Embed Size (px)
Citation preview

Journal of Molecular and Cellular Cardiology 49 (2010) 354–361
Contents lists available at ScienceDirect
Journal of Molecular and Cellular Cardiology
j ourna l homepage: www.e lsev ie r.com/ locate /y jmcc
Highlighted Article
Nitrative inactivation of thioredoxin-1 increases vulnerability of diabetic hearts toischemia/reperfusion injury
Tao Yin a,1, Rongrong Hou b,1, Shaowei Liu a,1, Wayne Bond Lau c, Haichang Wang a,⁎, Ling Tao a,⁎a Department of Cardiology, Xijing Hospital, The Fourth Military Medical University, 15 Changle West Road, Xi'an 710032, Chinab Department of Endocrinology, Xijing Hospital, The Fourth Military Medical University, 15 Changle West Road, Xi'an 710032, Chinac Department of Emergency Medicine, Thomas Jefferson University, Philadelphia, PA 19107, USA
⁎ Corresponding authors. L. Tao is to be contacted at t84775183; fax: +86 29 84771024. H. Wang, tel./fax: +
E-mail addresses: [email protected] (H. Wang)(L. Tao).
1 The first three authors contributed equally to this w
0022-2828/$ – see front matter © 2010 Elsevier Ltd. Aldoi:10.1016/j.yjmcc.2010.05.002
a b s t r a c t
a r t i c l e i n f oArticle history:Received 15 April 2010Received in revised form 4 May 2010Accepted 5 May 2010Available online 16 May 2010
Keywords:DiabetesThioredoxinProtein nitrationIschemia/reperfusion
Hyperglycemia (HG) significantly increases mortality after myocardial infarction (MI) in patients with andwithout established diabetes. The specific underlying mechanism remains unknown. The present studyattempted to determine whether nitrative inactivation of thioredoxin-1 (Trx-1) may contribute to theexaggerated myocardial ischemia/reperfusion (I/R) injury observed in the hyperglycemic condition. Diabeteswas induced by multiple intraperitoneal injections of low-dose streptozotocin (STZ) in mice. After 30 minischemia by slip-knot ligature of the left anterior descending coronary artery, the myocardium wasreperfused for 3 h after knot release (for apoptosis, Trx-1-activity, and -nitration determination) or 24 h (forcardiac function and infarct size determination). At 10 min before reperfusion, diabetic mice wererandomized to receive vehicle, EUK134 (a peroxynitrite scavenger), recombinant human Trx-1 (rhTrx-1),or SIN-1 (a peroxynitrite donor) nitrated Trx-1 (N-Trx-1) administration. Diabetes intensified I/R-inducedmyocardial injury, evidenced by further enlarged infarct size, increased apoptosis, and decreased cardiacfunction in diabetic mice. Trx-1 nitrative inactivation was elevated in the diabetic heart before I/R and wasfurther amplified after I/R. Treatment with EUK134 or rhTrx-1, but not N-Trx-1, before reperfusionsignificantly reduced Trx-1 nitration, preserved Trx-1 activity, attenuated apoptosis, reduced infarct size, andimproved cardiac function in diabetic mice. Taken together, our results demonstrated that HG increasedcardiac vulnerability to I/R injury by enhancing nitrative inactivation of Trx-1, suggesting that blockade ofTrx-1 nitration, or supplementation of exogenous rhTrx-1, might represent novel therapies to attenuatecardiac injury after MI in diabetic patients.
el.: +86 29 84771024, +86 2986 29 84773469., [email protected]
ork.
l rights reserved.
© 2010 Elsevier Ltd. All rights reserved.
1. Introduction
Ischemic heart disease (IHD) is the major cause of morbidity andmortality in diabetic patients [1,2]. Hyperglycemia, by itself, isassociated with increased risk of poor outcome after myocardialinfarction in patients with and without the formal diagnosis of diabetes[3]. Numerous experimental results have demonstrated that hypergly-cemia contributes to increased sensitivity of hearts to ischemia/reperfusion (I/R) injury. However, this phenomenon's underlyingmechanisms remain to be identified. IHD treatment of diabetic patientsis a great challenge, and the elucidation of the complex pathophysiologyunderlying diabetes myocardial ischemia/reperfusion (MI/R) injuryhas invaluable clinical applications worldwide.
Strong evidence exist that increased oxidative stress, reactivenitrogen species (RNS), and reactive oxygen species (ROS) productionare the common denominators in both I/R injury and diabetes [4,5].Indeed, modifications in oxidative stress and its downstream cellulartargets have altered the response of diabetic hearts to I/R injury [5,6].Posttranslational modification of proteins at nearly all molecular levelsby RNS and ROS has been demonstrated to be severely problematic,affecting the structure and function of key proteins related to geneexpression, signal transduction, and antioxidant defense [7]. Ofprominent note is the process of protein tyrosine residue nitration, awell-documentedpathologicmodificationbyRNS andROS,which altersprotein structure and function [8], the potential etiology of manycardiovascular diseases [9]. The hyperglycemic condition may exhibitpronounced myocardial injury as a result of rampant protein modifi-cation, but which specific responsible proteins are susceptible to whattypes of modification are completely unknown.
