Embed Size (px)
Citation preview

doi: 10.1111/ahg.12070
Novel ABCC8 (SUR1) Gene Mutations in Asian IndianChildren with Congenital Hyperinsulinemic Hypoglycemia
Suresh Jahnavi1, Varadarajan Poovazhagi2, Sekar Kanthimathi1, Kandasamy Balamurugan1,Dhanasekaran Bodhini1, Jaivinder Yadav3, Vandana Jain3, Rajesh Khadgawat3, Mahuya Sikdar1,Ayurchelvan Bhavatharini4, Ashok Kumar Das5, Tanvir Kaur6, Viswanathan Mohan1 andVenkatesan Radha1∗1Madras Diabetes Research Foundation, ICMR Advanced Centre for Genomics of Type 2 Diabetes and Dr. Mohan’s Diabetes SpecialitiesCentre, WHO Collaborating Centre for Non-Communicable Diseases Prevention & Control, IDF Centre of Education, Gopalapuram,Chennai, India2Institute of Child Health and Hospital for Children, Chennai, India3All India Institute of Medical Sciences, New Delhi, India4SRC Diabetes Care Centre, Erode, India5Jawaharlal Institute of Postgraduate Medical Education and Research, Puducherry, India6Indian Council of Medical Research, New Delhi, India
Summary
Congenital hyperinsulinemic hypoglycemia (HI) is a heterogeneous genetic disorder of insulin secretion characterized bypersistent hypoglycemia, most commonly associated with inactivating mutations of the β-cell ATP-sensitive K+ channel(KATP channel) genes ABCC8 (encoding SUR1) and KCNJ11(encoding Kir6.2). This study aimed to screen the mu-tations in the genes associated with congenital HI in Asian Indian children. Recessive mutations of these genes causehyperinsulinism that is unresponsive to treatment with channel agonists like diazoxide. Dominant KATP mutations havebeen associated with diazoxide-responsive disease. The KCNJ11, ABCC8, GCK, HNF4A, and GLUD1 genes were ana-lyzed by sequence analysis in 22 children with congenital HI. We found 10 novel mutations (c.1delA, c.61delG, c.267delT,c.619–629delCCCGAGGACCT, Gln444∗, Leu724Pro, Ala847Thr, Trp898∗, IVS30–2A>C, and Leu1454Arg) and twoknown mutations (Gly111Arg and Arg598∗) in the ABCC8 gene. This study describes novel and known ABCC8 genemutations in children with congenital HI. This is the first large genetic screening study on HI in India and our resultswill help clinicians in providing optimal treatment for patients with hyperinsulinemia and in assisting affected familieswith genetic counseling.
Keywords: KCNJ11 gene, ABCC8 gene, GLUD1 gene, hyperinsulinemia
Introduction
Congenital hyperinsulinemic hypoglycemia (HI) is a geneticdisorder of pancreatic β-cell characterized by persistent hy-poglycemia due to unregulated secretion of insulin. Inap-
∗Corresponding author: Venkatesan Radha, Madras Diabetes Re-search Foundation, ICMR Advanced Centre for Genomics of Type2 Diabetes, 4, Conran Smith Road, Gopalapuram, Chennai 600086, India. Tel: (91) 444 740 5900, (91) 444 396 8888; Fax: (91)442 835 0935; E-mail: [email protected]
propriate management may result in seizures, brain damageand death, therefore, the disorder requires immediate detec-tion and treatment to maintain adequate blood glucose lev-els (Hussain, 2008) . The incidence of HI varies from 1 in35,000–40,000 in the general population (Bruining, 1990)to a considerably higher frequency of 1 in 2500 in com-munities where consanguinity is more prevalent (Mathewet al., 1988). Germ line mutations in six genes have beenassociated with HI, the most common cause being muta-tions in the ABCC8 gene, coding for the sulfonylurea recep-tor1 (SUR1) subunit and an inward rectifying potassium ion
Annals of Human Genetics (2014) 78,311–319 311C© 2014 John Wiley & Sons Ltd/University College London

S. Jahnavi et al.
channel Kir6.2, encoded by the KCNJ11 gene. Other lessfrequent causes of HI are mutations in the genes coding forthe metabolic enzymes glucokinase (GCK; Christesen et al.,2008), glutamate dehydrogenase 1 (GLUD1; Stanley, 2004),and short-chain hydroxyacyl-CoA dehydrogenase (HADH;Molven et al., 2004), and mutations in the gene encoding thetranscription factor hepatocyte nuclear factor 4-α (HNF4A;Pearson et al., 2007). The β-cell KATP channel complex is het-erooctameric, composed of four inward-rectifying potassiumchannel pore forming (Kir6.2) subunits and four high affinitySUR1 subunits (Inagaki et al., 1995), which function only ifthey are assembled and correctly transported to the surface ofcell membrane. Mutations in KCNJ11 or ABCC8 result in re-duction or loss of KATP channel function which leads to con-stant depolarization of the cell membrane and persistent in-sulin secretion even at very low plasma glucose concentrations(Aguilar-Bryan & Bryan, 1999; Huopio et al., 2002; Dunneet al., 2004; Ashcroft, 2005). Recessive mutations of thesegenes cause hyperinsulinism that is unresponsive to treatmentwith channel agonists like diazoxide, while dominant KATP
mutations have been associated with diazoxide-responsivedisease.
