Embed Size (px)
Citation preview

BIOPHARMACEUTICS & DRUG DISPOSITIONBiopharm. Drug Dispos. (2014)Published online in Wiley Online Library(wileyonlinelibrary.com) DOI: 10.1002/bdd.1889
Nonclinical pharmacokinetics and metabolism of EPZ-5676, anovel DOT1L histone methyltransferase inhibitor
Aravind Basavapathruni, Edward J. Olhava, Scott R. Daigle, Carly A. Therkelsen, Lei Jin, P. Ann Boriack-Sjodin,Christina J. Allain, Christine R. Klaus, Alejandra Raimondi, Margaret Porter Scott, Angelos Dovletoglou,Victoria M. Richon†, Roy M. Pollock, Robert A. Copeland, Mikel P. Moyer, Richard Chesworth,Paul G. Pearson, and Nigel J. Waters*Epizyme Inc., 400 Technology Square, Cambridge, MA, USA
*CorrespFloor, CE-mail: n†Present02139, U
Copyrig
ABSTRACT: (2R,3R,4S,5R)-2-(6-Amino-9H-purin-9-yl)-5-((((1r,3S)-3-(2-(5-(tert-butyl)-1H-benzo[d]imidazol-2-yl)ethyl)cyclobutyl)(isopropyl)amino)methyl)tetrahydrofuran-3,4-diol (EPZ-5676) is anovel DOT1L histone methyltransferase inhibitor currently in clinical development for the treatmentof MLL-rearranged leukemias. This report describes the preclinical pharmacokinetics andmetabolism of EPZ-5676, an aminonucleoside analog with exquisite target potency and selectivitythat has shown robust and durable tumor growth inhibition in preclinical models. The in vivopharmacokinetics in mouse, rat and dog were characterized following i.v. and p.o. administration;EPZ-5676 had moderate to high clearance, low oral bioavailability with a steady-state volume ofdistribution 2–3 fold higher than total body water. EPZ-5676 showed biexponential kineticsfollowing i.v. administration, giving rise to a terminal elimination half-life (t1/2) of 1.1, 3.7 and13.6 h in mouse, rat and dog, respectively. The corresponding in vitro ADME parameters were alsostudied and utilized for in vitro–in vivo extrapolation purposes. There was good agreement betweenthe microsomal clearance and the in vivo clearance implicating hepatic oxidative metabolism as thepredominant elimination route in preclinical species. Furthermore, low renal clearance was observedin mouse, approximating to fu-corrected glomerular filtration rate (GFR) and thus passive glomerularfiltration. The metabolic pathways across species were studied in liver microsomes in which EPZ-5676 was metabolized to three monohydroxylated metabolites (M1, M3 and M5), one N-dealkylatedproduct (M4) as well as an N-oxide (M6). Copyright © 2014 John Wiley & Sons, Ltd.
Key words: MLL-rearranged leukemia; nonclinical pharmacokinetics; in vitro–in vivo extrapolation;metabolite identification
Introduction
Recent advances in the understanding of cancerincidence have implicated epigenetics and epige-netic targets as potential avenues for therapeutic
ondence to: Epizyme Inc., 400 Technology Square, 4thambridge, MA 02139, [email protected]: Sanofi, 270 Albany Street, Cambridge, MA,SA
ht © 2014 John Wiley & Sons, Ltd.
intervention. Epigenetic modifications that mayplay a hand in cancer development range fromchanges in chromatin remodeling, DNA methyla-tion or post-translational modifications of histones[1]. One such histone modification is strongly tiedto a specific form of leukemia, in which transloca-tion of the mixed lineage leukemia (MLL) generesults in MLL-fusion proteins that can aberrantlyassociate with the histone methyltransferaseDOT1L (disruptor of telomeric silencing-1 like),resulting in ectopic DOT1L-catalysed methylation
Received 5 August 2013Revised 27 November 2013Accepted 31 December 2013

A. BASAVAPATHRUNI ET AL.
of lysine 79 of histone H3 (H3K79) [2–8]. AberrantH3K79 methylation serves to drive the expressionof MLL target genes and an oncogenic phenotype.The strong causality between the H3K79 meth-
ylation mark and a cancer phenotype providesan opportunity for small molecule interventionof DOT1L catalytic activity. We have reportedpreviously structure-guided medicinal chemistryefforts that yielded a potent DOT1L inhibitor,EPZ004777, demonstrating the first meaningfulproof of concept in histone methyltransferase (HMT)inhibition [9,10]. Further expansion of our medicinalchemistry efforts generated the potent moleculeEPZ-5676 ((2R,3R,4S,5R)-2-(6-amino-9H-purin-9-yl)-5-((((1r,3S)-3-(2-(5-(tert-butyl)-1H-benzo[d]imidazol-2-yl)ethyl)cyclobutyl)(isopropyl)amino)methyl)tetrahy-drofuran-3,4-diol), an aminonucleoside analogwithimproved inhibition versus DOT1L in in vitrobiochemical and cellular assays [11]. EPZ-5676inhibits DOT1L with a Ki of≤ 80pM and displays37000-fold selectivity over a panel of other HMTs.The potency is further exemplified by treatmentin a rat xenograft model of MLL-rearrangedleukemia with EPZ-5676, in which continuousintravenous (i.v.) infusion of EPZ-5676 causedcomplete tumor regressions that were sustainedbeyond the compound infusion period with nosignificant weight loss or signs of toxicity [11].This report describes the preclinical pharmaco-
kinetics and metabolism of EPZ-5676, a novelDOT1L inhibitor and the first member of the novelHMTi class to enter clinical development as a po-tential therapeutic agent in MLL-rearrangedleukemia. The objectives of this work were tocharacterize the pharmacokinetics following i.v.and p.o. administration in mouse, rat and dog, toassess the cross-species in vitro–in vivo correlationand to identify the primary metabolic and elimi-nation pathways involved in the clearance ofEPZ-5676. Understanding the pharmacokineticproperties along with the remarkable potencyof EPZ-5676 both in vitro and in vivo promotedthe development of this molecule for acute leu-kemias bearing MLL-rearrangements. EPZ-5676is currently in Phase I evaluation and representsnot only the first reported histonemethyltransferase inhibitor to enter human clin-ical trials, but a further step towards under-standing the link between epigenetic processesand the pathophysiology of cancer.
Copyright © 2014 John Wiley & Sons, Ltd.
Materials and Methods
Chemicals and reagents
EPZ-5676 was synthesized by Epizyme [11]. Allother reagents were purchased from sources asdescribed below.
In vivo pharmacokinetics
All animal studies were conducted as perapproved IACUC protocols.
Pharmacokinetic study in mouse. The pharmacoki-netics of EPZ-5676 was evaluated in male CD1-mice (28–29 g, male, n= 21, purchased from BKLaboratory Animal Co. Ltd) following i.v. bolusadministration of doses of 5mg/kg and oraladministration at doses of 20mg/kg. Oral gavageand i.v. tail vein injection doses were administeredin a 10% ethanol and 90% saline vehicle. For i.v.administration, blood samples were taken (n= 3per time-point; two time-points per mouse) at0.05, 0.167, 0.5, 1, 2, 4, 6 and 24 h post-dose intopre-chilled K2-EDTA tubes. For p.o. dosing, bloodsamples were taken (n= 3 per time-point; twotime-points per mouse) at 0.167, 0.5, 1, 2, 4 and6 h post-dose into pre-chilled K2-EDTA tubes.Blood samples were put on wet ice andcentrifuged at 4°C (2000 × g for 5min) to obtainplasma within 15min of sample collection. Plasmasamples were stored at �20 °C prior to LC-MS/MS analysis. CD-1 mice (n= 3) also received a sin-gle 5mg/kg i.v. administration of EPZ-5676followed by urine collection in metabolism cagesfor 240min post-dose. The urine aliquots werepooled, the total volume recorded and stored fro-zen at �20 °C prior to LC-MS/MS analysis.
Pharmacokinetic study in rat. The pharmacokineticsof EPZ-5676 was evaluated in male Sprague-Dawley rats (n=3 per dose route, 245–265g,purchased from SLAC Laboratory Animal Co.Ltd). For the i.v. bolus, 1mg/kg doses prepared in0.4% hydroxypropyl-beta-cyclodextrin (HPBCD)in saline were administered via foot dorsal veininjection. For p.o. administration, 10mg/kg dosesprepared in 10% ethanol: 5% Solutol HS15: 85%(5% of dextrose inwater) were administered by oralgavage. Serial blood sampling was employed ineach animal at each time-point, 0.05, 0.217, 0.5, 1,
Biopharm. Drug Dispos. (2014)DOI: 10.1002/bdd

NONCLINICAL PK AND METABOLISM OF EPZ-5676, A DOT1L INHIBITOR
2, 4, 8 and 24h following i.v. administration and0.25, 0.5, 1, 2, 4, 6, 8 and 24h following p.o. adminis-tration,with 150μl of blood collected via the tail veininto pre-chilled K2-EDTA tubes. Blood samples wereput on wet ice and centrifuged at 4 °C (2000 × g for5min) to obtain plasma within 15min of samplecollection. Plasma samples were stored at �20 °Cprior to LC-MS/MS analysis.
