Embed Size (px)
Citation preview

DOI: 10.1161/CIRCULATIONAHA.114.013215
1
Myocardial Stiffness in Patients with Heart Failure and a Preserved Ejection
Fraction: Contributions of Collagen and Titin
Running title: Zile et al.; Stiffness in HFpEF
Michael R. Zile, MD1; Catalin F. Baicu, PhD1; John Ikonomidis MD, PhD2; Robert E. Stroud,
MS2; Paul J. Nietert, PhD3; Amy D. Bradshaw, PhD1; Rebecca Slater, BS4; Bradley M. Palmer,
PhD5,6; Peter Van Buren, MD5,6; Markus Meyer, MD, PhD5; Margaret Redfield, MD7;
David Bull, MD8; Henk Granzier, PhD4; Martin M. LeWinter, MD5,6
1Division of Cardiology, Dept of Medicine, Medical University of South Carolina, and RHJ Department of Veterans Affairs Medical Center, Charleston, SC; 2Division of Cardiothoracic
Surgery, Dept of Surgery, Medical University of South Carolina, and RHJ Department of Veterans Affairs Medical Center, Charleston, SC; 3Dept of Public Health Sciences, Medical University of
South Carolina, Charleston, SC; 4Dept of Cellular and Molecular Medicine, University of Arizona, Tucson, AZ; 5Cardiology Unit, Dept of Medicine, University of Vermont, Burlington, VT; 6Dept of Molecular Physiology and Biophysics, University of Vermont, Burlington, VT; 7Division of
Cardiology, Mayo Clinic, Rochester, MN; 8Division of Cardiothoracic Surgery, Dept of Surgery, University of Utah Health Sciences Center, Salt Lake City, UT
Address for Correspondence:
Michael R. Zile, MD
Division of Cardiology, Department of Medicine
Medical University of South Carolina
Ashley River Towers, 25 Courtenay Drive, Room 7067
Charleston, SC 29425
Tel: 843-792-6866
Fax: 843-789-6850
E-mail: [email protected]
Journal Subject Codes: Heart failure:[110] Congestive, Hypertension:[15] Hypertrophy, Hypertension:[115] Remodeling, Myocardial biology:[104] Structure
1Division of Cardiology, Dept of Medicine, Medical University of South Caroolilinanana, anannddd RHRHRHJJ Department of Veterans Affairs Medical Center, Charleston, SC; 2Division of Cardiothoracic
Surgery,, Depept of SSurgery, Medical University of South Carolina, and RHJ J Department of VeteransAfAfAffafafairirirss MeMeMedidd caal ll CCeCenter, Charleston, SC; 3Dept oooff PPPublic Health ScScS iencncceseses, Medical University of
SoSooutututh h Caroliliinnana, , ChChChararleleeststs ononon, , , SCSCC;;; 44DeDeD ptptp oof f CeCellllulularar aannd MMMolololecececulararr MeMeM didiciciinenen , UnUnniviviverere sisityty oooff f ArArArizizizononaTuTuucsc on, AZ; 55CCaC rrrdioiolologygygy UUUnininitt,t, DDDepepept t oofof Mededdiccinee, UUUniiivveerersisiityyy oof f VeVermrmoonontt, BBBuruurlilingnggtotoon,n,n VVVT;T;T; 666DeDeDept offf Molecululara PPPhyyysioolooogy aananddd BiB oppphyhyysiics, UnUnUniveerrssiityty ooof f f VeVeV rrmmonntt,, Burrrlinngngtttonnn, VVTTT; nn 777DiDiiviisionnn ooof
CaCaCardrddioioiolologygygy, , MaMaayooo CCllilinniic,c, RRRococo hehehestststeerer, MNMNMN;; ; 888DiDiiviviv ssisioonn ooof f CaCaCarrrdioioioththhororracacacicicc SuSuurgrggerere y,y,y, DDDepepept ooof SSSururggegerryry, UnUniviversisityt ooff UtUtah Heaaltlth h ScScieenncess CCenenteter,r SSalt t LaLakeke Citty,y UUTT
by guest on June 1, 2018http://circ.ahajournals.org/
Dow
nloaded from
by guest on June 1, 2018http://circ.ahajournals.org/
Dow
nloaded from
by guest on June 1, 2018http://circ.ahajournals.org/
Dow
nloaded from

DOI: 10.1161/CIRCULATIONAHA.114.013215
2
Abstract:
Background—The purpose of this study was to determine whether patients with heart failure
and a preserved ejection fraction (HFpEF) have an increase in passive myocardial stiffness and
the extent to which discovered changes are dependent on changes in extracellular matrix fibrillar
collagen and/or cardiomyocyte titin.
Methods and Results—Seventy patients undergoing coronary artery bypass grafting underwent
an echocardiogram, plasma biomarker determination, and intra-operative left ventricular (LV)
epicardial anterior wall biopsy. Patients were divided into 3 groups: referent control (n=17, no
hypertension or diabetes), hypertension (HTN) without(-) HFpEF (n=31), and HTN with(+)
HFpEF (n=22). One or more of the following studies were performed on the biopsies: passive
stiffness measurements to determine total, collagen-dependent and titin-dependent stiffness
(differential extraction assay), collagen assays (biochemistry or histology), or titin isoform and
phosphorylation assays. Compared with controls, patients with HTN(-)HFpEF had no change in
LV end diastolic pressure (LVEDP), myocardial passive stiffness, collagen, or titin
phosphorylation but had an increase in biomarkers of inflammation (CRP, sST2, TIMP-1).
Compared with both control and HTN(-)HFpEF, patients with HTN(+)HFpEF had increased
LVEDP, left atrial volume, NT-proBNP, total, collagen-dependent and titin-dependent stiffness,
insoluble collagen, increased titin phosphorylation on PEVK S11878(S26), reduced
phosphorylation on N2B S4185(S469), and increased biomarkers of inflammation.
Conclusions—Hypertension in the absence of HFpEF, did not alter passive myocardial stiffness.
Patients with HTN(+)HFpEF had a significant increase in passive myocardial stiffness; collagen-
dependent and titin-dependent stiffness were increased. These data suggest that the development
of HFpEF is dependent on changes in both collagen and titin homeostasis.
Key words: heart failure, diastole, hypertension, hypertrophy, collagen, Titin
differential extraction assay), collagen assays (biochemistry or histology), or tittininn iiisosoofofoformrmrm aaandndnd
phosphorylation assays. Compared with controls, patients with HTN(-)HFpEF hahad dd nnono ccchahahangngngee iniin
LV end diastolic pressure (LVEDP), myocardial passive stiffness, collagen, or titin
phhosososphphphororylylylaatatiionn bububut t had an increase in biomarkeeersrr oof inflammatiionono (CRCRCRPPP, sST2, TIMP-1).
CCCommpmpared wwitiith h h bobobothh cccononntrtrololo aaandndnd HHHTNTNTN((-(-)H)H)HFpFpFpEFEFE , papapatienenntstss wwwititith h HTHHTN(N(N +)+)+)HFHFH pEpEEFFF hahahaddd ininnccrcreaeaasesesed d d
LLVLVEEEDP, lefft t ata riialal vollumumme,, NNNTTT-prp oBoBBNNPNP, totttall, coolllagagenenn-d-d-depeppeeendeeenntt anddd ttititiinnn-ddedepep nnndeenenttt ssttiffnnnesss,
nnsosolululublblbleee cococollllagaggenenen, , ininncrcrcreaeaeases d d titititititinn n phphososphphphoororylylylatatatioioonnn ononon PPPEVEVEVKKK S1SS118181878778(S(S(S262626),), rredededucucucededed
phosphorylatatioioion nn ononon NNN2B2B2B SSS41414185855(S(S(S464669)9), , anannd d d ininncrcrcreaeae sesesed d d bibiiomomomarararkekekersrsr ooof f f inininflflflamamammamamatititiononon..
by guest on June 1, 2018http://circ.ahajournals.org/
Dow
nloaded from

DOI: 10.1161/CIRCULATIONAHA.114.013215
3
Introduction
Patients with heart failure and a preserved ejection fraction (HFpEF) have been shown to have
abnormalities of left ventricular (LV) diastolic function including slowed/incomplete relaxation,
decreased suction/recoil, and increased passive chamber stiffness 1-5. However, the myocardial
basis for these changes remains incompletely understood. Some recent clinical studies have
suggested that development of HFpEF is accompanied by significant changes in the composition
and structure of the extracellular matrix (ECM), especially fibrillar collagen 6-8. Other studies
and editorials have emphasized the contribution of changes in titin (the giant molecular spring
protein that is one important factor responsible for cardiomyocyte passive stiffness) 9-20, 21;
specifically, changes in the phosphorylation of titin 22-32. However, in previous studies of patients
with HFpEF, the role of titin was examined in isolated, chemically demembranated
cardiomyocytes obtained from endocardial myocardial biopsies; these preparations did not
include surrounding ECM structures. The aforementioned changes in collagen and/or titin are
expected to increase passive myocardial stiffness; however, myocardial stiffness has never been
measured directly in myocardium from HFpEF patients. Thus, the magnitude of the assumed
increase in myocardial passive stiffness and therefore its importance as a determinant of diastolic
dysfunction are unknown. In addition, the role of increased passive stiffness in the development
and clinical course of patients with HFpEF and the relative contribution of changes in the ECM
versus titin to passive stiffness in HFpEF are also unknown. In the present study, we
hypothesized that changes in both collagen and titin occur during the development of HFpEF and
that these changes combine to cause a major increase in myocardial passive stiffness that
contributes to the development of HFpEF.
To determine the extent to which ECM collagen and titin contribute to changes in
pecifically, changes in the phosphorylation of titin 22-32. However, in previous ssttutudididiess oof ff papapattitiene ts
with HFpEF, the role of titin was examined in isolated, chemically demembranated
caardrddioioiommymyocococytytytes ooobbtbtaia ned from endocardial myoocacac rrddial biopsies; ttthehh see prprprepe arations did not
nncllluude surrouundndndinnng ECECE M MM stststruruructctururresess. ThThThe affforrremmenenentiononneeded ccchhaangnggesss iin n cccolllagaggenenn aandndnd/o/o/orr titt tiiinn n ararree
exxpepepectctctedede ttoo o ininincrcreeaeassese pppasasssisiveve mmmyoyoyocacacarrdrdiiaal l stststifififfnnneeessss;;; hhohowweweveveverr,r, mmmyoyoyocacaardrdrdiaiaal sststififffnfnf esese ss hahahas nnenevveverr bbbeeenen
measured dirrececectltlt y y y ininin mmmyoyoy cacacardrdrdiuuum m m frfrromomom HHHFpFpFpEFEFEF papap titit enene tsss.. ThThThususus, , thththe mamamagngngnitititududude e ofofof ttthehehe aaassumed
by guest on June 1, 2018http://circ.ahajournals.org/
Dow
nloaded from

DOI: 10.1161/CIRCULATIONAHA.114.013215
4
myocardial passive stiffness in patients with HFpEF certain experimental conditions must be
met. First, since several potential determinants of passive stiffness could change concurrently
during the development of HFpEF, the contribution of each must be determined in samples of
myocardium which include an integrated, intact composite structure. To satisfy this condition,
methods of differential extraction have been developed to determine the separate contributions of
collagen and titin to stiffness in demembranated myocardial strips 9, 10. In the current studies
these methods were applied to strips prepared from LV myocardial biopsies obtained during
coronary artery bypass grafting (CABG). Second, the effects of co-morbidities and antecedent
diseases must be distinguished from changes associated with the clinical syndrome of HFpEF.
Two of the most common antecedent disease processes that lead to HFpEF are arterial
hypertension (HTN) and diabetes mellitus (DM) 33-37. In the current study, patients with HTN (or
DM combined with HTN) without HFpEF [HTN(-)HFpEF] were compared to patients with HTN
(or DM combined with HTN) with HFpEF [HTN(+)HFpEF]. Third, an appropriate referent
control group must be studied. For this purpose, CABG patients with no history of HTN or DM
were chosen. There are limitations in using these patients as referent controls in that CAD could
have uncertain effects on myocardial stiffness that cannot be distinguished from those related to
hypertension. However, CAD is present in a majority of patients with HFpEF (38); thus, a CAD
“background” is quite representative of the HFpEF population. Using these methods, the purpose
of this study was to determine whether patients with HFpEF and an antecedent history of HTN or
HTN/DM have an increase in passive myocardial stiffness and whether changes in stiffness are
dependent on changes in collagen and/or titin. Additionally, we sought to determine the
relationship between changes in myocardial passive stiffness and echocardiographic measures of
LV structure and function and selected plasma biomarkers.
Two of the most common antecedent disease processes that lead to HFpEF are aarrrterrriaial l l
hypertension (HTN) and diabetes mellitus (DM) 33-37. In the current study, patients with HTN (or
DMDMM ccomomombibibinenen d d wiwiwiththth HTN) without HFpEF [HTN(N(N(-))HFpEF] weree coc mpmppt ararared to patients with HTN
oor DDDM combiineneed wiwiiththt HHHTNTNTN)) ) wiwiththh HHHFFppEEF [[[HHHTN((+++)HFHFFppEpEFFF].. ThThhirrd,d, aannn aappprprpropopririatatate rerer fefef rereentnt
coontntn rorooll l grgrg ouououppp mumuuststt bbee e ststtududieed.dd FFFororor ttthihiss s pupupurprprpossse,e,e CCCAABABG G papapattitienenentsts wwwititith h h nnno hihistststororry y y oofof HHHTTNTN oor r DMDMDM
were chosen.n. TTTheheh rerere aarerere llimmmititi ataa iooonsnsn ininin uuusisis ngngng ttthehehesesese pppatattieientntnts ss asasas rrefefeferere entntnt cccononontrtrtrololo s s ininin ttthahahat t CAC D couldd
by guest on June 1, 2018http://circ.ahajournals.org/
Dow
nloaded from

DOI: 10.1161/CIRCULATIONAHA.114.013215
5
Methods
Study Population
Recruitment
The study cohort consisted of 70 males and females recruited to undergo intraoperative LV
myocardial biopsy from amongst those scheduled for CABG at 1) Fletcher Allen Health Care in
Burlington, Vermont, the clinical facility of the University of Vermont College of Medicine
(UVM), 2) the Ralph H. Johnson Department of Veterans Administration Medical Center and the
Medical University of South Carolina Hospital Authority (MUSC) in Charleston, South Carolina,
and 3) selected NHLBI Heart Failure Research Network (HFRN) centers (University of Alberta
[Alberta, Canada], Intermountain Medical Center [Murray, UT], the Mayo Clinic [Rochester,
MN], Minnesota Heart Institute [Minneapolis, MN], University of Utah [Salt Lake City, UT] and
the Utah VA Medical Center [Salt Lake City, UT]) between October 1, 2008 and August 6, 2012
who satisfied the inclusion and exclusion criteria specified below. All patients signed consent
forms approved by their respective Institutional Review Boards.
Experimental Measurements
Demographic, medication and laboratory data and cardiac catheterization results (coronary
anatomy, LV end-diastolic pressure) were tabulated. The severity of coronary artery disease
(CAD) was graded based on the number of major vessels (left anterior descending, left circumflex,
right coronary arteries) with a stenosis >70%, with left main coronary stenosis considered as two
vessels. Patients recruited at UVM and MUSC underwent an echocardiographic-Doppler
examination to assess LV chamber structure and function. In addition, in these patients a 10 cc
plasma sample was obtained for measurement of biomarkers. Intra-operative LV anterior wall
epicardial biopsies were obtained as previously described (39, 40). Patients recruited to this
Alberta, Canada], Intermountain Medical Center [Murray, UT], the Mayo Clinic [[[RoRoRochchheseesteteter,r,r, r
MN], Minnesota Heart Institute [Minneapolis, MN], University of Utah [Salt Lake City, UT] and
hhe e UtUtUtahahah VVVA AA MeMedididiccacal Center [Salt Lake City, UTTT]])]) bbbetween Octobberee 1,, 222000008 and August 6, 2012
wwhoo o satisfied ththhee innnclclluususioioon nn ananand d d exexclclc uususioonnn crititterrria ssppeeecifffieieied d bbbellooww.. AAAllll ppaata iieentnttss ssisigngnnededed ccoonnsesesentntt
foormrmrms ss apapapprprprovovoveded bbby y y ththeeieir r rerespspecece tititiveveve IIInsnsttitituuutititionononalalal RReevevieiewww BoBoBoaarardsdsds..
Experimentalal MMMeaeaasususurereememm ntnttsss
by guest on June 1, 2018http://circ.ahajournals.org/
Dow
nloaded from

DOI: 10.1161/CIRCULATIONAHA.114.013215
6
protocol were part of an NIH grant (RO1HL089944) with multiple specific aims; therefore, biopsy
samples were allocated to several protocols, data from some of which have been published 39, 40.
Given the size of each biopsy, all protocols could not be performed on each biopsy. For the current
study, 25 biopsies were used to assess myocardial passive stiffness; 30 were used to measure tissue
collagen content; and 14 were used for titin phosphorylation studies (Table 1).
General Inclusion Criteria
Patients scheduled to undergo CABG over 21 years of age, with a preserved LVEF ( 50%),
normal wall motion and end-diastolic volume index (EDVi, < 75mL/m2), and without evidence of
previous myocardial infarction were eligible. Patients were categorized into three groups: control,
HTN(-)HFpEF and HTN(+)HFpEF.
Specific Patient Group Inclusion Criteria
Control patients fulfilled the general inclusion criteria, but did not have a history of HTN or DM.
HTN(-)HFpEF patients fulfilled the general inclusion criteria, and had a history of HTN
documented in their records and/or had been told of this diagnosis by a physician, and were
receiving medications for its treatment. These patients had no evidence of heart failure as defined
below.
HTN(+)HFpEF patients fulfilled the general inclusion criteria and had HTN and HFpEF as
specified by the European Society of Cardiology and Heart Failure Society of America criteria41,42.
These criteria require: 1) signs and/or symptoms of heart failure (Framingham or Boston criteria,
exercise testing, quality of life questionnaire), 2) LVEF 50 %, 3) LVEDVi < 75 mL/m2, 4)
evidence of diastolic LV dysfunction obtained invasively (cardiac catheterization) or non-
invasively (transmitral or tissue Doppler or left atrial size) and 5) exclusion of non-cardiac diseases
that could cause symptoms commonly present in patients with heart failure.
HTN(-)HFpEF and HTN(+)HFpEF.
Specific Patient Group Inclusion Criteria u
CoContntntroroolll papaatititienennts fffulululfif lled the general inclusion crritititereriia, but did not hahh vee aaa hhhistory of HTN or DM. f
HTN(--)H)HHFpppEFEFEF ppatatatieieienntntsss fufulflflfilillleleddd tthhe gggennneraaal iincllluussioionnn ccrcrititeereriaia, ananndd hahadd d aa a hihistststororryy ofofof HHHTNTNTN
dodocucucummementntededd iiinn ththheiiir r rereecocoordrds aanand/d/d/ororor hhhadadd bbeeeeeen n n toooldldld ooof f tthiisis dddiaiaiagngngnosososisiss bbbyyydd aa ppphyhyhysisiicicic ananan,, anananddd wwwerrere
eceiving meedididicacac titiiononons s fofofor ititts s s trtrtreaaatmtmtmeeentnn . ThThThesesese e e papapatitiienenntststs hhadadad nnno o o evevevidididennncecece ooof f f hehehearara t t t fafafailililururure e e asa defined
by guest on June 1, 2018http://circ.ahajournals.org/
Dow
nloaded from

