extinction of the photoflash, and thereby to evaluate the rate constant k4. The plot of the reciprocals of the experimental values of ab- sorbance A(A) against time is sensibly linear during the dark period of each experiment. Accordingly, the gradient of each plot yields a value for an apparent second-order rate constant defined by the equation k41= (€(A) 1/2)d [1/A( A)]/&. In practice, k 4 f / ~ ( A ) is the directly determined experi- mental quantity. At each wavelength the mean value of k 4 r / c ( X ) is multi- plied by the mean value of €(A) to give a value of k41. The set of such values is then examined statistically for wavelength dependence.
The procedure for the evaluation of k4 is successively: (i) to place f equal to either 0.50 or 0.33, (ii) to calculate a set of values of the methylperoxy radical concentration C at 50 equal intervals over the period of interest, (iii) to fit a regression line to the plot of Co/C against time, and to derive a value of k4‘ from its gradient, and (iv) to compare this value with the ex- perimental value of kQ, and to repeat the procedure for further trial values of k4 until the computed value of k4’ is equal to the experimental result.
The validity of this procedure is confirmed by the value of the correlation coefficient r2 associated with the regression line of step (iii). For example, r 2 = 0.99994 when [O,] = 3.23 X moleh. and r2 = 1.00000 when [O,] = 1.00 X mol/l., iff = 0.50 and the reaction time is 4 msec.
The influence of the bimolecular reaction
CH’, + 0 2 ---+ O H + HCHO
upon the course of the reaction and the calculation of k4 was investigated by performing parallel sets of computations with successive values of 7.4 X lo6 and 2.0 X lo5 l./mol sec for ,414 [23,20]; the remaining constants had the values of Table I, with f = 0.50 and [O,] = 3.23 X mol/l. The re- spective maximum values of the radical concentrations were [OH],,/C,,, = 0.0290 and 0.000808, each at 30 psec; [CH31ma,/Cm, = 0.0115 and 0.0118, each at 4 psec; [CH300.]m,x/Cma, = 0.958 and 0.988, each at 34 psec; [CHsO]m,/Cm, = 0.0145 and 0.0138, each at 240 psec; and [HOO],,/C,, = 0.124 at 1760 psec and 0.124 at 1840 psec. The values for the hydroxyl radical differ by a factor of 36, and the corresponding percentages of the
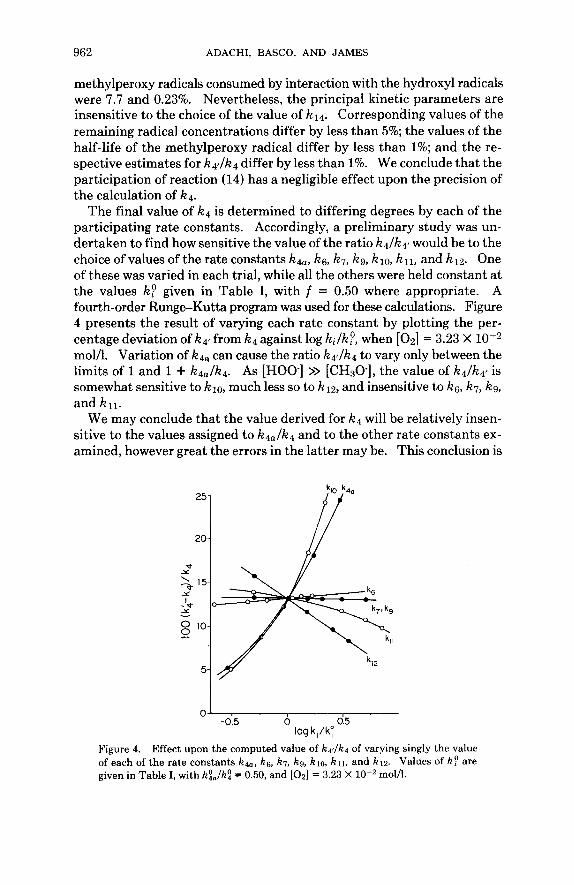
962 ADACHI, BASCO, AND JAMES
methylperoxy radicals consumed by interaction with the hydroxyl radicals were 7.7 and 0.23%. Nevertheless, the principal kinetic parameters are insensitive to the choice of the value of k14. Corresponding values of the remaining radical concentrations differ by less than 5%; the values of the half-life of the methylperoxy radical differ by less than 1%; and the re- spective estimates for k4llk4 differ by less than 1%. We conclude that the participation of reaction (14) has a negligible effect upon the precision of the calculation of k4.
The final value of k4 is determined to differing degrees by each of the participating rate constants. Accordingly, a preliminary study was un- dertaken to find how sensitive the value of the ratio k4Ikq would be to the choice of values of the rate constants k4a, ks, k7, kg, klo, k n , and k12. One of these was varied in each trial, while all the others were held constant at the values kP given in Table I, with f = 0.50 where appropriate. A fourth-order Runge-Kutta program was used for these calculations. Figure 4 presents the result of varying each rate constant by plotting the per- centage deviation Of k4f from k4 against log kilk:, when [ 0 2 ] = 3.23 X molh. Variation of k l a can cause the ratio k4flk4 to vary only between the limits of 1 and 1 + k4Jk4. As [HOO-] >> [CHSO.], the value of k41k4~ is somewhat sensitive to 1210, much less so to k12, and insensitive to ks, k7, kg, and k l l .
We may conclude that the value derived for k4 will be relatively insen- sitive to the values assigned to k4alk4 and to the other rate constants ex- amined, however great the errors in the latter may be. This conclusion is
251 P* I 20 Y
0' I
-0.5 0 0.5 log ki/kq
Figure 4. Effect upon the computed value of k4dk4 of varying singly the value of each of the rate constants k4,,, kg, k7, kg, k m , k l l , and k l z . Values of kp are given in Table I, with k9,lk: = 0.50, and [Oz] = 3.23 X loT2 mom.

METHYL AND METHYLPEROXY RADICAL INTERACTIONS 963
equally valid at the lower oxygen concentration of 1.0 X molh., at which [CH30.]/[HOO.] > 1. This is an advantage as the value of k4,/k4 is only approximately known, and the system'atic error from this source is unlikely to be the greatest of the errors in k4.
Measurement of € ( A ) and k4f/c(A) for the Methylperoxy Radical
The preliminary search for the absorption spectrum of the methylperoxy radical was conducted by recording the light transmitted between 200 and 320 nm on a photographic plate and evaluating the relative absorbance with a plate densitometer, following a procedure described previously [3]. This search revealed a weak, broad, structureless absorption band centered on 240 nm, which is included in Figure 8. This result was later confirmed by the photoelectric technique, and consequently the Beer-Lambert law may be applied with accuracy to the study of the methylperoxy radical. To obtain sufficient sensitivity for quantitative work, the study was continued with the photoelectric detection system described in [27], using a photoflash energy of 1080 J.
