Embed Size (px)
Citation preview

Newborn Screening for Duchenne Muscular Dystrophy
External review against programme appraisal criteria for the UK National Screening Committee (UK NSC)
Version: 4
Bazian Ltd
October 2011
The UK NSC advises Ministers and the NHS in all four UK countries about all aspects of screening policy. Its policies are reviewed on a 3 yearly cycle. Current policies can be found in the policy database at http://www.screening.nhs.uk/policies and the policy review process is described in detail at http://www.screening.nhs.uk/policyreview
Template v1.2, June 2010

UK NSC External Review
Page 2
Introduction
Duchenne Muscular Dystrophy
Duchenne Muscular Dystrophy (DMD) is a progressive genetic muscle wasting disorder. It an X-linked condition caused by a mutation in the DMD gene. This gene encodes the dystrophin protein and lies on the short arm of chromosome X (Xp21.2-p21.1). Mutations in this gene also give rise to a milder form of muscular dystrophy, with later onset, called Becker Muscular Dystrophy (BMD).
Various mutations can give rise to DMD, but the most common are large deletions of sections of the gene, which are reported to account for two thirds of cases. About 61% of cases are caused by deletions, 13% by duplications, and 26% by point mutations or other small mutations.1
The deletions that give rise to DMD are often those that result in a “frameshift”. That is, deletions that cause all the subsequent sections of the gene to be read incorrectly by the protein making machinery of the cell.
Women have two X chromosomes, and therefore if they have only one X chromosome with a mutation in the DMD gene, the other copy of the gene may be able to compensate. Up to about 20% of girls who carry a DMD mutation are thought to be affected to some extent.2,3 This is usually due to skewed X chromosome inactivation. The effects in girls are generally milder than those seen in boys, but a few girls have shown similar disease severity to boys.
Once a boy is identified as being affected by DMD or BMD, their mother can be assessed to see if she also carries the mutation, and if she proves to be a carrier, then other potential adult female carriers in her family can be tested, as well as any possibly affected male offspring (i.e. cascade screening).
About a quarter to a third of mutations are de novo.1 These cases occur in families with no known history of the disease, and would not be identifiable by cascade screening approaches.
A newborn DMD screening service is currently provided in Wales. This has its origins in a pilot study which continued as an NHS funded service following the pilot’s completion. It is not supported by the NSC.
Basis for current policy
Current UK NSC policy is that newborn screening for DMD is not recommended. The last review of this policy took place in 2004, and was carried out by the former Child Health Subgroup of the NSC.4
The report concluded that:
“Given that there is no available treatment, the benefits of screening appear to be modest. The greatest benefit would be that a very small number of births of second affected boys would be averted in cases where the index case would have presented late.”
It found that newborn screening for DMD:
met criteria 1, 2, 8, 17, 18 (the last criterion met in Wales only)
did not meet criteria 13
was uncertain as to whether it met criterion 15

UK NSC External Review
Page 3
made no explicit judgement or did not assess criteria 3, 4, 5, 6, 7, 9, 10, 11, 12, 14, 16, 19, 20, 21, 22.
It made these decisions based on two Health Technology Assessments (HTAs) on newborn screening from 1997,5,6 and publications regarding the DMD newborn screening programme in Wales.
This report
This paper uses evidence published from 2004 to 2011 to update the review of screening for DMD against the UK National Screening Committee (NSC) criteria for appraising the viability, effectiveness and appropriateness of a screening programme (National Screening Committee 2003). This update focuses on evidence relating to screening of newborns in the general population in the UK, as this is the policy under review. In particular, as the screening programme in Wales is for male newborns only, this is the approach focused on in this report. A comprehensive review of the evidence regarding alternative screening approaches e.g. cascade screening, is beyond the scope of this update, however, alternative approaches are mentioned where relevant.
We have referred to the 2004 report,4 and the most relevant 1997 HTA report5 where appropriate. The main barrier to screening identified in the 2004 was the lack of available treatment, so we have concentrated on this criterion (criterion 10), and also updated evidence available regarding other criteria where possible.
Experts in the field of DMD (Professor Francesco Muntoni and Dr Juliet Ellis) have also submitted a clinical vignette to the NSC describing their view on how newborn DMD screening matches the NSC criteria. Their view was that although the original priority for DMD newborn screening was to reduce the incidence of DMD, this has now changed to one of reducing disease progression/severity.
The vignette helped define the case for newborn screening in the UK and suggested that this approach would lead to an improvement in the standard of care by reducing the ‘diagnostic odyssey’ thereby facilitating:
timely initiation of interventions (e.g. physiotherapy, corticosteroids)
identification of behavioural and cognitive issues
avoidance of exposure to harmful interventions (e.g. general anaesthetics)
identification of cohorts for studies of novel therapies
provision of timely information to parents for subsequent reproductive decision making and to allow time to make emotional and practical preparations associated with the diagnosis.
A systematic search of literature published between January 2004 and 6 May 2011 yielded 1,523 studies addressing DMD (after removal of duplicates). Six ongoing trials were identified from trial registries. Overall, 213 references from this original search were judged as being potentially relevant to screening, diagnosis, management, prevalence, prognosis, treatment, cost or cost effectiveness of screening, attitudes and social ethical implications to screening for DMD, information provision, and the genetics of the condition (see appendix for study breakdown). Additional relevant references identified during the preparation of this report have also been included.

UK NSC External Review
Page 4
Due to the large number of references, a first pass appraisal at abstract level was followed by a retrieval of selected full text papers. An overview of the most informative and relevant references regarding the individual screening criteria is given below. Guidelines, systematic reviews of the evidence, and studies from the UK were prioritised, as were studies addressing key areas of uncertainty identified in the previous report.
Based on the evidence reviewed we have made provisional summary statements about whether each criterion is met, not met, partially met, not clear if met, or is not applicable. These judgements are provisional and should be reviewed by the Expert Panel in the context of all the evidence available.

Appraisal against UK NSC Criteria These criteria are available online at http://www.screening.nhs.uk/criteria.
1. The condition should be an important health problem
2004 report “Yes. Duchenne muscular dystrophy affects 1 in 4000 boys. It causes progressive weakness of the muscles, so that the boy becomes confined to a wheelchair by the age of 13 and death usually occurs by the age of 20. There is no proven long term treatment.”
Duchenne Muscular Dystrophy (DMD) is a progressive and currently incurable muscle weakness disorder that is ultimately fatal. The condition is genetic, and is caused by mutations in the DMD gene. This gene lies on the X chromosome, meaning that the vast majority of those affected are boys. For this reason, newborn screening for the condition would usually be carried out in male newborns only.
The DMD gene encodes the muscle protein dystrophin. Dystrophin links extracellular connective tissue to the internal scaffolding (cytoskeleton) of the muscle cells. This acts as a “shock absorber” protecting the muscles from repeated contraction and relaxation. If dystrophin is missing this link is broken and the muscle is weakened and becomes progressively damaged by normal activity. Eventually the muscle fibres die and are replaced by connective and fatty tissue, meaning that the muscles no longer function. This affects all muscles in the body, including the heart muscle.
Incidence and prevalence
DMD is the most common inherited muscular dystrophy, estimated to affect 1 in 3,600 to 1 in 6,000 male births.2 Data from twenty years of newborn screening has suggested an incidence of 1 in 5,266 male births in Wales.(S Moat, personal communication) In 1991 the estimated prevalence of DMD was 2·48/100,000 in the UK.7 With a population of about 60 million, this would suggest that there are just under 1,500 boys with the condition in the UK at any one time, a figure which is in line with estimates from the UK’s Muscular Dystrophy Campaign.8 The Muscular Dystrophy Campaign estimates that there are about 100 boys are born with the condition in the UK each year.
Some mutations in the DMD gene give rise to a milder form of muscular dystrophy called Becker muscular dystrophy (BMD). BMD is reported to affect about 1/18,450 male births in the UK, with a prevalence of 2.38 per 100,000 population.7 Mutations in the dystrophin gene can also cause X-linked dilated cardiomyopathy, where only the heart is affected.
One study suggested that the annual incidence of DMD in Canada had not changed significantly between 1969 to 2003 (1 in 3,745 to 1 in 7,711 male births; p value reported as not significant).9 The mean age at diagnosis also did not vary in that time (range of averages across the time period 2.8 to 6 years; p value reported as not significant). Age at diagnosis ranged from prenatal diagnosis to age 11 years. A study from the US found a trend towards earlier diagnosis between 1982 and 2000, but this trend was not statistically significant (p=0.055).10
The update search for this report did not identify publications since 2004 comparing age at diagnosis over time in the UK population. The 1997 HTA report on newborn screening cited the average age at diagnosis of DMD in Merseyside as 4.5 years (range 3 months to 8.5 years).5 A conference abstract from 2010 reported that the median age at diagnosis among ambulant boys with DMD in the UK was 4.1 years (range not reported).11

UK NSC External Review
Page 6
As boys are usually diagnosed with DMD between the ages of four to five their parents may have had other children, who may be affected or may be carriers.
Clinical course
DMD is characterised by slow motor development in early childhood, with the first signs appearing at the time when walking starts, between the ages of one and three years. Affected boys may also have cognitive or behavioural problems starting at this age.2 A meta analysis from 2001 suggested that the prevalence of low IQ (less than 70) in children with DMD is 34.8%.12 A narrative review also reported that other studies have found autistic spectrum disorders, ADHD, and obsessive compulsive disorders to be more common in children with DMD.13
Affected boys have difficulty in rising from the floor that requires them to use their arms for assistance – a classical sign of DMD called Gowers’ manoeuvre. Boys may also show an inability to jump or run as well as unaffected children, and slower walking speed. The boys continue to acquire motor skills, and then reach a “plateau”, usually around the age of 4-8 years old. After this plateau phase, their muscle strength and motor abilities deteriorate, and they lose the ability to walk in their early teens. Muscle weakness also gives rise to respiratory and cardiac problems, and without intervention life expectancy is reported to be about 19 years.
There have been improvements in life expectancy in this condition over time which have been attributed to improvements in treatment, particularly the provision of respiratory support in the advanced stages of the disease.14-16 Some affected individuals can now living into their 30s.2 A conference abstract published in 2007 reported a median age at death of patients with DMD in the South West of the UK as 19 years (range 14 to 32 years), and in DMD patients in the Trent region of 18 years 11 months (range 15 to 28 years).17 They reported that these findings supported an increase in life expectancy in DMD patients, but did not provide data for previous years in the UK.
The severity of BMD can range from borderline DMD to almost asymptomatic depending on the causative mutation.1 In BMD, boys remain ambulatory for longer, usually until after the age of 16 years. However, with the use of corticosteroids to prolong ability to walk, the differences between DMD and BMD are reported to be less distinct. Patients are sometimes classified based on age of wheelchair dependency, with those needing a wheelchair before age 13 classified as DMD, those needing a wheelchair at age 16 or over BMD, and those in between called “Intermediate Muscular Dystrophy”. BMD patients are reported to have near-normal life expectancy.18
Causative mutations
Various mutations in the DMD gene can give rise to DMD, but the most common are large deletions. In DMD, where the causative mutation is known, about 61% of cases are caused by large deletions, 13% by large duplications, and 26% by point mutations or other small mutations.1 About 5 to 10% of BMD/DMD cases are estimated to not have a mutation detectable.19
The size of the deletion does not appear to correlate well with the severity of the disease, but deletions that give rise to DMD are often those that result in a “frameshift”. That is, deletions which cause all the protein making instructions in subsequent sections of the gene to be incorrect. These mutations tend to lead to no dystrophin protein being produced, and a severe phenotype.

UK NSC External Review
Page 7
“In frame” deletions remove a section of the gene, but do not affect how the subsequent protein making instructions are read by the cell. In frame deletions lead to a dystrophin protein that is missing some of its internal sequence, but can often still perform some of its function. In frame deletions tend to result in BMD. BMD may also be caused by other types of mutations that reduce the amount of dystrophin the cells produce. About 81% of mutations in BMD patients are reported to be large deletions, 6% large duplications, and 13% point mutations.1
Carriers
Women have two X chromosomes, and therefore if they have only one X chromosome with a mutation in the DMD gene, the other copy of the gene may be able to compensate. Up to about 10-20% of girls who carry a DMD mutation are thought to be affected to some extent.2,3 The effects in girls are generally milder than those seen in boys, and may include cognitive and heart problems. However, a few girls have shown similar disease severity to boys.
At present it is not possible to predict which females who carry DMD mutations will show symptoms, and the extent to which they will be affected (see Criterion 4).
Once a boy is identified as being affected by DMD or BMD, their mother can be assessed to see if she also carried the mutation. If she proves to be a carrier, then other potential adult female carriers in her family can be offered testing and genetic counselling (i.e. cascade screening). In boys whose mother is a carrier, there is a 50% chance that any male child will be affected. There is also a 50% chance that any female child may be a carrier.
Not all boys with DMD have a mother who is a carrier; about a quarter to a third of mutations are reported to arise de novo in the egg before or after fertilisation.1,3 Mothers in whom DNA extracted from blood tests negative for DMD mutations may have germline mosaicism for the mutation. This means that some of their reproductive tissue including other eggs may carry the mutation. If this is the case there is still a risk of passing on the mutation to other offspring.
Summary: Criterion 1 met
DMD is debilitating and ultimately fatal. In Wales, about 1 in 5,266 male births are affected. This is towards the lower end of the range of incidence quoted in the literature (1 in 3,600 to 1 in 6,000 male births). The prevalence of DMD in the UK has been estimated at about 2·48 per 100,000 and the prevalence of BMD is estimated at 2.38 per 100,000 population.
2. The epidemiology and natural history of the condition, including development from latent to declared disease, should be adequately understood and there should be a detectable risk factor, disease marker, latent period or early symptomatic stage
2004 report: “Yes. The condition presents at age 18m to 6 years with progressive weakness, but is usually silent in the first year of life. Identification at an early stage allows the family to come to terms with the diagnosis and to make appropriate plans for housing, career moves, etc.. In addition, since the condition is inherited, they can be made aware of the risk that further boys might also be affected. This avoids the tragic situation where a family may have two or even three affected sons, before they discover the diagnosis. In a French study, it was hypothesised that neonatal screening might avoid the birth of 10 affected boys for 400,000 babies screened. Unpublished data from Scotland extrapolated to the whole of the UK population suggest that approximately 17 affected births could be avoided per year.”

UK NSC External Review
Page 8
Natural history
The natural history of the condition in boys diagnosed clinically and the progressive nature of the disease is generally well understood (see Criterion 1).2 The existence of newborn screening programmes has also allowed some prospective assessment of natural history at early ages. For example, the update search identified one study published since 2004 which described early progression in boys with DMD identified by the Welsh newborn screening programme (see Criterion 17 for description).20
However, some studies have noted the limited amount of population based longitudinal studies of DMD/BMD.19,21 Projects such as the Muscular Dystrophy Surveillance Tracking and Research Network (MD STARnet) in the US have been set up to facilitate such studies.19 There are also European registries which can also facilitate such studies, including those organised by Translational Research in Europe for the Assessment and Treatment of Neuromuscular Diseases (TREAT-NMD) network.22
One retrospective study from 2009, based on MD STARnet data, looked at the diagnostic process in 156 boys with DMD in the US, who had no known family history of the disease prior to their birth.10 The study only included children with first signs or symptoms before the age of 7 years, although it did not appear that any children were excluded on this basis. In this study:
first signs or symptoms were reported on average at age 2.5 years (range 0.2 to 6.1 years)
first evaluation of these signs or symptoms by a health specialist took place at an average age of 3.6 years (range 0.2 to 8.0 years)
first neurology/neuromuscular visit was at average age 4.6 (range 0.3 to 8.6)
first creatine kinase (CK) test was performed at average age 4.7 (range 0.3 to 8.6)
age at definitive diagnosis was an average age of 4.9 years (range 0.3 to 8.8).
There had been no change over time (1982 to 2000) in the age at which the first CK measurement took place (p=0.22). There was a non-significant trend towards reduction in age at definitive diagnosis (p=0.055).
In children aged under 1.5 years the most commonly reported first sign or symptom was gross motor delay (58.1%), among those aged 1.5 to 3 years it was trouble walking or running (54.6%), among those aged 3 to 5 it was muscle weakness (40.5%), and among those aged 5 and over it was inability to keep up with peers in motor activities (80%) and muscle weakness (80%).
Family members were the most likely to initiate medical investigation (58.3%), and a paediatrician or family practitioner were most likely to first evaluate the child (63.8%). In only two cases (1.6%) was the first evaluation by an emergency room/urgent care physician.
There is clearly a wide range of ages at which the different stages of the diagnostic process took place. What is not clear from these results is whether there was a correlation between the severity of symptoms and age at diagnosis. That is, whether those who are more severely affected tend to be diagnosed earlier, and those less severely affected later.
At a meeting between the European Medicines Agency (EMA) and the TREAT-NMD network in 2009, there was concern raised about the need to understand the natural history of the condition sufficiently to devise appropriate ways to measure the effects of new treatments in development.18

UK NSC External Review
Page 9
In particular it was noted that there was a need to collect information related to the natural history of the condition in neonates, up to the age that diagnosis is usually made based on symptoms. This was particularly to allow selection of outcome measures that would be able to capture any benefits of treatments for this age group.
The need for further understanding of the newborn period is of particular importance given the new treatments in development that may help to restore some level of dystrophin production. As the disease is progressive, the suggestion is that earlier treatment may improve prognosis, and therefore testing earlier in life is likely to be the goal if the treatments show sufficient safety and efficacy in older age groups.
A need to further understand disease progression and develop suitable ways of measuring outcome in boys who have lost the ability to walk was also highlighted at the meeting between TREAT-NMD and the regulatory authorities. A recent international guideline on DMD management also noted some areas where natural history needs further study, including the cardiac and gastrointestinal effects of the disease.2,23
There have been additional papers describing the natural history of the disease published since 2004. For example, the update search identified one study that prospectively assessed annual progression of 43 DMD in patients aged 5 to 35 years (average 15.3 years), who were followed for an average of 5 years.21 This paper reported that at that time longitudinal data on the progression of the disease in adults were sparse or outdated. In this study patients lost the ambulation at age 9.4 years on average (not further defined), became dependent on an electric wheelchair at an average age of 14.6 years, and started assisted ventilation at age 19.8 years on average. Three patients died in the study, at ages 22, 24, and 35 years. Median survival in this cohort was estimated to be 35 years (using Kaplan-Meier analysis).
There have also been other papers looking at DMD in adult patients, a group that is reported to be expanding due to improvements in treatment leading to extension in life expectancy.24,25
One of these studies in adults was from the UK. It looked at the 25 patients (24 male, 1 female) with DMD referred to an adult neuromuscular clinic in London between 1996 and 2003.25 Most of the patients were referred between the ages of 16 to 20 years, and their case notes were reviewed retrospectively. Mean age at death was 21.4 years (range 19 to 26). There were two sets of brothers. Mean age at diagnosis was 4.6 years (range 1 to 9 years). Three patients had a known family history of the disease, and they were diagnosed earlier (mean 2.0 years, range 1-3 years). Patients lost ability to walk at median age of ten years (range 5 to 13 years), and became totally wheelchair dependent at median age of 11 years (range 7 to 13). There was no correlation between age of wheelchair confinement and age of death. This paper describes the natural history of these patients as their disease progressed. For example, fractures were very common and were reported to often lead to sudden deterioration in mobility.
Another study has looked at the clinical heterogeneity of DMD, which is a challenge to interpreting results of clinical trials.26 It used an initial sample of 75 non-steroid using patients followed up for a median of 10.5 years, to identify predictors of outcome and another set of 34 patients (aged >12 years) to validate the findings. It suggested that prognosis could be predicted by severity of muscle and brain dysfunction.
The variability in how individuals are affected is thought to mainly relate to differences in genotype and how this affects dystrophin protein production.2 However, some level of uncertainty exists regarding genotype-phenotype correlation, with studies noting that phenotype cannot be predicted from genotype alone.27,28 Most of the mutations that cause

UK NSC External Review
Page 10
DMD are large deletions (about 61%). In most cases a deletion leading to a frameshift will have a more severe (Duchenne) phenotype and those not disrupting the reading frame to a milder (Becker) phenotype. One study reported that when applied to deletions that affect exons, the reading frame rule had 89.4% sensitivity, 54.3% specificity, 88.8% positive predictive value, and 55.9% negative predictive value for predicting a DMD phenotype.27
Point mutations in the DMD gene that give rise to premature stop codons would also be expected to give rise to dystrophinopathy. However, there have been reports of nonsense mutations leading to exon skipping and a variable phenotype, and nonsense mutation near to the end of the gene leading to a milder phenotype than expected.28,29 The effects of point mutations that lead to a change in a single amino acid in the very large dystrophin protein may also be difficult to predict, although these are reported to be rare.1
An audit of recorded DMD mutations from France noted that there can be inter- and intra-familial variability in the clinical presentation of the same or similar mutations.1 The examples provided were five deletions identified in asymptomatic males or in families where some males with the mutation had no symptoms, but others had BMD. In addition, complex mutations involving duplications may also complicate prediction of phenotype in some cases.30
Muscle biopsies can help to predict phenotype, as the absence of dystrophin predicts DMD, and a decrease in levels BMD.31
Detection
There are detectable disease markers that can be used in screening and subsequent diagnostic investigations. Raised levels of creatine kinase in the blood are the disease marker used in newborn screening, and further investigation would include genetic testing for mutations in the DMD gene and muscle biopsy to detect the presence or absence of dystrophin.
Summary: Criterion 2 partially met
There is generally a good understanding of the natural history of DMD, and this criterion was considered met in the 2004 report.4 However, this understanding has largely been derived from individuals diagnosed as a result of symptoms, rather than in a newborn screening population. Due to the lag period before symptoms first arise, and between first symptoms and diagnosis, the average age at diagnosis tends to be between ages four and five.
There are other areas of natural history which the literature has highlighted as needing further study, including older non-ambulant boys with DMD, and cardiac manifestations of the disease.
Research into novel genetic treatments that may be able to restore some level of dystrophin production is underway. These treatments, if proven to be effective and safe, could theoretically give greater benefit if started earlier in life.
The regulatory authorities have indicated that a better understanding of the natural history of the disease in newborns and non-ambulant boys is needed in order to be able to quantify the potential benefits of these new treatments in these groups.