DOI: 10.1161/CIRCULATIONAHA.114.013215
7
Exclusion Criteria
Patients were excluded if they had a previous transmural myocardial infarction, LVEF < 50%,
LVEDVi > 75 mL/m2, significant valvular or other non-coronary heart disease, severe chronic
pulmonary disease requiring oral steroids and/or oxygen therapy, any non- cardiac disease or
condition known to affect myocardial function, anemia (Hgb < 13.0 g/dl), serum creatinine > 2.0
mg/dL, poorly controlled hypertension (blood pressure > 140/90 mmHg), off-pump or
emergency CABG, morbid obesity, history of substance abuse, inability to provide informed
consent, poorly controlled diabetes (HbA1c >8.5% within the past 6 months), active malignancy,
severe connective tissue disease , severe liver disease, hypertrophic cardiomyopathy, restrictive
cardiomyopathy or constrictive pericarditis, HIV or active infection.
Myocardial Biopsy Procedure
Anterior LV free wall epicardial biopsies weighing ~25-50 mg were obtained during CABG
soon after the patient was placed on cardiopulmonary bypass, as previously described 39,40. The
biopsy was placed in oxygenated HEPES-based Krebs buffer containing 30 mmol/L 2,3-
butanedione monoximine (BDM) at room temperature 39, 40. Small samples (< 5 mg) were
removed and frozen for collagen and titin studies or placed in formalin for histology. The
remainder of the tissue was processed for stiffness studies. From the section of the biopsy that
remained in buffer, tissue was dissected into pieces < 2 mm in length and placed in skinning
solution containing Triton-X100 at 4 C. For samples obtained at MUSC and the HFRN centers,
the skinning period coincided with overnight transit to UVM at 4 C. After 18-24 hour skinning,
strips were dissected to 150- 200 μm diameter and 800-1200 μm length and then underwent
measurements of myocardial stiffness as described below.
All patients were followed until discharge, with particular attention to ventricular
cardiomyopathy or constrictive pericarditis, HIV or active infection.
Myocardial Biopsy Procedure
AnAnteteteriririooror LLLV VV ffreeee wwwala l epicardial biopsies weighihiingngn ~25-50 mg wweree e oobtbtbtaaiained during CABG
ooonnon after the ppataatieeentnt wwaasas ppplalaacceced d ononon ccaaardddioppulllmonnnaaary bybybypapaassss, asass pprereviviiouououslsly y y dededescscririribebebed dd 39,39,404040. ThThThe
biiopopopsysysy wwasass ppplalaceceed d inin oooxyxyygeg nananateteeddd HEHEHEPPEES-S-S-bababaseeed d d KKrKreebsss bububuffffffererr cccononntataainininininng g 30300 mmmmomomoll/l/LLL 2,2,,3---
butanedione momomononooxixiximimim nenn (((BDBDBDM)M)M) aatt t roroomomom ttememempepeperararatututurerer 3939,39, 404040. SmSmmalala l sasas mpmpmpleleles s s (<(<< 555 mmmg)g)g) wwere
by guest on June 1, 2018http://circ.ahajournals.org/
Dow
nloaded from

DOI: 10.1161/CIRCULATIONAHA.114.013215
8
arrhythmias and bleeding complications. No adverse effects or post-operative complications
ascribable to the biopsy procedure were detected and all patients were discharged alive.
Measurement of Passive Myocardial Stiffness
At time of study, aluminum T-clips were attached to the ends of each strip. The strip was
mounted between a piezoelectric motor (Physik Instrumente, Auburn, MA) and a strain gauge
(Kronex Technologies, Oakland, CA) and initially lowered into a 30 μL droplet of relaxing
solution maintained at 37 C. The composition of relaxing solution is specified in previous
reports 39, 40. Sarcomere length (SL) was measured by Fourier Transform of digital images
(IonOptix Corp, Milton, MA) 9. Measurements of total, collagen-dependent, and titin-dependent
stiffness were made using a previously published differential extraction protocol 9, 10. The
extraction method removes the anchors of titin within the myofilament, leaving only ECM-based
stiffness. This method, originally developed and validated by Granzier and colleagues 9, 10, has
been successfully used in other studies 43. In previous control studies in which titin’s stiffness
was eliminated by protease treatment and the muscle strip was then treated with KCL/KI 12,
ECM stiffness was shown to be unaffected by the KCL/KI treatment. Thus, while the differential
extraction protocol causes irreversible myocardial damage it does not affect the measurements
that are central to the questions addressed in this study.
Echocardiography
Echocardiographic studies performed at UVM and MUSC were interpreted by a core laboratory
at MUSC. Studies were de-identified, coded, and interpreted in a blinded fashion. Measurements
were made using American Society of Echocardiography criteria 44.
Collagen
Collagen was assessed using both biochemical and histologic methods. Soluble, insoluble, and
tiffness were made using a previously published differential extraction protocoll 99, 10101 . ThThThe e
extraction method removes the anchors of titin within the myofilament, leaving only ECM-based
ttififffnfnfneesess. TTThihhiss memeethththodo , originally developed and d d vavav llidated by Graaanznn ieer r r aanand colleagues 9, 10, has
beeenn successfufullllyyy uususededd iin n ototheheher ststudududieiesss 4433. Innn ppprevviioouus cococontntrrroll l ststuududieies ininn wwwhihihichchh ttititininin’ss’s ssttiffffffnenessss
wawasss elelelimimiminininatatateded bbby y prprproototeaeaasese ttrerereatattmemementnt aaandndd ttthhhe mmmusususclcllee ststs riririp p p wawawas s s ththhenenn tttrereeattedede wwwititithh h KCKCKCL/L//KIKIKI 1212,,,
ECM stiffnesesssss wawawas s s shshshowowown n tototo bbbe ee unununafafaffefectctctededed bbby y y thththe e e KCKCKCL/L//KIKIKI tttrerereatatatmemem ntntnt.. ThThThususus,, whwhwhililile e e thththe e e differentiaaal
by guest on June 1, 2018http://circ.ahajournals.org/
Dow
nloaded from

DOI: 10.1161/CIRCULATIONAHA.114.013215
9
total collagen content were determined using tissue samples sequentially extracted and assayed
directly using the microplate picrosirius red assay 45-47. Collagen volume fraction (CVF) was
measured using light microscopy with samples stained with picrosirius red (PSR) to detect
collagen and viewed with polarized light under dark field optics to detect birefringence of the
fibers 45.
Titin Studies
Titin isoform analysis was performed with 1% agarose gels using a vertical SDS-agarose gel
system as previously described 23, 48. LV myocardium co-expresses compliant N2BA titin and
stiffer N2B titin isoforms 9; their expression ratio was determined. We also measured titin
degradation as the ratio of T2 (~ 2 MDa degradation product of titin) to T1 (full length titin).
Titin phosphorylation levels were quantified via Western blotting 9, 25. Blots were stained
with Ponceau S (Sigma) to visualize the total protein transferred and then probed with phospho-
specific rabbit polyclonal antibodies against phosphorylated S11878(S26) and S12022(S170) of
the PEVK element 25. These sites are known to be phosphorylated by PKC. In addition, blots
were probed with phospho-specific rabbit polyclonal antibodies against phosphorylated
S4185(S469) of titin’s N2B element. This site is known to be phosphorylated by PKA and PKG
32. Membranes were labeled with secondary antibodies conjugated with fluorescent dyes with
infrared excitation spectra (CF680, goat anti-rabbit, Biotium Company, Hayward CA). Blots
were scanned using an Odyssey Infrared Imaging System (Li-Cor Biosciences, Lincoln NE) and
images analyzed using Li-Cor software. Ponceau S scans were analyzed in One-D scan to
normalize phosphorylation signal to protein loading.
Plasma Biomarkers
Biomarkers were chosen that reflect changes in ECM homeostasis, specifically matrix
degradation as the ratio of T2 (~ 2 MDa degradation product of titin) to T1 (full l llelengngngthhh tttitititininin).).).
Titin phosphorylation levels were quantified via Western blotting 9, 25. Blots were stained
wiwiththh PPPoononcececeauauau S (((SSiSiggma) to visualize the total prrotooteiin transferred anana d ththhenenen probed with phospho-r
ppecccifi ic rabbit t popoolyyyclcllooonalalal aaantntntibibbododieiei sss aaggaaainsttt pphossphphphorylylylatateedd SS111118778(8(S2SS 666) aaandndnd SS1212120202022(2(2 S1S1S170070) oof
hhe e e PEPEPEVKVKV eeelelelememeentntt 2525. . ThThhesese sisis tetetesss ararare e kknknowowownnn totoo bbeee pphphooosphphphoororylylylatata eeded bbby y y PKPKPKC.C IIIn nn adaddididittitionnn, bblblotottss
were probed d wiwiwiththt ppphohoh spspsphohoo-s-spepp cicicififific c rarar bbbbbbititt pppolololycycycloloonananall l annntititibobobodidid eseses aagagaainininststst ppphohohospspphohohoryryrylalalatetet d
by guest on June 1, 2018http://circ.ahajournals.org/
Dow
nloaded from

DOI: 10.1161/CIRCULATIONAHA.114.013215
10
metalloproteinases [MMPs] and their tissue inhibitors [TIMPs]. Four classes of MMPs,
gelatinases (MMP-2 and MMP-9), collagenase (MMP-1 and 8), stromelysin (MMP-3), and
matrilysin (MMP-7), and all 4 tissue inhibitors of MMPs (TIMP-1, -2, -3, -4) were assayed. In
addition, N-terminal propeptide of brain naturetic peptide (NT-proBNP) was measured. Finally,
biomarkers that reflect a proinflammatory and/or profibrotic state, specifically CRP, IL-6, IL-8,
TNF- , and sST2 were examined.
Statistical analysis
Data are reported as mean ± SD in tables and text and mean ± SE in figures. A t-test was used to
detect differences in continuous variables, Pearson’s Chi-square test was used to detect
differences in categorical variables for demographics, collagen-dependent and titin-dependent
tension, collagen content, and titin phosphorylation and isoforms and plasma biomarkers
amongst referent controls, HTN(-)HFpEF and HTN(+)HFpEF groups. A general linear mixed
model (GLMM) was also employed to compare the relationship between stress and sarcomere
length in the three groups (Figure 1); this type of model is ideal for handling repeated measures
obtained within the same study subjects 49. Within the GLMM, linear and quadratic relationships
between stress and sarcomere length were considered, and an unstructured covariance structure
was selected after comparing the model’s AIC value to those from models incorporating other
covariance structures (e.g. autoregressive, compound symmetry) 50. In addition, stress was
compared between the 3 groups at selected common values of SL using ANOVA. Linear
correlations were used to examine the relationship between echocardiographic measurements and
stiffness measurements and between echocardiographic measurements and plasma biomarkers
using a least squares best fit model and a Pearson’s correlation coefficient.
differences in categorical variables for demographics, collagen-dependent and tititiniin-d-d- epepepennendededentntn t
ension, collagen content, and titin phosphorylation and isoforms and plasma biomarkers ff
ammononongsgsgsttt rererefefeferrrentntt cccoontrols, HTN(-)HFpEF and HTHTHTNNN(+)HFpEF grrououo pss.. AA A general linear mixed
mmoddedel (GLMM)M)) wwwass aaalsssoo emememplplp oyoyededed ttooo ccompppaaare thhhee rellalattitiononnsshhipip bbetetweweenenn sstrtrresesss anannd dd sasarrrcoomomeeereee
eengngngththth iin n ththhe e e ththrereee grgrg oououppsps ((FiFiigugugurerere 111);); ttthihiisss tytytypeee ooff f mmmoddedelll isisis iidededealal ffororor hhaaanddldlininng g g rerer pepepeatatateded mmmeaeasusurrees s
obtained witthihiinnn ththhe e e sasasamemem ssstututudydyd sssububu jejejectttsss 494949. WiWiWithththininin ttheheh GGGLMLMLMM,M,M, lllinii eaeaear r r ananandd d ququq adadadrararatititicc c rerer lationshipsss
by guest on June 1, 2018http://circ.ahajournals.org/
Dow
nloaded from

DOI: 10.1161/CIRCULATIONAHA.114.013215
11
Results
Demographic and Echocardiographic Data
The mean age of the study group was 65 years; most subjects were male (Table 2). This age and
sex distribution is typical of CABG populations. By definition none of the controls had DM or
HTN. The prevalence of DM was comparable in both HTN groups [48% in HTN(-)HFpEF and
59% in HTN(+)HFpEF]. The extent of CAD was similar in the three groups (number of arteries
with a > 70% obstruction 2.5 ± 0.7). Compared with controls, blood pressure was higher in the two
HTN groups but not different from each other and within national guidelines. The mean creatinine
values were not statistically different between the 3 groups. There were significant differences in
the medications taken by each group (Table 2). As expected, both HTN groups were receiving
antihypertensive medications ( -blockers, ACE-I/ARBs and diuretics) more often than the
controls. Patients with HTN(+)HFpEF were taking diuretics and nitrates more often than the other
groups.
The structural and functional data obtained from the echocardiographic studies also
confirm the category definitions for each group (Table 3). By definition, LV volumes and EF were
normal in each group. Both HTN groups had increased LV mass and RWT. The number in each
HTN group with concentric LVH was comparable (39% in HTN(-)HFpEF and 41% in
HTN(+)HFpEF); the number with concentric remodeling was also comparable (32% in HTN(-
)HFpEF and 30% in HTN(+)HFpEF). Thus, the percentage with either concentric LVH or
concentric remodeling (i.e., increased RWT without increased LV mass) was ~70% in both groups.
Measurements that reflect diastolic function and filling pressure (E, E’, PCWP, LVEDP, LA
volume) were increased in HTN(+)HFpEF. With the exception of a small increase in LA volume,
these measurements were normal in HTN(-)HFpEF patients. LA enlargement was also more
he medications taken by each group (Table 2). As expected, both HTN groups wweeere e rer cecec ivivivinining g g
antihypertensive medications ( -blockers, ACE-I/ARBs and diuretics) more often than the
coontntntrororolslsls. PaPaPatititieeentstss wwwiti h HTN(+)HFpEF were takinnng gg dddiuretics and niniitrtt atesess mmmore often than the other
ggrouuupsp .
TThehee sssttrtrucucctutuuraral l anannd d fufunncnctititionononalalal ddaatata aa obobobtttaininineedd d frfrromomm ttthehehe eechchchococcararrdididiogoggraaaphphhicici ssstutuddidieees aaallsooo
confirm the cacaatetetegogogoryryry dddefeffinitititioioionsnsn fffororo eeeacacch hh grgrgrouououp p p (((TaTaTablblblee 333).).). BBBy y y dededefififininitititiononon, , LVLVLV vovoolululumememes s ana d EF weree
by guest on June 1, 2018http://circ.ahajournals.org/
Dow
nloaded from

DOI: 10.1161/CIRCULATIONAHA.114.013215
12
frequent in the HTN(+)HFpEF group (41%) vs. HTN(-)HFpEF (20%).
Myocardial Stiffness
Relationships between myocardial stress and SL between 2.0-2.6 m for the three groups, which
reflect total passive myocardial stiffness, are shown in Figure 1. Results of the general linear
mixed model (GLMM) indicated that a model assuming a quadratic relationship between stress
and SL provided a superior fit compared to a linear model. The GLMM indicated that as SL
increased, the slope increased most rapidly in the HTN(+)HFpEF group (p<0.0001 compared with
both control and HTN(-)HFpEF), and that the control and HTN(-)HFpEF curves were not
significantly different from one another. Significant (p<0.01) differences were noted between the
HTN(+)HFpEF group and the other groups at 2.1 m and at each SL assessed up to and including
2.6 m. Passive myocardial tension was also examined at SL 2.6μm before and after differential
extraction to estimate collagen-dependent and titin-dependent stiffness (Figure 2). There were no
significant differences in collagen- and titin-dependent tension between control and HTN(-
)HFpEF. However, both collagen- and titin-dependent tension was significantly increased in
HTN(+)HFpEF compared with the other groups. Collagen-dependent stiffness was increased by
220% and titin-dependent stiffness was increased by 92% in the HTN(+)HFpEF group.
There were significant correlations between in vitro measurements of passive myocardial
tension and in vivo echocardiographic measurements of diastolic function (Figure 3). Thus, there
was a statistically significant direct relationship between collagen-dependent tension and left atrial
diameter (r2 = 0.42, p = 0.006) and PCWP (r2 = 0.46, p = 0.002) and between titin-dependent
tension and left atrial diameter (r2 = 0.43, p = 0.006) but not PCWP (r2 = 0.16, p = 0.11).
Collagen
Myocardial collagen content was measured using both biochemical and histologic methods.
HTN(+)HFpEF group and the other groups at 2.1 m and at each SL assessed up totoo andndn iincncnclululudididingng
2.6 m. Passive myocardial tension was also examined at SL 2.6μm before and after differential
exxtrtrracacactititiononn ttto oo eeestiimamamatet collagen-dependent and tittininin-dddependent stifffnenn ss (((FiFiFigug re 2). There were no
iiignnnifi icant diffffererenenenceess innn cccoolollalalaggegen-n-- aaandndn tiittin-d-ddeeppenndedeent ttteennsisi nonon bbeeetwweweenen coonontrtrrololol aandndd HHHTNTNTN(-(--
HHHFpFpFpEFEFEF.. HoHoHowwewevveer,r,, bbootth h cocolllagagagenenn--- aaanndnd tttitttininin-d-d-depppenene ddedennt tttennnsisisioonon wwwaaass sisisigngng iiificccanantltlt y y y ininccrcreeeaseeed inin
HTN(+)HFpEpEpEF F F cocoompmpmparara edee wwwititith h thththeee otototheheerr r grgrgrouououpspsps. . CoCoColllllagagenenen-d-ddepepepenenendedd ntntnt ssstititiffffffnenenesssss wwwasasas iiincncncrer ased by
by guest on June 1, 2018http://circ.ahajournals.org/
Dow
nloaded from

