Embed Size (px)
Citation preview

Aus dem Institut für Mikrobiologie und Tierseuchen
der Tierärztlichen Hochschule Hannover
___________________________________________________________________
Untersuchungen zum intrazellulären Überleben vonMycobacterium avium ssp. paratuberculosis
in primären bovinen Makrophagen
INAUGURAL-DISSERTATION
Zur Erlangung des Grades einer
Doktorin der Veterinärmedizin
(Dr. med. vet.)
durch die Tierärztliche Hochschule Hannover
Vorgelegt von
Beatrice Schümann
aus Heidelberg
Hannover 2001

Wissenschaftliche Betreuung: Univ.-Prof. Dr. P. Valentin-Weigand
1. Gutachter: Univ.-Prof. Dr. P. Valentin-Weigand
2. Gutachter: Apl. Prof. Dieter Steinhagen
Tag der mündlichen Prüfung: 19.11.2001
Die Arbeit wurde gefördert durch ein Stipendium der Graduiertenförderung.

meinen Eltern


Inhaltsverzeichnis
A Einleitung........................................................................................................ 11
B Schrifttum ....................................................................................................... 12B.1 Die Paratuberkulose des Rindes.......................................................................12
B.1.1 Geschichte der Erkrankung ..................................................................12
B.1.2 Mycobacterium avium Subspezies paratuberculosis ............................12
B.1.3 Wirtsspektrum und Verbreitung ............................................................13
B.1.4 Pathogenese und Krankheitsbild..........................................................15
B.1.5 Immunologie der Erkrankung ...............................................................17
B.2 Makrophagen ....................................................................................................18
B.2.1 Makrophagenaktivierung und -deaktivierung........................................18
B.2.2 Allgemeine Probleme in der primären Makrophagenkultur...................20
B.3 Intrazelluläres Überleben von Bakterien der Gattung Mycobacterium in
Makrophagen ....................................................................................................21
B.4 Intrazelluläres Überleben von M. ptb in Makrophagen......................................23
C Material und Methoden .................................................................................. 26C.1 Material .............................................................................................................26
C.2 Methoden ..........................................................................................................26
C.2.1 Zellkultur...............................................................................................26
C.2.1.1 Mycobacterium avium Subspezies paratuberculosis .................26
C.2.1.2 Primäre bovine Makrophagen (PBMs) .......................................28
C.2.1.2.1 Gewinnung und Kultivierung................................................28
C.2.1.2.2 Bestimmung der Zellzahl .....................................................31
C.2.2 Infektionsversuche ...............................................................................32
C.2.2.1 Standardinfektionsversuch.........................................................32
C.2.2.2 Bestimmung der Infektionsraten ................................................32
C.2.2.3 Bestimmung der Überlebensraten von M. ptb in PBMs .............34

C.2.3 Mikroskopie ..........................................................................................34
C.2.3.1 Elektronenmikroskopie...............................................................34
C.2.3.2 Konfokale Laserscanning Mikroskopie.......................................35
C.2.4 FACS-Analyse......................................................................................37
C.2.5 Molekulargenetische Methoden............................................................39
C.2.5.1 RT-PCR .....................................................................................39
C.2.5.1.1 Gewinnung von RNA nach CHOMCZYNSKI u. SACCHI
(1987) ..................................................................................39
C.2.5.1.2 Bestimmung der RNA-Konzentration im Photometer ..........40
C.2.5.1.3 DNase-Behandlung von RNA..............................................41
C.2.5.1.4 Reverse Transkription .........................................................42
C.2.5.1.5 PCR.....................................................................................42
C.2.5.2 Agarosegel-Elektrophorese der PCR-Produkte .........................45
C.2.5.3 Quantifizierung der Genexpression............................................45
D Ergebnisse...................................................................................................... 47D.1 Eignung und Standardisierung des Teflon-Modells...........................................48
D.1.1 Makrophagenausbeute.........................................................................49
D.1.2 Infektion von PBMs aus der Teflonkultur mit M. ptb .............................49
D.1.3 Adhärenzraten nicht-infizierter sowie infizierter PBMs aus der
Teflonkultur...........................................................................................50
D.1.4 Stimulierbarkeit der PBMs aus der Teflonkultur ...................................52
D.1.5 Überleben von M. ptb in PBMs aus der Teflonkultur ............................53
D.2 Funktionelle Untersuchungen in PBMs aus der Teflonkultur.............................56
D.2.1 Elektronenmikroskopie .........................................................................56
D.2.2 Vitale M. ptb alkalisieren das endosomale Netzwerk............................57
D.2.3 Genexpression ausgewählter Gene bei M. ptb-Infektion......................61
D.2.3.1 Infektion mit vitalen bzw. hitzeinaktivierten M. ptb .....................61
D.2.3.2 Vergleich der Genexpression nach verschiedenen
Stimulationen .............................................................................64

D.2.4 Bafilomycin verhindert die Ansäuerung in der Infektion mit
hitzeinaktivierten M. ptb, verursacht jedoch keine entsprechenden
Genexpressionsmuster.........................................................................67
D.2.5 deaktivierende Effekte ..........................................................................70
E Diskussion...................................................................................................... 73E.1 Eignung und Standardisierung des Teflon-Modells...........................................73
E.2 Funktionelle Untersuchungen in PBMs aus der Teflonkultur.............................78
E.2.1 Vitale M. ptb alkalisieren das endosomale Netzwerk............................79
E.2.2 Modulation der Genexpression bei M. ptb-Infektion .............................81
E.2.2.1 Infektion mit vitalen bzw. hitzeinaktivierten M. ptb .....................81
E.2.2.2 Vergleich der Genexpression nach verschiedenen
Stimulationen .............................................................................85
E.2.3 deaktivierende Effekte ..........................................................................86
F Zusammenfassung ........................................................................................ 89
G Summary......................................................................................................... 91
H Literaturverzeichnis ....................................................................................... 93
I Anhang.......................................................................................................... 117I.1 Chemikalien und Verbrauchsmaterialien.........................................................117
I.2 weitere Reagenzien ........................................................................................120
I.3 Geräteverzeichnis ...........................................................................................121


Abkürzungsverzeichnis
� engl.: registered trademark (eingetragenes Warenzeichen)[v/v] engl.: volume per volume (Volumen pro Volumen)[w/v] engl.: weight per volume (Gewicht pro Volumen)ACD Acetat-Citrat-DextroseA. bidest. lat.: Aqua bidestillataA. dest. lat.: Aqua destillataAbb. AbbildungBCG Mycobacterium bovis Bacillus Calmette-Guérinbzw. beziehungsweiseca. zirkacDNA engl.: copy DNA (komplementäre DNA)cm2 QuadratzentimeterdATP 2‘-Desoxy-Adenosin-5‘-TriphosphatdCTP 2‘-Desoxy-Cytosin-5‘-TriphosphatdGTP 2‘-Desoxy-Guanosin-5‘-TriphosphatDNA engl.: desoxyribonucleic acid (Desoxyribonukleinsäure)dNTP 2‘-Desoxy-Nukleotid-5‘-TriphosphatEDTA Ethanoldiamintetraacetatet al. et aliiFCS engl.: fetal calf serum (fötales Kälberserum)FITC Fluorescein IsothiocyanatgRNA gesamte RNAGM-CSF engl.: granulocyte macrophage colony stimulating factor
(Granulozyten-Makrophagen Kolonie-Stimulations-Faktor)
IFN-� Interferon gammaiNOS induzierbare NO-SynthaseIL InterleukinKBE Kolonie-bildende EinheitenLAM LipoarabinomannanLPS Lipopolysaccharid

M Molarität (mol/l)M. avium Mycobacterium avium Subspezies aviummin Minute(n)mM millimolar (mmol/l)M. ptb Mycobacterium avium Subspezies paratuberculosismRNA engl.: messenger RNA (Boten-RNA)M. tbc Mycobacterium tuberculosisNO NitritoxidOD Optische DichteRG ReaktionsgefäßPBM primärer boviner MakrophagePBMs primäre bovine MakrophagenPBS engl.: phosphate buffered saline (phosphatgepufferte Saline)PCR engl.: polymerase chain reaction (Polymerase-Ketten-Reaktion)Pen/Strep Penicillin/StreptomycinRNA engl.: ribonucleic acid (Ribonukleinsäure)rRNA ribosomale RNART Reverse TranskriptionRTase Reverse Transkriptasesek Sekunde(n)SDS SodiumdodecylsulfatSSTE Sodium-SDS-Tris-EDTA-PufferTaq Thermus aquaticus
TNF-� Tumornekrosefaktor alphaTris Trihydroxymethylaminomethanu. undupm Umdrehungen pro MinuteUV UltraviolettV Voltz. B. zum BeispielZK Zellkultur

Einleitung
11
A Einleitung
Mycobacterium avium Subspezies paratuberculosis (M. ptb) verursacht eine
chronische, granulomatöse und nicht therapierbare Enteritis bei Wiederkäuern, die
Paratuberkulose. Die Tiere infizieren sich in der Regel in den ersten Lebensmonaten
und haben danach oft über Jahre keinerlei klinische Symptome. Mit Ausbruch der
Krankheit zeigen vor allem Rinder starken intermittierenden Durchfall. Allgemein ist
eine fortschreitende Abmagerung bei erhaltenem Appetit bis hin zur Kachexie zu
beobachten. Die Krankheit endet für gewöhnlich tödlich.
Die Pathogenese der Erkrankung ist bislang ungeklärt. Als fakultativ intrazellulärer
Erreger ist M. ptb in der Lage, in professionellen Phagozyten zu überleben und sich
zu vermehren. Es gilt als gesichert, daß M. ptb durch die M-Zellen im Epithel der
Peyer‘schen Platten die intestinale Mukosa überwindet und anschließend von
Gewebemakrophagen aufgenommen wird. Unbekannt sind jedoch die Strategie und
die Mechanismen, die M. ptb das intrazelluläre Überleben in diesen Zellen
ermöglichen.
Von anderen intrazellulär überlebenden Mykobakterien ist bekannt, daß sie durch
Modulation der Makrophagen deren Effektorfunktionen so beeinflussen, daß die
phagozytierten Erreger nicht abgetötet werden. Eine solche Modulation wird auch für
M. ptb angenommen. Daher sollten in diesem Projekt die Auswirkungen des
intrazellulären Überlebens von M. ptb auf die Genexpression primärer boviner
Makrophagen untersucht werden. Zu diesem Zweck wurde das Modell der
Teflonkultivierung primärer Makrophagen verwendet, welches das größte Problem,
die Inhomogenität in der Zellpopulation, minimiert. Ziel war es, eine Modulation der
Effektorfunktionen des Makrophagen durch den Erreger als Folge des Einwirkens
sezernierter oder struktureller Bestandteile, also als aktiv oder passiv, zu
diskriminieren.
Durch den auf diesem Gebiet bisher nicht eingesetzten experimentellen Ansatz sollte
ein Beitrag zur Aufklärung der Pathogenese der Paratuberkulose beim Rind geleistet
werden.

Schrifttum
12
B Schrifttum
B.1 Die Paratuberkulose des Rindes
B.1.1 Geschichte der Erkrankung
Die klinischen Symptome und das pathologisch-anatomische Erscheinungsbild einer
Infektion mit M. ptb bei einem Rind wurden zuerst im 19. Jahrhundert beschrieben.
Im Jahre 1895 stellten JOHNE und FROTHINGHAM erstmalig eine Verbindung
zwischen der Anwesenheit säurefester Mikroorganismen und chronischer Enteritis
bei Rindern fest. Elf Jahre später unterschied BANG (1906) diese Form der Enteritis
von der tuberkulösen und nannte sie pseudotuberkulöse Enteritis. Die Identifizierung
und Anzucht des Erregers gelang schließlich TWORT u. INGRAM (1912), und nach
eingehender Charakterisierung wurde der Erreger Mycobacterium enteritidis
chronicae pseudotuberculosis bovis johnei genannt. Seit dieser Zeit wurde er
mehrfach umbenannt. Aufgrund seiner hohen genetischen Verwandtschaft von 98 %
zu M. avium heißt er heute Mycobacterium avium Subspezies paratuberculosis
(THOREL et al. 1990).
B.1.2 Mycobacterium avium Subspezies paratuberculosis
Mycobacterium avium Subspezies paratuberculosis (M. ptb) ist ein Gram-positives,
unbewegliches, obligat aerobes Stäbchen mit einer Größe von 0,3-0,5 x 0,3-2 µm
(BISPING u. AMTSBERG 1988). Die Zellwand besteht aus zwei quervernetzten
Polymeren, dem Arabinogalactanmycolat-Komplex und Peptidoglycan. Zusätzlich
enthält sie einen großen Teil langkettiger Mykolsäuren, wodurch das Bakterium eine
außerordentliche Resistenz gegenüber Säuren und Laugen besitzt. Diese
Eigenschaft wird in der Ziehl-Neelsen-Färbung ausgenutzt, eine Methode zur
selektiven Färbung von säurefesten Bakterien. Weitere Möglichkeiten der
Darstellung sind Färbungen mit Fluoreszenzfarbstoffen wie Auramin O-Rhodamin
(TRUANT et al. 1962) und Phenol-Acridinorange (SMITHWICK et al. 1995).

Schrifttum
13
Im mikroskopischen Bild zeigen sich die Mykobakterien in nesterartigen
Gruppierungen, die vermutlich durch die Ausbildung interzellulärer Filamente
zustande kommen (MERKAL et al. 1973).
M. ptb besitzt aufgrund seiner Zellwandstruktur eine hohe Tenazität. Der Erreger
überlebt im Weidekot infizierter Tiere mehrere Monate (ROSENBERGER 1978) und
ist noch nach 270 Tagen aus stehenden Gewässern zu isolieren (LARSEN et al.
1956). Durch direkte Sonneneinstrahlung wird das Bakterium innerhalb von 100
Stunden abgetötet. Selbst aus Milch, die mit M. ptb inokuliert und anschließend
entweder pasteurisiert (63,5°C für 30 min) oder für 15 sek auf 71,7°C erhitzt wurde,
konnten in 85-96 % aller Fälle noch lebende Bakterien isoliert und kultiviert werden
(GRANT et al. 1996). In Milch aus England und Wales, die sich bereits im Verkauf
befand, war es sogar möglich, mittels PCR das M. ptb-spezifische Insertionselement
IS900 in 7-25 % aller untersuchten Proben nachzuweisen (MILLAR et al. 1996).
Charakteristisch für M. ptb ist ein extrem langsames Wachstumsverhalten. Die
Generationszeit liegt bei 1,3 - 4,4 Tagen, wodurch bei der Kultivierung erst nach 8-12
Wochen mit dem Auftreten von sichtbaren Kolonien zu rechnen ist (LAMPRECHT et
al. 1988). Ein weiteres Charakteristikum ist das Mycobactin-abhängige Wachstum.
Mycobactin ist ein Zellwand-assoziiertes Siderophor, welches von vielen
Mykobakterien, jedoch nicht M. ptb, synthetisiert wird (FRANCIS et al. 1953;
BARCLAY et al. 1985). Unter bestimmten Wachstumsbedingungen kann M. ptb
seine Mycobactin-Abhängigkeit verlieren (LAMBRECHT u. COLLINS 1992;
AADURIZ et al. 1995); deshalb müssen für die Abgrenzung gegen andere
Subspezies entweder weitere biochemische Verfahren herangezogen oder das
M. ptb-spezifische Insertionselement IS900 detektiert werden, welches selbst in der
nahe verwandten Subspezies M. avium nicht vorkommt (VARY et al. 1990).
B.1.3 Wirtsspektrum und Verbreitung
Die Paratuberkulose ist eine spezifisch bei Wiederkäuern auftretende Krankheit. Sie
kommt nicht nur bei den Hauswiederkäuern vor, sondern ist ebenso bei
Wildwiederkäuern wie Bisons, Antilopen, Kamelen und vielen anderen zu finden. Sie

Schrifttum
14
stellt daher auch ein Problem bei der Zootierhaltung dar (THOEN et al. 1977; Weber
et al. 1991). Monogastrische Tiere erkranken nicht an Paratuberkulose, obwohl sie
sich mit M. ptb infizieren können.
Geographisch gesehen ist die Paratuberkulose in den gemäßigten und
subtropischen Zonen weltweit verbreitet (MERKAL 1984a; THOEN u. BAUM 1988;
KREEGER 1991; COCITO et al. 1994; BEHYMER et al. 1991). Die wirtschaftliche
Bedeutung dieser Erkrankung ist vor allem in der milchproduzierenden, aber auch in
der fleischproduzierenden Industrie zu sehen. Milchleistung und Gewichtszunahme
der betroffenen Tiere sinken schon in der subklinischen Phase. Neben der deutlich
geringeren Lebenserwartung zeigen die Tiere eine verminderte Fertilität sowie eine
hohe Anfälligkeit gegenüber anderen Erkrankungen. Allein in den USA betrugen die
jährlichen Verluste nach den letzten Schätzungen ca. 1,5 Milliarden US-Dollar
(BUERGELT u. DUNCAN 1978; CHIODINI u. VAN KRUININGEN 1986;
BENEDICTUS et al. 1987; KREEGER 1991).
Am Beispiel von Schweden wird deutlich, welche Bedeutung der Paratuberkulose
beigemessen wird. Hier wurden unter behördlicher Kontrolle alle M. ptb-infizierten
Herden getötet (VISKE et al. 1996). In anderen Ländern werden Programme zur
Verhinderung der Ausbreitung bzw. zur Erlangung eines Paratuberkulose-freien
Status diskutiert (KENNEDY 1996; ROSSITER u. BURHANS 1996). Bislang ist die
Paratuberkulose eine meldepflichtige Erkrankung. Es ist zu diskutieren, ob sie, wie in
Österreich zur Zeit geplant, in den Status einer anzeigepflichtigen Erkrankung
erhoben werden sollte, da dann eine bessere Eradikation möglich wäre (HOMUTH
2001).
Ob M. ptb eine Gefahr für den Menschen darstellt, ist noch sehr umstritten. Es
existieren Hinweise, die M. ptb mit der Enteritis Regionalis Crohn (Morbus Crohn)
des Menschen in Verbindung bringen. Schon 1913 beschrieb DALZIEL Ähnlichkeiten
zwischen beiden Erkrankungen. Außerdem konnte aus dem Darmbereich von an
Morbus Crohn erkrankten Menschen M. ptb entweder direkt isoliert oder durch
Nachweis des IS900 identifiziert werden (CHIODINI 1989; LISBY et al. 1994;
SUANEGA et al. 1995).

Schrifttum
15
B.1.4 Pathogenese und Krankheitsbild
Die Pathogenese der Paratuberkulose ist noch weitgehend ungeklärt. Es herrscht
Einvernehmen darüber, daß die orale Infektion durch Haltung von Tieren auf M. ptb-
kontaminierten Weiden und das Saugen von Jungkälbern an kotverschmierten Zitzen
die häufigsten Infektionswege darstellen. Vitale Erreger konnten auch aus den
Reproduktionsorganen infizierter Tiere und Föten isoliert werden; die Möglichkeit
eines intrauterinen Infektionsweges ist jedoch noch nicht geklärt (CLARKE 1997;
COCITO et al. 1994; GERLACH u. VALENTIN-WEIGAND 1998; KREEGER 1991).
Symptomatisch unterscheidet man drei unterschiedliche klinische Stadien:
Das erste, subklinische Stadium ist ohne Symptome und geprägt von einer starken
zellulären Immunantwort; die humorale Immunantwort ist kaum entwickelt. Ein
Nachweis von Mykobakterien im Kot ist selten möglich. Die darauf folgende zweite
Phase wird als subklinische Exkretionsphase bezeichnet. Es ist ein Anstieg in der
humoralen Immunantwort erkennbar, Mykobakterien können im Kot nachweisbar
sein. Klinische Symptome sind nur bei einer sehr kleinen Anzahl von Tieren zu
beobachten (LEPPER et al. 1989). In der Mukosa kommt es zur kontinuierlichen
Vermehrung und Ausbreitung von M. ptb. Die dritte Phase ist die klinische oder
exkretorische Phase. Leitsymptom sind nicht therapierbare wässrige Durchfälle, die
zur Abmagerung bis hin zur völligen Auszehrung führen können, sowie zu einer
Reduktion der Milchleistung, diffusen Ödemen, Anämien und auch Infertilität. Die
Tiere sterben schließlich in einem katarrhalischen Stadium (COCITO et al. 1994).
Die Infektion erfolgt meist in den ersten sechs Lebensmonaten. Spätere Infektionen
sind möglich, jedoch selten (LARSEN et al. 1975). Über die orale Aufnahme
gelangen die Bakterien in den Darm, wo sie durch die M-Zellen im Epithel der
Peyer‘schen Platten die intestinale Mukosa überwinden (CHIODINI et al. 1984;
MOMOTANI et al. 1988). Bei adulten Tieren ist die Zahl der M-Zellen stark verringert,
wodurch vermutlich weniger Mykobakterien ins Epithel gelangen. Dies könnte die
Ursache für die geringere Infektionsanfälligkeit von adulten Tieren sein (CHIODINI
1991). Nach Aufnahme in die M-Zellen werden die Mykobakterien von sub- und
intraepithelialen Makrophagen, in denen sie lange Zeit überleben und sich
vermehren können, internalisiert. Bedingt durch das intrazelluläre Wachstum von

Schrifttum
16
M. ptb kommt es immer wieder zum Absterben einiger Makrophagen. Die frei
gewordenen Mykobakterien werden entweder mit dem Kot ausgeschieden, wodurch
es zur intermittierenden Erregerausscheidung bereits in der subklinischen Phase
kommt, oder von weiteren, zum Ort des Geschehens angelockten Phagozyten
aufgenommen. Dies führt zur Bildung von Granulomen in der Darmmukosa sowie in
den nahe gelegenen Lymphknoten (MOMOTANI et al. 1988; ZURBRICK u.
CZUPRYNSKI 1987). Die Reaktion im Tier ist eine chronische, katarrhalische
Entzündung, begleitet von Hyperplasie und einer intensiven Histiozyten-Infiltration
der Lamina propria (MERKAL et al. 1970). Histologisch sind in der Darmwand große
Mengen an epitheloiden Zellen und Makrophagen sowie einzelne Riesenzellen vom
Langerhanstyp und eine mäßige Lymphoplasmainfiltration zu beobachten. Die
Zellinfiltrate können die Krypten verdrängen und sich in Mukosa und Submukosa
ausdehnen. Im Innern von Makrophagen und Riesenzellen sind mittels Ziehl-
Neelsen-Färbung massenhaft säurefeste Stäbchen nachweisbar. Der Erreger
persistiert über Monate in den Makrophagen der Darmmukosa; der Ausbruch der
Erkrankung erfolgt selten vor dem zweiten Lebensjahr (POHLENZ 1991a).
Die Infektion breitet sich schließlich durch Aufnahme von M. ptb an unterschiedlichen
Stellen der Peyer‘schen Platten sowie durch graduelle Vermehrung von bereits
aufgenommenen Mykobakterien im Dünndarm aus. Im Verlaufe der Erkrankung
können großflächigere Darmregionen und angrenzende Körperbereiche wie Leber,
Niere, Milz, Lunge und Uterus befallen werden (TRAUTWEIN 1991). Neben der
diffusen granulomatösen Reaktion kommt es zu einer Enteropathie mit Proteinverlust
und schließlich zur Hypoproteinämie, verbunden mit Ödemen, die durch eine
Abnahme des osmotischen Drucks bei gleichzeitiger kontinuierlicher Verdickung des
Darmgewebes hervorgerufen werden (PATTERSON et al. 1967, 1968 und 1969).
Später werden die überfüllten epitheloiden Zellen ins Darmlumen abgestreift und es
kommt zur massiven Ausscheidung von Mykobakterien mit dem Kot. Die gefundenen
Zellzahlen liegen bei etwa 108 Zellen/g Kot bzw. 1012 Zellen/Tag (CHIODINI et al.
1984).

Schrifttum
17
Bei der Paratuberkulose des Rindes sind äußerlich eine Kachexie, sowie starke,
durch Hypoproteinämie bedingte Ödeme, besonders an Triel und Unterkiefer,
auffällig. Bei der Sektion stellen sich das Mesenterium und die intestinalen
Lymphknoten stark ödematisiert dar. In der Darmwand sind typischerweise
hirnwindungsartige Schleimhautfalten und eine Verdickung auffallend. Submukös
zeigt sich ebenfalls ein starkes Ödem mit hochgradiger Stauung der Lymphgefäße
(POHLENZ 1991b; ROSENBERGER 1978). Die mesenterialen Lymphknoten
erscheinen leicht vergrößert und zeigen eine granulomatöse Lymphadenitis
(TRAUTWEIN 1991).
B.1.5 Immunologie der Erkrankung
Der Krankheitsverlauf der Paratuberkulose ähnelt aus immunologischer Sicht dem
Krankheitsverlauf der Lepra des Menschen. Bei beiden Erkrankungen können eine
hyperreaktive und eine anergische Phase festgestellt werden (BENDIXEN 1978;
MERKAL et al. 1970). Am Anfang der Erkrankung steht die hyperreaktive Phase im
Vordergrund. Sie ist geprägt von starker, zellvermittelter Immunreaktion, die einer
Ausbreitung der Erreger entgegen wirkt. Diese Phase der Paratuberkulose entspricht
möglicherweise der tuberkulösen Form der Lepra. Gegen Ende der Erkrankung
wandelt sich das immunologische Bild hin zur Anergie. Diese Phase entspricht
wahrscheinlich der lepromatösen Form der menschlichen Lepra. Sie ist geprägt vom
Versagen der Immunabwehr und steht kurz vor dem Tod des erkrankten Tieres. Die
beiden Stadien werden verbunden durch eine Phase, die vom Erscheinungsbild der
„Borderline“ Form der menschlichen Lepra entspricht. Sie ist charakterisiert durch
eine kontinuierliche Schwächung der zellvermittelten Immunität und ansteigende
Antikörper-Titer gegen M. ptb (BENDIXEN 1978; CHIODINI et al. 1984; DAVIES et
al. 1974; DE LISLE u. DUNCAN 1981; LEPPER et al. 1989; PALIWAL et al. 1985).
Erklärbar wäre dies dadurch, daß die Bakterien nach oraler Infektion im Darm von M-
Zellen aufgenommen werden und weiter ins Innere des Gewebes gelangen. Hier
werden sie von Gewebemakrophagen phagozytiert, und es kommt zur zellulären
Immunantwort.

Schrifttum
18
Nach dem Absterben des Makrophagen werden die Bakterien freigesetzt, so daß es
im folgenden auch zu einer humoralen Immunreaktion kommt (CHIODINI et al. 1984;
BLOOD u. RADOSTITS 1989). Diese nimmt mit Fortschreiten der Krankheit zu und
bestimmt im Endstadium das immunologische Bild. KOETS et al. (2001) stellten fest,
daß die Höhe von humoraler bzw. zellvermittelter Antwort in Abhängigkeit zu den
verwendeten Antigenen und Isotypen steht.
Die zelluläre Immunantwort ist essentiell für einen umfassenden Schutz und wird
primär durch CD4+ Th1-Zellen vermittelt (BASSEY u. COLLINS 1997); nach der
Infektion mit M. ptb tritt zunächst eine starke CD4-Expression ein (CHIODINI u.
DAVIS 1992). Der protektive Effekt der T-Zellen wird möglicherweise durch IFN-�
vermittelt, ein Zytokin, welches eine herausragende Rolle in der antimykobakteriellen
Abwehr des Makrophagen spielt (CHAN u. KAUFMANN 1994). Es wird vermutet,
daß T-Zellen, die durch mykobakterielle Bestandteile aktiviert werden, für die
eintretende Anergie mit verantwortlich sind (BASSEY u. COLLINS 1997; HEIN u.
MACKAY 1991).
B.2 Makrophagen
B.2.1 Makrophagenaktivierung und -deaktivierung
Hämatopoetische Stammzellen reifen im Blut über Monoblasten, Promonozyten und
Monozyten zu Promakrophagen heran (ZEMBALA u. ASHERSON 1989). Diese
verlassen das Blutgefäßsystem, wandern in verschiedene Gewebe ein und
differenzieren dort zu Gewebemakrophagen aus. Besonders große Mengen an
Makrophagen finden sich im Bindegewebe, im Bereich bestimmter Blutgefäße, sowie
in Leber und Milz (LEONHARDT 1990). Der Begriff Makrophage bedeutet frei
übersetzt „der große Fresser“ und macht bereits deutlich, daß diesen Zellen eine
Schlüsselrolle in der Immunabwehr zukommt. Sie sind zu Phagozytose und
Pinozytose "großer" Partikel, wie Bakterien und anderer Mikroorganismen,
Fremdkörper, Zelltrümmer oder polymerisierter löslicher Moleküle und deren

Schrifttum
19
Elimination (durch enzymatischen Abbau bzw. Abtötung) oder deren Speicherung
befähigt. Makrophagen sind amöboid bewegliche, mononukleäre Zellen und besitzen
reichlich Zytoplasma, das zahlreiche endozytotische Vesikel und Lysosomen enthält.
Ihre mikrobizide Potenz und antitumoröse Zytotoxizität ist nach Aktivierung z. B.
durch bakterielle Stoffwechselprodukte oder Zytokine besonders ausgeprägt.
Makrophagen synthetisieren eine Vielzahl von Substanzen, die sie zum Teil
kontinuierlich, nach Phagozytose oder im aktivierten Zustand sezernieren (WECKER
1990). Dazu gehören Enzyme (z. B. Kollagenase, Hyaluronidase, Lysozym) und
andere an Entzündung und unspezifischen Abwehrmechanismen beteiligte Proteine
(z. B. Prostaglandine, Interleukin 1), Faktoren, die die Funktion anderer Zellen bzw.
Zellsysteme modulieren (z. B. mitogenes Protein, TNF-�), sowie Gerinnungsfaktoren.
Hauptfunktionen der Makrophagen sind: Induktion und Regulation von Entzündung;
Gewebeorganisation und Organheilung; Immuninduktion und Stimulation von
Lymphozyten als antigenverarbeitende und antigenpräsentierende Zelle; mikrobizide,
zytotoxische und Entzündungszelle von zentraler Bedeutung für die zellvermittelte
Immunität (LEONHARDT 1990).
Makrophagen sind die Zielzellen verschiedener Mikroorganismen, welche
intrazellulär überleben können. Die Makrophagenaktivierung ist essentiell für eine
verstärkte antimikrobielle Aktivität während dieser Infektionen. Allgemein wird die
antimikrobielle Aktivtät in zwei Kategorien unterteilt: Sauerstoff-abhängige und
Sauerstoff-unabhängige Systeme. Zu den Sauerstoff-abhängigen Systemen gehören
die reaktiven Sauerstoff-Intermediate, vermittelt durch den „oxidativen burst“, und die
reaktiven Nitrogen-Intermediate, Nitrid-Oxide, welche durch Oxidation von Arginin
entstehen. Die Sauerstoff-unabhängigen Mechanismen sind ebenfalls multifaktoriell
und können beinhalten: 1) Ansäuerung der phagolysosomalen Vakuole, 2) Wirkung
hydrolytischer lysosomaler Enzyme, 3) Nährstoffentzug (z. B. Eisen) und 4)
antimikrobielle Proteine (REINER 1994).