3. All the cost-effective primary prevention interventions should have been implemented as far as practicable
2004 report: Judged N/A
Strictly speaking there are no primary prevention interventions for the affected individual, as the disorder is genetic and therefore present from conception. Cascade screening of potential carriers in affected families could potentially reduce the risk of additional children being born with the condition. However, as diagnosis with DMD occurs on average at around age 4.1 years in the UK,11 additional children may already have been born with the condition before the index diagnosis is made. As about a quarter to a third of boys with DMD have no family history of DMD these cases would not be identifiable by cascade screening.1,5
The search did not identify any studies published since 2004 describing the extent to which cascade screening is offered and accepted in families with DMD in the UK, or to which this information leads the families to take measures to avoid having further affected children.
Studies from the UK prior to 2004 have suggested that a high proportion of families affected by DMD would be happy for information to be passed on to relatives either by themselves (98% of probands and 94% of at risk relatives agreed) or via the genetics clinic (78% of probands and 90% of at risk relatives agreed).32
One recent study suggested that in the Netherlands, about a third of adult women (aged over 16) at risk of being a carrier in all known DMD/BMD families (222 DMD/63 BMD families) had not received mutation testing.33 This included women who were at 50% risk (siblings of boys with a mutation whose mothers were carriers), and women at lower risk (estimated as 4.3% risk) due to the possibility of germline mutations (siblings of boys with a mutation whose mothers were not carriers based on lymphocyte DNA testing, and maternal aunts of boys with a mutation whose mothers were carriers but the carrier status of the grandmother was not known). In addition, 4% of mothers of boys with DMD had not been tested, and 22% of mothers of boys with BMD had not been tested. Some mothers had not been tested but were assumed to be carriers due to having more than one child with the condition (2% of DMD mothers, and 11% of BMD mothers). In addition, 42% of maternal grandmothers at risk of being carriers in DMD families had not been tested and 57% in BMD families. The study notes that the reasons for not having genetic testing were not investigated, and could include, for example, having been tested using creatine kinase levels before genetic testing was available, not wishing to have (more) children, or having died before testing could be carried out (particularly for grandmothers).
An audit of a mutational database from France suggested that carrier status of probands’ mothers was not known in 32.8% of cases.1
Summary: Unclear if Criterion 3 is directly applicable
Strictly speaking primary prevention of DMD is not possible, as the disease is genetic and present from conception. Cascade screening of potential carriers in affected families could potentially reduce the risk of additional children being born with the condition. The extent to which cascade screening for DMD is currently offered and accepted in the UK, and the influence on subsequent reproductive choices is not clear.
Cascade screening would not fully remove the possibility of additional affected children. This is due to three factors: the lag in clinical diagnosis of the first affected child, which means that

UK NSC External Review
Page 12
other affected children may have been born in the interim; the relatively high proportion of de novo DMD mutations (about a quarter to a third of cases); the fact that even when offered testing some relatives at risk of being carriers decline further investigation.
4. If the carriers of a mutation are identified as a result of screening the natural history of people with this status should be understood, including the psychological implications.
2004 report: Judged N/A
If screening was restricted to male newborns, as is the case in most newborn DMD screening programmes, this would not identify newborn female carriers. However, the identification of an affected male newborn would suggest that his mother is likely to be a carrier, and his female siblings may be carriers.
Natural history of carriers
Up to about 10-20% of carrier females are reported to show some of the symptoms of the disease, sometimes called “manifesting carriers”.2,3 A study from Wales published in 1989 found that 2.5% (3/119) of mothers of boys with DMD or BMD who were thought to be carriers (based on bloodspot creatine kinase levels and pedigree analysis) were manifesting carriers. This led the prevalence of manifesting carriers in Wales to be estimated as 1 in 100,000 women.34 The update search did not identify any more recent estimations published since 2004.
In women, one copy of the X chromosome is usually inactivated in most cells, as only one copy is needed. Women who are affected by DMD are thought to usually have skewed X chromosome inactivation, where the X chromosome carrying the non-mutated form of the DMD gene is inactivated in a much greater proportion of cells than the X chromosome carrying the mutation. This means that the non-mutated form of the DMD gene cannot compensate in these cells. In some cases, the girl may be affected due to chromosomal rearrangements.2 Affected women may also carry two mutated copies of the DMD gene, but this is likely to be rare.
Manifesting carriers may have only mild muscle weakness, but can also be as severely affected as boys.2,35 In one sample of 15 manifesting female carriers from the US aged between 6.5 and 68 years old, age at onset of symptoms ranged between 2 and 47 years (median 8 years, mean 14.9 years).35 Those with earlier symptoms were not necessarily more severely affected. Eight of these women had a family history of DMD, and none had a family history of BMD. Women who have a relative with BMD are reported to be less likely to be manifesting carriers and likely to be less severely affected than women with a family history of DMD. Eight of the women had a deletion or duplication within the DMD gene, and six had point mutations; one woman carried two DMD mutations. There was skewed X inactivation in 38% of the women where this was tested.
The women most commonly presented with muscle weakness (80% of cases), followed by muscle pain (myalgia) and/or muscle cramps (60% of cases). One woman was affected as severely as a typical boy with DMD, while the others showed characteristics similar to mild to severe BMD. Five of the women had dilated cardiomyopathy, and another had some cardiac symptoms but not cardiomyopathy.
There is concern that female carriers may be at increased risk of dilated cardiomyopathy, but there is reported to be no consensus among experts about whether these women need regular cardiac surveillance.36 One study from Scotland looked at life expectancy and cardiomyopathy in

UK NSC External Review
Page 13
397 female DMD/BMD carriers (319 DMD, 74 BMD, 4 IMD).36 The women were identified from known DMD/BMD families, and followed up to 2004 using death certificate data. The women were born from 1860 onwards and during follow up 94 women died. They found no differences between observed and expected numbers surviving at different ages. For example, 281 women were expected to be alive by age 40 based on Scottish population data, and 286 were actually alive; 35 were expected to be alive at age 80 and 38 were actually alive. There were two deaths from dilated cardiomyopathy among the carriers (2.1% of all deaths), and 19 deaths overall from cardiac causes (20% of deaths). The number of deaths from cardiac causes among carriers was less than the number expected based on the age and gender matched general population in Scotland (standardised mortality ratio 0.53, 95% CI 0.32 to 0.82). On this basis the authors concluded that although previous studies have suggested that carriers may have clinical features of cardiomyopathy, this does not appear to be associated with reduced life expectancy or increased risk of cardiac death. They therefore decided that routine cardiac surveillance of obligate carriers was probably unnecessary.
This contrasts somewhat with advice from the US. The American Academy of Pediatrics made recommendations about cardiac care in carriers of DMD or BMD in 2005, with a reaffirmation of this policy in 2009.37 They recommend that carriers of DMD or BMD:
should be made aware of the risk of developing cardiomyopathy and educated about the signs and symptoms of heart failure
should be referred for evaluation by a cardiac specialist with experience in the treatment of heart failure and/or neuromuscular disorders
should undergo initial complete cardiac evaluation in late adolescence or early adulthood or at the onset of cardiac signs and symptoms, if these signs or symptoms appear earlier
should be screened with a complete cardiac evaluation at a minimum of every 5 years starting at 25 to 30 years of age.
They note that treatment of cardiac disease in carriers is similar to that for boys with DMD or BMD. They say that there is a there is a need to research the natural history and outcome of treatment in female carriers. A survey published in 2009 suggested that 62.9% of carriers in the US were aware of their heart risk, and 64.4% had ever had a heart test.38
Based on current knowledge, if carrier females were identified it would not be possible to predict with a high level of certainty which might be affected and which not. It seems unlikely that mutation testing would be carried out in female siblings of an affected child unless they themselves were showing symptoms, or had reached an age where they wanted to have children. Mothers of affected children might be more willing to be tested if they are intending to have further children, and wish to avoid them being affected.
Effect on reproductive behaviour
Based on available studies, two narrative review articles suggested that the information provided by newborn screening may have limited impact on reproductive choices.3,39 A similar finding was reported by the 1997 Health Technology Assessment on the cost, yield, and outcome of neonatal screening.5 It suggested that at the time, low uptake of prenatal screening of pregnancies following neonatal diagnosis of DMD might relate to the fact that the child still only had mild symptoms at the time of the subsequent pregnancy.

UK NSC External Review
Page 14
One of the narrative reviews cites a Welsh publication from 1998 reporting that most families take up genetic counselling and prenatal screening, but that other countries report less uptake.39 The other narrative review cites a later Welsh publication from 2002 reporting that only 20% of families decided against a future pregnancy and 55% only delayed pregnancy.3 However, the review did not mention whether the families that did have a subsequent pregnancy used prenatal testing.
The update search did not identify any additional papers published since 2004 that reported on the reproductive choices made by women identified as carriers through newborn DMD screening.
Other effects
The update search did not identify any additional papers published since 2004 that reported on the other effects on women who are identified as carriers as a result of newborn DMD screening, for example any psychological effects.
Summary: Criterion 4 partially met
Newborn DMD screening is likely to be offered to male newborns, meaning that newborn female carriers will not be identified. However, a high proportion of mothers of boys with DMD will be carriers. The natural history of female carriers is less well understood than that of males with DMD, particularly with regard to predicting which carriers will themselves show symptoms. However, the proportion of carriers that do show symptoms is relatively low (10-20%), and although they may be at increased risk of cardiomyopathy, one study suggests that this may not reduce their life expectancy. Identification of carriers would allow reproductive choice to be made in subsequent pregnancies. However, some studies question whether reproductive behaviour is influenced in women identified as carriers through newborn screening.
5. There should be a simple, safe, precise and validated screening test
1997 HTA report: “The primary screen is creatine kinase (CK) activity, which is usually measured by a bioluminescence test or a fluorescence assay. For babies with increased levels this may then followed-up by assay on a second dried blood sample or on liquid blood. Depending on the exact cut-off level and probably other factors, the reported rates for repeat sampling range between 0.02–0.8%, typically 0.2%.” (page 62)
“Because of the long delay before the development of clear clinical symptoms, it is difficult to estimate the false negative rate.” (page 62)
2004 report: “The test involves the measurement of an enzyme [CK] using the neonatal blood spot collected for the phenylketonuria test. The results have been reported from a Welsh programme of screening. Parents appreciate the benefits of early diagnosis. Another proposed approach was to screen all late walking boys using [CK], but this did not prove to be effective or feasible and one attempt at implementation was abandoned.”
The update search identified one conference abstract describing the performance of the DMD screening test in a newborn screening programme, that took place in Antwerp in Belgium.40 The following discussion focuses mainly on data from a presentation on the Welsh newborn DMD screening programme.(S Moat, personal communication)
The test for elevated levels of creatine kinase (CK) in newborn heel prick blood spot samples is still the test used for newborn DMD screening. This test was reportedly first used to screen for

UK NSC External Review
Page 15
muscular dystrophy in 1975.3 It is relatively simple to perform as it is carried out using the newborn heel prick bloodspots that are currently collected for other newborn tests. However, its use alongside testing heel prick samples for treatable diseases such as phenylketonuria (PKU) may give rise to concerns about differentiating treatable and less treatable conditions and gaining truly informed consent (see Criterion 7).
CK is elevated in the blood of boys with a DMD mutation due to the muscle damage that occurs as a result of the lack of dystrophin. Muscle damage from other causes can also increase CK levels in the blood. For example, birth trauma and the method of delivery can lead to transient increases in CK levels, which may increase the risk of false positives when testing newborn blood samples.39
The results for the Welsh newborn screening programme over 20 years have been presented by Dr Stuart Moat, and results from this have been incorporated into this report.(S Moat, personal communication) The test is an opt-in test, and is described as an “extra” test in the literature provided to the parents. Parents need to explicitly consent to having the DMD screening test and provide a signature on the blood spot card to confirm this. If the parent consents, the test is carried out on blood spots taken between five and eight days after birth as part of other newborn screening tests. Those samples showing a bloodspot creatine kinase (CK) value of 200U/L or above are re-assayed again in duplicate, and those with an average value of 250U/L or greater across the three samples receive a follow up serum CK test at six to eight weeks. If serum CK levels are normal, DMD is not suspected. If serum CK is raised, DMD is suspected, and the child goes on to have mutational analysis.
Between 1990 and 2010 in Wales, 337,045 boys were born and 312,073 (92.6%) screened for elevated CK levels. Of these boys 123 showed an average blood spot CK level ≥250U/L, and had CK levels tested at 6-8 weeks. At this point 58 boys (47.2%) were found to still have elevated CK levels, and 65 to have normal CK levels (52.8%).
Of the 58 boys with persistently elevated CK levels, all were found to have a muscular dystrophy: 50 were found to have DMD, 5 BMD, and 3 other dystrophies. Ten boys who had a normal CK level on blood spot screening went on to develop DMD. In addition, four boys whose parents had declined DMD screening had DMD.
There is a concern that if a screening programme is in place, this could lead to false reassurance in false negatives, which could influence the likelihood of receiving a later correct diagnosis.5 The age at diagnosis of the false negatives in Wales was not reported in the presentation, but could be compared with the average age of diagnosis in the UK.
Using the figures from the Welsh programme gives the screening test a:
sensitivity of 83.33%
specificity of 99.98%
false positive rate of 0.02%
false negative rate of 16.67%
positive predictive value of 40.65%
negative predictive value of 99.997%
As screening is aimed at identifying DMD, cases of BMD and other dystrophies have been considered as false positives in these calculations.
UK NSC External Review
Page 16
The relatively high level of false negatives suggests that the threshold CK level that triggers a need for further testing could potentially be reduced (although this threshold already appears low in comparison to that used in other programmes, see below). Lowering the threshold might identify the previously missed cases; however, it would be likely to have a knock on effect of increasing the number of newborns on whom confirmatory testing is needed, and the number of false positives.
As the average age for clinical DMD diagnosis is around four to five years, the identification of false negative results will lag this far behind the newborn screen, so can only be judged several years after the newborn screen.
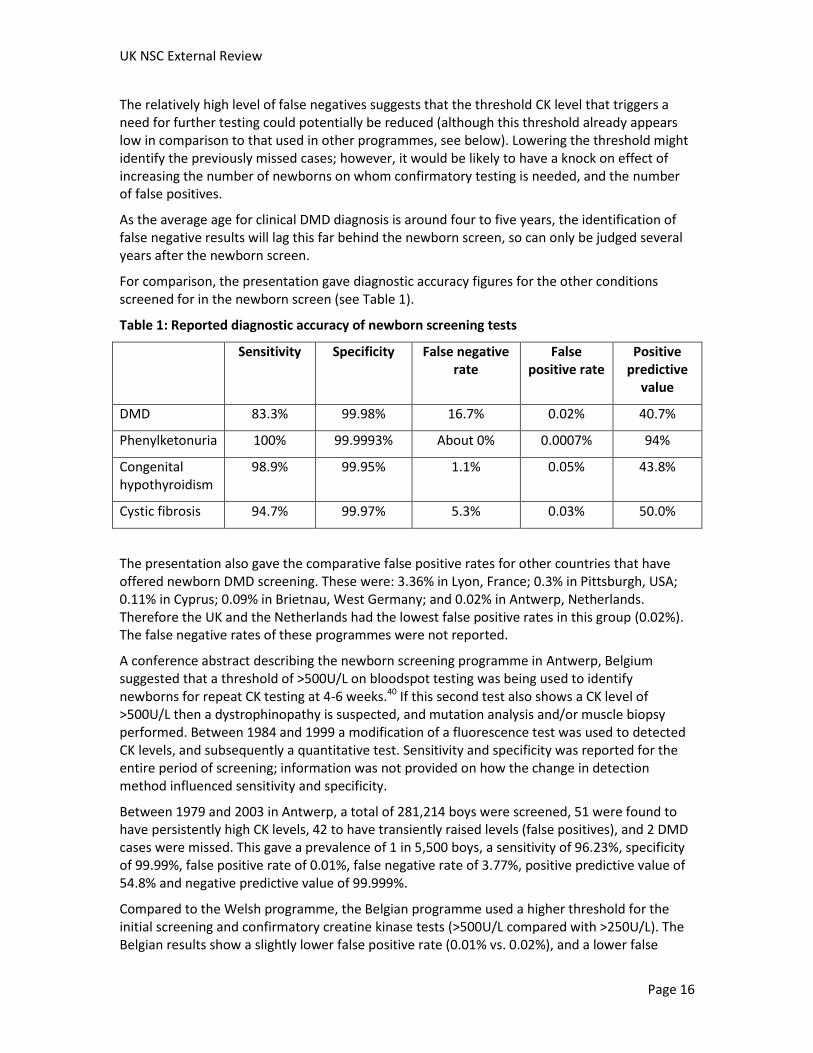
For comparison, the presentation gave diagnostic accuracy figures for the other conditions screened for in the newborn screen (see Table 1).
Table 1: Reported diagnostic accuracy of newborn screening tests
Sensitivity Specificity False negative rate
False positive rate
Positive predictive
value
DMD 83.3% 99.98% 16.7% 0.02% 40.7%
Phenylketonuria 100% 99.9993% About 0% 0.0007% 94%
Congenital hypothyroidism
98.9% 99.95% 1.1% 0.05% 43.8%
Cystic fibrosis 94.7% 99.97% 5.3% 0.03% 50.0%
The presentation also gave the comparative false positive rates for other countries that have offered newborn DMD screening. These were: 3.36% in Lyon, France; 0.3% in Pittsburgh, USA; 0.11% in Cyprus; 0.09% in Brietnau, West Germany; and 0.02% in Antwerp, Netherlands. Therefore the UK and the Netherlands had the lowest false positive rates in this group (0.02%). The false negative rates of these programmes were not reported.
A conference abstract describing the newborn screening programme in Antwerp, Belgium suggested that a threshold of >500U/L on bloodspot testing was being used to identify newborns for repeat CK testing at 4-6 weeks.40 If this second test also shows a CK level of >500U/L then a dystrophinopathy is suspected, and mutation analysis and/or muscle biopsy performed. Between 1984 and 1999 a modification of a fluorescence test was used to detected CK levels, and subsequently a quantitative test. Sensitivity and specificity was reported for the entire period of screening; information was not provided on how the change in detection method influenced sensitivity and specificity.
Between 1979 and 2003 in Antwerp, a total of 281,214 boys were screened, 51 were found to have persistently high CK levels, 42 to have transiently raised levels (false positives), and 2 DMD cases were missed. This gave a prevalence of 1 in 5,500 boys, a sensitivity of 96.23%, specificity of 99.99%, false positive rate of 0.01%, false negative rate of 3.77%, positive predictive value of 54.8% and negative predictive value of 99.999%.
Compared to the Welsh programme, the Belgian programme used a higher threshold for the initial screening and confirmatory creatine kinase tests (>500U/L compared with >250U/L). The Belgian results show a slightly lower false positive rate (0.01% vs. 0.02%), and a lower false

UK NSC External Review
Page 17
negative rate (3.77% vs. 16.67%). However, as mentioned above, false negatives will take time to identify, and therefore may be missed if follow up is not long enough.
A narrative review reported that in large scale DMD screening projects from the 1980s and 1990s positive predictive values have ranged from 17% to 56%.3 They suggest that variations could be due to differences in testing method, cutoff value, or protocol for testing and follow up.
Summary: Criterion 5 partially met
The CK screening tests used for screening for DMD have been in use for over 30 years. The test is simple to perform in that it can be used on the heel prick bloodspots collected for screening for other conditions, reducing the need for additional blood collection. However, use on newborn blood samples may increase the risk of false positives, as birth trauma and the method of delivery can lead to transient increases in CK levels.
The Welsh newborn screening programme has found a sensitivity of about 83%, meaning that a relatively high proportion, about 17%, of DMD cases are missed (false negatives). Specificity is high (99.98%), in part due to the low prevalence of the condition. However, the positive predictive value is about 41%, meaning that for every true positive there is roughly one false positive. The second step in those identified by screening is a second blood test at age 6-8 weeks. All of the babies found to have persistently raised levels of CK at this stage were found to have some form of dystrophy on further investigation (86.2% DMD, 8.6% BMD, and 5.2% other dystrophies). BMD and other dystrophies are not the targets of the screening programme under review, and are therefore considered false positives for the purposes of this report.
The negative predictive value of the test is high at 99.997%. Varying the threshold level for further investigations will affect the balance of sensitivity and specificity of the test.
6. The distribution of test values in the target population should be known and a suitable cut-off level defined and agreed
2004 report: “Data from the Welsh study show that half the babies referred with a high [CK] value turn out to be false positives and the level returns to normal by 5-6 weeks.”
The Welsh newborn screening programme has tested 312,073 newborn boys between 1990 and 2010, potentially providing a large amount of data on test values in the target population.(S Moat, personal communication)
The Welsh screening programme uses an initial CK threshold of 200U/L or more on the bloodspot to trigger further duplicate testing of the bloodspots. Infants who have an average CK level of 250U/L or higher have a blood sample taken at age 6-8 weeks for re-testing CK levels. If this test confirms raised levels of CK, then DMD is suspected, and the child has mutation testing.
As suggested in the 2004 screening update for the NSC, just over half (52.8%) of children with an initially raised level of CK in their newborn blood spot test did not have elevated CK levels on the 6-8 week test.
The average plasma CK level at 6-8 weeks in children with DMD was 7,872U/L (n=52), but were quite variable (range 2,442 to 14,200U/L). However, all values in this group were in excess of 2000U/L. The average plasma CK level at 6-8 weeks was much lower in healthy infants (n=47; mean 81U/L, range 73 to 167U/L) and in infants who only had a transient increase in CK levels (n=61; mean 116U/L, range 48 to 239U/L).(S Moat, personal communication)

UK NSC External Review
Page 18
The Welsh screening programme has identified five children with BMD and three children with other dystrophies, but missed 10 children with DMD.
Re-testing CK levels at 6-8 weeks in those above the threshold on the newborn screen reduces the need for mutational testing or muscle biopsy in false positives. The identification of half of those going forward to this second test as false positives could suggest an increase in the CK threshold. However, the fact that some DMD cases were missed argues for reducing the threshold. It would be of interest to look at what the CK levels were in the false negatives, and what effect reducing the threshold in order to capture more of them would have on overall diagnostic accuracy of the test. The update search identified no studies reporting this type of analysis.
A conference abstract describing a pilot newborn screening programme in Ohio reported that they determined reference values in the population by testing 40,000 anonymous newborn male blood spots.41 It reported a mean CK value of 250U/L±111. They decided on a threshold of >3SD above the mean (about 583U/L) to trigger DNA testing of the bloodspot. A second conference abstract from the same group suggested that a threshold of >600U/L was being used to identify newborns for DNA testing.42 They say that of 7,000 newborns tested, 95 had levels between 600-999U/L, seven had levels between 1000-1499U/L, one had levels between 1500 -1999U/L and two had levels above 2000U/L. The two boys with levels above 2000U/L were reported to have DMD mutations.
A conference abstract describing the newborn screening programme in Belgium suggested that a threshold of >500U/L on bloodspot testing was being used to identify newborns for repeat CK testing at 4-6 weeks.40 If this second test also shows a CK level of >500U/L then a dystrophinopathy is suspected, and mutation analysis and/or muscle biopsy performed. The method used to detect CK changed during the programme. Between 1979 and 2003 in Antwerp, a total of 281,214 boys were screened, 51 were found to have persistently high CK levels, 42 to have transiently raised levels (false positives), and 2 DMD cases were missed (false negatives).
What was not clear from these US and Belgian publications was whether they use the same detection methods as the Welsh newborn screening programme.
A publication from 2007 reported CK levels in a random sample of adults from the general population in the Netherlands.43 It found that the variation in CK activity was wider than that found in previous non-random samples, and above the assay manufacturer’s reference limits in some cases. This lead to a suggestion that higher upper reference limits might be needed. However, this sample only included adults, and it is not clear to what extent this reflects levels in babies and infants.
The update search identified no publications since 2004 describing levels in babies in the general population in the UK or those with DMD or BMD. A recent international guideline on DMD diagnosis and management suggests testing for “markedly increased” CK levels as a first step in diagnostic investigations, but it does not specify what level of CK elevation it considers to be an appropriate threshold.2
A report from a working group convened by the US Centers for Disease Control and Prevention (CDC) to inform lay people about their pilots of newborn screening suggest that different programmes may have used different laboratory techniques and different definitions of “elevated” results.44 This report also says that the results of CK level screening are difficult to interpret in females, with some female carriers detected by the test but not all. Most screening programmes were reported to have targeted male newborns only. It also suggests that false

UK NSC External Review
Page 19
positives may be reduced if the test is carried out a few months after birth. The report notes that this approach may not reach all infants, as opposed to newborn screening, but that it may be easier to obtain informed consent at this stage than directly after birth. The CDC is reported to be investigating both newborn and early infancy DMD screening in pilot programmes.
Summary: Criterion 6 not met
The CK test for DMD has been available since the 1970s, and programmes such as the Welsh newborn screening should have produced a large amount of data on CK levels in newborns.
The Welsh screening programme uses a CK threshold of ≥200U/L on the initial bloodspot test to prompt re-testing of the newborn bloodspots in duplicate, and those with an average value of 250U/L or greater across the three assays receive a follow up CK test at six to eight weeks. If serum CK is raised in this test, DMD is suspected, and the child goes on to have mutational analysis. This threshold is associated with a relatively high false negative rate (16.7%); the CK test values for these false negatives have not been published.
There is variation in the threshold level for triggering further investigations in different screening programmes. For example, the newborn DMD screening programme from Antwerp in Belgium reports using a CK threshold of >500U/L, and a US pilot programme in Ohio >600U/L. This may relate to different methods for detection of CK levels, or the precise timing of the test.
7. The test should be acceptable to the population
1997 HTA report: “Parents’ experiences of diagnostic delay and misdiagnosis have been found to result in their widespread support for neonatal screening. Of the parents of boys with Duchenne muscular dystrophy, 75% were in favour of neonatal screening. A major reason for this support was that it would avoid the anxiety involved in diagnostic delay.” (page 87)
2004 report: “The initial blood sample would be part of the routine newborn bloodspot screening already in place. The confirmatory sample at 6 weeks is additional. Some mothers of babies with a false positive result are likely to be upset, though the published Welsh data suggests this is usually of little significance.”
The population being screened is newborns, so their parents are required to consent on their behalf. The update search did not identify any studies published since 2004 that specifically addressed the attitudes of individuals identified as having DMD by newborn screening, rather than their parents or families.
The update search identified four publications on newborn screening in the UK published since 2004,45-48 and we received one presentation regarding the Welsh newborn screening programme.(S Moat, personal communication) The update search also identified two additional publications describing population attitudes to newborn screening in other countries.49,50 These are discussed below.
The newborn DMD screen is optional in Wales, as is testing for the other conditions on the bloodspot card. However, parental consent for DMD screening is sought separately from the other conditions, with a signature required on the bloodspot test card to confirm consent. Between 1990 and 2010, parents of 92.6% of parents of boys accepted newborn screening, 6.1% declined, and 1.3% defaulted.(S Moat, personal communication) Defaults were cases where parents did not consent or decline and the lab was not able to gain consent despite repeated attempts to do so.