HI is heterogeneous with respect to its clinical presenta-tion, histology, genetics, and response to treatment (De Leon& Stanley, 2007; Hussain, 2008; Palladino et al., 2008). Basedon histopathology, the disease may be a focal form, whichconsists of abnormal pancreatic β-cells restricted to a limitedpancreatic area or the diffuse form, consisting of abnormalpancreatic β-cells all over the pancreas. The focal form canbe cured by targeted removal of a pancreatic area whereasthe diffuse form can only be managed by near total pancre-atectomy, if the patients are unresponsive to intensive medi-cal treatment. Pharmacological correction of defective chan-nels has the highest potential in clinical application (Partridgeet al., 2001; Yan et al., 2004). Generally, recessive KATP muta-tions are unresponsive to the KATP channel agonist diazoxide,whereas the dominant KATP mutations are responsive to thisdrug.
This study included 22 children of Asian Indian originwith congenital HI and is the first, to our knowledge, on thegenetics of HI from India.
Materials and Methods
The study group comprised 22 children who were identi-fied as HI by pediatricians mostly from the local GovernmentInstitutes of Child Health and had been referred for genetictesting to Dr. Mohan’s Diabetes Specialties Centre, a largetertiary diabetes centre in South India. The diagnosis of HIwas based on the following criteria, that is, requirement of
glucose infusion of >8 mg/kg/min and laboratory blood glu-cose < 54 mg/dl with a detectable serum C-peptide or insulinlevels, in the absence of ketosis of ketonuria, with or withoutelevated ammonia and/or a positive response to diazoxide oroctreotide. Diazoxide responsiveness was defined as the abil-ity to maintain normoglycemia (blood glucose > 70 mg/dlor 3.88 mmol/l) after 12–18 hours of fasting with a doseof diazoxide 15 mg/kg/day (Snider et al., 2013) and with-out the need for any parenteral support of glucose. For thepurpose of this study, any child who had significant reduc-tion in the hypoglycemic episodes but was not absolutely freefrom hypoglycemia while on a dose 15 mg/kg/day of dia-zoxide was termed as “partial response”. “No response” wasdefined as follows: despite a diazoxide dose of 15–20 mg/kg,the child continued to have hypoglycemia needing glucose orother drugs. Children at the time of recruitment were eval-uated for gross motor, fine motor, and other developmentalmile stones and those children who had delayed milestones(motor and mental) and microcephaly were considered tohave developmental delay. Clinical information (birth weight,age at presentation, and treatment details of HI) was col-lected from the case records. Blood samples from the parentswere collected wherever possible to check the co-segregationof the mutations (identified in this study) in the respec-tive families. For mutation screening in the control popu-lation, we studied 100 subjects selected randomly from theChennai Urban Rural Epidemiological Study (CURES;Deepa et al., 2003; Rema et al., 2004) who were normalglucose tolerant (fasting value < 100 mg/dl and 2-hourvalue <140 mg/dl) and 100 type 1 diabetic children fromDr. Mohan’s Diabetes Specialties Centre who were GAD an-tibody positive (Euroimmun, Lubeck, Germany) and had on-set of diabetes above 1 year of age. DNA from the parents andthe control population was also sequenced wherever possi-ble. For all children, informed consent was obtained from theparents.
DNA was extracted from whole blood by the phenol-chloroform method. Direct sequencing was carried out onan ABI 3500 Genetic Analyzer (Applied Biosystems, FosterCity, CA, USA) using the Big Dye terminator V3.1 (Ap-plied Biosystems) chemistry and the sequences were com-pared with the public databases. Published primer sequences(Boutin et al., 2001; Ellard et al., 2007; Kapoor et al., 2009;Stoy et al., 2010) were used to amplify the DNA for theKCNJ11, ABCC8, GCK, HNF4A, and GLUD1 (exons 6,7, 10, 11, and 12) genes. Mutation screening of the KCNJ11,ABCC8, GCK, and HNF4A genes was performed on all 22children while GLUD1 screening was performed on the sam-ple from a child with hyperammonemia. In silico predictionsof pathogenicity were carried out using the online bioinfor-matics tools sift, Polyphen-2, and mutation t@ster.