Experiments of series [A] were designed to determine the absolute ab- sorption spectrum of the methylperoxy radical. Values of A(X) were measured over a period of 4 msec with concentrations of azomethane and oxygen of 1.64 X and 3.60 X mol/l., respectively; extrapolation to zero time gave the corresponding value of Amax(A). Absolute values of the extinction coefficient were evaluated from the equation €(A) = Amax(A)/Cmax 1. C,,, was equated to twice the yield of nitrogen in the absence of oxygen, but under otherwise identical conditions. Such con- ditions were obtained by substituting 1.64 X lop3 molh. n-pentane for oxygen, and gave a mean value C,,, = 2[N2] = 6.55 X mol/l. The primary yield of methyl radicals has been assumed to be the same in the presence and absence of oxygen. Computer simulation showed that the chosen oxygen concentration was sufficient to ensure that the generation of methylperoxy radicals was 99.9% complete within 50 psec, whereas the half-life of the latter radical was approximately 2000 psec in a typical ex- periment. The evaluation of A,,,( A) by short-range linear extrapolation of the values of A (A) to zero time is therefore precise.
Experiments of series [B] were designed to allow the determination of a set of values of the apparent second order rate constant k4' from the equation: k 4 f / ~ ( A ) = l/&d [1/A( A)]/dt. The concentrations of azomethane and oxygen were either (a) 4.42 X mol/l. or (b) 4.15 X mol/l., respectively, and the period of observztion was 4 msec.
Experiments of series [C] were designed to discover whether the values of k4' and k4 derived from the kinetic measurements would be changed significantly by a thirtyfold reduction in the oxygen concentration. Ac-
and 3.32 X and 3.11 X

964 ADACHI, BASCO, AND JAMES
cordingly the experimental conditions of series [B] were duplicated, except that the value [O,] = (1.15 f 0.05) X mol/l. replaced the former higher value. A set of 17 values of kq/~f240) were measured, and the corre- sponding values of kql were obtained by multiplication by the factor (4240)) derived from series [A].
Results and Discussion
Beer-Lambert Law for the Methyl Radical
The validity of the application of this law at the finite band pass used in the present apparatus was confirmed by the results of two independent sets of experiments.
Variable band pass experiments. The effective value of the extinction coefficient for a band pass of AA nm centered on 216.36 nm was defined as ~eff[AA] = A/[CH;]l. Pairs of values of ~eff[AA] and kz/~eff[AA] were measured at similar values of the signal-to-noise ratio with AA = 0.06 and 0.16 nm. The results are summarized under series 1 and 2, respectively, in Table 11. The mean of ks/f,ff[0.06] and kz/~,ff[0.16] are in close agree- ment, and the values of ~,ff[0.06] and ~,ff[0.16] would not differ significantly even if the ratio of each standard deviation to its mean value were as low as 0.03. On this basis there is no statistical evidence that ~,ff[0.16] differs from the true maximum value ~(216.4).
This conclusion was confirmed by values of E,,[AA] which were calculated from a theoretical equation proposed recently by Ghzer , Quack, and Troe [39]. In particular, Quack has computed values of E,~~[AA] at 300 K as- suming that the true maximum extinction coefficient at infinite resolution ceff[O] = 1.12 X lo4 l./mol cm [40]. Interpolation gives negative percentage errors of 1.6% at 0.06 nm and of 6% at 0.16 nm, supporting our conclusion that there is no significant difference between corresponding pairs of results of series 1 and 2. Measurements of the main series were conducted with
TABLE 11. Variable hand pass experiments and the evaluation of c,ff[AX] = A,,,/C,,, 1 for the methyl radical.
series slit pulse kinrcic measurements extinction coefficient
nvmber width voltage 10-6k2/seff[AA] no. of loe4 ceff[Ah] no. of
(vm) (nm) (V) (cm sec-3 expts. (amole-' cm-') expts.
AA
le 75 0.06 140 3.45 '. 0 . 4 3 11 1.05 6
2' - -200 0.16 60 3.54 t 0.37 a 0 . 9 9 3
3 200 0.16 140 3.83 ? 0.15 15 1.08 8
mean value from the three sets: 3.64 t 0.35 34
a Series 1 and 2 have a similar signal-to-noise ratio.

METHYL AND METHYLPEROXY RADICAL INTERACTIONS 965
Ax = 0.06 nm, and the corresponding error of 1.6% in the extinction coef- ficient would be insignificant in itself, and in its influence upon the derived estimates of k2.
Hochanadel and co-workers used a band pass of 0.6 nm and claimed that they observed no significant deviation from the Beer-Lambert law [13]. However, this band pass is large enough to overlap parts of both the diffuse R (215.76 nm) and P + Q (216.36 nm) branches, and their mean value ceff[0.60] = (9.0 f 0.8) X lo3 l./mol cm is correspondingly (but not signifi- cantly) lower than our mean values quoted above.
Table I1 also includes the results of the experiments of series 3, for which the signal-to-noise ratio was greater than for series 1 and 2. The precision of measurement shows a corresponding increase. The results of all three series are in agreement with each other and with the results of the main series of this investigation, which are k2/~(216.4) = (3.31 f 0.46) X lo6 cm/sec and 4216.4) = (9.5 f 0.4) X lo3 l./mol cm.
Variable path length experiments. One-half of the illuminated length of the cell was masked in all experiments. A single mask was used in pat- tern (i); experiments were conducted in pairs, with the cell covered from its center to either end, to allow for asymmetry of illumination. This mask was successively cut into three and 15 equal segments and placed sym- metrically along the cell in patterns (ii) and (iii), respectively. A band pass of 0.06 cm centered on 216.36 nm was used throughout this study.
We have assumed that the absorbance A of the methyl radical may be related to its concentration C by the usual empirical equation A = E ~ ~ ~ ( C Z ) ~ . When one-half of the length of the reaction cell is masked, the corre- sponding absorbance A112 is given by the equation A112 = t,,p(CZ/2)n only if the illumination of the reaction vessel is uniform in the absence of the mask. Here C is defined as the ratio of the number of moles of the methyl radical to the volume of the vessel, as the actual concentration varies along its length. Deviations from the uniformity of illumination are likely to have a greater effect upon the magnitude of A112 for pattern (i) than for the other patterns. Let (A:/2, C + , n + ) and (AT12, C - , n - ) be the sets of values ob- tained when the mask is placed on the anode and cathode sides of the photoflash tube, respectively. Then n+ and n- may be evaluated by an iterative method from the equations
No significant difference was found between the estimates of n+ and n- for each period of reaction, showing that the effect of asymmetry is negli- gible. Accordingly, the simpler equation n = log(Al,z/A)/log 2 was used for masking patterns (ii) and (iii).