UK NSC External Review
Page 20
Between 2002 and 2010, monitoring of the Welsh newborn screening programme showed that 12 out of 18 parents whose babies had tested positive for DMD were in favour of newborn DMD screening (66.7%), and six undecided (33.3%). None reported being against screening. Among the 22 families where the baby was identified as having transiently increased CK levels (i.e. false positives on the initial screen), 72.7% were in favour of newborn DMD screening, 13.6% were against, and 13.6% were undecided.
The presentation reported that an assessment of anxiety showed an average score within the normal range (9 to 12) for families with transiently elevated CK levels (i.e. false positives; average 10.38, range 6 to 18), and elevated anxiety scores among those families where DMD had been diagnosed (average score 14.70, range 8 to 24). It was not possible to draw firm conclusions based on this limited data.
One publication described the opinions of 18 mothers whose babies had recently received newborn screening in Wales (about 6-9 weeks previously).47 Ten of the women (55.6%) had accepted DMD screening, and all had accepted screening for cystic fibrosis, phenylketonuria, and congenital hyperthyroidism. Only women whose babies had normal newborn screening tests were included, so the results may not be representative of women whose babies were found to have DMD or other condition on newborn screening.
The mothers were reported to generally perceive screening as a “relatively unimportant routine procedure”, with little recognition of the differences between the different diseases being screened for in effect or treatability. They also saw newborn screening as something that was imperative and had to be done, in contrast to antenatal screening which they felt was a choice. The paper concluded that “the nature of consent required for each test needs to be clarified”, so that “mothers are aware that some tests are advisable whereas others, for less treatable diseases, are a matter of individual choice”. It also noted that information regarding newborn screening needs to be given during pregnancy.
Another study from Wales compared two different protocols for gaining consent for newborn screening.45 The study was carried out due to concerns that the addition of DMD screening to the screening panel of more treatable diseases using the same heel prick sample card does not distinguish DMD as a less treatable condition. This is a concern that has been echoed by other articles discussing screening.39,44
The intervention involved placing the newborn bloodspot for DMD on a separate card to the other bloodspots (e.g. for phenylketonuria) to emphasize the different nature of the test. The control group used the practice of placing all the bloodspots on one card, but requiring an opt-in signature for the DMD screening test, which is the approach used in the Welsh screening programme.
During the study 3,648 boys were born and 37% of mothers (n=1,347) filled in the study questionnaire. The majority of mothers in both groups accepted the test, although uptake in the intervention group was lower (91% vs. 96% in the control group; significance of difference not reported). Mothers in the intervention group were more likely to have high satisfaction levels with the information they received than those in the control group (p=0.001). Mothers in the intervention group were also more likely to know that the test was an extra test that they could choose to have (85% intervention vs. 79% control; p=0.002). More women in the intervention group also felt that their midwives had given them a choice about having the test (96% intervention vs. 93% control; p=0.001). There was no difference in the level of worry reported by the two groups. Women in the intervention group who accepted the test were less likely to give

UK NSC External Review
Page 21
a reason relating to the test being “routine” than women in the control group (13% intervention vs. 19% control; p≤0.01). The fact that some women in the intervention group still did not realise the test was optional, suggested that even with a rigorous protocol for obtaining consent, the decision to screen may not be fully “consented”.
A second publication on a similar intervention study focused on mothers’ reasons for accepting or refusing the newborn DMD screening.46 The study was carried out in three areas in the UK. A total of 2,124 women were offered newborn DMD screening, and 73% returned a sample card and returned a short questionnaire about their reasons for accepting/refusing the test.
Of the 1,363 who gave a reason for their decision, 82% had agreed to the test and 18% declined. Among the 1,655 reasons provided there were three themes. Firstly, most reasons given related to the ability of the test to detect an abnormality (1,135 reasons; 74% for, 26% against). Secondly, that the test was taken or not taken for reasons of reassurance, i.e. without acknowledging the possibility of an abnormality being detected (318 reasons; 96% for, 4% against). Thirdly, that the test was “routine” (202 of the reasons; 93% for, 7% against), and not something different that required more specific thought because the disease was untreatable. For those who accepted the test, the benefits were reported to be knowledge, time to prepare and get early help, and choice in future pregnancies. For those who refused the test, an early diagnosis was seen as being potentially harmful as there was no cure, as having the potential for causing stress, and as having an impact on other family members.
Overall, 72% of women gave an answer that indicated that they knew the test might detect DMD, and 9% gave an answer that indicated that the test was routine. Women who accepted the test were more likely to see the test as routine or as providing reassurance that those who refused it. Women who refused the test were more likely to give a reason that indicated that they understood that it might detect an abnormality (p≤0.001). Women with a professional occupation were more likely to give a reason that related to abnormality detection (78%) than those with a skilled occupation (69%) or other occupation (64%). Women with a skilled occupation (21%) or other occupation (30%) were more likely to give a reason that related to reassurance than those with a professional occupation (12%; p≤0.001).
A systematic review from 2004 looked at psychosocial aspects of genetic screening of pregnant women and newborns.51 It identified 28 publications regarding newborn screening, three of which related to DMD and were from Wales and Canada. The paper from Canada reported that there were seven subsequent pregnancies and prenatal diagnosis was only performed in two of these pregnancies. There were two affected boys born from these pregnancies. The review reported that in the Welsh screening programme the guiding principles were the provision of informed parental choice and reduction in parent distress; this approach was reported to be successful in terms of parental satisfaction.
The review’s main findings (not limited to DMD screening) included that generally levels of knowledge adequate for decision making were not being achieved despite information leaflets and videos having some effect, and that informed consent for neonatal screening had been little studied. The more recent findings from the Welsh programme above suggest that gaining fully informed consent is still challenging.
A third study from Wales investigated whether there was a link between uptake of newborn DMD screening and social deprivation.48 It studied uptake in a three month period across Wales. Of the 4,000 boys born in this period the researchers selected a sample of 1,069 who accepted the test and 567 who refused the test (2:1 ratio), to allow a reasonably sized sample of those

UK NSC External Review
Page 22
who refused. There was a weak association between total deprivation (based on electoral ward) and uptake that was of borderline significance (p=0.05). Uptake was higher than non-uptake in the most deprived areas, and the least deprived areas showed the greatest non-uptake (figures not reported).
A study from 2010 surveyed the acceptability of newborn screening for treatable and untreatable disorders in 1,372 prospective parents (96.5% women) in the Netherlands (participants had to be pregnant or planning to have a child in the near future).49 They found that 88% of the prospective parents showed a positive attitude to newborn screening for less treatable childhood onset disorders, and 73% reported that tests for untreatable childhood onset disorders such as DMD should be added to the newborn screening programme if a valid test was available. However, 34.8% of people stated that this was because the child would live longer if diagnosed early, even though they had been told that the question related to conditions that were untreatable. A similarly common reason (34.3%) was to prevent a long “diagnostic quest”. The main reason for saying that there should not be screening was that the disease could not be prevented or cured (58.5% of reasons). The positive response to screening for untreatable conditions led to the authors suggesting that there should be active debate about the possibility of including such disorders in newborn screening programmes, and that parental views should be given more weight.
Another study used focus groups to assess the opinions of 36 parents and ‘parents-to-be’ in the Netherlands on expanding newborn screening.50 The groups included parents of healthy children and parents of children affected by diseases including cystic fibrosis, phenylketonuria, DMD, and celiac disease. With regards to DMD, most of the arguments for screening related to the child and family, including reproductive choice and reducing diagnostic delay. The arguments against screening included that it could lead to a loss of normality and a carefree approach to life, prevent the normal development of the child, and lead to over-protectiveness on the part of the parents. None of the participants expressed concerns about parent-child bonding. About half of the participants reported that they would be willing to take part in a newborn DMD screening programme. Some participants said that improvements in diagnostic procedure to reduce diagnostic delay would be preferable to newborn screening. Generally, willingness to take part in screening for diseases where no treatment was available was reported to be much lower than if there was a treatment.
Summary: Criterion 7 partially met
The test appears to be acceptable to parents in Wales, where uptake of newborn screening for DMD is high (92.6% over 20 years). Surveys of families found to have an affected baby and also false positives in the initial screen still found a high proportion in favour of screening (66.7% and 72.7% respectively), although 13.6% of surveyed families which had experienced a false positive were against screening.
A study in mothers who were offered newborn screening tests in Wales and whose tests did not identify any problems suggested that they may not fully understand that newborn DMD screening is optional, and may accept screening because they feel that it is something that has to be done. Another larger study also suggested that some women accept newborn screening because they feel that it is routine or provides reassurance, rather than that because it could detect abnormalities. Another study in the UK also suggested that some mothers’ reasons for their decisions about newborn DMD screening tests are related to the tests being considered routine. These findings raise concern about whether consent to have the screening test is truly informed, at least in a proportion of women.

UK NSC External Review
Page 23
Surveys from the Netherlands suggested that most prospective parents would be in favour of screening even for untreatable childhood onset disorders.
8. There should be an agreed policy on the further diagnostic investigation of individuals with a positive test result and on the choices available to those individuals
2004 report: “Yes”
The DMD Care Considerations Working Group guideline from 2009 does not make recommendations regarding screening, but does make recommendations for diagnosis.2
They recommend initial testing for DMD in suspected cases by testing creatine kinase levels. If elevated levels are found then genetic testing for DMD mutations and/or muscle biopsy to look for the presence or absence of dystrophin are recommended. A muscle biopsy showing absence of dystrophin needs confirmation by genetic testing. Identification of the mutation provides information for genetic counselling and prenatal diagnosis, and may help to predict the likely severity of the phenotype. The nature of the mutation also determines eligibility for certain new treatments in development (see Criterion 10). The guideline describes the techniques commonly used for genetic diagnosis, but note that these are not universally available.
An open muscle biopsy is recommended if differential diagnosis includes DMD and other possibilities, to ensure adequate tissue is obtained. A muscle biopsy is not considered essential for those with a genetic diagnosis, especially as a muscle biopsy may be viewed by the family as traumatic. A muscle biopsy can be used to distinguishing DMD from milder phenotypes. If creatine kinase is elevated and there are signs or symptoms consistent with DMD, but no genetic mutation can be identified, then a muscle biopsy is needed. A muscle biopsy is also recommended if there is a family history of DMD and a suspicion of DMD in the patient, but no family mutation known.
Once a diagnosis is confirmed referral to a multidisciplinary team should be made, and patient and family support and contact with patient organisations offered. Genetic counselling is highly recommended for any at risk female family members.
In addition, in 2010 best practice guidelines for molecular testing for DMD/BMD were released.29 These guidelines were developed during a meeting of 29 international scientists and clinicians from 21 countries including the UK and other European countries, USA, India and Australia. The guideline outlines the procedures for molecular testing in male patients, carrier detection for a known familial mutation, carrier detection for an unknown mutation, diagnosis of manifesting carriers, prenatal diagnosis, pre-implantation genetic diagnosis, and haplotype analysis. It also discusses interpretation of results, and result reporting.
Both of these guidelines are endorsed by the European TREAT-NMD group.52,53 The European Federation of Neurological Societies (EFNS) also published guidelines on the molecular diagnosis of neurogenetic disorders such as DMD.31
The Welsh newborn screening programme has a set process of investigation following identification of a raised CK level on bloodspot testing (see Criterion 5 for details).

Summary: Criterion 8 met
There are international guidelines on appropriate diagnostic investigations, including molecular diagnosis. The Welsh programme has a set process of investigation following identification of a raised CK level on bloodspot testing.
9. If the test is for mutations the criteria used to select the subset of mutations to be covered by screening, if all possible mutations are not being tested, should be clearly set out
2004 report: N/A
Although DMD is a genetic disorder, the screening test is for elevated levels of creatine kinase in the blood. Follow up genetic testing is part of the subsequent diagnostic investigations. The mutations that cause DMD and BMD are very varied, and the gene is very large.1 This would make screening based only on mutation analysis very challenging based on currently available technology.
Summary: Criterion 9 not applicable
10. There should be an effective treatment or intervention for patients identified through early detection, with evidence of early treatment leading to better outcomes than late treatment
2004 report: “No treatment has been shown to have long term benefits for the child with the condition. There is some work to suggest that treatment with corticosteroids produces improvement in function, but long term studies are awaited. The main benefits of early diagnosis are avoidance of further affected births and a chance for the family to plan their future life style.”
There is still no curative treatment for DMD, although the existing treatments have been reported to improve outcomes and extend lifespan.2 There are also a number of genetic treatments in development which may provide further improvements.
There are few large RCTs of treatments for DMD. A recent international guideline produced by the DMD Care Considerations Working Group (DCCWG) on the diagnosis and management of DMD acknowledged this and therefore developed recommendations using the RAND Corporation-University of California Los Angeles Appropriateness Method (RAM).2,23 This method is reported to combine research evidence with the collective judgement of experts to decide on the appropriateness and necessity of assessments and interventions. The guideline included a systematic search for research published up to 2006, and also incorporated major studies published up to mid-2009 highlighted by expert discussions. We have used this guideline as the basis for our discussions of treatments for DMD.
The guideline reports that current treatment of DMD is multidisciplinary, and that “anticipatory and preventive clinical management of DMD is essential”.
We identified other guidelines regarding specific aspects treatment for DMD,16,37,54-56 but have focussed on the DWCC guideline as it is the most recent international guideline and covers all areas of diagnosis and management.

UK NSC External Review
Page 25
Corticosteroid treatment
The guideline reports that the only treatment reported to slow the decline in muscle strength and function in DMD is glucocorticoid treatment.2 It also says that glucocorticoid treatment reduces risk of scoliosis and stabilises lung function, and may improve heart function. As such we have focussed largely on this aspect of treatment in this Criterion, particularly the age at which this treatment is recommended to be started. The aim of corticosteroid treatment is to maintain the ability to walk, and minimise risk of other later complications.
The DCCWG guideline recommends considering glucocorticoid therapy in all patients with DMD. It suggests a daily regimen of prednisone or deflazacort, with the decision of which steroid based on availability, cost, formulation, and side effect profile. The recommended starting dose is 0.75mg/kg daily for prednisone and 0.9mg/kg daily for deflazacort.
The potential benefits of corticosteroid treatment need to be balanced against the side effects associated with long term glucocorticoid use. These side effects include: growth retardation, obesity, bone demineralisation and increased fracture risk, adverse behavioural changes, cataracts, Cushingoid features, hirsutism, acne, tinea, warts, delayed puberty, immune/adrenal suppression, hypertension, glucose intolerance, gastritis/gastroesophageal reflux disease, peptic ulcer disease, and myoglobinuria. Fractures pose particular problems as they can further limit ambulation.
The guideline outlines recommendations for monitoring and dealing with these side effects. It recommends that a reduction in glucocorticoid dose is required if side effects are not tolerable or manageable. If initial reduction in dose is not successful, further reduction or change in regimen should be attempted, and if this is not successful then termination of steroid treatment may be required.
They say that there are “no generally accepted guidelines” about when it is best to start glucocorticoid therapy. They recommended that the decision is made on an individual basis, based on functional state, and considering the child’s age and presence of risk factors for side effects. The say that in all cases, the national immunisation schedule should be completed and varicella (chicken pox) immunity should be established before starting glucocorticoids. In the UK, childhood vaccinations are mostly carried out before the age of 13 months, with some additional vaccinations given at 3 years and 4 months of age or soon after. Varicella vaccination is not routinely provided to all children in the UK. This would mean that in children diagnosed clinically with DMD, initiation of corticosteroids would have to wait until varicella vaccination had been given and immunity established.
The guideline says that starting glucocorticoids:
is not recommended in children still gaining motor skills, particularly when they are under the age of two
should be proposed once the plateau phase of motor skill development has been clearly identified as being reached (usually at age 4 to 8 years) unless there are substantial reasons for delaying until the decline phase, such as major pre-existing risk factors for side effects
is still recommended if the full decline phase has been reached or when ambulation is more marginal, but might be of less benefit.

UK NSC External Review
Page 26
They say that these recommendations should be seen as a “minimum threshold”. They say that some doctors favour earlier intervention when clinical symptoms first appear, but that there is no published data to support this, so the panel did not feel it was appropriate to endorse earlier steroid treatment.
As identification of the motor skill plateau stage is based on serial assessment and parental report, they recommend caution in starting glucocorticoid treatment on a first visit or second opinion consultation, particularly in children aged less than 6 years. A note in the guideline suggests that glucocorticoids are typically started at age 6 years (±2 years). Data from a national UK audit on 240 ambulant boys was in agreement with this figure, reporting a median age of steroid initiation of 6.3 years, supporting this as the typical age of initiation.11
These recommendations suggest that initiation of glucocorticoid therapy would not be likely to be started from the time of identification at newborn screening. It would be likely to start when child has reached the plateau phase, usually between the ages of 4 to 8 years. A national UK audit on corticosteroid practice from 2010 suggested that median age of DMD diagnosis in the UK is age 4.1 years.11 Therefore, although early diagnosis (e.g. by newborn screening) would allow routine assessment of motor function to start earlier in life, it would not necessarily result in earlier glucocorticoid treatment than opportunistic diagnosis. Any studies looking at the effect of earlier diagnosis need to avoid lead-time bias, which could make ambulation and survival duration appear to be prolonged due to earlier diagnosis.
Any benefits of earlier treatment would need to be weighed up against increased risks of side effects from prolonged glucocorticoid treatment.
Update on research evidence on corticosteroids
The most recent Cochrane systematic review on corticosteroids (search date 2006) identified six placebo controlled RCTs (n=303) in boys with DMD that met inclusion criteria.14 Five of the studies assessed prednisolone and one deflazacort. Four of the studies assessed a daily corticosteroid regimen. The studies included mostly ambulant boys (79%), and one trial included boys with BMD (4 boys, about 10% of participants). The boys included in the studies ranged in age from 4 to 19.4 years.
Only one RCT (n=28) looked at prolongation of walking. It found a 13 month improvement in ambulation time with deflazacort, with a mean interval for becoming wheelchair bound of 33.2 months with deflazacort, and 20.5 months with placebo. However, this analysis did not take into account that some boys were still walking at the end of the study (13/17 in the deflazacort group, and 5/11 in the placebo group), so the data for this outcome is incomplete. Also, the boys in both groups lost ambulation at a similar age (108 months with deflazacort and 104 months with placebo), and ages of the boys still walking was not reported. Therefore it is not possible to rule out that the extension in walking in this study could be related to differences in the age of the boys in the groups.
Glucocorticoids improved muscle strength and function over 6 months. For example, prednisolone 0.75mg/kg/day improved average MRC muscle score by 0.50 points (95% CI 0.35 to 0.66; 3 RCTs, n=147). They also reduced time take to rise to a standing position by 2.43 seconds (95% CI -3.30 to -1.56; 3 RCTs, n=147), and 9 metre walking time by 2.64 seconds (95% CI -3.70 to -1.58). In terms of side effects, at 6 months this dose of prednisolone increased weight gain (9.27% increase, 95% CI 6.87% to 11.68%; 2 RCTs, n=126), and increased risk of behavioural changes (RR 1.38, 95% CI 1.04 to 1.83), Cushingoid features (RR 2.46, 95% CI 1.58 to

UK NSC External Review
Page 27
3.84), excessive hair growth (RR 2.66, 95% CI 1.50 to 4.72), and increased appetite (RR 1.80, 95% CI 1.09 to 2.99).
One small trial (n=28) suggested that glucocorticoids stabilised muscle strength and function for up to two years. The most effective prednisolone regimen appeared to be 0.75mg/kg/day. The trials were short term (6 months to 2 years), so could not provide evidence about the efficacy or safety of glucocorticoids in the long term. The potential benefits associated with corticosteroid treatment mean that it is unlikely that long term placebo controlled trials will be carried out in DMD.
The Cochrane review also tabulated and discussed the studies it excluded, mainly observational studies. It lists 31 non-randomised studies (some controlled some un-controlled), in 1,425 boys aged between 2 and 30 years (there may have been overlap in some studies). These studies reported on periods of treatment ranging from 3 weeks to 18 years. Only six studies included children aged less than 4 years. These six studies ranged in size from two to 111 children (total n=205); what proportion of the total were under the age of 4 was not stated. The children in these six studies received treatment for between 1 month and over 5 years.
Only one small study (Merlini 2002) exclusively included children aged 4 and under. It included eight boys aged 2 to 4 years, and the five treated boys received treatment for about 4 to 5 years. It found that ability to rise from the floor was prolonged with corticosteroids, but that ability to climb stairs and 10 metre walking time was similar in treated and untreated boys. There was growth rate decline in the treated boys. Another uncontrolled study (Kinali 2002) included four children aged 3 years and 10 months to 4.5 years old who were treated with an intermittent low dosage steroid regimen (0.75 mg/kg per day for 10 days per month, or 10 days on and 10 days off). The study reported that the boys gained the ability to rise from the floor without Gowers' manoeuvre, hop on one or both legs, and run without a waddle. It found that there was stabilisation of motor function for up to 5 years. One boy lost the ability to walk at the age of 9 years, after 5 years of treatment. Bone mineral density was reported to be normal on DEXA scans at 1-6 years of treatment. The study was reported to suggest “that the beneficial effects of glucocorticoids appear to be greater when treatment is initiated at a younger age, in the early ambulant phase”. However, this study is very small, and did not include a control group of children not receiving treatment until a later age for comparison of long term results. This limits the conclusions that can be drawn from it.
Overall the Cochrane review reported that the excluded, non-randomised cohort studies suggested long-term (five years) functional benefit from corticosteroids, at least in some of the patients. They reported that a prolongation of time to loss of walking ability with corticosteroids was reported in many studies. They also noted that long-term pulmonary and cardiac benefits had been reported in some of these studies, but needed to be confirmed. The review also suggested that the increase in muscle strength occurs over the first six months after starting treatment, but this increase does not appear to continue indefinitely. They say that there may be stabilization for up to two years, followed by a decline, which may be slower than that in untreated controls.
The review stated that only one non-randomised study reported the impact of corticosteroids on survival (Biggar 2006),57 and was the first to their knowledge to do so. It studied boys aged 10 to 18 years, with those who received treatment doing so for a mean of 5.5 years. It found that 5% of the deflazacort treated boys died during follow up (2/40; at ages 13 and 18 years), compared with 35% of the untreated boys (12/34; mean age at death 17.6 years). No statistical comparisons were provided in the review.

UK NSC External Review
Page 28
The review notes that the studies and expert option support the need for close monitoring of adverse effects, with dose reduction needed in the long term to ensure tolerability. Although intermittent steroid regimens may potentially reduce the risk of side effects, there was only one RCT of an intermittent regimen, which was short term, did not look at prolongation of walking, and did not present data in a way that could be compared with other regimens.
The review concluded that RCTs have shown that glucocorticoid corticosteroids improve muscle strength and function for six months to two years, and respiratory muscle strength and function for six months in patients with DMD. The most effective prednisolone regime was concluded to probably be 0.75mg/kg daily; there was not enough data to compare the efficacy of prednisone and deflazacort. The long-term benefits and harms were not clear but non-randomised studies suggested that clinically significant prolongation of time to loss of walking was possible. However, potential harms are significant, including weight gain, behavioural changes, vertebral fractures and cataracts.
The review also concluded that RCTs are still needed to determine the ideal age or functional stage for starting and discontinuing corticosteroid treatment, as well as the optimal treatment regimen. This conclusion is also supported by another recent narrative review discussing the long term benefits of corticosteroid treatment.15
We assessed the additional papers we identified as being published since 2004 that had not been described in the Cochrane review. We focused on those studies solely in patients with DMD/BMD, with non-historical control groups of patients who also had these conditions but who did not receive steroid treatment.
The update search identified two abstracts describing an RCT in 64 boys aged 4 to 10 years, comparing daily steroids (0.75mg/kg prednisone) with intermittent high dose steroids (10mg/kg divided over two days each week) over 12 months.58,59 It found similar improvements in muscle strength in both groups, with the intermittent group showing greater growth (p=0.004) and less weight gain initially (p=0.04 at 3 months; p=0.07 at 12 months).58 However, the daily steroid group showed improvements in behaviour compared with the intermittent group (p≤0.01 for total problems, externalising, attention, and aggressive behaviours on the Child Behaviour Checklist).59
The UK studies identified are described separately below. The remaining six controlled studies included 779 children whose average age at starting steroid treatment was between about 5.4 and 9.1 years.60-65 Where stated, most studies reported the lowest age of starting treatment of about age 4, with one study reporting an age of 1.9 years as the lower 95% confidence interval for the mean age at initiation (7.3 years).64
These papers generally supported that steroid treatment was associated with a reduced need for spinal surgery; better preservation of cardiac function; delayed wheelchair dependency and possibly ventilator dependence.61-65 The studies also reported on the side effects of corticosteroids. One study was mainly an audit of corticosteroid use in the US, and did not quantify benefit.60 It reported that the most common reasons for discontinuing steroids included weight gain (19%), behavioural side effects (16%), and loss of ambulation resulting in full time wheelchair use (13%). They noted that there was substantial variation in clinical practise between sites.
The observational design of most of the studies of corticosteroids cannot preclude the possibility that factors other than corticosteroid treatment differences could be contributing to differences seen.

