Embed Size (px)
Citation preview

American Journal of Medical Genetics 9:43-53 (1981)
Nonprogressive Cerebellar Disorder With Mental Retardation and Autosomal Recessive Inheritance in Hutteri tes Verena Schurig, Alan Van Orman, and Peter Bowen
Division of Medical Genetics, Department of Pediatrics, University of Alberta, Edmonton, Alberta, Canaab
We report a nonprogressive neurological disorder in at least 11 Hutterites with healthy but consanguineous parents. In several of the affected, hypotonia was noted at birth. Retarded motor and mental development became apparent dur- ing the first year of life. The age of unsupported walking varied from 5-21 years. Consistent signs were unsteady, broadly based gait and stance, exaggerated deep tendon reflexes mainly in the lower limbs, and mild to moderate mental retarda- tion. Variable signs were extensor plantar reflexes (9/1 l), short stature (-2SD in 8/1 l), strabismus (7/1 l ) , small muscle mass (6/1 l ) , mild intention tremor (3/1 l ) , cataracts (1 / 1 l), and epilepsy (1/11). CAT scans in two affected sisters showed slight enlargement of the fourth ventricle in one and hypoplasia of the cerebellum in both. The disorder is probably the same as that described earlier under the heading, dysequilibrium syndrome.
Key words: Hutterites, consanguinity, dysequitibrium syndrome, autosomal recessive inheritance, hypo- tonia, mental retardation, cerebellar ataxia, Marinesco-Sjogren syndrome, cerebral palsy
INTRODUCTION
The approximately 7,000 Hutterites in the Province of Alberta comprise a genetic isolate whose ancestors, numbering about 400, came to North America between 1874 and 1877. Of three groups of colony Hutterites that settled in North America (Dariusleut, Lehrerleut, and Schmiedeleut), only the Dariusleut and Lehrerleut are represented in Alberta. They live in rural colonies of about 60 to 125 persons, and are endogamous, marrying almost always within the same leut. The average relationship of Hutterite spouses is slightly closer than that of 2nd cousins, and the average completed sibship size approaches 11 [Eaton and Mayer, 19531. These conditions favour the occurrence of homozygosity by descent from a common ancestor [McKusick, 19691, and it is not uncommon to find mul- tiple occurrences of an otherwise rare recessive disorder in related Hutterite sibships.
Here, we describe a recessively inherited neurological syndrome affecting several Dariusleut. Although the disorder was reported at least as early as 1905 [Batten, 19051
Received for publication August 29, 1980; revision received November 7, 1980.
Address reprint requests to Dr. Verena Schurig, Department of Pediatrics, 4-1 20 Clinical Sciences Building, Edmonton, Alberta, Canada T6G 2G3.
0148-7299/81/0901-0043$03.50 @ 1981 Alan R. Liss, Inc.

44 Schurig, Orman, and Bowen
and has been studied intensively in Sweden [Hagberg et al, 19721, it is essentially unknown in North America.
MATERIALS AND METHODS
One of us (A.V.) noted the occurrence of mental retardation and disturbance of equilibrium in Hutterites around Cardston, Alberta. This predominantly Mormon town is within 40 km of about 16 Hutterite colonies. Suspected additional cases were confirmed on visits to the respective colonies. Anamnestic and genealogic data were obtained from the families. The pedigrees were verified by reference to genealogical records kept by one of the Ddriusleut preachers. Affected individuals and members of their families were exam- ined in the colonies, and two affected sisters (VIII-17,21) were admitted to the University of Alberta Hospital for further studies. None of the affected individuals had been previously investigated for the disorder in question.