Schrifttum
20
Um intrazelluläre Erreger zu eliminieren, muß der Makrophage zur Aktivierung seiner
Sauerstoff-abhängigen und -unabhängigen antimikrobiellen Mechanismen angeregt
werden. Deshalb kann eine Makrophagendeaktivierung als Prozeß definiert werden,
in dem die Zelle teilweise oder vollständig unempfänglich wird für aktivierende
Stimuli, die normalerweise antimikrobielle Aktivität wie auch Zytokinausschüttung und
Antigenpräsentation ausgelöst hätten.
Hinweise für die Dysfunktion des Makrophagen durch eine Infektion mit
intrazellulären Erregern gibt es bei verschiedenen Infektionen, wie z. B. mit
Leishmania (REINER et al. 1990), Mycobacterium (SIBLEY et al. 1990, CHAN et al.
1991), Yersinia (BLISKA et al. 1993) oder dem humanen Immundefizienzvirus 1
(HIV-1) (BALDWIN et al. 1990). Die Mechanismen, welche zur Dysfunktion des
Makrophagen während der intrazellulären Infektion beitragen können, sind entweder
gegen die Sauerstoff-abhängigen oder gegen die Sauerstoff-unabhängigen
antimikrobiellen Systeme des Makrophagen gerichtet.
Negativ beeinflußt werden: 1) Transkription der „major histocompatibility complex“
(MHC) Klasse II-Gene, 2) Antigenprozessierung und -präsentation, 3)
Zytokinproduktion, 4) Expression transkriptioneller Regulatorproteine, 5) „oxidativer
burst“, 6) Phagozytose. Verstärkt wird die Produktion immunsuppressiver Moleküle,
wie z. B. Prostaglandin E2 oder IL-10 (REINER 1994).
B.2.2 Allgemeine Probleme in der primären Makrophagenkultur
Hauptproblem primärer Makrophagen in der Zellkultur ist die Inhomogenität der
Zellen und daraus folgende Schwankungen der Untersuchungsergebnisse (GOFF et
al. 1998). Dieses Problem kann durch das Verwenden der Methode nach JUNGI et
al. (1996) überwunden werden. Dabei wird ein Reifungsschritt im Teflonbeutel, an
den die Zellen nicht adhärieren können, vorgenommen: nach der Isolierung reifen die
monozytären Zellen unterschiedlicher Stadien im Teflonbeutel zu Promakrophagen
aus. Damit soll die Ausbeute erhöht und die Zellpopulation synchronisiert werden.

Schrifttum
21
B.3 Intrazelluläres Überleben von Bakterien der GattungMycobacterium in Makrophagen
Das intrazelluläre Überleben von Bakterien der Gattung Mycobacterium ist zentrales
Problem bei Erkrankungen mit diesen Erregern. Ist das Bakterium in der Lage, im
Makrophagen zu überleben, ist es vor der humoralen Abwehr geschützt. Schon bei
der Internalisierung kann das Umgehen der Abwehr beginnen: Bei M. avium und
M. tbc ist die Erkennungsstruktur zur Internalisierung des Erregers das
Lipoarabinomannan (LAM), welches von nicht-opsonierenden Makrophagen-
rezeptoren, wie z. B. CD14 oder dem Mannoserezeptor, erkannt wird. Dadurch
entkommt der Erreger der Opsonierung und somit dem Auslösen des „oxidativen
burst“ in der Zelle (CHAN u. KAUFMANN 1994).
Die genauen Mechanismen, die zur Umgehung der nach der Internalisierung
folgenden phagozytischen Abwehr führen, sind weitgehend unbekannt. Es gibt aber
Hinweise auf eine Reihe von Mechanismen, die M. avium ein intrazelluläres
Überleben ermöglichen. BERMUDEZ et al. (1991) fanden heraus, daß M. avium
nicht über Fc-Rezeptoren aufgenommen wird und damit keine Aktivierung des
Makrophagen in Form von reaktiven Sauerstoff-Intermediaten auslöst.
Die Hemmung der Ansäuerung ist ein weiterer Mechanismus, der es pathogenen
Mykobakterien ermöglicht, die Effektorfunktionen des Makrophagen in ihrem Sinne
zu verändern. STURGILL-KOSZYCKI et al. (1994) stellten fest, daß M. avium-haltige
Phagosomen keine Protonen-ATPase in ihrer Membran besitzen bzw. diese kurz
nach der Phagozytose aus der Membran entfernt wird. Diese Protonen-ATPase ist
ein Enzymkomplex, der für die Ansäuerung von intrazellulären Kompartimenten
zuständig ist und im besonderen den pH-Wert in Phagosomen absenkt, was einen
wichtigen Schritt in der Reifung des Phagosoms darstellt (NELSON 1987;
STURGILL-KOSZYCKI et al. 1994). Das Fehlen der ATPase ist eine mögliche
Erklärung für den fehlenden pH-Abfall in M. avium-haltigen Phagosomen.
Die bisherigen Erkenntnisse legen nahe, daß Mykobakterien aktiv an der Umgehung
der phagozytischen Abwehr beteiligt sind.

Schrifttum
22
In Infektionsversuchen mit verschiedenen Mykobakterien konnte außerdem die Rolle
einiger Zytokine in der Makrophagenabwehr teilweise charakterisiert werden. So
führte z. B. eine starke Interleukin-6- (IL-6-) Expression in Infektionen mit M. avium
zu einem immunosuppressorischen Effekt auf umliegende Makrophagen und die T-
Zell-Aktivierung (VAN HEYNINGEN et al. 1997). Alleine gegeben, änderte IL-6
jedoch nichts an der Abtötungsrate von M. bovis Bacillus Calmette-Guérin (BCG) in
humanen Makrophagen (BONAY et al. 1999) bzw. von Mycobacterium bovis in
murinen Knochenmarksmakrophagen (FLESCH u. KAUFMANN 1991). In denselben
Versuchen hatten sowohl Tumornekrosefaktor-alpha (TNF-�) als auch IL-10 alleine
ebenfalls keinen Effekt auf das intrazelluläre Überleben des Erregers. Mit M. avium
infizierte Makrophagen konnten jedoch nach Behandlung mit rekombinantem IL-6
das Abtöten der intrazellulären Erreger um 68 % erhöhen. Gleichzeitig wurde die
Expression von aktivierenden TNF-Rezeptoren um 78 % vermindert (BERMUDEZ et
al. 1992). In Lipopolysaccharid- (LPS-) oder LPS- und Interferon-gamma- (IFN-�-)
stimulierten humanen Makrophagen war IL-10, ein deaktivierendes Zytokin, in der
Lage, die Expression von IL-1, IL-6 und TNF-� zu inhibieren (FIORENTINO et al.
1991). In Versuchen mit M. avium in humanen Makrophagen führte IL-10 zu einer
verminderten Expression von TNF-� bei gleichzeitig vermehrter Ausschüttung des
TNF-Rezeptors Typ 2, welcher eine Inaktivierung von TNF-� zur Folge hat
(BALCEWICZ-SABLINSKA et al. 1999). Außerdem waren IL-10-defiziente Mäuse
wesentlich schneller in der Lage, eine BCG-Infektion zu eliminieren. In den
Makrophagen der Lungengranulome dieser Mäuse waren ungewöhnlich hohe Level
von TNF-� und der induzierbaren NO-Synthase (iNOS) zu detektieren (JACOBS et
al. 2000a). TNF-defiziente Mäuse starben nach 8 bis 12 Wochen mit atypischen
Granulomen in der Lunge (JACOBS et al. 2000b). Eine Vorinkubation mit
„granulocyte macrophage colony stimulating factor“ (Granulozyten-Makrophagen
Kolonie-Stimulations-Faktor; GM-CSF) führte in der Infektion humaner Makrophagen
mit BCG zu einem schnelleren Abtöten des Erregers. Spätere Gaben von GM-CSF
hatten keinen Einfluß mehr (BONAY er al. 1998). Sowohl TNF-� als auch GM-CSF
hatten in der Infektion humaner Makrophagen mit BCG hemmenden wie auch
verstärkenden Effekt, unterschiedlich in den jeweiligen Makrophagenchargen (HOAL-

Schrifttum
23
VAN HELDEN et al. 2001). Stimulation mit TNF-�, IFN-� oder GM-CSF führten in der
Infektion muriner Intestinalmakrophagen mit M. avium-Komplex zu einer erhöhten
mykobakteriostatischen und/oder mykobakterioziden Aktivität (HSU et al. 1995).
Murine Knochenmarksmakrophagen waren nach Stimulation mit IFN-� in der Lage,
das intrazelluläre Wachstum von Mycobacterium bovis zu unterbinden. Dies konnte
durch Zugabe eines spezifischen Inhibitors der Nitrit- und Nitrat-Synthese aus
L-Arginin, dessen erster Schritt die iNOS-Expression darstellt, wieder aufgehoben
werden (FLESCH u. KAUFMANN 1991).
NO spielt eine herausragende Rolle in der Abwehr intrazellulärer Parasiten und ist
notwendig für die Aktivierung des Makrophagen (JAMES 1995; GARCIA et al. 2000).
Die NO-Produktion von Makrophagen wird durch IFN-� in Kombination mit TNF-�
oder anderen sekundären Aktivierungssignalen stimuliert und durch eine Reihe von
Zytokinen (vor allem IL-4, IL-10 und „transforming growth factor beta“), anderen
Mediatoren und autoinhibitorischen Mechanismen reguliert (JAMES 1995). INOS-
defiziente Mäuse waren nicht in der Lage, BCG zu eliminieren und starben innerhalb
von 8 bis 12 Wochen, wohingegen die Kontrollgruppe überlebte (GARCIA et al.
2000). Ein experimentelles Medikament, CRL-1072, löste in humanen Makrophagen
eine verstärkte iNOS-Produktion und folgend ein höheres NO-Level aus und führte
so zum Abtöten von M. avium (JAGANNATH et al. 2000).
B.4 Intrazelluläres Überleben von M. ptb in Makrophagen
M. ptb ist nicht in der Lage, der Phagozytose durch den Makrophagen zu entgehen.
Wie die Internalisierung des Erregers ausgelöst wird, ist noch weitgehend ungeklärt.
Andere Mykobakterien werden von nicht-opsonierenden Rezeptoren erkannt und
entgehen so dem „oxidativen burst“ in der Zelle (CHAN u. KAUFMANN 1994). Dieser
Mechanismus kann für M. ptb jedoch nur spekuliert werden. Es wurde beobachtet,
daß die Zugabe von Serum frühe Aufnahmeraten von M. ptb in bovine Makrophagen
erhöht, ein Hinweis auf Rezeptoren, die einer Opsonierung des Erregers bedürfen
(ZURBRICK u. CZUPRYNSKI 1987). Außerdem konnte gezeigt werden, daß M. ptb

Schrifttum
24
Fibronektin bindet, welches möglicherweise als Ligand bei der Internalisierung dient
(VALENTIN-WEIGAND u. MORIARTY 1992).
Zur effektiven Eliminierung von Mykobakterien benötigt der Makrophage das
koordinierte Zusammenspiel des IFN-� der Th1-Zellen (eine Subpopulation der
Helfer-T-Lymphozyten) sowie von IL-1, IL-6, TNF-� und GM-CSF des Makrophagen
selbst (VALENTIN-WEIGAND u. GOETHE 1999). Aus diesem Grunde wurden in
letzter Zeit verschiedene Studien durchgeführt, welche die Rolle dieser Zytokine in
der Pathogenese boviner und oviner Paratuberkulose aufklären sollten (ADAMS u.
CZUPRYNSKI 1994; ADAMS et al. 1996; BEGARA-MCGORUM et al. 1998; STABEL
1995; ZHAO et al. 1997; ZURBRICK et al. 1988). Die Untersuchungen wurden mit
Gewebematerial experimentell infizierter Tiere bzw. Vollblut sowie als in vitro-
Infektionen mit Blutmonozyten, primären Makrophagen und Zellinien durchgeführt.
Die Ergebnisse dieser Studien geben Hinweise darauf, daß M. ptb in der Lage ist,
eine verstärkte Expression der proinflammatorischen bzw. aktivierende Zytokine
IL-1�, IL-6, TNF-� und GM-CSF hervorzurufen. Diese Effekte wurden sowohl mit
lebenden, als auch mit hitzeinaktivierten Bakterien und mit gereinigtem LAM
beobachtet. Eine Vorinkubation von J774-Mausmakrophagen mit TNF-� beeinflußte
das intrazelluläre Überleben von M. ptb: nach niedriger Dosis war das Überleben
verbessert, nach hoher Dosis jedoch war das Abtöten schneller und effizienter
(BALCEWICZ-SABLINSKA et al. 1999).
Wie sich die oben genannten Zytokine sowie die iNOS in der Infektion mit M. ptb
verhalten und welchen Einfluß sie auf das intrazelluläre Überleben von M. ptb in
primären bovinen Makrophagen haben, ist bislang nicht vollständig geklärt. Es wird
über M. ptb-spezifische Mechanismen spekuliert, da im Gewebe experimentell
infizierter Lämmer eine signifikant geringere Menge IFN-� mRNA gefunden wurde
(BEGARA-MCGORUM et al. 1998) und IFN-� nur geringe Effekte auf das
intrazelluläre Überleben von M. ptb in bovinen Monozyten hatte (ZHAO et al. 1997).
Dieser geringe antibakterielle Effekt von IFN-� in vitro steht im Gegensatz zu
Untersuchungen anderer Mykobakterien und wurde durch ungenügende Produktion
von NO erklärt, welches im zellfreien System zum Abtöten der Bakterien führen kann
(ZHAO et al. 1991).

Schrifttum
25
Die Hemmung der Ansäuerung (siehe auch B.3) ist ein weiterer möglicher
Mechanismus, die Abwehr des Makrophagen zu umgehen. KUEHNEL et al. (2001)
konnten zeigen, daß M. ptb in murinen Mausmakrophagen in der Lage ist, die
Ansäuerung des endosomalen Netzwerkes fast vollständig zu verhindern. Ob dieses
Phänomen auch in bovinen Makrophagen stattfindet, wurde bislang nicht untersucht.

Material und Methoden
26
C Material und Methoden
C.1 Material
Die verwendeten Chemikalien, Verbrauchsmaterialien und Reagenzien sind im
Anhang aufgeführt (siehe I.1 und I.2). Alle verwendeten Geräte sind im
Geräteverzeichnis unter der entsprechenden Hochzahl zu finden (siehe I.3).
C.2 Methoden
C.2.1 Zellkultur
C.2.1.1 Mycobacterium avium Subspezies paratuberculosis
Benötigte Lösungen:Watson-Reid-Medium: L-Asparagin 5 g
Glucose 10 gDiammoniumcitrat 2 gEisenammoniumcitrat 75 mgNaCl 2 gKaliumdihydrogenphosphat 2 gMgSO4 x 7 H2O 1 gZnSO4 x 7 H2O 10 mgCaCl2 x 2 H2O 2 mgGlycerin 63 mlA. dest ad. 1 l
pH-Wert mit NaOH auf 5,6 einstellen; nach dem Autoklavieren 2 mg Mycobactin und4 g Natriumpyruvat dazugeben
In sämtlichen Versuchen wurde der M. ptb-Stamm 6783 verwendet. Dabei handelt es
sich um einen Feldstamm, der 1996 von SCHEIBL erstmalig beschrieben wurde.
Zur langen Aufbewahrung wurden die Mykobakterien in Watson-Reid-Medium als
Glycerinstocks in 50 % Glycerin bei -80°C eingefroren. Die Kultivierung erfolgte in
20 ml Watson-Reid-Medium in ZK-Flaschen bei 37°C im Brutschrank 4) über mehrere
Monate.

Material und Methoden
27
Zur Infektion von Makrophagen wurden nur solche Kulturflaschen verwendet, die ein
dichtes Bakterienwachstum in Kolonien mit einem Durchmesser von 3-15 mm an der
Oberfläche des Mediums aufwiesen.
Mit einer sterilen Einmalpipette wurden die Kolonien vorsichtig von der Oberfläche
des Mediums abgehoben und in ein 50-ml-RG mit 10 ml Iscove’s Medium ohne
Pen/Strep und 30 sterilen Glasperlen überführt. Dieses Gemisch wurde für
15 min augerüttelt 28), wodurch die Mykobakterien vereinzelt wurden (SILVA et al.
1987). Um noch verbliebene große Bakterienklumpen zu entfernen, erfolgte eine
Zentrifugation bei 1000 Upm in der Hermle Zentrifuge 10) für 5 min. Der Überstand mit
den vereinzelten Bakterien wurde weiterverwendet, das Pellet mit den größeren
Bakterienklumpen wurde verworfen.
Die Vitalität der so gewonnenen Bakterien wurde mit der BacLight�-Färbung
überprüft.
Vitale Mykobakterien stellen sich in dieser Färbung grün dar, da nur der
membrangängige grüne Farbstoff Syto 9� in der Lage ist, in die Zelle einzudringen
und die DNA zu färben. Tote Mykobakterien stellen sich zusätzlich rot dar, da bei
diesen die Membranen teilweise zerstört sind und somit auch der nicht
membrangängige rote Farbstoff Propidiumjodid in den Zellkern eindringt und die
DNA färbt.
Hierzu wurden 100 µl Bakteriensuspension in einer Tischzentrifuge 26) bei
12.000 Upm für 5 min zentrifugiert, der Überstand abgesaugt und das Pellet in 100 µl
BacLight�-Lösung (nach Angaben des Herstellers gemischt) resuspendiert. Nach
einer Inkubation von 15 min bei Raumtemperatur erfolgte eine Zentrifugation wie
oben. Der Überstand wurde abgesaugt, das Pellet in 100 µl A. bidest. resuspendiert
und erneut wie oben zentrifugiert. Dies wurde wiederholt und das Pellet schließlich in
10 µl A. bidest. aufgenommen. 3 µl dieser Suspension wurden auf einen Objektträger
aufgetragen. Ein Deckgläschen wurde aufgelegt und mit Nagellack versiegelt, um ein
Austrocknen zu verhindern.

Material und Methoden
28
Die mikroskopische Beurteilung des prozentualen Verhältnisses vitaler bzw. toter
Mykobakterien erfolgte durch Fluoreszenzmikroskopie 8) bei 1000facher Ver-
größerung. Die Mykobakterien wurden nur verwendet, wenn mehr als 70 % der 200
gezählten Zellen grün, also vital waren.
C.2.1.2 Primäre bovine Makrophagen (PBMs)
C.2.1.2.1 Gewinnung und Kultivierung
Hämatopoetische Stammzellen reifen im Blut über Monoblasten, Promonozyten und
Monozyten zu Promakrophagen, die in Körpergeweben schließlich zu
unterschiedlichen Makrophagen ausdifferenzieren. Im Blut befinden sich also
monozytäre Zellen verschiedener Reifungsstadien, von denen nur die späteren
Stadien zur Adhärenz befähigt sind.
Die Methode der Teflonkultivierung beruht auf dem Prinzip, sämtliche
Reifungsstadien zu Promakrophagen ausdifferenzieren zu lassen, damit die
Ausbeute möglichst hoch und die Zellpopulation möglichst synchron ist. Am
Teflonbeutel können die Zellen nicht adhärieren, so daß ihnen zur endgültigen
Ausdifferenzierung dieses essentielle Signal fehlt. Dem Medium im Teflonbeutel wird
jedoch fötales Kälberserum (FCS) zugegeben, welches zur Reifung bis zum
Promakrophagen führt.
Aus dem „buffy coat“, dieser entsteht durch starke Zentrifugation von Blut zwischen
dem Pellet aus Erythrozyten und dem Überstand mit Thrombozyten und besteht aus
der Fraktion der Leukozyten, werden durch Waschen bzw. Lyse Thrombozyten und
Erythrozyten möglichst vollständig entfernt. Durch Dichtezentrifugation werden die
Leukozyten isoliert und nach weiterem Waschen ausgezählt. Mit einer
Zellkonzentration von 6 Mio. Leukozyten pro ml Medium wird die Suspension für
8 Tage bei 37°C und 8 % CO2 in Teflonbeuteln inkubiert. Anschließend werden die
ausgereiften Promakrophagen gewaschen und gezählt. Sie sind leicht anhand ihrer
Göße und Granularität von den verbliebenen Lymphozyten und Zelltrümmern zu
unterscheiden.

Material und Methoden
29
Benötigte Lösungen:ACD-Puffer (pH 7,3-7,35) 140 mM C6H8O7 x H2O, 200 mM C6H5Na3O7x 2H2O,
220 mM C6H12O6 (wasserfrei)Citratpuffer (pH 7,3-7,35) 30,0 mM C6H8O7 x H2O, 4,5 mM C6H12O6, 3,0 mM KCl,
100,0 mM NaClLysepuffer (pH 7,3-7,35) 155,0 mM NH4Cl, 10,0 mM KHCO3, 0,1 mM EDTA (pH 8,0)PBS (pH 7,3-7,35) 137,0 mM NaCl, 2,7 mM KCl, 7,4 mM Na2HPO4 x 2H2O,
1,5 mM KH2PO4PBS-EDTA 150,00 µl 0,5 M EDTA (pH 8,0), 250,00 ml PBS pH 7,3 – 7,35Iscove’s Medium ergänzt mit 10 % FCS, 100 µM Glutamin, mit bzw. ohne je 250 U/ml
Penicillin und Streptomycin
Durchführung:
Vor der Blutentnahme wurde der Boden des Raumes gründlich mit Wasser
abgespritzt, um den Staubgehalt in der Luft zu senken. Die Einstichstelle am Hals
des Tieres wurde großzügig mit 70%igem Alkohol desinfiziert; die Blutentnahme
erfolgte mit einer Braunüle (G12, 8 cm). Das Blut wurde unter Schwenken bis zu
einer Gesamtmenge von 500 ml in eine sterile 1000-ml-Schottflasche mit 50 ml ACD-
Puffer aufgefangen. Alle von hier an durchgeführten Arbeiten fanden unter sterilen
Bedingungen unter einer Lamina 14) statt.
Nach einer Stunde Ruhephase bei Raumtemperatur wurden 400 ml des
Blutgemisches auf acht 50-ml-Reaktionsgefäße (RG) verteilt und bei 2800 Upm und
10°C in der Hermle Zentrifuge 10) 25 min zentrifugiert. Deren Bremse wurde bei
sämtlichen Schritten ausgestellt, da sonst Verwirbelungen der „buffy coats“ bzw. der
Pellets auftreten. Nach der Zentrifugation wurde ein Teil des Überstandes abgesaugt
und die „buffy coats“ mit einer sterilen Transferpipette entnommen und in eine
1000-ml-Schottflasche mit 500 ml Citratpuffer überführt. Die Reste des
zentrifugierten Blutes wurden mit den übrig gebliebenen 100 ml Blut vermischt,
erneut auf acht 50-ml-RG verteilt und wie oben zentrifugiert. Die hier entstandenen
„buffy coats“ wurden den ersten hinzugefügt, das Gemisch mit Citratpuffer auf 800 ml
aufgefüllt und auf 16 50-ml-RG verteilt. Diese Röhrchen wurden in der Hermle
Zentrifuge 10) bei 1350 Upm und 10°C für 12 min zentrifugiert. Anschließend wurden
die Überstände, welche ein Gemisch aus Citratpuffer und Thrombozyten darstellten,
abgesaugt, die Röhrchen erneut mit Citratpuffer gefüllt und die Zellpellets
suspendiert. Dieser Waschvorgang wurde vier- bis fünfmal wiederholt, bis die
abgesaugten Überstände klar waren, also nur noch wenige Thrombozyten enthielten.

Material und Methoden
30
Jetzt wurden die Zellpellets jeweils in 20 ml Citratpuffer suspendiert, und es erfolgte
die Zugabe von 30 ml Lysepuffer. Nach 10 min Inkubation waren die Erythrozyten
lysiert, was an einer Farbänderung nach klarem Rot-Schwarz zu erkennen war.
Nach einer Zentrifugation für 10 min bei 4°C und 1350 Upm wurden die Überstände
abgesaugt. Die verbliebenen Zellpellets waren wenige Millimeter dick und, je nach
Effektivität der Lyse, beige bis dunkelrot gefärbt. Die Pellets aller RG wurden in
insgesamt 40 ml eiskaltem PBS-EDTA aufgenommen und je 10 ml dieses
Gemisches auf 4 ml Ficoll 400 in 15-ml-RG überschichtet. Nun erfolgte eine
Zentrifugation in der Hermle Zentrifuge 10) bei 2100 Upm und 4°C für 25 min. Danach
wurden alle Arbeiten auf Eis durchgeführt. Die bei der Zentrifugation entstandene
intermediäre Phase enthielt sowohl Monozyten als auch Lymphozyten. Sie wurde mit
einer sterilen Transferpipette in ein 50-ml-RG überführt, das Gemisch mit eiskaltem
PBS in 50 ml suspendiert und in der Hermle Zentrifuge 10) bei 4°C und 1350 Upm für
10 min zentrifugiert. Nach dem Absaugen des Überstandes wurde das Pellet in
eiskaltem Iscove’s Medium mit Pen/Strep resuspendiert und die Monozyten gezählt.
Anschließend wurden je 25 ml des Gemisches bei einer Zellkonzentration von 6 Mio.
Leukozyten pro ml Medium in Teflonbeutel gefüllt und dieser mit einem
Schweißgerät 23) verschweißt. Die Teflonbeutel wurden nun für 8 Tage bei 8 % CO2
und 37°C im Brutschrank 3) inkubiert.
Zum Ernten wurden die nur locker adhärierten Zellen durch Schwenken von der
Beutelwand gelöst und vermischt, die Suspension steril aus den Beuteln entnommen
und in 50-ml-RG überführt. Nach einer Zentrifugation in der Hermle Zentrifuge 10) bei
4°C und 1200 Upm für 8 min wurden die Überstände entfernt, die Pellets mit
eiskaltem PBS suspendiert und in vier 50-ml-RG gepoolt, die anschließend mit
eiskaltem PBS auf 50 ml aufgefüllt wurden. Nach erneuter Zentrifugation wie oben
wurden die Überstände abgesaugt, die Pellets mit Iscove’s Medium ohne Pen/Strep
suspendiert, in einem 50-ml-RG gepoolt und auf 10 ml mit Iscove’s Medium ohne
Pen/Strep aufgefüllt. In dieser Zellsuspension wurden nun die Promakrophagen
gezählt. Diese waren morphologisch anhand ihrer Größe und Granularität leicht von
den übrigen Zellen zu unterscheiden. Anschließend wurden die Zellen in einer
Konzentration von 2,5 Mio. Promakrophagen pro ml Medium ausgesät, so daß sich

Material und Methoden
31
folgende Zellzahlen pro Well bzw. Schale ergaben: 1,25 Mio. in 24 Well Platte
(1,9 cm2), 3,75 Mio. in 6 Well Platte (9,6 cm2), 5 Mio. in kleiner ZK-Schale (21 cm2).
Nach einer Adhärenz von drei bis vier Stunden bei 8 % CO2 und 37°C wurden die
ZK-Schalen viermal gründlich mit 37°C warmem PBS gewaschen, um die nicht-
adhärenten Zellen zu entfernen. Nach erneuter Inkubation von zwei Tagen in
Pen/Strep-freiem Iscove’s Medium wurden die PBMs wiederum mit warmem PBS
gewaschen, einen Tag in frischem Medium weiter inkubiert und schließlich für die
Infektion verwendet.
Die Stimulation sowie die Sekundärstimulation mit LPS bzw. IFN-� (freundlicherweise
überlassen von Prof. T. W. Jungi, Institut für Veterinärvirologie, Unviersität Bern,
Schweiz) erfolgte wie in D.1.4 und D.2.5 beschrieben.
C.2.1.2.2 Bestimmung der Zellzahl
Das Verwenden der PBMs für Infektionsversuche setzt eine Stabilität der Zellkultur
zumindest für den Zeitraum der Versuche, also für 72 Stunden, voraus. Aus diesem
Grunde wurde die Zahl adhärenter Zellen nicht-infizierter sowie infizierter PBMs in
Kultur gemessen.
Hierzu wurden die PBMs enzymatisch durch das Enzymgemisch Akutase
(Enzymgemisch; nach Angaben des Herstellers PAA eingesetzt) wie folgt abgelöst.
Durchführung:
Die Zellen wurden zweimal gründlich mit 37°C warmem PBS gewaschen und das
PBS möglichst restlos entfernt. Anschließend wurden 750 µl ebenfalls auf 37°C
temperierter Akutase pro Well aufpipettiert. Nach einer Inkubation von 10 min bei
37°C und 8 % CO2 war der größte Teil der PBMs bereits vom Boden gelöst. Die
restlichen, noch adhärenten PBMs ließen sich durch leichtes Tippen des Fingers
gegen den Boden der ZK-Schale lösen. Mit Zugabe von 750 µl Iscove’s Medium
ohne Pen/Strep pro Well wurde die Akutase durch das enthaltene FCS inaktiviert.
Die Zellen wurden mit Hilfe der Neubauerkammer ausgezählt (LINDL 1987).

