Embed Size (px)
Citation preview

New pyrrole-based ligand for first row
transition metal complexes
Degree Project C in Chemistry (1KB010) thesis by:
Ricardo J. Fernández Terán
Supervisor: Dr. Anders Thapper
Subject Specialist: Dr. Lukasz Pilarski
Examiner: Prof. Dr. Helena Grennberg
May 2015

2
To the loving memory of Prof. Dr. Isaura De Sanctis,
who passed away in December 2014,
for all what I learned from her,
including the love and passion for
Inorganic Chemistry.

3
LIST OF ABBREVIATIONS
acac Acetylacetonate (Pentane-2,4-dionate) ligand
Boc tert-butyloxycarbonyl (protecting group)
bpy 2,2’-bipyridine
DEPT Distortionless Enhancement by Polarisation Transfer (NMR pulse sequence)
DMAP 4-(dimethylamino)pyridine
Et2O Diethyl ether
Et3N Triethylamine
EtOH Ethanol
HTMP 2,2,6,6-tetramethylpiperidine
LiTMP Lithium 2,2,6,6-tetramethylpiperidide
MeOH Methanol
NADP+ Nicotinamide adenine dinucleotide phosphate
NADPH Nicotinamide adenine dinucleotide phosphate (protonated)
n-BuLi n-butyllithium
PCET Proton coupled electron transfer
PY Pyridine
PY5 2,6-bis(methoxydi(pyridin-2-yl)methyl)pyridine
PY5-OH Pyridine-2,6-diylbis(di(pyridin-2-yl)methanol)
PyPY2 5-(methoxydi(pyridin-2-yl)methyl)-pyrrole-2-carboxylate
PyPY4 (1H-pyrrole-2,5-diyl)bis(di(pyridin-2-yl)methanol)
RC Radical coupling
t-BuLi tert-butyllithium
THF Tetrahydrofuran
TON Turnover number
TOF Turnover frequency
WNA Water nucleophilic attack
WOC Water oxidation catalyst

4
ABSTRACT
The negative impact of the use of traditional fossil fuels in the environment and their limited
nature has prompted research in new, alternative sources of energy. Artificial photosynthesis,
which comprises the splitting of water into hydrogen and oxygen, can be used as clean source of
energy in many applications, especially since the main energy source is the sun. The oxidation
of water into dioxygen by transition metal complexes invokes the necessity for new catalysts to
be synthesized and studied, looking forward to achieving catalytic activities closer to those
shown by the oxygen evolving complex in Photosystem II. In this project, a new tridentate
pyrrole-based ligand and its metal complexes with Copper(II) and Nickel(II) were synthesized.
The obtained ligand, methyl 5-(methoxydi(pyridin-2-yl)methyl)-1H-pyrrole-2-carboxylate, was
synthesized in moderate yields (38%). Differences in reactivity between pyridine and pyrrole
diesters were also explored, since the designed pentadentate ligand, (1H-pyrrole-2,5-
diyl)bis(di(pyridin-2-yl)methanol)), could not be obtained. Also, structural differences where
observed in the products when following similar synthetic schemes for both ester substrates.
RESUMEN
El impacto negativo sobre el ambiente que tiene el uso de combustibles fósiles y su naturaleza
limitada han promovido la investigación en fuentes nuevas y alternativas de energía. La
fotosíntesis artificial, que involucra la ruptura de la molécula de agua en oxígeno e hidrógeno,
puede ser usada como una fuente limpia de energía para muchas aplicaciones, especialmente
debido a que la principal fuente de energía es el sol. La oxidación de agua en oxígeno molecular
mediada por complejos de metales de transición invoca la necesidad de sintetizar y estudiar
nuevos catalizadores, teniendo como meta la obtención de actividades catalíticas que se
acerquen a las exhibidas por el complejo liberador de oxígeno en el Fotosistema II. En este
proyecto se sintetizó un nuevo ligando tridentado basado en pirrol y sus complejos con Cobre(II)
y Níquel(II).
El ligando obtenido, metil 5-(metoxidi(2-piridinil)metil)-1H-pirrol-2-carboxilato, fue sintetizado
con un rendimiento moderado (38%) mediante el uso de reactivos de organolitio. Las diferencias
en la reactividad de los diésteres de piridina y pirrol fueron exploradas, debido a que el ligando
pentadentado diseñado, (1H-pirrol-2,5-diil)bis(di(2-piridil)metanol), no pudo ser obtenido. De
igual forma, se pudieron observar algunas diferencias estructurales en los productos cuando se
siguieron esquemas sintéticos similares para ambos sustratos.

5
TABLE OF CONTENTS
New pyrrole-based ligand for first row transition metal complexes ............................................ 1
LIST OF ABBREVIATIONS .................................................................................................. 3
ABSTRACT ............................................................................................................................... 4
RESUMEN ................................................................................................................................ 4
TABLE OF CONTENTS ......................................................................................................... 5
Introduction ..................................................................................................................... 7
The global energy problem .......................................................................................... 7
Natural and artificial photosynthesis ......................................................................... 7
Nature’s choice: Mn4Ca redox core in Photosystem II ................................................ 8
First attempts at metal-catalysed water oxidation..................................................... 9
Water oxidation with mononuclear Iron and Cobalt complexes ............................... 10
Previous catalysts based in pentadentate polypyridine ligands............................... 12
Synthetic routes for polypyridine ligands: considerations ........................................ 13
Proposed modifications to the ligand scaffold ........................................................... 14
Synthetic approach to the proposed ligand ............................................................... 15
Aim of the project .......................................................................................................... 16
Experimental .................................................................................................................. 16
Chemicals and solvents ............................................................................................. 16
Spectroscopic characterisation .................................................................................. 16
Synthesis of pyrrole-2,5-dicarboxylic acid diesters (dimethyl, diethyl) .................... 16
Synthesis of Di(n-butyl) pyrrole-2,5-dicarboxylate ................................................... 17
Synthesis of N-Boc-pyrrole (tert-butyl pyrrole-1-carboxylate) .................................. 17
Synthesis of 1-(tert-butyl) 2,5-dimethyl pyrrole-1,2,5-tricarboxylate ....................... 17
Synthesis of methyl 5-(methoxydi(pyridin-2-yl)methyl)-pyrrole-2-carboxylate
(PyPY2) ................................................................................................................................. 18
Synthesis of Nickel(II) and Copper(II) complexes with PyPY2 ................................ 19
Results and Discussion ................................................................................................ 19
Synthesis of pyrrole-2,5-dicarboxylic acid diesters (dimethyl, diethyl) .................... 19

6
Synthesis of Di(n-butyl) pyrrole-2,5-dicarboxylate ................................................... 20
Synthesis of N-Boc-pyrrole ........................................................................................ 21
Synthesis of 1-(tert-butyl) 2,5-dimethyl pyrrole-1,2,5-tricarboxylate ....................... 21
Synthesis of methyl 5-(methoxydi(pyridin-2-yl)methyl)-pyrrole-2-carboxylate
(PyPY2) ................................................................................................................................. 22
Attempted synthesis of Nickel(II) and Copper(II) complexes of the PyPY2 ligand . 23
Conclusions .................................................................................................................... 25
Acknowledgements ....................................................................................................... 26
References ...................................................................................................................... 27
Appendixes ..................................................................................................................... 31
1H NMR spectrum of N-Boc pyrrole .......................................................................... 32
1H, 13C and DEPT135 NMR spectra of 1-(tert-butyl) 2,5-dimethyl pyrrole-1,2,5-
tricarboxylate ....................................................................................................................... 33
1H, 13C, DEPT135 and DEPT90 NMR spectra of PyPY2 .......................................... 35
1H NMR spectrum of methyl 1H-pyrrole-2-carboxylate ........................................... 39

7
INTRODUCTION
The global energy problem
The current global energy problem, the need for alternative sources of energy and the fact that
traditional use of fossil fuels and other non-renewable energy sources have a negative impact
on the climate and the environment1-4, has promoted the development of new renewable and
cleaner energy sources. Among them, one of the most theoretically promising is the conversion
of solar energy into chemical energy through the light-driven splitting of water, also known as
artificial photosynthesis, which generates oxygen gas and hydrogen, a carbon-free fuel with the
highest energy output relative to molecular weight.
Artificial photosynthesis thus presents an environmentally friendly alternative to the use of
fossil fuels, and the development of new molecular catalysts for artificial photosynthesis is an
active field of research with possible many applications.
Natural and artificial photosynthesis
The natural photosynthetic process consists of two coupled processes: water oxidation and CO2
fixation via proton reduction, to form NADPH, which is then used to synthesize sugars and more
complex biomolecules.
The light-dependent reactions of photosynthesis occur mainly on two protein complexes:
Photosystem II (containing the oxygen-evolving complex) and Photosystem I (responsible for the
reduction of NADP+ to NADPH). This process is shown in Fig. 1:
+1.2
+0.8
+0.4
0.0
-0.4
-0.8
-1.2
2H O2
Mn Ca4
TyrZ
O + 4H2+ 4e−
ChlP680
ExcitedChl P680*
Photo
syste
m II Pheo
QAQB
PQFeS
Cyt.fPC
Cyt. b & b6H 6L
2H+
2H+ ChlP700
ExcitedChl P700*
Photo
syste
m I
A0A1
FX
F /FA B
FDFNR
NADPH
NADP+
LightEnergy
LightEnergy
Fig. 1: Light-dependent reactions of photosynthesis (Z-scheme): roles of Photosystems I and II in the different
processes. Orange arrows illustrate the electron transfer chains, formed by various cofactors.
Adapted from ref. 5 with permission.