Pharmacokinetic study in dog. The intravenous (i.v.)pharmacokinetics of EPZ-5676 was evaluated inbeagle dogs (male, n= 3, 7.5–8 kg purchased fromBeijing Marshall Biotechnology Co. Ltd) followinga single i.v. administration at a dose of 1mg/kg.The i.v. doses were administered by a singleintravenous infusion over 1min into the cephalicvein in a 10% ethanol and 90% saline vehicle. Atdesignated time-points (pre-dose, 0.083, 0.25, 0.5,1, 2, 4, 8 and 24 h post-dose), the animals wererestrained manually, and approximately 0.5mlblood per time point was collected from the non-injected cephalic vein into pre-chilled K2-EDTAtubes. Blood samples were put on wet ice andcentrifuged at 4 °C (2000 × g for 5min) to obtainplasma within 15min of sample collection. Plasmasamples were stored at �20 °C prior to LC-MS/MS analysis.
LC-MS/MS bioanalysis and pharmacokinetic dataanalysis
EPZ-5676 was extracted from K2-EDTA plasma orurine by protein precipitation using an acetoni-trile-containing internal standard (a structural an-alog of EPZ-5676 at a concentration of 5 ng/ml).Typically, samples were injected onto an LC-MS/MS system using a Waters BEH phenyl column.The aqueous mobile phase was water with 0.1%NH4OH (A), and the organic mobile phase wasacetonitrile with 0.1% NH4OH (B). The gradientwas as follows: 37% B for the first 0.2min,increased to 44% B from 0.2 to 0.6min, maintainedat 44% B for 0.5min, and decreased to 37% Bwithin 0.05min. The injection volume was 2μl,and the total run time was 1.5min with a flow rateof 0.6ml/min. The retention time of EPZ-5676was 0.85min. The ionization was conducted inthe positive ion mode using the multiple reactionmonitoring (MRM) transition [M+H]+ m/z 563.5parent ion tom/z 326.3 daughter ion, incorporating
Copyright © 2014 John Wiley & Sons, Ltd.
a turbo-ionspray interface. Eight to ten calibrationstandards were prepared in blank plasma or urineof the relevant species providing a typical standardcurve concentration range of 0.5–1000 ng/ml.Calibration curves were performed in duplicate ineach analytical run togetherwith low,mid and highconcentration QCs in duplicate. All standard andQC measured concentrations fell within 85–115%of the nominal concentration.
Pharmacokinetic parameters were calculated bynoncompartmental methods using WinNonlin(version 5.3; Pharsight, St Louis, Missouri). Termi-nal t1/2 values were determined by regression ofat least three data-points in the later phase of thetime–concentration profile. The volume of distri-bution at steady state was calculated as below:
VDss¼Dose · AUMC0� inf
AUC0� infð Þ2
Parameters are presented as mean±SDwhere ap-plicable. Parent excretion in urine was calculated asthe % dose excreted= (urine concentration * urinevolume)/dose, accounting for the sample poolingacross three animals. The renal clearance, CLr, wascalculated as the amount in urine to time t/AUC0-t.
In vitro stability assays in liver microsomes andhepatocytes
Liver microsomes (final protein concentration0.5mg/ml), 0.1M phosphate buffer at pH7.4 andEPZ-5676 (final concentration of 3μM; finaldimethylsulfoxide (DMSO) concentration of0.25%) were pre-incubated at 37 °C prior to theaddition of NADPH (final concentration of 1mM)to initiate the reaction. The final incubationvolume was 50μl. Control incubations wereincluded for each species where 0.1M phosphatebuffer pH7.4 was added instead of NADPH(minus NADPH). Positive control compounds(diazepam and diphenhydramine for rodent,verapamil and dextromethorphan for human,testosterone in all species) were incubated inparallel to confirm microsomal activity. Theintrinsic clearance values obtained were withinthe range of historical data. EPZ-5676 and controlswere incubated for 0, 5, 15, 30 and 45min. The con-trol (minus NADPH) was incubated for 45minonly. The reactions were stopped by transferring
Biopharm. Drug Dispos. (2014)DOI: 10.1002/bdd

A. BASAVAPATHRUNI ET AL.
25μl of incubate to 50μl methanol at the appropriatetime points. The incubation plates were centrifugedat 1640 × g for 20min at 4 °C to aid proteinprecipitation.Human, Beagle dog, Sprague-Dawley rat and
CD-1 mouse cryopreserved hepatocytes wereobtained from XenoTech and stored at �150 °Cuntil use. The hepatocytes were thawed and pre-pared according to the vendor’s instructions,pooled into Krebs Henseleit buffer (KHB,pH7.4), and kept on ice prior to initiating theexperiment. The hepatocyte suspensions werepre-incubated in a shaking water bath at 37 °Cfor 3min, and then the reaction was initiated bythe addition of EPZ-5676 into the hepatocytesuspensions (1.5 × 106 cells/ml) at a final concen-tration of 3μM, and a DMSO content of 0.1%.The reaction mixture was incubated in a shakingwater bath at 37 °C. Aliquots of the incubation so-lutions were sampled at 0, 15, 30, 60 and 120min.The reaction was immediately terminated by theaddition of three volumes of ice-cold acetonitrilecontaining 0.1% formic acid and internalstandards. After centrifugation at 1640 × g for10min, the supernatants were transferred intoHPLC vials, and the test compound was analysedby LC-MS/MS. Testosterone (20μM) and7-hydroxycoumarin (100μM), were performed inparallel to confirm the enzyme activities of thehepatocytes used.The in vitro t1/2 values were determined by plot-
ting the natural logarithm of the analyte/IS peakarea ratios versus time, with the slope of the linearregression (�k) converted to in vitro t1/2 values byin vitro t1/2 =�0.693/k. Experimental half-liveswere transformed to the corresponding scaled in-trinsic clearance values (in units of ml/min/kg)as below:
CL int ¼ 0:693in vitro t1=2
·mL incubationmg microsones
· MPPGL · LWPBW
CL int ¼ 0:693in vitro t1=2
·mL incubationmillioncells
· HPGL · LWPBW
where MPPGL is microsomal protein per gramliver, HPGL is hepatocellularity (million cells pergram liver) and LWPBW is grams liver per kgbody weight.The scaled intrinsic clearance values were then
subsequently scaled to predicted in vivo clearancevalues, using the well-stirred venous equilibration
Copyright © 2014 John Wiley & Sons, Ltd.
model as described previously [12–14] and asshown below;
CLH ¼ QH�CL int
QH þ CL intCLH ¼
QH�f up�CL int
QH þ f up�CL int
where QH is the blood flow, fup is the fractionunbound in plasma and CLint is the scaled intrinsicclearance. The appropriate species-specific scalingfactors including MPPGL, HPGL, LWPBW andhepatic blood flows were used throughout [15–17].Since the microsomal incubational binding of EPZ-5676 was measured close to unity (Table 2), the twoversions of the well-stirred model that were appliedto the data are as shown above; (i) scaled CLwith nocorrection for binding parameters and (ii) scaled CLwith correction for fraction unbound in plasma(fup) only. These data are presented in Table 2.