Material und Methoden
32
C.2.2 Infektionsversuche
C.2.2.1 Standardinfektionsversuch
Durchführung:
Für die Versuche mit hitzeinaktivierten Mykobakterien erfolgte ein Erhitzen der
Bakterien für 15 min bei 85°C im Wasserbad 29). Die jeweilige Bakteriensuspension
wurde in Iscove’s Medium auf eine OD660 von 0.1 eingestellt und im Wasserbad 29)
auf 37°C temperiert.
Das Medium wurde von den PBMs entfernt und durch die Infektionssuspension
ersetzt. Verwendet wurden folgende Mengen: 500 µl für 24 Well-, 1500 µl für 6 Well-,
2000 µl für kleine ZK-Schale. Nach einer Inkubation von zwei Stunden bei 37°C und
8 % CO2 wurde die Infektionssuspension entfernt und die Schalen dreimal mit
warmem PBS gründlich gewaschen, um nicht-phagozytierte Mykobakterien zu
entfernen. Anschließend erfolgte die weitere Inkubation der Zellen in Iscove’s
Medium bei 37°C und 8 % CO2. Nicht infizierte und als Kontrolle behandelte PBMs
wurden bezüglich der Medienwechsel sowie der Waschschritte identisch behandelt.
Die Verwendung von Bafilomycin, einem ATPase-Blocker, erfolgte wie in D.2.4
beschrieben.
Das Durchführen der Reinfektionsversuche mit vitalen sowie hitzeinaktivierten M. ptb
erfolgte wie in D.2.5 beschrieben.
C.2.2.2 Bestimmung der Infektionsraten
Nun sollte überprüft werden, wieviele Makrophagen die Mykobakterien der
Infektionssuspension phagozytierten, also wie hoch die Infektionsrate war.
Durchführung:
Zur Bestimmung der Infektionsraten der PBMs wurde die Infektion in 24 Well Platten
durchgeführt, in denen Glasdeckgläschen lagen, so daß ein Teil der PBMs am Glas
adhärierte. Die Infektionen wurden ansonsten wie in C.2.2.1 beschrieben

Material und Methoden
33
durchgeführt. Zu den jeweiligen Zeitpunkten wurden die Deckgläschen entnommen,
luftgetrocknet, hitzefixiert und anschließend nach Ziehl-Neelsen gefärbt (WAGENER
1966). Hierfür wurde das Deckgläschen auf einen Objektträger gelegt, dieser mit
Karbolfuchsin überschichtet und über dem Bunsenbrenner erhitzt, bis das
Karbolfuchsin Blasen schlug. Nach 2 - 3 min Kochen wurde die Färbelösung
abgegossen. Anschließend wurde der Objektträger in 3%igem salzsaurem Alkohol
geschwenkt, bis er farblos war (ca. 30 sek). Hierbei entfärben sich alle nicht
säurefesten Zellbestandteile, die säuerefesten Mykobakterien behalten den roten
Farbstoff. Nach Abspülen mit Wasser erfolgte die Gegenfärbung mit Methylenblau für
15 sek. Schließlich wurde der Objektträger mit Wasser abgespült und abgetrocknet.
Die Zellen stellen sich unter dem Mikroskop 16) bei 800facher Vergrößerung in dieser
Färbung blau dar, Mykobakterien sind rot gefärbt (siehe Abb. 1). Es wurden 80 bis
110 Zellen nach drei Kriterien ausgezählt: a) Zellen ohne Mykobakterien, b) Zellen
mit 1-50 Mykobakterien und c) Zellen mit über 50 Mykobakterien.
Abbildung 1: vitale Mykobakterien in PBMs, 8 h p. i., gefärbt nach Ziehl Neelsen
Mykobakterien stellen sich rot, Zellen blau dar
10 µm

Material und Methoden
34
C.2.2.3 Bestimmung der Überlebensraten von M. ptb in PBMs
Für die Tauglichkeit des Modells war neben dem Überleben der PBMs das
Überleben der Mykobakterien unerläßlich. Deshalb wurden auch die
Überlebensraten phagozytierter M. ptb, also die Zahl Kolonie-bildender Einheiten
(KBE) pro Makrophage, gemessen.
Durchführung:
Hierfür wurden PBMs wie in C.2.1.2.1 bzw. C.2.2.1 beschrieben in kleinen ZK-
Schalen kultiviert und infiziert. Zu den jeweiligen Zeitpunkten wurden die Zellen in
eiskaltem SSTE-Puffer (100,0 mM NaCl, 2,0 mM Tris pH 7,5, 10,0 mM EDTA pH 7,5,
0,5 % SDS mit einem Zellschaber von der Unterlage gekratzt und die PBMs durch
Scheren mit Kanüle (G19) und 1-ml-Spritze zerstört. Durch das Zerstören der Zellen
lagen die phagozytierten Mykobakterien nun wieder frei in der Suspension vor. Die
Suspension wurde in vier Schritten jeweils 1:10 verdünnt, und je 100 µl der
Verdünnungen 10-3, 10-4 und 10-5 wurden auf Watson-Reid-Agarplatten aufgebracht.
Die Petrischalen wurden sorgfältig mit mehreren Lagen Parafilm sowie Kreppband
verschlossen und bei 37°C mindestens vier Monate oder aber, bis eine zählbare
Anzahl sichtbarer Kolonien gewachsen waren, im Brutschrank 4) inkubiert. Die KBE
wurden durch Auszählen der Kolonien ermittelt.
C.2.3 Mikroskopie
C.2.3.1 Elektronenmikroskopie
Um zu bestätigen, daß sich M. ptb in primären bovinen Makrophagen aus der
Teflonkultur in phaogosomalen Vakuolen und nicht etwa frei im Zytoplasma befindet,
wurden elektronenmikroskopische Bilder angefertigt.
Benötigte Lösungen:Cacodylatpuffer (pH 6,9) 100 mM Cacodylat, 90 mM Saccharose, 10mM MgCl2,
10 mM CaCl2

Material und Methoden
35
Durchführung:
Für die Transmissions-Elektronenmikroskopie, kurz Elektronenmikroskopie, wurden
Zellen wie beschrieben in kleinen ZK-Schalen kultiviert und nach dem
Standardinfektionsversuch infiziert. Zu den jeweiligen Zeitpunkten wurden die Zellen
in 1 ml eiskaltem Cacodylatpuffer mit 3 % [w/v] Glutaraldehyd und
5 % [w/v] Formaldehyd fixiert und anschließend dreimal mit Cacodylatpuffer
gewaschen. Schließlich wurden die Zellen mit 1 % [w/v] Osmiumtetroxid in
Cacodylatpuffer für 1 Stunde bei Raumtemperatur inkubiert, anschließend erneut mit
Cacodylatpuffer gewaschen und schließlich in 1 ml Cacodylatpuffer mit einem
Zellschaber abgekratzt. Diese Suspension wurde in ein 1,5-ml-RG überführt und bis
zur weiteren Verwendung im Kühlschrank gelagert.
Nach einer Zentrifugation für 3 min bei 2.000 Upm wurden die Zellen in 1,5%igem
Agar eingebettet. Kleine Agarstücke wurden mit einer Reihe Acetongradienten
dehydriert und schließlich in Spurr resin eingebettet. Ultradünnschnitte wurden mit
Glasmessern 9) durchgeführt. Die Schnitte wurden 30 min bei 40°C in 0,5%igem
Uranylacetet (pH 4,5) und 30 min bei 20°C in 1%igem Bleicitrat im Ultrostainer 27)
kontrastiert. Die Schnitte wurden im Elektronenmikroskop 5) bei 80 kV
durchgemustert.
C.2.3.2 Konfokale Laserscanning Mikroskopie
Zur Bestimmung des intraendosomalen pH-Wertes mit FITC-Dextran mußten die
FITC-Dextran-Partikel und die Mykobakterien in den Makrophagen größtenteils
kolokalisiert sein. Aufgrund der Dicke der Makrophagen und der damit verbundenen
Streulichtproblematik wurde diese Kolokalisierung in der konfokalen Laserscanning
Mikroskopie, kurz Konfokalmikroskopie, untersucht. Die Optik des Konfokal-
mikroskops erlaubt es, optische Schnitte mittels Laser in relativ dicken Präparaten zu
erstellen. Somit kann die Lokalisierung des Farbstoffes in Relation zu den Bakterien
eindeutig bestimmt werden.

Material und Methoden
36
Durchführung:
Hierfür wurden die Zellen in 24 Well Platten kultiviert, in denen sich
Glasdeckgläschen befanden, so daß ein Teil der Zellen am Glas adhärierte (siehe
C.2.2.2). Die zu verwendenden Mykobakterien wurden in der Nacht zuvor mit 1 µg/ml
Sulpho-NHS-Biotin in PBS bei Raumtemperatur inkubiert. Sulpho-NHS-Biotin
markiert Proteine an der Bakterienoberfläche durch die Bildung einer
Succinyldiesterbindung. Um überschüssiges Sulpho-NHS-Biotin zu binden und zu
entfernen, wurden die Bakterien 30 min. in 1 mol Glycin-PBS inkubiert und
anschließend zweimal darin gewaschen. Dann wurden sie in der jeweilig benötigten
Menge Medium aufgenommen. Die Infektion erfolgte für zwei Stunden als
Koinkubation der markierten Mykobakterien mit 1 mg/ml FITC-Dextran. Dann wurden
die Zellen dreimal mit PBS gewaschen und für 15 min mit 2%igem Paraformaldehyd
bei Raumtemperatur inkubiert. Anschließend erfolgte ein dreimaliges Waschen mit
PBS mit 5 % FCS. Schließlich wurde das FCS-haltige PBS durch PBS mit 1 µg/ml
TexasRed-Streptavidin ersetzt, welches an Biotin bindet und bei 546 nm Anregungs
Wellenlänge eine Rotfluoreszenz zeigt. Nach weiteren 20 min wurden die Zellen mit
PBS gewaschen und mit der Zellseite nach unten auf einen Objektträger in 10 µl
Mowiol� mit 10 mg/ml DABCO� gelegt, um ein Austrocken und Ausbleichen zu
verhindern. Die Deckgläschen wurden mit Nagellack versiegelt und bis zur
Untersuchung bei 4°C gelagert.
Die Konfokalmikroskopie wurde mit einem Leica DM-IRBE konfokalen Laserscanning
Mikroskop durchgeführt, ausgestattet mit einem Plan-Apo 100x/1.4 n.a.
Ölimmersions-Objektiv und einem Argon/Krypton-Laser (Leica, Bensheim). Die
Auswertung der Bilder erfolgte mit der Standardsoftware von Leica für
Konfokalmikroskopie und Adobe Photoshop (Adobe Systems Incorporated,
Amsterdam, Holland). Eine Kolokalisation der Grünfluoreszenz des FITC mit der
Rotfluoreszenz des TexasRed resultierte in einer Gelbfluoreszenz und bedeutete
eine Kolokalisierung des FITC-Dextran mit den Mykobakterien. Es wurden 200
Mykobakterien in Form von roten und gelben Fluoreszenzen ausgezählt.

Material und Methoden
37
C.2.4 FACS-Analyse
Nach der Phagozytose befinden sich Bakterien in Phagosomen, welche Marker
früher Endosomen tragen. Sie reifen innerhalb Minuten bis weniger Stunden heran
und tragen dann Marker später Endosomen. Hauptunterschiede dieser ineinander
übergehender Endosomentypen sind zum einen die Oberflächenmarker, die erst eine
Zuordnung in frühe und späte Typen erlauben, zum anderen der intraendosomale
pH-Wert. Dieser liegt nach der Phagozytose zwischen 6,3 und 6,5, in reifen
Phagosomen mit Markern später Endosomen jedoch etwa bei pH 4,5-5,5. In diesem
sauren pH-Wert haben Enzyme das optimale Mileu zum Verdau des phagozytierten
Materials. Somit stellt die Ansäuerung des Phagosoms einen wichtigen Schritt in der
Phagosomenreifung dar.
Als Mittel der Wahl wurde die FACS-("fluorescence activated cell sorting"-)Analyse
verwendet. FACS besitzt den Vorteil, daß eine sehr große Zellzahl (ca. 300 - 500
Zellen pro Sekunde) anhand von physikalisch-morphologischen und Fluoreszenz-
eigenschaften qualitativ und quantitativ charakterisiert werden kann. Im Verlauf der
Messung im FACS-Gerät werden die Makrophagen einzeln durch einen Laserstrahl
geführt und dabei das Durchlicht, zur Messung der Zellgröße, das 90°-Streulicht zur
Bestimmung der Granularität sowie bis zu drei unterschiedliche Fluoreszenzen (hier:
490 nm sowie 540 nm) detektiert.
FITC ist ein Fluorochrom, welches bei einer Anregungswellenlänge von 488 nm pH-
abhängig Fluoreszenz emmitiert (SCHLESINGER 1994), je niedriger der pH-Wert,
desto schwächer die Fluoreszenz. Die internalisierte Menge an FITC-Dextran kann
zwischen den Proben anhand der nicht-pH-abhängigen Fluoreszenz bei 546 nm
abgeglichen werden. Dextran wird von der Zelle aufgenommen, wo es anfänglich in
frühe Endosomen, in der weiteren Endosomenreifung in späte Endosomen und
Lysosomen gelangt. Eine eventuelle Änderung des pH-Wertes durch die
Mykobakterien wird als Änderung der Fluoreszenzintensität angezeigt.

Material und Methoden
38
Benötigte Lösungen:pH-Eichpuffer 10 mM Nigericin, 1 mM MgCl2, 105 mM KCl
pH-Werte wurden mit unterschiedlichen Mengen an KH2PO4 sowie NaHPO4 auf5,0 bis 8,0 in 0,5-Schritten eingestellt
Durchführung:
Die Zellen wurden wie oben beschrieben in kleinen ZK-Schalen kultiviert und
anschließend mit M. ptb OD660 0,1 und 5 mg/ml FITC-Dextran inkubiert. Nach zwei
Stunden wurden die PBMs durch dreimaliges Waschen mit warmem PBS von nicht
internalisierten Mykobakterien bzw. FITC-Dextran befreit und wie in C.2.1.2.2
beschrieben durch Akutase (Enzymgemisch; wurde nach Angaben des Herstellers
PAA eingesetzt) abgelöst. Die Zellsuspension wurde zu Portionen à 300 µl auf acht
1,5-ml-RG verteilt, in einer Tischzentrifuge 26) bei 2.000 Upm für 1 min zentrifugiert
und die Überstände entfernt. Sieben Pellets wurden verwendet, um eine pH-
Eichkurve zu erstellen, das achte wurde in 300 µl eiskaltem PBS aufgenommen.
Zum Erstellen der Eichkurve wurden sieben Eichpuffer mit unterschiedlichen pH-
Werten (5,0 bis 8,0) verwendet. Je ein Zellpellet wurde in je 300 µl eines der
eiskalten pH-Eichpuffer aufgenommen. Die Proben wurden sofort per FACS-Analyse
im FACS-Scan-Gerät 7) gemessen. Insgesamt 10.000 Ereignisse wurden gezählt.
Die durchschnittliche Fluoreszenzintensität der Zellen in den Eichpuffern nach
Intensitätsabgleich anhand der Fluoreszenz bei 546 nm wurde verwendet, um die
Eichkurve des jeweiligen Experimentes zu erstellen. Die Fluoreszenzemission der
Zellen in den Puffern der pH-Werte 5,0-7,5 wurden als prozentuale
Fluoreszenzintensität der Zellen im Eichpuffer pH 8,0 dargestellt. Der pH-Wert des
Testers wurde anhand dessen Fluoreszenzintensität im Vergleich zur Eichkurve
errechnet. Die Auswertung der Daten erfolgte mit dem Softwareprogramm
WinMDI 2.8 (Joseph Trotter, USA).

Material und Methoden
39
C.2.5 Molekulargenetische Methoden
C.2.5.1 RT-PCR
Die RT-PCR bietet eine Möglichkeit, die exprimierte mRNA eines Gens und somit
dessen Aktivität zu quantifizieren.
C.2.5.1.1 Gewinnung von RNA nach CHOMCZYNSKI u. SACCHI (1987)
Bei dieser Methode werden die Zellen in Guanidinthiozyanat homogenisiert und
anschließend einer saueren Phenolextraktion unterzogen. Nach Zugabe von
Natriumacetat, Phenol sowie Chloroform/Isoamyalkohol und der anschließenden
Zentrifugation ist die DNA in der organischen Phase gelöst, die Proteine befinden
sich in der Interphase, die RNA in der wässrigen, oberen Phase. Nach
Isopropanolpräzipitation wird das RNA-Pellet erneut in Guanidinthiozyanat gelöst, in
Isopropanol präzipitiert und mit Ethanol gewaschen.
Benötigte Lösungen:Denaturierungspuffer 4,0 M Guanidinthiocyanat, 25,0 mM Natriumcitrat,
0,5 % Sarkosyl, 100,0 mM Mercaptoethanol
Durchführung:
Die PBMs wurden je nach Versuchsaufbau wie in Abschnitt C.2.1 und C.2.2
beschrieben kultiviert und infiziert. Die Gewinnung der mRNA erfolgte bei allen
Versuchen in kleinen ZK-Schalen wie folgt:
Das Medium wurde restlos abgesaugt, die ZK-Schalen auf Eis inkubiert und die
Zellen einmal mit eiskaltem PBS gewaschen. Ab hier erfolgten sämtliche Arbeiten auf
Eis, um die RNA vor dem Abdauen durch RNasen zu schützen. Es wurde 1 ml
eiskaltes PBS aufpipettiert, die Zellen mit einem Zellschaber vom Boden gekratzt und
die Zellsuspension in ein 2-ml-RG überführt. Nun erfolgte eine Zentrifugation in der
Tischzentrifuge 26) bei 13.000 Upm für 3 min. Anschließend wurde der Überstand
abgesaugt und das Zellpellet für mindestens 2 Stunden, höchstens aber 3 Tage bei
-70°C eingefroren. Zur RNA-Präparation wurden die Zellen auf Eis aufgetaut und
750 µl Denaturierungspuffer aufpipettiert. Mit Kanüle (G19) und 1-ml-Spritze wurden

Material und Methoden
40
die Zellen durch zehnmaliges Aufziehen geschert, somit Zellen, Zellkerne und DNA
zerstört und die RNA freigesetzt.
Zu den zerstörten Zellen wurden 75 µl 2 M Natriumacetatlösung (pH 4,0) gegeben,
als Ansäuerung zur sauren Phenolextraktion. Nach kurzem Aufrütteln wurden 750 µl
Phenol zur Proteinextraktion hinzugefügt und erneut aufgerüttelt. Nach Zugabe von
150 µl Chloroform:Isoamylalkohol (49:1) und Aufrütteln wurde das Gemisch milchig.
Nun erfolgte eine Inkubation auf Eis für 15 min. Die anschließende Zentrifugation bei
4°C und 11.000 Upm in der Kühlzentrifuge 13) für 20 min trennte die phenolische
Phase mit Proteinen und DNA von der wässrigen Phase mit RNA. Die wässrige
obere Phase wurde vorsichtig aufgenommen, in ein neues 2-ml-RG überführt, mit
derselben Menge Isopropanol (ca. 750 µl) versetzt und aufgerüttelt. Bei der
anschließenden Inkubation bei -20°C für eine Stunde fiel die RNA aus. Durch eine
Zentrifugation bei 4°C und 11.000 Upm für 20 min wurde die RNA pelettiert, der
Überstand abgesaugt. Das Pelett wurde erneut in 300 µl Denaturierungspuffer
aufgenommen und in ein 1,5-ml-RG überführt. Nach Zugabe von 300 µl Isopropanol
wurden die Gefäße erneut für 30 min bei -20°C inkubiert und anschließend bei 4°C
und 12.000 Upm für 10 min zentrifugiert. Der Überstand wurde verworfen und nach
kurzem Anzentrifugieren vollständig entfernt. Das Pellet wurde in 400 µl 80%igem
Ethanol aufgenommen und für 15 min auf Eis inkubiert. Durch mehrmaliges
Aufrütteln wurden die Salze gelöst. Nach Zentrifugation bei 4°C und 12.000 Upm für
10 min wurde der Überstand abgesaugt und das Pellet getrocknet. Das getrocknete
Pellet wurde in 22 µl eiskaltem A. bidest. (SIGMA) aufgenommen und 2 µl zur
Konzentrationsmessung der RNA im Photometer 20) verwendet. Die gewonnene RNA
wurde bis zur weiteren Verwendung bei -70°C aufbewahrt.
C.2.5.1.2 Bestimmung der RNA-Konzentration im Photometer
Um annähernd gleiche RNA-Mengen in die Reverse Transkription und damit später
cDNA in die PCR einsetzen zu können, muß die Konzentration der RNA festgestellt
werden. Dies erfolgt anhand der Lichttransmission bei definierten Wellenlängen im
Photometer. Hier kann gleichzeitig die Reinheit der RNA errechnet werden.
Gemessen wird die Transmission bei 260 sowie 280 nm. Anhand der Werte bei

Material und Methoden
41
260 nm wird abhängig vom eingesetzten Verdünnungsgrad die RNA-Konzentration
festgestellt (z. B. Wert bei 260 nm mal 2 bei einer Verdünnung von 1:50). Die Werte
bei 280 nm dividiert durch jene bei 260 nm ergeben die Reinheit der Probe. Dieser
Wert sollte zwischen 1,7 und 2,2 liegen.
C.2.5.1.3 DNase-Behandlung von RNA
Um auch die letzten eventuell vorhandenen DNA-Verunreinigungen zu entfernen,
wurden die Proben mit DNase (verdaut DNA) behandelt.
Benötigte Lösungen:DNase-Mastermix 12,5 mM Tris, 62,5 mM KCl, 7,5 mM MgCl2, 2,0 U/µl RNase Inhibitor,
2,5 U/µl DNase IPräzipitationsmix 7 µg/µl Glykogen, 840 mM Natriumacetat pH 5,2, 86 % [v/v] Ethanol
Durchführung:
Zu den je 20 µl gRNA wurden je 80 µl DNase-Mastermix pipettiert und für 30 min bei
37°C im Wasserbad inkubiert. Die folgenden Schritte fanden wiederum auf Eis statt.
Zur Phenolextraktion der Ansätze wurden 100 µl equilibriertes Phenol zugegeben,
anschließend aufgerüttelt und bei 4°C und 12.000 Upm in der Kühlzentrifuge 13) für
5 min zentrifugiert. Die obere, wässrige Phase wurde vorsichtig abpipettiert und in
ein neues 1,5-ml-RG überführt. Zum Phenol wurden 100 µl A. bidest. (SIGMA)
pipettiert und erneut aufgerüttelt sowie zentrifugiert. Die obere, wässrige Phase
wurde wiederum vorsichtig abgenommen und der ersten hinzugefügt. Zu diesen
200 µl wurden für die Ethanolpräzipitation der RNA mit Natriumacetat je 675 µl
Präzipitationsmix gegeben, aufgerüttelt und für 2 Stunden bei -70°C oder über Nacht
bei -20°C inkubiert. An diesen Inkubationsschritt schloß sich eine Zentrifugation bei
4°C und 12.000 Upm für 15 min an. Der Überstand wurde verworfen, das Pelett in
400 µl eiskaltem 80%igem Ethanol aufgenommen und erneut bei 4°C und 12.000 für
10 min zentrifugiert. Der Überstand wurde wiederum verworfen, das Pelett
getrocknet und anschließend in einer Konzentration von 0,5 µg gRNA/µl in A. bidest.
(SIGMA) aufgenommen. Bis zur weiteren Verwendung wurde die RNA bei -70°C
aufbewahrt.

Material und Methoden
42
C.2.5.1.4 Reverse Transkription
Bei der Reversen Transkription wird die vorhandene RNA durch das Enzym Reverse
Transkriptase in cDNA umgeschrieben. Durch das Verwenden eines oligo-dT-
Primers wird fast ausschließlich mRNA transkribiert, da sich diese durch eine
poly(A)-Sequenz auszeichnet.
Benötigte Lösungen:RT-Mastermix 1 x RT-Puffer (GIBCO), 15 mM Dithiothreitol (DTT), 0,04 µM oligo dT Primer,
0,03 mM dNTP each
Durchführung:
Je Ansatz wurden 2 µg der DNase-verdauten RNA mit 14 µl RT-Mastermix auf Eis
vermischt und im Thermocycler 25) erst 5 min bei 65°C, dann 10 min bei 37°C
inkubiert. Das RG wurde sofort auf Eis gestellt, 50 U RTase zupipettiert und
gemischt. Anschließend erfolgte eine weitere Inkubation bei 37°C für 50 min zur
Durchführung der eigentlichen Reversen Transkription. Das folgende Erhitzen für
5 min auf 75°C diente der Inaktivierung der RTase. Zur Vorbereitung für die PCR
wurde zu jedem Ansatz von 20 µl cDNA-Mix 80 µl A. bidest zupipettiert und
vermischt. Bis zur weiteren Verwendung wurde die hergestellte cDNA bei -70°C
gelagert.
C.2.5.1.5 PCR
Die PCR dient der Amplifikation der hergestellten cDNA zur anschließenden
Auftrennung und Quantifizierung in der Gelelektrophorese.
Bei der PCR synthetisiert Taq, eine spezielle DNA-Polymerase, einen neuen DNA-
Strang an einer einzelsträngigen Nukleinsäurematritze (in diesem Fall die cDNA).
Dazu werden Startermoleküle ("Primer") benötigt, die an die dentaurierte Matrizen-
DNA binden (hybridisieren). Von deren 3'-Ende aus synthetisiert die Polymerase den
neuen DNA-Strang.

Material und Methoden
43
1. Denaturierung des zu amplifizierenden DNA-Doppelstranges: Bei etwa
90-94°C wird die Matrizen-DNA aufgeschmolzen. Es entstehen einzelsträngige DNA-
Moleküle.
2. Primer-Hybridisierung an die einzelsträngige DNA (sog. "Annealing"): Die
synthetischen Oligonukleotide binden bei Temperaturen von etwa 50-60°C an die
komplementären Sequenzen der Matrizen-DNA und leiten auf diesem Weg die
Amplifikation des dazwischen liegenden Sequenzabschnittes ein.
3. Synthese des Gegenstranges (sog. "Extension"): Vom 3'-Ende der Primer
ausgehend wird der Gegenstrang der Matrizen-DNA durch eine DNA-Polymerase
synthetisiert. Die hier gewählte Temperatur entspricht in der Regel dem
Temperaturoptimum der verwendeten Polymerase.
Durch die Wahl eines gegenläufig orientierten Primerpaares kann die DNA-Sequenz
zwischen den Primern geziehlt vervielfältig werden. Das entscheidende Prinzip der
PCR ist die zyklische Wiederholung der einzelnen Reaktionsschritte, wodurch die
Ausgangsmenge an DNA bis zu einem Faktor von etwa 107 amplifiziert werden kann.
Die Sequenzen der verwendeten Primer sowie die aus der RT-PCR resultierenden
Fragmentgrößen sind in Tabelle 1 aufgeführt.
Benötigte Lösungen:PCR-Mastermix: 1 x PCR-Puffer, 1,88 mM MgCl2, 0,25 mM dNTP each, 0,25 µM Primer,
0,05 U/µl Taq-Polymerase
Durchführung:
Je 10 µl der verdünnten cDNA wurden mit je 40 µl PCR-Mastermix in einem 200-µl-
RG gemischt und im Thermocycler 25) amplifiziert. Nach der initialen Denaturierung
für 3 min bei 91°C erfolgte die Amplifikation je nach Produkt in 30-40 Ampli-
fikationsschritten, bestehend aus Denaturierung (1 min bei 94°C), „Annealing“ und
Synthese (2 min bei 72°C). Die „Annealing“-Temperaturen sowie die Anzahl der
Amplifikationszyklen sind ebenfalls Tabelle 1 zu entnehmen. Abschließend folgten
als terminale Synthese 7 min bei 72°C. Danach wurden die PCR-Produkte bis zur
weiteren Verwendung bei -20°C gelagert.

Material und Methoden
44
Tabelle 1: verwendete Primer, Quellen und Produktgrößen der ausgewählten Gene,
verwendete Annealingtemperatur und Zyklenzahl
�-Actin IL-1� IL-6 TNF-� GM-CSF IL-10 iNOS
ITO
u. K
OD
AMA
1996
405
bp
ITO
u. K
OD
AMA
1996
394
bp
ITO
u. K
OD
AMA
1996
251
bp
ADLE
R e
t al.
1995
309
bp
ITO
u. K
OD
AMA
1996
429
bp
KEEF
E et
al.
1997
471
bp
ADLE
R e
t al.
1995
372
bp
5‘ A
CG
TCG
CC
TTG
GAC
TTC
GAG
CAG
G
5‘ G
CTG
GAA
GG
TGG
ACAG
GG
AGG
CC
AGG
A
5‘ A
AAC
AGAT
GAA
GAG
CTG
CAT
CC
AA
5‘ C
AAAG
CTC
ATG
CAG
AAC
ACC
ACTT
5‘ G
ACG
GAT
GC
TTC
CAA
TCTG
5‘ A
CC
CAC
TCG
TTTG
AAG
ACTG
CAT
CTT
5‘ A
GC
TGG
CG
GAG
GAG
GTG
CTC
5‘ C
CAT
GAG
GG
CAT
TGG
CAT
AC
5‘ A
TGTG
GC
TGC
AGAA
CC
TGC
TTC
TCC
5‘ C
TTC
TGG
GC
TGG
TTC
CC
AGC
AGTC
A
5‘ G
TTG
CC
TGG
TCTT
CC
TGG
CTG
5‘ T
ATG
TAG
TTG
ATG
AAG
ATG
TC
5‘ T
AGAG
GAA
CAT
CTG
GC
CAG
G
5‘ T
GG
CAG
GG
TCC
CC
TCTG
ATG
55 °C
Ann
ealin
g
31 Z
ykle
n
60 °C
Ann
ealin
g
33 Z
ykle
n
60 °C
Ann
ealin
g
30 Z
ykle
n
60 °C
Ann
ealin
g
37 Z
ykle
n
60 °C
Ann
ealin
g
32 Z
ykle
n
60 °C
Ann
ealin
g
40 Z
ykle
n
60 °C
Ann
ealin
g
37 Z
ykle
n

Material und Methoden
45
C.2.5.2 Agarosegel-Elektrophorese der PCR-Produkte
In der Gelelektrophorese wird die amplifzierte DNA elektrophoretisch aufgetrennt und
mit Ethidiumbromid unter UV-Licht sichtbar gemacht.
Benötigte Lösungen:10x Tris-Borsäure-EDTA-Puffer (TBE) 900,0 mM Tris, 890,0 mM Borsäure, 5,8 mM EDTA6x Gel-Ladepuffer 15,0 mg Bromphenolblau, 1,5g Ficoll 400, 9,5 ml A. bidest
Durchführung:
In einer Elektrophoresekammer 6) wurden 95 ml 1,5%iges Agarosegel in TBE mit
1,4 µg/µl Ethidiumbromid gegossen und das ausgehärtete Gel mit 1xTBE
übergossen. Je 20 µl des PCR-Ansatzes wurden mit je 5 µl des 6x Gel-Ladepuffers
gemischt und auf das Gel aufgetragen. Als Größenmarker diente ein 100-Basenpaar-
Leiter, von dem 1 µl mit 5 µl 6x Gel-Ladepuffer und 19 µl A. dest. gemischt
aufgetragen wurde. Die elektrophoretische Auftrennung der Proben erfolgte bei 80 V
(Stromversorgungsgerät 24)) und wurde gestoppt, wenn die Bromphenolblau-Bande
das letzte Drittel des Abschnitts erreicht hatte. Anschließend wurde das Gel unter
UV-Licht fotografiert (Kamera digital 11)) und das Bild im Computer gespeichert.
Durch Ethidiumbromid wird DNA im UV-Licht sichtbar gemacht.
C.2.5.3 Quantifizierung der Genexpression
Das mit UV-Licht bestrahlte Gel, und somit die sichtbaren Banden, wurde
photographiert. Die Auswertung der PCR erfolgte anhand der Intensität der einzelnen
Banden. Um diese miteinander vergleichen zu können, muß bekannt sein, welche
RNA-Mengen im Vergleich aufgetragen wurden. Dies ist durch sogenannte
Haushaltsgene möglich. Diese Gene sind dadurch charakterisiert, daß die Regulation
ihrer Expression in der jeweiligen Zelle unabhängig von einem Stimulus erfolgt, sie
sind konstitutiv exprimiert. Außerdem sind sie für das Überleben der Zelle essentiell.
Die Bandenintensität der Haushaltsgene ist also genau dann gleich stark, wenn
exakt die gleichen RNA-Mengen eingesetzt wurden.

Material und Methoden
46
Dies ist technisch nicht möglich, daher ist es notwendig, die Bandenintensität der
untersuchten Gene anhand der Bandenintensität der Haushaltsgene abzugleichen:
IC – IH wobei IC = Intensität der Bande des zu untersuchenden Zytokins,
Ihg – IH IH = Hintergrundintensität, Ihg = Intensität der Bande des housekeeping genes
Um die Reproduzierbarkeit der Ergebnisse zu gewährleisten, wurden die
Untersuchungen in drei bzw. vier PCR-Ansätzen durchgeführt. In einem zweiten
Schritt ist es notwendig, die Intensität der Gele untereinander anzugleichen, zu
normalisieren. Hierzu wurde die Summe der Bandenflächen der einzelnen PCR-
Ansätze untereinander gleichgesetzt. Danach wurden die Werte als Durchschnitt des
X-fachen des jeweiligen Kontrollwertes mit Standardabweichung im Diagramm
dargestellt.
Die Auswertung der Daten erfolgte mit Microsoft� Excel 97, Microsoft Corporation,
USA, und Sigma Plot for Windows 5.00, SPSS, Incorporation, USA.

Ergebnisse
47
D Ergebnisse
M. ptb verursacht eine chronisch-progressive, nicht therapierbare Enteritis bei
Wiederkäuern. Die Pathogenese der Erkrankung ist weitgehend unbekannt. Es gilt
als gesichert, daß die Persistenz des Erregers in den Makrophagen der Darmwand
eine Schlüsselrolle spielt (COCITO et al. 1994). Von anderen pathogenen
Mykobakterien, die ebenfalls in Makrophagen überleben, ist bekannt, daß sie die
Effektorfunktionen der Makrophagen modulieren, um nicht abgetötet zu werden
(siehe B.3).
Im Blut vorkommende Monozyten sind keine einheitliche Zellfraktion, sie befinden
sich in unterschiedlichen Reifungsstadien von Promonozyten über Monozyten bis hin
zu Promakrophagen (OPPENHEIM u. LEONARD 1989). Wird diese Zellmischung in
die Zellkultur verbracht, erhält sie zwei wichtige Differenzierungssignale: zum einen
die Adhärenz an das feste Substrat (das Plastik der ZK-Schale), zum anderen das im
Medium befindliche Serum (DOUGHERTY u. MCBRIDE 1989). Aufgrund dieser
Signale differenzieren die Zellen zu Makrophagen aus.
Bisherige Arbeiten mit M. ptb wurden mit Gewebematerial experimentell infizierter
Tiere bzw. Vollblut sowie als in vitro-Infektionen mit Blutmonozyten, primären
Makrophagen und Zellinien durchgeführt. Zellinien werden im Umgang mit anderen
Erregern häufig verwendet, da sie deutlich einfacher in der Handhabung sind,
nahezu beliebig viele Zellen zur Verfügung stehen und die einzelnen Zellen
annähernd identische Eigenschaften besitzen. In Ermangelung einer bovinen
Makrophagenzellinie wurden Arbeiten in bovinen Makrophagen bislang mit frisch
isolierten, also primären Makrophagen durchgeführt, wobei hier das Problem der
hohen Diversität der Zellpopulation entsteht (siehe auch C.2.1).
In dieser Arbeit wird die Eignung des Teflon-Modells primärer boviner Makrophagen
(PBMs) nach JUNGI et al. (1996) getestet. Zudem werden die Auswirkungen des
intrazellulären Überlebens von M. ptb auf PBMs aus der Teflonkultur untersucht.