Figure 5 shows that the apparent value of n may be depressed below unity under certhin reaction conditions. Nevertheless, we shall show below that this pattern of values is consistent with a true value of unity for n.

966 ADACHI, BASCO, AND JAMES
Figure 5. Variable path length method for the evaluation of the exponent in the equation. A = C ~ ~ , , ( C ~ ) ~ ; variation of estimates of n with the time of reaction and the degree of subdivision of the original mask.
Two main characteristics emerge. First, each curve may be extrapolated to a value of n very close to unity for a zero delay time; the direct mea- surement a t this time is impossible. Second, each pattern of masking has its own curve, and the more the masking cylinder is subdivided, the greater is the deviation of n from unity. This deviation is due to the penumbra effect, which caused partial illumination of the zones of the cell that would have been completely dark if the shadows of the masks had been sharply defined, and partial transfer of the generation of methyl radicals to these zones, where their rate of consumption is comparatively slow. Conse- quently, the excess of the average concentration in the partially masked cell over the theoretical value for a sharply defined set of shadows became progressively greater as the reaction time increased. The magnitude of this excess increased with the subdivision of the masking cylinder and the consequent increase in the number of boundaries between the zones. The mathematical analysis is based upon two equations of the type A:/,/A = [C+/(C+ + C-)] R+, with no correction for the penumbra effect which raises the value of C+ in the numerator progressively above the values of C+ and C- in the denominator. The mathematical procedure compensates for this by reducing the apparent value of n below unity progressively as the delay time or the subdivision of the mask is increased. Extrapolation to zero delay time yields a true value of n which does not differ significantly from unity. A value of unity was also claimed by Hochanadel and co-workers [13] and van den Berg and Callear [4] on the basis of variable path length experiments, using a band pass of 0.6 nm and an unspecified value, re- spectively.
We conclude that the use of the Beer-Lambert law to derive the con- centration of the methyl radical from its absorbance under normal condi- tions in the present apparatus is valid.
Evaluation of €(A) and kz for the Methyl Radical General principles. Experiments of the main series 4 and 5 were con-
ducted under conditions chosen to increase the precision of measurement. These included a low band pass of 0.06 nm, a high spectroflash energy to

METHYL AND METHYLPEROXY RADICAL INTERACTIONS 967
increase the signal-to-noise ratio, and a low photoflash energy of 360 J. Series 4 comprises 24 pairs of measurements of ~(216.4) and kd~(216 .4 ) and nine extra measurements of ~(216.4) alone, and is designed to furnish an accurate value of k2. Series 5 comprises 15 measurements of k2/~(A) for a range of wavelength from 215.2 to 217.0 and is intended to characterize the absorption spectrum in absolute units with the aid of the mean value
The experiments of series 4 yield the following of kz.
Evaluation of k2. values:
~(216.4) = (9.5 f 0.4) X lo3 l./mol cm from 33 experiments
k2/~(216.4) = (3.3 f 0.5) X lo6 cm/sec from 24 experiments
The value of the extinction coefficient is in good agreement with the results reviewed by Callear and Metcalfe, which range from 9.6 X lo3 to 1.02 X lo4 l./mol cm 15). The product of each pair of values of kJt(216.4) and ~(216.4) yielded a value of k2; statistical analysis of the resultant set yielded, with one exclusion, the mean value
k2 = (3.2 f 0.4) X 1O1O l./mol sec from 23 experiments
which also agrees with the product of the mean values quoted above. Hochanadel and co-workers 1131 have reported ~ ( 2 1 6 ) = (9.0 f 0.8) X lo3 l./mol cm and k2 = (3.1 f 0.6) X 1O’O l./mol sec, in close agreement with these values. Our value of the rate constant falls into the middle of the “low” values in the review [5], supporting them against the “high” value of Bass and Laufer [41]. Our value also agrees well with the result of (3.37 f 0.46) X 1O’O l./mol sec reported by James and Simons [7]. This agree- ment is the more remarkable because those authors evaluated their result using the empirical equation A = E ~ ~ ~ ( C ~ ) , O . ~ ~ with AA = 0.20 nm at 216.4 nm, whereas both theory [40] and experiment indicate a negligible deviation of the exponent from unity.
T h e absorption spectrum of the methyl radical. The reciprocals of the 15,values of K ~ / E ( X ) from the experiments of series 5 were plotted against wavelength in Figure 6 to yield an absorption spectrum in arbitrary units. The absolute spectrum was derived by scaling the right-hand ordinate by the factor ( k 2 ) = 3.2 X 1O1O l./mol sec. For comparison, the relative spectrum obtained by the photographic technique is included in Figure 6 with arbitrary units. Both spectra show resolved maxima at wavelengths close to the values of 215.76 and 216.36 nm found by Herzberg and Shoos- mith [l].
We may conclude that the value of k2 and the characterization of the absorption spectrum derived from the present work are in full agreement with the most precise literature values.
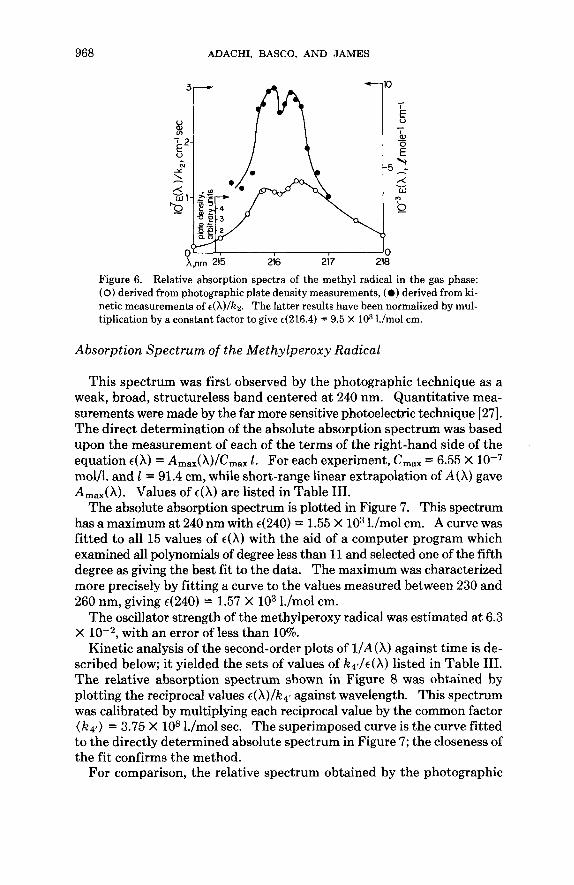
968 ADACHI, BASCO, AND JAMES
Figure 6. Relative absorption spectra of the methyl radical in the gas phase: (0) derived from photographic plate density measurements, (0 ) derived from ki- netic measurements of t(X)/kz. The latter results have been normalized by mul- tiplication by a constant factor to give 4216.4) = 9.5 X lo3 I./mol cm.