Thioredoxin (Trx, 12-kDa) is an important cellular survival andantioxidant protein ubiquitously expressed in all living cells. Of theTrx protein family, Trx-1 has been investigated most intensively inregard to human disease. Recent evidence shows that Trx-1 functions

355T. Yin et al. / Journal of Molecular and Cellular Cardiology 49 (2010) 354–361
as a critical myocardial protective factor directly via antioxidanteffects and indirectly by protein–protein interaction with keysignaling molecules [10]. Its activity is susceptible to the posttrans-lational modification processes of oxidation, nitrosylation, andglutathionylation [11]. We have previously demonstrated the nitra-tive modification of Trx-1, resulting in the irreversible inhibition ofactivity during the MI/R condition [12]. In a recent in vitro study usingcultured cardiomyocytes, we have demonstrated that high-glucosesensitization of cardiomyocytes to hypoxia/reoxygenation injuryinvolves nitrative inactivation of Trx-1 [13]. However, whethercardiac Trx-1 nitration is increased in vivo in diabetic heart andcontributes to enhanced vulnerability of diabetic hearts to I/R injuryremains completely unknown.
Therefore, the aims of the present study were (1) to examinewhether Trx-1 activity is reduced in diabetic heart, (2) to determinewhether nitrative modification is a cause of Trx-1 inactivation in thediabetic heart, (3) to establish a causative link between increased Trx-1 nitration and increased vulnerability of diabetic hearts to I/R injury,and (4) to determine the signaling mechanism by which increasedTrx-1 nitration exacerbates I/R-induced myocardial injury underhyperglycemic conditions.
2. Materials and methods
2.1. Experimental protocols
All experiments were performed in adherence with the NationalInstitutes of Health Guidelines on the Use of Laboratory Animals, andwere approved by the Fourth Military Medical University Committee
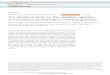
Fig. 1. (A) Myocardial infarct size assessed by Evans blue/TTC double staining. Represenonischemic/normal regions; red-stained areas indicate ischemic/reperfused but viable rQuantification of infarct size was expressed as the ratio of infarct area (Inf) to total ischechocardiography. DM, diabetic mice; MI/R, myocardial ischemia/ reperfusion; LVEF, left venvs. control+MI/R; n=6 to 8 hearts/group.
on Animal Care. Swiss mice at the age of 6–8 weeks were used forpresent study. Mice were rendered diabetic by 5 days of dailyintraperitoneal injection with 40 mg/kg STZ (Sigma) in 0.05 Msodium citrate, pH 4.5. Blood glucose was measured 5 days after thefinal injection, and diabetic condition was confirmed by markedlyelevated whole-blood glucose levels in STZ-treated mice were (meanvalue: 368±17 mg/dL, range 328–416 mg/dL vs. 134±8 mg/dL,range 104–166 mg/dL in control mice). Two days after successfulestablishment of diabetes (7 days after the first STZ injection), micewere subjected to myocardial ischemia as described below.
MI/R was produced by temporarily exteriorizing the heart via leftthoracic incision and placing a 6-0 silk suture slipknot at the distal 1/3of the left anterior descending coronary artery. After 30 min ofischemia, the slipknot was released, and the myocardium wasreperfused for 3 h (for apoptosis, Trx activity, and nitration assays)or 24 h (for cardiac function and infarct size assay). At 10 min beforereperfusion, mice were randomized to receive either vehicle (0.9%NaCl), EUK134 (a peroxynitrite scavenger, 5 mg/kg; Cayman Chem-ical), recombinant human Trx-1 (rhTrx-1, 2 mg/kg; Sigma), ornitrated Trx-1 (please see detail in Section 2.8) by intraperitonealinjection. Sham-operated mice underwent the same surgical proce-dures except that the suture placed under the left coronary artery wasnot tied.
2.2. Determination of cardiac function and myocardial infarct size
At the end of the 24-h reperfusion period,micewere reanesthetized,and cardiac function was determined by echocardiography. Afterassessment, the coronary artery ligature was retied, and myocardial
ntative photographs of heart sections are shown (top). Blue-stained areas indicateegions; negatively stained areas indicate ischemic/reperfused and infarcted regions.emic/reperfused area (area-at-risk, AAR) (bottom). (B) Cardiac function assessed bytricular ejection fraction. *Pb0.05 vs. control+sham; #Pb0.05 vs. DM+sham; **Pb0.05

356 T. Yin et al. / Journal of Molecular and Cellular Cardiology 49 (2010) 354–361
infarct size was determined by the Evans blue/2,3,5-triphenyl tetrazo-lium chloride (TTC) double staining method as described previously[14].
2.3. Determination of myocardial apoptosis
Myocardial apoptosis was determined by terminal deoxynucleoti-dyltransferase-mediated dUTP nick end labeling staining (TUNEL;Roche) and caspase-3 activity assay (Chemicon) as previously described[15]. In brief, using a 20× objective, the tissue slide (4 slides/heartsample) was digitally photographed with a QICAM-Fast Digital Cameramounted onto an Olympus BX51 Fluorescence Microscope to cover theentire area. Total nuclei and the TUNEL-positive nuclei were counted byIP Lab Imaging Analysis Software (Version 3.5; Scanalytics, Fairfax, VA).The index of apoptosis (number of TUNEL positive nuclei/total numberof nuclei×100)was automatically calculated and exported toMicrosoftExcel for further analysis. Results from different fields taken from thesame animal were averaged and counted as 1 sample.