Ergebnisse
48
D.1 Eignung und Standardisierung des Teflon-Modells
Hauptproblem bisher verwendeter primärer boviner Makrophagen war die geringe
Infektionsrate (ca. 40 %), die als Ausdruck der Inhomogenität der Zellpopulation
gewertet wurde. Daneben war der Anteil verunreinigender Lymphozyten so hoch,
daß eine wesentliche Beeinflussung der Makrophagen durch die Lymphozyten und
damit eine Verfälschung der Ergebnisse nicht ausgeschlossen werden konnte.
Bei dem hier verwendeten Teflon-Modell nach JUNGI et al. (1996) werden die
Makrophagen 7-10 Tage in Teflonbeuteln in FCS-haltigem Medium kultiviert. FCS
dient als erstes Differenzierungssignal für die Makrophagenvorläuferzellen und führt
zur Differenzierung bis zum Promakrophagen (ZEMBALA u. ASHERSON 1989). An
der Teflonoberfläche kann keine Adhärenz stattfinden, die Promonozyten und
Monozyten differenzieren zu Promakrophagen aus und bleiben in diesem Stadium
stehen. Durch das Verbringen auf ZK-Schalen erhalten die Promakrophagen das
zweite Signal, die Adhärenz, und differenzieren innerhalb weniger Stunden zu
Makrophagen aus (JUNGI et al. 1996).
Zur Kultivierung von PBMs im Teflonbeutel wurde das Blut semisteril entnommen. In
mehreren Waschschritten wurden die Thrombozyten entfernt, die Erythrozyten
anschließend osmotisch zerstört. Durch die nun folgende Dichtezentrifugation
wurden die restlichen Erythrozyten und anderen Zellen von den Lymphozyten und
Monozyten in verschiedenen Reifungsstadien getrennt. Diese leukozytären Zellen
wurden für 8 Tage in Teflonbeuteln kultiviert und anschließend auf ZK-Schalen
gegeben. Hier adhärierten die gereiften Promakrophagen, die übrigen Zellen wurden
durch Waschen entfernt. Nach weiteren drei Tagen Inkubation wurden die nun reifen
Makrophagen in die Versuche eingesetzt.

Ergebnisse
49
D.1.1 Makrophagenausbeute
Aus einem halben Liter Blut wurden zwischen 800 Mio. und 1,4 Mrd. leukozytäre
Zellen isoliert. Nach 8 Tagen im Teflonbeutel wurden die Promakrophagen gezählt;
sie waren aufgrund ihrer Größe und Granularität leicht von Lymphozyten und
anderen Zellen zu unterscheiden. 40-80 Mio. Promakrophagen wurden gezählt, also
etwa 5 % der ursprünglich isolierten monozytären Zellen.
Da im Blut etwa 10 % der monozytären Zellen Monozyten sind (LEONHARDT 1990),
betrug die Ausbeute bei der hier gewählten Teflon-Methode rund 50 %.
D.1.2 Infektion von PBMs aus der Teflonkultur mit M. ptb
In Infektionsversuchen ist es wichtig, daß möglichst jede Zelle mindestens ein
Bakterium phagozytiert hat, da ansonsten die Ergebnisse eine Mischung aus
infizierten und nicht-infizierten Zellen darstellen. Um eine mögliche Auswirkung der
Bakterien auf die Makrophagen als Folge sezernierter oder struktureller Bestandteile
identifizieren zu können, wurden in sämtlichen Versuchen neben vitalen M. ptb auch
hitzeinaktivierte Erreger eingesetzt.
Zur Bestimmung der Infektionsrate, d. h. der Anzahl phagozytierter Mykobakterien
pro PBM, wurden die Makrophagen mit vitalen bzw. hitzeinaktivierten M. ptb infiziert
und nach zwei Stunden nicht phagozytierte Bakterien durch Waschen entfernt. Zu
drei Zeitpunkten (2, 8 und 24 Stunden p. i.) wurden die Zellen hitzefixiert, nach Ziehl-
Neelsen gefärbt und infizierte sowie nicht-infizierte Zellen gezählt (siehe C.1.6).
In Abb. 2 sind die Infektionsraten von PBMs aus der Teflonkultivierung mit vitalen
und hitzeinaktivierten M. ptb im Verlauf von 24 Stunden prozentual als
Balkendiagramm mit Standardabweichung dargestellt. Zum Zwecke der
Übersichlichkeit sind auf der Y-Achse nur die Werte zwischen 92,5 und 100 %
aufgetragen. Es wurden Infektionsraten zwischen 94 und 98 % erreicht.
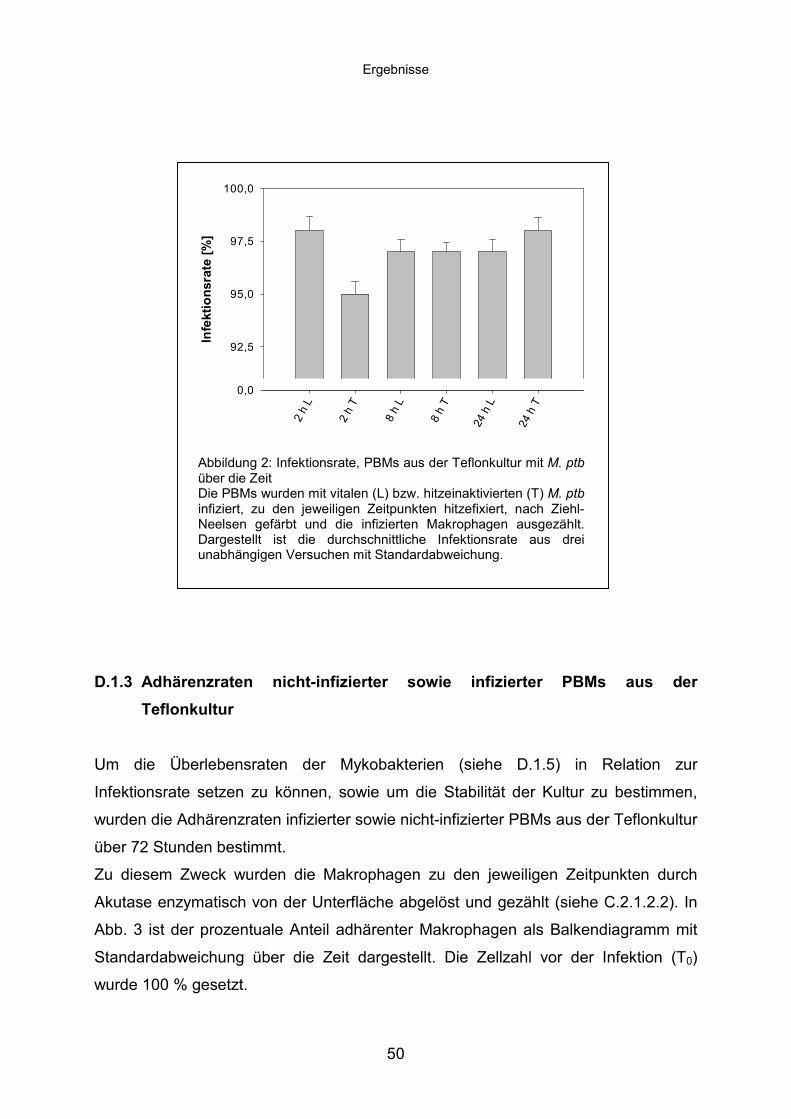
Ergebnisse
50
Abbildung 2: Infektionsrate, PBMs aus der Teflonkultur mit M. ptbüber die ZeitDie PBMs wurden mit vitalen (L) bzw. hitzeinaktivierten (T) M. ptbinfiziert, zu den jeweiligen Zeitpunkten hitzefixiert, nach Ziehl-Neelsen gefärbt und die infizierten Makrophagen ausgezählt.Dargestellt ist die durchschnittliche Infektionsrate aus dreiunabhängigen Versuchen mit Standardabweichung.
D.1.3 Adhärenzraten nicht-infizierter sowie infizierter PBMs aus derTeflonkultur
Um die Überlebensraten der Mykobakterien (siehe D.1.5) in Relation zur
Infektionsrate setzen zu können, sowie um die Stabilität der Kultur zu bestimmen,
wurden die Adhärenzraten infizierter sowie nicht-infizierter PBMs aus der Teflonkultur
über 72 Stunden bestimmt.
Zu diesem Zweck wurden die Makrophagen zu den jeweiligen Zeitpunkten durch
Akutase enzymatisch von der Unterfläche abgelöst und gezählt (siehe C.2.1.2.2). In
Abb. 3 ist der prozentuale Anteil adhärenter Makrophagen als Balkendiagramm mit
Standardabweichung über die Zeit dargestellt. Die Zellzahl vor der Infektion (T0)
wurde 100 % gesetzt.
2 h
L
2 h
T
8 h
L
8 h
T
24 h
L
24 h
T
Infe
ktio
nsra
te [%
]
0,0
92,5
95,0
97,5
100,0

Ergebnisse
51
Nach 72 Stunden waren noch 98 % der nicht infizierten PBMs adhärent; bei
infizierten Makrophagen lag diese Rate zwischen 60 und 90 %. Offensichtlich ist hier
ein deutlicher Unterschied zwischen der Infektion mit vitalen bzw. hitzeinaktivierten
M. ptb: in der Infektion mit vitalen M. ptb lag die Adhärenzrate zu den Zeitpunkten 48
bzw. 72 Stunden um 24 bzw. 18 % höher als in der Infektion mit hitzeinaktivierten
M. ptb.
Sowohl infizierte als auch nicht-infizierte PBMs zeichnen sich in diesem System
durch eine Adhärenzrate zwischen 60 und 98 % über einen Zeitraum von 72 Stunden
aus. Die Kultur ist also über den Untersuchungszeitraum stabil und damit für die
Versuche geeignet.
Abbildung 3: Adhärenzraten nicht-infizierter bzw. infizierterPBMs aus der TeflonkulturDie PBMs wurden mit Akutase enzymatisch abgelöst undgezählt.0 = vor der Infektion; 2, 8, 24, 48 und 72 sind die Zeitpunktepost infectionem.Dargestellt ist der Durchschnitt aus drei unabhängigenVersuchen mit Standardabweichung.
Zeit [h ]0 2 8 24 48 72
adhä
rent
e PB
Ms
[%]
0
20
40
60
80
100
nicht infiziertvita le M . ptbhitzeinaktivierte M . p tb

Ergebnisse
52
D.1.4 Stimulierbarkeit der PBMs aus der Teflonkultur
Als Kriterium für die Homogenität der Zellpopulation sollten Reproduzierbarkeit und
Gleichmäßigkeit der Stimulierbarkeit der PBMs getestet werden. Dies sollte auf Basis
der Expression ausgewählter, proinflammatorischer Zytokine durchgeführt werden.
Zu diesem Zweck wurden die Zellen mit LPS bzw. IFN-� behandelt. LPS ist
Bestandteil der Zellhülle gramnegativer Bakterien und ein potenter
Makrophagenstimulator; IFN-� wird von T-Zellen sezerniert und trägt wesentlich zur
endogenen Makrophagenaktivierung bei.
Überprüft wurden drei proinflammatorische Zytokine (IL-1�, IL-6 und TNF-�), GM-
CSF als Makrophagenaktivator, IL-10 als deaktivierendes Zytokin sowie die iNOS.
Die Makrophagen wurden wie in C.2.1 beschrieben isoliert und kultiviert. Nach drei
Tagen Adhärenz wurde frischem Medium 5 µg/ml LPS bzw. 10 U/ml IFN-� zugesetzt.
Nach weiterer Inkubation bei 37°C und 8 % CO2 wurden die PBMs zu den jeweiligen
Zeitpunkten abgekratzt und die RNA gewonnen. Nach Aufreinigung der RNA wurden
RT-PCRs mit ausgewählten Primern durchgeführt (siehe C.2.5.1)
Der zeitliche Verlauf der Expression ausgewählter Gene sowie die Höhe der
Expression innerhalb 24 Stunden unterscheiden sich wesentlich bei Stimulation mit
LPS oder mit IFN-�.
In den Abb. 4 und 5 ist die relative Extinktion der Genexpression als
Balkendiagramm mit Standardabweichung als Vielfaches der Kontrolle (K) über die
Zeit dargestellt.
Die Genexpression der proinflammatorischen Zytokine ist bei LPS-Stimulation durch
einen Verlauf mit einem „Peak“ nach 2 bzw. 8 Stunden gekennzeichnet. Bei
Stimulation mit IFN-� ist neben der geringeren Höhe der Genexpression ein früherer
„Peak“ (IL-1�), ein schnelleres Abfallen (IL-6) bzw. ein langsames Ansteigen ohne
„Peak“ (TNF-�) zu erkennen.
Auch die Expression der übrigen Gene zeichnet sich bei Stimulation mit LPS durch
einen Verlauf mit einem „Peak“ bei 2 bzw. 8 Stunden aus. Bei Stimulation mit IFN-�
ist wiederum die Höhe der Genexpression wesentlich geringer. Der zeitliche Verlauf
der IL-10-Genexpression ähnelt der nach LPS-Stimulation. Die Expression von GM-

Ergebnisse
53
CSF sowie der iNOS erreicht jedoch bei IFN-�-Stimulation nach 30 min einen leicht
erhöhten Wert, der als Plateau bestehen bleibt.
Die hier verwendeten PBMs aus der Teflonkultur sind also sowohl durch LPS, als
auch durch IFN-� stimulierbar. Die Expression der ausgewählten Gene unterscheidet
sich nach diesen beiden Stimulationen in der Höhe sowie im zeitlichen Verlauf.
D.1.5 Überleben von M. ptb in PBMs aus der Teflonkultur
Aus den vorherigen Versuchen leitet sich ab, daß PBMs aus der Teflonkultur unter
den beschrieben Bedingungen eine relativ homogene Zellpopulation darstellen.
Im Folgenden wurden die KBE (Kolonie-bildende Einheiten) pro Makrophage nach
Infektion von PBMs aus der Teflonkultivierung mit vitalen M. ptb ermittelt. Hierzu
wurden PBMs wie oben beschrieben mit M. ptb infiziert. Die Makrophagenzahl wurde
durch Auszählen festgehalten und in einem Paralellansatz die PBMs zerstört, so daß
die phagozytierten Bakterien frei wurden. Diese wurden anschließend in drei
Verdünnungsstufen ausplattiert und die KBE nach 8-10 Wochen gezählt. Durch
Verrechnen dieser beiden Werte ergab sich die Anzahl kolonie-bildender Einheiten
pro Makrophage.
In Abb. 6 ist das Verhältnis KBE/PBM als Balkendiagramm mit Standardabweichung
über die Zeit dargestellt.
Die KBE/PBM liegt durchschnittlich zwischen 0,3 und 0,4; pro 10 Makrophagen
entstanden also 3 bzw. 4 kolonie-bildende Einheiten. Während des hier untersuchten
Zeitraumes von 72 Stunden ändert sich dieses Verhältnis nicht.
Die Mykobakterien können in den PBMs aus der Teflonkultur überleben.

Ergebnisse
54
Abbildung 4: Expression der proinflammatorischen Gene IL-1� (oben), IL-6 (Mitte) undTNF-� (unten)Die PBMs wurden wie in C.1.1 beschrieben isoliert und kultiviert. Nach drei TagenAdhärenz wurde frischem Medium 5 µg/ml LPS (jeweils links) bzw. 10 U/ml IFN-� (jeweilsrechts) zugesetzt. Nach weiterer Inkubation bei 37°C und 8 % CO2 wurden die PBMs zuden jeweiligen Zeitpunkten abgekratzt und die RNA gewonnen. Nach Aufreinigung derRNA wurden RT-PCRs mit ausgewählten Primern durchgeführt. K ist die nicht stimulierteKontrolle.Dargestellt ist der Durchschnitt aus vier Reversen Transkriptionen zweier unabhängigerVersuche als Vielfaches von K über die Zeit.
LPS: IL-1 beta
K 0,5 h 2 h 8 h 24 h
rel.
Gen
expr
essi
on
0
20
40
60
80
100
120
IFN-gamma: IL-1 beta
K 0,5 h 2 h 8 h 24 h
rel.
Gen
expr
essi
on
0,0
0,5
1,0
1,5
2,0
2,5
3,0
LPS: IL-6
K 0,5 h 2 h 8 h 24 h
rel.
Gen
expr
essi
on
0
10
20
30
40
50
60
IFN-gamma: IL-6
K 0,5 h 2 h 8 h 24 h
rel.
Gen
expr
essi
on
02468
10121416
LPS: TNF-alpha
K 0,5 h 2 h 8 h 24 h
rel.
Gen
expr
essi
on
0,0
0,5
1,0
1,5
2,0
2,5
3,0
3,5IFN-gamma: TNF-alpha
K 0,5 h 2 h 8 h 24 h
rel.
Gen
expr
essi
on
0,0
0,5
1,0
1,5
2,0
2,5
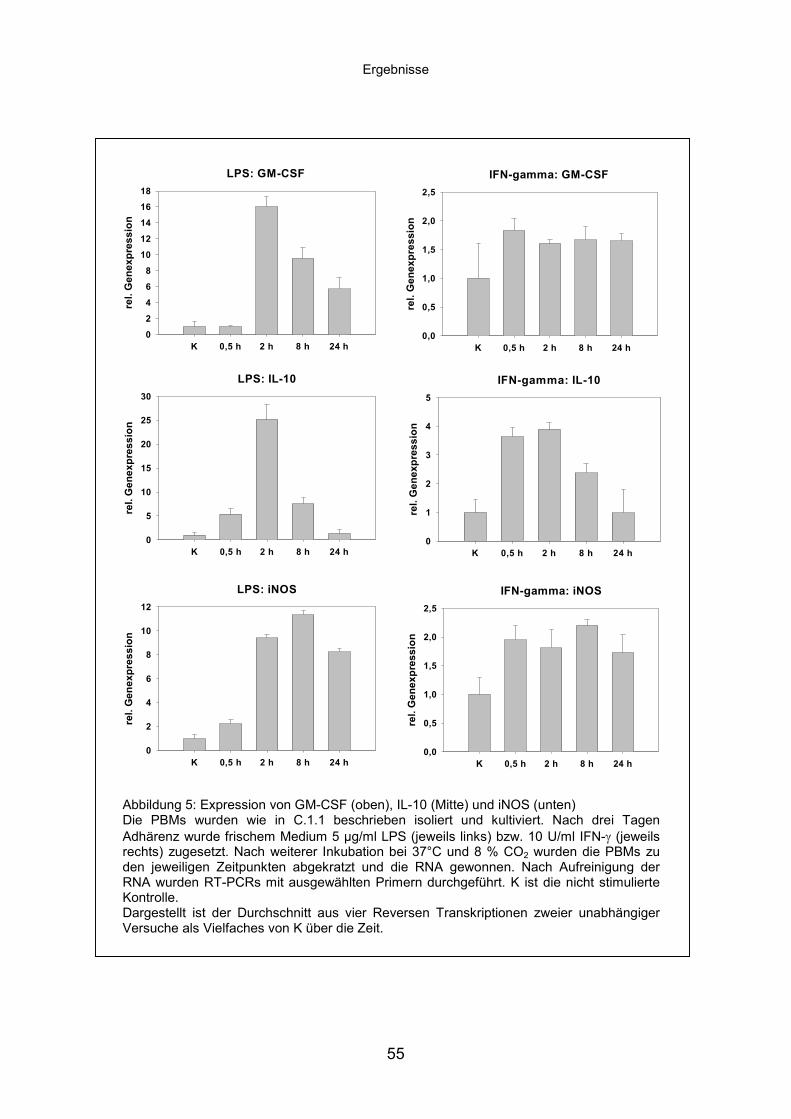
Ergebnisse
55
Abbildung 5: Expression von GM-CSF (oben), IL-10 (Mitte) und iNOS (unten)Die PBMs wurden wie in C.1.1 beschrieben isoliert und kultiviert. Nach drei TagenAdhärenz wurde frischem Medium 5 µg/ml LPS (jeweils links) bzw. 10 U/ml IFN-� (jeweilsrechts) zugesetzt. Nach weiterer Inkubation bei 37°C und 8 % CO2 wurden die PBMs zuden jeweiligen Zeitpunkten abgekratzt und die RNA gewonnen. Nach Aufreinigung derRNA wurden RT-PCRs mit ausgewählten Primern durchgeführt. K ist die nicht stimulierteKontrolle.Dargestellt ist der Durchschnitt aus vier Reversen Transkriptionen zweier unabhängigerVersuche als Vielfaches von K über die Zeit.
LPS: GM-CSF
K 0,5 h 2 h 8 h 24 h
rel.
Gen
expr
essi
on
02468
1012141618
IFN-gamma: GM-CSF
K 0,5 h 2 h 8 h 24 h
rel.
Gen
expr
essi
on
0,0
0,5
1,0
1,5
2,0
2,5
LPS: IL-10
K 0,5 h 2 h 8 h 24 h
rel.
Gen
expr
essi
on
0
5
10
15
20
25
30IFN-gamma: IL-10
K 0,5 h 2 h 8 h 24 h
rel.
Gen
expr
essi
on
0
1
2
3
4
5
LPS: iNOS
K 0,5 h 2 h 8 h 24 h
rel.
Gen
expr
essi
on
0
2
4
6
8
10
12IFN-gamma: iNOS
K 0,5 h 2 h 8 h 24 h
rel.
Gen
expr
essi
on
0,0
0,5
1,0
1,5
2,0
2,5

Ergebnisse
56
Abbildung 6: Anzahl kolonie-bildender Einheiten pro PBMDie PBMs aus der Teflonkultur wurden nach demStandardinfektionsmodell infiziert. Die Anzahl PBMs wurdegezählt, die PBMs zerstört und die so frei gewordenen Bakterien indrei Verdünnungsstufen ausgesät. Nach sechs bis acht Wochenwurde die Zahl kolonie-bildender Einheiten festgehalten.Dargestellt ist das Verhältnis KBE pro PBM über die Zeit alsDurchschnitt aus drei unabhängigen Versuchen mitStandardabweichung.
D.2 Funktionelle Untersuchungen in PBMs aus der Teflonkultur
D.2.1 Elektronenmikroskopie
Anhand der elektronenmikroskopischen Aufnahmen sollte bestätigt werden, daß
M. ptb in primären bovinen Makrophagen aus der Teflonkultur in phagosomalen
Vakuolen liegt. Zu diesem Zweck wurden die PBMs wie beschrieben kultiviert und
infiziert (siehe C.2.1 und C.2.2). Die infizierten Makrophagen wurden mit
Formaldehyd und Osmiumtetroxid fixiert, in Aceton dehydriert und schließlich in
Spurr resin eingebettet. Mit Glasmessern wurden Ultradünnschnitte der
Makrophagen angefertigt und im Elektronenmikroskop durchgemustert (siehe
C.2.3.1).
Zeit [h ]0 2 8 24 48 72
KB
E/PB
M
0,0
0,1
0,2
0,3
0,4
0,5
0,6
0,7

Ergebnisse
57
In Abb. 7 ist beispielhaft eine elektronenmikroskopische Aufnahme eines infizierten
Makrophagen abgebildet. Wie deutlich zu erkennen (Pfeil) ist der Erreger von einer
Membran umschlossen. Dies konnte bei sämtlichen Aufnahmen bestätigt werden.
M. ptb liegt in primären bovinen Makrophagen also in Phagosomen.
Abb 7: Elektronenmikroskopische Aufnahme von M. ptb in PBMs ausder Pfeil weist auf die deutlich zu erkennende vakuoläre Membran; Makrophagen
D.2.2 Vitale M. ptb alkalisieren das endosomale Netzwerk
Nach der Phagozytose befinden sich Bakterien in Phagosomen m
Wert (pH 6,3-6,5), welche Marker früher Endosomen tragen. Sie
Minuten bis wenigen Stunden heran und tragen dann Marker
Hier liegt der pH-Wert bei 4,5-5,5 (CLEMENS 1996). In dies
m
0,1 µder Teflonkultur;Ausschnitt eines
it leicht saurem pH-
reifen innerhalb von
später Endosomen.
em sauren pH-Wert

Ergebnisse
58
haben Enzyme das optimale Mileu zum Verdau des phagozytierten Materials. Somit
stellt die Ansäuerung des Phagosoms einen wichtigen Schritt in der
Phagosomenreifung dar (siehe auch C.2.3).
SIBLEY et al. stellten 1985 ein Verhinderung der Ansäuerung in Toxoplasma gondii-
haltigen Phagosomen fest. STURGILL-KOSZYCKI et al. konnten dies 1994 für
M. avium-haltige Phagosomen zeigen; auch andere Bakterien und Pilze sind dazu in
der Lage (MALIK et al. 2000, KAPOSZTA et al. 1999, BLACK et al. 1996). Eine
mögliche Erklärung ist das Fehlen der Protonen-ATPase in der Phagosomen-
membran. Diese ist ein Enzymkomplex, der für die Ansäuerung von intrazellulären
Kompartimenten zuständig ist und im besonderen den pH-Wert in Phagosomen
absenkt.
Bisher ist nicht bekannt, ob M. ptb in bovinen Makrophagen ebenfalls, wie in murinen
Mausmarkophagen bereits von KUEHENEL et al. (2001) beschrieben, die
Ansäuerung verhindert. Um dies zu untersuchen wurde eine von KUEHNEL et al.
(2001) etablierte und für diese Arbeit modifizierte Methode der intraphagosomalen
pH-Wert-Messung mit FITC-Dextran verwendet. FITC ist ein Fluorochrom, welches
bei einer Anregungswellenlänge von 488 nm pH-abhängig fluoresziert
(SCHLESINGER 1994), je niedriger der pH-Wert, desto geringer die Fluoreszenz.
Die internalisierte Menge an FITC-Dextran wurde zwischen den Proben anhand der
nicht-pH-abhängigen Fluoreszenz bei 546 nm normalisiert. FITC-Dextran wird von
der Zelle aufgenommen, wo es anfänglich in frühe Endosomen, in der weiteren
Endosomenreifung in späte Endosomen und Lysosomen gelangt (SCHLESINGER
1994). Eine Änderung des pH-Wertes wird als Änderung der relativen Fluoreszenz-
intensität angezeigt.
Anhand der konfokalen Laserscanning Mikroskopie, kurz Konfokalmikroskopie,
wurde geklärt, ob FITC-Dextran mit den Mykobakterien in Phagosomen kolokalisiert
ist, Voraussetzung für eine korrekte Erfassung des intraendosomalen pH-Wertes.
Um dies zu zeigen, wurden die Mykobakterien mit Sulfo-NHS-Biotin markiert. Die
PBMs wurden mit diesen Mykobakterien sowie FITC-Dextran für zwei Stunden
koinkubiert. Anschließend wurde TexasRed-Streptavidin zugegeben, welches an

Ergebnisse
Biotin bindet und bei 546 nm Anregungswellenlänge eine Rotfluoreszenz zeigt. Nach
dreimaligem Waschen wurden die Präparate fixiert und unter dem
Konfokalmikroskop mikroskopiert. Die Mykobakterien stellten sich rot dar, FITC-
Dextran grün. Eine Kolokalisierung der Mykobakterien mit dem FITC-Dextran wurde
durch Gelbfluoreszenz angezeigt.
Wie in Abb. 8 beispielhaft zu erkennen, war sowohl in den Infektionen mit vitalen, als
auch in den Infektionen mit hitzeinaktivierten M. ptb ca. 70-80 % der FITC-Dextran-
Partikel mit den Mykobakterien kolokalisiert. Das System zur intraendosomalen pH-
Messung konnte dementsprechend verwendet werden.
Ab g 8: Kolokalisierung von FITC-Dextran mit Mykobakterien in PBMs.DiehitzDieGe
Zur Be
FACS
durchg
Die P
koinku
bildun10 µm
59
PBMs aus der Teflonkultur wurden mit Teinaktivierten M. ptb (rechts) infiziert, bei gleic Mykobakterien fluoreszieren rot, FITC-Dexlbfluoreszenz angezeigt.
stimmung des intraendosomalen pH-W
-Analyse die Messung anhand der ber
eführt.
BMs wurden mit vitalen bzw. hitzein
biert, nach zwei Stunden gewasch
10 µm
10 µmexasRed-markierten vitalen (links) bzw.hzeitiger Koinkubation mit FITC-Dextran.tran grün; Kolokalisierung wird durch
ertes nach einer Infektion wurde in der
eits erwähnten Fluoreszenz von FITC
aktivierten M. ptb und FITC-Dextran
en, mit Akutase enzymatisch vom