Absorption Spectrum of the Methylperoxy Radical
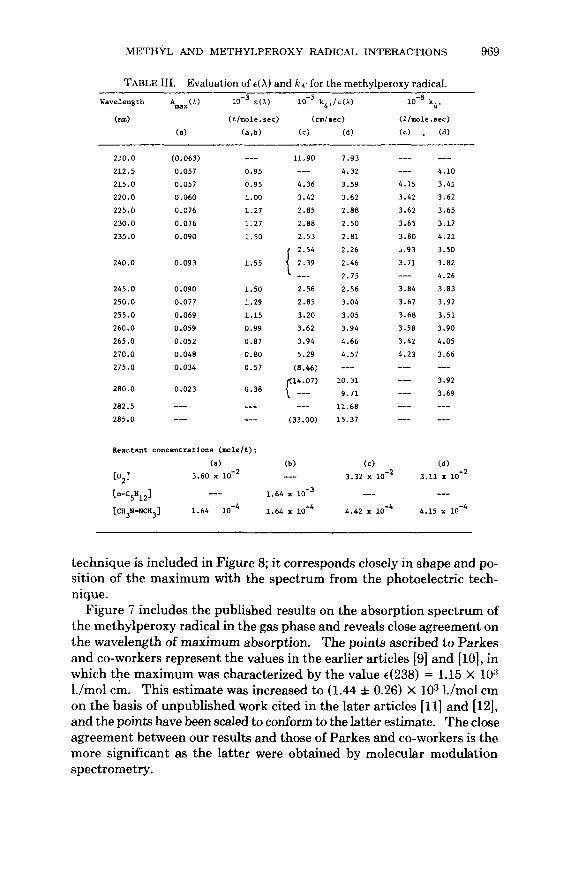
This spectrum was first observed by the photographic technique as a weak, broad, structureless band centered at 240 nm. Quantitative mea- surements were made by the far more sensitive photoelectric technique [27]. The direct determination of the absolute absorption spectrum was based upon the measurement of each of the terms of the right-hand side of the equation €(A) = Amax(A)/Cmax 1 . For each experiment, C,, = 6.55 X molfl. and I = 91.4 cm, while short-range linear extrapolation of A(X) gave Amax(A). Values of €(A) are listed in Table 111.
The absolute absorption spectrum is plotted in Figure 7. This spectrum has a maximum at 240 nm with 4240) = 1.55 X lo3 l./mol cm. A curve was fitted to all 15 values of €(A) with the aid of a computer program which examined all polynomials of degree less than 11 and selected one of the fifth degree as giving the best fit to the data. The maximum was characterized more precisely by fitting a curve to the values measured between 230 and 260 nm, giving ~(240) = 1.57 X lo3 l./mol cm.
The oscillator strength of the methylperoxy radical was estimated at 6.3 X lov2, with an error of less than 10%.
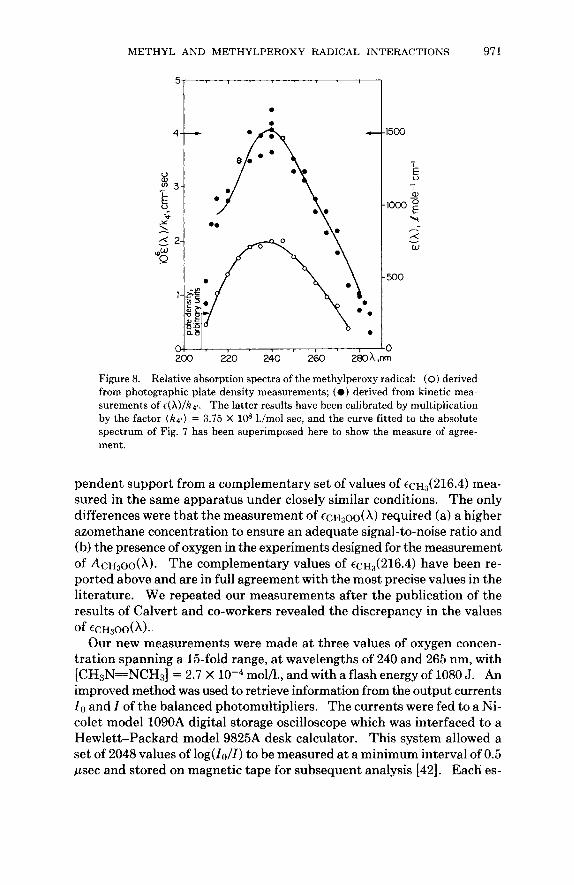
Kinetic analysis of the second-order plots of l/A(A) against time is de- scribed below; it yielded the sets of values of IZ4r/t(A) listed in Table 111. The relative absorption spectrum shown in Figure 8 was obtained by plotting the reciprocal values E ( A)/k4r against wavelength. This spectrum was calibrated by multiplying each reciprocal value by the common factor ( k 4 ~ ) = 3.75 X lo8 l./mol sec. The superimposed curve is the curve fitted to the directly determined absolute spectrum in Figure 7; the closeness of the fit confirms the method.