2.4. Determination of nitrotyrosine content in cardiac tissues
Paraformaldehyde-fixed cardiac tissues were cut into semithinsections 4 to 5 μm thick and stained with a primary antibody againstnitrotyrosine (Upstate). Immunostaining was done with VectastainABC kit (Vector Laboratories), and slides were analyzed by lightmicroscopy. Quantification of cardiac tissue nitrotyrosine content wasperformed by Millipore nitrotyrosine assay kit per manufacturer's
Fig. 2. (A) Myocardial apoptosis determined by TUNEL staining. Representative photographcounterstaining (blue) indicates total nuclei. (B) Quantification of apoptotic nuclei. TUNEL-pcounted and calculated by Image-Pro Plus software. (C) Myocardial apoptosis determined by csham; **Pb0.05 vs. control+MI/R; n=6 to 8 hearts/group.
instruction. Results are presented micrograms per milligram (μg/mg)protein.
2.5. Trx-1 activity assay
Trx-1 activity was determined by previously described insulindisulfide reduction assay [16,17]. In brief, 40 μg of cellular proteinextractwas preincubatedwith 2 μL of activation buffer (100 mMHEPES,2 mMEDTA, 1 mg/mL BSA, and 2 mMDTT) at 37 °C for 15 min for Trx-1reduction. Samples were mixed with 20 μL of reaction buffer (100 mMHEPES, 2 mM EDTA, 0.2 mM NADPH, and 140 μM insulin). The reac-tion was initiated by mammalian Trx reductase (1 μL, 15 mU, Sigma).Samples were incubated for 30 min at 37 °C. The reaction wasterminated by 125 μL of stopping solution (0.2 M Tris–HCl, 10 Mguanidine–HCl, and 1.7 mM 3-carboxy-4-nitrophenyl disulfide) fol-lowed by absorption measurement at 412 nm. Trx-1 activity wasexpressed as oxidized NADPH micromol per minute per milligram(μmol/min/mg) of protein.
2.6. Immunoprecipitation and immunoblotting
Trx-1 nitration and Trx-1/ASK1 interaction detection were per-formed as described by Vadseth et al. [18] and used in our previousstudy [12]. In brief, Trx-1 in homogenized cardiac tissues wasimmunoprecipitated with a monoclonal antibody against Trx-1(Redox Bioscience). After sample separation, Trx-1 nitration and Trx-1/ASK1 interaction were determined, respectively, by immunoblottingusing a primary antibody against nitrotyrosine (Upstate) or against
s of heart sections are shown. TUNEL staining (green) indicates apoptotic nuclei; DAPIositive nuclei are expressed as a percentage of the total number of nuclei, automaticallyaspase-3 activity by colorimetric assay kit. *Pb0.05 vs. control+sham; #Pb0.05 vs. DM+

357T. Yin et al. / Journal of Molecular and Cellular Cardiology 49 (2010) 354–361
ASK1 (Upstate). Following incubation with horseradish peroxidase–conjugated secondary antibody (Cell Signaling Technology), the blotwas developed with an ECL-Plus chemiluminescence reagent kit(Amersham) and visualized with UVP Bio-Imaging Systems. Blotdensities were analyzed with VisionWorks LS Acquisition and AnalysisSoftware.
2.7. p38 MAPK activity assay
p38MAPK activity assaywas performed by p38MAPK assay kit permanufacturer's instructions (Cell Signaling Technology) [14,17].
2.8. In vitro nitration of Trx-1
Human Trx-1 (the cytosolic form of Trx; Sigma Chemical Company,St Louise, MO) was subjected to in vitro nitration with a modifiedprocedure recently described by Cohen et al. for MnSOD nitration [19].In brief, purified human Trx-1 (dissolved in 0.1 µM phosphate buffer,pH 7.4, final concentration of 50 μM) was incubated with SIN-1 (finalconcentration of 100 µM; Cayman Chemical, Ann Arbor, MI) at 37 °Cfor 30 min. Unreacted SIN-1 was removed by ultrafiltration overmembranes with a 5-kDa cutoff.
2.9. Statistical analysis
All values in the text and figures are presented as mean±SD of nindependent experiments. All data (except immunoblotting density)were subjected to two-way ANOVA followed by Bonferroni correctionfor post hoc t test. Immunoblotting densities were analyzed with the
Fig. 3. (A)Nitrotyrosine content inmyocardiumdeterminedby immunohistochemistry (top) annitration determined by immunoprecipitation (IP). Representative immunoblot (IB) graphs a4=DM+MI/R. (C) Trx-1 activity determined by insulin disulfide reduction assay. Nty, nitrotyn=6 to 8 hearts/group.
Kruskal–Wallis test followed by Dunn post hoc test. Probabilities of0.05 or less were considered to be statistically significant.
3. Results
3.1. I/R-induced myocardial injury was exacerbated in diabetic mice
Thirty minutes of ischemia followed by 24 h of reperfusionresulted in a significant myocardial infarction in both control anddiabetic mice, evidenced by Evans blue staining describing area at risk(AAR) and TTC staining describing the infarct area (Fig. 1A). The ratioof infarct region to AAR after MI/R in diabetic mice was furtherincreased (Fig. 1A) compared to control. Left ventricular ejectionfraction (LVEF) by echocardiography was assessed to examine theeffect of hyperglycemia on cardiac function. Diabetic mice subjectedto MI/R exhibited significantly lower LVEF than control mice (Fig. 1B),and basal condition statistics was similar between the two groups. Insummation, I/R enlarged infarct size and depressed cardiac function indiabetic mice to a greater degree than in control.