8
The oxidation of water is catalysed in Photosystem II by a redox-active structure that contains
a tetramanganese-calcium cluster (Mn4Ca), bound with a Tyrosine residue (named TyrZ); this
oxygen-evolving complex binds two water molecules and stores the four oxidizing equivalents
that are required to drive the oxidation of water. In the light-independent reactions, CO2 is
reduced by NADPH to ultimately yield sucrose, starch and cellulose after a series of reactions
(known as the Calvin-Benson cycle). These sugars are the main compounds responsible for
natural solar energy storage5. Both water oxidation and proton reduction (H+ to H2(g)) can be
achieved by means of artificial catalysts (for a very complete and recent review about WOCs see
ref. 6). These reactions can be carried out both by using a chemical redox agent or
photochemically, by using a photosensitizer, which acts as a redox mediator by photon induced
electron transfer processes.
In a process mimicking natural photosynthesis, the use of solar energy to extract electrons and
release protons from water is called photosynthetic water oxidation or oxygen evolution, and
development of new catalysts for this half reaction is of great interest for the field, since water
oxidation is a complicated reaction involving proton-coupled electron transfer (PCET) of four
electrons, yielding four protons and two oxygen molecules from two water molecules (Eq. 1).
2H2O ⟶ O2 + 4H+ + 4e− (𝐸0 = −1.23 𝑉 vs. SHE) Eq. 1
This reaction is thermodynamically uphill and pH dependant (since protons are being
produced), and to be carried out separately from proton reduction or other catalytic reduction
systems, a sacrificial electron acceptor is required: typically peroxydisulfate anion (S2O82−) is
used for this purpose.
Nature’s choice: Mn4Ca redox core in Photosystem II
The natural catalyst, a Mn4Ca reactive centre inside Photosystem II is shown in Fig. 2:
Fig. 2: Structure of the Mn4Ca reactive centre in Photosystem II. Adapted from ref. 7 with permission.

9
The structure of this reactive centre was determined by several X-ray diffraction and absorption
spectroscopic techniques7-11. This Mn4Ca core has been the base for many artificial catalysts,
aimed to imitate, model and understand the structure-activity relationship and the role of the
different components in the high catalytic activity of the natural catalyst, with the ultimate goal
of reproducing this catalytic activity in a synthetic complex that can be later inserted into a
water-splitting device.
Very recently, a synthetic catalyst mimicking the oxygen-evolving centre of photosynthesis was
reported by the group of Zhang et al.12, 13. This synthetic analogue consists of a Mn4Ca core with
ligands derived from carboxylic acids and has a very close structural resemblance to the natural
one. However, its catalytic activity is much lower than the natural catalyst, which is a matter
that is still under investigation. The structure of this artificial catalyst is shown in Fig. 3:
Fig. 3: Structures of the natural and artificial (synthetic) catalysts. See caption in figure.
Reprinted from ref. 13 with permission.
The natural turnover number (TON) and turnover frequency (TOF) of the PSII catalyst have
been estimated to be around 600.000 mol O2 (mol cat)−1 and 40 s−1, respectively14, 15.
First attempts at metal-catalysed water oxidation
The first successful attempts at metal-catalysed water oxidation date back to a publication from
1982, on which Gersten et al.16 reported the catalytic oxidation of water by an oxo-bridged
ruthenium dimer, cis,cis-[(bpy)2(OH2)RuIII-O-RuIII(OH2)(bpy)2]4+, later known in the field as the
“blue dimer”, which was the only reported molecular catalyst for water oxidation for more than
a decade.

10
Many other catalysts based on polynuclear manganese and ruthenium systems have been
reported, and it was in 2012 when Duan et al. reported a pair of molecular ruthenium catalysts
with water-oxidation activities comparable to that of the natural Photosystem II17. Several other
complexes have been reported in the last decade, with the notable examples of new cobalt and
iridium-based systems18. The structures of these catalysts are summarized in Fig. 4, which
illustrates the chronological evolution of representative water oxidation catalysts among the
past decades.
Fig. 4: Chronological listing of representative WOCs reported over the past three decades.
Reprinted from ref. 18 with permission.
Water oxidation with mononuclear Iron and Cobalt complexes
Despite the progress made in the synthesis of highly active polynuclear metal complexes for
water oxidation catalysis, the fact that ruthenium is a fairly expensive and non-abundant metal
limits the future application of these catalysts as a possible solution to the global energy
problem. The use of earth-abundant metals like iron, cobalt and manganese for water oxidation
catalysis has been an active field of investigation, on which fewer catalytic systems have been
reported so far. Apart from the use of earth-abundant metals, there are two other important
aspects to consider: the first downside aspect of polynuclear metal complexes that makes them
unattractive for a big scale application is atom economy: several metallic sites involve a higher
cost in production and waste disposal. The second downside aspect of polynuclear metal
complexes is that the fine tuning of the spectroscopic and chemical properties is usually harder
to achieve than for mononuclear catalysts, on which direct substitutions and changes in the
ligand environment make it easier to establish key structure-activity relationships and to study
mechanistic aspects of the catalysis: studies are greatly simplified if only one active metal site
is involved.

11
Ruthenium catalysts constitute the most studied family of catalysts for water oxidation, with
several mechanisms proposed to explain the observed behaviour.
Two of them, namely the water nucleophilic attack (WNA) mechanism and the radical coupling
(RC) mechanism, constitute the most widely accepted and supported according to experimental
evidence.
The water nucleophilic attack mechanism postulates that ligands with at least one labile
coordination site allow for the formation of a high valent [RuV=O]2+ species through several
PCET steps after initial coordination of a water molecule. This high valent species is then
attacked by the lone pair of the oxygen of another water molecule, forming a short lived
[RuIII-OOH]2+ intermediate, which loses one proton and one electron to form a [RuIV-OO]2+
intermediate, which then releases gaseous oxygen after electronic rearrangement and the
catalytic cycle is again started by coordination of a new water molecule. Key aspects of the water
nucleophilic attack mechanism have also been identified in iron, iridium and cobalt complexes14,
18, 19, as shown in Fig. 5.
Fig. 5: Summary of WNA-type mechanisms for single site catalysts based on ruthenium, iridium and cobalt.
Adapted from ref. 18 with permission.

12
The radical coupling mechanism (Fig. 6) postulates the formation via PCET of a high valent
oxyl radical, [RuIV-O•]2+, which is a resonance form of the [RuV=O]2+ species identified in the
WNA mechanism. This oxyl radical couples with another unit to yield a [RuIV-(O2)-RuIV] complex
that then releases dioxygen. The particular bonding mode of the oxygen and the last mechanistic
steps will depend on particular ligand geometry and environment, but the rate law has been
found to be second order with respect to the concentration of the catalyst, which strongly
supports the bimolecular nature of the mechanism.
Fig. 6: Radical coupling mechanism for single site catalysts based on ruthenium.
Adapted from ref. 18 with permission.
Previous catalysts based in pentadentate polypyridine ligands
Of the published systems, the one studied by Goldsmith et al.20, 21 and Wasylenko et al.22 was
chosen as a basis for this project. The original ligand framework consists of a five-coordinated
pentapyridine system: a central 2,6-disubstituted pyridine ring with four 2-pyridyl arms and
hydroxyl-terminated carbon linkers. Fig. 7 shows the crystal structure of PY5-OH,
pyridine-2,6-diylbis(di(pyridin-2-yl)methanol), closely related to the original PY5 ligand (with
MeOH groups). The crystal geometry does not differ significantly between both ligands.
Fig. 7: Crystal structure of PY5-OH ligand. Colour code: O (red), N (purple), C (grey), H (white).
Hydrogen bonds in light blue. X-Ray single-crystal diffraction data obtained from ref. 23 with permission.