For comparison, the relative spectrum obtained by the photographic
METHYL AND METHYLPEROXY RADICAL INTERACTIONS 969
TABLE 111. Evaluation of c(X) and kq, for the methylperoxy radical. Uaveleng th AWx ( A ) ~(i) k4,/E(A) lo-’ k4,
(nm) (klmole.sec) (cmlsec) (I/mole.sec)
(6) (a.b) (C) (d) (c) . (d)
210.0 (0.063) --- 212.5 0.057 0.95 215.0 0.057 0.95 220.0 0.060 1.00 225.0 0.076 1.27 230.0 0.076 1.27 235.0 0.090 1.50
240.0 0.093 1.55
245.0 0.090 1.50 250.0 0.077 1.29 255.0 0.069 1.15 260.0 0.059 0.99 265.0 0.052 0.87 270.0 0.048 0.80 275.0 0.034 0.57
280.0 0.023 0.38
282.5 --- --- 285.0 --- ---
Reactant concentrations (mole/a):
lo, 1 3.60 x lo-’ (a)
11.90 __- 4.36 3.42 2.85 2.88 2.53 2.54 { 2.39 2.56 2.85
3.20 3.62
3.94 5.29
(8 .46)
-__
c Y7) _--
(33.00)
(b) ---
--- 7.93
4.32 3.59 4.15 3.62 3.42 2.88 3.62
2.50 3.65 2.81 3.80 2.26 i.93 2.46 3.71
2.75 2.56 3.84 3.04 3.67 3.05 3.68 3.94 3.58 4.66 3.42 4.57 4.23
---
___
--- --- 10.31 ---
___ 9.71 11.68 --- 15.37 ---
--- 4.10 3.41 3.62 3.65 3.17 4.21 3.50 3.82 4.26 3.83 3.92 3.51 3.90 4.05 3.66 _-_ 3.92 3.69 -- ---
(a) 3.32 x 3.11 x lo-’
--- --- 4.42 x 4.15 x l f 4
technique is included in Figure 8; it corresponds closely in shape and po- sition of the maximum with the spectrum from the photoelectric tech- nique.
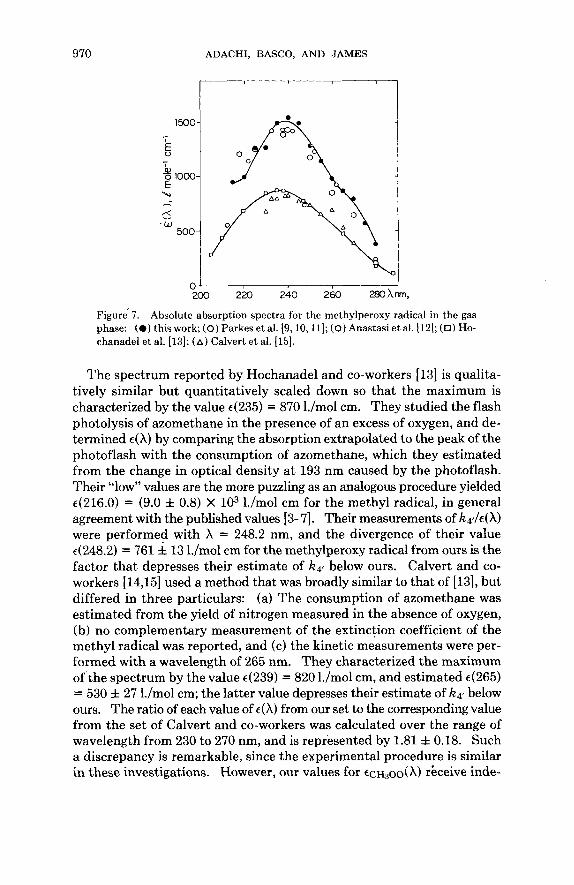
Figure 7 includes the published results on the absorption spectrum of the methylperoxy radical in the gas phase and reveals close agreement on the wavelength of maximum absorption. The points ascribed to Parkes and co-workers represent the values in the earlier articles [9] and [lo], in which the maximum was characterized by the value ~(238) = 1.15 X 103 l./mol cm. This estimate was increased to (1.44 f 0.26) X lo3 l./mol cm on the basis of unpublished work cited in the later articles [ll] and [12], and the points have been scaled to conform to the latter estimate. The close agreement between our results and those of Parkes and co-workers is the more significant as the latter were obtained by molecular modulation spectrometry.
970
1500- - I
5 & 1000-
- E
x a,
' W 500 -
ADACHI, BASCO, AND JAMES
I' P*
0 0 0 A 01 200 220 240 260 28oxm,
Figure' 7. Absolute absorption spectra for the methylperoxy radical in the gas phase: (0 ) this work; (0) Parkes et al. [9,10,11]; (0) Anastasi et al. [12]; (n) Ho- chanadel et al. [13]; ( A ) Calvert et al. [15].
The spectrum reported by Hochanadel and co-workers [13] is qualita- tively similar but quantitatively scaled down so that the maximum is characterized by the value 4235) = 870 l./mol cm. They studied the flash photolysis of azomethane in the presence of an excess of oxygen, and de- termined e(X) by comparing the absorption extrapolated to the peak of the photoflash with the consumption of azomethane, which they estimated from the change in optical density a t 193 nm caused by the photoflash. Their low^' values are the more puzzling as an analogous procedure yielded e(216.0) = (9.0 f 0.8) X lo3 l./mol cm for the methyl radical, in general agreement with the published values [3-71. Their measurements of k 4 4 4 X ) were performed with X = 248.2 nm, and the divergence of their value ~(248.2) = 761 i 13 l./mol cm for the methylperoxy radical from ours is the factor that depresses their estimate of kql below ours. Calvert and co- workers [14,15] used a method that was broadly similar to that of [13], but differed in three particulars: (a) The consumption of azomethane was estimated from the yield of nitrogen measured in the absence of oxygen, (b) no complementary measurement of the extinction coefficient of the methyl radical was reported, and (c) the kinetic measurements were per- formed with a wavelength of 265 nm. They characterized the maximum of the spectrum by the value ~(239) = 820 l./mol cm, and estimated 4265) = 530 f 27 l./mol cm; the latter value depresses their estimate of k41 below ours. The ratio of each value of 4) from our set to the corresponding value from the set of Calvert and co-workers was calculated over the range of wavelength from 230 to 270 nm, and is represented by 1.81 f 0.18. Such a discrepancy is remarkable, since the experimental procedure is similar in these investigations. However, our values for CCH~OO(X) rkceive inde-
METHYL AND METHYLPEROXY RADICAL INTERACTTONS 97 1
5
Figure 8. Relative absorption spectra of the methylperoxy radical: (0) derived from photographic plate density measurements; ( 0 ) derived from kinetic mea- surements of c(X)/hl , . The latter results have been calibrated by multiplication by the factor ( k q ) = 3.75 X lo8 l./mol sec, and the curve fitted to the absolute spectrum of Fig. 7 has been superimposed here to show the measure of agree- ment.
pendent support from a complementary set of values of ~ ~ ~ ~ ( 2 1 6 . 4 ) mea- sured in the same apparatus under closely similar conditions. The only differences were that the measurement of E C H ~ O O ( X ) required (a) a higher azomethane concentration to ensure an adequate signal-to-noise ratio and (b) the presence of oxygen in the experiments designed for the measurement of AcH~oo(X). The complementary values of ~ ~ ~ ~ ( 2 1 6 . 4 ) have been re- ported above and are in full agreement with the most precise values in the literature. We repeated our measurements after the publication of the results of Calvert and co-workers revealed the discrepancy in the values
Our new measurements were made at three values of oxygen concen- tration spanning a 15-fold range, a t wavelengths of 240 and 265 nm, with [CHsN=NCHs] = 2.7 X molh., and with a flash energy of 1080 J. An improved method was used to retrieve information from the output currents I0 and I of the balanced photomultipliers. The currents were fed to a Ni- colet model 1090A digital storage oscilloscope which was interfaced to a Hewlett-Packard model 9825A desk calculator. This system allowed a set of 2048 values of log(Io/I) to be measured at a minimum interval of 0.5 gsec and stored on magnetic tape for subsequent analysis [42]. Each es-
of ECH300(X).