3.2. I/R-induced myocardial apoptosis also exacerbated in diabetic mice
Substantial evidence exists that apoptosis plays a critical role incardiomyocyte loss and the subsequent development of cardiacdysfunction after MI/R [20]. To investigate whether the observedexacerbated myocardial injury in diabetic mice was associated withincreased apoptosis, the TUNEL staining and caspase-3 activity assayswere performed. Fig. 2 reveals that hyperglycemia alone did notelevate cardiomyocyte apoptosis significantly (sham operation
dELISA assay (bottom). Representative photographs of heart sections are shown. (B) Trx-1re shown. Lane 1=control+sham; lane 2=DM+sham; lane 3=control+MI/R; lanerosine. *Pb0.05 vs. control+sham; #Pb0.05 vs. DM+sham; **Pb0.05 vs. control+MI/R;

Fig. 4. (A) Trx-1/ASK1 binding in myocardium determined by immunoprecipitation.(B) p38 MAPK activity determined by assay kit. Representative immunoblot graphs areshown. Lane 1=control+sham; lane 2=DM+sham; lane 3=control+MI/R; lane4=DM+MI/R. *Pb0.05 vs. control+sham; #Pb0.05 vs. DM+sham; **Pb0.05vs. control+MI/R; n=6 to 8 hearts/group.
358 T. Yin et al. / Journal of Molecular and Cellular Cardiology 49 (2010) 354–361
caused no statistical difference in the percentage of TUNEL-positivenuclei or caspase-3 activity between diabetic and nondiabetic mice).However, apoptosis significantly increased after MI/R to a greaterextent in diabetic mice (Fig. 2).
3.3. I/R-induced protein nitration exacerbated in diabetic mice
Nitrotyrosine has been used extensively as a footprint of proteinnitration. Using immunohistochemistry techniques, we quantifiednitrotyrosine content by enzyme-linked immunosorbent assay(ELISA) kit. Both MI/R and diabetic groups manifested nitrotyrosineformation, while it was absent in the control group (Fig. 3A). Themostprominent staining occurred in diabetic mice following MI/R.Quantitative analysis confirmed MI/R-induced nitrotyrosine produc-tion amplification in diabetic mice (Fig. 3A).
3.4. I/R-induced Trx-1 nitration increased and Trx-1 activity decreased indiabetic mice
Emerging evidence indicates that Trx-1 acts as a key regulator ofcardiovascular homeostasis, with numerous defensive functionsagainst oxidative stress under different pathological conditions. In arecent study, we demonstrated both the susceptibility of Trx-1 tonitrative modification and its irreversible inhibition by this post-translational modification [12]. Therefore, Trx-1 was a likely andsuitable target protein for heightened nitration in the hyperglycemiccondition. Trx-1 nitration was not detected in control mice cardiactissues (Fig. 3B), while clear Trx-1 nitration was consistently detectedin diabetic cardiac tissues, even before MI/R. Consistent with ourprevious study, MI/R induced significant Trx-1 nitration, exacerbatedin diabetic mice (Fig. 3B). With increased nitration, Trx-1 activity wasinhibited in parallel (Fig. 3C).
3.5. The diabetic condition further inhibited Trx-1/ASK1 interaction andincreased p38 MAPK activity
Previous studies from others as well as ours have shown that thebinding of Trx-1 with ASK1 (causing inhibition of downstreamproapoptotic kinases) is an important mechanism by which Trx-1exerts antiapoptotic effects under oxidative stress [12,21]. Weinvestigated the signaling pathway by which enhanced nitration ofTrx-1 increased MI/R-induced myocardial apoptosis in diabetic mice.Trx-1 was physically associated with ASK1 in cardiac tissues isolatedfrom control mice (Fig. 4A), and p38 MAPK activity was inhibited(Fig. 4B). In both the diabetic and MI/R groups, this protein–proteininteraction was decreased (Fig. 4A), and p38 MAPK activity wasincreased (Fig. 4B). The disassociation of Trx-1 and ASK1, andconsequent p38 MAPK activation, was enhanced in the diabetic MI/Rgroup (Fig. 4). These results suggest that increased activation of theASK1–p38 MAPK signaling pathway may contribute to the increasedmyocardial apoptosis after reperfusion in the diabetic state.