13
When forming complexes with first row transition metals, the four pyridine arms bind to the
metal core with the four pyridine nitrogen atoms forming a plane, while the central metal-
pyridine bond lies perpendicular to this plane, leaving the sixth coordination position free for
the coordination of labile ligands (such as MeCN, Cl− and H2O). Furthermore, the metal-nitrogen
bond trans to this coordination site is greatly stabilized by means of the chelate effect22, making
the complex more stable and further improving the catalytic activity. The geometry is clearly
illustrated in the crystal structure of the complex [FeII(PY5-OH)Cl](CF3SO3) (Fig. 8):
Fig. 8: Two views of the crystal structure of [FeII(PY5-OH)Cl](CF3SO3). Colour code: Fe (orange), Cl (green), O (red),
N (purple), C (grey). Hydrogens and counter ions omitted for clarity, except for OH groups.
X-Ray single-crystal diffraction data obtained from ref. 24 with permission.
Synthetic routes for polypyridine ligands: considerations
Two main synthetic routes for PY5-type ligands have been proposed in the literature20-23, 25. Both
involve the addition of 2-lithiopyridine to carbonyl-containing pyridine moieties (i.e. dipicolinic
acid) in two to four steps. The advantages and disadvantages of each method are discussed
below.
1.7.1. Synthesis of pentapyridine ligands from 1,5-dipicolinic acid
The original synthesis of the PY5 ligand25 (Scheme 1) comprises the formation of the diacid
chloride derivative by reaction with SOCl2, subsequent reaction at low temperature with four
equivalents of 2-lithiopyridine (generated in-situ by lithium-halogen exchange at low
temperature) and further permethylation with methyl iodide. This synthetic route has been
reported to lead to several purification problems19, 20 and average overall yields of less than 50%.
Scheme 1: Original synthesis of PY5 ligand from dipicolinic acid

14
1.7.2. Synthesis of pentapyridine ligands from 1,5-dipicolinic acid diesters
A second synthetic route to the PY5 ligand (Scheme 2) was employed recently in the
literature20, 21 to improve the yield, facilitate the separation of the desired product and broaden
the scope of modifications to this ligand scaffold. In this synthetic route, dipicolinic acid alkyl
diesters are directly reacted with two equivalents of 2-lithiopyridine, from which a crystalline
solid intermediate diketone (PY3, see Scheme 2) is easily isolated by recrystallization in very
high yield (85%). This intermediate is further reacted with two more equivalents of 2-
lithiopyridine to yield the PY5-OH precursor, which is then permethylated to yield the PY5
ligand in greater overall yield (64%) while totally avoiding the need for purification via column
chromatography.
Scheme 2: Synthesis of PY5 ligand via PY3 diketone intermediate
The formation of the PY3 intermediate opens the ligand system for possible modifications, which
might consist, for example, on the introduction of two different “arms” for the ligand, such that
it would have two pyridine arms and two different arms (which might also be substituted
pyridines or other rings). The colour coding in Scheme 1 and Scheme 2 illustrates this
additional difference in the synthetic routes.
Proposed modifications to the ligand scaffold
The PY5 and PY5-OH ligands do not differ significantly in their geometry and metal binding
ability, and first row transition metal complexes from these ligands have been shown to be very
active as catalysts for water oxidation22, oxidation of unsaturated compounds20, 21, proton
reduction26 and even as cytotoxic anti-tumour agents24.
It has been shown22, 26 that the metal-ligand bond trans to the sixth coordination site (occupied
by water in the catalytic cycle) strongly affects the performance of the catalyst. If it is too weak,
then the catalyst might decompose into nanoparticles, while if it is too strong then the high-
valent oxo intermediate might not be electrophilic enough to promote the attack of water, or
might interfere with radical coupling. The trans influence can be accounted as the second major
source and explanation for this change in stability and reactivity.

15
The proposed modification to the ligand system consists in the replacement of the central
pyridine ring from PY5-OH for a five membered pyrrole ring to obtain the ligand shown in Fig.
9, (1H-pyrrole-2,5-diyl)bis(di(pyridin-2-yl)methanol) (PyPY4).
Fig. 9: Structure of the proposed ligand, (1H-pyrrole-2,5-diyl)bis(di(pyridin-2-yl)methanol)
This ligand is expected to introduce both a steric and electronic modification with respect to the
PY5 ligand. The NH group in pyrrole is to be deprotonated upon complexation, to form a N−(N)4
pentacoordinate ligand. The negative charge in the nitrogen from the pyrrole ring will then be
situated trans to the expected coordination site for water. Since the M+-N− has a more polar
character it is also expected to be stronger than the regular N→M dative bond, and is thus
expected to improve the stability of the complexes and increase the catalytic activity.
Synthetic approach to the proposed ligand
The two routes reported in literature for the preparation of PY5-OH and PY5 are, in principle,
viable. However, it has been reported27-31 that pyrrole-2-carboxylic acid readily undergoes
decarboxylation and is fairly unstable even under mildly acidic conditions, and therefore the
pyrrole-2,5-dicarboxylic acid and the diacid chloride derivative are also expected to be fairly
unstable. Furthermore, unprotected pyrrole is a substrate prone to polymerisation, which
suggests avoiding the diacid chloride route and the use of a protecting group for NH to prevent
polymerisation of the intermediate diketone and/or the starting pyrrole 2,5-diester. Boc was
chosen as the protecting group due to its easy introduction and removal32-35. Synthesis without
this protecting group was also attempted as the first choice. The proposed synthetic route for
the PyPY4 ligand is then a modification of the one reported for the PY5-OH ligand starting from
pyrrole-2,5-diester (Scheme 3).
Scheme 3: Proposed synthetic route for the new PyPY4 ligand

16
AIM OF THE PROJECT
The aim of this project was to synthesize a new ligand (PyPY4, (1H-pyrrole-2,5-
diyl)bis(di(pyridin-2-yl)methanol)), which would then be reacted with first row transition metal
salts to form new complexes that could show potential catalytic activity for water oxidation.
EXPERIMENTAL
Chemicals and solvents
Chemicals and solvents were obtained from commercial sources (Sigma-Aldrich) and were used
without further purification unless otherwise stated. Pyrrole was freshly distilled under vacuum
before use. Dry methanol, ethanol and CCl4 were obtained after storing the corresponding
solvent overnight under 15 wt.% freshly activated molecular sieves (4 Å). Dry THF and diethyl
ether were obtained by distillation under inert atmosphere from sodium/benzophenone still. All
glassware for water-sensitive reactions was stored overnight in the oven at 120 ºC.
Spectroscopic characterisation
NMR spectra were acquired on JEOL Eclipse+ ECP400 (1H 400 MHz, 13C 100 MHz) or Agilent
Mercury 400 (1H 400 MHz, 13C 100 MHz) spectrometers. All spectra were processed with
appropriate referencing to the solvent residual signal. For the measuring of small coupling
constants, where needed, the spectra were multiplied by the appropriate weighting functions
(exponential and gaussian) to resolve the peaks. DEPT135 and DEPT90 spectra (DEPT135
shows protonated CH/CH3 and CH2 carbons with opposite phases, and DEPT90 shows CH carbons
only, allowing for differentiation) were acquired for some of the compounds and used for the
assignment of the 13C resonances. LC-MS analysis were performed on a Dionex UltiMate 3000
LC system coupled to a Thermo Finnigan LCQ Deca XP Max ion trap mass spectrometer.
Synthesis of pyrrole-2,5-dicarboxylic acid diesters (dimethyl, diethyl)
In a dry microwave vial with a magnetic stir bar were added under argon Fe(acac)3 (3.2 mg, 0.01
mmol), pyrrole (60 mg, 0.9 mmol), dry CCl4 (1 g, 7 mmol) and dry MeOH (2 g, 60 mmol) or dry
EtOH (2.8 g, 60 mmol), which was then sealed hermetically with exclusion of oxygen by repeated
freeze-pump-thaw cycles, and reacted in a Biotage Initiator microwave reactor under different
reaction conditions, but the reaction yielded a black mixture of polypyrrole and less than 5% of
the desired product, among with an important (11%) fraction of the monoester.