DOI: 10.1161/CIRCULATIONAHA.114.013215
13
Biochemical studies assessed soluble, insoluble and total collagen (Figure 4A). CVF was
estimated by light microscopy of PSR stained sections (Figure 4B-F). There were no differences
in soluble collagen across the three groups. There was a significant increase in insoluble and total
collagen (by~100%) and CVF (by ~130%) in the HTN(+)HFpEF group compared with the
controls and HTN(-)HFpEF groups.
Titin
Titin N2B and N2BA isoforms and phosphorylation were examined in the 3 patient groups.
Three phosphorylation sites in titin’s spring region were examined: S11878(S26)and
S12022(S170) of the PEVK element, sites phosphorylated by protein kinase C (PKC) and
calcium/calmodulin dependent protein kinase II (CaMKII), and S4185(S469) of the N2B
element, a site phosphorylated by protein kinase A (PKA) and protein kinase G (PKG) (Figure
5). There were no differences in the N2BA/N2B titin ratio between the three groups (0.53±0.14
in control, 0.50±0.13 in HTN(-)HFpEF and 0.59±0.14 in HTN(+)HFpEF) and no changes in T2
(titin degradation product): T1 (full length titin) ratio amongst the groups. Patients with
HTN(+)HFpEF had a 31% higher phosphorylation value at the S11878(S26) PKC/CaMKII site,
no change at the S12022(S170) PKC CaMKII site and a 28% lower value at the S4185(S469)
PKA/PKG site compared with HTN(-)HFpEF. There were no significant differences between
HTN(-)HFpEF and control patients at any of the phosphorylation sites.
Plasma Biomarkers
Of the 16 biomarkers measured, four were significantly altered in the HTN patients compared
with the controls (Table 4). CRP, sST2, TIMP-1 and NT-proBNP were higher in the
HTN(-)HFpEF patients compared with the controls. Levels of sST2, TIMP-1 and NT-proBNP
increased further in the HTN(+)HFpEF group. There were no differences in MMP-1, 2, 3, 7, 9 or
calcium/calmodulin dependent protein kinase II (CaMKII), and S4185(S469) off ttthehehe NNN2B2B2B
element, a site phosphorylated by protein kinase A (PKA) and protein kinase G (PKG) (Figure
5).). TTTheheherere wwwererre nonoo ddifi ferences in the N2BA/N2B BB ttit ttiin ratio betweeenene thhe ee thththree groups (0.53±0.14
nn cccono trol, 0.5050±±0± ..1333 inin HHHTNTNTN(-(-(-)H)HFpFpFpEFEFF aandd 0..59±±±00..144 iinnn HHTHTN(N(N +++)HHFHFpEpEpEFFF) aaandndnd nno o chchhananngeeess innin TTT2
ttitittininin dddegegegraraadadadatitiononn ppprorodduductctc ):) TTT11 (f(f(fululull l l lelengngngththth titititiin)n)n) rratatioioi aaamomomonngngststst tthhehe gggroror uupupss.s PPPatata ieieientnttsss wwwiththth
HTN(+)HFpEpEpEF F F hahaad d d a a 313131% % % hihihighghhererer ppphohh spspsphohohoryryrylalalatitit ononon vvvala ueueue aaat t t thththee e S1SS 181818787878(S(S(S262626) ) PKPKPKC/C/C/CaCaCaMKII site,
by guest on June 1, 2018http://circ.ahajournals.org/
Dow
nloaded from

DOI: 10.1161/CIRCULATIONAHA.114.013215
14
TIMP-2, 3, 4 or IL-6, 8 or TNF- between groups.
There were significant correlations between plasma biomarkers and echocardiographic
measurements of diastolic function (Figure 6); there was a statistically significant direct
relationship between PCWP and NT-proBNP (r2 = 0.32, p = 0.001) sST2 (r2 = 0.26, p = 0.005)
and TIMP-1 (r2 = 0.36, p < 0.001).
Correlations between NTpro-BNP and titin phosphorylation at S4185(S469),
S11878(S26) and S12022 (S170) and NTpro-BNP and titin-dependent stiffness were tested.
While the sample size was limited, there was no clear relationship between NTpro-BNP and
S4185(S469) (r2=0.009) or S12022 (S170) phosphorylation (r2= 0.04). However, there was a
trend with S11878(S26) (r2 = 0.32, p=0.2). There was also a trend between NTpro-BNP and titin-
dependent stiffness, r2 =0.22, p=0.08.
Finally, we also tested for correlations between sST2 and CVF and collagen-dependent
stiffness. There were trends for both relationships, with sST2 increasing with increased CVF,
r2=0.58, p=0.047 and increased collagen-dependent stiffness, r2=0.40, p=0.011.
Effects of Diabetes Mellitus
In the two HTN groups, the presence of DM did not affect echocardiographic parameters of
hypertrophy or diastolic function, in vitro measures of stiffness, collagen or titin assays or
biomarkers. DM also did not alter the differences in any of these parameters between HTN
without or with HFpEF.
Discussion
The results of this study support several novel conclusions. The current study showed for the first
time that patients with HTN(+)HFpEF have a significant increase in passive myocardial stiffness
rend with S11878(S26) (r2 = 0.32, p=0.2). There was also a trend between NTprprooo-BNBNBNPPP ananand d d titittin
dependent stiffness, r2 =0.22, p=0.08.
FFFinininalalalllyly, wewewe aalso tested for correlations beeetwtwtweeen sST2 andd CCCVFF aaannnd collagen-dependent
ttiffffnfness. Therere wwweeeree ttrt enenendsdsds fffooorr bobooththh reeelaaationnnshhhipss, wwiththh ssSTSTT222 ininccrcreeaeasisiinnng wwititthh h inincrcrreaeaasesed dd CVCVCVF,FF
22=0=0=0.5558,8,8, ppp=0=0=0.0.047477 aaandnd iincnccrereasssededed cccololollalalaggenenen-d-d-depepepenenendededentntt sttiifffffnenenesss, rrr22==0=0.44.40,0,0 pp==0=0.0.0011111..
Effects of DDiaiaabebeb tetetesss MeMeMelll itttususus
by guest on June 1, 2018http://circ.ahajournals.org/
Dow
nloaded from

DOI: 10.1161/CIRCULATIONAHA.114.013215
15
measured directly in left ventricular myocardial strips. Although consistent with previous studies
in animal models of HFpEF 43, 45, 51, 52, this has never previously been documented in patients
with HFpEF. Previous studies in HFpEF patients have examined the passive properties of the
composite LV chamber 4 or the passive properties of isolated cardiomyocytes 19, 20, but not the
passive properties of the intact myocardium. Making direct measurements in intact myocardium
was a pivotal step for several reasons, including defining the relative mechanistic contributions
of changes in titin and collagen to the increased passive myocardial stiffness seen in patients
with HFpEF and the ability to differentiate the effects of co-morbid conditions on myocardial
properties from the effects of HFpEF itself. The techniques used to make direct measurements of
passive myocardial stiffness in left ventricular myocardial strips were developed using samples
from patients with heart failure and a reduced ejection fraction (HFrEF) but these techniques
have not been previously applied to patients with HFpEF.
Previous studies in patients with HFpEF examined the contribution of changes in titin to
the resting tension (passive stiffness) of isolated cardiomyocytes obtained from LV
endomyocardial biopsies; these studies also showed that collagen volume fraction was increased
19, 20. However, no previous study in patients with HFpEF has shown the relative contributions of
changes in both collagen and titin to passive myocardial stiffness. The current study showed for
the first time that the increase in myocardial stiffness in HFpEF patients was caused by changes
in both ECM fibrillar collagen and cardiomyocyte titin. Specifically, HFpEF patients had
increased collagen-dependent stiffness in association with increased fibrillar collagen content.
HFpEF patients also had increased titin-dependent stiffness in association with significant
changes in the phosphorylation state of titin, with decreased phosphorylation of a PKA/PKG site
in the N2B element and increased phosphorylation of one of the PKC sites in the PEVK element.
passive myocardial stiffness in left ventricular myocardial strips were developedd uusisiinggg ssamamamplplplese
from patients with heart failure and a reduced ejection fraction (HFrEFd ) but these techniques
haaveveve nnnototot bbbeeeee nnn prrrevevevioiously applied to patients witthh h HHHFpEF.
Previououss stttudddieiei s ininin ppatatatiieientnttss s wiwiw thth HFpFppEEEF eexaamiinineded ttthhee ccoonontrtribibbuuutiioon n ofofof cchahaangngngeses in n n tititititin n to
hhe e e rererestststiningg g tetetensnsioioon n (p(paaasssiiveve sstitit ffffffnenenessssss)) oofof iiisososollalateteed d d ccacarrdrdioioomymymyooocytytytesess ooobtbtbtaiainnened d frfrfromomom LLLVVVf
endomyocarrdidiialalal bbbioioiopspspsieieies;s ttthehehesese ssstutut dididiesss aaalslslsooo shshshowowowedede tthahaatt t cococolllllagagagenenen vvvolololumumumee e frfrfracacctititiononon wwwasaa increaseddd
by guest on June 1, 2018http://circ.ahajournals.org/
Dow
nloaded from

DOI: 10.1161/CIRCULATIONAHA.114.013215
16
While all of the HFpEF patients examined in the current study had antecedent HTN and ~
50% had DM, the presence of these co-morbid antecedent disease processes alone, in the absence
of heart failure, did not alter total myocardial stiffness or its titin and collagen-dependent
components. This is the first clinical study in patients with HFpEF to examine and differentiate
the effects of a major antecedent disease substrate, such as hypertension, on myocardial structure
and function before the clinical syndrome of HFpEF has actually developed. There has been
significant controversy regarding the ability to differentiate or distinguish patients with
hypertension from those with HFpEF. It has been proposed that HFpEF is simply the aggregate
of a number of co-morbid factors such as hypertension, diabetes, CAD, obesity, etc. The current
study clearly demonstrates that there are specific structural and functional differences between
patients with hypertension without and with HFpEF. Similar conclusions are likely to be
applicable to other co-morbid or antecedent disease processes.
The current study also shows for the first time that the development of collagen and titin
based changes in diastolic function in patients with HFpEF occur in association with
proinflammatory and profibrotic stimuli as measured by plasma biomarkers. These data provide
additional support for the overall schematic hypotheses proposed by Paulus and Tschope (i.e.,
that proinflammatory and profibrotic signaling play a significant mechanistic role in the
development of HFpEF) and constitute new mechanistic insights into the pathophysiology
underlying HFpEF 13.
Hypertensive Heart Disease
HTN results in LV pressure-overload (PO) and causes a spectrum of structural, functional and
clinical outcomes that have collectively been called hypertensive heart disease (HHD). Data
from animal models of PO suggest that the myocardium responds to increased load by
tudy clearly demonstrates that there are specific structural and functional differereencnccesee bbbetetetweweweenen
patients with hypertension without and with HFpEF. Similar conclusions are likely to be
appplplplicicicababablelele tttoo otheheherrr cco-morbid or antecedent diseeeasasa eee processes. r
The cururrereenntt sstutuudydyy aaalslslso o shshowowowss ffoorr thee fifiirst tttimmme ttthhahatt ththt ee e dedeevveelolopmpmmenent t ofofof ccololllalalagegeg nnn ananandd tit tttin
baasesesed dd chchc ananngegeges s ininn dddiaiaststtolllicic fununnctctctioioion n n iinin pppatatatieieiennnts ss wiwiw tthth HHFpFpFpEFEFEF ooccccccuurur iiinn n asasssosociciatatatioioon n n wwiwitththff
proinflammatatororory y y ananand dd prprprofofibibibrororotiiccc ststs imimmulululi ii asasas mmmeaeaeasusuurrrededd by y y plplplasasasmamama bbbiooomamamarkrkrkererers.s.s TTThehehesesese dddata a provide
by guest on June 1, 2018http://circ.ahajournals.org/
Dow
nloaded from

DOI: 10.1161/CIRCULATIONAHA.114.013215
17
undergoing cardiomyocyte hypertrophy which results in an increase in myofibrillar content 9, 10,
45, 51. In addition to the myofibrils, other structures and processes within both the cardiomyocyte
and the ECM are altered in response to increased load. In animal models these events have a
temporal sequence in which the additional changes in the cardiomyocyte and ECM occur only
after myofibrillar hypertrophy is well under way or even complete 45, 51. For example, in murine
and feline models PO during the first 1-2 weeks of PO, there are no significant changes in CVF
or diastolic filling pressures and no evidence of heart failure; however, after 4-8 weeks CVF and
filling pressures are increased and evidence of heart failure develops. Data from the current study
appear to parallel these temporal changes. Patients with HHD comprise a spectrum: HTN
without LVH, HTN with LVH/concentric remodeling but no heart failure [HTN(-)HFpEF], and
HTN with heart failure [HTN(+)HFpEF]. In the current study HHD patients were not followed in
a serial manner; data were obtained by cross-sectional analysis. However, it was only in the
patients with HTN(+)HFpEF that changes in collagen and titin were detected. Although in the
absence of sequential data in the same patients we cannot prove the concept that sequential
temporal changes occur in the cellular and molecular mechanisms that underlie HHD, our data
clearly support the idea that changes in collagen and titin constitute mechanisms are associated
with the transition from HTN to HFpEF.
Overall, there is good concordance between the current study and previous clinical
studies in pressure-overload and HFpEF 16, 19, 20, 26, 28. Van Heerebeck et al 19 and Borbely et al 20
showed that patients with pressure-overload induced HFpEF had increased cardiomyocyte
stiffness; however, because these mechanical studies were performed in isolated cardiomyocytes
with the ECM removed, the relative contribution of these changes in collagen and titin to
myocardial stiffness could not be determined.
without LVH, HTN with LVH/concentric remodeling but no heart failure [HTN(N(-)--)HFHFHFpEpEpEF]F]F],,, anana d
HTN with heart failure [HTN(+)HFpEF]. In the current study HHD patients were not followed in
a seseeriririalalal mmananannneer; dddaatataa were obtained by cross-sectctctioionnal analysis. HoHoH weeveveverrr, it was only in the
patiiiene ts with HTHTTNNN(+)+))HFHFFpEpEpEFFF thththatat cchhahanngngees innn cccollaaaggen anannd d ttit ttiin n wewwerere dddeete eececteteed.d.d. AAltltthohohouguggh ininin ttthehehe
abbbsesesencncncee ofoff ssseeqequeueentnttiaial l dadaatata inn ththheee sasasammeme ppatatatieieiennntstss wweee ccacannnnnototot ppproroovevev tthehehe cccoononcceceptptt tthahahat t seseseqququeeenttitialal
emporal chaangngngesese oooccccc ururur in n thththeee cececelllll ulululara aaandndnd mmmololo ecececulululararar mmmececechahahanininismsmsms thththatatat uuundndnderere liiie e e HHHHHHD,D,D our data
by guest on June 1, 2018http://circ.ahajournals.org/
Dow
nloaded from

DOI: 10.1161/CIRCULATIONAHA.114.013215
18
Our results indicate that at SL 2.6 m increases in ECM collagen account for more than
2/3 of the increase in resting tension in HFpEF. However, the relative contributions of titin and
collagen to resting tension are SL-dependent; collagen accounts for a larger proportion at longer
SLs and titin for a larger proportion at short SLs. Their relative contributions are therefore
ultimately determined by the actual operating SL range in our patients, which is unknown. Based
on studies performed in pigs 53, we do know that in large mammals the upper end of the
operating range extends to 2.5-2.6 m. To the extent that the actual operating range is lower, the
increase in total resting tension in HFpEF will be more evenly divided between the two.
Data from the current study significantly advance our understanding of how these
mechanisms contribute to changes in myocardial stiffness by examining intact myocardial
samples that allowed integrated physiologic studies to selectively examine the individual
contributions of collagen and titin. The current study showed hypophosphorylation of PKG/PKA
sites on titin in HFpEF patients, consistent with the reduction in passive tension detected when
cardiomyocytes from HFpEF patients are treated with PKA/PKG 19, 20 and consistent with
phosphorylation studies in animal models 43, 54. In contrast to Van Heerebeck et al 19 and Borbely
et al 20, we did not detect changes in the N2BA/N2B ratio in HFpEF patients. In some but not all
of the studies from these investigators, the N2BA/N2B ratio was increased in HFpEF, which
would be expected to result in a potentially compensatory decrease in titin stiffness 16-20, 26, 28. It
is worth noting that the HFpEF patients studied by these investigators had overtly
decompensated heart failure and were therefore very likely at a more advanced stage of the
syndrome. Perhaps more advanced disease is required to elicit such a compensatory change. The
current study also showed for the first time that patients with HFpEF have hyperphosphorylation
of one of the PKC/CaMKII sites on titin. Previous animal studies have demonstrated that
mechanisms contribute to changes in myocardial stiffness by examining intact mmyoyoyocacc rdrdrdiaiaal ll
amples that allowed integrated physiologic studies to selectively examine the individual
coontntntririribububutititiononons off cccoolollal gen and titin. The current stststudududy showed hypppopoo hoospspsphhorylation of PKG/PKA
iiiteess on titin inn HFHFHFpEEFFF ppapatititienenntststs,, cocoonnsnsisssteeent wwwitth thhehe reddduucuctitiononon iin n paassssivivive tetennsnsioioon n dededetetetectcttededd wwhheh nnn
caardrdrdioioiomymymyococcytytyteses ffrooom m HFHFFpEp FF F papaatititieeentntn s s ararare ee trtrtreaeae teteted d wiwiththh PPKAKAKA/PPPKGKGKG 19,19, 2020 anndnd ccconononsisiststtenenent wwwitthth
phosphorylatatioioion n n stststudududieiees s s inn aaaninin mamamall l momom dededelslss 434343, 545454. InInn ccconono trrrasasa tt t totoo VVVanana HHHeeeeeerererebebebeckckc eeet t t alalal 191919 aand Borbelyyy
by guest on June 1, 2018http://circ.ahajournals.org/
Dow
nloaded from

DOI: 10.1161/CIRCULATIONAHA.114.013215
19
hyperphosphorylation at these sites results in increased titin stiffness 23; however, the current
study is the first clinical study to show that this contributes to increased myocardial stiffness in
patients with HFpEF. In addition, while not yet studied in HFpEF, studies in HFrEF have
suggested that CaMKII-dependent phosphorylation of S12022 (S170) may reduce passive
stiffness 54.
The finding that both changes in collagen and titin may play a pivotal role in the
development of HFpEF in HHD suggests the possibility that there may be common upstream
mechanisms resulting in both changes 13. The presence of a HTN induced proinflammatory,
profibrotic, and/or redox stress state is supported by the plasma biomarker profiles found in the
current study. The increase in CRP, sST2 and TIMP-1 in the HFpEF patients supports this
hypothesis. Clearly, more investigation is needed in this area.
Diabetes Mellitus
Diabetes mellitus has been shown in both animal studies and clinical disease to cause abnormal
diastolic function that may contribute to the development of HFpEF 14, 16, 18. DM is reported to
cause changes in collagen homeostasis (by altering cross-linking) and titin phosphorylation.
Approximately 50% of the patients in the current study had DM as well as HTN. The presence of
DM in addition to HTN did not appear to alter myocardial stiffness, collagen or titin to an extent
greater than that of HTN alone. Because of their rarity in the CABG population, we were unable
to identify sufficient numbers of patients with DM without HTN for analysis. This lack of effect
differs from previous studies of pressure-overload (caused by aortic stenosis) in which the
presence of DM caused a significant increase in cardiomyocyte stiffness over and above that
caused by pressure-overload alone. This may be explained by patient selection differences. Thus,
previous studies did not specifically identify whether patients with pressure-overload or DM had
current study. The increase in CRP, sST2 and TIMP-1 in the HFpEF patients suppppopoorrtss thththisisis
hypothesis. Clearly, more investigation is needed in this area.
DiDiabababeteteteses MMMelele litutuusss
DDiaababetes mellilitutuss hhahas s bebeeenenn sshohohownwnn iinn n bbottth annnimmmal sttuudiieiesss ananndd clcliinniiccalal dddisseeasassee ttoto ccaauauseses aaabnbnnorormammal
didiasasastotoolililicc fufuuncncnctitiononn ttthahatt mmamayy cocoontntntririribububutete tttoo thththe ee dedeevevev llolopmpmpmenennttt ofoof HHHFpFppEFEFEF 1414,, 1666, 1, 1888. DMDMDM iiis s rreppporrtrtededd too o
cause changegesss ininn cccololollalaagegeen hohohomememeososostataasiss s s s (b(b(by yy alalaltetet ririr ngngng cccror ssssss-l-llinininkikikingngng)) ananand d d titititititin n n phphp osososphphphorororylyy ation.
by guest on June 1, 2018http://circ.ahajournals.org/
Dow
nloaded from