3.6. Blockade of Trx-1 nitrative inactivation, or Trx-1 supplement, isprotective of diabetic hearts from I/R injury
Peroxynitrite (ONOO−), the diffusion-controlled reaction productof nitric oxide and superoxide radical, is a potent pathologicallyrelevant nitrative molecule [22]. To obtain more direct evidence tosupport our hypothesis that increased nitrative inactivation of Trx-1 iscausatively related to increased MI/R injury in diabetic mice, EUK134(an ONOO− scavenger), or exogenous rhTrx-1 (which has beenshown to translocate into living cells, thus increasing intracellularTrx-1 content) [14], was administrated before reperfusion. Asillustrated in Figs. 5 and 6, treatment with EUK134 significantlyreduced nitrotyrosine content and Trx-1 nitration, preserved Trx-1activity, restored Trx-1/ASK1 interaction, and inhibited p38 MAPK
activity in diabetic mouse cardiac tissues after MI/R, suggesting thatEUK134 could reduce oxidative/nitrative stress and subsequentnitrative inactivation of Trx-1 in our animal model. Treatment withEUK134 or rhTrx-1 significantly reduced infarct size (Fig. 7A) andattenuated myocardial apoptosis in diabetic mice subjected to MI/R,evidenced by both decreased caspase-3 activity (Fig. 7C) and TUNELstaining (Fig. 7D). These treatments also improved cardiac function indiabetic mice after MI/R as evidenced by improved LVEF (Fig. 7B).Finally, in vitro incubation of Trx-1 (50 μM) with a peroxynitritedonor, SIN-1 (100 µM) for 30 min markedly inhibited Trx-1 activity(117±12 µmol/min/mg Trx-1 vs. 879±65 µmol/min/mg Trx-1 invehicle-incubated Trx-1, Pb0.001). This result is consistent with thatreported in our previous study [12]. Administration of this nitrativelyinactivated Trx-1 neither reduced MI/R-induced apoptosis (TUNELand caspase-3) and infarct size nor improved cardiac functionrecovery after MI/R (Figs. 7A–D).
4. Discussion
In recent years, enormous efforts have been made to identify themechanisms underlying diabetes-induced vascular injury, responsiblefor increased IHD risk in diabetic patients. However, many clinicalstudies have overwhelmingly demonstrated worse cardiac functionand poorer outcome after IHD in diabetic patients even afterrevascularization [2], suggesting diabetes may have directly adverse

Fig. 5. Effect of EUK134 on MI/R-induced nitrotyrosine content production (A) and Trx-1nitrative inactivation (B, C) in diabetic mouse myocardium. *Pb0.05 vs. DM+sham;#Pb0.05 vs. DM+MI/R; n=6 to 8 hearts/group.
Fig. 6. Effect of EUK134 on MI/R-induced dissociation of Trx-1 from ASK1 (A) and p38MAPK activation (B) in diabetic mouse myocardium. *Pb0.05 vs. DM+sham; #Pb0.05vs. DM+MI/R; n=6 to 8 hearts/group.
359T. Yin et al. / Journal of Molecular and Cellular Cardiology 49 (2010) 354–361
impact on ischemic myocardium beyond the coronary vasculature.Furthermore, hyperglycemia at the time of myocardial infarctionrepresents an independent risk factor for in-hospital mortality, even innondiabetic patients [3]. Therefore, identifying the mechanismsunderlying the direct association between hyperglycemia and ischemicmyocardium is equally important. Although our recent study demon-strated that preculture with high-glucose sensitized cardiomyocytes tosimulated I/R injury via Trx-1 nitrative inactivation [13], moreconvincing in vivo studies needed to be done.
In contrast to relatively consistent clinical study results, contro-versy exists as to the susceptibility of diabetic hearts to I/R injury inanimal models [6,23]. Our present study clearly demonstratedexacerbated MI/R infarct size injury in STZ-induced diabetic mice.We also revealed MI/R injury decreased left ventricular (LV) systolicperformance, consistent with the reports of others [24,25], a findingexaggerated in diabetic mice, despite the utilization of multiple low-dose STZ administration to minimize nonspecific toxic STZ effects[26]. However, while DM alone did not significantly affect LV function
before MI/R in the present study, others have shown diabetes by itselfis responsible for LV systolic and diastolic dysfunction [27]. Our resultsare partly supported by another study, which suggested that normalsystolic function was preserved in diabetic mice before MI/R [24]. Apossible explanation for these inconsistent results may be theemployment of differing experimental conditions such as differentanimal subject types, the duration and severity of the diabetic state,the degree of ischemia incurred, etc [23]. Since hyperglycemiaincreased the risk of congestive heart failure or cardiogenic shockafter myocardial infarction in patients with and without diabetes [3],the currently utilized animal model mimics the clinical situation tosome extent.
Apoptosis plays an important role in I/R- or hyperglycemia-inducedmyocardial injury [20,28]. Although our previous studies and those ofothers [29,30] indicate that I/R-induced myocardial apoptosis isexacerbated in the diabetic condition (contributive to left ventricularremodeling and dysfunction), the exact mechanism contributing to theobserved increased apoptosis remains unclear. Growing evidencesupports the role of pathologic protein nitration in cardiovasculardisease and apoptosis. In our present study, MI/R-induced proteinnitration, earmarked by nitrotyrosine content, was markedly potenti-ated indiabeticmice. Others have demonstrated increasednitrotyrosinelevels in hearts subjected toMI/Runderhigh-glucose conditions [31,32].Furthermore, from ventricular myocardial biopsies from diabeticpatients, Frustaci et al. [33] reported that apoptotic nucleiwere detectedonly in cells containing nitrated proteins (i.e., those nitrotyrosinepositively staining). Moreover, we have shown that blockade of protein

Fig. 7. Effect of EUK134, exogenous rhTrx-1, or nitrated Trx-1 (N-Trx-1) on infarct size (A), cardiac function (B), and myocardial apoptosis (C, D) in diabetic mice after MI/R. *Pb0.05vs. DM+sham; #Pb0.05 vs. DM+MI/R; n=6 to 8 hearts/group. Insert:Western blots showing that Trx is nitratively modified by SIN-1 and that this modification is blocked by a SODmimetic. Recombinant human Trx-1 was incubated with vehicle or SIN-1 at 37 °C for 30 min in the presence and absence of MnTE-2-PyP5+ (500 μM, 5-times of SIN-1 concentration),a cell-permeable SOD mimetic (SODm). Unreacted SIN-1 was removed by ultrafiltration over membranes with a 5-kDa cutoff. Samples were electrophoretically size fractionated onSDS–PAGE and transferred onto a polyvinylidene difluoride (PVDF)-plus membrane, and nitrated Trx-1 was detected with antinitrotyrosine antibody.