17
Isolation of the reaction material from entry 9 in Table 1 by column chromatography (Silica;
0.01% Et3N in dichloromethane) yielded the monoester, 2-methyl-pyrrole-2-carboxylate (62 mg,
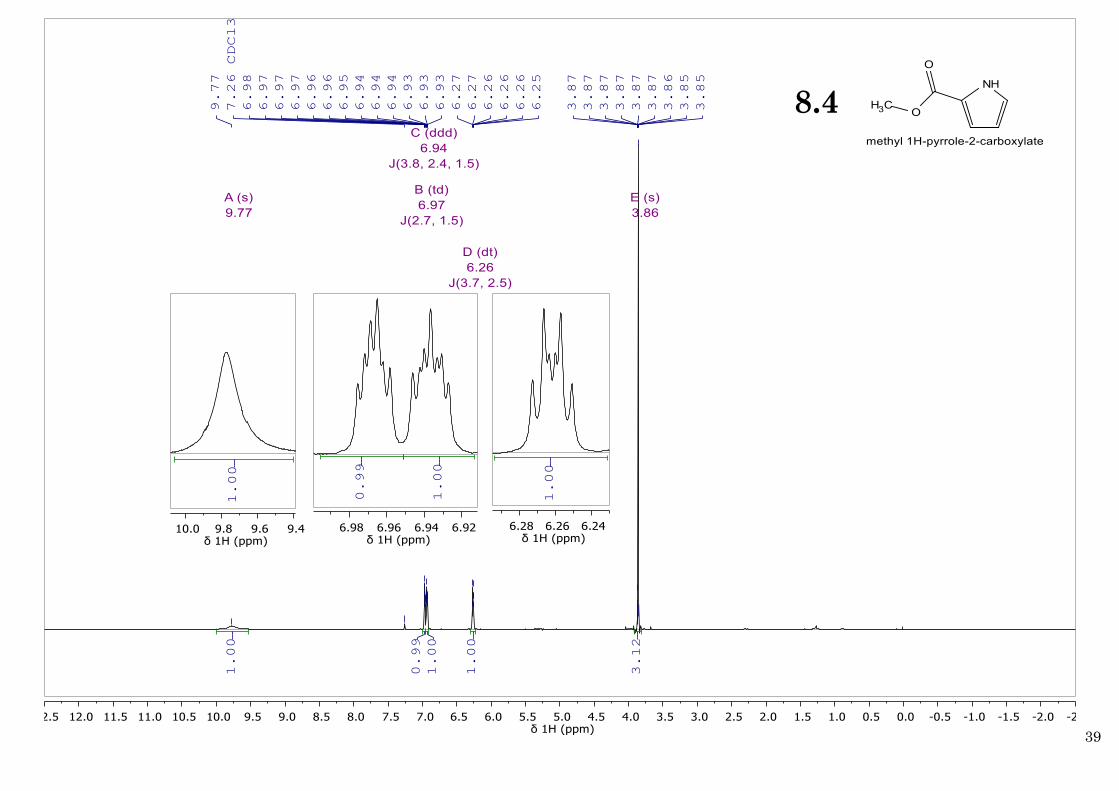
11%) as an off-white solid which readily decomposes upon storing for >1 week at RT. 1H NMR
(400 MHz, CDCl3, 25ºC) δ 9.77 (br. s, 1H; NH), 6.97 (td, J = 2.7, 1.5 Hz, 1H; H3 pyrrole), 6.94
(ddd, J = 3.7, 2.4, 1.5 Hz, 1H; H5 pyrrole), 6.26 (dt, J = 3.7, 2.5 Hz, 1H; H4 pyrrole), 3.86 (s, 3H;
OCH3). These results are in agreement with previously reported data for this compound36.
The reaction was also attempted with the same molar amount of CBr4 and some other catalysts,
and even without the use of microwave irradiation, yielding the same results (see discussion
below). This procedure was adapted from the reported by Khusnutdinov et al.37, where the
reaction yield is reported to be 99%.
Synthesis of Di(n-butyl) pyrrole-2,5-dicarboxylate
In an attempt to understand the reactivity of the system and get some insights into the
reactivity of pyrrole under the previous conditions, CBr4 (2.32 g, 7 mmol), pyrrole (60 mg, 0.9
mmol) and n-butanol (4.5 g, 60 mmol) were refluxed for 5 hours under inert atmosphere and
monitored by TLC and LC-MS, but only trace amounts of the expected di(n-butyl) pyrrole-2,5-
dicarboxylate were detected, while the reaction resulted in a black tar of polymeric material (see
discussion below).
Synthesis of N-Boc-pyrrole (tert-butyl pyrrole-1-carboxylate)
N-Boc pyrrole was synthesized as reported38. To a round bottomed flask containing pyrrole (2.0
g, 30 mmol) and 4-(dimethylamino)pyridine, DMAP, (500 mg, 4.5 mmol) in acetonitrile (30 mL)
was added di-tert-butyl dicarbonate, Boc2O, (7.8 g, 36 mmol) and the mixture stirred under argon
at room temperature for 2.5 hours. Evaporation of the solvent under reduced pressure and
purification by flash column chromatography (Al2O3; pentane) afforded 4.83 g (96%) of N-Boc-
pyrrole as a colourless liquid.
1H NMR (400 MHz, CDCl3, 25 ºC) δ 7.25 (t, J = 2.4 Hz, 2H; H2, H5), 6.22 (t, J = 2.4 Hz, 2H; H3,
H4), 1.61 (s, 9H; O-C(CH3)3).
Synthesis of 1-(tert-butyl) 2,5-dimethyl pyrrole-1,2,5-tricarboxylate
1-(tert-butyl) 2,5-dimethyl pyrrole-1,2,5-tricarboxylate was obtained from a modification of the
reported synthetic procedure39. In a 250 mL round-bottomed flask a 2.5 M solution of n-
butyllithium in hexanes (14.22 ml, 35.6 mmol) was slowly added to a solution of 2,2,6,6-
tetramethylpiperidine (6 ml, 35.6 mmol) in dry THF (50 mL) cooled to -78 °C under argon, to
give a light yellow solution, to which N-Boc-pyrrole (2.50 g, 15 mmol) in THF (10 ml) was added

18
dropwise and was left to stir for 3 hours at -78°C. The mixture was then transferred into a
dropping funnel and added dropwise into methyl chloroformate (3.5 ml, 45 mmol) and left to stir
at -78 ºC for an additional 30 minutes.
A saturated solution of ammonium chloride (10 mL) was then added and the mixture was
allowed to warm to room temperature. The mixture was then extracted with diethyl ether (25
mL) before being washed with 10% HCl solution (5 mL) and saturated sodium chloride solution
(20 mL).
The organic extracts were dried with MgSO4, filtered and concentrated under reduced pressure
to yield an orange solid, which was purified by flash column chromatography (Silica; 1:1
dichloromethane:pentane) and dried under vacuum overnight to yield pure
1-(tert-butyl) 2,5-dimethyl pyrrole-1,2,5-tricarboxylate as a white solid (3.56 g, 84%).
1H NMR (400 MHz, CDCl3, 25 ºC) δ 6.83 (s, 2H; H3, H4), 3.86 (s, 6H; 2xO-CH3), 1.66 (s, 9H; O-
C(CH3)3); 13C NMR (100 MHz, CDCl3) δ 160.02 (C=O(OMe)), 148.97 (C=O-O(t-Bu)), 126.82 (C2,
C5), 115.91 (C3, C4), 86.38 (O-C(CH3)3), 52.09 (O-CH3), 27.43 (t-Bu CH3).
Synthesis of methyl 5-(methoxydi(pyridin-2-yl)methyl)-pyrrole-2-
carboxylate (PyPY2)
To a 250 mL three-necked round-bottomed flask was added 2-bromopyridine (1.22 g, 7.8 mmol,
6 eq.) in dry THF (75 mL) to give a colourless solution, which was cooled in a methanol/liquid
nitrogen bath at -98 °C under argon. A 2.5 M solution of n-butyllithium in hexanes (3.10 mL,
7.8 mmol, 6 eq.) was added dropwise, keeping the reaction temperature below -80 ºC. A solution
of 1-(tert-butyl) 2,5-dimethyl pyrrole-1,2,5-tricarboxylate (365.4 mg, 1.3 mmol) in dry THF (20
mL) was added dropwise, keeping the temperature below -80 ºC. The reaction mixture was
stirred at -98 °C for 2 hours, after which the temperature was raised to RT and the reaction was
continuously stirred for 10 min. The reaction was quenched with MeOH (25 mL) and 5% aqueous
HCl (5 mL) was added, then the solution was basified with a saturated sodium hydroxide
solution (3 drops). The organic solvents were removed and the residue was extracted with
dichloromethane (5x10 mL), washed with brine and dried over Na2SO4. The organic extracts
were combined and dried under reduced pressure. An intensely colored deep red oil was then
purified by column chromatography (Al2O3; 0.1% Et3N in dichloromethane) to yield methyl 5-
(methoxydi(pyridin-2-yl)methyl)-pyrrole-2-carboxylate (PyPY2) as a deep red solid (243 mg,
38%). Further reaction of PyPY2 with two equivalents of 2-lithiopyridine did not afford the
desired PyPY4 product. See discussion below.