PEDIGREE
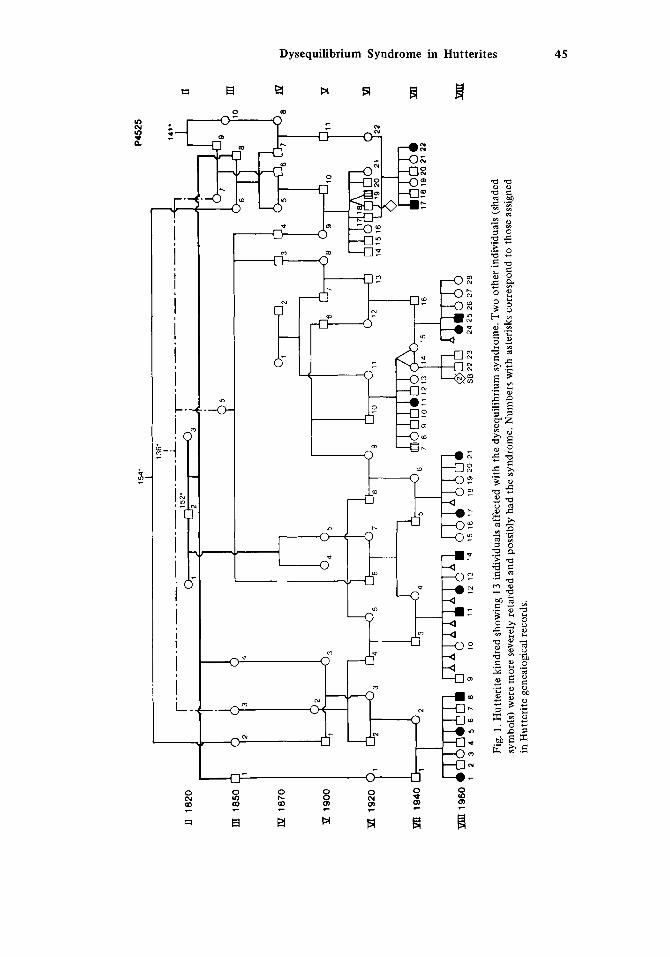
Fifteen individuals in seven interrelated sibships (Fig. 1) have a similar disorder that is described in the following section. Parents are clinically normal but consanguineous. Relationships could often be traced through more than one line of descent, and only the most immediate connections are shown in the pedigree.
walked and has wasting and contractures of lower limbs. It was not possible to determine whether or not he had a disturbance of equilibrium. The same applies to another relative (VI-19) who had severe mental retardation, epilepsy, and hemiparesis. These, and two other persons (VII-17,22) on whom documentation is incomplete, are not included in our subse- quent characterization of the condition.
One typically affected woman had a severely retarded brother (VII-7) who has never
CLINICAL DATA
The perinatal hstories were negative for factors known to be associated with cerebral palsy except for one instance of prematurity (VIII:21, birthweight, 1,400 g). The ages of affected individuals ranged from 8-40 years.
In all cases, the syndrome probably had a congenital onset. Delayed motor develop- ment and hypotonia were usually noted during the first year of life. None of the affected individuals walked before three years, and all pushed a four-wheel appliance (eg, a stroller) for support for 3-15 years. Most learned simple, self-care tasks, but none was able to complete elementary grades in the English school. Several responded to commands in German or English but they had virtually no expressive language. One 40-year-old woman had epileptic seizures that began at age 37 years.
most characteristic signs were: wide stance; broadly based, unsteady gait; falling without compensatory reflexes, reminiscent of a clown or mannequin; normal or reduced muscle tone; exaggerated (sometimes clonic) tendon jerks in the lower limbs; and flat, everted feet (Fig. 2). Nystagmus was not present and intention tremor could be demonstrated in only three individuals. Sensation was intact.
Height was below the third centile in 8/11 and was between the 3rd and 25th cen- tiles in the others. The occipital-frontal head circumference (OFC) was normal. Similarity
Major manifestations in older children and adults are summarized in Table I. The
154'
P
4525
II 1820
III 1
850
El
1870
Y 1
900
H 1
920
11
J
1 3
4
5 6
7 8
9
Pn 1
940
42
1960
1 2 3
4 5
6 7
8 9
10
11
12
13
14
15
16
17
18 1
92
0 2
1 Ab
irssb
S8 2
2 2
3
24 2
5 26
27
26
"&
17 1
6 1
9 2
0 21
22
Fig.
1. H
utte
rite
kin
dred
sho
win
g 13
indi
vidu
als a
ffec
ted
with
the
dys
equi
libri
um s
yndr
ome.
Tw
o ot
her
indi
vidu
als (
shad
ed
sym
bols
) wer
e m
ore
seve
rely
reta
rded
and
pos
sibl
y ha
d th
e sy
ndro
me.
Num
bers
with
ast
eris
ks c
orre
spon
d to
thos
e as
sign
ed
in H
utte
rite
gene
alok
jcal
reco
rds.
n m nl
P
PI
m
m
P
v,

fA e s.
Incr
ease
d D
TRs
3.