972 ADACHI, BASCO, AND JAMES
timate quoted below was based upon at least five experiments. The measurements a t 240 nm were made with values of the oxygen concentra- tion of 0.036, 0.0071, and 0.0022 mol/l. The corresponding estimates of 1 0 - 3 c c ~ 3 ~ ~ ( 2 4 0 ) were 1.46, 1.47, and 1.44 l./mol cm, respectively, and do not differ significantly from our previous value 10-3~c~300(240) = 1.55 l./mol cm measured with [O,] = 0.036 mol/l. The corresponding values of 10-%4( were 3.3,3.8, and 4.4 l./mol sec, and they do not differ significantly from our previous values 10-8k4j = 3.76 f 0.27 l./mol sec measured at [O,] = 0.032 f 0.001 mol/l. and 10-%4. = 4.67 f 0.65 l./mol sec measured at [O,] = 0.00115 f 0.0005 mol/l., which are discussed in a later section. The measurements a t 265 nm with [Oz] = 0.036 mol/l. yielded the estimates 10-36c~300(265) = 0.87 l./mol cm and 1O-'k4. = 4.9 l./mol sec; the former is in full agreement with our previous value 10-3~~~300(265) = 0.87 l./mol cm measured at [ 0 2 ] = 0.036 mol/l. The accuracy of the new measurements was confirmed by corroborative values of ~ ~ ~ ~ ( 2 1 6 . 4 ) from complementary experiments. Accordingly, the new experiments have confirmed the principal values of €(A), k 4 r / ~ ( X ) , and k 4 ~ reported for the methylperoxy radical in this investigation, and the disparity between these values and those of [ 13-15] remains unresolved.
The absorption spectrum of the methylperoxy radical in aqueous solution is similarly broad and structureless, with a maximum at 250 nm and ~(250) = 1.14 X lo3 l./mol cm [43]. The shift of 10 nm in the position of the maximum has been ascribed tentatively to complex formation between the methylperoxy radical and the water molecule by hydrogen bonding to the nonterminal oxygen atom of the radical [14].
Values of k4t/c(X)
Each value of k4j/~(X) a t a given wavelength X was calculated from the gradient of the corresponding plot of l/A(X) against time Statistical analysis of such plots yielded correlation coefficients ranging from 0.990 to 0.999, confirming the validity of the procedure. The principal set of values was measured with [ 0 2 ] = (3.22 f 0.10) X mol/l. at wavelengths varying from 210 to 285 nm, and is listed in Table 111. A minimum was observed at 240 nm, and the mean value k4!/~(240) = (2.48 f 0.18) X lo5 cm/sec is calculated from five measurements. A second set of values was measured with [ 0 2 ] = (1.15 f 0.05) X mol/l. at 240 nm only; the mean value k4,/e(240) = (3.01 f 0.42) X lo5 cm/sec is calculated from 17 mea- surements. The two sets are in reasonable agreement, particularly as we shall show below that k4' increases slightly as [O,] is decreased. Both sets of measurements were made at room temperature, with [CH3N=NCH31 = (4.28 f 0.14) X
Hochanadel and co-workers [13] made similar measurements at 248 nm with [O,] = 1.4 x mole/l. and [CH3N=NCH3] = 6.3 X lov5 molil., and
mol/l. and a photoflash energy of 1080 J.

METHYL AND METHYLPEROXY RADICAL INTERACTIONS 973
found that k4(/4248) = 3.0 X lo5 cm/sec, in good agreement with our mean value k4t/€(250) = 2.9 X lo5 cm/sec. The concordant estimate k4~/~(245) = 3.2 X 105 cm/sec is implied by the results of Kan and Calvert [14]. An- astasi and co-workers [12] employed the concentration [OZ] = 8.8 X mol/l., [CH3N=NCH3] = 5.0 X mol/l. and estimated that k4’/~(240) = (1.84 f 0.23) X lo5 cm/sec, which is three-quarters of our principal value. The divergence of their estimate of k4‘ from ours is due to this factor.
mol/l., and [Nz] = 1.2 X
Evaluation of k4‘
Complementary values of k 4 ’ / ~ ( A) and E ( A) were measured at 14 wave- lengths from 212.5 to 280 nm at the higher oxygen concentration. The 30 corresponding values of their product k~ are listed with them in Table 111. Statistical analysis gave the coefficient of correlation between kq‘ and X as r2 = 0.11, showing that an estimate Of k4’ is independent of the wavelength used for its measurement. The complete set of 30 values of k4’ may be represented by the mean and standard deviation
k q ~ = (3.76 f 0.27) X lo8 l./mol sec
The set of five values measured at 240 nm may be represented similarly:
k4/ = (3.84 f 0.28) X los l./mol sec
There is no significant difference between the respective values of either the mean or the standard deviation, confirming independence of the wavelength of measurement.
The 17 values of k4‘/~(240) were measured at the lower oxygen concen- tration of (1.15 f 0.05) X mol/l, and are represented by k4//6(240) = (3.01 f 0.42) X lo5 cm/sec, corresponding to k4’ = (4.67 f 0.65) X 108 l./mol sec, as ~(240) = 1.55 X lo5 I./mol cm.
The method of molecular modulation spectrometry has yielded the value k41 = (3.3 f 0.6) X lo8 l./mol sec [ll], in good agreement with our principal value of (3.8 f 0.3) X lo8 l./mol sec. Using the technique of flash photolysis, Anastasi and co-workers [12], Hochanadel and co-workers [13] and Kan and Calvert [14] reported respective values for k4r of (2.65 f 0.6) X lo8, (2.3 f 0.4) X 108, and 2.5 X lo8 l./mol sec, but the agreement is superficial. These values are lower than the two quoted above because “low” values of the respective factors k4’/~(240) = (1.84 f 0.23) X lo5 cm/sec, 4248.2) = 761 f 13 l./mol cm, and ~(245) = 770 l./mol cm are multiplied by “high” values of the corresponding conjugate factors to give the estimates of k 4‘.