312 Annals of Human Genetics (2014) 78,311–319 C© 2014 John Wiley & Sons Ltd/University College London

ABCC8 Mutations in Indian CHI Children
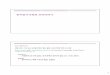
Family 1
ABCC8,c.1delA, M/N
NormalNT
NormalNT
NormalNT
ABCC8C.61 delG, M/NNormal
Family 2ABCC8C.61 del G , N/NNormal
NT Normal
ABCC8C.61 del G, M/N
3 months
Family 3
ABCC8c.267delT, M/Nc.619-629del11bases, M/N
ABCC8c.267delTM/N, Normal
ABCC8c.619-629del11basesM/N, Normal
NTNormal
Family 4
ABCC8Gly111Arg, M/M
ABCC8,Gly111Arg, M/N, Normal
ABCC8Gly111Arg, M/N,Normal
Pedigrees of HI children harbouring mutations in the genes studied
Family 5
ABCC8Gln444*, M/N Normal
ABCC8Gln444*, M/N
ABCC8Gln444*, N/NNormal
Family 6
NT Normal
ABCC8Arg598*, M/N
ABCC8Arg598*, M/NNormal
ABCC8Arg598*, N/N, Normal
Family 9
3 monthsNT ABCC8
IVS30-2A>C, M/M
ABCC8IVS30-2A>C, M/N, Normal
ABCC8IVS30-2A>C, M/N,Normal
Family 10
NTNormal
18TH DayNT
ABCC8Leu1454Arg,, M/N
ABCC8Leu1454Arg, M/NNormal
Family 7
ABCC8Leu724Pro, M/M
3 months
ABCC8Leu724Pro, M/NNormal
ABCC8Leu724Pro, M/NNormal
Family 8
ABCC8,Ala847Thr, M/M Trp898*, M/M
NTNormal
NTNormal
Figure 1 Pedigrees of children with HI harboring ABCC8 mutations. Pedigrees of the familiesshowing genetic and phenotypic status of each member: M/M (homozygous mutant), M/N(heterozygous), N/N (homozygous normal), NT (not tested). Hatched symbols represent carriers.Shaded symbols represent affected individuals.
Annals of Human Genetics (2014) 78,311–319 313C© 2014 John Wiley & Sons Ltd/University College London

S. Jahnavi et al.
Table 1 Summary of mutations identified in children with HI.
Mutation at Mutation at No. of Nature Prediction (SIFT/polyphenS. no. Gene Region protein level cDNA level subjects (n) of mutation Zygosity 2 and mutation t@ster)
1 ABCC8 Exon 1 A1fs∗38 c.1delA 1 Novel Hetero Disease causing2 Exon 1 V21fs∗77 c.61delG 1 Novel Disease causing3 Exon 2 I89fs∗981 c.267del T 1 Novel Disease causing4 Exon 3 Gly111Arg3 c.331G>A 1 Known Homo Disease causing5 Exon 5 P207fs∗2671 c.619–629del11 1 Novel Hetero Disease causing6 Exon 8 Gln444∗ c.1330C>T 1 Novel Disease causing7 Exon 12 Arg598∗4 c.1792C>T 1 Known Disease causing8 Exon 16 Leu724Pro c.2171T>C 1 Novel Homo Disease causing9 Exon 21 Ala847Thr2 c.2539G>A 1 Novel Disease causing10 Exon 22 Trp898∗2 c.2694G>A 1 Novel Disease causing11 IVS 30 IVS30–2A>C c.3754–2A>C 1 Novel Disease causing12 Exon 36 Leu1454Arg c.4361T>G 1 Novel Hetero Disease causing
1These two mutations were found in same patient.2These two mutations are found in same patient.3Fernandez-Marmiesse et al., 2006.4Suchi et al., 2003.
Results
Molecular abnormality was identified in 45.4% of studychildren. Twelve different ABCC8 mutations were iden-tified in 10 probands. Of these, 10 mutations (c.1delA,c.61delG, c.267delT, c.619–629delCCCGAGGACCT,Gln444∗, Leu724Pro, Ala847Thr, Trp898∗, Leu1454Arg,and IVS30–2A>C) were novel and two were previouslyreported (Gly111Arg [Fernandez-Marmiesse et al., 2006];Arg598∗ [Suchi et al., 2003]). Of the 10 novel mutations,four were deletions while six were point mutations. Onechild (patient no. 4) was a compound heterozygote withtwo novel deletion mutations, namely c.267delT andc.619–629delCCCGAGGACCT, and another (patient no.8) was a compound homozygote with two novel pointmutations (Ala847Thr and Trp898∗). Mutations identifiedin the study are summarized in Table 1. These eight novelmutations were not seen in the chromosomes of normalglucose tolerant subjects or in children with type1 diabetes.Family members of the affected children were screenedto check co-segregation of the mutation with the diseasewherever possible (Fig. 1). Clinical characteristics of childrenwho were harboring mutations in the ABCC8 gene aresummarized in Table 2. No mutation was identified in theKCNJ11, GCK, HNF4A, and GLUD1 genes.
Mutations c.61delG, Gln444∗, Arg598∗, Gly111Arg,Leu724Pro, Ala847Thr, and Trp898∗ were identified in chil-dren with onset of hypoglycemia on day one of life and theremaining mutations were identified in children with on-set of hypoglycemia before three days of life. Children whohad compound heterozygous and Leu1454Arg mutations re-
sponded to diazoxide partially with a dose of 12.5 mg/kg/dayand 15 mg/kg/day, respectively. The child with the IVS30–2A>C mutation was treated with a dose of 20 mg/kg/daydiazoxide, and therefore, was not completely diazoxide re-sponsive. Six children did not respond to diazoxide therapyand of these, four children (those with mutations c.61delG,Gly111Arg, Arg598∗, and Leu724Pro) underwent subto-tal pancreatectomy. Children with mutations c.1delA andGln444∗ were treated with a combination therapy of dia-zoxide and octreotide to maintain normoglycemia. Childrenwho were not responding to diazoxide were started on injec-tion octreotide as per protocol and those children who wererefractory to these medications were planned for pancreatec-tomy. Children with Gly111Arg and Leu724Pro mutation hadthe diffuse form of HI as revealed by postsurgical histopatho-logical sections of pancreas. In the case of the child with thec.61delG mutation, 18FDOPA PET/CT scan and postsurgicalhistopathological sections revealed the focal form of HI. Thetype of HI was not known in the case of children who didnot undergo FDOPA scan or pancreatectomy (Tables 2 and 3).Microcephaly was noted in those children with Arg598∗ andLeu724Pro mutations.