19
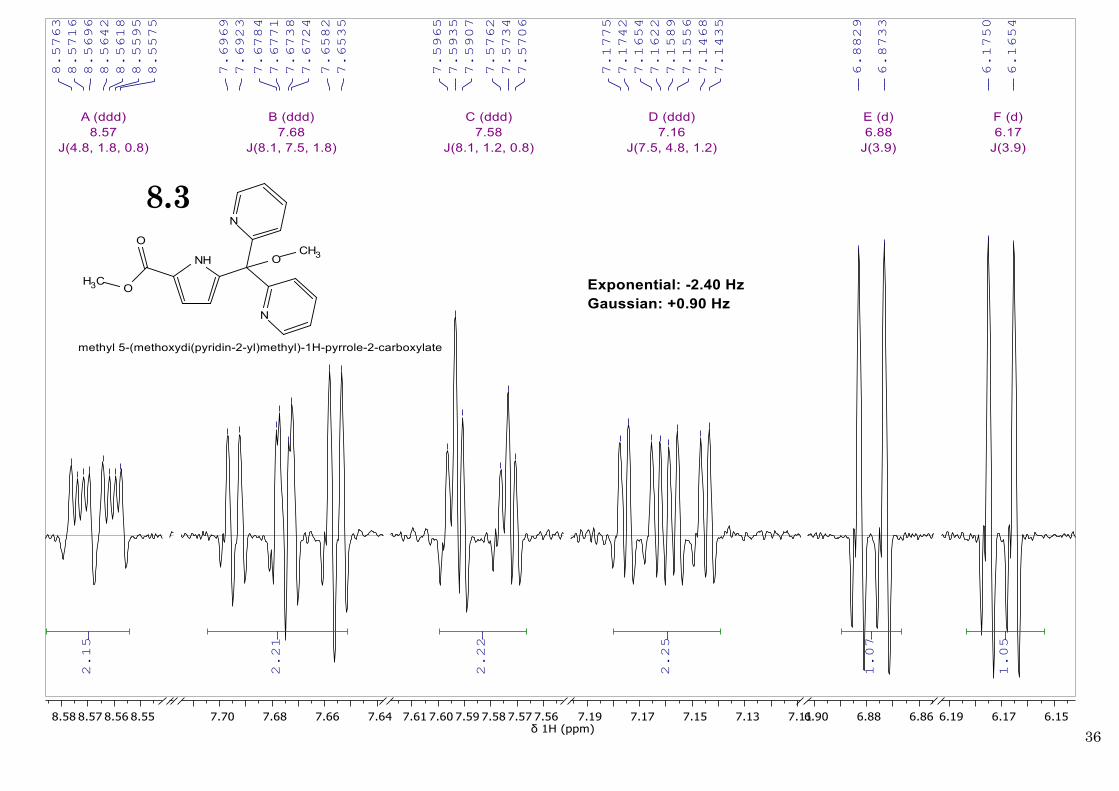
MS (ESI, m/z) 323.6 [M+] (Calc. for C18H17N3O3 323.1). 1H NMR (400 MHz, CDCl3, 25 ºC) δ 8.57
(ddd, J = 4.8, 1.8, 0.8 Hz, 2H; pyridine arms H6,H6’), 7.68 (ddd, J = 8.1, 7.5, 1.8 Hz, 2H; pyridine
arms H3,H3’), 7.58 (ddd, J = 8.1, 1.2, 0.8 Hz, 2H; pyridine arms H4,H4’), 7.16 (ddd, J = 7.5, 4.8,
1.2 Hz, 2H; pyridine arms H5,H5’), 6.88 (d, J = 3.9 Hz, 2H; pyrrole H3), 6.17 (d, J = 3.9 Hz, 2H;
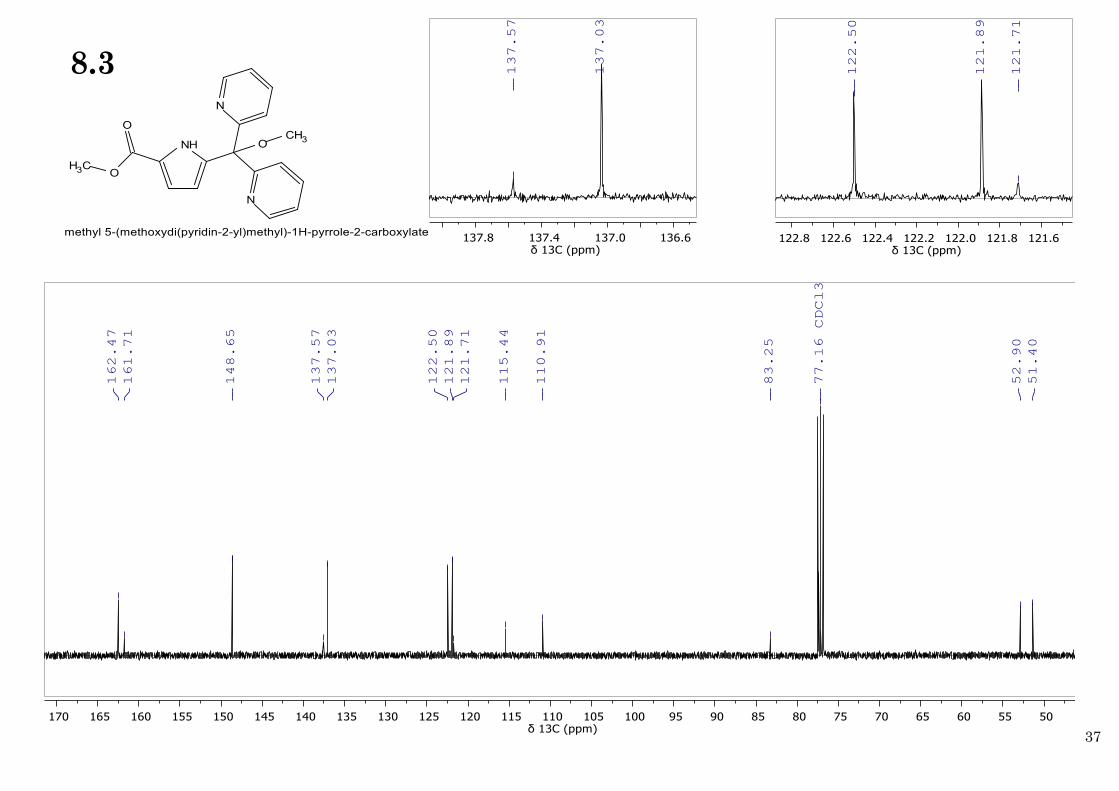
pyrrole H4), 3.85 (s, 3H; OCH3 ester), 3.19 (s, 3H; OCH3). 13C NMR (100 MHz, CDCl3, 25 ºC) δ
162.47 (C=O ester), 161.71 (C2,C2’ pyridine arms), 148.65 (C6,C6’ pyridine arms), 137.57 (C5
pyrrole), 137.03 (C4,C4’ pyridine arms), 122.50 (C5,C5’ pyridine arms), 121.89 (C3,C3’ pyridine
arms), 121.71 (C2 pyrrole), 115.44 (C3 pyrrole), 110.91 (C4 pyrrole), 83.25 (C(OCH3)(PY)2),
52.90 (OCH3 ester), 51.40 (OCH3).
Synthesis of Nickel(II) and Copper(II) complexes with PyPY2
To a solution of PyPY2 (60.0 mg for Cu(II); 16.3 mg for Ni(II)) in methanol (0.5 mL) was added
under argon a solution of the corresponding metal bromide (CuBr2: 41.3 mg, 1 eq.; NiBr2 · 𝑥H2O:
13.4 mg, ~1 eq) in 0.5 mL MeOH. The reaction mixtures were stirred at room temperature for 4
hours after which LC-MS analysis were performed from the crude solutions, which turned deep
red for Ni(II) and dark green for Cu(II). No attempt was made to isolate the products. See
discussion below.
RESULTS AND DISCUSSION
Synthesis of pyrrole-2,5-dicarboxylic acid diesters (dimethyl, diethyl)
The attempted synthesis of pyrrole-2,5-dicarboxylic acid dimethyl and diethyl esters (Scheme
4) was not successful, even though special care was taken in drying the solvents and reagents
used for the reaction. Furthermore, the microwave vials were sealed and all reagents were
handled under argon atmosphere. Table 1 summarized the different reaction conditions that
were explored in an attempt to reproduce the results by Khusnutdinov et al.37 by using
microwave irradiation instead of thermal activation.
Scheme 4: Synthesis of pyrrole-2,5-dicarboxylic acid diesters

20
Trial Temp.
(ºC) Time
Pressure
(bar)
mmol
pyrrole
mmol
MeOH
mmol
𝐂𝐂𝐥𝟒
mmol
cat. Catalyst
Yield of
diester
1 115 5 min 10 0.9 60 7 0 none N.R.
2 120 5 min 10 0.9 60 7 0.01 Fe(acac)3 trace
3 150 5 min 18 0.9 60 7 0.01 Fe(acac)3 polymer
4 60 5 min 5 0.9 60 7 0.01 Fe(acac)3 N.R.
5 120 30 min 12 0.9 60 7 0.01 Fe(acac)3 trace
6 150 30 min 18 0.9 60 7 0.01 Fe(acac)3 polymer
7 120 1 h 18 4.5 350 35 0.05 Fe(acac)3 polymer
8 150 1 h 21 4.5 350 35 0.05 Fe(acac)3 polymer
9 120 30 min 12 4.5 350 35 0.05 FeCl2 <5%
10 150 30 min 18 4.5 350 35 0.05 FeCl2 trace
Table 1: Tested reaction conditions and yields for the synthesis of pyrrole-2,5-dicarboxylic acid dimethyl ester
under microwave irradiation. N.R = no reaction; polymer = polypyrrole was the main product as a black tar;
trace = only trace amounts were detected (by LC-MS).
This reaction was also attempted with EtOH and CBr4 under the same conditions as entries
1-5 in Table 1, but the desired product was detected only in trace amounts by LC-MS.
It is interesting to note that the reaction yield improved slightly by using FeCl2 as the catalyst
instead of Fe(acac)3. This fact can be explained due to the proven catalytic activity of Iron(III)
salts for the synthesis of polypyrrole40-43, and thus the use of Iron(II) salts might lower the yield
for the undesired polypyrrole, but is still inadequate because Iron(II) can be oxidized in solution
to Iron(III) probably by trace amounts of oxygen, thus generating the catalytic species that
promotes the undesired polymerisation.
The reaction mechanism for the esterification of pyrrole with CCl4 or CBr4 is thought to proceed
via radical addition to form a pyrrole-CX3 (X = Cl, Br) intermediate, which then undergoes
alcoholysis to afford the monoester. This product undergoes yet another radical addition and
alcoholysis in the 5 position of the ring, thus affording the diester. The mechanism is discussed
in more detail by Khusnutdinov et al.37
From these experiments, it is important to note that the formation of polymeric material, the
instability of the monoester, the difficulty in successful separation of the desired product, added
to the fact that the major product was the monoester instead of the diester, hindered further
attempts to synthesize the NH unprotected dimethyl pyrrole-2,5-dicarboxylate, and thus this
synthetic route was abandoned.
Synthesis of Di(n-butyl) pyrrole-2,5-dicarboxylate
A reaction with CBr4, n-butanol and pyrrole refluxed under inert atmosphere was carried out to
test conditions where the reagents were kept below their boiling point and without the use of
microwave irradiation (Scheme 5).