360 T. Yin et al. / Journal of Molecular and Cellular Cardiology 49 (2010) 354–361
nitration inhibited MI/R-induced apoptosis and myocardial injuryin diabetic mice. In summation, these results suggest a plausiblerelationship between protein nitration and enhanced susceptibility ofdiabetic hearts to I/R injury caused by increased apoptosis.
Accumulating evidence indicates that hyperglycemia reducesantioxidant defenses and increases oxidative stress, which initiatesseveral complex molecular events contributive to diabetic tissuedamage, and subsequent complication development [34]. Both ex vivoand in vivo studies have reported that hyperglycemia enhancedinducible nitric oxide synthase (iNOS) expression and ROS/RNSproduction in reperfused hearts [31,35]. Furthermore, as other studieshave revealed, the ability of antioxidant treatment or iNOS knockoutto reduce the adverse effects of hyperglycemia-exacerbated MI/Rinjury suggests the important role of ROS and RNS in these responses[32,36–38]. Therefore, significant ROS and RNS generation, a conditionthat would favor ONOO− formation, may be responsible for theincreased protein nitration observed in the diabetic state [39].
We attempted to identify a specific protein target susceptible andrelevant for nitration. Trx, a small ubiquitous protein, fulfills a varietyof biological functions, and it has been shown to act as a key regulatorof cardiovascular homeostasis [10]. Trx-1 exerts its potent antiapop-totic effect by direct (Trx-1/ASK1 interaction) and indirect (antiox-idative) mechanisms [14]. ASK1, a member of the mitogen-activatedprotein kinase kinase kinase (MAP3K) family, induces apoptosis byactivating downstream p38 MAPK and JNK signaling pathways. Inresting conditions, ASK1 is inhibited by Trx-1 binding. In the presentstudy, we provided the first evidence that Trx-1 was nitrativelyinactivated in the diabetic mouse myocardial tissues even before I/R,and its effects were magnified after ischemia/reperfusion. In parallelwith the nitration of Trx-1, ASK1 disassociated from Trx-1, with
consequent activation of downstream signal molecule p38 MAPK.Based on these above data, Trx-1 is likely one of the nitration targetsthat account for amplified MI/R injury in diabetic mice.
To provide further cause-to-effect evidence, we determinedwhether blockade of Trx-1 nitrative inactivation or supplementationwith exogenous Trx-1 could protect diabetic mice against MI/R injury.We demonstrated that either treatment reduced myocardial apopto-sis by counteracting MI/R injury to some extent in diabetic mice.These results provide the first evidence that Trx-1 nitrative inactiva-tion plays a causative role in rendering the diabetic heart moresensitive to I/R injury. To the best of our knowledge, we link thenitration of a certain protein to increased vulnerability of the diabeticheart to worsened I/R injury in vivo for the first time.
Our results also reveal that increased nitrative Trx-1 modificationaccounts for, at least partly, further ASK1 releasing/activation-inducedapoptosis in diabetic mice when subjected to MI/R injury. Previousstudies have demonstrated that ASK1 binding requires the presence ofan intramolecular disulfide bridge between two critical cysteine (Cys)residues (Cys32 and Cys35) at the catalytic site of Trx-1 becauseoxidized Trx-1 cannot bind and inhibit ASK1 [21,40]. However,administration of oxidized Trx-1 still exerts significant antiapoptoticand cytoprotective effects, unless intracellular Trx reductase is inhibitedconcomitantly [41], indicating that oxidative modification of Trx-1 isreversible and, therefore, may not play a principal role in increasingapoptosis (via ASK-1 binding release) in vivo. In comparison, nitrativemodification of Trx-1 is irreversible [12]; however, the specificmechanism bywhich this form of posttranslational modification resultsin ASK1 disassociation remains to be investigated in our further studies.
It should be indicated that besides the nitrative inhibitory effect of Trxreported here, Trx activity is also inhibited by excessive S-nitrosylation

361T. Yin et al. / Journal of Molecular and Cellular Cardiology 49 (2010) 354–361
when exposed to high levels of nitric oxide donor [42,43]. However, inthe current study, Trx was modified either in vivo by ischemia/reperfusion (where nitric oxide level is much lower than those usedfor in vitro Trx S-nitrosylation) or in vitro by SIN-1, a chemical that onlyproduces peroxynitrite but not nitric oxide unless high concentrationsof SOD are present. Therefore, it is highly unlikely that excessive Trx S-nitrosylation occurs either in vivo or in vitro under our experimentalsetting. Moreover, in our previously published study [12], we havedemonstrated that at 100 μM concentration, SIN-1 caused significantTrx nitration. No Trx S-nitrosylation is detected until SIN-1 concentra-tion is increased to 1 mM. This result indicates that SIN-1 is highlyefficient in inducing Trx nitrative modification but is rather weak inS-nitrosylative modification of Trx. Therefore, the SIN-1 and ONOO−
inhibitory effects of Trx observed in this study could not be attributedto its S-nitrosylative inhibition.