Plasma protein binding, blood partitioning andplasma stability assays
Plasma protein binding was assessed by equilib-rium dialysis, utilizing the HT-dialysis cell formatwith a cellulose semi permeable membrane(molecular weight cut-off of 5000Da). Plasma waswarmed to 37 °C and adjusted to pH7.4 beforeuse. Male Sprague-Dawley rat, male Beagle dog,male CD-1 mouse and mixed sex human plasma(Harlan Sera-Lab Ltd, Loughborough, UK) wereused for the studies. A 5μM test compound solutionwas prepared in isotonic phosphate buffer and rat,dog, mouse and human plasma (final DMSOconcentration of 0.5%). The plasma-containingsolution was introduced to one side of themembrane, and the plasma-free on the other.Incubations were performed for 16h in duplicatein order to allow the compound to reach equilib-rium. Mass balance and recovery were assessedpost-incubation and were > 85% in all cases.Haloperidol was incubated in parallel as the controlcompound for each species. At the end of the equil-ibration time the cells were emptied. Followingprotein precipitation, the samples were centrifugedand analysed by LC-MS/MS. The samples from theprotein-containing compartment were quantifiedusing calibration standards prepared in plasmaand the protein-free compartments were quantifiedusing calibration standards prepared in dialysisbuffer. Using a similar methodology, the incubationalbinding of 3μM EPZ-5676 to liver microsomes
Biopharm. Drug Dispos. (2014)DOI: 10.1002/bdd

NONCLINICAL PK AND METABOLISM OF EPZ-5676, A DOT1L INHIBITOR
(0.5mg/ml) from mouse, rat, dog and human wasassessed, with amitriptyline as a positive controlcompound (fuinc 0.35–0.4).For blood partitioning, male Sprague Dawley
rat, male Beagle dog and male CD-1 mouse bloodwas sourced from Harlan Sera-Lab Ltd, Loughbor-ough, UK. Mixed sex human blood was obtainedfrom in-house healthy donors. The hematocritwas measured using a Hettich Hematokrit 210and calculated as the percentage of packed cellvolume compared with the total volume of wholeblood. EPZ-5676 (final test compound concentra-tion 0.5μM, final DMSO concentration 0.05%) wasincubated separately with fresh heparinized wholeblood, reference red blood cells and referenceplasma for 60min at 37 °C in triplicate. Followingincubation, the whole blood cell samples werecentrifuged for 5min at 5000 × g at 4 °C. The spikedreference plasma was stored on ice during thisperiod. The spiked reference red blood cells werefreeze-thawed quickly three times to assist inlysing the red blood cells. Following centrifugationof the whole blood experimental sample, an aliquotwas sampled from the plasma and red blood celllayers for analysis. As before, the red blood celllayer was freeze-thawed quickly three times to lysethe red blood cells. After protein precipitation andcentrifugation, the supernatants for the experi-mental samples and reference samples wereanalysed by LC-MS/MS. Blood-to-plasma ratioswere calculated as described previously [18].Chlorthalidone was used as a positive controlin this assay (rat B:P ratio of 73).For plasma stability, EPZ-5676 (1μM) was incu-
bated with pooled lots of human, Beagle dog,Sprague Dawley rat and CD-1 mouse plasma for0, 15, 30, 60 and 120min at 37 °C. Samples werequenched in methanol and analysed by LC-MS/MS analysis.
MDCK cell permeability assays
Confluent monolayers of Madin-Darby caninekidney (MDCK) or MDCK-MDR1 (P-glycoprotein)cells, 7–14days old, in Transwell® dual-chamberplates, with apical and basolateral compartmentsbuffered at pH7.4, were dosed on the apical side(A-to-B) or basolateral side (B-to-A) with EPZ-5676 (10μM) and incubated at 37 °C with 5% CO2
in a humidified incubator. Samples were taken
Copyright © 2014 John Wiley & Sons, Ltd.
from the donor and receiver chambers at 120min.Each determination was performed in duplicate.The co-dosed lucifer yellow fluxwas alsomeasuredfor each monolayer to ensure cell monolayersremained intact during the incubation. The recov-ery of EPZ-5676 in donor and recipient wells post-incubation was> 90% for all replicates. All sampleswere assayed by LC-MS/MS.
Metabolite profiling and identification
EPZ-5676 was incubated with liver microsomesof various species (mouse, rat, dog or human).In vitro metabolite profiling and identification wereconducted after incubating EPZ-5676 (final concen-tration of 10μM) with mouse, rat, dog or humanliver microsomes (final protein concentration of0.5mg/ml) at 37 °C in 100mM potassiumphosphate buffer containing 2mMMg2+ in thepresence of 2mM NADPH and 2mM uridinediphosphoglucuronic acid (UDPGA) (with the ad-dition of 0.1mg/ml alamethacin to human and ratmicrosomes). For all liver microsomal incubations,samples were taken at 0 and 20min. All sampleswere quenched by using the acetonitrile/methanolsolution and analysed using an LC-MS/MSQ-Trapsystem (AB Sciex, Framingham, MA).
The major metabolites of EPZ-5676 in terms ofthe mass spectrometry response were identifiedby comparison of the LC-MS total ion chromato-grams (TIC) of 0min and 20min samples in fullscan mode using LightSight™ 2.0 software. Thecorresponding product ion tandem mass spectraof EPZ-5676 and its metabolites were obtainedby using enhanced product ion (EPI) scans duringpositive ion electrospray. The possible chemicalstructures of the metabolites were deduced basedon their MS1 and MS2 spectra. In addition, thehydroxylated t-butyl analog of EPZ-5676 wassynthesized to aid metabolite structure elucidation.
Results
In vivo pharmacokinetics
The pharmacokinetics of EPZ-5676 was studiedfollowing i.v. bolus administration to mouse, ratand dog as well as following p.o. administrationto mouse and rat. The time–concentration dataare shown in Figure 1 and the parameters derived
Biopharm. Drug Dispos. (2014)DOI: 10.1002/bdd
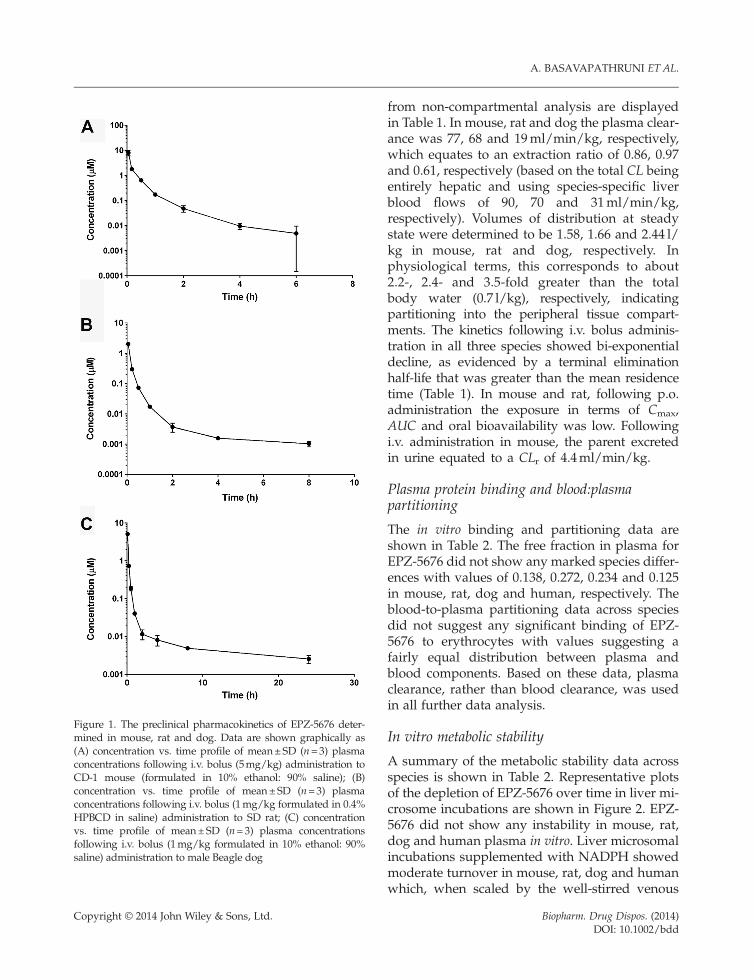
Figure 1. The preclinical pharmacokinetics of EPZ-5676 deter-mined in mouse, rat and dog. Data are shown graphically as(A) concentration vs. time profile of mean±SD (n=3) plasmaconcentrations following i.v. bolus (5mg/kg) administration toCD-1 mouse (formulated in 10% ethanol: 90% saline); (B)concentration vs. time profile of mean±SD (n=3) plasmaconcentrations following i.v. bolus (1mg/kg formulated in 0.4%HPBCD in saline) administration to SD rat; (C) concentrationvs. time profile of mean±SD (n=3) plasma concentrationsfollowing i.v. bolus (1mg/kg formulated in 10% ethanol: 90%saline) administration to male Beagle dog
A. BASAVAPATHRUNI ET AL.
Copyright © 2014 John Wiley & Sons, Ltd.
from non-compartmental analysis are displayedin Table 1. In mouse, rat and dog the plasma clear-ance was 77, 68 and 19ml/min/kg, respectively,which equates to an extraction ratio of 0.86, 0.97and 0.61, respectively (based on the total CL beingentirely hepatic and using species-specific liverblood flows of 90, 70 and 31ml/min/kg,respectively). Volumes of distribution at steadystate were determined to be 1.58, 1.66 and 2.44 l/kg in mouse, rat and dog, respectively. Inphysiological terms, this corresponds to about2.2-, 2.4- and 3.5-fold greater than the totalbody water (0.7 l/kg), respectively, indicatingpartitioning into the peripheral tissue compart-ments. The kinetics following i.v. bolus adminis-tration in all three species showed bi-exponentialdecline, as evidenced by a terminal eliminationhalf-life that was greater than the mean residencetime (Table 1). In mouse and rat, following p.o.administration the exposure in terms of Cmax,AUC and oral bioavailability was low. Followingi.v. administration in mouse, the parent excretedin urine equated to a CLr of 4.4ml/min/kg.