UK NSC External Review
Page 29
One of the additional studies identified was a conference abstract describing a case series in four boys who started steroid treatment at the mean age of 3.4 years (range 2.4 to 4 years).66 There may be overlap between these boys and eight boys described in the paper in the Cochrane review by the same authors (Merlini et al). These boys received daily corticosteroids at a dose of 0.75mg/kg for the first month, and then 1.25mg/kg. At last follow up at average age 13.15 years (range 12.2 to 14.9 years), all boys were still ambulant, and the two youngest could still run 10 metres and climb four stairs without using the banister. The two oldest could also climb four steps, but needed support from the bannister. The oldest had lost the ability to rise from the floor at age 12.5 years, while the others could still perform this task. They gradually became overweight with treatment and their final BMIs were in the 75th to 90th percentile. Two of the children had heights below the 3rd percentile, and the other two were in the 10th and 25th percentiles. Puberty was delayed in all four boys, and three had started on testosterone. The boys were reported not to have: compression fractures, high blood pressure, diabetes, gastrointestinal bleeding, psychosis, or cataracts. The authors reported that the corticosteroids had increased ambulation by 3-4 years on average from what would be expected. However, without a control group who received corticosteroids at a later age, it is not possible to judge the relative benefits of starting corticosteroids early. Also, the number of boys was very small and the findings were not fully published in a peer reviewed journal, appearing in abstract form only.
The update search also identified a case series of 73 patients from the USA (average age at starting steroids 9.2 years, range 4.5 to 19.6 years),15 a case series of 90 boys from Brazil with a historical control group (average age at starting steroids 7.6 years, range 4.1 to 10.9 years),67 and a case series of 35 boys from the Netherlands with a historical control group (average age at starting steroids 6.5 years, range 3.5 to 9.7 years).68
UK corticosteroid practice
The update search identified one abstract describing a national audit of corticosteroid practice, based on 17 UK centres.11 It reported that data from 240 ambulant boys aged 3 to 18 years was available. Most of these boys (92.9%) were receiving corticosteroids, and the median age for starting treatment was 6.3 years. Most boys had started on prednisolone (203), with ten starting on deflazacort. Over half the boys started on an intermittent corticosteroid regimen (117 boys), and 83 started on a daily regimen.
Vertebral fractures occurred in 30 steroid treated boys (13.5%), at an average age of 11.5 years (range 7.1 to 15.5 years). The fractures were symptomatic in 78% of boys. The average time between starting steroids and the vertebral fracture was 4.1 years (range 0.7 to 7.4 years).
The update search identified other abstracts describing a UK population, but these were all smaller or published earlier than the national audit above.69-72 Therefore these other abstracts may include individuals also included in the national audit described above.
Other drug treatments
The guideline does not recommend oxandrolone (an anabolic steroid). It also noted that botulinum toxin had not been tested for contractures in DMD, and its use is thought to be inappropriate. It made no recommendations regarding the use of creatine, and noted that an RCT of creatine had not shown clear benefit. They also made no recommendations about supplements (e.g. amino acids, coenzyme Q10, anti-inflammatories/antioxidants) as there was no supportive data from the literature and no expert opinion consensus from the panels. They

UK NSC External Review
Page 30
did not assess potential disease modifying drugs (e.g. pentoxifylline) as this was an area where additional research was needed.
Update on research evidence on other drug treatments
One systematic review identified in the update search suggested the potential for some short to medium term benefit in muscle strength with creatine, but other systematic reviews and RCTs did not identify conclusive evidence of benefit with other treatments. Most of these RCTs would have been available at the time of the evidence review for the DCCWG.
One Cochrane review of creatine for muscle disorders (search date 2010) included 14 RCTs in various conditions (n=364 participants).73 This included five RCTs in DMD, DMD and BMD or DMD and other conditions (147 with DMD/BMD). Two of these RCTs were in steroid naïve boys (aged 3 to 12 or 4 to 10 years); therefore they may not be representative of the current clinical practise where steroids are recommended as the standard of care. The other RCTs included boys with DMD whose average age was about 10 years, and one included BMD patients with average age 23 years.
When looking at the pooled results for the trials including DMD/BMD only, there was an improvement in maximum voluntary contraction with creatine, but no significant effect on change in MRC muscle score. Overall the review concluded that short and medium term creatine increased muscular strength in muscular dystrophies, but that there is a need for long-term studies of creatine in muscle diseases. No recommendations regarding creatine are currently made by the DCCWG.2
A Cochrane review of calcium antagonists for DMD identified 5 RCTs or quasi-randomised studies (n=196).74 It found that there was no evidence to show a significant beneficial effect of these drugs on muscle function.
One double blind RCT in 26 boys with DMD suggested that both oral glutamine and amino acid supplementation over ten days equally inhibited whole body protein degradation.75 However, a follow up double blind crossover RCT from 2009 found no difference in improvement in walking speed or other muscle function measures with four months of oral glutamine supplementation compared with placebo.76 Another double blind placebo controlled RCT (n=50, three groups, one creatine, one glutamine, one placebo) also reported no significant effect of glutamine on muscle strength in ambulant boys with DMD.77
A double blind placebo controlled RCT (n=153) failed to show any effect of ciclosporin A, either alone or as an adjunct to steroid treatment, on muscle strength or functional ability in ambulant boys aged 5 and over with DMD.78 A double blind placebo controlled crossover RCT (n=14) found no effect of 12 weeks treatment with extended release albuterol (a beta-2 agonist) on manual muscle tests or knee moments in ambulatory boys with DMD.79 There was a small but significant difference in change in time taken to walk or run 30 feet: no change was seen while using albuterol, but an increase of 0.3 seconds with placebo (p=0.025). There was also a significantly greater increase in lean body mass with albuterol (5.6kg, 95% CI 1.1 to 10.1; p=0.018), and a reduction in fat mass (-6.4kg, 95% CI -12.1 to +0.7, p=0.03). The boys in this study were aged 6 to 11 years and had never received steroid treatment. Further RCTs would be needed to confirm these findings.
Another placebo controlled RCT of the ACE inhibitor perindopril in 57 children (average age 10.7 years, range 9.5 to 13 years) with DMD with normal cardiac examination and left ventricular ejection fraction (LVEF) >55%.80 The children received perindopril or placebo in a double blind

UK NSC External Review
Page 31
fashion for three years, followed by two years of open label perindopril use in all participants. At both three and five years there was no difference between the groups in average LVEF. However, at five years the proportion of participants with LVEF<45% was lower in the group that initially received perindopril than in the group that initially received placebo (p=0.02). The DCCWG guideline cited this study, but stated that further confirmatory studies are needed before firm recommendations can be made.
The update search also identified a cohort study looking at the benefits of using bisphosphonates in boys with DMD who had taken steroids for at least 1 year (n=44).81 Sixteen boys used bisphosphonates (69% pamidronate, 6% pamidronate followed by alendronate, 19% alendronate, and 6% clodronate). At the time of the study boys had been treated with bisphosphonates for between 0.5 and 12 years, and 63% were still receiving bisphosphonates. The study found that boys who used bisphosphonates survived longer than those who did not (p=0.005). The median age at diagnosis in this cohort was 5 years, steroids were started at age 7 years, and bisphosphonates were started at median age 12.5 years (range 7-23 years). The DCCWG guideline reports that intravenous bisphosphonates such as pamidronate for vertebral fracture are indicated, but oral bisphosphonates as a treatment or prophylactic measure remains controversial.2,23
Other non-drug treatments:
The DCCWG guideline lays out the care considerations for the different stages of DMD.2,23 In the presymptomatic stage (the stage at which newborns would be diagnosed), the measures that are recommended include:
diagnostic examination and genetic counselling
planning for future neuromuscular developments
ensuring that the immunisation schedule is complete and includes 23-valent pneumococcal vaccine (for those aged 2 and over) and influenza vaccine (annual immunisation with trivalent inactivated vaccine for those aged 6 months and over)
rehabilitative management such as preventive measures to maintain muscle extensibility, encouragement and support for appropriate exercise/activity and function, provision of adaptive devices as appropriate, and education on rehabilitation management
an echocardiogram at diagnosis or by age 6
monitoring of weight and nutritional assessment for those who are over or under weight
provision of psychosocial support for the family
early assessment for development, learning, and behaviour, with intervention as needed.
With regard to psychosocial problems, the guideline reports that these problems may lead to more stress for many parents than the physical aspects of the disease. The guideline suggests that the emphasis should be on prevention and early intervention to maximise potential outcome. They recommend that these problems should be treated with the same evidence-based interventions used in the general population. This suggests that children with psychosocial problems would be treated in the same way regardless of whether they had yet received a DMD diagnosis.

UK NSC External Review
Page 32
Similarly, some other aspects of the recommended assessments and interventions might be offered in the absence of a DMD diagnosis, e.g. for children who are over and underweight, or who present with muscle problems. Some seem less likely to be offered in the absence of a diagnosis, for example, the pneumococcal and flu vaccinations, echocardiogram, and some rehabilitation measures.
No specific pieces of evidence were provided to illustrate whether children with DMD identified through newborn screening have better outcomes than children diagnosed after the onset of symptoms.
In the early ambulatory phase similar rehabilitation activities and psychosocial support continue. Weight, and neuromuscular, pulmonary, and cardiac function are monitored, with a maximum of 2 years between cardiac investigations up to the age of ten years. Promotion of independence and social development also starts.
If diagnosis is opportunistic the guideline reports that the child is likely to be diagnosed by the late ambulatory stage. At this stage surgical interventions may need to be considered for tendo-Achilles contractures in certain situations. Rehabilitation management continues as before, with additional provision of appropriate aids and adaptations (such as a wheelchair) to allow maximum independence, function and participation. Intervention may be needed if there are cardiac problems, even if asymptomatic. Once the boy becomes non-ambulatory monitoring is needed for complications such as scoliosis, respiratory complications, and cardiac problems, and interventions for these as appropriate.
Update on research evidence on non-drug treatments:
One Cochrane review (search date 2009) on interventions to increase ankle range of motion (ROM) in patients with neuromuscular disease identified two RCTs.82 One was an RCT of steroid which was included in the Cochrane review of steroids described above; it found no effect of steroids on ankle ROM. The second RCT compared early surgery in ambulant boys aged 4 to 6 years with DMD with routine management. This RCT found that although there was increased ankle dorsiflexion range at 12 months with surgery, functional outcomes were better in boys in the control group. By 24 months many of the surgically treated boys were reported to have had relapse of Achilles tendon contractures. The review concluded that there was no evidence of significant benefit from any intervention for increasing ankle range of motion in DMD.
The RCT of surgery described above was also the only study of DMD in a Cochrane review of rehabilitation interventions for foot drop in neuromuscular disease Search date 2009).83 Again the authors concluded no significant effect of the surgery.
A Cochrane review on surgery for scoliosis in DMD (search date 2010) identified no RCTs of this procedure.84 It noted that patients should be informed of the uncertainty of benefits and potential risks of surgery.
The update search identified a systematic review of exercise for DMD was reported as a conference abstract, and we identified the full review available as a dissertation.85,86 This review only included studies in boys aged 5 and over. It identified 15 studies, most of which were case series. It found that muscle strength increased early in the intervention, but then decreased, although this was as a rate lower than the normal decline. Respiratory exercises improved muscle endurance but not muscle strength. Low frequency electrical stimulation led to a low level of muscle strength improvement in younger patients. However, they note that the low quality of the included studies makes drawing firm conclusions difficult. The DCCWG also notes

UK NSC External Review
Page 33
that limited research has been carried out on the type, frequency, and intensity of exercise that is optimum in DMD.
One conference abstract from 2006 described the comparison of outcomes of a group of eight DMD patients who had long term rehabilitation exercise and another group of eight DMD patients who had no rehabilitation exercise.87 It found that those who had rehabilitation exercise had no obvious spinal deformity or extremity atrophy, compared to 75% of the untreated group. There was reported to be significant differences in some measures of lung function (for example FVC and FEV1) but not in the ratio of FEV1/FVC. The study did not appear to be randomised, was very small, was not a full peer reviewed publication, and provided very limited details of its methods. For these reasons there is uncertainty about the reliability of these results.
A publication from 2010 has described the protocol for an ongoing trial in the Netherlands looking at the effects of physical training in boys with DMD.88 This report noted that the current recommended voluntary exercises are aimed at maintaining comfort and symmetry, and are largely based on theory. This trial is reported to be the first clinical trial assessing the effects of low-intensity physical training on muscle endurance and functional abilities in boys with DMD.
The trial described (the No Use is Disuse [NUD] trial) includes an exploratory unblinded RCT, which will include 30 ambulant and recently wheelchair bound boys with DMD. It is comparing dynamic arm and leg exercise training versus usual care for 6 months, and the primary outcomes are muscle endurance and functional abilities. Results of the study were reported to be expected in 2011.
The update search identified no studies published since 2004 that compared the outcomes of any treatment (steroids, other drugs, or other non-drug treatments) or package of treatments in a newborn screened population with the treatment in a clinically diagnosed population.
New drug treatments
Development of genetic treatments has been slowed due to the large size of the DMD gene, and the fact that it is widely expressed in all muscle types and in the central nervous system.89 The large size of the DMD gene limits its ability to be treated by gene therapy, as the currently available vectors for carrying genes into cells cannot carry this much DNA. The potential for stem cell approaches has also been investigated, but these have not yet been perfected.
Therefore new treatments have focussed on targeting the mutated messenger RNA (mRNA). The most advanced potential new treatments have used two different approaches:
1. Antisense oligonucleotides that restore the normal reading frame of the DMD gene by causing the skipping of an exon affected by the mutation.
2. Drugs that promote the protein making machinery to “read through” premature stop codons, thus making the full length dystrophin protein.
These methods are described briefly here, along with the results of their most advanced trials. The ongoing and completed trials of the most advanced drugs in these groups are summarised in Table 2 and discussed below.
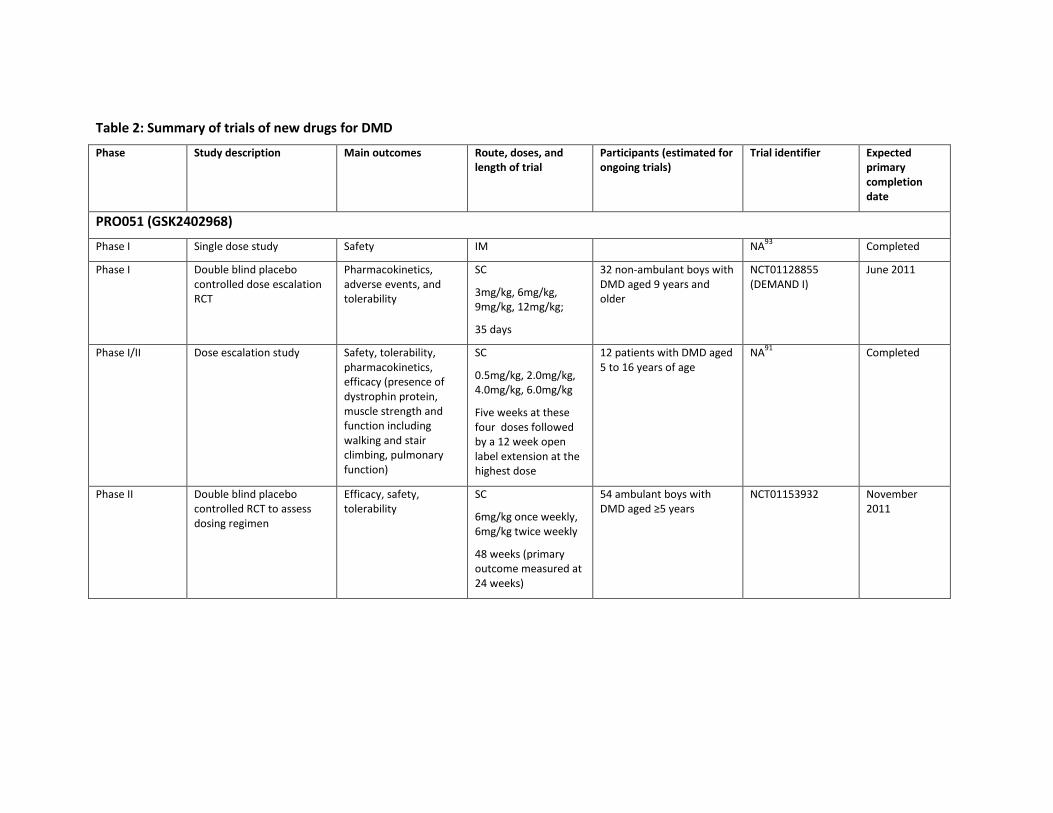
Table 2: Summary of trials of new drugs for DMD
Phase Study description Main outcomes Route, doses, and length of trial
Participants (estimated for ongoing trials)
Trial identifier Expected primary completion date
PRO051 (GSK2402968)
Phase I Single dose study Safety IM NA93
Completed
Phase I Double blind placebo controlled dose escalation RCT
Pharmacokinetics, adverse events, and tolerability
SC
3mg/kg, 6mg/kg, 9mg/kg, 12mg/kg;
35 days
32 non-ambulant boys with DMD aged 9 years and older
NCT01128855 (DEMAND I)
June 2011
Phase I/II Dose escalation study Safety, tolerability, pharmacokinetics, efficacy (presence of dystrophin protein, muscle strength and function including walking and stair climbing, pulmonary function)
SC
0.5mg/kg, 2.0mg/kg, 4.0mg/kg, 6.0mg/kg
Five weeks at these four doses followed by a 12 week open label extension at the highest dose
12 patients with DMD aged 5 to 16 years of age
NA91
Completed
Phase II Double blind placebo controlled RCT to assess dosing regimen
Efficacy, safety, tolerability
SC
6mg/kg once weekly, 6mg/kg twice weekly
48 weeks (primary outcome measured at 24 weeks)
54 ambulant boys with DMD aged ≥5 years
NCT01153932 November 2011
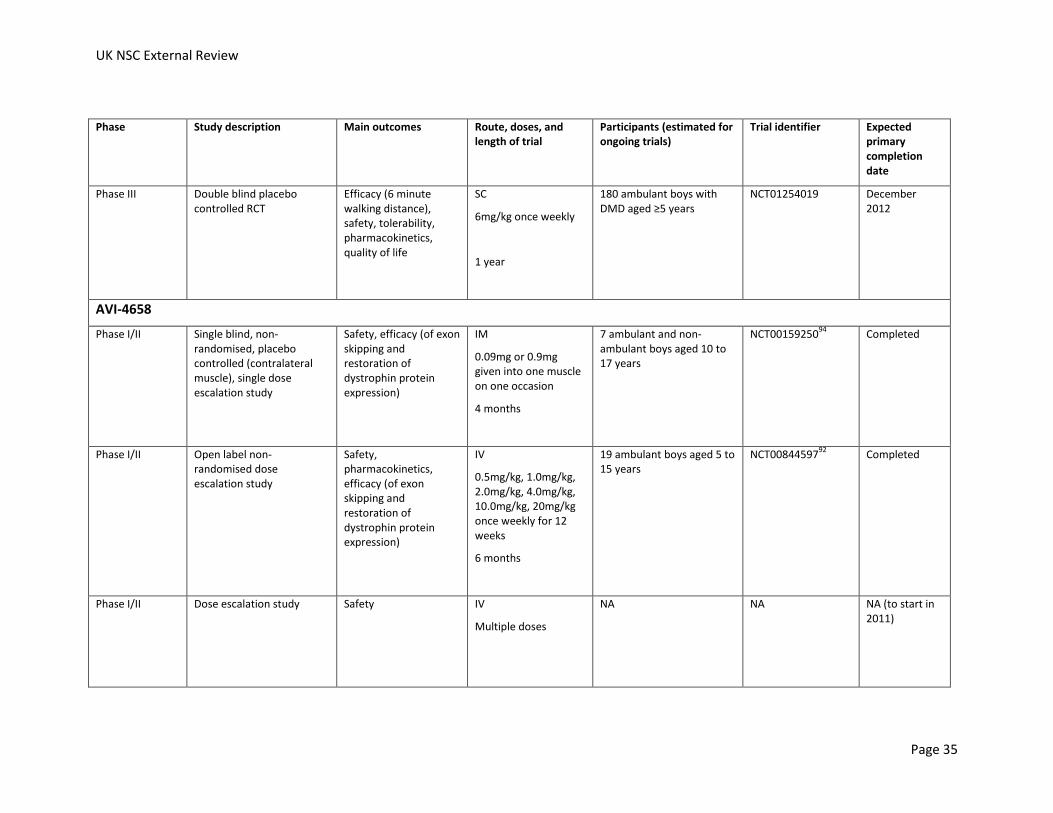
UK NSC External Review
Page 35
Phase Study description Main outcomes Route, doses, and length of trial
Participants (estimated for ongoing trials)
Trial identifier Expected primary completion date
Phase III Double blind placebo controlled RCT
Efficacy (6 minute walking distance), safety, tolerability, pharmacokinetics, quality of life
SC
6mg/kg once weekly
1 year
180 ambulant boys with DMD aged ≥5 years
NCT01254019 December 2012
AVI-4658
Phase I/II Single blind, non-randomised, placebo controlled (contralateral muscle), single dose escalation study
Safety, efficacy (of exon skipping and restoration of dystrophin protein expression)
IM
0.09mg or 0.9mg given into one muscle on one occasion
4 months
7 ambulant and non-ambulant boys aged 10 to 17 years
NCT0015925094
Completed
Phase I/II Open label non-randomised dose escalation study
Safety, pharmacokinetics, efficacy (of exon skipping and restoration of dystrophin protein expression)
IV
0.5mg/kg, 1.0mg/kg, 2.0mg/kg, 4.0mg/kg, 10.0mg/kg, 20mg/kg once weekly for 12 weeks
6 months
19 ambulant boys aged 5 to 15 years
NCT0084459792
Completed
Phase I/II
Dose escalation study Safety IV
Multiple doses
NA NA NA (to start in 2011)
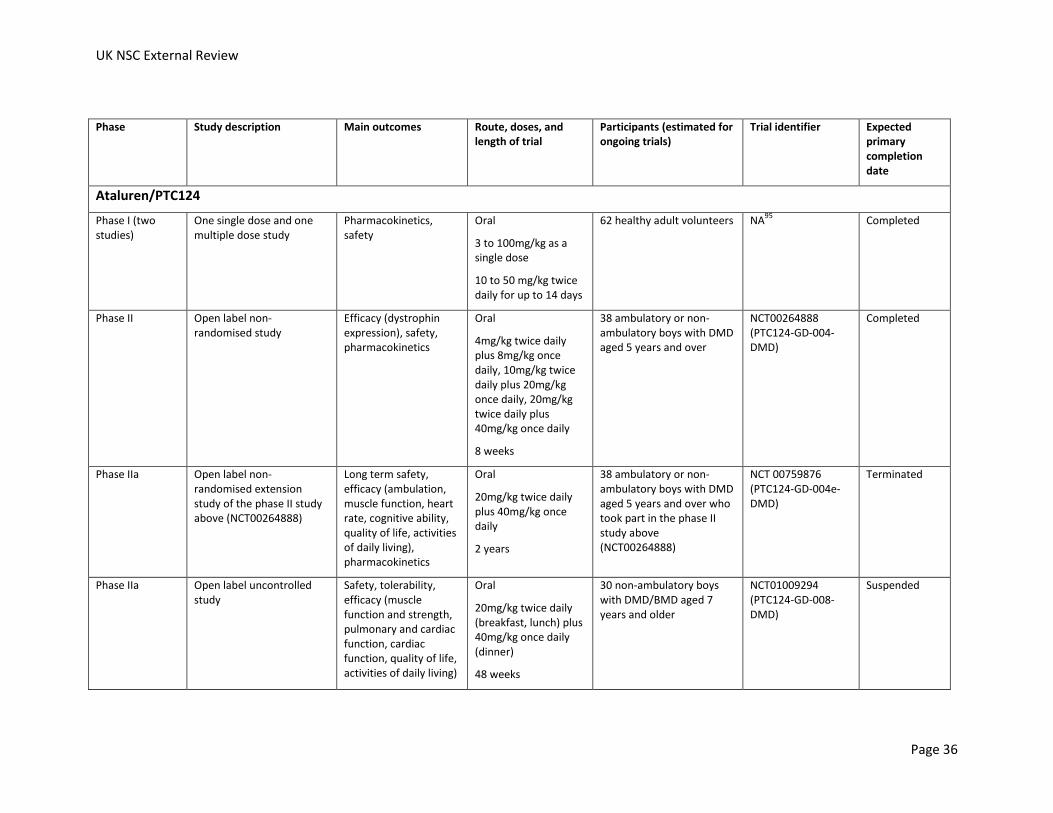
UK NSC External Review
Page 36
Phase Study description Main outcomes Route, doses, and length of trial
Participants (estimated for ongoing trials)
Trial identifier Expected primary completion date
Ataluren/PTC124
Phase I (two studies)
One single dose and one multiple dose study
Pharmacokinetics, safety
Oral
3 to 100mg/kg as a single dose
10 to 50 mg/kg twice daily for up to 14 days
62 healthy adult volunteers NA95
Completed
Phase II Open label non-randomised study
Efficacy (dystrophin expression), safety, pharmacokinetics
Oral
4mg/kg twice daily plus 8mg/kg once daily, 10mg/kg twice daily plus 20mg/kg once daily, 20mg/kg twice daily plus 40mg/kg once daily
8 weeks
38 ambulatory or non-ambulatory boys with DMD aged 5 years and over
NCT00264888 (PTC124-GD-004-DMD)
Completed
Phase IIa Open label non-randomised extension study of the phase II study above (NCT00264888)
Long term safety, efficacy (ambulation, muscle function, heart rate, cognitive ability, quality of life, activities of daily living), pharmacokinetics
Oral
20mg/kg twice daily plus 40mg/kg once daily
2 years
38 ambulatory or non-ambulatory boys with DMD aged 5 years and over who took part in the phase II study above (NCT00264888)
NCT 00759876 (PTC124-GD-004e-DMD)
Terminated
Phase IIa Open label uncontrolled study
Safety, tolerability, efficacy (muscle function and strength, pulmonary and cardiac function, cardiac function, quality of life, activities of daily living)
Oral
20mg/kg twice daily (breakfast, lunch) plus 40mg/kg once daily (dinner)
48 weeks
30 non-ambulatory boys with DMD/BMD aged 7 years and older
NCT01009294 (PTC124-GD-008-DMD)
Suspended
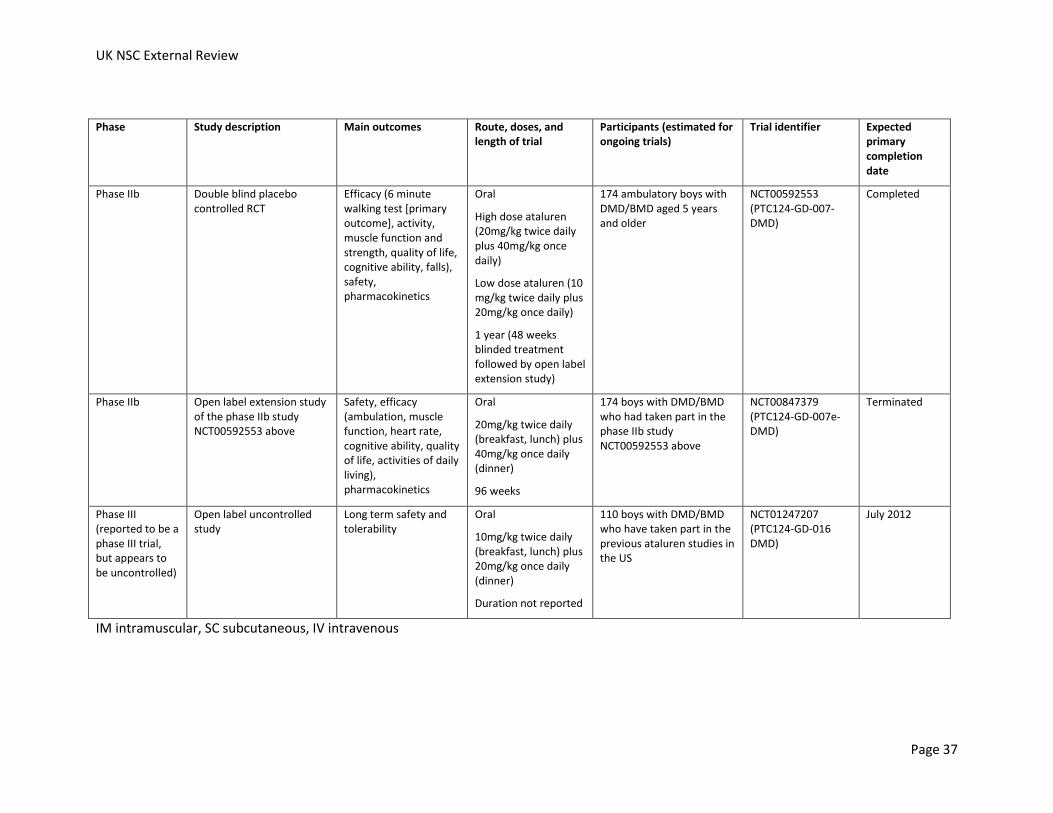
UK NSC External Review
Page 37
Phase Study description Main outcomes Route, doses, and length of trial
Participants (estimated for ongoing trials)
Trial identifier Expected primary completion date
Phase IIb Double blind placebo controlled RCT
Efficacy (6 minute walking test [primary outcome], activity, muscle function and strength, quality of life, cognitive ability, falls), safety, pharmacokinetics
Oral
High dose ataluren (20mg/kg twice daily plus 40mg/kg once daily)
Low dose ataluren (10 mg/kg twice daily plus 20mg/kg once daily)
1 year (48 weeks blinded treatment followed by open label extension study)
174 ambulatory boys with DMD/BMD aged 5 years and older
NCT00592553 (PTC124-GD-007-DMD)
Completed
Phase IIb Open label extension study of the phase IIb study NCT00592553 above
Safety, efficacy (ambulation, muscle function, heart rate, cognitive ability, quality of life, activities of daily living), pharmacokinetics
Oral
20mg/kg twice daily (breakfast, lunch) plus 40mg/kg once daily (dinner)
96 weeks
174 boys with DMD/BMD who had taken part in the phase IIb study NCT00592553 above
NCT00847379 (PTC124-GD-007e-DMD)
Terminated
Phase III (reported to be a phase III trial, but appears to be uncontrolled)
Open label uncontrolled study
Long term safety and tolerability
Oral
10mg/kg twice daily (breakfast, lunch) plus 20mg/kg once daily (dinner)
Duration not reported
110 boys with DMD/BMD who have taken part in the previous ataluren studies in the US
NCT01247207 (PTC124-GD-016 DMD)
July 2012
IM intramuscular, SC subcutaneous, IV intravenous