In summary, our current study's novel findings strongly suggest thathyperglycemia in diabetic patients promotes ONOO− formation, facilita-tive of advanced ROS and RNS production, enhancing nitrative inactiva-tion of Trx-1, and thereby sensitizing hearts to I/R injury by elevatedASK1–p38 MAPK signaling pathway-mediated cardiomyocyte apoptosis.
Acknowledgments
This study was supported by the following grants: NationalNatural Science Foundation of China 30670879 (L.T.) and 30770784(W.H.), National 863 Project of China 2009AA02Z104 (L.T.) andSubject Boosting Project of Xijing Hospital XJZT08Z02 (L.T.).
References
[1] Mazzone T. Hyperglycaemia and coronary heart disease: the meta picture. Lancet2009;373:1737–8.
[2] Donahoe SM, Stewart GC, McCabe CH, Mohanavelu S, Murphy SA, Cannon CP, et al.Diabetes and mortality following acute coronary syndromes. JAMA 2007;298:765–75.
[3] Capes SE, Hunt D, Malmberg K, Gerstein HC. Stress hyperglycaemia and increasedrisk of death after myocardial infarction in patients with and without diabetes: asystematic overview. Lancet 2000;355:773–8.
[4] Zweier JL, Talukder MA. The role of oxidants and free radicals in reperfusion injury.Cardiovasc Res 2006;70:181–90.
[5] Haidara MA, Yassin HZ, Rateb M, Ammar H, Zorkani MA. Role of oxidative stress indevelopment of cardiovascular complications in diabetes mellitus. Curr VascPharmacol 2006;4:215–27.
[6] Ferdinandy P, Schulz R, Baxter GF. Interaction of cardiovascular risk factors withmyocardial ischemia/reperfusion injury, preconditioning, and postconditioning.Pharmacol Rev 2007;59:418–58.
[7] Wattanapitayakul SK, Bauer JA. Oxidative pathways in cardiovascular disease: roles,mechanisms, and therapeutic implications. Pharmacol Ther 2001;89:187–206.
[8] Turko IV, Murad F. Protein nitration in cardiovascular diseases. Pharmacol Rev2002;54:619–34.
[9] Greenacre SA, Ischiropoulos H. Tyrosine nitration: localisation, quantification,consequences for protein function and signal transduction. Free Radic Res2001;34:541–81.
[10] Yamawaki H, Haendeler J, Berk BC. Thioredoxin: a key regulator of cardiovascularhomeostasis. Circ Res 2003;93:1029–33.
[11] Kondo N, Nakamura H, Masutani H, Yodoi J. Redox regulation of human thioredoxinnetwork. Antioxid Redox Signal 2006;8:1881–90.
[12] Tao L, Jiao X, Gao E, Lau WB, Yuan Y, Lopez B, et al. Nitrative inactivation ofthioredoxin-1 and its role in postischemic myocardial apoptosis. Circulation2006;114:1395–402.
[13] Luan R, Liu S, Yin T, Lau WB, Wang Q, Guo W, et al. High glucose sensitizes adultcardiomyocytes to ischaemia/reperfusion injury through nitrative thioredoxininactivation. Cardiovasc Res 2009;83:294–302.
[14] Tao L, Gao E, Bryan NS, Qu Y, Liu HR, Hu A, et al. Cardioprotective effects ofthioredoxin in myocardial ischemia and reperfusion: role of S-nitrosation. PNAS2004;101:11471–6.
[15] Tao L, Gao E, Jiao X, Yuan Y, Li S, Christopher TA, et al. Adiponectin cardioprotectionafter myocardial ischemia/reperfusion involves the reduction of oxidative/nitrativestress. Circulation 2007;115:1408–16.
[16] Yamamoto M, Yang G, Hong C, Liu J, Holle E, Yu X, et al. Inhibition of endogenousthioredoxin in the heart increases oxidative stress and cardiac hypertrophy. J ClinInvestig 2003;112:1395–406.
[17] Zhang H, Tao L, Jiao X, Gao E, Lopez BL, Christopher TA, et al. Nitrative thioredoxininactivation as a cause of enhanced myocardial ischemia/reperfusion injury in theaging heart. Free Radic Biol Med 2007;43:39–47.
[18] Vadseth C, Souza JM, Thomson L, Seagraves A, Nagaswami C, Scheiner T, et al. Pro-thrombotic state induced by post-translational modification of fibrinogen byreactive nitrogen species. J Biol Chem 2004;279:8820–6.
[19] GuoW, Adachi T, Matsui R, Xu S, Jiang B, Zou MH, et al. Quantitative assessment oftyrosine nitration of manganese superoxide dismutase in angiotensin II-infusedrat kidney. Am J Physiol Heart Circ Physiol 2003;285:H1396–403.
[20] Eefting F, Rensing B, Wigman J, Pannekoek WJ, Liu WM, Cramer MJ, et al. Role ofapoptosis in reperfusion injury. Cardiovasc Res 2004;61:414–26.
[21] Bishopric NH, Webster KA. Preventing apoptosis with thioredoxin: ASK me how.Circ Res 2002;90:1237–9.