Plasma protein binding and blood:plasmapartitioning
The in vitro binding and partitioning data areshown in Table 2. The free fraction in plasma forEPZ-5676 did not show any marked species differ-ences with values of 0.138, 0.272, 0.234 and 0.125in mouse, rat, dog and human, respectively. Theblood-to-plasma partitioning data across speciesdid not suggest any significant binding of EPZ-5676 to erythrocytes with values suggesting afairly equal distribution between plasma andblood components. Based on these data, plasmaclearance, rather than blood clearance, was usedin all further data analysis.
In vitro metabolic stability
A summary of the metabolic stability data acrossspecies is shown in Table 2. Representative plotsof the depletion of EPZ-5676 over time in liver mi-crosome incubations are shown in Figure 2. EPZ-5676 did not show any instability in mouse, rat,dog and human plasma in vitro. Liver microsomalincubations supplemented with NADPH showedmoderate turnover in mouse, rat, dog and humanwhich, when scaled by the well-stirred venous
Biopharm. Drug Dispos. (2014)DOI: 10.1002/bdd
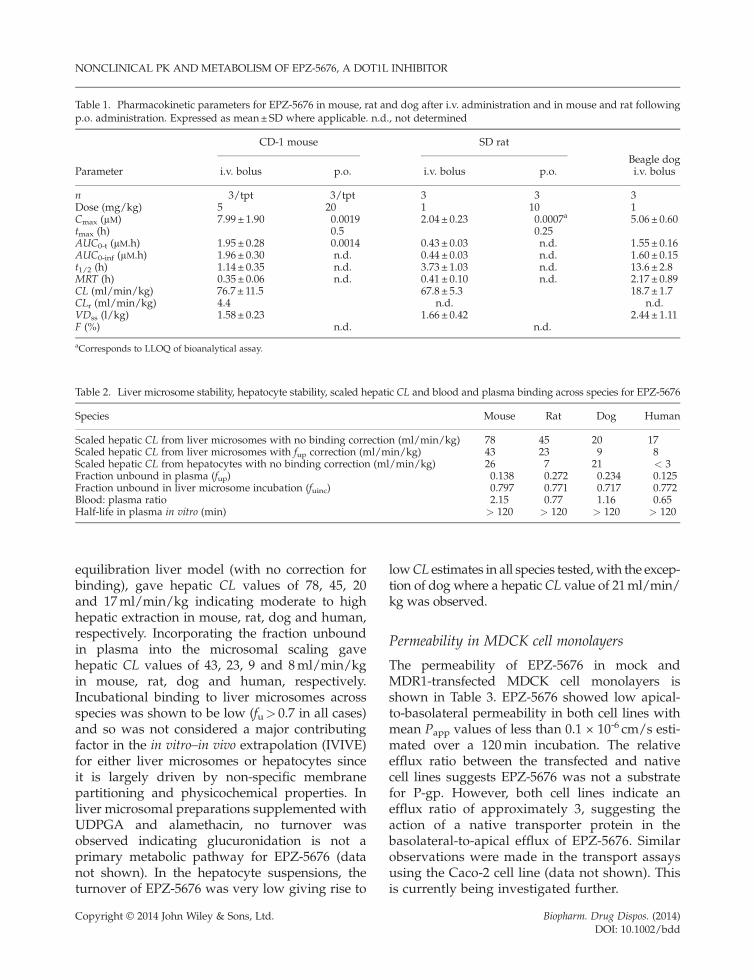
Table 1. Pharmacokinetic parameters for EPZ-5676 in mouse, rat and dog after i.v. administration and in mouse and rat followingp.o. administration. Expressed as mean±SD where applicable. n.d., not determined
Parameter
CD-1 mouse SD rat
Beagle dogi.v. bolus p.o. i.v. bolus p.o. i.v. bolus
n 3/tpt 3/tpt 3 3 3Dose (mg/kg) 5 20 1 10 1Cmax (μM) 7.99± 1.90 0.0019 2.04 ± 0.23 0.0007a 5.06 ± 0.60tmax (h) 0.5 0.25AUC0-t (μM.h) 1.95± 0.28 0.0014 0.43 ± 0.03 n.d. 1.55 ± 0.16AUC0-inf (μM.h) 1.96± 0.30 n.d. 0.44 ± 0.03 n.d. 1.60 ± 0.15t1/2 (h) 1.14± 0.35 n.d. 3.73 ± 1.03 n.d. 13.6 ± 2.8MRT (h) 0.35± 0.06 n.d. 0.41 ± 0.10 n.d. 2.17 ± 0.89CL (ml/min/kg) 76.7 ± 11.5 67.8 ± 5.3 18.7 ± 1.7CLr (ml/min/kg) 4.4 n.d. n.d.VDss (l/kg) 1.58± 0.23 1.66 ± 0.42 2.44 ± 1.11F (%) n.d. n.d.
aCorresponds to LLOQ of bioanalytical assay.
Table 2. Liver microsome stability, hepatocyte stability, scaled hepatic CL and blood and plasma binding across species for EPZ-5676
Species Mouse Rat Dog Human
Scaled hepatic CL from liver microsomes with no binding correction (ml/min/kg) 78 45 20 17Scaled hepatic CL from liver microsomes with fup correction (ml/min/kg) 43 23 9 8Scaled hepatic CL from hepatocytes with no binding correction (ml/min/kg) 26 7 21 < 3Fraction unbound in plasma (fup) 0.138 0.272 0.234 0.125Fraction unbound in liver microsome incubation (fuinc) 0.797 0.771 0.717 0.772Blood: plasma ratio 2.15 0.77 1.16 0.65Half-life in plasma in vitro (min) > 120 > 120 > 120 > 120
NONCLINICAL PK AND METABOLISM OF EPZ-5676, A DOT1L INHIBITOR
equilibration liver model (with no correction forbinding), gave hepatic CL values of 78, 45, 20and 17ml/min/kg indicating moderate to highhepatic extraction in mouse, rat, dog and human,respectively. Incorporating the fraction unboundin plasma into the microsomal scaling gavehepatic CL values of 43, 23, 9 and 8ml/min/kgin mouse, rat, dog and human, respectively.Incubational binding to liver microsomes acrossspecies was shown to be low (fu> 0.7 in all cases)and so was not considered a major contributingfactor in the in vitro–in vivo extrapolation (IVIVE)for either liver microsomes or hepatocytes sinceit is largely driven by non-specific membranepartitioning and physicochemical properties. Inliver microsomal preparations supplemented withUDPGA and alamethacin, no turnover wasobserved indicating glucuronidation is not aprimary metabolic pathway for EPZ-5676 (datanot shown). In the hepatocyte suspensions, theturnover of EPZ-5676 was very low giving rise to
Copyright © 2014 John Wiley & Sons, Ltd.
lowCL estimates in all species tested,with the excep-tion of dog where a hepatic CL value of 21ml/min/kg was observed.
Permeability in MDCK cell monolayers
The permeability of EPZ-5676 in mock andMDR1-transfected MDCK cell monolayers isshown in Table 3. EPZ-5676 showed low apical-to-basolateral permeability in both cell lines withmean Papp values of less than 0.1 × 10-6 cm/s esti-mated over a 120min incubation. The relativeefflux ratio between the transfected and nativecell lines suggests EPZ-5676 was not a substratefor P-gp. However, both cell lines indicate anefflux ratio of approximately 3, suggesting theaction of a native transporter protein in thebasolateral-to-apical efflux of EPZ-5676. Similarobservations were made in the transport assaysusing the Caco-2 cell line (data not shown). Thisis currently being investigated further.