Children with homozygous mutations were born to con-sanguineous parents who were heterozygous for the condi-tion. Co-segregation of the mutation with disease could notbe tested in case of patient no.1 as the parents’ samples werenot available. In the case of the child with compound het-erozygous mutations, c.267delT was found in the heterozy-gous state in the proband’s mother, whereas the 11 base pairdeletion mutation was found in the heterozygous state inthe father; the parents were not consanguineous. Healthy
314 Annals of Human Genetics (2014) 78,311–319 C© 2014 John Wiley & Sons Ltd/University College London

ABCC8 Mutations in Indian CHI Children
Tab
le2
Clin
ical
char
acte
rist
ics
ofH
Ipa
tient
sha
rbor
ing
mut
atio
nsin
the
gene
sst
udie
d(n
=9)
.
Clin
ical
para
met
ers
Patie
nt1
Patie
nt2
Patie
nt3
Patie
nt4
Patie
nt5
Patie
nt6
Patie
nt7
Patie
nt8
Patie
nt9
Patie
nt10
Sex
Mal
eFe
mal
eFe
mal
eFe
mal
eM
ale
Fem
ale
Mal
eM
ale
Fem
ale
Fem
ale
Age
aton
set
ofsy
mpt
oms
Day
3D
ay1
Day
3D
ay1
Day
1D
ay1
Day
1D
ay2
Day
2D
ay3
Cur
rent
age
6m
onth
s5
year
s3
mon
ths
16ye
ars
5m
onth
s7
mon
ths
Die
dD
ied
10m
onth
s11
mon
ths
Ges
tatio
nala
ge(in
wee
ks)
4239
3838
3732
3837
3939
Bir
thw
eigh
t(in
kg)
4.0
4.4
3.75
3.3
2.8
2.75
3.5
–3.
22.
8G
row
thce
ntile
90th
75th
90th
50th
25th
25th
50th
–50
th25
thB
lood
gluc
ose
leve
lsat
pres
enta
tion
(mg/
dl)
3530
2540
3032
25–
1830
Peak
insu
linle
vels
22.9
355
19.3
149
2119
50–
30.1
32.7
1D
iazo
xide
resp
onsiv
enes
sN
oN
oPa
rtia
lN
oN
oN
oN
oN
oN
oPa
rtia
l
Panc
reat
ecto
my
Not
done
Don
eN
otdo
neD
one
Not
done
Don
eD
one
Not
done
Not
done
Not
done
Type
ofH
I–
Foca
l–
Diff
use
–Fo
cal
Diff
use
––
–St
atus
ofch
ild–
Impr
oved
Impr
oved
Impr
oved
but
deve
lope
dD
M
On
octr
eotid
eO
noc
treo
tide
Dev
elop
eddi
abet
esm
ellit
usan
ddi
ed
Die
dIm
prov
edIm
prov
ed
Hyp
eram
mon
emia
Non
eN
one
Non
eN
one
Non
eN
one
Non
eN
one
Non
eN
one
Dev
elop
men
tal
dela
yN
oYe
sN
oYe
sN
oYe
sYe
sN
oN
oN
o
Rep
orte
dco
nsan
guin
ityN
oN
oN
oN
oN
oYe
sYe
s–
Yes
Yes
Mut
atio
nid
entifi
edin
AB
CC
8
Mut
atio
nC
.1de
lAc.
61de
lGc.
267d
elT,
c.61
9–62
9del
11ba
ses
Gly
111A
rgG
ln44
4∗A
rg59
8∗Le
u724
Pro
Ala
847T
hrT
rp89
8∗IV
S30–
2A>
CLe
u145
4Arg
Zyg
osity
Het
ero
Het
ero
Com
poun
dhe
tero
Hom
oH
eter
oH
eter
oH
omo
Com
poun
dho
mo
Hom
oH
eter
o
Annals of Human Genetics (2014) 78,311–319 315C© 2014 John Wiley & Sons Ltd/University College London

S. Jahnavi et al.
Tab
le3
Clin
ical
char
acte
rist
ics
ofch
ildre
nw
hodi
dno
tha
rbor
mut
atio
nsin
the
gene
sst
udie
d(n
=12
).