OI] 9 B
VII
I-5
20
Mild
-
-
Mod
erat
e 15
1.5
(3rd
) 53
.5
Left
eso-
-
+ +
+ ph
oria
a F
VII
I-8
15
Seve
re
? -
Mod
erat
e 15
7 (<
3rd)
54
.5
-
-
1 V
III-
11
21
Mild
+
-
Mod
erat
e 17
0(25
th)
56.5
-
-
- +
VII
I-12
19
M
od.
-
-
Mod
erat
e 15
1 (3
rd)
53.0
R
ight
eso
- -
-
+ +
s
VII
I-14
14
Se
vere
-
-
Mod
erat
e 14
8 (3
rd)
54.0
-
-
-
i-
+ T
Pr
r 1
- 32
M
od.
+ -
Mod
erat
e 14
6.5
(<3r
d)
54.0
A
ltern
at.
+ -
+ +
'1 20
M
od.
+ -
Mild
14
6 (<
3rd)
52
.0
Alte
rnat
. -
-
+ +
-
-
+ (c
lonu
s)
-
VII
I-24
9
Mild
-
VII
I-25
8
Seve
re
-
-
Mod
erat
e 11
4.5
(3rd
) 50
.0
Stra
bism
us
-
-
+ +
VII
-11
40
Mod
. ?
+ M
oder
ate
156.
5 (1
0th)
53
.0
Stra
bism
us,
+ ?
?
TABL
E I.
Man
ifest
atio
ns
Age
at
Dys
equi
- In
tent
. M
enta
l H
eigh
t (cm
) O
FC
Upp
er
Low
er
Indi
vidu
al
exam
(yr
) lib
rium
tr
emor
Se
izur
es
reta
rd.
(cen
tile)
(c
m)
Eye
sign
s H
ypot
onia
lim
bs
limbs
B
abin
ski
-
-
+ +
+ *E
VII
I-1
26
Mild
-
-
Mod
erat
e 15
1.5
(3rd
) 56
.0
i-
+ +
(clo
nus)
-
phor
ia
esot
ropi
a
esot
ropi
a -
Mild
12
8 (2
5th)
53
.5
Stra
bism
us
It+,
lt-
cata
ract
s

Dysequilibrium Syndrome in Hutterites 47
Fig. 2. Feet of VIII: 13, illustrating characteristic deformity.
in facial appearance may have been coincidental. Individual VIII-11 had undescended testes; otherwise, the genitalia were normal in every affected person. Strabismus was present in 7/11 and extensor plantar responses in 911 1. Only one individual (VII-1 l), the oldest affected member examined, had cataracts. These were of unknown duration and did not appear to interfere with her vision.
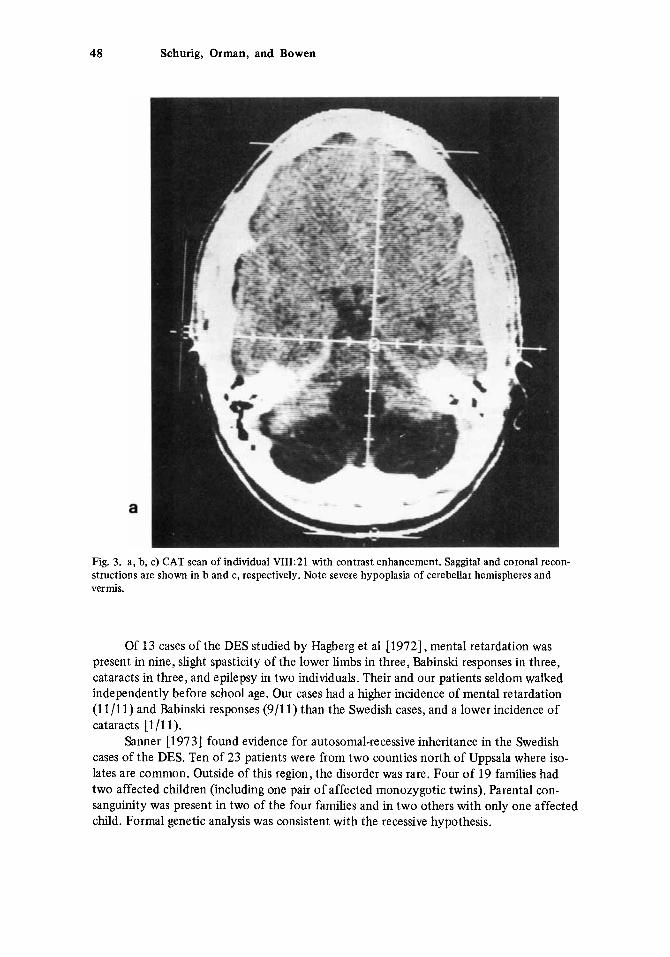
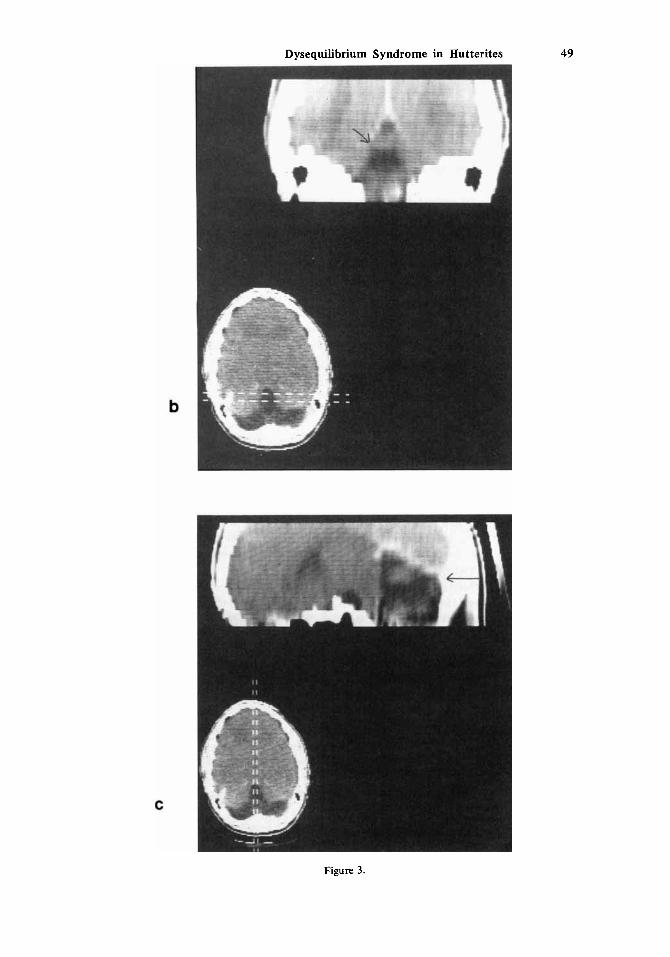