974 ADACHI, BASCO, A N D JAMES
Value of k4 for Mutual Interaction of Methylperoxy radicals
The absolute rate constant k4 was evaluated with the aid of the computer program described in a previous section, using its capacity to calculate k4rlk4. For each experiment a succession of trial values of k4 is entered in the input until the output corresponds to the measured value of k4'. The input also includes the fixed values of other participating rate constants given in Table I, the initial concentration CO = 6.65 X mol/l., the re- action time of 4 msec, and the appropriate value of the oxygen concentration from Table IV. Two values of 0.50 and 0.33 were selected for k4Jk4 to correspond to the literature values of 0.48 [18] and 0.34 [lo]. Table IV gives the respective pairs of values k4t and [O,] required for the input and lists the sets of four possible values of k4f lk4 and k4 from the output. The preferred value of k4 was obtained from the four mean values of the output by weighting each in proportion to the reciprocal of its variance. The result is
k4 = (3.5 f 0.3) X lo* l./mol sec
The method of molecular modulation spectrometry has yielded a cor- responding set of values: k4' = (3.3 f 0.6) X los l./mol sec, k4Jk4 = 0.34, k41/k4 = 1.18, and k4 = (2.8 f 0.7) X lo8 l./mol sec [Ill, which is in general agreement with our results. We may conclude that our present knowledge of k4 is best represented by the value
k4 = 10(8.5*0.1) l./mol sec
TABLE IV. Evaluation of k4 for mutual interaction of methylperoxy radicals.
1 0 ~ ~ 0 ~ 1 (mole ~ - 3 ~ 32.2 f 1.0 1.15 t 0.05
4.67 t 0.65 10-'k4, (E.mole-lsec-') 3.76 f 0.27
number of values 30 17
k4a/k4 (postulated) 0.50 0.33 0.50 0.33
k4, /k4 (computed) 1.126 1.092 1.143 1.125
10-8kI, (*mole-'sec-') 3.34 f 0.24 3.44 2 0.25 4.09 2 0.58 4.21 t 0.58
Preferred value: k4 - (3.5 f 0.3) x 10' e mole-' sec-l.
a Oxygen concentration used for the measurement of k4, /c (X) .

METHYL AND METHYLPEROXY RADICAL INTERACTIONS 975
Analytical Evaluation of k4 from the Stationary State Approximation
Analytical expressions for k4rlk4 were derived and evaluated at oxygen concentrations of 3.23 X and 1.0 X mol/l., respectively and compared with the output of the computer program. The rate constants were given the values listed in Table I, with k14 = 2.0 X lo5 l./mol sec
At the higher oxygen concentration and the representative reaction time of 2000 Fsec, the computed value of 29.1 for the ratio [HOO.]/[CH30-] in- dicates that the only significant reaction of the methoxy radical is its oxi- dation to the hydroperoxy radical; in fact, the weighted sum R6 + R7 + 2R11 of the rates of the competing reactions (6), (7), and (11) amounts to only 0.064 d [CH300.]/dt, and accordingly the latter reactions were treated as negligible. The stationary state approximation is justified by the low values of the ratios ( d [ C H 3 0 ] d t ) / ( d [ C H 3 0 O - ] / d t ) = 0.0982 and (d[HOO.]/dt)/ (d[CH300.]/dt) = 0.0217 and yields the equations
l201.
[HOO.]/[CH300] = [(k:, + 16k4ak12)”2 - kio]/4kl2
k4,/k4 = 1/(1 - (k10/2k4’)[HOO.]/[CH300.])
and the values of k4tlk4 calculated for k4Jk4 = 0.50 and 0.33 exceed the corresponding computed values by less than 1%.
At the lower oxygen concentration and the reaction time of 2000 psec, the computed value of the weighted sum Rlo + 2Rll + Rl2 of the rates of reactions (lo), (ll), and (12) of the hydroperoxy radical amounts to only 0.161 d [CH300]/dt, and, accordingly, the latter reactions were treated as negligible. The stationary state approximation is justified by the low values of the ratios ( d [ C H s O ] / d t ) / ( d [ C H 3 0 0 . ] d t ) = 0.0737 and (d[HOO-/dt)/ (d[CH300.]/dt) = -0.0580 and yields the equations
[CH30]/[CH300] = [(kz + 16k4ak7)1’2 - k6]/4k7
k41lk4 = 1/(1 - (k6/2k4() [CHRO.]/[CH~OO-])
an$ the values of 124‘1124 calculated for k4a/k4 = 0.50 and 0.33 are less than 2% lower than the corresponding computed values.
An alternative mechanism for the interaction of methylperoxy radicals has been proposed very recently by Nangia and Benson [44]. The relevant reactions are
(21) (22) (23) (24)
2CH300 - CH:<OOH + H2COO H2COO + CH3OO. - HCHO + CH30. + 0 2
CH30 + CH300 + CH30H + HACOO CH30 + CHL300. + CH3000CH3
On this basis, k4‘ = 2k21(1 + k23/k24) and consequently k21= k4d4 = 0.94 X lo8 and 1.17 X los l./mol sec when [ 0 2 ] = 0.0322 and 0.0011 mol/l., re- spectively. However, it should be noted that these authors did not include

976 ADACHI, BASCO, AND JAMES
the reaction of the methoxy radical with oxygen in their overall mecha- nism.
Conclusions
Our value for the rate constant for the mutual combination of methyl radicals, k z = (3.2 f 0.4) X 1O1O l./mol sec, has been measured under con- ditions for which the application of the Beer-Lambert Law is valid, and is in full agreement with the most precise literature values listed in the reviews [5 ] and [39].
Good agreement exists between our results on the methylperoxy radical and those of Parkes [ll] in relation to our principal themes:
(i) The quantitative characterization of the absorption spectrum of the methylperoxy radical in the gas phase
(ii) The evaluation of the rate constant for the mutual interaction of methylperoxy radicals in the gas phase at room temperature; the value loglok4(l./mol sec) = 8.5 f 0.1 is consistent with the results of both inves- tigations.
Agreement with the results of three independent studies by flash pho- tolysis is good in relation to the values of kqj/t(X) [13,14] and to €(A) [12], but otherwise it is quantitatively only moderate.
Acknowledgment
This work was supported by the National Research Council of Canada.