DOI: 10.1161/CIRCULATIONAHA.114.013215
20
HFpEF or selectively examine patients with LVH without HFpEF 14, 16, 18. Additionally, the
current study may not have been powered sufficiently to examine the separate effects of DM,
there may be differences between pressure-overload caused by HTN versus aortic stenosis, and
the duration, severity and management of DM may differ between studies. The current study
does not necessarily bring into question the importance of DM in HFpEF, but it does raise the
question of whether HTN and DM together have a combinatorial effect on passive stiffness. In
addition, we did not examine the effects of DM on other determinants of diastolic function that
could affect passive stiffness such as calcium homeostasis and contractile proteins, key
determinants of relaxation rate and extent.
Effects of changes in collagen and titin homeostasis on diastolic function
Changes in collagen content, geometry, and composition are associated with abnormal diastolic
function in HHD and other comorbidities common in patients with HFpEF 55. Collagen content
is the product of the balance between collagen synthesis, post-synthetic processing, post-
translational modification and degradation. The current study was not designed to examine the
determinants of collagen homeostasis or mechanisms that alter it. However, the changes in
plasma biomarkers detected in the HFpEF patients may provide some insights 56. Increases in
sST-2 and TIMP-1 suggest that a profibrotic stimulus was present that would be expected to
increase collagen synthesis and decrease collagen degradation. TIMP-1 has been shown to
inhibit MMPs, the major degradation enzymes present in the ECM. ST2 is a member of the
interleukin 1 receptor family. Its functional ligand is interleukin 33 (IL-33), a cardiac fibroblast
protein. Binding of IL-33 to membrane ST2 elicits an antihypertrophic and antifibrotic response.
This cardioprotective effect is negated by soluble ST2 which prevents binding of IL-33 to
membrane bound ST2. Thus, increased TIMP-1 and sST-2 provide two potential mechanisms for
Effects of changes in collagen and titin homeostasis on diastolic function
Changes in collagen content, geometry, and composition are associated with abnormal diastolic
fuuncncctititioonon iiinnn HHHHH DDD aanand other comorbidities commmmonono iin patients witith hh HFFFpEpEpEF 55. Collagen content
ss thhehe product oof f f thhhe babab lalaancncnce e bebebetwtweeeeeenn cocollaggennn synntnthhessisiss,s, ppoosost-t-sysyynnththeteticici ppprorocececessssininng,g,g ppoost-t--
rranannslsllatatatioionanaal l l momodddiffificacattioonon andndnd dddeeegrgrgradaadaatatiooonn.n. TThehehe ccurururrrenntnt ssstututudydydy wwwaaas s nononot t dededesisigngnnededed ttooo eexexamammiinine e tttheee
determinantss ooof f f cocoollllllagagagenenn hhhomomomeooostststasassisisi ooor rr mememechchhanana isisismsmsms thahahat t alalalteteterr r ititit.. HoHoHowewewevevever,r,r, tthehehe ccchahahangngnges in
by guest on June 1, 2018http://circ.ahajournals.org/
Dow
nloaded from

DOI: 10.1161/CIRCULATIONAHA.114.013215
21
the profibrotic state in HHD.
Titin’s I-band segment serves as a molecular spring that develops passive force when
extended57. Alternative splicing results in either the shorter and stiffer N2B or the longer and
more compliant N2BA isoforms, which are co-expressed within the sarcomere. When the
N2BA/N2B isoform ratio increases, cardiomyocyte and myocardial stiffness decrease as reported
in heart failure with reduced EF 58, 59. In contrast, Borbely et al reported an increase in the
N2BA/N2B isoform ratio in HFpEF patients but cardiomyocyte stiffness was increased 28,
implying that changes in phosphorylation outweigh the isoform shift. The N2BA/N2B ratio did
not change in the current study. As discussed above, it is possible that the isoform switch occurs
at a later stage of HFpEF. In addition, the N2BA:N2B ratio did not change in at least one study
of human HCM and DCM 60.
The phosphorylation state of 2 elements within titin’s spring segment also modulates
cardiomyocyte stiffness. Sites within the N2B element (e.g., S4185[S469]) are phosphorylated
by PKA and PKG, which decreases passive force 25, 32. Sites within the PEVK element
(S11878[S26] and S12022 [S170]) are phosphorylated by PKC (other PKC isoforms have not
yet been studied), which increases passive force. Additionally our data and the data of other
investigators suggest that these sites on titin are also a target of CaMKII 54, 61, 62. In both the
current and previous studies in HFpEF patients 28, hypophosphorylation of the N2B element has
been observed. Moreover, treatment of skinned cardiomyocytes from HFpEF patients with PKG
decreased cardiomyocyte stiffness 28, indicating that hypophosphorylation of PKA/PKG sites
contributes to elevated passive force. Importantly, PKG treatment did not lower stiffness to the
level present in control cardiomyocytes. It has been speculated 22 that this residual elevation in
tension is due to hyperphosphorylation of PKC sites. In the current study we demonstrate that
at a later stage of HFpEF. In addition, the N2BA:N2B ratio did not change in at t leleleasasstt onono e ee stststuduudy y
of human HCM and DCM 60.
TTThehehe pphhospspsphhohorylation state of 2 elements wwwittthin titin’s sprrininng seseegmgmgment also modulates
caardddioi myocyttee stsstiffffnneesss... SSSitititeseses wwititthihihinn n tthheee N222BBB elememmentt t ((e(e.g.gg.., SS414118855[S[SS4644 99]9])) arararee phphhosossphphphororrylyylatatateddd
byby PPPKAKAKA aandndnd PPPKGKGG,, whwhhicch h dedecrcrc eaeaaseseses ss papaasssssiviviveee fooorcrcr ee 255,5, 3222.. SiSiSitetetes s wiwiw ththhininn ttthehee PPPEVEVVK K K elelemememennttt
S11878[S26]6]6] aaandndnd SSS121220200 22 [S[S[S171770]0]0]) ) arara e e phphphosososphphphororo ylylylatatatedede bbby y y PKPKPKCCC (otototheheher r r PKPKPKCCC isisisofofofororormsmsm have not
by guest on June 1, 2018http://circ.ahajournals.org/
Dow
nloaded from

DOI: 10.1161/CIRCULATIONAHA.114.013215
22
hyperphosphorylation of the S11878(S26) PKC/CaMKII site in the PEVK segment is associated
with increased myocardial stiffness in HFpEF patients. This constitutes a novel mechanism of
increased stiffness in HFpEF.
The increased PKC activity that is suggested by the increased S26 phosphorylation did
not result in phosphorylation of S12022 (S170). Previous studies indicate that PKC is
preferably active at sites with N-terminal and C-terminal basic residues 63. These studies also
indicate that PKC preferentially phosphorylates serines with basic residues within three amino
acids of both the C-and N-termini. Based on these results, PKC should have a stronger affinity
for S11878(S26) than S170 because of the closer proximity of the neighboring basic amino acids
(lysine [K] and arginine [R]). Finally, increased PKC activity has been shown to directly
increase protein phosphatase inhibitor-1 (PP1) activity 64, an effect which should facilitate
hypophosphorylation of PKA/PKG sites in the N2B element (its effect on PKC sites might be
negated by the increased PKC activity). Thus, increased PKC levels might increase stiffness
directly through phosphorylation of PEVK S11878(S26) and indirectly through PP1 activation
causing hypophosphorylation of PKA/PKG sites.
Limitations
The demographic characteristics of the current study population were typical of a CABG
population. However, our HFpEF patients were slightly younger and more often male than
typical patients in epidemiologic studies or randomized clinical trials. From the viewpoint of LV
structure and function, however, they were typical of previous HFpEF studies.
What constitutes an appropriate control group is a challenging issue in all research using
human myocardium. The degree of angiographic CAD was similar in all three study groups;
therefore, CAD was not likely to be a confounding variable across groups. Alternative choices
lysine [K] and arginine [R]). Finally, increased PKC activity has been shown ttooo didid reectctctlylyly
ncrease protein phosphatase inhibitor-1 (PP1) activity 64, an effect which should facilitate
hyhypopopophphphososphphphoooryllatatatiioion of PKA/PKG sites in the NN2N2BBB element (its eeefff ecct tt oonon PKC sites might be
nnegagated by thee iiinncncrereasasseddd PPPKCKCKC aacctititivivivityty)). Thuuus,,, inccreeeasededed PPKCKCKC llevvvelels mimim ghght t t ininncrcreaeaeasesee sstitt ffffffnenesssss
didirereectcttlylyly tthrhrrouououghgh phhohospspphohooryrylaaatitt ononon ofofof PPEVEVEVK KK SS1S118181 77878(S(S( 22626))) ananand d ininindddirereectctc lylyy tthrhrh ouououghghgh PPPP1P1P1 aaccctiivvatatiiionnn
causing hypopophphphosossphphphororrylyly attioioion n n ofoff PPPKAKAKA/P/PPKGKGKG sssititi esese ..
by guest on June 1, 2018http://circ.ahajournals.org/
Dow
nloaded from

DOI: 10.1161/CIRCULATIONAHA.114.013215
23
for controls are limited and less than ideal. Unused explanted donor hearts are usually from
young subjects who have been subjected to high levels of stress for varying durations. Open
heart procedures in patients without CAD or LV remodeling (e.g., ASD closure) are now rare
and usually performed in younger patients. Samples from endomyocardial biopsies cannot be
used for mechanical tissue studies because of tissue trauma. On the other hand, CAD, much of
which is likely sub-clinical, is very common in HHD and HFpEF patients. Indeed, a recent report
indicates that a majority of HFpEF patients have epicardial coronary stenosis 38. Thus, the
presence of CAD can be considered a “real world” background in HTN patients and controls.
The limitation imposed by the presence of CAD, however, is that we cannot exclude possible
CAD effects on myocardial stiffness and/or distinguish them from the effects of hypertension or
diabetes.
The current study focused specifically on the contribution of collagen and titin to the
development of HFpEF. As in heart failure with a reduced ejection fraction (HFrEF), in HFpEF
other mechanisms also contribute to the development of heart failure. Thus, changes in calcium
homeostasis, energetics, and actin-myosin cross-bridge dynamics, which govern the speed and
completeness of relaxation, all may play a role in HFpEF in addition to changes in passive
stiffness.
Titin is a large and complex molecule with many phosphorylation sites outside the
mechanically relevant spring region; however, assessing all of them may not be required in the
context of the current study. The current study focused on three sites in the N2B and PEVK
elements that have been documented in several studies for which we have made and
characterized phospho-site specific antibodies. Two additional PKA/PKG sites in the N2B
element were published recently by Kotter et al (S4010 and S4099).In their recent study Kotter et
CAD effects on myocardial stiffness and/or distinguish them from the effects off hhhypypperrrteteensnsnsioioion n or
diabetes.
TTThehehe cuuurrereentntnt study focused specifically onnn tthhe contributionn oof cocoollllllaagen and titin to the
ddedevvevelopment ofoff HHHFFpFpEFEE .. AsAsAs iiin nn hheaeaartrtt ffaaailuure wwwiiith aaa rreeduuucceed d ejejecectitiionnn ffraacctiioion n (H(H(HFrFrEFEFEF),),), iiinn HFHFHFpEpEpEF
ottheheher r memem chchhanananisismmms aalslsooo ccocontn ririribubuutetete tttooo ththhe e dededevvveloloopmpmpmeennt ofoff hhheeaeartrtt ffaaiailuuurerere. ThThThusus, chchhanangegegess innn cccalalciciiummm
homeostasis,s,, eeenenen rgrgrgetete iciccs,s,, andndnd aaactcttininin-m-mmyoyoosisis nn n crcrcrososo s-s-s brbrbrididdgegeg dddynynynamamamicicics,s wwwhihihichchch gggovovo ererern n n thththe e e sps eed and
by guest on June 1, 2018http://circ.ahajournals.org/
Dow
nloaded from

DOI: 10.1161/CIRCULATIONAHA.114.013215
24
al showed that all three sites on the N2B element are hyophosphorylated in heart failure,
indicating that they change in concert 60. Thus, assessing the established Ser4185/485 site is
likely to reveal the general phosphorylation status of the N2B element.
It is important to acknowledge other limitations of the current study. We did not
systematically study all titin phosphorylation sites. The protein has at least 70 phosphorylation
sites, as shown by in-vivo phosphoproteomics using a SILAC approach 54. Moreover, recent
work has suggested that the N2B domain contains several more conserved serines which are
differentially phosphorylated in human HFrEF and in animal models of HFpEF 32, 43, 52.
Phosphospecific antibodies against the respective N2Bus sites were used in human HCM and
DCM hearts 60, 54. These studies underscore the complexity of titin phosphorylation schemes and
the need for further research in patients with HFpEF as well as other forms of heart failure that
will allow a more complete understanding of the contribution of titin to passive stiffness.
The current study also did not focus on upstream regulatory mechanisms effecting titin
phosphorylation such as changes in the expression, abundance and activity of relevant protein
kinases (e.g., PKA, PKG, CaMKII, PKC) and phosphatases (PP1, PP2a). Changes in kinase and
phosphatase activity have been shown to be important in other forms of heart failure 26, 43;
changes in HFpEF may represent important targets for novel therapies and important areas for
future research.
Several other important mechanisms influencing the ECM were not examined in the
current study, e.g., collagen isoform expression and content, troponin I and T, and galectin-3.
Unfortunately, the size of the LV biopsy limited the total number of experimental measurements
obtainable from each sample. Each of these additional mechanisms represents important
directions for future research.
DCM hearts 60, 54. These studies underscore the complexity of titin phosphorylattioioon n scchehehemememesss anand
he need for further research in patients with HFpEF as well as other forms of heart failure that
wiwilllll aallllllowoww aaa mmmorre ee ccocomplete understanding of theee ccooontribution of ttititi in ttooo pppassive stiffness. f
The cururrereenntt sstutuudydyy aaalslslso o didid d nononot t fofoccus onnn upppsttrreaaammm rereguguulalattoorryy mmeeechhahanininissmsmss efefeffefefectcttinnngg g tiititit nnn
phphhososo phphphororylylylatatatioionn suuuchch aass s chchannngegegesss ininin tthehee eeexpxpxprrer ssssssioionn,n, aabbubundndndaanancecece aandndnd acaca titiiviiityty ooof f rerer leleevavavantt prroroteteeinin
kinases (e.gg.,.,, PPPKAKAKA, , PKPKPKG,GG CCCaMaMaMKIKIKII,I, PPPKCKCKC) ) ) ananand d d phphphosososphphphatttasasaseseses (((PPPPPP1,11 PPPP2P2P2a)a)a .. ChChChananngegeges s s ininin kinase and
by guest on June 1, 2018http://circ.ahajournals.org/
Dow
nloaded from

DOI: 10.1161/CIRCULATIONAHA.114.013215
25
Finally, the effects of medications taken by the patients prior to surgery on titin
phosphorylation and ECM homeostasis could not be assessed.
Clinical Implications
HTN is commonly a progressive process that leads to adverse cardiovascular remodeling,
abnormal diastolic function, and the development of heart failure, particularly HFpEF 65-67. Once
HFpEF has developed, subsequent morbidity and mortality rates are very high 67-76. Treatment of
HHD is thus a critical unmet challenge. One clear answer is prevention of HHD by early
treatment of high blood pressure, but prevention alone is insufficient. LV hypertrophy,
concentric remodeling, and diastolic dysfunction commonly develop without concomitant
symptoms, often before HTN has been detected. Furthermore, once LV remodeling and/or
HFpEF develop, even guideline prescribed blood pressure control does not reduce morbidity and
mortality. Randomized clinical trials in HFpEF of beta-blockers, angiotension converting
enzyme inhibitors, angiotension II receptor blockers, aldosterone blockers and PDE-5 inhibitors,
in which >90% of the patients had hypertension, have failed to show improvement in morbidity
or mortality 70-76. Therefore, there is a need for novel treatments beyond blood pressure control.
The development of novel therapies must overcome several critical barriers. Most
importantly, the cellular, extracellular, and molecular mechanisms that cause the progression
from HHD to HFpEF must be defined in man. Although animal models are useful, they do not
capture all of the key elements of complex and chronic human disease processes. Once proven
operative in patients, each mechanism can serve as a target for successful therapy. The current
study identifies two of these mechanisms; changes in collagen content and titin phosphorylation,
each of which is a potential target. In addition, plasma biomarkers that reflect these changes in
collagen, titin and the profibrotic milieu could be used to improve diagnostic criteria for HFpEF
ymptoms, often before HTN has been detected. Furthermore, once LV remodellininnggg annndd/d/ororor
HFpEF develop, even guideline prescribed blood pressure control does not reduce morbidity and
momortrtrtalalalititity.y. RRRaanndooommimizzed clinical trials in HFpEF oofof bbbeta-blockers, aaangioiootetetennsion converting
ennzyyzyme inhibititororrs,, aangnggioooteetensnsnsioioion n IIIII rrrececepeptor blbloockkeerrss, aalalddodoststterrrononee bblblocockekek rsrs aaandndnd PPDEDEDE-5-5-5 iinhnhhibibibitii ooors,
nn wwwhihihichchc >>909090% % oofof tthehe paatatieientntts s hahahaddd hyhyh ppepertrtrtenenenssiononon, , hahahaveve ffaiaiaileleledd tototo ssshohoow w w imimmpprprovovvememmenennt t iiin mmmoororbibididitytyty
or mortalityy 707070-767676. ThThThererrefefforre,e,e, tttheheererere iis s s a a nenen ededed fffororor nnovovoveeell l trtreaeaeatmtmtmenenentststs bbeyeyyononond d d blblblooooo d d d prprpresesessususurer control.
by guest on June 1, 2018http://circ.ahajournals.org/
Dow
nloaded from

DOI: 10.1161/CIRCULATIONAHA.114.013215
26
and prognostic assessment. Thus, changes in biomarkers such as TIMP-1 and sST-2 may detect
the earliest transition from HTN to HFpEF. Finally, changes in these biomarkers could possibly
be used to monitor treatment efficacy before changes in myocardial structure and function or
clinical status are evident.
Conclusions
HTN in the absence of HFpEF was not associated with increased passive myocardial stiffness.
However, we show for the first time that patients with hypertension and HFpEF have markedly
increased passive myocardial stiffness due to increases in the contribution of both collagen and
titin. These results suggest that the development of HFpEF is linked to a major increase in
passive stiffness caused by changes in both collagen and titin homeostasis.
Funding Sources: This work was supported by National Institutes of Health grants
R56HL123478 (Zile), RO1HL089944 (LeWinter), U10 HL110342 (LeWinter), R01HL06288
(Granzier), and UL1TR000062 (Nietert), Merit Awards from the Veterans’ Affairs Health
Administration and the NHLBI Heart Failure Research Network. Dr. Michael Zile is supported
by the Research Service of the Department of Veterans Affairs (5101CX000415-02 and
5101BX000487-04).
Conflict of Interest Disclosures: None
References: 1. Shah AM, Shah SJ, Anand IS, Sweitzer NK, O'Meara E, Heitner JF, Sopko G, Li G, Assmann SF, McKinlay SM, Pitt B, Pfeffer MA, Solomon SD, and TOPCAT Investigators. Cardiac structure and function in heart failure with preserved ejection fraction: baseline findings from the echocardiographic study of the treatment of preserved cardiac function heart failure with an aldosterone antagonist trial. Circ Heart Fail. 2014;7:104-115. 2. Zile MR, Gottdiener JS, Hetzel SJ, McMurray JJ, Komajda M, McKelvie R, Baicu CF, Massie BM, Carson PE. Prevalence and significance of alterations in cardiac structure and function in
itin. These results suggest that the development of HFpEF is linked to a major ininncrreaee sesese iiinn n
passive stiffness caused by changes in both collagen and titin homeostasis.
FFunndnding Sourrcecees:: ThThThisis wwoororkk k wwawasss sususuppppooorteddd bbby NNNaattionananall InInnststtitituuttess oof ff HeHeHealalththth ggrarannntss
R5R556H6HHL1L 2347474788 ((ZZiiile)e),, RRORO1H1HHL0L0L0898994949444 (LLLeWeWWinnnteter)r)), U1U110 0 LHLHL1103033422 (((LeLeeWWiWinntntererr),), RR010101 LHLL006062828888
Graranznziier)r), anndd ULL1T1TR0R00000060622 (N(Niei tertt),), MMereritit AAwarardsds ffrorom m tht e e VeVeteterar nsns’ AfAffafairi s HeHeallthth
Administratitiiononon aaandndnd ttthehehe NNNHLHLHLBBIBI HHHeearara t FaFaFailililurururee e ReReResesearararchchc NNNetetwowoworkrkrk. DrDrDr. MiMiMichchchaeael l ZiZiZilelele iiiss ssupppported
by guest on June 1, 2018http://circ.ahajournals.org/
Dow
nloaded from