Electroencephalographs on two sisters (VIII-17,2 1) showed generalized grade-I1 dysrhythmia. CAT scans on the same two individuals (Fig. 3) showed variable reduction in size of the cerebellar hemispheres and vermis. VIII-17 also had mild enlargement of the lateral and third ventricles. No evidence of a myopathy could be demonstrated by electro- myography on either individual. They had normal serum amino acids. Both had low renal thresholds for glucose.
DISCUSSION
Similar cases have been reported earlier under different headings, including the atonic- astatic syndrome [Foerster, 1909; Van Rossum, 1959; Kramer and Vojta, 19691, cerebellar atrophy [Norman, 19401, congenital atrophy of the granular layer, type Norman [Goulon et al, 19681, and dysequilibrium syndrome (DES) [Alajouanine et al, 1943; Hagberg et al, 19721. The latter authors defined the DES as “a non-progressive neurological condition dominated throughout childhood by incapability of or pronounced difficulty in maintain- ing an upright body position and in experiencing the position of the body in space, ie, a lack of sense of equilibrium.” They emphasize the distinction between the DES and the syndrome of congenital ataxia (CA), defined by them as a “non-progressive neurological condition dominated by incoordination of voluntary movements, ie, signs of dyssynergia such as dysmetria, unsteady gait and marked intention tremor of the upper extremities.” Anatomical abnormality of the cerebellar vermis could be demonstrated by pneumoen- cephalography in a majority of cases of the DES, but was not present in individuals with CA [Bergstrom and Sanner, 19741.
48 Schurig, Orman, and Bowen
Fig. 3. a, b, c) CAT scan of individual VIII:21 with contrast enhancement. Saggital and coronal recon- structions are shown in b and c, respectively. Note severe hypoplasia of cerebellar hemispheres and vermis.