Antisense oligonucleotides
Frameshift mutations result in all the downstream protein coding sequence in the messenger RNA (mRNA) being incorrect, and introducing premature stops in the sequence. This mRNA is unstable and is broken down by the cell, resulting in no functional dystrophin being produced. Antisense oligonucleotides aim to alter the splicing of the DMD precursor mRNA by targeting exons affected by the deletion. They cause skipping of the targeted exon from the mature mRNA and stop the deletion from causing a frameshift. This method results in a protein which is missing a section of its sequence, but its sequence is otherwise normal (i.e. is “in frame”). The resulting protein should be able to perform some of its normal functions as long as the exon skipped does not code for crucial parts of the dystrophin protein (mainly near the beginning and end of the protein).
A low level of exon skipping occurs naturally in many DMD patients, allowing production of some shortened dystrophin protein.
As patients with Becker muscular dystrophy (BMD) have in frame deletions and are less severely affected than patients with DMD, the treatment should be able to make patients with DMD have a disease course more like that of a BMD patient. Whether a mutation is amenable to antisense oligonucleotide treatment is likely to depend on the nature of the mutation, and whether exon skipping would affect the crucial parts of the dystrophin protein. The level of improvement achievable may also vary depending on the exact mutation being treated. The success of this treatment depends on muscle tissue still being present, as it would not be expected to be able to reverse the loss of muscle. As muscle tissue is replaced by fibroadipose tissue in the advanced stages of DMD, it is predicted that the treatment will be less effective at these very advanced stages.89 However, there is some suggestion that the antisense oligonucleotides may be more easily taken up by muscles with some level of membrane damage caused by DMD.
Two main antisense oligonucleotides being tested for DMD, called AVI-4658 and PRO051/GSK2402968. Both target exon 51 of the DMD gene, as skipping this exon will be applicable to the largest group of DMD mutations. Patients with deletions of exons 50, 52, 52-63, 45-50, 48-50 or 49-50 can be treated with this antisense oligomer. Deletions of these exons are reported to account for about 13% of DMD mutations.90 A recent narrative review reported that four clinical studies on these treatments have been completed and four are underway.89
AVI-4658 is being developed by the MDEX consortium and AVI BioPharma, and PRO051 by Prosensa in partnership with GlaxoSmithKline. The antisense oligonucleotides have different sequences, with AVI-4658 having 30 nucleotides and PRO051 21 nucleotides. They also have different chemical backbones, with AVI-4658 being a phosphorodiamidate morpholino antisense oligonucleotide (PMO), and PRO051 a 2’-O-methyl phoshorothioate (2OMe) antisense oligonucleotide. Neither drug currently has marketing authorisation for use for the treatment of DMD in Europe. These drugs have thus far been tried only in boys aged 5 years and over with DMD.
Clinical trials of PRO051 are the most advanced, with a phase III trial ongoing, and due to be completed in 2012. In addition, a phase IIb study assessing two different dosing regimens of PRO051 (once weekly or twice weekly) is ongoing, and a phase I study of PRO051 in non-ambulant boys with DMD.
The most advanced completed trial of PRO051 is a phase I/IIa dose escalation study reported in 2011.91 It included 12 patients with an average age of 9.2 years (range 5 to 13 years old).

UK NSC External Review
Page 39
Participants had all been receiving a stable dose of glucocorticoids for at least 1 year when they enrolled in the trial. Abdominal subcutaneous weekly injections of PRO051 at doses of 0.5, 2.0, 4.0, and 6.0mg/kg bodyweight were given to three participants each, for five weeks. Six to 15 months after the final dose there was a 12 week extension where all participants received the highest dose.
No serious adverse effects were reported. The most common adverse effects were mild reactions at the injection site (between 25% and 75% experienced different types of irritation including itching, tenderness, bruising, redness and inflammation), elevated protein in the urine (100% of participants) and elevated urinary alpha 1 microglobulin levels (91.7%). The two highest doses (4.0 and 6.0mg/kg) resulted in production of DMD mRNA missing exon 51 in all three participants in each group. One out of three participants receiving the 2.0mg/kg dose showed dystrophin mRNA missing exon 51, and none out of the three receiving the 0.5mg/kg dose. At two weeks after the last dose, between 56% and 100% of muscle fibres in biopsies were found to contain dystrophin in ten participants; the other two participants had poor muscle biopsy specimens which showed up to 20% dystrophin positive muscle fibres.
There were no clear clinically important improvements in muscle strength, timed functional tests, or pulmonary function after five weeks at any of the doses. After 12 weeks of open label treatment at 6mg/kg/week there was an improvement in the distance that participants could walk in 6 minutes (mean improvement 35.2±28.7 metres from baseline mean of 384±121 metres; range of change -6m to +69m; 10 participants assessed). This contrasted with the reduction seen in 6 minute walking distance in the 6 to 15 months between the dose escalation phase and the subsequent extension study (mean reduction 37m). The authors note that without a placebo control the results should be interpreted carefully, but that there was no improvement in the dose escalation part of the trial suggesting a true drug effect in the extension phase, and learning effects should be minimal as all participants were familiar with the test before the trial began.
No improvement on muscle strength tests was reported to be seen. Results from the other clinical measures were not reported for the extension phase (such as ten metre walk, four stair climb, time to raise from the floor, and pulmonary function tests). The boys in this trial are reported to be taking part in an open label extension to determine the efficacy and tolerability of the treatment over a 12 month period.89
Intramuscular and intravenous AVI-4658 has been tested in phase I/II trials, and a further phase I/II dose escalation trial of this drug (intravenous delivery) in the US is due to start in 2011.89
The most advanced completed trial of AVI-4658 is a phase I/II dose escalation study reported in 2011.92 It included 19 patients with DMD with average age 8.7 years (range 6 to 13 years old). It tested intravenous weekly doses of 0.5, 1.0, 2.0, 4.0, 10.0, and 20.0mg/kg bodyweight over 12 weeks, each in two to four participants. All but one participant were taking corticosteroids.
There were no serious adverse events that were considered to be drug related. Adverse events were mostly mild (63%) or moderate (32%), were consistent with disease related complications, and did not show a dose-related increase in severity or frequency.
AVI-4658 treatment led to exon 51 skipping at all doses, and resulted in new dystrophin protein production in a dose dependent manner from the 2mg/kg dose upwards. Between 0% and 55% of muscle fibres showed dystrophin protein. Seven of the 19 participants were found to have responded to treatment by showing production of exon 51 skipped mRNA and an increase in

UK NSC External Review
Page 40
dystrophin in the muscle post treatment; these participants received doses from 2mg/kg upwards.
Pulmonary function (forced vital capacity) did not change significantly during the study. Muscle function did not show any dose dependent changes; this included the six minute walking test and muscle strength tests. Most patients were reported to remain stable during the study. Four participants lost their ability to walk during the study, which was reported to have been expected based on their ambulatory stage at study entry.
Prosensa are carrying out a pilot open label dose escalation non-randomised phase I/IIa study (NCT 01037309) on another antisense oligonucleotide (PRO044) which targets exon 44. The study is assessing the effect of PRO044 on expression of dystrophin, as well as its safety, tolerability and pharmacokinetics in boys aged 5 to 16 years. This study is due to have full data on the primary outcome collected by December 2011, and to be completed in April 2012.
Different chemical backbones for these antisense oligonucleotides are being investigated to try to improve their uptake by muscle cells.89 Safety and efficacy of new formulations would need to be tested.
The exon skipping method could theoretically be adapted to target frameshift DMD mutations arising in other parts of the gene, and is reported to potentially be possible for 83% of all DMD mutations, including deletions, small mutations and duplications.90 This would require 120 different single or double exon skipping oligonucleotide combinations, with only ten of these 120 applying to more than 2% of the patient population. Just under a fifth (19%) of DMD patients would need to have skipping of two exons to create an in frame dystrophin mRNA.
The number of children who would be treatable with each of the individual single or double exon skipping oligonucleotide combinations is low. This could hamper the ability to perform clinical trials of the sort that are normally required for licensing of drugs by the EMA and FDA for each antisense oligonucleotide. Performing these tests for each antisense oligonucleotide would also have economic implications for the manufacturer. There is a suggestion that antisense oligonucleotide toxicity is “class-dependent” and that a thorough analysis of the toxicity of one or two antisense oligonucleotides with a given chemistry in animal study and clinical trials may allow testing of new antisense oligonucleotide sequences with the same chemistry in humans, after a short 2 to 4 week trial to ensure that the safety profile looks to be similar to the previously tested oligonucleotides.89
The European TREAT-NMD clinical network held a workshop with the EMA regarding this issue in 2009.18 This highlighted the fact that even a slowing of progression of the disease would be of value to the patients and their families, as well as the importance of acceptance by the regulators of appropriate outcome measures to judge the effects of these treatments in non-ambulant boys as well as ambulant boys, and the potential problems of normal regulatory hurdles for antisense oligonucleotides for patients with DMD.
Members of the regulatory bodies were reported to express concern about the lack of neonatal screening and appropriate outcome measures for assessing the effects of treatment in individuals younger than 4 or 5 years. The lack of screening was explained to be related to the lack of early treatments for DMD, and that appropriate outcome measures would be developed if permission was granted for testing of antisense oligonucleotides in neonates. It was also noted that it would be important to gain information about the natural history of the condition in this age group, as this was reportedly not well described in the literature, as diagnoses based on symptoms are usually made later in life.18

UK NSC External Review
Page 41
The EMA noted the importance of identifying the optimal antisense oligonucleotide dosing regimen that would maximise benefit and minimise risk, determining longer term safety profiles (as the drug may be used over a lifetime), and showing a functional benefit of the treatment for the patients.
Stop codon readthrough
Stop codon readthrough treatments induce the protein making machinery to “read through” premature stop codons, allowing the full length protein to be made. This approach would only be appropriate for patients with a point mutation that leads to a premature stop codon (nonsense mutations). About 7% of DMD mutations are reported to be nonsense mutations.89
This group of treatments includes a drug called ataluren (or PTC 124), and the antibiotic gentamicin.
A trial of gentamicin published in 2010 tested the drug in 26 patients with DMD caused by nonsense mutations and 8 controls with DMD caused by frameshift mutations (all participants aged 5 to 15 years).96 Gentamicin was given intravenously at 7.5mg/kg daily for 14 days in 18 patients (including the control group), and at the same dose given once weekly (n=12) or twice weekly (n=4) for 6 months. The patients with nonsense mutations treated for 14 days showed a drop in creatine kinase levels in their blood, while the controls did not. In the groups treated for 6 months, dystrophin levels increased significantly (p=0.027) and creatine kinase levels in the blood were reduced. Muscle strength was reported to stabilise (i.e. was not significantly different at the end of trial, although a decline had been expected). There was also no significant change in muscle function (e.g. time to arise from chair, 9 metre walking test), or lung function (forced vital capacity), although the trials was small and not designed to assess efficacy. In one patient an immune response to dystrophin was identified.
Gentamicin does have side effects, and there is the possibility of kidney toxicity and ototoxicity. In addition, gentamicin appears to be better at promoting read through of some stop codons than others (there are three different codons that act as stop codons). As an antibiotic, extended use could lead to increasing antibiotic resistance.
An oral alternative to gentamicin called ataluren (or PTC 124) was developed, which allows nonsense mutation readthrough, but is reported not to have differing efficacy for the different stop codons, and not to affect normal stop codons. It has been suggested that the fact that genes often have multiple stop codons in the untranslated region at the end of the mRNA after the normal stop codon, should also reduce the risk of readthrough of real stop codons.
Although the results of two phase I studies of ataluren in healthy individuals have been published,95 full results of ongoing phase II trials have not. In March 2010, the manufacturer reported that a phase IIb placebo controlled study (NCT00592553) had not shown any improvement in the primary outcome with ataluren (six minute walk distance, 6MWD).97 At 48 weeks, the placebo group and the high dose ataluren group’s 6MWD had reduced by 42 metres from baseline, while the low dose ataluren group’s 6MWD had only reduced by 13 metres from baseline. This difference was not statistically significant when using the prespecified methods of analysis.
However, further analyses using a different statistical method did show that the difference was significant. Pre-specified analyses also showed that the low dose ataluren group had a longer time to experience 10% worsening of their 6MWD. At 48 weeks, 26% of the low dose ataluren

UK NSC External Review
Page 42
group had showed 10% worsening of their 6MWD compared with 44% in the placebo group (p=0.039), and 48% in the high dose ataluren group (p value not reported).98 The improvements with low dose ataluren seemed to be similar across different ages, corticosteroid use, and walking ability at baseline.
Ataluren was reported to be well tolerated at high and low dose, with no discontinuations due to adverse events. No serious adverse events were thought to be related to ataluren. The most common adverse events included vomiting, headache, and diarrhoea. The relative rates of these events in placebo and ataluren groups were not reported in the manufacturer’s press release, although it did say that rates were similar.
These results should be interpreted with caution as full details have yet to be published in a peer reviewed journal.
As the ongoing phase II clinical trials were all using high dose ataluren, which was found not to be effective, these were all suspended or terminated (see Table 2). A new open label safety study of low dose ataluren has been initiated for all the boys who have taken part in previous ataluren studies in the US, and is expected to be completed in 2012. This study is recorded in the ClinicaTrials.gov database as a phase III trial, but appears to be an uncontrolled study.
Ataluren also does not have European marketing authorisation for use in DMD/BMD.
There are also ongoing trials of other treatments for DMD, including a phase I/II study of IGF-1 in boys aged over five years (NCT01207908); a phase III study of idebenone in boys with DMD aged 10 to 18 years old (NCT01027884); and a phase II/III placebo controlled trial of epigallocatechin-gallate in DMD patients aged 5 to 10 years old (NCT01183767).
Summary: Criterion 10 not met
There are no curative treatments currently available for DMD. None of the currently available treatments have been shown to have greater benefit when delivered earlier to a screen detected children compared to current practice.
Steroids have been shown to improve function, and are the current standard of care. However, the current consensus is that they are not recommended to be started before a child reaches the plateau of their motor development, which usually occurs between the age of 4 and 8 years. In 2010, the UK median age at diagnosis was reported to be 4.1 years, and median age at initiation of corticosteroids 6.3 years. Therefore, although early diagnosis (e.g. by newborn screening) would allow routine assessment of motor function to start earlier in life, it would not necessarily result in much earlier glucocorticoid treatment than clinical diagnosis, based on current recommendations. Any benefits of earlier treatment would need to be weighed up against increased risks of side effects from prolonged glucocorticoid treatment.
There is uncertainty about the optimal age of initiating corticosteroids, and the most recent Cochrane review and other sources state that studies are needed to determine both the optimum age of initiation, and the optimum treatment regimen.
Earlier diagnosis could allow other interventions to be initiated, such as physiotherapy and intervention for behavioural problems. However, no studies were identified in the post-2004 search that addressed the benefits of initiation of these interventions earlier. (This section relates only to the benefits of early diagnosis for treatment; other potential benefits of early diagnosis are considered elsewhere in this document).

UK NSC External Review
Page 43
There are new genetic treatments in development (AVI4658, PRO051, ataluren) which may be able to restore some level of dystrophin production, and therefore improve outcomes in boys with DMD. None of these treatments have yet been given marketing authorisation for use in DMD by the European Medicines Authority (EMA). These exon skipping and stop codon readthrough treatments only apply to patients with specific types of mutations, and would not work for all DMD patients. AVI4658, PRO051, and ataluren would between them be likely to be appropriate for about 20% of DMD patients. The exon skipping method is theoretically applicable to over 80% of patients, but would need different antisense oligonucleotides to be designed for different mutations. This may pose some regulatory challenges, but these are being discussed between research groups and the EMA.
The exon skipping treatments hope to improve the outlook of DMD to resemble the milder BMD. These treatments are not curative, and would need to be given throughout the boys’ lives. Therefore long term safety will need to be determined; existing trials have all lasted less than a year.
As yet these trials have been tested in phase I and II trials, with a phase III trial reported to be ongoing for at least one of these treatments (PRO051/GSK2402968; ClinicalTrials.gov trial identifier NCT01254019). These treatments have only thus far been used in boys aged 5 and over, therefore the effects in younger boys are not known. It is likely that permission will be sought to carry out trials in newborns if the trials in older boys show sufficient efficacy and safety.
11. There should be agreed evidence based policies covering which individuals should be offered treatment and the appropriate treatment to be offered
2004 report: N/A
All individuals with DMD would be likely to be offered treatment after diagnosis.
The update search identified one recent international guideline on the diagnosis and management of DMD from the DMD Care Considerations Working Group, under the auspices of the US Centers for Disease Control and Prevention (CDC).2,23 This guideline is currently seeking accreditation from NHS Evidence, and the provisional recommendation from June 2011 was that this guideline is given accreditation.99
The guideline provides recommendations about appropriate treatments and at stage in the disease they should be offered. It includes recommendations that extend into the pre-symptomatic period, which would include those diagnosed at birth. However, as described in Criterion 10 above, this guideline is largely based on consensus, as there is little RCT evidence. This means that there is some uncertainty as to the most effective treatment regimens, for example the optimum steroid treatment regimen, including the optimum age at initiation.
It is not clear to what extent current practice in the UK follows these guidelines.
The guideline does not specifically describe what if any alterations to the approach might be needed for girls showing symptoms of DMD, or boys with BMD. One piece of older guidance from the US does address cardiac care in female carriers and in boys with BMD.37

UK NSC External Review
Page 44
Summary: Criterion 11 partially met
There is a recent international guideline on the diagnosis and management of DMD from the DMD Care Considerations Working Group. The guideline is currently seeking accreditation from NHS Evidence, and the provisional recommendation from June 2011 was that this guideline is given accreditation. The guideline does not explicitly address treatment in boys with BMD or female manifesting carriers.
The guideline is largely based on consensus as there is limited RCT evidence available. The lack of RCT evidence means that there is uncertainty as to the most effective treatment regimens, for example the optimum steroid treatment regimen, including the optimum age at initiation.
12. Clinical management of the condition and patient outcomes should be optimised in all health care providers prior to participation in a screening programme
2004 report: “This has been done in Wales. It would need extra resources elsewhere.”
The search did not identify any publications specifically describing the level of compliance in the UK with the multidisciplinary care recommendations from the DCCWG from 2010, which provide the most up to date standard of care. The guideline states that variable and inconsistent health care provision for people with DMD has been reported.
A national UK audit reported that the majority of boys with DMD (93%) are receiving steroid treatment.11
The Muscular Dystrophy Campaign national survey in 2008 found that families affected by neuromuscular illnesses such as DMD reported that there was a shortage of specialist and respite care.100 Half of those surveyed reported no access to a specialist neuromuscular consultant, and over 75% reported no access to respite care. Also, almost half of patients and their families had to fund their wheelchair themselves or from charity grants, and half of patients did not see a physiotherapist. A similar survey was conducted in 2010, with results yet to be released.
One study from 2005 also suggested that between 1996 and 2003, for some adults with DMD some areas of care provision had not been optimised.25
This study looked at the 25 patients with DMD referred to an adult neuromuscular clinic in London between 1996 and 2003 to assess whether there were areas where service provisions might not be adequate.25 Most of the patients were referred between the ages of 16 to 20 years, and their case notes were reviewed retrospectively. The clinic provided multidisciplinary care, including consultants in respiratory medicine, orthopaedics, cardiology, and rehabilitation, as well as physiotherapists, a family care officer, and a specialist nurse.
All the patients lived at home some or all of the time, and only four of the 24 male patients received support from external carers other than their parents. In some cases this was attributed to failure of social services to provide adequate assessment or understand the patients’ complex needs. In other cases support was refused by the parents or patients, this was reported to be generally due to a concern that the carers would not be adequately trained. The study reports that “there was often a reluctance from local health provision to support the funding of such carers”. The authors note that “this emphasizes the importance of both providing adequate funding for carers but also of ensuring carers are appropriately trained in techniques of managing patients disabled by neuromuscular weakness”.

UK NSC External Review
Page 45
The study reported that the greatest difficulties were in provision of practical aids such as hoists and belts, feeding and toileting aids, and conversion of accommodation. The wait for more complex needs such as appropriate beds was reported to be in excess of two years. This was despite most patients receiving full provision of disability allowances, and access to the local muscular dystrophy care workers, who provided considerable practical support in accessing available information.
The authors say that their findings suggested that “in the UK, society is lagging behind in its ability to provide appropriate support to allow these patients to remain independent in the community and to afford their families the necessary back-up.” Due to the improvements in life expectancy in DMD patients providing care and support for this group is likely to become more important.15
The update search did not identify any further studies published since 2004 describing provision of services within the UK.
Summary: Criterion 12 not met
The search did not identify any publications specifically describing the level of compliance with the most recent DCCWG recommendations from 2010 in the UK. A study from 2005 described difficulties in obtaining practical aids for adult patients with DMD. It is not clear whether this has since been remedied. A survey from the Muscular Dystrophy Campaign in 2008 suggested that there are areas in which service provision may not be optimised for people with disorders such as DMD, for example, in the provision of wheelchairs, specialist neuromuscular care, respite care and physiotherapy.
As was the case in 2004, although there is likely to be provision for management of newborn screening cases of DMD in Wales, this would require additional resources in the rest of the UK.
13. (a) There should be evidence from high quality Randomised Controlled Trials that the screening programme is effective in reducing mortality or morbidity.
2004 report: “No.”
The update search identified no RCTs assessing the effects of screening for DMD on mortality or morbidity published since the 2004 report.
There is a DMD newborn screening programme in place in Wales and there are reported to be similar programmes in Belgium, Germany, Australia, and pilot programmes in the US.(S Moat, personal communication) In 2006 the Belgian and German DMD screening programmes were reported to be regional rather than nationwide programmes.101 One of the two pilot US programmes is assessing screening later in infancy while the other assesses newborn screening.39,44
There have been reported to be newborn screening for DMD in the past in other countries including Cyprus, France, and New Zealand.44 A report from the CDC in 2004 stated that the most common reason for discontinuing these programmes was reported to be that there was no evidence that early diagnosis improved medical outcomes. However, it did note that non-medical benefits to boys and their families, such as reducing the “diagnostic odyssey” and enabling reproductive choice, was the reason for the continuing newborn screening programmes.