[22] Szabo C, Ischiropoulos H, Radi R. Peroxynitrite: biochemistry, pathophysiologyand development of therapeutics. Nat Rev Drug Discov 2007;6:662–80.
[23] GalinanesM, Fowler AG. Role of clinical pathologies inmyocardial injury followingischaemia and reperfusion. Cardiovasc Res 2004;61:512–21.
[24] Shiomi T, Tsutsui H, Ikeuchi M, Matsusaka H, Hayashidani S, Suematsu N, et al.Streptozotocin-induced hyperglycemia exacerbates left ventricular remodelingand failure after experimental myocardial infarction. J Am Coll Cardiol 2003;42:165–72.
[25] Shishido T, Woo CH, Ding B, McClain C, Molina CA, Yan C, et al. Effects of MEK5/ERK5 association on small ubiquitin-relatedmodification of ERK5: implications fordiabetic ventricular dysfunction after myocardial infarction. Circ Res 2008;102:1416–25.
[26] Hsueh W, Abel ED, Breslow JL, Maeda N, Davis RC, Fisher EA, et al. Recipes forcreating animal models of diabetic cardiovascular disease. Circ Res 2007;100:1415–27.
[27] Joffe II, Travers KE, Perreault-Micale CL, Hampton T, Katz SE, Morgan JP, et al.Abnormal cardiac function in the streptozotocin-induced non-insulin-dependentdiabetic rat: noninvasive assessment with doppler echocardiography andcontribution of the nitric oxide pathway. J Am Coll Cardiol 1999;34:2111–9.
[28] Cai L, Li W, Wang G, Guo L, Jiang Y, Kang YJ. Hyperglycemia-induced apoptosis inmouse myocardium: mitochondrial cytochrome C-mediated caspase-3 activationpathway. Diabetes 2002;51:1938–48.
[29] Yue TL, Bao W, Gu JL, Cui J, Tao L, Ma XL, et al. Rosiglitazone treatment in zuckerdiabetic Fatty rats is associated with ameliorated cardiac insulin resistance andprotection from ischemia/reperfusion-induced myocardial injury. Diabetes2005;54:554–62.
[30] Backlund T, Palojoki E, Saraste A, Eriksson A, Finckenberg P, Kyto V, et al. Sustainedcardiomyocyte apoptosis and left ventricular remodelling after myocardialinfarction in experimental diabetes. Diabetologia 2004;47:325–30.
[31] Ceriello A, Quagliaro L, D'Amico M, Di Filippo C, Marfella R, Nappo F, et al. Acutehyperglycemia induces nitrotyrosine formation and apoptosis in perfused heartfrom rat. Diabetes 2002;51:1076–82.
[32] Marfella R, Di Filippo C, Esposito K, Nappo F, Piegari E, Cuzzocrea S, et al. Absenceof inducible nitric oxide synthase reduces myocardial damage during ischemiareperfusion in streptozotocin-induced hyperglycemic mice. Diabetes 2004;53:454–62.
[33] Frustaci A, Kajstura J, Chimenti C, Jakoniuk I, Leri A, Maseri A, et al. Myocardial celldeath in human diabetes. Circ Res 2000;87:1123–32.
[34] Brownlee M. Biochemistry and molecular cell biology of diabetic complications.Nature 2001;414:813–20.
[35] Bucciarelli LG, Ananthakrishnan R, Hwang YC, Kaneko M, Song F, Sell DR, et al.RAGE and modulation of ischemic injury in the diabetic myocardium. Diabetes2008;57:1941–51.
[36] Marfella R, D'AmicoM, Di Filippo C, Piegari E, Nappo F, Esposito K, et al. Myocardialinfarction in diabetic rats: role of hyperglycaemia on infarct size and earlyexpression of hypoxia-inducible factor 1. Diabetologia 2002;45:1172–81.
[37] Ceradini DJ, Yao D, Grogan RH, Callaghan MJ, Edelstein D, Brownlee M, et al.Decreasing intracellular superoxide corrects defective ischemia-induced newvessel formation in diabetic mice. J Biol Chem 2008;283:10930–8.
[38] Heusch G, Boengler K, Schulz R. Cardioprotection: nitric oxide, protein kinases,and mitochondria. Circulation 2008;118:1915–9.
[39] Schulz R, Kelm M, Heusch G. Nitric oxide in myocardial ischemia/reperfusioninjury. Cardiovasc Res 2004;61:402–13.
[40] Saitoh M, Nishitoh H, Fujii M, Takeda K, Tobiume K, Sawada Y, et al. Mammalianthioredoxin is a direct inhibitor of apoptosis signal-regulating kinase (ASK) 1.EMBO J 1998;17:2596–606.
[41] Andoh T, Chock PB, Chiueh CC. The roles of thioredoxin in protection againstoxidative stress-induced apoptosis in SH-SY5Y cells. J Biol Chem 2002;277:9655–60.
[42] Hashemy SI, Holmgren A. Regulation of the catalytic activity and structure ofhuman thioredoxin 1 via oxidation and S-nitrosylation of cysteine residues. J BiolChem 2008;283:21890–8.
[43] Sumbayev VV. S-nitrosylation of thioredoxin mediates activation of apoptosissignal-regulating kinase 1. Arch Biochem Biophys 2003;415:133–6.