Of 13 cases of the DES studied by Hagberg et a1 [ 19721, mental retardation was present in nine, slight spasticity of the lower limbs in three, Babinski responses in three, cataracts in three, and epilepsy in two individuals. Their and our patients seldom walked independently before school age. Our cases had a higher incidence of mental retardation (1 111 1) and Babinski responses (911 1) than the Swedish cases, and a lower incidence of cataracts [1/11).
Sanner [1973] found evidence for autosomal-recessive inheritance in the Swedish cases of the DES. Ten of 23 patients were from two counties north of Uppsala where iso- lates are common. Outside of this region, the disorder was rare. Four of 19 families had two affected children (including one pair of affected monozygotic twins). Parental con- sanguinity was present in two of the four families and in two others with only one affected child. Formal genetic analysis was consistent with the recessive hypothesis.
Dysequilibrium Syndrome in Hutterites 49
Figure 3.

50 Schurig, Orman, and Bowen
All affected persons in the present study belong t o the same inbred population group. Multiple occurrences were observed in at least five sibshps with unaffected, consanguineous parents. Regardless of whether ascertainment was considered complete or incomplete, the proportion of affected children was that expected for an autosomal recessive by several dif- ferent tests [Emery, 19761.
have been reported earlier [Beyermann, 1917; Norman, 1940; Alajouanine et al, 1943; Jervis, 1950; Goulon et al, 19681.
esco-Sjogren syndrome (MSS) [Marinesco et al, 1931; Sjogren, 1947, 1950; Alter et al, 19621, which is also an autosomal recessive trait. Progressive muscle weakness develops in many patients with this disorder [Alter et al, 19621, and electromyographic changes con- sistent with a myopathy have been reported [Chaco, 19691. Follow-up on the Swedish DES patients [Sanner, 19791 showed evidence of a myopathy after about age 15 years in those with cataracts but not in those without cataracts. It is possible that those with cataracts may have had the MSS. In the case of the Hutterites, it is unlikely that we are dealing with more than a single entity, for affected members are interrelated. The absence of evidence of a myopathy in the two patients investigated is consistent with the DES rather than the MSS. The cataracts in one of the 11 patients studied may have been coincidental or, altema- tively, an uncommon manifestation of the DES.
Gross abnormalities of the brain in patients of the DES have been demonstrated frequently by appropriate radiographic techniques, but are not a constant finding. Pneumoencephalography (PEG) showed dilatation of the fourth ventricle in’two affected sisters studied by Goulon et a1 [1968]. Bergstrom and Sanner [1974] studied 21 DES pa- tients by PEG. The findings ranged from no abnormality (3/21) to marked reduction in size of all cerebellar parts. The most consistent finding was small size of the lingula and central lobule of the vermis, demonstrated by saggital tomography. One or both lateral ventricles were enlarged in eight patients and the third ventricle was enlarged in four. Four patients with cataracts could not be differentiated from the remainder of the DES patients on the basis of their PEG findings. In the present study, two affected sisters had CAT scans that showed abnormally small size of the cerebellar hemispheres and vermis. One also had dilatation of the third and lateral ventricles.
Several neuropathological studies have been performed on individuals with the DES. Case 1 of Norman [1940], a 20-year-old man, had cataracts attributed to long-stand-
ing iritis. The brain was small (853 g) with especially marked reduction in size of the cere- bellum. The most marked histopathological changes were in the lateral lobes and superior part of the vermis, which showed almost complete absence of the granular layer. Granule neurons were present on the surface of the cortex of the inferior vermis and the left floc- culus, which were less severely involved than other areas. Purkinje cells were relatively normal, except when heterotopic in the molecular layer. These latter ones had distorted cell bodies, cactus-like expansions of the dendrites, and numerous large axonal “torpedoes.” The basket fibres were preserved. The Bergmann fibrils in the molecular layer were “thick- ened and proliferated.” Massive fibrillary gliosis was present in all layers of the cerebellar cortex, the medullary lamellae of the folia, the central white matter, and around the den- tate nuclei (which contained a normal number of nerve cells). The nerve cells of the inferior olives in the medulla were shrunken, partially filled with lipochrome deposits, and surrounded by dense fibrillary ghosis.