Biopharm. Drug Dispos. (2014)DOI: 10.1002/bdd
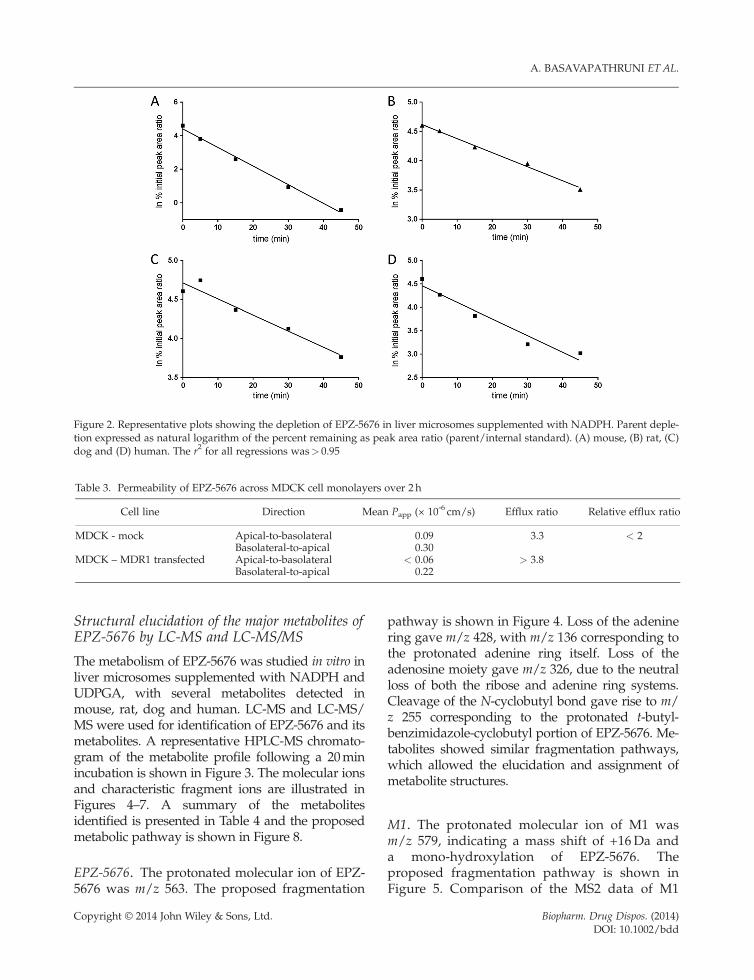
Figure 2. Representative plots showing the depletion of EPZ-5676 in liver microsomes supplemented with NADPH. Parent deple-tion expressed as natural logarithm of the percent remaining as peak area ratio (parent/internal standard). (A) mouse, (B) rat, (C)dog and (D) human. The r2 for all regressions was> 0.95
Table 3. Permeability of EPZ-5676 across MDCK cell monolayers over 2 h
Cell line Direction Mean Papp (× 10-6 cm/s) Efflux ratio Relative efflux ratio
MDCK - mock Apical-to-basolateral 0.09 3.3 < 2Basolateral-to-apical 0.30
MDCK – MDR1 transfected Apical-to-basolateral < 0.06 > 3.8Basolateral-to-apical 0.22
A. BASAVAPATHRUNI ET AL.
Structural elucidation of the major metabolites ofEPZ-5676 by LC-MS and LC-MS/MS
The metabolism of EPZ-5676 was studied in vitro inliver microsomes supplemented with NADPH andUDPGA, with several metabolites detected inmouse, rat, dog and human. LC-MS and LC-MS/MS were used for identification of EPZ-5676 and itsmetabolites. A representative HPLC-MS chromato-gram of the metabolite profile following a 20minincubation is shown in Figure 3. The molecular ionsand characteristic fragment ions are illustrated inFigures 4–7. A summary of the metabolitesidentified is presented in Table 4 and the proposedmetabolic pathway is shown in Figure 8.
EPZ-5676. The protonated molecular ion of EPZ-5676 was m/z 563. The proposed fragmentation
Copyright © 2014 John Wiley & Sons, Ltd.
pathway is shown in Figure 4. Loss of the adeninering gave m/z 428, with m/z 136 corresponding tothe protonated adenine ring itself. Loss of theadenosine moiety gave m/z 326, due to the neutralloss of both the ribose and adenine ring systems.Cleavage of the N-cyclobutyl bond gave rise to m/z 255 corresponding to the protonated t-butyl-benzimidazole-cyclobutyl portion of EPZ-5676. Me-tabolites showed similar fragmentation pathways,which allowed the elucidation and assignment ofmetabolite structures.
M1. The protonated molecular ion of M1 wasm/z 579, indicating a mass shift of +16Da anda mono-hydroxylation of EPZ-5676. Theproposed fragmentation pathway is shown inFigure 5. Comparison of the MS2 data of M1
Biopharm. Drug Dispos. (2014)DOI: 10.1002/bdd
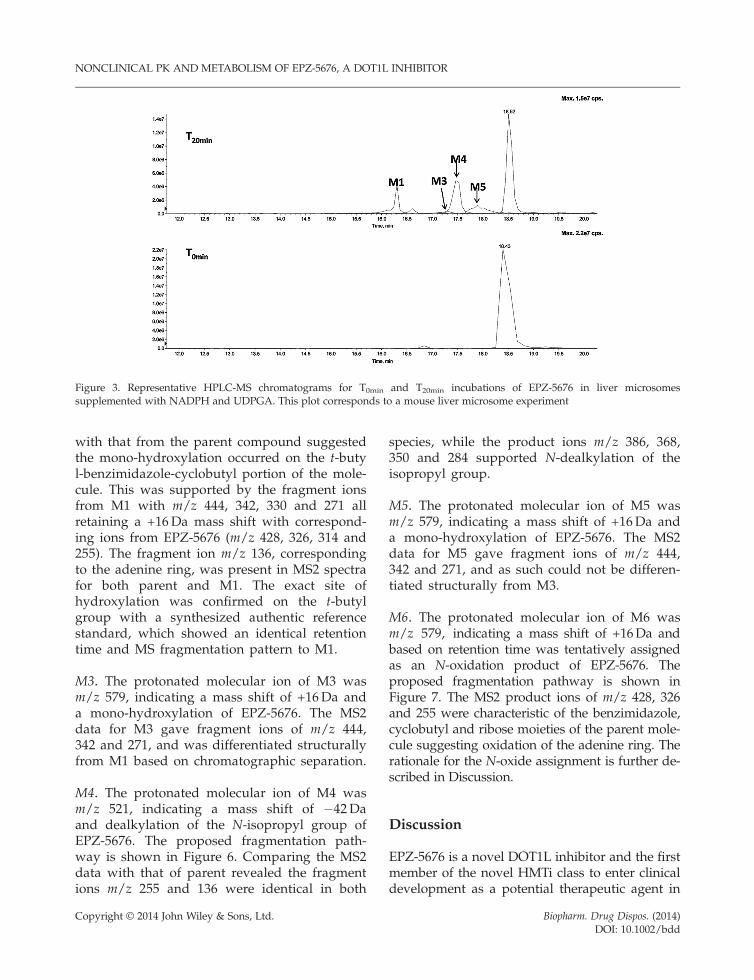
Figure 3. Representative HPLC-MS chromatograms for T0min and T20min incubations of EPZ-5676 in liver microsomessupplemented with NADPH and UDPGA. This plot corresponds to a mouse liver microsome experiment
NONCLINICAL PK AND METABOLISM OF EPZ-5676, A DOT1L INHIBITOR
with that from the parent compound suggestedthe mono-hydroxylation occurred on the t-butyl-benzimidazole-cyclobutyl portion of the mole-cule. This was supported by the fragment ionsfrom M1 with m/z 444, 342, 330 and 271 allretaining a +16Da mass shift with correspond-ing ions from EPZ-5676 (m/z 428, 326, 314 and255). The fragment ion m/z 136, correspondingto the adenine ring, was present in MS2 spectrafor both parent and M1. The exact site ofhydroxylation was confirmed on the t-butylgroup with a synthesized authentic referencestandard, which showed an identical retentiontime and MS fragmentation pattern to M1.
M3. The protonated molecular ion of M3 wasm/z 579, indicating a mass shift of +16Da anda mono-hydroxylation of EPZ-5676. The MS2data for M3 gave fragment ions of m/z 444,342 and 271, and was differentiated structurallyfrom M1 based on chromatographic separation.
M4. The protonated molecular ion of M4 wasm/z 521, indicating a mass shift of �42Daand dealkylation of the N-isopropyl group ofEPZ-5676. The proposed fragmentation path-way is shown in Figure 6. Comparing the MS2data with that of parent revealed the fragmentions m/z 255 and 136 were identical in both
Copyright © 2014 John Wiley & Sons, Ltd.
species, while the product ions m/z 386, 368,350 and 284 supported N-dealkylation of theisopropyl group.
M5. The protonated molecular ion of M5 wasm/z 579, indicating a mass shift of +16Da anda mono-hydroxylation of EPZ-5676. The MS2data for M5 gave fragment ions of m/z 444,342 and 271, and as such could not be differen-tiated structurally from M3.
M6. The protonated molecular ion of M6 wasm/z 579, indicating a mass shift of +16Da andbased on retention time was tentatively assignedas an N-oxidation product of EPZ-5676. Theproposed fragmentation pathway is shown inFigure 7. The MS2 product ions of m/z 428, 326and 255 were characteristic of the benzimidazole,cyclobutyl and ribose moieties of the parent mole-cule suggesting oxidation of the adenine ring. Therationale for the N-oxide assignment is further de-scribed in Discussion.