21
Scheme 5: Synthesis of di(n-butyl) pyrrole-2,5-dicarboxylate
However, the formation of the expected di(n-butyl) pyrrole-2,5-dicarboxylate was achieved only
in trace quantities, while the reaction mixture turned deep black after 2.5 hours of refluxing,
indicating the presence of a big amount of polypyrrole. TLC and LC-MS analysis indicated the
formation of other side products besides polypyrrole, which constitute a final confirmation that
raw pyrrole is not a good substrate to start with in order to get 2,5-diesters.
Synthesis of N-Boc-pyrrole
In this reaction (Scheme 6), the excellent yield, stability and ease of purification of the product
contrast with the behaviour of deprotected pyrrole, thus suggesting that the protection of the
NH group of pyrrole was, in fact, needed for this synthetapproach to succeed. Boc2O is a cheap
reagent and the reaction conditions are straightforward. Note however that N-Boc-pyrrole is
also available commercially.
Scheme 6: Synthesis of N-Boc-pyrrole
Synthesis of 1-(tert-butyl) 2,5-dimethyl pyrrole-1,2,5-tricarboxylate
The modification of the reported synthesis introduced in this work consisted of using a dropping
funnel instead of a cannula to transfer the lithiated pyrrole intermediate into methyl
chloroformate. This reaction is a model reaction for the attack of N-Boc 2,5-dilithiopyrrole to an
electrophilic substrate, methyl chloroformate, according to Scheme 7.
Scheme 7: Synthesis of 1-(tert-butyl) 2,5-dimethyl pyrrole-1,2,5-tricarboxylate
An important aspect of this reaction is that the N-Boc 2,5-dilithiopyrrole intermediate is
stabilised by the action of the Boc protecting group, which has an impact on the stability of this
organolithium reagent, as illustrated in Fig. 10.

22
Fig. 10: Stabilisation of 2,5-dilithiopyrrole by the N-Boc protecting group
Synthetic routes to the formation of N-Boc 2,5-dilithiopyrrole also include the lithium-halogen
exchange, but in this particular case, the reaction of n-BuLi with HTMP (2,2,6,6-
tetramethylpiperidine) to afford LiTMP (lithium 2,2,6,6-tetramethylpiperidide) and subsequent
reaction with N-Boc pyrrole enable for the synthesis of the 2,5-dimethyl ester. A final remark
from this reaction is that the Boc protecting group was not cleaved or attacked under these
reaction conditions.
Synthesis of methyl 5-(methoxydi(pyridin-2-yl)methyl)-pyrrole-2-
carboxylate (PyPY2)
Initial attempts to synthesize an intermediate analogous to PY3 (Scheme 2) by using only two
equivalents of 2-lithiopyridine in dry THF at -78 ºC were unsuccessful, and only the starting
triester was isolated from the reaction mixture.
The reaction was also attempted in dry Et2O at -78 ºC, but this solvent promotes decomposition
of the 2-lithiopyridine reagent, and for that particular case, no traces of the starting material
could be isolated, polymeric material being the only product. When four equivalents of 2-
lithiopyridine were used, the reaction yield of PyPY2 improved slightly to ~15%, but still the
desired PyPY4 product was not observed.
To improve the stability of the 2-lithiopyridine, very dilute solutions and much lower
temperature (-98 ºC) were tested, which improved the reaction yield and allowed the successful
synthesis of PyPY2, according to Scheme 8.
Scheme 8: Synthesis of PyPY2

23
A remarkable feature of this reaction is that the obtained product is an asymmetrically
substituted pyrrole, which contrasts with the symmetric pyridine diketone intermediate (PY3)
reported when pyridine 2,6-diesters are used as the starting materials. This suggests that
pyrrole 2,5-diesters have a very different reactivity trend towards addition of 2-lithiopyridine.
Also, methoxy groups were found in the final product instead of the expected OH groups.
Furthermore, when six equivalents of 2-lithiopyridine were used, PyPY2 was the only product
observed and the reaction yield improved significantly (38%). Only polymeric material was
observed when PyPY2 was reacted again with two equivalents of 2-lithiopyridine, which
confirms that six equivalents is a fair excess.
The 13C chemical shift in CDCl3 of the methoxy ester carbonyl in 1-(tert-butyl)-2,5-dimethyl
pyrrole-1,2,5-tricarboxylate is 160.08 ppm, while that of the pyridine analogue (dimethyl
dipicolinate) is 164.6 ppm44, thus suggesting that the two carbonyls do not differ significantly
in electrophilicity, while still it can be deduced that the carbonyl from the pyrrole system is
more shielded (and therefore less electrophilic) because the pyrrole ring is more electron-rich
than the pyridine ring. A final interesting aspect of this reaction is that the Boc protecting group
is removed from the pyrrole system during workup, as determined by LC-MS, even though the
NH resonance was not found in the 1H NMR spectrum due to the sample being too dilute (thus
making the signal-to-noise ratio for the broad NH resonance too low to be detected). We
therefore propose that the Boc protecting group is too bulky to allow attack of the 2-
lithiopyridine on the second carbonyl once the first pyridine has been introduced.
Attempted synthesis of Nickel(II) and Copper(II) complexes of the
PyPY2 ligand
The fact that PyPY2 is a new tridentate ligand itself prompted for an attempt to synthesize
complexes with first row transition metals. Among them Nickel(II) and Copper(II) were chosen
since they are known to form stable tetracoordinated complexes.
The attempted synthesis of a Copper(II) complex yielded a mixture of complexes, identified by
mass spectrometry, but whose structural characteristics such as the disposition of the ligands
around the metal centre need to be further investigated, and to improve the reaction yield the
mixture should be refluxed and a catalytic amount of a weak base added to promote
deprotonation of the pyrrole NH group. Fig. 11 illustrates the experimental and calculated mass
spectrum of a Cu(II) complex with candidate formula [Cu(PyPY2−)2], resulting from the
coordination of two ligand molecules to a single metal site. A candidate structure for this
complex is shown in Fig. 12, but further spectroscopic studies (XRD, NMR) are needed in order
to confirm the structures and oxidation states.

24
Fig. 11: Mass spectrum of the complex ion with formula [Cu(PyPY2−)2], C36H33CuN6O6.
Top: experimental mass spectrum; Bottom: calculated isotopic distribution for C36H34CuN6O6+, [M+H]+.
Fig. 12: Candidate structure for complex [Cu(PyPY2−)2] based on mass spectrometric results
Also, another mass pattern was recognized (Fig. 13), corresponding to an oxo bridged Cu(III)
dimer, [Cu(PyPY2−)(μ-O)2(PyPY2−)Cu]. The candidate structure is shown in Fig. 14.
Fig. 13: Mass spectrum of the complex ion with formula [Cu(PyPY2−)(μ-O)2(PyPY2−)Cu], C36H32Cu2N6O8.
Top: experimental mass spectrum; Bottom: calculated isotopic distribution for C36H33Cu2N6O8+, [M+H]+.
700 705 710 715 720
m/z
0
20
40
60
80
100
0
20
40
60
80
100
Re
lative
Ab
un
da
nce
708.99
710.94
712.02
712.93704.25700.85 715.30 717.61706.59
709.18
711.18
712.18
713.19 715.19 718.20
NL:1.75E8
RF24A-Cu-after4h#211 RT: 5.64 AV: 1 T: + c ESI Full ms [80.00-2000.00]
NL:4.51E5
C 36 H 34 CuN 6 O 6: C 36 H 34 Cu 1 N 6 O 6
pa Chrg 1
790 795 800 805 810
m/z
0
20
40
60
80
100
0
20
40
60
80
100
Rela
tive A
bundance
802.92
804.98
806.01
808.19
796.24 801.92 813.70811.53799.62787.97 790.48
803.09805.09
806.10
807.09
808.09810.10 813.11
NL:3.80E7
RF24A-Cu-after4h#255 RT: 6.81 AV: 1 T: + c ESI Full ms [80.00-2000.00]
NL:3.10E5
C 36 H 33 Cu 2 N 6 O 8+: C 36 H 33 Cu 2 N 6 O 8
pa Chrg 1