Other examples of what is probably the DES, with autosomal-recessive inheritance,
The triad of ataxia, mental retardation, and congenital cataracts occurs in the Marin-

Dysequilibrium Syndrome in Hutterites 5 1
Case 2 of Norman [ 19401, a 44-year-old woman (unrelated to Case 1). had similar neuropathological findings to those of Case 1, except for greater variabillry in tne degree of reduction of the granular layer of the cerebellar cortex. The nodule was one of the more normal areas examined whereas the flocculi were grossly abnormal, some folia showirg a reduction in the number of Purkinje cells and absence of granule cells. The nerve cells in the olives appeared normal.
ganule neurons, with secondary changes of the F’urkinje and glia cells. He interpreted the persistence of granule cells on the surface in some areas as evidence of retarded development.
Jervis [1950] reported three affected sibs and was impressed by the similarities to Norman’s cases. In the one who was autopsied, a 20-year-old girl, there was severe degenera- tion of granule cells and also moderate to severe degeneration of F’urkinje cells. The hemi- spheres were more severely involved than the vermis and flocculi. Proliferation of Berg- mann’s &a was noted. The olives were also affected.
who noted a selective reduction in granule cells, consistent with Norman’s observations.
[1965] reported a 4-year-old boy with the MSS who had generalized cerebellar atrophy, especially marked in the vermis. There was thinning of the molecular layer with fine fibril- lary gliosis, marked deficiency of Purkinje cells, and diffuse thinning of the granular layer. Golgi cells seemed more numerous than in a control case, possibly due to the cortical atrophy. The nodulus, flocculus, and paraflocculus were relatively spared, and the dentate nuclei and inferior olives were not affected. Mahloudji et a1 [1972] also noted a generalized (rather than a selective) loss of neurons in the cerebellar cortex in a 13-year-old girl with MSS, and observed a normal layer of Bergmann glial cells. In contrast with the former case, they also noted marked loss of neurons in the inferior olives and the pontine nuclei.
The selective reduction of granule neurons described in the DES is reminiscent of the defect in the weaver mutant in the mouse [Sidman, 19681. The homozygous wv/wv mouse has a nonprogressive disturbance of equilibrium, fine tremor, and hypotonia. The cerebellum is about one fourth the normal size and is almost completely lacking in granule cells. Purkinje cells are present in relatively normal numbers. Raluc and Sidman [1973] have proposed that the migration of granule cells from the surface of the cerebellar cortex to their definitive location deep to the F’urkinje cells may depend on the presence of normal Bergmann glial fibres. Their studies of heterozygous (+/wv) weaver mice have shown that these fibres are abnormal. They postulate that the Bergmann glial defect may be closer to the primary action of the mutant gene, and that degeneration of the granule cells may be a consequence of their failure to migrate and establish contacts with the Purkinje cells.
We have adopted the term, dysequilibrium syndrome, as it serves the important func- tion of differentiating t h s disorder from the more familiar type of (nonprogressive) cere- bellar ataxia [Hagberg et al, 19721. The main limitation of the term is that it denotes only one manifestation of a disorder that results in apparent widespread dysfunction of the ner- vous system. Of interest in this regard is the finding of low serum dopamine-beta-hydroxyl- ase activity in eight patients with the DES [Gustavson et al, 19771.
Since the present study was begun, we recognized what is probably the DES in a 14- year-old, mentally retarded, non-Hutterite girl. Her parents are not consanguineous and her two sisters are unaffected. The affected child had been seen by consultants in several university medical centers without a specific diagnosis having been made. This confirms our
Norman [1940] suggested that the primary defect in his cases was a degeneration of
The only other autopsy on a probable case of the DES was by Goulon et a1 [ 19681,
It is of interest t o compare the pathological findings in the DES and the MSS. Todorov

52 Schurig, Orman, and Bowen
suspicion that the disorder is unfamiliar t o those who are most likely to encounter it. As the basis for most, if not all, cases of the DES is autosomal-recessive inheritance,
it is important to differentiate the disorder from other types of cerebral palsy for the pur- pose of genetic counselling.
ACKNOWLEDGMENTS
Dr. Brian Biederman and David Williams gave valuable assistance during part of the study. The CAT scan shown in Figure 3 was done at the Cross Cancer Institute, Edmonton, Alberta, courtesy of Dr. W. R. Castor, Director, Department of Radiology.
Dr. Verena Schurig is recipient of a grant from the Deutsche Forschungsgemeinschaft.