Discussion
EPZ-5676 is a novel DOT1L inhibitor and the firstmember of the novel HMTi class to enter clinicaldevelopment as a potential therapeutic agent in
Biopharm. Drug Dispos. (2014)DOI: 10.1002/bdd

Figure 4. LC-MS chromatogram, MS1 and MS2 spectra (A) with proposed fragmentation pathways of EPZ-5676 (B)
A. BASAVAPATHRUNI ET AL.
MLL-rearranged leukemia. The discovery ofEPZ-5676 was facilitated by a structure-guidedmedicinal chemistry approach [10] and hasshown superior efficacy in preclinical modelsof MLL-rearranged leukemia [11]. The aims ofthis work were to characterize the pharmacoki-netics following i.v. and p.o. administration inmouse, rat and dog, to assess the cross-speciesin vitro–in vivo correlation to gain insight intothe primary elimination pathways involved in theclearance of EPZ-5676, and to determine the majormetabolic pathways.The in vivo time–concentration profiles in mouse,
rat and dog following i.v. bolus administrationshowed biexponential kinetics that was moreapparent as the body size of the species increased.
Copyright © 2014 John Wiley & Sons, Ltd.
This resulted in terminal half-lives increasing from1.1 h in mouse, 3.7 h in rat and 13.6 h in dog. Inaddition, terminal t1/2 was longer than meanresidence time (MRT) (3–9 fold) further supportingmulti-exponential kinetics in the animal species[19]. As for many drugs that exhibit multiphasicconcentration vs time profiles, the MRT will be abetter indicator than t1/2 of the potential dosingfrequency needed and expected accumulation ratiothat will occur with repeated administration. TheCL in all species was moderate to high withestimated hepatic extraction ratios of 0.80, 0.97and 0.61 in mouse (accounting for the measuredrenal component), rat and dog, respectively.Expressing CL in its unbound or blood form didnot change the interpretation of the cross-species
Biopharm. Drug Dispos. (2014)DOI: 10.1002/bdd
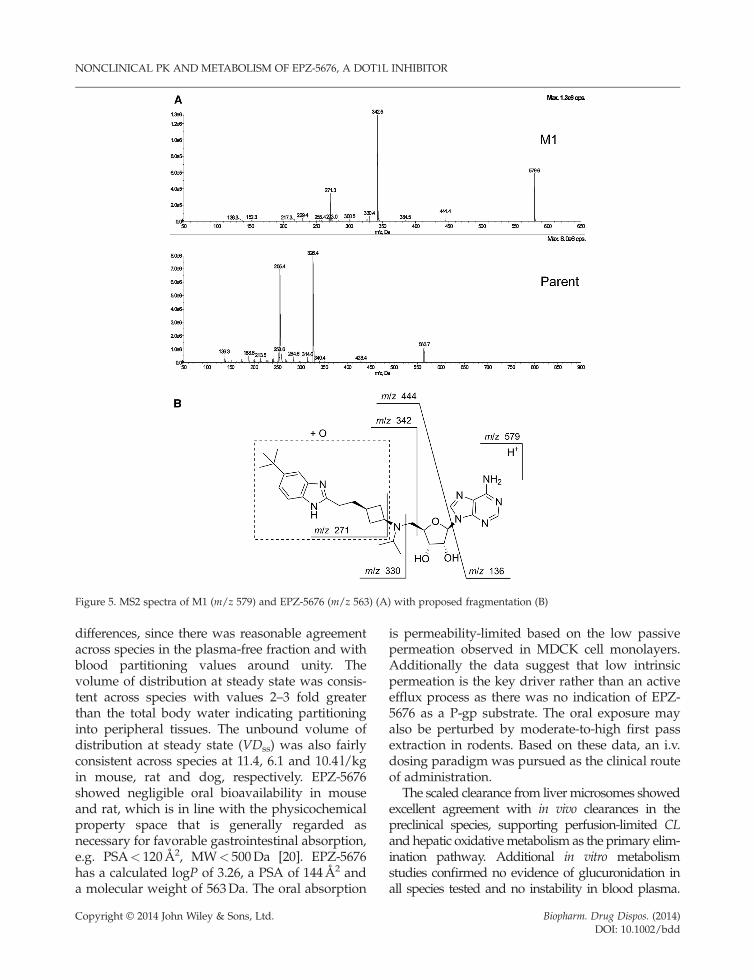
Figure 5. MS2 spectra of M1 (m/z 579) and EPZ-5676 (m/z 563) (A) with proposed fragmentation (B)
NONCLINICAL PK AND METABOLISM OF EPZ-5676, A DOT1L INHIBITOR
differences, since there was reasonable agreementacross species in the plasma-free fraction and withblood partitioning values around unity. Thevolume of distribution at steady state was consis-tent across species with values 2–3 fold greaterthan the total body water indicating partitioninginto peripheral tissues. The unbound volume ofdistribution at steady state (VDss) was also fairlyconsistent across species at 11.4, 6.1 and 10.4 l/kgin mouse, rat and dog, respectively. EPZ-5676showed negligible oral bioavailability in mouseand rat, which is in line with the physicochemicalproperty space that is generally regarded asnecessary for favorable gastrointestinal absorption,e.g. PSA< 120Å2, MW< 500Da [20]. EPZ-5676has a calculated logP of 3.26, a PSA of 144Å2 anda molecular weight of 563Da. The oral absorption
Copyright © 2014 John Wiley & Sons, Ltd.
is permeability-limited based on the low passivepermeation observed in MDCK cell monolayers.Additionally the data suggest that low intrinsicpermeation is the key driver rather than an activeefflux process as there was no indication of EPZ-5676 as a P-gp substrate. The oral exposure mayalso be perturbed by moderate-to-high first passextraction in rodents. Based on these data, an i.v.dosing paradigm was pursued as the clinical routeof administration.
The scaled clearance from livermicrosomes showedexcellent agreement with in vivo clearances in thepreclinical species, supporting perfusion-limited CLand hepatic oxidativemetabolism as the primary elim-ination pathway. Additional in vitro metabolismstudies confirmed no evidence of glucuronidation inall species tested and no instability in blood plasma.
Biopharm. Drug Dispos. (2014)DOI: 10.1002/bdd
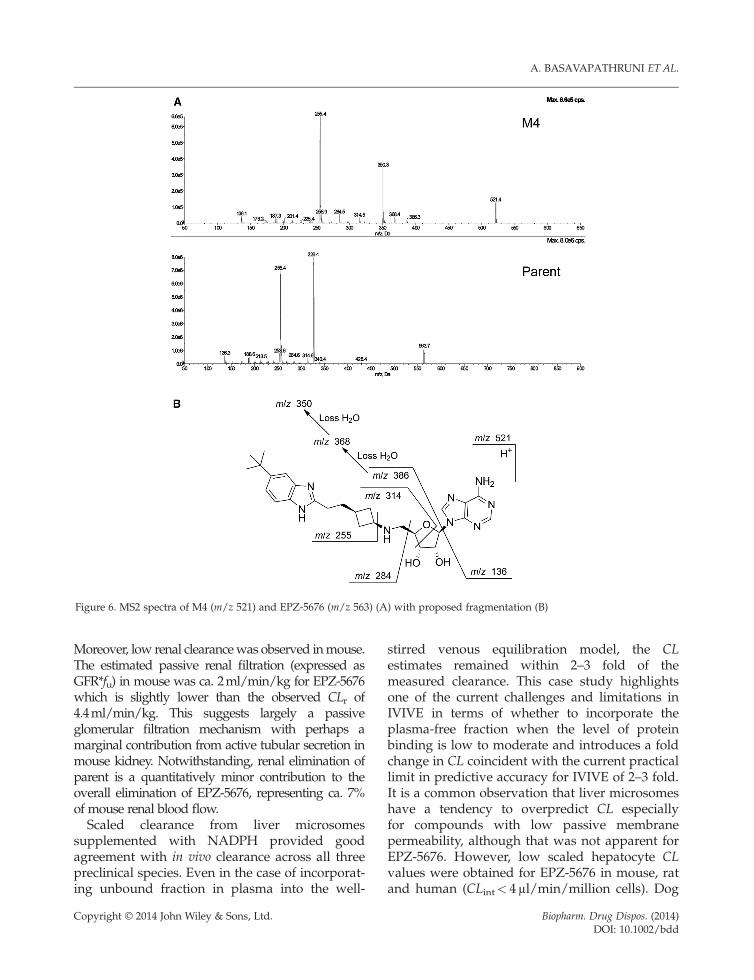
igure 6. MS2 spectra of M4 (m/z 521) and EPZ-5676 (m/z 563) (A) with proposed fragmentation (B)
A. BASAVAPATHRUNI ET AL.
F
Moreover, low renal clearancewas observed inmouse.The estimated passive renal filtration (expressed asGFR*fu) in mouse was ca. 2ml/min/kg for EPZ-5676which is slightly lower than the observed CLr of4.4ml/min/kg. This suggests largely a passiveglomerular filtration mechanism with perhaps amarginal contribution from active tubular secretion inmouse kidney. Notwithstanding, renal elimination ofparent is a quantitatively minor contribution to theoverall elimination of EPZ-5676, representing ca. 7%of mouse renal blood flow.Scaled clearance from liver microsomes
supplemented with NADPH provided goodagreement with in vivo clearance across all threepreclinical species. Even in the case of incorporat-ing unbound fraction in plasma into the well-
Copyright © 2014 John Wiley & Sons, Ltd.