Clin
ical
para
met
ers
Patie
nt11
Patie
nt12
Patie
nt13
Patie
nt14
Patie
nt15
Patie
nt16
Patie
nt17
Patie
nt18
Patie
nt19
Patie
nt20
Patie
nt21
Patie
nt22
Sex
Mal
eM
ale
Fem
ale
Mal
eFe
mal
eFe
mal
eFe
mal
eM
ale
Fem
ale
Mal
eFe
mal
eM
ale
Age
atdi
agno
sisD
ay1
Day
1D
ay1
Day
1D
ay1
Day
1D
ay2
Day
3D
ay3
3m
onth
s3
mon
ths
3.5
mon
ths
Cur
rent
age
1Ye
ar11
mon
ths
3m
onth
s2.
3ye
ars
7m
onth
s6
mon
ths
5m
onth
s1.
7ye
ars
5ye
ars
4yea
rs1.
5ye
ars
2ye
ars
Ges
tatio
nala
ge(in
wee
ks)
3835
3737
3837
3837
3835
3939
Gro
wth
cent
ile2n
d50
th50
th75
th75
th75
th75
th2n
d5t
h10
th5t
h10
thPe
akin
sulin
leve
ls19
.516
2022
2019
36.5
96.
322
6.2
544.
9R
espo
nse
todi
azox
ide
Yes
No
Yes
No
No
Yes
Yes
No
No
Yes
Yes
Yes
Panc
reat
ecto
my
Not
done
Not
done
Not
done
Don
eN
otdo
neD
one
Not
done
Not
done
Not
done
Not
done
Not
done
Not
done
Type
ofH
I–
––
Diff
use
–D
iffus
e–
––
––
–C
urre
ntst
atus
Impr
oved
On
octr
eotid
eIm
prov
edIm
prov
edO
noc
treo
tide
Impr
oved
Impr
oved
On
octr
eotid
eIm
prov
edIm
prov
edIm
prov
edIm
prov
edH
yper
amm
onem
iaN
one
Non
ePr
esen
tN
one
Non
eN
one
Non
eN
one
Non
eN
one
Non
eN
one
Dev
elop
men
tal
dela
yN
one
Non
eN
one
Non
eN
one
Non
eYe
sYe
sYe
sYe
sYe
sYe
s
Rep
orte
dco
nsan
guin
ityN
oN
oN
oN
oN
oN
oN
oN
oN
oYe
sN
oN
o
siblings of the children under study did not undergo genetictesting.
One child (Table 3, patient no. 13) had hyperammonemiawith a serum ammonia concentration of 3.8 μg/ml (normalrange: 0.7–1.35 μg/ml) and was screened for exons 6, 7, 10,11, and 12 of the GLUD1 gene, but no mutation was detectedin these exons. Occurrence of mutations in the other exonsof the GLUD1 and HADH genes cannot be ruled out in thispatient and this is a limitation of this study.
Twelve out of 22 children (54.5%) did not harbor mutationsin any of the genes studied (Table 3). Of these, significantresponse to pharmacological therapy (diazoxide or octreotide)was present in 10 children (83.3%).
Discussion
Here we report on the molecular characterization of 22 AsianIndian children with congenital HI. In this study, mutationanalysis was successful in 45.4% of the recruited children (10out of 22). All the mutations identified in this study are presentin the important domains of the SUR1 subunit of the KATP
channel and also are in highly conserved regions. We identi-fied six heterozygous mutations (five novel and one known)and five homozygous mutations (four novel and one known)in 10 children.
The deletion mutations identified in this study shift thenormal reading frame and introduce a stop signal which couldabruptly terminate the synthesis of protein, resulting in a trun-cated protein product. Of these deletion mutations, the mu-tation c.1delA which is located at the initiation codon ATGis likely to create a disturbance in the consensus sequence andmay fail to start the normal translation process. The otherdeletion mutations identified at residues 61, 267, and 619–629, are located in the transmembrane domain 0 (TMD0)and CL3 domains of SUR1. Previous studies have shown thatthe TMD0 domain of SUR1 has a strong association betweenSUR1 and Kir6.2 which modulates trafficking and gating ofthe KATP channel (Chan et al., 2003).
The mutation Arg598∗ identified in this study was previ-ously found in children with HI (Suchi et al., 2003). Func-tional studies have shown that the mutant (Arg598∗) KATP
channels were defective with no response to diazoxide (Kaneet al., 1996; de Vroede et al., 2004). In the case of residue444, two previously published missense mutations had beendetected in HI patients with diazoxide unresponsive diffuse(Gln444His) and focal (Gln444Arg) forms of HI (Hardy et al.,2007; Damaj et al., 2008). The child with Gln444∗ mutationin this study was also unresponsive to diazoxide and this non-sense mutation (Gln444∗) introduces a stop signal leading toabnormal termination of protein synthesis, which could re-sult in a shorter protein. The novel mutation Leu1454Arg
316 Annals of Human Genetics (2014) 78,311–319 C© 2014 John Wiley & Sons Ltd/University College London

ABCC8 Mutations in Indian CHI Children
detected in patient no.10 is located in the nucleotide bindingdomain (NBD) of SUR1.