REFERENCES
Alajouanine T, Aubrey M, Nehlil J (1943): Sur une affection familiale caracteriste par un syndrome de dbskquilibration avec importantes perturbations vestibulaires centrales. Rev Neurol 75 :252 -254.
Alter M, Talbert 0, Croffead G (1962): Cerebellar ataxia, congenital cataracts, and retarded somatic and mental maturation. Neurology 12:836-847.
Batten F (1905): Ataxia in childhood. Brain:484-505. Bergstrom K, Sanner G (1974): Pneumoencephalography in non-progressive ataxic syndromes. Acta
Beyermann (1 917): Ueber angeborene Kleinhirnsttrrungen. Arch Psychiat Nervenkr 57:610-658. Chaco J (1969): Marinesco-Sjtigren syndrome with myopathy. Conf Neurol31:349-351. Faton JW, Mayer AJ (1953): The social biology of very high fertility among the Hutterites. The
Emery AE (1976): Methodology in Medical Genetics. Edinburgh: Churchill Livingstone, pp 36-49. Foerster 0 (1 909): Der atonisch-astatische Typus der infantilen Cerebralliihmung. Deutsch Arch Klin
Goulon M, Escourolle R, Barois A, Grosbuis S (1968): Atrophie conghitale de la couche des grains, type
Gustavson K, Ross S , Sanner G (1977): Low serum dopamine-beta-hydroxylase activity in the dysequi-
Hagberg B, Sanner G, Steen M (1972): The dysequilibrium syndrome in cerebral palsy. Acta Paediatr
Jervis G (1950): Early familial cerebellar degeneration. J New Ment Dis 111:398-407. Gamer S , Vojta V (1969): Zur Problematik und Diagnostik der Kleinhirn Symptomatologie im frUhen
Mahloudji M, Amirhakimi G, Haghighi P, Khodadoust A (1972): Marinesco-Sjcigren syndrome. Brain 95:
Marinesco G, Draganesco S, Vasiliu D (1931): Nouvelle maladie familiale caracterisbe par une cataracte
McKusick VA (1969): Human Genetics. 2nd Ed., Englewood Cliffs, NJ: Prentice-Hall, Inc. Norman R (1940): Primary degeneration of the granular layer of the cerebellum: An unusual form of
Rakic P, Sidman R (1973): Weaver mutant mouse cerebellum: Defective neuronal migration secondary
Sanner G (1973): The dysequilibrium syndrome. A genetic study. Neuropaediatrie 4:403-413. Sanner G (1979): Pathogenetic and preventive aspects of non-progressive ataxic syndromes. Dev Med
Sidman R (1968): Development of intemeuronal connections in brains of mutant mice. In Carlson FD
Paediatr Scand 63:732-742.
demography of a unique population. Human Biol25:206-262.
Med 98:216-244.
Norman. Observation de deux soeurs: Examen neuro-pathologique d’un cas. Rev Neurol 118:87-88.
librium syndrome. Clin Genet 11 :270-272.
Scand 61 (Suppl) 226:l-63.
Kindesalter. Z Kinderheilk 105:80-87.
675-680.
conghitale et un arr&t du developpement somato-neuro-psychique. Encbphale 26:97-109.
familial cerebellar atrophy occurring in early life. Brain 63:365 -379.
to abnormality of Bergmann glia. Proc Natl Acad Sci USA 70:240-244.
Child Neurol21:663-671.
(ed): “Physiological and Biochemical Aspects of Nervous Integration. Englewood Cliffs, NJ: Pren- tice-Hall, Inc., pp 163-193.
Sjtigren T (1947): Hereditary congenital spinocerebellar ataxia with congenital cataract and oligophrenia. Acta Psychiatr Neurol S a n d 46(Suppl) 286-289.

Dysequilibrium Syndrome in Hutterites 53
SjUgren T (1950): Hereditary congenital spinocerebellar ataxia accompanied by congenital cataract and
Todorov A (1965): Le syndrome de Marinesco-Sjogren. Premitre Btude anatomo-clinique. J Genet Hum
Van Rossum A (1959): Foerster’s atonic-astatic syndrome. In Biemonds A (ed): “Recent Neurological
oligophrenia. Conf Neurol 10:293-308.
14: 197 -234.
Research.” Amsterdam: Elsevier Publishing Company, pp 157-167.
Edited by John M. Opitz





