stirred venous equilibration model, the CLestimates remained within 2–3 fold of themeasured clearance. This case study highlightsone of the current challenges and limitations inIVIVE in terms of whether to incorporate theplasma-free fraction when the level of proteinbinding is low to moderate and introduces a foldchange in CL coincident with the current practicallimit in predictive accuracy for IVIVE of 2–3 fold.It is a common observation that liver microsomeshave a tendency to overpredict CL especiallyfor compounds with low passive membranepermeability, although that was not apparent forEPZ-5676. However, low scaled hepatocyte CLvalues were obtained for EPZ-5676 in mouse, ratand human (CLint< 4μl/min/million cells). Dog
Biopharm. Drug Dispos. (2014)DOI: 10.1002/bdd
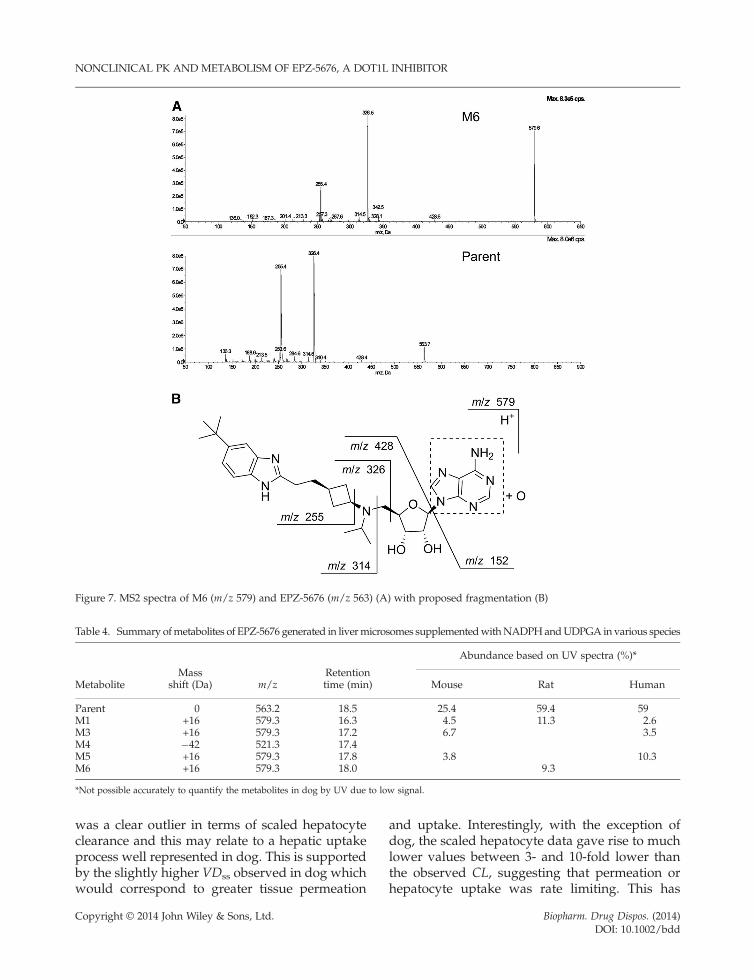
Figure 7. MS2 spectra of M6 (m/z 579) and EPZ-5676 (m/z 563) (A) with proposed fragmentation (B)
Table 4. Summary ofmetabolites of EPZ-5676 generated in livermicrosomes supplementedwithNADPHandUDPGA in various species
MetaboliteMass
shift (Da) m/zRetentiontime (min)
Abundance based on UV spectra (%)*
Mouse Rat Human
Parent 0 563.2 18.5 25.4 59.4 59M1 +16 579.3 16.3 4.5 11.3 2.6M3 +16 579.3 17.2 6.7 3.5M4 �42 521.3 17.4M5 +16 579.3 17.8 3.8 10.3M6 +16 579.3 18.0 9.3
*Not possible accurately to quantify the metabolites in dog by UV due to low signal.
NONCLINICAL PK AND METABOLISM OF EPZ-5676, A DOT1L INHIBITOR
was a clear outlier in terms of scaled hepatocyteclearance and this may relate to a hepatic uptakeprocess well represented in dog. This is supportedby the slightly higher VDss observed in dog whichwould correspond to greater tissue permeation
Copyright © 2014 John Wiley & Sons, Ltd.
and uptake. Interestingly, with the exception ofdog, the scaled hepatocyte data gave rise to muchlower values between 3- and 10-fold lower thanthe observed CL, suggesting that permeation orhepatocyte uptake was rate limiting. This has
Biopharm. Drug Dispos. (2014)DOI: 10.1002/bdd
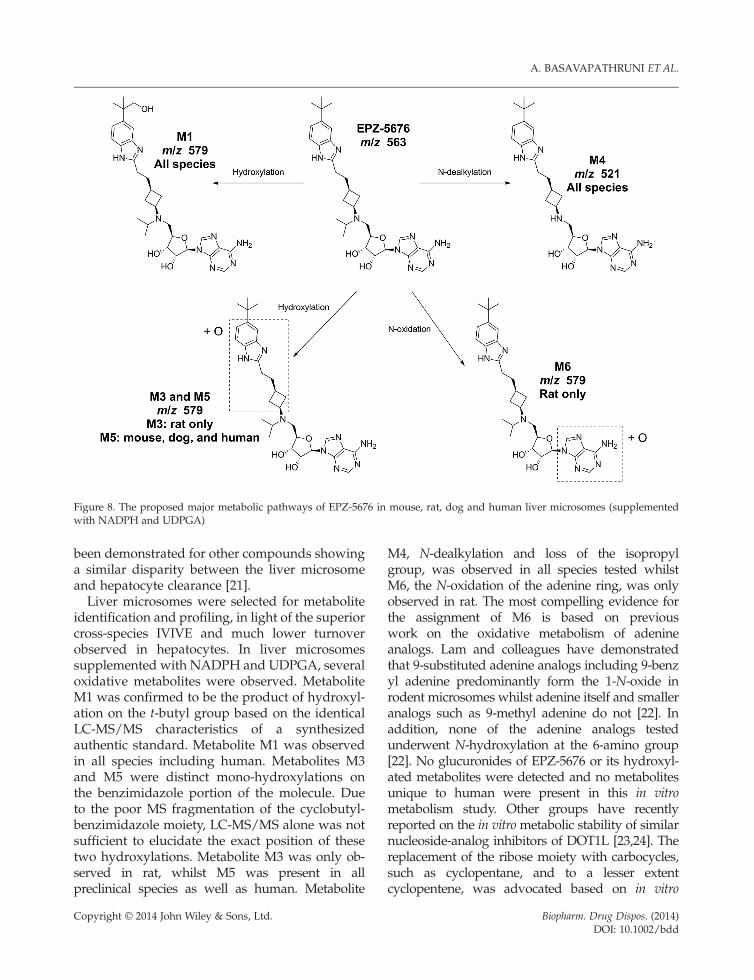
Figure 8. The proposed major metabolic pathways of EPZ-5676 in mouse, rat, dog and human liver microsomes (supplementedwith NADPH and UDPGA)
A. BASAVAPATHRUNI ET AL.
been demonstrated for other compounds showinga similar disparity between the liver microsomeand hepatocyte clearance [21].Liver microsomes were selected for metabolite
identification and profiling, in light of the superiorcross-species IVIVE and much lower turnoverobserved in hepatocytes. In liver microsomessupplemented with NADPH and UDPGA, severaloxidative metabolites were observed. MetaboliteM1 was confirmed to be the product of hydroxyl-ation on the t-butyl group based on the identicalLC-MS/MS characteristics of a synthesizedauthentic standard. Metabolite M1 was observedin all species including human. Metabolites M3and M5 were distinct mono-hydroxylations onthe benzimidazole portion of the molecule. Dueto the poor MS fragmentation of the cyclobutyl-benzimidazole moiety, LC-MS/MS alone was notsufficient to elucidate the exact position of thesetwo hydroxylations. Metabolite M3 was only ob-served in rat, whilst M5 was present in allpreclinical species as well as human. Metabolite
Copyright © 2014 John Wiley & Sons, Ltd.