In the case of the child with Gln444∗ mutation, the typeof HI might be likely to be focal in accordance with the cal-culations on the prevalence of focal HI in previous studies(Snider et al., 2013). The proband with the paternally inher-ited Arg598∗ mutation, who was classified as having the dif-fuse form of HI, may have actually had the focal form whichcould have been missed. The type of HI was not known inthe case of the child with the heterozygous c.1delA mutation,as the parents’ DNA samples were not available and the childdid not undergo pancreatectomy.
The mutation Leu1454Arg in the ABCC8 gene was foundin the heterozygous state in the DNA of the proband’smother. In this case, a parental history of hypoglycemiahelps in identifying the dominant phenotypes but unfortu-nately the mother (heterozygous for Leu1454Arg) of thischild was not available for biochemical investigations. Thereis also a possibility that the child could be a compoundheterozygote with an unidentified mutation in the otherchromosome which could be responsible for the diseasephenotype.
Recessive missense mutations that impair trafficking pre-vent the assembly of channels on the plasma membrane result-ing in complete absence of channel activity. An infant withhomozygous Leu724Pro mutation developed diabetes withina few weeks after surgery. Further clinical evaluation was notpossible as the child unfortunately developed bronchopneu-monia/sepsis and subsequently disseminated intravascular co-agulation (DIVC) during subsequent hospitalization and died.Previous studies showed that the KATP channel activity wasseen to be decreased (Tornovsky et al., 2004; Yorifuji et al.,2011) in individuals with Gly111Arg mutant SUR1 protein.This was also demonstrated in electrophysiological studies ofcultured islets which revealed defects in the electrical activityof KATP channels (De Vroede et al., 2004). The child with theGly111Arg mutation in this study (patient no.5) underwentnear total pancreatectomy at the age of 2 months and pro-gressively developed diabetes mellitus by the age of 13 years.The risk of recurrence is 25% for subsequent pregnancies asthe disease is recessively inherited (Arnoux et al., 2011) inchildren born in nonconsanguineous marriages. Patient no. 8was a compound homozygote for mutations Ala847Thr andTrp898∗. This child had severe hypoglycemic episodes andwas not responsive to diazoxide or octreotide. Unfortunately,the child with compound homozygous mutations died ofmultiorgan failure and further evaluation was not possible. Inspite of the prevalence of consanguinity in the Indian popula-tion, most of the mutations identified in the study are novel,which is similar to the scenario in some outbred populationsin the world (Nestorowicz et al., 1996; Kapoor et al., 2013).Of note, a founder effect of the Gly111Arg mutation was
reported recently in patients of Indian ethnic origin (Kapooret al., 2013).
Patient no. 13 who had a serum ammonia level of3.8 μg/ml (normal range: 0.7–1.35 μg/ml) was not har-boring mutations in exons 6, 7, 10, 11, and 12 of GLUD1.However, the other less frequently mutated exons were notscreened, which is a limitation of this study. Five children whowere unresponsive to diazoxide did not harbor mutations inany of the genes studied and, therefore, will require furthergenetic analyses to test for intragenic mutations that are missedby direct sequencing (Snider et al., 2013).
In conclusion, molecular abnormality was identified in45.4% of the study population and all the mutations werepresent only in the KCNJ11 and ABCC8 genes. Mutationswere identified in 88.9% of diazoxide unresponsive cases andin 11.1% of children who were treated with diazoxide. Mostof the children (58.3%) who were not harboring mutations inany of the genes studied responded well to pharmacologicaltherapy, whereas true diazoxide responsiveness was not seenin children with mutations. Genetic testing assists in under-standing the nature of the molecular abnormality and in mostcases the timely prediction of the type of hyperinsulinemia islikely to aid in avoiding hypoglycemia related brain damage.
Acknowledgements
This study was supported by the Indian Council for MedicalResearch (ICMR) through the project “Genetic Analysis ofMaturity Onset diabetes of young (MODY) and Neonataldiabetes in India” awarded to RV.
ReferencesAguilar-Bryan, L. & Bryan, J. (1999) Molecular biology of adenosine
triphosphate-sensitive potassium channels. Endocr Rev 20, 101–135.
Arnoux, J. B., Verkarre, V., Saint-Martin, C., Montravers, F.,Brassier, A., Valayannopoulos, V., Brunelle, F., Fournet, J. C.,Robert, J. J., Aigrain, Y., Bellanne-Chantelot, C. & de Lonlay,P. (2011) Congenital hyperinsulinism: Current trends in diagnosisand therapy. Orphanet J Rare Dis 6, 63–76.
Ashcroft, F. M. (2005) ATP-sensitive potassium channelopathies:Focus on insulin secretion. J Clin Invest 115, 2047–2058.
Boutin, P., Vasseur, F., Samson, C., Wahl, C. & Froguel, P. (2001)Routine mutation screening of HNF-1α and GCK genes inMODY diagnosis: How effective are the techniques of DHPLCand direct sequencing used in combination? Diabetologia 44, 775–778.
Bruining, G. J. (1990) Recent advances in hyperinsulinism and thepathogenesis of diabetes mellitus. Curr Opin Pediatr 2, 758–65.
Chan, K. W., Zhang, H. & Logothetis, D. E. (2003) N-terminaltransmembrane domain of the SUR controls trafficking and gatingof Kir6 channel subunits. EMBO J 22, 3833–3843.