DOI: 10.1161/CIRCULATIONAHA.114.013215
27
patients with heart failure and a preserved ejection fraction. Circulation. 2011;124:2491-2501. 3. Zile MR, Bennett TD, St. John Sutton M, Cho YK, Adamson PB, Aaron MF, Aranda JM Jr, Abraham WT, Smart FW, Warner-Stevenson L, Kueffer FJ, Bourge RC. Transition from chronic compensated to acute decompensated heart failure: pathophysiologic insights obtained from continuous monitoring of intracardiac pressures. Circulation. 2008;118:1433-1441. 4. Zile MR, Baicu CF, Gaasch WH: Diastolic heart failure-abnormalities in active relaxation and passive stiffness of the left ventricle. N Engl J Med. 2004;350:1953-1959. 5. Zile MR, Gaasch WH, Carroll JD, Feldman MD, Aurigemma GP, Schaer GL, Ghali JK, Liebson PR: Heart failure with a normal ejection fraction. Is measurement of diastolic function necessary to make the diagnosis of diastolic heart failure? Circulation. 2001;104:779-782. 6. Kasner M1, Westermann D, Lopez B, Gaub R, Escher F, Kühl U, Schultheiss HP, Tschöpe C. Diastolic tissue Doppler indexes correlate with the degree of collagen expression and cross-linking in heart failure and normal ejection fraction. J Am Coll Cardiol. 2011;57:977-985. 7. Aoki T1, Fukumoto Y, Sugimura K, Oikawa M, Satoh K, Nakano M, Nakayama M, Shimokawa H. Prognostic impact of myocardial interstitial fibrosis in non-ischemic heart failure. -Comparison between preserved and reduced ejection fraction heart failure. Circ J. 2011;75:2605-2613. 8. Azevedo CF1, Nigri M, Higuchi ML, Pomerantzeff PM, Spina GS, Sampaio RO, Tarasoutchi F, Grinberg M, Rochitte CE. Prognostic significance of myocardial fibrosis quantification by histopathology and magnetic resonance imaging in patients with severe aortic valve disease. JAm Coll Cardiol. 2010;56:278-287. 9. Chung CS, Hutchinson KR, Methawasin M, Saripalli C, Smith JE, Hidalgo CG, Luo X, Labeit S, Guo C, Granzier HL; Shortening of the elastic tandem immunoglobulin segment of titin leads to diastolic dysfunction. Circulation. 2013;128:19-28. 10. Chung CS, Granzier HL; Contribution of titin and extracellular matrix to passive pressure and measurement of sarcomere length in the mouse left ventricle. J Mol Cell Cardiol. 2011;50:731-739. 11. Wu Y, Peng J, Campbell KB, Labeit S, Granzier H; Hypothyroidism leads to increased collagen-based stiffness and re-expression of large cardiac titin isoforms with high compliance. JMol Cell Cardiol. 2007;42:186-95. 12. Wu Y, Cazorla O, Labeit D, Labeit S, Granzier H; Changes in titin and collagen underlie diastolic stiffness diversity of cardiac muscle. J Mol Cell Cardiol. 2000;32:2151-2162. 13. Paulus WJ, Tschöpe C. A novel paradigm for heart failure with preserved ejection fraction: comorbidities drive myocardial dysfunction and remodeling through coronary microvascular endothelial inflammation. J Am Coll Cardiol. 2013;62:263-271.
7. Aoki T1, Fukumoto Y, Sugimura K, Oikawa M, Satoh K, Nakano M, Nakayamamma aa M,M,M, Shimokawa H. Prognostic impact of myocardial interstitial fibrosis in non-ischemmicicic hhheaeaartrtrt fffaiaailullurerereComparison between preserved and reduced ejection fraction heart failure. Circ J.
2011;7;75:2605-2613.
8.8. AAAzzevedo CCCF11,,, NiNiN grgrii i M,M,M, HHHigiggucucuchihih MML,L,, PPomomererananttzzeeff PPPM,M,M, SSpiinanana GGS,S, SSSamama papaioioo RRRO,O, TTarararasasa ouououtctchiFF, GGGrir nberg M,M RRRooochihihittteee CECECE.. PrPProgoggnononoststicic signinin fficancncnce oofof mmyyyoccacardrddiaalal ffibibibrooosiis s s quqquanantititififificacatitt ononon bbbyy hihihisttoopopatholoogygygy aaandndd magagnnetiticc rrresoonaaancnce iimaaggiinng iinin ppatattieieientntntsss wwwithhh sseeverree aaaororrticc c vav lvvve didiseseaase.. JJJAmAmm CCCololo ll CaCaardrdrdioiolll. 220201100;5556:6:27778-8-2828287.7.7.
9. Chung CS,S, HHHututtchchchininnsososon KRKRKR, MeMeMeththhawawwasasasininin MMM, , SaSaaririr papapalllii i C,C,C, SSSmimimiththth JJJE,E,E HHHidididalalalgogog CCCG,G,G, LLLuou X, LabeiiitSS GGuouo CC GrGrananzizierer HHL;L; SShohortrteneniningg ofof tthehe eelalaststicic ttanandedemm imimmmununogoglolobubulilinn sesegmgmenentt ofof ttititinin lleaeadsds
by guest on June 1, 2018http://circ.ahajournals.org/
Dow
nloaded from

DOI: 10.1161/CIRCULATIONAHA.114.013215
28
14. Hamdani N, Franssen C, Lourenço A, Falcão-Pires I, Fontoura D, Leite S, Plettig L, López B, Ottenheijm CA, Becher PM, González A, Tschöpe C, Díez J, Linke WA, Leite-Moreira AF, Paulus WJ; Myocardial titin hypophosphorylation importantly contributes to heart failure with preserved ejection fraction in a rat metabolic risk model. Circ Heart Fail. 2013;6:1239-1249. 15. Hamdani N, Paulus WJ; Myocardial titin and collagen in cardiac diastolic dysfunction: partners in crime. Circulation. 2013;128:5-8.
16. Falcão-Pires I, Hamdani N, Borbély A, Gavina C, Schalkwijk CG, van der Velden J, van Heerebeek L, Stienen GJ, Niessen HW, Leite-Moreira AF, Paulus WJ; Diabetes mellitus worsens diastolic left ventricular dysfunction in aortic stenosis through altered myocardial structure and cardiomyocyte stiffness. Circulation. 2011;124:1151-1159. 17. Borbély A, Papp Z, Edes I, Paulus WJ; Molecular Determinants of heart failure with normal left ventricular ejection fraction. Pharmacol Rep. 2009;61:139-145. 18.van Heerebeek L, Hamdani N, Handoko ML, Falcao-Pires I, Musters RJ, Kupreishvili K, Ijsselmuiden AJ, Schalkwijk CG, Bronzwaer JG, Diamant M, Borbély A, van der Velden J, Stienen GJ, Laarman GJ, Niessen HW, Paulus WJ; Diastolic stiffness of the failing diabetic heart: importance of fibrosis, advanced glycation end products, and myocyte resting tension. Circulation. 2008;117:43-51. 19. van Heerebeek L, Borbély A, Niessen HW, Bronzwaer JG, van der Velden J, Stienen GJ, Linke WA, Laarman GJ, Paulus WJ. Myocardial structure and function differ in systolic and diastolic heart failure. Circulation. 2006;113:1966-1973. 20. Borbély A, van der Velden J, Papp Z, Bronzwaer JG, Edes I, Stienen GJ, Paulus WJ; Cardiomyocyte stiffness in diastolic heart failure. Circulation. 2005;111:774-781. 21. Makarenko I, Opitz CA, Leake MC, Neagoe C, Kulke M, Gwathmey JK, del Monte F, Hajjar RJ, Linke WA; Passive stiffness changes caused by upregulation of compliant titin isoforms in human dilated cardiomyopathy hearts. Circ Res. 2004;95:708-716. 22. LeWinter MM, Granzier HL; Titin is a major human disease gene. Circulation. 2013;127:938-944. 23. Hudson BD, Hidalgo CG, Gotthardt M, Granzier; Excision of titin's cardiac PEVK spring element abolishes PKCalpha-induced increases in myocardial stiffness. J Mol Cell Cardiol. 2010;48:972-978. 24. Granzier HL, Labeit S; The giant protein titin: a major player in myocardial mechanics, signaling, and disease. Circ Res. 2004;94:284-295. 25.Yamasaki R, Wu Y, McNabb M, Greaser M, Labeit S, Granzier H; Protein kinase A phosphorylates titin's cardiac-specific N2B domain and reduces passive tension in rat cardiac myocytes. Circ Res. 2002;90:1181-1188.
jsselmuiden AJ, Schalkwijk CG, Bronzwaer JG, Diamant M, Borbély A, van der r VeVeVeldldenen JJ, , Stienen GJ, Laarman GJ, Niessen HW, Paulus WJ; Diastolic stiffness of the faililingnng ddiaiaabebebetititic c cheart: importance of fibrosis, advanced glycation end products, and myocyte resttininnggg tetensnsnsioioionnn. Circulation. 2008;117:43-51.
199. . vavavannn HeHeHeerere eeebeeeeekkk L,L Borbély A, Niessen HW, BBrBrooonzwaer JG, vaaan nn deer rr VVVelden J, Stienen GJ,LiLiinknkke WAWA, LaLL ararmamam n n GJGJGJ, , , PaPaPaululususs WWWJ.J. MMyoyoy cacardrdiaial l ststrructtururureee anana d fufufunncnctitiononn dddiffefer r ininin sssysysy totoliliiccc ananndd dddidiassstot lic heart t fafafailuurure.e. CiCiCircrculululatatatioion.n.n. 2220000066;1133:3 1196666--1977733.3.
2000. BoBoBorbrbr élély y y A,AA, vvvannn ddeerer VVVele dedeen n JJJ, PPPaapapppp Z,Z,Z BBBroror nznznzwwawaeere JJJG,G,G EEEdedees s s I,I,, SSStititienenenenenn GGJ,J,J, PPPauaulululusss WWJWJ;;;Cardrdioiomyyococyttee stififfnfneess inin didiasastotolilic hearartt fafaililurre.e Circrcululatatioion.n. 200005;5;11111:77774-4-78781.1
2121 MaMakakarerenknkoo II OOpipitztz CCAA LLeaeakeke MMCC NNeaeagogoee CC KKululkeke MM GwGwatathmhmeyey JJKK ddelel MMonontete FF HaHajjjjaaar
by guest on June 1, 2018http://circ.ahajournals.org/
Dow
nloaded from

DOI: 10.1161/CIRCULATIONAHA.114.013215
29
26. van Heerebeek L1, Hamdani N, Falcão-Pires I, Leite-Moreira AF, Begieneman MP, Bronzwaer JG, van der Velden J, Stienen GJ, Laarman GJ, Somsen A, Verheugt FW, Niessen HW, Paulus WJ. Low myocardial protein kinase G activity in heart failure with preserved ejection fraction. Circulation. 2012;126:830-839.
27. Borbély A, van Heerebeek L, Paulus WJ; Transcriptional and posttranslational modifications of titin: implications for diastole. Circ Res. 2009;104:12-14.
28. Borbély A, Falcao-Pires I, van Heerebeek L, Hamdani N, Edes I, Gavina C, Leite-Moreira AF, Bronzwaer JG, Papp Z, van der Velden J, Stienen GJ, Paulus WJ; Hypophosphorylation of the stiff N2B titin isoform raises cardiomyocyte resting tension in failing human myocardium. Circ Res. 2009;104:780-786. 29. Hamdani N, de Waard M, Messer AE, Boontje NM, Kooij V, van Dijk S, Versteilen A, Lamberts R, Merkus D, Dos Remedios C, Duncker DJ, Borbely A, Papp Z, Paulus W, Stienen GJ, Marston SB, van der Velden J; Myofilament dysfunction in cardiac disease from mice to men. J Muscle Res Cell Motil. 2008;29:189-201. 30. van der Velden J, Narolska NA, Lamberts RR, Boontje NM, Borbély A, Zaremba R, Bronzwaer JG, Papp Z, Jaquet K, Paulus WJ, Stienen GJ; Functional effects of protein kinase C-mediated myofilament phosphorylation in human myocardium. Cardiovasc Res. 2006;69:876-887. 31. Linke WA, Hamdani N. Gigantic business: titin properties and function through thick and thin. Circ Res. 2014;114:1052-1068. 32. Krüger M, Kötter S, Grützner A, Lang P, Andresen C, Redfield MM, Butt E, dos Remedios CG, Linke WA; Protein kinase G modulates human myocardial passive stiffness by phosphorylation of the titin springs. Circ Res. 2009;104:87-94. 33. Brouwers FP1, de Boer RA, van der Harst P, Voors AA, Gansevoort RT, Bakker SJ, Hillege HL, van Veldhuisen DJ, van Gilst WH. Incidence and epidemiology of new onset heart failure with preserved vs. reduced ejection fraction in a community-based cohort: 11-year follow-up of PREVEND. Eur Heart J. 2013;34:1424-1431. 34. Shah SJ1, Heitner JF, Sweitzer NK, Anand IS, Kim HY, Harty B, Boineau R, Clausell N, Desai AS, Diaz R, Fleg JL, Gordeev I, Lewis EF, Markov V, O'Meara E, Kobulia B, Shaburishvili T, Solomon SD, Pitt B, Pfeffer MA, Li R. Baseline characteristics of patients in the treatment of preserved cardiac function heart failure with an aldosterone antagonist trial. CircHeart Fail. 2013;6:184-192. 35. Komajda M, Carson PE, Hetzel S, McKelvie R, McMurray J, Ptaszynska A, Zile MR, Demets D, Massie BM. Factors associated with outcome in heart failure with preserved ejection fraction: findings from the Irbesartan in heart failure with preserved ejection fraction study (I-PRESERVE). Circ Heart Fail. 2011;4:27-35.
30. van der Velden J, Narolska NA, Lamberts RR, Boontje NM, Borbély A, Zarereembmbmba R,R,R, Bronzwaer JG, Papp Z, Jaquet K, Paulus WJ, Stienen GJ; Functional effects of prprotototeieiein n kikikinananasesse CCC-mediated myofilament phosphorylation in human myocardium. Cardiovasc Res. 2006;69:876-887.
31311. LLiLinke WAWAWA, HaHaHamdmdananani i N.N.N GGigigigananantiticc bubusisinenessss:: tit tttinnn propopopererertitit ese aaandndnd ffununctctctioion n ththrororougugugh h ththicicck k ananand d hhhinnn.. Circ Ress. 20202 1114;11;11444:1:1:10505052-2-2-1010686868.
3222. KrKrKrügügügererr MMM, , KKöKöttttterer SS, , GrGrütüttznznnererer AAA,, LLaLangngng PPP, AAnAndddreeseseenen CCC, , ReReRedfdfd iieieldldd MMMM,M,M, BBututu t tt E,E,, dddooos RRRememmededdiooos sCGG, , LiLinknke e WAWA; ; Prrototeie n kikinaasese GG modululatateses hhumuman mmyoyocacarrdidiala ppasassisiveve stitiffffnenessss bby phosphorylatatioioion nn ofofof ttthehee tttiti innn sssprpp ininingsgsg .. CiCiircrcr RRReseses.. 222000009;9;9 10101 4:4:4:878787-9-994.4.4.
by guest on June 1, 2018http://circ.ahajournals.org/
Dow
nloaded from

DOI: 10.1161/CIRCULATIONAHA.114.013215
30
36. McMurray JJ, Carson PE, Komajda M, McKelvie R, Zile MR, Ptaszynska A, Staiger C, Donovan JM, Massie BM. Heart failure with preserved ejection fraction: clinical characteristics of 4133 patients enrolled in the I-PRESERVE trial. Eur J Heart Fail. 2008;10:149-156 37. Lee DS1, Gona P, Vasan RS, Larson MG, Benjamin EJ, Wang TJ, Tu JV, Levy D. Relation of disease pathogenesis and risk factors to heart failure with preserved or reduced ejection fraction: insights from the Framingham heart study of the national heart, lung, and blood institute. Circulation. 2009;119:3070-3077. 38. Hwang SJ, Melenovsky V, Borlaug BA; Implications of coronary artery disease in heart failure with preserved ejection fraction. J Am Coll Cardiol. 2014;63:2817-2827. 39. Donaldson C, Taatjes DJ, Zile MR, Palmer B, VanBuren P, Spinale F, Maughan D, Von Turkovich M, Bishop N, LeWinter MM; Combined immunoelectron microscopic and compter-assisted image analyses to detect advanced glycation end-products in human myocardium. Histochem Cell Biol. 2010;134:23-30. 40. Donaldson C, Palmer BM, Zile MR, Maughan DW, Ikonomidis JS, Granzier H, Meyer M, VanBuren P, LeWinter MM; Myosin cross-bridge dynamics in patients with hypertension and concentric left ventricular remodeling. Circ Heart Fail. 2012;5:803-811. 41. Paulus WJ, Tschöpe C, Sanderson JE, Rusconi C, Flachskampf FA, Rademakers FE, Marino P, Smiseth OA, De Keulenaer G, Leite-Moreira AF, Borbély A, Edes I, Handoko ML, Heymans S, Pezzali N, Pieske B, Dickstein K, Fraser AG, Brutsaert DL. How to diagnose diastolic heart failure: A consensus statement on the diagnosis of heart failure with normal left ventricular ejection fraction by the Heart Failure and Echocardiography Associations of the European Society of Cardiology. Eur Heart J. 2007;28:2539. 42. Heart Failure Society of America, Lindenfeld J, Albert NM, Boehmer JP, Collins SP, Ezekowitz JA, Givertz MM, Katz SD, Klapholz M, Moser DK, Rogers JG, Starling RC, Stevenson WG, Tang WH, Teerlink JR, Walsh MN. HFSA 2010 Comprehensive heart failure practice guideline. J Card Fail. 2010;16:e1-194. 43. Hamdani N, Bishu KG, von Frieling-Salewsky M, Redfield MM, Linke WA; Deranged myofilament phosphorylation and function in experimental heart failure with preserved ejection fraction. Cardiovasc Res. 2013;97:464-471. 44. Lang RM, Bierig M, Devereux RB, Flachskampf FA, Foster E, Pellikka PA, Picard MH, Roman MJ, Seward J, Shanewise JS, Solomon SD, Spencer KT, John Sutton M, Stewart WJ. Recommendations for chamber quantification: a report from the American Society of Echocardiography’s guidelines and standards committee and the chamber quantification writing group, developed in conjunction with the European Association of Echocardiography, a branch of the European Society of Cardiology. J Am Soc Echocardiogr. 2005;18:1440-1463. 45. Baicu CF, Li J, Zhang Y, Kasiganesan H, Cooper G, Zile MR, Bradshaw AD. Time course of right ventricular pressure-overload induced myocardial fibrosis: relationship to changes in
40. Donaldson C, Palmer BM, Zile MR, Maughan DW, Ikonomidis JS, Granzier HHH, , , MeMeM yeyey r r M,M,, VanBuren P, LeWinter MM; Myosin cross-bridge dynamics in patients with hyppeeertetetennssioioionn n ananandd concentric left ventricular remodeling. Circ Heart Fail. 2012;5:803-811.
41. Paulus WWJ, Tschöpe C, Sanderson JE, Rusconi C, Flachskampf FA,, Rademakers FE, MarinoP,P, SSSmimimisseseththth OOOAA, DDDeee KeK ulenaer G, Leite-Moreiraa AAAFFF, Borbély A, EdEdE ess III, , HHHandoko ML, HeymansaaS,S,, PPeeezzali NN, , Piiesesskekek BB,,, DiDiickckc ststeieiinnn K,K,K FFraraseserr AGAG,, BBrBruutsaaerererttt DLDD . HHoHow w toto dddiai gngnososseee didid asastotolililic c c hehehearara t tffaailurure: A connsesennnsusus ssstatatettememementntnt oon nn ththhe e didiaagnooossis ooff hhheaartrtr ffaiaiiluluurere wwiwithth nnooormmamal l l leleeftft vvvenenntrtricici ululularar ejejjeccctit on fractctioi n byy thhee HHeaarrt FFaili ururre ee aanandd Eccchhoocardrdrdioiogrgrgrapapaphyhyh AAssssocciatiooonnsns ooof ththe e EEuEurroropppeaaan SoSocicic etetetyyy ofof CCCaarardidiolologogo yyy. EEuEur r HHeHearararttt J.J.J. 22000007;7;7;282828:22253535 99.9.
42. Heart Faailillururure e SoSoSociciietetety y ofofof AAAmememeririr cacaca, LiLiL ndndndenenenfffelele d dd J,J,J AAAlbbbererertt t NMNMNM,,, BoBoBoehehehmememerr r JPJPJP, CoCoColllll inininsss SP,ffffEzEzekekowowititzz JAJA GiGivevertrtzz MMMM KaKatztz SSDD KKlalaphphololzz MM MMososerer DDKK RRogogererss JGJG StStararlilingng RRCC
by guest on June 1, 2018http://circ.ahajournals.org/
Dow
nloaded from