Ergebnisse
60
Untergrund abgelöst und auf acht Reaktionsgefäße verteilt. In sieben dieser Gefäße
wurden zur Erstellung der Eichkurve mit Nigericin, einem Ionophor, pH-Werte von 5,5
bis 8,0 angelegt. Im achten Gefäß wurde der intrazelluläre pH-Wert in PBS
gemessen. Die ermittelten pH-abhängigen Fluoreszenzen bei 480 nm wurden
anhand der pH-unabhängigen Fluoreszenz bei 546 nm normalisiert.
Abb. 9 zeigt die Eichkurve als pH-Wert in Abhängigkeit zur relativen Fluoreszenz.
Dargestellt ist der Durchschnitt aus sechs Versuchen als Kurvendiagramm mit
Standardabweichung. Die relative Fluoreszenz kommt zustande, indem der jeweils
höchste Wert 100 % gesetzt und die übrigen Werte entsprechend verrechnet
wurden. Die ermittelten relativen Fluoreszenzen der Proben konnten nun anhand der
Eichkurve als pH-Werte abgelesen werden und sind in Tab. 2 aufgeführt.
In der Infektion mit vitalen M. ptb lag der durchschnittliche pH-Wert aus sechs
Versuchen bei 6,97, in der Infektion mit hitzeinaktivierten M. ptb bei pH 5,19.
Das heißt, vitale M. ptb sind nicht nur in der Lage, die intraphagosomale Ansäuerung
in PBMs vollständig zu verhindern, sie führen sogar zu einer leichten Alkalisierung,
da der normale pH-Wert früher Endosomen bei 6,3-6,5 liegt (FUCHS et al. 1989).
Hitzeinaktivierte M. ptb können die Ansäuerung nicht verhindern.
Tab. 2: relative Fluoreszenz und sich daraus ergebende pH-Werte bei Infektionmit vitalen bzw. hitzeinaktivierten PBMs aus der Teflonkultur
rel. Fluoreszenz [%] errechneter pH-Wert
vitale M. ptb 90,22 6,97
hitzeinaktivierte M. ptb 61,36 5,19
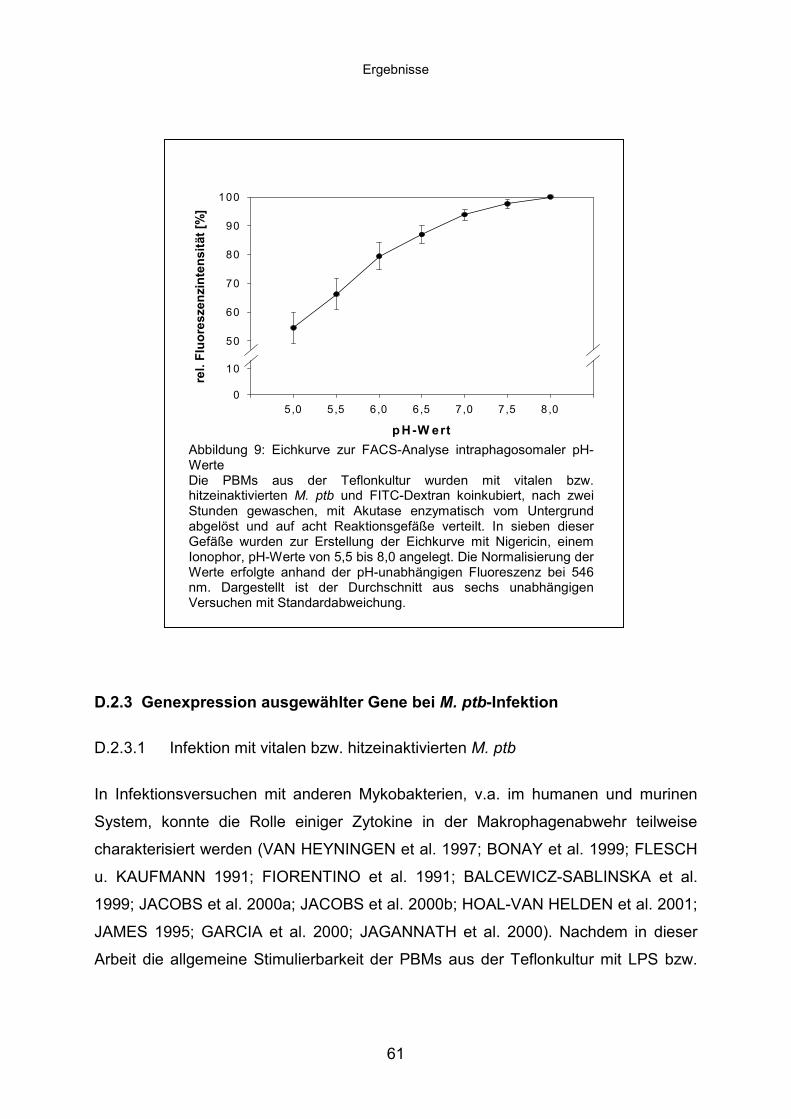
Ergebnisse
61
Abbildung 9: Eichkurve zur FACS-Analyse intraphagosomaler pH-WerteDie PBMs aus der Teflonkultur wurden mit vitalen bzw.hitzeinaktivierten M. ptb und FITC-Dextran koinkubiert, nach zweiStunden gewaschen, mit Akutase enzymatisch vom Untergrundabgelöst und auf acht Reaktionsgefäße verteilt. In sieben dieserGefäße wurden zur Erstellung der Eichkurve mit Nigericin, einemIonophor, pH-Werte von 5,5 bis 8,0 angelegt. Die Normalisierung derWerte erfolgte anhand der pH-unabhängigen Fluoreszenz bei 546nm. Dargestellt ist der Durchschnitt aus sechs unabhängigenVersuchen mit Standardabweichung.
D.2.3 Genexpression ausgewählter Gene bei M. ptb-Infektion
D.2.3.1 Infektion mit vitalen bzw. hitzeinaktivierten M. ptb
In Infektionsversuchen mit anderen Mykobakterien, v.a. im humanen und murinen
System, konnte die Rolle einiger Zytokine in der Makrophagenabwehr teilweise
charakterisiert werden (VAN HEYNINGEN et al. 1997; BONAY et al. 1999; FLESCH
u. KAUFMANN 1991; FIORENTINO et al. 1991; BALCEWICZ-SABLINSKA et al.
1999; JACOBS et al. 2000a; JACOBS et al. 2000b; HOAL-VAN HELDEN et al. 2001;
JAMES 1995; GARCIA et al. 2000; JAGANNATH et al. 2000). Nachdem in dieser
Arbeit die allgemeine Stimulierbarkeit der PBMs aus der Teflonkultur mit LPS bzw.
p H -W ert5,0 5,5 6,0 6,5 7,0 7,5 8 ,0
rel.
Fluo
resz
enzi
nten
sitä
t [%
]
0
10
50
60
70
80
90
100

Ergebnisse
62
IFN-� bereits anhand ausgewählter Gene untersucht wurde, wurde nun die
Genexpression derselben Gene nach Infektion mit M. ptb untersucht. Um Folgen
sezernierter bzw. struktureller Bestandteile voneinander unterscheiden zu können,
wurden auch hier hitzeinaktivierte M. ptb neben vitalen eingesetzt.
Die Mykobakterien und Makrophagen wurden wie in C.2.1 beschrieben kultiviert. Die
Infektion erfolgte als Standardinfektion wie in C.2.2.1 beschrieben. Zu den jeweiligen
Zeitpunkten wurden die Makrophagen abgekratzt, die RNA isoliert und eine RT-PCR
mit verschiedenen spezifischen Primern durchgeführt. Mit Ethidiumbromid wurden
die PCR-Produkte sichtbar gemacht und die Bandenintensität im Biorad-System
festgehalten. Dann erfolgte eine Normalisierung anhand des Haushaltsgenes
�-Actin. Zur weiteren Berechnung siehe C.2.5.3.
In Abb. 10 ist die relative Genexpression ausgewählter Gene als Vielfaches der nicht
stimulierten Kontrolle (K) über die Zeit als Balkendiagramm mit Standardabweichung
dargestellt.
Der auffallendste Unterschied ist die höhere Expression aller untersuchter Gene in
der Infektion mit vitalen M. ptb im Vergleich zur Infektion mit hitzeinaktivierten M. ptb.
Die Unterschiede liegen zwischen 25 und 700 %.
Außerdem unterscheidet sich bei zwei Zytokinen der zeitliche Verlauf der
Expression:
Die GM-CSF-Genexpression erreicht in der Infektion mit vitalen M. ptb bereits nach
2 Stunden den höchsten Wert („Peak“) und fällt dann langsam ab. In der Infektion mit
hitzeinaktierten M. ptb ist der „Peak“ erst nach 8 Stunden erreicht.
Noch deutlicher ist der Unterschied in der IL-6-Genexpression: in der Infektion mit
vitalen M. ptb ist ein biphasischer Verlauf mit dem ersten „Peak“ nach 2, dem zweiten
nach 24 Stunden zu erkennen. In der Infektion mit hitzeinaktivierten M. ptb handelt
es sich um einen monophasischen Verlauf mit gleichhohen Werten nach 2 sowie 8
Stunden.
Es gibt also deutliche Unterschiede in der Genexpression der ausgewählten Gene
bei Infektionen mit vitalen im Vergleich zu hitzeinaktivierten M. ptb. Dies spricht
dafür, daß zumindest ein Teil der Effekte auf von vitalen Bakterien sezernierten
Bestandteilen beruht.

Ergebnisse
63
Abb. 10: Expression ausgewählter Gene der nicht stimulierten Kontrolle (K) sowie beiStimulation mit vitalen (schwarz) und hitzeinaktivierten (grau) M. ptb als Vielfaches derKontrolle über die Zeit.Die PBMs wurden wie in C.1.1 beschrieben isoliert und kultiviert. Nach drei TagenAdhärenz erfolgte die Infektion mit den Mykobakterien für zwei Stunden. Nach Waschenund weiterer Inkubation bei 37°C und 8 % CO2 wurden die PBMs zu den jeweiligenZeitpunkten abgekratzt und die RNA gewonnen. Nach Aufreinigung der RNA wurden RT-PCRs mit ausgewählten Primern durchgeführt.Dargestellt ist der Durchschnitt aus vier Reversen Transkriptionen zweier unabhängigerVersuche als Vielfaches von K über die Zeit.
IL-1 beta
K 2 h 8 h 24 h
rel.
Gen
expr
essi
on
0
5
10
15
20
25
30
35
40lebendtot
GM-CSF
K 2 h 8 h 24 h
rel.
Gen
expr
essi
on
0
5
10
15
20
25lebendtot
IL-6
K 2 h 8 h 24 h
rel.
Gen
expr
essi
on
0
1
2
3
4
5
6
7
8lebendtot
IL-10
K 2 h 8 h 24 h
rel.
Gen
expr
essi
on
0,0
0,5
1,0
1,5
2,0
2,5
3,0lebendtot
TNF-alpha
K 2 h 8 h 24 h
rel.
Gen
expr
essi
on
0
2
4
6
8
10
12
14
16lebendtot
iNOS
K 2 h 8 h 24 h
rel.
Gen
expr
essi
on
0
1
2
3
4
5lebendtot

Ergebnisse
64
D.2.3.2 Vergleich der Genexpression nach verschiedenen Stimulationen
Es stellte sich die Frage, ob die ermittelten Zytokinmuster Hinweise auf dieSignaltransduktion im Makrophagen liefern.LPS ist ein potenter Makrophagenaktivator mit bekannten Signalwegen. NachBindung von LPS an CD14, TLR („toll like receptor“) oder andere LPS-Rezeptoren isteine Tyrosin-Phosphorylierung an der weiteren Signaltransduktion beteiligt(KOVARIK et al. 1998; MEANS et al. 1999). Es ist jedoch noch unklar, welcheTyrosinkinasen diese Phosphorylierung durchführen.Bekannt ist außerdem, daß LAM, also das LPS der Mykobakterien, ebenfalls anCD14, aber auch an andere Rezeptoren als LPS bindet (MEANS et al. 1999). LPSkann TLR nur in Anwesenheit von CD14 aktivieren, LAM aktviert TLR auchunabhängig von CD14. M. tuberculosis aktiviert TLR2 und TLR4, M. avium nur TLR2.Um Hinweise auf die Aktivierung von Signalwegen durch M. ptb zu finden, wurdendie zeitlichen Verläufe der Expression der ausgewählten Gene nach Stimulation mitLPS und Infektion mit Mykobakterien miteinander verglichen.In Abb. 11 sind die Verläufe der Expression der ausgewählten Gene infolge aller vierStimulationen im Vergleich aufgetragen. Benutzt wurden die Durchschnittswerte der
Abb. 4, 5 und 10. Die Höhe der Expression nach LPS- und IFN-�-Stimulation hängtvon der verwendeten Konzentration der beiden Stoffe ab. Um die Werte unabhängigvon der Höhe nur im zeitlichen Verlauf miteinander vergleichen zu können, wurdedaher der jeweils höchste Wert 100 % gesetzt und die übrigen entsprechend demDreisatz berechnet.Die zeitlichen Verläufe der Genexpression von IL-10 bzw. der iNOS ist nach allenvier Stimulation ähnlich, mit einem „Peak“ nach 2 bzw. 8 Stunden.Die Genexpression der vier übrigen Zytokine weist jedoch Unterschiede zwischenden verschiedenen Stimulationen auf:
Nach Stimulation mit LPS bzw. Infektion mit Mykobakterien steigt die IL-1�-Genexpression von unter 10 % auf 100 % nach 8 Stunden an und fällt dann auf rund50 % (LPS und vitale M. ptb) bzw. 12 % (hitzeinaktivierte M. ptb) ab. Nach
Stimulation mit IFN-� erfolgt ein rascher Anstieg von 74 % auf 100 % nach 2 Stundenund ein schnelles Abfallen auf 19 % nach 8 Stunden. Nach 24 Stunden war keineGenexpression mehr detektierbar.

Ergebnisse
65
Die Expression des IL-6-Gens verläuft in den ersten beiden Stunden nach allen
Stimulationen ähnlich, mit einem Anstieg von 2-30 % auf 100 %. Nach IFN-�-Stimulation erfolgt dann ein schnelles Abfallen zum Zeitpunkt 8 h auf 6 %; nach24 Stunden ist keine Genexpression mehr detektierbar. Nach LPS-Stimulation fälltdie Expression von IL-6 auf 42 % nach 8 Stunden und 30 % nach 24 Stunden. BeiInfektion mit hitzeinaktivierten M. ptb bleibt die Expression über 8 Stundenannähernd konstant und fällt dann nach 24 Stunden auf 77 % ab. Bei Infektion mitvitalen M. ptb sinkt die Genexpression von IL-6 nach 8 Stunden auf 57 % und steigtnach 24 Stunden wieder auf 91 % an, so daß sich ein biphasischer Verlauf mit zwei„Peaks“ ergibt.
Nach LPS-Stimulation steigt die TNF-�-Genexpression schnell von 32 % auf 100 %nach 2 Stunden an und sinkt anschließend langsam nach 8 Stunden auf 81 %, nach24 Stunden auf 62 %. Nach Infektion mit vitalen bzw. hitzeinaktivierten M. ptb erfolgtebenfalls ein schnelles Ansteigen: von 7 bzw. 14 % auf 100 %. Anschließend sinktdie Genexpression jedoch rasch auf 28 bzw. 9 % und ist nach 24 Stunden nur noch
gering mit 1 % bzw. nicht mehr zu detektieren. Nach Stimulation mit IFN-� steigt die
Expression des TNF-�-Gens langsam von 56 % auf 66 % nach 2 Stunden, 76 %nach 8 Stunden und erreicht nach 24 Stunden schließlich 100 %.
Nach Stimulation mit IFN-� steigt die Expression des GM-CSF-Gens von 60 % auf96 % nach 2 Stunden an und bleibt bis 24 Stunden annähernd gleich (100 % bzw.99 %). Bei Infektion mit hitzeinaktivierten M. ptb erfolgt ein Ansteigen von 8 % auf68 % nach 2 Stunden und 100 % nach 8 Stunden. Anschließend fällt die Expressionauf 71 % nach 24 Stunden. Bei Stimulation mit LPS bzw. Infektion mit vitalen M. ptbsteigt die Genexpression von 6 bzw. 5 % auf 100 % nach 2 Stunden. Nach LPS-Stimulation fällt sie dann nach 8 Stunden auf 60 %, nach 24 Stunden auf 36 %. NachInfektion mit vitalen M. ptb erfolgt das Absinken langsamer auf 94 % nach 8 Stundenund 88 % nach 24 Stunden.Die zeitlichen Verläufe der Genexpression von IL-10 und der iNOS sind also nach
Stimulation mit LPS oder IFN-� bzw. nach Infektion mit vitalen oder hitzeinaktiviertenM. ptb ähnlich. Die Expression der übrigen untersuchten Gene unterscheidet sichmehr oder weniger stark (siehe oben). Besonders auffällig ist hier der biphasischeVerlauf der IL-6-Expression nach Infektion mit vitalen M. ptb.
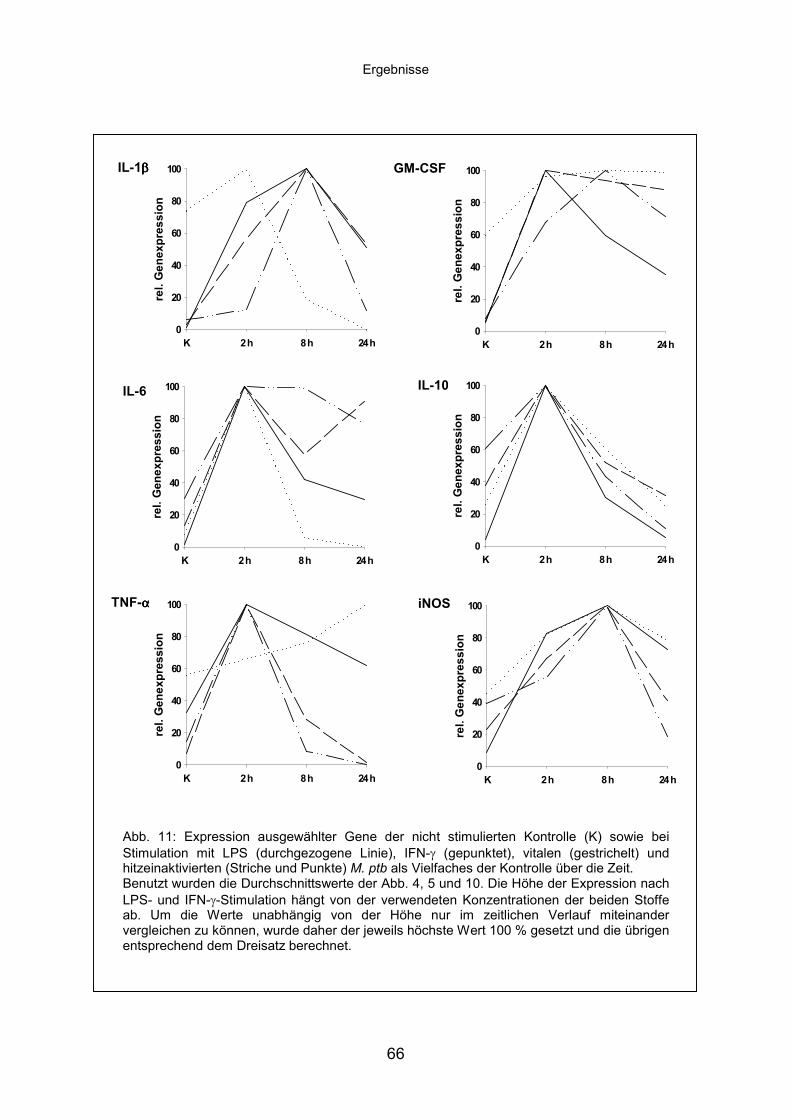
Ergebnisse
66
Abb. 11: Expression ausgewählter Gene der Stimulation mit LPS (durchgezogene Linie), IFhitzeinaktivierten (Striche und Punkte) M. ptb als Benutzt wurden die Durchschnittswerte der Abb.LPS- und IFN-�-Stimulation hängt von der verwab. Um die Werte unabhängig von der Höhvergleichen zu können, wurde daher der jeweils hentsprechend dem Dreisatz berechnet.
IL-1����
IL-6
TNF-����
GM-CSF
K 2 h 8 h 24 h
rel.
Gen
expr
essi
on
0
20
40
60
80
100
K 2 h 8 h 24 h
rel.
Gen
expr
essi
on
0
20
40
60
80
100
K 2 h 8 h 24 h
rel.
Gen
expr
essi
on
0
20
40
60
80
100 100
K 2 h 8 h 24 h
rel.
Gen
expr
essi
on
0
20
40
60
80
100
IL-10
nicht stimulierten Kontrolle (K) sowie beiN-� (gepunktet), vitalen (gestrichelt) und
Vielfaches der Kontrolle über die Zeit. 4, 5 und 10. Die Höhe der Expression nachendeten Konzentrationen der beiden Stoffee nur im zeitlichen Verlauf miteinanderöchste Wert 100 % gesetzt und die übrigen
iNOS
K 2 h 8 h 24 h
rel.
Gen
expr
essi
on
0
20
40
60
80
K 2 h 8 h 24 h
rel.
Gen
expr
essi
on
0
20
40
60
80
100

Ergebnisse
67
D.2.4 Bafilomycin verhindert die Ansäuerung in der Infektion mithitzeinaktivierten M. ptb, verursacht jedoch keine entsprechendenGenexpressionsmuster
Wie in D.2.2 gezeigt sind vitale M. ptb in der Lage, die Ansäuerung des Phagosoms
fast vollständig zu verhindern, wohingegen hitzeinaktivierte M. ptb dies nicht können.
Es konnte ebenfalls gezeigt werden, daß der zeitliche Verlauf der Expression
ausgewählter Gene sich nach Infektion mit vitalen M. ptb bzw. mit hitzeinaktivierten
M. ptb unterschied (IL-6 und GM-CSF). Außerdem lag die Expression sämtlicher
untersuchter Gene nach Infektion mit vitalen M. ptb zwischen 25 und 700 % über der
nach Infektion mit hitzeinaktivierten M. ptb.
Nun wurde untersucht, ob diese beiden durch vitale M. ptb verursachten Phänomene
im Zusammenhang stehen.
Die Ansäuerung des Phagosoms erfolgt durch in der Membran befindliche ATPasen,
also ATP-betriebene Protonenpumpen, die einen Wasserstoff-Gradienten aufbauen
und so den intraphagosomalen pH-Wert verändern (FUCHS et al. 1989). Protonen-
ATPasen können durch den Wirkstoff Bafilomycin blockiert werden (BOWMAN et al.
1988). Es stellte sich die Frage, ob bei einer Infektion mit hitzeinaktivierten M. ptb
und gleichzeitiger Gabe von Bafilomycin der Effekt vitaler M. ptb, also die
Verhinderung der Ansäuerung sowie die erhöhte bzw. im zeitlichen Verlauf
veränderte Genexpression imitiert werden kann. Dies sollte wie in D.2.2 beschrieben
durch FACS-Analyse der mit FITC-Dextran koinkubierten, infizierten PBMS (siehe
unten) untersucht werden.
Zu diesem Zweck wurden die PBMs aus der Teflonkultur 15 min vor der Infektion mit
100 µM Bafilomycin inkubiert. Auch während der Infektion mit den hitzeinaktivierten
M. ptb wurde neben dem FITC-Dextran 100 µM Bafilomycin zugesetzt. Nach zwei
Stunden Infektion wurden die PBMs gewaschen, mit Akutase enzymatisch abgelöst
und in der FACS-Analyse (siehe auch D.2.2.2) eingesetzt.
Wie in D.2.2 beschrieben, wurde der intraphagosomale pH-Wert durch Unterschiede
der Fluoreszenzintensität anhand einer Eichkurve (Abb. 9) abgelesen.

Ergebnisse
68
Bei Infektion mit hitzeinaktivierten M. ptb und gleichzeitiger Gabe von Bafilomycin
wurde eine relative Fluoreszenintensität von 96,58 % gemessen, was anhand der
Eichkurve als pH 7,41 abgelesen werden konnte.
Bafilomycin ist somit in der Lage, die Ansäuerung des Phagosoms bei Infektion mit
hitzeinaktivierten M. ptb zu verhindern und somit den Effekt vitaler M. ptb zu
imitieren.
Bezüglich des intraphagosomalen pH-Wertes konnte mit Bafilomycin also der Effekt
vitaler M. ptb in der Infektion mit hitzeinaktivierten M. ptb imitiert werden. Ob dies
auch bezüglich der Expression ausgewählter Gene möglich ist, sollte anhand der RT-
PCR überprüft werden.
Der prinzipielle Versuchsaufbau folgt dem in D.2.3.1 beschriebenen Prinzip. Die
Zugabe von Bafilomycin erfolgte auch hier zum einen 15 min vor der Infektion mit
den hitzeinaktivierten M. ptb, zum anderen während der Infektion in einer
Konzentration von 100 µM. Zur Kontrolle wurden nicht infizierte PBMs in derselben
Konzentration mit Bafilomycin behandelt.
Die PBMs wurden wie in D.2.3.1 beschrieben abgkratzt, die RNA aufgereinigt und
RT-PCRs mit spezifischen Primern für IL-6, TNF-� und iNOS durchgeführt.
In Abb. 12 ist die relative Expression der drei genannten Gene als Balkendiagramm
mit Standardabweichung dargestellt. Bafilomycin alleine hat nur einen sehr geringen
Effekt auf die Expression der drei Gene. Die Unterschiede zwischen der Infektion mit
vitalen bzw. hitzeinaktivierten M. ptb wurden reproduziert: in der Infektion mit vitalen
M. ptb lag die Expression um ca. 40-50 % über der bei Infektion mit hitzeinaktivierten
Bakterien. Bei gleichzeitiger Gabe von Bafilomycin zur Infektion mit hitzeinaktivierten
M. ptb lag die Expression der drei Gene jedoch unter den Werten bei Infektion mit
hitzeinaktivierten M. ptb ohne die Gabe von Bafilomycin.
Die Gabe von Bafilomycin zur Infektion mit hitzeinaktivierten M. ptb konnte die
Expressionsmuster der Infektion mit vitalen M. ptb also nicht imitieren.
Die verhinderte Ansäuerung des Phagosoms und die veränderten Zytokin-
expressionsmuster scheinen also zwei voneinander unabhängige Beeinflussungen
des Makrophagen durch vitale M. ptb zu sein.

Ergebnisse
69
Abbildung 12: Expression des Il-6- (oben), TNF-�- (Mitte)und iNOS-Gens (unten) nach 2 Stunden als Vielfaches derKontrolle. K = nicht-stimulierte Kontrolle; KB = PBMs, die nurmit Bafilomycin stimuliert wurden; L = Infektion mit vitalenM. ptb; T = Infektion mit hitzeinaktivierten M. ptb; TB =Infektion mit hitzeinaktivierten M. ptb bei gleichzeitigerStimulation mit Bafilomycin.Die Zugabe von Bafilomycin erfolgte zum einen 15 min. vordem Infektionszeitpunkt, zum anderen während derInfektionsphase in einer Konzentration von 100 µM (sieheauch D.2.3.1). Nach 2 Stunden wurde die PBMs gewaschenund abgekratzt, die RNA aufgereinigt und RT-PCRs mitspezifischen Primern durchgeführt.Dargestellt ist der Durchschnitt aus vier ReversenTranskriptionen zweier unabhängiger Versuche.
K K B L T T B
rel.
Gen
expr
essi
on
0
2
4
6
8
1 0
K K B L T T B
rel.
Gen
expr
essi
on
0 , 0
2 ,5
5 ,0
7 ,5
1 0 ,0
1 2 ,5
1 5 ,0
1 7 ,5
K K B L T T B
rel.
Gen
expr
essi
on
0
2
4
6
8
1 0

Ergebnisse
70
D.2.5 deaktivierende Effekte
LPS führt zur Aktivierung von Makrophagen und im Folgenden u. a. zur
Ausschüttung von TNF-�, IL-6 und IL-12. Eine Vorinkubation mit LPS führte jedoch
zu einer reduzierten Sensitivtiät auf weitere LPS-Gaben. Dieses Phänomen wird
LPS-Toleranz, „LPS-hyporesponsiveness“ oder Refraktärität genannt. In vivo wurden
hierbei eine verminderte Fieberantwort und geringere Letalität vermerkt. In vitro ging
mit dem Phänomen eine verminderte Produktion proinflammatorischer Zytokine nach
Sekundärstimulation mit LPS einher (NOMURA et al. 2000).
Ob eine solche Refraktärität auch in bovinen Makrophagen nach Infektion mit vitalen
bzw. hitzeinaktivierten M. ptb auftritt, wurde in dieser Arbeit untersucht.
Hierzu wurden Makrophagen mit vitalen bzw. mit hitzeinaktivierten M. ptb für zwei
Stunden infiziert (t=0). Nach weiteren 20 Stunden (t=22) wurden die Makrophagen
einem zweiten Stimulus in Form von a) LPS, b) vitalen M. ptb, c) hitzeinaktivierten
M. ptb oder d) IFN-� ausgesetzt. Nachdem dieser zweite Stimulus 2 Stunden
einwirkte, wurden die Makrophagen wie in D.2.3.1 beschreiben abgekratzt, die RNA
gewonnen und RT-PCRs mit spezifischen Primern durchgeführt. Als Kontrollen
dienten nicht infizierte PBMs sowie PBMs, die nur mit vitalen bzw. hitzeinaktivierten
M. ptb infiziert, jedoch keinem Sekundärstimulus ausgesetzt wurden.
In Abb. 13 ist die relative Expression der Gene IL-6, TNF-�, GM-CSF und iNOS als
Balkendiagramm mit Standardabweichung dargestellt. In der integrierten Tabelle ist
aufgeführt, zu welchem Zeitpunkt welcher Stimulus gesetzt wurde. Im linken Teil
jedes Diagrammes ist die Genexpression der Kontrollen sowie nach Zweitstimulation
mit LPS bzw. Infektion mit vitalen und hitzeinaktivierten M. ptb aufgetragen; im
rechten Teil wiederum die Expression der Kontrollen sowie nach Zweitstimulation mit
IFN-�. Ist der Balken nach Sekundärstimulus höher als der Vergleichsbalken der
nicht-zweit-stimulierten Makrophagen, so konnten die Zellen auf den zweiten
Stimulus reagieren, waren also nicht deaktivert. Ist der Balken in etwa gleich hoch,
konnten die Makrophagen mit keiner weiteren Genexpression reagieren, sie waren
also bezüglich der Expression dieses Gens refraktär.

Ergebnisse
71
Ist der Balken wesentlich niedriger, so führte der Sekundärstimulus sogar zum
Abschalten des jeweiligen Gens.
Nach Infektion mit hitzeinaktivierten M. ptb führten die bakteriellen Sekundärstimuli
zu einer verstärkten Genexpression aller vier untersuchten Gene. Nach Infektion mit
vitalen M. ptb waren die Makrophagen jedoch bei den bakteriellen Sekundärstimuli
bezüglich der Genexpression von TNF-� sowie der iNOS refraktär: die Balken sind
genauso hoch wie die Vergleichsbalken. Bezüglich der Genexpression von IL-6
sowie GM-CSF besteht keine Refraktärität.
Nach Infektion mit vitalen M. ptb und IFN-� als Sekundärstimulus geht die
Genexpression von IL-6, GM-CSF und der iNOS auf unter 5 %, die von TNF-� auf
50 % zurück. Nach Infektion mit hitzeinaktivierten M. ptb und IFN-� als
Sekundärstimulus geht die Genexpression von IL-6, GM-CSF und der iNOS auf
20 %, 37 % bzw. 30 % zurück, die von TNF-� jedoch steigt um 25 %.
Die Infektion mit M. ptb führt also tatsächlich zu einer graduellen Deaktivierung des
Makrophagen, wobei vitale Mykobakterien eine wesentlich stärkere Deaktivierung
verursachen, als hitzeinaktivierte. Tatsächlich führt der körpereigene Makrophagen-
aktivator IFN-� sogar zu einer Deaktivierung anstelle einer Aktivierung der
Makrophagen.

Ergebnisse
72
t=0 Lebend - + + + + - - - - - + +Tot - - - - - + + + + - - -
t=22 LPS - - + - - - + - - - - -Lebend - - - + - - - + - - - -Tot - - - - + - - - + - - -IFN-� - - - - - - - - - - - +
Abbildung 13: Expression der Gene (von oben nach unten) IL-6, TNF-�, GM-CiNOS als Vielfaches der Kontrolle; in der Tabelle ist angegeben, welchen StimPBMs aus der Teflonkultur zu den Zeitpunkten t=0 (Beginn des Versuches) und Stunden nach Beginn des Versuches und 2 Stunden vor dem Ernten der Zellen) Nach insgesamt 24 Stunden wurden die PBMs wie in D.2.3.1 beschreiben abgekRNA gewonnen und RT-PCRs mit spezifischen Primern durchgeführt. DargesteDurchschnitt aus drei unabhängigen Versuchen.
re
l.
Ge
ne
xp
re
ss
ion
0
5
1 0
1 5
2 0
2 5
0 , 0
0 , 5
1 , 0
1 , 5
2 , 0
2 , 5
3 , 0
3 , 5
0
5
1 0
1 5
2 0
2 5
3 0
0 , 0
2 , 5
5 , 0
7 , 5
1 0 , 0
1 2 , 5
1 5 , 0
6
IL-S
����
iNO
TNF-
GM-CSF
- -+ +- -- -- -- +
SF undulus diet=22 (22
erhielten.ratzt, diellt ist der

Diskussion
73
E Diskussion
Die Pathogenese der Paratuberkulose ist bislang ungeklärt. Als fakultativ
intrazellulärer Erreger ist M. ptb in der Lage, in professionellen Phagozyten zu
überleben und sich zu vermehren. Es gilt als gesichert, daß M. ptb durch die
M-Zellen im Epithel der Peyer‘schen Platten die intestinale Mukosa überwindet und
anschließend von Gewebemakrophagen aufgenommen wird. Unbekannt sind jedoch
die Strategie und die Mechanismen, die M. ptb das intrazelluläre Überleben in diesen
Zellen ermöglichen.
In der vorliegenden Arbeit wurde ein Modell entwickelt und standardisiert, um das
Überleben der Bakterien in den Makrophagen des Darmes in vitro zu imitieren. In
dem etablierten Modell wurden die Auswirkungen des intrazellulären Überlebens von
M. ptb auf die Genexpression der Makrophagen untersucht. Verwendet wurde die
Teflonkultivierung primärer Makrophagen. Durch den Einsatz sowohl vitaler als auch
hitzeinaktivierter M. ptb sollten Ergebnisse als Folge des Einwirkens sezernierter
oder struktureller Bestandteile, also als aktiv oder passiv, eingeordnet werden.
Erster Schritt der Arbeit waren die Etablierung und Standardisierung des Modells der
Teflonkultivierung primärer boviner Makrophagen.
E.1 Eignung und Standardisierung des Teflon-Modells
Es wird häufig angenommen, periphäre Blutmonozyten seien im Gegensatz zu
Gewebemakrophagen eine relativ homogene Zellpopulation (COHN 1968).
Tatsächlich besitzen zwar alle Monozyten gewisse einheitliche morphologische,
biochemische und funktionelle Charakteristika (MORAHAN 1980), es muß jedoch
betont werden, daß sie in keiner Hinsicht völlig uniform sind. In den vergangen
Jahren wurden verschieden Untersuchungen durchgeführt, um die Art und das
Ausmaß der Heterogenität zu charakterisieren und mögliche Ursprünge für diese
Unterschiede herauszustellen (TREVES 1984; DOUGHERTY u. MCBRIDE 1984).