Christesen, H. B., Tribble, N. D., Molven, A., Siddiqui, J., Sandal, T.,Brusgaard, K., Ellard, S., Njølstad, P. R., Alm, J., Brock Jacobsen,
Annals of Human Genetics (2014) 78,311–319 317C© 2014 John Wiley & Sons Ltd/University College London

S. Jahnavi et al.
B., Hussain, K. & Gloyn, A. L. (2008) Activating glucokinase(GCK) mutations as a cause of medically responsive congenitalhyperinsulinism: Prevalence in children and characterisation of anovel GCK mutation. Eur J Endocrinol 159, 27–34.
Damaj, L., le Lorch, M., Verkarre, V., Werl, C., Hubert, L.,Nihoul-Fekete, C., Aigrain, Y., de Keyzer, Y., Romana, S.P., Bellanne-Chantelot, C., de Lonlay, P. & Jaubert, F. (2008)Chromosome 11p15 paternal isodisomy in focal forms ofneonatal hyperinsulinism. J Clin Endocrinol Metab 93, 4941–4947.
De Leon, D. D., Stanley, C. A. (2007) Mechanisms of disease: Ad-vances in diagnosis and treatment of hyperinsulinism in neonates.Nat Clin Pract Endocrinol Metab 3, 57–68.
de Vroede, M., Bax, N. M., Brusgaard, K., Dunne, M. J. & Groenen-daal, F. (2004) Laparoscopic diagnosis and cure of hyperinsulinismin two cases of focal adenomatous hyperplasia in infancy. Pediatrics114, e520–e522.
Deepa, M., Pradeepa, R., Rema, M., Mohan, A., Deepa, R., Shan-thirani, S. & Mohan, V. (2003) The Chennai Urban Rural Epi-demiology Study (CURES)-study design and methodology (Ur-ban Component) (CURES 1). J Assoc Physicians India 51, 863–870.
Dunne, M. J., Cosgrove, K. E., Shepherd, R. M., Aynsley-Green, A. & Lindley, K. J. (2004) Hyperinsulinism in infancy:From basic science to clinical disease. Physiol Rev 84, 239–275.
Ellard S., Flanagan, S. E., Girard, C. A., Patch, A. M., Harries, L.,Parrish, A., Edghill, E., Mackay, D. J. G., Proks, P., Shimomura,K., Haberland, H., Carson, D. J., Shield, J. P. H., Hattersley, A.T. & Ashcroft, F. M. (2007) Permanent neonatal diabetes causedby dominant, recessive, or compound heterozygous SUR1 muta-tions with opposite functional effects. Am J Hum Genet 81, 375–382.
Fernandez-Marmiesse, A., Salas, A., Vega, A., Fernandez-Lorenzo,J. R., Barreiro, J. & Carracedo, A. (2006) Mutation spectra ofABCC8 gene in Spanish patients with hyperinsulinism of infancy(HI). Hum Mutat 27, 214–227.
Hardy, O. T., Hernandez-Pampaloni, M., Saffer, J. R., Suchi, M.,Ruchelli, E., Zhuang, H., Ganguly, A., Freifelder, R., Adzick,N. S., Alavi, A. & Stanley, C. A. (2007) Diagnosis and localizationof focal congenital hyperinsulinism by 18F-fluorodopa PET scan.J Pediatr 150, 140–145.
Huopio, H., Shyng, S. L., Otonkoski, T. & Nichols, C. G. (2002)K(ATP) channels and insulin secretion disorders. Am J PhysiolEndocrinol Metab 283(2), E207–E216.
Hussain K. (2008) Diagnosis and management of hyperinsulinaemichypoglycaemia of infancy. Horm Res 69, 2–13.
Inagaki, N., Gonoi, T., Clement, J. P. 4th, Namba, N., Inazawa,J., Gonzalez, G., Aguilar-Bryan, L., Seino, S. & Bryan, J. (1995)Reconstitution of IKATP: An inward rectifier subunit plus thesulfonylurea receptor. Science 270, 1166–1170.
Kane, C., Shepherd, R. M., Squires, P. E., Johnson, P. R., James, R.F., Milla, P. J., Aynsley-Green, A., Lindley, K. J. & Dunne, M. J.(1996) Loss of functional KATP channels in pancreatic beta-cellscauses persistent hyperinsulinemic hypoglycemia of infancy. NatMed 2, 1344–1347.
Kapoor, R. R., Flanagan, S. E., Fulton, P., Chakrapani, A., Chade-faux, B., Ben-Omran, T., Banerjee, I., Shield, J. P., Ellard,S. & Hussain, K. (2009) Hyperinsulinism-hyperammonaemiasyndrome: Novel mutations in the GLUD1 gene andgenotype-phenotype correlations. Eur J Endocrinol 161, 731–735.
Kapoor, R. R., Flanagan, S. E., Arya, V. B., Shield, J. P., Ellard, S.& Hussain, K. (2013) Clinical and molecular characterisation of300 patients with congenital hyperinsulinism. Eu J Endocrinol 168,557–564.