25
Fig. 14: Candidate structure for complex [Cu(PyPY2−)(μ-O)2(PyPY2−)Cu]+ based on mass spectrometric results
The high valent Cu(III) structure is suggested from oxo bridges, which are favoured for higher
oxidation states.
The attempted synthesis of a Nickel complex yielded new species in the mass chromatogram,
but no recognizable mass patterns were detected. As for the case of Copper, the reaction mixture
should be refluxed and a catalytic amount of a weak base added to promote deprotonation of the
pyrrole NH group, and after obtaining the corresponding complexes, further spectral
characterisation is needed.
These complexes were not tested for water oxidation, but Ni(II) and Cu(II) complexes have also
been shown very recently to possess catalytic activity towards water oxidation.
CONCLUSIONS
In conclusion, the synthesis of a new pyrrole-based ligand, methyl 5-(methoxydi(pyridin-2-
yl)methyl)-pyrrole-2-carboxylate (PyPY2), was achieved under moderate yield (38%) by addition
of six equivalents of 2-lithiopyridine at -98ºC in THF to 1-(tert-butyl) 2,5-dimethyl pyrrole-1,2,5-
tricarboxylate. Deprotection of the pyrrole ring was observed during workup, thus making a
deprotection step unnecessary. The ligand was properly characterized by 1H and 13C NMR
spectroscopy and mass spectrometry. Attempted synthesis of pyrrole 2,5-dicarboxylic acid
diesters from reported methods were not successful under the tested conditions, and thus the
Boc protecting group was shown to be necessary for the success of the reaction. Attempted
synthesis of the original proposed ligand, (1H-pyrrole-2,5-diyl)bis(di(pyridin-2-yl)methanol)),
was not successful. The obtained PyPY2 compound could instead be used as a tridentate ligand,
tentatively forming coordination compounds with Ni(II) and Cu(II), which were identified by
LC-MS in the case of Cu(II). Further characterization and analysis of the ligand and its metal
complexes are a work still on progress, and in the future these complexes should be tested for
their catalytic activity towards water oxidation.

26
ACKNOWLEDGEMENTS
I would like to thank Dr. Anders Thapper and Dr. Sascha Ott for accepting me in their research
group, thus allowing me to carry out this project and enabling me to learn more about transition
metal chemistry and water oxidation catalysis in the labs. I would also like to thank Dr.
Biswanath Das for his great help in developing this project, ranging from the introduction to
the PY5-OH system to key discussions, help and encouragement throughout the whole project.
Many thanks have to be given as well to all the administrative and support personnel in House
7 at Ångström laboratory.
I wish to thank my family, especially my mother, whose unconditional support, even throughout
the distance, helped me to be strong even in the toughest moments.
To my best friend, Estefanía Sucre, and the rest of my friends from the Laboratory for
Photochemistry and Photobiology at Universidad Simón Bolívar in Venezuela: María Lourdes
Nóvoa, Carlos Gámez, Prof. Nieves Canudas and Prof. Sara Pekerar for their support and
encouragement, many thanks.
To my professors from the Department of Chemistry at Universidad Simón Bolívar in
Sartenejas, Venezuela, who formed me during four years and enabled me to lift my wings for
this dream which is now a reality. Thank you all.
Finally, I wish to thank the great academic composers: Sergei Rachmaninoff, Franz Liszt, Pyotr
Ilyich Tchaikovsky, Frédéric Chopin, Wolfgang Amadeus Mozart, Charles Camille Saint-Saëns
and many others, whose wonderful music was essential for the writing of this manuscript and
has accompanied me during almost my whole life.
“Success is not final, failure is not fatal: it is the courage to continue that counts.”
Winston Churchill

27
REFERENCES
1. Panwar, N.L., S.C. Kaushik, and S. Kothari, Role of renewable energy sources in
environmental protection: A review. Renewable and Sustainable Energy Reviews, 2011.
15(3): p. 1513-1524, DOI: 10.1016/j.rser.2010.11.037.
2. Pérez-Lombard, L., J. Ortiz, and C. Pout, A review on buildings energy consumption
information. Energy and Buildings, 2008. 40(3): p. 394-398, DOI:
10.1016/j.enbuild.2007.03.007.
3. Solomon, S., G.K. Plattner, R. Knutti, and P. Friedlingstein, Irreversible climate change
due to carbon dioxide emissions. Proc Natl Acad Sci U S A, 2009. 106(6): p. 1704-9, DOI:
10.1073/pnas.0812721106.
4. Kasting, J.F. and J.L. Siefert, Life and the evolution of Earth's atmosphere. Science, 2002.
296(5570): p. 1066-8, DOI: 10.1126/science.1071184.
5. Collings, A.F. and C. Critchley, Artificial photosynthesis: from basic biology to industrial
application. 2005, Weinheim: Wiley-VCH.
6. Kärkäs, M.D., O. Verho, E.V. Johnston, and B. Åkermark, Artificial photosynthesis:
molecular systems for catalytic water oxidation. Chem Rev, 2014. 114(24): p. 11863-2001,
DOI: 10.1021/cr400572f.
7. Najafpour, M.M., A.N. Moghaddam, S.I. Allakhverdiev, and Govindjee, Biological water
oxidation: lessons from nature. Biochim Biophys Acta, 2012. 1817(8): p. 1110-21, DOI:
10.1016/j.bbabio.2012.04.002.
8. Yano, J., J. Kern, K. Sauer, M.J. Latimer, Y. Pushkar, J. Biesiadka, B. Loll, W. Saenger,
J. Messinger, A. Zouni, and V.K. Yachandra, Where water is oxidized to dioxygen:
structure of the photosynthetic Mn4Ca cluster. Science, 2006. 314(5800): p. 821-5, DOI:
10.1126/science.1128186.
9. Ferreira, K.N., T.M. Iverson, K. Maghlaoui, J. Barber, and S. Iwata, Architecture of the
photosynthetic oxygen-evolving center. Science, 2004. 303(5665): p. 1831-8, DOI:
10.1126/science.1093087.
10. Yano, J. and V. Yachandra, Mn4Ca cluster in photosynthesis: where and how water is
oxidized to dioxygen. Chem Rev, 2014. 114(8): p. 4175-205, DOI: 10.1021/cr4004874.
11. Tran, R., J. Kern, J. Hattne, S. Koroidov, J. Hellmich, R. Alonso-Mori, N.K. Sauter, U.
Bergmann, J. Messinger, A. Zouni, J. Yano, and V.K. Yachandra, The Mn4Ca
photosynthetic water-oxidation catalyst studied by simultaneous X-ray spectroscopy and
crystallography using an X-ray free-electron laser. Philos Trans R Soc Lond B Biol Sci,
2014. 369(1647): p. 20130324, DOI: 10.1098/rstb.2013.0324.
12. Zhang, C., C. Chen, H. Dong, J.R. Shen, H. Dau, and J. Zhao, A synthetic Mn4Ca-cluster
mimicking the oxygen-evolving center of photosynthesis. Science, 2015. 348(6235): p. 690-
3, DOI: 10.1126/science.aaa6550.
13. Sun, L., A closer mimic of the oxygen evolution complex of photosystem II. Science, 2015.
348(6235): p. 635-6, DOI: 10.1126/science.aaa9094.