Diskussion
74
Frisch isolierte Monozyten unterschieden sich u. a. bezüglich Größe, Dichte,
Expression von Oberflächenproteinen und funktionellen Kapazitäten (TREVES
1984).
Hauptproblem bei Arbeiten mit primären Makrophagen in der Zellkultur ist daher die
Inhomogenität der Zellen aufgrund der Diversität im Blut befindlicher monozytärer
Zellen und daraus folgende Schwankungen der Untersuchungsergebnisse (GOFF et
al. 1998). Das Verwenden einer bovinen Makrophagenzellinie wäre vorteilhaft, kann
jedoch in Ermangelung einer solchen Zellinie nicht durchgeführt werden.
Hämatopoetische Stammzellen reifen im Blut über Monoblasten, Promonozyten und
Monozyten zu Promakrophagen, die in Körpergeweben schließlich zu unter-
schiedlichen Makrophagen ausdifferenzieren (OPPENHEIM u. LEONARD 1989). Im
Blut befinden sich also monozytäre Zellen verschiedener Reifungsstadien, von denen
nur die späteren Stadien zur Adhärenz befähigt sind. Wird diese Zellmischung in die
Zellkultur verbracht, erhält sie zwei wichtige Differenzierungssignale: zum einen die
Adhärenz an das feste Substrat (das Plastik der ZK-Schale), zum anderen das im
Medium befindliche Serum (DOUGHERTY u. MCBRIDE 1989). Aufgrund dieser
Signale differenzieren die Zellen in vitro zu Makrophagen aus.
In Vorarbeiten von KUEHNEL (nicht veröffentlicht) wurden die Probleme der
Kultivierung primärer boviner Makrophagen deutlich. Die durch Dichtezentrifugation
gewonnenen Blutmonozyten wurden hier direkt auf ZK-Schalen gegeben, nach drei
Stunden Adhärenz wurden nicht-adhärente Zellen durch Waschen mit PBS entfernt
und die Makrophagen in die Versuche eingesetzt. Zum einen war die Ausbeute bei
dieser Methode mit nur 10 % der im Blut befindlichen Makrophagenvorläuferzellen
sehr gering, da ein Teil der Monozyten noch nicht reif genug war, um adhärieren zu
können und somit beim Waschen entfernt wurde. Zum anderen betrug die Infektions-
rate der Zellen nur etwa 40 %, da wiederum ein Teil der adhärenten Zellen noch
nicht vollständig zu Makrophagen ausdifferenziert und somit nicht in der Lage war,
Bakterien zu phagozytieren. Die Zellpopulation war inhomogen.

Diskussion
75
Eine möglichst homogene Zellpopulation ist jedoch Voraussetzung für eine geringe
Standardabweichung zwischen den einzelnen Versuchen und somit für
reproduzierbare und aussagekräftige Ergebnisse.
Ziel der Etablierung des Teflonmodells war das Überwinden des Problems der
Inhomogenität. Ursprünglich wurde die Kultivierung von Makrophagen und anderen
Zellen in Teflonbeuteln verwendet, um Untersuchungen durchzuführen, nach denen
die Zellen möglichst vollständig wiedergewonnen werden sollten. Dies war in der
Teflonkultur möglich, da die Zellen nicht an der Teflonoberfläche adhärieren (VAN
DER MEER et al. 1978). Bereits CAMPBELL u. ADAMS (1992) verwendeten primäre
bovine Makrophagen und Teflon-Beutel. In ihren Arbeiten mit Brucella abortus
wurden die Makrophagen jedoch über den gesamten Untersuchungszeitraum in der
Teflonkultur gehalten, um Adhärenz zu verhindern und um die Makrophagen in
einem relativ unstimulierten Zustand zu belassen. In der vorliegenden Arbeit wurde
eine Methode von JUNGI et al. (1996) leicht modifiziert, um sie an die
Gegebenheiten des Labors anzupassen. Hierbei diente die Kultivierung im
Teflonbeutel über mehrere Tage der Synchronisation der Zellen; sie wurden
anschließend auf Plastik verbracht, um sie als synchronisierte und ausdifferenzierte,
adhärente Makrophagen in die Versuche einzusetzen.
In der Differenzierung humaner Monozyten zu Makrophagen wurden zwei
Differenzierungssignale erkannt (ZEMBALA u. ASHERSON 1989). Die Inkubation mit
Serum führte zur Differenzierung bis zum Promakrophagen; die endgültige
Differenzierung zum Makrophagen wurde durch Adhärenz erreicht. Diesen
Erkenntnissen folgend, wurden die isolierten Leukozyten für 8 Tage in FCS-haltigem
Medium in Teflonbeuteln kultiviert (siehe C.2.1.2.1). Durch das FCS erhielten sie das
erste Differenzierungssignal und entwickelten sich zu Promakrophagen. Die
Zellpopulation war jetzt synchronisiert und wurde auf ZK-Schalen gegeben. Hier
adhärierten die vitalen Promakrophagen und differenzierten innerhalb weniger
Stunden zu Makrophagen aus (JUNGI et al. 1996). Die Makrophagenausbeute
erhöhte sich bei dieser Methode auf 50 % der im Blut befindlichen
Makrophagenvorläuferzellen (siehe D.1.1).

Diskussion
76
Für die Infektionsversuche wurden die Makrophagen für 2 Stunden mit einer
definierten Zahl Mykobakterien inkubiert, anschließend gewaschen und bis zum
Erntezeitpunkt weiterinkubiert (siehe C.2.2.1). Die Infektionsrate wurde durch
Auszählen der infizierten Makrophagen unter dem Mikroskop festgestellt (siehe
D.1.2); sie lag über 72 Stunden zwischen 94 und 98 %. In anderen Untersuchungen
lag die Infektionsrate zwischen 40 und 80 % (BERMUDEZ et al. 1997; BONAY et al.
1999). Die hier erreichte hohe Infektionsrate spricht für eine homogene und vitale
Zellkulur.
Ein Problem der Kultivierung primärer Zellen ist die geringe Stabilität in der Zellkultur.
Geringe Stabilität ist hier definiert als abnehmende Vitalität verbunden mit dem
Verlust der Adhärenz. Zur Bestimmung der Stabilität der Kultur wurden die
Adhärenzraten der Makrophagen durch Auszählen adhärenter Zellen in der
Neubauerkammer untersucht (siehe C.2.1.2.2). Die Adhärenzraten über 72 Stunden
unterschieden sich deutlich zwischen den Versuchsansätzen: nicht-infizierte
Makrophagen waren nach 72 Stunden noch zu 98,6 % adhärent. Mit vitalen M. ptb
infizierte Makrophagen waren nach 72 Stunden noch zu 79,7 % adhärent, mit
hitzeinaktivierten M. ptb infizierte noch zu 65,8 %.
Bemerkenswert ist hier vor allem der Unterschied zwischen der Infektion mit vitalen
und hitzeinaktivierten Bakterien. Ursache hierfür könnte die Zahl der phagozytierten
Erreger sein: In der Mikroskopie wurde deutlich, daß Makrophagen, die mit
hitzeinaktivierten M. ptb infiziert wurden, zu einem höheren Prozentsatz über 50
Bakterien pro Zelle phagozytierten (durchschnittlich 14 % gegenüber 6 %). Dieses
„Überfressen“ könnte Ursache für den schnelleren Verlust der Adhärenz sein.
Eine weitere mögliche Erklärung für die Abnahme adhärenter Zellen, ist die
Apoptose. Die Infektion humaner Makrophagen mit M. avium führte zu 20 %
(BALCEWICZ-SABLINSKA et el. 1999), in anderen Untersuchungen zu bis zu 31 %
apoptotischen Zellen (BERMUDEZ et al. 1997). Es ist allgemein akzeptiert, daß
intrazelluläre Mykobakterien Apoptose auslösen können. Vermutlich ist die Apoptose
ein Verteidigungsmechanismus der Makrophagen gegen die Mykobakterien, der
durch pathogene Mykobakterien zum Teil ausgeschaltet werden kann (FRATAZZI et
al. 1999). Da es im Interesse intrazellulärer Erreger liegt, die Wirtszelle am Leben zu

Diskussion
77
erhalten, ist ein Mechanismus möglich, der Makrophagen daran hindert, vitale
Erreger in letaler Zahl zu phagozytieren oder aber apoptotisch zu werden. Das
Entdecken eines solchen Mechanismus war nicht Ziel der vorliegenden Arbeit,
könnte jedoch in Zukunft ein interessanter Untersuchungsgegenstand sein.
Um die Stimulierbarkeit der Makrophagen sowie die geringe Standardabweichung im
Sinne einer homogenen Zellpopulation zu überprüfen, wurden die Zellen mit LPS
bzw. IFN-� behandelt und anschließend die Expression ausgewählter Gene durch
RT-PCR überprüft (siehe D.1.4). LPS ist Bestandteil der Zellhülle gramnegativer
Bakterien und ein potenter Makrophagenaktivator; IFN-� wird von T-Zellen sezerniert
und trägt wesentlich zur endogenen Makrophagenaktivierung bei. Überprüft wurden
drei proinflammatorische Zytokine (IL-1�, IL-6 und TNF-�), GM-CSF als
Makrophagenaktivator, IL-10 als deaktivierendes Zytokin sowie die iNOS. In nicht-
stimulierten primären bovinen Makrophagen war eine Genexpression von IL-1 und
der iNOS nicht zu detektieren (GOFF et al. 1998). IL-6, GM-CSF sowie TNF-� waren
in nicht-stimulierten Zellen nur gering exprimiert (ADLER et al. 1995; KEEFE et al.
1997).
Wie erwartet waren die primären bovinen Makrophagen aus der Teflonkultur
stimulierbar. Die durchschnittliche Standardabweichung sämtlicher Versuche lag bei
0,97 (siehe D.1.4), was ein weiterer Beleg für die Homogenität der Zellpopulation und
damit für die Aussagekraft der Versuche ist.
Die vorliegende Arbeit sollte ein in vitro-Modell für das Überleben der Mykobakterien
im Darm infizierter Tiere etablieren und verwenden. Dafür war es notwendig, daß die
Bakterien in den Makrophagen aus der Teflonkultur überleben. Um dies zu
überprüfen wurde die Zahl Kolonie-bildender Einheiten (KBE) pro Makrophagen über
einen Zeitraum von 72 Stunden gemessen (siehe C.2.2.3). Diese änderte sich im
Untersuchungszeitraum nicht. Die Mykobakterien konnten also in den primären
bovinen Makrophagen überleben.

Diskussion
78
Die KBE pro Makrophagen lag hier durchschnittlich zwischen 0,3 und 0,4. Dies
stimmt mit Untersuchungen in anderen Arbeitsgruppen überein, in denen Werte
zwischen 0,1 und 0,4 (BONAY et al. 1997; KEMPER at al. 1998) festgestellt wurden.
Die Abweichungen zwischen der KBE (0,3-0,4) und der im Mikroskop festgestellten
Infektionsrate (zwischen 1 und mehr Bakterien pro Zelle) ist durch die Methode der
KBE-Ermittlung selbst zu erklären. Es ist allgemein akzeptiert, daß beim Ermitteln
der KBE nur etwa 10 % der tatsächlich vorhandenen Zahl vitaler Bakterien erfaßt
wird (BISPING u. AMTSBERG 1988). Mögliche Gründe hierfür sind das Verklumpen
der Bakterien, deren Absterben durch die Reisolierung aus den Makrophagen oder
die Kultivierung auf den Platten selbst. M. ptb wächst innerhalb 6-8 Wochen zu
sichtbaren Kolonien heran; ein Verlust eines Teils der vitalen Erreger in dieser Zeit ist
nicht überraschend. Der entstehende Fehler ist jedoch in allen Versuchen gleich und
daher zu vernachlässigen. Wichtig war hier der Vergleich zwischen den einzelnen
Versuchen und nicht die absoluten Zahlen, da diese durch Mikroskopie ermittelt
wurden (siehe C.2.2.2).
Zusammenfassend läßt sich sagen, daß die hier verwendete Methode der
Teflonkultivierung das Hauptproblem der Inhomogenität der Zellpopulation
überwunden hat. Die Kultur war außerdem stabil, die Makrophagen aktivierbar und
die Mykobakterien in den Makrophagen überlebensfähig. Das Modell der
Teflonkultivierung war somit geeignet für die durchzuführenden Versuche.
In der vorliegenden Arbeit wurde also ein standardisiertes Infektionsmodell mit
charakterisierten Zytokinmustern etabliert, welches für zukünftige Arbeiten im
bovinen Modell weiter Verwendung finden kann.
E.2 Funktionelle Untersuchungen in PBMs aus der Teflonkultur
Makrophagen sind die Zielzellen verschiedener intrazellulärer Mikroorganismen. Die
Aktivierung des Makrophagen ist essentiell für eine verstärkte antimikrobielle Aktivität
während dieser Infektionen. Allgemein wird die antimikrobielle Aktivtät in Sauerstoff-
abhängige (z. B. reaktive Stickstoff-Intermediate, Nitrid-Oxide) und Sauerstoff-

Diskussion
79
unabhängige (z. B. Ansäuerung der phagolysosomalen Vakuole) Systeme unterteilt
(REINER 1994). Um intrazelluläre Erreger zu eliminieren, muß der Makrophage zur
Aktivierung dieser Mechanismen angeregt werden. Deshalb kann eine Makropha-
gendeaktivierung als Prozeß definiert werden, in dem die Zelle teilweise oder
vollständig unempfänglich wird für aktivierende Stimuli. Hinweise für die Dysfunktion
des Makrophagen aufgrund intrazellulärer Erreger gibt es bei verschiedenen
Infektionen, wie z. B. mit Leishmania (REINER et al. 1990), Mycobacterium (SIBLEY
et al. 1990, CHAN et al. 1991), Yersinia (BLISKA et al. 1993) oder dem humanen
Immundefizienzvirus 1 (HIV-1) (BALDWIN et al. 1990).
E.2.1 Vitale M. ptb alkalisieren das endosomale Netzwerk
Phagozytierte Partikel liegen in Makrophagen meist in Vakuolen, den Phagosomen,
die Teil des endosomalen Netzwerkes sind. In der vorliegenden Arbeit wurde anhand
elektronenmikroskopischer Aufnahmen bestätigt, daß M. ptb in primären bovinen
Makrophagen aus der Teflonkultur ebenfalls in Vakuolen liegt (siehe Abb. 7). Diese
Bestätigung war essentiell für die folgenden Untersuchungen am endosomalen
Netzwerk der Makrophagen.
Ein Weg, die Sauerstoff-unabhängige Abwehr zu umgehen, ist das Verhindern der
Ansäuerung des Phagosoms (REINER 1994). Nach der Phagozytose befinden sich
Bakterien in Phagosomen mit leicht saurem pH-Wert (pH 6,3-6,5), welche Marker
früher Endosomen tragen. Sie reifen innerhalb von Minuten bis wenigen Stunden
heran und akkumulieren dann Marker später Endosomen. Hier liegt der pH-Wert bei
4,5-5,5 (CLEMENS 1996). In diesem sauren pH-Wert haben proteolytische Enzyme
des späten endosomalen Netzwerkes, die an Abbauvorgängen beteiligt sind, das
optimale Mileu zum Verdau des phagozytierten Materials. Somit stellt die
Ansäuerung des Phagosoms einen wichtigen Schritt in der Phagosomenreifung dar
(siehe auch C.2.3).
SIBLEY et al. stellten 1985 ein Verhinderung der Ansäuerung in Toxoplasma gondii-
haltigen Phagosomen fest. STURGILL-KOSZYCKI et al. konnten dies 1994 für
M. avium-haltige Phagosomen zeigen; auch andere Bakterien und Pilze sind dazu in

Diskussion
80
der Lage (MALIK et al. 2000, KAPOSZTA et al. 1999, BLACK et al. 1996). In J774-
Mausmakrophagen waren vitale M. ptb in der Lage, die Ansäuerung des Phagosoms
fast vollständig zu verhindern (KUEHNEL et a. 2001). Eine mögliche Erklärung ist
das Fehlen der Protonen-ATPase in der Phagosomenmembran. Diese ist ein
Enzymkomplex, der für die Ansäuerung von intrazellulären Kompartimenten
zuständig ist und im besonderen den pH-Wert in Phagosomen absenkt.
In der vorliegenden Arbeit wurde untersucht, ob vitale oder auch hitzeinaktivierte
M. ptb in primären bovinen Makrophagen ebenfalls die Ansäuerung des Phagosoms
verhindern.
Zu diesem Zweck wurde eine von KUEHNEL et al. (2001) etablierte und für diese
Arbeit modifizierte Methode der intraendosomalen pH-Wert-Messung mit FITC-
Dextran verwendet (siehe C.2.4). Eine Änderung des pH-Wertes wurde hier als
Änderung der relativen Fluoreszenzintensität angezeigt (siehe D.2.2). KUEHNEL et
al. (2001) führten Untersuchungen zum intraendosomalen pH-Wert in M. ptb-
infizierten J774 Mausmakrophagen durch. Hier waren vitale M. ptb in der Lage, die
Ansäuerung des Phagosoms zu verhindern, führten jedoch nicht zu einer
Alkalisierung. Der pH-Wert lag nach 2 Stunden bei pH 6,3.
Vitale M. ptb waren in primären bovinen Makrophagen aus der Teflonkultur in der
Lage, die Ansäuerung des Phagosoms und endosomalen Netzwerkes nicht nur zu
verhindern, sondern leiteten eine leichte Alkalisierung ein: der intraendosomale pH-
Wert lag 2 Stunden post infectionem bei 6,97. Der endosomale pH-Wert von
Makrophagen, die mit hitzeinaktivierten M. ptb infiziert waren, lag bei 5,19. Das
Verhindern der Ansäuerung ist also ein Prozeß, der nur von vitalen M. ptb
durchgeführt wird. Durch das Blockieren der Protonen-ATPasen mit dem Wirkstoff
Bafilomycin (siehe D.2.2) wurde die Ansäuerung auch in der Infektion mit
hitzeinaktivierten M. ptb verhindert. Möglicherweise handelt es sich also auch bei
dem Effekt durch vitale M. ptb um eine Beeinflussung der Protonen-ATPasen.

Diskussion
81
E.2.2 Modulation der Genexpression bei M. ptb-Infektion
E.2.2.1 Infektion mit vitalen bzw. hitzeinaktivierten M. ptb
Zur effektiven Eliminierung von Mykobakterien benötigt der Makrophage das
koordinierte Zusammenspiel des IFN-� der Th1-Zellen (eine Subpopulation der
Helfer-T-Lymphozyten) sowie der eigenen proinflammatorischen Zytokine IL-1, IL-6
und TNF-� wie auch des Makrophagenaktivators GM-CSF (VALENTIN-WEIGAND u.
GOETHE 1999). Auch NO spielt eine herausragende Rolle in der Abwehr
intrazellulärer Parasiten und ist Bestandteil der Aktivierung des Makrophagen
(JAMES 1995; GARCIA et al. 2000). Die NO-Produktion wird durch IFN-� in
Kombination mit TNF-� oder anderen sekundären Aktivierungssignalen über die
iNOS (induzierbare NO-Synthase) stimuliert und durch eine Reihe von Zytokinen
(vor allem IL-4, IL-10 und transforming growth factor beta), anderen Mediatoren und
autoinhibitorischen Mechanismen reguliert (JAMES 1995).
In Infektionsversuchen mit verschiedenen Mykobakterien konnte die Rolle einiger
Zytokine in der Makrophagenabwehr teilweise charakterisiert werden (FLESCH u.
KAUFMANN 1991; FIORENTINO et al. 1991; JAMES 1995; VAN HEYNINGEN et al.
1997; BONAY et al. 1999; BALCEWICZ-SABLINSKA et al. 1999; JACOBS et al.
2000a; JACOBS et al. 2000b; GARCIA et al. 2000; JAGANNATH et al. 2000; HOAL-
VAN HELDEN et al. 2001). Auch mit M. ptb wurden Arbeiten, v. a. im murinen, aber
auch im bovinen System durchgeführt (ZURBRICK et al. 1988; ADAMS u.
CZUPRYNSKI 1994; STABEL 1995; ADAMS et al. 1996; ZHAO et al. 1997;
BEGARA-MCGORUM et al. 1998; VALENTIN-WEIGAND u. GOETHE 1999).
Bisherige Arbeiten geben Hinweise darauf, daß eine Infektion mit vitalen oder
hitzeinaktivierten M. ptb sowie eine Inkubation mit gereinigtem LAM zu einer
verstärkten Expression von IL-1�, IL-6, TNF-� und GM-CSF führen. Wie sich die
genannten Zytokine sowie IL-10 als deaktivierendes Zytokin und die iNOS in der
Infektion mit M. ptb exakt verhalten und welchen Einfluß sie auf das intrazelluläre
Überleben von M. ptb in primären bovinen Makrophagen haben, ist jedoch nicht
vollständig geklärt. Es wird über M. ptb-spezifische Mechanismen spekuliert (siehe
unten).

Diskussion
82
In der vorliegenden Arbeit wurde der Einfluß von vitalen sowie hitzeinaktivierten
M. ptb auf die Expression ausgewählter Gene in primären bovinen Makrophagen aus
der Teflonkultur untersucht. Überprüft wurde die Expression von IL-1�, IL-6, TNF-�,
GM-CSF, IL-10 und der iNOS durch RT-PCR im zeitlichen Verlauf von 24 Stunden
(siehe C.2.5).
Es bestanden deutliche Unterschiede in der Genexpression von Makrophagen, die
mit vitalen M. ptb und solchen, die mit hitzeinaktivierten M. ptb infiziert wurden (siehe
D.2.3.1). Der auffallendste Unterschied war die höhere Expression aller untersuchter
Gene in der Infektion mit vitalen M. ptb im Vergleich zur Infektion mit
hitzeinaktivierten M. ptb. Die Unterschiede lagen zwischen 25 und 700 %. Außerdem
unterschied sich bei zwei Zytokinen der zeitliche Verlauf der Expression: die GM-
CSF-Genexpression stieg in der Infektion mit vitalen M. ptb schneller an, die IL-6-
Genexpression nahm einen biphasischen Verlauf.
Diese Unterschiede sprechen dafür, daß zumindest ein Teil der Effekte auf von
vitalen Bakterien sezernierten Bestandteilen beruht. Es wäre auch möglich, daß die
Hitzeinaktivierung der Mykobakterien eine Denaturierung der strukturellen
Bestandteile bewirkt und somit die beobachteten Effekte nicht durch sezernierte,
sondern duch strukturelle Bestandteil zu erklären wären. Dagegen sprechen
Vorversuche mit hitzeinaktivierten sowie strahleninaktivierten Mykobakterien, die
keine Unterschiede in den Ergebnissen zeigten. Bei einer Inaktivierung mit Strahlen
bleiben strukturelle Bestandteile intakt.
Alle untersuchten Zytokine und die iNOS wurden nach Infektion mit vitalen M. ptb
stärker exprimiert als nach Infektion mit hitzeinaktivierten Bakterien. Dies spricht auf
den ersten Blick für eine stärkere Aktivierung des Makrophagen. Trotzdem ist der
Makrophage nicht in der Lage, die intrazellulären Erreger abzutöten (siehe D.1.5).
Eine erhöhte Expression des proinflammatorischen IL-6 muß nicht zwingend einen
inaktivierenden Einfluß auf die Mykobakterien nehmen. Sie kann sogar zu einem
immunsuppressiven Effekt auf die Makrophagen führen und damit einen
inaktivierenden Einfluß auf die Mykobakterien verhindern. Die Zugabe von IL-6
änderte nichts an der Abtötungsrate von M. bovis Bacillus Calmette-Guérin (BCG) in
humanen Makrophagen (BONAY et al. 1999) bzw. von Mycobacterium bovis in

Diskussion
83
murinen Knochenmarksmakrophagen (FLESCH u. KAUFMANN 1991). Bei M. avium-
infizierten Makrophagen wurde eine verminderte Expression von aktivierenden TNF-
Rezeptoren um 78 % verzeichnet (BERMUDEZ et al. 1992). In einer anderen Arbeit
führte die starke IL-6-Expression in Infektionen mit M. avium zu einem
immunosuppressorischen Effekt auf umliegende Makrophagen und die T-Zell-
Aktivierung (VAN HEYNINGEN et al. 1997).
Auch eine erhöhte Expression von TNF-� muß nicht zwingend einen
mykobakteriziden Effekt nach sich ziehen. Eine Vorinkubation von Maus-
makrophagen mit geringen Dosen TNF-� verbesserte sogar das intrazelluläre
Überleben von M. ptb (BALCEWICZ-SABLINSKA et al. 1999). Die in dieser Arbeit
detektierte starke Erhöhung der TNF-�-Expression kann zwar nicht als geringe Dosis
bezeichnet werden. Doch die mRNA-Menge korreliert nicht zwingend mit der
wirksamen Menge an TNF-�. So führte IL-10 in M. avium-infizierten humanen
Makrophagen zu einer vermehrten Ausschüttung des TNF-Rezeptors Typ 2. Dieser
hat eine Inaktivierung von TNF-� zur Folge (BALCEWICZ-SABLINSKA et al. 1999).
Wie bereits erwähnt wurde bei M. avium-infizierten Makrophagen eine verminderte
Expression von aktivierenden TNF-Rezeptoren um 78 % verzeichnet (BERMUDEZ et
al. 1992). Alleine gegeben hatte TNF-� außerdem keinen Einfluß auf das Überleben
von M. bovis in murinen Knochenmarksmakrophagen (FLESCH u. KAUFMANN
1991; BONAY et al. 1999). In humanen Makrophagen, die mit BCG infiziert waren,
hatte TNF-� hemmenden wie auch verstärkenden Effekt, unterschiedlich in den
jeweiligen Makrophagenchargen (HOAL-VAN HELDEN et al. 2001). Der fehlende
mykobakterizide Effekt trotz hoher mRNA-Menge könnte also durch verminderte
Mengen aktivierender TNF-Rezeptoren oder erhöhte Mengen inaktivierender TNF-
Rezeptoren zu erklären sein.
Auch GM-CSF hatte nicht in allen bisherigen Arbeiten einen mykobakteriziden Effekt.
Eine Vorinkubation mit GM-CSF führte in der Infektion humaner Makrophagen mit
BCG zwar zu einem schnelleren Abtöten des Erregers. Spätere Gaben von GM-CSF
hatten jedoch keinen Einfluß mehr (BONAY er al. 1998). In humanen Makrophagen,
die mit BCG infiziert waren, hatte GM-CSF hemmenden wie auch verstärkenden
Effekt, unterschiedlich in den jeweiligen Makrophagenchargen (HOAL-VAN HELDEN

Diskussion
84
et al. 2001). Außerdem könnte die Aufregulation der GM-CSF-Genexpression zur
Rekrutierung weiterer Makrophagen dienen (RIEDEL u. KAUFMANN 2000). Diese
können dann im folgenden ebenfalls infiziert und deaktiviert werden.
Da es sich bei IL-10 um ein deaktivierendes Zytokin handelt, ist eine verstärke
Expression hier nicht überraschend. Wie bereits erwähnt führte IL-10 in M. avium-
infizierten humanen Makrophagen zu einer vermehrten Ausschüttung des
inaktivierenden TNF-Rezeptors Typ 2 (BALCEWICZ-SABLINSKA et al. 1999).
Außerdem waren IL-10-defiziente Mäuse wesentlich schneller in der Lage, eine
BCG-Infektion zu eliminieren. In den Makrophagen der Granulome waren
ungewöhnlich hohe Spiegel von TNF-� und der iNOS zu detektieren (JACOBS et al.
2000a).
INOS-defiziente Mäuse waren nicht in der Lage, BCG zu eliminieren und starben
innerhalb von 8 bis 12 Wochen, wohingegen die Kontrollgruppe überlebte (GARCIA
et al. 2000). Ein experimentelles Medikament, CRL-1072, löste in humanen
Makrophagen eine verstärkte iNOS-Produktion und folgend einen höheren NO-Level
aus und führte so zum Abtöten von M. avium (JAGANNATH et al. 2000).
Die erhöhte iNOS-Expression korreliert ebenso wie bei TNF-� nicht zwingend mit
den tatsächlich freigesetzten Mengen an NO. Inwiefern dies durch veränderte
Rezeptormengen oder eine schnellere Inaktvierung bzw. Zersetzung von NO zu
erklären ist, bleibt in Zukunft zu untersuchen.
Bezüglich der erhöhten Expression von IL-1� kann nur über ähnliche Mechanismen
oder Gründe spekuliert werden. Es gibt in bisherigen Arbeiten keine Hinweise auf
eine besondere Rolle von IL-1� in der Eliminierung von intrazellulären
Mykobakterien.
Die hier detektierten Veränderungen in der Genexpression v. a. durch vitale, aber
auch durch hitzeinaktivierte M. ptb sind möglicherweise Teil des Pathogenitäts-
mechanismus. Die oben erwähnten weiterführenden Untersuchungen könnten
Aufschluß über die Bedeutung eines solchen Mechanismus geben.

Diskussion
85
Die Zugabe von Bafilomycin verhinderte die Ansäuerung des Phagosoms in der
Infektion mit hitzeinaktivierten M. ptb und imitierte damit den Effekt vitaler
Mykobakterien. Bei der Untersuchung der Genexpression von IL-6, TNF-� und der
iNOS ließ sich ein solcher Effekt auf die Genexpression nicht nachweisen: die
Expression der Gene lag sogar unter der nach Infektion mit hitzeinaktivierten M. ptb
und nicht um 25 bis 700 % höher wie nach Infektion mit vitalen M. ptb. Dies legt
nahe, daß es sich bei der Verhinderung der Ansäuerung und der Veränderung der
Genexpression durch vitale M. ptb um zwei voneinander unabhängige
Pathogenitätsmechanismen handelt.
E.2.2.2 Vergleich der Genexpression nach verschiedenen Stimulationen
Es stellte sich die Frage, ob die ermittelten Zytokinmuster Hinweise auf die
Signaltransduktion im Makrophagen liefern.
LPS ist ein potenter Makrophagenaktivator mit bekannten Signalwegen. Nach
Bindung von LPS an CD14, TLR („toll like receptor“) oder andere LPS-Rezeptoren ist
eine Tyrosin-Phosphorylierung an der weiteren Signaltransduktion beteiligt
(KOVARIK et al. 1998; MEANS et al. 1999). Es ist jedoch noch unklar, welche
Tyrosinkinasen diese Phosphorylierung durchführen.
Bekannt ist außerdem, daß LAM, also das LPS der Mykobakterien, ebenfalls an
CD14, aber auch an andere Rezeptoren als LPS bindet (MEANS et al. 1999). LPS
kann TLR nur in Anwesenheit von CD14 aktivieren, LAM aktviert TLR auch
unabhängig von CD14.
Um Hinweise auf die Aktivierung von Signalwegen durch M. ptb zu finden, wurden
die zeitlichen Verläufe der Expression der ausgewählten Gene nach Stimulation mit
LPS und Infektion mit Mykobakterien miteinander verglichen (siehe D.2.3.2).
Die zeitlichen Verläufe der Genexpression von IL-1�, IL-10, GM-CSF und der iNOS
unterschieden sich nach Stimulation mit LPS bzw. nach Infektion mit vitalen oder
hitzeinaktivierten M. ptb nur wenig.