UK NSC External Review
Page 46
The update search did not identify any publications since 2004 that compared the clinical outcomes (for example, time to loss of ambulation, timed function tests etc.) of newborns identified through these programmes and those identified later in life by clinical diagnosis based on symptoms.
(b) Where screening is aimed solely at providing information to allow the person being screened to make an “informed choice” (eg. Down’s syndrome, cystic fibrosis carrier screening), there must be evidence from high quality trials that the test accurately measures risk. The information that is provided about the test and its outcome must be of value and readily understood by the individual being screened
2004 report: “Screening for DMD is primarily, but not solely, to allow “informed choice”. The test accurately measures risk and appropriate information is available.”
Newborn screening aims to identify boys with raised CK levels at risk of being affected by DMD, and testing would need to be accepted by the parents. Screening and subsequent genetic testing to confirm results can facilitate the parents making an “informed choice” regarding their subsequent reproduction.
There is evidence from the Welsh screening programme about the performance of the test (see Criteria 5 and 6). The screening and later diagnostic tests are likely to accurately measure risk of DMD. However, there is some variability in how individuals are affected due in part to the wide range of mutations that can cause DMD (see Criterion 2). Some individuals with BMD and other dystrophies may also be identified by the test, and these conditions may also have variable phenotypes.
There is some suggestion that the information available is not always provided during pregnancy, when the women has more time to think about her options (see Criterion 20). Also, there is evidence to suggest that even when women accept optional DMD screening they may not fully understand that they do have a choice, and the implications of accepting screening (see Criteria 7 and 20). There is also some suggestion that women’s reproductive behaviour may not be largely influenced by the results of newborn screening (see Criterion 4). The update search did not identify any publications since 2004 which assessed the impact of DMD screening information on reproductive behaviour.
Women who have an affected child do have options regarding future pregnancies. DMD and BMD are conditions that can be tested for using prenatal diagnosis or preimplantation genetic diagnosis (PGD).102 Testing for specific mutations could be carried out if the causative mutation is known, or, as the condition is X linked, identification of embryo sex could be carried out and only female embryos selected for implantation to reduce the chances of an affected child.103 However, there is a chance that 10-20% of girls may be affected to some level. Some of the PGD is done using preimplantation genetic haplotyping (PGH), which uses genetic markers to identify which chromosome the mutated gene lies on, and selects embryos not carrying this chromosome. This means that unaffected male embryos and non-carrier female embryos can be selected, although female carriers could be selected for implantation if there are no other viable embryos. This form of testing is being offered by Guy’s and St Thomas’ Centre for Preimplantation Genetic Diagnosis, and may also be offered at other centres.104 They report that the chance of the PGH being wrong is 1 in 100, although it was not clear if this figure pertains to

UK NSC External Review
Page 47
both false positives and false negatives. Follow up amniocentesis or chorionic villus sampling is offered to confirm the results of the PGD.
One study from the UK surveyed 420 white and Pakistani background women who had recently had a baby about whether they would want a prenatal test or termination for 30 conditions which were described according to their effects rather than by name.105 It found that the majority of women were in favour of prenatal testing for a condition with the effects of DMD (about 80% overall, as estimated from a graph). Roughly 40% of white women were in favour if termination of a pregnancy affected by DMD, and roughly 25% of Pakistani women (figures estimated from graphs). However, as these decisions were only hypothetical, and the women were not known carriers of any condition, or known to have affected family members it is not clear to what extent these results reflect what women who know they are carriers would do. Women who are carriers might prefer the option of preimplantation genetic diagnosis if available.
Summary: Criterion 13 not met
The update search identified no RCTs assessing the effects of screening for DMD on mortality or morbidity published since the 2004 report. It did not identify any other studies published since 2004 that compared the clinical outcomes of newborns identified through these programmes and those identified later in life by clinical diagnosis (for example, time to loss of ambulation, timed function tests etc.)
The screening and later diagnostic tests are likely to accurately measure risk of DMD. However, there is some variability in how individuals are affected due in part to the wide range of mutations that can cause DMD (see Criterion 2). Some individuals with BMD will also be identified by the test, and BMD mutations can also lead to a variable phenotype.
There is some evidence that despite the availability of information regarding the test, this may not be fully understood by the women who receive it. In particular, they may not fully understand that the test is optional, and the full implications of having the test. These issues would need to be addressed by a screening programme.
14. There should be evidence that the complete screening programme (test, diagnostic procedures, treatment/ intervention) is clinically, socially and ethically acceptable to health professionals and the public
1997 HTA report: “There is increasing acceptance of the programme by relevant health professionals, with 79% in favour of screening and only 1.5% wishing to see it discontinued.” (page 63)
2004 report: “It is reported that parents welcome the early diagnosis.”
We have addressed the acceptability of the screening programme to parents under Criterion 7. Here we address health professionals and the wider public.
Another study from the US in 2005 surveyed attitudes to newborn screening in 600 paediatricians, with 232 eligible paediatricians responding.106 Paediatricians were given a small amount of information on DMD, including that steroids may retard muscle weakness. Of those responding, 26% supported mandatory newborn screening for DMD, 58% did not support mandatory newborn screening, and 16% did not answer or were not sure. Of those in favour of mandatory newborn screening, 45% were in favour of screening boys only, and 55% boys and girls. The survey did not ask about support for voluntary newborn screening. When asked about

UK NSC External Review
Page 48
screening children infants for DMD at a later date (mandatory or voluntary) 31% were for and 57% against; 77% were for testing asymptomatic infants whose mother was a known carrier in the first 6 weeks of life and 12% against.
A similar study surveyed attitudes to newborn screening in 267 genetic counsellors in the US.107 Over two thirds (68%) of counsellors supported testing high risk infants for DMD (infants whose parents were carriers), while 17% were against, and 15% unsure or missing an answer. Screening after the newborn period was favoured over mandatory newborn screening for DMD (p≤0.01). Only 14% supported mandatory newborn DMD screening, while 25% supported voluntary or mandatory screening later in infancy. About two thirds were against newborn testing (65%), and 57% were against screening later in infancy. Of those who supported screening for DMD in later infancy, 93% thought it should be voluntary and 7% mandatory. Of those who supported newborn screening, 53% thought it should be for both boys and girls, and 47% only boys. Of those who supported later infancy screening, half thought it should be for both boys and girls, and half only boys. Of those who supported high risk infant screening, 35% thought it should be for both boys and girls, and 65% only boys.
Opinions on voluntary newborn screening were not canvassed in these two surveys.
A newborn DMD screening programme is currently being provided in Wales, but the update search did not identify any publications since 2004 on the acceptability of newborn DMD screening to the healthcare professionals in Wales or other parts of the UK, or in the general public (i.e. not just prospective parents).
Summary: Unclear if Criterion 14 met
A newborn DMD screening programme is currently being provided in Wales, but the update search did not identify any publications since 2004 on the acceptability of newborn DMD screening to the healthcare professionals in Wales or other parts of the UK, or in the general public (i.e. not just prospective parents).
Two studies from the US suggested that mandatory newborn screening for DMD is not supported by the majority of paediatricians and genetic counsellors.
15. The benefit from the screening programme should outweigh the physical and psychological harm (caused by the test, diagnostic procedures and treatment)
1997 HTA report: “The systematic review of the research surrounding the psychological impact of neonatal screening indicates that the psychological benefits of neonatal screening outweigh the costs.”(page 100)
“At present galactosaemia, congenital adrenal hyperplasia, Duchenne muscular dystrophy, and biotinidase deficiency do not merit high priority as stand-alone screening programmes. Though all produce benefits of various kinds, the cost–benefit ratio is lower than for the other screens recommended here.” (page 164)
2004 report: “Uncertain”
The potential benefits and harms of screening are discussed largely in the questions above and below.
Two narrative reviews discussed the pros and cons of newborn DMD screening.3,39

UK NSC External Review
Page 49
One of these from 2006 concluded that while screening in infancy was a “valid moral option” ambiguity in the balance of benefit and harm from newborn DMD screening meant that state funding for such a screening programme in the US was not “morally obligatory”.39 They concluded that voluntary screening after the newborn period would be morally preferable as it would allow greater ability to gain truly informed consent. The second from 2007 concluded that there was insufficient data on which to recommend routine newborn DMD screening, and that studies were needed to assess the potential benefits and harms of screening, including incremental costs.
One potential benefit not discussed thus far in this report is the fact that children with DMD are at greater risk of complications when they are sedated or under general anaesthesia.54 The update search did not identify any publications since 2004 which described the number of children with DMD who experience complications of general anaesthesia before being diagnosed.
At present, there remain no curative treatments, and any benefits to the child’s clinical outcomes arising from newborn diagnosis are not clear.
Potential benefits from screening include:
reproductive choice for parents
forward planning for parents and clinicians, including financial planning for future needs
removal of the delay between first symptoms and diagnosis
avoiding subsequent affected children being born before diagnosis of the index child
early detection improving the timeliness of interventions
ability to provide appropriate sedation and general anaesthesia if required
Potential harms include:
anxiety caused by screening and false positives
harm to the parent-child bonding from a positive diagnosis
risk of false reassurance in those missed by the screening (false negatives)
changes in family and other people’s attitudes to children diagnosed with the condition
changes in the child’s early life experience as a result of the diagnosis
possibility of side effects from early treatment.
Summary: Unclear if Criterion 15 met
Quantifying the balance of benefits and harms for newborn screening for DMD is difficult. This is because of the lack of evidence in key areas, for example, about the effects of treatment in screen detected populations. As such any additional clinical benefits from newborn screening are unclear. Perceptions of this balance may vary from different perspectives.
16. The opportunity cost of the screening programme (including testing, diagnosis and treatment, administration, training and quality assurance) should

UK NSC External Review
Page 50
be economically balanced in relation to expenditure on medical care as a whole (ie. value for money). Assessment against this criteria should have regard to evidence from cost benefit and/or cost effectiveness analyses and have regard to the effective use of available resource
2004 report: “No cost data available yet.”
No evaluations of the cost-effectiveness of screening published since 2004 were identified. The test could be added to the newborn bloodspot tests, reducing the need for additional blood taking. Additional resources would be needed to provide information about the test (including any written materials and midwife time), follow up creatine kinase tests, genetic tests, and muscle biopsies, as well as subsequent management of screen identified cases.
One paper discussing the possibility of newborn DMD screening made the point that screening could paradoxically limit the resources available for care after diagnosis.3 This narrative review identified one cost-effectiveness analysis of newborn DMD screening from Canada in 1988. This study found that the additional cost of the CK test would be $4.88 per infant screened, and for follow up testing would be $20.01 per positive test. The cost per case of DMD avoided was modelled at $172,000. However, these costs are over 20 years old and may not apply to the current UK health system. The also did not take into account the costs, benefits or risks to the affected individual or family.
Boys with DMD are reported to be living longer, and there is evidence to suggest that their needs in later life may not be fully met (see Criteria 12 and 17). Resources for screening might be directed towards providing for the care needs of affected boys later in life. This and other options are discussed in Criterion 17.
Summary: Criterion 16 not met
No evaluations of the cost-effectiveness or opportunity cost of screening published since 2004 were identified.
17. All other options for managing the condition should have been considered (eg. improving treatment, providing other services), to ensure that no more cost effective intervention could be introduced or current interventions increased within the resources available
2004 report: “There is evidence that the age at clinical diagnosis has not decreased in the last 20 years in spite of much publicity to clinicians in training.”
Alternatives to newborn screening
Alternatives to newborn screening for the identification of carriers and children with DMD might be cascade screening or improved clinical diagnosis to reduce the time from first identification of symptoms to diagnosis.
One alternative to newborn screening would be to try to improve clinical diagnosis. One study from Wales addressed the issue of diagnostic delay by looking at whether suggested methods for earlier clinical diagnosis would have picked up those children diagnosed as newborns.20 The study reviewed the medical records and assessments of 18 boys diagnosed as part of the newborn screening programme. The study did not have a control group without DMD, and compared outcomes with normative data where possible. They also interviewed 14 mothers

UK NSC External Review
Page 51
whose children had been diagnosed clinically between 1993 and 1997 about their child’s symptoms, but did not assess these children’s medical records for verification.
They found that between 35% and 89% of the boys with newborn-diagnosed DMD reached individual developmental locomotor or language milestones late (for example, 35% began cruising late, and 89% began walking alone late). All boys had at least one late locomotor milestone by age 24 months. In addition, the early symptoms reported were variable in nature and age at onset. Up to the age of 24 months most paediatricians noted only minor symptoms which might have been missed if the boys were not known to have DMD. A full Gower sign was recorded at age 26 to 60 months, and early contractures were reported at age 54 to 65 months.
Families reported first noticing symptoms from the age of 8 to 39 months, although this may again have been influenced by knowledge of the child’s diagnosis. The main presenting features noted were late development (e.g. of walking or talking), enlargement of calves, falling over or an odd walk. Other symptoms as boys got older were toe walking, flat feet, painful legs, difficulty walking up and down hills, and muscle cramps. Four of the 18 boys had problems with concentration in an educational context. Also, 43% of boys had difficulty gaining bladder and bowel control.
Features that have been suggested for early indicator of DMD include not walking by age of 18 months, by itself or plus other criteria (for boys): not using ≥10 recognisable words by 2 years, unexplained global developmental delay, awkward or clumsy gait under 4 years, unable to run or jump by 4 years, referral for painful hips or legs under 4 years. The individual criteria would have picked up between 22% (not walking by 18 months, global delay at 24 months, painful hips and legs under 4 years) and 94% of newborn screened cases (awkward gait under 4 years). However, the study did not look at how effective combining any of these criteria might be, or assess their diagnostic accuracy if used within the wider population.
Mothers of children who were clinically diagnosed reported first noticing something wrong between the age of 8 and 50 months. The first symptoms noticed included late walking, toe walking, poor balance, late speech development, or inability to run or climb stairs. In some cases boys had shown no symptoms up to age 4, and in other cases symptoms had not been identified as a cause for concern by medical professionals, or been attributed to other causes. The mean age at diagnosis in this group was 53.5 months (4.5 years), with an average delay of 25.5 months (2.1 years) after symptoms were first noticed. Of the total delay across families (357 months), 12% was reported to occur within the family (e.g. in raising concerns initially, or in re-raising concerns after initial reassurance), 57% at the primary care level, and 31% at the specialist level (ENT, orthopaedics, or paediatrics).
The paper concluded that the implementation of a clinical screening protocol to identify children with DMD would be complex. Other evidence that has been cited to support the difficulties in improving clinical diagnosis are studies that have shown no change in mean age of DMD diagnoses over time, despite increasing knowledge of the condition.39
One study from 2010 found that age at diagnosis did not change significantly between 1969 and 2003 in Canada (range of averages across the time period 2.8 to 6 years; p value reported as not significant).9 One US study found a trend towards earlier diagnosis between 1982 and 2000, but this trend was not statistically significant (p=0.055).10
The update search for this report did not identify publications describing similar figures for the UK population. The 1997 HTA report on newborn screening cited the average age at diagnosis of DMD in Merseyside as 4.5 years (range 3 months to 8.5 years).5 A conference abstract from

UK NSC External Review
Page 52
2010 reported that the median age at diagnosis among ambulant boys with DMD in the UK was 4.1 years (range not reported).11
Cascade screening would be performed irrespective of whether newborn screening is provided. The update search did not identify any publications since 2004 that report on the extent to which it is currently offered and accepted in the UK. If cascade screening is not yet optimal, funds could be directed towards this. However, about a quarter to a third of DMD mutations are reported to be de novo.1,3 These cases would not be identifiable by cascade screening.
Service provision
Other options include using available resources for existing treatment, development of new treatments, and then provision of these new treatments if they prove to be effective and safe.
The update search did not identify any publications since 2004 that described to what extent service provision is in line with the multidisciplinary care recommended by the DCCWG 2010 guideline. This information may be available through DMD clinical networks in the UK.
The Muscular Dystrophy Campaign reported that a national survey in 2008 found that families affected by neuromuscular illnesses such as DMD reported that there was a shortage of specialist and respite care.100 Half of those surveyed reported no access to a specialist neuromuscular consultant, and over 75% reported no access to respite care. Also, almost half of patients and their families had to fund their wheelchair themselves or from charity grants, and half of patients did not see a physiotherapist. A similar survey was conducted in 2010, with results yet to be released.
One study from 2005 suggested that provision of practical aids for adults with DMD in the UK has not been optimised at that point.25 If care has not yet been optimised, resources could be channelled towards optimising care.
There is also limited research available on treatments for DMD, including important areas such as when best to start steroid treatment, or the optimal treatment regimen. In addition there are new experimental treatments in trials (see Criterion 10). Resources could be funnelled towards this type of research.
If the new experimental treatments are proven to be sufficiently effective and safe, they are likely to need to be provided lifelong. Due to the prevalence of DMD being less than five in 10,000 drugs for this condition would be considered “orphan drugs”. The costs of such drugs are usually high. Therefore resources could be put towards purchasing these drugs for the existing population rather than for newborn screening, at least until efficacy and safety is sufficiently established in the older populations and sample newborn populations.
Summary: Unclear if Criterion 17 met
There are other options for available resources, including optimisation of cascade screening; survey and improvement of current service provision; funding for robust studies looking at effects of existing and new treatments; and funding for new treatments if they are proven to be safe and effective. Some studies have suggested that reducing the lag in clinical diagnosis is not likely to be easy.
18. There should be a plan for managing and monitoring the screening programme and an agreed set of quality assurance standards
2004 report: “Yes”

UK NSC External Review
Page 53
The update search did not identify any publications since 2004 on the managing and monitoring of newborn DMD screening programmes. It has been reported that the US CDC, which used to provide an external quality assessment (EQA) programme for bloodspot creatine kinase testing has discontinued this programme as there are not enough participants. There are reported to be no other EQA programmes or sources of QA materials for this component of the screening programme.(NSC, personal communication)
In terms of the subsequent genetic diagnosis of DMD, we identified a guideline on the best practice procedures for the molecular diagnosis of DMD/BMD.29 The guideline lays out recommendations for internal and external quality control. These include:
Following good laboratory practice
Ideally demonstrating compliance with internationally standards for laboratory testing (e.g. ISO standards) by achieving formal accreditation with a member of the International Laboratory Accreditation Cooperation (ILAC) or a national equivalent
Verifying/validating all tests in each individual laboratory prior to implementation
Collecting control samples of all mutation types to facilitate test validation/verification
Participating in appropriate external quality assessment (EQA) schemes for DMD testing if possible, or exchanging samples between laboratories to compare and validate tests.
NHS laboratories providing genetic testing would be working to these standards.
Summary: Unclear if Criterion 18 met
The update search did not identify any publications on the managing and monitoring of newborn DMD screening programmes. It has been reported that the US CDC, which used to provide an EQA programme for bloodspot creatine kinase testing has discontinued this programme, as there are not enough participants. There are reported to be no other EQA programmes or sources of QA materials for this component of the screening programme.
There are recommended QA standards for molecular diagnosis. NHS laboratories will be working to achieve national quality control standards.
19. Adequate staffing and facilities for testing, diagnosis, treatment and programme management should be available prior to the commencement of the screening programme
2004 report: “Yes in Wales. Would need extra resources elsewhere”
The update search did not identify any publications since 2004 that addressed this question in the UK. As in 2004, it is likely that the provision of newborn DMD screening in the rest of the UK would require additional resources.
Summary: Criterion 19 not met
20. Evidence-based information, explaining the consequences of testing, investigation and treatment, should be made available to potential participants to assist them in making an informed choice
2004 report: “Via leaflets provided for mothers.”

UK NSC External Review
Page 54
As a newborn screening test, the choice is made by the parents rather than the child being tested. In Wales, all mothers of baby boys are reported to be given verbal and written information about the DMD test to assist them in making an informed choice.45
However, one study from 2007 that interviewed 18 mothers found that few women reported receiving information on newborn screening during pregnancy (17%), and most received it after the birth (83%).47 Mothers often reported that it was difficult to read the leaflet and absorb the details with a newborn baby. The mothers all said they would have preferred to receive the information during pregnancy.
The majority of mothers (78%) felt that the process of being given the information was not satisfactory, mainly due to a lack of information, literature overload, or lack of time spent by the midwife. Four mothers (22%) were satisfied with the process and that they had been given enough information.
A study from the Netherlands used focus groups to assess the opinions of 36 parents and parents to be in the Netherlands on information and informed consent for existing and expanded newborn screening.108 The groups included 9 parents of healthy children and 12 parents of children affected by diseases including cystic fibrosis, phenylketonuria, DMD, and celiac disease. With regards to the existing heel prick test (which at that time screened for three disorders, not including DMD), parents in the group had agreed to the heel prick without really knowing what it was for. Most stated that they had not been given explicit information about the heel prick. They saw the heel prick as a routine test, and most (20/21) did not realise they could refuse it. Future parents were also unaware that they could refuse the heel prick. Two parents said that the wait for a possible result had caused them some concern. However, few parents in the group objected to the fact that screening tended to be more or less automatic.
Opinions were divided on whether additional screening test (such as the one for DMD) should be compulsory or optional. They felt that more detailed information should be provided if the screening programme was expanded to include disorders such as DMD, and most thought that this information should be given in pregnancy rather than after birth. The researchers concluded that considerable work would be needed to do justice to the requirement that informed consent should be obtained, and that parental preference should be incorporated in new screening programmes.
The TREAT-NMD group, along with other muscular dystrophy organisations have produced a guide for families based on the international DMD Care Considerations Working Group Guidelines.109 Although these guidelines do not address screening, they do address diagnostic testing, investigation and treatment. They could be used to help explain the condition and potential treatments to families, but this detailed information may be more appropriate for parents whose children are identified as being affected through screening rather than as pre-screening information.
Summary: Criterion 20 partially met
There is information available, but some studies have suggested that in practice this information is not always distributed during pregnancy, which might allow women time to make an informed choice. This led to dissatisfaction with the process and the information available.
21. Public pressure for widening the eligibility criteria for reducing the screening interval, and for increasing the sensitivity of the testing process, should be

UK NSC External Review
Page 55
anticipated. Decisions about these parameters should be scientifically justifiable to the public
2004 report: N/A
Screening in Wales is offered to male newborns only, as the condition mainly affects boys. This has also been reported to be the case for most other DMD newborn screening programmes.
Using CK levels to screen for female carriers has been reported to be difficult, as the test is reported to be less sensitive in this group than in affected boys.39,44 This is because CK levels in carrier girls are reported to not be as high as in boys, and may decrease to normal levels in infancy.39
Some females will show symptoms of muscular dystrophy during their life, and the severity may range from only mildly affected to as severe as DMD in males. Carriers who show symptoms are called “manifesting carriers”. Up to 20% of carriers are estimated to be affected, and in most cases this is due to non-random X chromosome inactivation. If newborn female carriers are identified it would not be possible to identify those who would be affected or to what extent they would be affected.
Although most newborn DMD screening programmes do not screen girls for these reasons, if boys are to be screened for DMD mainly to give their parents information for reproductive planning, then identification of a carrier daughter would be equally informative.39,44 It would provide the ability to give this information before the birth of an affected boy. In addition, these girls would be at risk of having affected children themselves. However, there is concern that knowledge of being a carrier from birth could be psychologically damaging.39 An alternative to newborn screening could be later screening for women, possibly prenatally. However, testing CK levels is reportedly not an accurate DMD carrier screen in adult women.39
Newborn screening programmes will either not identify manifesting carriers if they are applied to boys only, or miss more female carriers than males due the difficulties with CK level testing in females.44 This would mean that manifesting carriers would not receive the same potential benefits of early diagnosis as male newborns with DMD. For example, diagnostic delay in females without an affected male family member may be more prolonged than for an affected boy.
Replacing CK screening with DNA analysis would have similar efficacy for boys and girls, but the size of the DMD gene, and the wide variety of mutations would be likely to make this approach unfeasible using current technology.
Summary: Criterion 21 met
Newborn DMD screening is generally restricted to males. There are reasons why there could be pressure for screening in girls as well, particularly if parental reproductive choice is a main reason for screening.
Practically, CK screening is likely to be most effective for screening in male newborns. Therefore, on the basis of current testing methods and limited evidence of benefit of the information for girls, it would be scientifically justifiable to offer screening to newborn boys only. The potential limitations to the screening process in boys are discussed throughout this document.