Bibliography
[l] G. Herzberg and J. Shoosmith, Can. J . Phys., 34,523 (1956). [2] G. Herzberg, Proc. R. SOC. London Ser. A, 262,291 (1961). [3] N. Basco, D. G. L. James, and R. D. Suart, Znt. J . Chem. Kinet., 2,215 (1970). [4] H. E. van den Berg and A. B. Callear, Trans. Faraday Sac., 67,2017 (1971). [5] A. B. Callear and M. P. Metcalfe, Chem. Phys., 14,275 (1976). [6] D. A. Parkes, D. M. Paul, and C. P. Quinn, J. Chem. SOC. FQradQy Trans. 1, 72,1935
[7] F. C. James and J . P. Simons, Int. J . Chem. Kinet., 6,887 (1974). [8] F. C. James, J. A. Kerr, and J. P. Simmons, J. Chem. SOC. Faraday Trans. 1 69,2124
[9] D. A. Parkes, D. M. Paul, C. P. Quinn, and R. C. Robson, Chem. Phys. Lett., 23,425
[lo] D. A. Parkes, in “Proceedings of the 15th Symposium on Combustion” (Tokyo, 19741,
[ l l ] D. A. Parkes, Int. J . Chem. Kinet., 9,451 (1977). [12] C. Anastasi, I. W. M. Smith, and D. A. Parkes, J. Chem. SOC. Faraday Trans. 1, 74,1693
[13] C. J. Hochanadel, J . A. Ghormley, J . W. Boyle, and P. J. Ogren, J. Phys. Chem., 81,3
(1976).
(1973).
(1973).
Combustion Institute, Pittsburgh, PA, 1975, p. 795.
(1 978).
(1977).

METHYL AND METHYLPEROXY RADICAL INTERACTIONS 977
[14] C. S. Kan and J. G. Calvert, Chem. Phys. Lett., 63,111 (1979). [15] C. S. Kan, R. D. McQuigg, M. R. Whitbeck, and J . G. Calvert, Znt. J . Chem. Kinet., 11,
(161 R. A. Cox and G. S. Tyndall, Chem. Phys. Lett., 65,357 (1979). (171 J. Weaver, R. Shortridge, J. Meagher, and J. Heicklen, J . Photochem., 4,109 (1975). [18] W. G. Alcock and B. Mile, Combust. Flame, 24,125 (1975). 1191 J. R. Barker, S. W. Benson, and D. M. Golden, Int. J . Chem. Kinet., 9,31 (1977). [20] (a) N. Basco, D. G. L. James, and F. C. James, Int. J . Chem. Kinet., 4,129 (1972); (b)
0. Klais, P. C. Anderson, A. H. Laufer, and M. J. Kurylo, Chem. Phys. Lett., 66, 598 (1979).
[21] D. M. Golden, G. N. Sokes, and S. W. Benson, Angew. Chem. (Int. Ed. Engl.), 12,540 (1973).
[22] R. W. Walker, “Reaction Kinetics,” vol. 2, Spec. Periodical Rep., Chemical Society London, 1977, chap. 7, p. 324.
[23] N. Washida and K. D. Bayes, Int. J . Chem. Kinet., 8,777 (1976). [24] N. Washida, in “Proceedings of the Yamada Conference I11 on Free Radicals” (14th Int.
[25] A. C. Baldwin and D. M. Golden, Chem. Phys. Lett., 55,350 (1978). [26] E. Sanhueza, R. Simonaitis, and J. Heicklen, Znt. J Chem. Kinet., 11,907 (1979). [27] H. Adachi, N. Basco, and D. G. L. James, Int. J . Chem. Kinet., 11,1211 (1979). [28] H. Adachi, N. Basco, and D. G. L. James, Int. J . Chem. Kinet., 11,995 (1979). [29] R. E. Berkeley, J . Safarik, 0. P. Strauss, and H. E. Gunning, J. Phys. Chem., 77,1741
(1973). [30] K. Selby and D. J. Waddington, “Proceedings of the 5th International Symposium on
Gas Kinetics,” Manchester, 1977, p. 61. (311 H. Radford, J. Burrows, and W. Brune, in “Proceedings of the Yamada Conference I11
on Free Radicals” (14th Int. Symp.), Sanda, Hyogo-ken, Japan, Sept. 1979, p. 123. [32] A. C. Lloyd, Int. J . Chem. Kinet., 6,169 (1974). [33] E. J. Hamilton, Jr., and R.-R. Lii, Int. J . Chem. Kinet., 9,875 (1977). [34] D. W. Trainor and C. W. von Rosenberg, Jr., in “Proceedings of the 15th Symposium
[35] NASA Ref. Publ. 1010, Aug. 1977, chap. 1. [36] A. C. Hindmarsh, UCID-30059-1, Lawrence Livermore Laboratory, University of Cali-
fornia/Livermore, 1975. [37] C. W. Gear, “Numerical Initial Value Problems in Ordinary Differential Equations,”
Prentice-Hall, Englewood Cliffs, N J , 1971. [38] N. Basco, A. B. Callear, and R. G. W. Norrish, Proc. R. Soc. London, Ser. A. 260,459
(1961). 1391 K. Glanzer, N. Quack, and J. Troe, in “Proceedings of the 16th Symposium on Com-
bustion,” (M.I.T., Cambridge, MA 1976). Combustion Institute, Pittsburgh, PA, 1977, p. 949.
921 (1979).
Symp.), Sanda, Hyogo-ken, Japan, Sept. 1979, p. 173.
on Combustion” (Tokyo, 1974), Combustion Institute, Pittsburgh, PA 1975, p. 755.
[40] M. Quack, private communication. [41] A. M. Bass and A. H. Laufer, Int. J . Chem. Kinet., 5,1053 (1973). 1421 H. Adachi and N. Basco, Chem. Phys. Lett., 63,490 (1979). [43] B. Hickel, J. Phys. Chem., 79,1054 (1975). [44] P. S. Nangia and S. W. Benson, Int. J. Chem. Kinet., 12,43 (1980).
Received December 27,1979 Accepted May 2,1980







![Impacts of aerosols and clouds on photolysis frequencies and ... of aerosols and cloud… · [2] Photolysis reactions play a very important role in atmospheric chemistry. Ozone photolysis](https://img.pdfslide.net/doc/110x75/5f07e35b7e708231d41f41d6/impacts-of-aerosols-and-clouds-on-photolysis-frequencies-and-of-aerosols-and.jpg)










![SHOCK TUBE MEASUREMENTS OF ELEMENTARY OXIDATION … · iv 306 nm, respectively. CH radicals were generated by shock-heating highly dilute mixtures of ethane [C2H6], or methyl iodide](https://img.pdfslide.net/doc/110x75/5f0840837e708231d4211674/shock-tube-measurements-of-elementary-oxidation-iv-306-nm-respectively-ch-radicals.jpg)