Mathew, P. M., Young, J. M., Abu-Osba, Y. K., Mulhern, B. D.,Hammoudi, S., Hamdan, J. A. & Sa’di, A. R. (1988) Persistentneonatal hyperinsulinism. Clin Pediatr (Phila) 27, 148–151.
Molven, A., Matre, G. E., Duran, M., Wanders, R. J., Rishaug,U., Njølstad, P. R., Jellum, E. & Søvik, O. (2004) Familial hy-perinsulinemic hypoglycemia caused by a defect in the SCHADenzyme of mitochondrial fatty acid oxidation. Diabetes 53, 221–227.
Nestorowicz, A., Wilson, B. A., Schoor, K. P., Inoue, H., Glaser,B., Landau, H., Stanley, C. A., Thornton, P. S., Clement, J. P.4th, Bryan, J., Aguilar-Bryan, L. & Permutt, M. A. (1996) Mu-tations in the sulonylurea receptor gene are associated with famil-ial hyperinsulinism in Ashkenazi Jews. Hum Mol Genet 5, 1813–1822.
Palladino A. A., Bennett M. J. & Stanley C. A. (2008) Hyperinsulin-ism in infancy and childhood: When an insulin level is not alwaysenough. Clin Chem 54, 256–263.
Partridge, C. J., Beech, D. J. & Sivaprasadarao, A. (2001) Identifi-cation and pharmacological correction of a membrane traffick-ing defect associated with a mutation in the sulfonylurea recep-tor causing familial hyperinsulinism. J Biol Chem 276, 35947–35952.
Pearson, E. R., Boj, S. F., Steele, A. M., Barrett, T., Stals, K., Shield,J. P., Ellard, S., Ferrer, J. & Hattersley, A. T. (2007) Macrosomiaand hyperinsulinaemic hypoglycaemia in patients with heterozy-gous mutations in the HNF4A gene. PLoS Med 4, e118–e127.
Rema, M., Mohan, V., Deepa, R. & Ravikumar, R.; Chen-nai Urban Rural Epidemiology Study-2. (2004) Association ofcarotid intima-media thickness and arterial stiffness with dia-betic retinopathy: The Chennai Urban Rural Epidemiology Study(CURES-2). Diabetes Care 27, 1962–1967.
Snider, K. E., Becker, S., Boyajian, L., Shyng, S. L., MacMullen,C., Hughes, N., Ganapathy, K., Bhatti, T., Stanley, C. A. &Ganguly, A. (2013) Genotype and phenotype correlations in 417children with congenital hyperinsulinism. J Clin Endocrinol Metab98, E355–E363.
Stanley, C. A. (2004) Hyperinsulinism/hyperammonemia syn-drome: Insights into the regulatory role of glutamate dehydro-genase in ammonia metabolism. Mol Genet Metab 81(Suppl. 1),S45–S51.
Stoy, J., Steiner, D. F., Park, Y., Ye, H., Philipson, L. & Bell, G.(2010) Clinical and molecular genetics of neonatal diabetes dueto mutations in the insulin gene. Rev Endocr Metab Disord 11,205–215.
Suchi, M., MacMullen, C., Thornton, P. S., Ganguly, A., Stanley,C. A. & Ruchelli, E. D. (2003) Histopathology of congenitalhyperinsulinism: Retrospective study with genotype correlations.Pediatr Dev Pathol 6, 322–333.
Tornovsky, S., Crane, A., Cosgrove, K. E., Hussain, K., Lavie, J.,Heyman, M., Nesher, Y., Kuchinski, N., Ben-Shushan, E., Shatz,O., Nahari, E., Potikha, T., Zangen, D., Tenenbaum-Rakover,Y., de Vries, L., Argente, J., Gracia, R., Landau, H., Eliakim,A., Lindley, K., Dunne, M. J., Aguilar-Bryan, L. & Glaser, B.(2004) Hyperinsulinism of infancy: Novel ABCC8 and KCNJ11mutations and evidence for additional locus heterogeneity. J ClinEndocrinol Metab 89, 6224–6234.
318 Annals of Human Genetics (2014) 78,311–319 C© 2014 John Wiley & Sons Ltd/University College London

ABCC8 Mutations in Indian CHI Children
Yan, F., Lin, C. W., Weisiger, E., Cartier, E. A., Taschenberger,G. & Shyng, S. L. (2004) Sulfonylureas correct traffickingdefects of ATP-sensitive potassium channels caused by muta-tions in the sulfonylurea receptor. J Biol Chem 279, 11096–11105.
Yorifuji, T., Kawakita, R., Nagai, S., Sugimine, A., Doi, H., No-mura, A., Masue, M., Nishibori, H., Yoshizawa, A., Okamoto,S., Doi, R., Uemoto, S. & Nagasaka, H. (2011) Molecular and
clinical analysis of Japanese patients with persistent congenital hy-perinsulinism: Predominance of paternally inherited monoallelicmutations in the KATP channel genes. J Clin Endocrinol Metab 96,E141–E145.
Received: 30 January 2014Accepted: 20 April 2014
Annals of Human Genetics (2014) 78,311–319 319C© 2014 John Wiley & Sons Ltd/University College London