DOI: 10.1161/CIRCULATIONAHA.114.013215
31
fibroblast dependent post-synthetic procollagen processing. Am J. Physiol. 2012;303:H1128-34. 46. Walsh B.J., Thornton S.C., Penny R., Breit S.N. Microplate reader-based quantitation of collagens. Anal Biochem. 1992;203:187-190. 47. Marotta M., Martino G. Sensitive spectrophotometric method for the quantitative estimation of collagen. Anal Biochem. 1985;150:86-90. 4. Warren CM, Krzesinski PR, Greaser ML; Vertical agarose gel electrophoresis and electroblotting of high-molecular-weight proteins. Electrophoresis. 2003;24;1695-1702. 49. McCulloch C & Searle SR. Generalized, linear, and mixed models. New York: John Wiley & Sons, Inc. 2001. 50. Akaike, Hirotugu. A new look at the statistical model identification, IEEE Transactions on Automatic Control. 1974;19:716-723. 51. Zile MR, Baicu CF, Stroud RE, Van Laer A, Arroyo J, Mukherjee R, Jones JR, Spinale FG. Pressure-overload dependent membrane-type 1 matrix metalloproteinase induction: relationship to lv remodeling and fibrosis. Am J Physiol. 2012;302:H1429-37. 52. Bishu K, Hamdani N, Mohammed SF, Kruger M, Ohtani T, Ogut O, Brozovich FV, Burnett JC Jr, Linke WA, Redfield MM; Sildenafil and B-type natriuretic peptide acutely phosphorylate titin and improve diastolic distensibility in vivo. Circulation. 2011;124:2882-2891. 53. Lewinter MM, Popper J, McNabb M, Nyland L, Bell SB, Granzier H. Extensible behavior of titin in the miniswine left ventricle. Circulation. 2010;121:768-774. 54. Hamdani N, Krysiak J, Kreusser MM, Neef S, dos Remedios CG, Maier LS, Krüger M, Backs J, Linke WA; Crucial role for Ca2+/calmodulin-dependent protein kinase-II in regulating diastolic stress of normal and failing hearts via titin phosphorylation. Circ Res. 2013;112:664-674. 55. Spinale FG, Zile MR. Integrating the myocardial matrix into heart failure recognition and management. Circ Res. 2013;113:725-738. 56. Zile MR, Baicu CF. Biomarkers of diastolic dysfunction and myocardial fibrosis: application to heart failure with preserved ejection fraction. J Cardiovasc Trans Res. 2013;6:501-515. 57. Granzier H1, Wu Y, Siegfried L, LeWinter M. Titin: physiological function and role in cardiomyopathy and failure. Heart Fail Rev. 2005;10:211-223. 58. Nagueh SF1, Shah G, Wu Y, Torre-Amione G, King NM, Lahmers S, Witt CC, Becker K, Labeit S, Granzier HL. Altered titin expression, myocardial stiffness, and left ventricular function in patients with dilated cardiomyopathy. Circulation. 2004;110:155-162.
51. Zile MR, Baicu CF, Stroud RE, Van Laer A, Arroyo J, Mukherjee R, Jones JR,R, SSSpipinanalele FFGG. Pressure-overload dependent membrane-type 1 matrix metalloproteinase inductioioon:: reelelatatatioioionsnsnshihippo lv remodeling and fibrosis. Am J Physiol. 2012;302:H1429-37.
52. Bishu K,, Hamdani N, Mohammed SF, Krugeer M, Ohtani T, Ogut O,O, Brozovich FV, Burnett JCC JJJr,r,r, LLLinininkekeke WWWA,A,A RRRedfield MM; Sildenafil and BBB-t-tyype natriureticc pep ptpttidididee acutely phosphorylate iittitin n and imprprprovve e e didid asastotooliliccc ddisisteteensnsnsibibi ililitityy y inin vvivivo.o CCiCirrcullatatatioioionn.n 220101011;1;1;12124:4::2822 822-2-228989891.1.
53533. LLeL winterer MMM,M,M, Pooppppper J,J,, MMMcNNababbbb MMM, Nyylylaand LL,, BBelelell l l SBSBSB,,, Grrrannnzierrr HHH. EExExtetensn ibbbleee bbbehhhaviioorrr ofiititiin nn ininin tthehe mmmiininisiswiwiinene llefefft t veventntn riririclclcle.e.e. CiCircrcrculululatatatiooon.n.n 22200110;0;;12221:1:1:7676768-8-8 777774.44.
54. Hamdani i N,N,N, KKKryryrysisiiakakak J,,, KrKrKreuuussssssererr MMMM,M,M NNNeeeeeef f f S,S,S dddososo RRRemememedededioioios s s CGCGCG, , MaMaMaieieer r r LSLSLS,,, KrKrKrügügüger M, BaBackckss JJ LLininkeke WWA;A; CCrurucicialal rrololee foforr CaCa2+2+/c/calalmmododululinin d-depepenendedentnt pproroteteinin kikinanasese I-III inin rregegululatatiningg
by guest on June 1, 2018http://circ.ahajournals.org/
Dow
nloaded from

DOI: 10.1161/CIRCULATIONAHA.114.013215
32
59. Neagoe C, Kulke M, del Monte F, Gwathmey JK, de Tombe PP, Hajjar RJ, Linke WA; Titin isoform switch in ischemic human heart disease. Circulation. 2002;106:1333-1341. 60. Kötter S, Gout L, Von Frieling-Salewsky M, Müller AE, Helling S, Marcus K, Dos Remedios C, Linke WA, Krüger M; Differential changes in titin domain phosphorylation increase myofilament stiffness in failing human hearts. Cardiovasc Res. 2013;99:648-656. 61. Hidalgo CG, Chung CS, Saripalli C, Methawasin M, Hutchinson KR, Tsaprailis G, Labeit S, Mattiazzi A, Granzier HL; The multifunctional Ca2+/calmodulin-dependend protein kinase II delta (CaMKII ) phosphorylates cardiac titin’s spring elements. J Mol Cell Cardiol. 2013;54:90-97. 62. Gladden JD, Linke WA, Redfield MM; Heart failure with preserved ejection fraction. Eur J Physiol. 2014;466:1037-1053. 63. House C, Wettenhall RE, Kemp BE. The influence of basic residues on the substrate specificity of protein kinase C. J Biol Chem. 1987;262:772-777. 64. Braz JC, Gregory K, Pathak A, Zhao W, Sahin B, Klevitsky R, Kimball TF, Lorenz JN, Nairn AC, Liggett SB, Bodi I, Wang S, Schwartz A, Lakatta EG, DePaoli-Roach AA, Robbins J, Hewett TE, Bibb JA, Westfall MV, Kranias EG, Molkentin JD. PKC-alpha regulates cardiac contractility and propensity toward heart failure. Nat Med. 2004;10:248-254. 65. Levy D, Larson MG, Vasan RS, Kannel WB, Ho KKL. The progression from hypertension to congestive heart failure. JAMA. 1996;275:1557-1562. 66. Lloyd-Jones DM, Larson MG, Leip EP, Beiser A, D’Agostino RB, Kannel WB, Murabito JM, Vasan RS, Benjamin EJ, Levy D. Lifetime risk for developing congestive heart failure. The Framingham heart study. Circulation. 2002;106:3068-3072. 67. Kane GC, Karon BL, Mahoney DW, Redfield MM, Roger VL, Burnett JC, Jacobsen SJ, Rodeheffer RJ. Progression of left ventricular diastolic dysfunction and risk of heart failure. JAMA. 2011;306:856-863. 68. Bhatia RS, Tu JV, Lee DS., Austin PC, Fang J, Haouzi A, Gong Y, Liu PP. Outcome of heart failure preserved ejection fraction in a population-based study. N Engl J Med. 2006;355:260-269. 69. Owan TE, Hodge DO, Herges RM, Jacobsen SJ, Roger VL, Redfield MM. Trends in prevalence and outcome of heart failure with preserved ejection fraction. N Engl J Med. 2006;355:251-259. 70. Yusef S, Pfeffer MA, Swedberg K, Granger CB, Held P, McMurray JJV, Michelson EL, Olofsson B, Ostergren J, for the CHARM Investigators and Committees. Effects of candesartan in patients with chronic heart failure and preserved left-ventricular ejection fraction: the CHARM-Preserved trial. Lancet. 2003;362:777-781.
64. Braz JC, Gregory K, Pathak A, Zhao W, Sahin B, Klevitsky R, Kimball TF, LLoLorerr nznznz JJJN,N,N, Nairn AC, Liggett SB, Bodi I, Wang S, Schwartz A, Lakatta EG, DePaoli-Roachh AAAAAA, RoRoRobbbbbbinninsss JJ,JHewett TE, Bibb JA, Westfall MV, Kranias EG, Molkentin JD. PKC-alpha regulates cardiaccontractility y and prp opo ensity toward heart failure. Nat Med. 2004;10:248-254.
65655. LLeLevy DD,, LLaLarsrsonono MMG,G,G, VVVasasanann RRRS,S,S KKanannenell WBWB, HHHo KKKLKLKL.. ThThe e e prprprogoggreressssssioi n n frfrromomom hhypyppererrtetet nsnsnsioioi nnoo cccono gestive hehearara tt fafailii ururure.ee JAJAJAMMAMA.. 111999996;;27555:11155777-1156662.2.
6666. LlLlLloyoyoyd-d-JoJoJoneness DMDM, , LaLaarsrsonnn MMMGG,G, LLLeieippp EPEPEP,,, BeBeeisisi eeer AA, D’D’D’AgAgAgososostitit nnono RRRB,B,, KKKanannenen ll l WBWBWB,, MMMurrarabibitoto rJMM,, VaVasaan n RSRS, BeBenjnjamaminn EEJ, LLevevy y D. LLififetetimime ririsk fforor ddevevelelopo inng g cocongn esestit veve hheae rt ffaia luurere.. ThThe eFraminghamm hhheaeae rtrtrt ssstututudydydy. CiCiCircrcrculullatatatioioon.nn 222000002;2;2;1010106:6:6 30303068686 -3330707072.2.2.
by guest on June 1, 2018http://circ.ahajournals.org/
Dow
nloaded from