Diskussion
86
Die Expression von IL-6 fiel nach Stimulation mit LPS relativ schnell wieder ab, blieb
nach Infektion mit hitzeinaktivierten M. ptb auf einem höheren Level bestehen und
nahm nach Infektion mit vitalen M. ptb einen biphasischen Verlauf. Das hohe Level
von IL-6 zu einem späten Zeitpunkt der Infektion wurde auch bei der Infektion von
J774 mit M. ptb beobachtet (GOETHE, unveröffentlicht). Möglicherweise handelt es
sich hier um einen M. ptb-spezifischen Mechanismus, der einen immunsuppressiven
Effekt zur Folge hat. Es kann spekuliert werden, daß vitale M. ptb einen anderen
Signaltransduktionsweg als hitzeinaktivierte M. ptb und LPS nehmen. Diese
Möglichkeiten werden in Zukunft in der Arbeitsgruppe untersucht werden.
Die Expression von TNF-� blieb nach Stimulation mit LPS auf relativ hohem Niveau,
fiel jedoch nach Infektion sowohl mit vitalen als auch mit hitzeinaktivierten M. ptb
bereits nach 8 Stunden auf unter 30 %, nach 24 Stunden auf Null ab. Dieser schnelle
Abfall könnte ein weiterer Grund für die geringe Wirksamkeit von TNF-� gegen
M. ptb sein. Auch dies steht möglicherweise im Zusammenhang mit
unterschiedlichen Signaltransduktionwegen und wird ebenfalls zukünftiger
Untersuchungsgegenstand sein.
Inwieweit M. ptb Signaltransduktionswege beschreitet, die IFN-� oder andere
Aktivatoren verwenden, bleibt zu klären.
E.2.3 deaktivierende Effekte
Das Deaktivieren der Wirtszelle ist eine Möglichkeit für das intrazelluläre Überleben
von Erregern. Das Phänomen der Refraktärität ist ein Weg einer solchen
Deaktivierung: LPS führt zur Aktivierung von Makrophagen und im Folgenden u. a.
zur Ausschüttung von TNF-�, IL-6 und IL-12. Eine Vorinkubation mit geringen
Mengen LPS führte jedoch zu einer reduzierten Sensitivtiät auf weitere LPS-Gaben
(NOMURA et al. 2000). Dieses Phänomen wird LPS-Toleranz, „LPS-
hyporesponsiveness“ oder Refraktärität genannt. In vivo wurden hierbei eine
verminderte Fieberantwort und geringere Letalität vermerkt. In vitro ging mit dem
Phänomen eine verminderte Produktion proinflammatorischer Zytokine nach

Diskussion
87
Sekundärstimulation mit LPS einher (NOMURA et al. 2000). Auch LAM ist in der
Lage, Refrakterität in Makrophagen auszulösen (RIEDEL u. KAUFMANN 2000).
Um zu untersuchen, ob eine solche Refraktärität auch in bovinen Makrophagen nach
Infektion mit M. ptb auftritt, wurden Makrophagen aus der Teflonkultur, die mit vitalen
bzw. hitzeinaktivierten M. ptb infiziert waren, einem Zweitstimulus ausgesetzt. Bei
diesem handelte es sich entweder um eine Stimulation mit LPS bzw. IFN-� oder um
eine Reinfektion mit vitalen bzw. hitzeinaktivierten M. ptb. (siehe D.2.5).
Anschließend wurde die mRNA gewonnen und in der RT-PCR eingesetzt.
Untersucht wurde die Genexpression von IL-6, TNF-�, GM-CSF sowie der iNOS.
Nach Infektion mit hitzeinaktivierten M. ptb führten die Zweitstimulation mit LPS
sowie mit vitalen oder hitzeinaktivierten Mykobakterien zu einer verstärkten
Genexpression aller vier untersuchten Gene. Mit vitalen M. ptb infizierte
Makrophagen jedoch waren bei Sekundärstimulation mit weiteren vitalen, aber auch
mit hitzeinaktivierten M. ptb bezüglich der Genexpression von TNF-� sowie der iNOS
refraktär. Eine Zweitstimulation dieser Makrophagen mit LPS zeigte bei der
Expression von TNF-� die Refraktärität des Makrophagen. Vitale M. ptb waren also
in der Lage, im Gegensatz zu hitzeinaktivierten M. ptb, den Makrophagen in einen
refraktären Zustand zu versetzen.
Dies könnte einen wichtigen Pathogenitätsmechanismus darstellen. Im Tier kann
ebenfalls eine Zweitstimulation mit vitalen M. ptb erfolgen. Zum einen erfolgt durch
wiederholte Aufnahme ein ständiger Erregereintrag in die Darmwand. Zum anderen
stirbt ein Teil der infizierten Makrophagen ab und gibt dabei die phagozytierten
Erreger wieder frei, so daß sie von umliegenden Makrophagen phagozytiert werden.
Somit kommt es vermutlich häufig zu Reinfektionen einzelner Makrophagen mit
vitalen Erregern. Befindet sich der Makrophage durch die Erstinfektion in einem
refraktären Zustand, kann er auf die Reinfektion nicht in gewohnter Weise reagieren;
die Überlebenswahrscheinlichkeit des phagozytierten Bakteriums steigt.
Die Zweitstimulation mit IFN-� führte zu den weitaus interessantesten Ergebnissen.
IFN-� wird hauptsächlich von TH-1-Zellen ausgeschüttet und trägt wesentlich zur
Aktivierung des Makrophagen bei. Murine Knochenmarksmakrophagen waren nach
Stimulation mit IFN-� in der Lage, das intrazelluläre Wachstum von M. bovis zu

Diskussion
88
unterbinden (FLESCH u. KAUFMANN 1991). In der Infektion muriner
Intestinalmakrophagen mit M. avium-Komplex führte die Stimulation mit IFN-� zu
einer erhöhten mykobakteriostatischen und/oder mykobakteriziden Aktivität (HSU et
al. 1995). Es wird jedoch über M. ptb-spezifische Mechanismen spekuliert, da in
Gewebe experimentell infizierter Lämmer eine signifikant geringere Menge IFN-�
mRNA gefunden wurde (BEGARA-MCGORUM et al. 1998) und IFN-� nur geringe
Effekte auf das intrazelluläre Überleben von M. ptb in bovinen Monozyten hatte
(ZHAO et al. 1997). Dieser geringe antibakterielle Effekt von IFN-� in vitro steht im
Gegensatz zu Untersuchungen anderer Mykobakterien und wurde durch
ungenügende Produktion von NO erklärt, welches im zellfreien System zum Abtöten
der Bakterien führen kann (ZHAO et al. 1991).
In der vorliegenden Arbeit wurde die Genexpression M. ptb-infizierter primärer
boviner Makrophagen aus der Teflonkultur nach Sekundärstimulation mit IFN-�
gemessen.
Nach Infektion mit vitalen M. ptb und IFN-� als Sekundärstimulus ging die
Genexpression von IL-6, GM-CSF und der iNOS auf unter 5 %, die von TNF-� auf
50 % zurück. Nach Infektion mit hitzeinaktivierten M. ptb und IFN-� als
Sekundärstimulus ging die Genexpression von IL-6, GM-CSF und der iNOS auf
20 %, 37 % bzw. 30 % zurück, die von TNF-� jedoch stieg um 25 %.
Die Infektion mit M. ptb führte also nicht nur zu einer graduellen Deaktivierung des
Makrophagen. Tatsächlich führte der körpereigene Makrophagenaktivator IFN-�,
sogar zu einer Deaktivierung anstelle einer Aktivierung der Makrophagen. Diese
Deaktivierung war nach Infektion mit vitalen M. ptb deutlich höher als nach Infektion
mit hitzeinaktivierten Bakterien und ist ein Hinweis, daß es sich hier zumindest
teilweise um einen durch sezernierte Bestandteile der Mykobakterien hervor-
gerufenen Effekt handelt.
Der intrazelluläre Erreger M. ptb verleitet seine Wirtszelle, den Makrophagen, also
scheinbar dazu, auf einen ansonsten aktivierenden Stimulus mit einer Deaktivierung
zu reagieren. Da der Makrophage das Ausschütten von IFN-� durch die erhöhte
TNF-�-Expression selbst ausgelöst hat, kann man hier von einem suiziden Effekt
sprechen. Der Makrophage selbst führt zu seiner eigenen Deaktivierung.

Zusammenfassung
89
F Zusammenfassung
Mycobacterium avium Subspezies paratuberculosis (M. ptb) verursacht einechronische, granulomatöse und nicht therapierbare Enteritis bei Wiederkäuern, dieParatuberkulose. Die Tiere infizieren sich in der Regel in den ersten Lebensmonatenund haben danach oft über Jahre keinerlei klinische Symptome. Mit Ausbruch derKrankheit zeigen vor allem Rinder starken intermittierenden Durchfall undfortschreitende Abmagerung bis hin zur Kachexie. Die Krankheit endet fürgewöhnlich tödlich, die Pathogenese ist weitgehend unbekannt. Es gilt als gesichert,daß die Persistenz des Erregers in den Makrophagen der Darmwand eineSchlüsselrolle spielt. Ungeklärt sind jedoch die Strategie und die Mechanismen, dieM. ptb das intrazelluläre Überleben in diesen Zellen ermöglichen. Von anderenpathogenen Mykobakterien, die ebenfalls in Makrophagen überleben, ist bekannt,daß sie die Effektorfunktionen der Makrophagen modulieren, um nicht abgetötet zuwerden. Eine solche Modulation wird auch für M. ptb angenommen.Im Umgang mit anderen Erregern werden häufig Zellinien verwendet, da dieseeinfach in der Handhabung sind, nahezu beliebig viele Zellen zur Verfügung stehen,und die einzelnen Zellen annähernd identische Eigenschaften besitzen. InErmangelung einer bovinen Makrophagenzellinie werden Arbeiten in bovinenMakrophagen mit frisch isolierten, also primären Makrophagen durchgeführt, wobeihier das Problem der hohen Diversität der Zellpopulation entsteht.In der vorliegenden Arbeit wurde ein Modell der Teflonkultivierung primärer bovinerMakrophagen etabliert, charakterisiert und standardisiert. Das Modell ist aufgrundder hohen Reproduzierbarkeit für Arbeiten im bovinen Modell sehr gut geeignet.In dem etablierten und standardisierten Modell wurden verschiedeneUntersuchungen zur Modulation der Makrophageneffektorfunktionen durchintrazelluläre M. ptb durchgeführt.Die Ansäuerung des Phagosoms ist Teil der bakteriziden bzw. bakteriostatischenEffektorfunktionen des Makrophagen und damit ein wichtiger Schritt in derPhagosomenreifung. Andere intrazelluläre Erreger, wie z. B. M. avium, verhinderndie Ansäuerung des Phagosomes, um im Makrophagen überleben zu können. In dervorliegenden Arbeit wurde gezeigt, daß vitale M. ptb, im Gegensatz zuhitzeinaktivierten, die Ansäuerung des Phagosoms nicht nur verhindern, sondern zu

Zusammenfassung
90
einer Alkalisierung der Vakuole führen. Dies ist möglicherweise ein wichtigerPathogenitätsmechanismus vitaler M. ptb.Um intrazelluläre Erreger abtöten zu können, benötigt der Makrophage daskoordinierte Zusammenspiel verschiedener Zytokine und anderer Enzyme, wie z. B.der induzierbaren NO-Synthase (iNOS). In Untersuchungen mit anderenMykobakterien konnte die Rolle einiger Zytokine teilweise charakterisiert werden.Welche Bedeutung diese Zytokine und die iNOS in der Infektion von M. ptb inprimären bovinen Makrophagen haben, wurde in der vorliegenden Arbeit untersucht.Zu diesem Zweck wurde die Genexpression von Interleukin (IL) -1�, IL-6,
Tumornekrosefaktor-alpha (TNF-�), IL-10, Granulozyten-Makrophagen Kolonie-Stimulationsfaktor (GM-CSF) und der iNOS in der Infektion mit vitalen sowie mithitzeinaktivierten M. ptb gemessen. Sie lag in der Infektion mit vitalen Erregerndeutlich über der mit hitzeinaktivierten Bakterien und unterschied sich im zeitlichenVerlauf. Auch diese Modifikation der Genexpression durch vitale M. ptb istmöglicherweise Teil der Pathogenitätsmechanismen, da eine Koordinierung derZytokinexpression und damit ein Abtöten der Erreger nicht mehr möglich ist.Ein dritter Weg, den Makrophagen auszuschalten, ist das Auslösen der sogenanntenRefraktärität. Sie bedeutet, daß der Makrophagen durch die erste Infektion desErregers nicht mehr in der Lage ist, auf eine Sekundärinfektion mit den bakterizidenbzw. bakteriostatischen Effektorfunktionen zu antworten. In der vorliegenden Arbeitwurde diese Refraktärität anhand der Expression der genannten Gene untersucht.VitaleM. ptb waren in der Lage, die Zelle in einen teilweise refraktären Zustand zuversetzen: auf eine Reinfektion mit vitalen M. ptb war keine Erhöhung der TNF-�-und iNOS-Genexpression zu verzeichnen. Besonders ausgeprägt war dieRefraktärität nach Stimulation mit Interferon-gamma (IFN-�). Dieses wird im Körpervon T-Zellen ausgeschüttet und dient der Aktivierung des Makrophagen, soll also dasAbtöten der intrazellulären Erreger unterstützen. In der vorliegenden Arbeit führte dieStimulation infizierter Makrophagen mit IFN-� jedoch zum genauen Gegenteil: die
Expression von IL-6, TNF-�, GM-CSF und der iNOS ging um 50 bis über 95 %
zurück. Die zur Aktivierung des Makrophagen gedachte IFN-�-Ausschüttung könnteim Körper also zu einer weiteren Deaktvierung des Makrophagen führen. Dies istmöglicherweise ein weiterer Pathogenitätsmechanismus vitaler M. ptb.

Summary
91
G Summary
Investigation of intracellular survival of Mycobacterium avium ssp. paratuberculosis in
primary bovine macrophages
Beatrice Schuemann
Mycobacterium avium Subspezies paratuberculosis (M. ptb) is the causative agent ofa chronic, granulomatous and non-treatable enteritis of ruminants, paratuberculosis.The infection normally occurs within the first few months, and the animals do notshow any clinical signs for years. With the outbreak of the disease cattle, above all,show severe intermittant diarrhoea and progressive wasting leading to cachexia. Thedisease usually ends lethally; the pathogenesis is largely unknown. It is widelyaccepted that the persistence of the pathogen within the macrophages of theintestinal wall plays a key role. The strategy and the mechanism of the intracellularsurvival of M. ptb within these cells is yet unknown. From work with other intracellularpathogenic mycobacteria it is known that they modulate the macrophage’s effectorfunctions with the aim not to be killed. Such a modulation is assumed for M. ptb aswell.Cell lines, since they are easy to handle and to multiply, are often used working withother intracellular pathogens. In the case of M. ptb the absence of a suitable cellculture system necessitates working with primary bovine macrophages. Such cellsare more difficult to standardise than cultured cell lines.In the present study a model of teflon cultivation of primary bovine macrophages wasestablished, characterised and standardised. Because of the high reproducibility themodel is well suited for the investigation in bovine cells.With the established and standardised model investigation of the modulation ofmacrophage’s effector functions by intracellular M. ptb were carried out in the presentstudy.The acidification of the phagosome is part of the bactericidal and bacteriostaticeffector functions of the macrophage and therefore an important part of thephagosomal maturation. Other intracellular pathogens, such as M. avium, prevent theacidification of the phagosome in order to survive within the macrophage.

Summary
92
In the present study it was shown, that live M. ptb, in contrast to heat-inactivatedM. ptb, do not only prevent acidification but lead to partial alcalization of the vacuole.This may be an important mechanism of pathogenicity of live M. ptb.In order to kill intracellular pathogens the macrophage needs the co-ordinatedexpression of different cytokines and other enzymes, like the inducable NO-synthase(iNOS). In studies with other mycobacteria the role of some cytokines was partiallycharacterised. The role of these cytokines, and of the iNOS, in M. ptb-infected bovinemacrophages was investigated in the present study. For this purpose the expression
of interleukin (IL-) 1�, IL-6, tumor necrosis factor alpha (TNF-�), IL-10, granulocytemacrophage colony stimulating factor (GM-CSF) and iNOS was measured inmacrophages infected with live and heat-inactivated M. ptb. In macrophages infectedwith live M. ptb the expression of all these genes was much higher and differences inthe kinetics of these expression levels between these products were observed. Thismodulation of gene expression by live M. ptb might well be part of the mechanisms ofpathogenicity, since a co-ordinated cytokine expression and, as a consequence, thekilling of the pathogen is prevented.A third possibility for the pathogen to deactivate the macrophage is to trigger the socalled ‘refractory state’. This means that after a primary stimulus the macrophage isunable to react with its bacteriocidal and bacteriostatic effector functions in responseto a secondary stimulus with the same stimulant. In the present study this refractorystate was investigated by means of the expression of the mentioned genes. Live M.ptb were able to cause a partial refractory state: after the re-infection with live M. ptb
there was no increase in TNF-� nor iNOS gene expression.
After stimulation with interferon gamma (IFN-�) this refractory state was especially
distinct. IFN-� is released in vivo by T-cells in order to activate the macrophage andthereby to kill intracellular pathogens. In the present study the stimulation of infected
macrophages with IFN-� led to the contrary: the gene expression of IL-6, TNF-�, GM-CSF and iNOS was decreased to between 50 and 5 percent. Therefore release ofIFN-� in vivo, meant to activate the macrophage, might instead lead to thedeactivation of the macrophage. This might be another mechanism by which M. ptbleads to pathogenicity.

Literaturverzeichnis
93
H Literaturverzeichnis
AADURIZ, J. J., R. A. JUSTE u. N. CORTABARRIA (1995):
Lack of mycobactin dependence of mycobacteria isolated on Middlebrook 7H11 from
clinical cases of ovine paratuberculosis.
Vet. Microbiol. 45, 211-217
ADAMS, J. L. u. C. J. CZUPRYNSKI (1994):
Mycobacterial cell wall components induce the production of TNF-alpha, IL-1, and IL-
6 by bovine monocytes and the murine macrophage cell line RAW 264.
Microb. Pathog. 16, 401-411
ADAMS, J. L., M. T. COLLINS u. C. J. CZUPRYNSKI (1996):
Polymerase chain reaction analysis of TNF-� and IL-6 mRNA levels in whole blood
from cattle naturally or experimentally infected with Mycobacterium paratuberculosis.
Can. J. Vet. Res. 60, 257-262
ADLER, H., B. FRECH, M. THÖNY, H. PFISTER, E. PETERHANS u. T. W. JUNGI
(1995):
Inducible Nitric Oxide Synthase in Cattle.
J. Immunol. 154, 4710-4718
ALPUCHE-ARANDA, C. M., J. A. SWANSON, W. P. LOOMIS u. S. I. MILLER
(1992):
Sallmonella typhimurium activates virulence gene transcription within acidified
macrophage phagosomes.
Proc. Natl. Acad. Sci. USA 89, 10079-10083

Literaturverzeichnis
94
BALCEWICZ-SABLINSKA, M. K., H. X. GAN u. H. G. REMOLD (1999):
Interleukin 10 Produced by Macrophages Inoculated with Mycobacterium avium
Attenuates Mycobacteria-Induced Apoptosis by Reduction of TNF-� Activity.
J. Infect. Dis. 180, 1230-1237
BALDWIN, G. C., J. FLEISCHMANN, Y. CHUNG, Y. KOYANAGI, I. S. Y. CHEN u. D.
W. GOLDE (1990):
Human immunodeficiency virus causes mononuclear phagocyte dysfunction.
Proc. Natl. Acad. Sci. U.S.A. 90, 3442-3446
BANG, B. (1906):
Chronische pseudotuberkulöse Darmentzündung beim Rinde.
Berl. Tierärztl. Wochenschr. 8, 759-763
BARCLAY, R., D. F. EWING u. C. RATLEDGE (1985):
Isolation identification and structural analysis of the mycobactins of Mycobacterium
avium, Mycobacterium intracellulare, Mycobacterium scrofulaceum and
Mycobacterium paratuberculosis.
J. Bacteriol. 164, 896-903
BASSEY, E. O. E. u. M. T. Collins (1997):
Study of T-Lymphocyte subsets of healthy and Mycobacterium avium subsp.
paratuberculosis-infected cattle.
Infect. Immun. 65, 4869-4872
BEGARA-MCGORUM, I., L. A. WILDNLOOD, C. J. CLARKE, K. M. CONNOR, K.
STEVENSON, C. J. MCINNES, J. M. SHARP u. D. G. JONES (1998):
Early immunopathological events in experimental ovine paratuberculosis.
Vet. Immunol. Immunopathol. 63, 265-287

Literaturverzeichnis
95
BEHYMER, D., H. RIEMANN, W. UTTERBACK, C. ELMI u. C. FRANTI (1991):
Mass screening of cattle sera against 14 infectious desease agents, using an ELISA
system for monitoring health in livestock.
Am. J. Vet. Res. 52, 1699-1704
BENDIXEN, P. H. (1978):
Immunologic reactions caused by infection with Mycobacterium paratuberculosis.
Nord. Vet. Med. 30, 163-168
BENEDICTUS, G., A. A. DIJKHUIZEN u. J. STELWAGEN (1987):
Economic losses due to paratuberculosis in dairy cattle.
Vet. Rec. 121, 142-146
BERMUDEZ, L. E., L. S. YOUNG u. H. ENKEL (1991):
Interaction of Mycobacterium avium complex with human macrophages: roles of
membrane receptors and serum proteins.
Infect. Immun. 59, 1697-1702
BERMUDEZ, L. E., M. WU, M. PETROFSKY u. L. S. YOUNG (1992):
Interleukin-6 antagonizes tumor necrosis factor-mediated mycobacteriostatic and
mycobactericidal activities in macrophages.
Inf. Immun. 60, 4245-4252
BISPING, W. u. G. AMTSBERG (1988):
Farbatlas zur Diagnose bakterieller Infektionserreger der Tiere.
Verlag Parey, Berlin, Hamburg, 119-120

Literaturverzeichnis
96
BLACK, C. M., M. PALIESCHESKEY, B. L. BEAMAN, R. M. DONOVAN u. E.
GOLDSTEIN (1986):
Acidification of phagosomes in murine macrophages: blockage by Nocardia
asteroides.
J. Infect. Dis. 154, 952-8
BLISKA, J. B., J. E. GALÁN u. S. FALKOW (1993):
Signal transduction in the mammalian cell during bacterial attachment and entry.
Cell 73, 903-920
BLOOD, D.C. u. O. M. RADOSTITIS (1989):
Veterinary Medicine. 7. Aufl.
Baillière, Tindall, London, 722-729
BONAY, M., F. BOUCHONNET, V. PELICIC, B. LAGIER, M. GRANDSAIGNE, D.
LECOSSIER, A. GRODET, M. VUKORKA, B. GICQUEL u. A. J. HANCE (1999):
Effect of Stimulation of Human Macrophages on Intracellular Survival of
Mycobacterium bovis Bacillus Calmette-Guerin.
Am. J. Respir. Crit. Care Med. 159, 1629-1637
BOWMAN, E. J., A. SIEBERS u. K. ALTENDORF (1988):
Bafilomycins: a class of inhibitors of membrane ATPases from microorganisms,
animal cells, and plant cells.
Proc. Natl. Acad. Sci. U.S.A. 85,7972-6
BUERGELT, C. D. u. J. R. DUNCAN (1978):
Age and milk production data of cattle culled from a dairy herd with paratuberculosis.
J. Am. Vet. Med. Ass. 173, 478-480

Literaturverzeichnis
97
BUCHMEIER, N. A. u. F. HEFFRON (1991):
Inhibition of macrophage phagosome-lysosome fusion by Salmonella typhimurium.
Infect. Immun. 59, 2232-2238
CAMPBELL, G. A. u. L. G. ADAMS (1992):
The long-term culture of bovine monocyte-derived macrophages and their use in the
study of intracellular proliferation of Brucella abortus.
Vet. Immunol Immunopathol. 34, 291-305
CARROL, M. E., P. S. JACKETT, V. R. ABER u. D. B. LOWRIE (1979):
Phagolysosome formation, cyclic adenosine 3' :5'-monophosphate and the fate of
Salmonella typhimurium within mouse peritoneal macrophages.
J. Gen. Microbiol. 110, 421-429
CHAN, J., X. FAN, S. W. HUNTER, P. J. BRENNAN u. B. R. BLOOM (1991):
Lipoarabinomannan, a possible virulence factor involved in persistence of
Mycobacterium tuberculosis within macrophages.
Infect. Immun. 59, 1755-1761
CHAN, J. u. S. H. E. KAUFMANN (1994):
Immune mechanisms of protection
in: B. R. BLOOM (Hrsg.): Tuberculosis: pathogenesis protection and control, ASM
Press, Washington DC, 389-415
CHEN, L. M., K. KANIGA u. J. E. GALAN (1996):
Salmonella spp. are cytotoxic for cultured macrophages.
Mol. Microbiol. 21, 1101-1115

Literaturverzeichnis
98
CHIODINI, R. J., H. J. VAN KRUININGEN u. R. S. MERKAL (1984):
Ruminant paratuberculosis (Johne’s disease): the current status and future
prospects.
Cornell. Vet. 74, 218-262
CHIODINI, R. J. u. H. J. VAN KRUININGEN (1986):
The prevalence of paratuberculosis in culled New England cattle.
Cornell Vet. 76, 91-104
CHIODINI, R. J. (1989):
Crohn’s disease and the mycobacteriosis: a review and comparison of two disease
entities.
Clin. Microbiol. Rev. 2, 90-117
CHIODINI, R. J. (1991):
Cellular immunology of intracellular infections.
in: R. J. CHIODINI u. J. M. KREEGER (Hrsg.): Proceedings of the 3rd International
Colloquium on paratuberculosis, Orlando, USA, 269-283
CHIODINI, R. J. u. W. C. DAVIS (1992):
The cellular immunology of bovine paratuberculosis: the predominant response is
mediated by cytotoxic gamma / delta T-lymphocytes which prevent CD4 activity.
Microbial. Pathog. 13, 447-463
CHOMCZYNSKI, P. u. N. SACCHI (1987):
Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-
chloroform extraction.
Anal Biochem 162, 156-9

Literaturverzeichnis
99
CLARKE, C. J. (1997):
The pathology and pathogenesis of paratuberculosis in ruminants and other species.
J. Comp. Path. 116, 217-261
CLEMENS, D. L. (1996):
Characterization of the Mycobacterium tuberculosis phagosome.
Trends Microbiol. 4, 113-8
COCITO, C., P. GILOT, M. COENE, M. DE KESEL, P. POUPART u. P. VANUFFEL
(1994):
Paratuberculosis.
Clin. Microbiol. Rev. 7, 328-345
COHN, Z. A. (1968):
The structure and function of monocytes and macrophages.
Adv. Immunol. 9, 163-214
DALZIEL, T. K. (1913):
Chronic interstitial enteritis.
Br. Med. J. 2, 1068-1070
DAVIES, D., L. CORBEIL, D. WARD u. J. DUNCAN (1974):
A humoral suppressor of in vitro lymphocyte transformation responses in cattle with
Johne’s disease.
Proc. Soc. Exp. Biol. Med. 145, 1372-1377
DOUGHERTY, G. J. u. W. H. MCBRIDE (1984):
Macrophage heterogeneity.
J. Clin. Lab. Immunol. 14, 1-11

Literaturverzeichnis
100
DOUGHERTY, G. J. u. W. H. MCBRIDE (1989):
Monocyte differentiation in vitro.
in: M. ZEMBLA u. G. L. ASHERSON (Hrsg.): Human Monocytes,
Verlag Academic Press, 49-58
FINLAY, B. B. u. S. FALKOW (1997):
Common themes in microbial pathogenicity revisited.
Microbiol. Mol. Biol. Rev 61, 136-169
FIORENTINO, D. F., A. ZLOTNIK, T. R. MOSMANN, M. HOWARD u. A. O’GARRA
(1991):
IL-10 inhibits cytokine production by activated macrophages.
J. Immunol. 147, 3815-3822
FLESCH, I. E. u. S. H. KAUFMANN (1991):
Mechanisms involved in mycobacterial growth inhibition by gamma interferon-
activated bone marrow macrophages: role of reactive nitrogen intermediates.
Infect. Immun. 59, 3213-3218
FRANCIS, J., H. M. MACTURK, J. MADINAVEITIA u. G. A. SNOW (1953):
Mycobactin a growth factor for Mycobacterium johnei. I. Isolation from
Mycobacterium phlei.
Biochem. J. 55, 596-607
FRATAZZI, C., R. D. ARBEIT, C. CARINI, M. K. BALCEWICZ-SABLINSKA, J.
KEANE, H. KORNFELD u. H. G. REMOLD (1999):
Macrophage apoptosis in mycobacterial infections.
J. Leukoc. Biol. 66, 763-764

Literaturverzeichnis
101
FUCHS, R., S. SCHMID u. I. MELLMAN (1989):
A possible role for Na+-K+-ATPase in regulating ATP-dependent endosome
acidification.
Proc. Natl. Acad. Sci. U.S.A. 2, 539-543
GARCIA, I., R. GULER, D. VESIN, M. L. OLLEROS, P. VASSALLI, Y. CHVATCHKO,
M. JACOBS u. B. RYFFEL (2000):
Lethal Mycobacterium bovis Bacillus Calmette Guerin infection in nitric oxide
synthase 2-deficient mice: cell-mediated immunity requires nitric oxide synthase 2.
Lab. Invest. 80, 1385-1397
GARCIA-DEL PORTILLO, F. u. B. B. FINLAY (1995):
Targeting of Salmonella typhimurium to vesicles containing lysosomal membrane
glycoproteins bypasses compartements with mannose 6-phosphate receptors.
J. Cell Biol. 129, 81-97
GARCIA-DEL PORTILLO, F., M. G. PUCCIARELLI, W. A. JEFFRIES u. B. B.
FINLAY (1994):
Salmonella typhimurium induces selective aggregation and internalization of host cell
surface proteins during invasion of epithelial cells.
J. Cell Sci. 107, 2005-2020
GERLACH, G. F. u. P. VALENTIN-WEIGAND (1998):
Bovine paratuberculosis: History and results of new efforts to control an old disease.
Berl. Munch. Tierarztl. Wschr. 111, 386-373
GOFF, W. L., K. I. O’ROURKE, W. C. JOHNSON, P. A. LACY, W. C. DAVIS u. C. R.
WYATT (1998):
The role of IL-10 in iNOS and cytokine mRNA expression during in vitro
differentiation of bovine mononuclear phagocytes.
J Interferon Cytokine Res 18, 139-149