UK NSC External Review
Page 56
22. If screening is for a mutation the programme should be acceptable to people identified as carriers and to other family members
2004 report: N/A
The screening test is not a genetic test, but DMD is a genetic disease, and genetic testing would be part of the diagnostic follow up. The identification of an affected male would identify his mother as a potential carrier, although in some cases the mutation arises de novo. Female siblings of the affected boy and female relatives on the mother’s side may also be carriers if the mother is a carrier. Criterion 7 discusses the acceptability of the test to mothers, who may be found to be at risk of being carriers as a result of newborn screening. Criterion 3 discusses the uptake of carrier testing in mothers of affected boys.
The update search identified no publications since 2004 that have reported the acceptability of newborn DMD screening to other family members, including those found to be carriers.
Summary: Unclear if Criterion 22 met
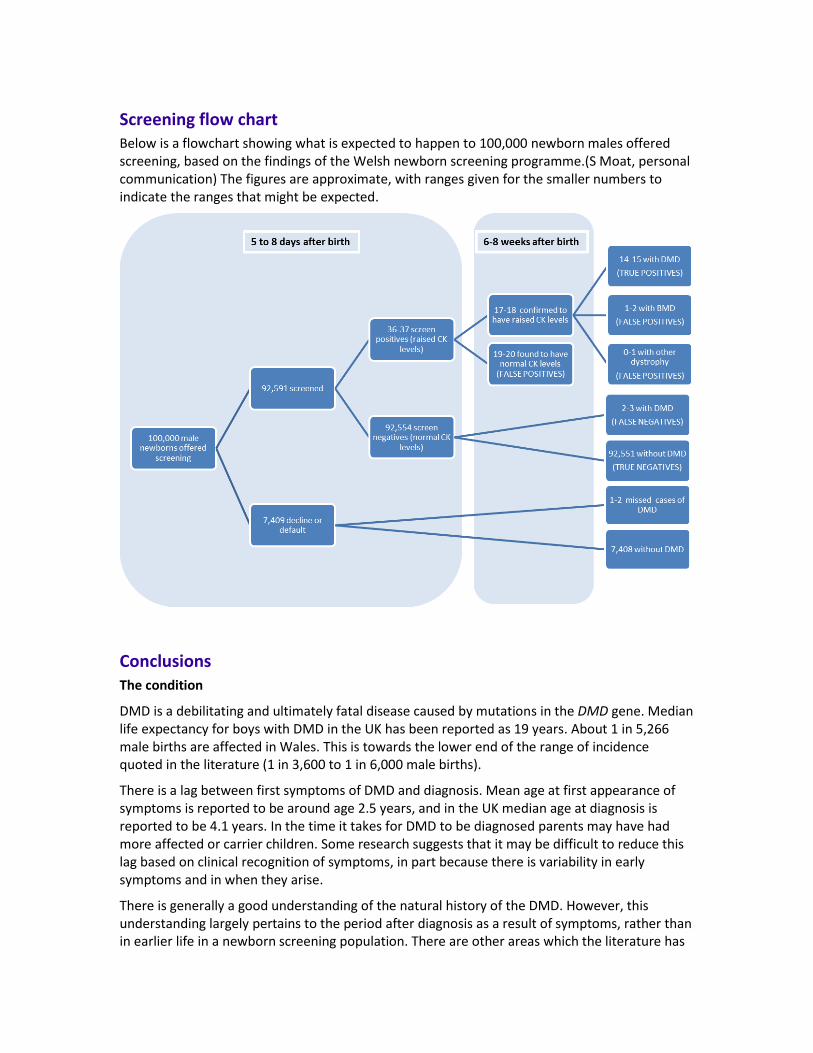
Screening flow chart Below is a flowchart showing what is expected to happen to 100,000 newborn males offered screening, based on the findings of the Welsh newborn screening programme.(S Moat, personal communication) The figures are approximate, with ranges given for the smaller numbers to indicate the ranges that might be expected.
Conclusions The condition
DMD is a debilitating and ultimately fatal disease caused by mutations in the DMD gene. Median life expectancy for boys with DMD in the UK has been reported as 19 years. About 1 in 5,266 male births are affected in Wales. This is towards the lower end of the range of incidence quoted in the literature (1 in 3,600 to 1 in 6,000 male births).
There is a lag between first symptoms of DMD and diagnosis. Mean age at first appearance of symptoms is reported to be around age 2.5 years, and in the UK median age at diagnosis is reported to be 4.1 years. In the time it takes for DMD to be diagnosed parents may have had more affected or carrier children. Some research suggests that it may be difficult to reduce this lag based on clinical recognition of symptoms, in part because there is variability in early symptoms and in when they arise.
There is generally a good understanding of the natural history of the DMD. However, this understanding largely pertains to the period after diagnosis as a result of symptoms, rather than in earlier life in a newborn screening population. There are other areas which the literature has

UK NSC External Review
Page 58
highlighted as needing further study, including natural history in older non-ambulant boys with DMD, and of the cardiac manifestations of the disease.
Newborn DMD screening is likely to be offered to male newborns, meaning that newborn female carriers will not be identified. However, a high proportion of mothers of boys with DMD will be carriers (about two thirds). The natural history of carriers is less well understood than that of males with DMD, and it is not possible to predict which carriers will themselves show symptoms, or how severe these symptoms will be.
The proportion of carriers that do show symptoms is relatively low (10-20%), and although they may be at increased risk of cardiomyopathy, one study suggests that this may not reduce their life expectancy. It is suggested that identification of carriers would allow reproductive choice to be made in subsequent pregnancies. However, some studies question whether reproductive behaviour is influenced in women identified as carriers through newborn screening.
The test
The creatine kinase test used for screening for DMD test is simple to perform and can be used on the heel prick bloodspots collected for screening for other conditions, reducing the need for additional blood collection. Programmes such as the Welsh newborn screening should have produced a large amount of data on creatine kinase levels in newborns. There does seem to be variation in the threshold level for triggering further investigations in different screening programmes internationally. This may relate to different methods for creatine kinase detection, or the precise timing of the test. In addition, the only existing external quality assessment programme for the test is reported to have been discontinued.
The Welsh newborn screening programme showed a sensitivity of about 83%, meaning that there is a relatively high rate of false negatives, with about 17% of DMD cases missed. Specificity is high (99.98%). However, the positive predictive value is about 41%, meaning that for every true positive there is roughly one false positive. The negative predictive value of the test is high at 99.997%. The test picked up mainly newborns with DMD, but also a smaller number of newborns with BMD and other dystrophies.
The test appears to be acceptable to parents in Wales, where uptake of newborn screening for DMD is high (92.6% over 20 years). Surveys of families found to have an affected baby and those with babies who were false positives in the initial screen still found a high proportion in favour of screening (66.7% and 72.7% respectively). However, some studies have suggested that mothers may accept screening because they feel that it is something routine that has to be done.
Treatments
There is a recent international guideline on the diagnosis and management of DMD from the DMD Care Considerations Working Group. This is largely based on consensus as there is limited RCT evidence available. This guideline is currently seeking accreditation from NHS Evidence, and the provisional recommendation from June 2011 was that this guideline is given accreditation.
There is little RCT evidence regarding existing treatments, and most research has been carried out in children aged over the age of four or five, which is the average age at which they would be diagnosed clinically. There is not yet conclusive evidence that existing treatments will improve outcome as a consequence of early detection through newborn screening.

UK NSC External Review
Page 59
Steroids are the mainstay of medical treatment, as they have been shown to improve muscle function. There is uncertainty about the optimal age of initiating corticosteroids.
Current consensus is that steroids are not recommended to be started before a child reaches the plateau of their motor development, which usually occurs between the age of 4 and 8 years. In 2010, the UK median age at diagnosis was reported to be 4.1 years, and median age at initiation of corticosteroids 6.3 years. Therefore, although early diagnosis (e.g. by newborn screening) would allow routine assessment of motor function to start earlier in life, it would not necessarily result in much earlier glucocorticoid treatment than clinical diagnosis, based on current recommendations. Any benefits of earlier treatment would need to be weighed up against increased risks of side effects from prolonged glucocorticoid treatment, which include weight gain, growth restriction, osteoporosis and the risk of fractures, and behavioural changes.
If newborn screening were instituted, education and support for parents could be offered earlier, and planning for future needs could also be initiated. Earlier diagnosis might also allow earlier monitoring and better detection of the plateau phase, and physiotherapy or monitoring and intervention for any psychosocial problems could also be started earlier. However, the impact of early initiation of these aspects of care on clinical outcomes has not been studied.
There are new exon skipping and stop codon readthrough treatments in development (AVI4658, PRO051, ataluren) which may be able to restore some level of dystrophin production. None of these treatments have yet been given marketing authorisation for use in DMD by the European Medicines Authority (EMA). These treatments only apply to patients with specific types of mutations, and would not work for all DMD patients. AVI4658, PRO051, and ataluren would between them be likely to be appropriate for about 20% of DMD patients. The exon skipping method is theoretically applicable to over 80% of patients, but would need different antisense oligonucleotides to be designed for different mutations. This may pose some regulatory challenges, but these are being discussed between research groups and the EMA.
Theoretically, these treatments could be more effective if started earlier, before symptoms appear. The regulatory authorities have indicated that a better understanding of the natural history of the disease in newborn and non-ambulant boys is needed in order to be able to quantify the potential benefits of these new treatments in these groups.
Opportunity costs
Some evidence suggests that care has not yet been optimised for existing DMD patients. This may particularly be the case for older, non-ambulant adolescents and adults.
The opportunity costs of instituting a newborn DMD screening programme would have to be weighed up against other options for improving care. There are other options for available resources, including improvement of current service provision; funding for robust studies looking at effects of existing and new treatments; and funding for new treatments if they are proven to be safe and effective.
Implications for policy
The updated evidence published since 2004 does not support a change in national policy regarding newborn screening for DMD.

Implications for research
The evidence update highlights areas where additional research could assist future NSC reviews:
Further research into the screening test for DMD
Research to assess the optimal age of steroid initiation and optimum steroid regimen, as well as similar research regarding other treatments
Comparison of outcomes between individuals identified by newborn screening for DMD in Wales and those identified at later ages from the remainder of the UK to identify any benefits and harms associated with earlier diagnosis
Audit of healthcare provision in the UK for patients with DMD to identify areas where provision needs to be optimised to meet the recommendations in the most recent guidance
Further study of the natural history of newborns with the condition, potentially via the Welsh DMD screening programme
More formal assessment of whether age at diagnosis could be lowered by improved clinical diagnosis
Continued research into the new treatments for DMD, including long term follow up to assess efficacy and safety
Cost effectiveness analysis of newborn DMD screening

Methodology
Search strategy
BACKGROUND: The Child Health Sub-Group of the UK National Screening Committee last reviewed Duchenne muscular dystrophy against the criteria for a screening programme in 2004.
The conclusion, accepted by the UK NSC, was that screening should not be recommended mainly because there was no available treatment to offer affected children.
More recently, a proposal for screening has been received and this provides the stimulus for the current review. The proposal was drafted by Professor Francesco Muntoni and Dr Juliet Ellis as part of the development of a series of vignettes relating to candidates to expand the number of conditions in the newborn bloodspot screening programme.
An update search on newborn screening for DMD was carried out by the UK NSC to cover the period January 2004 to May 2011. The details of this search are provided below.
SOURCES SEARCHED: Medline (OvidSP), Embase, PsychINFO, Cinahl, Web of Science and the
Cochrane Library.
SEARCH STRATEGY: Medline (OvidSP)
1. Muscular Dystrophy, Duchenne/ (2314)
2. DMD.tw. (3622)
3. Duchenne muscular dystrophy.tw. (5275)
4. 1 or 2 or 3 (7050)
5. Infant, Newborn/ (445773)
6. neonat$.tw. (164613)
7. newborn$.tw. (109871)
8. 5 or 6 or 7 (556466)
9. Mass screening/ (70033)
10. screen$3.tw. (357438)
11. detect$3.tw. (1227167)
12. (test or tests or testing).tw. (1219682)
13. 9 or 10 or 11 or 12 (2503049)
14. Neonatal Screening/ (5695)
15. 4 and 8 and 13 (139)
16. 4 and 14 (32)
17. 15 or 16 (142)
18. Prevalence/ (145817)
19. Incidence/ (142521)

UK NSC External Review
Page 62
20. (prevalen$ or inciden$).tw. (784292)
21. exp Epidemiological Studies/ (1302837)
22. 18 or 19 or 20 or 21 (1931284)
23. "Predictive Value of Tests"/ (109696)
24. "Sensitivity and Specificity"/ (233062)
25. ((positive or negative) adj predictive value$).tw. (30436)
26. (false adj (positive$ or negative$)).tw. (46612)
27. (sensitiv$ or specific$).tw. (2315581)
28. 23 or 24 or 25 or 26 or 27 (2516457)
29. Creatine Kinase/an, bl [Analysis, Blood] (14150)
30. ((CK or creatine kinase) and (assay or serum or elevated or level or blood)).tw. (11748)
31. 29 or 30 (21071)
32. 28 and 31 (4109)
33. corticosteroid$.tw. (64472)
34. Prednisone/ad, ae, tu [Administration & Dosage, Adverse Effects, Therapeutic Use] (28725)
35. Prednisolone/ad, ae, tu [Administration & Dosage, Adverse Effects, Therapeutic Use] (20663)
36. (prednisone or prednisolone or deflazacort).tw. (35359)
37. ((experimental or novel) adj (therap$ or treatment$)).tw. (26312)
38. antisense oligonucleotide$.tw. (7640)
39. exon skipping.tw. (1022)
40. (AVI-4658 or PRO051 or GSK2402968).tw. (6)
41. (ataluren or PTC124).tw. (27)
42. gentamicin.tw. (17322)
43. physiotherapy.tw. (8597)
44. 33 or 34 or 35 or 36 or 37 or 38 or 39 or 40 or 41 or 42 or 43 (183243)
45. Early diagnosis/ (7441)
46. Delayed diagnosis/ (473)
47. Disease progression/ (75972)
48. Prognosis/ (300667)
49. "Quality of Life"/ (89776)
50. exp Treatment outcome/ (486031)
51. Morbidity/ (20626)
52. Mortality/ (30732)

UK NSC External Review
Page 63
53. Reproductive behavior/ (417)
54. Genetic counseling/ (10647)
55. Parents/px [Psychology] (13513)
56. Decision making/ (55939)
57. Adaptation, psychological/ (63526)
58. Health Knowledge, Attitudes, Practice/ (54481)
59. Attitude to health/ (64990)
60. 45 or 46 or 47 or 48 or 49 or 50 or 51 or 52 or 53 or 54 or 55 or 56 or 57 or 58 or 59 (1149209)
61. 13 and 60 (167226)
62. 22 or 32 or 44 or 61 (2179117)
63. 4 and 62 (1280)
64. 17 or 63 (1365)
65. limit 64 to yr="2004 -Current" (674)
Similar searches were also carried out in Embase, PsychINFO, Cinahl, Web of Science and the
Cochrane Library. All searches carried out on 6 May 2011.
Source Number of records
Medline 674
Embase 881
Cochrane Library 57
Web of Science 321
PsycINFO 157
Cinahl 337
Total 2,427
Inclusions and exclusions
The above search strategies retrieved 2,427 references in total. After duplicate references were removed a total of 1,523 potentially relevant references were left. The title and abstracts of the remaining citations were scanned for relevance to newborn screening for Duchenne muscular dystrophy, focussing on the NSC criteria, particularly:
• The condition (especially prevalence in different ethnic groups and by socioeconomic group)
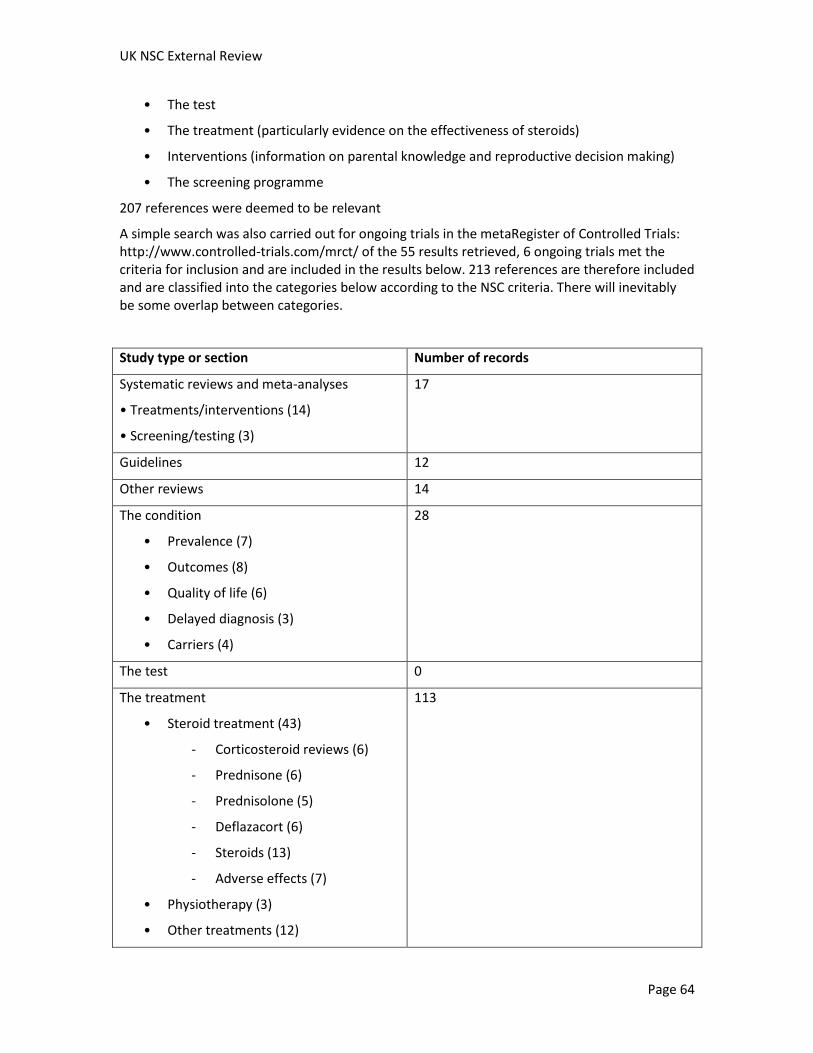
UK NSC External Review
Page 64
• The test
• The treatment (particularly evidence on the effectiveness of steroids)
• Interventions (information on parental knowledge and reproductive decision making)
• The screening programme
207 references were deemed to be relevant
A simple search was also carried out for ongoing trials in the metaRegister of Controlled Trials: http://www.controlled-trials.com/mrct/ of the 55 results retrieved, 6 ongoing trials met the criteria for inclusion and are included in the results below. 213 references are therefore included and are classified into the categories below according to the NSC criteria. There will inevitably be some overlap between categories.
Study type or section Number of records
Systematic reviews and meta-analyses
• Treatments/interventions (14)
• Screening/testing (3)
17
Guidelines 12
Other reviews 14
The condition
• Prevalence (7)
• Outcomes (8)
• Quality of life (6)
• Delayed diagnosis (3)
• Carriers (4)
28
The test 0
The treatment
• Steroid treatment (43)
- Corticosteroid reviews (6)
- Prednisone (6)
- Prednisolone (5)
- Deflazacort (6)
- Steroids (13)
- Adverse effects (7)
• Physiotherapy (3)
• Other treatments (12)
113
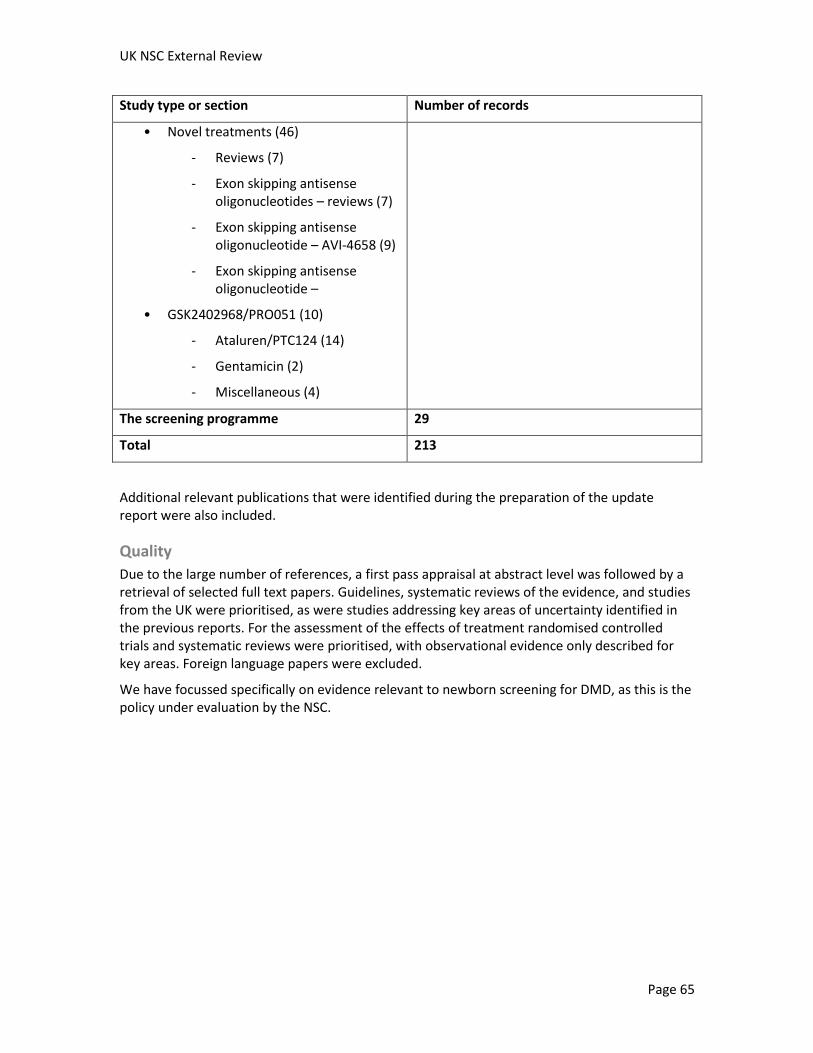
UK NSC External Review
Page 65
Study type or section Number of records
• Novel treatments (46)
- Reviews (7)
- Exon skipping antisense oligonucleotides – reviews (7)
- Exon skipping antisense oligonucleotide – AVI-4658 (9)
- Exon skipping antisense oligonucleotide –
• GSK2402968/PRO051 (10)
- Ataluren/PTC124 (14)
- Gentamicin (2)
- Miscellaneous (4)
The screening programme 29
Total 213
Additional relevant publications that were identified during the preparation of the update report were also included.
Quality
Due to the large number of references, a first pass appraisal at abstract level was followed by a retrieval of selected full text papers. Guidelines, systematic reviews of the evidence, and studies from the UK were prioritised, as were studies addressing key areas of uncertainty identified in the previous reports. For the assessment of the effects of treatment randomised controlled trials and systematic reviews were prioritised, with observational evidence only described for key areas. Foreign language papers were excluded.
We have focussed specifically on evidence relevant to newborn screening for DMD, as this is the policy under evaluation by the NSC.

Glossary
Antisense oligonucleotide
An oligonucleotide that carries a sequence complementary to a specific target sequence in the precursor mRNA, and therefore allows it to bind to this complementary target sequence.
Codon
A sequence of three nucleotides that codes for a specific amino acid or tells the cell’s protein making machinery that the protein coding part of the sequence has stopped.
Exon
A sequence in the gene that contains protein-making instructions and forms part of the mRNA that is sent to the cell’s protein making machinery. Genes are made up of exons interspersed with non-coding sequences called introns. The introns are removed when the pre-mRNA is spliced to make the mature mRNA transcript which will be read by the cell’s protein making machinery.
Frameshift mutation
A mutation that causes all of the genetic code downstream to be read incorrectly, by shifting the reading “frame” of codons.
Intron
A sequence in the gene that does not contain protein-making instructions. Genes are usually made up of exons (coding sequences) interspersed with introns. The introns are removed when the pre-mRNA is spliced to make the mature mRNA transcript which will be read by the cell’s protein making machinery.
mRNA
Messenger RNA is the ribonucleic acid molecule that carries the gene’s protein making instructions to the cell’s protein making machinery to be translated into a protein.
Oligonucleotide
A short single strand of several nucleotides joined together.
Phenotype
The characteristics that result from a particular genotype.

UK NSC External Review
Page 67
Point mutation
A mutation that changes one nucleotide in the DNA sequence. This change could have no effect on the protein sequence (silent mutation), change one amino acid to another (missense mutation), or give rise to a premature stop codon (nonsense mutation).
Splicing
The processing of the pre-mRNA transcribed from a gene to remove all the non-coding sequences.
Stop codon
A codon that tells the cell’s protein making machinery that the protein coding part of the sequence has stopped.