DOI: 10.1161/CIRCULATIONAHA.114.013215
33
71. Ahmed A, Rich MW, Fleg JL, Zile MR, Young JB, Kitzman DW, Love TE, Aronow WS, Adams KF Jr, Gheorghiade M. Effects of digoxin on morbidity and mortality in diastolic heart failure: The ancillary digitalis investigation group trial. Circulation. 2006;114:397-403. 72. Cleland JG, Tendera M, Adamus J, Freemantle N, Polonski L, Taylor J. The perindopril in elderly people with chronic heart failure (PEP-CHF) study. Eur Heart J. 2006;27:2338-2345. 73.Massie BM, Carson PE, McMurray JJ, Komajda M, McKelvie R, Zile MR, Anderson S, Donovan M, Iverson E, Staiger C, Ptaszynska A, I-PRESERVE Investigators. Irbesartan in patients with heart failure and preserved ejection fraction. N Engl J Med. 2008;359:2456-2467.
74. van Veldhuisen DJ, Cohen-Solal A, Böhm M, Anker SD, Babalis D, Roughton M, Coats AJ, Poole-Wilson PA, Flather MD, SENIORS Investigators. Beta-blockade with nebivolol in elderly heart failure patients with impaired and preserved left ventricular ejection fraction: Data from SENIORS (Study of Effects of Nebivolol Intervention on Outcomes and Rehospitalization in Seniors with heart failure). J Am Coll Cardiol. 2009;53:2150-2158. 75. Pitt B, Pfeffer MA, Assmann SF, Boineau R, Anand IS, Claggett B, Clausell N, Desai AS, Diaz R, Fleg JL, Gordeev I, Harty B, Heitner JF, Kenwood CT, Lewis EF, O'Meara E, Probstfield JL, Shaburishvili T, Shah SJ, Solomon SD, Sweitzer NK, Yang S, McKinlay SM, TOPCAT Investigators. Spironolactone for heart failure with preserved ejection fraction. N Engl J Med. 2014;370:1383-1392 76. Redfield MM, Chen HH, Borlaug BA, Semigran MJ, Lee KL, Lewis G, LeWinter MM, Rouleau JL, Bull DA, Mann DL, Deswal A, Stevenson LW, Givertz MM, Ofili EO, O'Connor CM, Felker GM, Goldsmith SR, Bart BA, McNulty SE, Ibarra JC, Lin G, Oh JK, Patel MR, Kim RJ, Tracy RP, Velazquez EJ, Anstrom KJ, Hernandez AF, Mascette AM, Braunwald E; RELAX Trial. Effect of phosphodiesterase-5 inhibition on exercise capacity and clinical status in heart failure with preserved ejection fraction: a randomized clinical trial. JAMA. 2013;309:1268-1277.
75. Pitt B, Pfeffer MA, Assmann SF, Boineau R, Anand IS, Claggett B, Clausell NN, , , DeDeD sasaii ASAS,,,Diaz R, Fleg JL, Gordeev I, Harty B, Heitner JF, Kenwood CT, Lewis EF, O'Meeaaara aa EE,E, Probstfield JL, Shaburishvili T, Shah SJ, Solomon SD, Sweitzer NK, Yang S, MMcKcKKinininlalaayy SMSMSM, TOPCAT Investigators. Spironolactone for heart failure with preserved ejection fraction. h N Engl J Med. 2014;3;3700:1383-1392
76766. RReRedfieldld MMMM,M,M, CChehenn n HHHHHH,, BoBoorlrlrlauaua g g g BABA, , SeSemimigrrraann MMJ,J,J, LLLeeee KKKLL,L, LLewewisisis G,, LeLeeWiWiW ntnterer MMMM,M,M, RRouululeau JL, BuBulllll DDDA,A,A MMManannn n DLDLD , DeDeDeswswwaaal AA,, SSStevenenensonnn LWLWLW, GiGivvverrtrtz z MMMMMM, , OfOfOfiilili i EOEOEO,, OO'O'CoCoConnnnnorrr CMCMCM, , , Felker GGM,, GGGoldddsmmmithth SSR,R Bararrtt BABABA, MMcMcNullltyyy SESEE, , IbIbIbaaarrrra JJC,C Linn GGG, OOhOh JJK,K PPPaatatelell MMMR, KKKimRJRJJ, TrTrTracaca yy y RPRPRP,, VVeelalaazqzqqueuez z EJEJ, , AnAnAnstststrororom m KJKJKJ,, HHeHernrnrnanannddedez z AFAFAF,, MaMaMascsccetettetete AAAM,M,M, BBBrarar uununwawawalldld EEE; RERER LALALAX XTriaal.l EEffffecect t ofof pphoospsphoh didiese teterarasee-55 inhhibibititioion n onon exeercrcisise e cacapap ciityty aandnd clilininicacall ststatusus in n heheara t failure with ppprerereseses rvrvrvededed eeejejej ctctctioioion nn frfrfracaca tititiononn:: : a a a rararandndndomomomizizzedede cclililinininicacacal l trtrtriaiai l... JAJAJAMAMAMA.. 222010113;3;3;3030309:9:9 1268-1277..
by guest on June 1, 2018http://circ.ahajournals.org/
Dow
nloaded from
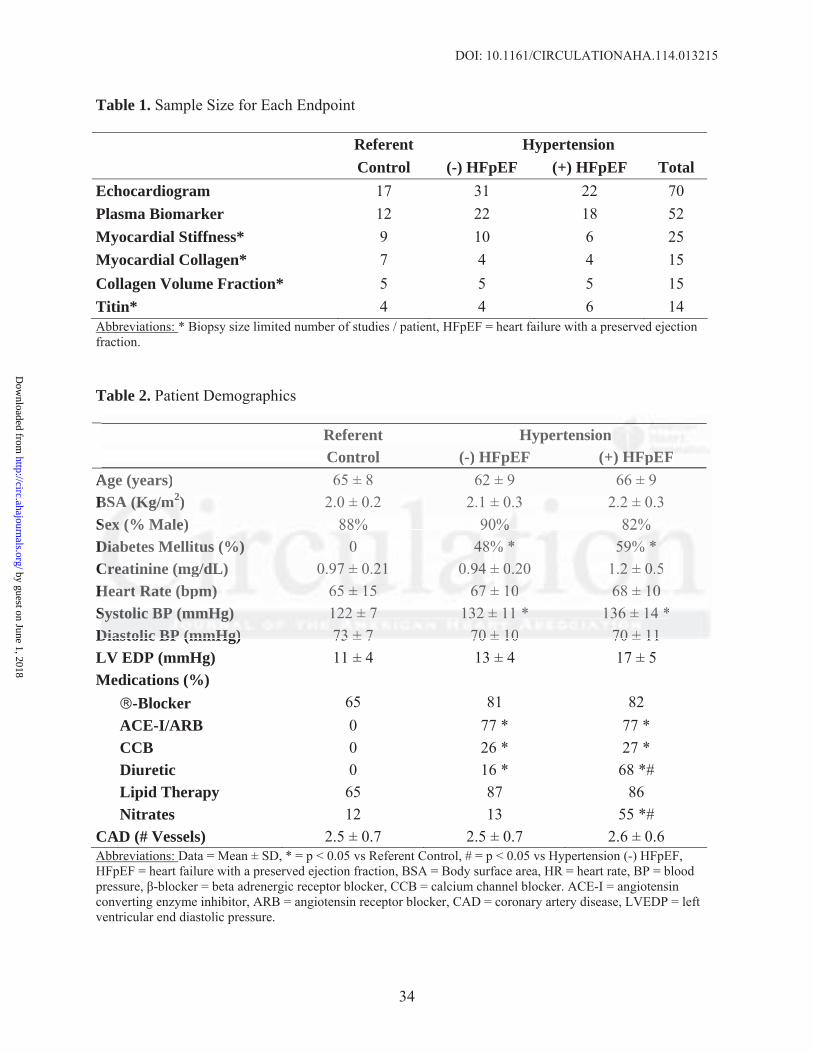
DOI: 10.1161/CIRCULATIONAHA.114.013215
34
Table 1. Sample Size for Each Endpoint
Referent Hypertension Control (-) HFpEF (+) HFpEF TotalEchocardiogram 17 31 22 70 Plasma Biomarker 12 22 18 52 Myocardial Stiffness* 9 10 6 25 Myocardial Collagen* 7 4 4 15 Collagen Volume Fraction* 5 5 5 15 Titin* 4 4 6 14 Abbreviations: * Biopsy size limited number of studies / patient, HFpEF = heart failure with a preserved ejection fraction.
Table 2. Patient Demographics Referent Hypertension Control (-) HFpEF (+) HFpEFAge (years) 65 ± 8 62 ± 9 66 ± 9 BSA (Kg/m2) 2.0 ± 0.2 2.1 ± 0.3 2.2 ± 0.3 Sex (% Male) 88% 90% 82% Diabetes Mellitus (%) 0 48% * 59% * Creatinine (mg/dL) 0.97 ± 0.21 0.94 ± 0.20 1.2 ± 0.5 Heart Rate (bpm) 65 ± 15 67 ± 10 68 ± 10Systolic BP (mmHg) 122 ± 7 132 ± 11 * 136 ± 14 *Diastolic BP (mmHg) 73 ± 7 70 ± 10 70 ± 11LV EDP (mmHg) 11 ± 4 13 ± 4 17 ± 5Medications (%)
-Blocker 65 81 82ACE-I/ARB 0 77 * 77 *CCB 0 26 * 27 *Diuretic 0 16 * 68 *#Lipid Therapy 65 87 86Nitrates 12 13 55 *#
CAD (# Vessels) 2.5 ± 0.7 2.5 ± 0.7 2.6 ± 0.6Abbreviations: Data = Mean ± SD, * = p < 0.05 vs Referent Control, # = p < 0.05 vs Hypertension (-) HFpEF, HFpEF = heart failure with a preserved ejection fraction, BSA = Body surface area, HR = heart rate, BP = blood pressure, -blocker = beta adrenergic receptor blocker, CCB = calcium channel blocker. ACE-I = angiotensin converting enzyme inhibitor, ARB = angiotensin receptor blocker, CAD = coronary artery disease, LVEDP = left ventricular end diastolic pressure.
Referent Hypertensionn Control (-) HFpEF (++)) HHFHFpEpEFF
Age (years) 65 ± 8 62 ± 9 66 ± 9 BSA A (K(K(Kg/g/g/mmm22)) 2.0 ± 0.2 2.1 ± 0.3 2.2 ± 0.3 SeSeex ((%(% MMalalleee) 8888% % 900% % % 8282% % DDiaababetes Melllilitutuuss (%%%) 00 4888% % * 595959% % *CrCrCreaeaatinine (((mgmgm //d/dL)L) 0.0.0 97977 ±±± 0.22211 0.0.949494 ± 00.2220 1..22 ±± 000.555HeHeararrttt RaRaRatetete ((bpbppm)m)m) 65655 ±±± 11155 5 676767 ±±± 111000 686868 ±±± 111000SSySy tstst lololiicic BBBPPP (m(m(m HmHmHg)g)g) 121212222 ±±± 777 131313222 ±±± 111111 *** 111363636 ±±± 111444 ***DiDiasastotolilicc BPBP ((mmmmHgHg)) 7373 ±± 77 7700 ±± 1010 7700 ±± 1111
by guest on June 1, 2018http://circ.ahajournals.org/
Dow
nloaded from
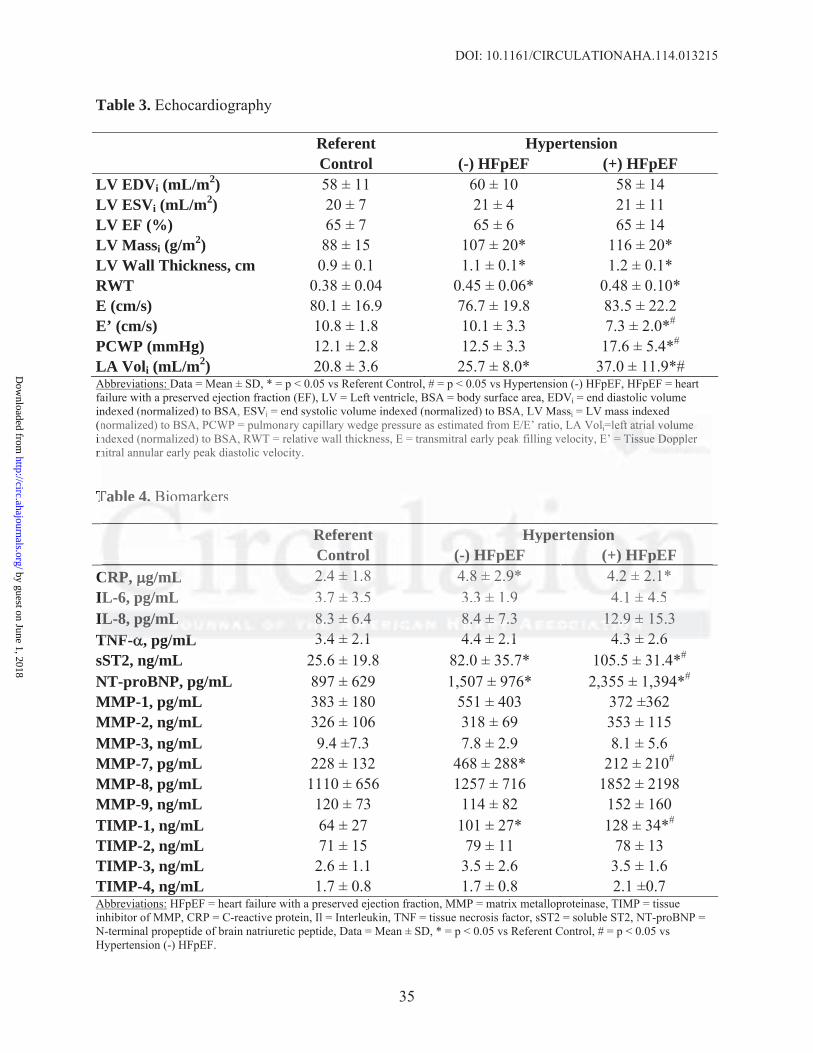
DOI: 10.1161/CIRCULATIONAHA.114.013215
35
Table 3. Echocardiography
Referent Hypertension Control (-) HFpEF (+) HFpEFLV EDVi (mL/m2) 58 ± 11 60 ± 10 58 ± 14 LV ESVi (mL/m2) 20 ± 7 21 ± 4 21 ± 11 LV EF (%) 65 ± 7 65 ± 6 65 ± 14 LV Massi (g/m2) 88 ± 15 107 ± 20* 116 ± 20* LV Wall Thickness, cm 0.9 ± 0.1 1.1 ± 0.1* 1.2 ± 0.1* RWT 0.38 ± 0.04 0.45 ± 0.06* 0.48 ± 0.10*E (cm/s) 80.1 ± 16.9 76.7 ± 19.8 83.5 ± 22.2E’ (cm/s) 10.8 ± 1.8 10.1 ± 3.3 7.3 ± 2.0*#
PCWP (mmHg) 12.1 ± 2.8 12.5 ± 3.3 17.6 ± 5.4*# LA Voli (mL/m2) 20.8 ± 3.6 25.7 ± 8.0* 37.0 ± 11.9*# Abbreviations: Data = Mean ± SD, * = p < 0.05 vs Referent Control, # = p < 0.05 vs Hypertension (-) HFpEF, HFpEF = heart failure with a preserved ejection fraction (EF), LV = Left ventricle, BSA = body surface area, EDVi = end diastolic volume indexed (normalized) to BSA, ESVi = end systolic volume indexed (normalized) to BSA, LV Massi = LV mass indexed (normalized) to BSA, PCWP = pulmonary capillary wedge pressure as estimated from E/E’ ratio, LA Voli=left atrial volume indexed (normalized) to BSA, RWT = relative wall thickness, E = transmitral early peak filling velocity, E’ = Tissue Doppler mitral annular early peak diastolic velocity.
Table 4. Biomarkers
Referent Hypertension Control (-) HFpEF (+) HFpEFCRP, g/mL 2.4 ± 1.8 4.8 ± 2.9* 4.2 ± 2.1* IL-6, pg/mL 3.7 ± 3.5 3.3 ± 1.9 4.1 ± 4.5IL-8, pg/mL 8.3 ± 6.4 8.4 ± 7.3 12.9 ± 15.3 TNF- , pg/mL 3.4 ± 2.1 4.4 ± 2.1 4.3 ± 2.6sST2, ng/mL 25.6 ± 19.8 82.0 ± 35.7* 105.5 ± 31.4*# NT-proBNP, pg/mL 897 ± 629 1,507 ± 976* 2,355 ± 1,394*# MMP-1, pg/mL 383 ± 180 551 ± 403 372 ±362 MMP-2, ng/mL 326 ± 106 318 ± 69 353 ± 115 MMP-3, ng/mL 9.4 ±7.3 7.8 ± 2.9 8.1 ± 5.6 MMP-7, pg/mL 228 ± 132 468 ± 288* 212 ± 210# MMP-8, pg/mL 1110 ± 656 1257 ± 716 1852 ± 2198MMP-9, ng/mL 120 ± 73 114 ± 82 152 ± 160 TIMP-1, ng/mL 64 ± 27 101 ± 27* 128 ± 34*# TIMP-2, ng/mL 71 ± 15 79 ± 11 78 ± 13 TIMP-3, ng/mL 2.6 ± 1.1 3.5 ± 2.6 3.5 ± 1.6 TIMP-4, ng/mL 1.7 ± 0.8 1.7 ± 0.8 2.1 ±0.7 Abbreviations: HFpEF = heart failure with a preserved ejection fraction, MMP = matrix metalloproteinase, TIMP = tissue inhibitor of MMP, CRP = C-reactive protein, Il = Interleukin, TNF = tissue necrosis factor, sST2 = soluble ST2, NT-proBNP = N-terminal propeptide of brain natriuretic peptide, Data = Mean ± SD, * = p < 0.05 vs Referent Control, # = p < 0.05 vs Hypertension (-) HFpEF.
ndexed (normalized) to BSA, ESVi end systolic volume indexed (normalized) to BSA, LV Massi LV maasss iindndexexed normalized) to BSA, PCWP = pulmonary capillary wedge pressure as estimated from E/E’ ratio, LA Volii=llefefeftt t atatatririr alalal vvololo umumume endexed (normalized) to BSA, RWT = relative wall thickness, E = transmitral early peak filling velocity, EE’’ = TiTT sssueueue DDopoppplplp erer k
mitral annular early peak diastolic velocity.
Tablble e 4.4.4. BiBiomomomararrkekekersr
RRRefefefererenennt tt HyHyHyppeperrtenennsisisiononon CCCononnttrrol (-(-(-))) HHHFppEEFF (+))) HHFHFpEEEFF
CRCRRP,P,P, g/g/mLmLmL 2.2.2.44 ±±± 1.1.88 4.4.888 ±±± 2.2.9*9** 4.4..22 ±± 222.11*1* L-6-6, , pgpg//m/mLL 3.3 7 ±± 33.3.555 3.3.333 ±± 1.999 444.1 ±± 444.555L-8, pg/mLL 8.8..33 ±±± 6.6.6 444 8.8.8.4 4 4 ±±± 7.7.7.3 121212.9.99 ±± 15.3
TNTNFF pg//mLL 33 44 ±± 22 11 44 44 ±± 22 11 44 33 ±± 22 66
by guest on June 1, 2018http://circ.ahajournals.org/
Dow
nloaded from

DOI: 10.1161/CIRCULATIONAHA.114.013215
36
Figure Legends:
Figure 1. Total myocardial stiffness expressed as the relationship between myocardial stress
(mN/mm2) versus cardiomyocyte sarcomere length ( m) for referent control patients (open
circle, solid line), patients with hypertension but without heart failure and a preserved ejection
fraction (HTN(-)HFpEF, closed circle, dashed line), and patients with hypertension and HFpEF
(HTN(+)HFpEF closed squares, dotted line). As sarcomere length increases, the slope increases
most rapidly in the HTN(+)HFpEF group (p<0.0001 when compared to both the control and
HTN(-)HFpEF groups). Overall, the curves for the control and HTN(-)HFpEF groups were not
significantly different from one another. Patients with HTN(+)HFpEF had an increase in total
myocardial stiffness as indicated by a leftward shift in the stress vs. sarcomere length
relationship; for any given sarcomere length 2.1 m, stress was higher in the HTN(+)HFpEF
vs. HTN(-)HFpEF or referent control patients. There were no significant differences between
HTN(-)HFpEF vs. referent control patients. * = p < 0.01 vs. referent control, # = p < 0.01 vs.
HTN(-)HFpEF.
Figure 2. Collagen-dependent and titin-dependent myocardial stress at a sarcomere length of 2.6
m for referent control patients (white column), patients with hypertension but without heart
failure and a preserved ejection fraction (HTN(-)HFpEF, cross-hatched column), and patients
with hypertension and HFpEF (HTN(+)HFpEF black column). Total stress is the numerical sum
of collagen and titin specific data. Patients with HTN(+)HFpEF had an increase in collagen-
dependent and titin-dependent myocardial stress. There were no significant differences between
HTN(-)HFpEF or referent control patients. * = p < 0.01 vs. referent control, # = p < 0.01 vs.
HTN(-)HFpEF.
ignificantly different from one another. Patients with HTN(+)HFpEF had an increreasasasee ininin tttotototalala
myocardial stiffness as indicated by a leftward shift in the stress vs. sarcomere leengnggththth
elaatiiononshshipip;; fof r r anana y y given sarcomere length 22.1. m, stress was highherere in the HTN(+)HFpEF
vsvss. HHHTN(-)HFHFFpEpEpEF F F ororr rrrefefeferererenene t t cococontntntroror l l l papapatitienenentststs.. ThThherrre wewewererere nnno oo sisisigngngnififificicicananant t t diffffferererenenencececes s bebebetwtwtweeeeeenn n
HTHTHTNNN(-)HFpEpEEFFF vvs.. reffeerreentt ccconnntrtrol pppatatieiennts. * = pp << 0.0.010101 vvsss. rrrefeeereeent cooonntntrorool, ## = ppp <<< 00.0011 vsss.
HTHTN(N((-)-))HFHFpEpEp F.F.
by guest on June 1, 2018http://circ.ahajournals.org/
Dow
nloaded from

DOI: 10.1161/CIRCULATIONAHA.114.013215
37
Figure 3. Relationship between in vivo echocardiographic derived assessment of LV diastolic
dysfunction (left atrial diameter and echocardiographically estimated pulmonary capillary wedge
pressure [PCWP]) and in vitro measures of myocardial diastolic dysfunction (collagen-dependent
and titin-dependent myocardial stiffness) for all patients studied with both measures available.
There was a statistically significant direct relationship between collagen-dependent stiffness and
left atrial diameter (r2 = 0.42, p = 0.006) and PCWP (r2 = 0.46, p = 0.002) and between titin-
dependent stiffness and left atrial diameter (r2 = 0.43, p = 0.006) but not PCWP (r2 = 0.16, p =
0.11).
Figure 4. Myocardial collagen content in patients with hypertension (HTN) with(+) and
without(-) heart failure with a preserved ejection fraction (HFpEF). Referent control patients
(white column), patients with HTN(-)HFpEF (cross-hatched column), and patients with
HTN(+)HFpEF (black column). * = p < 0.01 vs. referent control, # = p < 0.01 vs.
HTN(-)HFpEF. Panel A: Soluble, insoluble, and total collagen measured biochemically. Patients
with HTN(+)HFpEF had an increase in insoluble and total collagen. There were no significant
differences between HTN(-)HFpEF or referent control patients. Panel B: Examples of picrosirius
stained myocardial sections. Patients with HTN(+)HFpEF had an increase in collagen. There
were no significant differences between HTN(-)HFpEF or referent control patients. Panel C:
Collagen volume fraction (CVF) measured from histologic sections. Patients with
HTN(+)HFpEF had an increase in CVF. There were no significant differences between
HTN(-)HFpEF or referent control patients..
Figure 5. Titin phosphorylation state in referent control patients (white column), patients with
Figure 4. Myocardial collagen content in patients with hypertension (HTN) witth(h((+)+)) aandndnd
without(-) heart failure with a preserved ejection fraction (HFpEF). Referent control patients
wwhihihitetete ccolololumumumnn), papapatitients with HTN(-)HFpEF (crororossss-hatched columummn)),, ananand d patients with
HHTNNN(+)HFpEEF F (b(b( lalackckck cccolololumumumn)nn). *** == p p << 0.0011 vvvs. rrrefferenenent t cocoonnttrorolll, ### == ppp <<< 00.0.00111 vsvs.
HTHTTN(N(N(-)-))HFHFFpEpEpEF.F. PPPaananelel AAA:: SoSolululublblbleee, iiinnsnsoololububublelele,, ananand d ttototatat l cocoollllllagagagenenen mmmeaeaasusus rrredd d bibiiococo heheh mmimicccalllly.y. PPatattieentntn s
with HTN(++)H)H)HFpFpFpEFEFEF hhhadadad aaan n n ininincrcrreaeaeasesese in n n ininnsososoluluublblb ee e ananand d d toootatatall l cococollllllagagagennn.. ThThThererere e e wewewererere nnnooo sisis gnificant
by guest on June 1, 2018http://circ.ahajournals.org/
Dow
nloaded from