Literaturverzeichnis
102
GRANT, I. R., H. J. BALL, S. D. NEILL u. M. T. ROWE (1996):
Inactivation of Mycobacterium paratuberculosis in Cow’s Milk at pasteurization
temperatures.
Appl. Environ. Microbiol. 62, 631-636
HEIN, W. R. u. C. R. MACKAY (1991):
Prominence of gamma/delta T cells in the ruminant immune systeme.
Immunol. Today 12, 30-34
HOAL-VAN HELDEN, E. G., L. A. STANTON, R. WARREN, M. RICHARDSON u. P.
D. VAN HELDEN (2001):
Diversity of in vitro cytokine responses by human macrophages to infection by
Mycobacterium tuberculosis strains.
Cell Biol. Int. 25, 83-90
HOMUTH, M. (2001):
persönliche Mitteilung
HSU, N., L. S. YOUNG u. L. E. BERMUDEZ (1995):
Response to stimulation with recombinant cytokines and synthesis of cytokines by
murine intestinal macrophages infected with Mycobaterium avium complex.
Inf. Immun. 63, 528-533
ISHIBASHI, Y., u. T. ARAI (1990)
Specific inhibition of phagosome-lysosome fusion in murine macrophages mediated
by Salmonella typhimurium infection.
FEMS Microbiol. Immunol. 2, 35-43

Literaturverzeichnis
103
ITO, T. u. M. KODAMA (1996):
Demonstration by reverse transcription-polymerase chain reaction of multiple
cytokine mRNA expression in bovine alveolar macrophages and peripheral blood
mononuclear cells.
Res. Vet. Sci. 60, 94-96
JACOBS, M., N. BROWN, N. ALLIE, R. GULERT u. B. RYFFEL (2000a):
Increased resistance to mycobacterial infection in the absence of interleukin-10.
Immunology 100, 494-501
JACOBS, M., M. W. MARINO, N. BROWN, B. ABEL, L. G. BEKKER, V. J.
QUESNIAUX, L. FICK u. B. RYFFEL (2000b):
Correction of defective host response to Mycobacterium bovis BCG infection in TNF-
deficient mice by bone marrow transplantation.
Lab. Invest. 80, 901-914
JAGANNATH, C., E. SEPULVEDA, J. K. ACTOR, F. LUXEM, M. R. EMANUELE u.
R. L. HUNTER (2000):
Effect of poloxamer CRL-1072 on drug uptake and nitric-oxide-mediated killing of
Mycobacterium avium by macrophages.
Immunopharmacology 48, 185-197
JAMES S. L. (1995):
Role of nitric oxide in parasitic infections.
Microbiol. Rev. 59, 533-547
JOHNE, H.A. u. L. FROTHINGHAM (1895):
Ein eigenthümlicher Fall von Tuberculose beim Rind.
Dtsch. Z. Thiermed. Vergl. Path. 21, 438-454

Literaturverzeichnis
104
JUNGI, T. W., M. THÖNY, M. BRCIC, B. ADLER, U. PAULI u. E. PETERHANS
(1996):
Induction of nitric oxide synthase in bovine mononuclear phagocytes is differentation
stage-dependent.
Immunobiology 195, 385-400
KAPOSZTA, R., L. MARODI, M. HOLLINSHEAD, S. GORDON u. R. P. Da SILVA
(1999):
Rapid recruitment of late endosomes and lysosomes in mouse macrophages
ingesting Candida albicans.
J. Cell Sci. 112, 3237-48
KEEFE, R. G., Y. CHOI, D. A. FERRICK u. J. L. SCOTT (1997):
Bovine cytokine expression during different phases of bovine leukemia virus
infection.
Vet. Immunol. Immunopathol. 56, 39-51
KEMPER, C. A., L. E. BERMUDEZ u. S. C. DERESINSKI (1998):
Immunomodulatory treatment of Mycobacterium avium complex badteremia in
patients with AIDS by use of recombinant granulocyte-macrophage colony
stimulating factor.
J. Infect. Dis. 177, 914-920
KENNEDY, D. J. (1996):
The Australian national Johne’s Disease Market Assurance Program.
The Paratuberculosis News Lett. 8, 28
KISHIMA, M., Y. YOKOMIZO u. N. GOTO (1991):
Suppressive effect of nonviable Mycobacterium paratuberculosis on the delayed-type
hypersensitivity reaction to sheep erytrocytes in mice.
Lab. Anim. 25, 310-318

Literaturverzeichnis
105
KOETS, A. P., V. P. M. G. RUTTEN, M. DE BOER, D. AKKER, P. VALENTIN-
WEIGAND u. W. VAN EDEN (2001):
Differential changes in heat shock protein-, lipoarabinomannan-, and purified protein
derivated-specific immunoglobulin G1 and G2 isotype responses during bovine
Mycobacterium avium subsp. paratuberculosis infection.
Infect. Immun. 69, 1492-1498
KOPECKY, K. E. (1977):
Distribution of paratuberculosis in Wisconsin, by soil regions.
J. Am. Vet. Med. Assoc. 170, 320-324
KREEGER, J. M. (1991):
Ruminant paratuberculosis - a century of progress and frustration.
J. Vet. Diagn. Invest. 3, 373-383
KUEHNEL, M. P., R. GOETHE, A. HABERMANN, E. MUELLER, M. ROHDE, G.
GRIFFITHS, P. VALENTIN-WEIGAND (2001):
Characterization of the intracellular survival of Mycobacterium avium ssp.
paratuberculosis: phagosomal pH and fusogenicity in J774 macrophages compared
with other mycobacteria.
Cell. Microbiol. 3, 551-566
LAMBRECHT, R. S., J. F. CARRIERE u. M. T. COLLINS (1988):
A model for analyzing growth kinetics of a slowly growing Mycobacterium sp..
Appl. Environ. Microbiol. 54, 910-916
LAMBRECHT, R. S. u. M. T. COLLINS (1992):
Mycobacterium paratuberculosis. Factors that influence mycobactin dependence.
Diagn. Microbiol. Infect. Dis. 15, 239-246

Literaturverzeichnis
106
LARSEN, A. B., R. S. MERKAL u. R. C. CUTLIP (1975):
Age of cattles as related to resistance to infection with Mycobacterium
paratuberculosis.
Am. J. vet. Res. 36, 255-257
LARSEN, A. B., R. S. MERKAL u. T. H. VARDAMAN (1956)
Survival time of Mycobacterium paratuberculosis.
Am. J. Vet. Res. 17, 549-551
LEONHARDT, H. (1990):
Histologie, Zytologie und Mikroanatomie des Menschen.
Verlag Thieme
LEPPER, A. W., C. R. WILKS, M. KOTIW, J. T. WITHEHEAD u. K. S. SWART
(1989):
Sequential bacteriological observations in relation to cell-mediated and humoral
antibody response of cattle infected with Mycobacterium paratuberculosis and
maintained on normal or high iron intake.
Aust. Vet. J. 66, 50-55
LINDL, T. (1987):
Zell- und Gewebekultur.
Gustav Fischer Verlag, 76-80
LISBY, G., J. ANDERSEN, K. ENGBAEK u. V. BINDER (1994):
Mycobacterium paratuberculosis in intestinal tissue from patients with Crohn’s
disease demonstrated by a nested primer polymerase chain reaction.
Scand. J. Gastroenterol. 10, 923-929

Literaturverzeichnis
107
LIU, S. L. u. K. E. SANDERSON (1995):
Rearragements in the genome of the bacterium Salmonella typhi.
Proc. Natl. Acad. Sci. USA 92, 1018-1022
MALIK, Z. A., G. M. DENNING u. D. J. KUSNER (2000):
Inhibition of Ca(2+) signaling by Mycobacterium tuberculosis is associated with
reduced phagosome-lysosome fusion and increased survival within human
macrophages.
J. Exp. Med. 191, 287-302
MEANS, T. K., S. WANG, E. LIEN, A. YOSHIMURA, D. T. GOLENBOCK u. M. J.
FENTON (1999):
Human toll-like receptors mediate cellular activation by Mycobacterium tuberculosis.
J. Immunol. 163, 3920-3927
MERKAL, R. S., K. KOPECKY, A. LARSEN u. R. NESS (1970):
Immunologic mechanisms in bovine paratuberculosis.
Am. J. Vet. Res. 31, 475-485
MERKAL, R. S., K. R. RHOADES, J. E. GALLAGHER u. A. E. RICHIE (1973):
Scanning electron microscopy of mycobacteria.
Am. Rev. Resp. Dis. 108, 382-387
MERKAL, R. S., u. B. J. CURRAN (1974):
Growth and metabolic characteristics of Mycobacterium paratuberculosis.
Appl. Microbiol. 28, 276-279
MERKAL, R. S. (1984 a):
Paratuberculosis.
in: G. P. KUBICA u. L. G. WAYNE (Hrsg.): The mycobacteria: a source book.
Verlag Dekker, New York, Basel, 1237-1249

Literaturverzeichnis
108
MERKAL, R. S. (1984 b):
Paratuberculosis: Advances in cultural, serologic and vaccination methods.
J. Am. Vet. Med. Assoc. 184, 939-943
MILLAR, D., J. FORD, J. SANDERSON S. WITHEY, M. TIZARD, T. DORAN u. J.
HERMON-TAYLOR (1996):
IS900 PCR to detect Mycobacterium paratuberculosis in retail supplies of whole
pasteurized cows' milk in England and Wales
Appl. Environ. Microbiol. 62, 3446-52
MOMOTANI, E., D. L. WHIPPLE, A. B. THIERMANN u. N. F. CHEVILLE ( 1988):
Role of M-cells and macrophages in the entrance of Mycobacterium paratuberculosis
into domes of ileal Peyer‘s patches in calves.
Vet. Pathol. 25, 131-137
MONACK, D. M., B. RAUPACH, A. E. HROMOCKY u. S. FALKOW (1996):
Salmonella typhimurium invasion induces apoptosis in infected macrophages.
Proc. Natl. Acad. Sci. USA 93, 9833-9838
MORAHAN, P. S. (1980):
Macrophage nomenclature: where are we going?
J. Reticuloendothelial Soc. 27, 223-245
NELSON, N. (1987):
The vacular proton-ATPase of eucaryotic cells.
Bioessays 7, 251-254

Literaturverzeichnis
109
NOMURA, F., S. AKASHI, Y. SAKAO, S. SATO, T. KAWAI, M. MATSUMOTO, K.
NAKANISHI, M. KIMOTO, K. MIYAKE, K. TAKEDA u. S. AKIRA (2000):
Cutting Edge: Endotoxin tolerance in mouse peritoneal macrophages correlates with
down-regulation of surface toll-like receptor 4 expression.
J. Immunol. 164, 3476-3479
OPPENHEIM, J. J. u. E. J. LEONARD (1989):
Introduction.
in: M. ZEMBLA u. G. L. ASHERSON (Hrsg.): Human Monocytes,
Verlag Academic Press, 3-6
PALIWAL, O. P., R. KUMAR u. R. SOMVANSHI (1985):
The immune spectrum of Mycobacterium johnei infections in goats.
Indian Vet. J. 69, 743-747
PATTERSON, D. S., W. M. ALLEN u. M. K. LLOYD (1967):
Clinical Johne’s disease as a protein losing enteropathy.
Vet. Rec. 81, 717-718
PATTERSON, D. S. u. S. BERRETT (1968):
Malabsorption in Johne’s disease of cattle: depressed in vitro amino-acid uptake by
isolated intestinal tissue preparations.
Vet. Rec. 81, 55-56
PATTERSON, D. S. u. S. BERRETT (1969):
Malabsorption in Johne’s disease in cattle: an in vitro study of L-histidine uptake by
isolated intestinal tissue preparations.
J. Med. Microbiol. 2, 327-334

Literaturverzeichnis
110
POHLENZ, J. (1991a):
Krankheiten der Verdauungsorgane.
in: L. C. SCHULZ (Hrsg.): Pathologie der Haustiere Band 1, 1. Auflage,
Verlag Gustav Fischer Jena, 312-313
POHLENZ, J. (1991b):
Krankheiten und Syndrome der Wiederkäuer.
in: L. C. SCHULZ (Hrsg.): Pathologie der Haustiere Band 2, 1. Auflage,
Verlag Gustav Fischer Jena, 76-77
REINER, N. E., W. NG, C. B. WILSON, W. R. MCMASTER u. S. K. BURCHETT
(1990):
Modulation of in vitro monocyte cytokine responses to Leishmania donovani.
Interferon-gamma prevents parasite-induced inhibition of interleukin 1 production and
primes monocytes to respond to Leishmania by producing both tumor necrosis
factor-alpha and interleukin 1.
J. Clin. Invest. 85, 4330-4334
REINER, N. E. (1994):
Altered cell signaling and mononuclear phagocyte deactivation during intracellular
infection.
Immunol. Today 15, 374-381
RIEDEL, D. D. u. S. H. E. KAUFMANN (2000):
Differential tolerance induction by lipoarabinomannan and lipopolysaccharide in
human macrophages.
Microb. Infect. 2, 463-471

Literaturverzeichnis
111
RICHARDS, W. D. (1988):
The apparent effect of environmental acidity on the incidence of paratuberculosis.
In: Second international Colloquium on Paratuberculosis, Maisons-Alfort, 342-349
ROSENBERGER, G. (1978):
Krankheiten des Rindes.
2. Aufl. Verlag Parey, Berlin, Hamburg, 756-760
ROSSITER, C. A. u. W. S. BURHANS (1996):
Farm specific approach to paratuberculosis (Johne’s disease) control.
The Paratuberculosis News Lett. 8, 28
SCHEIBL, P. (1996):
Differenzierung klinischer Mycobacterium paratuberculosis-Isolate mittels 16S-23S
rDNS-Spacer-Analyse und Random Amplified Polymorphic DNA-PCR (RAPD-PCR).
Dissertation, Tierärztliche Hochschule Hannover
SCHLESINGER, P. H. (1994):
Measuring the pH of Pathogen-Containing Phagosomes.
Meth. Cell Biol. 45, 289-311
SIBLEY, L. D., L. B. ADAMS u. J. L. KRAHENBUHL (1990):
Inhibition of interferon-gamma-mediated activation in mouse macrophages treated
with lipoarabinomannan.
Clin. Exp. Immunol. 80, 141-148
SIBLEY, L. D., E. WEIDNER u. J. L. KRAHENBUHL (1985):
Phagosome acidification blocked by intracellular Toxoplasma gondii.
Nature 315, 416-429

Literaturverzeichnis
112
SILVA, M. T., R. APPELBERG, M. N. SILVA u. P. M. MACEDO (1987):
In vivo killing and degradation of Mycobacterium aurum within mouse peritoneal
macrophages.
Infect. Immun. 55,2006-16
SMITHWICK, R. W., M. R. BIGBIE, R. B. FERGUSON, M. A. KARLIX u. C. K.
WALLIS (1995):
Phenolic acridine orange fluorescent stain for mycobacteria.
J. Clin. Microbiol. 33, 2763-2764
SPURR, A. R. (1969):
A low-viscositty epoxy resin embedding medium for electron microscopy.
J. Ultrastruct. Res. 26, 31-43
STABEL, J. R. (1995):
Temporal effects of tumor necrosis factor-alpha on intracellular survival of
Mycobacterium paratuberculosis.
Vet. Immunol. Immunopathol. 45, 321-332
STREETER, R. N., G. F. HOFFSIS, S. BECH-NIELSEN, W. P. SHULAW u. D. M.
RINGS (1995):
Isolation of Mycobacterium paratuberculosis from colostrum and milk of subclinically
infected cows.
Am. J. Vet. Res. 56, 1322-1324
STURGILL-KOSZYCKI, S., P. H. SCHLESINGER, P. CHAKRABORTY, P. L.
COLLINS, A. K. FOK, R. D. ALLEN, S. L. GLUCK, J. HEUSER u. D. G. RUSSEL
(1994):
Lack of acidification in Mycobacterium phagosomes produced by exclusion of the
vesicular proton ATPase.
Science 263, 678-681

Literaturverzeichnis
113
SUANEGA, K., Y. YOKOYAMA, K. OKAZAKI u. Y. YAMAMOTO (1995):
Mycobacteria in the intestine of Japanese patients with inflammatory bowel disease.
Am. J. Gastroenterol. 1, 76-80
THOEN, C. O. u. K. H. BAUM (1988):
Current knowledge on paratuberculosis.
J. Am. Vet. Med. Assoc. 192, 1609-1611
THOEN, C. O., W. D. RICHARDS u. J. L. JARNAGIN (1977):
Mycobacteria isolated from exotic animals.
J. Am. Med. Vet. Assoc. 170, 987-990
THOREL, M.-F., M. KRICHEVSKY u. V. V. LEVY-FREBAULT (1990):
Numerical taxonomy of mycobactin-dependent mycobacteria, emended description
of Mycobacterium avium, and description of Mycobacterium avium subsp. avium
subsp. nov., Mycobacterium avium subsp. paratuberculosis subsp. nov., and
Mycobacterium avium subsp. silvaticum subsp. nov..
Int. J. Syst. Bacteriol. 40, 254-260
TRAUTWEIN, G. (1991):
Krankheiten der Leber.
in: L. C. SCHULZ (Hrsg.): Pathologie der Haustiere Band 1, 1. Auflage,
Verlag Gustav Fischer Jena, 393
TREVES, A. J. (1984):
The origin of monocyte-macrophage heterogeneity: possible alternatives.
Med. Hypoth. 14, 335-346
TRUANT, J. P., W. A. BRETT u. W. THOMAS (1962):
Fluorescence microscopy of tubercle bacilli stained with auramine and rhodamine.
Henry Ford. Hosp. Med. Bull. 10, 287-296

Literaturverzeichnis
114
TSUKANO, H., F. KURA, S. INOUE, S. SATO, H. IZUMIYA, T. YASUDA u. H.
WATANABE (1999):
Yersinia pseudotuberculosis blocks the phagosomal acidification of B10.A mouse
macrophages through the inhibition of vacuolar H(+)-ATPase activity.
Microb. Pathog. 27, 253-63
TWORT, F. W., u. G. L. Y. INGRAM (1912):
A method for isolating and cultivating the Mycobacterium enteritidis chronicae
pseudotuberculosae johne and some experiments on the preparation of a diagnostic
vaccine for pseudotuberculous enteritis of bovines.
Proc. Roy. Soc. London 84, 517-543
VALENTIN-WEIGAND, P. u. R. GOETHE (1999):
Pathogenesis of Mycobacterium avium Subspezies paratuberculosis infections in
ruminants: still more questions than answers.
Microb. Infect. 1, 1121-1127
VALENTIN-WEIGAND, P. u. K. M. MORIARTY (1992):
Mycobacterium paratuberculosis binds fibronectin.
Res. Microbiol. 143, 75-79
VAN DER MEER, J. W., D. BULTERMAN, T. L. VAN ZWET, I. ELZENGA-CLAASEN
u. R. VAN FURTH (1978):
Culture of mononuclear phagocytes on a teflon surface to prevent adherence.
J. Exp. Med. 147, 271-276
VAN HEYNINGEN T. K., H. L. COLLINS u. D. G. RUSSELL (1997):
IL-6 produced by macrophagees infected with Mycobacterium species suppresses T
cell responses.
J. Immunol. 158, 330-337

Literaturverzeichnis
115
VARY, P. H., P. R. ANDERSEN, E. GREEN, J. HERMON-TAYLOR u. J. J.
MACFADDEN (1990):
Use of highly specific DNA probes and the polymerase chain reaction to detetct
Mycobacterium paratuberculosis in Johnes disease.
J. Clin. Microbiol. 28, 933-937
VISKE, D., B. LARSSON, A. ENGVALL u. G. BOELSKE (1996):
Paratuberculosis in Sweden.
The Paratuberculosis News Lett. 8, 29
WAGENER, K. (1966):
Kursus der veterinärmedizinischen Mikrobiologie.
Verlag Paul Parey, 104-105
WEBER, A., M. ROTH u. R. GUERKE-WERNER (1991):
Paratuberkulose beim Damwild.
VET 9-91, 21-23
WECKER, E. (1990):
Immunologie kurzgefasst.
Wissenschaftsverlag
ZEMBALA, M. u. G. L. ASHERSON (1989):
Human Monocytes.
Verlag Academic Press
ZHAO, B., M. T. COLLINS u. C. J. CZUPRYNSKI (1997):
Effects of gamma interferon and nitric oxide on the interaction of Mycobacterium
avium subsp. paratuberculosis with bovine monocytes.
Infect. Immun. 65, 1761-1766

Literaturverzeichnis
116
ZURBRICK, B. G. u. C. J. CZUPRYNSKI (1987):
Ingestion and intracellular growth of Mycobacterium paratuberculosis within blood
monocytes and monocyte-derived macrophages.
Infect. Immun. 116, 1588-1593
ZURBRICK, B. G., D. M. FOLLETT u. C. J. CZUPRYNSKI (1988):
Cytokine regulation of the intracellular growth of Mycobacterium paratuberculosis in
bovine monocytes.
Infect. Immun. 56, 1692-1697

Anhang
117
I Anhang
I.1 Chemikalien und Verbrauchsmaterialien
A. bidest. (SIGMA) SIGMA, Deisenhofen
Aceton BIESTERFELD, Harsum
Agar Agar MERCK, Darmstadt
Agarose GIBCO, Eggenstein
Akutase PAA, Cölbe
Ammoniumchlorid SIGMA, Deisenhofen
Ammoniumpersulfat SIGMA, Deisenhofen
Bafilomycin CALBIOCHEM, Bad Soden
Bleicitrat SIGMA, Deisenhofen
Borsäure MERCK, Darmstadt
Braunüle (G12, 8 cm) BRAUN, Melsungen
Bromphenolblau MERCK, Darmstadt
Butanol MERCK, Darmstadt
Cacodylat MERCK, Darmstadt
Calciumchlorid MERCK, Darmstadt
Calciumchlorid-Dihydrat MERCK, Darmstadt
Chloroform MERCK, Darmstadt
Citronensäure-Monohydrat MERCK, Darmstadt
DABCO� FLUKA, Neu-Ulm
Diammoniumcitrat MERCK, Darmstadt
Diethanolamin MERCK, Darmstadt
Dinatriumhydrogenphosphat-Dihydrat MERCK, Darmstadt
Dithiothreitol (DTT) SIGMA, Deisenhofen
Eisenammoniumcitrat SIGMA, Deisenhofen
Essigsäure (Eisessig) ROTH, Karlsruhe
Ethanol absolut ROTH, Karlsruhe
Ethanol (96%) BIESTERFELD, Harsum
Ethanoldiamintetraacatat (EDTA) SIGMA, Deisenhofen

Anhang
118
Ethidiumbromid SIGMA, Deisenhofen
Ficoll 400 MERCK, Darmstadt
Fluorescein Isothiocyanat (FITC)-Dextran SIGMA, Deisenhofen
fötales Kälberserum (FCS) PAA, Cölbe
Formaldehyd (37%) ROTH, Karlsruhe
Glasperlen MERCK, Darmstadt
Glukose (wasserfrei) MERCK, Darmstadt
Glutamin GIBCO, Eggenstein
Glutaraldehyd FLUKA, Neu-Ulm
Glycerin ROTH, Karlsruhe
Glykogen BOEHRINGER, Mannheim
Guanidinthiocyanat SIGMA, Deisenhofen
Harnstoff SIGMA, Deisenhofen
Iscove's Medium GIBCO, Eggenstein
Isoamylalkohol MERCK, Darmstadt
Isopropanol SIGMA, Deisenhofen
Kaliumacetat MERCK, Darmstadt
Kaliumchlorid MERCK, Darmstadt
Kaliumdihydrogenphosphat MERCK, Darmstadt
Kaliumhydrogencarbonat MERCK, Darmstadt
Kanüle (G19) BRAUN, Melsungen
Karbolfuchsin MERCK, Darmstadt
Kryoröhrchen SARSTEDT, Nümbrecht
L-Asparagin SIGMA, Deisenhofen
Lipopolysaccharid (LPS, Salmonella) SIGMA, Deisenhofen
Magnesiumchlorid GIBCO, Eggenstein
Magnesiumsulfat MERCK, Darmstadt
Magnesiumsulfat-Heptahydrat MERCK, Darmstaddt
Mercaptoethanol MERCK, Darmstadt
Methanol BIESTERFELD, Harsum
Methylenblau MERCK, Darmstadt

Anhang
119
Middlebrook OADC-Enrichment BECTON DICKINSON, Sparks (USA)
Mikroliterküvetten (1 ml) RATIOLAB, Dreieich
Mowiol MERCK, Darmstadt
Mycobactine J SYNBIOTICS, Lyon (Frankreich)
Natriumacetat MERCK, Darmstadt
Natriumchlorid ROTH, Karlsruhe
Natriumcitrat ROTH, Karlsruhe
Natriumhydroxid Plätzchen MERCK, Darmstadt
Natriumthiosulfat MERCK, Darmstadt
Nigericin SIGMA, Deisenhofen
Nukleotide (dATP, dCTP, dGTP, dTTP) GIBCO, Eggenstein
Osmiumtetroxid SERVA, Heidelberg
Paraformaldehyd SIGMA, Deisenhofen
PCR-Zusätze GIBCO, Eggenstein
Penicillin GIBCO, Eggenstein
Phenol ROTH, Karlsruhe
Phosphorsäure (89%) MERCK, Darmstadt
Propidiumjodid MOLECULAR PROBES, Eugene (USA)
Reaktionsgefäß, 200 µl BIOZYM, Hess. Oldendorf
Reaktionsgefäß, 1,5 ml BRAND, Wertheim
Reaktionsgefäß, 2 ml SARSTEDT, Nümbrecht
Reaktionsgefäß, 15 ml GREINER, Frickenhausen
Reaktionsgefäß, 50 ml SARSTEDT, Nümbrecht
Saccharose SERVA, Heidelberg
Salzsäure SIGMA, Deisenhofen
Sarkosyl (Natrium-N-Laurylsarcosine) SIGMA, Deisenhofen
Sodiumdodecylsulfat (SDS) ROTH, Karlsruhe
Sterilfilter (0,2 µm und 0,45 µm) MILLIPORE, Bedford (USA)
Streptomycin GIBCO, Eggenstein
Sulpho-NHS-Biotin PIERCE, Rockford (USA)
Syto 9 MOLECULAR PROBES, Eugene (USA)

Anhang
120
Teflonfolie ANGST UND PFISTER, Zürich (Schweiz)
Tetramethyl-ethylendiain (TEMED) SIGMA, Deisenhofen
TexasRed-Streptavidin PIERCE, Rockford (USA)
Transferpipette BRAUN, Melsungen
Trichloressigsäure (TCA) ROTH, Karlsruhe
Tri-Natriumcitrat-Dihydrat ROTH, Karlsruhe
Tris (hydroxymethyl) aminomethan MERCK, Darmstadt
Triton X 100 SERVA, Heidelberg
Uranylacetet MERCK, Darmstadt
Zellschaber NUNC GmBH, Wiesbaden
Zinksulfat-Heptahydrat MERCK, Darmstadt
ZK-Flasche, mittel GREINER, Frickenhausen
ZK-Schale, 24-well NUNC GmBH, Wiesbaden
ZK-Schale, 6-well NUNC GmBH, Wiesbaden
ZK-Schale, klein GREINER, Frickenhausen
I.2 weitere Reagenzien
5 x Reverse Transkriptase Puffer GIBCO, Eggenstein
10 x PCR-Puffer GIBCO, Eggenstein
50 mM Magnesiumchlorid GIBCO, Eggenstein
100 Basenpaar Leiter (100 bp ladder) GIBCO, Eggenstein
DNase I (RNase frei, 10 U/µl) GIBCO, Eggenstein
DTT (0,1 M) GIBCO, Eggenstein
RNase Inhibitor (40 U/µl) GIBCO, Eggenstein
Super ScriptTM II-Reverse Transkriptase (200 U/µl) GIBCO, Eggenstein
Taq DNA Polymerase GIBCO, Eggenstein

Anhang
121
I.3 Geräteverzeichnis
1) Analysenwaage, (80-400 g), SARTORIUS über Landgraf, Hannover2) Analysenwaage, BA 61 (0-60 g), SARTORIUS über Landgraf, Hannover3) Brutschrank, CO2-Auto-Zero, HERAEUS-CHRIST, Osterode4) Brutschrank, MEMMERT GmbH & Co., Schwabach5) Elektronenmikroskop, Transmissions-EM 910, ZEISS, Göttingen6) Elektrophoresekammer horizontal für DNA, Easy-Cast Electrophoresis
System, OWL SCIENTIFIC, Inc. Woburn, MA USA7) FACS-Scan-Gerät, BECTON DICKINSON, Heidelberg8) Fluoreszenzmikroskop, LEICA, Bensheim9) Glasmesser, FCS Kryomikrotom, LEICA, Bensheim10) Hermle Zentrifuge, ZK 380, HERMLE, Wehingen11) Kamera digital, BIO-RAD, MultiAnalyst-Sytem, München12) Konfokalmikroskop, DM-IRBE, invertiert, LEICA, Bensheim13) Kühlzentrifuge, Hermle Z 400 K, HERMLE, Wehingen14) Lamina, HB 2448 S, HERAEUS, Hanau15) Magnetrührer, LR 12, METTLER über Landgraf, Hannover16) Mikroskop, Orthoplan, LEITZ, Wetzlar17) Mikrowelle, PANASONIC, Hannover18) Multi-Screen-Systems, BIO RAD, München19) pH-Meter, WTW, Weilheim20) Photometer und Zubehör, Spektratphotometer Ultrospec III, PHARMACIA,
Freiburg21) Photometer für Mikrotiterplatten, BCA-Reader, DINATEC, Guernsey, Island22) Pipettboy, TECNOMARA, Fernwald23) Schweißgerät, polystar� 100 GE, RISCHE UND HERFURTH, Hamburg24) Stromversorgungsgerät, PowerPac 300, BIO RAD, München25) Thermocycler, Primus, MWG-BIOTECH, Ebersberg26) Tischzentrifuge, EPPENDORF, Tespe27) Ultrostainer, LEICA, Bensheim28) Vortex/Aufrüttler, HEIDOLPH über Landgraf, Hannover29) Wasserbad, GFL, Burgwedel

122
An dieser Stelle möchte ich mich bedanken
bei Prof. Peter Valentin-Weigand für die Überlassung des Themas und die Betreuungder Arbeit,
bei Dr. Ralph Goethe für sein stetes Interesse, seine Ideen, sein Fachwissen, seineUnterstützung bei der Durchführung der Experimente, aber vor allem bei derAnfertigung der Dissertation - und in den letzten Wochen in Hannover sogar fürseinen Humor und das angenehme Arbeitsklima,
bei den Arbeitsgruppen von Manfred Rohde, Thomas Jungi, Hans-JoachimSchuberth und Beate Sodeik,
und bei den Mitarbeitern und Doktoranden des Institutes für Mikrobiologie undTierseuchen der Tierärztlichen Hochschule Hannover für die Hilfsbereitschaft und diestets nette und freundliche Zusammenarbeit. Mein besonderer Dank gilt hier:
Mark Kühnel für die Arbeiten am Konfokalmikroskop und viele andere Arbeitenund Hilfestellungen,Jörg Matusch für seine Hilfe bei allen Computerfragen (egal ob amWochenende oder spät Abends, auf Dich konnte ich immer zählen),Herrn Richter für das Zähmen und liebevolle Pflegen von Felix (ohne Sie wäreich nie an sein Blut gekommen),Herrn Kipri und Werner Scharnhorst für die Hilfe bei Felix (alleine ist es ebenfast unmöglich),Herrn Wernheimer für das Sterilfiltrieren der Unmengen an Citratpuffer (wennauch noch so kurzfristig, Sie haben’s immer möglich gemacht)und Felix für sein Blut (möge er ewig im Rinder-Himmel herumtollen).
Mein allerherzlichster Dank gilt aber meinen Eltern, die mir das Studium und dieDoktorarbeit erst ermöglicht haben,und Mark, der mich privat und auch fachlich immer unterstützt und die Zeit im Institutversüßt hat.





