M4, N-dealkylation and loss of the isopropylgroup, was observed in all species tested whilstM6, the N-oxidation of the adenine ring, was onlyobserved in rat. The most compelling evidence forthe assignment of M6 is based on previouswork on the oxidative metabolism of adenineanalogs. Lam and colleagues have demonstratedthat 9-substituted adenine analogs including 9-benzyl adenine predominantly form the 1-N-oxide inrodent microsomes whilst adenine itself and smalleranalogs such as 9-methyl adenine do not [22]. Inaddition, none of the adenine analogs testedunderwent N-hydroxylation at the 6-amino group[22]. No glucuronides of EPZ-5676 or its hydroxyl-ated metabolites were detected and no metabolitesunique to human were present in this in vitrometabolism study. Other groups have recentlyreported on the in vitrometabolic stability of similarnucleoside-analog inhibitors of DOT1L [23,24]. Thereplacement of the ribose moiety with carbocycles,such as cyclopentane, and to a lesser extentcyclopentene, was advocated based on in vitro
Biopharm. Drug Dispos. (2014)DOI: 10.1002/bdd

NONCLINICAL PK AND METABOLISM OF EPZ-5676, A DOT1L INHIBITOR
stability in human plasma and liver microsomes.Differences in human liver microsome turnoverwere observed between these two carbocyclicanalogs with the implication that the 5-memberedring system was a metabolic liability. Our data donot support the ribose moiety as being a majormetabolic soft-spot but rather suggest that P450-mediated metabolism elsewhere on the molecule isthe major metabolic pathway for the DOT1Lnucleoside analog chemotype.
Conclusion
EPZ-5676 showed biexponential kinetics followingi.v. administration, giving rise to a terminal t1/2 of1.1, 3.7 and 13.6 h in mouse, rat and dog, respec-tively. Steady state VD was 2–3-fold greater thantotal body water with a high clearance in rodentandmoderate clearance in dog. EPZ-5676 exhibiteda low oral bioavailability in rodent. In vitro scalingof liver microsome clearance data showed goodagreement with the in vivo clearance across speciesindicating P450-mediated metabolism as a primaryelimination pathway. Moreover, low renal clear-ance was observed in mouse mediated largelyby passive glomerular filtration. Hepatocyte clear-ance suggested permeation- or hepatic uptake-limitations. The metabolic pathways for EPZ-5676across species included three monohydroxylatedmetabolites (M1, M3 and M5), one N-dealkylatedproduct (M4) as well as an N-oxide (M6). EPZ-5676is afirst-in-classDOT1L inhibitor and is currentlyunderclinical investigation for MLL-rearranged leukemias.Further work is underway at present to characterizethe metabolism and disposition of EPZ-5676.
Conflict of Interest
The authors have declared that there is no conflictof interest. All authors are employees of, and/orhold equity in, Epizyme, Inc.
References
1. Portela A, Esteller M. Epigenetic modifications andhuman disease.Nature Biotechnol 2010; 28: 1057–1068.
Copyright © 2014 John Wiley & Sons, Ltd.
2. Tamai H, Inokuchi K. 11q23/MLL acute leukemia:update of clinical aspects. J Clin Exp Hematopathol2010; 50: 91–98.
3. Ayton PM, Chen EH, Cleary ML. Binding tononmethylated CpG DNA is essential for targetrecognition, transactivation, and myeloid transfor-mation by an MLL oncoprotein. Mol Cell Biol 2004;24: 10470–10478.
4. Milne TA, Briggs SD, Brock HW, et al. MLL targetsSET domain methyltransferase activity to Hoxgene promoters. Mol Cell 2002; 10: 1107–1117.
5. Nakamura T, Mori T, Tada S, et al. ALL-1 is ahistone methyltransferase that assembles asupercomplex of proteins involved in transcrip-tional regulation. Mol Cell 2002; 10: 1119–1128.
6. Slany RK, Lavau C, Cleary ML. The oncogeniccapacity of HRX-ENL requires the transcriptionaltransactivation activity of ENL and the DNA bindingmotifs of HRX. Mol Cell Biol 1998; 18: 122–129.
7. Zeleznik-Le NJ, Harden AM, Rowley JD. 11q23translocations split the ‘AT-hook’ cruciformDNA-binding region and the transcriptional re-pression domain from the activation domain ofthe mixed-lineage leukemia (MLL) gene. Proc NatlAcad Sci U S A 1994; 91: 10610–10614.
8. Biswas D,Milne TA, Basrur V, et al. Function of leuke-mogenic mixed lineage leukemia 1 (MLL) fusion pro-teins through distinct partner protein complexes. ProcNatl Acad Sci U S A 2011; 108: 15751–15756, S/1–S/7.
9. Daigle SR, Olhava EJ, Therkelsen CA, et al. Selectivekilling of mixed lineage leukemia cells by a potentsmall-molecule DOT1L inhibitor. Cancer Cell 2011;20: 53–65.
10. Basavapathruni A, Jin L, Daigle SR, et al. Conforma-tional adaptation drives potent, selective and durableinhibition of the human protein methyltransferaseDOT1L. Chem Biol Drug Design 2012; 80: 971–980.
11. Daigle SR, Olhava EJ, Therkelsen CA, et al. Potentinhibition of DOT1L as treatment for MLL-fusionleukemia. Blood 2013; 122(6): 1017–1025.
12. Pang KS, Rowland M. Hepatic clearance of drugs. I.Theoretical considerations of a ‘well-stirred’ modeland a ‘parallel tube’ model. Influence ofhepatic blood flow, plasma and blood cellbinding, and the hepatocellular enzymatic ac-tivity on hepatic drug clearance. J PharmacokinetBiopharm 1977; 5: 625–653.
13. Obach RS. Prediction of human clearance oftwenty-nine drugs from hepatic microsomal intrin-sic clearance data: an examination of in vitro half-life approach and nonspecific binding to micro-somes. Drug Metab Dispos 1999; 27: 1350–1359.
14. Houston JB. Utility of in vitro drug metabolismdata in predicting in vivo metabolic clearance.Biochem Pharmacol 1994; 47: 1469–1479.
15. Barter ZE, Bayliss MK, Beaune PH, et al. Scalingfactors for the extrapolation of in vivo metabolicdrug clearance from in vitro data: reaching a con-sensus on values of human microsomal protein
Biopharm. Drug Dispos. (2014)DOI: 10.1002/bdd

A. BASAVAPATHRUNI ET AL.
and hepatocellularity per gram of liver. Curr DrugMetab 2007; 8: 33–45.
16. Davies B, Morris T. Physiological parameters inlaboratory animals and humans. Pharm Res 1993;10: 1093–1095.
17. Ring BJ, Chien JY, Adkison KK, et al. PhRMACPCDC Initiative on Predictive Models of HumanPharmacokinetics, Part 3: Comparative assessmentof prediction methods of human clearance. J PharmSci 2011; 100: 4090–4110.
18. Hinderling PH. Red blood cells: a neglectedcompartment in pharmacokinetics and pharmaco-dynamics. Pharmacol Rev 1997; 49: 279–295.
19. Sahin S, Benet LZ. The operational multiple dosinghalf-life: a key to defining drug accumulation inpatients and to designing extended release dosageforms. Pharm Res 2008; 25: 2869–2877.
20. van deWaterbeemd H. Physicochemical Approachesto Drug Absorption. In Drug Bioavailability:
Copyright © 2014 John Wiley & Sons, Ltd.
Estimation of Solubility, Permeability, Absorptionand Bioavailability, van de Waterbeemd H, Testa B(eds). Wiley-VCH Verlag GmbH & Co. KGaA:Weinheim, Germany, 2009; 40: 71–99. doi: 10.1002/9783527623860.ch5.
21. Di L, Keefer C, Scott DO, et al. Mechanistic insightsfrom comparing intrinsic clearance values betweenhuman liver microsomes and hepatocytes to guidedrug design. Eur J Med Chem 2012; 57: 441–448.
22. Lam SP, Devinsky F, Gorrod JW. Biological N-oxidation of adenine and 9-alkyl derivatives. Eur J DrugMetab Pharmacokinet 1987; 12: 239–243.
23. Deng L, Zhang L, Yao Y, et al. Synthesis, activityand metabolic stability of non-ribose containinginhibitors of histone methyltransferase DOT1L.Med Chem Comm 2013; 4: 822–826.
24. Anglin JL, SongY.Amedicinal chemistry perspectivefor targeting histone H3 lysine79 methyltransferaseDOT1L. J Med Chem 2013; 56(22): 8972–8983.
Biopharm. Drug Dispos. (2014)DOI: 10.1002/bdd





