DOI: 10.1161/CIRCULATIONAHA.114.013215
38
hypertension but without heart failure and a preserved ejection fraction (HTN(-)HFpEF, cross-
hatched column), and patients with hypertension and HFpEF (HTN(+)HFpEF black column).
Three titin sites were examined: S11878(S26) and S12022(S170), sites known to be
phosphorylated by protein kinase c (PKC) and S4185(S469), a site known to be phosphorylated
by protein kinase a (PKA). Patients with HTN(+)HFpEF had an increase in S11878(S26), no
change in S12022(S170) and a decrease in S4185(S469). There were no significant differences
between HTN(-)HFpEF or referent control patients at any of the three sites. Insert: examples of
the phospho-blot (top) and total protein (bottom) for each of the patient groups. There are two
bands, the top band represents N2BA and the bottom band represents N2B. Data from both N2B
and N2BA bands were summed. * = p = 0.05.
Figure 6. Relationship between echocardiographically derived assessment of LV diastolic
dysfunction (estimated pulmonary capillary wedge pressure [PCWP]) and plasma biomarkers of
filling pressure (NT-proBNP, N-terminal propeptide of brain natriuretic peptide) and profibrotic
factors (soluble ST2 [sST2], tissue inhibitor of matrix metalloproteinase 1 [TIMP-1]). There was
a statistically significant direct relationship between PCWP and NT-proBNP (r2 = 0.32, p =
0.001), sST2 (r2 = 0.26, p = 0.005), and TIMP-1 (r2 = 0.36, p < 0.001).
and N2BA bands were summed. * = p = 0.05.
Fiigugugurerere 666. RRRelelelatioioonnsnshih p between echocardiograpphhihicaally derived asssssessmsmmeenent of LV diastolic
ddydysfsffunu ction (eeststimimimattedede ppp lululmomom nnanaryryy ccacappillllary wwwedggge preesesssusureree [[[PCPCCWPWWP]]))) ananand d plplplaasasmamaa bbbioioiomamamarkrkrkererrs s of
fiilllllinining gg prprpresesssususurere (((NTNTT-p-pproroBNBNP,P,P NNN-t-t-tererermmiminananalll prprpropoppepepe ttitiddee ooof f brbrbraiaain n nanan tttriuiuurerer titiic pepep ptptptididde)e)) aaanndnd ppprooofifibrbrrottticic
factors (solubublelele SST2T2T2 [[sSsSsST222],],] tttisissususuee inininhihiibibibitototorr r ofofo mamamatrtrt ixixi mmmetetetalalallololoprprprotoo eieieinananasesese 111 [[[TITIIMPMPMP-1-11])])]).. There wasss
by guest on June 1, 2018http://circ.ahajournals.org/
Dow
nloaded from
0
2
4
6
8
10
12
14
16
18
20
22
2.0 2.1 2.2 2.3 2.4 2.5 2.6
Referent ControlHTN(-)HFpEFHTN(+)HFpEF
Stre
ss (m
N/m
m2 )
Sarcomere Length ( m)
**
*
*
*
*
#
#
#
#
#
Figure 1
44
66
8
101
12
14
******
*
*
###
#
#
by guest on June 1, 2018http://circ.ahajournals.org/
Dow
nloaded from
0
2
4
6
8
10
12
14
16
18
20
22
Collagen dependent Titin dependent Total
Referent ControlHTN(-)HFpEFHTN(+)HFpEF
LV M
yoca
rdia
l Stif
fnes
s(S
tres
s at
SL
= 2.
6 m
)
Collagendependent
Titindependent
Total
*
#*
#*
#
Figure 2
4
66
8
010
12
14
(SSSttrrr
eeessssss
aaattt SSS
L =
2.6 #
#
by guest on June 1, 2018http://circ.ahajournals.org/
Dow
nloaded from
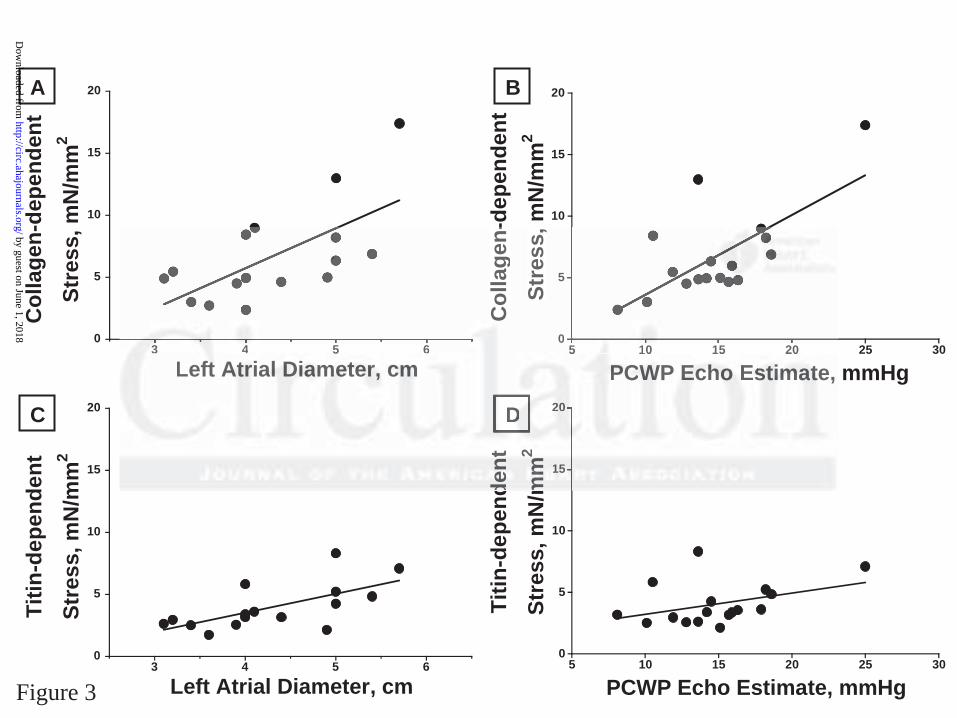
0
5
10
15
20
5 10 15 20 25 30
Col
lage
n-de
pend
ent
Stre
ss, m
N/m
m2
LADPCWP Echo Estimate, mmHg
0
5
10
15
20
3 4 5 6
Col
lage
n-de
pend
ent
Stre
ss, m
N/m
m2
LADLeft Atrial Diameter, cm
0
5
10
15
20
3 4 5 6
Titin
-dep
ende
ntSt
ress
, mN
/mm
2
LADLeft Atrial Diameter, cm0
5
10
15
20
5 10 15 20 25 30
Titin
-dep
ende
ntSt
ress
, mN
/mm
2
LADPCWP Echo Estimate, mmHg
A
D
B
C
Figure 3
0
5
5 110 10 5 20
Col
lage
n-St
ress
,
PPPCCCWWWP EEEccchhhooo EEEssstttiiimmmaaattte, ADLADADLADLAD
3 43 4 5 6
Leeffttt AAAtttrrriiaaalll DDDiiiaammmeeettteeerrr,, cccmmmLADD
151515
2022
ddeeenntt
mmmmmm
22
DDD
by guest on June 1, 2018http://circ.ahajournals.org/
Dow
nloaded from
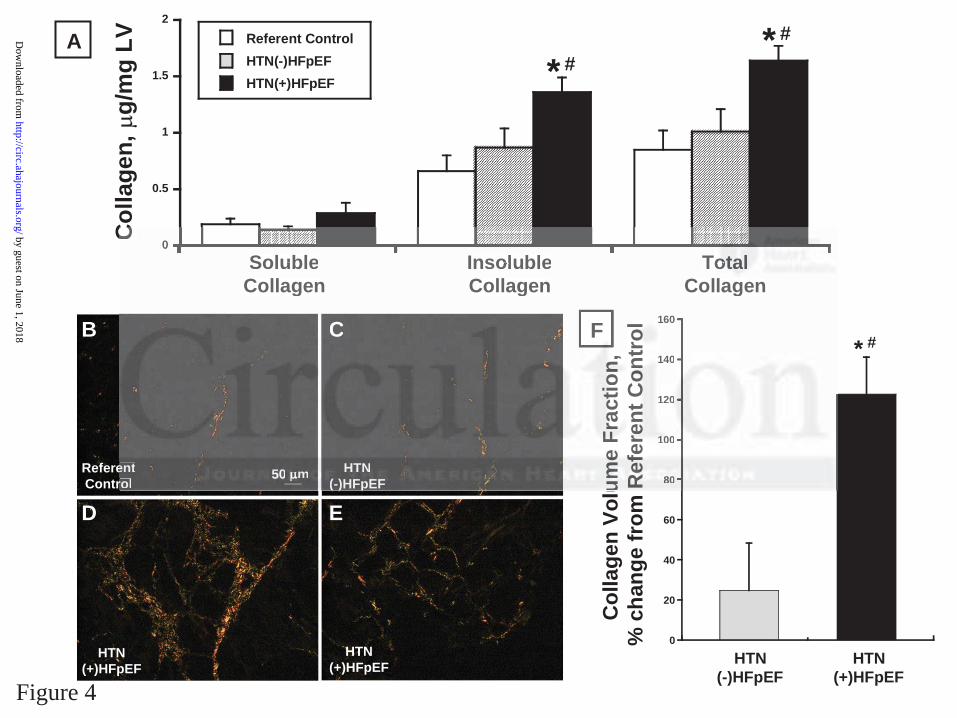
0
0.5
1
1.5
2
Soluble Collagen Insoluble Collagen Total Co llagen
Referent ControlHTN(-)HFpEFHTN(+)HFpEF
Col
lage
n,
g/m
g LV
SolubleCollagen
InsolubleCollagen
TotalCollagen
**
##
A
F
0
20
40
60
80
100
120
140
160
*
Col
lage
n Vo
lum
e Fr
actio
n,
% c
hang
e fr
om R
efer
ent C
ontr
ol #
HTN(-)HFpEF
HTN(+)HFpEF
B
ED
C
ReferentControl
HTN(-)HFpEF
HTN(+)HFpEF
HTN(+)HFpEF
50 m
Figure 4
0C
SolubleSoluble Collagenle Colla
CollagenInsolubleInsoluble Collagenble Collagen
CollagenTToootttaaallTotal Co llagenagen
Collagen
F
80808
10010010
0120
1401
160
uummmeee
FFFrrraaaccc
tttiiiooonnn,,
RRR
eeefffeerrr
eeennnttt CCC
ooonnnttrr
ooollC
entrol
HTHTNN(-( )H)HFpFpEFEF
5050 mm
by guest on June 1, 2018http://circ.ahajournals.org/
Dow
nloaded from
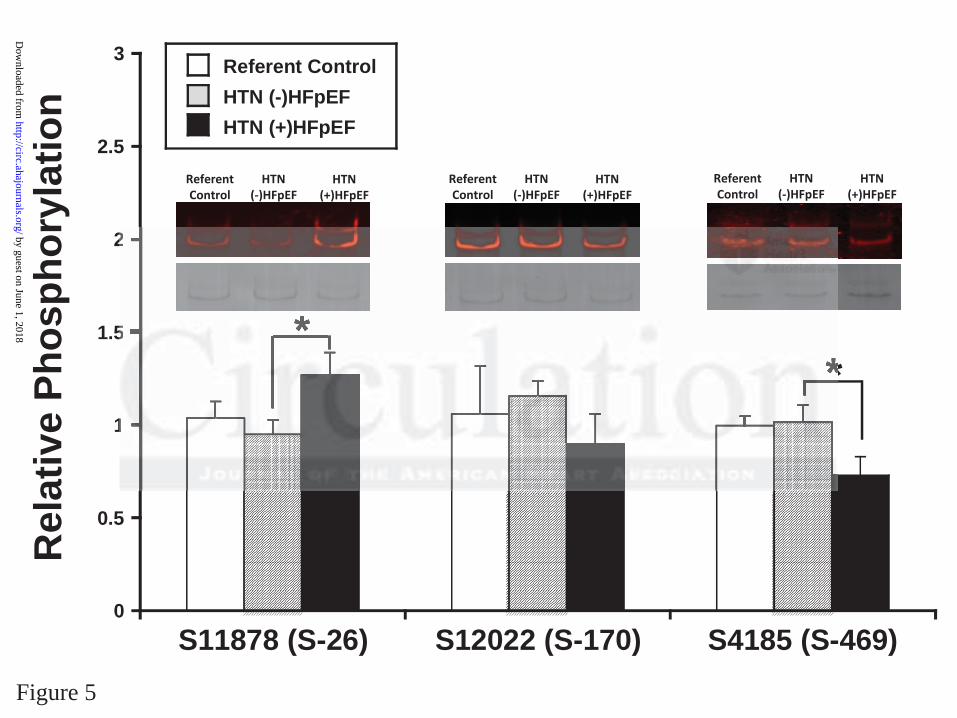
0
0.5
1
1.5
2
2.5
3
S11878 (S-26) S12022 (S-170) S4185 (S-469)
Referent ControlHTN (-)HFpEFHTN (+)HFpEF
*
Rel
ativ
e Ph
osph
oryl
atio
n
*
HTN(+)HFpEF
HTN( )HFpEF
ReferentControl
HTN(+)HFpEF
HTN( )HFpEF
ReferentControl
HTN(+)HFpEF
HTN( )HFpEF
ReferentControl
Figure 5
1
5
2 by guest on June 1, 2018http://circ.ahajournals.org/
Dow
nloaded from
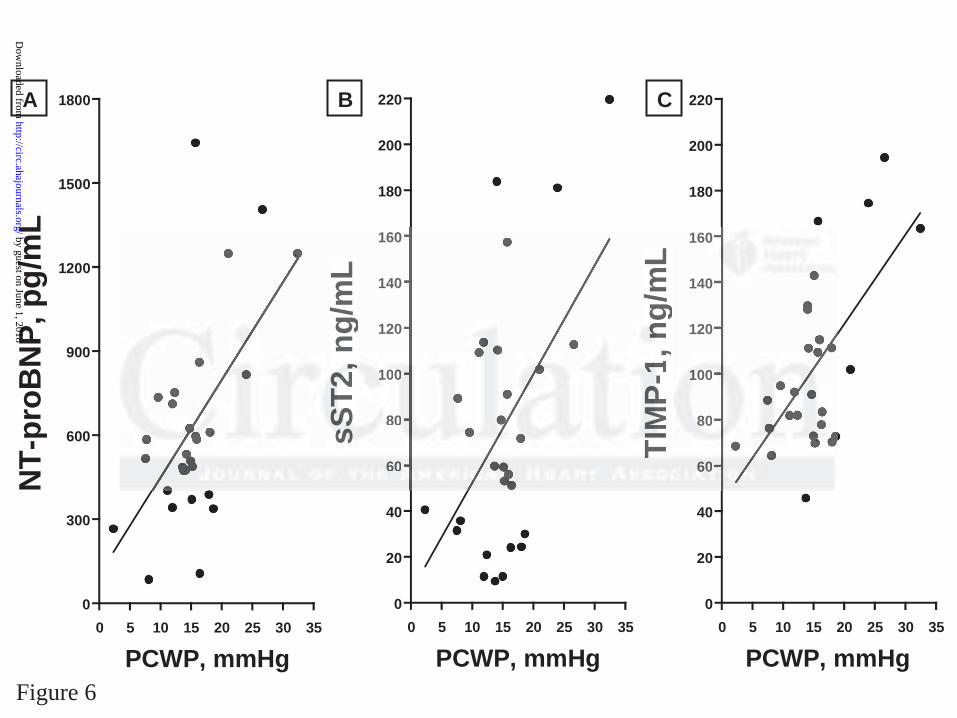
0
300
600
900
1200
1500
1800
0 5 10 15 20 25 30 35
NT-
proB
NP,
pg/
mL
PCWP, mmHg
0
20
40
60
80
100
120
140
160
180
200
220
0 5 10 15 20 25 30 35
sST2
, ng/
mL
PCWP, mmHg
0
20
40
60
80
100
120
140
160
180
200
220
0 5 10 15 20 25 30 35
TIM
P-1,
ng/
mL
PCWP, mmHg
A B C
Figure 6
606
08080
11010000
120
140
160
sssSSSTTT222
,,, ng/
mL
606060
80808
10101000
120
140
160
TTIIMMM
PPP--111,
nnng/
mL
by guest on June 1, 2018http://circ.ahajournals.org/
Dow
nloaded from

David Bull, Henk Granzier and Martin M. LeWinterBradshaw, Rebecca Slater, Bradley M. Palmer, Peter Van Buren, Markus Meyer, Margaret Redfield,
Michael R. Zile, Catalin F. Baicu, John Ikonomidis, Robert E. Stroud, Paul J. Nietert, Amy D.Contributions of Collagen and Titin
Myocardial Stiffness in Patients with Heart Failure and a Preserved Ejection Fraction:
Print ISSN: 0009-7322. Online ISSN: 1524-4539 Copyright © 2015 American Heart Association, Inc. All rights reserved.
is published by the American Heart Association, 7272 Greenville Avenue, Dallas, TX 75231Circulation published online January 30, 2015;Circulation.
http://circ.ahajournals.org/content/early/2015/01/30/CIRCULATIONAHA.114.013215World Wide Web at:
The online version of this article, along with updated information and services, is located on the
http://circ.ahajournals.org/content/suppl/2016/12/29/CIRCULATIONAHA.114.013215.DC1Data Supplement (unedited) at:
http://circ.ahajournals.org//subscriptions/
is online at: Circulation Information about subscribing to Subscriptions:
http://www.lww.com/reprints Information about reprints can be found online at: Reprints:
document. Permissions and Rights Question and Answer available in the
Permissions in the middle column of the Web page under Services. Further information about this process isOnce the online version of the published article for which permission is being requested is located, click Request
can be obtained via RightsLink, a service of the Copyright Clearance Center, not the Editorial Office.Circulation Requests for permissions to reproduce figures, tables, or portions of articles originally published inPermissions:
by guest on June 1, 2018http://circ.ahajournals.org/
Dow
nloaded from

129129
초록
배경
본 연구의 목적은 좌심실 수축기능이 보존된 심부전(heart
failure with preserved ejection fraction, HFpEF) 환자에서 수
동 심근 경직도가 증가하는지 여부와 이러한 현상이 세포외기질
의 섬유성 콜라겐과 심근세포 내 티틴의 변화와 상관관계가 있는
지 알아보고자 하였다.
방법 및 결과
관상동맥 우회술을 시행 받는 70명의 환자에서 심초음파, 혈
중 바이오마커 분석 및 수술 중 좌심실 심외막 전벽에서 생검
을 시행하였다. 환자들을 고혈압이나 당뇨병이 없는 대조군(17
명), HFpEF가 없는 고혈압군[HTN(-)HFpEF, 31명], HFpEF
가 있는 고혈압군[HTN(+)HFpEF, 22명]의 세 개 군으로 분류하
고, 검체를 이용하여 수동 경직도 측정(differential extraction
assay), 콜라겐 분석(biochemistry or histology) 및 티틴의 아형
및 인산화 분석 등을 시행하였다. 대조군과 비교했을 때 HTN(-)
HFpEF군의 좌심실 확장기말 압력, 심근의 수동 경직도, 콜라겐
이나 티틴의 인산화 분석에서는 차이가 없었으나, 염증과 관련
된 바이오마커(C-reactive protein, soluble ST2, tissue inhibitor
of metalloproteinase 1)는 증가되어 있었다. HTN(+)HFpEF군
에서는 대조군이나 HTN(-)HFpEF군과 비교하여, 좌심실 확장
기말 압력, 좌심방 용적, NTproBNP(N-terminal propeptide of
brain natriuretic peptide), 수동 경직도, 불용성 콜라겐 등이 증
가되었으며, 티틴의 아형분석에서 PEVK S11878(S26)의 인산
화 증가와 N2B S4185(S469) 인산화 감소 및 염증과 관련된 바이
오마커의 증가가 관찰되었다.
결론
HTN(-)HFpEF는 수동 심근 경직도를 변화시키지 않았으나,
HTN(+)HFpEF 환자에서는 콜라겐과 티틴에 의존적으로 수동
심근 경직도가 유의하게 증가한다. 이는 HFpEF의 발생 과정에
서 콜라겐과 티틴의 항상성 변화가 연관이 있음을 시사한다.
좌심실 수축기능이 보존된 심부전 환자에서 보이는 심근 경직도는 콜라겐과 티틴의 항상성 변화 때문이다
박 훈 준 교수 가톨릭대학교 서울성모병원 순환기내과
Heart Failure





