28
14. Limburg, B., E. Bouwman, and S. Bonnet, Molecular water oxidation catalysts based on
transition metals and their decomposition pathways. Coordination Chemistry Reviews,
2012. 256(15-16): p. 1451-1467, DOI: 10.1016/j.ccr.2012.02.021.
15. Cady, C.W., R.H. Crabtree, and G.W. Brudvig, Functional Models for the Oxygen-
Evolving Complex of Photosystem II. Coord Chem Rev, 2008. 252(3-4): p. 444-455, DOI:
10.1016/j.ccr.2007.06.002.
16. Gersten, S.W., G.J. Samuels, and T.J. Meyer, Catalytic oxidation of water by an oxo-
bridged ruthenium dimer. Journal of the American Chemical Society, 1982. 104(14): p.
4029-4030, DOI: 10.1021/ja00378a053.
17. Duan, L., F. Bozoglian, S. Mandal, B. Stewart, T. Privalov, A. Llobet, and L. Sun, A
molecular ruthenium catalyst with water-oxidation activity comparable to that of
photosystem II. Nat Chem, 2012. 4(5): p. 418-23, DOI: 10.1038/nchem.1301.
18. Wasylenko, D.J., R.D. Palmer, and C.P. Berlinguette, Homogeneous water oxidation
catalysts containing a single metal site. Chem Commun (Camb), 2013. 49(3): p. 218-27,
DOI: 10.1039/c2cc35632e.
19. Kärkäs, M.D., T. Åkermark, E.V. Johnston, S.R. Karim, T.M. Laine, B.L. Lee, T.
Åkermark, T. Privalov, and B. Åkermark, Water oxidation by single-site ruthenium
complexes: using ligands as redox and proton transfer mediators. Angew Chem Int Ed
Engl, 2012. 51(46): p. 11589-93, DOI: 10.1002/anie.201205018.
20. Goldsmith, C.R. and T.D. Stack, Hydrogen atom abstraction by a mononuclear ferric
hydroxide complex: insights into the reactivity of lipoxygenase. Inorg Chem, 2006. 45(15):
p. 6048-55, DOI: 10.1021/ic060621e.
21. Goldsmith, C.R., R.T. Jonas, and T.D.P. Stack, C−H Bond Activation by a Ferric
Methoxide Complex: Modeling the Rate-Determining Step in the Mechanism of
Lipoxygenase. Journal of the American Chemical Society, 2002. 124(1): p. 83-96, DOI:
10.1021/ja016451g.
22. Wasylenko, D.J., C. Ganesamoorthy, J. Borau-Garcia, and C.P. Berlinguette,
Electrochemical evidence for catalytic water oxidation mediated by a high-valent cobalt
complex. Chem Commun (Camb), 2011. 47(14): p. 4249-51, DOI: 10.1039/c0cc05522k.
23. Görlitz, J.J., P. Nielsen, H. Toftlund, and A.D. Bond, 2,6-Bis[bis(2-
pyridyl)hydroxymethyl]pyridine. Acta Crystallographica Section E Structure Reports
Online, 2004. 60(8): p. o1319-o1320, DOI: 10.1107/s1600536804016368.
24. Wong, E.L., G.S. Fang, C.M. Che, and N. Zhu, Highly cytotoxic iron(II) complexes with
pentadentate pyridyl ligands as a new class of anti-tumor agents. Chem Commun (Camb),
2005(36): p. 4578-80, DOI: 10.1039/b507687k.
25. Jonas, R.T. and T.D.P. Stack, C−H Bond Activation by a Ferric Methoxide Complex: A
Model for the Rate-Determining Step in the Mechanism of Lipoxygenase. Journal of the
American Chemical Society, 1997. 119(36): p. 8566-8567, DOI: 10.1021/ja971503g.
26. Bachmann, C., M. Guttentag, B. Spingler, and R. Alberto, 3d element complexes of
pentadentate bipyridine-pyridine-based ligand scaffolds: structures and photocatalytic
activities. Inorg Chem, 2013. 52(10): p. 6055-61, DOI: 10.1021/ic4004017.
27. Alvarez-Builla, J., J.J. Vaquero, and J. Barluenga, Modern heterocyclic chemistry. 2011,
Weinheim: Wiley-VCH.

29
28. Dunn, G.E. and G.K.J. Lee, Kinetics and Mechanism of the Decarboxylation of Pyrrole-2-
carboxylic Acid in Aqueous Solution. Canadian Journal of Chemistry, 1971. 49(7): p.
1032-1035, DOI: 10.1139/v71-172.
29. Liang, J., B. Wang, and Z. Cao, The Mechanism of Acid-Catalyzed Decarboxylation of
Pyrrole-2-Carboxylic Acid: Insights from Cluster-Continuum Model Calculations.
Journal of Theoretical and Computational Chemistry, 2013. 12(04): p. 1350017, DOI:
10.1142/s021963361350017x.
30. Cheng, X., J. Wang, K. Tang, Y. Liu, and C. Liu, Decarboxylation of pyrrole-2-carboxylic
acid: A DFT investigation. Chemical Physics Letters, 2010. 496(1-3): p. 36-41, DOI:
10.1016/j.cplett.2010.07.052.
31. Mundle, S.O. and R. Kluger, Decarboxylation via addition of water to a carboxyl group:
acid catalysis of pyrrole-2-carboxylic acid. J Am Chem Soc, 2009. 131(33): p. 11674-5,
DOI: 10.1021/ja905196n.
32. Wang, J., Y.L. Liang, and J. Qu, Boiling water-catalyzed neutral and selective N-Boc
deprotection. Chem Commun (Camb), 2009(34): p. 5144-6, DOI: 10.1039/b910239f.
33. Gibson, F.S., S.C. Bergmeier, and H. Rapoport, Selective Removal of an N-BOC
Protecting Group in the Presence of a tert-Butyl Ester and Other Acid-Sensitive Groups.
The Journal of Organic Chemistry, 1994. 59(11): p. 3216-3218, DOI:
10.1021/jo00090a045.
34. Han, G., M. Tamaki, and V.J. Hruby, Fast, efficient and selective deprotection of the tert-
butoxycarbonyl (Boc) group using HCl/dioxane (4 M). Journal of Peptide Research, 2001.
58(4): p. 338-341, DOI: 10.1034/j.1399-3011.2001.00935.x.
35. Ravinder, K., A.V. Reddy, K.C. Mahesh, M. Narasimhulu, and Y. Venkateswarlu, Simple
and Selective Removal of t-Butyloxycarbonyl (Boc) Protecting Group on Indoles, Pyrroles,
Indazoles, and Carbolines. Synthetic Communications, 2007. 37(2): p. 281-287, DOI:
10.1080/00397910601033724.
36. Schmuck, C., V. Bickert, M. Merschky, L. Geiger, D. Rupprecht, J. Dudaczek, P. Wich,
T. Rehm, and U. Machon, A Facile and Efficient Multi-Gram Synthesis of N-Protected 5-
(Guanidinocarbonyl)-1H-pyrrole-2-carboxylic Acids. European Journal of Organic
Chemistry, 2008. 2008(2): p. 324-329, DOI: 10.1002/ejoc.200700756.
37. Khusnutdinov, R.I., A.R. Baiguzina, R.R. Mukminov, I.V. Akhmetov, I.M. Gubaidullin,
S.I. Spivak, and U.M. Dzhemilev, New synthesis of pyrrole-2-carboxylic and pyrrole-2,5-
dicarboxylic acid esters in the presence of iron-containing catalysts. Russian Journal of
Organic Chemistry, 2010. 46(7): p. 1053-1059, DOI: 10.1134/s1070428010070158.
38. Salman, H., Y. Abraham, S. Tal, S. Meltzman, M. Kapon, N. Tessler, S. Speiser, and Y.
Eichen, 1,3-Di(2-pyrrolyl)azulene: An Efficient Luminescent Probe for Fluoride.
European Journal of Organic Chemistry, 2005. 2005(11): p. 2207-2212, DOI:
10.1002/ejoc.200500012.
39. Donohoe, T.J., C.E. Headley, R.P. Cousins, and A. Cowley, Flexibility in the partial
reduction of 2,5-disubstituted pyrroles: application to the synthesis of DMDP. Org Lett,
2003. 5(7): p. 999-1002, DOI: 10.1021/ol027504h.
40. Armes, S.P., Optimum reaction conditions for the polymerization of pyrrole by iron(III)
chloride in aqueous solution. Synthetic Metals, 1987. 20(3): p. 365-371, DOI:
10.1016/0379-6779(87)90833-2.

30
41. Kang, E.T., K.G. Neoh, Y.K. Ong, K.L. Tan, and B.T.G. Tan, X-ray photoelectron
spectroscopic studies of polypyrrole synthesized with oxidative iron(III) salts.
Macromolecules, 1991. 24(10): p. 2822-2828, DOI: 10.1021/ma00010a028.
42. Chitte, H.K., G.N. Shinde, N.V. Bhat, and V.E. Walunj, Synthesis of Polypyrrole Using
Ferric Chloride (FeCl<sub>3</sub>) as Oxidant Together with Some Dopants
for Use in Gas Sensors. Journal of Sensor Technology, 2011. 01(02): p. 47-56, DOI:
10.4236/jst.2011.12007.
43. Vernitskaya, T.y.V. and O.N. Efimov, Polypyrrole: a conducting polymer; its synthesis,
properties and applications. Russian Chemical Reviews, 1997. 66(5): p. 443-457, DOI:
10.1070/RC1997v066n05ABEH000261.
44. Jokela, R., Interpretation of the 13C NMR spectra of thirteen methoxycarbonylpyridines
and some of their derivatives. Magnetic Resonance in Chemistry, 1992. 30(7): p. 681-683,
DOI: 10.1002/mrc.1260300720.

31
APPENDIXES
1H NMR spectrum of N-Boc pyrrole
1H, 13C and DEPT135 NMR spectra of 1-(tert-butyl) 2,5-dimethyl
pyrrole-1,2,5-tricarboxylate
1H, 13C, DEPT135 and DEPT90 NMR spectra of PyPY2
1H NMR spectrum of methyl 1H-pyrrole-2-carboxylate

32
8.1

33
8.2

34
8.2

35
8.3
36
8.3
37
8.3

38
8.3
39
8.4





