References
1. Tuffery-Giraud S, Beroud C, Leturcq F, Yaou RB, Hamroun D, Michel-Calemard L, et al. Genotype-phenotype analysis in 2,405 patients with a dystrophinopathy using the UMD-DMD database: a model of nationwide knowledgebase. Hum Mutat. 2009;30(6):934-45.
2. Bushby K, Finkel R, Birnkrant DJ, Case LE, Clemens PR, Cripe L, et al. Diagnosis and management of Duchenne muscular dystrophy, part 1: diagnosis, and pharmacological and psychosocial management. Lancet Neurol. 2010;9(1):77-93.
3. Kemper AR, Wake MA. Duchenne muscular dystrophy: issues in expanding newborn screening. Curr Opin Pediatr. 2007;19(6):700-4.
4. National Screening Committee Child Health Sub-Group. Report on Duchenne Muscular Dystrophy. London: National Screening Committee; 2004. Available from: http://tinyurl.com/3esvk4j.
5. Pollitt R, Green A, McCabe C, Booth A, Cooper N, Leonard J, et al. Neonatal screening for inborn errors of metabolism: cost, yield and outcome. Health Technol Assess. 1997;1(7).
6. Seymour C, Thomason M, Chalmers R, Addison G, Bain M, Cockburn F, et al. Neonatal screening for inborn errors of metabolism: a systematic review. Health Technol Assess. 1997;1(11).
7. Bushby KM, Thambyayah M, Gardner-Medwin D. Prevalence and incidence of Becker muscular dystrophy. Lancet. 1991;337(8748):1022-4.
8. Muscular Dystrophy Campaign. Duchenne muscular dystrophy. London: MDC; 2011. Available from: http://tinyurl.com/ybnklh4.
9. Dooley J, Gordon KE, Dodds L, MacSween J. Duchenne muscular dystrophy: a 30-year population-based incidence study. Clin Pediatr (Phila). 2010;49(2):177-9.
10. Ciafaloni E, Fox DJ, Pandya S, Westfield CP, Puzhankara S, Romitti PA, et al. Delayed diagnosis in duchenne muscular dystrophy: data from the Muscular Dystrophy Surveillance, Tracking, and Research Network (MD STARnet). J Pediatr. 2009;155(3):380-5.
11. Manzur A, Scott E, Munot P, Vijaykumar K, Muntoni F, UK NorthStar Clinical Network. UK NorthStar Neuromuscular Clinical Network (NSCN): National audit results in Duchenne muscular dystrophy (DMD) corticosteroid practice, vitamin D status and bone health. Neuromuscul Disord. 2010;20:S8.
12. Cotton S, Voudouris NJ, Greenwood KM. Intelligence and Duchenne muscular dystrophy: full-scale, verbal, and performance intelligence quotients. Dev Med Child Neurol. 2001;43(7):497-501.
13. Maalouf FT, Hatoum C, Atwi M, Boustany RMN. Psychiatric comorbidities in common genetic disorders with physical disability. Ped Health. 2010;4(6):591-601.

UK NSC External Review
Page 69
14. Manzur AY, Kuntzer T, Pike M, Swan A. Glucocorticoid corticosteroids for Duchenne muscular dystrophy. Cochrane Database Syst Rev. 2008;(1):CD003725.
15. Moxley RT, III, Pandya S, Ciafaloni E, Fox DJ, Campbell K. Change in natural history of Duchenne muscular dystrophy with long-term corticosteroid treatment: implications for management. J Child Neurol. 2010;25(9):1116-29.
16. Moxley RT, III, Ashwal S, Pandya S, Connolly A, Florence J, Mathews K, et al. Practice parameter: corticosteroid treatment of Duchenne dystrophy: report of the Quality Standards Subcommittee of the American Academy of Neurology and the Practice Committee of the Child Neurology Society. Neurology. 2005;64(1):13-20.
17. Shah S, Baxter P, Brooke A, Casselld J, Chowe G, Jardine P. Age at death in Duchenne muscular dystrophy. British Paediatric Neurology Association Annual Meeting 2007, 17th-19th January. Dev Med Child Neurol. 2007;49:27-8.
18. Muntoni F. The development of antisense oligonucleotide therapies for Duchenne muscular dystrophy: report on a TREAT-NMD workshop hosted by the European Medicines Agency (EMA), on September 25th 2009. Neuromuscul Disord. 2010;20(5):355-62.
19. Mathews KD, Cunniff C, Kantamneni JR, Ciafaloni E, Miller T, Matthews D, et al. Muscular Dystrophy Surveillance Tracking and Research Network (MD STARnet): case definition in surveillance for childhood-onset Duchenne/Becker muscular dystrophy. J Child Neurol. 2010;25(9):1098-102.
20. Parsons EP, Clarke AJ, Bradley DM. Developmental progress in Duchenne muscular dystrophy: lessons for earlier detection. Eur J Paediatr Neurol. 2004;8(3):145-53.
21. Kohler M, Clarenbach CF, Bahler C, Brack T, Russi EW, Bloch KE. Disability and survival in Duchenne muscular dystrophy. J Neurol Neurosurg Psychiatr. 2009;80(3):320-5.
22. TREAT-NMD Neuromuscular Network. TREAT-NMD global patient registries. Newcastle upon Tyne: University of Newcastle upon Tyne; 2011. Available from: http://www.treat-nmd.eu/resources/patient-registries/overview/
23. Bushby K, Finkel R, Birnkrant DJ, Case LE, Clemens PR, Cripe L, et al. Diagnosis and management of Duchenne muscular dystrophy, part 2: implementation of multidisciplinary care. Lancet Neurol. 2010;9(2):177-89.
24. Rahbek J, Werge B, Madsen A, Marquardt J, Steffensen BF, Jeppesen J. Adult life with Duchenne muscular dystrophy: observations among an emerging and unforeseen patient population. Pediatr Rehabil. 2005;8(1):17-28.
25. Parker AE, Robb SA, Chambers J, Davidson AC, Evans K, O'Dowd J, et al. Analysis of an adult Duchenne muscular dystrophy population. QJM. 2005;98(10):729-36.

UK NSC External Review
Page 70
26. Desguerre I, Christov C, Mayer M, Zeller R, Becane HM, Bastuji-Garin S, et al. Clinical heterogeneity of duchenne muscular dystrophy (DMD): definition of sub-phenotypes and predictive criteria by long-term follow-up. PLoS One. 2009;4(2):e4347.
27. Flanigan KM, Dunn DM, von NA, Soltanzadeh P, Gappmaier E, Howard MT, et al. Mutational spectrum of DMD mutations in dystrophinopathy patients: application of modern diagnostic techniques to a large cohort. Hum Mutat. 2009;30(12):1657-66.
28. Flanigan KM, Dunn DM, von NA, Soltanzadeh P, Howard MT, Sampson JB, et al. Nonsense mutation-associated Becker muscular dystrophy: interplay between exon definition and splicing regulatory elements within the DMD gene. Hum Mutat. 2011;32:299-308.
29. Abbs S, Tuffery-Giraud S, Bakker E, Ferlini A, Sejersen T, Mueller CR. Best practice guidelines on molecular diagnostics in Duchenne/Becker muscular dystrophies. Neuromuscul Disord. 2010;20(6):422-7.
30. White SJ, Aartsma-Rus A, Flanigan KM, Weiss RB, Kneppers AL, Lalic T, et al. Duplications in the DMD gene. Hum Mutat. 2006;27(9):938-45.
31. Burgunder JM, Schols L, Baets J, Andersen P, Gasser T, Szolnoki Z, et al. EFNS guidelines for the molecular diagnosis of neurogenetic disorders: motoneuron, peripheral nerve and muscle disorders. Eur J Neurol. 2011;18(2):207-17.
32. Newson AJ, Humphries SE. Cascade testing in familial hypercholesterolaemia: how should family members be contacted? Eur J Hum Genet. 2005;13(4):401-8.
33. Helderman-van den Enden AT, van den Bergen JC, Breuning MH, Verschuuren JJ, Tibben A, Bakker E, et al. Duchenne/Becker muscular dystrophy in the family: have potential carriers been tested at a molecular level? Clin Genet. 2011;79(3):236-42.
34. Norman A, Harper P. A survey of manifesting carriers of Duchenne and Becker muscular dystrophy in Wales. Clin Genet. 1989;36(1):31-7.
35. Soltanzadeh P, Friez MJ, Dunn D, von NA, Gurvich OL, Swoboda KJ, et al. Clinical and genetic characterization of manifesting carriers of DMD mutations. Neuromuscul Disord. 2010;20(8):499-504.
36. Holloway SM, Wilcox DE, Wilcox A, Dean JC, Berg JN, Goudie DR, et al. Life expectancy and death from cardiomyopathy amongst carriers of Duchenne and Becker muscular dystrophy in Scotland. Heart. 2008;94(5):633-6.
37. American Academy of Pediatrics. Cardiovascular health supervision for individuals affected by Duchenne or Becker muscular dystrophy. Pediatrics. 2005;116(6):1569-73.
38. Bobo JK, Kenneson A, Kolor K, Brown MA. Adherence to american academy of pediatrics recommendations for cardiac care among female carriers of duchenne and becker muscular dystrophy. Pediatrics. 2009;123(3):e471-e475.

UK NSC External Review
Page 71
39. Ross LF. Screening for conditions that do not meet the Wilson and Jungner criteria: the case of Duchenne muscular dystrophy. Am J Med Genet A. 2006;140(8):914-22.
40. Eyskens F, Philips E. Newborn screening for Duchenne muscular dystrophy. The experience in the province of Antwerp. Neuromuscul Disord. 2006;16.(9-10):721-21.
41. Al-Dahhak R, Shilling CJ, Iyer M, Dunn D., Roush K, Gailey S, et al. Implementation of a Newborn Screening Program for Duchenne Muscular Dystrophy (DMD). Neurology. 2008;70(11):A311.
42. Al-Dahhak R, Gailey S, Roush K, Schueller L, Beauman J, Lilly H, et al. Newborn Screening (NBS) in Duchenne Muscular Dystrophy: A Practical, Comprehensive and Affordable Approach. Neurology. 2009;72(11):A306--A06.
43. Brewster LM, Mairuhu G, Sturk A, van Montfrans GA. Distribution of creatine kinase in the general population: implications for statin therapy. Am Heart J. 2007;154(4):655-61.
44. Newborn Screening for Duchenne Muscular Dystrophy Workgroup: Lay Report. Atlanta (GA): US Centers for Disease Prevention and Control; 2004. Available from: www.cdc.gov/NCBDDD/duchenne/documents/NBS_Lay_Report.pdf.
45. Parsons EP, Moore C, Israel J, Hood K, Clarke AJ, Bradley DM. Emphasizing parental choice on newborn screening. Br J Midwifery. 2005;13(3):165-9.
46. Parsons EP, Israel J, Hood K, Bradley DM. Optional screening for DMD: reasons given by mothers for opting in or out. Br J Midwifery. 2006;14(12):710-5.
47. Parsons EP, King JT, Israel JA, Bradley DM. Mothers' accounts of screening newborn babies in Wales (UK). Midwifery. 2007;23(1):59-65.
48. Joomun L, Parsons E, Bradley D. Uptake of optional newborn screening and social distribution in Wales. Br J Midwifery. 2008;16(12):815-8.
49. Plass AM, van El CG, Pieters T, Cornel MC. Neonatal screening for treatable and untreatable disorders: prospective parents' opinions. Pediatrics. 2010;125(1):e99-106.
50. Detmar S, Dijkstra N, Nijsingh N, Rijnders M, Verweij M, Hosli E. Parental Opinions about the Expansion of the Neonatal Screening Programme. Community Genet. 2008;11(1):11-7.
51. Green JM, Hewison J, Bekker HL, Bryant LD, Cuckle HS. Psychosocial aspects of genetic screening of pregnant women and newborns: a systematic review. Health Technol Assess. 2004;8(33):iii, ix-iii,109.
52. TREAT-NMD Neuromuscular Network. Care Standards for DMD. Newcastle upon Tyne: University of Newcastle upon Tyne; 2011. Available from: http://www.treat-nmd.eu/care/dmd/care-standards/.

UK NSC External Review
Page 72
53. TREAT-NMD Neuromuscular Network. Best practice guidelines for molecular testing published in Neuromuscular Disorders. Newcastle upon Tyne: University of Newcastle upon Tyne; 2011. Available from: http://www.treat-nmd.eu/care/genetic-testing/dmd-molecular-testing/
54. Birnkrant DJ. The American College of Chest Physicians consensus statement on the respiratory and related management of patients with Duchenne muscular dystrophy undergoing anesthesia or sedation. Pediatrics. 2009;123 Suppl 4:S242-S244.
55. Finder JD, Birnkrant D, Carl J, Farber HJ, Gozal D, Iannaccone ST, et al. Respiratory care of the patient with Duchenne muscular dystrophy: ATS consensus statement. Am J Respir Crit Care Med. 2004;170(4):456-65.
56. Bushby K, Bourke J, Bullock R, Eagle M, Gibson M, Quinby J. The multidisciplinary management of Duchenne muscular dystrophy. Curr Paediatr. 2005;15(4):292-300.
57. Biggar WD, Harris VA, Eliasoph L, Alman B. Long-term benefits of deflazacort treatment for boys with Duchenne muscular dystrophy in their second decade. Neuromuscul Disord. 2006;16(4):249-55.
58. Escolar D, McDonald C, Kornberg AJ. Randomized, double-blind, controlled study to compare efficacy and tolerability of standard daily prednisone regime with a novel intermittent high dose regime in ambulamt boys with Duchenne muscular dystrophy. Neurology. 2008;(11 Suppl 1)(Abstract no: S05.004):A109-A110.
59. Fridman A, Arrieta A, Hu F, Hache L, Weimer J, Abdel-Hamid H, et al. Behavioral outcomes of a CINRG multi-center, double-blind, controlled study comparing high weekly versus low daily dose prednisone therapy in boys with Duchenne muscular dystrophy. Neurology. 2009;11(Suppl 3):A305-A306.
60. Matthews DJ, James KA, Miller LA, Pandya S, Campbell KA, Ciafaloni E, et al. Use of corticosteroids in a population-based cohort of boys with duchenne and becker muscular dystrophy. J Child Neurol. 2010;25(11):1319-24.
61. Markham LW, Kinnett K, Wong BL, Woodrow BD, Cripe LH. Corticosteroid treatment retards development of ventricular dysfunction in Duchenne muscular dystrophy. Neuromuscul Disord. 2008;18(5):365-70.
62. Fournier A, Therien J, Schram G, Dahdah N. Steroids and vascular resistance reducing agents improve cardiac outcome and survival in patients with duchenne muscular dystrophy. Cardiol Young. 2010;20((S70)):1047-9511.
63. Houde S, Filiatrault M, Fournier A, Dube J, D'Arcy S, Berube D, et al. Deflazacort use in Duchenne muscular dystrophy: an 8-year follow-up. Pediatr Neurol. 2008;38(3):200-6.
64. Dooley JM, Gordon KE, MacSween JM. Impact of steroids on surgical experiences of patients with duchenne muscular dystrophy. Pediatr Neurol. 2010;43(3):173-6.

UK NSC External Review
Page 73
65. Bach JR, Martinez D, Saulat B. Duchenne muscular dystrophy: the effect of glucocorticoids on ventilator use and ambulation. Am J Phys Med Rehabil. 2010;89(8):620-4.
66. Merlini L, Malaspina E, Gennari M, Cicognani A, Franzoni E, Talim B. 10 years follow-up of early corticosteroid treatment of Duchenne muscular dystrophy. 2. Italy: 2007. Available from: http://www.distrofiamuscular.net/abstracts13.htm.
67. Parreira SL, Resende MB, Zanoteli E, Carvalho MS, Marie SK, Reed UC. Comparison of motor strength and function in patients with Duchenne muscular dystrophy with or without steroid therapy. Arq Neuro-Psiquiat. 2010;68(5):683-8.
68. Straathof CS, Overweg-Plandsoen WC, van den Burg GJ, van der Kooi AJ, Verschuuren JJ, de Groot IJ. Prednisone 10 days on/10 days off in patients with Duchenne muscular dystrophy. J Neurol. 2009;256(5):768-73.
69. Munot P, Robb SA, Muntoni F, Manzur A. A multicentre audit of vertebral fractures in corticosteroid treated Duchenne muscular dystrophy (DMD). Eur J Paediatr Neurol. 2009;13.
70. Kulshrestha R, Hoyle E, Davie M, Haddaway M, Quinlivan R. An audit of bone density and vertebral fractures during steroid treatment in Duchenne muscular dystrophy. Neuromuscul Disord. 2010;20.
71. Ambegaonkar GP, Kinali M, Muntoni F, Robb SA, Manzur AY. Vertebral fractures in corticosteroid-treated boys with Duchenne muscular dystrophy. Dev Med Child Neurol. 2009;51(44):0012-1622.
72. Ambegaonkar GP, Kinali M, Muntoni F, Robb SA, Manzur AY. Vertebral fractures in corticosteroid-treated boys with Duchenne muscular dystrophy. Neuromuscular Disord. 2008;18(9):825-6.
73. Kley RA, Tarnopolsky MA, Vorgerd M. Creatine for treating muscle disorders. Cochrane Database Syst Rev. 2011;(2):CD004760.
74. Phillips MF, Quinlivan R. Calcium antagonists for Duchenne muscular dystrophy. Cochrane Database Syst Rev. 2008;(4):CD004571.
75. Mok E, Eleouet-Da VC, Daubrosse C, Gottrand F, Rigal O, Fontan JE, et al. Oral glutamine and amino acid supplementation inhibit whole-body protein degradation in children with Duchenne muscular dystrophy. Am J Clin Nutr. 2006;83(4):823-8.
76. Mok E, Letellier G, Cuisset JM, Denjean A, Gottrand F, Alberti C, et al. Lack of functional benefit with glutamine versus placebo in Duchenne muscular dystrophy: a randomized crossover trial. PLoS One. 2009;4(5):e5448.
77. Escolar DM, Buyse G, Henricson E, Leshner R, Florence J, Mayhew J, et al. CINRG randomized controlled trial of creatine and glutamine in Duchenne muscular dystrophy. Ann Neurol. 2005;58(1):151-5.

UK NSC External Review
Page 74
78. Kirschner J, Schessl J, Schara U, Reitter B, Stettner GM, Hobbiebrunken E, et al. Treatment of Duchenne muscular dystrophy with ciclosporin A: a randomised, double-blind, placebo-controlled multicentre trial. Lancet Neurol. 2010;9(11):1053-9.
79. Skura CL, Fowler EG, Wetzel GT, Graves M, Spencer MJ. Albuterol increases lean body mass in ambulatory boys with Duchenne or Becker muscular dystrophy. Neurology. 2008;70(2):137-43.
80. Duboc D, Meune C, Lerebours G, Devaux JY, Vaksmann G, Becane HM. Effect of perindopril on the onset and progression of left ventricular dysfunction in Duchenne muscular dystrophy. J Am Coll Cardiol. 2005;45(6):855-7.
81. Gordon KE, Dooley JM, Sheppard KM, MacSween J, Esser MJ. Impact of bisphosphonates on survival for patients with Duchenne muscular dystrophy. Pediatrics. 2011;127(2):e353-e358.
82. Rose KJ, Burns J, Wheeler DM, North KN. Interventions for increasing ankle range of motion in patients with neuromuscular disease. Cochrane Database Syst Rev. 2010;(2):CD006973.
83. Sackley C, Disler PB, Turner-Stokes L, Wade DT, Brittle N, Hoppitt T. Rehabilitation interventions for foot drop in neuromuscular disease. Cochrane Database Syst Rev. 2009;(3):CD003908.
84. Cheuk DK, Wong V, Wraige E, Baxter P, Cole A, N'Diaye T, et al. Surgery for scoliosis in Duchenne muscular dystrophy. Cochrane Database Syst Rev. 2007;(1):CD005375.
85. Maugere E, Townsend E, Watkins MP. Abstract. Role of exercise in the management of patients with Duchenne muscular dystrophy: a systematic review. Pediatr Phys Ther. 2008;20(1):116-7.
86. Maugere E. Role of exercise on the management of Duchenne Muscular dystrophy: A systematic review [PhD dissertation]. Buenos Aires: University of Buenos Aires; 2003.
87. Li Z, Huang J, Zhong Y, Yao X, Zhang C. Effect of rehabilitation exercise on the extremity function and lung function in patients with Duchenne's muscular dystrophy. 2219. Guangzhou, China: European Respiratory Society; 2006. Available from: http://tinyurl.com/3rh7r9t.
88. Jansen M, de Groot IJ, van AN, Geurts AC. Physical training in boys with Duchenne Muscular Dystrophy: the protocol of the No Use is Disuse study. BMC Pediatr. 2010;10:55.
89. Muntoni F, Wood MJ. Targeting RNA to treat neuromuscular disease. Nat Rev Drug Discov. 2011;10(8):621-37.
90. Aartsma-Rus A, Fokkema I, Verschuuren J, Ginjaar I, van DJ, van Ommen GJ, et al. Theoretic applicability of antisense-mediated exon skipping for Duchenne muscular dystrophy mutations. Hum Mutat. 2009;30(3):293-9.

UK NSC External Review
Page 75
91. Goemans NM, Tulinius M, van den Akker JT, Burm BE, Ekhart PF, Heuvelmans N, et al. Systemic administration of PRO051 in Duchenne's muscular dystrophy. N Engl J Med. 2011;364(16):1513-22.
92. Cirak S, Arechavala-Gomeza V, Guglieri M, Feng L, Torelli S, Anthony K, et al. Exon skipping and dystrophin restoration in patients with Duchenne muscular dystrophy after systemic phosphorodiamidate morpholino oligomer treatment: an open-label, phase 2, dose-escalation study. Lancet. 2011;378(9791):595-605.
93. van Deutekom JC, Janson AA, Ginjaar IB, Frankhuizen WS, Aartsma-Rus A, Bremmer-Bout M, et al. Local dystrophin restoration with antisense oligonucleotide PRO051. N Engl J Med. 2007;357(26):2677-86.
94. Kinali M, Arechavala-Gomeza V, Feng L, Cirak S, Hunt D, Adkin C, et al. Local restoration of dystrophin expression with the morpholino oligomer AVI-4658 in Duchenne muscular dystrophy: a single-blind, placebo-controlled, dose-escalation, proof-of-concept study. Lancet Neurol. 2009;8(10):918-28.
95. Hirawat S, Welch EM, Elfring GL, Northcutt VJ, Paushkin S, Hwang S, et al. Safety, tolerability, and pharmacokinetics of PTC124, a nonaminoglycoside nonsense mutation suppressor, following single- and multiple-dose administration to healthy male and female adult volunteers. J Clin Pharmacol. 2007;47(4):430-44.
96. Malik V, Rodino-Klapac LR, Viollet L, Wall C, King W, Al-Dahhak R, et al. Gentamicin-induced readthrough of stop codons in Duchenne muscular dystrophy. Ann Neurol. 2010;67(6):771-80.
97. Muscular Dystrophy Campaign. Ataluren clinical trial for Duchenne - is less more? London: 2011. Available from: http://www.muscular-dystrophy.org/research/news/2250_ataluren_clinical_trial_for_duchenne-is_less_more.
98. PTC Therapeutics. Pivotal data presented at the World Muscle Society Congress suggest ataluren slows the loss of walking ability in patients with nonsense mutation Duchenne/Becker muscular dystrophy. 2010. Available from: http://ptct.client.shareholder.com/releasedetail.cfm?ReleaseID=518941.
99. NHS Evidence. Diagnosis and Management of Duchenne Muscular Dystrophy guideline: Draft Accreditation Report - for consultation. London: NICE; 24-6-2011.
100. Muscular Dystrophy Campaign. State of the Nation: The 2008 National Survey. London: MDC; 2008. Available from: http://www.muscular-dystrophy.org/assets/0000/6266/State_of_the_Nation_Survey_Report.pdf.
101. Javaher P, Nyoungui E, Kaariainen H, Kristoffersson U, Nippert I, Sequeiros J, et al. Genetic screening in Europe. Public Health Genomics. 2010;13(7-8):524-37.
102. Human Fertility and Embryology Authority. PGD conditions licensed by the HFEA. London: HFEA; 2011. Available from: http://www.hfea.gov.uk/cps/hfea/gen/pgd-screening.htm.

UK NSC External Review
Page 76
103. Human Fertility and Embryology Authority. Sex Selection. London: HFEA; 2009. Available from: http://www.hfea.gov.uk/pgd-sex-selection.html.
104. Preimplantation Genetic Diagnosis PGH- Embryo Sexing for X linked conditions Supplementary Leaflet. London: Guys & St Thomas NHS Foundation Trust; 2008. Available from: http://www.pgd.org.uk/resources/tests/pgh_general.pdf
105. Hewison J, Green JM, Ahmed S, Cuckle HS, Hirst J, Hucknall C, et al. Attitudes to prenatal testing and termination of pregnancy for fetal abnormality: a comparison of white and Pakistani women in the UK. Prenat Diagn. 2007;27(5):419-30.
106. Acharya K, Ackerman PD, Ross LF. Pediatricians' attitudes toward expanding newborn screening. Pediatrics. 2005;116(4):e476-e484.
107. Hiraki S, Ormond KE, Kim K, Ross LF. Attitudes of genetic counselors towards expanding newborn screening and offering predictive genetic testing to children. Am J Med Genet A. 2006;140(21):2312-9.
108. Detmar S, Hosli E, Dijkstra N, Nijsingh N, Rijnders M, Verweij M. Information and informed consent for neonatal screening: opinions and preferences of parents. Birth. 2007;34(3):238-44.
109. TREAT-NMD Neuromuscular Network. The diagnosis and management of Duchenne Muscular Dystrophy: A guide for families. Newcastle upon Tyne: University of Newcastle upon Tyne; 2011. Available from: http://www.treat-nmd.eu/downloads/file/standardsofcare/dmd/us_english/dmd_us_familyguide_june2010.pdf





























